
Submitted:
17 April 2025
Posted:
18 April 2025
You are already at the latest version
Abstract
There has been extensive research in the field of KSHV/HHV8 virus, which has led to the development in the understanding of KSHV/HHV8 transmission, viral pathogenesis, how the virus drives neoplastic lesions, and how we can combat these processes. On extensive review of literature, only two true KSHV/HHV8 positive lymphoid neoplasms are described: Primary effusion lymphoma (PEL), which can also present as solid or extra-cavitary primary effusion lymphoma (EC-PEL) and diffuse large B-cell lymphoma (DLBCL). Two lymphoproliferative disorders have also been described, and while they are not true monotypic neoplasms, these lesions can transform into neoplasms: KSHV/HHV8 positive germinotropic lymphoproliferative disorder (GLPD) and Multicentric Castleman's disease (MCD). This review provides a somewhat concise overview of information related to KSHV/HHV8 positive lymphoid neoplasms and pertinent associated lymphoproliferative lesions.
Keywords:
KSHV
; HHV8
; Lymphoma
; Lymphoproliferative
; Neoplasm
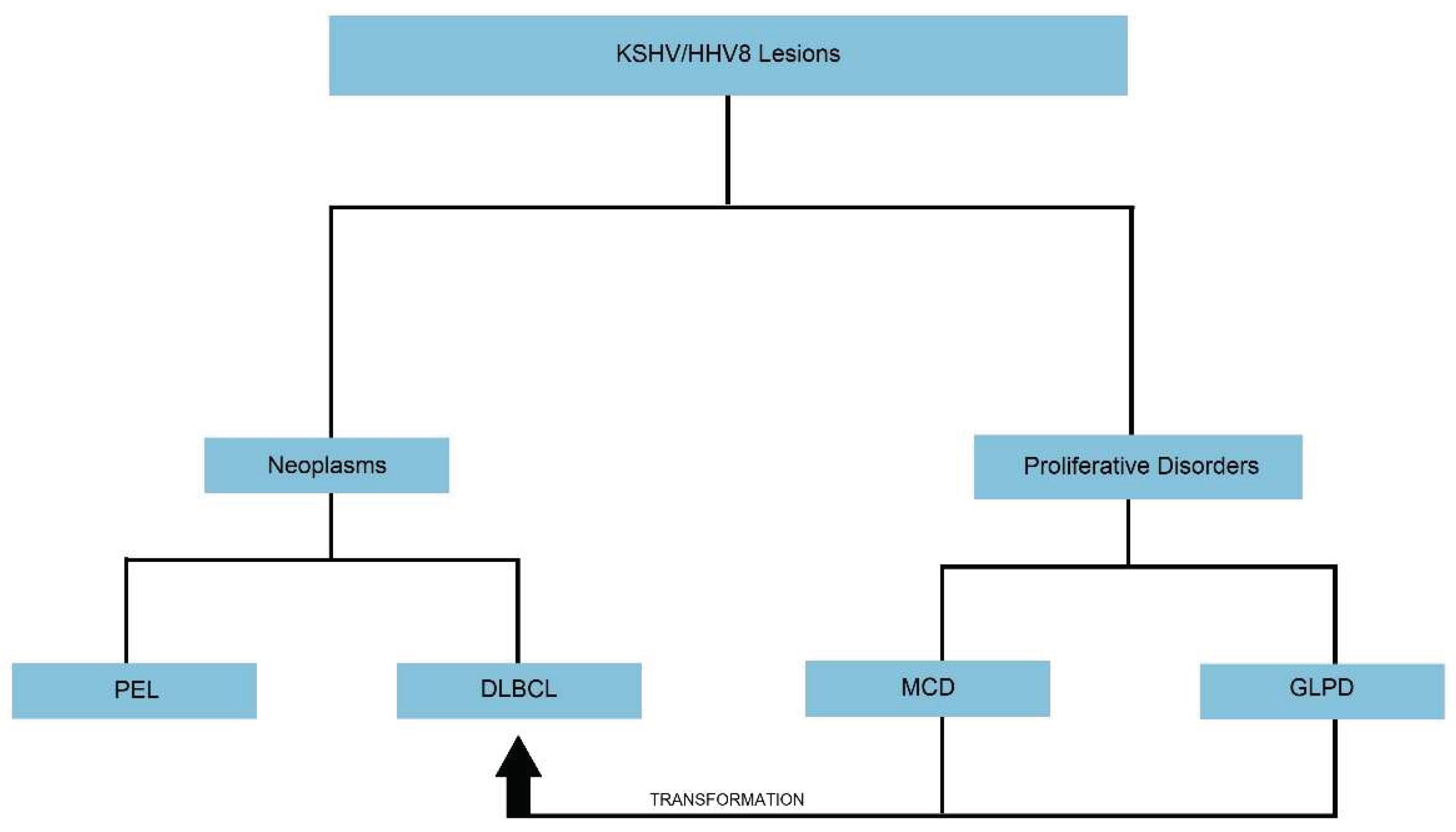
1. Introduction
Human herpesvirus 8 or Kaposi sarcoma-associated herpesvirus (KSHV/HHV8) was initially identified in 1994 after it was found in the tissues of Kaposi’s sarcoma lesions of AIDS patients. The newly discovered human herpesvirus was subsequently implicated in the pathogenesis of Primary Effusion Lymphoma (PEL) and Multicentric Castleman Disease (MCD) [1,2]. KSHV/HHV8 was detected in infected B-cells in these lymphoproliferative and neoplastic lesions, although it was unclear how the virus was related.
Since then, the virus has also been detected in Diffuse large B-cell lymphoma (DLBCL) and Germinotropic lymphoproliferative disorder (GLPD), and the pathogenesis in these lesions is better understood.
2. Biology of HHV8
KSHV/HHV8 is a double-stranded DNA gammaherpesvirus virus with an icosahedral capsid and an outer lipid bilayer for a structure between 120 – 150 nm [2]. Its genome consists of a 140.5-kb-long unique coding region flanked by multiple Guanine and Cytosine-rich 801-bp terminal repeat sequences [2,3]. The coding region is comprised of 85-87 open reading frames (ORFs) [3,5] encoding for many pathogenically significant genes. The most notable genes code for homologs to our own immunologically active proteins, including interferon regulatory factors, complement-binding proteins, macrophage inflammatory proteins, bcl-2, interleukin 6, interleukin 8 receptor [3], and Fas-associated beta-convertase enzyme [37]. Many of these homologs inhibit cellular immune responses, the cytokine cascade, and apoptosis [2]. Other genes code for viral proteins like Kaposins or latency-associated nuclear antigens (LANAs), which play a role in cell transformation and oncogenesis, affecting cytokine function and maintaining the latent phase [37].
The most common KSHV/HHV-8 viral mode of transmission depends on geographic location. Prevalence of infection in healthy individuals is high in certain regions of sub-Saharan Africa (36-90%), Brazil (16-53%), Ecuador (20-100%), the Mediterranean (11 – 54%), Taiwan (40%), and northwestern China (47%), more specifically the Uygur population. In all these regions, the prevalence is higher in HIV-positive individuals [30]. In endemic regions, direct contact with saliva is the most common mode of transmission [2]. In regions where prevalence is low, sexual contact is often the most common mode of transmission, and so sexual practices also play a role in infection and transmission. Blood-borne transmission is rare but has been documented [30].
Entry of KSHV/HHV8 into B cells is related to DC-SIGN-mediated endocytosis [6]. DC-SIGN, or dendritic cell-specific ICAM-3-grabbing nonintegrin, is a receptor on the surface of dendritic cells and macrophages that plays a role in immune responses [7]. This same receptor was found to be on activated B-cells [6], which explains how KSHV/HHV8 infected B-cells become the driving force in each of these lesions.
Following invasion into the cell, the viral genome circularizes and enters a latent phase, where only a few genes are expressed [1,2]. The most important of which is LANA1, or Latency-associated nuclear antigen 1, which binds to Kaposi’s sarcoma-associated herpesvirus (KSHV) terminal repeat (TR) DNA to mediate episome replication and persistence [8]. LANA1 is also responsible for maintaining the latent state, altering cell proliferation and apoptosis by interacting with p53 regulatory protein [37]. LANA1 also binds to retinoblastoma (RB), leading to cell proliferation through the RB / E2F pathway [80,81]. So, just this one viral product assists in viral genome replication and taking over cellular mechanisms to control the cell cycle.
Other important viral products made during latency include viral interferon regulatory factor 3 (vIRF3; also called LANA2), viral interleukin-6 (vIL-6), viral cyclin (vCYC), and viral FLICE-inhibitory protein (vFLIP) [1]. These products, in particular, are implicated in the progression of HHV8-associated malignancies [80]. IL-6 and vIL-6 induce vascular endothelial growth factor (VEGF), which stimulates the growth of new vessels, increases vascular permeability, and plays a role in forming PEL-related effusions. vIL-6 also suppresses proapoptotic cathepsin D, preventing apoptosis [80]. vFLIP is a homolog to cellular Fas-associated beta-convertase enzyme and inhibits apoptosis by blocking caspase activation mediated by FAS and tumor necrosis factor (TNF) [81]. It also plays a role in LANA activities, cell transformation, and tumor induction [37]. vCyclin and v-FLIP contribute to tumor growth by constitutively activating cyclin-dependent kinase 6 and the transcription factor nuclear factor kappa B (NF-κB) pathway, respectively. Collectively, the actions of viral gene products lead to tumor proliferation and inhibition of apoptosis while maintaining viral latency.
All of these latent gene products are transcribed from a single multicistronic locus. The other viral genes that encode components of the lytic cycle are kept silent through a bivalent epigenetic histone modification, which keeps chromatin in a quiescent state [35]. Under specific conditions, the latently infected cells can be induced to enter the lytic cycle through a not yet fully characterized process that involves the Replication and Transcriptional Activator (RTA), which serves as a major regulator that orchestrates the expression of viral lytic genes [36].
Certain KSHV/HHV8 genes that are typically expressed during lytic cycles, including vIL-6, vMIR1, and vMIR2, can be activated in the absence of full lytic activation, independent of viral RTA [46], suggesting that KSHV/HHV8 may not be confined to the concepts of latent and lytic cycles as we understand them.
The lytic and reproductive phase of HHV8 leads to lysis and death of the infected cell; therefore, it benefits the virus to remain in the latent phase to promote tumor growth and cell death [80].
Kaposins are another viral gene product present during the viral phase. The isoform Kaposin A regulates oncogenesis through cytokesin 1. Kaposin B stabilizes cytokine expressions such as interleukin 6 (IL6) and granulocyte-macrophage colony-stimulating factor (GM-CSF) by stabilizing cytokine mRNA containing adenylate uridylate-rich elements [76]. The viral homolog of cyclin D (vCyclin) binds to cyclin-dependent kinase 6 (CDK6), which inactivates RB protein and leads to dysregulation of the cell cycle [76]. LANA2 / viral interferon regulatory factor 3 (vIRF3) induces drug resistance by binding to polymerized microtubules, decreasing their stability [76].
Studies show that KSHV/HHV-8 maintains an undetectable latent infection in circulating B-lymphocytes and monocytes. When these circulating monocytes are exposed to inflammatory cytokines, they undergo lytic transformation and transmission of the virus to other cells [2,30]. Several different cell types can be infected by KSHV/HHV-8, including oral epithelial cells, endothelial cells, and, as mentioned, B-lymphocytes and monocytes.
3. HHV8-Associated B-Cell Lymphoid Proliferations and Lymphomas
3.1. Primary Effusion Lymphoma (PEL) and Extra-Cavitary Primary Effusion Lymphoma (EC-PEL)
Primary effusion lymphoma (PEL) is a clinically aggressive large B-cell lymphoma presenting as a serous effusion involving the pleural cavity, the peritoneal cavity, or pericardial cavity, without nodal involvement or tumor masses [14,15]. PEL typically involves a single serous cavity, but more than one cavity can be involved [17]. A subset of cases presents with nodal involvement or extranodal solid tumor masses. This variant is called extra-cavitary primary effusion lymphoma (EC-PEL). EC-PEL can occur with or without a lymphomatous effusion but most frequently occurs after PEL has developed [17]. Extra cavitary sites include the gastrointestinal tract, lung, skin, central nervous system, or bone marrow [14,15,16,22]. Lymphoma cells are coinfected by both KSHV/HHV8 and EBV [WHO].
PEL occurs most often in HIV-positive adults with low CD4+ T-cell counts but has been documented in other immunocompromised individuals, including transplant (immunosuppressed) patients or elderly (immunosenescence) individuals living in endemic regions [14,15,16,22]. Other manifestations of KSHV/HHV8 infection, including Kaposi sarcoma and multicentric Castleman disease, are present in approximately 50% of patients at presentation [15]. Approximately 80% of PEL cases are EBV-positive. The co-infection is seen predominantly in HIV-positive individuals and occasionally in HIV-negative individuals living in endemic regions [15,23]. The age of diagnosis is dependent on location and HIV status; HIV-positive individuals were diagnosed at the median age of 42, while HIV-negative individuals were diagnosed at the median age of 73 [14,22]. PEL occurs almost exclusively in males [14,25].
Clinical presentation depends on the cavity /cavities or extra-cavitary sites affected. Those with pericardial effusions often present with chest pain, shortness of breath, and hemodynamic instability in the event of cardiac tamponade [18,19]. Pleural effusion can also cause shortness of breath and chest pain but may be accompanied by cough [20]. Effusion of the peritoneal cavity will present as ascites, which causes abdominal distension, sensation of fullness, abdominal discomfort or pain, and occasionally shortness of breath [21]. General symptoms can include weight loss, fever, and night sweats. Depending on the extent and severity of lymphoproliferation, some patients develop lymphadenopathy and/or splenomegaly [16].
Lab abnormalities range from hypoalbuminemia, thrombocytopenia, and anemia to elevated IL-6 and elevated KSHV viral load that is relatively unique amongst HIV-associated lymphomas [69].
Since these are effusions, fluid is generally collected by aspiration and assessed on smear (Papanicolaou stain and/or Diff-Quik) and cell block (H&E). The effusion specimen will likely be more cellular when compared to an individual without lymphomatous effusion. Lesional cells can range from plasmablastic to anaplastic in appearance. Plasmablasts are large neoplastic cells with round to irregular nuclei, prominent nucleoli, and basophilic cytoplasm. Vacuolation is variable. Anaplastic cells are poorly differentiated or completely undifferentiated and may exhibit variable size and pleomorphism. Anaplastic cells may occasionally resemble the Reed-Sternberg cells found in classical Hodgkin lymphoma. The proliferation rate is usually high, and numerous mitotic figures are seen. [70] Figure 1
For patients with EC-PEL, specimens are typically resected and evaluated on standard hematoxylin and eosin-stained sections. Lymph nodes and masses may or may not share features grossly. Lesional cells appear similar to those seen on cytology but may appear less pleomorphic on tissue sections [70].
In PEL and EC-PEL, lymphoma cells stain positive for HHV8, EBER, CD45, CD30, CD38, CD71, EMA, HLA-DR, and CD138. The cells are typically negative (or dim/patchy) for B-cell markers, including CD19, CD20, and CD79a. Cells are also negative for T-cell markers like CD3, CD4, and CD8. BCL-6 is also typically negative [70]. Figure 2
Lymphoma cells are post-GC B cells that have undergone an intense somatic mutation process on the immunoglobulin gene hypervariable region, which is demonstratable on molecular testing [17,70].
Unusual features for solid primary effusion lymphoma are the presence of multiple lymphadenopathies, negativity for CD138, and expression of IgM. Although primary effusion lymphoma frequently occurs in patients with HHV8-associated multicentric Castleman disease, these are considered separate entities, and there is currently no definitive evidence of evolution of multicentric Castleman disease to primary effusion lymphoma [32]. This is an important distinction between PEL and DLBCL, which will be discussed in the following section.
Depending on HIV status, treatment of PEL may begin with antiretroviral therapy. In fact, complete remission of PEL has been documented in a few cases after the sole use of HAART [72]. Other treatments may include prophylaxis against opportunistic infections such as Pneumocystis jirovecii and cytomegalovirus (CMV), as well as granulocyte-macrophage colony-stimulating factor (GM-CSF) to prevent complications of chemotherapy-induced neutropenia [72,73,74].
NCCN (National Comprehensive Cancer Network) recommends R-EPOCH (rituximab, etoposide phosphate, prednisone, vincristine sulfate (Oncovin), cyclophosphamide, and doxorubicin hydrochloride (hydroxydaunorubicin) or R-CHOP (rituximab, cyclophosphamide, and doxorubicin, vincristine sulfate (Oncovin), prednisone) as first-line therapy, which is commonly used in the treatment of other non-Hodgkin lymphomas (NHL). Research is still ongoing in the exploration and development of novel treatments for HHV8 lesions that target HHV8 and/or associated pathways. These include but are not limited to, antiviral drugs, monoclonal antibodies, immunomodulatory drugs, and small molecular inhibitors [40].
Prognosis is influenced by viral status, extent of viral infection, cytokine production, protein expression, performance status, and body cavity involvement. The strongest predictor of improved survival is the use of HAART - effective viral replication control is known to allow greater tolerance to therapies, including autologous stem cell transplantation - though notably, the impact of antiretroviral therapy is lower in PEL than other HIV-1 related lymphomas such as DLBCL [23,72,73,74]. EBV positivity in PEL patients is weakly associated with improved survivability and has a more indolent course [16,17,25]. High levels of IL-6, CD47 positivity, and CD20 negativity are all associated with aggressive behavior, poor prognosis, and decreased overall survival [16,77]. Involvement of the pericardial cavity, in particular, as opposed to the pleural and peritoneal cavities, or involvement of multiple cavities concurrently portends a poor prognosis [16]. Variants of PEL such as EC-PEL (extracavitary PEL) and PEL-LL (primary effusion-like Lymphoma) may have a better prognosis, though drawing comparisons remains difficult [75,78,79]. Meanwhile, PT-PEL (post-transplant PEL) has a similarly poor prognosis of less than a year [23].
PEL / EC PEL prognosis is poor, and median survival, even with treatment, remains < 24 months [76].
3.2. KSHV/HHV8-Positive Diffuse Large B-Cell Lymphoma (DLBCL)
KSHV/HHV8-positive DLBCL is a rare form of DLBCL that is associated with KSHV/HHV8 infection. This large B-cell lymphoma is often seen in individuals with severe immunodeficiency and who have been diagnosed with MCD [17,25]. Patients may have other KSHV/HHV-8 infection manifestations, including Kaposi sarcoma (KS). Most cases of KSHV/HHV8-positive DLBCL are seen in HIV-positive men ages 30 – 40 years of age [25]. Rarely does this lymphoma develop in the absence of MCD; however, these cases arise in immunosenescent individuals in KSHV/HHV-8 highly endemic regions. KSHV/HHV8-positive DLBCL occurs more than three times in men than in women (3.3:1) [28].
Patients usually present with generalized lymphadenopathy, which is often accompanied by splenomegaly [26,32]. These manifestations are often accompanied by B-type symptoms [33,35,40] and may involve peripheral blood and bone marrow. KSHV/HHV8-positive DLBCL can also disseminate to organs, including the lungs, liver, or gastrointestinal tract [36]. In a rare case, lymphoma involvement was confined to the spleen [27,32]. In another rare instance, one HIV-negative elderly male with a history of MCD developed massive ascites that was immunophenotypically consistent with KSHV/HHV8-positive DLBCL [34]. These cases highlight the presentation overlap and difficulty distinguishing KSHV/HHV8-positive DLBCL from other HHV8-positive lesions. Labs may show cytopenias in one or all three cell hematopoietic lineages, especially when bone marrow is involved. LDH, in particular, is a useful prognostic factor in all DLBCLs, reflecting disease severity, response to treatment, and relapse. Hypercalcemia may also be a biomarker for more aggressive disease and/or high-risk features. Metabolic panels may show changes in renal or hepatic function, as well as changes in electrolytes. Some of these changes, however, reflect treatment effects and/or tumor lysis syndrome [67].
Fine needle aspiration and cytology can suggest a diagnosis of KSHV/HHV8-positive DLBCL; however, a definitive diagnosis should be made on excisional lymph node biopsy to evaluate background MCD and GLPD changes [26,68].
Grossly, lymph nodes are firm and fleshy in consistency and demonstrate homogeneous white cut surfaces. Hilar obliteration may be appreciated, and necrosis is variable. Extranodal sites may show a confluent appearance or discrete nodules. Histologically, lymph node architecture is effaced by diffuse or coalescing sheets of large, immature-appearing lymphoid cells [26]. The cells are called plasmablasts with classic immunoblastic morphology, showing vesicular and eccentrically placed nuclei with one to two prominent nucleoli, and a moderate amount of amphophilic cytoplasm [26,36,38]. Occasionally, cells can appear scattered, which may coalesce in small clusters once termed microlymphomas. Clonal studies, however, showed that these clusters are usually polyclonal and do not necessarily progress to lymphoma, so the term was eliminated. These KSHV/HHV-8 positive blasts produce a cytoplastic IgM lambda and rarely kappa. While DLBCL and MCD share the feature of lesions with lambda-restricted plasmablasts infected by KSHV/HHV-8, the distinction is in their clonality. MCD lesions have polyclonal plasmablasts, while frank lymphoma is a monoclonal process [38]. Evidence suggests that DLBCL arises in the same plasmablasts of MCD lesions when DLBCL develops in the setting of MCD [33,39]. This is an important distinction between the two neoplasms; PEL can arise in the setting of MCD but does not develop directly from these lesions. Figure 3
Lymphomatous cells of KSHV/HHV8-positive DLBCL are naïve, non-mutated, pre-germinal center B cells [17]; they are positive for LANA, vIL-6, and IRF4/MUM1 [17,26] and negative for plasma cell markers, including CD38 and CD138. They show variable staining for B-cell markers like CD45 and CD20 but are negative for CD79a [26,32,33]. Molecular studies reveal monoclonal IGH rearrangement with a lack of somatic mutations of the IGH variable regions [32], which is shown in molecular studies.
For hematopathologists making this diagnosis, it should be noted that the World Health Organization (WHO) describes this entity in Hematolymphoid Tumours (5th ed.) under the category of KSHV/HHV-associated B-cell lymphoid proliferations and lymphomas as “KSHV/HHV8-positive diffuse large B-cell lymphoma”. International consensus classification (ICC) categorizes this lesion similarly under HHV-8–associated lymphoproliferative disorders; however, the diagnosis “HHV-8–positive diffuse large B-cell lymphoma, NOS” is used [ICC].
Similar to PEL/EC-PEL, NCCN recommends R-EPOCH or RCHOP as first-line therapy. KSHV/HHV8-positive DLBCL is particularly challenging to treat due to poor response to conventional chemotherapy. A retrospective study on 67 KSHV/HHV8-positive DLBCL patients found no significant association between chemotherapy use and improved survival outcomes [40].
KSHV/HHV8-positive DLBCL is both rare and aggressive. The prognosis is poorest amongst HIV-positive individuals in which HDN arises in the setting of MCD [26]. Research has suggested that age is the most important predictor of survival in DLBCL patients. A 2023 study found older age to be significantly associated with a lower overall survival rate. This same study also showed that survival was significantly better in nodal rather than extranodal HDN and that marital status of single had worse overall survival [40]. Most recent research in 2024 shows that survivors have an increased risk of malignancy within the first year of diagnosis, and the 1-year, 3-year, and 5-year Overall survival was 63.6%, 59.7%, and 54.4%, respectively [41]. The WHO reported an overall median survival of a few months.
3.3. KSHV/HHV8-Associated Multicentric Castleman Disease (MCD)
Castleman disease was initially described in 1956 in asymptomatic patients with lymphoid proliferations arising in the mediastinum. Several morphological and clinical variants were subsequently identified [17,33]. Clinically, Castleman disease is divided into unicentric (involving a single lymph node) or multicentric (presenting in two or more lymph nodes). The multicentric form was described in 1980, and in its early stages, MCD was subcategorized into HIV-positive and HIV-negative cases. Later research showed that KSHV/HHV-8 status was more significant in classifying these lesions [33]. HIV status is still used in subclassifying KSHV/HHV8-associated MCD, which is a systemic, polyclonal lymphoproliferative disorder that is driven by KSHV/HHV8 infection [33,34,44]. MCD that arises in the absence of KSHV/HHV-8 infection is termed idiopathic multicentric Castleman disease (iMCD) and generally has a less aggressive clinical course than KSHV/HHV8-associated MCD [32].
The majority of KSHV/HHV8+MCD cases occur in HIV-positive patients with low CD4+ T-cell counts, predominantly in men (M:F; 8:1) and with a median age of 40 – 45 years. In HIV-negative individuals, patients tend to be older, with a median age of 65 years, and with a lower male-to-female ratio (2.4:1) [43].
Patients present with fever and other constitutional symptoms, including fatigue, weight loss, and myalgias. MCD is a relapsing-remitting disease, and symptoms often come and go in “flares.” The flares correlate with proinflammatory hypercytokinaemia and viral load [43].
The most common clinical manifestation in MCD is generalized lymphadenopathy, frequently involving the axillary, abdominal, pelvic, mediastinal, and cervical nodes, although any lymph nodes can be involved, and lymphadenopathy is often diffuse. Lymphadenopathy is often accompanied by hepatosplenomegaly and rarely involves bone marrow [33,42,43,44]. Other symptoms documented include effusions, edema, pulmonary and/or gastrointestinal symptoms, autoimmune hemolytic anemia, or hemophagocytic lymphohistiocytosis (HLH) [43,44]. Lab findings are often significant for polyclonal hypergammaglobulinemia, hypoalbuminemia, elevated C-reactive protein, thrombocytopenia, and anemia [17,32,33,43,44]. The most notable lab finding is high serum levels of IL-6. This cytokine, as well as IL-10 and vIL-6, contribute to the systemic inflammatory symptoms experienced in this condition.
The majority of patients also have a diagnosis of Kaposi sarcoma (KS), and a smaller percentage of patients have concurrent or will have a subsequent diagnosis of lymphoma [43,44].
Core biopsies are inadequate for diagnosing MCD, and so an excisional lymph node biopsy is preferred. Grossly, lymph nodes are enlarged and show a range of features, from pinkish-gray and well-encapsulated [48], gray-white with ill-defined borders and cut surfaces with a multinodular appearance [45], to diffuse nodularity with prominent vasculature [47]. Calcification may also be variable [49].
Microscopically, lymph nodes show lymphocyte-depleted germinal centers, vascular proliferation, and plasmacytosis [32]. The lesions are caused by the same elevated cytokines and growth factors that cause systemic inflammatory symptoms. [33].
All forms of Castleman disease can present morphologically as hyaline-vascular, plasma cell, or mixed-pattern variants [50]; however, MCD tends to be a plasma cell/plasmacytic variant or mixed-pattern [32,43,51]. KSHV/HHV8-associated MCD, in particular, demonstrates plasmacytic histology with plasmablastic features. Figure 4
Follicles can be small with regressed germinal centers [32] and hypocellular in appearance [50]. There is marked vascular proliferation [27,33], usually with some degree of hyalinization [32,33]. Plasmacytosis in the interfollicular zones is marked [27], with some histology showing sheets of polytypic plasma [33]. These plasma cells are CD138 positive [43]. Plasmablasts are seen primarily in mantle zones but can also be seen in perifollicular and interfollicular zones [33,43]. Plasmablasts are B-cells with plasmacytic differentiation [32], range from medium to large [33,43], and show vesicular chromatin with prominent nucleoli. Although these cells produce a monotypic IgM that is lambda-restricted [27,38], these immature cells are polyclonal, according to molecular studies [38,43]. Plasmablasts are HHV8 infected, as evidenced by the presence of LANA staining on immunohistochemistry. These cells are also positive for OCT2 [46], BLIMP1, IRF4 (MUM1) and negative for CD 20 and CD138 [43,46]. Some sources show that CD20 and CD79a staining is variable in up to 50% of cases [33,52]. Sources showed mixed reporting on PAX5 staining [17,32,33]. Rarely, plasmablasts are co-infected with EBV, and are EBER positive [32].
While ICC describes all HHV-8–associated lymphoproliferative disorders together, including “HHV-8–associated multicentric Castleman disease”, WHO categorizes “KSHV/HHV8-associated multicentric Castleman disease” under Tumour-like lesions with B-cell predominance.
In the most recent iteration of the National Comprehensive Cancer Network (NCCN) guidelines, there is guidance for the treatment of all forms of Castleman disease, including “HHV8-positive MCD.” Rituximab is first-line therapy of KSHV/HHV8-associated MCD and effectively depletes the reservoir of HHV8-infected B-cells, reducing cytokine levels and the systemic inflammatory response in MCD flares. Even in relapse, rituximab improves survival and makes KSHV/HHV8-associated MCD a more manageable lymphoproliferative disorder [55]. Per NCCN guidelines, rituximab may be combined with prednisone and or liposomal doxorubicin, which causes reductions in lymphadenopathy and viral load [56]. Other treatments include antivirals, IL-6 inhibitors [57], and chemotherapy, depending on the severity of the disease [NCCN]. Second-line therapy and subsequent therapy for relapsed/refractory or progressive disease are also available [NCCN]. However, no gold standard therapy has been established [56]. Castleman Disease Collaborative Network (CDCN) offers an algorithm that takes into consideration KS status and includes recommendations for specific treatments, numbers of cycles and timelines, as well as surveillance [62].
KSHV/HHV8-associated MCD has an increased risk of developing lymphoma as well as other KSHV/HHV8-related disease [43]. In the past, the prognosis was poor, with an overall survival rate equal to or less than 2 years [43]. With the introduction of rituximab and antiretroviral therapy, the overall survival rate at 5 years has increased to 90-92% [43,53,54].
3.4. Germinotropic Lymphoproliferative Disorder (GLPD)
Germinotropic lymphoproliferative disorder, first described in 2002 [12], is one of two entities resulting from co-infection by KSHV/HHV8 and Epstein-Barr Virus (EBV). The role of EBV in this lesion is unclear, so it is suggested that KSHV/HHV8 and vIL-6 must be primary drivers [11].
GLPD is rare and tends to affect the elderly (median age: 60 years, range: 20–86 years), although it can be seen at any age and has a slight male predominance [11]. HIV status is variable, although the majority of cases are in HIV-negative individuals [13]. It is unclear from the literature whether or not there is an increased incidence of GLPD in endemic regions of KSHV/HHV8, however a number of cases from these areas have been described.
GLPD is typically indolent. The most common clinical presentation is lymphadenopathy. This can involve one or multiple lymph nodes, often seen as enhancing nodes on computed tomography (CT) or as fluorodeoxyglucose (FDG) avid nodes on positron emission tomography (PET). Other reported findings included B-type symptoms (weight loss, night sweats, fevers), effusions, leg swelling, paresthesias, abdominal pain, autoimmune hemolytic anemia, and splenomegaly. No lab abnormalities were specifically reported. The majority of patients, however, are healthy and asymptomatic [13].
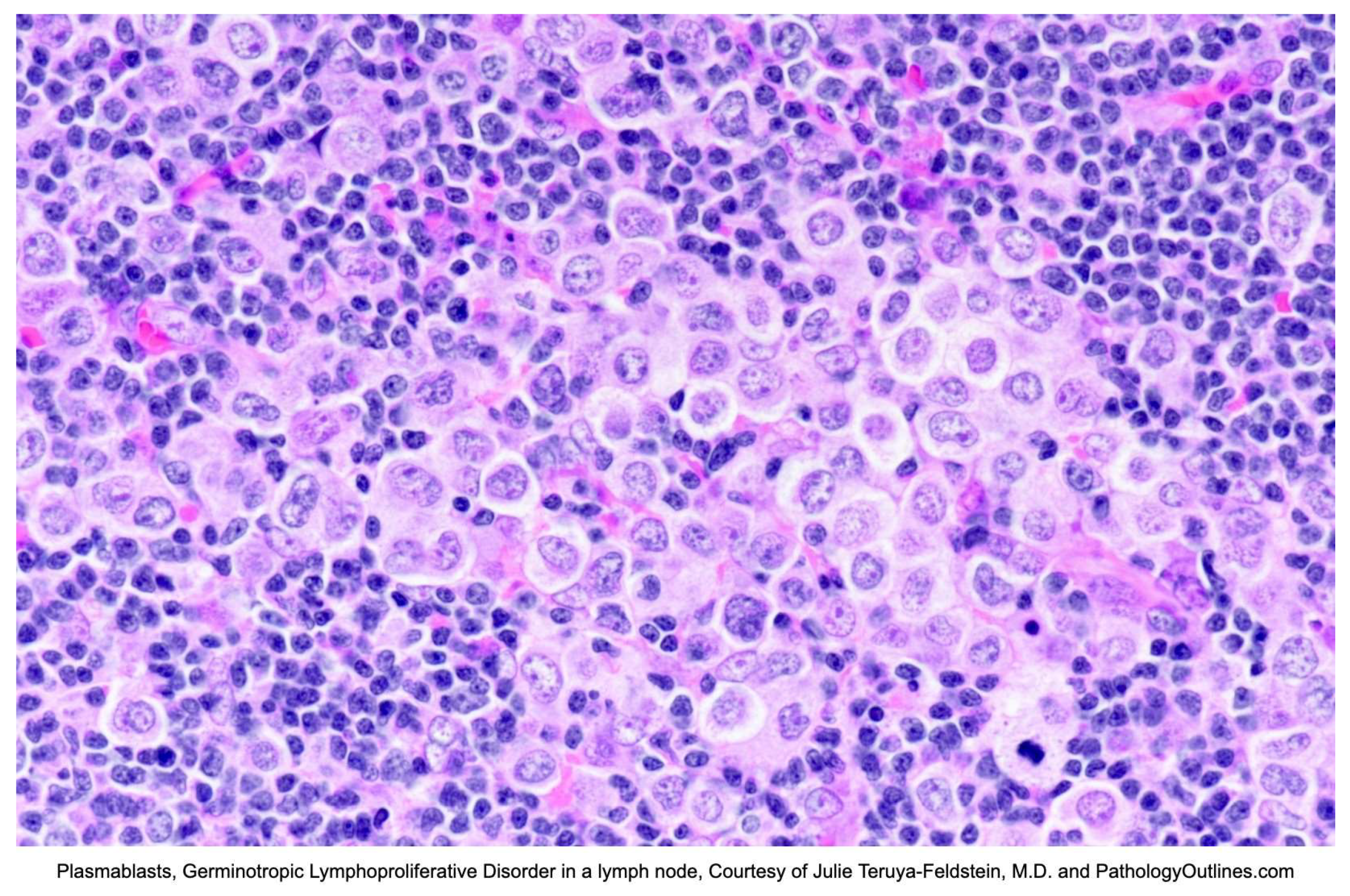
On excisional biopsy, lymph nodes are enlarged, ranging from 1.5 - 26 cm [11,13]. Grossly, the nodes are white, with cut surfaces showing focal nodularity [13]. Microscopically, there are often aggregates of plasmablasts replacing germinal centers, and in some cases, germinal centers are completely replaced by plasmablasts. These large, atypical cells are polyclonal to oligoclonal and are coinfected with KSHV/HHV8 and EBV [11]. Occasionally, there can be scattered plasmablasts in mantle zones, sinuses, and interfollicular areas [11,13]. The background can share features with KSHV/HHV8-associated MCD, including marked plasmacytosis [11,32] with atrophic and hyalinized follicles and vascular proliferation [11,13]. Lesional cells are positive for EBER, HHV8/LANA, vIL-6, MUM1/IRF4, PDL1 and show monotypic light chain restriction [path outlines, seer, 58,32]. Other plasma cell markers like CD38 and CD138 are variable [32]. T-cell markers may occasionally be positive. The cells are usually negative for B-cell markers, including CD20, CD79a, PAX5, BCL6, CD10, and CD30 [32]. The proliferation index is usually high [11]. Given the possible overlap with KSHV/HHV8 MCD, it should be noted that plasmablastic cells in MCD are not (or rarely) EBV positive and are almost always lambda restricted. Figure 5
While NCCN offers guidance on the treatment of other KSHV/HHV8-associated lesions described in this review, in its current iteration, there is no guidance for the treatment of GLPD. In general, there is no standard care for these lesions.
Available data suggests that there are a variety of treatments to which GLPD often responds well, including chemotherapy, surgical excision, and radiotherapy [63]. Cases report that some patients on CHOP or DA-EPOCH-based therapy achieved remission [64]. In fewer cases, patients underwent surgical excision, with some undergoing subsequent radiotherapy. The reported remission rate in these cases was 100% [66]. It is thought that overall, GLPD is indolent in nature and can be observed, but has the risk of transforming into aggressive lymphoma [63].
An interesting potential therapeutic role of PD1/PD-L1 immunotherapy has been suggested by Ronaghy et al. due to the PD-L1 expression found in their case but this still requires additional research [13]
4. Conclusions
KSHV/HHV8 is a virus that infects B-cells resulting in lymphomas (PEL/EC-PEL and DLBCL), and lymphoproliferative disorders (MCD and GLPD) that can transform into aggressive lymphoma. B-cell invasion occurs via cell adhesion molecules, and once inside the cell, viral gene products control the cell cycle, evade apoptosis and induce oncogenesis to promote viral proliferation and infection. While infection can occur in endemic regions and in immunocompetent patients, most patients with these lesions are immunocompromised, and so these lesions often portend a poor prognosis. While new treatments are becoming available, and overall survival times are improving, with the exception of KSHV/HHV8-associated GLPD, life expectancy is still short ranging from a few months to a few years. This review provides a collection of the most up-to-date information on KSHV/HHV8-associated lymphoid disorders, from viral infection to treatment and prognosis.
References
- Cesarman, E.; Chadburn, A.; Rubinstein, P.G. KSHV/HHV8-mediated hematologic diseases. Blood 2022, 139, 1013–1025. [Google Scholar] [CrossRef]
- De Paoli, P. Human herpesvirus 8: an update. Microbes Infect. 2004, 6, 328–335. [Google Scholar] [CrossRef]
- Russo, J.J.; Bohenzky, R.A.; Chien, M.-C.; Chen, J.; Yan, M.; Maddalena, D.; Parry, J.P.; Peruzzi, D.; Edelman, I.S.; Chang, Y.; et al. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. 1996, 93, 14862–14867. [Google Scholar] [CrossRef]
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef]
- Dourmishev, L.A.; Dourmishev, A.L.; Palmeri, D.; Schwartz, R.A.; Lukac, D.M. Molecular Genetics of Kaposi's Sarcoma-Associated Herpesvirus (Human Herpesvirus 8) Epidemiology and Pathogenesis. Microbiol. Mol. Biol. Rev. 2003, 67, 175–212. [Google Scholar] [CrossRef]
- Rappocciolo, G.; Hensler, H.R.; Jais, M.; Reinhart, T.A.; Pegu, A.; Jenkins, F.J.; Rinaldo, C.R. Human Herpesvirus 8 Infects and Replicates in Primary Cultures of Activated B Lymphocytes through DC-SIGN. J. Virol. 2008, 82, 4793–4806. [Google Scholar] [CrossRef]
- McGreal, E.P.; Miller, J.L.; Gordon, S. Ligand recognition by antigen-presenting cell C-type lectin receptors. Curr. Opin. Immunol. 2005, 17, 18–24. [Google Scholar] [CrossRef]
- Komatsu, T.; E Ballestas, M.; Barbera, A.J.; Kelley-Clarke, B.; Kaye, K.M. KSHV LANA1 binds DNA as an oligomer and residues N-terminal to the oligomerization domain are essential for DNA binding, replication, and episome persistence. Virology 2004, 319, 225–236. [Google Scholar] [CrossRef]
- L. Jeffrey Medeiros, Roberto N. Miranda, Primary Effusion Lymphoma and Solid Variant of Primary Effusion Lymphoma, In Diagnostic Pathology: Lymph Nodes and Extranodal Lymphomas (Second Edition), Elsevier, 2018, Pages 510-517, ISBN 9780323477796. [CrossRef]
- Naresh, K. (2024). KSHV/HHV8-associated B-cell lymphoid proliferations and lymphomas: Introduction. World Health Organization. https://tumourclassification.iarc.who.int/ chaptercontent/63/173.
- Chadburn, A. , Said, J., Du, M., & Vega, F. (2024). KSHV/HHV8-positive germinotropic lymphoproliferative disorder. World Health Organization. https://tumourclassification.iarc.who. Int/chaptercontent/63/177.
- Du, M.-Q.; Diss, T.C.; Liu, H.; Ye, H.; Hamoudi, R.A.; Cabeçadas, J.; Dong, H.Y.; Harris, N.L.; Chan, J.K.C.; Rees, J.W.; et al. KSHV- and EBV-associated germinotropic lymphoproliferative disorder. Blood 2002, 100, 3415–3418. [Google Scholar] [CrossRef] [PubMed]
- Zanelli, M.; Zizzo, M.; Bisagni, A.; Froio, E.; De Marco, L.; Valli, R.; Filosa, A.; Luminari, S.; Martino, G.; Massaro, F.; et al. Germinotropic lymphoproliferative disorder: a systematic review. Ann. Hematol. 2020, 99, 2243–2253. [Google Scholar] [CrossRef]
- Shimada, K.; Hayakawa, F.; Kiyoi, H. Biology and management of primary effusion lymphoma. Blood 2018, 132, 1879–1888. [Google Scholar] [CrossRef]
- Said J, Cesarman E. Primary effusion lymphoma. In: Swerdlow SH, Campo E, Harris NL, et al., eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 2017.
- Gathers, D.A.; Galloway, E.; Kelemen, K.; Rosenthal, A.; Gibson, S.E.; Munoz, J. Primary Effusion Lymphoma: A Clinicopathologic Perspective. Cancers 2022, 14, 722. [Google Scholar] [CrossRef]
- Calabrò, M.L.; Sarid, R. Human herpesvirus 8 and lymphoproliferative disorders. Mediterr. J. Hematol. Infect. Dis. 2018, 10, e2018061. [Google Scholar] [CrossRef]
- Albugami, S.; Al-Husayni, F.; AlMalki, A.; Dumyati, M.; Zakri, Y.; AlRahimi, J. Etiology of Pericardial Effusion and Outcomes Post Pericardiocentesis in the Western Region of Saudi Arabia: A Single- center Experience. Cureus 2020, 12, e6627. [Google Scholar] [CrossRef]
- Sagristà-Sauleda, J.; Mercè, A.S.; Soler-Soler, J. Diagnosis and management of pericardial effusion. World J. Cardiol. 2011, 3, 135–243. [Google Scholar] [CrossRef]
- Krishna R, Antoine MH, Alahmadi MH, et al. Pleural Effusion. [Updated 2024 Aug 31]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK448189/.
- Chiejina M, Kudaravalli P, Samant H. Ascites. [Updated 2023 Aug 8]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK470482/.
- Zanelli, M.; Sanguedolce, F.; Zizzo, M.; Palicelli, A.; Bassi, M.C.; Santandrea, G.; Martino, G.; Soriano, A.; Caprera, C.; Corsi, M.; et al. Primary effusion lymphoma occurring in the setting of transplanted patients: a systematic review of a rare, life-threatening post-transplantation occurrence. BMC Cancer 2021, 21, 1–13. [Google Scholar] [CrossRef]
- Lurain, K.; Polizzotto, M.N.; Aleman, K.; Bhutani, M.; Wyvill, K.M.; Gonçalves, P.H.; Ramaswami, R.; Marshall, V.A.; Miley, W.; Steinberg, S.M.; et al. Viral, immunologic, and clinical features of primary effusion lymphoma. Blood 2019, 133, 1753–1761. [Google Scholar] [CrossRef]
- Arga, K.Y.; Kori, M. Chapter 10 - Current status of viral biomarkers for oncogenic viruses, Editor(s): Moulay Mustapha Ennaji, Oncogenic Viruses, Academic Press, 2023, Pages 221-252, ISBN 9780128241561. [CrossRef]
- Hu, Z.; Pan, Z.; Chen, W.; Shi, Y.; Wang, W.; Yuan, J.; Wang, E.; Zhang, S.; Kurt, H.; Mai, B.; et al. Primary Effusion Lymphoma: A Clinicopathological Study of 70 Cases. Cancers 2021, 13, 878. [Google Scholar] [CrossRef]
- Vega F, Chadburn A. KSHV/HHV8-positive diffuse large B-cell lymphoma. In: de Jong D, Naresh K, et al., eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 2022.
- Gonzalez-Farre, B.; Martinez, D.; Lopez-Guerra, M.; Xipell, M.; Monclus, E.; Rovira, J.; Garcia, F.; Lopez-Guillermo, A.; Colomo, L.; Campo, E.; et al. HHV8-related lymphoid proliferations: a broad spectrum of lesions from reactive lymphoid hyperplasia to overt lymphoma. Mod. Pathol. 2017, 30, 745–760. [Google Scholar] [CrossRef] [PubMed]
- Sukswai, N.; Lyapichev, K.; Khoury, J.D.; Medeiros, L.J. Diffuse large B-cell lymphoma variants: an update. Pathology 2020, 52, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Oksenhendler, E.; Boulanger, E.; Galicier, L.; Du, M.-Q.; Dupin, N.; Diss, T.C.; Hamoudi, R.; Daniel, M.-T.; Agbalika, F.; Boshoff, C.; et al. High incidence of Kaposi sarcoma–associated herpesvirus–related non-Hodgkin lymphoma in patients with HIV infection and multicentric Castleman disease. Blood 2002, 99, 2331–2336. [Google Scholar] [CrossRef]
- Edelman, D.C. Human herpesvirus 8 – A novel human pathogen. Virol. J. 2005, 2, 78–78. [Google Scholar] [CrossRef]
- Monini, P.; Colombini, S.; Stürzl, M.; Goletti, D.; Cafaro, A.; Sgadari, C.; Buttò, S.; Franco, M.; Leone, P.; Fais, S.; et al. Reactivation and persistence of human herpesvirus-8 infection in B cells and monocytes by Th-1 cytokines increased in Kaposi's sarcoma. . 1999, 93, 4044–58. [Google Scholar] [PubMed]
- Vega, F.; Miranda, R.N.; Medeiros, L.J. KSHV/HHV8-positive large B-cell lymphomas and associated diseases: a heterogeneous group of lymphoproliferative processes with significant clinicopathological overlap. Mod. Pathol. 2020, 33, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Chadburn, A.; Said, J.; Gratzinger, D.; Chan, J.K.C.; de Jong, D.; Jaffe, E.S.; Natkunam, Y.; Goodlad, J.R. HHV8/KSHV-Positive Lymphoproliferative Disorders and the Spectrum of Plasmablastic and Plasma Cell Neoplasms. Am. J. Clin. Pathol. 2017, 147, 171–187. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.-C.; Fu, P.-A.; Wang, S.-H.; Chang, K.-C.; Hsu, Y.-T. Kaposi sarcoma herpesvirus/human herpesvirus 8-positive diffuse large B-cell lymphoma characterized by malignant ascites: A case report. Pathol. - Res. Pr. 2024, 255, 155185. [Google Scholar] [CrossRef]
- Dave, A.; Schwartz, M.; Van, J.; Owczarzak, L.; Miller, I.; Jain, S. A Challenging Diagnosis of HHV-8-Associated Diffuse Large B-Cell Lymphoma, Not Otherwise Specified, in a Young Man with Newly-Diagnosed HIV. Am. J. Case Rep. 2024, 25, e945162–e945162. [Google Scholar] [CrossRef]
- Fenu, E.M.; Beaty, M.W.; O’neill, T.E.; O’neill, S.S. Cardiac Involvement by Human Herpesvirus 8-Positive Diffuse Large B-Cell Lymphoma: An Unusual Presentation in a Patient with Human Immunodeficiency Virus. Case Rep. Pathol. 2022, 2022, 1–5. [Google Scholar] [CrossRef]
- Angius, F.; Ingianni, A.; Pompei, R. Human Herpesvirus 8 and Host-Cell Interaction: Long-Lasting Physiological Modifications, Inflammation and Related Chronic Diseases. Microorganisms 2020, 8, 388. [Google Scholar] [CrossRef]
- Carbone, A.; Cesarman, E.; Spina, M.; Gloghini, A.; Schulz, T.F. HIV-associated lymphomas and gamma-herpesviruses. Blood 2009, 113, 1213–1224. [Google Scholar] [CrossRef]
- Dupin, N.; Diss, T.L.; Kellam, P.; Tulliez, M.; Du, M.-Q.; Sicard, D.; Weiss, R.A.; Isaacson, P.G.; Boshoff, C. HHV-8 is associated with a plasmablastic variant of Castleman disease that is linked to HHV-8–positive plasmablastic lymphoma. Blood 2000, 95, 1406–1412. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Du, Z. Demographic characteristics and prognosis of HHV8-positive diffuse large B-cell lymphoma, not otherwise specified: Insights from a population-based study with a 10-year follow-up. Medicine 2023, 102, e36464. [Google Scholar] [CrossRef]
- Bharathi, V.; Farooq, A. Epidemiological insights and survival patterns of HHV8-positive diffuse large B-cell lymphoma (HDN) associated with multicentric Castleman disease (CD): A comprehensive analysis using SEER database. J. Clin. Oncol. 2024, 42, e19080. [Google Scholar] [CrossRef]
- van Rhee, F.; Fajgenbaum, D. Insights into the etiology of Castleman disease. Blood 2024, 143, 1789–1790. [Google Scholar] [CrossRef]
- Chadburn A, Cesarman E. KSHV/HHV8-associated multicentric Castleman disease. In: Ferry J, Naresh K, et al., eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 2022.
- Bower, M.; Carbone, A. KSHV/HHV8-Associated Lymphoproliferative Disorders: Lessons Learnt from People Living with HIV. Hemato 2021, 2, 703–712. [Google Scholar] [CrossRef]
- Fetica, B.; Pop, B.; Lisencu, C.; Rancea, A.C.; Coman, A.; Cucuianu, A.; Petrov, L. Castleman disease. A report of six cases. Med. Pharm. Rep. 2014, 87, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-W.; Pittaluga, S.; Jaffe, E.S. Multicentric Castleman disease: Where are we now? Semin. Diagn. Pathol. 2016, 33, 294–306. [Google Scholar] [CrossRef]
- The spectrum of Castleman’s disease: Mimics, radiologic pathologic correlation and role of imaging in patient management. Madan, Rachna et al. European Journal of Radiology, Volume 81, Issue 1, 123 - 131.
- Abdul-Rahman, I.S.; Al-Amri, A.M.; Ghallab, K.Q. Castleman's Disease: A Study of a Rare Lymphoproliferative Disorder in a University Hospital. Clin. Med. Blood Disord. 2009, 2, CMBD.S2161–36. [Google Scholar] [CrossRef]
- Venkateswaran, J. D., & Balakrishna, J., M. D. (2023, October 4). Lymph nodes-inflammatory / reactive disorders: Castleman Disease (E. Courville M. D. & R. N. Miranda M. D., Eds.). Lymph Nodes & Spleen, Nonlymphoma. Retrieved February 1, 2025, from https://www.pathologyoutlines.com/topic/lymphnodescastleman.html.
- Carbone, A.; Borok, M.; Damania, B.; Gloghini, A.; Polizzotto, M.N.; Jayanthan, R.K.; Fajgenbaum, D.C.; Bower, M. Castleman disease. Nat. Rev. Dis. Prim. 2021, 7, 84. [Google Scholar] [CrossRef]
- Bacon, C.M.; Miller, R.F.; Noursadeghi, M.; McNamara, C.; Du, M.; Dogan, A. Pathology of bone marrow in human herpes virus-8 (HHV8)-associated multicentric Castleman disease. Br. J. Haematol. 2004, 127, 585–591. [Google Scholar] [CrossRef]
- Bacon, C.M. , Miller, R.F., Noursadeghi, M., McNamara, C., Du, M.-Q. and Dogan, A. (2004), Pathology of bone marrow in human herpes virus-8 (HHV8)-associated multicentric Castleman disease. British Journal of Haematology, 127: 585-591. [CrossRef]
- Pria, A.D.; Pinato, D.; Roe, J.; Naresh, K.; Nelson, M.; Bower, M. Relapse of HHV8-positive multicentric Castleman disease following rituximab-based therapy in HIV-positive patients. Blood 2017, 129, 2143–2147. [Google Scholar] [CrossRef] [PubMed]
- Bower, M.; Newsom-Davis, T.; Naresh, K.; Merchant, S.; Lee, B.; Gazzard, B.; Stebbing, J.; Nelson, M. Clinical Features and Outcome in HIV-Associated Multicentric Castleman's Disease. J. Clin. Oncol. 2011, 29, 2481–2486. [Google Scholar] [CrossRef] [PubMed]
- Ramaswami, R.; Uldrick, T.S. Remission after rituximab for HHV8+ MCD: what next? Blood Adv. 2023, 7, 5661–5662. [Google Scholar] [CrossRef] [PubMed]
- Dunn, R.; Jariwal, R.; Venter, F.; Mishra, S.; Bhandohal, J.; Cobos, E.; Heidari, A. HHV-8-Associated Multicentric Castleman Disease, a Diagnostic Challenge in a Patient With Acquired Immunodeficiency Syndrome and Fever. J. Investig. Med. High Impact Case Rep. 2022, 10. [Google Scholar] [CrossRef]
- Bower, M. How I treat HIV-associated multicentric Castleman disease. Blood 2010, 116, 4415–4421. [Google Scholar] [CrossRef]
- Bacha, D.; Chelly, B.; Kilani, H.; Charfi, L.; Douggaz, A.; Chatti, S.; Chelbi, E. HHV8/EBV Coinfection Lymphoproliferative Disorder: Rare Entity with a Favorable Outcome. Case Rep. Hematol. 2017, 2017, 1578429. [Google Scholar] [CrossRef]
- Guerrero, C.; Jain, T.; Kelemen, K. HHV-8-Associated Lymphoproliferative Disorders and Pathogenesis in an HIV-Positive Patient. Case Rep. Hematol. 2019, 2019, 1–6. [Google Scholar] [CrossRef]
- Wu, D.; Lim, M.S.; Jaffe, E.S. Pathology of Castleman Disease. Hematol. Clin. North Am. 2018, 32, 37–52. [Google Scholar] [CrossRef]
- SEER Hematopoietic and Lymphoid Neoplasm Database. SEER. https://seer.cancer.gov/seertools/hemelymph/5a7d9f261ef55751804cbb20/.
- Castleman Disease Collaborative Network. Treatment - CDCN. CDCN. Published January 22, 2025. https://cdcn.org/treatment/.
- Du, M.-Q.; Diss, T.C.; Liu, H.; Ye, H.; Hamoudi, R.A.; Cabeçadas, J.; Dong, H.Y.; Harris, N.L.; Chan, J.K.C.; Rees, J.W.; et al. KSHV- and EBV-associated germinotropic lymphoproliferative disorder. Blood 2002, 100, 3415–3418. [Google Scholar] [CrossRef]
- Cesarman, E.; Chadburn, A.; Rubinstein, P.G. KSHV/HHV8-mediated hematologic diseases. Blood 2022, 139, 1013–1025. [Google Scholar] [CrossRef]
- Bhavsar, T.; Lee, J.C.; Perner, Y.; Raffeld, M.; Xi, L.; Pittaluga, S.; Jaffe, E.S. KSHV-associated and EBV-associated Germinotropic Lymphoproliferative Disorder. Am. J. Surg. Pathol. 2017, 41, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Ghoneima, A.; Cooke, J.; Shaw, E.; Agrawal, A. Human herpes virus 8-positive germinotropic lymphoproliferative disorder: first case diagnosed in the UK, literature review and discussion of treatment options. BMJ Case Rep. 2020, 13, e231640. [Google Scholar] [CrossRef] [PubMed]
- Dpt MALP. Diffuse large B-Cell lymphoma diagnosis. Rare Disease Advisor. Published October 4, 2022. https://www.rarediseaseadvisor.com/disease-info-pages/diffuse-large-b-cell-lymphoma-diagnosis/.
- HHV8 positive DLBCL, NOS. https://www.pathologyoutlines.com/topic/lymphomaHHV8DLBCL.html.
- Gonçalves, P.H.; Uldrick, T.S.; Yarchoan, R. HIV-associated Kaposi sarcoma and related diseases. AIDS 2017, 31, 1903–1916. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Xiao, P. Primary Effusion Lymphoma. Arch. Pathol. Lab. Med. 2013, 137, 1152–1154. [Google Scholar] [CrossRef]
- Kim, Y.; Leventaki, V.; Bhaijee, F.; Jackson, C.C.; Medeiros, L.J.; Vega, F. Extracavitary/solid variant of primary effusion lymphoma. Ann. Diagn. Pathol. 2012, 16, 441–446. [Google Scholar] [CrossRef]
- Carbone, A.; Gloghini, A. PEL and HHV8-unrelated effusion lymphomas. Cancer 2008, 114, 225–227. [Google Scholar] [CrossRef]
- Chen, Y.-B.; Rahemtullah, A.; Hochberg, E. Primary Effusion Lymphoma. Oncol. 2007, 12, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Goto, H.; Yotsumoto, M. Current status of treatment for primary effusion lymphoma. Intractable Rare Dis. Res. 2014, 3, 65–74. [Google Scholar] [CrossRef]
- Guillet, S.; Gérard, L.; Meignin, V.; Agbalika, F.; Cuccini, W.; Denis, B.; Katlama, C.; Galicier, L.; Oksenhendler, E. Classic and extracavitary primary effusion lymphoma in 51 HIV-infected patients from a single institution. Am. J. Hematol. 2016, 91, 233–237. [Google Scholar] [CrossRef]
- Primary effusion lymphoma. (n.d.). https://www.pathologyoutlines.com/topic/lymphomaeffusion.html.
- Panaampon, J.; Okada, S. Promising immunotherapeutic approaches for primary effusion lymphoma. Explor. Target. Anti-tumor Ther. 2024, 5, 699–713. [Google Scholar] [CrossRef]
- Hayashino, K.; Meguri, Y.; Yukawa, R.; Komura, A.; Nakamura, M.; Yoshida, C.; Yamamoto, K.; Oda, W.; Imajo, K. Primary Effusion Lymphoma-like Lymphoma Mimicking Tuberculous Pleural Effusion: Three Case Reports and a Literature Review. Intern. Med. 2023, 62, 2531–2537. [Google Scholar] [CrossRef] [PubMed]
- de Goes, V.A.; Cortez, A.C.; Morbeck, D.L.; Costa, F.D.; da Silveira, T.B. The role of autologous bone marrow transplantation in primary effusion lymphoma: a case report and literature review. Hematol. Transfus. Cell Ther. 2024, 46, S316–S321. [Google Scholar] [CrossRef] [PubMed]
- Narkhede, M.; Arora, S.; Ujjani, C. Primary effusion lymphoma: current perspectives. OncoTargets Ther. 2018, 11, 3747–3754. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
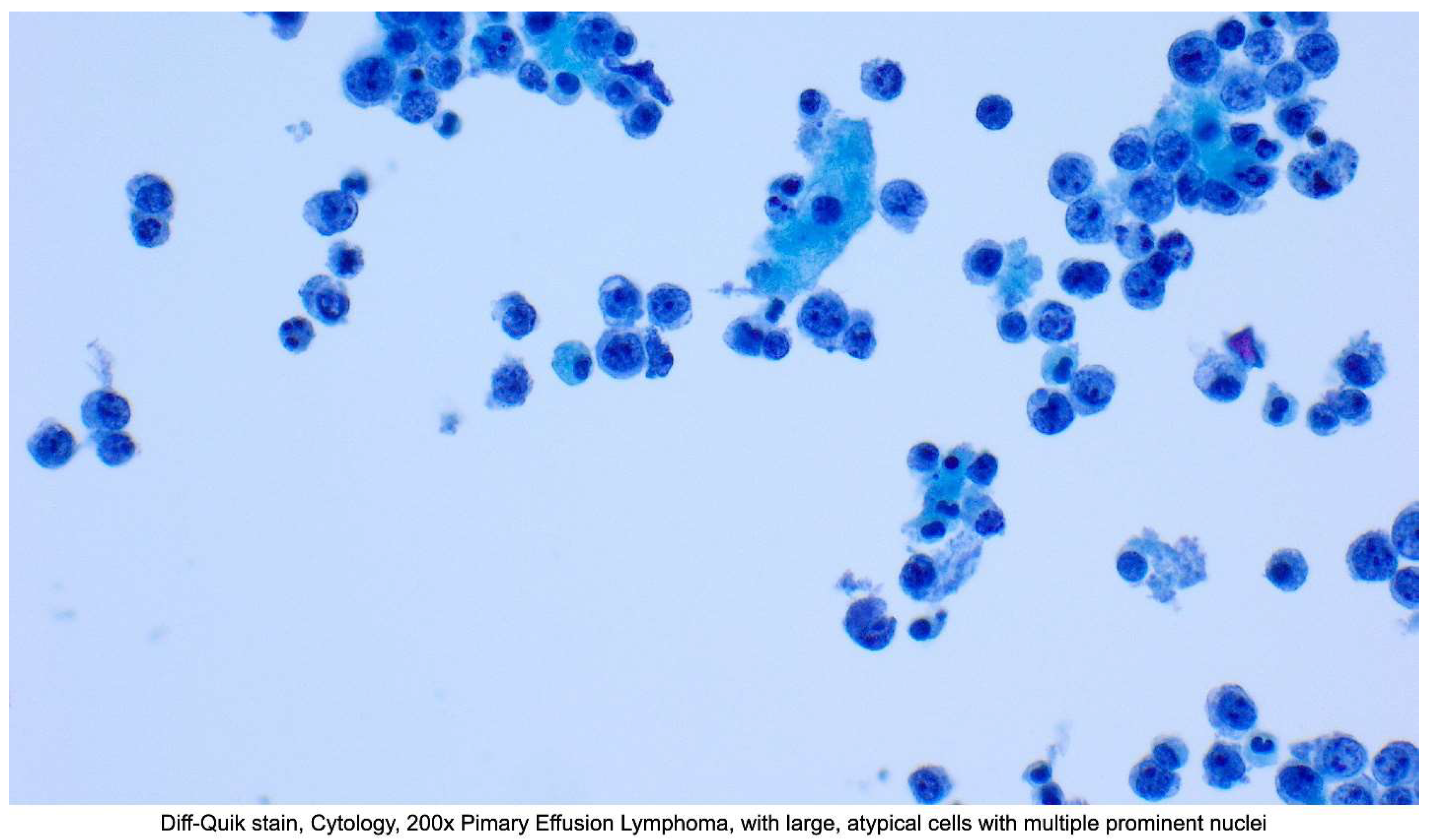
Figure 2.
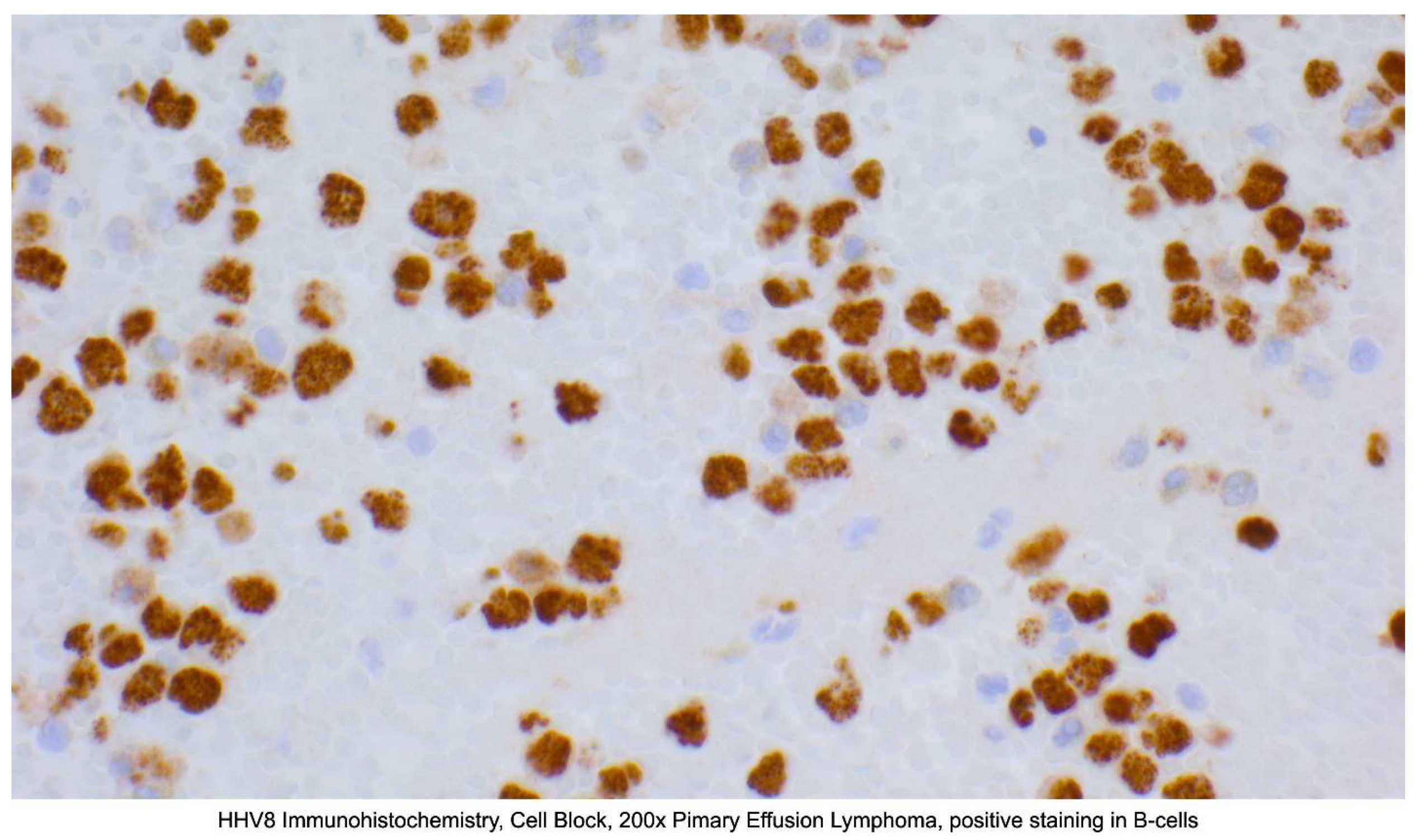
Figure 3.
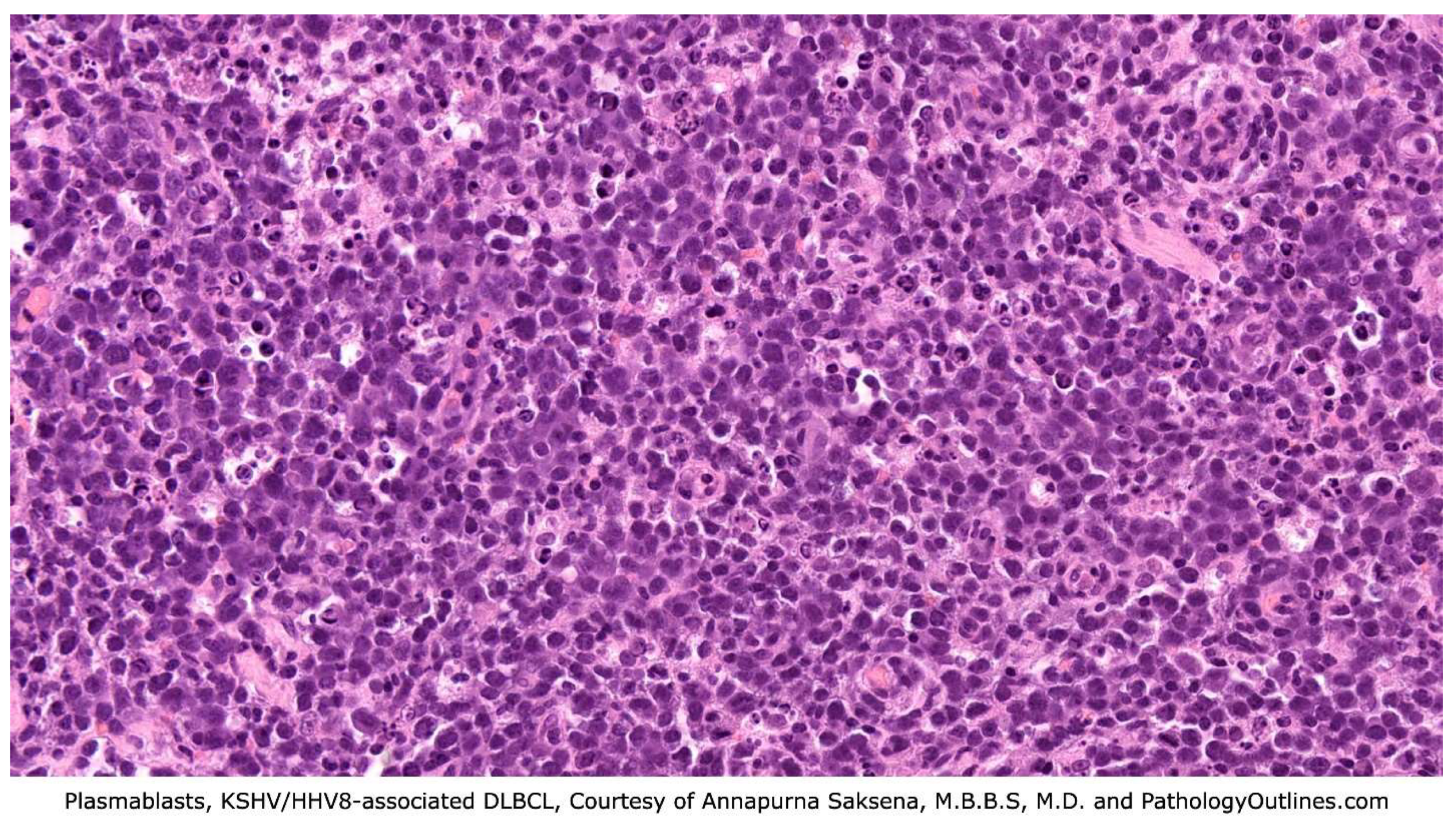
Figure 4.
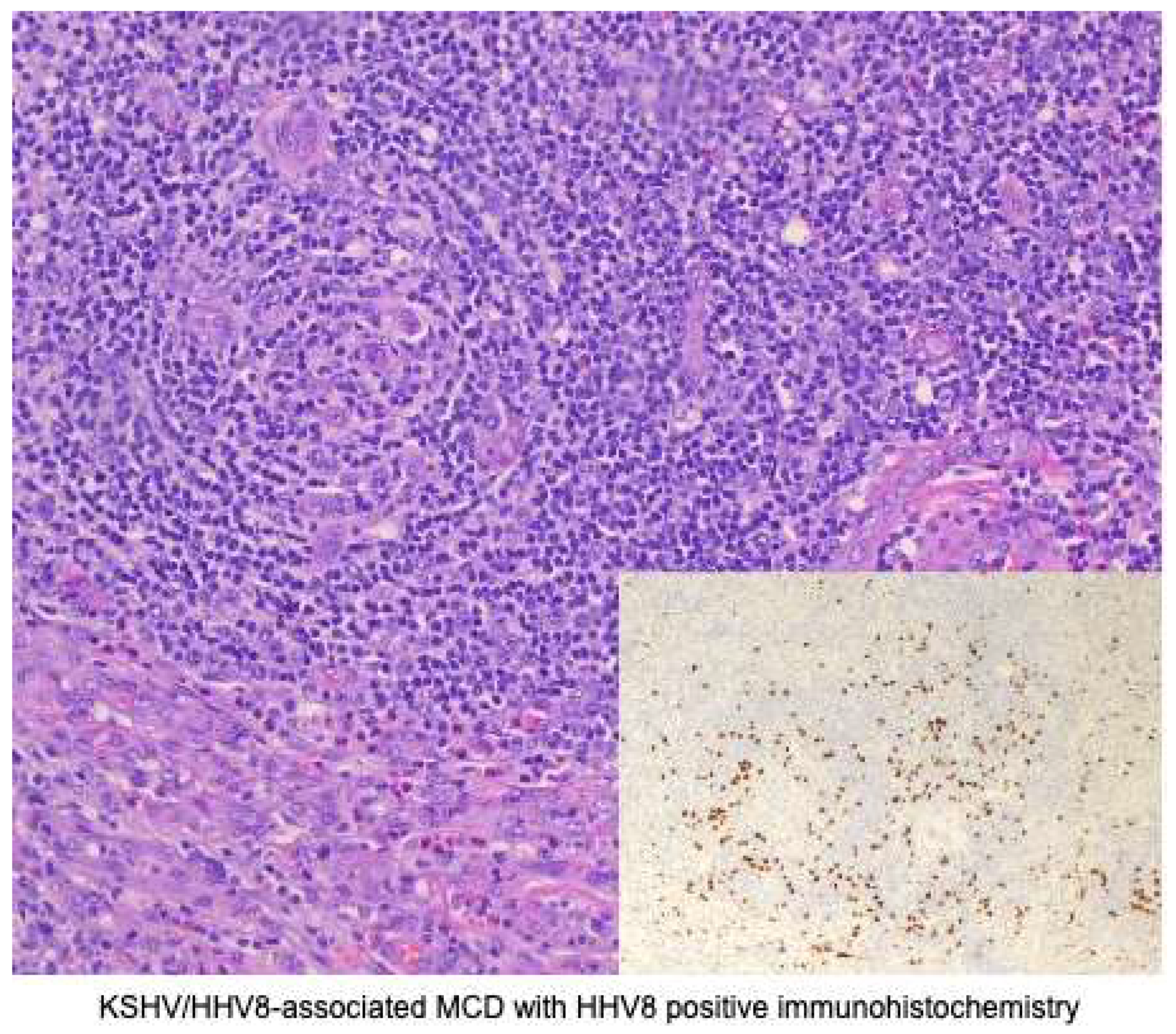
Figure 5.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



