
Submitted:
06 April 2025
Posted:
07 April 2025
You are already at the latest version
Abstract
Background: Currently, there are two major theories for the pathogenesis of sepsis: hyperimmune and hypoimmune. Hyperimmune theory suggests that cytokine storm causes the symptoms of sepsis. On the contrary, hypoimmune theory suggests that immunosuppression causes the manifestations of sepsis. Methods: By using microarray study on peripheral leukocytes from septic patients, this study implies that hyperactivity of TH17 immunity are noted in sepsis patients. Results: I find out that innate immunity related genes are significantly up-regulated including CD14, TLR1,2,4,5,8, HSP70, CEBP proteins, AP1(JUNB, FOSL2), TGF-β, IL-6, TGF-α, CSF2 receptor, TNFRSF1A, S100A binding proteins, CCR2, formyl peptide receptor2, amyloid proteins, pentraxin, defensins, CLEC5A, whole complement machinery, CPD, NCF, MMP, neutrophil elastase, caspases, IgG and IgA Fc receptors(CD64, CD32), ALOX5, PTGS, LTB4R, LTA4H, and ICAM1. Majority of adaptive immunity genes are down-regulated including MHC related genes, TCR genes, granzymes/perforin, CD40, CD8, CD3, TCR signaling, BCR signaling, T & B cell specific transcription factors, NK killer receptors, and TH17 helper specific transcription factors(STAT3, RORA, REL). In addition, Treg related genes are up-regulated including TGFβ, IL-15, STAT5B, SMAD2/4, CD36, and thrombospondin. Conclusions: Thus, Th17 with Treg over-presentation plays important roles in the pathophysiology of sepsis.
Keywords:
sepsis
; Th17
; innate immunity
; adaptive immunity
; Treg
Introduction
Despite of the discovery of antibiotics, mortality rate of sepsis is still very high. Most important of all, the exact pathophysiology of sepsis is still unclear. Currently, there are two dominant theories to explain the etiology of sepsis: hyperimmune theory and hypoimmune theory. However, these two theories are contrary with each other. Hyperimmune theory was proposed by Dr. Lewis Thomas. In his classical paper in NEJM 1972, he proposed that hyperactivation of proinflammatory cytokines, the cytokine storm, is the actual cause of sepsis symptoms. These uncontrolled cytokines destruct and cause multiple organ failure. His theory is the mainstream theory of sepsis etiology. Based on this theory, therapeutic strategy such as antibody neutralizing TNF-α was tested in septic patients in clinical trials. However, these antibodies did not improve the survival rate of septic patients. Further, anti-TNF-α increased the mortality rate of septic patients in several clinical trials. That makes people to doubt the hyperimmune theory. Thus, another theory-hypoimmune theory emerges. Based on the observation that immunosuppressive patients are prone to get sepsis, hypoimmune status was suggested to be the etiology of sepsis. However, the hypoimmune theory cannot successfully explain the proinflammatory cytokines storm noted in sepsis. Both hyperimmune theory and hypoimmune theory have clinical and experimental evidences. However, they are contrary with each other. In my previous study, I proposed a whole framework of host immunological pathway including eradicable and tolerable immune reactions[1,2,3,4,5,6,7,8]. Here, I use the microarray study of whole blood of septic patients to propose a new theory: Sepsis is a syndrome of Th17 immunity with over-presentations of pro-inflammatory cytokines as well as Treg cells. This new theory solves the above controversy.
Material and Methods
Microarray Dataset
According to Dr. J. A. Howrylak’s research in Physiol Genomics 2009, he collected total RNA from whole blood in sepsis and sepsis induced ARDS patients.[9] Patients were recruited from the Medical Intensive Care Unit of the University of Pittsburgh Medical Center from February 2005 to June 2007. Patients admitted to the Medical Intensive Care Unit for less than 48 hours who were intubated and receiving mechanical ventilation were eligible for the study. Patients were classified as septic patients if they met the criteria for sepsis as defined by the Society of Critical Care Medicine Consensus statement. He tried to find out molecular signature of ARDS compared to sepsis patients. His dataset is available in Gene Expression Omnibus (GEO) www.ncbi.nlm.nih.gov/geo (assession number GSE 10474). I use his samples of sepsis patients from this dataset to do the further microarray analysis. The sample size is 21 patients with 35% mortality rate. The second dataset is from GSE20189 of Gene Expression Omnibus. This dataset was collected by Dr. Melissa Rotunno in Cancer Prevention Research 2011.[10] Molecular signature of early stage of lung adenocarcinoma was studied by microarray. I use the healthy control (sample size 21) whole blood RNA from this dataset to compare the septic patients. In this study, I perform further analysis to study peripheral leukocyte gene expression profiles of sepsis compared to those of healthy controls.
Statistical Analysis
Affymetrix HG-U133A 2.0 genechip was used in both samples. RMA express software(UC Berkeley, Board Institute) is used to do normalization and to rule out the outliners of the above dataset. I rule out the potential outliners of samples due to the following criteria:
- Remove samples which exceed 99% line in RLE-NUSE T2 plot
Then, Genespring XI software was done to analysis the significant expressed genes between ARDS and healthy control leukocytes. P value cut-off point is less than 0.05. Fold change cut-off point is >2.0 fold change. Benjamini-hochberg corrected false discovery rate was used during the analysis. Totally, a gene list of 3277 genes was generated from the HGU133A2.0 chip with 18400 transcripts including 14500 well-characterized human genes.
RT-PCR Confirmation
Dr. J. A. Howrylak performed real time PCR for selected transcripts (cip1, kip2) by using TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA). In the second dataset, Dr. Melissa Rotunno also performed qRT-PCR test to validate the microarray results. RNA quantity and quality was determined by using RNA 600 LabChip-Aligent 2100 Bioanalyzer. RNA purification was done by the reagents from Qiagen Inc. All real-time PCRs were conducted by using an ABI Prism 7000 Sequence Detection System with the designed primers and probes for target genes and an internal control gene-GAPDH. This confirms that their microarray results are convincing compared to RT-PCR results.
Results
RMA Analysis of Whole Blood from Healthy Normal Control
The RMA analysis was performed for RNA samples from whole blood of healthy control of the lung adenocarcinoma dataset. Raw boxplot, NUSE plot, RLE value plot, RLE-NUSE multiplot, and RLE-NUSE T2 plot were generated. Then, sample was included and excluded by using these graphs Because of the strong deviation in the T2 plot, the sample GSM506435 was removed for the further analysis.
RMA Analysis of Whole Blood from Septic Patients
The RMA analysis was performed for RNA samples from whole blood of sepsis patients dataset. Raw boxplot, NUSE plot, RLE value plot, RLE-NUSE multiplot, and RLE-NUSE T2 plot were generated. Then, sample was included and excluded by using these graphs. GSM265024 and GSM265030 are removed due to above criteria.
Toll-like Signaling and Heat Shock Protein Expression in Septic Patients
According to the microarray analysis, Toll-like receptors 1, 2, 4, 5, 8 are up-regulated in sepsis. (Table 1) CD14 molecule and downstream signaling such as IRAK4 and TAB2 are also up-regulated. TLR1, 2, 4, 5, 8 are mediating anti-bacterial immune response. Thus, TH17-like proinflammatory cytokines such as IL-6 will be triggered. However, the negative TLR regulator-IRAK3 is 21 fold up-regulated. Thus, TLR 1, 2, 4, 5, 8 signaling may not successfully trigger proinflammatory cytokines. Other pathway such as CD14 may act as an important alternative pathway to trigger IL-6 and other TH17-like cytokines. Other pattern recognition receptors such as formyl peptide receptors (FPR) which can recognize specific bacterial antigen to trigger innate immunity are also differentially expressed. FPR1 is 7.6 fold down-regulated, but FPR2 is 4.7 fold up-regulated.
Antigen Processing and Antigen Presentation Genes in Sepsis
In table 2, we can see all MHC related genes are down-regulated in leukocytes of septic patients. These down-regulated genes include HLA-DPB, HLA-DQA, HLA-DRB, HLA-DOB, HLA-DRA, HLA-DQB, Tapasin, MHC-related transcripts, HLA-B, and HLA-DPA. Among them, HLA-B is more than 11 fold down-regulated. MHC genes are keys to the antigen presentation to trigger adaptive immune reaction such as B cell or T cell activation. Since all the MHC related genes are down-regulated, antigen presentation during sepsis is likely to be impaired. This matches previous observations[11].
TH17-like Innate Immune Transcription Factors in Sepsis
In table 3, many immune related transcription factors are differentially regulated during sepsis. First of all, many innate immunity related transcription factors are up-regulated in septic patients. These include AP1 (JunB and FosL2), NFIL3, ARNT, and CEBP (CEBPA, CEBPG, and CEBPD) genes. Aryl hydrocarbon receptor nuclear translocator (ARNT) plays an important role in the activation of TH17-like innate immunity. CEBP family genes are related to the activity of myeloid cells and granulocytes. CEBP genes are also related to the activation of acute response proteins. In addition, the inhibitor of NF-κB, NFKBIA, is down-regulated in sepsis. It means that the activity of NF-κB, a key innate immunity mediator, is up-regulated in septic patients. It is worth noting that two important transcription factors: High Mobility Group Box (HMGB) and Hypoxia inducible factor alpha (HIF-α) are also up-regulated during sepsis. HMGB, a vital innate immunity mediator, is greater than nine fold up-regulation.
STAT1, a key transcription factor for TH1 and THαβ immunity, is down-regulated in sepsis. In addition, TBX21 (T-bet), a key TH1 immune response driver, is also down-regulated. In addition, MafB which can suppress IFNαβ in THαβ immunity is up-regulated [12]. Other TH2 related key transcription factors such as GATA3 and C-MAF are also down-regulated [13]. It means that TH1, TH2, and THαβ are down-regulated in sepsis. Surprisingly, key TH17 related transcription factors are also down-regulated including REL, STAT3, and RORA [14]. Besides, SOCS3, a negative regulator of the central TH17 transcription factor STAT3, is up-regulated. It means that TH17 helper cells cannot be successfully triggered. On the other hand, Treg and TGFβ signaling are up-regulated including STAT5B, IL-15, SMAD2, and SMAD4 [15,16]. TH17 and Treg associated aryl hydrocarbon receptor nuclear translocator (ARNT) is also up-regulated in sepsis [17]. Thus, Treg cells are likely to be activated in sepsis. This matches the previous observations that Treg cells are up-regulated during sepsis.
Down-regulated genes include B cell stimulatory transcription factor (PAX5), BCR signaling (FYN and LYN), and PI3K signaling (PIK3CB, PIK3IP1, PIK3CG, and PIK3R1) [18,19,20]. The negative regulator of PI3K signaling, PTEN, is 4.6 fold up-regulated. BCL6 is the key transcription factor for the follicular helper T cell for IgM producing B cells. IBTK can inhibit B cell differentiation and activation. PI3K signaling is the downstream stimulatory pathway of B cell activation. Thus, BCR signaling appears to be suppressed during sepsis.
TH17-like and Treg Related Cytokines Are Up-Regulated During Sepsis
In table 4, many TH17-like and Treg related cytokines are up-regulated in septic patients. The whole TGFβ activation machinery is up-regulated including thrombospondin, CD36, and TGFβ itself. TGFA and IL-15 are also up-regulated. Besides, IL-6 is also up-regulated in sepsis. Thus, both key TH17 driven cytokines, TGFβ and IL-6, are activated in septic patients. However, full activation of TH17 helper cells also need a TCR signaling. IL-32, a TH1 related macrophage differentiation factor, is down-regulated [21]. TH22 mediators, IL1A is down-regulated and IL1RN (IL1 receptor antagonist is up-regulated. It means that TH22 is not activated in sepsis.
In table 5, cytokine receptors are differentially regulated in sepsis. On the contrary with cytokine, cytokine receptor in a certain immunological pathway is usually down-regulated. Thus, since TH17-like immunity is activated. TGFBR3, IL6R, and IL17RA are all down-regulated. TGF-β receptor 3 is greater than 11 fold down-regulated, and interleukin 6 receptor is greater than 16 fold down-regulated. Treg is also triggered in sepsis, so TGFBR3, IL2RB, and IL7R are also down-regulated. TH1 related cytokine receptors, IFNGR1 and IFNGR2, are up-regulated. TH2 cytokine receptor, IL4R, is also up-regulated. As for TH-αβ immunity, IFNAR1 is up-regulated but IFNAR2 is down-regulated. TH22 cytokine receptors, IL1R1 and IL1R2, are up-regulated. Thus, TH1, TH2, TH-αβ, and TH22 are not activated during sepsis.
Th17-like Innate Immunity Related Effector Molecule Up-Regulation in Sepsis
In table 6, many acute response proteins are up-regulated. These acute phase proteins are up-regulated by IL-6 and CEBP proteins. These genes include amyloid proteins (APP and APLP2), pentraxin(PTX3), transferrin receptor (TFRC), CLEC (CLEC5A and CLEC1B), and defensins (DEFA1, DEFA1B, DEFA3, and DEFA4). These above proteins are innate immunity effector proteins to attack bacterial antigens non-specifically. Defensin A4 is greater than 6 fold up-regulated.
In table 7, the whole set complement machinery, an important effector component of innate immunity, is up-regulated. These include CD59, CD55, C1QB, ITGAM, CR1, CD46, C3AR1, ITGAX, C1QA, C1RL, C5AR1, and CD97. Thus, complement molecules are activated during sepsis. These complement molecules attack bacterial cell walls and membranes to cause their damage. However, complements may also cause harmful effect to the host.
In table 8, PMN matrix metallopeptidases(MMP) and elastase are up-regulated. These protein enzymes can digest bacterial antigens as well as extracellular matrix. These genes include MMP8, MMP9, MMP25, and ELANE (elastase). In addition, tissue inhibitor of MMP, TIMP2, and serum inhibitors of elastase or proteinase, SERPINA1, SERPINB1, and SERPINB2, are also up-regulated. It means that PMN proteinases are dysregulated. It is worth noting that MMP8 is 32 fold up-regulated and MMP9 is 10 fold up-regulated.
Coagulation, Glycolysis, Acidosis, and Vasodilation Gene Dysregulation in Sepsis
In Table 9, many coagulation related genes are dysregulated during sepsis. Actually, disseminated intracellular coagulopathy is a common manifestation of sepsis. Up-regulated coagulation genes include F13A1, F5, F8, GP1BB, PROS1, PLAUR, MCFD2, TFPI, F2RL1, ITGA2B, PDGFC, ITGB3, and THBD. Both coagulation factors and inhibitors are dysregulated in sepsis.
The whole glycolytic pathway enzymes are up-regulated during sepsis.(Table 10) These include lactate dehydrogenase A, phosphoglycerate kinase 1, pyruvate kinase, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3, hexokinase 2, glycogen phosphorylase, 2,3-bisphosphoglycerate mutase, hexokinase 3, glucose-6-phosphate isomerase, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 2, glyceraldehyde-3-phosphate dehydrogenase, enolase 1, and phosphoglycerate kinase 1. In addition, the enzyme, pyruvate dehydrogenase kinase, which can stop pyruvate to form acetyl-CoA is up-regulated. The enzyme, pyruvate dehyrogenase phosphatase, which can facilitate pyruvate to form acetyl-CoA to enter aerobic citric acid cycle is down-regulated in sepsis. Thus, pyruvate can keep on forming lactate in anaerobic pathway during sepsis.
Concurrently, H+-ATPases are also up-regulated during sepsis (Table 11). In my previous article(paper in press), I find out the coupling between glycolytic enzymes and H+-ATPases during falciparum malarial infection. Here, I also find out up-regulated H+-ATPases including ATP6V0B, ATP6V0E1, ATP6AP2, ATP6V1C1, TCIRG1, ATP6V1D, ATP11B, and ATP11A. Besides, carbonic anhydrase IV & II, which can produce H2CO3, are up-regulated in sepsis. Thus, this can help to explain the acidosis during sepsis.
Failure of T Lymphocyte Adaptive Immunity During Sepsis
Lymphocytes play important roles in adaptive immunity. In sepsis, major lymphocyte populations: T cells, and B cells are all down-regulated. Thus, lymphocyte adaptive immunity fails to induce during sepsis. This is very important is sepsis pathogenesis.
In table 12, many T cell related genes are also down-regulated. These down-regulated genes include TCR genes (TRAC, TARP, TRBC1/C2, TRD@, TRGC2, and TRDV3), CD costimulatory molecules(CD3E, CD8A, CD3G, LY9, CD3D,CD2), T cell specific transcription factors(IKZF1, TCF7, NFAT5, NFATC3, TCF7L2, NFATC2IP, TBX21, ID2, and ID2B), granzyme/perforin (GZMA, GNLY, GZMK, GZMB, GZMH, and PRF1), and TCR downstream signaling(ZAP70 and LCK) [22]. Thus, the whole-set of T cell activation machinery is suppressed. Both CD4 helper T cells and CD8 cytotoxic T cells are inactivated and down-regulated in septic patients.
Results of Ingenuity Pathway Analysis of Sepsis Patients Compared to Control
In the network analysis, top 1 over-represented network is HIF1A (Hypoxia induced factor 1A) centered network and the top 2 over-represented network is PTEN centered network. (Figure 1 and Figure 2) Sepsis is related to tissue hypoxia and PTEN is related to immunosuppression. In figure 3 and figure 4, top regulator effector networks are shown including ITGB3, IL1B, and FOXO1. ITGB3 and IL1B play important roles in innate immunity. And, FOXO1 can play a role in immunosuppression. TGFB is in the center position of the regulator effector network. Thus, both innate immunity and immunosuppression are important in the pathogenesis of sepsis. In figure 5, the identified upstream regulator in sepsis is TNF, and it also suggests that innate immunity is key to the pathophysiology of sepsis.
Discussion
Despite of current antibiotics treatment, sepsis still causes a very high mortality. The pathophysiology of sepsis is still unclear [23,24]. The most dominant theory for sepsis mechanism is hyperimmune [25]. Hyperimmunity with cytokine storm was observed in sepsis by Dr. Lewis Thomas [26]. He suggested the symptom and sign in sepsis is due to the overactivity of pro-inflammatory cytokines in a NEJM paper. His theory is widely accepted. Based on his hyperimmune theory, many therapeutic strategies were developed. Most famous approach is the anti-TNF agent in sepsis clinical trials. Because the pro-inflammatory cytokine TNFα is up-regulated in sepsis, use of anti-TNF agent should help to control sepsis. However, the result is opposite. Usage of anti-TNF agent increase the sepsis mortality rate [27,28,29]. Thus, the sepsis-hyperimmune theory is doubtful.
The other sepsis pathophysiology theory emerged. This is the hypoimmune theory. Because immunocompromised patients are prone to develop sepsis, hypoimmune should be related to the cause of sepsis [30]. In addition, massive effector lymphocyte apoptosis, depletion of dendritic cells, and elevated Treg cells are noted during sepsis [31,32,33,34,35]. In previous reports, down-regulation of co-stimulatory molecules and MHC are noted in septic patients [36]. In addition, B cells play important roles in recovery from sepsis status [37]. This hypothesis is not accepted by most scientists because it cannot explain the observed cytokine storm during sepsis. Thus, both theories have some evidence support and both are only partially correct.
Thus, the third theory proposed. This is the sequence theory. There is hyperimmune first during sepsis, and then hypoimmune follows. This theory tried to incoperate both theories. However, it is not clear why there will be such sequential immune response. There is no existing immunological mechanism to explain this sequential effect. Why does the hyperimmune happen first? Why does the hyperimmune become to be hypoimmune? In addition, immunodeficiency patients are easily got sepsis. Then, why do these immunodeficiency patients easily develop hyperimmune status first? Current sepsis theory cannot well explain this.
In this study, I use microarray analysis to demonstrate that sepsis is actually a hyperactivity of innate immunity and hypoactivity of adaptive immunity. Thus, it can well explain the co-existence of hyperimmune and hypoimmune. The hypoimmune adaptive immunity explains why immunocompromised patients tend to suffer from sepsis easily. The hyperimmune innate immunity explains why pro-inflammatory cytokine storm is observed at sepsis. The adaptive immune dysfunction with lack of T helper cells is the key to sepsis pathogenesis. TH22 is the eradicable immunity against extracellular bacteria, and TH17 is the tolerable immunity against extracellular bacteria. Thus, block TH22 related cytokines such as TNFα can further stop the successful generation of TH22 helper cells to initiate adaptive immunity to combat or to eradicate extracellular bacteria. Thus, it can explain why TNF blockade increase the mortality rate of sepsis patients. Previous studies pointed out that TH22 immunity can successfully combat sepsis[38,39,40]. TH17 immunity has the components of both interleukin-17 dominant pro-inflammatory cytokines and the TGF-β dominant regulatory T cells. Thus, sepsis cannot activate host eradicable immunity to completely kill the bacteria. On the other hand, sepsis triggers a host tolerable immunity with hyperimmune cytokine storms and hypoimmune TGF-β. The up-regulation of TGF-β will cause the multi-organ failure with promoting tissue fibrosis[7]. This explains why sepsis is usually related to multi-organ failure. There is a misunderstanding that TH17/Treg ratio determines the severity of sepsis[41,42]. It is wrong because TH17 immune response itself already includes Treg cell component. TH17 is initiate by TGF-β plus interleukin-6 or other pro-inflammatory cytokines. It is possible these pathogenic bacteria trigger the host TH17 immunity instead of TH22 immunity. The key point is we need to successfully induce eradicable TH22 immunity in order to completely destroy the extracellular bacteria.
In the microarray study, I find out evidences to support my theory. The whole blood from septic patients can reflect the leukocyte expression patterns. I find out that innate immunity related genes are significantly up-regulated. These genes include CD14, TLR1,2,4,5,8, HSP70, CEBP proteins, AP1(JUNB, FOSL2), TGF-β, IL-6, TGF-α, CSF2 receptor, formyl peptide receptor2, amyloid proteins, pentraxin, defensins, CLEC5A, whole complement machinery, CPD, NCF, MMP, and neutrophil elastase. I also find out that majority of adaptive immunity genes are down-regulated including MHC related genes, TCR genes, granzymes/perforin, CD40, CD8, CD3, TCR signaling, BCR signaling, T & B cell specific transcription factors, and TH22 helper specific transcription factors (STAT3, RORA, REL). In addition, Treg related genes are up-regulated including TGFβ, IL-15, STAT5B, SMAD2/4, CD36, and thrombospondin. Up-regulated regulatory cells during sepsis are also shown in other previous studies. Up-regulated Treg related genes can also suppress the adaptive immunity in sepsis. These all support my sepsis pathogenesis. Our analysis confirmed a two-hit model of sepsis. The first hit is to trigger over-activated innate immunity. The second hit is to suppress MHC and T helper cells to up-regulate immunosuppression by regulatory T cells. This study provides further pathophysiology of sepsis.
Sepsis is also related to several complications such as disseminated intravascular coagulation(DIC), hypotension/shock, and lactate acidosis [43]. In this microarray analysis, I find out that many coagulation related genes are up-regulated during sepsis including factor5, factor8, facto13, protein S, plasminogen receptor, ITGA2B, ITGB3, and thrombomodulin. Thus, it can help to explain the mechanism of DIC during sepsis. The whole set of glycolytic enzymes are up-regulated during sepsis including LDHA, PGK1, PKM2, PFKFB3, HK2, PYGL, BPGM, HK3, PDK3, GPI, PFKFB2, GAPDH, and ENO1. In addition, glycolytic enzyme coupled H+-ATPase genes are also up-regulated. These can explain the lactate acidosis noted during sepsis.
Bacteria have strategies to suppress host immunity for their survival, especially the adaptive immunity [44]. In conclusion, after knowing the pathogenesis of sepsis, we can develop better preventive and therapeutic agents to control sepsis. The impairment of adaptive immunity could be more important than the overactivation of innate immunity during sepsis. Thus, I may use medications to activate host adaptive immunity such as T helper cells to combat sepsis. In addition, we can also develop therapeutic strategies to cope with sepsis related complications such as DIC and lactate acidosis. Hopefully, the detrimental illness-sepsis will be overcome one day.
Acknowledgement
Many thanks to Dr. J. A. Howrylak for the sepsis microarray dataset as well as to the publicly available Gene Expression Omnibus website. I am also very grateful for National Taiwan University microarray core for the service of Ingenuity Pathway Analysis.
Competing interests
The authors declare that they have no any financial interest relating to this study.
References
- Hu, W.C. A Framework of All Discovered Immunological Pathways and Their Roles for Four Specific Types of Pathogens and Hypersensitivities. Front Immunol 2020, 11, 1992. [CrossRef]
- Hu, W.C. Human immune responses to Plasmodium falciparum infection: molecular evidence for a suboptimal THαβ and TH17 bias over ideal and effective traditional TH1 immune response. Malar J 2013, 12, 392. [CrossRef]
- Chu, Y.T.; Liao, M.T.; Tsai, K.W.; Lu, K.C.; Hu, W.C. Interplay of Chemokines Receptors, Toll-like Receptors, and Host Immunological Pathways. Biomedicines 2023, 11. [CrossRef]
- Huang, Y.M.; Shih, L.J.; Hsieh, T.W.; Tsai, K.W.; Lu, K.C.; Liao, M.T.; Hu, W.C. Type 2 hypersensitivity disorders, including systemic lupus erythematosus, Sjögren's syndrome, Graves' disease, myasthenia gravis, immune thrombocytopenia, autoimmune hemolytic anemia, dermatomyositis, and graft-versus-host disease, are THαβ-dominant autoimmune diseases. Virulence 2024, 15, 2404225. [CrossRef]
- Lee, Y.H.; Tsai, K.W.; Lu, K.C.; Shih, L.J.; Hu, W.C. Cancer as a Dysfunctional Immune Disorder: Pro-Tumor TH1-like Immune Response and Anti-Tumor THαβ Immune Response Based on the Complete Updated Framework of Host Immunological Pathways. Biomedicines 2022, 10. [CrossRef]
- Lu, K.C.; Tsai, K.W.; Wang, Y.K.; Hu, W.C. Types of cell death and their relations to host immunological pathways. Aging (Albany NY) 2024, 16, 11755-11768. [CrossRef]
- Lu, K.C.; Tsai, K.W.; Hu, W.C. Role of TGFβ-producing regulatory T cells in scleroderma and end-stage organ failure. Heliyon 2024, 10, e35590. [CrossRef]
- Wen, T.H.; Tsai, K.W.; Wu, Y.J.; Liao, M.T.; Lu, K.C.; Hu, W.C. The Framework for Human Host Immune Responses to Four Types of Parasitic Infections and Relevant Key JAK/STAT Signaling. Int J Mol Sci 2021, 22. [CrossRef]
- Howrylak, J.A.; Dolinay, T.; Lucht, L.; Wang, Z.; Christiani, D.C.; Sethi, J.M.; Xing, E.P.; Donahoe, M.P.; Choi, A.M. Discovery of the gene signature for acute lung injury in patients with sepsis. Physiol Genomics 2009, 37, 133-139. [CrossRef]
- Rotunno, M.; Hu, N.; Su, H.; Wang, C.; Goldstein, A.M.; Bergen, A.W.; Consonni, D.; Pesatori, A.C.; Bertazzi, P.A.; Wacholder, S., et al. A gene expression signature from peripheral whole blood for stage I lung adenocarcinoma. Cancer Prev Res (Phila) 2011, 4, 1599-1608. [CrossRef]
- Docke, W.D.; Randow, F.; Syrbe, U.; Krausch, D.; Asadullah, K.; Reinke, P.; Volk, H.D.; Kox, W. Monocyte deactivation in septic patients: restoration by IFN-gamma treatment. Nat Med 1997, 3, 678-681.
- Kim, H.; Seed, B. The transcription factor MafB antagonizes antiviral responses by blocking recruitment of coactivators to the transcription factor IRF3. Nat Immunol 2010, 11, 743-750. [CrossRef]
- Ho, I.C.; Lo, D.; Glimcher, L.H. c-maf promotes T helper cell type 2 (Th2) and attenuates Th1 differentiation by both interleukin 4-dependent and -independent mechanisms. J Exp Med 1998, 188, 1859-1866.
- Ruan, Q.; Kameswaran, V.; Zhang, Y.; Zheng, S.; Sun, J.; Wang, J.; DeVirgiliis, J.; Liou, H.C.; Beg, A.A.; Chen, Y.H. The Th17 immune response is controlled by the Rel-RORgamma-RORgamma T transcriptional axis. J Exp Med 2011, 208, 2321-2333. [CrossRef]
- Asanuma, S.; Tanaka, J.; Sugita, J.; Kosugi, M.; Shiratori, S.; Wakasa, K.; Shono, Y.; Shigematsu, A.; Kondo, T.; Kobayashi, T., et al. Expansion of CD4(+)CD25 (+) regulatory T cells from cord blood CD4(+) cells using the common gamma-chain cytokines (IL-2 and IL-15) and rapamycin. Ann Hematol 2011, 90, 617-624. [CrossRef]
- Cohen, A.C.; Nadeau, K.C.; Tu, W.; Hwa, V.; Dionis, K.; Bezrodnik, L.; Teper, A.; Gaillard, M.; Heinrich, J.; Krensky, A.M., et al. Cutting edge: Decreased accumulation and regulatory function of CD4+ CD25(high) T cells in human STAT5b deficiency. J Immunol 2006, 177, 2770-2774.
- Quintana, F.J.; Basso, A.S.; Iglesias, A.H.; Korn, T.; Farez, M.F.; Bettelli, E.; Caccamo, M.; Oukka, M.; Weiner, H.L. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008, 453, 65-71. [CrossRef]
- Gonda, H.; Sugai, M.; Nambu, Y.; Katakai, T.; Agata, Y.; Mori, K.J.; Yokota, Y.; Shimizu, A. The balance between Pax5 and Id2 activities is the key to AID gene expression. J Exp Med 2003, 198, 1427-1437. [CrossRef]
- Horikawa, K.; Nishizumi, H.; Umemori, H.; Aizawa, S.; Takatsu, K.; Yamamoto, T. Distinctive roles of Fyn and Lyn in IgD- and IgM-mediated signaling. Int Immunol 1999, 11, 1441-1449.
- Kirstetter, P.; Thomas, M.; Dierich, A.; Kastner, P.; Chan, S. Ikaros is critical for B cell differentiation and function. Eur J Immunol 2002, 32, 720-730.
- Netea, M.G.; Azam, T.; Lewis, E.C.; Joosten, L.A.; Wang, M.; Langenberg, D.; Meng, X.; Chan, E.D.; Yoon, D.Y.; Ottenhoff, T., et al. Mycobacterium tuberculosis induces interleukin-32 production through a caspase- 1/IL-18/interferon-gamma-dependent mechanism. PLoS Med 2006, 3, e277. [CrossRef]
- Yang, C.Y.; Best, J.A.; Knell, J.; Yang, E.; Sheridan, A.D.; Jesionek, A.K.; Li, H.S.; Rivera, R.R.; Lind, K.C.; D'Cruz, L.M., et al. The transcriptional regulators Id2 and Id3 control the formation of distinct memory CD8+ T cell subsets. Nat Immunol 2011, 12, 1221-1229. [CrossRef]
- Hotchkiss, R.S.; Karl, I.E. The pathophysiology and treatment of sepsis. N Engl J Med 2003, 348, 138-150. [CrossRef]
- Oberholzer, A.; Oberholzer, C.; Moldawer, L.L. Sepsis syndromes: understanding the role of innate and acquired immunity. Shock 2001, 16, 83-96.
- Girardin, E.; Grau, G.E.; Dayer, J.M.; Roux-Lombard, P.; Lambert, P.H. Tumor necrosis factor and interleukin-1 in the serum of children with severe infectious purpura. N Engl J Med 1988, 319, 397-400. [CrossRef]
- Thomas, L. Germs. N Engl J Med 1972, 287, 553-555. [CrossRef]
- Warren, H.S. Strategies for the treatment of sepsis. N Engl J Med 1997, 336, 952-953. [CrossRef]
- Fisher, C.J., Jr.; Agosti, J.M.; Opal, S.M.; Lowry, S.F.; Balk, R.A.; Sadoff, J.C.; Abraham, E.; Schein, R.M.; Benjamin, E. Treatment of septic shock with the tumor necrosis factor receptor:Fc fusion protein. The Soluble TNF Receptor Sepsis Study Group. N Engl J Med 1996, 334, 1697-1702. [CrossRef]
- Zeni, F.; Freeman, B.; Natanson, C. Anti-inflammatory therapies to treat sepsis and septic shock: a reassessment. Crit Care Med 1997, 25, 1095-1100.
- Ward, P.A. Immunosuppression in sepsis. JAMA 2011, 306, 2618-2619. [CrossRef]
- Kasten, K.R.; Tschop, J.; Adediran, S.G.; Hildeman, D.A.; Caldwell, C.C. T cells are potent early mediators of the host response to sepsis. Shock 2010, 34, 327-336. [CrossRef]
- Hein, F.; Massin, F.; Cravoisy-Popovic, A.; Barraud, D.; Levy, B.; Bollaert, P.E.; Gibot, S. The relationship between CD4+CD25+CD127- regulatory T cells and inflammatory response and outcome during shock states. Crit Care 2010, 14, R19. [CrossRef]
- Ayala, A.; Chung, C.S.; Xu, Y.X.; Evans, T.A.; Redmond, K.M.; Chaudry, I.H. Increased inducible apoptosis in CD4+ T lymphocytes during polymicrobial sepsis is mediated by Fas ligand and not endotoxin. Immunology 1999, 97, 45-55.
- Hotchkiss, R.S.; Tinsley, K.W.; Swanson, P.E.; Grayson, M.H.; Osborne, D.F.; Wagner, T.H.; Cobb, J.P.; Coopersmith, C.; Karl, I.E. Depletion of dendritic cells, but not macrophages, in patients with sepsis. J Immunol 2002, 168, 2493-2500.
- Unsinger, J.; Herndon, J.M.; Davis, C.G.; Muenzer, J.T.; Hotchkiss, R.S.; Ferguson, T.A. The role of TCR engagement and activation-induced cell death in sepsis-induced T cell apoptosis. J Immunol 2006, 177, 7968-7973.
- Manjuck, J.; Saha, D.C.; Astiz, M.; Eales, L.J.; Rackow, E.C. Decreased response to recall antigens is associated with depressed costimulatory receptor expression in septic critically ill patients. J Lab Clin Med 2000, 135, 153-160. [CrossRef]
- Kelly-Scumpia, K.M.; Scumpia, P.O.; Weinstein, J.S.; Delano, M.J.; Cuenca, A.G.; Nacionales, D.C.; Wynn, J.L.; Lee, P.Y.; Kumagai, Y.; Efron, P.A., et al. B cells enhance early innate immune responses during bacterial sepsis. J Exp Med 2011, 208, 1673-1682. [CrossRef]
- Hammer, A.M.; Morris, N.L.; Cannon, A.R.; Khan, O.M.; Gagnon, R.C.; Movtchan, N.V.; van Langeveld, I.; Li, X.; Gao, B.; Choudhry, M.A. Interleukin-22 Prevents Microbial Dysbiosis and Promotes Intestinal Barrier Regeneration Following Acute Injury. Shock 2017, 48, 657-665. [CrossRef]
- Liu, E.H.; Zheng, Z.N.; Xiao, C.X.; Liu, X.; Lin, X.Q. IL-22 relieves sepsis-induced liver injury via activating JAK/STAT3 signaling pathway. J Biol Regul Homeost Agents 2020, 34, 1719-1727. [CrossRef]
- Yu, C.; Ling, Q.; Jiao, J.; Liu, J.; Huang, Z.; Wang, F.; Sun, X.; Kong, X. Interleukin-22 protects from endotoxemia by inducing suppressive F4/80(+)Ly6G(hi)Ly6C(hi) cells population. BMC Immunol 2022, 23, 45. [CrossRef]
- Liu, X.; Chen, L.; Peng, W.; Deng, H.; Ni, H.; Tong, H.; Hu, H.; Wang, S.; Qian, J.; Liang, A., et al. Th17/Treg balance: the bloom and wane in the pathophysiology of sepsis. Front Immunol 2024, 15, 1356869. [CrossRef]
- Wu, Y.; Wu, G.; Li, M.; Chang, Y.; Yu, M.; Meng, Y.; Wan, X. Prediction of Th17/Treg cell balance on length of stay in intensive care units of patients with sepsis. J Intensive Med 2024, 4, 240-246. [CrossRef]
- Naylor, J.M.; Kronfeld, D.S. In vivo studies of hypoglycemia and lactic acidosis in endotoxic shock. Am J Physiol 1985, 248, E309-316.
- Hornef, M.W.; Wick, M.J.; Rhen, M.; Normark, S. Bacterial strategies for overcoming host innate and adaptive immune responses. Nat Immunol 2002, 3, 1033-1040. [CrossRef]
Figure 1.
HIF centered network in sepsis via Ingenuity pathway analysis.
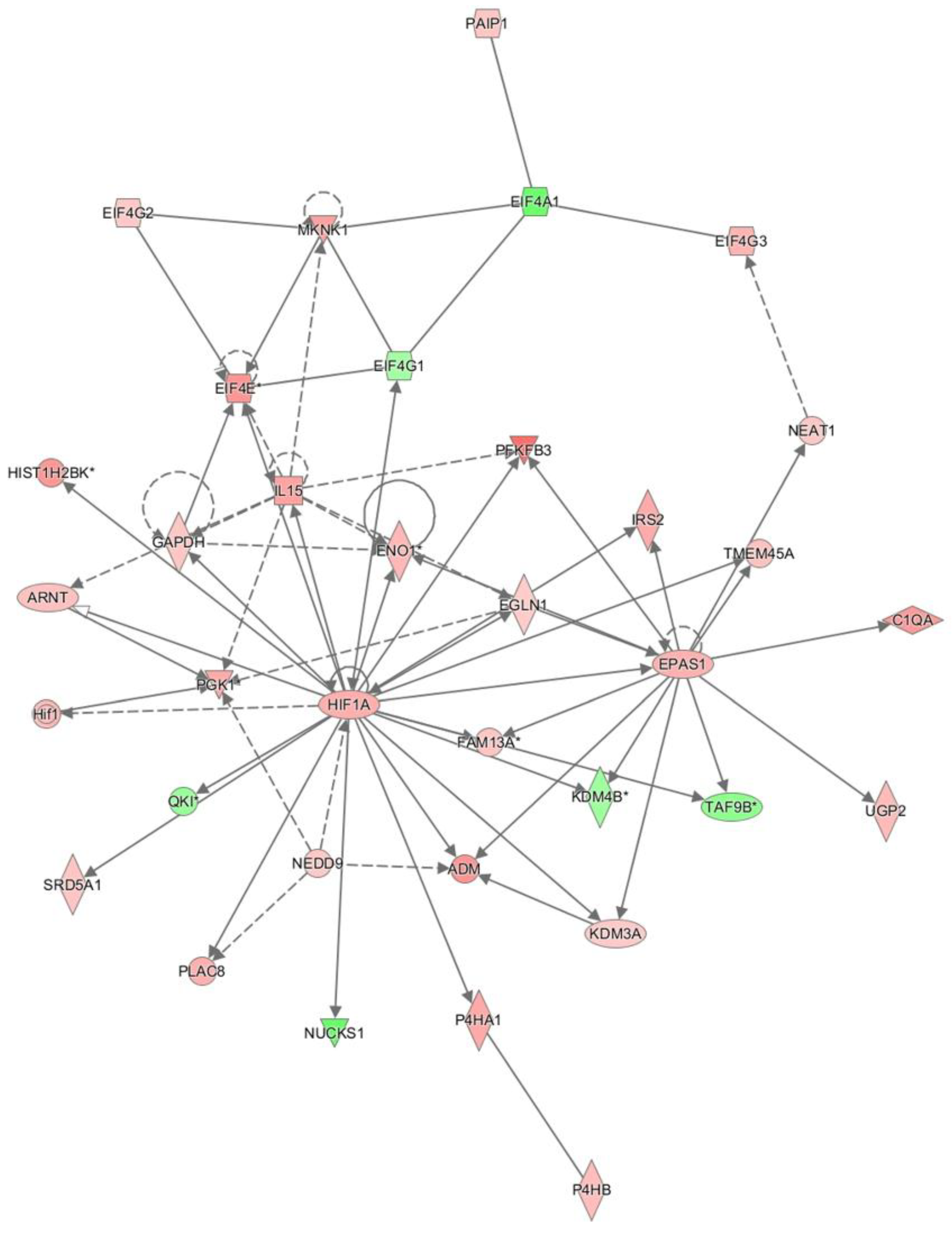
Figure 2.
PTEN centered network is sepsis via Ingenuity pathway analysis.
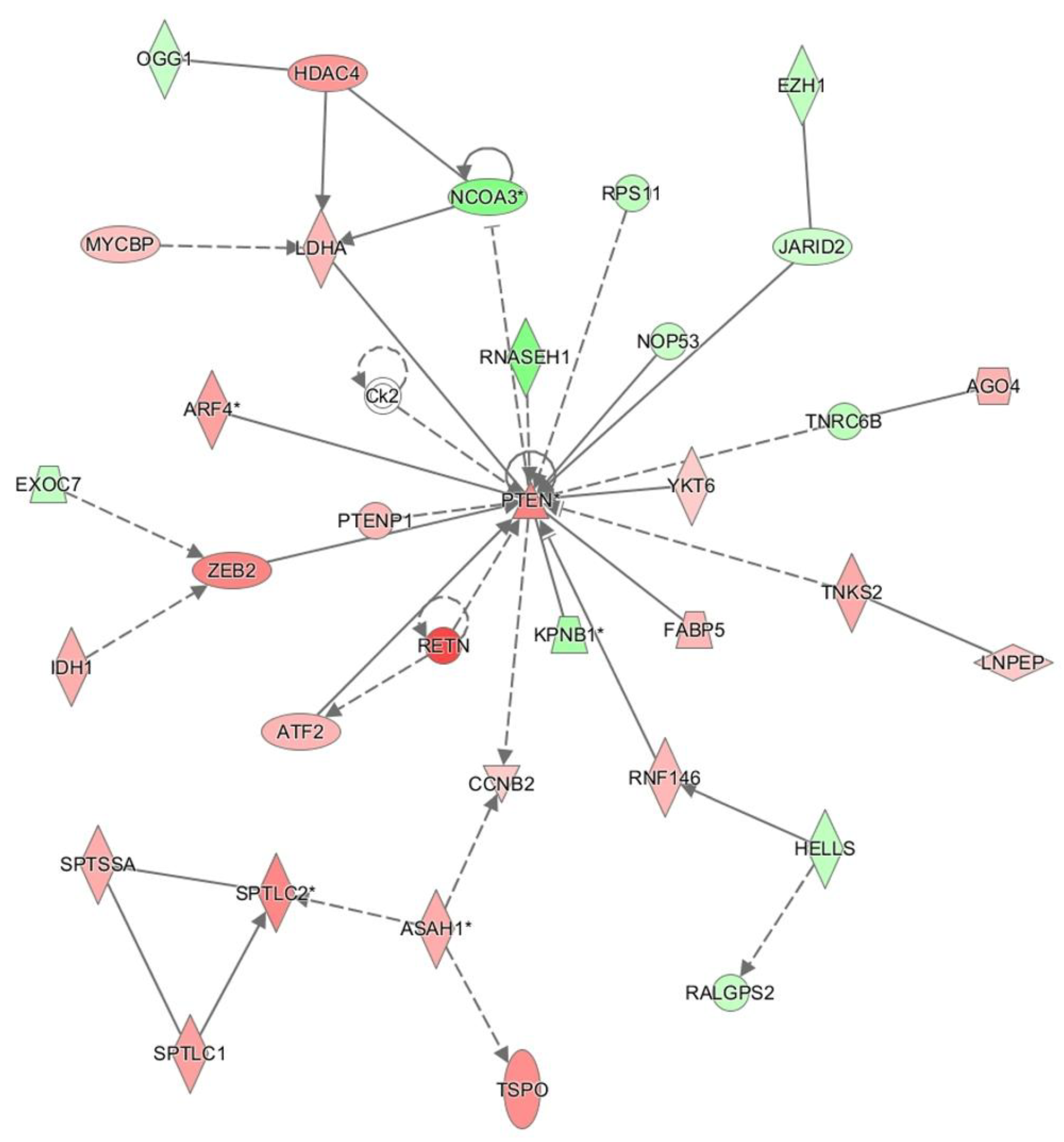
Figure 3.
ITGB3 and MMP9 dominant regulatory pathways in sepsis via Ingenuity pathway analysis.
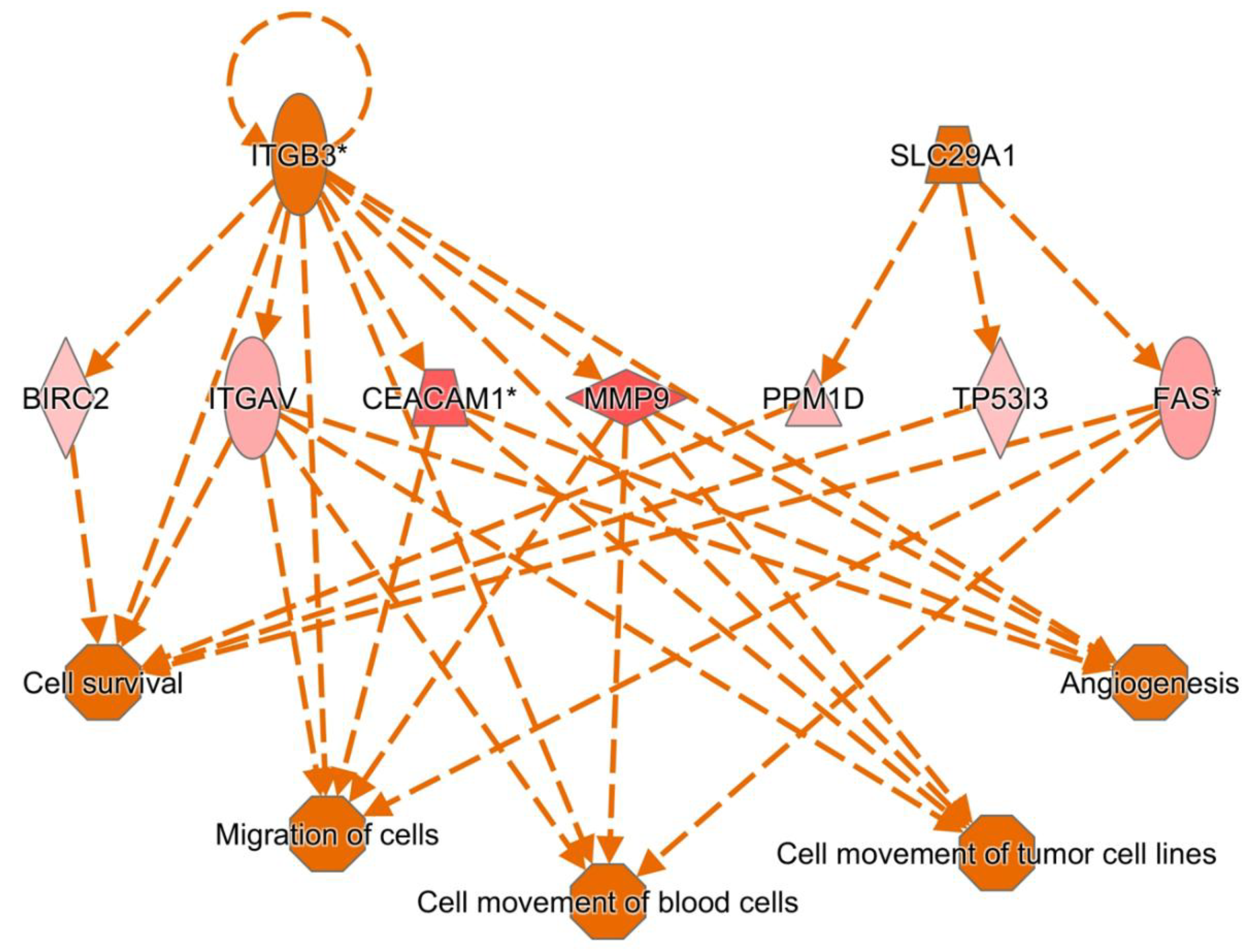
Figure 4.
IL1B, TGFB and TGM2 dominant regulatory pathways in sepsis via Ingenuity pathway analysis.
Figure 4.
IL1B, TGFB and TGM2 dominant regulatory pathways in sepsis via Ingenuity pathway analysis.
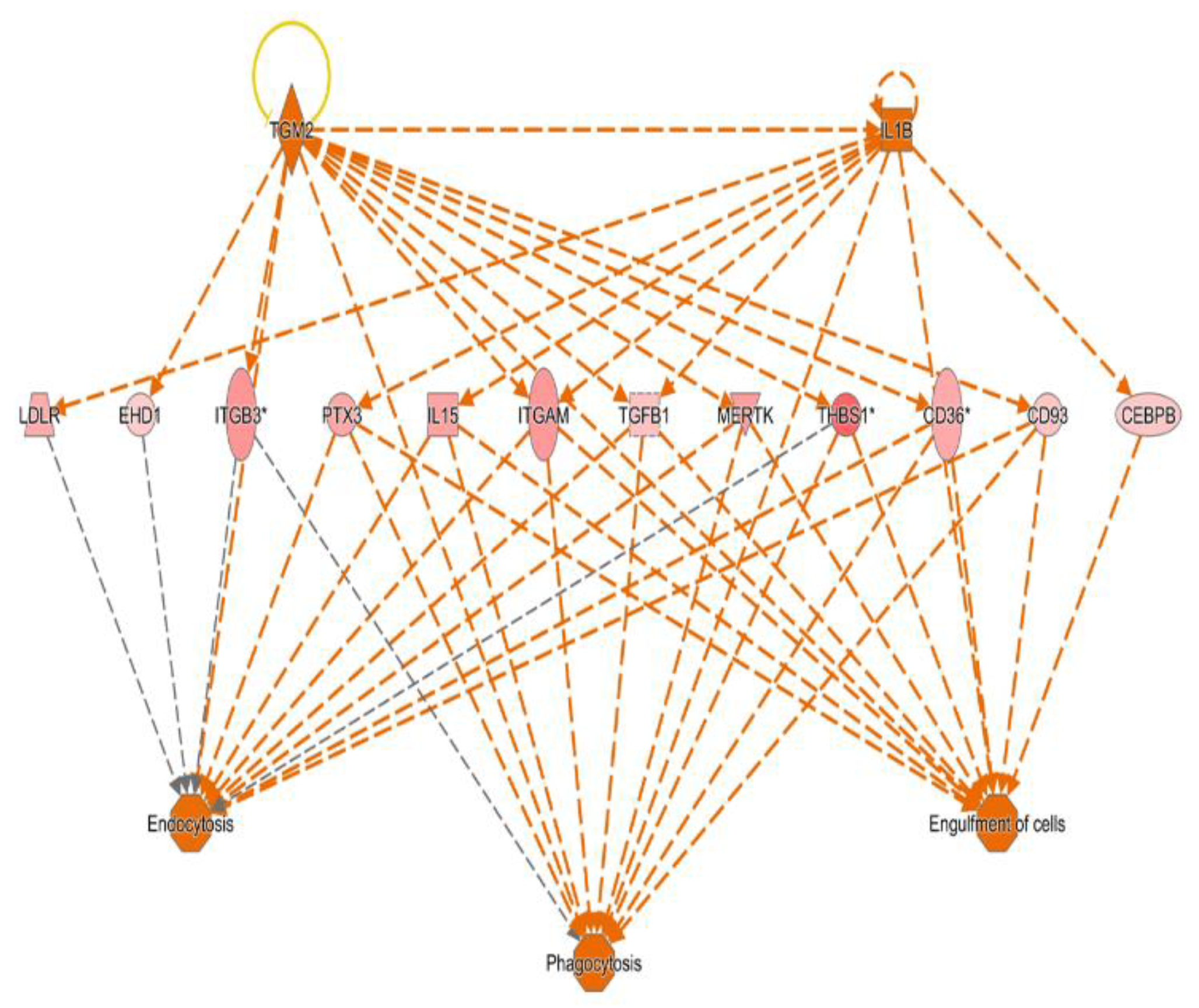
Figure 5.
TNF is the key upstream mediator during sepsis via Ingenuity pathway analysis.
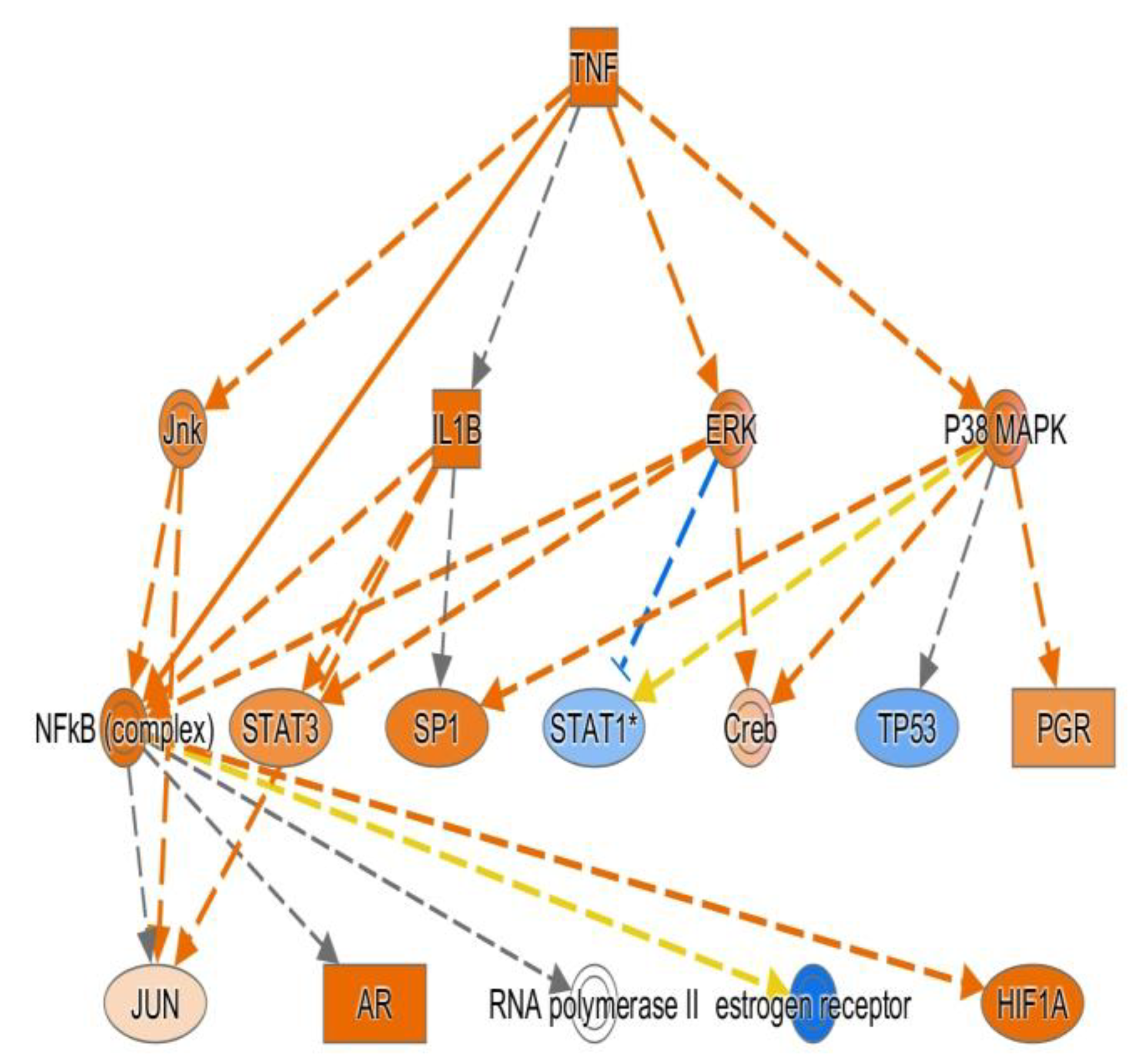

Table 1.
TLR.
| Probe ID | Pvalue | Arrow | Fold | Gene |
| 201743_at | 1.37E-04 | up | 2.18 | CD14 |
| 204924_at | 1.45E-10 | up | 3.38 | TLR2 |
| 210166_at | 9.16E-08 | up | 2.40 | TLR5 |
| 210176_at | 0.001131 | up | 2.07 | TLR1 |
| 213817_at | 3.14E-13 | up | 21.04 | IRAK3 |
| 219618_at | 1.89E-09 | up | 2.69 | IRAK4 |
| 220832_at | 4.76E-09 | up | 5.16 | TLR8 |
| 221060_s_at | 6.62E-07 | up | 3.33 | TLR4 |
| 212184_s_at | 2.03E-05 | up | 2.61 | TAB2 |
| 221705_s_at | 8.46E-10 | down | 2.08 | SIKE1 |
| 205118_at | 1.05E-10 | down | 7.61 | FPR1 |
| 210772_at | 2.06E-08 | up | 4.78 | FPR2 |
Table 2.
MHC.
| Probe ID | Pvalue | Arrow | Fold | Gene |
| 201137_s_at | 5.80E-04 | down | 2.09 | HLA-DPB1 |
| 203290_at | 2.56E-08 | down | 5.19 | HLA-DQA1 |
| 204670_x_at | 6.77E-08 | down | 2.85 | HLA-DRB1/B4 |
| 205671_s_at | 1.27E-04 | down | 2.02 | HLA-DOB |
| 208306_x_at | 1.53E-06 | down | 2.44 | HLA-DRB1 |
| 208894_at | 8.06E-07 | down | 2.76 | HLA-DRA |
| 209312_x_at | 1.24E-06 | down | 2.67 | HLA-DRB1/B4/B5 |
| 209823_x_at | 8.65E-04 | down | 2.08 | HLA-DQB1 |
| 210294_at | 7.08E-10 | down | 2.25 | TAPBP |
| 210528_at | 1.28E-05 | down | 2.57 | MR1 |
| 211948_x_at | 3.66E-28 | down | 11.75 | BAT2L2 |
| 211990_at | 5.10E-06 | down | 3.19 | HLA-DPA1 |
| 212384_at | 8.83E-15 | down | 2.98 | HLABAT1 |
| 212671_s_at | 0.002545 | down | 2.27 | HLA-DQA1/A2 |
| 214055_x_at | 1.16E-24 | down | 9.42 | BAT2L2 |
| 215193_x_at | 2.90E-06 | down | 2.52 | HLA-DRB1/B3/B4 |
| 221491_x_at | 1.50E-06 | down | 2.25 | HLA-DRB1/B3/B4/B5 |
Table 3.
Transcription factor.
| Probe ID | Pvalue | Arrow | Fold | Gene |
| 201473_at | 3.65E-09 | up | 2.46 | JUNB |
| 201502_s_at | 9.16E-07 | down | 2.36 | NFKBIA |
| 202527_s_at | 5.77E-09 | up | 3.24 | SMAD4 |
| 203077_s_at | 4.90E-07 | up | 2.37 | SMAD2 |
| 203574_at | 4.37E-10 | up | 5.18 | NFIL3 |
| 204039_at | 4.62E-08 | up | 2.06 | CEBPA |
| 204203_at | 9.92E-07 | up | 2.17 | CEBPG |
| 205841_at | 1.02E-13 | up | 4.66 | JAK2 |
| 206036_s_at | 8.46E-12 | down | 4.75 | REL |
| 206359_at | 3.22E-07 | up | 2.09 | SOCS3 |
| 206363_at | 9.68E-06 | down | 2.26 | MAF |
| 208991_at | 1.49E-13 | down | 3.22 | STAT3 |
| 209604_s_at | 2.74E-19 | down | 6.55 | GATA3 |
| 209969_s_at | 2.12E-08 | down | 4.75 | STAT1 |
| 210479_s_at | 5.21E-15 | down | 7.85 | RORA |
| 212501_at | 1.73E-07 | up | 2.17 | CEBPB |
| 212550_at | 7.19E-10 | up | 2.52 | STAT5B |
| 213006_at | 6.03E-10 | up | 4.21 | CEBPD |
| 218221_at | 1.49E-11 | up | 2.35 | ARNT |
| 218559_s_at | 9.49E-07 | up | 3.35 | MAFB |
| 218880_at | 5.34E-11 | up | 3.75 | FOSL2 |
| 208808_s_at | 1.07E-11 | up | 9.12 | HMGB |
| 200989_at | 1.17E-06 | up | 3.00 | HIF1A |
| 221969_at | 9.96E-13 | down | 4.20 | PAX5 |
| 203140_at | 3.09E-10 | up | 3.69 | BCL6 |
| 210105_s_at | 9.37E-10 | down | 3.32 | FYN |
| 210754_s_at | 2.98E-10 | down | 3.55 | LYN |
| 217620_s_at | 4.31E-12 | down | 2.79 | PIK3CB |
| 221756_at | 5.52E-10 | down | 2.62 | PIK3IP1 |
| 204054_at | 2.56E-10 | up | 5.51 | PTEN |
| 206370_at | 2.95E-09 | down | 2.44 | PIK3CG |
| 212240_s_at | 4.29E-13 | down | 4.51 | PIK3R1 |
Table 4.
Cytokine.
| Probe ID | Pvalue | Arrow | Fold | Gene |
| 201110_s_at | 2.02E-09 | up | 8.27 | THBS1 |
| 203085_s_at | 1.57E-08 | up | 2.33 | TGFB1 |
| 203828_s_at | 7.88E-05 | down | 2.13 | IL32 |
| 205016_at | 8.33E-10 | up | 4.86 | TGFA |
| 205992_s_at | 4.40E-06 | up | 3.58 | IL15 |
| 208114_s_at | 7.75E-20 | down | 5.85 | ISG20L2 |
| 208200_at | 3.06E-11 | down | 4.80 | IL1A |
| 212195_at | 3.90E-06 | up | 2.67 | IL6ST |
| 209555_s_at | 2.87E-05 | up | 3.18 | CD36 |
| 212657_s_at | 2.96E-07 | up | 2.31 | IL1RN |
Table 5.
Cytokine receptor.
| Probe ID | Pvalue | Arrow | Fold | Gene |
| 201642_at | 1.42E-09 | up | 2.32 | IFNGR2 |
| 202948_at | 5.77E-10 | up | 6.46 | IL1R1 |
| 203233_at | 2.36E-10 | up | 3.27 | IL4R |
| 204191_at | 2.98E-07 | up | 2.06 | IFNAR1 |
| 204731_at | 7.48E-21 | down | 11.93 | TGFBR3 |
| 204786_s_at | 5.23E-19 | down | 6.86 | IFNAR2 |
| 205227_at | 2.89E-05 | up | 2.68 | IL1RAP |
| 205291_at | 2.89E-08 | down | 2.44 | IL2RB |
| 205707_at | 1.73E-09 | down | 2.41 | IL17RA |
| 205798_at | 2.48E-24 | down | 31.79 | IL7R |
| 205926_at | 1.06E-09 | down | 2.19 | IL27RA |
| 205945_at | 1.49E-22 | down | 16.69 | IL6R |
| 206618_at | 4.70E-09 | up | 12.92 | IL18R1 |
| 207072_at | 5.22E-08 | up | 4.93 | IL18RAP |
| 211372_s_at | 1.76E-08 | up | 10.68 | IL1R2 |
| 211676_s_at | 6.66E-09 | up | 4.61 | IFNGR1 |
| 205159_at | 1.17E-06 | up | 2.51 | CSF2RB |
| 210340_s_at | 4.36E-10 | up | 2.30 | CSF2RA |
Table 6.
Acute Response Protein.
| Probe ID | Pvalue | Arrow | Fold | Gene |
| 200602_at | 3.75E-12 | up | 4.38 | APP |
| 206157_at | 8.31E-08 | up | 3.27 | PTX3 |
| 208691_at | 0.001264 | up | 2.49 | TFRC |
| 208703_s_at | 1.26E-07 | up | 3.05 | APLP2 |
| 219890_at | 1.43E-12 | up | 7.83 | CLEC5A |
| 220496_at | 2.59E-07 | up | 3.33 | CLEC1B |
| 205033_s_at | 1.17E-05 | up | 4.79 | DEFA1/A1B/A3 |
| 207269_at | 2.87E-05 | up | 6.67 | DEFA4 |
| 201943_s_at | 7.91E-12 | up | 6.94 | CPD |
| 204961_s_at | 7.26E-08 | up | 2.02 | NCF1/1B/1C |
| 207677_s_at | 5.88E-10 | up | 2.66 | NCF4 |
| 214084_x_at | 1.31E-08 | up | 2.25 | NCF1C |
Table 7.
Complement.
| Probe ID | Pvalue | Arrow | Fold | Gene |
| 200985_s_at | 4.85E-11 | up | 6.59 | CD59 |
| 201925_s_at | 2.14E-07 | up | 5.61 | CD55 |
| 202953_at | 7.01E-06 | up | 2.53 | C1QB |
| 205786_s_at | 5.02E-13 | up | 4.05 | ITGAM |
| 206244_at | 6.06E-12 | up | 6.76 | CR1 |
| 208783_s_at | 0.004769 | up | 2.21 | CD46 |
| 209906_at | 7.48E-09 | up | 4.34 | C3AR1 |
| 210184_at | 1.17E-06 | up | 2.07 | ITGAX |
| 218232_at | 1.52E-08 | up | 3.97 | C1QA |
| 218983_at | 7.83E-08 | up | 2.64 | C1RL |
| 220088_at | 9.13E-08 | up | 2.49 | C5AR1 |
| 202910_s_at | 3.42E-07 | up | 2.26 | CD97 |
Table 8.
MMP.
| Probe ID | Pvalue | Arrow | Fold | Gene |
| 203167_at | 1.02E-13 | up | 3.14 | TIMP2 |
| 203936_s_at | 2.89E-16 | up | 10.59 | MMP9 |
| 206871_at | 1.04E-06 | up | 5.39 | ELANE |
| 207329_at | 3.41E-11 | up | 32.06 | MMP8 |
| 207890_s_at | 1.30E-11 | up | 3.11 | MMP25 |
| 202833_s_at | 2.83E-09 | up | 2.78 | SERPINA1 |
| 204614_at | 5.64E-08 | up | 3.07 | SERPINB2 |
| 212268_at | 8.64E-11 | up | 5.64 | SERPINB1 |
Table 9.
Coagulation.
| Probe ID | Pvalue | Arrow | Fold | Gene |
| 203305_at | 2.16E-04 | up | 2.18 | F13A1 |
| 204714_s_at | 1.87E-08 | up | 3.93 | F5 |
| 205756_s_at | 2.79E-05 | up | 2.08 | F8 |
| 205871_at | 7.54E-07 | down | 3.12 | PLGLA/B1/B2 |
| 206655_s_at | 2.25E-08 | up | 5.37 | GP1BB/SEPT5 |
| 207808_s_at | 6.30E-08 | up | 2.88 | PROS1 |
| 211924_s_at | 5.53E-07 | up | 2.33 | PLAUR |
| 212245_at | 6.18E-07 | up | 2.30 | MCFD2 |
| 213258_at | 1.07E-06 | up | 2.35 | TFPI |
| 213506_at | 0.002877 | up | 2.35 | F2RL1 |
| 214415_at | 1.30E-09 | down | 5.54 | PLGLB1/B2 |
| 216956_s_at | 4.64E-05 | up | 2.39 | ITGA2B |
| 218718_at | 2.79E-10 | up | 9.39 | PDGFC |
| 204627_s_at | 1.30E-06 | up | 4.18 | ITGB3 |
| 203887_s_at | 8.66E-09 | up | 4.53 | THBD |
Table 10.
Glycolysis.
| Probe ID | Pvalue | Arrow | Fold | Gene |
| 200650_s_at | 2.02E-09 | up | 2.71 | LDHA |
| 200737_at | 2.94E-11 | up | 3.17 | PGK1 |
| 201030_x_at | 9.45E-05 | down | 2.02 | LDHB |
| 201251_at | 2.51E-10 | up | 2.67 | PKM2 |
| 202464_s_at | 6.45E-09 | up | 7.30 | PFKFB3 |
| 202934_at | 9.80E-14 | up | 4.77 | HK2 |
| 202990_at | 2.15E-12 | up | 4.20 | PYGL |
| 203502_at | 1.24E-04 | up | 3.67 | BPGM |
| 205936_s_at | 5.17E-12 | up | 4.99 | HK3 |
| 206348_s_at | 9.53E-11 | up | 2.60 | PDK3 |
| 208308_s_at | 3.92E-09 | up | 2.22 | GPI |
| 209992_at | 3.99E-09 | up | 11.77 | PFKFB2 |
| 213453_x_at | 2.13E-12 | up | 2.18 | GAPDH |
| 217294_s_at | 3.28E-06 | up | 2.62 | ENO1 |
| 218273_s_at | 1.01E-07 | down | 2.25 | PDP1 |
Table 11.
H+-ATPase.
| Probe ID | Pvalue | Arrow | Fold | Gene |
| 200078_s_at | 6.15E-13 | up | 2.53 | ATP6V0B |
| 201171_at | 4.49E-10 | up | 2.48 | ATP6V0E1 |
| 201443_s_at | 5.84E-06 | up | 2.33 | ATP6AP2 |
| 201971_s_at | 4.45E-13 | down | 5.21 | ATP6V1A |
| 202872_at | 1.95E-10 | up | 6.18 | ATP6V1C1 |
| 202874_s_at | 6.99E-10 | up | 5.72 | ATP6V1C1 |
| 204158_s_at | 5.14E-08 | up | 2.07 | TCIRG1 |
| 208898_at | 2.66E-09 | up | 2.41 | ATP6V1D |
| 213587_s_at | 1.13E-08 | down | 2.07 | ATP6V0E2 |
| 206208_at | 1.00E-11 | up | 3.51 | CA4 |
| 206209_s_at | 4.18E-15 | up | 7.98 | CA4 |
| 209301_at | 2.78E-06 | up | 3.42 | CA2 |
| 212536_at | 4.38E-09 | up | 4.21 | ATP11B |
| 213582_at | 1.89E-08 | up | 2.24 | ATP11A |
Table 12.
T cell.
| Probe ID | Pvalue | Arrow | Fold | Gene |
| 205255_x_at | 3.09E-08 | down | 2.96 | TCF7 |
| 205456_at | 5.31E-08 | down | 2.88 | CD3E |
| 205488_at | 1.01E-05 | down | 2.87 | GZMA |
| 205495_s_at | 5.33E-10 | down | 4.38 | GNLY |
| 205758_at | 1.20E-07 | down | 3.26 | CD8A |
| 206666_at | 1.84E-07 | down | 3.45 | GZMK |
| 206804_at | 1.10E-15 | down | 5.12 | CD3G |
| 207460_at | 3.78E-09 | down | 2.50 | GZMM |
| 208003_s_at | 5.52E-18 | down | 12.04 | NFAT5 |
| 209671_x_at | 3.58E-08 | down | 2.77 | TRAC |
| 209813_x_at | 1.49E-09 | down | 4.42 | TARP |
| 210164_at | 8.91E-09 | down | 3.76 | GZMB |
| 210321_at | 8.94E-10 | down | 5.80 | GZMH |
| 210370_s_at | 1.34E-07 | down | 2.48 | LY9 |
| 210556_at | 4.68E-08 | down | 2.85 | NFATC3 |
| 210972_x_at | 1.78E-07 | down | 2.88 | TRAC/J17/V20 |
| 211796_s_at | 6.35E-06 | down | 2.93 | TRBC1/C2 |
| 212759_s_at | 3.98E-16 | down | 3.93 | TCF7L2 |
| 213193_x_at | 2.53E-06 | down | 2.92 | TRBC1 |
| 213539_at | 1.00E-08 | down | 3.19 | CD3D |
| 214617_at | 2.22E-06 | down | 2.65 | PRF1 |
| 216191_s_at | 4.71E-07 | down | 4.76 | TRDV3 |
| 216920_s_at | 2.28E-10 | down | 5.34 | TARP/TRGC2 |
| 217143_s_at | 1.26E-08 | down | 6.06 | TRD@ |
| 217527_s_at | 2.12E-13 | down | 5.80 | NFATC2IP |
| 220684_at | 7.39E-09 | down | 2.08 | TBX21 |
| 220704_at | 2.15E-10 | down | 5.69 | IKZF1 |
| 214032_at | 6.60E-08 | down | 2.52 | ZAP70 |
| 204891_s_at | 4.58E-08 | down | 3.31 | LCK |
| 205831_at | 4.40E-10 | down | 3.93 | CD2 |
| 201565_s_at | 8.13E-13 | down | 4.17 | ID2 |
| 213931_at | 7.33E-08 | down | 3.55 | ID2/2B |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



