
Submitted:
29 April 2025
Posted:
30 April 2025
You are already at the latest version
Abstract
With the rapid development of radical initiation technologies such as photocatalysis and electrocatalysis, radical reactions have become an increasingly attractive approach for constructing target molecules. However, determining how to efficiently synthesize a specific target molecule using radical reactions remains a challenging task, requiring years of education, training, and extensive trial-and-error by synthetic chemists. The advancement of computer-aided synthesis planning (CASP) has opened new possibilities for accelerating the optimization of radical reaction pathways through artificial intelligence. In this study, we collected 21.6K entries of radical reaction data to build the Radical Reaction Database (RadicalDB). After pre-training with ZINC-15 and USPTO database, we fine-tuned the Chemformer model using RadicalDB, resulting in the development of RadicalRetro, a retrosynthetic prediction model specialized in radical reactions. RadicalRetro achieved a top-1 prediction accuracy of 69.3% for radical retrosynthesis, outperforming similar models such as LocalRetro and Mol-Transformer by 23.0% and 25.4%, respectively. Case studies and attention weight analyses confirm that RadicalRetro effectively captures the unique characteristics of radical reactions.
Keywords:
radical reactions
; retrosynthesis
; deep learning
; drug synthesis
1. Introduction
Since E. J. Corey[1] introduced the concept of retrosynthesis, polar disconnections (two-electron reactions) have been widely adopted due to their intuitive and easily understandable nature. Classical cross-coupling reactions, such as the Suzuki[2], Negishi[3], or Heck[4] reactions, as well as polar addition reactions like the Aldol reaction[5], Michael addition[6,7], or Grignard reaction[8], are mainstream methods for constructing pharmaceutical molecules. In contrast, the inherent instability and uncontrollability of radicals have historically hindered the development of radical chemistry (single-electron reactions)[9,10]. Nonetheless, recent advancements, particularly in chemical oxidation[11,12], photocatalysis[13,14], and electrocatalysis[15,16], have brought exciting progress. Compared to two-electron processes, radical reactions exhibit unique advantages, including sequential bond formation (such as cascade cyclization), high stereoselectivity and regioselectivity, and excellent functional group compatibility[17,18,19]. These features make radical chemistry an increasingly valuable tool in modern synthesis, playing a crucial role in the development of drugs for cancer, viral infections, malaria, hyperlipidemia, and depression[17].
This raises the pivotal question: can we design synthetic routes for known drugs or candidate compounds by leveraging the unique properties of radical reactions? Furthermore, how can reactants be designed to utilize radical chemistry effectively for drug development? Addressing these challenges requires not only deep theoretical knowledge but also extensive laboratory experience and iterative experimentation to ensure reliable and practical solutions. With the rapid advancement of pharmaceutical big data technologies, AI models for retrosynthesis prediction[20,21,22] targeting specific reaction types, such as biocatalysis[23,24] and carbohydrate chemistry[25], are continually being developed. However, to the best of our knowledge, there has yet to be a deep learning-based retrosynthesis AI model specifically designed for radical reactions. One likely reason for this is the lack of dedicated databases for radical reactions. Training AI models using general chemical reaction databases (such as USPTO[26], Reaxys[27], ChEMBL[28], SciFinder[29]) may lead to the unique characteristics of radical reactions being overlooked.
This study collected high-impact literature on radical reactions, with a particular focus on coupling and cyclization reactions involving radicals for drug molecule construction. By analyzing reaction mechanisms, we built the first-ever database dedicated to radical reactions, named RadicalDB (Radical reaction database). Following model screening and optimization of training strategies with deep learning, we developed a retrosynthesis model capable of capturing the unique characteristics of radical reactions, which we named RadicalRetro. Test results demonstrated that RadicalRetro effectively recognizes radical reaction features and utilizes radical-involved coupling and cyclization methods to design synthetic routes for drugs. Attention weight analysis showed that RadicalRetro exhibits strong interpretability in single-step retrosynthesis design for drug molecules. By developing this AI model for retrosynthetic analysis of radical reactions, this study advances the application of radical reactions in drug synthesis.
2. Results and Discussion
2.1. Construction and Analysis of the Radical Reaction Database (RadicalDB)
2.1.1. Data Collection Strategy for RadicalDB
Leveraging our research group's expertise in radical reactions[30] and AI-assisted drug synthesis[31], this study manually curated hundreds of high-impact journal articles on radical reactions, gathering a total of 21.6K radical reactions. Special attention was given to the mechanisms of these reactions during the literature review to avoid missing any reactants. The chemical reactions in both data sources are represented using SMILES notation[38], with reactants separated by a "." and reactants and products separated by ">>". Each reaction entry is accompanied by its literature source. RadicalDB is an open database.
2.1.2. Composition and Distribution of RadicalDB Data
Multi-component radical reactions (green) account for 47.3% of the total in RadicalDB, making them the primary reaction type in the database. These reactions are characterized by high atom economy and operational simplicity[39,40], reflecting the data value of RadicalDB through their significant proportion. From the perspective of product molecular weight distribution, Figure 1a shows that both multi-component radical reactions (green) and bi-component reactions (pink) exhibit a normal distribution of product molecular weights. The overall molecular weight distribution of products in the dataset (Figure 1b) ranges from 80 to 965 and also follows a normal distribution, indicating that the data in RadicalDB are homogeneous. This suggests that the database avoids excessive collection of similar radical reactions, maintaining a diverse dataset[41].
To further analyze the dataset, we utilized T-map[42], a tree-based unsupervised learning algorithm, to visualize RadicalDB. Overall, the data does not exhibit excessive clustering, providing additional evidence that RadicalDB has not over-collected similar types of radical reactions. As shown in Figure 1c, RadicalDB contains classic named radical reactions such as Barton-McCombie[43], Birch[44], Giese[45], Keck[46], and Minisci[47] reactions. These named reactions are well-classified in the chemical space, forming distinct clusters, demonstrating that T-map effectively distinguishes the characteristic types of these reactions.
We also analyzed the rapidly developing photocatalytic/electrocatalytic reactions in recent years, labeling them separately from other radical reactions. From Figure 1d, it is evident that photocatalytic/electrocatalytic reactions (red) account for a significant portion of RadicalDB, representing 31.6%. Unlike the typical clustering of named reactions, the red points in the lower-left corner of Figure 1c are evenly distributed in space, blending smoothly with general radical reactions (purple). This indicates that photocatalytic/electrocatalytic radical reactions do not exhibit distinct characteristic differences from other general radical reactions. In other words, there is a high potential for general radical reactions to be optimized through photocatalytic or electrocatalytic synthetic methods.
2.2. Training and Testing of the Radical Reaction Retrosynthesis Prediction Model (RadicalRetro)
To select the deep learning model suitable for the task of radical reaction retrosynthesis, we chose three models that have achieved state-of-the-art (SOTA) performance in single-step retrosynthesis on the USPTO-50K dataset. These models include the graph neural network-based LocalRetro[48], the natural language processing-based Mol-Transformer[49], and Chemformer[50]. Among them, LocalRetro is a template-based deep learning model, while Mol-Transformer and Chemformer are template-free deep learning models.
- (1)
- Training Strategy for Mol-Transformer. Mol-Transformer typically employs transfer learning[25,51] to enhance its performance. In this study, a multi-task strategy[52] was used by combining the USPTO dataset with the target dataset (RadicalDB) for training, allowing the Transformer model to learn both the USPTO dataset (containing 1M reactions) and the chemical reaction features of RadicalDB. The ratio for mixed sampling was set at 9:1 (USPTO: RadicalDB).
- (2)
- Training Strategy for LocalRetro. First, DGL-LifeSci (https://github.com/awslabs/dgl-lifesci) was used to initialize the features of atoms and bonds, with the molecules in RadicalDB represented as graphs where vertices denote atoms and edges denote bonds. The message passing neural network (MPNN)[53] was applied to update the features of each atom, considering its neighboring atoms and bonds. Local reaction templates were then extracted by comparing the atomic mapping differences between products and reactants. This process resulted in the identification of 2,877 radical reaction retrosynthesis templates, including 2,227 bond-changing templates and 1,342 atom-changing templates. LocalRetro applies a global reaction attention mechanism (GRA)[48] to account for non-local effects in chemical reactions, using template classifiers to score the templates. During retrosynthesis analysis, the model predicts a set of local reaction templates for each chemical center, and these predicted templates are ranked by score to derive the final reactants.
- (3)
- Training Strategy for Chemformer. First, the ZINC-15 dataset[54] (containing 100 million molecules) was used for molecular pretraining. The pretraining process involved masking molecular SMILES codes, primarily through a span-masking algorithm, where short sequences within the SMILES were randomly replaced with a single "<MASK>" token to help the model better understand the combination patterns of atoms and bonds. Next, reaction pretraining was conducted using the USPTO dataset[26] (containing 1M reactions), allowing the model to learn chemical reaction patterns and features. Finally, fine-tuning was performed on the RadicalDB to help the model grasp the specifics and patterns of radical reactions. The resulting retrosynthesis Chemformer model, after molecular pretraining, reaction pretraining, and RadicalDB fine-tuning, is named RadicalRetro (Figure 2).
3. Test Results and Analysis
This section analyzes the test results of the radical reaction retrosynthesis model. To minimize testing errors, 5-fold cross-validation was used to evaluate the results. For each cross-validation experiment, RadicalDB was split into a ratio of 8:1:1 for training, validation, and testing, respectively. The model's performance in predicting radical reactions was compared using Top-K accuracy (K=1, 3, 5, 10) as the evaluation metric.
In the task of retrosynthesis prediction for radical reactions, the performance of the three models is shown in Figure 3. As illustrated in Figure 3a, RadicalRetro, trained using the Chemformer model, achieved the best performance in this task, with an average Top-1 accuracy of 69.31%. The average Top-K (K=3, 5, 10) accuracies were 76.11%, 78.24%, and 80.07%, respectively, demonstrating that the training strategy of RadicalRetro is scientifically sound and effective. The second-best performing model was LocalRetro, with an average Top-1 accuracy of 46.35%, and average Top-K (K=3, 5, 10) accuracies of 55.63%, 59.21%, and 61.42%. MolTransformer's performance was slightly lower than that of LocalRetro, achieving a Top-1 accuracy of 43.91%, and average Top-K (K=3, 5, 10) accuracies of 55.89%, 60.36%, and 63.54%.
It is worth noting that in the single-step retrosynthesis task on the USPTO-50K dataset, the prediction performance of the three models, Chemformer, LocalRetro, and MolTransformer, is quite close, with Top-1 accuracies of 54.3%[50], 53.4%[48], and 51.4%[55], respectively. The difference in prediction performance in this task may be attributed to the fact that radical reactions represent a newer type of reaction, particularly driven by the rapid development of photocatalytic/electrocatalytic synthesis technologies over the past decade[40]. The USPTO dataset contains relatively few radical reaction data[41], which likely limited MolTransformer's ability to fully extract the characteristic features of radical reactions during training. This hypothesis is further supported by the proportion of invalid SMILES generated by MolTransformer and RadicalRetro (Figure 3b). Additionally, the relatively small number of reactions in RadicalDB (26,000 reactions) may not have been sufficient for LocalRetro to fully extract relevant reaction templates, contributing to the larger performance gap between LocalRetro and RadicalRetro.
RadicalRetro, on the other hand, benefited from molecular pretraining, which enabled the model to learn atomic composition features, and from reaction pretraining, which helped it capture bond change patterns. Moreover, RadicalDB's reaction data was collected using methods that explored reaction mechanisms, ensuring consistent data quality, thereby enabling RadicalRetro to achieve higher retrosynthesis prediction accuracy for radical reactions compared to USPTO-50K.
(2) Analysis of RadicalRetro’s Retrosynthesis Prediction Results for Different Types of Radical Reactions
To investigate the predictive performance of RadicalRetro across different reaction types, this study analyzed the first set of data from the 5-fold cross-validation experiments, and the results are shown in Figure 4. From the perspective of radical initiation methods (Figure 4a), the retrosynthesis prediction accuracy for photocatalytic/electrocatalytic radical reactions was 71.22%, higher than the 60.96% accuracy for other types of radical reactions.
The reason for this may lie in the composition of the RadicalDB dataset: while the number of photocatalytic/electrocatalytic radical reactions (11,900 reactions) is slightly lower than the number of other radical reactions (14,100 reactions), photocatalytic/electrocatalytic reactions have experienced exponential growth in recent years, with research focused more intensively on these reactions. This likely made it easier for the model to extract reaction patterns. This hypothesis is further supported by the data distribution characteristics of photocatalytic/electrocatalytic reactions in RadicalDB, as shown in Figure 1d.
From the perspective of named radical reactions, RadicalRetro achieved the highest retrosynthesis prediction accuracy in the Faterno-Biichs and Meerwein arylation reactions, with Top-1 accuracies of 92.31% and 84.12%, respectively. The prediction accuracy for Birch, Giese, and Minisci reactions was also close to or even exceeded the average accuracy of RadicalRetro. However, the retrosynthesis prediction accuracy for the Barton-McCombie deoxygenation radical reaction was the lowest, with a Top-1 accuracy of only 41.13%, significantly below the average accuracy of RadicalRetro. A possible reason for this lower performance lies in the nature of the reaction itself: in the Barton-McCombie reaction, a hydroxyl group in an organic compound is replaced by a hydrogen atom[43]. Since drug molecules typically have an abundance of C-H bonds, the model struggles to determine which hydrogen atom in the precursor was originally part of a hydroxyl group. From this perspective, the achieved prediction accuracy for the Barton-McCombie reaction can still be considered relatively ideal.
In summary, RadicalRetro demonstrated robust retrosynthesis prediction capabilities from the perspectives of both different radical initiation methods and named reactions, further confirming the robustness of the Transformer model with molecular and reaction pretraining in the domain of chemical reactions.
Interpretability of RadicalRetro
Good interpretability is crucial for users to trust and manage AI tools. It not only helps users understand the decision-making process of AI but also enhances their confidence in its use. Furthermore, clear interpretability is essential for monitoring and adjusting AI applications to ensure they meet the specific needs of a given field[56].
In the drug synthesis process, the general steps of a radical reaction usually include the following[9]: (1) Radical generation. The reaction begins with the formation of radicals, which can be achieved through various means, such as photocatalysis[14], electrocatalysis[57], mechanochemistry[34], or the use of specific chemical reagents[58]; (2) Radical addition or abstraction. Radicals react by adding to unsaturated bonds or by abstracting an atom (such as a hydrogen atom) from a saturated molecule; (3) Chain propagation: Radical reactions often proceed through a chain mechanism. In the chain propagation step, newly formed radicals continue to react with other molecules, generating more radicals and perpetuating the reaction; (4) Chain termination: When two radicals meet, they may combine to form a stable molecule, terminating the reaction chain. Therefore, retrosynthetic analysis of radical reactions must consider not only the global and local structures of the target molecule (such as functional group substitutions) but also the generation, propagation, and termination of radicals.
In this section, we explore the interpretability of RadicalRetro by selecting a phenylindene scaffold molecule, which has been shown to exhibit various biological activities[59]. Specifically, we analyzed the retrosynthetic prediction of 2-(2-phenyl-1H-ind-1-yl)acetonitrile 1. RadicalRetro predicted the precursors to be 3-(2-phenylethynylphenyl)acrylonitrile 2 and tributyltin 3 (Figure 5a). The rationale behind this retrosynthetic route has been confirmed by the work of the Alabugin group[60], and the reaction process shown in the figure was computationally validated.
The attention weight heatmap (Figure 5c) reveals several insights: On a global scale, the predicted precursor structures (on the vertical axis) focused on the overall structure of 1 (on the horizontal axis). When generating tributyltin 3, which only serves as a radical initiator, the model paid attention to the entire structure of 1. However, when generating precursor 2, the model focused on the detailed structure of 1. Locally, when generating the radical receptor alkene structure "C=C" in 2, the model paid attention to the chemical environment of the α and β carbons of the electron-withdrawing nitrile group (region 1') and the structural information of the phenyl ring (region 2'). When generating the radical receptor alkyne structure "C≡C" in 2, the model focused on the chemical structure surrounding the indene (region 3') and the structural information of the unsaturated bonds within the indene scaffold (region 4'). This attention mechanism, from global to local focus, mirrors the way chemists approach retrosynthetic analysis.
This study also selected a fused-ring scaffold molecule as a case for retrosynthetic analysis to explore the interpretability of RadicalRetro. Azapolycyclic aromatic hydrocarbons (PAHs) 4, due to the electron-donating nitrogen atom, are prone to oxidative decomposition, making their synthesis particularly challenging[61]. When RadicalRetro was used for the retrosynthetic analysis of 6-4 (Figure 5b), it predicted the precursor to be a diyne 5, which undergoes a radical cascade cyclization reaction. The validity of this retrosynthetic route was confirmed by the Xu group's work on electrocatalytic synthesis of PAHs[62]. From the attention heatmap of RadicalRetro's retrosynthetic analysis (Figure 5d), it can be seen that the structure of diyne 5 was generated based on the structure of the PAH. The structural information of the precursor can be correspondingly traced back to 4. Interestingly, the two alkyne groups "C≡C" formed through the "cleavage" characteristic of radical reactions were generated based on the structural information of the two aromatic rings in 4 (regions 5' and 6'), which mirrors the thought process of human chemists. These cases demonstrate that RadicalRetro shows good interpretability when performing retrosynthetic analysis based on the characteristics of radical reactions. Further examples are shown in Figure S1–S3 (ESI†)
4. Application of RadicalRetro in Synthesis
Retrosynthetic Analysis Using Radical Reaction Characteristics
In this section, we explore whether RadicalRetro has effectively learned the principles of radical reactions through specific retrosynthetic analysis cases. In this study, the Chemformer model, trained with molecular and reaction pretraining but without fine-tuning on the RadicalDB dataset, is referred to as GeneralRetro, which serves as the baseline model for RadicalRetro. Several drug intermediates were selected for retrosynthetic analysis, and both GeneralRetro and RadicalRetro were used to analyze them. The results of the retrosynthetic outputs were then compared (Figure 7).
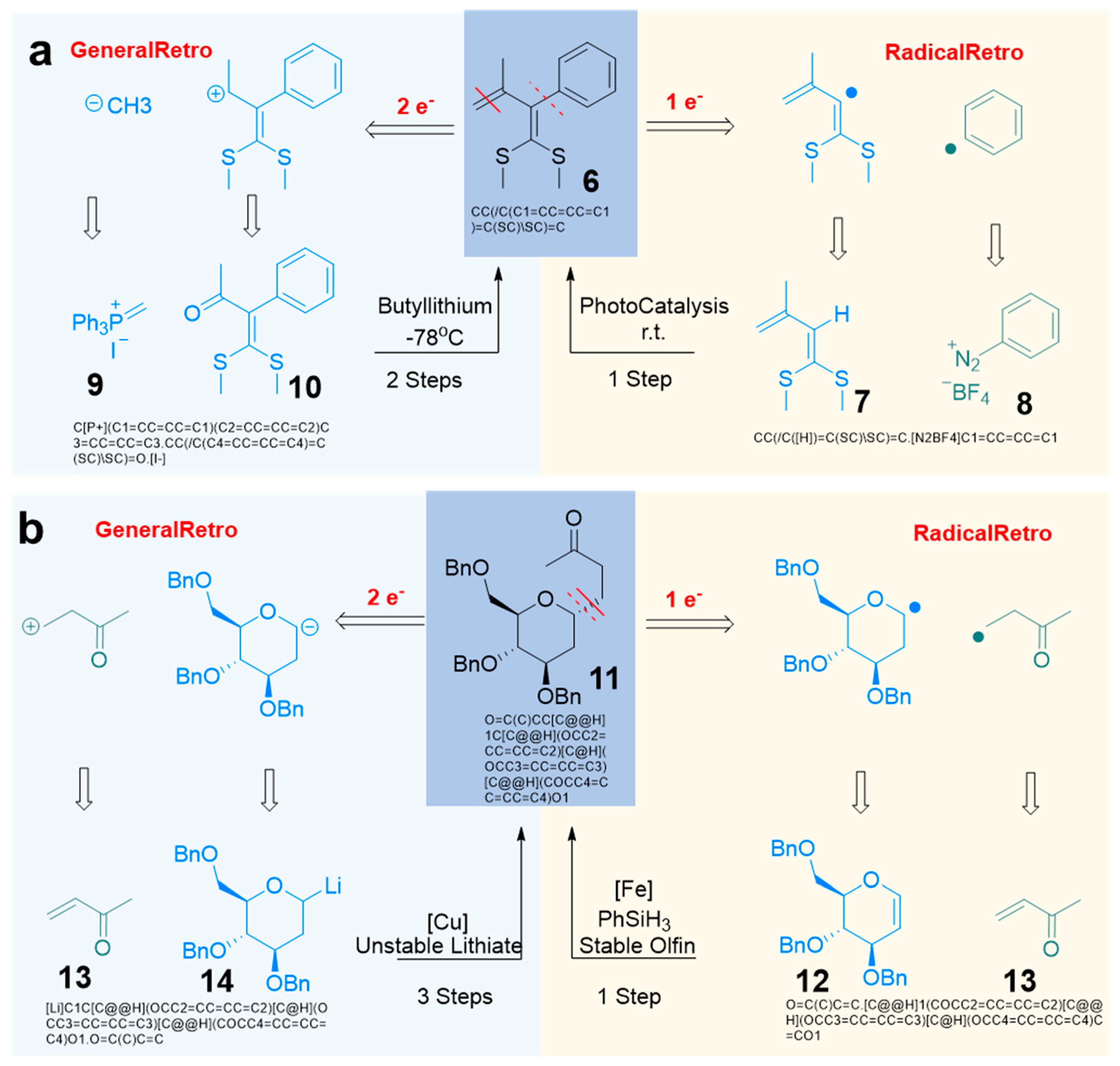
Through case analysis, it was found that RadicalRetro can leverage the characteristics of radical reactions to design synthetic routes involving radical-mediated coupling reactions. For example, 1,1-S,S-functionalized tetrasubstituted alkene 6 is the starting material for the estrogen receptor modulator Tamoxifen[63] (Figure 6a). When retrosynthetically analyzing this molecule using the two AI models trained in this study, two different synthetic strategies were provided: GeneralRetro suggested the starting materials for a Wittig reaction, namely triphenylphosphine 9 and 4,4-bis(methylthio)-3-phenylbut-3-en-2-one 10. This route is one that would commonly come to mind for chemists, and it has been used in recent synthesis efforts by Zhang's group and Monfette's group[63,64]. In contrast, RadicalRetro proposed a strategy involving radical coupling, with the predicted precursors being (3-methylbut-1,3-diene-1,1-diyl)bis(methylthio) 7 and phenyl diazonium salt 8. Upon reviewing the literature, we found that this route's rationale was supported by recent photocatalytic synthesis work by Yu's group[65]. Comparing the two retrosynthetic strategies, The strategy proposed by GeneralRetro involves the use of unstable ylides, requiring low temperatures (-78°C) and strong bases (e.g., tert-butyllithium) across three steps[63]. In contrast, the photocatalytic coupling strategy suggested by RadicalRetro only requires a small amount of photosensitizer and can proceed under mild conditions, such as room temperature and light exposure, making the reaction conditions milder and the process simpler[65].
Another case is the synthesis of glycoside derivative 11 (Figure 6b). Glycoside derivatives have been shown to possess biological activity, including anticancer and anti-Alzheimer's effects[66]. When both AI models were used to analyze the retrosynthesis of glycoside derivative 11, they proposed two different synthetic strategies: GeneralRetro suggested a Michael addition route with the precursors being organolithium compound 14 and the Michael acceptor but-3-en-2-one 13, consistent with the strategy employed by Suginome's group[67]. In contrast, RadicalRetro proposed a hydrogen atom transfer-initiated radical coupling retrosynthesis, with the precursors being the more stable alkene derivative 12 and radical acceptor 13. This approach aligns with the synthesis method reported by Baran's group[68]. Of the two retrosynthesis routes, RadicalRetro's strategy avoids the use of unstable precursors like organolithium compounds and reduces the number of steps by two compared to GeneralRetro, offering a more efficient radical-mediated coupling strategy.
These retrosynthesis cases demonstrate that, compared to GeneralRetro, which was not fine-tuned on RadicalDB, RadicalRetro is better able to utilize the characteristics of radical reactions and propose radical-based synthetic strategies in drug retrosynthesis predictions. This highlights the significant impact that fine-tuning with RadicalDB has had on the performance of the deep learning model.
5. Conclusions
Deep learning models have been applied to retrosynthetic analysis across various types of reactions, but the analysis of single-electron transfer (SET) retrosynthesis has not received sufficient attention or in-depth study. This studyr presents a data-driven model, RadicalRetro, designed to leverage the characteristics of radical reactions for retrosynthetic analysis of drug molecules. After model selection, the natural language processing-based Chemformer was found to be suitable for the task of predicting radical reaction retrosynthesis in drug synthesis. RadicalRetro learned atomic arrangement features through molecular pretraining, organic synthesis reaction features through reaction pretraining, and radical reaction characteristics through fine-tuning. It effectively utilizes features such as radical-mediated coupling and cyclization reactions to design synthetic routes, achieving an average Top-1 accuracy of 69.31% and a Top-10 accuracy of 80.07% in radical reaction retrosynthesis prediction. RadicalRetro demonstrated consistent performance across different types of radical reaction retrosynthesis tasks. Attention weight analysis of the model indicates that the output of products considers both the global structural information and local functional group details of the target molecule, showcasing the model's interpretability. This AI-driven approach helps advance the application of radical reactions in drug synthesis.
6. Method
6.1. Deep Learning Models and Parameters
6.1.1. Chemformer
Chemformer is a deep learning model based on BART (Bidirectional and Auto-Regressive Transformers), commonly used for generative tasks such as text summarization and machine translation. The main parameter settings for Chemformer in this study are as follows:
python -m molbart.fine_tune \
-dataset uspto_50 \
-data_path data/radicals/radicals_fold${fold}.pkl \
-task backward_prediction \
-n_epochs 100 \
-lr 0.001 \
-schedule cycle \
-batch_size 64 \
-acc_batches 4 \
-augmentation_strategy all \
-aug_prob 0.5
6.1.2. Mol-Transformer
Mol-Transformer is a natural language processing model specifically designed for chemical reactions and is based on the Transformer architecture. This study utilized the multi-task learning method provided by OpenNMT. The mixed sampling ratio for training was set at 9:1 (USPTO: RadicalDB). Other parameters were consistent with the research by Probst's group, with the main parameter settings as follows:
onmt_train -data ${DATASET} \
-save_model ${OUTDIR} \
-data_ids uspto transfer --data_weights 9 1 \
-seed 42 -gpu_ranks 0 \
-train_steps 50000 -param_init 0 \
-param_init_glorot -max_generator_batches 32 \
-batch_size 4096 -batch_type tokens \
-normalization tokens -max_grad_norm 0 -accum_count 4 \
-optim adam -adam_beta1 0.9 -adam_beta2 0.998 -decay_method noam \
-warmup_steps 800 -learning_rate 2 -label_smoothing 0.0 \
-layers 2 -rnn_size 384 -word_vec_size 384 \
-encoder_type transformer -decoder_type transformer \
-dropout 0.1 -position_encoding -share_embeddings \
-global_attention general -global_attention_function softmax \
-self_attn_type scaled-dot -heads 4 -transformer_ff 1024 \
-tensorboard --tensorboard_log_dir ${LOGDIR}
6.1.3. LocalRetro
LocalRetro is a template-based deep learning model that achieved 53.4% accuracy on the single-step retrosynthesis task in the USPTO-50K dataset and was once a state-of-the-art (SOTA) model in this field. The main parameter settings are as follows:
python Train.py --gpu cuda:0 --dataset RadicalDB
-config default_config.json
-batch-size 16 \
-num-epochs 50 \
-patience 5 -max-clip 20 -learning-rate 1e-4 \
-weight-decay 1e-6 -schedule_step 10 -num-workers 0 \
-print-every 20
6.2. Training Strategy
This study employed a multi-task transfer learning approach, using a convex weighting scheme for the USPTO and fine-tuning datasets, with weights of 9 and 1, respectively. This approach was based on the method used by Pesciullesi's group in their study on predicting glycosylation reactions.
The transfer learning data comes from the USPTO dataset, originally derived from Lowe's dataset, which was extracted from USPTO patent data. The dataset was preprocessed to remove reagents, solvents, temperatures, and other reaction conditions, and then filtered to eliminate duplicate, incorrect, or incomplete reactions. This dataset contains 1 million reaction records.
6.3. Testing Method
Cross-validation is a widely used machine learning evaluation technique that divides the dataset into mutually exclusive subsets for training and validation, allowing for a comprehensive assessment of a model's performance and generalization ability. This method effectively addresses overfitting and underfitting, providing a thorough understanding of the model's actual performance.
In this study, all models were constructed and evaluated using 5-fold cross-validation, ensuring the robustness of the results. During the 5-fold cross-validation, the study also balanced the different categories of radical reactions by adopting a stratified sampling strategy. Each category, including photocatalytic/electrocatalytic synthesis reactions and named radical reactions, was randomly sampled. This approach ensured that the models were exposed to various types of radical reactions, thereby enhancing their prediction capabilities.
Author Contributions
J. X. designed the research project; J. D., J. P., W.L. and K. D. collected literature and established RadicalDB; J. X. designed and trained the models; J. X. and W. Y. analyzed data and wrote the manuscript. All authors discussed the results and approved the manuscript.
Funding
Data Availability
The RadicalDB and supplementary datasets used in this study are available at https://github.com/MolAstra/RadicalRetro.
Code Availability
The source code of RadicalRetro and associated data preparation python scripts are available at https://github.com/MolAstra/RadicalRetro.
Acknowledgments
This research was supported by the General Research Project of the Zhejiang Provincial Department of Education No. Y202456962 and Zhejiang Provincial Research Project on Chinese Vocational Education No. ZJCV2024B31.
Competing Interests
The authors declare no competing interests.
References
- Corey, E. J. Robert Robinson Lecture. Retrosynthetic Thinking—Essentials and Examples. Chem. Soc. Rev. 1988, 17, 111–133. [Google Scholar] [CrossRef]
- Niwa, T.; Uetake, Y.; Isoda, M.; Takimoto, T.; Nakaoka, M.; Hashizume, D.; Sakurai, H.; Hosoya, T. Lewis Acid-Mediated Suzuki–Miyaura Cross-Coupling Reaction. Nature Catalysis 2021, 4, 1080–1088. [Google Scholar] [CrossRef]
- Zhao, B.; Rogge, T.; Ackermann, L.; Shi, Z. Metal-Catalysed C–Het (F, O, S, N) and C–C Bond Arylation. Chem. Soc. Rev. 2021, 50, 8903–8953. [Google Scholar] [CrossRef]
- Bhoyare, V. W.; Sosa Carrizo, E. D.; Chintawar, C. C.; Gandon, V.; Patil, N. T. Gold-Catalyzed Heck Reaction. J. Am. Chem. Soc. 2023, 145, 8810–8816. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, Y.; Yasukawa, T.; Yoo, W.-J.; Kitanosono, T.; Kobayashi, S. Catalytic Enantioselective Aldol Reactions. Chem. Soc. Rev. 2018, 47, 4388–4480. [Google Scholar] [CrossRef]
- Wang, J.; Young, T. A.; Duarte, F.; Lusby, P. J. Synergistic Noncovalent Catalysis Facilitates Base-Free Michael Addition. J. Am. Chem. Soc. 2020, 142, 17743–17750. [Google Scholar] [CrossRef]
- Földes, T.; Madarász, Á.; Révész, Á.; Dobi, Z.; Varga, S.; Hamza, A.; Nagy, P. R.; Pihko, P. M.; Pápai, I. Stereocontrol in Diphenylprolinol Silyl Ether Catalyzed Michael Additions: Steric Shielding or Curtin–Hammett Scenario? J. Am. Chem. Soc. 2017, 139, 17052–17063. [Google Scholar] [CrossRef]
- Guisán-Ceinos, M.; Martín-Heras, V.; Tortosa, M. Regio- and Stereospecific Copper-Catalyzed Substitution Reaction of Propargylic Ammonium Salts with Aryl Grignard Reagents. J. Am. Chem. Soc. 2017, 139, 8448–8451. [Google Scholar] [CrossRef]
- Smith, J. M.; Harwood, S. J.; Baran, P. S. Radical Retrosynthesis. Acc. Chem. Res. 2018, 51, 1807–1817. [Google Scholar] [CrossRef]
- Petzold, D.; Giedyk, M.; Chatterjee, A.; König, B. A Retrosynthetic Approach for Photocatalysis. Eur. J. Org. Chem. 2020, 2020, 1193–1244. [Google Scholar] [CrossRef]
- Mandal, S.; Bera, T.; Dubey, G.; Saha, J.; Laha, J. K. Uses of K2S2O8 in Metal-Catalyzed and Metal-Free Oxidative Transformations. ACS Catal. 2018, 8, 5085–5144. [Google Scholar] [CrossRef]
- Hinz, A.; Bresien, J.; Breher, F.; Schulz, A. Heteroatom-Based Diradical(Oid)s. Chem. Rev. 2023, 123, 10468–10526. [Google Scholar] [CrossRef] [PubMed]
- Pitre, S. P.; Overman, L. E. Strategic Use of Visible-Light Photoredox Catalysis in Natural Product Synthesis. Chem. Rev. 2022, 122, 1717–1751. [Google Scholar] [CrossRef]
- Wang, H.; Tian, Y.-M.; König, B. Energy- and Atom-Efficient Chemical Synthesis with Endergonic Photocatalysis. Nat. Rev. Chem. 2022, 6, 745–755. [Google Scholar] [CrossRef]
- Novaes, L. F. T.; Liu, J.; Shen, Y.; Lu, L.; Meinhardt, J. M.; Lin, S. Electrocatalysis as an Enabling Technology for Organic Synthesis. Chem. Soc. Rev. 2021, 50, 7941–8002. [Google Scholar] [CrossRef] [PubMed]
- Rein, J.; Zacate, S. B.; Mao, K.; Lin, S. A Tutorial on Asymmetric Electrocatalysis. Chem. Soc. Rev. 2023, 52, 8106–8125. [Google Scholar] [CrossRef]
- Gupta, A.; Laha, J. K. Growing Utilization of Radical Chemistry in the Synthesis of Pharmaceuticals. Chem. Rec. 2023, 23, e202300207. [Google Scholar] [CrossRef]
- Fischer, H. The Persistent Radical Effect: A Principle for Selective Radical Reactions and Living Radical Polymerizations. Chem. Rev. 2001, 101, 3581–3610. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, P.; Li, W. Synthesis of BCP Nitriles Enabled by a Metallaphotoredox-Based Multi-Component Reaction. Chem Catal. 2023, 3, 100618. [Google Scholar] [CrossRef]
- Yu, T.; Boob, A. G.; Volk, M. J.; Liu, X.; Cui, H.; Zhao, H. Machine Learning-Enabled Retrobiosynthesis of Molecules. Nat. Catal. 2023, 6, 137–151. [Google Scholar] [CrossRef]
- Zhong, Z.; Song, J.; Feng, Z.; Liu, T.; Jia, L.; Yao, S.; Hou, T.; Song, M. Recent Advances in Deep Learning for Retrosynthesis. WIREs Comput. Mol. Sci. 2024, 14, e1694. [Google Scholar] [CrossRef]
- Han, Y.; Xu, X.; Hsieh, C.-Y.; Ding, K.; Xu, H.; Xu, R.; Hou, T.; Zhang, Q.; Chen, H. Retrosynthesis Prediction with an Iterative String Editing Model. Nat. Commun. 2024, 15, 6404. [Google Scholar] [CrossRef] [PubMed]
- Probst, D.; Manica, M.; Nana Teukam, Y. G.; Castrogiovanni, A.; Paratore, F.; Laino, T. Biocatalysed Synthesis Planning Using Data-Driven Learning. Nat. Commun. 2022, 13, 964. [Google Scholar] [CrossRef]
- Finnigan, W.; Hepworth, L. J.; Flitsch, S. L.; Turner, N. J. RetroBioCat as a Computer-Aided Synthesis Planning Tool for Biocatalytic Reactions and Cascades. Nat. Catal. 2021, 4, 98–104. [Google Scholar] [CrossRef]
- Pesciullesi, G.; Schwaller, P.; Laino, T.; Reymond, J.-L. Transfer Learning Enables the Molecular Transformer to Predict Regio- and Stereoselective Reactions on Carbohydrates. Nat. Commun. 2020, 11, 4874. [Google Scholar] [CrossRef]
- Beliveau, S.; Ma, J. Recent Developments in AI and USPTO Open Data, 2022.
- Radestock, S. Optimising Chemical Information Workflows: Integrating Reaxys - Use Cases and Applications. J. Cheminform. 2013, 5, P39. [Google Scholar] [CrossRef]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A. P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L. J.; Cibrián-Uhalte, E.; Davies, M.; Dedman, N.; Karlsson, A.; Magariños, M. P.; Overington, J. P.; Papadatos, G.; Smit, I.; Leach, A. R. The ChEMBL Database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Somerville, A. N. SciFinder Scholar (by Chemical Abstracts Service). J. Chem. Educ. 1998, 75, 959. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, Y.; Han, J.; Su, A.; Qiao, H.; Zhang, C.; Tang, J.; Shen, X.; Sun, B.; Yu, W.; Zhai, S.; Wang, X.; Wu, Y.; Su, W.; Duan, H. Providing Direction for Mechanistic Inferences in Radical Cascade Cyclization Using a Transformer Model. Org. Chem. Front. 2022, 9, 2498–2508. [Google Scholar] [CrossRef]
- Xu, J.; Yu, W.; Luo, Y.; Liu, T.; Su, A. Developing Lead Compounds of eEF2K Inhibitors Using Ligand–Receptor Complex Structures. Processes 2024, 12. [Google Scholar] [CrossRef]
- Zhong, Z.; Song, J.; Feng, Z.; Liu, T.; Jia, L.; Yao, S.; Wu, M.; Hou, T.; Song, M. Root-Aligned SMILES: A Tight Representation for Chemical Reaction Prediction. Chem. Sci. 2022, 13, 9023–9034. [Google Scholar] [CrossRef]
- Guo, H.-M.; Wang, J.-J.; Xiong, Y.; Wu, X. Visible-Light-Driven Multicomponent Reactions for the Versatile Synthesis of Thioamides by Radical Thiocarbamoylation. Angew. Chem. Int. Ed. 2024, 63, e202409605. [Google Scholar] [CrossRef]
- Coppola, G. A.; Pillitteri, S.; Van der Eycken, E. V.; You, S.-L.; Sharma, U. K. Multicomponent Reactions and Photo/Electrochemistry Join Forces: Atom Economy Meets Energy Efficiency. Chem. Soc. Rev. 2022, 51, 2313–2382. [Google Scholar] [CrossRef]
- de Almeida, A. F.; Moreira, R.; Rodrigues, T. Synthetic Organic Chemistry Driven by Artificial Intelligence. Nat. Rev. Chem. 2019, 3, 589–604. [Google Scholar] [CrossRef]
- Probst, D.; Reymond, J.-L. Visualization of Very Large High-Dimensional Data Sets as Minimum Spanning Trees. J. Cheminform. 2020, 12, 12. [Google Scholar] [CrossRef]
- Mahy, W.; Plucinski, P.; Jover, J.; Frost, C. G. Ruthenium-Catalyzed O- to S-Alkyl Migration: A Pseudoreversible Barton–McCombie Pathway. Angew. Chem. Int. Ed. 2015, 54, 10944–10948. [Google Scholar] [CrossRef]
- Nemirovich, T.; Kostal, V.; Copko, J.; Schewe, H. C.; Boháčová, S.; Martinek, T.; Slanina, T.; Jungwirth, P. Bridging Electrochemistry and Photoelectron Spectroscopy in the Context of Birch Reduction: Detachment Energies and Redox Potentials of Electron, Dielectron, and Benzene Radical Anion in Liquid Ammonia. J. Am. Chem. Soc. 2022, 144, 22093–22100. [Google Scholar] [CrossRef]
- Gant Kanegusuku, A. L.; Roizen, J. L. Recent Advances in Photoredox-Mediated Radical Conjugate Addition Reactions: An Expanding Toolkit for the Giese Reaction. Angew. Chem. Int. Ed. 2021, 60, 21116–21149. [Google Scholar] [CrossRef]
- Smith, M. W.; Snyder, S. A. A Concise Total Synthesis of (+)-Scholarisine A Empowered by a Unique C–H Arylation. J. Am. Chem. Soc. 2013, 135, 12964–12967. [Google Scholar] [CrossRef]
- Gu, Y.-J.; Luo, M.-P.; Yuan, H.; Liu, G.-K.; Wang, S.-G. Photocatalytic Enantioselective Radical Cascade Multicomponent Minisci Reaction of β-Carbolines Using Diazo Compounds as Radical Precursors. Adv. Sci. 2024, 11, 2402272. [Google Scholar] [CrossRef]
- Chen, S.; Jung, Y. Deep Retrosynthetic Reaction Prediction Using Local Reactivity and Global Attention. JACS Au 2021, 1, 1612–1620. [Google Scholar] [CrossRef] [PubMed]
- Schwaller, P.; Laino, T.; Gaudin, T.; Bolgar, P.; Hunter, C. A.; Bekas, C.; Lee, A. A. Molecular Transformer: A Model for Uncertainty-Calibrated Chemical Reaction Prediction. ACS Cent. Sci. 2019, 5, 1572–1583. [Google Scholar] [CrossRef]
- Irwin, R.; Dimitriadis, S.; He, J.; Bjerrum, E. J. Chemformer: A Pre-Trained Transformer for Computational Chemistry. Mach. Learn.: Sci. Technol. 2022, 3, 015022. [Google Scholar] [CrossRef]
- Zhang, C.; Zhai, Y.; Gong, Z.; Duan, H.; She, Y.-B.; Yang, Y.-F.; Su, A. Transfer Learning across Different Chemical Domains: Virtual Screening of Organic Materials with Deep Learning Models Pretrained on Small Molecule and Chemical Reaction Data. J. Cheminform. 2024, 16, 89. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Lin, L.; Liu, B.; Xiao, Y.; Xiong, X. A Multi-Task Transfer Learning Method with Dictionary Learning. Knowl.-Based Syst. 2020, 191, 105233. [Google Scholar] [CrossRef]
- X. Fan; M. Gong; Z. Tang; Y. Wu. Deep Neural Message Passing With Hierarchical Layer Aggregation and Neighbor Normalization. IEEE Trans. Neural Netw. Learn. Syst. 2022, 33, 7172–7184. [Google Scholar] [CrossRef]
- Tingle, B. I.; Tang, K. G.; Castanon, M.; Gutierrez, J. J.; Khurelbaatar, M.; Dandarchuluun, C.; Moroz, Y. S.; Irwin, J. J. ZINC-22─A Free Multi-Billion-Scale Database of Tangible Compounds for Ligand Discovery. J. Chem. Inf. Model. 2023, 63, 1166–1176. [Google Scholar] [CrossRef]
- Wang, X.; Li, Y.; Qiu, J.; Chen, G.; Liu, H.; Liao, B.; Hsieh, C.-Y.; Yao, X. RetroPrime: A Diverse, Plausible and Transformer-Based Method for Single-Step Retrosynthesis Predictions. Chem. Eng. J. 2021, 420, 129845. [Google Scholar] [CrossRef]
- Kovács, D. P.; McCorkindale, W.; Lee, A. A. Quantitative Interpretation Explains Machine Learning Models for Chemical Reaction Prediction and Uncovers Bias. Nat. Commun. 2021, 12, 1695. [Google Scholar] [CrossRef]
- Novaes, L. F. T.; Liu, J.; Shen, Y.; Lu, L.; Meinhardt, J. M.; Lin, S. Electrocatalysis as an Enabling Technology for Organic Synthesis. Chem. Soc. Rev. 2021, 50, 7941–8002. [Google Scholar] [CrossRef]
- Yang, X.; Wang, H.; Zhang, Y.; Su, W.; Yu, J. Generation of Aryl Radicals from in Situ Activated Homolytic Scission: Driving Radical Reactions by Ball Milling. Green Chem. 2022, 24, 4557–4565. [Google Scholar] [CrossRef]
- Liu, L.; Ward, R. M.; Schomaker, J. M. Mechanistic Aspects and Synthetic Applications of Radical Additions to Allenes. Chem. Rev. 2019, 119, 12422–12490. [Google Scholar] [CrossRef]
- Jabor Gozzi, G.; Bouaziz, Z.; Winter, E.; Daflon-Yunes, N.; Aichele, D.; Nacereddine, A.; Marminon, C.; Valdameri, G.; Zeinyeh, W.; Bollacke, A.; Guillon, J.; Lacoudre, A.; Pinaud, N.; Cadena, S. M.; Jose, J.; Le Borgne, M.; Di Pietro, A. Converting Potent Indeno[1,2-b]Indole Inhibitors of Protein Kinase CK2 into Selective Inhibitors of the Breast Cancer Resistance Protein ABCG2. J. Med. Chem. 2015, 58, 265–277. [Google Scholar] [CrossRef]
- Mondal, S.; Mohamed, R. K.; Manoharan, M.; Phan, H.; Alabugin, I. V. Drawing from a Pool of Radicals for the Design of Selective Enyne Cyclizations. Org. Lett. 2013, 15, 5650–5653. [Google Scholar] [CrossRef]
- Takase, M.; Narita, T.; Fujita, W.; Asano, M. S.; Nishinaga, T.; Benten, H.; Yoza, K.; Müllen, K. Pyrrole-Fused Azacoronene Family: The Influence of Replacement with Dialkoxybenzenes on the Optical and Electronic Properties in Neutral and Oxidized States. J. Am. Chem. Soc. 2013, 135, 8031–8040. [Google Scholar] [CrossRef]
- Hou, Z.-W.; Mao, Z.-Y.; Song, J.; Xu, H.-C. Electrochemical Synthesis of Polycyclic N-Heteroaromatics through Cascade Radical Cyclization of Diynes. ACS Catal. 2017, 7, 5810–5813. [Google Scholar] [CrossRef]
- Monfette, S.; Turner, Z. R.; Semproni, S. P.; Chirik, P. J. Enantiopure C1-Symmetric Bis(Imino)Pyridine Cobalt Complexes for Asymmetric Alkene Hydrogenation. J. Am. Chem. Soc. 2012, 134, 4561–4564. [Google Scholar] [CrossRef]
- Zhang, S.; Bedi, D.; Cheng, L.; Unruh, D. K.; Li, G.; Findlater, M. Cobalt(II)-Catalyzed Stereoselective Olefin Isomerization: Facile Access to Acyclic Trisubstituted Alkenes. J. Am. Chem. Soc. 2020, 142, 8910–8917. [Google Scholar] [CrossRef]
- Wang, Q.; Yang, X.; Wu, P.; Yu, Z. Photoredox-Catalyzed C–H Arylation of Internal Alkenes to Tetrasubstituted Alkenes: Synthesis of Tamoxifen. Org. Lett. 2017, 19, 6248–6251. [Google Scholar] [CrossRef]
- Alizadeh, S. R.; Ebrahimzadeh, M. A. O-Glycoside Quercetin Derivatives: Biological Activities, Mechanisms of Action, and Structure–Activity Relationship for Drug Design, a Review. Phytother. Res. 2022, 36, 778–807. [Google Scholar] [CrossRef]
- Noguchi, H.; Hojo, K.; Suginome, M. Boron-Masking Strategy for the Selective Synthesis of Oligoarenes via Iterative Suzuki−Miyaura Coupling. J. Am. Chem. Soc. 2007, 129, 758–759. [Google Scholar] [CrossRef]
- Lo, J. C.; Gui, J.; Yabe, Y.; Pan, C.-M.; Baran, P. S. Functionalized Olefin Cross-Coupling to Construct Carbon–Carbon Bonds. Nature 2014, 516, 343–348. [Google Scholar] [CrossRef]
Figure 1.
Statistical overview and distribution of data in RadicalDB. (a) Multi-component radical reactions account for 47.3% of the database, and the molecular weight of the products from different component radical reactions follows a normal distribution. (b) Overall, the molecular weight of radical reaction products in RadicalDB also exhibits a normal distribution. (c) RadicalDB includes classic named reactions, each well-clustered according to its reaction type. (d) Photocatalytic and electrocatalytic radical reactions constitute 31.6% of RadicalDB. However, some photo/electrocatalytic reactions (indicated by the red dots in the lower-left corner) show less distinct clustering among radical reactions.
Figure 1.
Statistical overview and distribution of data in RadicalDB. (a) Multi-component radical reactions account for 47.3% of the database, and the molecular weight of the products from different component radical reactions follows a normal distribution. (b) Overall, the molecular weight of radical reaction products in RadicalDB also exhibits a normal distribution. (c) RadicalDB includes classic named reactions, each well-clustered according to its reaction type. (d) Photocatalytic and electrocatalytic radical reactions constitute 31.6% of RadicalDB. However, some photo/electrocatalytic reactions (indicated by the red dots in the lower-left corner) show less distinct clustering among radical reactions.
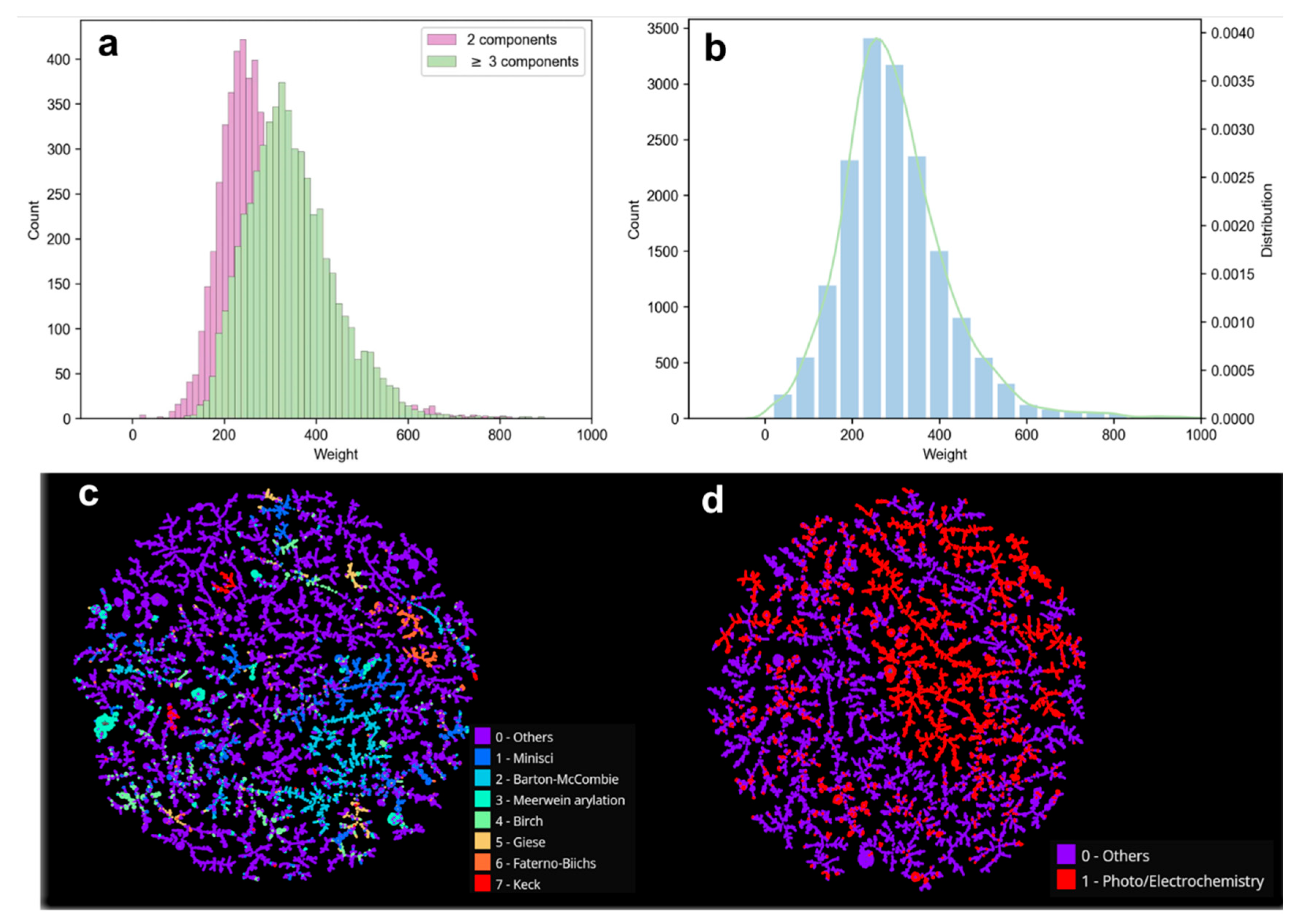
Figure 2.
Training strategy diagram of RadicalRetro. The Chemformer model, initially pre-trained on 100 million molecules and 1 million reactions, is fine-tuned using RadicalDB to develop the retrosynthesis model, RadicalRetro, specifically tailored for radical reactions.
Figure 2.
Training strategy diagram of RadicalRetro. The Chemformer model, initially pre-trained on 100 million molecules and 1 million reactions, is fine-tuned using RadicalDB to develop the retrosynthesis model, RadicalRetro, specifically tailored for radical reactions.
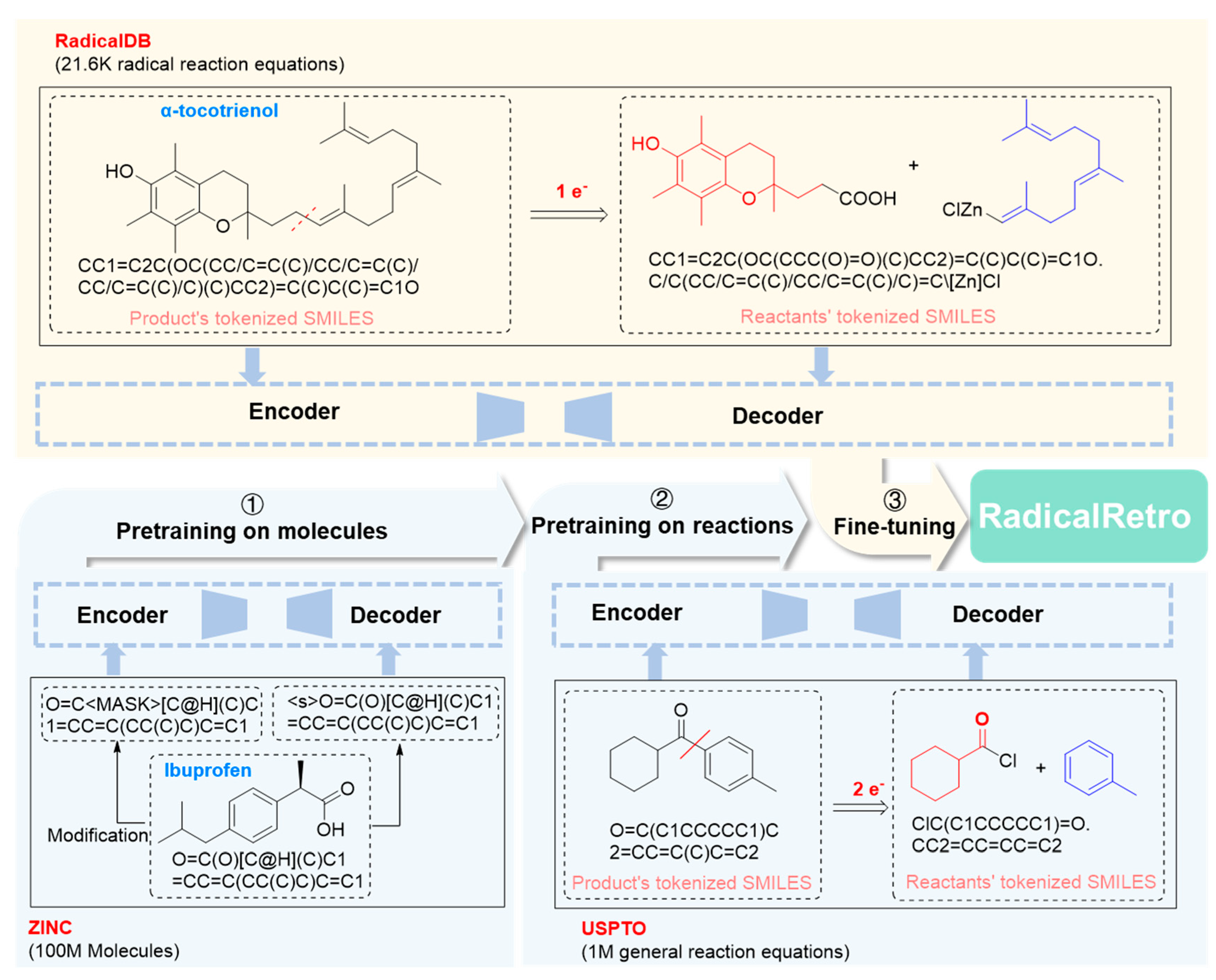
Figure 3.
Figure 3. Comparison of the prediction results of the three models. (a) RadicalRetro, based on the Chemformer model, achieved the highest prediction accuracy. (b) RadicalRetro outperformed MolTransformer in avoiding invalid SMILES predictions.
Figure 3.
Figure 3. Comparison of the prediction results of the three models. (a) RadicalRetro, based on the Chemformer model, achieved the highest prediction accuracy. (b) RadicalRetro outperformed MolTransformer in avoiding invalid SMILES predictions.
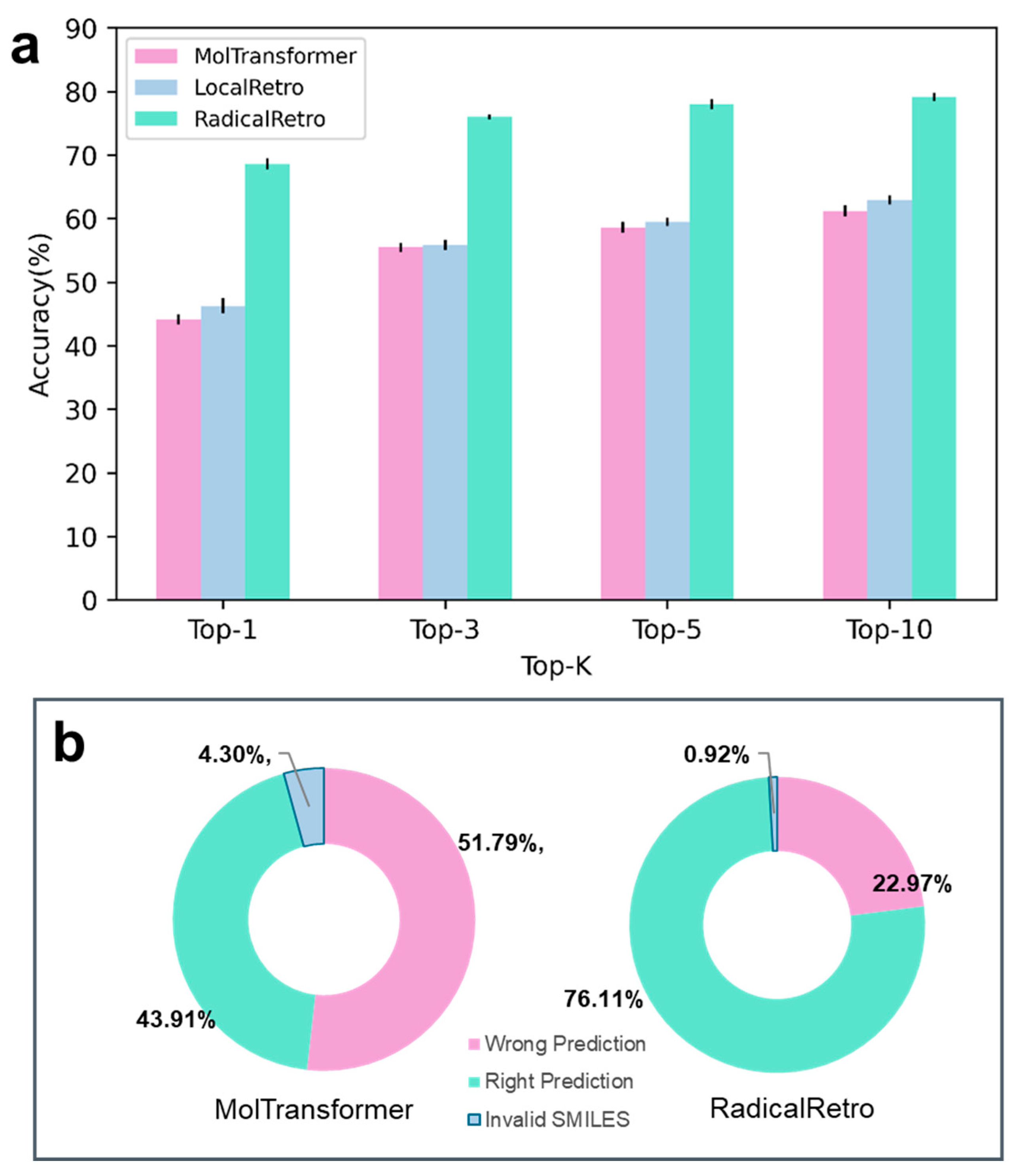
Figure 4.
Comparison of RetroSynthesis Accuracy of RadicalRetro across Different Types of Radical Reactions. (a) From the perspective of radical initiation, the retrosynthesis accuracy for photocatalytic/electrocatalytic radical reactions is slightly higher than that for other types of radical reactions. (b) In terms of name reactions, the Faterno Biichs reaction and the Meerwein arylation reaction exhibit the highest retrosynthesis prediction accuracy. However, due to the abundance of C-H bonds in molecules, the model struggles to determine which hydrogen atom in the precursor was originally part of a hydroxyl group. As a result, the Barton-McCombie deoxygenation radical reaction shows the lowest retrosynthesis prediction accuracy.
Figure 4.
Comparison of RetroSynthesis Accuracy of RadicalRetro across Different Types of Radical Reactions. (a) From the perspective of radical initiation, the retrosynthesis accuracy for photocatalytic/electrocatalytic radical reactions is slightly higher than that for other types of radical reactions. (b) In terms of name reactions, the Faterno Biichs reaction and the Meerwein arylation reaction exhibit the highest retrosynthesis prediction accuracy. However, due to the abundance of C-H bonds in molecules, the model struggles to determine which hydrogen atom in the precursor was originally part of a hydroxyl group. As a result, the Barton-McCombie deoxygenation radical reaction shows the lowest retrosynthesis prediction accuracy.
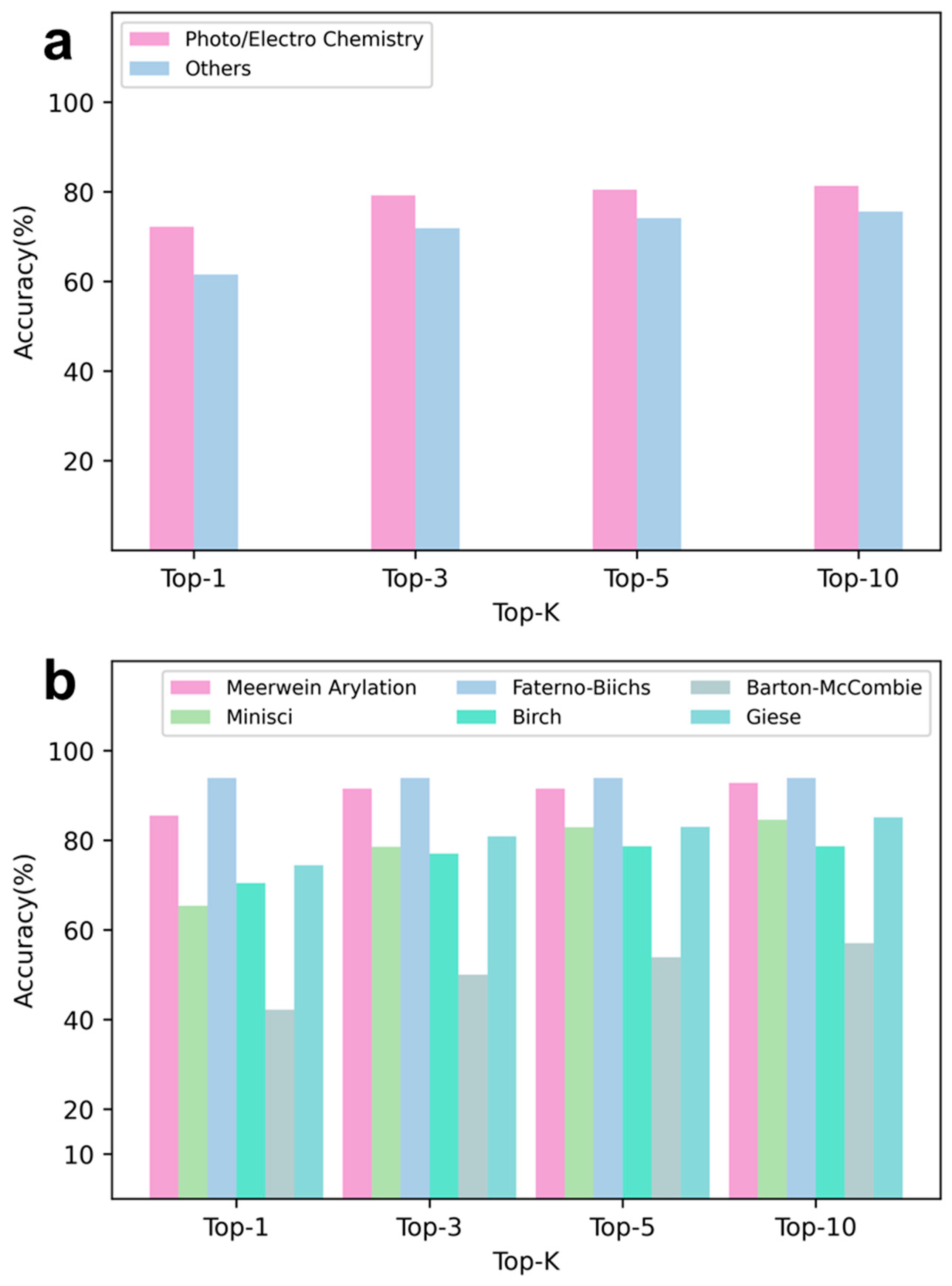
Figure 5.
Interpretability of LocalRetro. (a) and (b) demonstrate that RadicalRetro successfully identifies the appropriate radical reaction pathways for synthesizing biologically active phenylindane scaffold molecules and nitrogen-containing polycyclic aromatic hydrocarbons, respectively. (c) and (d) illustrate that, during the retrosynthesis process for these two compounds, RadicalRetro’s attention mechanism, from global to local, aligns closely with how chemists approach retrosynthetic analysis, confirming the interpretability of the model.
Figure 5.
Interpretability of LocalRetro. (a) and (b) demonstrate that RadicalRetro successfully identifies the appropriate radical reaction pathways for synthesizing biologically active phenylindane scaffold molecules and nitrogen-containing polycyclic aromatic hydrocarbons, respectively. (c) and (d) illustrate that, during the retrosynthesis process for these two compounds, RadicalRetro’s attention mechanism, from global to local, aligns closely with how chemists approach retrosynthetic analysis, confirming the interpretability of the model.
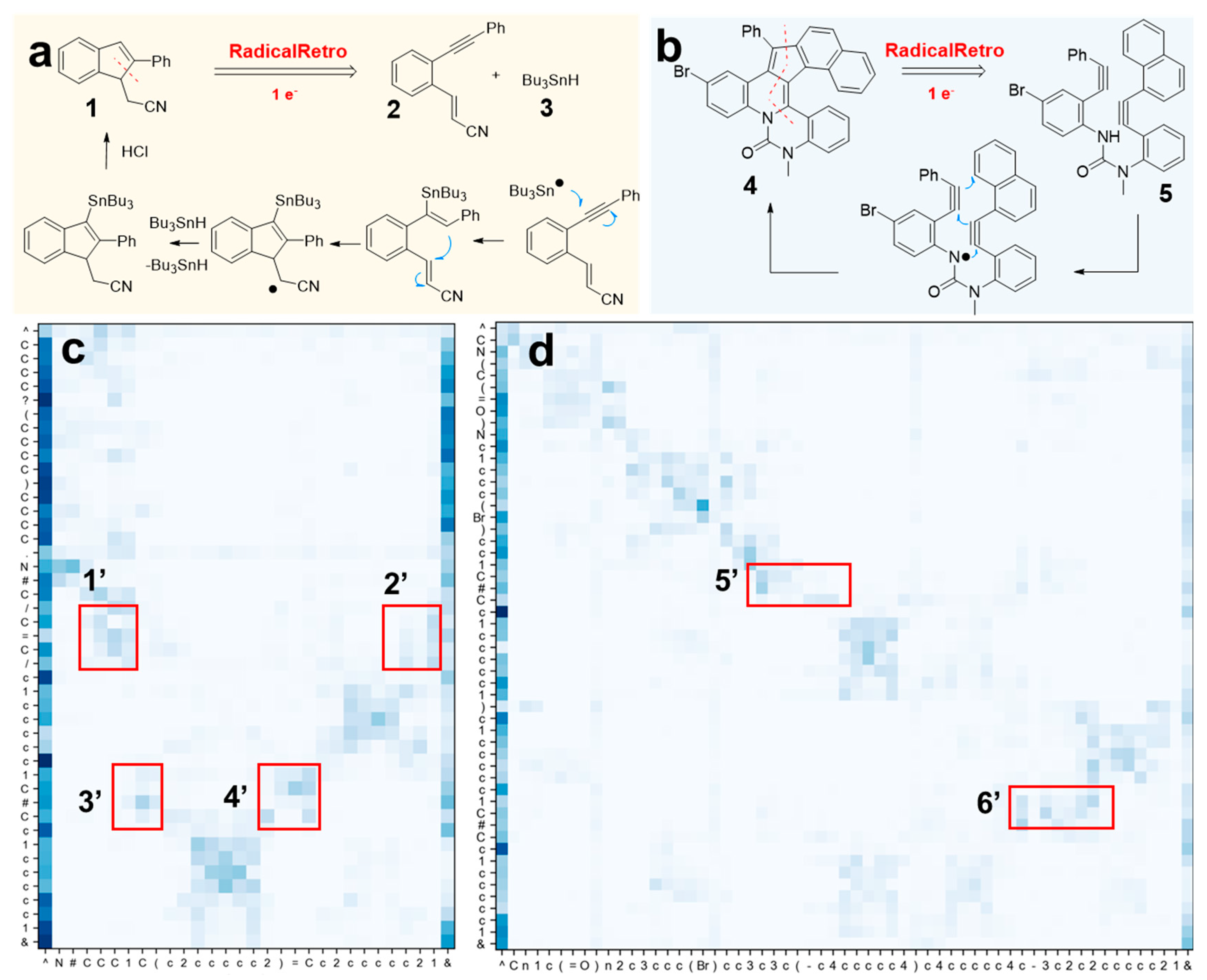
Figure 6.
Comparison of the retrosynthesis between RadicalRetro and GeneralRetro. For the Tamoxifen Precursor (a) and Glycoside Derivative (b): Compared to GeneralRetro without fine-tuning on RadicalDB, the synthesis pathways proposed by RadicalRetro demonstrate a better grasp of radical reaction characteristics. This approach shows significant potential in reducing reaction steps and optimizing reaction conditions.
Figure 6.
Comparison of the retrosynthesis between RadicalRetro and GeneralRetro. For the Tamoxifen Precursor (a) and Glycoside Derivative (b): Compared to GeneralRetro without fine-tuning on RadicalDB, the synthesis pathways proposed by RadicalRetro demonstrate a better grasp of radical reaction characteristics. This approach shows significant potential in reducing reaction steps and optimizing reaction conditions.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



