Submitted:
10 April 2025
Posted:
11 April 2025
You are already at the latest version
Abstract
In extensive production systems, where beef cattle are often raised unfenced and in remote areas, accurately determining their age is crucial for genomic selection to enhance genetic gain in important traits such as age at first calving, age at puberty and growth rate. Many studies have demonstrated a link between DNA methylation and aging in various species. In this study, methylation profiles of three yearling and three adult cattle were examined using Oxford Nanopore Technologies. A total of 1,044 differentially methylated genes (DMGs) were detected, with most differentially methylated regions located within 500 bp of transcription start sites. Among these DMGs, 27 genes were classified as age-related genes based on the Aging Atlas. Pathway enrichment analysis of these 27 age-related genes highlighted 61 pathways linked to aging, suggesting their biological significance in aging. Interestingly, many of these pathways are also known to be associated with the puberty process, which aligns with the context of this study where two years of age is recognised as a key transitional period into puberty in cattle. These findings underscore the potential of DNA methylation as a valuable biomarker for cattle aging and puberty, offering practical applications for livestock management and genetic selection in extensive production systems.
Keywords:
Age-related genes
; Beef cattle
; DNA methylation
; Differentially methylated genes
; Puberty
; Oxford Nanopore Technologies
1. Introduction
The beef cattle industry is one of Australia’s major agricultural industries, contributing significantly to national export earnings. In 2023-24, the total value of Australian agriculture exports fell to $72.4 billion, accounting for 13.5% of country’s total goods exports [1]. Currently, beef cattle breeding programs focus on increasing genetic gain rates for economically important traits by escalating selection accuracy at earlier ages. Age is particularly critical information for integrating into genomic evaluation systems, especially for low heritable traits such as female reproduction (age at puberty, age at calving or calving intervals). Improving the accuracy of age estimation is where genomic selection can propose substantial improvements in genetic gain rates for beef cattle in extensive production systems like Australian Northern Beef Industry. This is especially important because obtaining accurate age information in Northern Australia farms is challenging, as cattle are raised extensively unfenced and often mustered infrequently.
DNA methylation is one of the most studied epigenetic modifications that change gene expression but not the DNA sequence. In mammals, DNA methylation as an epigenetic regulation commonly occurs at CpG dinucleotide [2]. Remarkably, alterations in global DNA methylation patterns have been documented to be associated with age in a wide range of species, including humans. The first human epigenetic clock was developed using 353 CpG sites to predict age with a mean absolute deviation (MAD) of 3.6 years (r=0.96) [3]. Epigenetic clocks for dogs/wolves, mice, cats, and chickens were also established [4,5,6,7,8]. In cattle, the investigation of DNA methylation clock in cattle has focused on the reproductive aging using oocyte samples [9]. Recently, a study by Hayes and Nguyen et al. (2021) demonstrated the use of methylation profiles to predict the age of beef cattle [10]. The model achieved an accuracy of 0.71, with the difference between actual age and predicted age averaging 1.4 years for animals under 3 years old.
Here, we hypothesised that methylation profiles differ between yearling and adult Bos indicus cattle and that methylated genes in these groups may play a role in the aging and puberty process. To investigate this, differentially methylated regions and genes were identified using R package - DSS, followed by functional enrichment analysis to understand their biological significance. The identified age-related genes could serve as potential biomarkers for cattle aging, contributing to improved age prediction models and livestock management strategies.
2. Materials and Methods
2.1. Animal Ethics
Animal ethics approval was obtained from the University of Queensland ethics board. The approval Number is QAAFI/269/17.
2.2. Sample Information
A total of six unrelated Droughmaster beef cattle were used in this study (Table 1). Of these six animals, three were grouped as yearling group, with an average age of 1.48 years (SD = 0.05), while the other three were groupped as adult group, with an average age of 2.64 years (SD = 0.01). There was no significant difference in the body condition score among the six animals. However, there was a slightly significant difference in weight (p-value = 0.02) and hip height (p-value = 0.02) between the two groups. Out of these six samples, 4 samples were also sequenced and used in epigenetic clocks described by Hayes and Nguyen et al. 2021 [10]. The hairs were plucked and stored at room temperature for over a year before DNA extraction.
2.3. DNA Extraction
DNA was extracted from 30 - 40 hair follicles using Puregene Gentra DNA Tissue kit (QIAGEN, Australia) according to the manufacturer’s instructions with some modifications as previously described by [11] and [10]. The quantity and quality of extracted DNA were assessed using an Invitrogen Qubit 4.0 device (Thermo Fisher Scientific) and NanoDropND 1000 (v.3.5.2, Thermo Fisher Scientific). The size of extracted DNA samples was visualized by running on Pulsed-field Gel Electrophoresis system.
2.4. Oxford Nanopore Sequencing
Samples were prepared for sequencing following the protocol in the genomic sequencing kit SQK-LSK109 (Oxford Nanopore Technologies; ONT, Oxford, UK) with modifications as previous describe by [10]. Briefly, approximately 4ug DNA was end-repaired and deoxyadenosine (dA)-tailed using the Ultra II end-repair module (New England BioLabs; NEB). Sequencing adapters (Oxford Nanopore Technologies; ONT) were ligated using blund/T4 ligase (NEB). Libraries from End-repair reaction and ligation steps were clean-up with AMPureXP beads (Beckman Coulter). After cleaning, DNA was resuspended in 12ul of Elution Buffer (Oxford Nanopore Technologies; ONT) before being combined with 37.5ul of sequencing buffer (SQB - Oxford Nanopore Technologies; ONT) and 25.5ul of loading bead and loaded on a MinION SpotON flow cell (FLO-MIN106). Sequencing was performed for 48h with a MinION sequencer.
2.5. Methylation Calling
Guppy software (version 6.4.6, ONT, Oxford, UK) was used to basecall raw FAST5 data generated from each sequencing run. Briefly, all fastq files of each sample were merged into a single file. The basecalled reads were then aligned to the bovine reference genome ARS-UCD1.2 assembly [12] using minimap2 [13] with the parameters “–a –x map-ont –secondary=no”.
F5c software version 1.3 was used for indexing and methylation calling [14]. Briefly, an index file linking read IDs with their signal-level data in the FAST5 files was created using f5c index function. Methylated based (5-methylcytosine in a CpG context) were detected using f5c call-methylation command. Finally, methylation frequency at each reference position was calculated using “f5c calculate_methylation_frequency.py”.
2.6. Differentially Methylated Genes and Regions Identification
Differential gene expression analysis was conducted using the DSS (v 2.44.0) package [15] in R. DSS implements a dispersion shrinkage method, which follows the Wald statistical test to evaluate each CpG site for differential methylation. The “DMLtest” function in DSS was used to identify the differentially methylated loci under the statistical method ‘smoothing =True’. Loci were considered significant if the false discovery rate was below 0.05. The resulting differentially methylated loci (DML) were then filtered using a delta threshold (δ = 0.2), followed by the “CallDMR” function to detect differentially methylated regions (DMRs) from the DML table. The DNA methylation analysis results included the average methylation level of each age group (yearling versus adult) at each region, as well as DMRs length, and the number of CpG sites.
2.7. DMRs Annotation and DMRs Features Visualisation
The annotation of DMRs based on the reference ARS-UCD1.2 [12] was conducted by R package ChIPseeker version 1.32.1 [16]. The R function readPeakFiles from ChIPseeker was used to read the DMRs from DMR calling results. The R function annotatePeak was then used to annotate DMRs to differentially methylated genes (DMGs). The DMGs results included information on DMRs, gene ID, transcription ID and the distance to the transcription start site (TSS) within a range of ± 3000 bp from the TSS. DMR features were then plotted using plotAnnoBar. To visualize the distributions of DMRs around TSS, a heatmap was constructed using tagHeatmap.
2.8. Identifying of Age-Related Genes
To identify age-related genes from the list of identified DMGs, the human age-related dataset was first obtained from Aging Atlas (https://ngdc.cncb.ac.cn/aging/age_related_genes). The R package homologene version 1.4.68.19.3.2 was used to detect the cattle homologous genes from human data based on the Entrez IDs. After that, the list of age-related genes in cattle was compared to the identified DMGs to identified age-related DMGs. Additionally, gene sets used in the human-dog epigenetic aging clock [17] and cattle aging clocks [11] were also retrieved to identify overlapping genes with our DMGs.
2.9. Identifying the Age-Related Gene Ontology Terms and KEGG Pathways
Differentially methylated genes and overlap Age-related DMGs with Human Aging Atlas were used to perform the functional enrichment analysis for Gene Ontology (GO) terms and KEGG pathways using clusterProfiler version 4.4.4 [18]. Specifically, the function “enrichGO” and “enrichKEGG” with p.adjust < 0.05 under the Benjamini Hochberg False discovery rate (FDR) correction was used to identify significant GO terms and KEGG pathways. For visualising the results of the KEGG pathway analysis, the function “dotplot” from R package enrichplot version 1.16.2 [19] was used for visualising the top KEGG pathway, and the function “pathview” from the R package pathview version 1.36.1 [20] were used to display specific pathway.
3. Results
3.1. Sequencing Data Statistic
This study successfully sequenced 6 samples with an average coverage of 3.95 ± 1.12 and read length N50 of 1.94 ± 0.64 kilobase pairs (Table 1). The read sequence was mapped to the Bos taurus reference genome with a mapping efficiency of 90.98 ± 2.53%. A total of 18,105 ± 2,310 million raw CpG sites was identified. The CpG sites that were not assigned to chromosomes or had sequencing depths lower than 10X were removed, resulting in 1.21 ± 0.36 million CpG sites remaining in each sample.
3.2. Differentially Methylated Genes and Regions Identification
Using the DSS package, 1,724 differentially methylated regions were identified from a total of 34,351 methylated regions between the yearling and adult cattle groups (DMRs, FDR < 0.05 and delta ≥ 0.2; Supplementary Table S1). Annotation of 1,724 DMRs resulted in 1,044 genes identified as differentially methylated genes (DMGs; Supplementary Table S2). Among all DMGs, 27 were identified as age-related genes (Figure 1, Supplementary Table S3), based on a comparison with the age-related gene set retrieved from the Human Aging Atlas. When compared to the cattle aging clock [10], 9 genes overlapped between our DMGs and cattle aging clock, including Frizzled Class Receptor 10 (FZD10), Growth Factor Receptor-Bound Protein 10 (GRB10), LIM Homeobox 9 (LHX9), Leucine Rich Repeat Transmembrane Neuronal 3 (LRRTM3), NK6 Homeobox 1 (NKX6-1), SIX Homeobox 3 (SIX3), SRY-Box Transcription Factor 17 (SOX17), T Cell Leukemia Homeobox 2 (TLX2) and T Cell Leukemia Homeobox 3 (TLX3) (Figure 1). It is important to highlight that the list of 43 genes in the cattle aging clock was originally derived from human and dog epigenetic studies [17], and then tested for association with age in cattle. This explains why, out of 34 overlapping genes between our DMGs and the human-dog aging clock, all of these 9 genes were overlapped in all three datasets (our study, the human-dog aging clock and the cattle aging clock). Among these nine overlapping genes, six had methylated regions located in promoters (≤1 kb) and distal intergenic regions. TLX2 had methylation exclusively in the promoter region, while LRRTM3 had methylated sites only in the intron and distal intergenic regions. SIX3 showed methylated sites in the intron, promoter, and distal intergenic regions. When comparing the human-dog aging clock with the Human Aging Atlas gene set, only 13 genes overlapped. No gene was found to overlap across all four datasets.
3.3. Annotation of CpG Sites
Differentially methylated regions were compared against the protein-coding gene set annotated on bovine reference assembly (ARS-UCD1.2 assembly, [12]). This classified CpG sites as overlapping gene body, promoters and distal regulatory elements (Figure 2). Most of identified CpG sites fall within distal intergenic (64.85%), followed by promoter regions (12.64%). The percentage of CpG sites that fall within untranslated regions (UTR) was minimal (1.45%).
To further visualize the DMRs, the distribution of identified DMRs within 3000bp upstream and downstream of the transcription start site (TSS) was investigated. As shown in Figure 3, most identified DMRs were concentrated within ± 500 bp of TSS. The density of methylated regions at 1-2kb and 2-3kb downstream of the TSS (Figure 3) was slightly greater compared to the upstream regions, suggesting that these methylated sites may be situated within the gene body (eg, first intron or exon). This pattern may reflect the active transcriptional activation of these genes.
3.4. Functional Enrichment Analysis
Enrichment analyses of GO terms and KEGG pathways were performed using the 1044 DMGs as the target gene list, with Bos taurus genes used as a background list. No GO biological process terms or KEGG pathways were significantly enriched. However, seven GO cellular component terms were enriched after correction for multiple testing (FDR < 0.05), including nucleoplasm, cytoplasm, endoplasmic reticulum membrane, nucleus, Golgi apparatus, cytosol and exocyst. Enrichment of DMGs in nucleus and nucleoplasm suggests their potential role in transcriptional regulation, consistent with the well-documented evidence of DNA methylation in modulating gene expression. In addition, protein binding was only one molecular function GO term enriched, with 132 DMGs associated with this term (FDR = 0.001).
Enrichment analyses of GO terms and KEGG pathways were also performed using the 27 overlapping age-related DMGs (those overlapping with the Human Aging Atlas) as the target gene list, with Bos taurus genes used as a background. The results showed that 54 GO terms were significantly enriched in the biological process category, 5 GO terms in the cellular component category, and 11 GO terms in the molecular function category (FDR < 0.05, Supplementary Table S4).
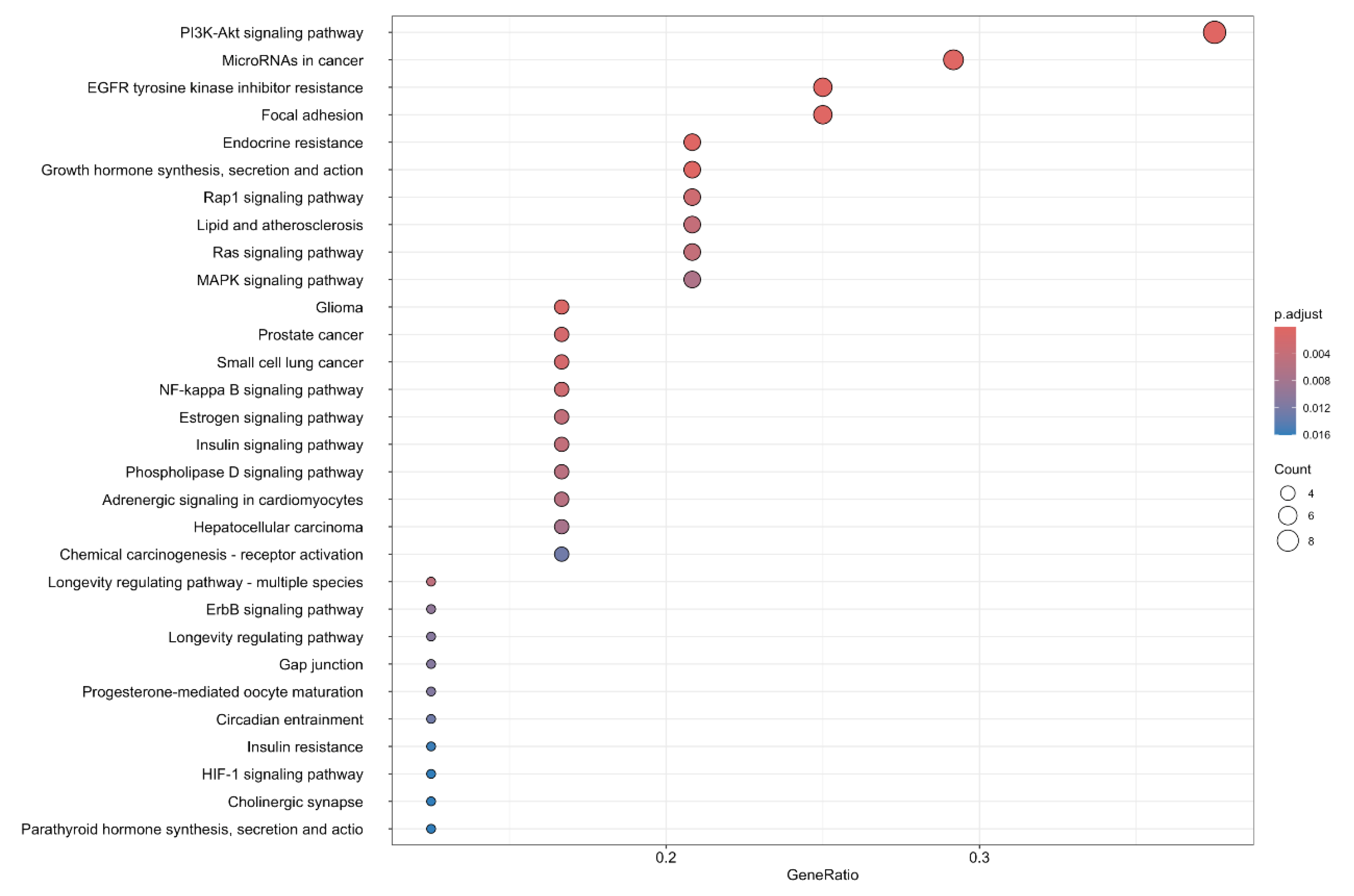
Pathway analysis of these 27 overlapping age-related DMGs revealed 60 significantly enriched KEGG pathways (Supplementary Table S5, FDR < 0.05). As expected, several aging-related pathways were among the enriched results, including longevity regulating pathway, PI3K-Akt signaling pathway, MAPK signaling pathway, Estrogen signaling pathway, Calcium signaling pathway, TNF signaling pathway, mTOR signaling pathway, GnRH signaling pathway, Wnt signaling pathway and Oxytocin signaling pathway. Interestingly, these pathways are also well-documented to be associated with puberty.
Figure 4.
KEGG pathway enrichment of differentially methylated genes overlapping with the Human Aging Atlas.
Figure 4.
KEGG pathway enrichment of differentially methylated genes overlapping with the Human Aging Atlas.
4. Discussion
This study successfully profiled methylation sites, identifying an average of over 18 million methylation sites at an average coverage of 3.95x in beef cattle hair samples (n=6). The identified CpG sites aligned with findings from a dairy cattle study using semen samples (n=6), which reported an average of 10 million CpG sites at 4x coverage [21]. The difference in the number of methylation sites between these two studies could be explained by the difference in tissue type, as hair is somatic tissue, whereas semen represents the germline as well as tissue-specific methylation characteristics.
Since the small sample size (n=3 per group) could potentially impact the robustness of our findings, CpG sites with fewer than 10 reads were excluded to reduce the influence of low sequencing depth in this study. Additionally, DMRs were identified using a false discovery rate (FDR) threshold of 0.05, ensuring that only regions passing multiple testing corrections were included in downstream analyses. An additional filtering step was applied to retain only DMRs with a methylation difference (delta) of ≥ 0.2, representing the minimum difference in mean methylation levels between the two groups. This filtering strategy is consistent with a recent study by López-Catalina et al. (2024), who investigated DMRs in dairy cattle using Oxford Nanopore Technology (ONT) at 4X coverage, with a similarly small sample size of n = 6 [21]. As a result, a total of 1,724 DMRs were identified after filtering out sites with a sequencing depth lower than 10x (FDR < 0.05, delta ≥ 0.2), further confirming the difference in methylation profiles between yearling and adult age groups, even with just a one-year age gap. The 1,724 DMRs identified in our study were also comparable with the 2,001 DMRs identified in the dairy cattle study [21].
As expected, promoter regions and TSS, which act as switches regulating gene expression, exhibited a high percentage of DMRs in our study. Consistent with the findings of López-Catalina et al. (2024), this study also detected a large number of CpGs identified in intergenic regions using ONT. The high level of DMRs in distal intergenic regions is biologically meaningful, as these regions tend to exhibit high methylation levels, especially in the context of puberty-related epigenetic changes in cattle. Methylation in these regions may play a regulatory role in developmental transitions. Additionally, unlike reduced-representation bisulphite sequencinng (RRBS), which is based toward CpG-dense regions, ONT enables genome-wide methylation detection, including in distal intergenic regions. The detection of higher metyhlation levels in intergenic regions by ONT provides valuable insights into genome-wide regulatory mechanisms. Study by López-Catalina et al. (2024) [21] also compared ONT with RRBS and concluded that ONT is an alternative for methylation profiling in livestock populations. Although our study did not aim to compare methylation sequencing techniques, ONT’s simple library preparation using native DNA without bisulfite treatment makes the dataset versatile for other genomic studies.
When comparing the 1,044 DMGs with the cattle epigenetic aging clock [10], 9 overlapping DMGs were identified. It is worth noting that although 4 out of 6 samples used in the current study were also included in the cattle epigenetic clock study by Hayes and Nguyen et al. (2021), their study employed a different approach. Specifically, Hayes and Nguyen et al. (2021) used all methylation information to build methylation relationship matrices for age prediction using the Best Linear Unbiased Prediction model. Additionally, the list of genes reported in their study was derived from Human-dog epigenetic clock, and not based on differentially methylation with age, as no differential methylation analysis was performed in that study.
When compared to the Human Aging Atlas, a total of 27 overlapping DMGs were identified out of 1024 DMGs, and their biological functions are known to be associated with the aging process. Importantly, among these overlapping DMGs, several have been shown to play a role in both aging and reproduction regulation. For instance, this study observed insulin-like growth factor 1 receptor (IGF1R), a gene essential for normal development and growth in IGF1/IGF1R signalling pathway, which has also been linked to age at puberty in cattle [22,23]. Additioanlly, IGF1R has been implicated in aging-related processes, as demonstrated by [24], who reported that IGF1R interacts with progerin in premature aging. Moreover, genetic variants in IGF1R have been related to human longevity [25], highlighting its role in lifespan regulation [26].
An interesting finding in this study was the identification of telomerase reverse transcriptase (TERT), a gene that plays a crucial role in maintaining telomere length [27,28]. The presence of TERT among the age-related DMGs aligns with the previous research, as telomere length has long been recognized as a biomarker of aging, with telomere shortening with age [28]. Moreover, a study in humans has reported an association between early pubertal timing and shorter telomere length in preadolescent girls [29].
Heat shock protein 90 Alpha Family class A member 1 (HSP90AA1), a key heat shock protein, was also identified among the age-related DMGs in our study. Notably, this gene was previously reported as a differentially abundant protein between pre- versus post-pubertal Brahman heifers and represented as a hub protein in the hypothalamus pubertal network [30]. Its role in aging is further supported by a study in rats, which found that hepatic Hsp90 levels significantly decreased in aged rats [31].
When analyzing the KEGG pathway of 27 overlaping age-related DMGs, several pathways are found to be associated not only with aging but also with the puberty process. Notably, the GnRH signaling pathway, calcium signaling pathway, estrogen signaling pathway, oxytocin signaling pathway, Wnt signaling pathway, PI3K-AKT signaling pathway and mTOR signaling pathway are all known to play significant roles in pubertal development.
Of particular importance, the GnRH signaling pathway is essential for puberty [32]. In a cattle puberty study, this pathway was enriched among differentially abundant proteins in the hypothalamus of heifers during pubertal transition [33]. Supporting this, a study in rats also confirmed the GnRH signaling pathway was enriched among their differentially methylated regions between prepuberty and puberty rats [34].
The enrichment of the calcium signaling pathway, estrogen signaling pathway and oxytocin signalling pathway was also observed in the hypothalamus and pituitary gland transcriptome studies of pre- versus post-puberty cattle [33]. Additionally, the Wnt signaling was enriched among differentially expressed genes in a liver transcriptomic study comparing pre- and post-pubertal heifers [35]. Furthermore, in an adipose proteomic analysis, the PI3K-AKT signaling pathway was enriched among differentially abundant proteins, further highlighting its role in pubertal-related metabolic regulation [30]. Finally, mTOR signaling pathway has been recognised as a key modulator of both aging [36] and puberty [37]. Notably, mTOR signaling pathway has been studied as a potential target for delaying aging in humans, further underscoring its relevance across developmental and aging processes.
Given that cattle in this study are at the age of puberty (1.5 years-old versus 2.6-years-old), the observed methylation patterns in the abovementioned genes and KEGG pathways may reflect important regulatory mechanisms linking aging and reproductive development. A study in rats has shown that DNA methylation changes occur between the pre- and post-pubertal stages [34]. In human studies, variations in methylation profiles between girls with normal and precocious puberty have demonstrated an association between specific epigenetic changes and the regulation of the pubertal process [38]. Additionally, recent research has highlighted the role of both environmental and genetics in pubertal timing and development, providing evidence that DNA methylation serves as a potential mechanistic link between genotypes and puberty [39]. Moreover, another human study reported that the adolescent transition is associated with DNA methyation changes [40].
Although limited research has directly compared methylation profiles at puberty onset in cattle, existing evidence still supports a link between epigenetic regulation and pubertal timing. For instance, research in Holstein-Friesian demonstrated that a higher plane of nutrition before puberty is associated with earlier pubertal onset, yet epigenetic modification persists in sperm DNA methylation after puberty [41]. Additionally, increased body weight gain before puberty in crossbred heifers has been shown to alter the methylation patterns of genes in the hypothalamic arcuate nucleus, a region known to regulate pubertal maturation.
In summary, age-related DMGs identified in this study hold promise as potential biomarkers for cattle aging and puberty onset, offering valuable insights for the development of age and genomic prediction models. Accurate genomic prediction of age at puberty or the identification of genes responsive to this crucial developmental stage is particularly valuable for the beef cattle industry, as achieving calving at two years of age can significantly enhance productivity and overall reproductive efficiency. A lifetime productivity study found that heifers calving at two years old produced 0.6 more calves by five years after first calving compared to those calving at three years old [42]. Future research should focus on validating these epigenetic markers across diverse cattle populations and integrating them with other omics approaches. One potential application could involve validating the role of these DMGs in aging by extracting their methylation profiles from 66 animals provided by Hayes et al. 2021 [10], and incorporating them into age prediction models to assess their potential as aging biomarkers.
5. Conclusion
This study identifies a list of potential aging biomarkers in cattle, with 27 differentially methylated genes overlapping with those in the Human Aging Atlas. Additionally, the findings highlight the link between DNA methylation changes, aging, and puberty in cattle. This study provides valuable insights for future research on the role of DNA methylation in regulating aging and pubertal development, contributing to the advancement of genomic prediction models and livestock management strategies.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Supplementary Table S1. List of differentially methylated regions between two groups. Supplementary Table S2. List of differentially methylated genes between two groups. Supplementary Table S3. List of age-related differentially methylated genes between two groups. Supplementary Table S4. Enriched GO terms of overlaping age-related differentially methylated genes. Supplementary Table S5. Enriched KEGG pathways of overlaping age-related differentially methylated genes
Author Contributions
Conceptualization, L.T.N, B.H., E.R.; Methodology, L.T.N, B.H., E.R.; Formal Analysis, Y.G.; Writing – Original Draft Preparation, Y.G.; Writing – Review & Editing, L.T.N.; Visualization, Y.G.; Supervision, L.T.N.; Project Administration, L.T.N.; Funding Acquisition, B.H., E.R.
Funding
This work was supported by Meat and Livestock Australia grants (L.GEN.1808 and P.PSH.1401).
Institutional Review Board Statement
The animal study was reviewed and approved by Animal ethics approval was obtained from the University of Queensland ethics board (Animal Ethics Approval Number QAAFI/269/17).
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI SRA BioProject, accession no: PRJNA770750.
Acknowledgments
We would also like to thank the 54 collaborators in the Northern genomics project and Russell Lyons (Agri-Genetics Consulting), Shannon Speight and Geoffrey Fordyce for their contribution in collecting tail hair samples used.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Agricultural Trade Report [https://www.ruralbank.com.au/knowledge-and-insights/publications/agricultural-trade/].
- Holliday R, Pugh JE: DNA modification mechanisms and gene activity during development. Science (New York, NY) 1975, 187(4173):226-232.
- Horvath S: DNA methylation age of human tissues and cell types. Genome Biology 2013, 14(10):3156.
- Raj K, Szladovits B, Haghani A, Zoller JA, Li CZ, Black P, Maddox D, Robeck TR, Horvath S: Epigenetic clock and methylation studies in cats. GeroScience 2021, 43(5):2363-2378.
- Thompson MJ, Chwiałkowska K, Rubbi L, Lusis AJ, Davis RC, Srivastava A, Korstanje R, Churchill GA, Horvath S, Pellegrini M: A multi-tissue full lifespan epigenetic clock for mice. Aging (Albany NY) 2018, 10(10):2832-2854.
- Thompson MJ, vonHoldt B, Horvath S, Pellegrini M: An epigenetic aging clock for dogs and wolves. Aging 2017, 9(3):1055-1068.
- Wagner W: Epigenetic aging clocks in mice and men. Genome Biology 2017, 18(1):107.
- Raddatz G, Arsenault RJ, Aylward B, Whelan R, Böhl F, Lyko F: A chicken DNA methylation clock for the prediction of broiler health. Communications biology 2021, 4(1):76.
- Kordowitzki P, Haghani A, Zoller JA, Li CZ, Raj K, Spangler ML, Horvath S: Epigenetic clock and methylation study of oocytes from a bovine model of reproductive aging. Aging cell 2021, 20(5):e13349.
- Hayes BJ, Nguyen LT, Forutan M, Engle BN, Lamb HJ, Copley JP, Randhawa IAS, Ross EM: An Epigenetic Aging Clock for Cattle Using Portable Sequencing Technology. Frontiers in genetics 2021, 12:760450.
- Lamb HJ, Ross EM, Nguyen LT, Lyons RE, Moore SS, Hayes BJ: Characterization of the poll allele in Brahman cattle using long-read Oxford Nanopore sequencing. J Anim Sci 2020, 98(5).
- Rosen BD, Bickhart DM, Schnabel RD, Koren S, Elsik CG, Tseng E, Rowan TN, Low WY, Zimin A, Couldrey C et al.: De novo assembly of the cattle reference genome with single-molecule sequencing. GigaScience 2020, 9(3):giaa021.
- Li HJB: Minimap2: pairwise alignment for nucleotide sequences. 2018, 34(18):3094-3100.
- Gamaarachchi H, Lam CW, Jayatilaka G, Samarakoon H, Simpson JT, Smith MA, Parameswaran S: GPU accelerated adaptive banded event alignment for rapid comparative nanopore signal analysis. BMC Bioinformatics 2020, 21(1):343.
- Feng H, Conneely KN, Wu H: A Bayesian hierarchical model to detect differentially methylated loci from single nucleotide resolution sequencing data. Nucleic Acids Res 2014, 42(8):e69-e69.
- Wang Q, Li M, Wu T, Zhan L, Li L, Chen M, Xie W, Xie Z, Hu E, Xu S et al.: Exploring Epigenomic Datasets by ChIPseeker. Current Protocols 2022, 2(10):e585.
- Wang T, Ma J, Hogan AN, Fong S, Licon K, Tsui B, Kreisberg JF, Adams PD, Carvunis AR, Bannasch DL et al.: Quantitative Translation of Dog-to-Human Aging by Conserved Remodeling of the DNA Methylome. Cell Syst 2020, 11(2):176-185.e176.
- Xu S, Hu E, Cai Y, Xie Z, Luo X, Zhan L, Tang W, Wang Q, Liu B, Wang R et al.: Using clusterProfiler to characterize multiomics data. Nature protocols 2024, 19(11):3292-3320.
- Yu G: enrichplot: Visualization of Functional Enrichment Result. In.: R package; 2025.
- Luo W, Brouwer C: Pathview: an R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics (Oxford, England) 2013, 29(14):1830-1831.
- López-Catalina A, Costes V, Peiró-Pastor R, Kiefer H, González-Recio O: Oxford nanopore sequencing as an alternative to reduced representation bisulphite sequencing for the identification of CpGs of interest in livestock populations. Livestock Science 2024, 279:105377.
- Fortes MR, Li Y, Collis E, Zhang Y, Hawken RJ: The IGF1 pathway genes and their association with age of puberty in cattle. Anim Genet 2013, 44(1):91-95.
- Fortes MRS, Nguyen LT, Porto Neto LR, Reverter A, Moore SS, Lehnert SA, Thomas MG: Polymorphisms and genes associated with puberty in heifers. Theriogenology 2016, 86(1):333-339.
- Jiang B, Wu X, Meng F, Si L, Cao S, Dong Y, Sun H, Lv M, Xu H, Bai N et al.: Progerin modulates the IGF-1R/Akt signaling involved in aging. Sci Adv 2022, 8(27):eabo0322.
- Tazearslan C, Huang J, Barzilai N, Suh Y: Impaired IGF1R signaling in cells expressing longevity-associated human IGF1R alleles. Aging cell 2011, 10(3):551-554.
- Mao K, Quipildor GF, Tabrizian T, Novaj A, Guan F, Walters RO, Delahaye F, Hubbard GB, Ikeno Y, Ejima K et al.: Late-life targeting of the IGF-1 receptor improves healthspan and lifespan in female mice. Nature Communications 2018, 9(1):2394.
- Savage SA, Chanock SJ, Lissowska J, Brinton LA, Richesson D, Peplonska B, Bardin-Mikolajczak A, Zatonski W, Szeszenia-Dabrowska N, Garcia-Closas M: Genetic variation in five genes important in telomere biology and risk for breast cancer. Br J Cancer 2007, 97(6):832-836.
- Shammas MA: Telomeres, lifestyle, cancer, and aging. Curr Opin Clin Nutr Metab Care 2011, 14(1):28-34.
- Koss KJ, Schneper LM, Brooks-Gunn J, McLanahan S, Mitchell C, Notterman DA: Early Puberty and Telomere Length in Preadolescent Girls and Mothers. J Pediatr 2020, 222:193-199.e195.
- Nguyen LT, Zacchi LF, Schulz BL, Moore SS, Fortes MRS: Adipose tissue proteomic analyses to study puberty in Brahman heifers. J Anim Sci 2018, 96(6):2392-2398.
- Nardai G, Csermely P, Söti C: Chaperone function and chaperone overload in the aged. A preliminary analysis. Exp Gerontol 2002, 37(10-11):1257-1262.
- Plant TM: Neuroendocrine control of the onset of puberty. Front Neuroendocrinol 2015, 38:73-88.
- Nguyen LT, Lau LY, Fortes MRS: Proteomic Analysis of Hypothalamus and Pituitary Gland in Pre and Postpubertal Brahman Heifers. Frontiers in genetics 2022, 13:935433.
- Luo L, Yao Z, Ye J, Tian Y, Yang C, Gao X, Song M, Liu Y, Zhang Y, Li Y et al.: Identification of differential genomic DNA Methylation in the hypothalamus of pubertal rat using reduced representation Bisulfite sequencing. Reproductive Biology and Endocrinology 2017, 15(1):81.
- Nguyen LT, Reverter A, Cánovas A, Venus B, Anderson ST, Islas-Trejo A, Dias MM, Crawford NF, Lehnert SA, Medrano JF et al.: STAT6, PBX2, and PBRM1 Emerge as Predicted Regulators of 452 Differentially Expressed Genes Associated With Puberty in Brahman Heifers. Frontiers in genetics 2018, 9:87.
- Johnson SC, Rabinovitch PS, Kaeberlein M: mTOR is a key modulator of ageing and age-related disease. Nature 2013, 493(7432):338-345.
- Roa J, Garcia-Galiano D, Varela L, Sánchez-Garrido MA, Pineda R, Castellano JM, Ruiz-Pino F, Romero M, Aguilar E, López M et al.: The mammalian target of rapamycin as novel central regulator of puberty onset via modulation of hypothalamic Kiss1 system. Endocrinology 2009, 150(11):5016-5026.
- Palumbo S, Palumbo D, Cirillo G, Giurato G, Aiello F, Miraglia Del Giudice E, Grandone A: Methylome analysis in girls with idiopathic central precocious puberty. Clin Epigenetics 2024, 16(1):82.
- Sehovic E, Zellers SM, Youssef MK, Heikkinen A, Kaprio J, Ollikainen M: DNA methylation sites in early adulthood characterised by pubertal timing and development: a twin study. Clin Epigenetics 2023, 15(1):181.
- Han L, Zhang H, Kaushal A, Rezwan FI, Kadalayil L, Karmaus W, Henderson AJ, Relton CL, Ring S, Arshad SH et al.: Changes in DNA methylation from pre- to post-adolescence are associated with pubertal exposures. Clinical Epigenetics 2019, 11(1):176.
- Perrier JP, Kenny DA, Chaulot-Talmon A, Byrne CJ, Sellem E, Jouneau L, Aubert-Frambourg A, Schibler L, Jammes H, Lonergan P et al.: Accelerating Onset of Puberty Through Modification of Early Life Nutrition Induces Modest but Persistent Changes in Bull Sperm DNA Methylation Profiles Post-puberty. Frontiers in genetics 2020, 11:945.
- Meaker HJ, Coetsee TPN, Lishman AW: The effects of age at first calving on the productive and reproductive performance of beef cows. South African Journal of Animal Science 1980, 10(2):105-113.
Figure 1.
Venn diagram showing the overlap between differentially methylated genes (DMGs) identified from ONT data and age-related gene sets retrieved from the Aging Atlas, the human-dog aging clock, and the cattle aging clock.
Figure 1.
Venn diagram showing the overlap between differentially methylated genes (DMGs) identified from ONT data and age-related gene sets retrieved from the Aging Atlas, the human-dog aging clock, and the cattle aging clock.
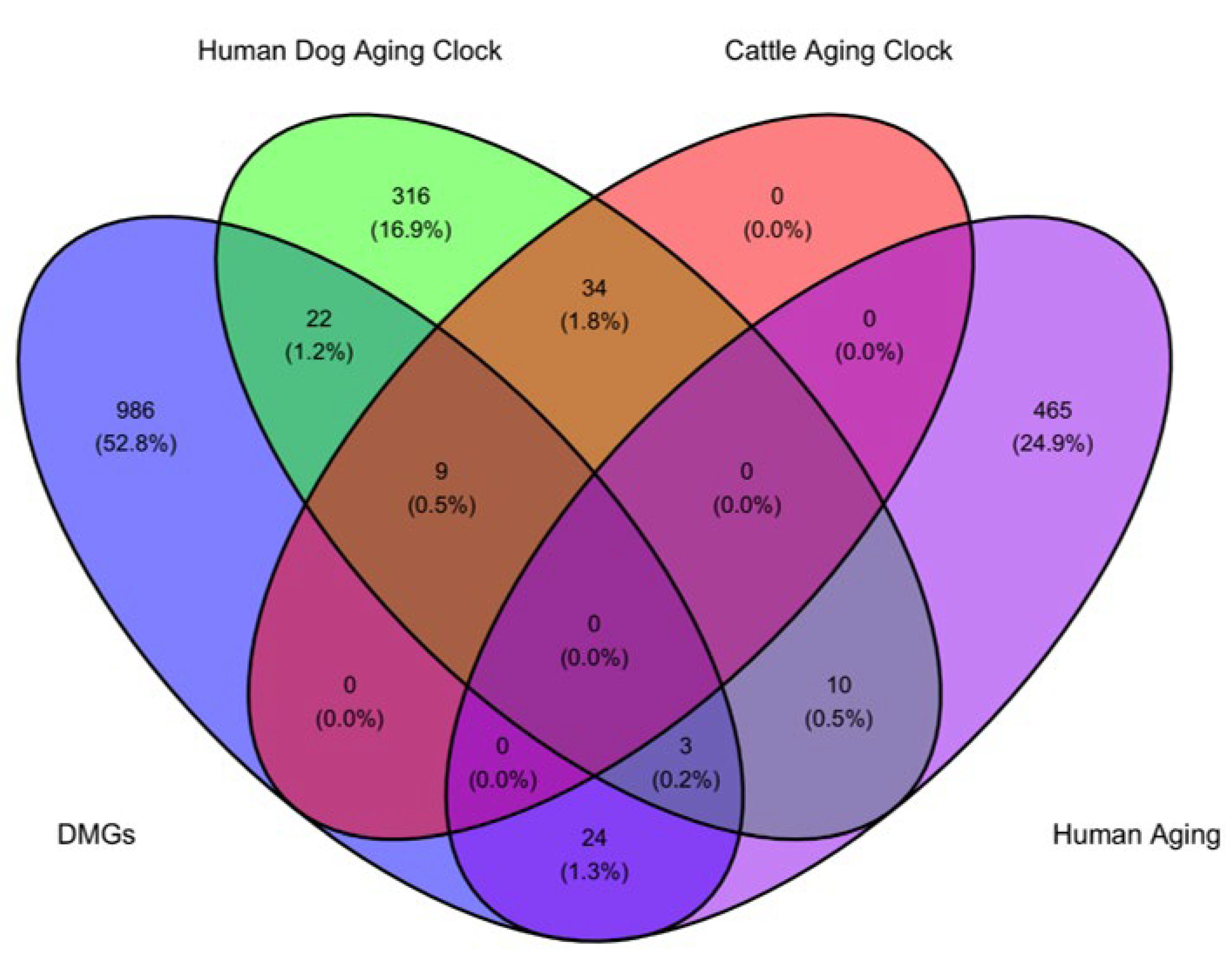
Figure 2.
The Feature Distribution of differentially methylated regions at 10x, considering promoter regions those closer than 3kb to the transcription start site.
Figure 2.
The Feature Distribution of differentially methylated regions at 10x, considering promoter regions those closer than 3kb to the transcription start site.
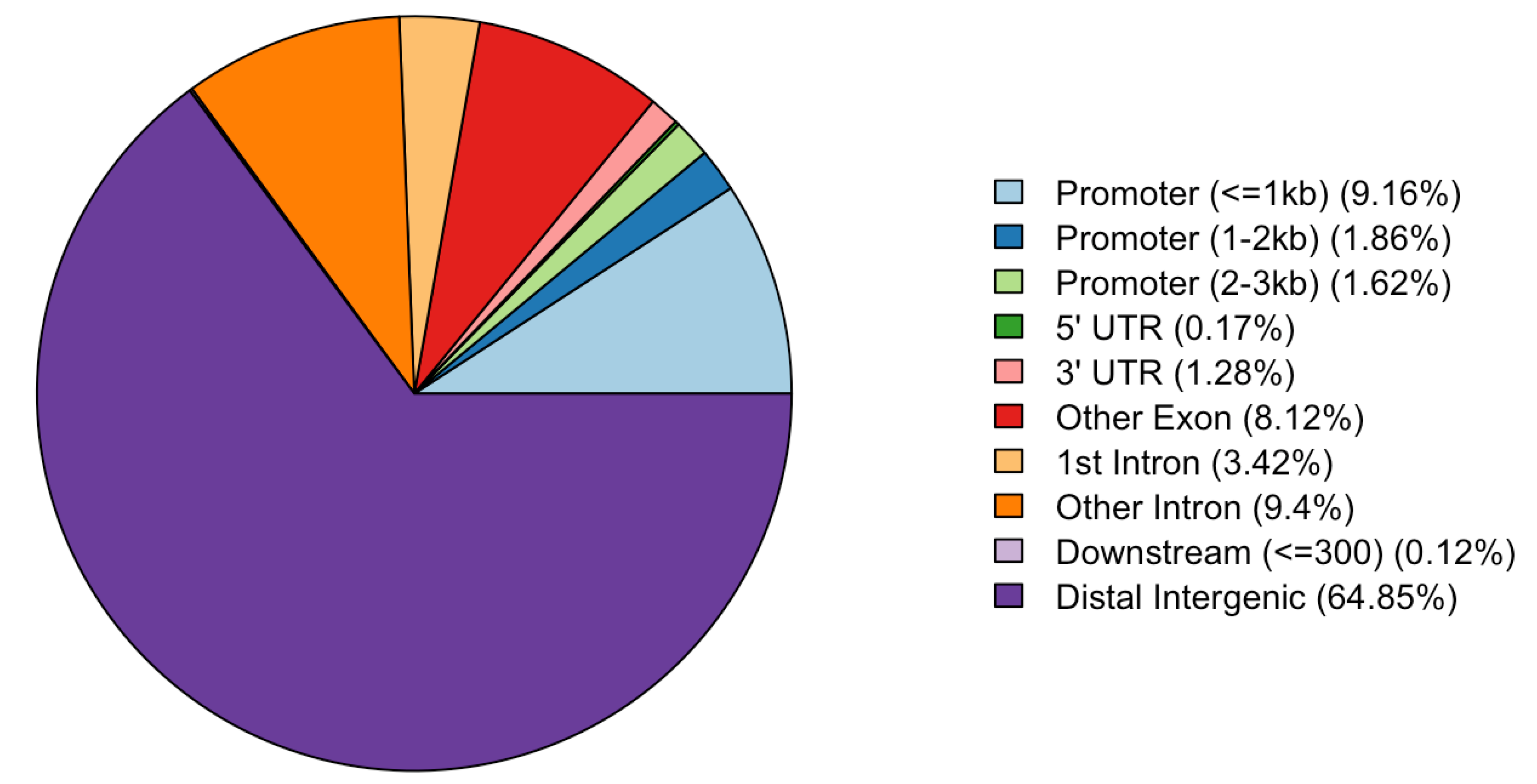
Figure 3.
Distribution of identified differentially methylated regions at 10x coverage overlaping with transcription start sites within a ± 3000 bp range.
Figure 3.
Distribution of identified differentially methylated regions at 10x coverage overlaping with transcription start sites within a ± 3000 bp range.
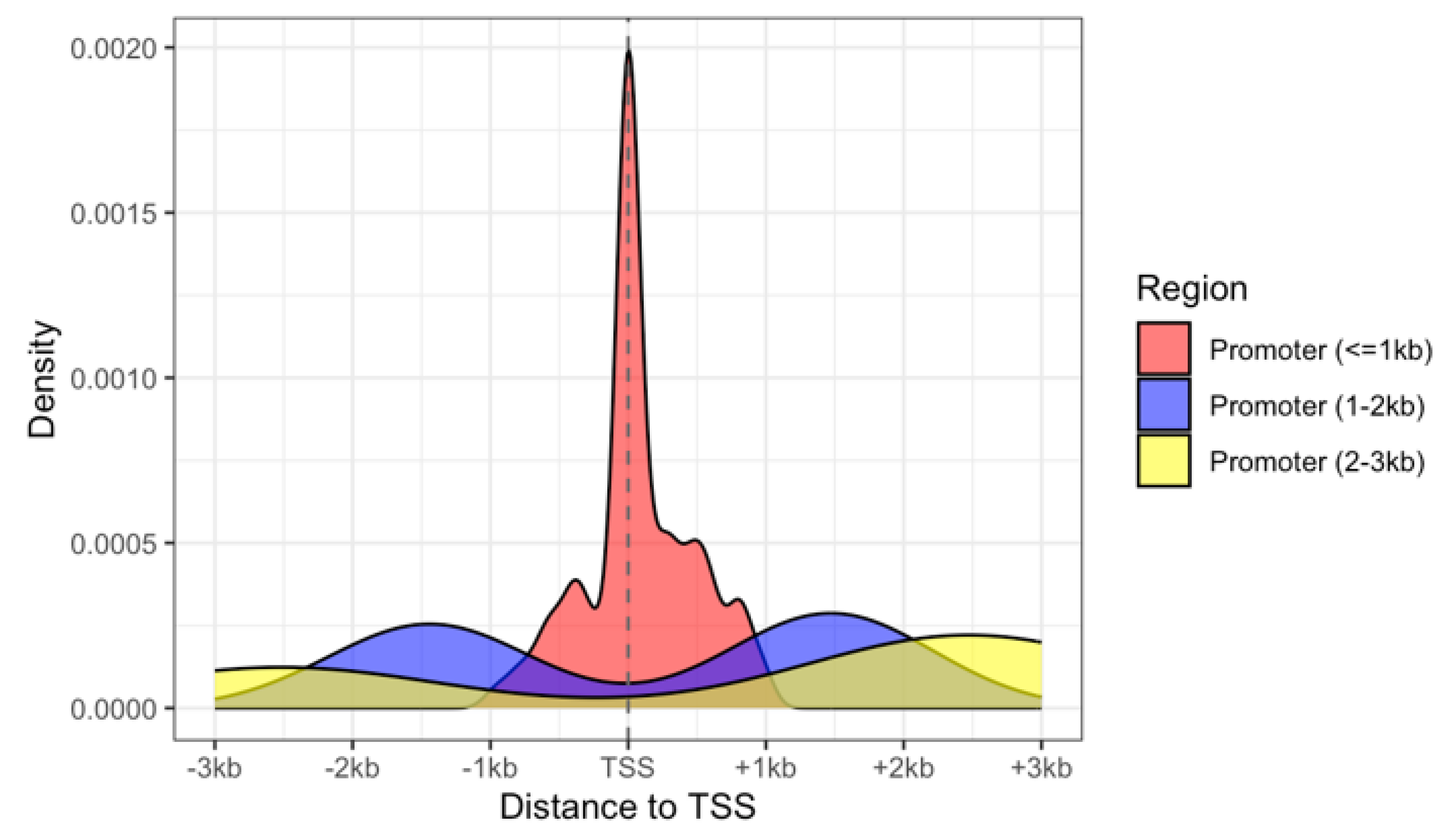

Table 1.
Information on studied animals and sequencing statistics.
| Sample | Age | Weight | Hip Height | Body Condition Score | N50 | Total Methylation site (M) | Filtered Methylation site (10X - M) |
|---|---|---|---|---|---|---|---|
| Adult_01 | 2.64 | 586 | 1430 | 4 | 1.25 | 15.5 | 0.77 |
| Adult_02 | 2.65 | 610 | 1410 | 4 | 2.45 | 14.7 | 0.79 |
| Adult_03 | 2.62 | 530 | 1340 | 5 | 2.33 | 17.8 | 1.17 |
| Mean | 2.64 | 575.33 | 1393.33 | 4.33 | 2.01 | 16.00 | 0.91 |
| SD | 0.02 | 41.05 | 47.26 | 0.58 | 0.66 | 1.61 | 0.23 |
| Yearling_01 | 1.5 | 281 | 1290 | 3 | 1.63 | 18.9 | 1.39 |
| Yearling_02 | 1.52 | 424 | 1290 | 4.5 | 1.27 | 20.7 | 1.63 |
| Yearling_03 | 1.42 | 414 | 1240 | 4.5 | 2.72 | 20.2 | 1.49 |
| Mean | 1.48 | 373.00 | 1273.33 | 4.00 | 1.87 | 19.93 | 1.50 |
| SD | 0.05 | 79.83 | 28.87 | 0.87 | 0.76 | 0.93 | 0.12 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



