
Submitted:
16 April 2025
Posted:
17 April 2025
You are already at the latest version
Abstract
Background/ aim:
Fibrillary glomerulonephritis (FGN) is a rare glomerular disease characterized by non-amyloid fibrillary deposits in the glomeruli, and positive staining for DNAJB9.
We aimed to investigate the clinical, pathological characteristics and the outcomes of FGN patients from a tertiary nephrology center.
Methods:
Retrospective cohort study of eleven patients with a diagnosis of FGN between 2016 and 2025.
Results:
At diagnosis, nine patients had nephrotic-range proteinuria and eight patients had microscopic haematuria. The mean serum creatinine was 1.6 mg/dL and the mean proteinuria was 3.78 g/24h. In terms of comorbidities, one patient had systemic lupus erythematosus (SLE), one had sarcoidosis and one had lung cancer. Histologically, the most common pattern was a mesangial proliferative pattern, observed in six patients. DNAJB9 staining was performed in 5 patients and was strongly positive. 10 out of 11 patients were positive for both IgG immunofluorescence and C3. All patients received renin-angiotensin-aldosterone system blockade and immunosuppression. In 80% of patients, proteinuria was reduced by more than 50% with stable renal function. Partial remission was observed in 73% of patients with a median follow-up of 24 months.
Conclusion:
The disease is very heterogeneous and associated with co-morbidities. There is no standard therapy and outcomes are questionable.
Keywords:
DNAJB9
; fibrillary glomerulonephritis
; kidney biopsy
1. Introduction
The term "fibrillary glomerulonephritis" (FGN) was first introduced in 1987 by Alpers et al [1] to describe a distinct entity of unknown aetiology. It was defined by the presence of randomly arranged non-amyloid fibrillar deposits 14-24 nm in diameter in the glomeruli, negative for Congo red staining and positive by immunofluorescence for IgG, C3 and both kappa and lambda light chains [2]. FGN has been observed in 0.4-1.4% [3,4] of kidney biopsies, predominantly affecting individuals aged 49-60 years, and is more common in white women [3,5].
FGN has been associated with a number of diseases, including autoimmune diseases (affecting 13-30% of patients) [3], cancers [6] and viral infections (hepatitis C 3% to 27% in non-whites) [7].
FGN is characterised by the manifestation of nephrotic syndrome (characterised by proteinuria, haematuria, renal insufficiency and hypertension) with rapidly progressive glomerulonephritis due to a crescentic phenotype in some patients [3,7]. Serum cryoglobulins and rheumatoid factor are often negative and hypocomplementemia is rare. Hypogammaglobulinemia is usually absent and serum IgG subclass levels are normal [3,7].
FGN typically presents with six distinct histological patterns on light microscopy. These patterns correlate with clinical features and prognosis and include a) mesangial proliferative, b) membranoproliferative, c) endocapillary hyperplasia, d) crescentic, e) membranous and f) diffuse sclerosis patterns. Mesangial proliferative is the most common, occurring in 21-78% of cases [5,8,9].
In 2018, two independent research teams from the Mayo Clinic and the University of Washington discovered the presence of DNAJB9 at high levels in the glomerular proteome [10,11,12]. The Mayo Clinic study showed that DNAJB9 was present in all 24 cases of FGN, whereas its absence was observed in 145 cases of amyloidosis, 72 cases of other GN and the 12 healthy controls. Co-localisation of DNAJB9 and IgG was also found in the mesangium and glomerular basement membrane, with DNAJB9 also found in FGN fibrils, but not in fibrils associated with amyloidosis or immunotactoid GN [11,12]. Similar results were documented by the University of Washington, where DNAJB9 was identified in all 11 glomeruli of patients with FGN, but was absent in the biopsy specimens of 31 patients without FGN [10].
These studies showed that DNAJB9 is one of the most abundant glomerular proteins in FGN patients. Its absence in biopsies from patients with amyloidosis, other GN or healthy controls established DNAJB9 as the first biomarker for FGN, allowing differentiation from amyloidosis and other glomerular diseases [13,14]. The role of DNAJB9 in the pathogenesis of FGN remains to be fully elucidated and further research is needed to determine whether it plays a causative role or is merely a marker of the disease [15].
As current treatment options remain largely empirical with variable efficacy, understanding the molecular mechanisms underlying FGN is critical for the development of targeted therapies [3].
As FGN is an uncommon disorder of unclear aetiology and low prevalence, we aimed to present our clinical experience of FGN from a tertiary nephrology centre combined with the expertise and knowledge of a referral pathology team. The study will attempt to integrate our current experience to address gaps in understanding of various aspects of the disease, improve management of the disease and contribute to data in the field. It is also hoped that the study will identify future directions for the unclear pathophysiology.
2. Methods
In this retrospective cohort study, a total of eleven patients (six female and five male) diagnosed with fibrillary glomerulonephritis (FGN) at the Nephrology Clinic of G. Gennimatas Hospital between 2016 and 2025 were evaluated. The study was approved by the Medical Scientific Review Board of GNA "G. Gennimatas" (protocol number 369; date of approval 5 January 2024).
2.1. Patients
Patients Inclusion criteria:
Patients who met the diagnostic criteria for FGN were included in the study.
The diagnosis of FGN is documented by the presence of at least one of the following criteria in renal biopsy specimens
1. Deposition of fibrils in the glomerulus by light electron microscopy. The fibrils must have the following characteristics:
a. Random arrangement
b. Diameter of 14 to 24 nm
c. Deposition in the mesangium and the glomerular basement membrane
2. Expression of DNAJB9 in immunohistochemistry
Eleven patients were included. Patient data extracted from medical records included demographic data [sex, race, age at diagnosis, follow-up period] and clinical data included the clinical presentation of the disease, including the presence of haematuria on urinalysis, proteinuria (>300 mg/24 h urine collection), nephrotic-range proteinuria (>3 g/24 h urine collection) and hypertension, defined as systolic blood pressure >140 mmHg, diastolic blood pressure >90 mmHg or the use of antihypertensive medication. A medical history was taken, focusing on possible comorbidities related to FGN.
Laboratory data collected at the time of diagnosis included the following: blood test values for urea, creatinine, albumin and total serum protein; urine test results (24-hour urine protein excretion and urinalysis); and immunological markers. ANCA, ANA and anti-dsDNA antibodies; serum and urine protein immunoelectrophoresis; and screening for hepatitis B, hepatitis C and HIV. Follow-up laboratory data included final recorded values for urea, creatinine and 24-hour urine protein excretion.
The renal biopsy findings which were subjected to assessment included the following: - the histological pattern, as revealed by light microscopy; - the degree of glomerulosclerosis, as determined by light microscopy; - the degree of interstitial fibrosis and tubular atrophy, as determined by light microscopy; - the results of immunofluorescence analysis; - fibril morphology, as determined by electron microscopy; - the expression of DNAJB9; and - Congo Red staining.
Each patient's treatment regimen and response to therapy were documented using renal function markers. Renal dysfunction was defined as serum creatinine >1.2 mg/dL and proteinuria as urinary protein excretion >300 mg over 24 hours.
Therapeutic response was classified as follows:
- Complete response (CR): proteinuria <0.5 g/day and normal renal function (serum creatinine <1.2 mg/dL)
- Partial response (PR): reduction of proteinuria by >50% from the peak recorded value, stable renal function
- Persistent renal dysfunction (PRD): failure to meet the criteria for CR or PR, or worsening renal function without progression to ESRD
- ESRD: eGFR <15 mL/min.
Data analysis was performed using IBM SPSS Statistics 17.0.
3. Results
3.1. Demographic, Clinical Features and Laboratory Data
The study included eleven patients, six females and five males, with a median age at diagnosis of 58 years (IQR: 50.5 – 62). All patients had proteinuria at renal biopsy, with a median 24-hour urine protein of 3.78 g (range: 3.2 - 5 g) and a median serum creatinine of 1.6 mg/dL (range: 1 - 2 mg/dL). At the time of diagnosis, nine patients (81%) had nephrotic-range proteinuria without full-blown nephrotic syndrome and eight patients had microscopic haematuria. Three patients had macroscopic haematuria without hypoalbuminemia and one patient had peripheral oedema. Hyperlipidaemia was observed in six patients.
Regarding the presence of comorbidities in the FGN patients, one patient had been diagnosed with systemic lupus erythematosus (SLE), one with sarcoidosis, and another with rheumatological symptoms without a confirmed diagnosis. Four patients were diagnosed with hypothyroidism and one patient was diagnosed with lung cancer two months before the diagnosis of FGN. All patients tested negative for HIV, HBV and HCV, and imaging studies were also negative.
Table 1 shows the demographic and clinical characteristics of patients with a diagnosis of FGN confirmed by renal biopsy. It should be noted that two patients were diagnosed after repeat biopsy.
All patients were tested for paraproteinemia. In one patient (patient 8), a bone marrow biopsy was performed because of the predominance of λ light chains on immunofluorescence despite negative serum and urine immunoelectrophoresis. The results showed a polyclonal plasma cell population with a κ/λ ratio of approximately 5/2. This finding is consistent with a diagnosis of plasma cell dyscrasia type MGUS. Six of the 11 patients were ANA positive.
3.2. Pathology Findings
Renal biopsy results showed that six patients had a mesangial proliferative pattern under light microscopy. Two patients had crescents and necrosis involving at least 50% of the glomeruli, consistent with crescentic glomerulonephritis. Another patient had sclerosing changes, another had a membranoproliferative pattern, another had a membranous pattern, and one patient had both mesangial hyperplasia and a membranoproliferative pattern , Figure 1. The mean percentage of globally sclerotic glomeruli was 37%. Crescents were reported in three cases, with interstitial fibrosis and tubular atrophy ranging from 20% to 35% (avg. 26%). Congo Red staining was negative.
Immunofluorescence results showed that 10 out of 11 cases were positive for IgG, with a mean intensity of 1.6+ (on a scale of 0 - 3+). The positivity was mainly observed in the mesangium and glomerular basement membrane and manifested as a granular or linear pattern. In addition, 27% of cases were positive for IgA (mean intensity 1.5+), while 45% were positive for IgM (mean intensity 1.5+). Glomerular C3 deposition was detected in 10 out of 11 patients (mean intensity 2.3+), while C1q was found in only two patients (mean intensity 1.5+). Glomerular deposits were positive for κ (three patients) and λ chains (six patients). Three patients had both light chains and three others had only the λ chain. Further testing (SPEP/IFE) revealed monoclonal IgG-λ in one patient.
Electron microscopy was performed on 10 of the 11 renal biopsies. The fibrils showed a random orientation, with a linear and unbranched configuration, an average size of 10 to 20 nanometres, and a predominant distribution within the mesangium and glomerular basement membrane, Figure 2. Fusion of the foot processes was observed in 8 of the 10 cases examined by electron microscopy.
Immunohistochemistry for DNAJB9 was performed in five patients and showed intense positivity in the mesangium and in two cases also in the glomerular basement membrane, Figure 3. In one patient (patient 3), the diagnosis of FGN was made on the basis of positivity for the marker, as electron microscopy was not available.
The histological findings for each patient are summarized in Table 2.
3.3. Treatment and Clinical Outcome
All patients received a renin-angiotensin-aldosterone axis inhibitor and immunosuppression.
Table 3 summarizes the therapeutic regimens and clinical responses of the patients. One patient was initially diagnosed with lupus nephritis and was diagnosed with FGN on repeat renal biopsy. Immunosuppressive therapy (glucocorticoids, cyclophosphamide and MMF) was administered for the treatment of lupus nephritis, which was subsequently discontinued following the diagnosis of FGN. The majority of patients received corticosteroids for six months and two doses of rituximab (1 g at 15-day intervals), with a repeat dose administered six months later. One patient (patient 2) received cyclophosphamide because of uncontrolled diabetes, while two patients (patients 10 and 11) received cyclophosphamide and rituximab because of rapidly progressive glomerulonephritis and the presence of crescents in ≥50% of the glomeruli. In another patient (patient 9), non-response to multiple repeated cycles of rituximab led to a change in treatment to mycophenolate mofetil (MMF).
The median duration of follow-up was 24 months (interquartile range: 9-42), during which time 73% of patients had a partial remission, 9% had a complete remission and 18% had persistent renal dysfunction.
In this study, the majority (eight out of eleven patients, 80%) of patients showed a reduction in proteinuria of more than 50% with stable renal function and partial remission. One patient (patient 5) showed complete remission after a total of four doses of rituximab administered every six months, resulting in normal renal function without proteinuria or haematuria. A significant decrease in proteinuria was observed after the first dose of rituximab (from 590 mg to 2.9 g/24 h).
Two patients (8 and 9) had persistent renal dysfunction. One of these patients died from complications of SARS-CoV-2 infection, while the other showed a significant reduction in proteinuria after treatment with both rituximab and MMF. However, due to an allergic reaction to rituximab and gastrointestinal discomfort from MMF, the patient discontinued immunosuppressive treatment, resulting in further worsening of proteinuria.
The vast majority of patients showed a significant reduction in proteinuria, with mean proteinuria reaching 1.9 g/24h after treatment compared to 4.6 g/24h at diagnosis. The mean serum creatinine level at the end of treatment was 1.4 mg/dL, the same as at diagnosis, so the maintenance of stable levels should not be interpreted as a diminished patient response. The mean rate of improvement in serum creatinine between pre- and post-treatment was 5.33%.
4. Discussion
Eleven patients diagnosed with FGN at our centre between 2016 and 2025 are included in this study. The clinical and histological characteristics appear to be consistent with those described in large retrospective studies. Most patients have nephrotic-range proteinuria without nephrotic syndrome at diagnosis, and the mesangial proliferative histological pattern predominates in patients.
Currently, there is no preferred treatment or established therapeutic protocol for the treatment of FGN, and there are no randomized clinical trials to guide therapeutic decisions [16]. Given the potential autoimmune nature of FGN, rituximab, a monoclonal antibody targeting CD20 on B lymphocytes, has been tested and is considered one of the main therapeutic options for FGN. The first and only prospective study on the use of rituximab in FGN patients was conducted by Erickson et al. in 2021 [17]. Drugs such as corticosteroids, cyclophosphamide, cyclosporine, mycophenolate, azathioprine, lenalidomide and sirolimus have also been shown to have no significant therapeutic benefit [5,8,16].
Until recently, the diagnosis of FGN was based on electron microscopy. However, this approach has evolved significantly since the identification of the DNAJB9 marker [13,18]. Despite the diagnostic limitations imposed by the inability to use electron microscopy, FGN was diagnosed using DNAJB9 staining. In a large clinical study that included biopsies from 84 patients with FGN [12], 21 with amyloidosis, 98 with other glomerular diseases and 11 healthy controls, staining for DNAJB9 was found to have a sensitivity of 98% and a specificity of 99% for the diagnosis of FGN. Recent studies had shown that the positive expression of the DNAJB9 marker is considered to be highly specific for FGN, as further data emerged [13,14].
DNAJB9, also known as ERdj4 or Mdg-1, is a member of the DNAJ protein family, which acts as a co-chaperone for the HSP70 heat shock protein family [19]. DNAJB9 assists in protein folding and degradation within the endoplasmic reticulum and is found at low levels in most body cells, with higher expression in tissues such as the liver and placenta [12,15]. In the kidney, DNAJB9 is present in tubular cells, podocytes, epithelial cells and endothelial cells [15].
The role of DNAJB9 in the pathogenesis of FGN is still under investigation [15]. It has been hypothesised that DNAJB9 acts as an autoantigen, triggering an autoimmune response when misfolded proteins accumulate in the glomerulus [20]. However, no circulating autoantibodies to DNAJB9 have been detected. An alternative hypothesis is that DNAJB9 binds to misfolded IgG and contributes to fibril formation [15]. Despite these hypotheses, many questions remain, including the formation of fibrils and why immunosuppressive therapy is not as successful.
Possible next steps in understanding the pathogenesis of FGN include the study of circulating levels of DNAJB9 and its antibodies and the possible development of animal models. Measuring plasma levels of DNAJB9 had been performed, according to which patients with FGN have higher levels of DNAJB9 compared to controls [21], possibly due to increased production and decreased clearance [21]. However, before potential clinical use, these initial findings need to be confirmed, studies in patients with preserved renal function and the development of simpler techniques.
This study described two rare cases of FGN associated with sarcoidosis and SLE. The rarity of documented cases of these co-morbidities in the available literature supports the hypothesis that these diseases and FGN are related and further research is recommended. One case involves a woman diagnosed with lupus nephritis and FGN. Fibrils in patients with FGN associated with SLE are significantly smaller, ranging in size from 8 to 15 nm, and may have a "fingerprint" pattern. A retrospective study of 185 lupus nephritis biopsies found these deposits in 17.3% of patients, but only 1% had fibrils resembling FGN [22]. The association of FGN with SLE has been sparsely documented in the literature. Examples included: a 12-year-old with class IV lupus nephritis who was diagnosed with FGN on repeat biopsy [23], a 28-year-old man with SLE and thrombotic microangiopathy [TMA] who was also diagnosed with FGN [24], and in a study of 66 FGN patients, two cases of SLE but no histologically confirmed lupus nephritis [5]. Rosenstock et al. reported one case of SLE in 67 FGN patients, none of whom had features of lupus nephritis [2]. A recently published case report described a woman who has had SLE for 20 years. The patient presented with proteinuria and a biopsy revealed FGN with DNAJB9 positivity [25]. Consequently, the association between GN and SLE remains poorly documented, with limited data available on their clinical course, therapeutic interventions and prognosis.
In addition, one patient was diagnosed with sarcoidosis immediately after the diagnosis of FGN and during the investigation of secondary causes. Kidney damage in patients with sarcoidosis is rare [10-20%] and is usually the result of calcium homeostasis disorders, manifesting as nephrolithiasis and nephrocalcinosis, and the rarer granulomatous tubulointerstitial nephritis. The literature on glomerular damage in patients with sarcoidosis is limited, and the pattern of glomerular damage is variable [26]. Cases of sarcoidosis associated with membranous nephropathy, minimal change disease, focal segmental glomerulosclerosis and IgA nephropathy have been documented [26]. The association of FGN with sarcoidosis is rare, with only one documented case in the literature to date. This particular study was retrospective and looked at cases of FGN associated with the expression of the DNAJB9 marker. The study reported two cases of sarcoidosis associated with FGN, and these cases were in the context of autoimmune diseases that may be associated with FGN [7].
5. Conclusions
FGN is a rare glomerular disease that presents with a variety of clinical and histological features, making the differential diagnosis complex. The identification of DNAJB9 as a precise and specific marker for FGN has fundamentally altered the diagnostic process for the disease, thereby decreasing the reliance on electron microscopy. Despite these advances, effective treatment options for FGN remain limited. Our experience as a tertiary nephrology centre highlights the evolving landscape of FGN management and emphasizes the importance of individualized treatment strategies.
Author Contributions
TP: Conceptualization, wrote manuscript, editing data curation, analysis. MD, Conceptualization, wrote manuscript. AF, MS, review and editing data curation, editing of the manuscript. PL Supervision, Contributed to data collection and analysis, validation. GL Contributed to data analysis and validation. GM supervision, review and editing data analysis.
Funding
no external funding.
Institutional Review Board Statement
The study was conducted in accordance with ethical regulations and the principles of the Declaration of Helsinki and with the approval of the Medical and Scientific Review Board of the G. Gennimatas Hospital (protocol number 369 - 5/1/2024).
Informed Consent Statement
Written informed consent was obtained from each patient.
Data Availability Statement
Upon request, the corresponding author can provide the data sets used and analysed in this study.
Acknowledgments
We would like to acknowledge and gratefully thank all the nephrologists and other clinicians, nurses and other members of the Department of Nephrology of Genimatas Hospital, the 1st Nephrology Clinic of Aristotle University Medical School and the Pathology Department of the Medical School of National and Kapodistrian University of Athens who were involved in the care of the patients but are not listed as authors. We would also like to express our gratitude to all the patients who consented to participate in the study and to the families who provided them with support.
Conflicts of Interest
The authors declare that they have no competing interests.
References
- Alpers, C.E.; Rennke, H.G.; Hopper, J.; Biava, CG. Fibrillary glomerulonephritis: an entity with unusual immunofluorescence features. Kidney Int. 1987, 31, 781–789. [Google Scholar] [CrossRef]
- Rosenstock, J.L.; Markowitz, G.S.; Valeri, A.M. Fibrillary and immunotactoid glomerulonephritis: Distinct entities with different clinical and pathologic features. Kidney Int. 2003, 63, 1450–1461. [Google Scholar] [CrossRef] [PubMed]
- Rosenstock, J.L.; Markowitz, G.S. Fibrillary Glomerulonephritis: An Update. Kidney Int Rep. 2019, 4, 917–922. [Google Scholar] [CrossRef] [PubMed]
- O’Shaughnessy, M.M.; Hogan, S.L.; Poulton, C.J.; et al. Temporal and Demographic Trends in Glomerular Disease Epidemiology in the Southeastern United States, 1986-2015. Clin J Am Soc Nephrol. 2017, 12, 614–623. [Google Scholar] [CrossRef]
- Nasr, S.H.; Valeri, A.M.; Cornell, L.D.; et al. Fibrillary glomerulonephritis: a report of 66 cases from a single institution. Clin J Am Soc Nephrol. 2011, 6, 775–784. [Google Scholar] [CrossRef]
- Normand, G.; Jolivot, A.; Rabeyrin, M.; et al. Paraneoplastic fibrillary glomerulonephritis associated with intrahepatic cholangiocarcinoma: When diagnosis of a rare kidney disease leads to successful hepatic cancer treatment. Clin Res Hepatol Gastroenterol. 2017, 41, e8–11. [Google Scholar] [CrossRef]
- Andeen, N.K.; Troxell, M.L.; Riazy, M.; et al. Fibrillary Glomerulonephritis: Clinicopathologic Features and Atypical Cases from a Multi-Institutional Cohort. Clin J Am Soc Nephrol. 2019, 6, 1741–1750. [Google Scholar] [CrossRef] [PubMed]
- Javaugue, V.; Karras, A.; Glowacki, F.; et al. Long-term kidney disease outcomes in fibrillary glomerulonephritis: a case series of 27 patients. Am J Kidney Dis. 2013, 62, 679–690. [Google Scholar] [CrossRef]
- Marinaki, S.; Tsiakas, S.; Liapis, G.; et al. Clinicopathologic features and treatment outcomes of patients with fibrillary glomerulonephritis: A case series. Medicine 2021, 100, e26022. [Google Scholar] [CrossRef] [PubMed]
- Andeen, N.K.; Yang, H.Y.; Dai, D.F.; et al. Homolog Subfamily B Member 9 Is a Putative Autoantigen in Fibrillary GN. J Am Soc Nephrol. 2018, 29, 231–239. [Google Scholar] [CrossRef]
- Dasari, S.; Alexander, M.P.; Vrana, J.A.; et al. DnaJ Heat Shock Protein Family B Member 9 Is a Novel Biomarker for Fibrillary GN. J Am Soc Nephrol. 2018, 29, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Nasr, S.H.; Vrana, J.A.; Dasari, S.; et al. DNAJB9 Is a Specific Immunohistochemical Marker for Fibrillary Glomerulonephritis. Kidney Int Rep. 2018, 3, 56–64. [Google Scholar] [CrossRef]
- Liang, S.; Chen, D.; Liang, D.; et al. Clinicopathological characteristics and outcome of patients with fibrillary glomerulonephritis: DNAJB9 is a valuable histologic marker. J Nephrol. 2021, 34, 883–892. [Google Scholar] [CrossRef]
- Gambella, A.; Pitino, C.; Barreca, A.; et al. DNAJB9 Is a Reliable Immunohistochemical Marker of Fibrillary Glomerulonephritis: Evaluation of Diagnostic Efficacy in a Large Series of Kidney Biopsies. Biomedicines. 2022, 27, 2102. [Google Scholar] [CrossRef]
- Daverkausen-Fischer, L.; Pröls, F. The function of the co-chaperone ERdj4 in diverse (patho-)physiological conditions. Cell Mol Life Sci. 2021, 79, 9. [Google Scholar] [CrossRef]
- 16 Attieh, R.M.; Yang, Y.; Rosenstock, J.L. Updates on the Diagnosis and Management of Fibrillary Glomerulonephritis. Adv Kidney Dis Health. 2024, 31, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Erickson, S.B.; Zand, L.; Nasr, S.H.; et al. Treatment of fibrillary glomerulonephritis with rituximab: a 12-month pilot study. Nephrol Dial Transplant. 2021, 36, 104–110. [Google Scholar] [CrossRef]
- Sabanis, N.; Liaveri, P.; Geladari, V.; et al. DNAJB9 Fibrillary Glomerulonephritis With Membranous-Like Pattern: A Case-Based Literature Review. Cureus. 2023, 28, e47862. [Google Scholar] [CrossRef] [PubMed]
- Behnke, J.; Mann, M.J.; Scruggs, F.L. Members of the Hsp70 Family Recognize distinct types of sequences to execute ER quality control. Mol cell. 2016, 1, 739–752. [Google Scholar] [CrossRef]
- Andeen, N.K.; Yang, H.Y.; Dai, D.F.; et al. DnaJ Homolog Subfamily B Member 9 Is a Putative Autoantigen in Fibrillary GN. J Am Soc Nephrol. 2018, 29, 231–239. [Google Scholar] [CrossRef]
- Nasr, S.H.; Dasari, S.; Lieske, J.C.; et al. Serum levels of DNAJB9 are elevated in fibrillary glomerulonephritis patients. Kidney Int. 2019, 95, 1269–1272. [Google Scholar] [CrossRef] [PubMed]
- Hvala, A.; Kobenter, T.; Ferluga, D. Fingerprint and other organised deposits in lupus nephritis. Wien Klin Wochenschr. 2000, 112, 711–715. [Google Scholar] [PubMed]
- Menon, S.; Zeng, X.; Valentini, R. Fibrillary glomerulonephritis and renal failure in a child with systemic lupus erythematosus. Pediatr Nephrol. 2009, 24, 1577–1581. [Google Scholar] [CrossRef]
- Bhat, Z.Y.; Zeng, X.; Hingorani, J. Fibrillary glomerulonephritis in a patient with systemic lupus erythematosus: a rare association. Int Urol Nephrol. 2013, 45, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Whelband, M.C.; Willingham, T.; Thirunavukkarasu, S. Fibrillar glomerulonephritis in a patient with systemic lupus erythematosus with no evidence of lupus nephritis. BMJ Case Rep. 2023, 16, e253388. [Google Scholar] [CrossRef]
- Stehlé, T.; Joly, D.; Vanhille, P.; et al. Clinicopathological study of glomerular diseases associated with sarcoidosis: a multicenter study. Orphanet J Rare Dis. 2013, 30, 65. [Google Scholar] [CrossRef]
Figure 1.
Patterns on light microscopy. (A) Diffuse sclerosing pattern (patient 1 – H&Ε x400). (B) Mesangial proliferative pattern (patient 4 – H&Ε x400). (C) Mesangial proliferative pattern (patient 5 – H&Ε x400). (D) Membranous pattetn (patient 7 – H&Ε x400).
Figure 1.
Patterns on light microscopy. (A) Diffuse sclerosing pattern (patient 1 – H&Ε x400). (B) Mesangial proliferative pattern (patient 4 – H&Ε x400). (C) Mesangial proliferative pattern (patient 5 – H&Ε x400). (D) Membranous pattetn (patient 7 – H&Ε x400).
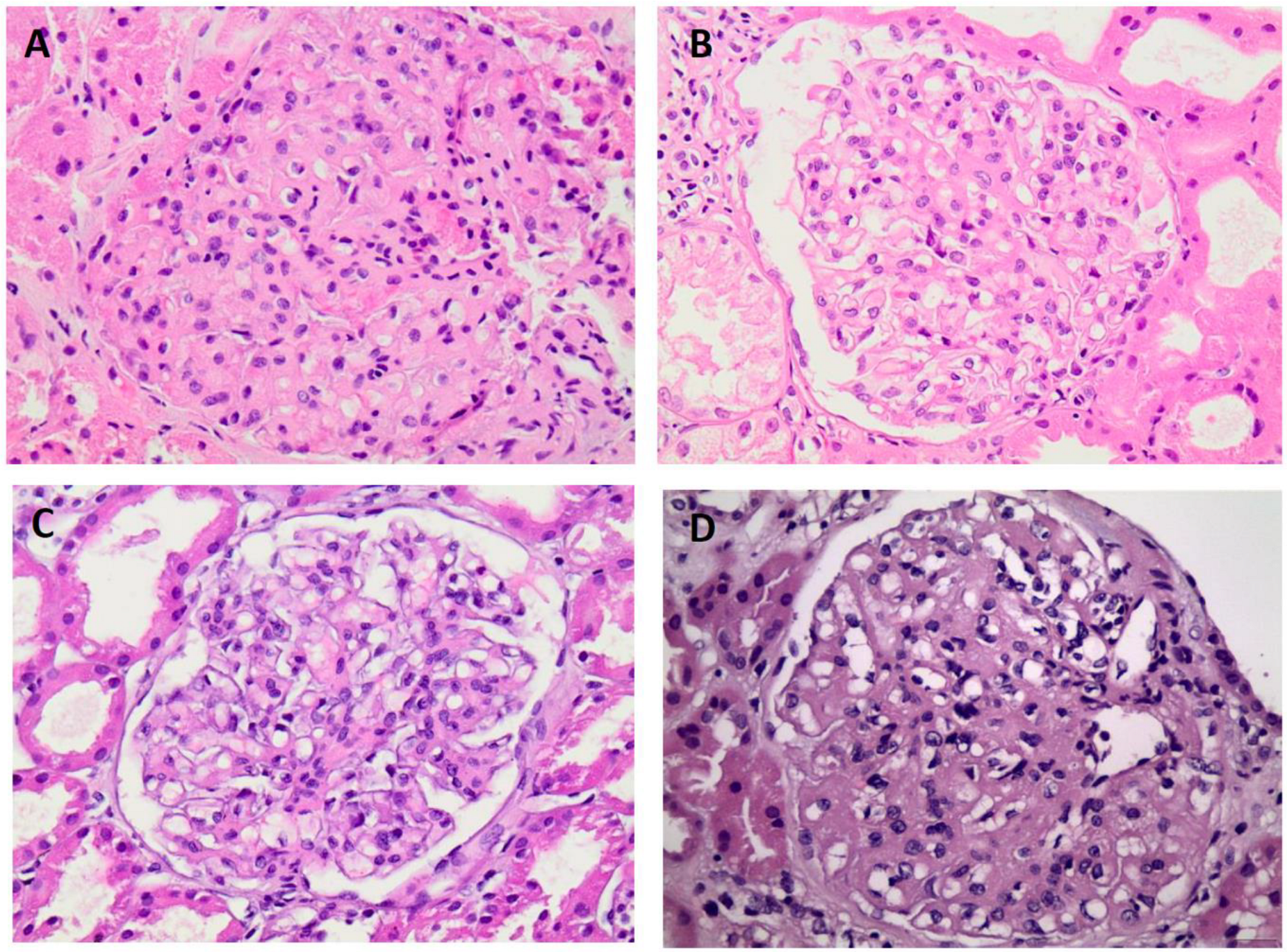
Figure 2.
Electron microscopy findings in FGN. (A) Fibril deposits with random orientation in the mesangium (patient 4 - uranyl acetate x28000). (B) Fibril deposits in the mesangium and glomerular membranes (patient 1 - uranyl acetate x18000).
Figure 2.
Electron microscopy findings in FGN. (A) Fibril deposits with random orientation in the mesangium (patient 4 - uranyl acetate x28000). (B) Fibril deposits in the mesangium and glomerular membranes (patient 1 - uranyl acetate x18000).
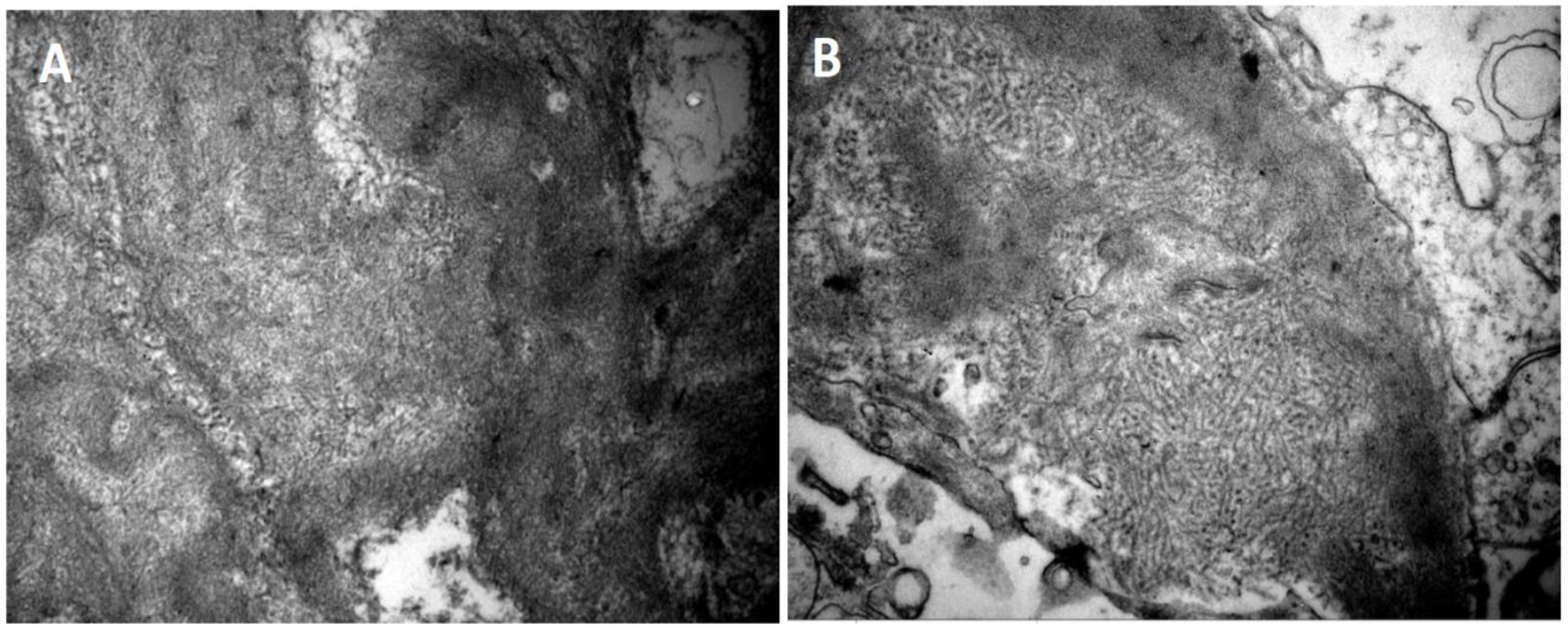
Figure 3.
Glomeruli showing positive staining for DNAJB9 (A) Patient 1 - DNAJB9 x400. (Β) Patient 4 - DNAJB9 x400.
Figure 3.
Glomeruli showing positive staining for DNAJB9 (A) Patient 1 - DNAJB9 x400. (Β) Patient 4 - DNAJB9 x400.
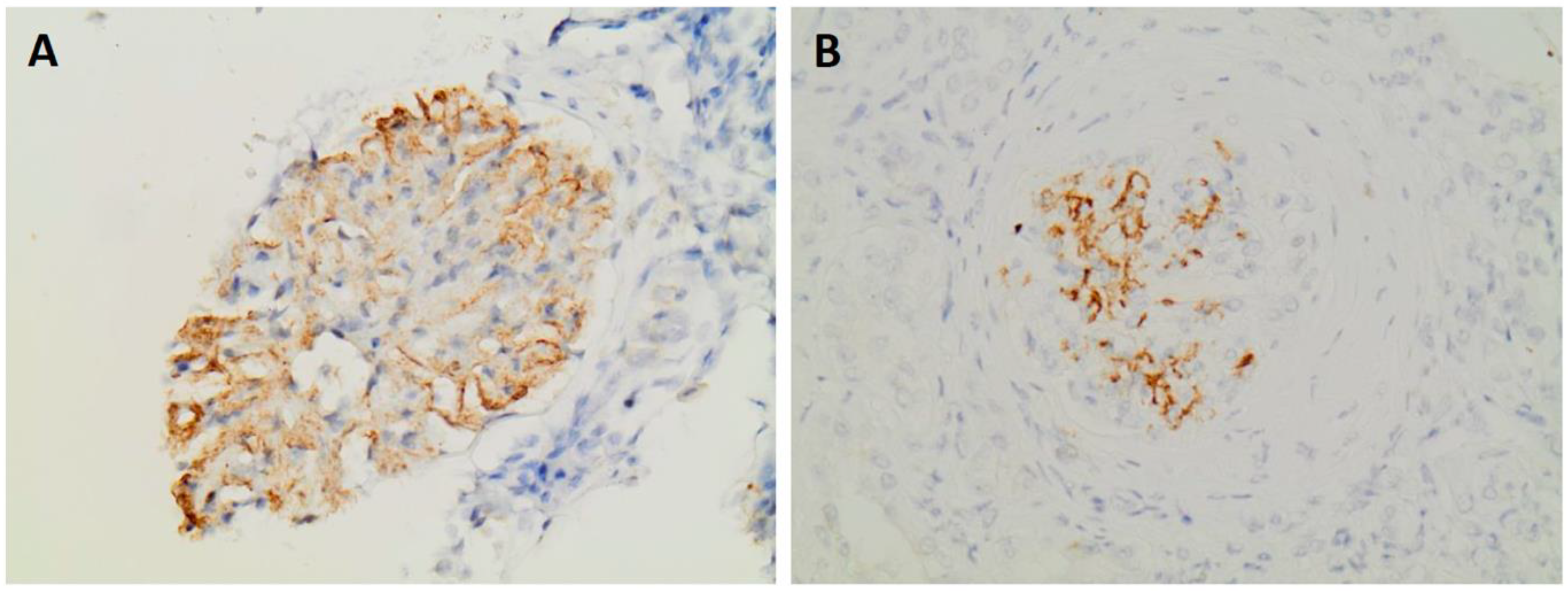

Table 1.
Demographic and clinical characteristics at biopsy. DM2: Type 2 diabetes mellitus, AF: atrial fibrillation, CKD: chronic kidney disease, SLE: systemic lupus erythematosus.
Table 1.
Demographic and clinical characteristics at biopsy. DM2: Type 2 diabetes mellitus, AF: atrial fibrillation, CKD: chronic kidney disease, SLE: systemic lupus erythematosus.
| Patients | Gender | Age | Serum Creatinine [mg/dl] | Proteinuria [gr/ 24h] | Hematuria | Hypertension | Comorbidities |
|---|---|---|---|---|---|---|---|
| 1 | Male | 45 | 1.7 | 3.70 | - | + | - |
| 2 | Female | 58 | 1.4 | 3.50 | - | + | DM2, rheumatologic disease |
| 3 | Female | 31 | 0.9 | 3.00 | + | + | Hypothyroidism, tonsillectomy |
| 4 | Female | 69 | 1.7 | 4.50 | + | + | DM2, hypothyroidism, AF, CKD, smoking |
| 5 | Male | 34 | 0.8 | 1.33 | + | - | smoking |
| 6 | Female | 60 | 2.4 | 3.78 | + | - | Thyroid nodules, dyslipidemia, SLE |
| 7 | Male | 63 | 1.2 | 11.68 | + | + | Obesity, smoking |
| 8 | Male | 61 | 1.6 | 8.00 | + | + | Colon polyps |
| 9 | Female | 56 | 0.8 | 1.80 | - | - | Hypothyroidism, dyslipidemia, sarcoidosis |
| 10 | Male | 74 | 4.5 | 5.60 | + | + | Hypothyroidism, CKD |
| 11 | Female | 57 | 3.6 | 4.00 | + | - | Lung cancer, dyslipidemia |
Table 2.
Histological findings (NA: not applicable).
| Patients | Histological pattern | Glomerulosclerosis | Tubular atrophy/ interstitial fibrosis | Crescents | DNAJB9* | Immunofluorescence |
|---|---|---|---|---|---|---|
| 1 | Diffuse sclerosing | 75% | 35% | - | NA | IgG 1+, IgM 1+, IgA 1+, C3 3+, C1q trace, κ negative, λ 1-2 + |
| 2 | Membranoproliferative | 66% | 25-30% | - | NA | IgG 1 -2+, IgM 1-2+, IgA trace, C3 1-2+, C1q trace, κ λ negative |
| 3 | Mesangial proliferative | 30% | 25% | - | + | IgG 2+, IgM 2-3+, IgA trace, C3 3-4+, C1q, κ, λ trace |
| 4 | Mesangial proliferative | 63% | 35% | - | + | IgG 1-2 +, IgM trace, ΙgA negative, C3 1-2+, C1q negative, λ 1-2+, κ negative |
| 5 | Mesangial proliferative | 18% | 20% | 1 cellular | NA | IgG 2-3 +, IgM trace, ΙgA 2-3+, C3 2+, C1q 2+, λ 3+, κ 2+ |
| 6 | Mesangial proliferative + membranoproliferative | 18% | 25% | - | NA | IgG 1-2+, IgM 1-2+ IgA negative, C3 3+, C1q, κ, λ trace |
| 7 | Membranous | 60% | 25% | - | + | IgG 1-2+, IgM 1+, IgA trace, C3 2-3+, C1q trace, κ trace, λ 2+ |
| 8 | Mesangial proliferative | 40% | 20% | - | NA | IgG 1+, IgM trace - 1+, IgA negative, C3 2-3+, C1q 1+, κ 1+, λ 2-3+ |
| 9 | Mesangial proliferative | 6% | 20% | - | NA | IgG 2-3+, IgM trace, IgA 1+, C3 2+, C1q trace, κ 1-2+, λ 3+ |
| 10 | Crescentic | 27% | 35% | 4 [2 cellular, 2 fibrocellular] |
+ | IgG 1+, IgM trace, IgA negative, C3 trace, C1q negative, κ, λ trace |
| 11 | Crescentic | 8% | 25% | 8 [5 cellular, 3 fibrocellular] | + | IgG, IgA negative, IgM trace, C3 1-2+, C1q trace, κ, λ negative |
Table 3.
Treatment and therapeutic response. GC: glucocorticoids, RTX: rituximab, CYC: cyclophosphamide, MMF: mycophenolate mofetil, PR: partial remission, CR: complete remission, PRD: persistent renal dysfunction, sCr: serum creatinine, Upr: urinary protein.
Table 3.
Treatment and therapeutic response. GC: glucocorticoids, RTX: rituximab, CYC: cyclophosphamide, MMF: mycophenolate mofetil, PR: partial remission, CR: complete remission, PRD: persistent renal dysfunction, sCr: serum creatinine, Upr: urinary protein.
| Patients | Follow up period [months] | Immunosuppression | sCr [mg/dl] diagnosis | sCr [mg/dl] last measurement | Upr [gr/ 24h] diagnosis | Upr [gr/ 24h] last measurement | Therapeutic response |
|---|---|---|---|---|---|---|---|
| 1 | 24 | GC + RTX | 1.7 | 1.7 | 3.7 | 0.8 | PR |
| 2 | 24 | CYC + RTX | 1.4 | 1.5 | 3.5 | 0.5 | PR |
| 3 | 6 | GC + RTX | 0.9 | 0.9 | 3 | 1.2 | PR |
| 4 | 12 | GC + RTX | 1.7 | 1.4 | 4.5 | 2.7 | PR |
| 5 | 48 | GC + RTX | 0.8 | 1 | 1.3 | 0.1 | CR |
| 6 | 36 | GC + CYC + MMF | 2.4 | 1.7 | 3.7 | 0.05 | PR |
| 7 | 12 | GC + RTX | 1.2 | 1.2 | 11.6 | 1.9 | PR |
| 8 | 60 | GC + RTX | 1.6 | 2.2 | 8 | 7.8 | PRD |
| 9 | 72 | GC + RTX + MMF | 0.8 | 0.7 | 1.8 | 2.5 | PRD |
| 10 | 6 | GC + CYC + RTX | 4.5 | 3.3 | 5.6 | 1.6 | PR |
| 11 | 2 | GC + CYC + RTX | 3.8 | 1.6 | 4 | 1.5 | PR |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



