Submitted:
14 April 2025
Posted:
15 April 2025
You are already at the latest version
Abstract
Bruton's tyrosine kinase (BTK) is a key signaling molecule involved in both hematological malignancies and solid tumors. In B-cell malignancies such as Chronic Lymphocytic Leukemia (CLL) and Non-Hodgkin Lymphoma (NHL), BTK mediates B-cell receptor signaling, promoting tumor survival and proliferation, leading to the development of BTK inhibitors like ibrutinib that improve patient outcomes. In solid tumors, BTK isoforms, particularly p65BTK, contribute to tumor growth and therapy resistance, with inhibition showing promise in cancers like colorectal, ovarian, and non-small cell lung cancer. BTK also influences the tumor microenvironment by modulating immune cells such as myeloid-derived suppressor cells and tumor-associated macrophages, aiding immune evasion. BTK inhibition can enhance anti-tumor immunity and reduce inflammation-driven tumor progression. Additionally, BTK contributes to tumor angiogenesis, with inhibitors like ibrutinib showing anti-angiogenic effects. Beyond cancer, BTK is linked to aging, where its modulation may reduce senescent cell accumulation and preserve cognitive function. This review explores BTK's dual role, focusing on its oncogenic effects and potential impact on aging processes. We also discuss the use of BTK inhibitors in cancer treatment and their potential to address age-related concerns, providing a deeper understanding of BTK as a therapeutic target and mediator in the complex relationship between cancer and aging.
Keywords:
BTK
; Cancer
; Aging
; Senescence
; Inflammaging
1. Introduction
Bruton’s Tyrosine Kinase (BTK) is a non-receptor cytoplasmic tyrosine kinase that plays a critical role in the development, activation, and survival of B cells [1,2]. Encoded by the BTK gene, it is a key component of the B-cell receptor (BCR) signaling cascade, mediating downstream pathways such as phospholipase C gamma 2 (PLCγ2), phosphatidylinositol-3-kinase (PI3K)/Akt, and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) [3,4]. These pathways regulate essential processes like B-cell proliferation, differentiation, and survival, underscoring BTK's indispensable role in adaptive immunity [5]. Beyond its classical role in B cells, BTK also contributes to innate immunity by regulating Toll-like receptor (TLR) signaling, inflammasome activation, and Fc receptor-mediated responses in macrophages and dendritic cells [4,5,6]. This broad immunological involvement highlights BTK as a central mediator in immune homeostasis and response [6].
BTK exhibits a dual nature as both a pro-tumorigenic factor and a therapeutic target [7,8]. Its overactivation has been implicated in the pathogenesis of various hematological malignancies, including chronic lymphocytic leukemia (CLL) and mantle cell lymphoma (MCL) [9,10]. In these cancers, BTK drives oncogenic signaling that supports tumor cell survival and proliferation [5,9]. The advent of BTK inhibitors such as ibrutinib has revolutionized the treatment landscape for B-cell malignancies [11]. Ibrutinib, a first-generation covalent BTK inhibitor, irreversibly binds to Cys481 in the ATP-binding domain of BTK, effectively blocking its activity [2]. Second-generation inhibitors like acalabrutinib were subsequently developed to improve specificity and reduce off-target effects [12]. These inhibitors have demonstrated remarkable efficacy in clinical settings, replacing traditional chemoimmunotherapy regimens for certain hematological cancers [10].
Interestingly, recent studies have expanded the oncogenic role of BTK beyond hematological malignancies to include solid tumors such as breast cancer, glioblastoma, and colorectal cancer [5]. Aberrant expression of BTK isoforms in these cancers has been linked to enhanced tumorigenesis through mechanisms such as immune modulation within the tumor microenvironment (TME), promotion of cancer stemness, and resistance to chemotherapy [13]. For example, ibrutinib has shown promising preclinical activity in solid tumors by targeting both tumor cells directly and immunosuppressive components within the TME [13,14]. These findings suggest that BTK may serve as a broader oncogenic driver than previously recognized.
In addition to its role in cancer biology, BTK is increasingly associated with aging-related processes [15,16]. Chronic inflammation and immune dysregulation hallmarks of aging [17], and they are influenced by BTK activity [18]. For instance, BTK regulates inflammatory cytokine production via TLR signaling and inflammasome activation, contributing to "inflammaging," a state of persistent low-grade inflammation observed during aging [15]. This suggests that targeting BTK could have therapeutic potential not only for cancer but also for mitigating age-related immune dysfunctions.
This review aims to provide a comprehensive exploration of the multifaceted roles of BTK in cancer and aging. By examining its contributions to oncogenesis and immune regulation, we aim to elucidate the therapeutic implications of targeting BTK. Understanding this kinase's dual role may pave the way for innovative strategies to address malignancies and age-related conditions alike.
2. BTK in Cancer
2.1. Role of BTK in Hematological Malignancies
BTK is a critical player in the development and function of B cells, especially through its role in the B-cell receptor (BCR) signaling pathway [19,20]. The BCR signaling cascade is crucial for normal B-cell development, activation, and survival, and it becomes dysregulated in various hematological malignancies, particularly those involving B cells [21]. BTK functions as a key mediator in the BCR signaling pathway, where it activates downstream signaling molecules like phospholipase C gamma 2 (PLCγ2), phosphatidylinositol-3-kinase (PI3K), and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), which regulate crucial processes like cell survival, proliferation, and differentiation [7,11,22]. Aberrant BTK signaling contributes significantly to the pathogenesis of B-cell malignancies, where it drives cancer cell proliferation, survival, and resistance to apoptosis [23].
In Chronic Lymphocytic Leukemia (CLL), a common B-cell malignancy, BTK is overactivated due to mutations or continuous stimulation through the BCR [24]. These mutations result in increased survival of malignant B cells, contributing to the progression of CLL [25]. CLL cells show enhanced BCR signaling and BTK-dependent survival signals [10]. As a result, therapeutic strategies targeting BTK have gained significant traction in recent years. Similarly, in Non-Hodgkin Lymphoma (NHL), BTK is also implicated in various subtypes of non-Hodgkin lymphoma, such as mantle cell lymphoma (MCL) and diffuse large B-cell lymphoma (DLBCL) [26,27]. In these cancers, BTK signaling is crucial for the survival and growth of malignant B cells [27]. In particular, MCL is often driven by constitutively activated BCR signaling, with BTK playing an essential role in the activation of downstream survival pathways [9]. Overactivation of BTK can lead to uncontrolled proliferation and resistance to cell death.
The development of BTK inhibitors has revolutionized the treatment of hematological cancers, especially in diseases like CLL and MCL [28]. Ibrutinib, the first-generation BTK inhibitor, irreversibly binds to the cysteine residue (Cys481) in the ATP-binding pocket of BTK, inhibiting its kinase activity and blocking BCR signaling. This leads to reduced B-cell proliferation, survival, and migration, thereby significantly improving patient outcomes in CLL and other B-cell malignancies [28,29]. Clinical trials have demonstrated that ibrutinib significantly enhances both progression-free survival (PFS) and overall survival (OS) in patients with chronic lymphocytic leukemia (CLL), including those with high-risk genetic features like TP53 mutations [30,31]. The RESONATE-2 study, which followed patients for up to 10 years, showed a sustained PFS benefit with ibrutinib compared to chlorambucil, with a median PFS of 8.9 years for ibrutinib versus 1.3 years for chlorambucil [32]. Although RESONATE-2 excluded patients with del(17p), which often correlates with TP53 mutations, ibrutinib has shown effectiveness in patients with other high-risk features such as del(11q) and unmutated IGHV [32]. Additionally, analyses have indicated that using ibrutinib as a first-line treatment reduces the risk of death compared to delaying its use until second-line treatment. Overall, these findings highlight the efficacy of ibrutinib in managing CLL across various risk profiles [31,32].
Subsequent generations of BTK inhibitors, such as acalabrutinib and zanubrutinib, have been developed to enhance selectivity and reduce off-target effects [33]. Acalabrutinib is a more selective inhibitor of BTK, showing lower rates of adverse effects such as atrial fibrillation compared to ibrutinib, while maintaining similar efficacy in CLL and MCL [33,34]. Zanubrutinib has shown comparable efficacy and safety profiles to ibrutinib, with a more favorable pharmacokinetic profile, offering an alternative to patients intolerant to other BTK inhibitors [33,35]. The development of these drugs has provided patients with safer and more effective treatment options.
2.2. BTK in Solid Tumors
The overproduction of BTK isoforms, especially p65BTK, has been recognized as a major oncogene in several solid tumors, such as colorectal cancer (CRC), non-small cell lung cancer (NSCLC), and ovarian cancer [36,37]. The p65BTK isoform has been identified as a potent oncogene that interacts with the RAS/MAPK pathway, driving tumor progression and drug resistance [36]. Studies have shown that p65BTK is highly expressed in colorectal cancer tissues and cell lines, as well as in more than 50% of NSCLC cases [36]. Furthermore, its inhibition using BTK-targeting drugs like ibrutinib has demonstrated efficacy in reducing tumor growth and overcoming resistance to standard chemotherapy and targeted therapies in these cancers [5].
Ovarian cancer has one of the highest recurrence rates among gynecological cancers, ranging from 60% to 85%, with a significant likelihood of developing drug resistance [38,39,40]. Elevated expression of the p65BTK isoform of BTK has been identified as a critical factor in therapy resistance, correlating with early relapse and reduced progression-free survival (PFS) [41,42]. This makes p65BTK a promising prognostic biomarker and therapeutic target.
Studies using in vitro (cell lines) and ex vivo (patient-derived xenografts and dissociated cancer cells) models have demonstrated that BTK inhibitors, such as Ibrutinib, significantly suppress ovarian carcinoma cell proliferation and survival [43]. Notably, Ibrutinib has shown greater efficacy in reducing cancer cell survival compared to standard-of-care (SOC) treatments like carboplatin, paclitaxel, and bevacizumab [44,45]. This highlights the potential of targeting p65BTK as a therapeutic strategy for ovarian tumors resistant to conventional therapies.
In CRC, the isoform p65BTK is highly expressed through a unique mechanism involving hnRNPK-dependent and IRES-driven translation from mRNA containing an alternative first exon in the 5′ untranslated region (UTR) [46]. This expression is further modulated by post-transcriptional regulation via the mitogen-activated protein kinase (MAPK) pathway, with hnRNPK playing a pivotal role [46,47]. p65BTK exhibits robust transforming activity that is critically dependent on ERK1/2, a key downstream effector of the MAPK pathway. Inhibiting ERK1/2 not only abrogates the transforming activity of p65BTK but also eliminates RAS-mediated oncogenic effects [37]. In colon cancer tissues, p65BTK overexpression is strongly correlated with ERK1/2 activation, and disrupting this pathway significantly impairs the growth and survival of colon cancer cells [46]. Moreover, blocking p65BTK can re-sensitize drug-resistant colon cancer cell lines, organoids, and xenografts to 5-Fluorouracil, highlighting its potential as a novel therapeutic target for CRC treatment [47,48]. Moreover, studies by Basile et al.[47] have demonstrated that p65BTK is particularly overexpressed in Stage III CRC, where it serves as an unfavorable prognostic factor.
Recent studies have highlighted p65BTK as a promising therapeutic target in NSCLC, particularly in KRAS-mutated/EGFR-wild type adenocarcinomas [36,49]. p65BTK is significantly overexpressed in EGFR-wild type adenocarcinomas from non-smoker patients, with its expression preserved at metastatic sites [49,50]. This isoform is more prevalent in adenocarcinomas than squamous carcinomas and is associated with KRAS mutations and components of the RAS/MAPK pathway [36]. The expression of p65BTK is also observed in tumor-infiltrating lymphocytes (TILs), suggesting a broader role in the tumor microenvironment [49,50,51]. BTK inhibitors including Ibrutinib, AVL-292, and RN486, have shown efficacy in reducing cancer cell viability and impairing proliferation and clonogenicity in NSCLC models [46].
Recently, Betzler at al.[52] reported BTK isoforms p80 and p65 have been identified as key players in the progression of head and neck squamous cell carcinoma (HNSCC). Accordingly, the overexpression of these isoforms contributes to tumor growth and poor prognosis in HNSCC patients [52]. Importantly, inhibiting BTK activity using drugs like ibrutinib or AVL-292 can suppress tumor progression by inducing cell cycle arrest, apoptosis, and autophagy [52]. BTK has also been suggested as a prognostic marker for poor survival in glioma patients [53,54]. In this context, high BTK expression is associated with increased glioma cell proliferation, migration, and invasion [54]. Significantly, The study shows that the BTK inhibitor ibrutinib can effectively inhibit glioma cell growth by inducing G1 cell-cycle arrest and modulating key cell cycle regulators [54]. Moreover, ibrutinib blocks EGFR-induced NF-κB activation, which is crucial for glioma cell survival and proliferation [54].
3. The Role of BTK in Tumor Microenvironment (TME) Modulation
BTK plays a critical role in the modulation of the tumor microenvironment (TME), influencing both immune and stromal components. While BTK is essential for B cell development, it is also expressed in various myeloid cells, including myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages (TAMs), which are central to the TME and contribute significantly to immune suppression and tumor progression [55,56]. These immune cells activate key signaling pathways, such as the NLRP3 inflammasome and NF-κB, that regulate inflammation and immune responses [56,57]. Through these pathways, BTK-activated MDSCs and TAMs promote an immunosuppressive environment that facilitates tumor growth and immune evasion [58,59].
MDSCs, in particular, contribute to immune suppression in the TME by producing immunosuppressive factors like arginase-1, indoleamine 2,3-dioxygenase (IDO), nitric oxide (NO), reactive oxygen species (ROS), and proinflammatory cytokines such as IL-10 and TGF-β [50,56,60]. These factors create an environment that inhibits the effective functioning of tumor-fighting immune cells, allowing the tumor to evade immune surveillance [37]. Moreover, BTK is involved in the production of proinflammatory cytokines like tumor necrosis factor-alpha (TNF-α) in response to lipopolysaccharide (LPS) stimulation, further contributing to the inflammatory milieu within the TME [59]. This cytokine production supports tumor progression by enhancing inflammation, which can promote tumor cell survival, proliferation, and metastasis [59].
In addition to its role in cytokine production, BTK also modulates the microRNA expression profile in myeloid cells, which further influences inflammation in the TME [58]. Specifically, BTK inhibition has been shown to reduce the levels of proinflammatory microRNAs, such as miR-155-5p, which is known to target negative regulators of immune responses [58]. The downregulation of miR-155-5p helps to attenuate proinflammatory signaling in myeloid cells, reducing the production of cytokines that perpetuate inflammation [58]. Simultaneously, BTK inhibition increases the levels of anti-inflammatory microRNAs, such as miR-223-3p[58], which plays a key role in limiting excessive inflammation by promoting a more regulatory, anti-inflammatory phenotype in immune cells [61]. This shift in the microRNA landscape from proinflammatory to anti-inflammatory markers fosters a more balanced immune response, which could have significant therapeutic implications in the context of tumor-associated inflammation[58,61].
By reprogramming myeloid cells towards a less proinflammatory phenotype, BTK inhibitors can help mitigate the chronic inflammation commonly associated with tumor progression [5,50]. Tumor-associated inflammation is well-established as a driver of cancer biology, contributing to tumor growth, metastasis, immune evasion, and resistance to therapy [29,62,63]. Therefore, the ability to modulate the inflammatory response through BTK inhibition presents a promising strategy for controlling inflammation in the TME and enhancing the effectiveness of cancer therapies. This approach could lead to better clinical outcomes in cancers characterized by inflammation-driven immune suppression and poor prognosis.
In various cancer types, including pancreatic cancer, inhibition of BTK has been shown to restore T-cell-mediated antitumor immunity [55]. BTK inhibition reduces immunosuppressive signals, enabling T-cells to more effectively target and attack tumor cells [64]. For example, in pancreatic ductal adenocarcinoma (PDAC), BTK inhibition with ibrutinib has been shown to reprogram TAMs from a T(H)2 to a T(H)1 phenotype, thereby enhancing CD8+ T-cell cytotoxicity and inhibiting tumor growth [65]. Beyond TAMs, BTK inhibition also affects other immune cells, such as MDSCs and dendritic cells, which are known to express BTK [65]. Modulating the immune microenvironment in this way improves antitumor responses by boosting T-cell function and reducing the immunosuppressive actions of myeloid cells [65]. Consequently, BTK inhibitors offer a promising strategy to not only reduce inflammation but also enhance the immune system's ability to fight cancer, improving overall therapeutic efficacy.
4. BTK Impact on Tumor Angiogenesis
Tumor angiogenesis refers to the formation of new blood vessels within a tumor, supplying it with oxygen and nutrients essential for growth and metastasis [66,67,68]. This process is pivotal for cancer progression, as tumors cannot grow beyond 1–2 mm³ without an adequate blood supply [69,70,71,72]. Hypoxia and nutrient deprivation in the tumor microenvironment trigger the release of pro-angiogenic factors such as VEGF, which initiate this process [72].
Recent studies have highlighted the significant role of Bruton's tyrosine kinase (BTK) in tumor angiogenesis [52,73]. For instance, Betzler et al. demonstrated that BTK inhibition disrupts tumor angiogenesis in vivo [52]. Similarly, Liu et al. found that Ibrutinib, a BTK inhibitor, acts as an angiogenesis inhibitor in ovarian and breast cancers by inducing endothelial cell dysfunction [73]. Furthermore, additional studies in breast cancer confirmed that Ibrutinib significantly inhibits tumor angiogenesis by suppressing the expression of key pro-angiogenic factors such as VEGF, MMP9, and CXCL1, which are critical regulators of angiogenesis [74]. This growing body of evidence underscores BTK's potential as a therapeutic target in anti-angiogenic cancer treatments.
5. Tumoricidal Effects of BTK Inhibition
In recent years, BTK inhibitors (BTKi) have garnered significant attention as potential therapeutic agents in treating malignancies, particularly hematological cancers like chronic lymphocytic leukemia (CLL), mantle cell lymphoma (MCL), and Waldenström's macroglobulinemia [75]. More recently, the antitumor potential of BTK inhibitors has been extended to solid tumors, sparking considerable interest in their broader applicability [75,76].
BTK inhibitors exert their tumoricidal effects through multiple mechanisms. The most direct mechanism is the disruption of BCR signaling [76,77]. In many B-cell malignancies, aberrant activation of BCR signaling leads to tumor cell survival, proliferation, and resistance to apoptosis [23]. BTK inhibition directly interrupts this signaling cascade, leading to decreased tumor cell viability and increased apoptosis [53,78]. Importantly, the use of BTKi has shown promising results in targeting both the tumor cells themselves and the supportive tumor microenvironment [5].
BTK inhibitors also modulate immune responses, which can have an indirect but powerful antitumor effect [24]. By inhibiting BTK, these agents impact the function of immune cells, such as T cells and macrophages, that play pivotal roles in antitumor immunity [79]. For example, BTK inhibitors have been shown to enhance T-cell activity, improving the immune system's ability to target and destroy tumor cells [24,79]. Additionally, BTKi can influence the macrophage-mediated inflammatory response in the tumor microenvironment, potentially reducing immune suppression that facilitates tumor growth [9,76].
5.1. Ibrutinib
Ibrutinib, the first-in-class BTK inhibitor, has shown significant therapeutic benefits in hematologic malignancies, particularly in chronic lymphocytic leukemia (CLL) and mantle cell lymphoma (MCL), where the B-cell receptor (BCR) pathway plays a central role in tumor survival [23,28]. Clinical trials have shown that ibrutinib treatment leads to prolonged progression-free survival and, in some cases, complete remission, underscoring its tumoricidal potential in these cancers [28].
Emerging studies have also investigated the role of ibrutinib in solid tumors [5]. Preclinical evidence suggests that ibrutinib can reduce tumor growth and enhance therapeutic responses when combined with other treatments, such as immune checkpoint inhibitors or chemotherapy [5,36]. For example, ibrutinib has demonstrated efficacy in preclinical models of breast, lung, and prostate cancers by directly inhibiting tumor cell proliferation and modulating the tumor microenvironment [5,36,55]. Additionally, ibrutinib has been shown to restore chemosensitivity in drug-resistant cancer cells and reduce the clonogenicity of cancer stem cells in colorectal cancer and ovarian cancer [29,43,44,73].
5.2. Acalabrutinib
Acalabrutinib, a second-generation Bruton tyrosine kinase (BTK) inhibitor, has demonstrated significant anti-tumor activity in B-cell malignancies and shows promise in solid tumors [80]. Acalabrutinib, with its improved specificity and reduced off-target effects compared to first-generation inhibitors including ibrutinib, offers a safer and more effective therapeutic option [81]. Clinically, acalabrutinib has demonstrated high overall response rates (ORRs) and improved progression-free survival (PFS) in patients with relapsed/refractory CLL and MCL [82,83]. Acalabrutinib was approved by the FDA in 2019 for the treatment of relapsed/refractory MCL and CLL [83].
Recent studies suggest that acalabrutinib may have anti-tumor effects in various solid tumors, including glioblastoma, breast cancer, and prostate cancer [29]. These effects are attributed to both direct inhibition of BTK in tumor cells and indirect immunomodulation within the tumor microenvironment [29,79]. However, various investigations suggested that acalabrutinib's anti-tumor activity is primarily mediated through the inhibition of BTK, which plays a crucial role in cell survival and proliferation pathways [84]. Further studies regarding the efficacy of acalabrutinib are needed to optimize dosing strategies and explore combination therapies that could enhance its anti-tumor effects in these settings. Additionally, understanding the mechanisms underlying variable responses across different tumor types will be crucial for maximizing its therapeutic potential.
5.3. Zanubrutinib
Zanubrutinib, a second-generation BTK inhibitor, has garnered attention for its therapeutic potential in both cancer and aging-related conditions due to its improved specificity and reduced off-target effects compared to earlier BTK inhibitors like ibrutinib [35]. Zanubrutinib selectively inhibits BTK by binding irreversibly to the Cys481 residue in the ATP-binding domain, effectively blocking downstream oncogenic signaling pathways such as NF-κB and PI3K/Akt [35,85]. Clinical studies have demonstrated zanubrutinib's efficacy in treating CLL and MCL [86], showing comparable or superior outcomes to ibrutinib [87]. For instance, zanubrutinib has a more favorable pharmacokinetic profile, leading to sustained BTK occupancy and reduced adverse effects like atrial fibrillation [85,87]. This makes it an attractive option for patients intolerant to first-generation inhibitors. By inhibiting BTK, zanubrutinib can reduce the production of pro-inflammatory cytokines (e.g., IL-10, TNF-α) and modulate microRNA profiles to foster an anti-inflammatory immune phenotype [88,89]. This reprogramming of the TME may enhance the efficacy of immunotherapies and other cancer treatments [87,88].
6. Resistance to BTK Inhibition and Challenges
Resistance to Bruton's Tyrosine Kinase (BTK) inhibitors has emerged as a significant challenge in the treatment of various hematologic malignancies, including chronic lymphocytic leukemia (CLL) and mantle cell lymphoma (MCL) [26,90,91]. This resistance can occur through a variety of mechanisms, which may be either tumor-intrinsic or extrinsic in nature [91]. Intrinsic mechanisms are primarily related to genetic mutations in the tumor cells, while extrinsic factors involve the tumor microenvironment and interactions with surrounding immune cells, stromal components, and cytokines [26,91]. These mechanisms can lead to treatment failure, making it crucial to understand and address them in clinical practice.
6.1. Tumor-Intrinsic Mechanisms of Resistance
The predominant mechanisms of resistance to BTK inhibitors involve mutations in the BTK gene itself or in phospholipase Cγ2 (PLCG2), both of which are crucial components in the signaling pathways that BTK inhibitors target [25].
The most common mutations linked to resistance are located in the BTK kinase domain, particularly the C481S mutation, which occurs at the active site of the enzyme [92]. This mutation hinders the ability of covalent BTK inhibitors, such as ibrutinib, to bind and irreversibly inhibit BTK, thus allowing the tumor cells to bypass BTK inhibition and continue proliferating [92,93]. This resistance mechanism has been well documented in clinical studies and presents a major obstacle to the effectiveness of covalent inhibitors [92,94].
Another critical resistance mechanism involves mutations in PLCG2, a downstream effector of BTK in the B-cell receptor (BCR) signaling pathway [95]. Mutations in PLCG2 allow tumor cells to maintain signaling even in the presence of BTK inhibition, effectively bypassing the block in the BCR signaling cascade [25,26,91,94]. These mutations are particularly concerning because they often lead to sustained activation of pathways that promote cell survival and proliferation, driving resistance to therapy [95,96].
The use of noncovalent BTK inhibitors can help to overcome resistance seen with covalent BTK inhibitors, specifically the C481S mutation [94]. However, research has shown that resistance to noncovalent BTK inhibitors can arise from mutations in the catalytic domain of BTK [94]. Some of these mutations can be kinase dead, which prevent inhibition of BTK through BTK inhibitors [94].
6.2. Extrinsic Mechanisms of Resistance
In addition to genetic mutations, the tumor microenvironment (TME) plays a crucial role in modulating resistance to Bruton's tyrosine kinase (BTK) inhibition [25,94,97]. Tumors are often supported by a complex network of stromal cells, immune cells, and extracellular matrix components, which can significantly influence the efficacy of therapeutic interventions [62,63,66,98].
One way in which the TME contributes to resistance is through the secretion of cytokines and growth factors that activate alternative signaling pathways [99,100]. For example, cytokines such as IL-4 and IL-10 have been shown to activate compensatory survival pathways in B-cells, rendering them less sensitive to BTK inhibition [19]. Moreover, interactions between tumor cells and immune cells, including T-cells and macrophages, can provide pro-survival signals that help tumors evade the effects of treatment [19,101].
BTK inhibitors are known to induce lymphocytosis in chronic lymphocytic leukemia (CLL) patients by drawing malignant cells out of their survival niches within the lymph nodes and into the circulation [102]. BTK also appears to play a role in CD38 and CD40 signaling, though the exact nature of its involvement in these pathways remains less well defined [103]. Studies suggest that BTK-deficient B-cells produce lower levels of IL-10 upon stimulation with TLR9 compared to their physiological counterparts [104]. Additionally, BTK-deficient B-cells respond more efficiently to CpG-DNA stimulation, generating significantly higher levels of pro-inflammatory cytokines while producing lower levels of the inhibitory cytokine IL-10 [19]. Interestingly, these BTK-deficient cells also express higher levels of TLR9 when compared with normal B-cells [104]. Moreover, the activation of the PI3K/mTOR/Akt pathway, NFκB pathway, and upregulation of chemokine or integrin signaling, contribute to resistance [105]. These findings highlight the complex role of the TME in modulating BTK inhibition resistance and underscore the need for further investigation into these extrinsic mechanisms.
7. The Role of BTK in Aging
BTK has been reported as a key player in the processes associated with aging, particularly in the accumulation of senescent cells and the decline of cognitive function as the organism ages [15]. BTK is an integral part of the signaling pathways that contribute to cellular stress responses, and its dysregulation has been implicated in several age-related conditions [15,16]. One of the primary pathways through which BTK influences aging is its involvement in the tumor suppressor protein p53 pathway [2,16]. p53 is a crucial transcription factor that regulates cellular processes such as cell cycle arrest, apoptosis, and senescence in response to cellular stress and damage, including DNA damage [2,3,106]. BTK modulates p53 activity by phosphorylating both p53 and its negative regulator, MDM2, which enhances p53's activity [1,3]. This increased activity of p53 can lead to the induction of cellular senescence, a state in which cells are permanently arrested in the cell cycle and no longer divide [16] (Figure 1). Senescent cells accumulate over time and contribute to aging-related pathologies, including tissue dysfunction and inflammation [107].
Interestingly, blocking BTK can impair p53-induced senescence, suggesting that sustained BTK inhibition may slow down aging processes by reducing the accumulation of senescent cells in tissues [15,16]. Studies using ibrutinib, a selective BTK inhibitor, have shown promising results in animal models of premature aging, indicating that inhibiting BTK could have therapeutic potential in aging-related conditions [15]. Ibrutinib has been shown to extend the lifespan of progeroid mice, a model of accelerated aging, and to mitigate age-related fitness loss [15]. In particular, ibrutinib administration preserves cognitive function in these mice by reducing anxiety-like behavior and improving spatial memory, indicating its potential to counteract age-related cognitive decline [15]. Furthermore, this preservation of cognitive function is associated with a decrease in the expression of senescence markers in the brain, which suggests that BTK inhibition can reduce the accumulation of senescent cells within the brain, potentially slowing down neurodegeneration and cognitive decline [15].
Studies using ibrutinib, a BTK inhibitor, have shown promising results in animal models of premature aging [15]. Accordingly, Ibrutinib has been shown to prolong the maximum lifespan of progeroid mice and reduce general age-related fitness loss [15]. Moreover, it preserves certain brain functions, reducing anxiety-like behavior and improving long-term spatial memory [15]. This preservation of cognitive function is associated with a decreased expression of senescence markers in the brain, confirming a lower accumulation of senescent cells after BTK inhibition [15].
BTK expression increases in various organs, including the brain, of old wild-type mice, along with the expression of p16, a marker of senescent cells [15]. This suggests that the brain may be particularly sensitive to BTK inhibitors in the context of aging [15].
It is also noteworthy that BTK expression increases is associated with brain injury [108]. In the brain of aged wild-type mice, BTK upregulation is accompanied by heightened expression of p16, a well-known marker of senescent cells [15]. These findings suggest that the brain may be particularly sensitive to BTK inhibition in the context of aging, and targeting BTK could help ameliorate the age-related accumulation of senescent cells in the brain.
Human aging is characterized by a chronic, low-grade inflammation, and this phenomenon has been termed as “inflammaging” [109]. Given that BTK is involved in inflammatory responses [59], it may play a significant role in inflammaging. However, more research is needed to fully understand the direct links between BTK and this phenomenon.
In summary, BTK plays a pivotal role in aging by regulating the p53 pathway and promoting cellular senescence. By inhibiting BTK, it may be possible to delay the onset of senescence despite the persistent DNA damage typically observed in aging, thereby protecting against age-related functional decline. BTK inhibitors hold significant potential for mitigating the loss of physical and cognitive function, particularly in the brain, by reducing the accumulation of senescent cells and preserving cognitive health.
8. Conclusions
BTK is a key regulator in both aging and cancer, influencing processes like cellular senescence, immune function, and tumor progression (Figure 2). In cancer, BTK dysregulation supports the survival of malignant cells in B-cell malignancies (e.g., CLL, NHL) and solid tumors (e.g., CRC, NSCLC), where it promotes tumor growth and resistance to treatment. BTK inhibitors have significantly improved clinical outcomes, offering new therapeutic options. Additionally, BTK's role in the tumor microenvironment and angiogenesis further emphasizes its potential as a therapeutic target to inhibit tumor progression and metastasis. In aging, BTK modulates the p53 pathway, driving cellular stress responses and senescence, contributing to age-related diseases. BTK inhibition, especially with ibrutinib, shows promise in delaying aging, preserving cognitive function, and reducing tissue dysfunction. Overall, targeting BTK offers a promising strategy for treating age-related diseases and cancer, though further research is needed to optimize these therapies and understand their long-term effects.
Authors contributions
MR and ZQ conceived of the manuscript’s concept. ZQ and MR did the literature search and wrote the manuscript. MR, reviewed and approved the manuscript.
Declaration of Competing Interest
The authors report no declarations of interest.
Funding
This research received no external funding.
Data Availability Statement
No new data were created or analyzed in this study.
References
- Rada, M.; Barlev, N.; Macip, S. BTK modulates p73 activity to induce apoptosis independently of p53. Cell Death Discov. 2018, 4, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Rada, M. M. Characterisation of Novel Post-translational Modulators of p53. (University of Leicester, 2016).
- Rada, M.; Barlev, N.; Macip, S. BTK: a two-faced effector in cancer and tumour suppression. Cell Death Dis. 2018, 9, 1064. [Google Scholar] [CrossRef] [PubMed]
- Rada, M.; Qusairy, Z.; Massip-Salcedo, M.; Macip, S. Relevance of the Bruton Tyrosine Kinase as a Target for COVID-19 Therapy. Mol. Cancer Res. 2020, 19, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Uckun, F.M.; Venkatachalam, T. Targeting Solid Tumors With BTK Inhibitors. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef]
- Campbell, R.; Chong, G.; Hawkes, E.A. Novel Indications for Bruton’s Tyrosine Kinase Inhibitors, beyond Hematological Malignancies. J. Clin. Med. 2018, 7, 62. [Google Scholar] [CrossRef]
- Maas, A.; Hendriks, R.W. Role of Bruton′s Tyrosine Kinase in B Cell Development. J. Immunol. Res. 2001, 8, 171–181. [Google Scholar] [CrossRef]
- Dobrovolsky, D.; Wang, E.S.; Morrow, S.; Leahy, C.; Faust, T.; Nowak, R.P.; Donovan, K.A.; Yang, G.; Li, Z.; Fischer, E.S.; et al. Bruton tyrosine kinase degradation as a therapeutic strategy for cancer. Blood 2019, 133, 952–961. [Google Scholar] [CrossRef]
- Papin, A.; Tessoulin, B.; Bellanger, C.; Moreau, A.; Le Bris, Y.; Maisonneuve, H.; Moreau, P.; Touzeau, C.; Amiot, M.; Pellat-Deceunynck, C.; et al. CSF1R and BTK inhibitions as novel strategies to disrupt the dialog between mantle cell lymphoma and macrophages. Leukemia 2019, 33, 2442–2453. [Google Scholar] [CrossRef]
- Patel, V. et al. Comparison of acalabrutinib, a selective Bruton tyrosine kinase inhibitor, with ibrutinib in chronic lymphocytic leukemia cells. Clinical Cancer Research 23, 3734–3743 (2017).
- Wang, Y.; Zhang, L.; Champlin, R.; Wang, M. Targeting Bruton's tyrosine kinase with ibrutinib in B-cell malignancies. Clin. Pharmacol. Ther. 2015, 97, 455–468. [Google Scholar] [CrossRef]
- Khan, Y.; O’brien, S. Acalabrutinib and its Use in Treatment of Chronic Lymphocytic Leukemia. Futur. Oncol. 2018, 15, 579–589. [Google Scholar] [CrossRef]
- Wang, X.; Wong, J.; Sevinsky, C.J.; Kokabee, L.; Khan, F.; Sun, Y.; Conklin, D.S. Bruton’s Tyrosine Kinase Inhibitors Prevent Therapeutic Escape in Breast Cancer Cells. Mol. Cancer Ther. 2016, 15, 2198–2208. [Google Scholar] [CrossRef] [PubMed]
- George, B.; Chowdhury, S.M.; Hart, A.; Sircar, A.; Singh, S.K.; Nath, U.K.; Mamgain, M.; Singhal, N.K.; Sehgal, L.; Jain, N. Ibrutinib Resistance Mechanisms and Treatment Strategies for B-Cell Lymphomas. Cancers 2020, 12, 1328. [Google Scholar] [CrossRef]
- Ekpenyong-Akiba, A.E.; Poblocka, M.; Althubiti, M.; Rada, M.; Jurk, D.; Germano, S.; Kocsis-Fodor, G.; Shi, Y.; Canales, J.J.; Macip, S. Amelioration of age-related brain function decline by Bruton's tyrosine kinase inhibition. Aging Cell 2019, 19, e13079. [Google Scholar] [CrossRef]
- Althubiti, M.; Rada, M.; Samuel, J.; Escorsa, J.M.; Najeeb, H.; Lee, K.-G.; Lam, K.-P.; Jones, G.D.D.; Barlev, N.A.; Macip, S. BTK Modulates p53 Activity to Enhance Apoptotic and Senescent Responses. Cancer Res. 2016, 76, 5405–5414. [Google Scholar] [CrossRef]
- Baechle, J.J.; Chen, N.; Makhijani, P.; Winer, S.; Furman, D.; Winer, D.A. Chronic inflammation and the hallmarks of aging. Mol. Metab. 2023, 74, 101755. [Google Scholar] [CrossRef]
- Tavakoli, G.M.; Yazdanpanah, N.; Rezaei, N. Targeting Bruton’s tyrosine kinase (BTK) as a signaling pathway in immune-mediated diseases: from molecular mechanisms to leading treatments. Adv. Rheumatol. 2024, 64, 1–23. [Google Scholar] [CrossRef]
- Vazquez, M.I.; Catalan-Dibene, J.; Zlotnik, A. B cells responses and cytokine production are regulated by their immune microenvironment. Cytokine 2015, 74, 318–326. [Google Scholar] [CrossRef]
- Hasan, M. et al. Defective Toll-like receptor 9-mediated cytokine production in B cells from Bruton’s tyrosine kinase-deficient mice. Immunology 123, 239–249 (2008).
- Profitós-Pelejà, N.; Santos, J.C.; Marín-Niebla, A.; Roué, G.; Ribeiro, M.L. Regulation of B-Cell Receptor Signaling and Its Therapeutic Relevance in Aggressive B-Cell Lymphomas. Cancers 2022, 14, 860. [Google Scholar] [CrossRef]
- McDonald, C.; Xanthopoulos, C.; Kostareli, E. The role of Bruton's tyrosine kinase in the immune system and disease. Immunology 2021, 164, 722–736. [Google Scholar] [CrossRef]
- Mouhssine, S.; Maher, N.; Matti, B.F.; Alwan, A.F.; Gaidano, G. Targeting BTK in B Cell Malignancies: From Mode of Action to Resistance Mechanisms. Int. J. Mol. Sci. 2024, 25, 3234. [Google Scholar] [CrossRef]
- Palma, M.; Mulder, T.A.; Österborg, A. BTK Inhibitors in Chronic Lymphocytic Leukemia: Biological Activity and Immune Effects. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef]
- Wiśniewski, K.; Puła, B. A Review of Resistance Mechanisms to Bruton’s Kinase Inhibitors in Chronic Lymphocytic Leukemia. Int. J. Mol. Sci. 2024, 25, 5246. [Google Scholar] [CrossRef]
- Nakhoda, S.; Vistarop, A.; Wang, Y.L. Resistance to Bruton tyrosine kinase inhibition in chronic lymphocytic leukaemia and non-Hodgkin lymphoma. Br. J. Haematol. 2022, 200, 137–149. [Google Scholar] [CrossRef]
- Nakhoda, S., Vistarop, A. & Wang, Y. L. Resistance to BTK inhibition in CLL and non-Hodgkin lymphoma. Br J Haematol 200, 137–149 (2023).
- Alu, A.; Lei, H.; Han, X.; Wei, Y.; Wei, X. BTK inhibitors in the treatment of hematological malignancies and inflammatory diseases: mechanisms and clinical studies. J. Hematol. Oncol. 2022, 15, 1–35. [Google Scholar] [CrossRef]
- Zhu, S.; Jung, J.; Victor, E.; Arceo, J.; Gokhale, S.; Xie, P. Clinical Trials of the BTK Inhibitors Ibrutinib and Acalabrutinib in Human Diseases Beyond B Cell Malignancies. Front. Oncol. 2021, 11, 737943. [Google Scholar] [CrossRef]
- Robak, T.; Doubek, M.; Ferrant, E.; Diels, J.; Andersone, L.; Wilbertz, S.; Healy, N.C.; Neumayr, L.; van Sanden, S. Overall survival of patients with CLL treated with ibrutinib in the first line compared to second-line ibrutinib after chemotherapy/chemoimmunotherapy. Curr. Med Res. Opin. 2024, 40, 1369–1378. [Google Scholar] [CrossRef]
- Barr, P.M.; Owen, C.; Robak, T.; Tedeschi, A.; Bairey, O.; Burger, J.A.; Hillmen, P.; Coutre, S.E.; Dearden, C.; Grosicki, S.; et al. Up to 8-year follow-up from RESONATE-2: first-line ibrutinib treatment for patients with chronic lymphocytic leukemia. Blood Adv. 2022, 6, 3440–3450. [Google Scholar] [CrossRef]
- Burger, J. et al. CLL-076 Final Analysis of the RESONATE-2 Study: up to 10 Years of Follow-Up of First-Line Ibrutinib Treatment in Patients With Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma. Clin Lymphoma Myeloma Leuk 24, S342–S343 (2024).
- Kittai, A.S.; Allan, J.N.; James, D.; Bridge, H.; Miranda, M.; Yong, A.S.M.; Fam, F.; Roos, J.; Shetty, V.; Skarbnik, A.P.; et al. An indirect comparison of acalabrutinib with and without obinutuzumab vs zanubrutinib in treatment-naive CLL. Blood Adv. 2024, 8, 2861–2869. [Google Scholar] [CrossRef]
- Patel, V. et al. Comparison of Acalabrutinib, A Selective Bruton Tyrosine Kinase Inhibitor, with Ibrutinib in Chronic Lymphocytic Leukemia Cells. Clinical Cancer Research 23, 3734–3743 (2017).
- Brown, J.R.; Eichhorst, B.; Hillmen, P.; Jurczak, W.; Kaźmierczak, M.; Lamanna, N.; O’brien, S.M.; Tam, C.S.; Qiu, L.; Zhou, K.; et al. Zanubrutinib or Ibrutinib in Relapsed or Refractory Chronic Lymphocytic Leukemia. New Engl. J. Med. 2023, 388, 319–332. [Google Scholar] [CrossRef]
- Grassilli, E.; Cerrito, M.G.; Bonomo, S.; Giovannoni, R.; Conconi, D.; Lavitrano, M. p65BTK Is a Novel Biomarker and Therapeutic Target in Solid Tumors. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef]
- Wang, X.; Kokabee, L.; Kokabee, M.; Conklin, D.S. Bruton’s Tyrosine Kinase and Its Isoforms in Cancer. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Rada, M.; Nallanthighal, S.; Cha, J.; Ryan, K.; Sage, J.; Eldred, C.; Ullo, M.; Orsulic, S.; Cheon, D.-J. Inhibitor of apoptosis proteins (IAPs) mediate collagen type XI alpha 1-driven cisplatin resistance in ovarian cancer. Oncogene 2018, 37, 4809–4820. [Google Scholar] [CrossRef] [PubMed]
- Rada, M.; Cha, J.; Sage, J.; Zhou, B.; Yang, W.; Orsulic, S.; Cheon, D.-J. Abstract A16: COL11A1 confers cisplatin resistance through fatty acid oxidation in ovarian cancer cells. Clin. Cancer Res. 2018, 24, A16–A16. [Google Scholar] [CrossRef]
- Nallanthighal, S.; Rada, M.; Heiserman, J.P.; Cha, J.; Sage, J.; Zhou, B.; Yang, W.; Hu, Y.; Korgaonkar, C.; Hanos, C.T.; et al. Inhibition of collagen XI alpha 1-induced fatty acid oxidation triggers apoptotic cell death in cisplatin-resistant ovarian cancer. Cell Death Dis. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Liu, J.; Ma, J.; Zhang, J.; Li, C.; Yu, B.; Choe, H.C.; Ding, K.; Zhang, L.; Zhang, L. Bibliometric and visualized analysis of drug resistance in ovarian cancer from 2013 to 2022. Front. Oncol. 2023, 13, 1173863. [Google Scholar] [CrossRef]
- Sala, L.; Cirillo, G.; Riva, G.; Romano, G.; Giussani, C.; Cialdella, A.; Todisco, A.; Virtuoso, A.; Cerrito, M.G.; Bentivegna, A.; et al. Specific Expression of a New Bruton Tyrosine Kinase Isoform (p65BTK) in the Glioblastoma Gemistocytic Histotype. Front. Mol. Neurosci. 2019, 12, 2. [Google Scholar] [CrossRef]
- Zucha, M.A.; Wu, A.T.; Lee, W.-H.; Wang, L.-S.; Lin, W.-W.; Yuan, C.-C.; Yeh, C.-T. Bruton's tyrosine kinase (Btk) inhibitor ibrutinib suppresses stem-like traits in ovarian cancer. Oncotarget 2015, 6, 13255–13268. [Google Scholar] [CrossRef]
- Szklener, K.; Michalski, A.; Żak, K.; Piwoński, M.; Mańdziuk, S. Ibrutinib in the Treatment of Solid Tumors: Current State of Knowledge and Future Directions. Cells 2022, 11, 1338. [Google Scholar] [CrossRef]
- Metzler, J.M.; Fink, D.; Imesch, P. Ibrutinib Could Suppress CA-125 in Ovarian Cancer: A Hypothesis. Appl. Sci. 2020, 11, 222. [Google Scholar] [CrossRef]
- Grassilli, E.; Pisano, F.; Cialdella, A.; Bonomo, S.; Missaglia, C.; Cerrito, M.G.; Masiero, L.; Ianzano, L.; Giordano, F.; Cicirelli, V.; et al. A novel oncogenic BTK isoform is overexpressed in colon cancers and required for RAS-mediated transformation. Oncogene 2016, 35, 4368–4378. [Google Scholar] [CrossRef]
- Basile, D.; Gerratana, L.; Buonadonna, A.; Garattini, S.K.; Perin, T.; Grassilli, E.; Miolo, G.; Cerrito, M.G.; Belluco, C.; Bertola, G.; et al. Role of Bruton’s Tyrosine Kinase in Stage III Colorectal Cancer. Cancers 2019, 11, 880. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.; Liu, Y.; Xu, Z.; Meng, C.; Yang, D.; Qian, J.; Deng, X.; Zhang, Y.; Ling, Y. Recent development of BTK-based dual inhibitors in the treatment of cancers. Eur. J. Med. Chem. 2022, 233, 114232. [Google Scholar] [CrossRef]
- Giordano, F.; Vaira, V.; Cortinovis, D.; Bonomo, S.; Goedmakers, J.; Brena, F.; Cialdella, A.; Ianzano, L.; Forno, I.; Cerrito, M.G.; et al. p65BTK is a novel potential actionable target in KRAS-mutated/EGFR-wild type lung adenocarcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 1–16. [Google Scholar] [CrossRef]
- Grassilli, E., Cerrito, M. G. & Lavitrano, M. BTK, the new kid on the (oncology) block? Front Oncol 12, (2022).
- Huang, J.; Yuan, Y.; Guo, L.; Xia, G.; Chen, Y.; Chen, Q.; Wang, M. The impact of BTK knockdown on lung adenocarcinoma growth and immune response. Cancer Sci. 2024. [Google Scholar] [CrossRef]
- Betzler, A.C.; Strobel, H.; Kors, T.A.; Ezić, J.; Lesakova, K.; Pscheid, R.; Azoitei, N.; Sporleder, J.; Staufenberg, A.-R.; Drees, R.; et al. BTK Isoforms p80 and p65 Are Expressed in Head and Neck Squamous Cell Carcinoma (HNSCC) and Involved in Tumor Progression. Cancers 2023, 15, 310. [Google Scholar] [CrossRef]
- Lim, S.; Kwak, M.; Kang, J.; Cesaire, M.; Tang, K.; Robey, R.W.; Frye, W.J.E.; Karim, B.; Butcher, D.; Lizak, M.J.; et al. Ibrutinib disrupts blood-tumor barrier integrity and prolongs survival in rodent glioma model. Acta Neuropathol. Commun. 2024, 12, 1–20. [Google Scholar] [CrossRef]
- Yue, C.; Niu, M.; Shan, Q.Q.; Zhou, T.; Tu, Y.; Xie, P.; Hua, L.; Yu, R.; Liu, X. High expression of Bruton’s tyrosine kinase (BTK) is required for EGFR-induced NF-κB activation and predicts poor prognosis in human glioma. J. Exp. Clin. Cancer Res. 2017, 36, 132–132. [Google Scholar] [CrossRef]
- Messex, J.K.; Liou, G.-Y. Targeting BTK Signaling in the Microenvironment of Solid Tumors as a Feasible Cancer Therapy Option. Cancers 2021, 13, 2198. [Google Scholar] [CrossRef]
- Good, L.; Benner, B.; Carson, W.E. Bruton’s tyrosine kinase: an emerging targeted therapy in myeloid cells within the tumor microenvironment. Cancer Immunol. Immunother. 2021, 70, 2439–2451. [Google Scholar] [CrossRef]
- Benner, B.; Scarberry, L.; Stiff, A.; Duggan, M.C.; Good, L.; Lapurga, G.; Butchar, J.P.; Tridandapani, S.; Carson, W.E. Evidence for interaction of the NLRP3 inflammasome and Bruton’s tyrosine kinase in tumor-associated macrophages: implications for myeloid cell production of interleukin-1beta. OncoImmunology 2019, 8, 1659704. [Google Scholar] [CrossRef]
- Benoit, R.Y.; Zagrodnik, J.L.; Carew, S.J.; Moore, C.S. Bruton Tyrosine Kinase Inhibition Decreases Inflammation and Differentially Impacts Phagocytosis and Cellular Metabolism in Mouse- and Human-derived Myeloid Cells. ImmunoHorizons 2024, 8, 652–667. [Google Scholar] [CrossRef] [PubMed]
- Reedquist, K.; Hartkamp, L.M.; Radstake, T.R. Bruton’s tyrosine kinase in chronic inflammation: from pathophysiology to therapy. Int. J. Interf. Cytokine Mediat. Res. 2015, 7, 27–34. [Google Scholar] [CrossRef]
- Stiff, A.; Trikha, P.; Wesolowski, R.; Kendra, K.; Hsu, V.; Uppati, S.; McMichael, E.L.; Duggan, M.; Campbell, A.; Keller, K.; et al. Myeloid-Derived Suppressor Cells Express Bruton’s Tyrosine Kinase and Can Be Depleted in Tumor-Bearing Hosts by Ibrutinib Treatment. Cancer Res. 2016, 76, 2125–2136. [Google Scholar] [CrossRef]
- Jiao, P.; Wang, X.-P.; Luoreng, Z.-M.; Yang, J.; Jia, L.; Ma, Y.; Wei, D.-W. miR-223: An Effective Regulator of Immune Cell Differentiation and Inflammation. Int. J. Biol. Sci. 2021, 17, 2308–2322. [Google Scholar] [CrossRef]
- Rada, M.; Lazaris, A.; Kapelanski-Lamoureux, A.; Mayer, T.Z.; Metrakos, P. Tumor microenvironment conditions that favor vessel co-option in colorectal cancer liver metastases: A theoretical model. Semin. Cancer Biol. 2020, 71, 52–64. [Google Scholar] [CrossRef]
- Mayer, T.Z.; Kim, D.H.; Rada, M.; Petrillo, S.; Lazaris, A.; Metrakos, P. Abstract 3331: Role of innate immune cells in the development of vessel co-opting CRC liver metastases. Cancer Res. 2020, 80, 3331–3331. [Google Scholar] [CrossRef]
- Tie, Y.; Tang, F.; Wei, Y.-Q.; Wei, X.-W. Immunosuppressive cells in cancer: mechanisms and potential therapeutic targets. J. Hematol. Oncol. 2022, 15, 1–33. [Google Scholar] [CrossRef]
- Gunderson, A.J.; Kaneda, M.M.; Tsujikawa, T.; Nguyen, A.V.; Affara, N.I.; Ruffell, B.; Gorjestani, S.; Liudahl, S.M.; Truitt, M.; Olson, P.; et al. Bruton Tyrosine Kinase–Dependent Immune Cell Cross-Talk Drives Pancreas Cancer. Cancer Discov. 2016, 6, 270–285. [Google Scholar] [CrossRef]
- Rada, M., Reynolds, A. R., Lazaris, A., Seidah, N. & Metrakos, P. Inhibition of proprotein convertase subtilisin-like kexin type 9 (PCSK9) potentiates anti-angiogenic therapy in colorectal cancer liver metastases. BioRxiv 1–12 (2023).
- Rada, M. et al. Angiopoietin-1 Upregulates Cancer Cell Motility in Colorectal Cancer Liver Metastases through Actin-Related Protein 2/3. Cancers (Basel) 14, 2540 (2022).
- Rada, M. et al. Vitamin D supplementation improves the prognosis of patients with colorectal cancer liver metastases. medRxiv 1–18 (2022).
- Rada, M.; Kapelanski-Lamoureux, A.; Petrillo, S.; Tabariès, S.; Siegel, P.; Reynolds, A.R.; Lazaris, A.; Metrakos, P. Runt related transcription factor-1 plays a central role in vessel co-option of colorectal cancer liver metastases. Commun. Biol. 2021, 4, 1–15. [Google Scholar] [CrossRef]
- Rada, M.; et al. High levels of serum cholesterol positively correlate with the risk of the development of vessel co-opting tumours in colorectal cancer liver metastases. MedRxiv ( 2022.
- Rada, M. et al. Cancer cells induce hepatocytes apoptosis in co-opted colorectal cancer liver metastatic lesions. bioRxiv 429243, (2021).
- Ibrahim, N. et al. Angiopoietin1 Deficiency in Hepatocytes A ff ects the Growth of Colorectal Cancer Liver. Cancers (Basel) 12, 1–18 (2020).
- Liu, J.; Liu, Z.; Zhang, J.; Chen, X.; Chen, J.; Sui, L.; Yu, J. Ibrutinib Inhibits Angiogenesis and Tumorigenesis in a BTK-Independent Manner. Pharmaceutics 2022, 14, 1876. [Google Scholar] [CrossRef] [PubMed]
- Varikuti, S.; Singh, B.; Volpedo, G.; Ahirwar, D.K.; Jha, B.K.; Saljoughian, N.; Viana, A.G.; Verma, C.; Hamza, O.; Halsey, G.; et al. Ibrutinib treatment inhibits breast cancer progression and metastasis by inducing conversion of myeloid-derived suppressor cells to dendritic cells. Br. J. Cancer 2020, 122, 1005–1013. [Google Scholar] [CrossRef]
- Rozkiewicz, D.; Hermanowicz, J.M.; Kwiatkowska, I.; Krupa, A.; Pawlak, D. Bruton’s Tyrosine Kinase Inhibitors (BTKIs): Review of Preclinical Studies and Evaluation of Clinical Trials. Molecules 2023, 28, 2400. [Google Scholar] [CrossRef]
- Fares, A.; Uribe, C.C.; Martinez, D.; Rehman, T.; Rondon, C.S.; Sandoval-Sus, J. Bruton’s Tyrosine Kinase Inhibitors: Recent Updates. Int. J. Mol. Sci. 2024, 25, 2208. [Google Scholar] [CrossRef]
- Estupiñán, H.Y.; Berglöf, A.; Zain, R.; Smith, C.I.E. Comparative Analysis of BTK Inhibitors and Mechanisms Underlying Adverse Effects. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef]
- Sharma, M.D.; Pacholczyk, R.; Shi, H.; Berrong, Z.J.; Zakharia, Y.; Greco, A.; Chang, C.-S.S.; Eathiraj, S.; Kennedy, E.; Cash, T.; et al. Inhibition of the BTK-IDO-mTOR axis promotes differentiation of monocyte-lineage dendritic cells and enhances anti-tumor T cell immunity. Immunity 2021, 54, 2354–2371.e8. [Google Scholar] [CrossRef]
- Zhu, S.; Gokhale, S.; Jung, J.; Spirollari, E.; Tsai, J.; Arceo, J.; Wu, B.W.; Victor, E.; Xie, P. Multifaceted Immunomodulatory Effects of the BTK Inhibitors Ibrutinib and Acalabrutinib on Different Immune Cell Subsets – Beyond B Lymphocytes. Front. Cell Dev. Biol. 2021, 9, 727531. [Google Scholar] [CrossRef]
- Abbas, H.A.; Wierda, W.G. Acalabrutinib: A Selective Bruton Tyrosine Kinase Inhibitor for the Treatment of B-Cell Malignancies. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef]
- Series, J.; Garcia, C.; Levade, M.; Viaud, J.; Sié, P.; Ysebaert, L.; Payrastre, B. Differences and similarities in the effects of ibrutinib and acalabrutinib on platelet functions. Haematologica 2019, 104, 2292–2299. [Google Scholar] [CrossRef]
- Abbas, H.A.; Wierda, W.G. Acalabrutinib: A Selective Bruton Tyrosine Kinase Inhibitor for the Treatment of B-Cell Malignancies. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef]
- Egyed, M.; Lueff, S.; Borbely, J.; Illes, A. Acalabrutinib and its use in the Treatment of Chronic Lymphocytic Leukemia. Futur. Oncol. 2022, 18, 755–769. [Google Scholar] [CrossRef] [PubMed]
- Herman, S.E.; Montraveta, A.; Niemann, C.U.; Mora-Jensen, H.; Gulrajani, M.; Krantz, F.; Mantel, R.; Smith, L.L.; McClanahan, F.; Harrington, B.K.; et al. The Bruton Tyrosine Kinase (BTK) Inhibitor Acalabrutinib Demonstrates Potent On-Target Effects and Efficacy in Two Mouse Models of Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2017, 23, 2831–2841. [Google Scholar] [CrossRef] [PubMed]
- Tam, C. S., Muñoz, J. L., Seymour, J. F. & Opat, S. Zanubrutinib: past, present, and future. Blood Cancer J 13, 154 (2023).
- Wolska-Washer, A.; Robak, T. Zanubrutinib for the treatment of lymphoid malignancies: Current status and future directions. Front. Oncol. 2023, 13. [Google Scholar] [CrossRef]
- Alsuhebany, N.; Pan, C.; Holovac, E.; Do, B.; McBride, A. Zanubrutinib in Mantle Cell Lymphoma Management: A Comprehensive Review. Blood Lymphat. Cancer: Targets Ther. 2023, ume 13, 67–76. [CrossRef]
- Li, X.; Wei, Y.; Li, S.; Liang, J.; Liu, Z.; Cui, Y.; Gao, J.; Yang, Z.; Li, L.; Zhou, H.; et al. Zanubrutinib ameliorates lipopolysaccharide-induced acute lung injury via regulating macrophage polarization. Int. Immunopharmacol. 2022, 111, 109138. [Google Scholar] [CrossRef]
- Li, W.; Zhu, S.; Liu, J.; Liu, Z.; Zhou, H.; Zhang, Q.; Yang, Y.; Chen, L.; Guo, X.; Zhang, T.; et al. Zanubrutinib Ameliorates Cardiac Fibrosis and Inflammation Induced by Chronic Sympathetic Activation. Molecules 2023, 28, 6035. [Google Scholar] [CrossRef]
- Nakhoda, S., Vistarop, A. & Wang, Y. L. Resistance to Bruton tyrosine kinase inhibition in chronic lymphocytic leukaemia and non-Hodgkin lymphoma. Br J Haematol 200, 137–149 (2023).
- Yang, Y.; Li, S.; Wang, Y.; Zhao, Y.; Li, Q. Protein tyrosine kinase inhibitor resistance in malignant tumors: molecular mechanisms and future perspective. Signal Transduct. Target. Ther. 2022, 7, 1–36. [Google Scholar] [CrossRef]
- Bödör, C.; Kotmayer, L.; László, T.; Takács, F.; Barna, G.; Kiss, R.; Sebestyén, E.; Nagy, T.; Hegyi, L.L.; Mikala, G.; et al. Screening and monitoring of the BTKC481S mutation in a real-world cohort of patients with relapsed/refractory chronic lymphocytic leukaemia during ibrutinib therapy. Br. J. Haematol. 2021, 194, 355–364. [Google Scholar] [CrossRef]
- Woyach, J.A.; Jones, D.; Jurczak, W.; Robak, T.; Illés, Á.; Kater, A.P.; Ghia, P.; Byrd, J.C.; Seymour, J.F.; Long, S.; et al. Mutational profile in previously treated patients with chronic lymphocytic leukemia progression on acalabrutinib or ibrutinib. Blood 2024, 144, 1061–1068. [Google Scholar] [CrossRef]
- Wang, E.; Mi, X.; Thompson, M.C.; Montoya, S.; Notti, R.Q.; Afaghani, J.; Durham, B.H.; Penson, A.; Witkowski, M.T.; Lu, S.X.; et al. Mechanisms of Resistance to Noncovalent Bruton’s Tyrosine Kinase Inhibitors. New Engl. J. Med. 2022, 386, 735–743. [Google Scholar] [CrossRef]
- Lampson, B. L. & Brown, J. R. Are BTK and PLCG2 mutations necessary and sufficient for ibrutinib resistance in chronic lymphocytic leukemia? Expert Rev Hematol 11, 185–194 (2018).
- Ondrisova, L.; Mraz, M. Genetic and Non-Genetic Mechanisms of Resistance to BCR Signaling Inhibitors in B Cell Malignancies. Front. Oncol. 2020, 10, 591577. [Google Scholar] [CrossRef]
- Rada, M. et al. Disruption of integrin alpha-5/beta-1-dependent transforming growth factor beta-1 signaling pathway attenuates vessel co-option in colorectal cancer liver metastases. bioRxiv 2003–2005 (2022). [CrossRef]
- Rada, M.; Krzywon, L.; Petrillo, S.; Lazaris, A.; Metrakos, P. A Retrospective Study on the Role of Metformin in Colorectal Cancer Liver Metastases. Biomedicines 2023, 11, 731. [Google Scholar] [CrossRef] [PubMed]
- Mayer, T.Z.; Kim, D.H.; Rada, M.; Petrillo, S.; Lazaris, A.; Metrakos, P. Abstract 3331: Role of innate immune cells in the development of vessel co-opting CRC liver metastases. Cancer Res. 2020, 80, 3331–3331. [Google Scholar] [CrossRef]
- Anderson, N. M. & Simon, M. C. The tumor microenvironment. Current Biology 30, R921–R925 (2020).
- Pernot, S.; Evrard, S.; Khatib, A.-M. The Give-and-Take Interaction Between the Tumor Microenvironment and Immune Cells Regulating Tumor Progression and Repression. Front. Immunol. 2022, 13, 850856. [Google Scholar] [CrossRef] [PubMed]
- Rey-Barroso, J.; Munaretto, A.; Rouquié, N.; Mougel, A.; Chassan, M.; Gadat, S.; Dewingle, O.; Poincloux, R.; Cadot, S.; Ysebaert, L.; et al. Lymphocyte migration and retention properties affected by ibrutinib in chronic lymphocytic leukemia. Haematologica 2023, 109, 809–823. [Google Scholar] [CrossRef]
- Bajpai, U.D.; Zhang, K.; Teutsch, M.; Sen, R.; Wortis, H.H. Bruton's Tyrosine Kinase Links the B Cell Receptor to Nuclear Factor κb Activation. J. Exp. Med. 2000, 191, 1735–1744. [Google Scholar] [CrossRef]
- Rip, J.; de Bruijn, M.J.W.; Appelman, M.K.; Singh, S.P.; Hendriks, R.W.; Corneth, O.B.J. Toll-Like Receptor Signaling Drives Btk-Mediated Autoimmune Disease. Front. Immunol. 2019, 10, 95. [Google Scholar] [CrossRef]
- Woyach, J.A.; Smucker, K.; Smith, L.L.; Lozanski, A.; Zhong, Y.; Ruppert, A.S.; Lucas, D.; Williams, K.; Zhao, W.; Rassenti, L.; et al. Prolonged lymphocytosis during ibrutinib therapy is associated with distinct molecular characteristics and does not indicate a suboptimal response to therapy. Blood 2014, 123, 1810–1817. [Google Scholar] [CrossRef]
- Rada, M.; Vasileva, E.; Lezina, L.; Marouco, D.; Antonov, A.V.; Macip, S.; Melino, G.; A Barlev, N. Human EHMT2/G9a activates p53 through methylation-independent mechanism. Oncogene 2016, 36, 922–932. [Google Scholar] [CrossRef]
- Mylonas, A.; O’loghlen, A. Cellular Senescence and Ageing: Mechanisms and Interventions. Front. Aging 2022, 3, 866718. [Google Scholar] [CrossRef]
- Ito, M.; Shichita, T.; Okada, M.; Komine, R.; Noguchi, Y.; Yoshimura, A.; Morita, R. Bruton’s tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat. Commun. 2015, 6, 7360. [Google Scholar] [CrossRef]
- Franceschi, C.; Campisi, J. Chronic Inflammation (Inflammaging) and Its Potential Contribution to Age-Associated Diseases. J. Gerontol. A Ser. Biol. Sci. Med. Sci. 2014, 69 (Suppl. 1), S4–S9. [Google Scholar] [CrossRef]
Figure 1.
The role of BTK in p53 and MDM2 modulation. BTK regulates p53 and MDM2 activity through phosphorylation. MDM2 typically downregulates p53 activity and stability by promoting its ubiquitination. However, phosphorylation of MDM2 by BTK impedes its negative effect on p53. Additionally, BTK-dependent phosphorylation of p53 enhances its stability and activity, ultimately leading to the induction of either apoptosis or cellular senescence.
Figure 1.
The role of BTK in p53 and MDM2 modulation. BTK regulates p53 and MDM2 activity through phosphorylation. MDM2 typically downregulates p53 activity and stability by promoting its ubiquitination. However, phosphorylation of MDM2 by BTK impedes its negative effect on p53. Additionally, BTK-dependent phosphorylation of p53 enhances its stability and activity, ultimately leading to the induction of either apoptosis or cellular senescence.
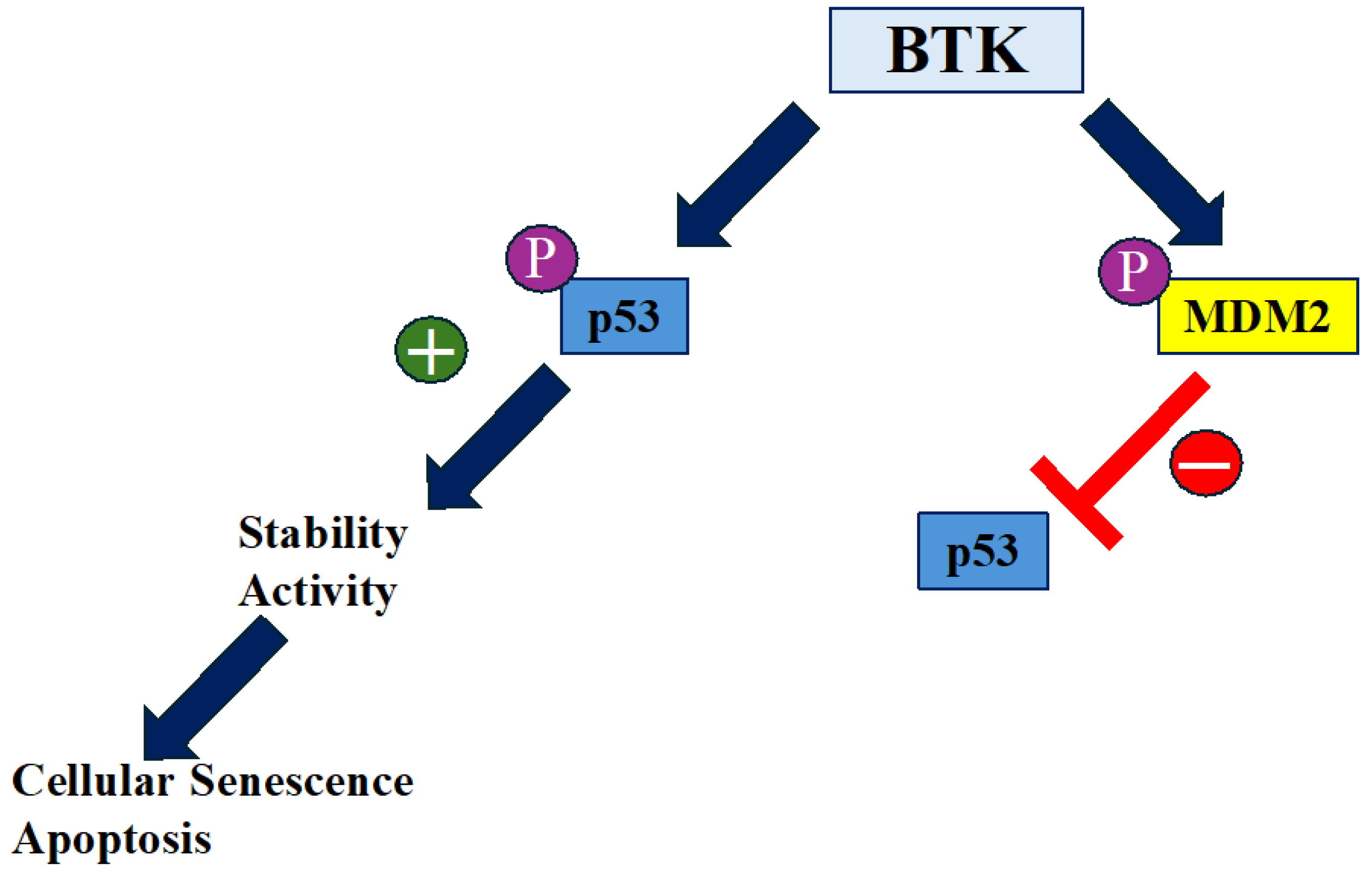
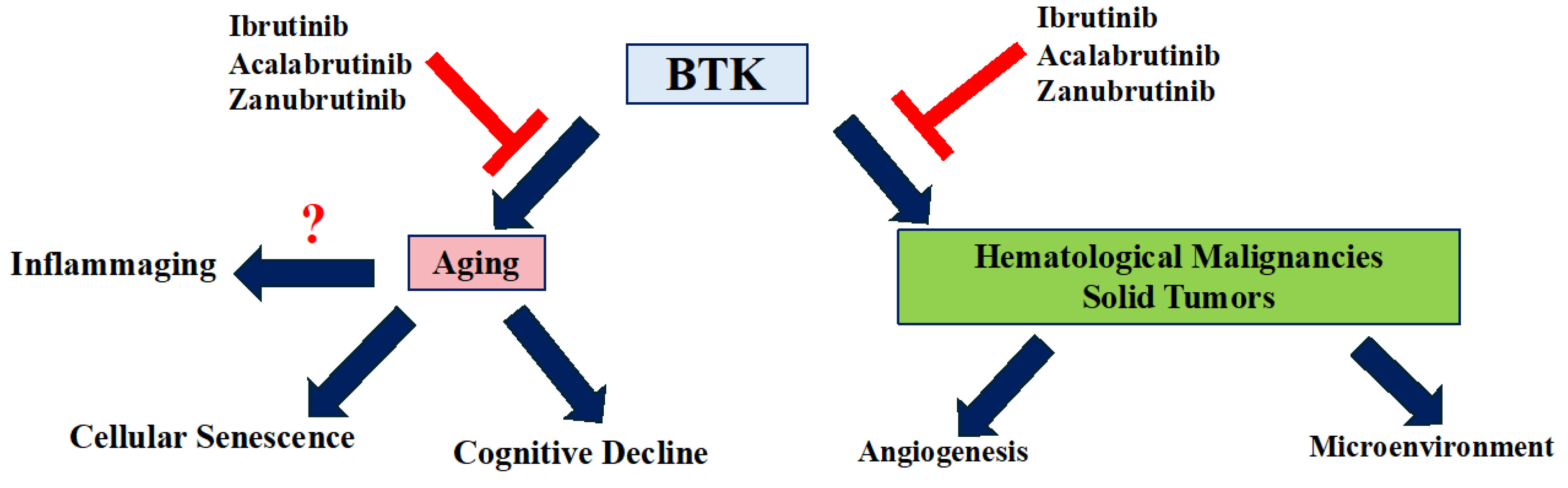
Figure 2.
The impact of BTK on cancer and aging. BTK plays a crucial role in the progression of cancer, including both hematological malignancies and solid tumors, primarily through its involvement in the tumor microenvironment and angiogenesis. Similarly, BTK contributes to aging, mainly by promoting cellular senescence and cognitive decline. Additionally, BTK may be implicated in inflammaging, a process that plays a critical role in the aging process. Both of these BTK-mediated activities can be inhibited by BTK inhibitors such as Ibrutinib, Acalabrutinib, and Zanubrutinib.
Figure 2.
The impact of BTK on cancer and aging. BTK plays a crucial role in the progression of cancer, including both hematological malignancies and solid tumors, primarily through its involvement in the tumor microenvironment and angiogenesis. Similarly, BTK contributes to aging, mainly by promoting cellular senescence and cognitive decline. Additionally, BTK may be implicated in inflammaging, a process that plays a critical role in the aging process. Both of these BTK-mediated activities can be inhibited by BTK inhibitors such as Ibrutinib, Acalabrutinib, and Zanubrutinib.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



