
Submitted:
06 August 2025
Posted:
08 August 2025
You are already at the latest version
Abstract
It is not widely known that the human genome retains ancestral emergency programs that can be activated in response to germline stress and irreparable DNA damage, freeing affected cells from the constraints of multicellularity. The reactivation of unicellular genome modules—via a process known as the multicellular-to-unicellular transition (MUT)—represents an ancient mechanism rooted in the gradual back-and-forth transition to multicellularity. MUT modules are preserved in a reactivatable state within the genomes of all metazoans and are activated in approximately 50% of humans, particularly at advanced stages of life. This inversion to a unicellular genome state gives rise to a subpopulation of precancerous cells that proliferate through defective symmetric cell division (DSCD). Once DSCD progeny acquire fusibility, they form hyperpolyploid genome repair syncytia (MGRS), which work to eliminate genomic defects and establish a unicellular cancer stemgermline capable of producing committed, non-proliferative cancer stem cells. Similar to the Ur-germline of unicellular ancestors, the cancer stemgermline, along with its clones and sublines, is hypoxic and becomes dysfunctional in tissues with oxygen levels above 6.0% O₂, a condition referred to as germline hyperoxia. Together with a somatic helper lineage that does not generate cancer stem cells, the stem-germline forms an autonomous cellular system that functions according to the principles of unicellular life. The stem-germline and its somatic counterpart work in concert to maintain and expand a stable, unicellularized stemgermline genome.
Keywords:
cancer
; Entamoeba
; stemgermline
; committed CSCs
; multicellular-unicellular transition
; genome evolution and stability
1. Introduction
In recent years, a growing number of researchers have questioned whether the predominant molecular approach to cancer research is the most effective means of addressing fundamental questions—such as what cancer truly is, how it originates, and how it might be prevented. Molecular studies have extensively analyzed tumors in search of so-called driver gene mutations, believed to be the primary initiators of carcinogenesis. At one point, it was widely assumed that just two or three driver mutations could be sufficient to transform a normal cell into a tumorigenic one. However, these efforts have yielded only limited success, and the role of replicative mutations now appears to have been significantly overestimated
1.1. Is Cancer a Mutation-Driven Deviation from Multicellularity or an Accidental Reversion to an Ancestral Cell System?
Most cancer researchers today still view cancer cells as deregulated, diseased multicellular entities that give rise to a chaotic population of both stem cells and non-stem variants, all marked by genomic instability and a loss of genomic integrity. This prevailing view [1] neglects a growing body of evidence accumulated over the past two decades, suggesting that cancer cells may retain and reactivate features characteristic of ancient unicellular life forms—particularly amoebozoans and fungi—and may rely on deeply conserved ancestral genes and mechanisms of unicellular cell control.
So, why has the evolutionary perspective, centered on the unicellularization of the cancer cell system, been largely overlooked and ignored? There are two primary reasons for this persistent oversight. First, there remains a widespread lack of foundational understanding in evolutionary biology and protistology—disciplines that are critical for interpreting the unicellular-like behaviors observed in cancer. Second, the field has shown a systematic disregard for early warnings issued by skeptical experts, who cautioned—more than a decade ago—that the exclusive reliance on rapidly evolving, molecular technologies would ultimately become a conceptual trap in cancer research.
For instance, Mariano Bizzarri [2] warned that the search for mutation-driven causes of cancer is an illusion. He argued that rather than focusing on isolated molecular or genetic events, researchers should investigate the complex, non-linear behavior of genomic networks. Yet despite these warnings, the dominant paradigm remains entrenched—centered on mutated oncogenes and tumor suppressor genes—while the deeper evolutionary logic of cancer as an inversion to unicellular autonomy remains underexplored.
Why do most researchers believe that a reversal to multicellularity is impossible? Because the prevailing view among these researchers is still shaped by Dollo’s law of evolutionary irreversibility, a paradigm established 135 years ago. In 1893, Louis Dollo [3] postulated that evolution cannot return to earlier states or to a previously realized state in an ancestral lineage. According to this dogma, once a more or less complex trait has been lost, it remains lost forever.
However, Dollo’s postulate has gradually lost its universal validity. The deeper we look into the early evolution of multicellularity, the less the postulate holds true. This is particularly evident in primitive facultative multicellular organisms such as social amoebae, yeasts, fungi, and others, as well as in primitive cell lineages like stem cells and cancer stem cells (CSCs), all of which are believed to descend from a common ancestor—often referred to as the common AMF ancestor of Amoebozoans, Metazoans, and Fungi).
1.2. Ancestral State Reconstruction (ASR)
Recent studies in phylogenomics and functional genomics have shown that evolution can inert to conserved genetic and developmental ancestral programs. [4,5,6]
As recently noted by Elmer and Clobert [7], comprehensive evolutionary reconstructions and comparative studies of multiple multicellular lineages provide evidence of both loss and regain of traits through Ancestral State Reconstruction (ASR). [8,9,10] Several compelling cases of “breaking Dollo’s law” have been reported in the recent literature, spanning diverse taxa, from bacteria to plants to animals. [11,12,13,14,15,16,17,18,19,20,21,22]
In this sense, Barerre et al. [11] described in 2023 life cycles in the budding yeast Saccharomyces cerevisiae, in which phases of multicellular growth are followed by dispersal into single cells. This alternation between unicellular and multicellular phases appears to be a constitutive feature of the organism’s life cycle. Clonal multicellularity offers inherent advantages, yet it retains the evolutionary capacity to revert quickly to a unicellular state.
Similar mechanisms have been observed in Dictyostelium discoideum and Myxococcus xanthus [12,13,14,15,16,17], where the transition between unicellular and multicellular states is environmentally dependent. Beyond fungi and yeasts, certain animal species also exhibit unicellular stages during their life cycles—for example, the miracidium stage of Trematoda. In 2023, Conlin and Ratcliff [18] termed such life strategies “facultative multicellularity,” noting that these organisms spend most of their life cycles in a unicellular state.
All these evolutionary findings clearly demonstrate that the transition from multicellularity to unicellularity—embedded within an ancient cellular program—is neither rare nor a speculative hypothesis, but rather an evolutionary reality rooted in the early stages of the transition to multicellular life.
Cancer is unequivocally an example of an ASR process. [23,24,25,26] It represents a functional genomic reversal within the evolutionary trajectory of a clade. [7] Importantly, ASR is neither a transient phenomenon nor merely a manifestation of cellular plasticity. [27,28,29] Rather, cancer illustrates that ASR can drive the reactivation of entire cellular systems—specifically, a shift from a multicellular to a unicellular state. In this re-acquired ancestral condition, intermediate genomic losses and gains emerge that were absent or suppressed in the original multicellular context. Further molecular investigations are necessary to functionally classify these events—whether as losses or recoveries —at the genomic and post-genomic levels. [7]
1.3. The Multicellular-to-Unicellular Transition (MUT) in Experimental Tumor Xenograft Models
Chen et al. [19] demonstrated in 2015 that the process of carcinogenesis involves a form of reverse evolution—from multicellularity back to unicellularity. The researchers proposed that complex multicellular organisms, including humans, have evolved sophisticated regulatory pathways—genetic mechanisms that suppress the fitness of individual cells in order to preserve the fitness of the whole organism.
In this context, cancer can be viewed as a reversal of the evolutionary transition to unicellular life. To explore this idea, the authors above studied the experimental evolution of human breast cell-derived xenograft tumors in mice, aiming to reconstruct the complete evolutionary history of a tumor. They observed a general loss-of-function strategy in cancer cells, which systematically dismantled the genetic constraints necessary for maintaining multicellularity throughout tumor evolution.
The findings by Chen et al. [19], who (i) xenographed human breast cells in mice; (II9 the 2025 Ancestral State Reconstruction study by Elmer and Clobertz [7]; and t(iii) the numerous examples of facultatively multicellular organisms that spend most of their life cycles in a unicellular state clearly demonstrate that MUTs (multicellular- to- unicellular transition processes) are an evolutionary reality rooted in the era of the unicellular transition to multicellularity (UMT), which occurred approximately 600 million years ago (Mya).
The only shortcoming of Chen et al.’s statement is that the authors were unaware that the origin of the ancestral cancer cell system dates back approximately one billion years to the hypoxic Mesoproterozoic era, when the common AMF ancestor and its stemgermline (Ur-germline) emerged. (Table 1).
1.4. How Much Recognition Has Early Evolutionary Research Received?
Unfortunately, the above evolutionary work has not received the attention they deserve. They have been largely ignored or, worse, dismissed as speculative by the majority of cancer researchers—many of whom remain committed to traditional cancer cell theories and uphold the dominance of the molecular-mutational theory of cancer. A significant reason for this lack of acceptance is a marked deficiency in evolutionary understanding. Within expert circles, it is repeatedly asserted that the central claim of the evolutionary theory of cancer—namely, the transition from multicellularity to unicellularity—embedded in ancient evolutionary programs is speculative and lacks direct empirical support.
Now one wonders why this is so. It’s hard to understand because traditional, “non-evolutionary” cancer research still faces numerous unresolved issues and remains unable to adequately address several fundamental questions, such as: (i) how cancer originates and develops; (ii) why it progresses so rapidly and often follows a seemingly fixed pattern; (iii) why CSC lineages—believed to possess dual potential—are consistently associated with extreme hypoxia; and (iv) why the cancer cell system evolves so rapidly and in a highly reproducible manner.
Most proponents of traditional theories argue that the concept of an evolutionary transition from multicellularity to unicellularity disregards decades of research and findings in molecular oncology, particularly those concerning driver mutations in tumor suppressor genes. In contrast, they continue to uphold interpretations and models that have arguably led cancer research into a conceptual dead end. Has molecular cancer research been on the wrong path? At times, it appears that researchers are reluctant to acknowledge that they may have been studying, for years, a cell system that is in fact unicellularized—rather than genuinely multicellular, as commonly assumed. Could it be that the so-called mutated cancer genes are actually unrecognized ancestral homologs of genes once essential for multicellular organization?
Even the ad hoc cancer evolution proposed by molecular cancer research occur far too rapidly to be considered true de novo evolution. Genuine evolutionary processes unfold over millions of years and are inherently non-linear—characterized by prolonged periods of fluctuation, with progress often following a “two steps forward, one or two steps back” pattern. In contrast, cancer-associated mutations—whether permanent or transient—arise with such speed and frequency that it is difficult to accept them as causal in the classical evolutionary sense. Furthermore, cancer cell hypoxia cannot be seen as an adaptation to human tissue environments, but rather as an intrinsic genetic trait of an ancestral cellular system that originally evolved under hypoxic conditions. The authentic genomic evolution that shaped the archaic genomic modules exploited by cancer today took place over the course of roughly a billion years (Table 1), and such deep evolutionary processes cannot simply be recapitulated in a rapid, ad hoc fashion.
1.5. Discovery of Deep Homologies Between Cancer and Entamoeba
The concept of the evolutionary cancer cell biology (ECCB) is based on well-substantiated experimental research on the biology of parasitic and free-living protists (Entamoeba, Giardia, Colpoda), conducted the author in earlier years. [30,31,32,33,34,35,36,37,38,39]. The findings revealed an archaic hypoxic cell system that exhibits striking similarities to the cancer cell system and its CSCs. Comparative data between these two systems have been previously tabulated. [40]
The initial breakthrough that led to the identification of a deep homology between the cancer cell system and that of Entamoeba —as well as their shared ancestral cell type, the AMF ancestor (Figure 1) — was the development of a novel hypoxic culture technique for Entamoeba by the author. Since the 1960s, Entamoeba had been cultivated exclusively in synthetic air cultures, which are hyperoxic for the stemgermline of Entamoeba. In contrast, the author’s technique utilized sediment cultures containing metabolically suppressed oxygen-consuming bacteria (OCB), in which active bacterial growth was deliberately inhibited. An active bacterial growth was metabolically suppressed. [30] These OCB cultures established a variable oxygen gradient that closely resembled the physiological gradient observed in the human intestine, particularly near the mucosal surface—ranging from ≤1% to approximately 5.7%.
OCB cultures facilitated the discovery of the previously unknown stemgermline of Entamoeba, its ACD (asymmetric cell division) phenotype and a population of committed stem cells that accumulate through cycles of cystic polyploidy and depolyploidization, generating progenitor cells that give rise to new stemgermline clones. Comparative analyses between hypoxic OCB cultures and and hyperoxic air cultures of Entamoeba revealed that the hypoxic, stemness-positive ACD phenotype was present exclusively under hypoxic conditions. In contrast, hyperoxic, bacteria-free cultures were dominated by a predominantly phagocytic somatic cell type, in addition to a minor fraction displaying a dysfunctional DSCD stemgermline phenotype. Restoration of the genomic integrity and stability of the stemgermline was only possible through the formation of hyperpolyploid multinucleated genom repair structures (MGRS). [40,41,42] The new findings on the oxygen-sensitive germline and its oxygen-resistant somatic sister line reveal their mutual plasticity in the bidirectional transitions between germline and soma (GST) and soma-to-germ (SGT).
The ECCB, the new evolutionary cancer cell biology, emerged from the deciphering of the deep homologies between the amoebic and cancer cell systems. It required the introduction of new terms and definitions as shown in Table 1.
1.6. Unicellularization and Its Consequences
Unicellularization precedes carcinogenesis and is initiated in senescent stemgermline cells during a prolonged phase of restorative senescence and subsequent senescence exit. [26] Multicellular cells harboring genomic instability and capable of bypassing apoptosis appear to seek alternative routes for DNA repair, genomic reorganization, and survival. Such mechanisms were present in ancient unicellular life forms and persist in certain cellular systems today. It cannot be ruled out that MUT is regulated by a specific set of MUT-associated genes that may represent an ancient class of as yet undiscovered carcinogenic driver genes. These MUT mechanisms likely played a crucial role during the transition to multicellularity (the UMT era), when early, unstable multicellular organisms that proved non-viable reverted to a unicellular state—choosing survival as unicellular entities over death as multicellular organisms.
MUT can be understood as a process of genomic inversion, wherein stable unicellular genome networks replace the unstable genomic networks of multicellular systems. This principle of early cellular evolution also applies to contemporary damaged stemgermline cells, which may undergo unicellularization within the context of a multicellular organism. However, the question arises: what prospects do unicellularized cells have in the complex and regulated environment of a multicellular host? If they are not eliminated by the immune system, these cells may persist and proliferate as dysfunctional symmetric cycling entities (DSCD phenotype. Following the logic of their shared ancestry with the AMF Ur-germline, they may subsequently generate progenitors for an emergent cancer stemgermline lineage through hyperpolyploid MGRS repair structures.
During its physiological expansion, the nascent cancer stemgermline remains continuously exposed to the host organism’s oxygen gradient. In this context, it exhibits behavior reminiscent of its ancestral relatives: hypoxic regions within the gradient function as inducers and effectors of the stemgermline, while hyperoxic conditions (particularly those exceeding 6.0% O2) act as stressors that temporarily impair stemgermline’s genomic integrity. In response, the unicellularized cancer cell system activates compensatory stemgermline repair and replacement mechanisms to restore genomic stability of the system. This dynamic represents a continuous struggle, one in which the cancer stemgermline ultimately prevails. The end products of this evolutionary and physiological trajectory are the foreign bodies we identify as tumors.
The concept of genomic instability, chaos, and mutations as propagated by current cancer research, arises largely from comparing the unicellular genome of cancer cell lineages with the multicellular genome of human cells—leading to misleading conclusions. Such comparisons overlook fundamental evolutionary differences and should be approached with caution. Even when the genome of the damaged stemgermline becomes temporarily unstable, the unicellularized cancer system actively engages in genomic repair, striving to restore integrity through hyperpolyploid genome repair mechanisms.
2. The Entamoeba Model of Cancer
Recent advancements by the evolutionary cancer cell biology ECCB [23,24,25,26,43,44,45,46] highlight the deep evolutionary homology existing between cancer cells and parasitic amoebae, drawing parallels with the common AMF ancestor. This perspective places ancestral state reconstruction at a higher explanatory level than mutational changes, which alone have not satisfactorily clarified the origin, genome and function of cancer.
2.1. Surprising Similarities Between the Parasitic Cell Systems of Cancer and Entamoeba Reveal Their Common Origin
The strikingly homologous lineages observed in evolutionarily distant species such as mammals and amoebozoans can only be explained by their descent from a common ancestral lineage. The nearly one-to-one correspondence between their life cycle structures—alongside complementary features unique to each system—helps complete the evolutionary picture of the shared AMF ancestor. This ancestral cell system laid the foundation for the stemgermline mechanisms observed across a wide range of descendants, including protists, invertebrates, vertebrates, mammals, and humans.
Furthermore, the ECCB has demonstrated that the primary driver of both unicellular cell systems—cancer and its Entamoeba model—is the stemgermline, previously referred to as the non-gametogenic (NG) germline [24,25] or the proliferative cancer stem cells. All modern days stemgermlines are phylogenetically related to the hypoxic Ur-germline of the AMF ancestor that give rise to non-proliferative committed stem cells and committed cancer stem cells (com-CSC). [23] (Figure 1)
Figure 1.
Cancer and Entamoeba share the same basic mechanisms of unicellular cell systems evolved by the common AMF ancestor. They are: enlarged DDR processes, genome damage repair by MGRS (PGCC) structures, polyploidization and hyperpolypolyploidization, homotypic cell fusion, ACD and DSCD phenotypes, stemness loss and recovery cycles, SGT/EMT processes, etc.
Figure 1.
Cancer and Entamoeba share the same basic mechanisms of unicellular cell systems evolved by the common AMF ancestor. They are: enlarged DDR processes, genome damage repair by MGRS (PGCC) structures, polyploidization and hyperpolypolyploidization, homotypic cell fusion, ACD and DSCD phenotypes, stemness loss and recovery cycles, SGT/EMT processes, etc.
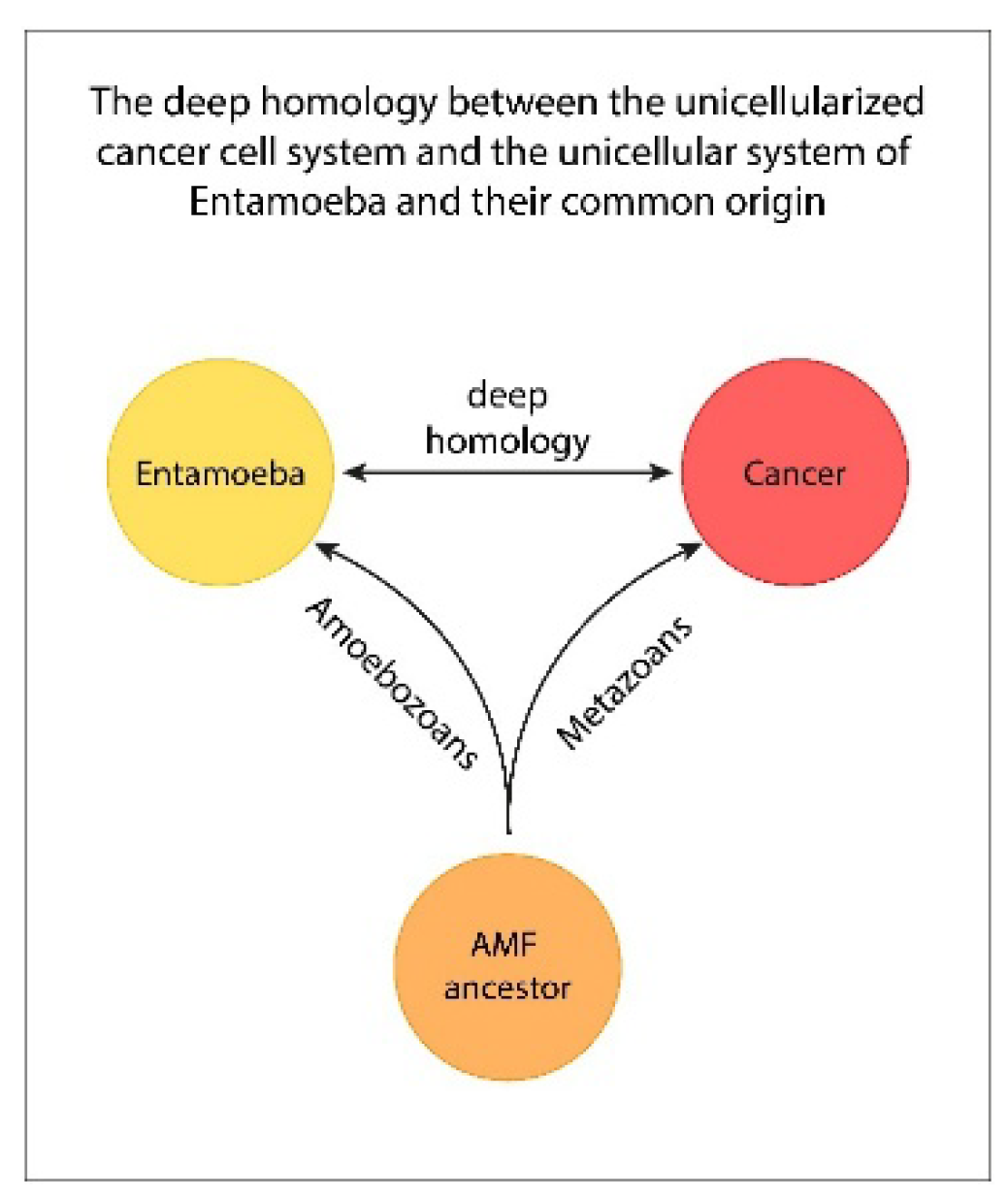
2.2. The Stem Cell Line and Its Ancestral Oxygen Sensitivity Dominate the Unicellular Cell Systems of Cancer and Amoebae
In host tissues, including the blood circulation, the oxygen content ranges from 0.1% to 15% (host’s oxygen gradient). However, the unicellular stemgermlines of cancer and parasitic amoebae function physiologically and differentiate progenitor cells via cystic-polyploidy only within the lower ranges of this O2 gradient, at about 1% O2, as found in less oxygenated OCB sediments (Entamoeba), or in low-hypoxic hematopoietic niches. [47] Oxygen levels exceeding the hypoxic threshold (≥5.7-6.0%) create a hyperoxic environment for the stemgermline (germline hyperoxia), leading to irreversible genome damage. However, both cell systems possess unicellular repair mechanisms, such as homotypic cell fusion and hyperpolyploid MGRS’ (multiple genome repair structures) or polyploid giant cancer cells (PGCCs), to restore genomic stability and full functional states.
For understanding historic AMF and stemgermline evolution it is necessary to understand the biological focus for the last 2100 My oxygen event [48] prior and during of the “big bang” moment in eukaryotic evolution and dispersion that did not occur until 1200 to 1000 Mya. [49] Since the Great Oxidation E, 2,5-2.3 giga years ago (Gya), oxygen has remained at low levels throughout the Meso- and Neoproterozoic. [50] (Table 2)
2.3. Deep Homologies to Giardia and Other Facultative Unicellular Parasites
We know that ancestors of the parasitic protists such as Giardia and Trichomonas existed—and evolved structurally and metabolically—in Archean environments containing significanly less than 1% PAL PO2 (Partial Oxygen Pressure). Geological data indicates that eukaryotes began to radiate and spread relatively late when PO2 was 1-2% of PAL. This was a time when ancestors of modern intestinal and vaginal parasites developed aerotolerance to toxic oxygen levels, as well as the ability to phagocytose (Table 2). For example, the somatic cell line of the AMF ancestor evolved at the beginning of the oxygen rise. There is evidence that a gradual rise in oxygen occurs over the Neoproterozoicum (The Neoproterozoic Oxygen Window, 1000-550 Mya) but well-oxygenated conditions appear delayed until the Paleozoic Oxygenation Event (550-250 Mya). [50]
According to ECCB, gaining deeper insight into the evolutionary history of the stemgermline, which drives cancer evolution is paramount to understanding cancer. [26] In recent years, significant progress in amoeba cell biology has provided a fortunate parallel [40,41,42]. Currently, Entamoeba represents the most evolutionarily relevant model for cancer, demonstrating that cancer exploits the ancient life cycle established by the common ancestor (Figure 1).
Cancer and Entamoeba possess both a unicellular oxygen-sensitive stemgermline with two distinct phenotypes. One is the ACD phenotype, which produces committed stem cells. The other is the dysfunctional DSCD phenotype, which have lost its stemness potential and proliferates through defective symmetric cycles. (Figure 2) Additionally, there is a somatic oxygen-resistant cell lineage consisting of numerous subtypes and clones, which play an essential role in maintaining and expanding the stemgermline genome (Table 2).
Both cancer and Entamoeba stemgermlines share deep homologous life cycle characteristics, including (1) germline hyperoxia, (2) stemness loss and recovery (3) dysfunctional genome reconstruction by the hyperpolyploid MGRS/ PGCC, and others. All these homologies strongly support the unicellular nature of cancer cell systems and demonstrate the common origin of cancer and amoebic stemgermlines.
The two autonomous cell systems—cancer and its parasitic Entamoeba model [23,24,25,26] –are vulnerable to oxygen excess and severely compromised in environments where oxygen levels exceed germline normoxia (≥ 6.0% O2). Such hyperoxic germline conditions of tissue and blood flow lead to the loss of DNA repair capabilities (homologous recombination, HR), resulting in irreparable double-strand break (DSB).
From an evolutionary perspective, committed non-proliferative human stem cells, such as adult stem cells (ASCs), originate from a proliferative human stemgermline historically closely related to the Ur-germline of the common AMF ancestor. Some of the genomically dysfunctional ASCs can bypass the barriers of multicellularity and switch to unicellularity. Understanding the evolutionary roadmap of this transition is essential to guide future molecular research toward solving long-standing questions in cancer biology.
3. The Cancer Stemgermline: Function, Dysfunction and Repair
3.1. Hypoxic ACD Phenotypes Produce com-CSCs or Quiescent Q Cells
Stem cells are germline stem cells (GSCs). During the ACD program, all descendants of the ancestral Ur-germline —whether in protists or metazoans— function as proliferative lines that generate two functionally distinct daughter cells, depending on the environmental oxygen concentration.
If oxygen levels remain near ~1% O2—consistent with the ancestral conditions of the Ur-stemgermline over its ≥800-million-year evolutionary history (Meso- and Neoproterozoic eras)- the stemgermline divides into (i) a self-renewing stemgermline cell that maintains stemness, and (ii) a terminal committed stem cell (com-SC or com-CSC). Committed stem cells are capable of homotypic, non-mitotic accumulation and can generate new stemgermline progenitors through processes such as “cystic polyploidization” and subsequent depolyploidization. This ancestral program persists in metazoan and human cancers.
However, if oxygen levels increase by approximately 2% or more—as occurred during the late Neoproterozoic period—a different outcome emerges: instead of forming a terminal com-SC or com-CSC, the stemgermline produces an intermediate, quiescent phenotype (Q cell). Q cells retains the capacity to revert either into a self-renewing stemgermline cell or to progress into a committed stem cell. (Figure 3)
This different differentiation path has often been overlooked, yet it plays a pivotal role in the emergence and progression of the cancer cell system and its stemgermline.
The widespread, long-held belief that com-CSCs perform both proliferation and differentiation functions is inaccurate. This misconception stems from a lack of knowledge about the distant origin of the AMF ancestor and its ancestral germline. Second, it makes our ability to understand the stemgermline biology much more difficult.
Recent studies on the biology of parasitic amoebae have shown that Entmoeba’s stemgermline produces com-SCs only in OCB sediments, where bacterial activity reduces the oxygen levels to approximately 1.0% O2 [26]. If oxygen consumption by the OCB sediment is insufficient and oxygen levels remain higher, the production of terminally differentiated polyploid structures (cysts) is replaced by the generation of Q cells instead. A comparable evolutionary response to varying degrees of hypoxia has also been observed in the hematopoietic cell system. [47]
3.2. Stemness Loss and Recovery Cycles: DSCD-ACD Transitions and Genome Repair Processes
Exposure to oxidative stress damages the genome of the ACD phenotype, leading—after a restorative senescence phase—to a dysfunctional DSCD cell state that lacks stemness and proliferates through defective mitotic cycles. The subsequent transition from this stemness-negative DSCD phenotype back into the functional stemness-positive ACD state is not possible. There is no simple bidirectional plasticity between the genomically dysfunctional DSCD phenotype and the functional ACD state that gives rise to com-CSCs. Rather, the transition from DSCD to ACD represents a one-way process embedded within the extended DNA damage response (DDR) observed in both cancer and amoebic cell systems.
Earlier cancer research, however, misinterpreted this unidirectional conversion as a bidirectional switch—from undifferentiated stem cells to more differentiated non-stem cells and vice versa. According to that view, ACD–DSCD–ACD cycles were seen merely as fluctuations in differentiation status. Yet this is incorrect. These cycles must be understood as cycles of stemness loss and recovery (see Figure 2), and the DSCD-ACD transition must be recognized as a mechanism including genomic repair, rather than spontaneous reprogramming. Crucially, both cell states—ACD and DSCD—belong to the dual stemgermline.
In recent years, a more nuanced discussion has emerged regarding the balance between asymmetric and symmetric division, with a focus on the roles of key signaling pathways, which regulate critical cell cycle components, including p53 and CDK inhibitors. [51] Within this framework, asymmetrically dividing cancer cells are classified as CSCs, whereas symmetrically dividing cells are labeled as non-CSCs or de-differentiating CSCs. These ambiguous classifications arose partly because, at the time, proliferative CSCs had not been functionally distinguished from non-proliferative CSCs.
Roughly a decade ago, it was assumed that CSCs (undergoing asymmetric division) and non-CSCs (dividing symmetrically) may be epigenetically stable and developed independently of one another. This perspective suggested that stemgermline’s cell fate decisions were binary—either differentiation of CSCs into non-CSCs, or de-differentiation of newly formed non-CSCs back into undifferentiated CSCs. Additionally, it was proposed that de-differentiation of non-CSCs into CSCs involved secretory factors, such as the cytokine IL-6. Indeed, Iliopoulos et al. [52] suggested that the CSC/non-CSC balance may be controlled by the concentration of secreted signaling molecules and their receptor availability.
Nonetheless, it was already apparent that asymmetric division depends on specific microenvironmental cues, and that the functional expression of the ACD phenotype requires precise external signals. As early as 2008, Knoblich emphasized the role of the extrinsic environment in determining whether hematopoietic stem cells undergo symmetric or asymmetric divisions. [53]
From today’s perspective, the interpretation of bidirectional plasticity—between stem and non-stem cells—is fundamentally flawed. Symmetrically cycling DSCD phenotypes represent intermediate, repair-oriented stages that are activated in response to hyperoxia-induced genomic damage. This response enriches the pool of DSCD cells, facilitating homotypic cell fusion and the formation of hyperpolyploid MGRS/PGCC genome repair structures. DSCD cells thus constitute a transient phase within an evolutionarily conserved unicellular DNA damage response (DDR) program. Understanding the mechanisms governing the transition from DSCD to ACD is therefore essential for deciphering the cell and molecular biology of the stemgermline system.
More recently, in 2024, Chao et al. [54] reported that the dysregulation and disruption of asymmetric division can lead to a significantly increased risk of cancer. [55,56,57] They regard this disruption as a major contributor to cancer initiation. This observation is consistent with statements made by the author of this article in his previous work [26].
Less accurate, however, is the role attributed to PGCCs by Chao et al. [54] and others [58,59,60], as their interpretations were developed without reference to evolutionary models. According to these authors, PGCC formation is regulated by a recompartmentalization of cell cycle regulatory proteins that are normally involved in the control of asymmetric division.
These researchers describe an “asymmetric cell division of giant cancer cells by meiosis-like depolyploidization.” However, MGRS/PGCC structures do not engage in conventional mitotic cell cycles, nor do they undergo asymmetric division. Rather, these hyperpolyploid structures arise through homotypic cell fusion or—under severe conditions such as chemotherapy or irradiation—de novo, likely from severely damaged stemgermline cells.
Hyperpolyploid MGRS/PGCC structures first undergo a cyst-like polyploidization phase, producing dysfunctional daughter nuclei. This is followed by a second phase, in which these nuclei fuse into one or more giant nuclei [25,26]. After genomic repair, these giant nuclei undergo reductive nuclear division, resulting in numerous daughter nuclei that cellularize into progenitor cells of the ACD stemgermline phenotype.
Note. Stemgermlines capable of producing committed stem cells in humans, vertebrates, invertebrates and protists, are evolutionarily interrelated. They all retain genes and modules derived from the ancestral genome, or at least significant portions of it. Over the course of evolution, sensitivity to oxygen—and vulnerability to excess oxygen (such as germline hyperoxia)—was inherited by all modern stem cell lineages found in contemporary metazoans and mammals. These lineages retain the ancestral Ur-germline’s sensitivity to oxygen, exhibit genome damage under hyperoxic conditions, and possess the ability to repair stress-induced damage in ways that stemgermline cells in multicellular organisms lack.
3.3. The “Uncontrolled” Proliferation of Cancer Cells as an Intermediate Mechanism for Restoring Genome Integrity
Non-cancerous cells in metazoans—including humans, stemgermlines, sublines, and clones—are prone to apoptotic death, as multicellularity has introduced numerous barriers that prevent the restoration of divergent genomes. In contrast to the cell populations of protists, normal multicellular stem germline or somatic cells cannot survive or proliferate indefinitely. Unlike unicellular organisms, multicellular organisms lack the capacity to repair stressor-damaged genomes via the hyperpolyploid MGRS/PGCC pathway. If such a mechanism were present, a multicellular organism could, in theory, live indefinitely—yet, as we know, this is not the case.
Pre-carcinogenic cells that do resume proliferation after a phase of restorative senescence are typically unicellularized DSCD cells [26]. Human cells capable of indefinite proliferation under air culture conditions, such as HeLa cells, represent cell populations that have escaped the regulatory constraints of multicellularity and apoptosis. Like all cancer cell populations, HeLa cells have undergone unicellularization and exhibit the DSCD phenotype. It is likely that HeLa cells represent a hybrid cancer cell population, expressing both unicellular (UG) and multicellular (MG) genes. Alternatively, they may represent a fractal epithelial–mesenchymal transition (EMT) clone characterized by genomic anomalies such as chromothripsis affecting the shattered chromosome 11. [61]
It cannot be excluded that manipulated cancer cells such as HeLa cells, under conditions of germline hypoxia conditions, may cease their otherwise unlimited DSCD proliferation and initiate a process of genomic repair. It is well established that low oxygen levels activate hypoxic signaling cascades through the stabilization of hypoxia-inducible factor α (HIFα), which in turn regulates the expression of numerous proteins, including enzymes involved in glycolysis and factors that stimulate local angiogenesis. [62] In certain stem cells and cancers, alterations in local oxygen concentration can induce profound phenotypic changes, including differentiation and reprogramming. [63,64] Edwald et al. [65] demonstrated that, in HeLa cells, hypoxic imaging conditions lead to significant alterations in the speed, confinement, and heterogeneity of plasma membrane protein dynamics.
As known, some genomically damaged stemgermline cells in multicellular organisms can overcome the usual multicellular constraints and escape apoptosis. These cells arrest proliferation and mitosis, entering a state of restorative senescence and unicellularization. Similar to parasitic unicellular behavior, they engage extended DNA damage response (DDR) circuits and attempt to repair the dysfunctional DSCD genome via MGRS/PGCC repair pathway. [26]
Note: from the evolutionary perspective, cancer is not uncontrolled, unlimited DSCD proliferation. Dysfunctional DSCD proliferation is limited and only a part of the extended DDR circuits and genomic repair pathways
4. MUT and Unicellularization
4.1. The Triumph of Cellular Autonomy over Cooperative Multicellularity
Following mitotic arrest, some DNA-damaged stem germline cells may find restorative senescence niches and favorable environmental conditions that enable them to bypass apoptotic barriers and prepare for the MUT (multicellular to unicellular transition) process. But why do these nearly apoptotic cells overcome the constraints of multicellularity and revert to a unicellular state?
Normally, dysfunctional, severely damaged stem cells with multiple replicative DNA damages, including multicellular DNA repair genes, await their apototic death in a state of senescence. The only viable repair pathway involves the activation of unicellular repair processes, including the extended unicellular DDR (DNA damage response) and hyperpolyploid MGRS’ (Figure 4). This critical cell fate decision for unicellularization occurs within the restorative senescence niche.It triggers the activation of unicellularity-associated MUT genes, which facilitate senescence exit, induce the DSCD phenotype, promote aberrant SCD proliferation, and initiate homotypic cell fusion leading to the formation of hyperpolyploid MGRS/PGCC genome repair structures.
In other words, the intrinsic cellular drive for survival—an ancient, evolutionarily conserved trait—compels life-threatening cells to seek repair mechanisms and survival opportunities. In this context, the transition from multicellularity to unicellularity (MUT) represents an escape strategy, allowing cells to circumvent the structural and functional constraints imposed by a multicellular system and adopt an alternative state better suited for repairing severe genomic damage. This shift can be seen as the triumph of cellular autonomy over cooperative multicellularity—an individual’s liberation from the “planned economy” of multicellular regulation and restrictions. It is a survival strategy that prioritizes individual persistence over collective homeostasis
But what are the consequences? A fundamental redefinition of “self” and “non-self” emerges. The newly formed cellular entity, now distinct from its original tissue community, behaves as a separate and autonomous system, potentially acting against the host organism.
4.2. The Causes of MUT Processes
Cancer research today stands at a crossroads between traditional molecular oncology and the emerging field of evolutionary oncology [23,24,25,26]. As Arun Upadhay noted in 2020 [66] “Cancer today is still an unknown territory that requires rethinking before moving forward.”
Current cancer research believe that cancer driver mutations may affect cell-cycle control, leading to insensitivity to growth inhibitory signals and escape from immune surveillance. [67] Recent studies attempting to reconstruct the evolutionary history of individual tumors showed that 50% of all early clonal driver mutations are located in only nine driver genes, whereas subclonal mutations occur in 35 different genes, pointing to a diverse set of drivers in later evolution. [68] Driver genes are usually classified into oncogenes and tumor-suppressor genes. Oncogenes usually harbor gain-of-function mutations, which activate the protein and lead to uncontrollable cell growth or proliferation. Tumor-suppressor genes (TSGs), on the other hand, are responsible for homeostasis during cell division and DNA replication, and there is strong positive selection in cancer for deactivating mutations. However, some genes can have both tumor-suppressor and oncogene characteristics under different circumstances
The ECCB interprets such driver and deactivating mutations as part of a broader evolutionary mechanism that downregulates MGs (multicellularity genes) and reactivates UGs (unicellularity genes) to initiate unicellularization, particularly through the activation of specific MUT genes.
4.3. Are Oncogenes and TSG the True Determinants of Cancer and MUT Processes?
If tumor-suppressor genes (TSGs) and oncogene are responsible for homeostasis during cell division and DNA replication, then these cell-cycle regulating genes should be very old genes that regulate the cell cycle of all eukaryotes.
MUT genes would be however, much younger genes that were only formed during the early UMT period. The age difference could be more than 800 My. MUT genes would have emerged during the transition to multicellularity, when early multicellular organisms developed unicellularity- suppression genes on the one hand, but also anti-suppression genes or MUT genes at the same time.
From an evolutionary perspective, therefore, the assumption of “mutated oncogenes” as the driver of carcinogens does not seem to apply. This idea obviously arose from the consideration that cancer would arise from the simple deregulation of the cell cycle, which, from an evolutionary point of view, is a much too narrow and not necessarily determinant cause.
However, it remains an open question why not all severely damaged stemgermline cells activate the MUT corridor and instead succumb to apoptotic death. This appears to parallel the broader evolutionary question of why not all damaged or nonviable early metazoans were able to revert to a unicellular state for survival. One possible explanation is (i) that not all damaged stemgermline cells—either then or now—are capable of reactivating MUT genes, or (ii) that the MUT pathway is simply not accessible or functional in the majority of multicellularity cells.
4.4. The Main Controversy Between Mutational and Evolutionary Approaches
Mutational and evolutionary viewpoints differ fundamentally in their understanding of the nature and origin of cancer cells. Previous oncology views cancer cells as divergent multicellular entities that retain their multicellularity. In the absence of a comprehensive biological model to explain the initial steps and causes of cancer, previous oncology adopts a retrospective strategy, drawing molecular inferences from final tumors to determine the causes and onset of cancer. In recent years, extensive tumor screenings have been conducted to identify specific driver genes, under the assumption that these genes are responsible for carcinogenic transformation. However, the results remain controversial and subject to ongoing debate.
The evolutionary perspective presents a markedly different view. Like Entamoeba, cancer represents an autonomous parasitic cell system that shares deep homology with the unicellular common AMF ancestor. Cancer cells follow the organizational logic of a unicellular system, not the regulatory constraints of the multicellular host organism. Key discoveries in evolutionary cell biology reveal that the stemgermline undergoes transient genomic disruption and phenotypic changes in response to stressors; however, it retains the ability to restore its functional genomic architecture and integrity through evolutionary mechanisms inherent to unicellular life. These repair mechanisms, lost in multicellular systems, remain preserved and reactivatable within the ancestral genome compartment of all metazoans, including humans.
4.5. The Unicellular Centric View of Cancer
The most relevant difference between classical r oncology and the more recent evolutionary ECCB is their mutual perspective: the molecular cancer research adheres to the multicellular-centric view of cancer, while the latter embraces a unicellular-centric paradigm. [69]
This unicellular-focused framework allows oncology to explain cancer not merely as a genetic or molecular aberration through mutations but as the reactivation of ancient unicellular survival strategies embedded within the genome of metazoans. Cancer is a biological anomaly and disease in which an archaic unicellular cell system develops within a multicellular organism and becomes active according to its own rules. It is not subject to regulation by the host organism and cannot be effectively combated by it.
5. The Traditional Cancer Concept Left Many Questions Unresolved
The unicellular nature of the cancer cell system, along with its genomically stable stem-germlines and clones, has received little attention and has been largely overlooked in modern cancer research. The concepts of genomic chaos and loss of genomic integrity, as postulated by earlier cancer research, may reflect reductionist thinking, an uncritical reliance on molecular biology, and a neglect of evolutionary and non-genetic regulatory processes.
5.1. Current Research Does Not Question the Multicellularity of the Cancer Cell System
Just a decade ago, the prevailing view was that there is no fixed cancer genome. Instead, cancer was thought to involve a progressive reorganization of multicellularity genes (MGs), which determines and defines the genetic network structure of cancer through a process of genome remodeling. Horne in 2015 [70] noted that the early theories of cancer genome remodeling arose on assertions from dedifferentiation and deprogramming processes. [70,71,72]
Such theories suggested that the genome acts as an “instruction manual,” guiding the assembly of new biological systems. According to this perspective, the genome serves as a blueprint that determines how genes and their encoded products combine to form the structure of new genetic and protein networks
Despite similar gene content, changes in genomic topology could drastically alter gene interaction networks, with significant consequences for cellular function and phenotype. Changes to individual genes could disrupt this organisation, leading to genomic instability and chaos.
Comparison of sequencing data from primary and metastatic tumors with somatic mutation catalogs [73] supported an earlier alternative hypothesis that the cancer cell genome is the results from multiple genomic aberrations accumulated throughout the tumor’s developmental history and even earlier. [74,75,76,77,78,79] Additionally, a proposed timeline for tumor development suggests that significant carcinogenic events may occur decades before diagnosis. Overall, it was suggested, that cancer genomes are shaped by a lifelong process of somatic evolution, which blurs the distinction between normal aging processes [80,81] and pre-cancerous evolution.
The evolutionary perspective does not deny that significant carcinogenic events may occur long before diagnosis. However, it interprets these events in terms of DSCD risk following unicellularization. DSCD cells may remain in a quiescent state within specific niches or slowly proliferate over extended periods through defective symmetric proliferation. This process may continue until DSCD proliferation is halted by homotypic agglutination and hyperpolyploidization.
5.2. Contemporary Views on Genome Reprogramming
But how important are dedifferentiation, reprogramming and redifferentiation of multicellularity cells for the development of cancer?
According to Hanahan [82] dedifferentiation converts adult somatic cells into a pluripotent ground state whereas reprogramming or redifferentiation involve the conversion of pluripotent cells into a differentiated cell state. In vitro, dedifferentiation is achieved through the overexpression of four key reprogramming factors: OCT4, SOX2, KLF4, and C-MYC. These factors induce the transformation of a differentiated somatic cell into an induced pluripotent stem cell (iPSC). The subsequent step in reprogramming involves re-differentiating these iPSCs into the desired cell type.
All these processes normally take place within the multicellular cell system and its development. Do they also occur during carcinogenesis? Can iPSCs differentiate into true CSCs? And vice versa? The results were discouraging.
Several researchers consider that CSCs share many molecular features with iPSCs. Malignant cells could be successfully reprogrammed into iPSC-like cancer cells (cancer iPSCs) [83] but also normal iPSCs could be transformed into CSC-like cells by genetic manipulation involving tumor microenvironment factors. Some cancer-derived iPSCs have been shown to exhibit reduced malignancy and can differentiate into benign cancer lineages. [84,85,86,87,88]
Shamsian et al. in 2022 [89] underline that cancer cells are however, largely resistant to reprogramming due to several biological barriers, including mutations, accumulated DNA damage, epigenetic changes, and the activation of cancer-related genes. Consequently, the reprogramming of cancer cells has been largely unsuccessful in regenerative medicine. While the aforementioned barriers typically permit only partial reprogramming into cancer- iPSCs, even fully reprogrammed iPSCs often revert to the original cancer cell traits over time. [90,91]
In short, the outcomes of such studies have not very promising, despite researchers’ hopes of gaining valuable insights into various oncogenic processes to facilitate their modulation.
Attempts at transdifferentiation have also been largely unsuccessful. Transdifferentiation is the process by which differentiated adult somatic cells are directly converted into cells of a different lineage without reverting to a pluripotent state. However, natural transdifferentiation is rare in mammals but it is more common in cancer and protists, where germlines undergo transdifferentiation through GST processes to form somatic cell lines [23,24,25,26]. According to Graham and Sottoriva [92], transdifferentiation have a lower risk for carcinogenesis compared to reprogramming as it bypasses the intermediate pluripotent state, which is a significant source of tumorigenic potential in iPS cells. Consequently, autologous cells obtained through transdifferentiation do not inherently acquire the ability to self-renew and proliferate uncontrollably as in reprogramming. Nevertheless, the risk of carcinogenesis cannot be entirely ruled out due to potential genetic and epigenetic changes that may occur during the transdifferentiation process.
5.3. Tumor Suppressor Genes and Oncogenes: Caretakers, Gatekeepers, and Landscapers
In the absence of solid evolutionary insights and a suitable cell biological model to explain MUT processes as induced by evolutionary MUT genes, the question of what causes uncontrolled somatic outgrowth in multicellular organisms has been debated for years. It was concluded that tumor suppressor genes, often referred to as caretaker genes, must play a critical role in preventing somatic outgrowth and, by extension, tumor development. These genes were believed to produce specific proteins responsible for maintaining genomic stability.
If these genes became defective due to replication errors (“mutations”) and lost their functional control, it was thought that the uncontrolled multicellular cell growth would lead to tumor formation. There was a strong belief that mutations in tumor suppressor genes were the primary drivers of cancer. [93]
The distinction between “caretakers” and “gatekeepers was introduced by Kinzler and Vogelstein. [94] Unlike caretaker genes, which would maintain genomic integrity, gatekeeper genes would encode products that actively prevent the growth of potential cancer cells and suppress the accumulation of mutations that directly lead to increased cellular proliferation. [95,96] Gatekeeper genes would directly regulate tumor growth by encoding dual-function proteins that either stimulate or inhibit proliferation, differentiation, or apoptosis in a dose-dependent manner. Key gatekeeper and caretaker genes of significant interest include APC, RB1, CDKN2A, TP53, BRCA genes.
Caretaker genes would ensure genomic stability by preventing the accumulation and transmission of replication defects (mutations). According to this dogma, the loss of function of mutant caretaker genes allows mutations in other genes to arise and persist, potentially driving the conversion of a normal cell into a neoplastic one. In this concept, caretaker genes do not directly regulate cell proliferation. Instead, they function by slowing the cell division process to allow DNA repair to be completed or by initiating apoptosis to eliminate damaged cells.
Additionally, Michor et al., [97] introduced the term “landscaper” genes. According to the researchers, landscaper genes encode products that, when mutated, would contribute to the neoplastic growth of cells by fostering a stromal environment that is conducive to unregulated cell proliferation.
This concept also has its limits. Notably, the restoration of a caretaker gene from its mutated form to the wild-type version does not appear to significantly limit tumorigenesis, raising questions about its role in halting or reversing cancer progression.
More recently, oncogenes and tumor suppressor genes are considered genes whose alterations—including intragenic mutations, chromosomal deletions and loss of expression—are involved in tumor processes, including cell cycle progression, differentiation, maintenance of genomic integrity, DNA (deoxyribonucleic acid) damage repair, and even apoptosis. According to Fanale et al. [98] “inactivation of both genes contributes directly to cancer development and progression”.
The author of the present work challenges this prevailing assumption of the past 20–30 years, which explain cancer solely through driver mutations in TSGs and oncogenes, and view cancer cells as remaining within the framework of multicellular life. At the time, this was a convenient explanation; however, it can no longer withstand the current evolutionary understanding of non-genetic mechanisms, genomic MUT processes, and the role of younger MUT genes, which evolved in early multicellular life forms under conditions of severe environmental stress and life-threatening challenges.
5.4. Genomic Alterations and the Ineffectiveness of the Multicellular DNA Damage Response (DDR)
As early as 1976, Nowell [99] proposed that tumor initiation and progression might result from acquired genomic changes within the “original normal cells,” triggered by exogenous DNA damage. These changes, and the accumulation and transmission of replication defects, were later interpreted as evidence of genomic instability, with tumor cell populations appearing to be genetically more unstable and more heterogenous (chaotic) than normal cell populations. [100] From this perspective, it was concluded that cancer cells evade the regulatory control of the multicellular organism. Additionally, their outgrowth proliferation through shorter cell cycles was considered a significant growth advantage. These factors contributed to the development of the concept of genomic instability in cancer and tumors, contrasting with the stable genome of normal multicellular cells. This discrepancy is considered one of the biggest problems in cancer. But is the assumption of cancer genome instability correct?
DNA damage, if left unchecked, is associated with an increased risk of tumor development. To counteract this, cells have evolved several conserved pathways that respond to such errors by initiating DNA repair processes and/or apoptosis. The process of DNA repair is tightly linked to the DNA damage response (DDR), which involves the recruitment and localization of DNA damage sensors, mediators, transducers, and effector proteins to distinct nuclear foci. [101]
From the evolutionary perspective, there is still confusion about the genomic stability and instability of the cancer stemgermline. However, several older hypotheses proposed the driving force behind tumour initiation and progression through the emergence of a “mutator”. These phenotypes would result from the loss of gene function due to DNA replication stress. [102,103,104] Mutator phenotypes have been observed in the early stages of tumour progression and were considerd to be the result of changes in genes that normally maintain genomic stability in multicellular organisms. The evolutionary cancer cell biology (ECCB) framework considers such earlier interpretations to be partially correct, but it challenges the underlying assumption that the process is purely mutational and confined within the logic of multicellularity.
Overall, current cancer research indicates that the transformation of a normal cell into a malignant one is driven by genetic, epigenetic, transcriptomic, and metabolic alterations. However, the predominant focus on genetic, genomic, and mutational studies—along with the century-old dogma of cancer as a purely multicellular phenomenon—has yielded limited success. This approach has often led to disappointing statements, leaving many aspects unclear or unresolved, which remains a significant challenge.
5.5. The Dogma of Reversible Cancer Cell Plasticity
A recent study by Warrier et al. in 2023 [105] provides a compelling illustration of this issue. The researchers outline three models of tumor heterogeneity: the clonal evolution model, the cancer stem cell (CSC) model, and the reversible cellular plasticity model. According to the researchers, the first two models aim to explain tumor initiation, maintenance, progression, and origin, [106] while the last model proposes a unified framework that integrates elements of both other proposals.
The cellular plasticity model emphasizes the dynamic ability of cancer cells to switch between several several distinct states—including (i) CSC and non-CSC, (ii) differentiated and stem-like, (iii) asymmetrically and symmetrically dividing, (iv) quiescent and proliferative, and (v) epithelial and mesenchymal phenotypes. Warrier et al. [105] present these transitions in an almost tabular manner, yet without adequately addressing the contextual and environmental conditions necessary for such conversions. The specific functional purposes of each transition are not individually explored, nor is there a distinction between transitions that are truly reversible and those that follow a unidirectional course.
These points have already been discussed in detail in the earlier sections of this paper. Consistent with the preceding analysis, most of Warrier’s examples of plasticity represent systemic, long-lasting processes that do not allow for immediate or direct reversibility. [25,26].
For example, com-CSC phenotypes and dysfunctional DSCD states (referred to by Warrier et al. [105] as symmetrically dividing stem-like cells) are irreversibly committed either to differentiation, including non-mitotic accumulation via cystic polyploidization, or to genome repair via hyperpolyploid MGRS/PGCC pahway These cell types cannot revert to their original phenotype.
Only EMT/MET transitions appear to retain true bimodal plasticity ability under specific environmental changes within defined oxygen gradient ranges, as observed in protist cultures [23,24]. Epithelial–mesenchymal transition (EMT) represents an irreversible soma-to-germ transition (SGT), whose evolutionary function is to compensate for the loss of functional ACD phenotypes and the depletion of com-CSCs. In contrast, mesenchymal–epithelial transition (MET) constitutes a germ-to-soma transition (GST), serving to prevent genomic damage within oxygen-resistant somatic cancer cells. GST–SGT–GST cycle represents an ancient unicellular plasticity mechanism evolved by the common AMF ancestor and used by cancer.
It should be noted that non-genetic, evolutionary perspectives do not support the broad and often overstreched model of cancer cell plasticity. [105] This model tends to restate previous assumptions and overlooks the structured, regulated progression of the cancer cell system. This overreliance on a generalized plasticity framework obscures the more precise, evolutionarily grounded understanding of cancer behavior and may misrepresent the true challenges of therapeutic intervention.
5.6. The Need for Non-Genetic Research
Numerous mutations found in healthy tissues exposed to carcinogens challenge the interpretation that cancer arises from premalignant clonal outgrowths. As recently reviewed by Nam et al. in 2022 [107], somatic mutations observed in clonal outgrowths sometimes regress or overlap with recurrent driver mutations. [108,109,110,111,112,113] This suggests that genetic mechanisms alone may not be sufficient to induce malignant transformation. [114]
According to Nam et al. [107], as malignant populations expand, cells undergo further genetic diversification, driving tumour progression, relapse and resistance to therapy. [115] This concept led to the assumption that integrating multiple layers of information from individual cancer cells using single-cell multi-omics would be essential for a comprehensive understanding of cancer evolution. However, clear genetic factors associated with cancer progression, metastasis and therapy resistance have been identified in only a limited number of tumors, further suggesting the significant involvement of non-genetic factors. Genetic mechanisms alone may not fully capture the complexity of intratumoural heterogeneity.
6. Two Decades of Phylostratigraphic Cancer Gene Research and Evolutionary Statements of this Time
The first consequential evolutionary concepts emerged at the end of the first decade of the present century when Pepper et al. in 2009 [116] thought that cancer involves complex evolutionary processes. He regards neoplastic progression as a process of somatic evolution through cell cycle arrest and senescence.
Regarding the previous term CSC, most researchers adopted the prevailing view—still widely accepted today—that CSCs are proliferative, a notion contradicted by the ECCB. Moreover, the claim that CSC proliferation serves to produce more CSCs is doubly incorrect. According to the framework of Evolutionary Cancer Cell Biology (ECCB), additional cancer stem cells (com-CSCs) cannot be generated through mitotic division of com-CSCs. Only the ACD stemgermline is capable of mitotic proliferation, giving rise to com-CSCs—but this does not result in mass cell proliferation. Large-scale cancer cell proliferation is driven instead by the symmetrically cycling DSCD phenotype, which produces further dysfunctional DSCDs that lack stemness potential and cannot generate com-CSCs. More com-CSCs expansion occurs only through cyst-like polyploidization and depolyploidization processes. The accumulated products serve as progenitors for the emergence of new functional clones and sublines.
In 2010, Mark D. Vincent [117] published a work on cancer speciation, a topic that was becoming fashionable at the time. Vincent addresses the somewhat older cancer-as-species thesis. He refers to Huxley 1956 [118] supposition, which proposed that “once the neoplastic process has crossed the threshold of autonomy, the resultant tumor can be logically regarded as a new biologic species” a view largely shared by Greaves, [119] Swanton, [120] and Duesberg and Rasnick. [121] In contrast, Vincent considers tumor cell heterogeneity a real obstacle to the cance- species concept and also questions how much certainty exists that cancer cells are indeed genetically heterogeneous.
For the time, it was unconventional; Vincent [117] mentions cancer as a separate cell system that lives off the host in a manner similar to destructive parasites, as described by Merlo et al. (2006). [122] They view cancer cells as a colony of loosely cooperating yet often competing and independently evolving individual cells—or, at worst, a collection of unicellular eukaryotic organisms that are fully capable of existing independently of each other.
Notably, as early as 2004 Gray et al. [123] and Rivera and Lake [124] saw cancer cells as exhibiting colonial attributes, with host organisms “literally being eaten inside out by a very primitive type of animal”, primitive in the sense of being minimized due to secondary losses of characteristics. This was a remarkably bold proposition for its time—when no suitable evolutionary models had yet been developed, and the evolution of multicellularity was not yet understood as a bidirectional process occurring along a two-way street.
Vincent believed that the correspondence between the mediators of multicellularity and those that “fail” during carcinogenesis strongly supports the notion that these two apparently disparate phenomena might actually lie on the same continuum.
In 2012, Vincent [125] added that cancer represents the de-repression of a default survival program inherent to all eukaryotic cells. He suggests that cancer arises from the reactivation of an ancestral program that prioritizes cellular survival above all else. To describe this phenomenon, he introduced the term “adaptive resilience,” referring to an “any-cost cellular survivalism” that is independent of identity and cell origin. The author characterizes cancer cells as a protozoan-like population with evolutionary roots in the Precambrian era. He argues that through the deconstruction of the metazoan phenotype, these cells acquire autonomous, parasitic traits, ultimately becoming entities that, while residing within the body, are no longer truly part of it.
Nevertheless, when molecular cancer research was gaining momentum, such evolutionary perspectives were met with little enthusiasm in the cancer research community and often went unnoticed. However, over the past three to four years, the ECCB has thoroughly validated Vincent’s earlier position, confirming its accuracy.
In the second decade of this century, evolutionary researchers such as Domazet-Lošo and Tautz, [126,127] Trigos, [128,129,130] and Lineweaver and Davis. [131] Zhou et al. [132] extend these evolutionary ideas and provided evidence that in cancer, young multicellular genes (yMGs) from the transition period are downregulated and older unicellular genes– untypical for multicellular cell systems -are upregulated. They proposed that tumor growth follows a branching evolutionary pattern, tracing back to a common ancestor from which cancer cell subclones with different fitness diverge and proliferate, supporting the idea that key features of cancer arise from the disruption of molecular networks that originally evolved during the transition to multicellularity.
In a recent review article, Rebolleda-Gomez and Travisano in 2018 [133] investigated whether ancestral unicellular wild forms capable of developing multicellular phenotypes could revert from multicellularity to unicellularity (MUT). They found that environmental changes and constant agitation and oxygenation of Saccaromyces cerevisiae cultures could induce multicellular structures really to revert to a unicellular state and demonstrate that the reversible switch from unicellularity to multicellularity and vice versa is still possible today.
Although the transition to multicellularity is believed to be evolutionarily stabilized by traits that prevent unicellular reversion [134,135,136,137]—this is not the case. MUT events provided a clear survival advantage in the evolutionary past and in cancer. Reverting cells evolved into independent populations with evident reproductive benefits. The ability to undergo genomic inversion is a key evolutionary factor of the transition period [138]
The aforementioned researchers recognized that the unicellularized cancer cell system exists within the host body but is no longer an integrated part of it. They provided valuable insights, particularly the idea that this system resembles a protozoan-like population with evolutionary roots in the Precambrian.
However, only ECCB research—leveraging the deep homology between the unicellular cancer cell system and the Entamoeba model—has been able to clarify the organization and evolution of the cancer cell system [23,24,25,26]. ECCB studies have demonstrated that MUT processes begin even in non-cancerous individuals, specifically in dysfunctional stemgermline cells with severe DNA DSB that cannot be repaired by multicellular mechanisms. The primary function of MUT is to eliminate irreparable DSB damage, which can no longer be resolved within a multicellular framework. Evolutionary genome inversion facilitates hyperpolyploid MGRS repair.
MUT processes and unicellularization begin within restorative senescence niches. However, neither MUT nor the subsequent proliferation of DSCD cells restores genomic integrity. Importantly, MUT-derived DSCD cells are not yet cancerous; rather, they are governed by the logic of a unicellular system. These cells can undergo homotypic fusion to form hyperpolyploid unicellular MGRS syncytia. Within these syncytia, giant nuclei initiate genome reconstruction—but according to unicellular principles. These hyperpolyploid nuclei eliminate DNA DSB damage of multicellular origin and reduce their nuclear mass by producing numerous haploid buds. These buds, having regained both genomic integrity and stemness, serve as precursors to a nascent stemgermline that establishes the autonomous unicellular cancer cell system. This, ultimately, is the fatal price the multicellular system pays: it enables MGRS-mediated genome repair and the survival of individually dysfunctional cells, at the cost of relinquishing multicellular control.
7.0. Intratumoral Evolution: Oxygen Gradients and Intra-Tumoral Heterogeneity (ITH)
7.1. Angiogenesis and Hypoxia: Contradictory Effects, Stemness Loss
Anatomically, the development of solid tumors progresses through several stages. [139] The first is hyperplasia, characterized by the rapid proliferation of genetically altered cells. This is followed by dysplasia, in which the overgrowing cells begin to undergo changes in cell type (Figure 5).
From the perspective of evolutionary cancer biology, hyperplastic overgrowth is driven by defective symmetric proliferation, associated with the DSCD phenotype. In contrast, dysplastic growth is mediated by progenitor cells and the nascent cancer stem-germline, which proliferate asymmetrically to form com-CSCs. This DSCD-to-ACD transition represents a key component of the stemness-regain cycle, in which DSCD cells (depicted as green cells) give rise to emerging stemgermline cells (brown cells) (Figure 5). Both hyperplastic and dysplastic cell states may reflect irreversible phenotypic transitions during the stemness recovery process.
Hyperplastic overgrowth progressively consumes oxygen from the tumor periphery toward the center, creating hypoxic conditions in the inner regions. These hypoxic zones, in turn, facilitate the dysplastic transition and promote ACD-driven growth. Both hyperplastic and dysplastic tumor layers develop simultaneously, influenced by gradients of oxygen availability and microenvironmental stimuli.
The resulting spectrum of intra-tumoral oxygen levels both accelerates and constrains ACD-driven growth. In particular, oxygen excess can temporarily halt the production of com-CSCs (com-CSCs) and increase the proportion of cells entering a quiescent state. This generates a compensatory physiological demand for enhanced oxygen supply. In response, angiogenic signals (Figure 6) are released from consequent hypoxic tumor core, stimulating nearby blood vessels and initiating the growth of new angiogenic capillaries toward the tumor mass. [140]
Some of the oxygen-sensitive, stemness-positive ACD cells located near blood vessels are exposed to too much oxygens. This leads dysfunction of DNA repair genes and DNA DSB damage, resulting in a phenotypic shift toward the dysfunctional, stemness-negative DSCD state, which no longer produces com-CSCs. To compensate for this loss of CSCs, these cells activate the MGRS/PGCC pathway to generate new functional stem germlines, sublines, or clones capable of reactivating com-pCSC production.
The dynamics of oxygen supply, consumption, and depletion within the tumor microenvironment drive continuous intra-tumoral cycles of stemness loss and recovery, enabling the regeneration of com-pCSC pools.
A second pathway for intratumoral com-CSC generation is the SGT/EMT process initiated within the primary tumor from somatic cancer cells that are oxygen- resistant and preserve the integrity and functionality of the stemgermline genome.
7.2. Further Evolution of the Cancer Genome: Lateral Gene Transfer and Genome Expansion
The molecular processes driving to genomic expansion are based on ancient lateral gene transfer (LGT) processes dating back to the transition period to multicellularity. It can be assumed that during the early transition period, the common AMF ancestor expanded its genome through heterotypic cell fusions with genomically distinct competitors. Functionally advantageous genes were retained, while non-beneficial ones were eliminated or repurposed.
Foreign gene hijacking serves as an evolutionary mechanism for expanding genomic functionality within the cancer cell system. This process likely enhances invasiveness and pathogenicity in evolutionarily related parasitic amoebae while also contributing to the emergence of new strains.
7.3. Hijacked Genes Are Active Multicellularity Genes (MGs) of the Host
Through conserved mechanisms from the transition period, a large number of hybrid genomes can be formed in primary tumors where heterosyncytia arise from the heterotypic fusion of unicellularizeds cancer cells with multicellular somatic host cells. Notably, many host macrophages undergo heterotypic fusions with cancer cells. It is hypothesized that some of these UG/MG hybrids remain non-proliferative, while others become proliferative and accumulate DNA replication defects (mutations).
As previously described, when stemgermlines lose their stemness and cease producing pCSCs, they send distress signals to their environment. These signals reach not only homotypic somatic cancer cells—genomically identical to the dysfunctional stemgermline—but also proliferative and non-proliferative hybrid cells that arise from heterotypic fusion with multicellular host cells and also contain functional multicellular host genes (MGs). In response to loss-of-stemness signaling, hybrid cells are also integrated into SGT/EMT processes, leading to the formation of secondary stemgermlines clones, and secondary sCSCs.
7.4. Intratumoral Heterogeneity (ITH) Reflects Phenotypic and Genomic Diversity Within Cancer Stemgerm Lineages
Each newly formed secondary germline clone differs genomically and phenotypically previous formed clones, which arise through fractal or non-fractal SGT/EMT processes [25,26]. Functional clones and sublines derived from heterosyncytia exhibit distinct proliferative capacities, forming secondary sCSC pools of varying sizes and potency. Additionally, their genomes may contain genes or gene modules associated with tissue-specific and teratoma-like profiles.
Each new secondary ACD clone and subline arising from heterotypic syncytia is oxygen-sensitive and susceptible to intra-tumoral oxygen fluctuations. In response to increased oxygen availability, secondary ACD cells also interrupt com-CSC production. The depletion of com-CSCs and the formation of sDSCD cells can subsequently activate the MGRS/PGCC-mediated genome repair program or initiate additional EMT processes.Through successive rounds of lateral gene transfer (LGT), MGRSs/PGCCs, and fractal EMT processes, new secondary stem-germline clones and sublines emerge—often exhibiting enhanced invasive potential. These evolutionary processes collectively contribute to intra-tumoral heterogeneity (ITH) and the dynamic genomic landscape observed in tumors.
The genomic expansions described here are rooted in evolutionary mechanisms, during transitional period to multicellularity, when the common AMF ancestor repeatedly accessed genes from genomically distinct competitors, facilitating the evolution toward multicellularity.
All secondary ACD clones responsible for sCSC production maintain genomic stability and preserve their integrity. If one were to refer to genomic chaos or instability, it would apply solely to the numerous somatic cancer cell lines and sublines, which accumulate irreparable replication defects (mutations), rather than to the bouquet of stable stemgermline clones.
At the end of this intra-tumoral evolution, secondary stemgermline cells with varying degrees of invasive potential exit the primary tumor structure and encounter damaging hyperoxic conditions within the bloodstream and surrounding tissues. However, most of these cells migrate in association with oxygen-resistant cells, forming multicellular clusters that enhance the viability and functional capacity of the migrating stemgermline cells. The most favorable niches for metastatic colonization are located in the bone microenvironment, particularly within its hypoxic niches, where metastases preferentially develop. [141,142]
8. Circulating Tumor Cells (CSCs) in Tissue and Bone Metastases
8.1. CTCs Originate from Cancer Stemgermlines, Sublines and Clones
Circulating tumor cells (CTCs) is a collective term that was introduced by Ashworth as early as 1869. [143] It refers to all types of cancer cells that leave the primary tumor and migrate through peripheral blood vessels and the bloodstream to secondary sites in various organs. [144,145,146,147,148] CTCs can have an epithelial, mesenchymal, or hybrid profile. Although the majority of CTCs are eliminated by the host immune system, a subset possesses high metastatic potential and can evade immune surveillance, leading to the formation of microscopic cancer foci, tumor recurrence, and metastasis.
8.2. Intrinsic Properties of CTCs
According to Gao et al., [149] only a limited number of disseminated tumor cells are capable of forming metastatic tumors. This capacity depends on both the intrinsic properties of CTCs and external regulatory factors. Previous studies by Lambert et al., [150] and Liu and Cao [151] highlighted genotypic and phenotypic characteristics as key determinants of CTC survival, invasion and metastatic efficiency. Gao et al. [149] also refer to the “temporal/spatial secrets” of metastatic tumor cells, proposing that “transient and bidirectional CSC differentiation occurs, which is strongly influenced by microenvironmental factors”. Accordingly, “metastatic cells inherit critical driver mutations from their parental tumors allowing the persistence of tumorigenic ability and on the other hand, metastatic cells develop new mutations to adapt to emerging challenges during metastasis”.
Current insights from Evolutionary Cancer Cell Biology do not support this proposal. From an evolutionary perspective, metastatic progression also involves recurring ACD–DSCD cycles and alternating phases of stemness depletion and restoration. Crucially, the reestablishment of stemgermline functionality and genomic integrity occurs exclusively through the MGRS/PGCC-mediated genome repair pathway. In contrast, EMT processes primarily function to replace damaged stemgermlines by generating new secondary ACD clones and com-sCSCs. According to ECCB, the behavior of stem-germline cells during migration, invasion, and metastasis is governed exclusively by ancestral mechanisms intrinsic to the unicellularized cancer cell system. In contrast, the current multicellular
8.3. Migrating CTC Clusters Escape Hyperoxic Damage
Cancer cells can detach from the primary tumor either as multicellular aggregates or as single cells that subsequently form heterotypic clusters within the peripheral blood circulation. [152] These CTC clusters arise through the aggregation of varying numbers of cells [148] mediated by intercellular adhesion molecules. [153] Both the size and number of CTC clusters have been shown to correlate significantly with metastatic potential and clinical outcomes. [154,155]
CTCs clusters that survive in the bloodstream are capable of initiating metastases. The prevailing view is that clustered CTCs exhibit a higher metastatic potential compared to single CTCs. Upon entering the circulation, tumor cells lose the protective hypoxic microenvironment of the primary tumor. Consequently, only those CTCs that can adapt to the hyperoxic conditions of the circulatory system are able to survive, disseminate to distant tissues, and ultimately form metastases.
According to Chen et al., [142] additional challenges in peripheral blood further threaten CTC survival. These include attacks by the host immune system and exposure to apoptotic signals, both of which limit the ability of migrating cancer cells to persist and colonize secondary sites.
Metastasis is a multistage process involving several intermediate steps. During this progression, CTC-protected stemgermline cells and must adapt to the unfamiliar hyperoxic environment of distant tissues in order to survive, colonize, and ultimately form secondary tumor lesions. Aggregation and clustering is the response of stemgermline cells that evade primary tumors.
8.4. The Advaage of Collective Migration
As revealed by Chen et al., [142] only those CTCs capable of adapting to the new microenvironment are able to successfully disseminate and establish metastatic lesions. Notably, CTC clusters are 50–100 times more likely to facilitate metastasis than individual tumor cells [156,157] As cancer progresses, the number of detectable CTC clusters increases, suggesting a strong association between cluster formation and metastatic progression. [158,159]
During migration and invasion, CTC clusters possess significant advantages over single CTCs. This advantage lies in the formation of hypoxic–normoxic gradients within the CTC clusters, along with a protective outer layer of surface cells that shields the oxygen-sensitive stemgermline cells at the core. This protective architecture enables stemgermline cells to maintain and recover stemness and ACD potential, thereby allowing the generation of sufficient numbers of com-CSCs. The oxygen-buffering and immunosuppressive microenvironment within the cluster [160] facilitates the continued progression of cancer by preserving the genomic integrity and functionality of the stemgermline, clones and subclones.
8.5. Homotypic and Heterotypic CTC Clusters
As cancer cells travel through the bloodstream, they are exposed to a variety of external stresses until they reach distant organs. [161] The collective migration by hyperoxia-resistant clusters has been identified as a key mechanism of invasion, whereas the migration of single, unprotected cancer cells appears to be largely ineffective or absent. [162]
In recent years, it was observed that CTCs are heterotypic aggregates with non-tumor cells, including neutrophils, platelets, myeloid cells, cancer-associated fibroblasts (CAFs), and tumor-associated macrophages (TAMs). [148] These heterotypic associations are widely believed to provide CTCs with enhanced protection against the hostile conditions of the circulatory system, enabling them to evade immune attack, resist shear stress, and survive transit. Ultimately, such interactions facilitate the successful establishment of metastatic lesions at distant sites but also heterotypic cell fusion. Heterotypic clusters show greater metastatic capability compared to homotypic clusters. [163]
Gu et al. [148] discussed recently the role of adhesion proteins in homotypic clustering and showed that tumor hypoxia can upregulate genes that produce such adhesion proteins, [164] and improve overexpression of stem-like traits, [165,166] survival and self-renewal ability.
The ECCB demostrates that homotypic clustering is an ancient evolutionary adhesion mechanism, also observed in amoebae. In Entamoeba, homotypic cell clustering leads to cluster hypoxia, a condition essential for initiating the generation of stemgermline replacement, through processes such as SGT and polyploidization/depolyploidization cycles. [24] Ultimately, this environmentally controlled pathway gives rise to progenitor cells capable of producing new stemgermline, sublines and clones. In this way, the unicellular cell system resolves genomic dysfunction and recovers new ACD clones and sublines capable of generating new committed, non-proliferative stem cells.
8.6. Fractal EMT Phenotypes and Polyclonal Dissemination
Recent studies reveal that the progression of cancer from the primary tumor to metastasis involves polyclonal dissemination and polyclonal transfer, characterized by individual traits related to growth. [167] Fractal EMT/SGT phenotypes play an important role in this process. Studies have shown that weakly migrating subsets tend to be more epithelial (E). [168] and thus, more somatic in nature, whereas strongly migrating subsets are more mesenchymal (M) and exhibit more stemness-like characteristics. Many CTC clusters display a partial EMT phenotype, [169] with some cells within the cluster acting as “leaders” (initiating migration) and others as “followers.” The alternation between mesenchymal “M-leader” phenotypes and epithelial E-followers is considered particularly advantageous for tumor progression. [170,171] This phenotypic M- and E-clonal diversity enables cells to more effectively adapt to changes in the tumor microenvironment cancer, thereby promoting tumor progression. [172]
8.7. Bone Metastases
Nearly all types of cancer have the potential to spread (metastasize) to the bones. However, certain cancers, such as breast and prostate cancer, are particularly prone to bone metastasis. Among various metastatic sites, bone is the most frequent target for tumors originating in the breast and prostate. Tumor cells can escape from the primary site and colonize hypoxic bone niches. [173]
Depending on the degree of hypoxia within the bone niche, disseminated tumor cells—including those associated with multiple myeloma—may enter a state of dormancy, remaining quiescent for years before reactivating. Upon resuming proliferation, these cells contribute to overt metastasis, leading to bone destruction through osteoclast-mediated osteolysis. [174]
9. The Dogma of Cancer Genome Instability
The current concept of genomic instability (GI) stems from the dogma that cancer is purely multicellular. Current cancer research often compares the multicellular genome of non-cancerous host cells genome with the unicellular genome of cancer cells, revealing significant differences (“mutations”)—which are not surprising from an ECCB perspective. In the multicellular cancer view, GI is a hallmark of the entire cancer population without distinguishing between stem and non-stem (ACD and DSCD) cell types. GI refers to an increased tendency to genomic alterations during the cell cycle and has been defined as an abnormally high frequency of errors in the mitotic progeny.
In 2011, Zhiyuan Shen [175] identified GI as the main driving force of tumorigenesis. According to this concept, the accumulation of genomic alterations—through mutations at specific genes, amplifications, deletions or rearrangements of chromosome segments—can lead to dysregulated cell division and imbalanced growth, eventually resulting in [multicellular] cancer. In contrast, non-cancerous cells were characterized by genomic stability (GS) which is maintained through high fidelity DNA replication, error-free repair of sporadic DNA, and tightly regulated cell cycle progression with checkpoint control.
This view remained dominant and has expanded year by year. In 1991, Loeb [102,103] proposed that an early step in tumor progression is the emergence of a mutator phenotype resulting from mutations in genes normally responsible for maintaining genetic stability. At that time, it was believed that the loss of such genes could lead to GI and thus promote cancer development. Consequently, the prevailing assumption was that the functional differences between healthy multicellularity and dysfunctional cancer multicellularity could only be explained by mutations, genomic instability, and chaos.
Many researchers supported this perspective. Yao and Dai in 2014 [100] recognized a heightened tendency for genome alteration during cancer cell division, attributing this to defects in surveillance mechanisms such as DNA damage checkpoints, repair systems, and mitotic checkpoints. They concluded that the malfunction these multicellular regulatory mechanisms predisposes cells to malignant transformation. Unfortunately, the authors do not acknowledge that the transformation of the dysfunctional multicellular unit is actually a MUT process.
In this vein, Salmaninejad et al. (2021) [176] linked aberrations in DNA repair mechanisms with GI and mutation, identifying the activation of oncogenes and/or inactivation of tumor suppressor genes as major consequences of this instability. This is why GI genomic instability is frequently considered a critical factor in carcinogenesis and a defining feature of many human malignancies.
Even in 2022, Chen et al. [177] and others pointed out that GI results from various genomic alterations, including germline or somatic defects in DNA repair, [158] oncogene-induced replication stress, [178] defective mitotic chromosome segregation, [179] and other genomic impairments. [180] They emphasize that cancer-associated GI is driven by multiple processes that regulate DNA replication and repair, including the formation of micronuclei due to defective DNA repair. [181]
9.1. Loss and Recovery of Genomic Stability in the Primary Stemgermline
Today, however, thanks to ECCB, we understand that the unicellularized cancer cell system—like all other related unicellular cell systems—has two mechanisms for generating genomically identical sublines and clones, with the nascent (primary) stemgermline. One is the MGRS/PGCC genome repair pathway; the other is the homotypic EMT replacement pathway, which ensues the formation of additional clones and sublines of the same genomic identitiy. Both mechanisms restore the previously intact genomic state that was lost in dysfunctional DSCD stemgermline phenotypes due to stress and hyperoxia. Cancer stemgerm lineages are therefore highly stable, not chaotic.
Thus, the previous assumption of instability and loss of genomic integrity is incorrect—at least for the overweight stemgermline (1–2% of tumor cells), which regulates cell fate in the remaining tumor cell population. Nevertheless, this new and still emerging understanding of ECCB has not yet been widely recognized. We continue to speak of genomic instability, loss of integrity, and chaos
9.2. Genome Expansion and Evolution: Heterotypic cell Fusion, Hybridization, Fractal SGT/EMT; Secondary Stemgermlines and com-sCSCs
According to the ECCB, the unicellularized cancer and tumor cell system operates as a dual cell system, comprising two distinct lineages: (i) genomically stable stemgermlines, which include multiple sublines and clones—the stemgerm lineage—capable of repairing genomic defects and maintaining genomic stability (GS) in all stemgermlines formed through heterotypic SGT from hybrid UG/MG cells, and (ii) secondary somatic cell lineages derived from the stemgermlines conversion, which accumulate more replicative DNA damage (mutations) due to their higher symmetric-proliferative activity. Secondary and primary (nascent) cancer stemgermlines are always repaired by MGRS/PGCCs repair pathway and hyperpolyploidy.
9.3. Primary and Secondary com-CSCs as Progenitor Reservoirs for Stemgermline Sublines and Clones (Progenitor Accumulation)
In the past, the stemgermline was mistakenly understood as a proliferative CSC line. In reality, “proliferative CSCs” are the self-renewing stemgermline cells, while “differentiating CSCs” are the non-proliferative com-CSCs. As long as the two synergistic cell types were not analyzed separately, a comprehensive understanding of cancer plasticity, carcinogenesis, and tumorigenesis remained elusive.
Due to this confusion, conventional multicellular cancer research has failed to resolve key questions about the origin of CSCs. Their definition remains ambiguous and is still based on the foundational assumption by Bonnet and Dick in 1997, [182] who proposed that CSCs are both proliferative and capable of differentiation. CSCs were also believed to exhibit phenotypic plasticity, enabling them to adapt to the tumor microenvironment and contribute to the genetic heterogeneity of tumors. [183,184,185]
According to the ECCB, committed pCSCs and sCSCs are non-proliferative and only capable of differentiation or pool accumulation. Only the stemgermline is proliferative, giving rise to com-CSCs. Each pCSCs and sCSCs fractions are genomically identical to their corresponding primary and secondary stemgermlines that generated them. The heterogeny observed in the CSC population reflects the heterogeny within in the stemgerm lineage of cancer, which include various genomically stable stemgermlines, sublines and clones. CSC evolution is essentially the evolution of genomically expanding stemgermlines.
Note From the perspective of the ECCB, there is no genomic instability in the all-determining, all-controlling stemgermlines of cancer. The differences observed between the multicellular genome of non-cancerous organisms and the unicellularized genome of cancer cells do not signify a loss of genomic stability or integrity. Similarly, genomic expansion through intra-tumoral lateral gene transfer does not represent chaos. Whether expanded or unexpanded, the stemgermline genome remains remarkably stable. It is repaired by the MGRS/PGCC pathway, which restore the integrity of the genome.
9.4. Stemgermlines Resilience to Mutational Dysregulation
A fundamental question remains: Why does the seemingly endless chain of mutations observed in molecular cancer research exert so little influence—and such minimal negative effect—on the organization and development of key stem-germ lineages, including the productive sublines, clones and sub-clones responsible for com-CSCs generation?
The author of this paper proposes that the answer lies in the hybrid nature of cancer cells. These cells, in addition to expressing upregulated unicellular genes (UGs), also more or less downregulated homologs of young multicellular genes (yMGs) from the historic transition period, and non-downregulated, hijacked MGs from the host organism. This hybrid genomic configuration gives rise to persistent conflicts within the cell’s regulatory network, resulting in widespread replicative mutations. However, many of these mutations may be eliminatedthrough hyperpolyploid genome repair mechanisms activated during DSCD–ACD cycles, ultimately restoring the genomic integrity and stemness of cancer stemgermlines.
Recent literature provides growing support for the idea that cancer cells actively engage mutation control mechanisms. For instance, a 2020 study by Zhou et al. [186] on DNA repair pathways during cancer evolution demonstrated that cancer cells often depend on compensating repair mechanisms to maintain a controlled level of genomic instability. Reactivation of homologous recombination (HR) pathways—such as through restoration of BRCA protein functionality [187] or withdrawal of mutagenic therapeutic agents—can lead to the rewiring of DNA repair pathways. In addition, DNA repair pathways could also be regulated under controlled environmental conditions
In line with this evidence, the author [26] posits that the genomic stability and integrity of cancer stemgermlines and their derivative clones is safeguarded by hyperpolyploid MGRS/PGCC mechanism. Thise act as a “guardian system” of stemgermline autonomy and genomic integrity, continuously purging the genome of deleterious mutations and restoring functional order. This mechanism is likely homologous to the ancient functions of the common AMF ancestor, which may have performed similar genome-stabilizing roles during the Neoproterozoic and Paleozoic eras (1000–500 million years ago). Notably, stressed or defective DSCD cells in modern amoebae such as Entamoeba still appear to fulfill this function.
Note: One could argue that the MGRS/PGCC mechanism is more than a genome repair program; it represents a critical system for mutation clearance, stemness restoration, and the maintenance of genetic integrity. This perspective may also explain the frequent detection of PGCCs in metastatic and highly invasive tumors, suggesting that the prevalence of PGCCs could serve as a prognostic indicator or staging tool in cancer progression.
10. Conclusions and Perspectives
Despite decades of intensive research and the accumulation of vast molecular mutational datasets, fundamental questions regarding cancer initiation and the origin of cancer stem cells remain unresolved. This disconnection between the sheer volume of data and the lack of conceptual clarity calls into question the continued dominance of classical, mutation-centric theories. Over the past 25 years, breakthroughs in non-genetic cancer research and evolutionary biology have increasingly exposed these contradiction—and, in many cases, the failures—of reductionist explanations of carcinogenesis and tumorigenesis solely in multicellular mutational theories.
Persistent theoretical impasses in cancer genomics reflect a deeper conceptual rift: the traditional view of cancer as a disease driven by the progressive accumulation of mutations continues to overshadow the evolutionary framework of cancer. Prevailing older hypotheses, which interpret cancer as a chaotic disorder and mutational overload, have proven inadequate in explaining fundamental questions such as the origin and evolution of cancer.
Given these enduring conceptual challenges and mounting inconsistencies, it is increasingly untenable to uphold mutation-driven narratives as the sole explanatory model. A paradigm shift—one that embraces evolutionary dynamics, non-genetic inheritance, and systems-level constraints—is urgently needed to advance a more coherent and predictive understanding of cancer biology.
Contrary to longstanding assumptions, the cancer cell is not a collection of persistent degenerating multicellular entitiesy undergoing progressive genomic and organizational disintegration. Rather, it arises through a multicellular-to-unicellular transition (MUT) process, giving rise to a self-organizing cell system that frequently demonstrates remarkable autonomy, adaptability, resilience, and regenerative potential. The prevailing portrayal of cancer as an aberrant, randomly evolving, and chaotic multicellular system fails to account for the coordinated and highly reproducible behaviors observed in many tumors, where organized hierarchies and lineage programs are both established and maintained.
Remarkably, the seemingly endless chain of mutations often has little relevance to the genomic stability of stemgermlines that sustain tumor proliferation. Cancer stemgermlines can repeatedly restore lost stemness and regain genomic and functional integrity through hyperpolyploid genome repair and reductive nuclear divisions, challenging the assumption that mutation accumulation inevitably leads to terminal disorganization. Importantly, transient states of genomic dysfunction should not be mistaken for irreversible chaos or instability
The historical absence of an evolutionary framework has compelled cancer biology to rely on explanatory models that once seemed rational but no longer withstand critical scrutiny. As cancer research moves forward, an evolutionarily informed perspective is essential for resolving the conceptual deadlocks that have long impeded progress.
Nevertheless, recent advances in molecular research have generated a wealth of valuable insights into cancer cell biology. However, in the absence of a comprehensive, biologically coherent system model, many of these findings remain disconnected—insightful in isolation, yet difficult to interpret within a unified conceptual framework. The ECCB, with its integrative and systems-level approach, offers the appropriate theoretical toolkit to contextualize and synthesize disparate molecular findings. By embedding molecular data within an evolutionary perspective, ECCB enables a more coherent interpretation of cancer’s complexity, dynamics, and emergent behaviors.
Previous mutational research, sought to identify the cause of carcinogenesis in mutations affecting oncogenes and tumor suppressor genes (TSGs)—and their driver mutations. From an evolutionary perspective, however, oncogenes and TSGs are ancient, deeply conserved genes that regulate cell cycle processes across all forms of life—including the cell cycles of protists, metazoans, and the unicellularized cell cycle of cancer. These regulatory genes are thought to have originated in the deeply hypoxic Mesoproterozoic era and continued to evolve during the more oxygen-rich Neoproterozoic, where they came to control both asymmetric ACD and dysfunctional- symmetric DSCD cell cycling.
In contrast, MUT genes— which emerged approximately 600 to 550 million years ago during the evolutionary transition to multicellularity (the UMT era)—are evolutionarily much younger. These genes initiate carcinogenic MUT processes and promote the regeneration of dysfunctional genomes. From the standpoint of effective cancer prevention, identifying and molecularly characterizing these MUT genes could offer a novel strategy: the targeted downregulation of such genes might prevent the onset of carcinogenic processes. Additional targets include the DSCD cells, the precarcinogenic and tumorigenic genome repair cycles, the phases of restorative senescence, and the stemness loss and repair cycles—particularly the transition from DSCD to ACD phenotypes.
All of these emerging research areas offer the promise of cancer prevention strategies, including the development of vaccines, as well as improved treatment options in the early stages of cancer evolution.
Acknowledgments
This work is likely the final contribution in a long series of articles dedicated to the field of Evolutionary Cancer Cell Biology (ECCB). I extend my deepest gratitude to the many scientific colleagues, friends, and mentors who have supported and inspired me throughout the development of this project, which remains very close to my heart. Above all, I thank my wife, microbiologist Dr. Eugenia R. Niculescu, for her tireless scientific support over the years. I am also especially grateful to Professor Dr. Dan L. Danielopol (University of Graz, Austria) for his insightful guidance and enduring influence on my understanding of evolutionary theory and conceptual change.
Abbreviations
ACD, asymmetric cell division; AMF, amoebozoans, metazoans and fungi;
CSC, cancer stem cells; pCSCs, primary CSCs; CTC, circulating tumor cells; DSCD, dysfunctional symmetric cell division; ECCB, evolutionary cancer cell biology; GI, genome instability; GS, genome stability; GST/MET, germ-to soma transition/mesenchymal-epithelial-transition; ITH, intra-tumoral heterogeneity; MGRS/PGCC, multinucleated genome repair structures/ polyploid giant cancer cells; MUT, multicellular to unicellular transition; SCD, symmetric cell division; SGT/EMT, soma to germ transition; UMT, unicellular to multicellular transition; UG/MG, hybrid unicellular cancer cell containing hijacked multicellular genes MGs (lateral gene transition)
References
- Bodmer WF. The evolutionary progression of cancers. Academia Oncology 2024; 1(2). [CrossRef]
- Bizzarri M. Systems Biology for Understanding Cancer Biology. Curr Synthetic Sys Biol 2013; 2:2. 10.4172/2332-0737.1000e103.
- Dollo L. Les lois de l’évolution/ The Laws of Evolution (translated by M. Carrano). Bull. Soc. Belge Geol. Paleontol. Hydrol. 1893; VII, 164–166.
- Gould SJ. Dollo on Dollo’s law: irreversibility and the status of evolutionary laws. J. Hist. Biol. 1970; 3, 189–212.
- Collin R, Miglietta MP. Reversing opinions on Dollo’s Law. Trends Ecol. Evol. 2008; 23, 602–609.
- Hall BK. Dollo’s Law. In Encyclopedia of Evolution, Pagel M, ed, 2002, 287–288, Oxford Univ Press.
- Elmer KR, Clobert J. Dollo’s law of irreversibility in the post-genomic age. Trends Ecol Evol. 2025; 40(2):136-146. [CrossRef]
- Forni G, Martelossi J, Valero P, Hennemann F, Conle O et al. Macroevolutionary analyses provide new evidence of phasmid wings evolution as a reversible process. Syst. Biol. 2022; 71, 1471–1486.
- Sadier A, Sears KE, Womack M. Unraveling the heritage of lost traits. J Exp Zool B Mol Dev Evol. 2022; 338(1-2):107-118. [CrossRef]
- Paluh DJ, Dillard WA, Stanley EL, Fraser GJ, Blackburn DC. Reevaluating the morphological evidence for the re-evolution of lost mandibular teeth in frogs. Evolution 2021; 75, 3203–3213.
- Barrere J, Nanda P, Murray AW. Alternating selection for dispersal and multicellularity favors regulated life cycles. Curr Biol. 2023; 33(9):1809-1817.e3. [CrossRef]
- Grosberg RK, and Strathmann RR. The Evolution of Multicellularity: A Minor Major Transition? Annual Review of Ecology, Evolution, and Systematics 2007;38, 621–654. 10.1146/annurev.ecolsys.36.102403.114735.
- Brunet T, King N. The Origin of Animal Multicellularity and Cell Differentiation. Developmental Cell 2017; 43, 124–140. 10.1016/j.devcel.2017.09.016.
- Velicer GJ, Yu YN. Evolution of novel cooperative swarming in the bacterium Myxococcus xanthus. Nature 2003;425:75–78. 10.1038/nature01908.
- Gilbert OM, Foster KR, Mehdiabadi NJ, Strassmann JE, Queller DC. High relatedness maintains multicellular cooperation in a social amoeba by controlling cheater mutants. PNAS 2007;104, 8913–8917. 10.1073/pnas.0702723104.
- Kuzdzal-Fick JJ, Fox SA, Strassmann JE, Queller DC. High relatedness is necessary and sufficient to maintain multicellularity in Dictyostelium. Science 2011;334, 1548–1551. 10.1126/science.1213272.
- Wielgoss S, Wolfensberger R, Sun L, Fiegna F, Velicer GJ. Social genes are selection hotspots in kin groups of a soil microbe. Science 2019;363, 1342–1345. [CrossRef]
- Conlin PL, Ratcliff WC. Evolution: Understanding the origins of facultative multicellular life cycles. Curr Biol. 2023; 33 (9), R356-R358.
- Chen H, Lin F, Xing K. et al. The reverse evolution from multicellularity to unicellularity during carcinogenesis. Nat Commun 2015; 6: 6367 . [CrossRef]
- Srivastava, M. et al. The Amphimedon queenslandica genome and the evolution of animal complexity. Nature 2010; 466, 720–726.
- Davies, PC, Lineweaver CH. Cancer tumors as Metazoa 1.0: tapping genes of ancient ancestors. Phys.Biol.2011;8,15001.
- Sanchez Alvarado A. Cellular hyperproliferation and cancer as evolutionary variables. Curr. Biol. 2012; 22, R772–R778.
- Niculescu VF. Reevaluating cancer stem cells and polyploid giant cancer cells from the evolutionary cancer cell biology perspective. Cancer Plus 2024, 6(4), 3970. [CrossRef]
- Niculescu VF. Understanding cancer from an evolutionarymperspective: High-risk reprogramming of genome-damaged stem cells. Acad Med. 2024;2.
- Niculescu VF, Niculescu ER. The enigma of cancer polyploidy as deciphered by evolutionary cancer cell biology (ECCB). Acad Med. 2024;1(2).
- Niculescu VF. Cancer Genomics: Restorative Senescence, Transition to Unicellularization, and Cycles of Stemness Recovery. Acad Mol Bol Gen. 2025;. [CrossRef]
- Bank S, Bradler S. A second view on the evolution of flight in stick and leaf insects (Phasmatodea). BMC Ecol. Evol. 2022; 22, 62.
- Visser B, Alborn HT, Rondeaux S, Haillot M, Hance T, Reba D. (2021) Phenotypic plasticity explains apparent reverse evolution of fat synthesis in parasitic wasps. Sci. Rep. 2021, 11, 7751 . [CrossRef]
- Lynch VJ. Is there a loophole in Dollo’s law? A DevoEvo perspective on irreversibility (of felid dentition). J. Exp. Zool. B Mol. Dev. Evol. 2023: 340, 509–517.
- Niculescu VF. Growth of Entamoeba invadens in sediments with metabolically repressed bacteria leads to multicellularity and redefinition of the amoebic cell system. Roumanian Archives of Microbiology and Immunology 2013;72(1):25-48.
- Niculescu VF. On the Origin of Stemness and Ancient Cell Lineages in Single-Celled Eukaryotes. SOJ Microbiol Infect Dis 2014; 2(2): 1-3. [CrossRef]
- Niculescu VF. Extrinsic and intrinsic signaling control cell fate specification in the stem cell system of Entamoeba invadens. Researchgate. 2014;. [CrossRef]
- Niculescu VF. The cell system of Giardia lamblia in the light of the protist stem cell biology. Stem Cell Biol Res.1:3. [CrossRef]
- Niculescu VF. Evidence for asymmetric cell fate and hypoxia induced differentiation in the facultative pathogen protist Colpoda cucullus.Microbiol Discov. 2014;2:3. [CrossRef]
- Niculescu VF. The stem cell biology of the protist pathogen Entamoeba invadens in the context of eukaryotic stem cell evolution. Stem Cell Biol Res. 2015; 2:2. [CrossRef]
- Niculescu VF (2015) Axenic stress leads the minor stem cell line of Entamoeba histolytica to defective mitosis and aberrant reversible endopolyploidy. Conference: XVIII Seminar of Amebiasis, Campeche, Mexico, 2015;13-16, 2015. [CrossRef]
- Niculescu VF. Low-oxygen environments slow down precursor cell development in the pathogen anaerobe protist Entamoeba invadens. J Stem Cell Res Med 2016;1(1): 27-35. [CrossRef]
- Niculescu VF. Developmental and Non- Developmental Polyploidy in Xenic and Axenic Cultured Stem Cell Lines of Entamoeba invadens and E. histolytica. Insights Stem Cells.2016; 2:1.
- Niculescu Vladimir F. Regulatory Mechanisms of Asymmetric/Symmetric Cell Division and Quiescence in the Primitive-/ Stem Progenitor Cell Lineage of Entamoeba. Stem Cells Regen Med. 2017;1(2): 1-7.
- Niculescu VF. The evolutionary cancer genome theory and its reasoning. Genetics in Medicin Open. 2023; 1(1):100809. [CrossRef]
- Krishnan D, Ghosh SK. Cellular events of multinucleated giant cell formation during the encystation of Entamoeba invadens. Front Cell Infect Microbiol 2018; 31(8):262.
- Hazra S, Kalyan Dinda S, Kumar Mondal N, Hossain SR, Datta P et al. Giant cells: Multiple cells unite to survive. Front Cell Infect Microbiol. 2023; 13:1220589. [CrossRef]
- Niculescu VF. The reproductive life cycle of cancer: Hypotheses of cell of origin, TP53 drivers and stem cell conversions in the light of the atavistic cancer cell theory. Med Hypoth. 2019; 123:19-23. [CrossRef]
- Niculescu VF. Carcinogenesis: recent insights in protist stem cell biology lead to a better understanding of atavistic mechanisms implied in cancer development. MOJ Tumor Research 2018; 1(1).
- Niculescu VF. aCLS cancers: Genomic and epigenetic changes transform the cell of origin of cancer into a tumorigenic pathogen of unicellular organization and lifestyle. Gene. 2020; 726:144174. [CrossRef]
- Niculescu VF. Cancer genes and cancer stem cells in tumorigenesis: Evolutionary deep homology and controversies. Genes Dis. 2022; 9(5):1234-1247. [CrossRef]
- Cheloni G, Poteti M, Bono S, Masala E, Mazure NM, Rovida E, Lulli M, Dello Sbarba P. The Leukemic Stem Cell Niche: Adaptation to “Hypoxia” versus Oncogene Addiction. Stem Cells Int. 2017;4979474. [CrossRef]
- Knoll AH, Holland HD. Oxygen and Proterozoic Evolution: An Update. In “ Panel on Effects of Past Global Change on Life. Book. 1995. National Research Council (US), Washington (DC): National Academies Press (US).
- Knoll AH. The early evolution of eukaryotes: A global perspective, Science 1992;256:622-627.
- Tostevin R, Mills BJW. Reconciling proxy records and models of Earth’s oxygenation during the Neoproterozoic and Palaeozoic. Interface Focus. 2020;10(4):20190137. [CrossRef]
- Yoo YD, Kwon YT. Molecular mechanisms controlling asymmetric and symmetric self-renewal of cancer stem cells. J Anal Sci Technol. 2015;6(1):28. [CrossRef]
- Iliopoulos D, Hirsch HA, Wang G, Struhl K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc Natl Acad Sci U S A. 2011;108(4):1397–402. [CrossRef]
- Knoblich JA. Mechanisms of asymmetric stem cell division. Cell. 2008;132(4):583–97 . [CrossRef]
- Chao, S., Yan, H, Bu, P. Asymmetric division of stem cells and its cancer relevance. Cell Regen 13, 5 (2024). [CrossRef]
- Bajaj J, Zimdahl B, Reya T. Fearful symmetry: subversion of asymmetric division in cancer development and progression. Cancer Res. 2015;75:792–7. [CrossRef]
- Choi HY, Siddique HR, Zheng M, Kou Y, Yeh DW, Machida T, Chen CL, Uthaya Kumar DB, Punj V, Winer P, et al. p53 destabilizing protein skews asymmetric division and enhances NOTCH activation to direct self-renewal of TICs. Nat Commun. 2020;11:3084. [CrossRef]
- Li Z, Zhang YY, Zhang H, Yang J, Chen Y, Lu H. Asymmetric cell division and tumor heterogeneity. Front Cell Dev Biol. 2022;10:938685. [CrossRef]
- Zhang S, Mercado-Uribe I, Xing Z, Sun B, Kuang J, Liu J. Generation of cancer stem-like cells through the formation of polyploid giant cancer cells. Oncogene. 2014;33:116–28. [CrossRef]
- Zhou X, Zhou M, Zheng M, Tian S, Yang X, Ning Y, Li Y, Zhang S. Polyploid giant cancer cells and cancer progression. Front Cell Dev Biol. 2022;10:1017588. [CrossRef]
- Granit RZ, Masury H, Condiotti R, Fixler Y, Gabai Y, Glikman T, Dalin S, Winter E, Nevo Y, Carmon E, et al. Regulation of Cellular Heterogeneity and Rates of Symmetric and Asymmetric Divisions in Triple-Negative Breast Cancer. Cell Rep. 2018;24(12):3237–50. [CrossRef]
- Mittelman D, Wilson JH. The fractured genome of HeLa cells. Genome Biology. 2013; 17;14(4):111. [CrossRef]
- Mohyeldin A., Garzón-Muvdi T., Quiñones-Hinojosa A. Oxygen in stem cell biology: a critical component of the stem cell niche. Cell Stem Cell. 2010;7:150–161. [CrossRef]
- Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21:297–308. [CrossRef]
- Semenza G.L. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu. Rev. Cell Dev. Biol. 1999;15:551–578. [CrossRef]
- Edwald E, Stone MB, Gray EM, Wu J, Veatch SL. Oxygen depletion speeds and simplifies diffusion in HeLa cells. Biophys J. 2014 Oct 21;107(8):1873-1884. 10.1016/j.bpj.2014.08.023.
- Upadhyay A. Cancer: An unknown territory; rethinking before going ahead. Genes Dis. 2020;8(5):655-661.
- Ostroverkhova D, Przytycka TM, Panchenko AR. Cancer driver mutations: predictions and reality. Trends Mol Med. 2023 Jul;29(7):554-566. [CrossRef]
- Gerstung, M. et al. (2020) The evolutionary history of 2,658 cancers. Nature 578, 122–128.
- Heng HH. The genome-centric concept: resynthesis of evolutionary theory. Bioessays 2009; 31:512-25.
- Horne SD, Ye CJ, Abdallah BY, Liu G, Heng HH. Cancer genome evolution. Transl Cancer Res 2015;4(3):303-313. [CrossRef]
- Heng HH. Bio-complexity: challenging reductionism. In: Sturmberg JP, Martin CM, editors. Handbook on Systems and Complexity in Health. New York: Springer 2013:193-208.
- Heng HH, Liu G, Stevens JB, Bremer SW, Ye KJ et al. Decoding the genome beyond sequencing: the new phase of genomic research. Genomics 2011; 98:242-52.
- Danovi S. The evolving cancer genome. Nat Genet 2023; 55; 1082 . [CrossRef]
- Jolly, C. & Van Loo, P. Timing somatic events in the evolution of cancer. Genome Biol. 2018; 19, 95.
- Mitchell TJ, Turajlic S, Rowan A, Nicol D, Farmery JHR, et al. TRACERx Renal Consortium. Timing the Landmark Events in the Evolution of Clear Cell Renal Cell Cancer: TRACERx Renal. Cell. 2018;173(3):611-623.e17. [CrossRef]
- Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JMC et al. The evolutionary history of lethal metastatic prostate cancer. Nature. 2015;520(7547):353-357. [CrossRef]
- Yates LR, Gerstung M, Knappskog S, Desmedt C, Gundem G et al. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat Med. 2015;21(7):751-9. [CrossRef]
- Brastianos PK, Carter SL, Santagata S, Cahill DP, Taylor-Weiner A et al. Genomic Characterization of Brain Metastases Reveals Branched Evolution and Potential Therapeutic Targets. Cancer Discov. 2015 Nov;5(11):1164-1177. [CrossRef]
- Landau DA, Tausch E, Taylor-Weiner AN, Stewart C, Reiter JG et al. Mutations driving CLL and their evolution in progression and relapse. Nature. 2015;526(7574):525-30. [CrossRef]
- European Molecular Biology Laboratory. Cancer mutations occur decades before diagnosis. Science Daily, 6 February 2020. www.sciencedaily.com/releases/2020/02/200206080451.htm.
- Cheek DM, Naxerova K. Mapping the long road to cancer. Cell. 2022;185(6):939-940. [CrossRef]
- Hanahan D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022;12(1):31-46. [CrossRef]
- Sarker DB, Xue Y, Mahmud F, Jocelyn JA, Sang QA. Interconversion of Cancer Cells and Induced Pluripotent Stem Cells. Cells. 2024; 13(2):125. [CrossRef]
- Gandre-Babbe S, Paluru P, Aribeana C, Chou ST, Bresolin S et al. Patient-derived induced pluripotent stem cells recapitulate hematopoietic abnormalities of juvenile myelomonocytic leukemia. Blood J. Am. Soc. Hematol. 2013; 121, 4925–4929.
- Oshima N, Yamada Y, Nagayama S, Kawada K, Hasegawa S et al. Induction of cancer stem cell properties in colon cancer cells by defined factors. PLoS ONE 2014; 9, e101735.
- Yu Z, Pestell TG, Lisanti MP, Pestell RG. Cancer stem cells. Int. J. Biochem. Cell Biol. 2012; 44, 2144–2151.
- Miyoshi N, Ishii H, Nagai K, Hoshino H, Mimori K. et al. Defined factors induce reprogramming of gastrointestinal cancer cells. Proc. Natl. Acad. Sci. USA 2010, 107, 40–45.
- Utikal J, Maherali N Kulalert W, Hochedlinger K. Sox2 is dispensable for the reprogramming of melanocytes and melanoma cells into induced pluripotent stem cells. J. Cell Sci. 2009; 122, 3502–3510.
- Shamsian A, Sahebnasagh R, Norouzy A, Hussein SH, Ghahremani MH, Azizi Z. Cancer cells as a new source of induced pluripotent stem cells. Stem Cell Res Ther. 2022;13(1):459. [CrossRef]
- Kim HJ, Jeong J, Park S, Jin YW, Lee SS et al. Establishment of Hepatocellular Cancer Induced Pluripotent Stem Cells Using a Reprogramming Technique. Gut Liver. 2017 Mar 15;11(2):261-269. [CrossRef]
- Mahalingam D, Kong CM, Lai J, Tay LL, Yang H, Wang X. (2012). Reversal of aberrant cancer methylome and transcriptome upon direct reprogramming of lung cancer cells. Scientific Reports, 2012; 2. [CrossRef]
- Graham TA, Sottoriva A. Measuring cancer evolution from the genome: Measuring cancer evolution. Journal of Pathology 2016.
- Levitt NC, Hickson ID. Caretaker tumour suppressor genes that defend genome integrity”. Trends in Mol. Medicine. 2002; 8 (4): 179–86. [CrossRef]
- Kinzler KW, Vogelstein B. Cancer-susceptibility genes. Gatekeepers and caretakers. Nature. 1997;386(6627):761, 763. [CrossRef]
- Frank SA. Somatic mutation: Early cancer steps depend on tissue architecture. Current Biology. 2003; 13 (7): R261–3. [CrossRef]
- Campisi J. Aging, tumor suppression and cancer: High wire-act! Mechanisms of Ageing and Development. 2005:126 (1): 51-58. [CrossRef]
- Michor F, Iwasa Y, Nowak M. Dynamics of cancer progression. Nat Rev Cancer 2004; 4:197–205 . [CrossRef]
- Fanale D, Maragliano R, Bazan V, Russo A. Caretakers and Gatekeepers. Genetics & Disease. eLS Book—first published 15.09.2017 .
- Nowell PC. The clonal evolution of tumor cell populations. Science. 1976; 194:23–28.
- Yao Y, Dai W. Genomic Instability and Cancer. J Carcinog Mutagen. 2014; 5:1000165. [CrossRef]
- Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 2011; 25:409–433.
- Loeb LA. Mutator phenotype may be required for multistage carcinogenesis. Cancer Res. 1991; 51:3075–3079.
- Loeb LA. A mutator phenotype in cancer. Cancer Res. 2001; 61:3230–3239.
- Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability-an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010; 11:220–228.
- Warrier NM, Kelkar N, Johnson CT, Govindarajan T, Prabhu V, Kumar P. Understanding cancer stem cells and plasticity: Towards better therapeutics. Eur J Cell Biol. 2023;102(2):151321. [CrossRef]
- Plaks V, Kong N, Werb Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells. Cell Stem Cell, 2015; 16, 225-238, 10.1016/j.stem.2015.02.015.
- Nam AS, Chaligne R, Landau DA. Integrating Genetic and Non-Genetic Determinants of Cancer Evolution by Single-Cell Multi-Omics. Nat. Rev. Genet. 2021, 22, 3–18.
- Martincorena I, Fowler JC, Wabik A, Lawson ARJ, Abascal F et al. Somatic mutant clones colonize the human esophagus with age. Science. 2018;362(6417) 911-917. [CrossRef]
- Yizhak K, Aguet F, Kim J, Hess JM, Kübler K et al. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science. 2019;7;364(6444): eaaw0726. [CrossRef]
- Yokoyama A, Kakiuchi N, Yoshizato T, Nannya Y, Suzuki H et al. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature. 2019 Jan;565(7739):312-317. [CrossRef]
- Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015;348(6237):880-6. [CrossRef]
- Yoshida K, Gowers KHC, Lee-Six H, Chandrasekharan DP, Coorens T et al. Tobacco smoking and somatic mutations in human bronchial epithelium. Nature. 2020;578(7794):266-272. [CrossRef]
- Teixeira VH, Pipinikas CP, Pennycuick A, Lee-Six H, Chandrasekharan D et al. Deciphering the genomic, epigenomic, and transcriptomic landscapes of pre-invasive lung cancer lesions. Nat Med. 2019;25(3):517-525. [CrossRef]
- Kaufman CK, Mosimann C, Fan ZP, Yang S, Thomas AJ et al. A zebrafish melanoma model reveals emergence of neural crest identity during melanoma initiation. Science. 2016;351(6272):aad2197. [CrossRef]
- Turajlic S, Sottoriva A, Graham T, Swanton C. Resolving genetic heterogeneity in cancer. Nat. Rev. Genet 2019: 20, 404–416 (2019) . [CrossRef]
- Pepper JW, Scott Findlay C, Kassen R, Spencer SL, Maley CC. Cancer research meets evolutionary biology. Evol Appl. 2009;2(1):62-70. [CrossRef]
- Vincent MD. The animal within: carcinogenesis and the clonal evolution of cancer cells are speciation events sensu stricto. Evolution. 2010;64(4):1173-83. [CrossRef]
- Huxley, J. 1956. Cancer biology: comparative and genetic. Biol. Rev. 31:474–514.
- Greaves M, Maley CC. Clonal Evolution in Cancer. Nature 2012, 481, 306–313.
- Swanton, C. Intratumor Heterogeneity: Evolution through Space and Time. Cancer Res. 2012;72, 4875–4882.
- Duesberg P, Rasnick D. Aneuploidy, the somatic mutation that makes cancer a species of its own. Cell Motil. Cytoskeleton 2000; 47:81–107.
- Merlo LMF, Pepper JW, Reid BJ, Maley CC. Cancer as an evolutionary and ecological process. Nat. Rev. Cancer 2006; 6:924–935.
- Gray MW, Lang BF, Burger G. Mitochondria of protists. Ann. Rev. Genet. 2004; 38:477–524.
- Rivera M, Lake J. The ring of life provides evidence for a genome fusion origin of eukaryotes. Nature 2004; 431:152–155.
- Vincent M. Cancer: a de-repression of a default survival program common to all cells? a life-history perspective on the nature of cancer. Bioessays. 2012 ;34(1):72-82. [CrossRef]
- Domazet-Lošo T, Tautz D. An ancient evolutionary origin of genes associated with human genetic diseases. Mol Biol Evol. 2008; 25:2699-2707. [CrossRef]
- Domazet-Loso T, Tautz D. Phylostratigraphic tracking of cancer genes suggests a link to the emergence of multicellularity in metazoa. BMC Biol. 2010; 8:66. [CrossRef]
- Trigos AS, Pearson RB, Papenfuss AT, Goode DL. Altered interactions between unicellular and multicellular genes drive hallmarks of transformation in a diverse range of solid tumors. Proc Natl Acad Sci U S A. 2017; 114:6406–6411. [CrossRef]
- Trigos AS, Pearson RB, Papenfuss AT, Goode DL. How the evolution of multicellularity set the stage for cancer. Br J Cancer. 2018; 118:145-152. [CrossRef]
- Trigos AS, Pearson RB, Papenfuss AT, Goode DL. Somatic mutations in early metazoan genes disrupt regulatory links between unicellular and multicellular genes in cancer. Elife. 2019; 8:e40947. [CrossRef]
- Lineweaver CH, Davies P. Comparison of the atavistic model of cancer to somatic mutation theory: Phylostratigraphic analyses support the atavistic model. In: Gerstman BS, editor. The Physics of Cancer: Research Advances. Singapore: World Scientific; 2020. p. 243-261. [CrossRef]
- Zhou J, Cisneros L, Knijnenburg T, Truhana K, Davies PWC, Hang S. Phylostratigraphic analysis of tumor and developmental transcriptomes reveals relationship between oncogenesis, phylogenesis and ontogenesis. Convergent Science Physical Oncology. 2018; 4(2). [CrossRef]
- Rebolleda-Gomez M, Travisano M. The cost of being big: local competition, importance of dispersal and experimental evolution of reversal to unicellularity. Am.Nat. 2018; 192,731–744. [CrossRef]
- Libby E, Ratcliff WC. 2014. Ratcheting the evolution of multicellularity. Science 2014; ce 346 (6208), 426–427. [CrossRef]
- Michod RE, Viossat Y, Solari CA, Hurand M, Nedelcu AM. Life-history evolution and the origin of multicellularity. J Theor Biol. 2006;239(2):257-72. [CrossRef]
- Michod RE. Evolution of individuality during the transition from unicellular to multicellular life. Proc Natl Acad Sci U S A. 2007 May 15;104 Suppl 1(Suppl 1):8613-8. [CrossRef]
- Folse HJ, Roughgarden J. What is an individual organism? A multilevel selection perspective. Q. Rev. Biol. 2010, 85 (4), 447–472.
- Rebolleda-Gómez M, Travisano M. Adaptation, chance, and history in experimental evolution reversals to unicellularity. Evolution. 2019 Jan;73(1):73-83. [CrossRef]
- Canval S. Effect of O-GlcNAcylation on tamoxifen sensitivity in breast cancer derived MCF-7 cells. [Master’s thesis]. Université René Descartes—Paris V; 2013.
- Liu, ZL., Chen, HH., Zheng, LL. et al. Angiogenic signaling pathways and anti-angiogenic therapy for cancer. Sig Transduct Target Ther 8, 198 (2023). [CrossRef]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11, Cell 2011;145, 529–542.
- Chen Q, Zou J, He Y, Pan Y, Yang G, et al. A narrative review of circulating tumor cells clusters: A key morphology of cancer cells in circulation promotes hematogenous metastasis. Front. Oncol. 2022; 12:944487. [CrossRef]
- Ashworth TR. A case of cancer in which similar cells similar to those in the tumour were seen in the blood after death. Australian Medical Journal. 1869;14: 146-147.
- Gwark S, Kim J, Kwon NJ, Kim KY, Kim Y, Lee CH et al. Publisher Correction: Analysis of the serial circulating tumor cell count during neoadjuvant chemotherapy in breast cancer patients. Sci Rep. 2021;11(1):6148. Erratum for: Sci Rep. 2020 Oct 15;10(1):17466. [CrossRef]
- Tellez-Gabriel M, Knutsen E, Perander, M. Current status of circulating tumor cells, circulating tumor DNA, and exosomes in breast cancer liquid biopsies. Int. J. Mol. Sci. 2020; 21, 9457.
- Vismara M, Reduzzi C, Daidone, MG, ,CappellettiV. Circulating tumor cells (CTCs) heterogeneity in metastatic breast cancer: different approaches for different needs. Adv. Exp. Med. Biol. 2020;1220, 81–91.
- Pinheiro R, Martínez-Pena I, López-López R. Relevance of CTC clusters in breast cancer metastasis. Adv. Exp. Med. Biol. 2020; 1220, 93–115.
- Gu X, Wei S, Lv X. Circulating tumor cells: from new biological insights to clinical practice. Signal Transduct Target Ther. 2024 Sep 2;9(1):226. [CrossRef]
- Gao D, Mittal V, Ban Y, Lourenco AR, Yomtoubian S, Lee S. Metastatic tumor cells—genotypes and phenotypes. Front Biol (Beijing). 2018;13(4):277-286. [CrossRef]
- Lambert A W, Pattabiraman D R, Weinberg R A (2017). Emerging Biological Principles of Metastasis. Cell 2017;168:670–691.
- Liu H, Patel MR, Prescher JA, Patsialou A, Qian D, Lin J et al. Cancer stem cells from human breast tumors are involved in spontaneous metastases in orthotopic mouse models. Proc Natl Acad Sci U S A 2010;107:18115–18120.
- Comaills V, Kabeche L, Morris R, Buisson R, Yu M et al. Genomic instability is induced by persistent proliferation of cells undergoing epithelial-to-mesenchymal transition, Cell Rep. 2016; 17, 2632–2647.
- Levine MS, A.J. Holland AJ. The impact of mitotic errors on cell proliferation and tumorigenesis. Genes Dev. 2018;32,620–638.
- Davoli T, Uno H, Wooten EC, Elledge SJ. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy, Science. 2017;355(6322). [CrossRef]
- Germano G, Lamba S, Rospo G, Barault L, Magrì A, et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth, Nature. 2017; 552, 116–120.
- Christensen S, Van der Roest B, Besselink N, Janssen R, Boymans RS et al. 5-fluorouracil treatment induces characteristic T>G mutations in human cancer, Nat. Commun. 2019; 10 , 4571.
- Schuster E, Taftaf R, Reduzzi C, Albert MK, Romero-Calvo I, Liu H. Better together: circulating tumor cell clustering in metastatic cancer. Trends Cancer. 2021;7(11):1020-1032. [CrossRef]
- Aguilera A, García-Muse T. Causes of genome instability, Annu. Rev. Genet. 2013;47, 1–32.
- Schlacher K, Wu H, Jasin M. A distinct replication fork protection pathway connects fanconi anemia tumor suppressors to RAD51-BRCA1/2, Cancer cell. 2012; 22, 106–116.
- Nassour J, Radford R, Correia A, Fust’e JM, Schoell B, Jauch A. Autophagic cell death restricts chromosomal instability during replicative crisis, Nature. 2019;565, 659–663.
- Ira G, Pellicioli A, Balijja A, Wang X, Fiorani S et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature. 2004;431(7011):1011-7. [CrossRef]
- Wyman C, Ristic D, Kanaar K. Homologous recombination-mediated doublestrand break repair, DNA repair. 2004; 3, 827–833.
- Piazza A, HeyeWD. Homologous recombination and the formation of complex genomic rearrangements. Trends Cell Biol. 2019; 29,135–149.
- Liu X, Taftaf R, Kawaguchi M, Chang YF, Chen W et al. Homophilic CD44 Interactions Mediate Tumor Cell Aggregation and Polyclonal Metastasis in Patient-Derived Breast Cancer Models. Cancer Discov. 2019;9(1):96-113. [CrossRef]
- Taftaf R, Liu X, Singh S, Jia Y, Dashzeveg NK et al. ICAM1 initiates CTC cluster formation and trans-endothelial migration in lung metastasis of breast cancer. Nat Commun. 2021;12(1):4867. [CrossRef]
- Chimonidou M, Strati A, Tzitzira A, Sotiropoulou G, Malamos N et al. DNA methylation of tumor suppressor and metastasis suppressor genes in circulating tumor cells. Clin Chem. 2011;57(8):1169-77. [CrossRef]
- Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541,321–330.
- Lara-Gonzalez P, Westhorpe FG, Taylor SS. The spindle assembly checkpoint, Curr. Biol. 2012;22, R966–R980.
- McClintock B. The production of homozygous deficient tissues with mutant characteristics by means of the aberrant mitotic behavior of ring-shaped chromosomes. Genetics. 1938; 23, 315–376.
- Drost J, van Jaarsveld RH, Ponsioen B, Zimberlin C, van Boxtel R, et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature. 2015;7;521(7550):43-7. [CrossRef]
- Davies AA, Masson JY, McIlwraith MJ, Stasiak AZ, Stasiak, A et al. Role of BRCA2 in control of the RAD51 recombination and DNA repair protein, Mol. Cell. 2001;7, 273–282.
- Petros JA, Baumann AK, Ruiz-Pesini E, Amin MB, Sun CQ et al. mtDNA mutations increase tumorigenicity in prostate cancer, Proc. Natl. Acad.Sci. U. S. A. 2005; 102, 719–724.
- Coleman RE, Croucher PI, Padhani AR, Clézardin P, Chow E et al. Bone metastases. Nat Rev Dis Primers. 2020;6(1):83. [CrossRef]
- Croucher P, McDonald M, Martin, T. Bone metastasis: the importance of the neighbourhood. Nat Rev Cance.r 2016; 16, 373–386 (2016). [CrossRef]
- Shen Z. Genomic instability and cancer: an introduction. J Mol Cell Biol. 2011 ;3(1):1-3. [CrossRef]
- Salmaninejad A, Ilkhani K, Marzban H, Navashenaq JG, Rahimirad S et al.. Genomic Instability in Cancer: Molecular Mechanisms and Therapeutic Potentials. Curr Pharm Des. 2021;27(28):3161-3169. [CrossRef]
- Chen M, Linstra R, van Vugt. Genomic instability, inflammatory signaling and response to cancer immunotherapy. Biochim Biophys Acta Rev Cancer. 2022;1877(1):188661. [CrossRef]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144, 646–674.
- Lawrence KS, Chau T, Engebrecht J. DNA damage response and spindle assembly checkpoint function throughout the cell cycle to ensure genomic integrity.PLoS Genet. 2015;11, e1005150.
- Hatch EM, Fischer AH, Deerinck TJ, Hetzer MW., Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell 2013; 154, 47–60.
- Chu X, Tian W, Ning J et al. Cancer stem cells: advances in knowledge and implications for cancer therapy. Sig Transduct Target Ther. 2024; 9, 170 . [CrossRef]
- Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchythat originates from a primitive hematopoietic cell. Nat. Med. 1997;3, 730–737.
- Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008 May 16;133(4):704-15. [CrossRef]
- Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymaltransitions in development and disease. Cell. 2009;139, 871–890.
- Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009 Dec;11(12):1487-95. [CrossRef]
- Zhou J, Zhou XA, Zhang N, Wang J. Evolving insights: how DNA repair pathways impact cancer evolution. Cancer Biol Med. 2020 Nov 15;17(4):805-827. [CrossRef]
- Barber LJ, Sandhu S, Chen L, Campbell J, Kozarewa I, et al. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J Pathol. 2013; 229: 422-9.
Figure 2.
Stemness loss and recovery cycles (DSCD-ACD cycles).
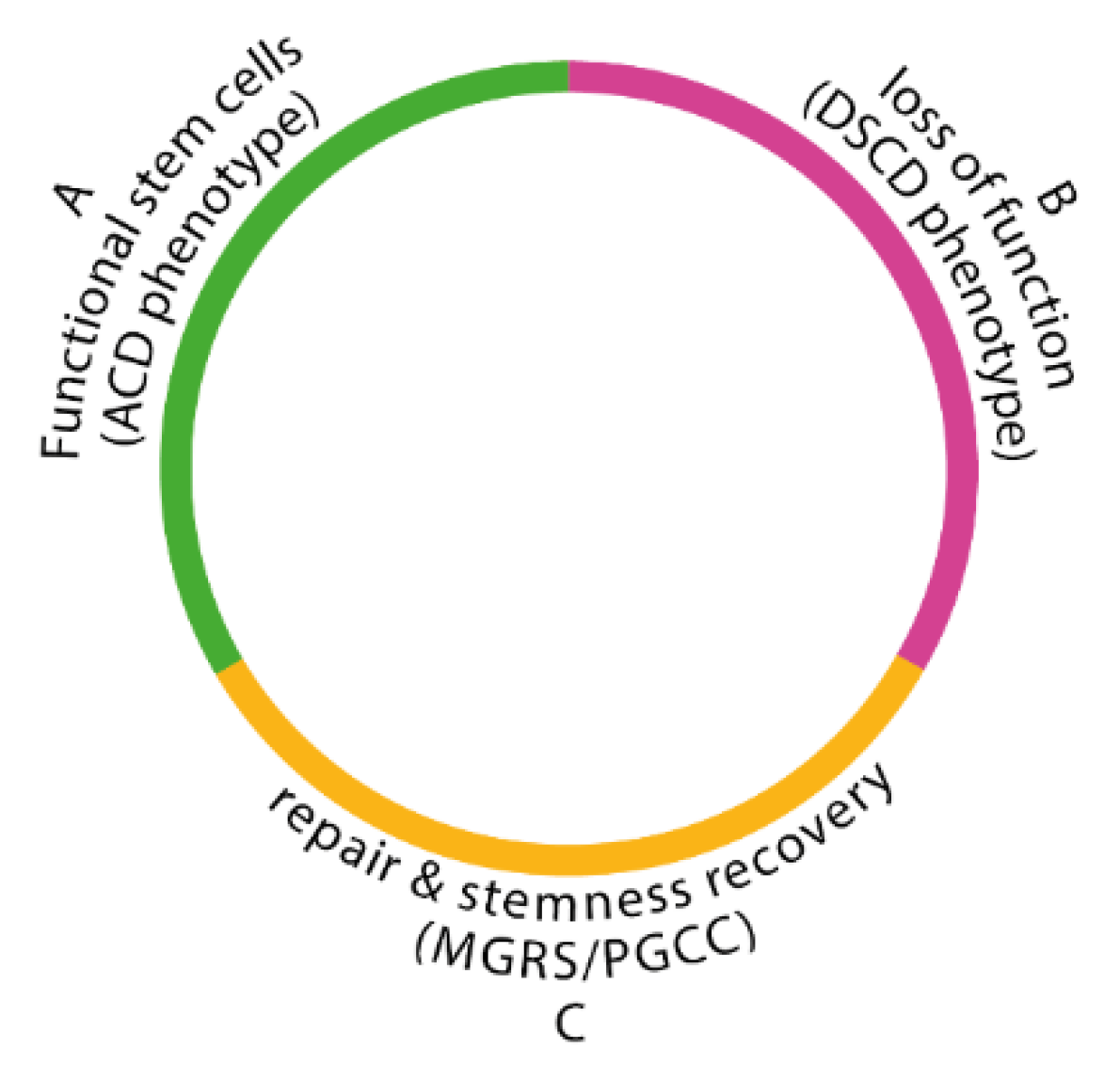
Figure 3.
The generation of committed CSC and uncommitted quiescent Q cells produced by the ACD phenotype of the stemgermline depending on the ambient oxygen levels. Oxygen concentrations around ~1% O2 permit commitment, while levels ≥2% O2 inhibit commitment and instead lead to the formation of an intermediate, quiescent Q cells.
Figure 3.
The generation of committed CSC and uncommitted quiescent Q cells produced by the ACD phenotype of the stemgermline depending on the ambient oxygen levels. Oxygen concentrations around ~1% O2 permit commitment, while levels ≥2% O2 inhibit commitment and instead lead to the formation of an intermediate, quiescent Q cells.
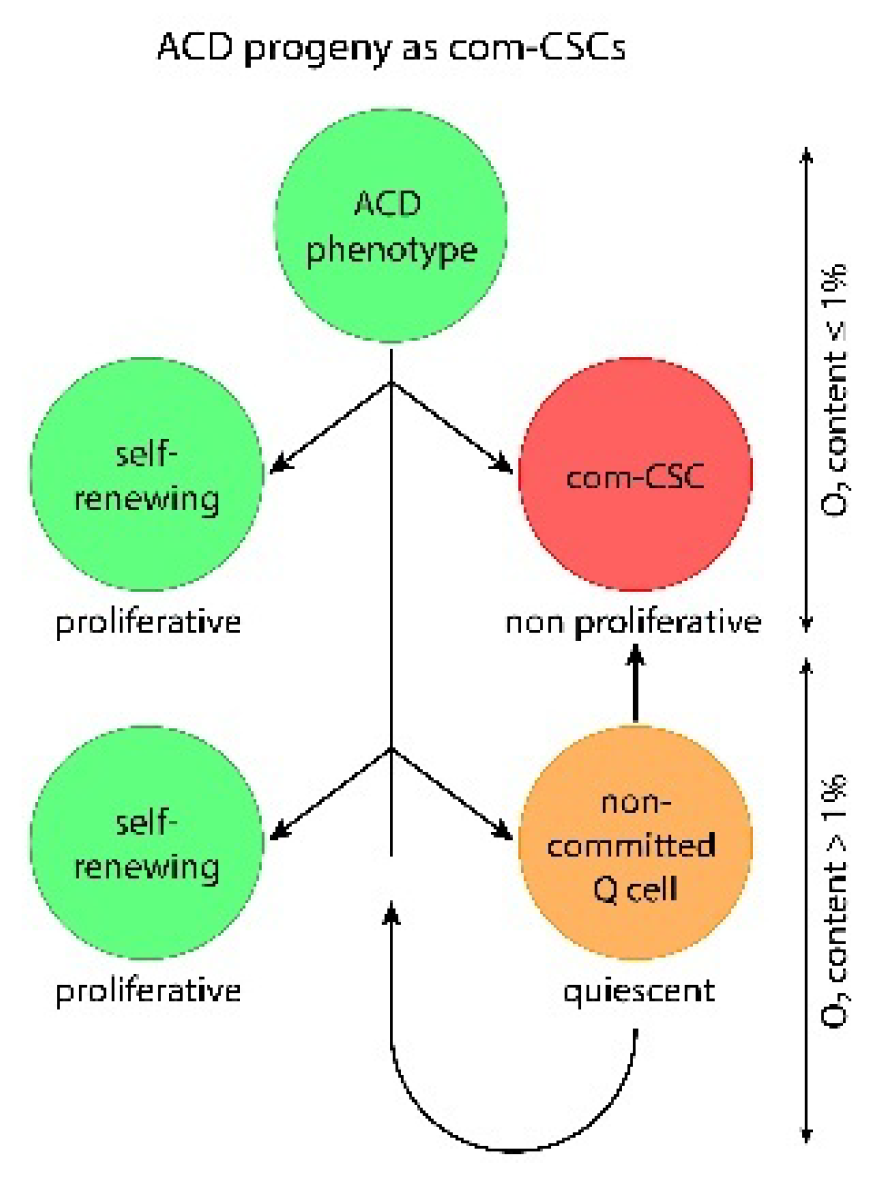
Figure 4.
Unicellularization and stemness loss and recovery cycles (DSCD-ACD-DSCD cycles); Generation of com-CSC by the ACD phenotype and by fractal EMT processes (SGT) that give rise to M- and E-cell fractions. [26].
Figure 4.
Unicellularization and stemness loss and recovery cycles (DSCD-ACD-DSCD cycles); Generation of com-CSC by the ACD phenotype and by fractal EMT processes (SGT) that give rise to M- and E-cell fractions. [26].
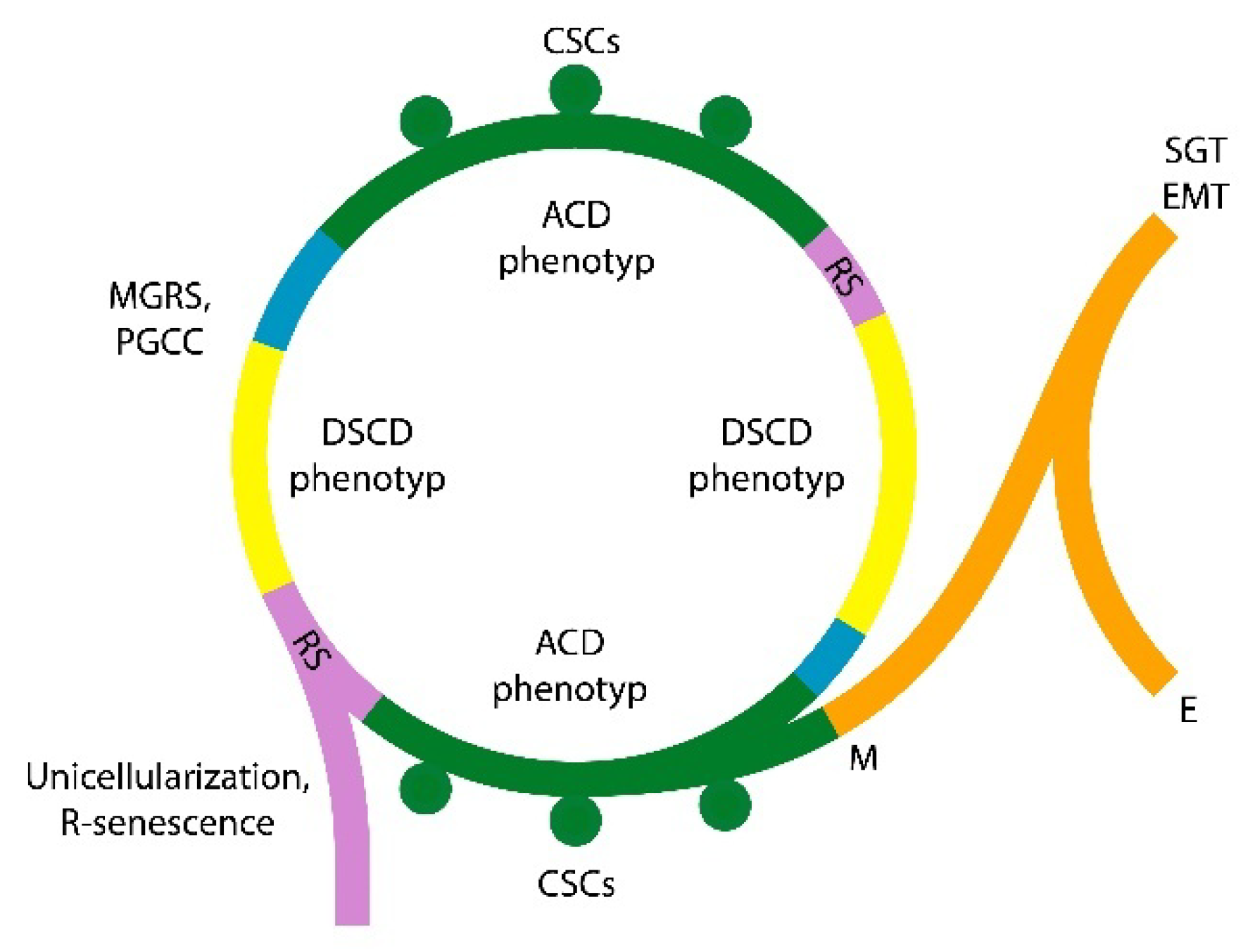
Figure 5.
From genetical altered epithelial cells to metastases—original legend [36]: Tumors development takes place in different steps. (A) Hyperplasia is the stage where genetically altered or abnormal cells show uncontrolled and rapid growth. (B) Dysplasia, stage of tumor development where overgrowing cells changes their original form. It consists of more immature cells than mature. (C) In situ cancer represent neoplastic lesion where cells do not go into the process of maturation, have lost their tissue identity and grow without regulation. (D) With malignant tumor, overgrowing cells invade other areas by rupturing basal membrane (E.) Metastases occur when cancer cells reach to the distant parts through lymphatic system and blood circulation. [This figure is uploaded from the HAL archive for the deposit and dissemination of scientific research documents. These documents come from teaching and research institutions in France orabroad, or from public or private research centers. It was uploaded by Shahzina Kanwal [139]]
Figure 5.
From genetical altered epithelial cells to metastases—original legend [36]: Tumors development takes place in different steps. (A) Hyperplasia is the stage where genetically altered or abnormal cells show uncontrolled and rapid growth. (B) Dysplasia, stage of tumor development where overgrowing cells changes their original form. It consists of more immature cells than mature. (C) In situ cancer represent neoplastic lesion where cells do not go into the process of maturation, have lost their tissue identity and grow without regulation. (D) With malignant tumor, overgrowing cells invade other areas by rupturing basal membrane (E.) Metastases occur when cancer cells reach to the distant parts through lymphatic system and blood circulation. [This figure is uploaded from the HAL archive for the deposit and dissemination of scientific research documents. These documents come from teaching and research institutions in France orabroad, or from public or private research centers. It was uploaded by Shahzina Kanwal [139]]
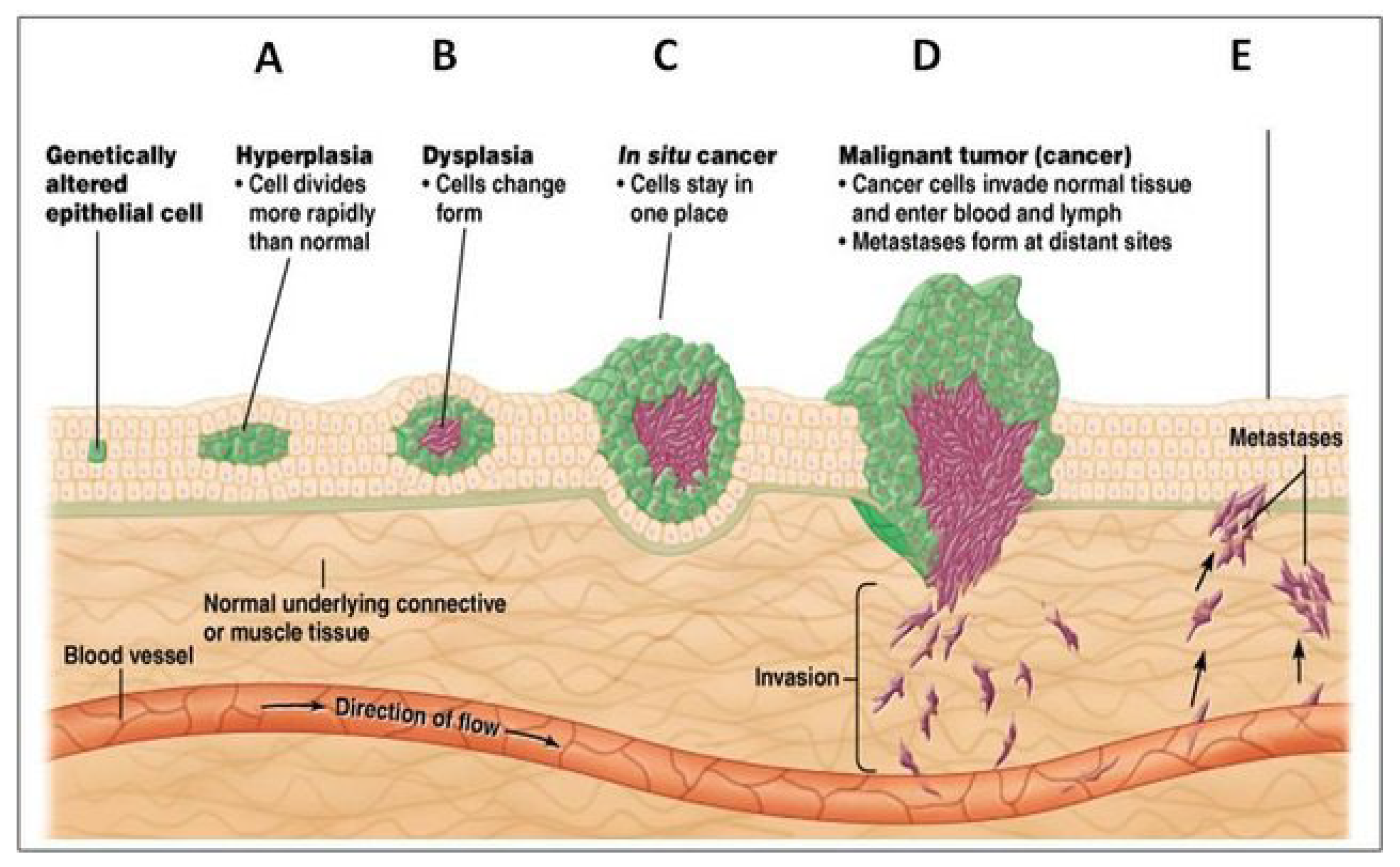
Figure 6.
The progression of the canceration through growth and angiogenesis. The rapid expansion of tumor results in a reduction in the oxygen supply. The consequent hypoxic tumor microenvironment stimulates excessive angiogenesis via increasing various angiogenic pro-factors including VEGF, PDGF, FGF, and angiopoietin. Later, new blood vessels facilitate the transportation of oxygen and nutrients to further support the survival, growth and proliferation of tumor cells. When tumor cells develop a more aggressive phenotype, they continue to proliferate, spread and induce angiogenesis, with the invasion and metastasis of tumor cells into distant tissues through blood circulation. Original legend, Liu et al. [140]; This article is licensed under the Creative Commons Attribution 4.0 International License (Request for Springer-Nature approval dated 21.07.2025).
Figure 6.
The progression of the canceration through growth and angiogenesis. The rapid expansion of tumor results in a reduction in the oxygen supply. The consequent hypoxic tumor microenvironment stimulates excessive angiogenesis via increasing various angiogenic pro-factors including VEGF, PDGF, FGF, and angiopoietin. Later, new blood vessels facilitate the transportation of oxygen and nutrients to further support the survival, growth and proliferation of tumor cells. When tumor cells develop a more aggressive phenotype, they continue to proliferate, spread and induce angiogenesis, with the invasion and metastasis of tumor cells into distant tissues through blood circulation. Original legend, Liu et al. [140]; This article is licensed under the Creative Commons Attribution 4.0 International License (Request for Springer-Nature approval dated 21.07.2025).
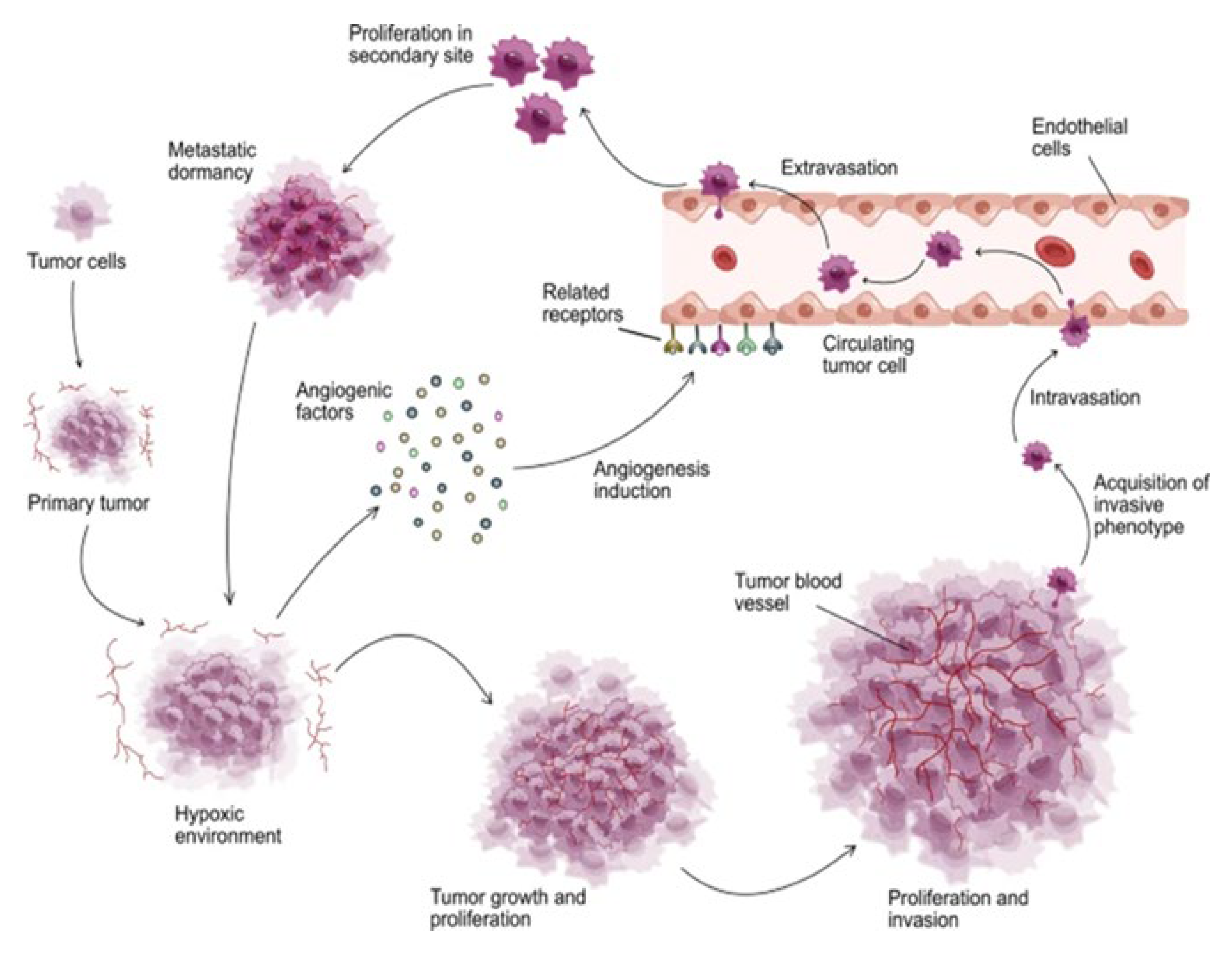

Table 1.
New Terms and Definitions Used in Evolutionary Cancer Cell Biology (ECCB).
| Term | Definition |
| Stemgermline |
|
| Nascent germline |
|
| com-CSCs |
|
| GST |
|
| p-CSCs |
|
| Somatic cell line |
|
| SGT |
|
| Fractal EMT |
|
| s-CSCs |
|
| ACD phenotype |
|
| DSCD phenotype |
|
| MGRS (PGCC) |
|
| Hypoxia |
|
| Hyperoxia |
|
| O2 gradient |
|
| Genome evolution |
|
| Unicellularization |
|
| MUT genes |
|
Table 2.
Oxygen dependant AMF evolution before and during the transition to multicellularity.
| Period | Age | Quality | O2 | Developments | Cell line and phenotypes |
|---|---|---|---|---|---|
| Mesoproterozoic | 1600-1000 Mya | hypoxic | ≤ 1% | AMF | Stemgermline’s |
| ancestor | ACD phenotype | ||||
| Produced com-SCs | |||||
| capable of cystic polyploidy; | |||||
| (progenitor accumulation) | |||||
| Neoproterozoic | 1000-800 Mya | less | ≥ 1% | Q cells | Q cells replaced com-SCs; |
| (early NP) | hypoxic | Q cell fate: | |||
| (i) reversion to self-renewal | |||||
| (ii) conversion to com-SCs | |||||
| (Q cell plasticity) | |||||
| (middle /late NP) | 800-550 Mya | NP | 1-2% | Somatic | Upgrade to dual cell system; |
| oxygen | cells | addition of a O2 -resistant | |||
| events | somatic cell line | ||||
| capable of phagocytosis; | |||||
| Cell plasticity: GST, SGT | |||||
| Paleozoic (PZ) | ≤550 Mya | PZ | ⟩2% | MGRS | Excess oxygen damaged |
| (early PZ) | oxygen | development | stemgermline’s genome; | ||
| events | The stemgermline developed | ||||
| genome repair mechanisms: | |||||
| DSCD proliferation, | |||||
| hyperpolyploid MGRS; | |||||
| recover genomic integrity | |||||
| Fig | |||||
| UMT | final | Colateral MUT processes and | |||
| transition period | evolution | genome expansion through | |||
| horizontal gene transfer UMT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



