Submitted:
31 March 2025
Posted:
01 April 2025
You are already at the latest version
Abstract
Autistic spectrum disorders (ASD) are the most common neurodevelopmental pathology, with a global trend of increasing prevalence. The biomarker-based diagnosis and signaling pathways based modulation could improve both clinical symptoms and long-term outcomes. In the review, the role of viruses in causing prenatal CNS damage and the development of autism is discussed. The distinct CNS involvement of the most common pathogens such as Rubella, CMV, HSV, Influenza and Covid-19 virus elucidated. The specific importance of a retinoid metabolism disorder in the brain in prenatal rubella virus infection and of low levels of IgF-1 as a neurotrophic factor in Covid-19 infection has been demonstrated. The role of ferroptosis and changes in ferritin and transferrin levels in the development of ASD-like symptoms have been shown. Altered serotonin signaling could related to early prenatal factors and result in negative ASD symptoms. Hypermethylation of the oxytocin receptor gene in ASD patients results in impaired oxytocin metabolism, discoordination between nucleus basalis and ventral pallidum, and impaired visual attention and motivated behavior. The role of NMDA receptors and SHANK scaffolding proteins in ASD cognitive signs demonstrated. Clinically relevant biomarkers and a combined modulation approach were proposed based on the analysed information.
Keywords:
autistic spectrum disorder
; children
; transferrin
; ferritin
; serotonin
; oxytocin
; combined modulation
1. Introduction
ASD is a complex neurobehavioral condition with pervasive signs of social communication deficits, repetitive patterns and restricted interests.[1] The prevalence of ASD has increased significantly over the last 10 years, from 2.47% to 3.14% in the USA, from 0.11% to 1.53% in the Middle East, and from 1.41% to 2.52% in Australia, and is more common in boys than girls.[2,3]. Latest research suggests that the median prevalence of ASD in the global population is 65 per 10,000 [4]. In adolescents the prevalence of ASD is 17,7 per 1000. In males 1 in 55 and 1 in 172 in females [5]. There has been a significant increase in ASD prevalence globally since 2010, as reported by Australia, the USA, Canada, Oman, Sweden, and Italy [6]. Thus, the global trend of increasing prevalence remains consistent despite differences in diagnostic approaches and methods of estimating ASD between countries.
Many genes associated with the development of ASD and its symptoms are under investigation [7,8]. The distinct genes involved in ASD symptoms such as social behavior, vocalization, synaptic interaction and transmission, memory and learning [9]. Children with autism often have comorbidities not typically associated with neuropsychiatric manifestations, due to the pleiotropic effects of the genes involved, requiring a multidisciplinary approach to identify opportunities for targeted therapy. These conditions include short stature and other growth disorders, obesity, and gaining weight difficulties [10]. The genetic risk variants and altered patterns of gene expression observed in individuals with ASD have been demonstrated to affect the functional and structural characteristics of the brain. [11]. Considering the above, it may be posited that there is potential for more targeted modulation of gene signalling pathways, contingent upon the specific clinical manifestations observed and the identification of pertinent metabolic and genomic expression markers in ASD. A preliminary analysis of the available literature data has identified the main factors and signalling pathways involved in the pathological manifestations of ASD. This analysis has also provided insights into the possibilities of selective pharmacological modulation.
1. Viruses. The impact of various viral infections on neurodevelopment in the intrauterine period has been a significant focus, including Rubella, Cytomegalovirus, Herpes Simplex virus, Varicella Zoster Virus, Influenza virus, Zika virus, and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). These viral infections have also been implicated in the subsequent postnatal emergence of ASD [12].
2. Disorders of iron metabolism. Children with autism have iron metabolism disorders, mainly due to low ferritin levels in 30-50% of patients. There is also a strong link between iron overload and the risk of depression. The link between elevated transferrin levels and the risk of autism spectrum disorders was also highlighted [13,14,15,16].
3. Serotonin. Serotonin is another biomarker that is important in ASD. Serum serotonin levels are elevated in 25% of children with ASD. Experimental data demonstrated altered social, communication, and repetitive behaviours in genetic variants of serotonin transporter gene, SERT [17].
4. Oxytocin. Oxytocin system plays important role in social interaction and helps regulate stress. Oxytocin receptor (OXTR) gene hyper-methylation was found in ASD individuals and directly correlated with social responsivity deficits. Oxytocin serotonin interaction with receptors located mainly in closely related brain structures (amygdalae and dorsal raphe nuclei) regulates social and emotional behaviour. It has been shown that the serotonergic system is modulated by oxytocin, and this leads to an inhibition of the serotonergic signalling pathway [18,19,20,21].
5. N-methyl-D-aspartate (NMDA) receptors. NMDA are the glutamate receptors responsible for excitatory neurotransmission and serve as a basis for forming memories. It was found the presence of anti -NMDA antibodies in ASD patients especially with stereotypes behaviours. There is evidence that suppression or stimulation of NMDA receptors may influence the symptoms of ASD [22,23,24].
6. SHANK. The SHANK gene family plays a crucial role in the integrity and composition of excitatory glutamatergic synapses with highest expression in the prefrontal cortex, striatum, hippocampus and cerebellum. Mutations in the SHANK genes lead to NMDA hypofunction and the phenotypes associated with ASD [25,26].
7. Solute carrier superfamily. The solute carrier (SLC) superfamily of transmembrane transporters plays a pivotal role in the cellular uptake and secretion of nutrients, electrolytes, and metabolites. It has been observed that 71 of these SLC genes are linked to a spectrum of human neurological disorders. Epilepsy, intellectual delay and autism represent a number of the neurological phenotypes that have been linked with SLC mutations. A correlation has been identified between the polymorphism of the serotonin transporter gene SLC6A4 and mood instability in children with ASD [27,28,29].
Considering the available preliminary data, it seems feasible to define an integral approach to diagnosis and targeted signaling pathways modulation in ASD, which was the objective of this review. Figure 1 shows the main metabolic and signaling pathways involved in the pathogenesis of ASD
As shown in Figure 1, at least 4 of the 7 pathways (oxytocin, serotonin, iron metabolism and NMDA) are involved in the pathological manifestations of ASD. These pathways are related to hippocampal structures and may be important in the approach to diagnosis and pharmacological modulation. In consideration of the aforementioned information, we have endeavored to elucidate the available evidence concerning the mechanisms by which ASD symptoms are developed.
2. The Role of Viruses
The role played by viruses in the development of ASD symptoms, in addition to the specific pathogenetic mechanisms of CNS impairment, remains to be fully elucidated. In the context of congenital rubella syndrome (CRS), the hypothesis has been proposed that the risk of ASD and related neurodevelopmental disorders is associated with alterations in retinoid metabolism and hepatic dysfunction, which are induced by the virus. Such damage could be extensive, with potential consequences for many organs, including the brain. It may be hypothesised that the underlying cause of this phenomenon is damage of enzyme responsible for converting retinol into retinoic acid, resulting in the accumulation of retinoid and subsequent severe liver damage [30]. On the other hand, neuroimaging studies have exhibited signs of encephalomalacia, periventricular calcifications, temporal lobe atrophy associated with gliosis and demyelination [31].
By the results of autopsy the rubella virus directly infected nerve cells in the cerebral cortex [32]. It is also important to note that retinoic acid plays a key role in the regeneration in the central nervous system, tissue differentiation and as a modulator of many signalling pathways in neurodegenerative diseases. In addition, retinoic acid is involved in the activation of protein kinases during neurodevelopment and in the formation of synapses [33]. The rubella virus has been demonstrated to infect the nervous system, resulting in progressive encephalitis. This condition has been shown to cause white matter destruction, perivascular inflammation, and degenerative processes. The consequences of these pathologies include progressive dysarthria and ataxia due to cerebellar atrophy[34]. It should be noted that these signs are highly consistent with those associated with ASD.
In the context of cytomegalovirus (CMV) infection, an association has been identified between maternal viral status and social responsiveness score (SRS) points in children diagnosed with ASD. [35]. In the other study, a sample of 74,872 children diagnosed with autism was analysed. The analysis revealed a significant risk of CMV with a hazard ratio of 2.5, on the basis of diagnosis code, in comparison with the control group [36]. Histological examination of fetuses affected by CMV revealed the presence of necrosis, microglial nodules, microglial activation, astrocytosis and vascular changes, which have been indicative of encephalitis [37]. By immunofluorescence and RNA-sequencing in CMV infected cerebral organoids the aberrant expression in excitatory neurons, inhibitory neurons, and more significant in intermediate progenitor cells was found. The alterations pertained to DYRK kinases (dual-specificity tyrosine-regulated kinases that phosphorylate proteins, regulate proliferation, cell resistance and programmed cell death), SHH (sonic hedgehog signaling molecules that regulate cell growth, differentiation and morphogenesis), pluripotency, neurodegeneration, axon guidance, Hippo signaling and dopaminergic synapse pathways. The infection with CMV led to an upregulation of 2208 out of 4029 genes, in addition to the dysregulation of 236 genes that were associated with ASD pathways [38].
In the context of herpes simplex virus, the high level of HSV-2 antibodies in midpregnancy and after delivery in mothers of children with ASD was established, in comparison to antibodies against Toxoplasma gondii, rubella, cytomegalovirus and HSV-1 [39]. However, the results of other study do not appear to support the association [40]. Nonetheless, it is to be noted that HSV, categorised as a neurotropic virus, has the potential to cause significant CNS damage. In the review conducted by Duarte L.F., Farias M.A. et al., it was observed that HSV-1 virus replication within neurons occurs within a latent episome, characterized by minimal virus protein expression. Simultaneously, acute HSV-1 infection can result in the development of herpetic simplex encephalitis (HSE), a condition characterized by the occurrence of apoptosis in affected cells. Autophagy serves as a protective factor against viral replication in HSV infection is a key regulator of neurogenesis supporting new neuron pools stem cells are enriched in the sub-ventricular zone of the lateral ventricle and in the hippocampus The presence of HSV-1 within infected neurons resulted in the degradation of DNA in mitochondria (mtDNA). Levels of ROS (reactive oxygen species) in microglia have been shown to increase after infection with HSV-1 and linked to neurodegeneration. The process appears to be dependent upon Toll-like receptor 2 (TLR2), p38 MAP kinase and the ERK1/2 signaling pathways. DNA damage response is beneficial for HSV-1 viral replication in non-differentiated cells and counteracted in neuronal cells.
In animal models, HSV-1 has been observed to modulate a pathway related to apoptosis, leading to cell death by activating mitogen-activated protein kinases, Jun N-terminal kinase (MAPK/JNK), which results in neuronal damage. HSV-1 has been demonstrated to induce mitochondrial dysfunction and to cause mitochondrial disbalance, which is associated with neurodegeneration. It was demonstrated that HSV-1 alters the DNA response to damage, which results in DNA mutations and constitutes a key factor in viral replication in undifferentiated neuronal cells, thereby contributing to HSV-1 latency [41].
In the context of influenza virus A (IVA), the RNA sequencing data derived from experimental models has indicated significant downregulation of genes including Apc2 (responsible for neuron cortical lamination), Mdga1 (radial migration of upper layer neurons), and Usp11 (cortical neurogenesis and migration) in fetal brains of maternal infected offspring. This study has demonstrated that the presence of IVA infection during pregnancy can result in dysregulation of cortical lamination and an imbalance between excitatory and inhibitory synapses during development and result in later neurodevelopmental disorders during the postnatal period [42]. As demonstrated by another experimental study, exposure to IVA in mice has been shown to induce symptoms analogous to those associated with ASD. These symptoms encompass aggressive behavior, increased locomotor activity, and reduced levels of oxytocin and serotonin in male offspring.
Additionally, the study revealed alterations in microglia density and catecholamine levels in male offspring exposed to the IVA [43]. In despite of contradictory data presence, postulating no association [44], a study of offspring from a samples of pregnant women revealed an autism rate incidence of approximately 8% among children exposed to influenza during gestation [45].
It has been demonstrated within the context of the CoVid-19 virus that it plays a specific role in the reduction of insulin-like growth factor (IGF-1). IGF-1 is the fundamental substance in the development of ASD, participating in neuromyelinisation during both the pre- and postnatal periods. The regulation of IGF-1 translation is controlled by the IGF-R/IRS1/PI3K/AKT/mTOR pathway (which regulates cell cycle, cell growth, proliferation and differentiation) [46]. A decline in serum IGF-1 concentrations was observed in children diagnosed with ASD, in correlation with the IGF-1 promoter polymorphism (rs12579108) and the predominance of the AA genotype [47].
In the review conducted by Jiamin S. et al., it was demonstrated that the levels of IGF-1 in the postmortem brains of individuals with ASD were significantly elevated in the cingulate gyrus in comparison with the other convolutions. Decreased level in the urine was also observed, which reflected its serum concentration. In addition, treatment with IGF-1 has been shown to be efficacious in improving ASD-like symptoms in a number of models, as well as in neurodevelopmental disorders associated with the core symptoms of ASD [48].
Consequently, in the event of an intrauterine infection, viruses exert a selective effect on the CNS, which results in impaired neuronal differentiation across various developmental stages. This, in turn, can lead to the manifestation of postnatal ASD symptoms. As illustrated in Figure 2, the specific virus-dependent CNS lesions that potentially result in postnatal ASD symptoms have been delineated.
Therefore, the combined action of viruses in intrauterine CNS lesions instigates neurodegenerative processes, impaired neurogenesis and demyelination, which may initiate postnatal neurodevelopmental disorders and symptoms associated with ASD. This, in turn, determines the possibilities for pharmacological modulation and rehabilitation.
3. The Role of Iron Metabolism
Several studies have demonstrated the role of ferroptosis in the pathogenesis of ASD. Ferroptosis is a regulated non-apoptotic cell death program that is involved in the initiation and progression of various diseases, including ASD. Increased expression of ferroptosis-related genes (FRG) was found in children with RAS compared to controls. Two distinct molecular clusters associated with ferroptosis have been identified in ASD. Immune infiltration analysis revealed immune heterogeneity between the two clusters. Cluster 2, characterised by a higher immune score and greater infiltrated immune cells, demonstrated a stronger immune response and was significantly enriched in signaling pathways associated with the immune response. Furthermore, the ferroptosis score model demonstrated the capacity to predict ASD subtypes and immunity.
Higher levels of ferroptosis scores were associated with immune system activation, as evidenced by Cluster 2. Conversely, lower ferroptosis scores were concomitant with relative immune regulation, as demonstrated by Cluster 1. The ferroptosis model predicted ASD subtypes and immunity. The following genes were found to be overexpressed in Cluster1: Fbxw7 (induces apoptosis and growth arrest and inhibits the epithelial-to-mesenchymal transition ), ALB (regulation of the colloidal osmotic pressure of blood. major zinc transporter in plasma, typically binds about 80% of all plasma zinc), MafG (plays a critical role in MC3T3-E1 cell apoptosis induced by simulated microgravity and radiation), AKR1C2 (acts as a targetable oncogene in esophageal squamous cell carcinoma via activating PI3K/AKT signaling pathway), RB1 (provides instructions for making a protein called pR,. regulates cell growth and keeps cells from dividing too fast or in an uncontrolled way), ARNTL (involved in the maintenance of circadian rhythms and other neurodevelopmental processes, acts as an important regulator of a wide array of physiological functions including metabolism, body temperature, blood pressure and associated with Th17-cell proinflammatory immunity at mucosal level ) gene. Cluster 2 was associated with enhanced expression of CS (citrate synthases, catalyzes the condensation of acetyl coenzyme A (AcCoA) with oxaloacetate (OAA) to form citrate and coenzyme A (CoA), the first step in the citric acid cycle) and TMBIM4 (transmembrane BAX inhibitor motif-containing protein 4, helps regulate apoptosis and involved in promoting cell migration) genes [49,50,51]. Ferroptosis is a distinct mode of cell death and is distinct from other regulated cell death (RCD) processes, including apoptosis, necrosis, autophagy and pyroptosis. Ferroptosis is triggered by Fe²⁺-dependent lipid peroxidation and is characterized by its unique biological properties and pathophysiological processes [52,53]. Pathways related to ferritin metabolism, such as the ATG5-ATG7-NCOA4 and p62-Keap1-NRF2 axis (ferritin autophagy pathways) are considered critical in the regulation of intracellular Fe 2+ metabolism and ROS formation, thus playing a regulatory role in ferroptosis. NCOA4 (nuclear receptor coactivator 4) is a selective receptor for ferritin autophagy (ferritinophagy) in ferroptosis. Overexpression of NCOA4 results in ferritin degradation and promotes ferroptosis [54,55]. It has also been posited that in cases of ASD, ferritin levels in children are significantly lower than those observed in the control group, particularly in patients where the condition has a more severe course [56]. In children diagnosed with ASD, the presence of single nucleotide polymorphisms (SNPS) in the Transferrin Receptor (TFRC) and Solute Carrier Family Member 1 (SLC40A1) genes was compared with that observed in the control group of unaffected patients. This finding has the potential to serve as an early biomarker of the condition. [57]. A negative correlation was identified between the parasomnias subscale and serum ferritin levels in the ASD group [58]. Total of 28.07% of the autistic children diagnosed with anemia exhibited low hemoglobin and ferritin levels [59]. Restless legs syndrome in ASD children was positively correlated to serum low ferritin level [60]. Inverse relationships have been observed between serum ferritin concentrations during pregnancy and DNA methylation levels at specific loci in offspring during the postnatal period [61]. In children diagnosed with ASD, reduced levels of transferrin and ceruloplasmin, and increased lipid peroxidation, were observed in comparison to healthy siblings. The changes have been shown to be strictly correlated with the loss of acquired language skills [62]. Transferrin mRNA has been identified as expressed by oligodendrocyte-like cells in the choroid plexus, as well as neurons located in the medulla, and the cortex, the hippocampus, and the basal ganglia. However, the expression levels observed in the hippocampus remained notably low. This observation has the potential to result in heightened vulnerability to iron-induced hyperoxidation within this specific brain region [63]. In an experimental mouse model of extensive amyloid pathology, cognitive impairment and neuroinflammation, deficits of potentiation and synaptic transmission, the activation of transferrin receptor (TfR1) expression in brain cortical tissue was observed to be associated with upregulation of HIF-1 (hypoxia- induced factor 1, a transcriptional activator dependent on oxygen, which regulates the expression of genes associated with adaptation to hypoxia) [64]. Transferin (Tf) has been shown to reduce cell death and promote neuronal differentiation with enhanced IL-10, making transferin a promising candidate for use in regenerative strategies for neurodegenerative diseases [65]. It has been demonstrated that, depending on their polarization state, brain microglia preferentially acquire iron from transferrin (Tf) or from non-Tf sources. Furthermore, the uptake of non transferin bound iron is enhanced by the proinflammatory response. In addition, under these conditions, microglia sequester both extra- and intracellular iron [66]. Low transferrin levels in cerebrospinal fluid (CSF), which varies depending on iron content in the brain, may reflects underlying pathophysiology of neurodegenerative proteinopathies [67].
Therefore, in children diagnosed with ASD, the ferroptosis may be associated with the severity of neurodegenerative processes caused by immune activation against immature neurons in specific brain regions. This is also accompanied by decreased serum levels of transferrin and ferritin. These data are summarised in Figure 3.
4. The Role of Serotonin Pathway
Serotonin (5 hydroxytryptamine; 5-HT) is a neurotransmitter whose action is mediated by various receptor interactions and consists of 14 subtypes. Serotonin, a monoamine neurotransmitter that functions within the central nervous system (CNS), is implicated in a multitude of processes, including cognition and memory. Psychological stress has also been demonstrated to decrease serotonin synthesis, thereby affecting numerous immunological processes and inducing a decline in pro-inflammatory cytokines, such as tumour necrosis factor (TNF-α). It is hypothesised that an imbalance in serotonin receptors 5HT1a and 5HT2a may have a role in the regulation of apoptosis in inflammatory cells. In such cases, this protein could be considered a valuable therapeutic target. Elevated whole blood serotonin levels, or hyperserotonemia, was the first biomarker identified for ASD and is present in more than 25% of affected children. Evidence from both neuroimaging and postmortem samples further suggests that alterations in the brain serotonin system are present in ASD. Genetic linkage and association studies of both whole blood serotonin levels and risk of ASD indicate a chromosomal region containing the serotonin transporter (SERT) gene in male subjects, but not in female subjects. In ASD families with evidence of linkage to this domain, the presence of multiple rare SERT amino acid variants has been observed to result in convergent increases in serotonin uptake in cellular models. A mouse model knockout of one of these variants, SERT Gly56Ala, reproduces a biomarker of hyperserotonemia and shows increased brain serotonin clearance, increased serotonin receptor sensitivity and altered social, communicative and repetitive behaviours. Evidence from other rodent models indicates that the serotonin system plays a significant role in the domains of social behaviour, cognitive flexibility, and sensory development. [68]. A variations in 5-HT levels has been demonstrated to exert an influence on the developmental trajectory of the brain, consequently giving rise to behavioral aberrations such as anxiety and depression, as well as diminished social capabilities. The 5HT2A receptor is predominantly located in glutamate neurons within the frontal and parietal cortices, as well as in the hippocampus. In addition, it is found in dopaminergic neurons of the midbrain and in the anterior pituitary's endocrine cells. Upon activation, the 5HT2A receptor has been observed to elicit a variety of effects, including the stimulation of glutamatergic neurons in the frontal/parietal cortex, the inhibition of dopamine release, and the augmentation of the secretion of neurohypophyseal peptide hormones such as growth hormone, luteinizing hormone, adrenocorticotropic hormone, and prolactin. These 5HT2 receptors are in different regions of the brain and are responsible for different functions. In mouse model of autism, when 5HT2A receptors antagonist be added into the dorsomedial striatum, the reduces grooming behavior and reversal learning deficits observed [69]. Hypomethylation of the human serotonin receptor 4 (HTR4) promoter has been identified as a significant marker for male ASD [70]. Evidence has demonstrated that the serotonin 1A receptor, which is expressed in the striatum, has the capacity to regulate social behaviour in individuals diagnosed with ASD [71].
Tryptophane (TRP) is a precursor of 5-HT. It has been demonstrated the significant decreased expression of TRP metabolic pathway genes in ASD children including MAOA (monoamine oxidase A is involved in the breakdown of the neurotransmitters serotonin, epinephrine, norepinephrine, and dopamine), HAAO (is a monomeric cytosolic protein belonging to the family of intramolecular dioxygenases containing nonheme ferrous iron, in the brain mainly located in glial cells, catalyzes the synthesis of quinolinic acid (QUIN) is an excitotoxin whose toxicity is mediated by its ability to activate glutamate N-methyl-D-aspartate receptors) and AADAT (Aminoadipate Aminotransferase, the precursor of kynurenic acid which has neuroprotective properties) [72]. A study of 65 youngsters diagnosed with ASD revealed elevated serum concentrations of Brain-Derived Neurotrophic Factor (BDNF), diminished levels of TRP and kynurenic acid in comparison with a matched control cohort [73]. In the review of Santana-Coelho D., the significance of the kynurenine pathway as a primary metabolic route for TRP is highlighted. This pathway is responsible for the metabolism of this amino acid and the generation of several neuroactive metabolites that exert neuromodulatory effects due to its actions in glutamatergic and cholinergic receptors. This pathway also has been identified as a possible therapeutic target for a number of disorders of inflammatory origin, including those caused by maternal immune activation during pregnancy [74].
The solute carrier transporters (SLCs) represent a large superfamily of over 450 proteins, which exhibit a range of functions, structures, and expression patterns. These proteins helps to remove the neurotransmitter (eg. dopamine, norepinephrine, serotonin) from the target compartment with receptors on cell surface and plays an essential role in blood brain barrier functioning [75]. It has been established that a number of neurological diseases may arise from abnormalities in the genes that encode the SLC transporters. SLC6A4, the gene responsible for coding the serotonin transporter (SERT), is pivotal in regulating serotonin, a neurotransmitter that plays a fundamental role in regulating emotion, mood, and social behavior. SLC6A4 gene variants can impact transporter protein structure and function and are linked to ASD [76]. SLC4 genes participate in neuronal modulation, maintenance of brain function, association with cellular growth and activation of PI3K/AKT/mTOR signaling pathway. The impact of SLC proteins has been demonstrated to result in intracellular acidosis and diminished nerve excitability. Alterations in exploratory behaviours or emotional abilities have also been observed, in addition to deficits in visual and acoustic faculties, altered perceptions of sensory cues and a reduction in locomotor activity [77].
Therefore, the dysregulation of serotonin pathways and the occurrence of variations in serotonin blood levels in individuals diagnosed with ASD is closely related to variations in the SERT gene, immune activation and consequent impaired tryptophan metabolic pathways. The data is represented in Figure 4.
5. The Role of Oxytocin
Oxytocin (OT), a neuropeptide, has been demonstrated to be involved in both the reproductive and social behavior of animals and humans. In the context of human social behavior, three oxytocin signaling genes have received frequent attention: OXT (the structural gene for oxytocin), OXTR (the oxytocin receptor), and CD38 (the oxytocin secretion gene). The expression of the three genes selected from the oxytocin pathway was found to be enriched in subcortical and olfactory regions. Furthermore, there was evidence of high co-expression with multiple dopaminergic and muscarinic acetylcholine genes, thereby indicating an anatomical basis for interactions between critical gene pathways. The distribution of oxytocin pathway gene maps has been found to correspond with the processing of anticipatory, appetitive, and aversive cognitive states. The oxytocin signalling system is interacting with dopaminergic and muscarinic acetylcholine signalling, thereby modulating cognitive processes involved in complex human behaviours. [78]. Genes that support the OT system have been observed to be overexpressed in specific regions of the cortex, most notably in the precentral gyrus. Concurrently, these genes have been found to be underexpressed in certain subcortical and central structures, including the basal ganglia, limbic structures and the brainstem [79]. OT and OXTR in the brain regulate neuronal excitability, network oscillations, synaptic plasticity and social recognition memory[80]. OXTR gene polymorphisms (rs2254298, rs53576) play an essential role in ASD development. [81].
In the case of individuals diagnosed with ASD, there is a higher level of OXTR binding in the nucleus basalis of Meynert which is responsible for visual attention, and a significantly lower level of OXTR binding in the ventral pallidum in the mesolimbic reward pathway is responsible for the processing and execution of motivated behaviors. However, in the case of patients diagnosed with ASD in early childhood, no binding in this region was found [82]. Oxytocin receptor (OXTR) gene hyper-methylation was found in ASD individuals and directly correlated with social responsivity deficits. Hypermethylation has been demonstrated to correlate with clinical symptoms and hypoconnectivity between cortico-cortical areas involved in theory of mind. Furthermore, methylation at a CpG site in exon 1 has been shown to be positively related to social responsiveness deficits in ASD and to hyperconnectivity between striatal and cortical brain areas [83]. A significant increase in serotonin and a decrease in OT levels in the blood of children with ASD have been identified [84]. A meta-analysis of a total of 31 studies, incorporating a sample of 1231 individuals diagnosed with ASD, has revealed a decrease in OT levels. The analysis also reveals a more significant decrease in oxytocin levels in children [85]. The interaction between serotonin and oxytocin has been demonstrated to play an essential role in the context of social behaviour. Oxytocin has been shown to induce the release of serotonin, which subsequently influences the serotonin 1A receptor system through the regulation of its availability. This phenomenon occurs in several key brain regions involved in social behaviour, including the amygdala and the insula [86]. It was found that presynaptic OT receptors on dorsal suture projections in the contiguous nucleus induce the release of 5-HT and, furthermore, that activation of postsynaptic 5-HT 1B receptors is required for the establishment of social preference [87].
The blunting of the OXTR protomers in 5-HT2AR, and especially in 5-HT2CR heteroreceptor complexes, has been demonstrated to contribute to the development of depression and other types of psychiatric diseases involving disturbances in social behaviors [88]. Rats treated with a 5-HT agonist (5-methoxytryptamine), which causes hyperserotonemia, exhibit a decline in OT in the paraventricular nucleus of the hypothalamus and a form of "autistic-like" behaviour [89]. These findings suggest the existence of mutual interactions between the 5-HT and OXT systems. It can therefore be concluded that changes in oxytocin signaling pathways, due to close co-expressor coupling with serotonin and dopaminergic regulatory systems, play a key role in the manifestation of cognitive syndromes and social maladaptation in autism. The data can be viewed in summary form in Figure 5.
6. The Role of NMDA and SHANK
Glutamate receptors are major mediators of excitatory neurotransmission at synapses in the central nervous system. The two major subtypes of glutamate receptors that cluster on the postsynaptic membrane are N-methyl-d-aspartate (NMDA) and α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptors. The NMDA glutamate receptor subtype NMDA is critical for synapse development, neuroplasticity and pathological neurotoxicity, where its levels at the synapse critically regulate brain function. N-methyl-d-aspartate receptors (NMDARs) are a distinct type of ligand-dependent ionotropic receptor that possess a unique combination of biophysical and functional characteristics. These receptors play a crucial role in the cellular mechanisms that regulate the formation of new neuronal networks and the strengthening of synapses. This role is critical to the development and maintenance of cognitive processes that are vital to higher cognitive functions in humans [90,91]. NMDARs have been demonstrated to exhibit high permeability to calcium and require an allosteric co-agonist that binds to glycine or D-serine in addition to glutamate for opening. The build-up of substantial concentrations of excitatory neurotransmitter amino acids has been shown to result in the overexcitation of glutamate receptors.
The NMDA receptor is a complex supramolecular entity that includes several regulatory sites, the mediator co-antagonist specific binding site (L-glutamic acid), specific binding site of the co-antagonist (glycine) and allosteric modulatory sites located both on the membrane and in the ion channel conjugated to the receptor (binding site of divalent cations and phencyclidine site is a binding site of non-competitive antagonists). [92,93].
Over-excitation of the NMDA receptors leads to an excessive influx of Ca2+ ions into the neurons and a sudden increase in their concentration [94,95]. Disrupted NMDAR function is associated with various psychiatric disorders, including ASD. Multiple genetic variants of the NMDAR gene family (GRIN) have been found in people with ASD [97]. ASD-associated mutations and rare variants were found in all NMDAR subunit genes, including GRIN1 encoding the GluN1 subunit, GRIN2A encoding the GluN2A subunit, GRIN2B encoding the GluN2B subunit, GRIN2C encoding the GluN2C subunit, and GRIN2D encoding the GluN2D subunit. Nevertheless, of all the human NMDAR subunit genes involved in ASD, the GRIN2B represents the most predominant ASD risk gene [98,99]. ASD is accompanied by changes in glutamate concentration, receptor desensitisation, channelopathies, charge transport, receptor trafficking, surface expression, dendrite growth, spike density and synaptic transmission [100].
Experimental animals with the GluN2B-C456Y mutation exhibited decreased GluN2B and GluN1 protein expression and impaired NMDAR currents in GluN2B-containing synapses of the Schaffer-CA1 pyramidal collateral (SC-CA1) in the hippocampus. Animals with Grin2b +/C456Y exhibited signs of hypoactivity, anxiolytic-like behaviour, and a moderately increased tendency to self-care in adulthood. However, social interaction and communication remained within normal parameters [101,102]. A mounting body of evidence suggests that impaired NMDA receptor signaling may represent a significant molecular mechanism underpinning the pathogenesis of idiopathic autism.
Many ASD risk genes have been found to be involved in the structural and functional integrity of synapses, in particular the SHANK gene family are major scaffolding proteins at postsynaptic densities. All three SHANK genes (SHANK1, SHANK2 and SHANK3) have been implicated in ASD. However, individuals diagnosed with autism who have mutations in the SHANK3 gene exhibit more severe behavioral deficits. The function of the SHANK3 protein is to act as a scaffold that supports the connections between neurons. In this way, it ensures that the signals sent by one neuron are received by another. Experimental Shank mutations associated with ASD exhibit typical behavioral phenotypes in animals, including impaired sociability, communication, repetitive behaviors and anxiety [103,104]. Experimental animals with human autism mutations also showed NMDAR dysfunction - impaired hippocampal long-term potentiation and reduced NMDA/AMPA ratio at corticostriate synapses in mice with a transcriptional stop cassette inserted upper of exon 13 is encoding the PDZ domain in Shank3 ( Shank3 +/E13 and Shank -/E13 ) [105,106].
It is evident that other postsynaptic adhesion molecules play a pivotal role in ASD, including a family of neuronal postsynaptic cell adhesion molecules known as neuroligins. The expression of neuroligins exhibits significant heterogeneity among distinct neuronal types, manifesting in an isoform-specific manner. The neuroligin 1 is predominantly expressed in excitatory synapses, neuroligin 2 is expressed in inhibitory synapses, and neuroligin 3 is expressed in both excitatory and inhibitory synapses. In addition, neuroligins interact transsynaptically with neurexins, cooperatively modulating synapse formation and specification as well as NMDAR regulation. Genetic alterations in the NLGN1 gene encoding neuroligin 1 are associated with ASD in humans. Mice deficient in neuroligin 1 (Nlgn1-/-) exhibited increased repetitive licking and impaired spatial memory [107,108].
Another high-risk ASD gene associated with synaptic cell adhesion is CNTNAP2, which encodes a contacting-associated protein 2 (CNTNAP2 or CASPR2). CNTNAP2 is a type I transmembrane protein, highly homologous to neurexins, that interacts with the postsynaptically localized membrane protein of cell adhesion, contactin 2, and plays an important role in the recruitment of K+ channels to juxtaparanodal regions in myelinated axons and transport of GluA1 AMPAR subunits in spines [109,110,111]. Therefore, dysmorphology in scaffolding SHANK proteins and synaptic adhesion molecules could play an accessory role in ASD symptoms by affecting post-synaptic NMDA and co-agonist receptor activity.
7. Discussion (Clinical Comparisons, Suggested Biomarkers and Modulation Possibilities)
In accordance with the findings of recent data analysis, it is becoming apparent that intrauterine CNS injury and maternal immune activation play a pivotal role in the development of ASD. Furthermore, there was an absence of compelling evidence pertaining to inheritance in ASD cases. In the same time, intrauterine viral influence was found to have a distinct effect on diverse neuronal components and pathways. In the event of an intrauterine rubella infection, the inhibition of the conversion of retinoids to retinoic acid (RA) leads to the accumulation and deposition of retinoids within the CNS. RA has been demonstrated to promote the growth of cellular dendrites and the neuronal differentiation of neuron stem cells, which in turn induce the functional maturation of differentiated neurons. RA increases the effects of BDNF and nerve growth factor, which appear to participate in RA-induced cell differentiation. [112,113]. In this context the retinoid acid deficiency affects the synapse development and neuronal differentiation, and retinoid status evaluation could be considered as important biomarker in rubella-induced ASD. Retinoid acid deficiency affects synapse development and neuronal differentiation. Consequently, the evaluation of retinoid status could be considered as a significant biomarker in cases of ASD induced by rubella in relation to the state of neuronal maturity. In this regard, it should also be noted that the use of retinoic acid preparations (all-trans retinoic acid) may be promising in ASD patients due to a history of confirmed prenatal rubella virus exposure. At present, a certain result of its use has been described in patients with certain types of haemoblastosis and is also associated with potent differentiation effects [114,115].
HSV induces more potent immune response in CNS resulting in encephalitis, cell apoptosis, neuronal mitochondrial DNA degradation and ROS elevation in microglia, leading to neurodegeneration. In this context, the oxidative stress indicators and markers of neuroinflammation could be utilized. CMV infection can have a detrimental effect on cerebral organoids, resulting in the activation of a number of pathways that have been linked to ASD. This activation of pathways ultimately leads to neurodegeneration and impaired dopamine signaling. It is posited that the neuroinflammatory and dopamine-linked markers could serve as useful indicators. Influenza virus more inhibits gene pathways related to cortical neurogenesis, neuron migration and cortical lamination, alters microglia density and catecholamine levels. The aforementioned summarized effects could result in cortical damage and ASD-like symptoms, with reduced levels of oxytocin and serotonin and determines an approach to biomarkers and to the modulation strategy.
SARS-CoV-2 virus has the specific capacity to inhibit IGF-1, thus precipitating dysregulation of myelinization and neuronal differentiation. At the same time, elevated levels have revealed in patients with major depressive disorders [116,117]. This may indicate a discrepancy in the neurotrophic regulation of IgF1, depending on the prevalence of degenerative processes or neuronal network activation. In this regard, IgF1 can be considered as a rather specific marker of neurodegeneration level in ASD as well as targeted modulation strategy.
Evidence was presented which suggests that impaired iron metabolism may have a significant role in the development of ASD. Low levels of ferritin and transferrin in patients diagnosed with ASD have been demonstrated to be strongly associated with the occurrence of ferroptosis and ferritinophagy. Heightened TfR1 expression in the brain cortex has the capacity to reduce cell death and to promote neuronal differentiation and antihypoxic adaptational reactions. A decline in ferritin levels has been observed in children diagnosed with ASD (autism spectrum disorder), with such decreases being associated with ferroptosis and hyperoxidation, a state of hypermethylation, the presence of parasomnias and restless legs syndrome. Additionally, low Tf levels have been linked to a loss of acquired language skills. In turn, ferroptosis and peroxidation related to immune response with upregulation of gene pathways related to apoptosis, cell growth control, colloidal osmotic pressure, cell migration and circadian rhythm. Concurrently, it was demonstrated that dopamine exerts a direct influence on cellular iron homeostasis by increasing the incorporation of iron into macrophages. This process consequently promotes intracellular oxidative stress responses [118]. Consequently, a correlation between the severity of subcortical vascular cognitive impairment and the expression of dopamine D2 receptors on peripheral blood lymphocytes, suggesting a potential underlying mechanism for cognitive impairment in patients with subcortical vascular deficits was demonstrated [119]. It is therefore hypothesized that immune activation resulting in ferroptosis could lead to more specific involvement of the dopaminergic system in patients diagnosed with ASD. On this basis, it can be posited that the detection of serum transferrin and ferritin levels, in conjunction with immune markers of inflammation and oxidative stress, may offer a means of gauging the acuity of the neurodegenerative process, as well as the extent to which the dopamine system and cognitive disorders are implicated in the ASD.
In view of the above, the selection of optimal biomarkers to characterize the immune activation and neuroinflammation in ASD is of clinical and prognostic significance. It is evident that immune biomarkers are absent in 13% of patients diagnosed with ASD, indicating that the aetiogenesis of the disease may be distinct in this subset of patients [120]. In this subgroup ASD symptoms are thought to be a secondary feature of other genetic diseases affecting CNS. The review by Stancioiu F., Bogdan R., Dumitrescu R. [121] cites several meta-analytic studies that have demonstrated an absence of reproducibility and reduced sensitivity in a range of genetic, biochemical, and neuroimaging biomarkers related to ASD. In the same time, inflammatory and immune biomarkers identified in 87% of ASD cases. The authors have revealed the significant elevation of serum neuron-specific enolase (NSE) in 97.5% of ASD patients, suggesting neuronal post-apoptotic release and considering this biomarker as indicative of neuronal damage severity in ASD.
Heat shock proteins (HSP) are ubiquitous in human cells and are critical for maintaining proper protein synthesis and function. These proteins may serve as targets for autoimmune lesions, with elevated levels of HSP90, a 90-kDa member of the HSP family, and antibodies to HSP90 detected in non-psychiatric disease. Levels of HSP90 are elevated in lupus patients with neuropsychiatric disease, with changes in protein levels highly sensitive to disease activity and in individuals with schizophrenia and in autistic individuals with compulsive symptoms. [122,123] HSP70 has the capacity to bind to misfolded proteins, thereby assisting them in adopting their native conformation. Consequently, an increase in the production of HSPs may promote cell survival in the presence of damaging stimuli. As a result of cell death, HSP70 is released into the extracellular space and acts as a signalling ‘danger molecule’ by interacting with TLR2 and TLR4 receptors of macrophages, glial and dendritic cells. In particular, HSP70 increase both at cytosolic and mitochondrial glutathione levels, which prevents the development of oxidative stress [124]. HSP72 has previously demonstrated its usefulness as an indicator of neuronal vulnerability with high sensitivity. In ASD patients the elevated HSP70 levels were found compared to those with attention deficit hyperactivity disorder (ADHD) with significantly lower levels [125]. Pharmacological modulation of HSP70 leads to improved synaptic function and long-range connectivity in ASD. HSP70 affects the expression of Nrf2 (nuclear respiratory factor 2) gene transcription, which is reduced in ASD and regulates Atp1a1 and Atp1b1 are important in mediating the energy generation and neuronal activity. Sulforaphane (SF), an isothiocyanate derived from broccoli sprouts, induces HSP70 and subsequently Nrf2 as well as ‘cell-protective’ responses that may benefit ASD through common cellular mechanisms underlying heterogeneous phenotypes. Sulforaphane (SFN: 1-isothiocyanato-4-methylsulfinylbutane), a potent antioxidant, is one of the HSP70 interventions that has been studied in ASD and other comorbidities with beneficial effects.
An increase in molecular and biochemical markers of mitochondrial dysfunction was found in the blood of ASD patients. A significant increase (all p < 0.01) in the prevalence of lactate, pyruvate, alanine and creatine kinase was found in individuals with ASD. Clinical signs such as speech delay, impaired social interaction, cognition, motor function, repetitive behaviors and gastrointestinal symptoms have been associated with lactate, pyruvate, lactate/pyruvate ratio, carnitine and acyl-carnitines. Treatment targeting mitochondria, particularly carnitine and ubiquinol, seem to be helpful in ASD. Further studies may help to understand the role of mitochondria in ASD by better defining subgroups and understanding the molecular mechanisms driving some of the unique mitochondrial changes seen in autistic people [127]. Thus, based on the close interrelationships between immune activation, ferroptosis, peroxidation levels and neuronal mitochondrial damage in ASD, it may be considered promising to identify biomarkers specific for these processes and apply targeted modulation. Individuals with ASD have increased expression of TLR-4 on T cells, which is associated with increased NOX-2 expression and ROS generation compared to typically developing children. [128,129]. Consequently, ASD may be associated with oxidative stress, and antioxidants are commonly used in the treatment of autistic young people. Antioxidants (N-acetylcysteine (NAC), other antioxidants) have been found to be more effective than placebo in improving irritability among symptoms in the aberrant behavior checklist and symptoms of impaired communication in the developing behavior checklist in patients with ASD. A favorable trend towards a reduction in symptoms of stereotypical behavior was observed. Treatment with NAC and antioxidants had a good tendency towards improvement of irritability and hyperactivity symptoms[130]. In a double-blind placebo-controlled study, the safety and efficacy of glutathione alone and treatment with glutathione, vitamin C and NAC were evaluated in children with autism . Improvements in both developmental skills and behavior were observed with both glutathione and combination therapy of glutathione, vitamin C and NAC compared to placebo therapy in children with ASD [131]. The analysis of this dataset suggests that NAC may be a beneficial addition to combined modulation in cases where ferroptosis-associated oxidative stress markers are present in individuals diagnosed with ASD.
Serotonin plays an essential role in neurodevelopment, cognition and memory. Polymorphisms or mutations in the SLC6A4 gene which results in damage to the SERT, leads to a series of cascade changes in the location of the relevant serotonin receptor sites. Consequently, there is hyperserotoninemia, decreased levels of the serotonin precursor TRP and downregulation of MAOA signaling pathways. These pathways are involved in the metabolism of both serotonin and other neurotransmitters. This ultimately creates a vicious cycle. Furthermore, the downregulated expression of other TRP pathways (HAAO, AADAT) has been demonstrated to influence NMDA activation and neuroprotection. The net result is the attenuation of excitation and the manifestation of a series of ASD symptoms. These are typically characterized by negative signs as reduced exploratory behaviors, diminished emotional abilities, deficits in visual and acoustic faculties, altered perceptions, and a decline in locomotor activity and accompanying to BDNF elevation. The BDNF, which is a constituent of the family of neurotrophins, plays a critical role not only in the growth, development and maintenance of the nervous system, but also in supporting the survival of neurons and facilitating their ability to proliferate. Furthermore, it is implicated in the processes of glutamatergic and gamma-aminobutyric acid (GABA)-ergic synaptic plasticity, in addition to influencing serotonergic and dopaminergic neurotransmission [132,133,134,135,136]. It is also interesting to note the contrary trend of a significant decrease in BDNF and elevation in IGF-1 in major depression disorder [137]. This could suggest that neurodegeneration is predominant in ASD, and that elevated levels of adaptive BDNF are directed towards stimulating neuron progenitor cells. In this regard, it is crucial to ascertain whether the rationale for detecting serotonin, tryptophan, IGF-1 and BDNF lies in the presence of multiple functional deficits associated with ASD. The above data could support the inclusion in the modulation of some substances that influence serotonin metabolism, in addition to the traditionally used serotonin reuptake inhibitors. Folate links to the production of neurotransmitters in the brain as 5-HT, dopamine (DA), norepinephrine (NA) and plays an antidepressant-like role in several pathways involving monoamines. In experimental model of chronic unpredictable mild stress the folic acid administration result in increased levels of dopamine and norepinephrine and modulates the levels of homocysteine, BDNF and β-endorphine [138]. Most people with ASD produce the folate receptor antibodies (FRA), which blocks the uptake of folate into the brain and results in cerebral folate deficiency (CFD) [139]. In 83% of cases, the etiology of CFD in ASD was attributed to FRA, and in 43% to mitochondrial dysfunction. By meta analysis the prevalence of FRA in ASD was 71%, with no significant variation observed across studies. Children diagnosed with ASD exhibited a 19.03-fold elevated likelihood of positivity for a FRA when compared with typically developing children. Administration of d,l-leucovorin (folinic acid) is associated with improvements in core and associated symptoms in 67% of ASD patients concerning irritability (58%), ataxia (88%), pyramidal signs (76%), movement disorders (47%), and epilepsy (75%) [140]. Vitamin B6 plays a crucial role in amino acid metabolism, acting as a rate-limiting factor in the synthesis of neurotransmitters such as DA, 5-HT, GABA, NE and the melatonin. Vitamin B6 levels affect neurotransmitter synthesis. Even mild deficiency could lead to reduce GABA and serotonin, which results in a loss of inhibition of a neural activity of GABA, and affecting sleep, behavior, and a loss of hypothalamus -pituitary control [141]. Supplementation with B6 reduced self-reported anxiety symptoms, with the effect being more pronounced for generalized anxiety disorder symptoms [142]. Hence, adding vitamin 6 and folic acid to serotoninergic modulation might be an option for ASD patients who have changes in serotonin, tryptophane and BDNF levels.
A considerable body of literature exists which describes the close interplay between OXT, 5-HT, GABA and DA. OXT exerts its influence over GABAergic neurons by promoting their function and development and participate in neurogenesis. Recent meta-analyses and review studies have emphasised that reduced levels of the peripheral OXT may be associated with ASD [143]. Concurrently, an inverse OXTR distribution between nucleus basalis and ventral pallidum result in altered cortico-striatal connectivity and could produce typical signs of sensorial hyporeactivity with unusual visual attention. In contrast, the presence of OXTR hypermethylation in children diagnosed with ASD could result in diminished levels of OXT, which may consequently precipitate impairment in 5-HT, DA and GABA. In this context, the study of methylation state (e.g. folic acid genes) and OXT levels may hold diagnostic significance for individuals diagnosed with ASD who also exhibit prevailing sensory deficits and attention disorders. In randomized, placebo control study four weeks of chronic OXT administration has been shown to stimulate the oxytocinergic system in children with ASD. Evidence for this stimulation has been found in the form of increased salivary oxytocin levels and associated decreased levels of oxytocin OXTR methylation with clinically evidenced improved feelings of secure attachment [144]. Meta analysis found the significant effect of OXT with the dosage of 48 IU per day on social impairments and repetitive behavior [145]. Thus, the most recent studies provide evidence for an option for intranasal OXT in patients with ASD.
NMDARs, which are widely distributed in the cortex, play a role in the process of synaptic plasticity, which includes phenomena such as long-term potentiation and long-term depression. These are the cellular mechanisms that underpin memory and learning. They are involved in the phenomenon of excitotoxicity. This is the process that results in neuronal death and is observed in several neurodegenerative diseases. NMDARs adjacent to axons with terminals and glial cells [146].
Voxel-wise GM-based spatial statistics analysis revealed reduced neurite density index in left prefrontal cortices associated with reduced empathy in children with ASD [147]. Consistent with this, using multivoxel proton magnetic resonance spectroscopy, total N-acetyl-aspartate concentration was significantly lower in the insula and putamen of ASD patients, whereas glutamate levels were significantly higher in the hippocampus, explained by reduced neuronal or axonal density and impaired mitochondrial metabolism. [148]. In postmortem analysis of human frozen tissue samples from individuals diagnosed with ASD, a significant increase in the density of neuron-positive polymorphic by morphology cell profiles within the subplate zone of the cerebral cortex has been revealed. This finding suggests that the retention of an immature state, which is characteristic of early development, has occurred [149]. Based on the above, it can be suggested that the increased neuronal density in the cortex of ASD patients, with the highest concentration of NMDARs, may be related to damaging factors in the intrauterine period that impact to neuronal “pruning” and disruption of cortico-cortical and cortico-striatal projections.
Prenatal zinc deficiency is another non-genetic model of ASD, causally linked to ASD behavior, learning and memory deficits and neuropsychological symptoms as demonstrated in animal models [150]. Transsynaptic mobilization of Zn rapidly rescues social interaction in two independent mouse models of ASD. In mice with lacking an excitatory postsynaptic scaffolding protein Shank2, the postsynaptic Zn elevation induced by clioquinol (a Zn chelator and ionophore) improves social interaction. Postsynaptic Zn is mainly released from presynaptic pools and activates NMDARs via postsynaptic tyrosine kinase activation [151,152]. Non-competitive NMDA receptor antagonists include cerestat, phencyclidine, ketamine, magnesium preparations, dextromethorphan, dextrorphan, disolcipine, and remacemide. These drugs bind to the phencyclidine site on NMDA-associated ion channels [153,154,155,156]. The excitor/inhibitory imbalance in mice with prenatally valproic acid induced ASD-like behavior was associated to 5-HT receptors hypo signaling and decreased by 5-HT1A receptor agonist [157].
Another substance that has been demonstrated to block NMDA-dependent channels in a potential-dependent manner is magnesium ions. The coordination of Mg²⁺ with the Zn²⁺ pocket on GluN2-NTD subunits of NMDARs results in the allosteric inhibition of channel activity. Magnesium is a key regulator of NMDARs, making them coincidence detectors for excitatory synaptic transmission, and essential for brain development, synaptic plasticity, and homeostasis [158]. Magnesium preserves neuronal integrity by reducing oxidative stress and inflammation. The direct correlation between magnesium intake and cognitive function in healthy individuals was demonstrated [159].
The role of glycine as an inhibitory neurotransmitter has been demonstrated in almost all parts of the CNS. In the brain, a high density of glycine receptors is found in the brainstem structures, cerebral cortex, striatum, hypothalamic nuclei, conduits from the frontal cortex to the hypothalamus, and cerebellum. A number of experimental studies have demonstrated that glycine, in submicromolecular concentrations, is an essential component for the normal functionality of glutamate NMDA receptors. NMDA receptors are only able to be activated by glycine binding to their specific glycine sites, i.e. glycine is required to function as their co-agonist. Glycine significantly reduced neuronal death as NMDA receptor agonists. By adding to the incubation medium with glutamate and NMDA, glycine not only reduced neuronal death but also affected the morphological type of death, switching it from necrosis to the less dangerous apoptosis. All this makes perspective from the point of view of ASD-targeted therapy [160,161,162]. Glycine biosynthesis for utilisation in neurotransmission is a process that is facilitated by serine hydroxymethyl transferase, an enzyme that functions as a cofactor in a reaction that utilises both pyridoxal phosphate and tetrahydrofolate. In the nervous system, glycine is synthesised by the glycine synthase (GCS) enzyme, which catalyzes a readily reversible reaction between carbon dioxide, ammonium ion, 5- and 10-methylene tetrahydrofolate, NADH and a proton. This reaction produces glycine, tetrahydrofolate and NAD+ [163]. In this context, it can be posited that folate could also function as a modulator of glycine in individuals diagnosed with ASD. Scaffolding SHANK proteins (e.g. SHANK3) play special role in the assembled prenatal development of nervous system. It has been established that SHANK3 fulfils a pivotal developmental function in regulating intrinsic neuronal excitability in human neurons. The substance has been demonstrated to be enriched in neuronal postsynaptic densities. In these locations, it functions as a scaffold, thereby orchestrating post-synaptic signaling processes. It has been demonstrated that mutations affecting SHANK3 expression or function cause neurodevelopmental deficits primarily by impairing synaptic activity. Increases in cortical activity occurring at an early stage in Shank3B-/- mutant animals are responsible for the premature maturation of striatal projection neurons and corticostriatal connectivity [164]. Patients who have been diagnosed with SHANK3 mutations are characterised by a severe cognitive deficit. SHANK3 heterozygous mutations has been demonstrated to induce alterations in the transportation of glutamate transmitters in synapses. These mutations in different exons induced ASD -like clinical signs as anxiety, social interaction deficits and impaired motor coordination SHANK mutant mice show specific ASD-like phenotypes due to NMDAR hypofunction [165]. Consequently, the presence of dysfunctional SHANK proteins may have a deleterious effect on postsynaptic transmission, synaptic connections and neuronal networks, affecting NMDA neurons, potentially exacerbating the cognitive impairments observed in ASD. Therefore, on the basis of the information analyzed, we have compared the clinical data characteristic of the ASD, corresponding biomarkers and modulating agents.
These data are presented in Table 1.
As shown in Table 1, there is a relatively new class of FDA-approved recombinant and synthetic IgF-1 derivative analogue biopreparations that may be applicable to several syndromes characteristic of ASD [166]. On the other hand, a safe and pathogenetically determined combination of certain substances could produce a potentiating effect. However, this depends both on the prevalence of certain ASD symptoms and on the level of biomarkers in a particular patient, which allows the dosage and regimen to be adjusted. It is also noteworthy that in pediatric cases, medication for ASD can be effective when administered in conjunction with remedial methods, such as sensory integration therapy. Sensory integration therapy has been proven to be effective in helping children diagnosed with ASD in many areas. It can improve motor, cognitive, social and communication skills. The quality of life and daily functioning of those receiving the intervention is also significantly improved [167]. Sensory hyperreactivity may occur in conjunction with other models of sensory modulation disorders. In terms of structure and function, the thalamus plays a crucial role in multimodal sensorimotor processing. Evidence has been found in the ASD which suggests increased thalamic connectivity and altered function of the auditory, visual and olfactory cortices [168].
Serotonin system is the earlier brain developing signal system. 5-HT has been demonstrated to play a critical role in the process of fetal programming, potentially establishing a sensitivity to both negative and positive life experiences. This sensitivity has been shown to predict long-term risks for behavioral and psychiatric disorders [169]. In this context, the early prenatal environment may exert an influence on 5-HT regulation, with potential implications for other signaling systems and thalamocortical connectivity. This, in turn, has the potential to exacerbate disorders in sensory integration, a core manifestation of ASD. It should also be emphasized, the efficiency of multidisciplinary approach in ASD management. The reversal of level 3 severity autism in dizygotic female twins was achieved through a parent-driven, multidisciplinary, therapeutic approach involving of licensed specialists, with a focus on lifestyle modification was demonstrated. This intervention was personalized to each twin's symptoms, laboratory results, and other outcome measures [170].
8. Conclusions
In this semantic review, we have shown the current possibilities for ASD diagnostic confirmation depending on the manifestation variants. The possibilities of targeted combined modulation with the use of relatively accessible in clinical practice substances, based on most common genes signaling pathways, are demonstrated. In this regard, further investigation is necessary to elucidate the potentiality for optimal combining of pathogenetic modulational effects with correctional rehabilitative programs.
Author Contributions
Conceptualization, A.K. and I.B. in consultation with co-authors; Writing—original draft preparation, A.K., I.B., A.P., V.O., and O.K. Writing—review and editing, A.K., I.B., V.O., and O.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Hodges, H.; Fealko, C.; Soares, N. Autism spectrum disorder: definition, epidemiology, causes, and clinical evaluation. Transl Pediatr. 2020 Feb;9 (Suppl 1):P.55-65. [CrossRef]
- Li Q, Li Y, Liu B, Qian Li,; Yanmei Li, ; Qingsong Chen, ; Xiaohui Xing, ; Guifeng Xu, ; Wenhan Yang. Prevalence of Autism Spectrum Disorder Among Children and Adolescents in the United States From 2019 to 2020. AMA Pediatr. [CrossRef]
- Shuang, Qiu,; Yuping, Lu:, Yan Li,; Jikang, Shi,; Heran, Cui,; Yulu, Gu,; Yong, Li.; Weijing, Zhong,; Xiaojuan, Zhu,; Yunkai, Liu,; Yi, Cheng,; Yawen, Liu,; Yichun, Qiao. Prevalence of autism spectrum disorder in Asia: A systematic review and meta-analysis. Psychiatry Research, Volume 284, 2020, 112679. [CrossRef]
- Zeidan, J,; Fombonne, E,; Scorah, J,; Ibrahim, A,; Durkin, MS,; Saxena, S,; Yusuf, A,, Shih, A,; Elsabbagh, M. Global prevalence of autism: A systematic review update. Autism Res. [CrossRef]
- Zahorodny, W.; Shenouda, J.; Sidwell, K.; et al. Prevalence and Characteristics of Adolescents with Autism Spectrum Disorder in the New York-New Jersey Metropolitan Area. J Autism Dev Disord. 2023. [Google Scholar] [CrossRef]
- Chiarotti, F.; Venerosi, A. Epidemiology of Autism Spectrum Disorders: A Review of Worldwide Prevalence Estimates Since 2014. Brain Sci. 2020, 10, 274. [Google Scholar] [CrossRef]
- Cardoso, IL.; Almeida, S. Genes Involved in the Development of Autism. Int Arch Commun. Disord. 2019. [Google Scholar]
- Rylaarsdam, L.; Guemez-Gamboa, A. Genetic Causes and Modifiers of Autism Spectrum Disorder. Front Cell Neurosci. 2019, Aug 20;13:385. [CrossRef]
- Madhavi, Apte.; Aayush, Kumar. Correlation of mutated gene and signalling pathways in ASD. IBRO Neuroscience Reports. [CrossRef]
- Khachadourian, V.; Mahjani, B.; Sandin, S.; Kolevzon, A.; Buxbaum, JD.; Reichenberg, A.; Janecka, M. ; Comorbidities in autism spectrum disorder and their etiologies. Transl Psychiatry. [CrossRef]
- Vilela, J.; Rasga, C.; Santos, J.X.; Martiniano, H.; Marques, A.R.; Oliveira, G.; Vicente, A.M. Bridging Genetic Insights with Neuroimaging in Autism Spectrum Disorder—A Systematic Review. Int. J. Mol. Sci. 2024, 25, 4938. [Google Scholar] [CrossRef]
- Al-Beltagi, M.; Saeed, NK.; Elbeltagi, R.; Bediwy, AS. ; Aftab, SAS.; Alhawamdeh, R; Viruses and autism: A Bi-mutual cause and effect. World J Virol. [CrossRef]
- De Giacomo, A.; Medicamento, S.; Pedaci, C.; Giambersio, D.; Giannico, O.V.; Petruzzelli, M.G.; Simone, M.; Corsalini, M.; Marzulli, L.; Matera, E. Peripheral Iron Levels in Autism Spectrum Disorders vs. Other Neurodevelopmental Disorders: Preliminary Data. Int. J. Environ. Res. Public Health. 2022, 19, 4006. [Google Scholar] [CrossRef]
- Rytel, A.; Ostrowska, S.; Marcinkiewicz, K.; Krawczuk-Rybak, M. ; & Sawicka-Żukowska,, M. Ferritin – novel uses of a well-known marker in paediatrics. Pediatria Polska - Polish Journal of Paediatrics, 2023 98(1), 57-65. [CrossRef]
- Bener., A.; Khattab, AO.; Bhugra, D. Bener., A.; Khattab, AO.; Bhugra, D. et al. Iron and vitamin D levels among autism spectrum disorders children. Ann Afr Med. [CrossRef]
- Li, Chen; Xingzhi, Guo; Chen Hou, Peng; Tang, Xin; Zhang, Li; Chong Rui Li; The causal association between iron status and the risk of autism: A Mendelian randomization study. Front. Nutr., 2022. [CrossRef]
- Muller, CL. ; Anacker, AMJ.; Veenstra-VanderWeele, J. The serotonin system in autism spectrum disorder: From biomarker to animal models. Neuroscience. [CrossRef]
- Yamasue, H.; Domes, G. ; Oxytocin and Autism Spectrum Disorders. Curr Top Behav Neurosci. [CrossRef]
- Nyffeler, J.; Walitza, S.; Bobrowski, E.; Gundelfinger, R.; Grünblatt, E. Association study in siblings and case-controls of serotonin- and oxytocin-related genes with high functioning autism. J Mol Psychiatry. [CrossRef]
- Raphaelle Mottolese, AB.; Jérôme Redouté, BC.; Nicolas Costes, C.; Didier Le Bars, BC. and Angela Sirigua, B. Switching brain serotonin with oxytocin. PNAS. [CrossRef]
- Zhao, Feng; Zhang, Hao; Wang, Peng; Cui, Wenjie; Xu, Kaiyong; Chen, Dan; Hu, Minghui; Li, Zifa; Geng, Xiwen; Wei, Sheng. Oxytocin and serotonin in the modulation of neural function: Neurobiological underpinnings of autism-related behavior. Front. Neurosci., 22 July 2022. Sec. NeurodevelopmentVolume 16 – 2022. [CrossRef]
- Elshahawi, H.H.; Taha, G.R.A.; Azzam, H.M.E.; et al. N-Methyl-d-aspartate (NMDA) receptor antibody in relation to autism spectrum disorder (ASD): presence and association with symptom profile. Middle East Curr Psychiatry, 2021. [Google Scholar] [CrossRef]
- Lee, E.; Choi, S.; Kim, E. NMDA receptor dysfunction in autism spectrum disorders. Curr Opin Pharmacol. [CrossRef]
- Ruu-Fen, Tzang.; Chuan-Hsin, Chang.; Yue-Cune, Chang.; Hsien-Yuan, Lane. Autism Associated With Anti-NMDAR Encephalitis: Glutamate-Related Therapy, Front. Psychiatry, 21 June 2019. Sec. Molecular Psychiatry, Volume 10, 2019. [CrossRef]
- Yukti, Vyas.; Juliette, E.; Cheyne, Kevin, Lee.; Yewon, Jung.; Pang Ying, Cheung.;, Montgomery, Johanna M.; Shankopathies in the Developing Brain in Autism Spectrum Disorders. Front. Neurosci.,. [CrossRef]
- Huang, M.; Qi, Q.; Xu, T. Targeting Shank3 deficiency and paresthesia in autism spectrum disorder: A brief review. Front Mol Neurosci. [CrossRef]
- Hu, C.; Tao, L.; Cao, X.; Chen, L. The solute carrier transporters and the brain: Physiological and pharmacological implications. Asian J Pharm Sci. 2020 Mar;15(2):131-144. [CrossRef]
- Mir, A.; Almudhry, M.; Alghamdi, F.; Albaradie, R.; Ibrahim, M.; Aldurayhim, F.; Alhedaithy, A.; Alamr, M.; Bawazir, M.; Mohammad, S.; Abdelhay, S.; Bashir, S.; Housawi, Y. SLC gene mutations and pediatric neurological disorders: diverse clinical phenotypes in a Saudi Arabian population. Hum Genet. [CrossRef]
- Schuch, J. B.; Müller, D.; Endres, R. G.; Bosa, C. A.; Longo, D.; Schuler-Faccini, L.; Ranzan, J.; Becker, M. M.; dos Santos Riesgo, R.; Roman, T. Psychomotor agitation and mood instability in patients with autism spectrum disorders: A possible effect of SLC6A4 gene? Research in Autism Spectrum Disorders,. [CrossRef]
- Shuid, A.N.; Jayusman, P.A.; Shuid, N.; Ismail, J.; Kamal Nor, N.; Mohamed, I.N. Association between Viral Infections and Risk of Autistic Disorder: An Overview. Int. J. Environ. Res. Public Health. 2021, 18, 2817. [Google Scholar] [CrossRef]
- Sawlani, V.; ShivaShankar, J. J.; White, C. Magnetic resonance imaging findins in a case of congenital rubella encephalitis. Can. J. Infect. Dis. Med. Microbiol. [CrossRef]
- Thong, V N.; Van, H Ph.; Kenji, A. Pathogenesis of Congenital Rubella Virus Infection in Human Fetuses: Viral Infection in the Ciliary Body Could Play an Important Role in Cataractogenesis. eBioMedicine, 2015, Volume 2, Issue 1, 59 – 63.
- Jonathan, P. C.; Jörg, M. The role of retinoic acid signaling in maintenance and regeneration of the CNS: from mechanisms to therapeutic targeting. Front. Mol. Neurosci., Sec. Molecular Signalling and Pathways, Volume 17 – 2024. [CrossRef]
- Oza, Y. R.; Mody, M.; Avula, M.; Allahudeen, S. D.; Appana, L. S. M.; Ali, M. A. Understanding progressive rubella pan encephalitis: a rare neurological disorder. International Journal of Research in Medical Sciences, 2024. [Google Scholar] [CrossRef]
- Slawinski, B.L.; Talge, N.; Ingersoll, B.; Smith, A.; Glazier, A.; Kerver, J.; Paneth, N.; Racicot K. Maternal cytomegalovirus sero-positivity and autism symptoms in children. Am J Reprod Immunol. 2018. May;79(5):e12840. [CrossRef]
- Pesch, M.H.; Leung, J.; Lanzieri, T.M.; Tinker, S.C.; Rose, C.E.; Danielson, M.L.; Yeargin-Allsopp, M.; Grosse, S.D. ; Autism Spectrum Disorder Diagnoses and Congenital Cytomegalovirus. Pediatrics. [CrossRef]
- Piccirilli, G. , Gabrielli, L., Bonasoni, M.P.et al. Fetal Brain Damage in Human Fetuses with Congenital Cytomegalovirus Infection: Histological Features and Viral Tropism. Cell Mol Neurobiol. [CrossRef]
- Egilmezer, E. , Hamilton, S.T., Foster, C.S.P.et al. Human cytomegalovirus (CMV) dysregulates neurodevelopmental pathways in cerebral organoids. Commun Biol. [CrossRef]
- Milada, M.; Siri, M.; Hege, M. B.; Gunnes, N. , et al. Maternal Immunoreactivity to Herpes Simplex Virus 2 and Risk of Autism Spectrum Disorder in Male Offspring. mSphere, 2017; 1. [Google Scholar] [CrossRef]
- Gentile, I.; Zappulo, E.; Bonavolta, R.; Maresca, R.; Ricci, M.P.; et al. Prevalence of herpes simplex virus 1 and 2 antibodies in patients with autism spectrum disorders. In Vivo. [PubMed]
- Duarte, L.F.; Farias, M.A.; Álvarez, D.M.; Bueno, S.M.; Riedel, C.A.; González, P.A. Herpes Simplex Virus Type 1 Infection of the Central Nervous System: Insights Into Proposed Interrelationships With Neurodegenerative Disorders. Front. Cell. Neurosci. 2019. [Google Scholar] [CrossRef]
- Otero, A.M.; Connolly, M.G.; Gonzalez-Ricon, R.J.; et al. Influenza A virus during pregnancy disrupts maternal intestinal immunity and fetal cortical development in a dose- and time-dependent manner. Mol Psychiatry, 2025. [Google Scholar] [CrossRef]
- Miller, V.M.; Zhu, Y.; Bucher, C.; McGinnis, W.; Ryan, L.K.; Siegel, A.; Zalcman, S. Gestational flu exposure induces changes in neurochemicals, affiliative hormones and brainstem inflammation, in addition to autism-like behaviors in mice. Brain, Behavior, and Immunity, Volume 33, 2013, Pages 153-163. [CrossRef]
- Becerra-Culqui, T.A.; Getahun, D.; Chiu, V.; Sy, L.S.; Tseng, H.F. Prenatal Influenza Vaccination or Influenza Infection and Autism Spectrum Disorder in Offspring. Clin Infect Dis. [CrossRef]
- Zerbo, O.; Qian, Y.; Yoshida, C.; Fireman, B.H.; Klein, N.P.; Croen, L.A. Association between influenza infection and vaccination during pregnancy and risk of autism spectrum disorder. JAMA Pediatr. [CrossRef]
- Steinman, G. COVID-19 and autism. Med Hypotheses. [CrossRef]
- Abedini, M.; Mashayekhi, F.; Salehi, Z. Analysis of Insulin-like growth factor-1 serum levels and promoter (rs12579108) polymorphism in the children with autism spectrum disorders, Journal of Clinical Neuroscience, V. 99, 2022. [CrossRef]
- Shen, Liu L.; Yang, Y.; Zhou, M.; Xu, S.; Zhang, W.; Zhang, C. Insulin-Like Growth Factor 1 Has the Potential to Be Used as a Diagnostic Tool and Treatment Target for Autism Spectrum Disorders. Cureus. [CrossRef]
- Lichun, L.; Yongxing, L.; Zhidong, Z.; Qingxian, F.; Yuelian, J. Identification of Ferroptosis-Related Molecular Clusters and Immune Characterization in Autism Spectrum Disorder. Frontiers in Genetics. Sec. Computational Genomics. 2022, 13. [Google Scholar] [CrossRef]
- Tang, R.; Xu, J.; Zhang, B.; Liu, J.; Liang, C.; Hua, J.; Meng, Q.; Yu, X.; Shi, S. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J Hematol Oncol. [CrossRef]
- Singh, S.; Parween, F.; Edara, N.; Zhang, H.; Gardina, P. ; Otaizo-Carrasquero, F, Myers, T,; Farber, J.M. The circadian clock gene ARNTL regulates the expression of CCR6 and Th17 related genes in human cells. J Immunol. [CrossRef]
- Zhang, Y.; Lu, X.; Tai, B.; Li, W.; Li, T. Ferroptosis and Its Multifaceted Roles in Cerebral Stroke. Front Cell Neurosci. [CrossRef]
- Friedmann Angeli, JP.; Krysko, DV.; Conrad, M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat Rev Cancer. [CrossRef]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M. T.; Zeh, H. J. Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. [CrossRef]
- Zhang, J.J.; Du, J.; Kong, N.; Zhang, GY.; Liu, MZ.; Liu, C. Mechanisms and pharmacological applications of ferroptosis: a narrative review. Ann Transl Med. [CrossRef]
- Prakash, P.; Kumari, R.; Sinha, N.; Kumar, S.; Sinha, P. Evaluation of Iron Status in Children with Autism Spectral Disorder: A Case-control Study. Journal of Clinical & Diagnostic Research,. [CrossRef]
- Rabaya, S. ;∙Nairat, S, ∙Bader K.; Mohammad M.; Herzallah MM.;∙ Darwish, HM. Iron metabolism in autism spectrum disorder; inference through single nucleotide polymorphisms in key iron metabolism genes. Journal of the Neurological Sciences. [CrossRef]
- Giambersio, D.; Marzulli, L.; Margari, L.; Matera, E.; Nobili, L.; De Grandis, E.; Cordani, R.; Barbieri, A.; Peschechera, A.; Margari, A.; et al. Correlations between Sleep Features and Iron Status in Children with Neurodevelopmental Disorders: A Cross-Sectional Study. J. Clin. Med. 2023, 12, 4949. [Google Scholar] [CrossRef] [PubMed]
- Castro, K.; Marchezan, J.; Faccioli, LS.; Riesgo, R..; Perry IS.; Anemia Associated with Autism Spectrum Disorder. Int J Pediat Health Care Adv. 2016 ,3(2), 17-20. ISSN 2572-7354.
- Kanney, ML. ; Durmer,; JS, Trotti,LM.; Leu, R. Rethinking bedtime resistance in children with autism: is restless legs syndrome to blame? J. Clin Sleep Med. [CrossRef]
- Taeubert, MJ.; de Prado-Bert, P.; Geurtsen, ML.; et al. Maternal iron status in early pregnancy and DNA methylation in offspring: an epigenome-wide meta-analysis. Clin Epigenet 2022, 14, 59. [Google Scholar] [CrossRef]
- Chauhan, A.; Chauhan, V.; Brown, WT.; Cohen, I. Oxidative stress in autism: Increased lipid peroxidation and reduced serum levels of ceruloplasmin and transferrin - the antioxidant proteins. Life sciences (1973). [CrossRef]
- Abe, E.; Fuwa, T.J.; Hoshi, K.; Saito, T.; Murakami, T.; Miyajima, M.; Ogawa, N.; Akatsu, H.; Hashizume, Y.; Hashimoto, Y.; et al. Expression of Transferrin Protein and Messenger RNA in Neural Cells from Mouse and Human Brain Tissue. Metabolites 2022, 12, 594. [Google Scholar] [CrossRef]
- Petralla, S.; Saveleva, L.; Kanninen, KM.; et al. Increased Expression of Transferrin Receptor 1 in the Brain Cortex of 5xFAD Mouse Model of Alzheimer’s Disease Is Associated with Activation of HIF-1 Signaling Pathway. Mol Neurobiol.
- Pérez, MJ.; Carden, TR.; dos Santos Claro, PA.; et al. Transferrin Enhances Neuronal Differentiation. ASN Neuro. [CrossRef]
- McCarthy, RC.; Sosa, JC.; Gardeck, AM.; Baez, AS, Lee CH., Wessling-Resnick, M. Inflammation-induced iron transport and metabolism by brain microglia. J Biol Chem. 2018 May 18;293(20):7853-7863. [CrossRef]
- Mensikova K.; Katerina Bucilova, K., Konickova, D.; Kurcova, S.; Otruba, P.; Grambalova, Z.; Kaiserova, M.; Petr Kanovsky, P. Transferrin as a Possible CSF Biomarker in Neurodegenerative Proteinopathies (P3-11.004). Neurology.2023. 100 (17, supplement 2). [CrossRef]
- Suri, D.; Teixeira, CM.; Cagliostro, MK.; Mahadevia, D. Ansorge, MS. Monoamine-sensitive developmental periods impacting adult emotional and cognitive behaviors. Neuropsychopharmacology. [CrossRef]
- Feng, Z.; Hao, Z.; Peng, W.; Wenjie, C.; Kaiyong, Xu. et.al. Oxytocin and serotonin in the modulation of neural function: Neurobiological underpinnings of autism-related behavior. Frontiers in Neuroscience. [CrossRef]
- Hu, Z.; Ying, X.; Huang, L.; Zhao, Y.; Zhou, D.; Liu, J.; et al. Association of human serotonin receptor 4 promoter methylation with autism spectrum disorder. Medicine. [CrossRef]
- Lefevre, A.; Richard, N.; Mottolese, R.; Leboyer, M.; Sirigu, A. An Association Between Serotonin 1A Receptor, Gray Matter Volume, and Sociability in Healthy Subjects and in Autism Spectrum Disorder. Autism Res 2020, 13, 1843–1855. [Google Scholar] [CrossRef]
- Higazi, A.M.; Kamel, H.M.; Abdel-Naeem, E.A.; et al. Expression analysis of selected genes involved in tryptophan metabolic pathways in Egyptian children with Autism Spectrum Disorder and learning disabilities. Sci Rep, 2021. [Google Scholar] [CrossRef]
- Ormstad, H.; Bryn, V.; Verkerk, R.; Skjeldal, O. H.; Halvorsen, B.; Saugstad, O. D.; Isaksen, J.; Maes, M. Serum Tryptophan, Tryptophan Catabolites and Brain-derived Neurotrophic Factor in Subgroups of Youngsters, with Autism Spectrum Disorders. Cns & Neurological Disorders-Drug Targets, 2018, 7, 626–639. [Google Scholar] [CrossRef]
- Santana-Coelho, D. Does the kynurenine pathway play a pathogenic role in autism spectrum disorder? Brain, Behavior, & Immunity - Health, 2024. [Google Scholar] [CrossRef]
- Sijben, HJ.; Superti-Furga, G.; IJzerman, AP.; Heitman, LH. Targeting solute carriers to modulate receptor–ligand interactions, Trends in Pharmacological Sciences, Volume 43, Issue 5, 2022, Pages 358-361, doi :10.1016/j.tips.2022.02.004.
- Bala, K.; Aran, Kh. R. Exploring the common genes involved in autism spectrum disorder and Parkinson's disease: A systematic review, Aging and Health Research, Volume 4, Issue 4, 2024. [CrossRef]
- Zhong, J.; Dong, J.; Ruan, W.; Duan, X. Potential Theranostic Roles of SLC4 Molecules in Human Diseases. Int. J. Mol. Sci. 2023, 4, 15166. [Google Scholar] [CrossRef] [PubMed]
- Quintana, D.S.; Rokicki, J.; van der Meer, D.; et al. Oxytocin pathway gene networks in the human brain. Nat Commun 10, 668, 2019. [CrossRef]
- Sartorius, A.M.; Rokicki, J.; Birkeland, S.; et al. An evolutionary timeline of the oxytocin signaling pathway. Commun Biol, 2024. [Google Scholar] [CrossRef]
- Lin, Y.T.; Hsu, K.S. Oxytocin receptor signaling in the hippocampus: Role in regulating neuronal excitability, network oscillatory activity, synaptic plasticity and social memory. Prog. Neurobiol. 2018, 171, 1–14. [Google Scholar] [CrossRef]
- Pierzynowska, K.; Gaffke, L.; Żabińska, M.; Cyske, Z.; Rintz, E.; Wiśniewska, K.; Podlacha, M.; Węgrzyn, G. Roles of the Oxytocin Receptor (OXTR) in Human Diseases. Int. J. Mol. Sci. 2023, 24, 3887. [Google Scholar] [CrossRef] [PubMed]
- Freeman, S.M.; Palumbo, M.C.; Lawrence, R.H.; Smith, A.L.; Goodman, M.M.; Bales, K.L. Effect of age and autism spectrum disorder on oxytocin receptor density in the human basal forebrain and midbrain. Transl. Psychiatry 2018, 8, 257. [Google Scholar] [CrossRef] [PubMed]
- Andari, E.,; Nishitani, S.; Kaundinya, G.; Caceres, GA.; Morrier, MJ.’ Ousley, O.; Smith, AK.; Cubells, JF.; Young, LJ. Epigenetic modification of the oxytocin receptor gene: implications for autism symptom severity and brain functional connectivity. Neuropsychopharmacology. 2020 Jun;45(7):1150-1158. [CrossRef]
- El-Ramly, Amal; Samy, Azza; Soliman, Eman; Oteify, Golnar. Evaluation of oxytocin and serotonin levels in autism spectrum disorder, Journal of Medicine in Scientific Research: 2018 Vol. 1: Iss. 1, Article 12. [CrossRef]
- John, S.; Jaeggi, A. V. Oxytocin levels tend to be lower in autistic children: A meta-analysis of 31 studies. Autism. [CrossRef]
- Lefevre, A.; Richard, N.; Jazayeri, M.; Beuriat, PA.; Fieux, S.; Zimmer, L.; Duhamel, JR. , Sirigu, A. Oxytocin and Serotonin Brain Mechanisms in the Nonhuman Primate. J Neurosci. [CrossRef]
- Dölen, G.; Darvishzadeh, A. ; Huang, KW; Malenka, RC. Social reward requires coordinated activity of nucleus accumbens oxytocin and serotonin. Nature. [CrossRef]
- Borroto-Escuela, D.O.; Ambrogini, P.; Chruścicka, B.; Lindskog, M.; Crespo-Ramirez, M.; Hernández-Mondragón, J.C.; Perez de la Mora, M.; Schellekens, H.; Fuxe, K. The Role of Central Serotonin Neurons and 5-HT Heteroreceptor Complexes in the Pathophysiology of Depression: A Historical Perspective and Future Prospects. Int. J. Mol. Sci. 2021, 22, 1927. [Google Scholar] [CrossRef]
- Whitaker-Azmitia, PM. Behavioral and cellular consequences of increasing serotonergic activity during brain development: A role in autism? Int J Dev Neurosci. 2005, 2005.23, 75–83. [Google Scholar] [CrossRef]
- Beaurain, M.; Salabert, A.-S.; Payoux, P.; Gras, E.; Talmont, F. NMDA Receptors: Distribution, Role, and Insights into Neuropsychiatric Disorders. Pharmaceuticals 2024, 17, 1265. [Google Scholar] [CrossRef]
- Hansen, KB.; Wollmuth, LP.; Bowie, D.; Furukawa, H.; Menniti, FS.; Sobolevsky, AI.; Swanson, GT.; Swanger, SA.; et al. Structure, Function, and Pharmacology of Glutamate Receptor Ion Channels. Pharmacol Rev. [CrossRef]
- Vyklicky, V.; Stanley, C.; Habrian, C.; et al. Conformational rearrangement of the NMDA receptor amino-terminal domain during activation and allosteric modulation. Nat Commun 2021, 2021 12, 2694. [Google Scholar] [CrossRef]
- Vieira, M. ; Yong,XLH.; Roche KW.; Anggono, V. Regulation of NMDA glutamate receptor functions by the GluN2 subunits. J Neurochem. [CrossRef]
- Sprengel, R.; Eltokhi, A. Ionotropic Glutamate Receptors (and Their Role in Health and Disease). In: Pfaff, D.W., Volkow, N.D., Rubenstein, J.L. (eds) Neuroscience in the 21st Century. Springer, Cham. 2022. [CrossRef]
- Egunlusi, AO.; Joubert, J. NMDA Receptor Antagonists: Emerging Insights into Molecular Mechanisms and Clinical Applications in Neurological Disorders. Pharmaceuticals 2024, 17, 639. [Google Scholar] [CrossRef]
- Yamamoto, H.; Hagino, Y.; Kasai S.; Ikeda K.; Specific Roles of NMDA Receptor Subunits in Mental Disorders. Curr Mol Med. 2015;15 (3):193-205. [CrossRef]
- Kysilov, B.; Kuchtiak, V.; Hrcka Krausova, B.; et al. Disease-associated nonsense and frame-shift variants resulting in the truncation of the GluN2A or GluN2B C-terminal domain decrease NMDAR surface expression and reduce potentiating effects of neurosteroids. Cell. Mol. Life Sci. 2024. [Google Scholar] [CrossRef]
- Myers, SJ.; Yuan, H.; Kang, JQ. ; Tan, FCK.; Traynelis SF.; Low CM. Distinct roles of GRIN2A and GRIN2B variants in neurological conditions. F1000Res. 2019 Nov 20;8: F1000 Faculty Rev-1940. [CrossRef]
- Tumdam, R.; Hussein, Y.; Garin-Shkolnik, T.; Stern, S. NMDA Receptors in Neurodevelopmental Disorders: Pathophysiology and Disease Models. Int. J. Mol. Sci. 2024, 25, 12366. [Google Scholar] [CrossRef] [PubMed]
- Montanari, M. , Martella, G.; Bonsi, P.; Meringolo, M. Autism Spectrum Disorder: Focus on Glutamatergic Neurotransmission. Int J Mol Sci. [CrossRef]
- Shin, W.; Kim, K.; Serraz, B.; Cho, YS.; Kim, D.; Kang, M.; Lee, EJ.; Lee, H.; Bae, YC.; Paoletti, P.; Kim, E. Early correction of synaptic long-term depression improves abnormal anxiety-like behavior in adult GluN2B-C456Y-mutant mice. PLoS Biol. [CrossRef]
- Sabo, SL.; Lahr, JM.; Offer, M.; Weekes, ALA.; Sceniak, MP. GRIN2B-related neurodevelopmental disorder: current understanding of pathophysiological mechanisms. Front. Synaptic Neurosci. 2023, 14:1090865. [CrossRef]
- Leblond, CS.; Nava, C.; Polge, A.; Gauthier, J.; Huguet, G.; Lumbroso, S.; et al. Meta-analysis of SHANK Mutations in Autism Spectrum Disorders: a gradient of severity in cognitive impairments. PLoS Genet. [CrossRef]
- Li, X. Unravelling the role of SHANK3 mutations in targeted therapies for autism spectrum disorders. Discov Psychol, 2024. [Google Scholar] [CrossRef]
- Jaramillo, TC.; Speed, HE.; Xuan, Z.; Reimers, JM.; Escamilla, CO.; Weaver, TP.; Liu, S.; Filonova, I.; Powell, CM. Novel Shank3 mutant exhibits behaviors with face validity for autism and altered striatal and hippocampal function. Autism Res. [CrossRef]
- Jung, S.; Park, M. Shank postsynaptic scaffolding proteins in autism spectrum disorder: Mouse models and their dysfunctions in behaviors, synapses, and molecules. Pharmacol Res. [CrossRef]
- Lisé, MF.; El-Husseini, A. The neuroligin and neurexin families: from structure to function at the synapse. Cell Mol Life Sci. [CrossRef]
- Altas, B.; Tuffy, LP.; Patrizi, A.; Dimova, K.; Soykan, T. et. al. Region-Specific Phosphorylation Determines Neuroligin-3 Localization to Excitatory Versus Inhibitory Synapses. Biol Psychiatry. [CrossRef]
- Peñagarikano, O.; Geschwind, DH. What does CNTNAP2 reveal about autism spectrum disorder? Trends Mol Med. [CrossRef]
- Vogt, D. ; Cho, KKA.; Shelton, SM.; Paul, A.; Huang, ZJ.; Sohal, VS.; Rubenstein, JLR. Mouse Cntnap2 and Human CNTNAP2 ASD Alleles Cell Autonomously Regulate PV+ Cortical Interneurons. Cereb Cortex. [CrossRef]
- Chalkiadaki, K.; Statoulla, E.; Zafeiri, M.; Voudouri, G. GABA/Glutamate Neuron Differentiation Imbalance and Increased AKT/mTOR Signaling in CNTNAP2-/- Cerebral Organoids. Biol Psychiatry Glob Open Sci. [CrossRef]
- Tan, BT.; Wang, L.; Li, S.; Long, ZY.; Wu, YM.; Liu, Y. Retinoic acid induced the differentiation of neural stem cells from embryonic spinal cord into functional neurons in vitro. Int J Clin Exp Pathol. [PubMed]
- Lazzeri, G.; Lenzi, P.; Signorini, G.; Raffaelli, S.; Giammattei, E.; Natale, G.; Ruffoli, R.; Fornai, F.; Ferrucci, M. Retinoic Acid Promotes Neuronal Differentiation While Increasing Proteins and Organelles Related to Autophagy. Int. J. Mol. Sci. 2025, 26, 1691. [Google Scholar] [CrossRef]
- Nguyen, CH.; Grandits, AM.; Purton, LE.; Sill, H.; Wieser, R. All-trans retinoic acid in non-promyelocytic acute myeloid leukemia: driver lesion dependent effects on leukemic stem cells. Cell Cycle. 2020 Oct;19(20):2573-2588. [CrossRef]
- Chen, Y.; Tong. X.; Lu, R.; Zhang, Z.; Ma, T. All-trans retinoic acid in hematologic disorders: not just acute promyelocytic leukemia. Front. Pharmacol. 2024, 15:1404092. [CrossRef]
- Levada, OA.; Troyan, AS. Insulin-like growth factor-1: a possible marker for emotional and cognitive disturbances, and treatment effectiveness in major depressive disorder. Ann General Psychiatry. [CrossRef]
- Levada, OA. ; Troya, AS; Pinchuk, IY. Serum insulin-like growth factor-1 as a potential marker for MDD diagnosis, its clinical characteristics, and treatment efficacy validation: data from an open-label vortioxetine study. BMC Psychiatry. [CrossRef]
- Dichtl, S.; Haschka, D.; Nairz, M.; Seifert, M.; Volani, C.; Lutz, O.; Weiss, G. Dopamine promotes cellular iron accumulation and oxidative stress responses in macrophages. Biochem Pharmacol. [CrossRef]
- Levada, OA.; Dobrodub, IV.; Trailin, AV. Reduced expression of dopamine d2-receptors on peripheral blood lymphocytes as a marker for progression of subcortical cognitive impairments. Zaporozhye Medical Journal, No 5, 2013, p.43-45.
- Gesundheit, B.; Zisman, PD.; Hochbaum, L.; Posen, Y.; Steinberg, A.; Friedman, G.; Ravkin, HD.; Rubin, E.; Faktor, O.; Ellis, R. Autism spectrum disorder diagnosis using a new panel of immune- and inflammatory-related serum biomarkers: A case-control multicenter study. Front Pediatr. [CrossRef]
- Stancioiu, F.; Bogdan, R.; Dumitrescu, R. Neuron-Specific Enolase (NSE) as a Biomarker for Autistic Spectrum Disease (ASD). Life (Basel). 2023 Aug 13;13(8):1736. [CrossRef]
- Evers, M. ; Cunningham-Rundles.; C.; Hollander, E. Heat shock protein 90 antibodies in autism. Mol Psychiatry. [CrossRef]
- Singh, VK. Phenotypic expression of autoimmune autistic disorder (AAD): a major subset of autism. Ann Clin Psychiatry. 2009 Jul-Sep;21(3):148-61. PMID: 19758536.
- Belenichev, IF.; Aliyeva, OG.; Popazova, OO.; Bukhtiyarova, NV. Involvement of heat shock proteins HSP70 in the mechanisms of endogenous neuroprotection: the prospect of using HSP70 modulators. Front Cell Neurosci. [CrossRef]
- Iorgu, AM.; Inta, D.; Peter, G. Inducible HSP72 protein as a marker of neuronal vulnerability in brain research: A potential biomarker for clinical psychiatry?, Biomarkers in Neuropsychiatry, V. 12, 2025. [CrossRef]
- Liu, H.; Talalay, P.; Fahey, JW. Biomarker-Guided Strategy for Treatment of Autism Spectrum Disorder (ASD). CNS Neurol Disord Drug Targets. 2016;15(5):602-13. [CrossRef]
- Frye, RE.; Rincon, N.; McCarty, PJ.; Brister, D.; Scheck, AC.; Rossignol, DA. Biomarkers of mitochondrial dysfunction in autism spectrum disorder: A systematic review and meta-analysis. Neurobiol Dis. [CrossRef]
- Manivasagam, T. ; Arunadevi. S.; Essa, M.; Saravana Babu, C.; Borah, A.; Thenmozhi, AJ.; Qoronfleh, MW. Role of Oxidative Stress and Antioxidants in Autism. Adv Neurobiol. [CrossRef]
- Nadeem, A.; Ahmad, SF.; Bakheet, SA.; Al-Harbi, NO.; Al-Ayadhi, LY.; Attia, SM. ; Zoheir, KMA. Toll-like receptor 4 signaling is associated with upregulated NADPH oxidase expression in peripheral T cells of children with autism. Brain Behav Immun. [CrossRef]
- Liu, Y.; Yang, Z.; Du, Y.; Shi, S.; Cheng, Y. Antioxidant interventions in autism spectrum disorders: A meta-analysis. Prog Neuropsychopharmacol Biol Psychiatry. [CrossRef]
- Manivasagam, T.; Arunadevi, S.; Essa, M. M.; SaravanaBabu, C.; Borah, A.; Thenmozhi, A. J.; Qoronfleh, M. W. Role of Oxidative Stress and Antioxidants in Autism.
- Colucci-D’Amato, L.; Speranza, L.; Volpicelli, F. Neurotrophic Factor BDNF, Physiological Functions and Therapeutic Potential in Depression, Neurodegeneration and Brain Cancer. Int. J. Mol. Sci. [CrossRef]
- Shu-Hui, D.; Yu, C.; Shu-Ming, H.; Bo, Zh. The Role of Brain-Derived Neurotrophic Factor Signaling in Central Nervous System Disease Pathogenesis. Frontiers in Human Neuroscience. V.16. 2022. [Google Scholar] [CrossRef]
- Komori, T.; Okamura, K.; Ikehara, M.; et al. Brain-derived neurotrophic factor from microglia regulates neuronal development in the medial prefrontal cortex and its associated social behavior. Mol Psychiatry. [CrossRef]
- Miranda, M.; Morici, JF.; Zanoni, MB.; Bekinschtein, P. Brain-Derived Neurotrophic Factor: A Key Molecule for Memory in the Healthy and the Pathological Brain. Frontiers in Cellular Neuroscience. V.13. 2019. [Google Scholar] [CrossRef]
- Bathina, S.; Das, UN. Brain-derived neurotrophic factor and its clinical implications. Arch Med Sci. [CrossRef]
- Troyan, AS.; Levada, OA. The Diagnostic Value of the Combination of Serum Brain-Derived Neurotrophic Factor and Insulin-Like Growth Factor-1 for Major Depressive Disorder Diagnosis and Treatment Efficacy. Front Psychiatry. [CrossRef]
- Zhou, Y.; Cong, Y.; Liu, H. Folic acid ameliorates depression-like behaviour in a rat model of chronic unpredictable mild stress. BMC Neurosci. [CrossRef]
- Ahmavaara, K.; Ayoub, G. Cerebral Folate Deficiency in Autism Spectrum Disorder Folate in Nervous System Development Crimson Publishers Wings to the Research. 2022. [Google Scholar] [CrossRef]
- Rossignol, DA.; Frye, RE. ; Cerebral Folate Deficiency, Folate Receptor Alpha Autoantibodies and Leucovorin (Folinic Acid) Treatment in Autism Spectrum Disorders: A Systematic Review and Meta-Analysis. J Pers Med. [CrossRef]
- Kennedy, DO. B Vitamins and the Brain: Mechanisms, Dose and Efficacy--A Review. Nutrients. [CrossRef]
- Field, DT.; Cracknell, RO.; Eastwood, JR.; Scarfe, P.; Williams, CM.; Zheng, Y.; Tavassoli, T. High-dose Vitamin B6 supplementation reduces anxiety and strengthens visual surround suppression. Hum Psychopharmacol. [CrossRef]
- Havranek, Т.; Bacova, Z. ; Bakos J. Oxytocin, GABA, and dopamine interplay in autism. Endocrine regulations, 2024; 2. [Google Scholar] [CrossRef]
- Moerkerke, M.; Daniels, N.; Tibermont, L.; et al. Chronic oxytocin administration stimulates the oxytocinergic system in children with autism. Nat Commun. [CrossRef]
- Yingying, Z.; Xiaolu, Z.; Linghong, H. Optimal dose of oxytocin to improve social impairments and repetitive behaviors in autism spectrum disorders: meta-analysis and dose–response meta-analysis of randomized controlled trials. Frontiers in Psychiatry, 2025; 15. [Google Scholar] [CrossRef]
- Beaurain, M.; Salabert, A.-S.; Payoux, P.; Gras, E.; Talmont, F. NMDA Receptors: Distribution, Role, and Insights into Neuropsychiatric Disorders. Pharmaceuticals 2024, 17, 1265. [Google Scholar] [CrossRef]
- Takashi, A.; Koji, K.; Wataru, U.; Christina, A.; Kaito, T. ; S. Yuya. et. al. Reduced neurite density index in the prefrontal cortex of adults with autism assessed using neurite orientation dispersion and density imaging. Frontiers in Neurology. [CrossRef]
- Dionísio, A.; Espírito, A.; Pereira; A.C. et al. Neurochemical differences in core regions of the autistic brain: a multivoxel 1H-MRS study in children. Sci Rep 14, 2024, 2374. [CrossRef]
- Avino, T.; Hutsler.J.J. Supernumerary neurons within the cerebral cortical subplate in autism spectrum disorders. Brain Research. V.1760, 2021, 147350. [CrossRef]
- Sauer, AK.; Hagmeyer, S.; Grabrucker, AM. Prenatal Zinc Deficient Mice as a Model for Autism Spectrum Disorders. Int J Mol Sci. [CrossRef]
- Lee, K.; Mills, Z.; Cheung, P.; Cheyne, JE.; Montgomery, JM. The Role of Zinc and NMDA Receptors in Autism Spectrum Disorders. Pharmaceuticals. [CrossRef]
- Schoen, M.; Asoglu, H.; Bauer, HF. ; Müller, H-P.; Abaei, A.; Sauer, AK.; Zhang, R. et al. Shank3 Transgenic and Prenatal Zinc-Deficient Autism Mouse Models Show Convergent and Individual Alterations of Brain Structures in MRI. Front. Neural Circuits. [CrossRef]
- Ortiz, DMD.; Kim, M.; Lee, HJ.; Botanas, CJ.; Custodio, RJP.; Sayson, LV.; Campomayor, NB.; Lee, C.; Lee, YS.; Cheong, JH.; Kim, HJ. 4-F-PCP, a Novel PCP Analog Ameliorates the Depressive-Like Behavior of Chronic Social Defeat Stress Mice via NMDA Receptor Antagonism. Biomol Ther (Seoul). [CrossRef]
- Dessus-Gilbert, ML. ; Nourredine. M.; Zimmer. L.; Rolland, B.; Geoffray, MM.; Auffret, M.; Jurek, L. NMDA antagonist agents for the treatment of symptoms in autism spectrum disorder: a systematic review and meta-analysis. Front Pharmacol. [CrossRef]
- Ralston, M.; Osman, A.; Suryadevara, P.; Cleland, E. Effect of Ketamine Treatment on Social Withdrawal in Autism and Autism-Like Conditions. Clin Neuropharmacol. [CrossRef]
- Harris, CP.; Jones, B.; Walker, K.; Berry, MS. Case report: Adult with bipolar disorder and autism treated with ketamine assisted psychotherapy. Front. Psychiatry. [CrossRef]
- Kurahashi, H.; Kunisawa, K.; Tanaka, K.F.; et al. Autism spectrum disorder-like behaviors induced by hyper-glutamatergic NMDA receptor signaling through hypo-serotonergic 5-HT1A receptor signaling in the prefrontal cortex in mice exposed to prenatal valproic acid. Neuropsychopharmacol. [CrossRef]
- Huang, X.; Sun, X.; Wang, Q.; Zhang, J.; We, H.; Chen, WJ.; Zhu, S. Structural insights into the diverse actions of magnesium on NMDA receptors, Neuron, 2025. [CrossRef]
- Veer, P.; Akimbekov, NS.; Grant, WB.; Dean, C.; Fang, X.; Razzaque, MS. Neuroprotective effects of magnesium: implications for neuroinflammation and cognitive decline. Frontiers in Endocrinology. [CrossRef]
- Ghanizadeh, A. Targeting of glycine site on NMDA receptor as a possible new strategy for autism treatment. Neurochem Res. [CrossRef]
- Chen, WX.; Chen, YR.; Peng, MZ.; et al. Plasma Amino Acid Profile in Children with Autism Spectrum Disorder in Southern China: Analysis of 110 Cases. J Autism Dev Disord. [CrossRef]
- Randazzo, M.; Prato, A.; Messina, M.; Meli, C.; Casabona, A.; Rizzo, R.; Barone, R. Neuroactive Amino Acid Profile in Autism Spectrum Disorder: Results from a Clinical Sample. Children (Basel). [CrossRef]
- Salceda, R. Glycine neurotransmission: Its role in development. Front Neurosci. [CrossRef]
- Panagiotakos, G.; Pasca, SP. A matter of space and time: Emerging roles of disease-associated proteins in neural development, Neuron,V. 2022; 2. [Google Scholar] [CrossRef]
- Huang, M.; Qi, Q.; Xu, T. Targeting Shank3 deficiency and paresthesia in autism spectrum disorder: A brief review. Front Mol Neurosci. [CrossRef]
- Shen, J.; Liu, L.; Yang, Y.; Zhou, M.; Xu, S.; Zhang, W.; Zhang, C. Insulin-Like Growth Factor 1 Has the Potential to Be Used as a Diagnostic Tool and Treatment Target for Autism Spectrum Disorders. Cureus. [CrossRef]
- Camino-Alarcón, J.; Robles-Bello, MA.; Valencia-Naranjo, N.; Sarhani-Robles, A. A Systematic Review of Treatment for Children with Autism Spectrum Disorder: The Sensory Processing and Sensory Integration Approach. Children (Basel). [CrossRef]
- Cerliani, L.; Mennes, M.; Thomas, RM.; Di Martino, A.; Thioux, M. Keysers, C. Increased functional connectivity between subcortical and cortical resting-state networks in autism spectrum disorder. JAMA Psychiatry. [CrossRef]
- Brummelte, S.; Mc Glanaghy, E.; Bonnin, A.; Oberlander, TF. Developmental changes in serotonin signaling: Implications for early brain function, behavior and adaptation. Neuroscience. [CrossRef]
- D’Adamo, C.R.; Nelson, J.L.; Miller, S.N.; Rickert Hong, M.; Lambert, E.; Tallman Ruhm, H. Reversal of Autism Symptoms among Dizygotic Twins through a Personalized Lifestyle and Environmental Modification Approach: A Case Report and Review of the Literature. J. Pers. Med. 2024, 14, 641. [Google Scholar] [CrossRef]
Figure 1.
Metabolic and signaling pathways involved in the pathogenesis of ASD and their brain locations. 1. Viruses (Rubella, Cytomegalovirus, Herpes Simplex virus, Varicella Zoster Virus, Influenza virus, Zika virus) can damage mitochondrial DNA in the brain during intrauterine development, which may subsequently result in the manifestation of ASD in the postnatal period. 2. A significant number of patients with ASD display iron metabolism disorders, transferrin receptors located in the cortex large neurons, hippocampus and striatum. 3. Serotonin levels elevated in ASD, serotonin receptors and pathways located in the brainstem raphe nucleus, hippocampus, pre and postganglionic autonomic neurons. 4. Oxytocin receptors hypermethylation observed in ASD, located in amygdala and hypothalamus. 5. Anti NMDA-antibodies found in ASD related stereotype behaviors, NMDA receptors are in excitatory synapses, predominantly in the hippocampus. 6. SHANK genes mutations found in ASD phenotypes and lead to NMDA hypofunction, with their expression in prefrontal cortex, brainstem and thalamus, hippocampus, striatum and cerebellum. 7. SLC gene mutations (SLC6A4) have been identified in children with ASD who display mood instability. These mutations affect the function of transporters that facilitate the movement of nutrients, ions, drugs and neurotransmitters to neurons and astrocytes in the brain.
Figure 1.
Metabolic and signaling pathways involved in the pathogenesis of ASD and their brain locations. 1. Viruses (Rubella, Cytomegalovirus, Herpes Simplex virus, Varicella Zoster Virus, Influenza virus, Zika virus) can damage mitochondrial DNA in the brain during intrauterine development, which may subsequently result in the manifestation of ASD in the postnatal period. 2. A significant number of patients with ASD display iron metabolism disorders, transferrin receptors located in the cortex large neurons, hippocampus and striatum. 3. Serotonin levels elevated in ASD, serotonin receptors and pathways located in the brainstem raphe nucleus, hippocampus, pre and postganglionic autonomic neurons. 4. Oxytocin receptors hypermethylation observed in ASD, located in amygdala and hypothalamus. 5. Anti NMDA-antibodies found in ASD related stereotype behaviors, NMDA receptors are in excitatory synapses, predominantly in the hippocampus. 6. SHANK genes mutations found in ASD phenotypes and lead to NMDA hypofunction, with their expression in prefrontal cortex, brainstem and thalamus, hippocampus, striatum and cerebellum. 7. SLC gene mutations (SLC6A4) have been identified in children with ASD who display mood instability. These mutations affect the function of transporters that facilitate the movement of nutrients, ions, drugs and neurotransmitters to neurons and astrocytes in the brain.
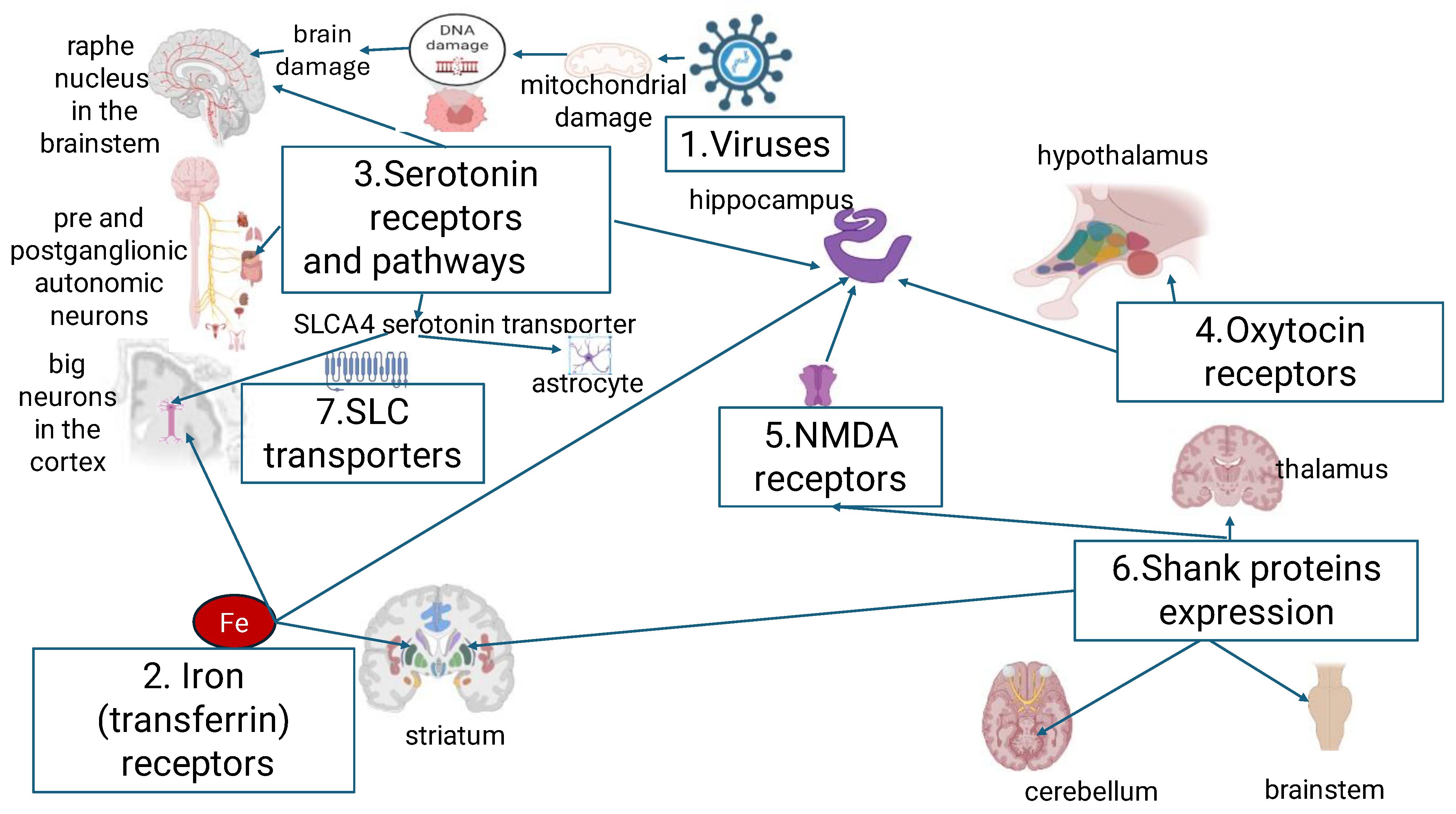
Figure 2.
The role of intrauterine viral CNS damage as a potential for ASD signs development. The Rubella virus has been found to inhibit the enzyme responsible for converting retinol to retinoid acid, thus leading to retinol tissue accumulation. This consequently results in impaired synapse development, block of neuronal differentiation and the potentiation of neurodegeneration. CMV infection results in gene hyperexpression in excitatory and inhibitory neurons, as well as in neuron progenitor cells (NPCs). Concurrently, CMV has been shown to inhibit DYRK kinases, thus potentiating programmed cell death and inhibiting NPCs. The inhibition of the SHH pathway by CMV has been demonstrated to result in the suppression of neuronal growth and the impairment of dopamine synapse pathways, ultimately leading to neurodegeneration. The result of an HSV infection is the increased production of reactive oxygen species (ROS) in microglia and DNA degradation in neurons. The consequence of this is also neuronal degeneration. The influenza virus A (IVA) has been observed to exert an inhibitory effect on the expression of the Apc2, Mdga1, and Usp11 genes. These genes have been identified as causative factors for neuronal cortical lamination, radial neuron migration, and cortical neurogenesis, respectively. IVA infection also results in altered microglia density. The CoVid-19 virus has been observed to decrease IGF-1 levels, which are regulated by IGF-R/IRS1/PI3K/AKT/mTOR pathway. This results in impaired myelinization. Potential biomarkers marked with “!”.
Figure 2.
The role of intrauterine viral CNS damage as a potential for ASD signs development. The Rubella virus has been found to inhibit the enzyme responsible for converting retinol to retinoid acid, thus leading to retinol tissue accumulation. This consequently results in impaired synapse development, block of neuronal differentiation and the potentiation of neurodegeneration. CMV infection results in gene hyperexpression in excitatory and inhibitory neurons, as well as in neuron progenitor cells (NPCs). Concurrently, CMV has been shown to inhibit DYRK kinases, thus potentiating programmed cell death and inhibiting NPCs. The inhibition of the SHH pathway by CMV has been demonstrated to result in the suppression of neuronal growth and the impairment of dopamine synapse pathways, ultimately leading to neurodegeneration. The result of an HSV infection is the increased production of reactive oxygen species (ROS) in microglia and DNA degradation in neurons. The consequence of this is also neuronal degeneration. The influenza virus A (IVA) has been observed to exert an inhibitory effect on the expression of the Apc2, Mdga1, and Usp11 genes. These genes have been identified as causative factors for neuronal cortical lamination, radial neuron migration, and cortical neurogenesis, respectively. IVA infection also results in altered microglia density. The CoVid-19 virus has been observed to decrease IGF-1 levels, which are regulated by IGF-R/IRS1/PI3K/AKT/mTOR pathway. This results in impaired myelinization. Potential biomarkers marked with “!”.
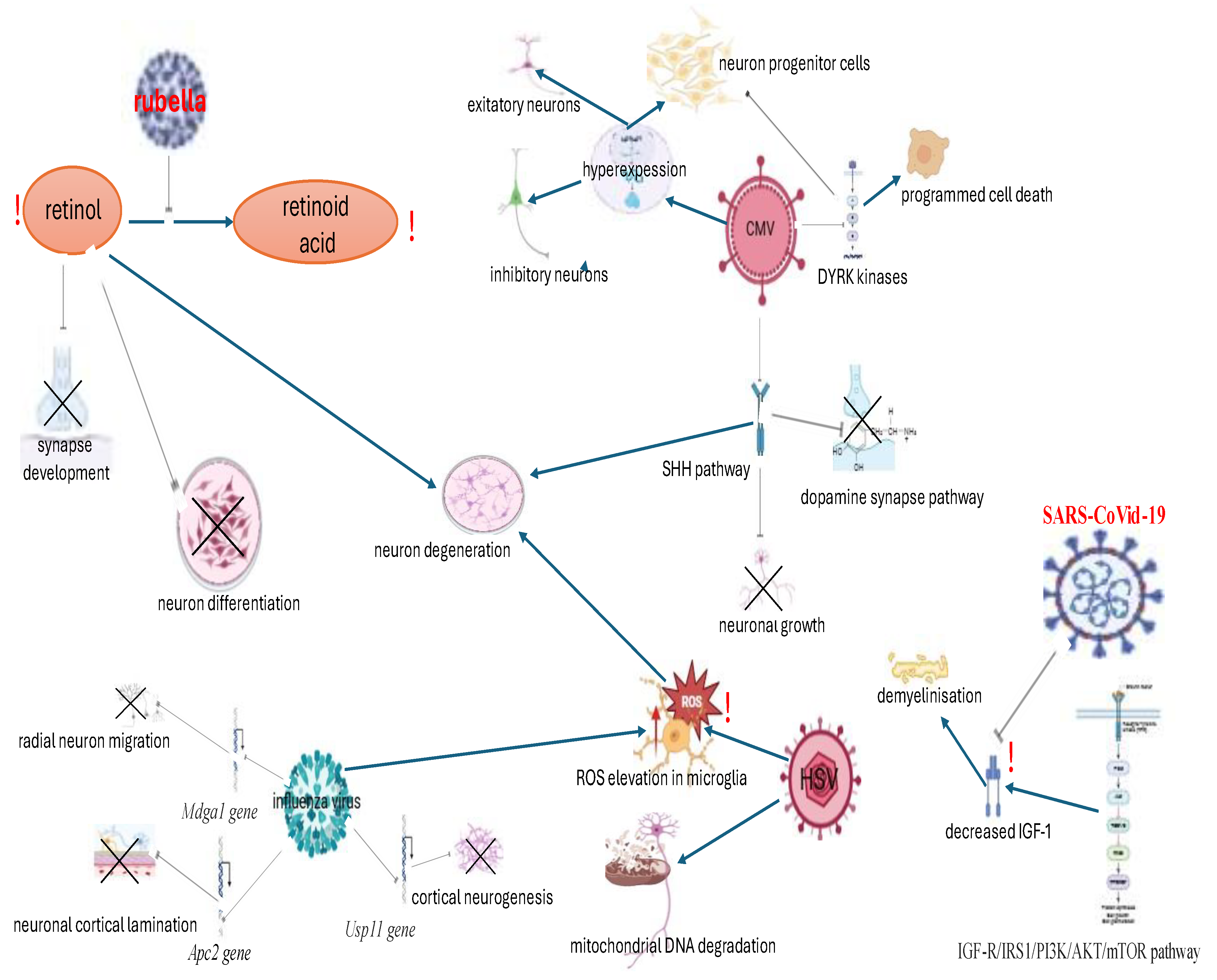
Figure 3.
Dysregulation of ferroptosis and iron metabolism as a potential contributing factor in ASD associated signs. It is hypothesised that the presence of neuronal immaturity in ASD may serve as a triggering factor for an immune reaction with subsequent activation of intracellular lipid peroxidation. On the one hand, ferroptosis instigates Fe²⁺-dependent lipid peroxidation and activates the ferritin autophagy pathways. This, in turn, results in the process of ferritinophagy and decreased ferritin levels. On the other hand, ferroptosis has been demonstrated to induce the expression of Fbxw7, MafG, AKR1C2, RB1, TMBIM4, ALB, CS, and ARNTL genes. This process has been shown to promote a number of cellular processes, including apoptosis, cell growth, cell migration, colloid oncotic pressure, zinc transmembrane transport, the citric acid cycle, and circadian rhythm regulation, as well as Th-17 cell activation with mucosal proinflammatory response. Simultaneously, active ferroptosis could reduce transferrin levels, thereby impaired neuronal differentiation. Potential biomarkers marked with “!”.
Figure 3.
Dysregulation of ferroptosis and iron metabolism as a potential contributing factor in ASD associated signs. It is hypothesised that the presence of neuronal immaturity in ASD may serve as a triggering factor for an immune reaction with subsequent activation of intracellular lipid peroxidation. On the one hand, ferroptosis instigates Fe²⁺-dependent lipid peroxidation and activates the ferritin autophagy pathways. This, in turn, results in the process of ferritinophagy and decreased ferritin levels. On the other hand, ferroptosis has been demonstrated to induce the expression of Fbxw7, MafG, AKR1C2, RB1, TMBIM4, ALB, CS, and ARNTL genes. This process has been shown to promote a number of cellular processes, including apoptosis, cell growth, cell migration, colloid oncotic pressure, zinc transmembrane transport, the citric acid cycle, and circadian rhythm regulation, as well as Th-17 cell activation with mucosal proinflammatory response. Simultaneously, active ferroptosis could reduce transferrin levels, thereby impaired neuronal differentiation. Potential biomarkers marked with “!”.
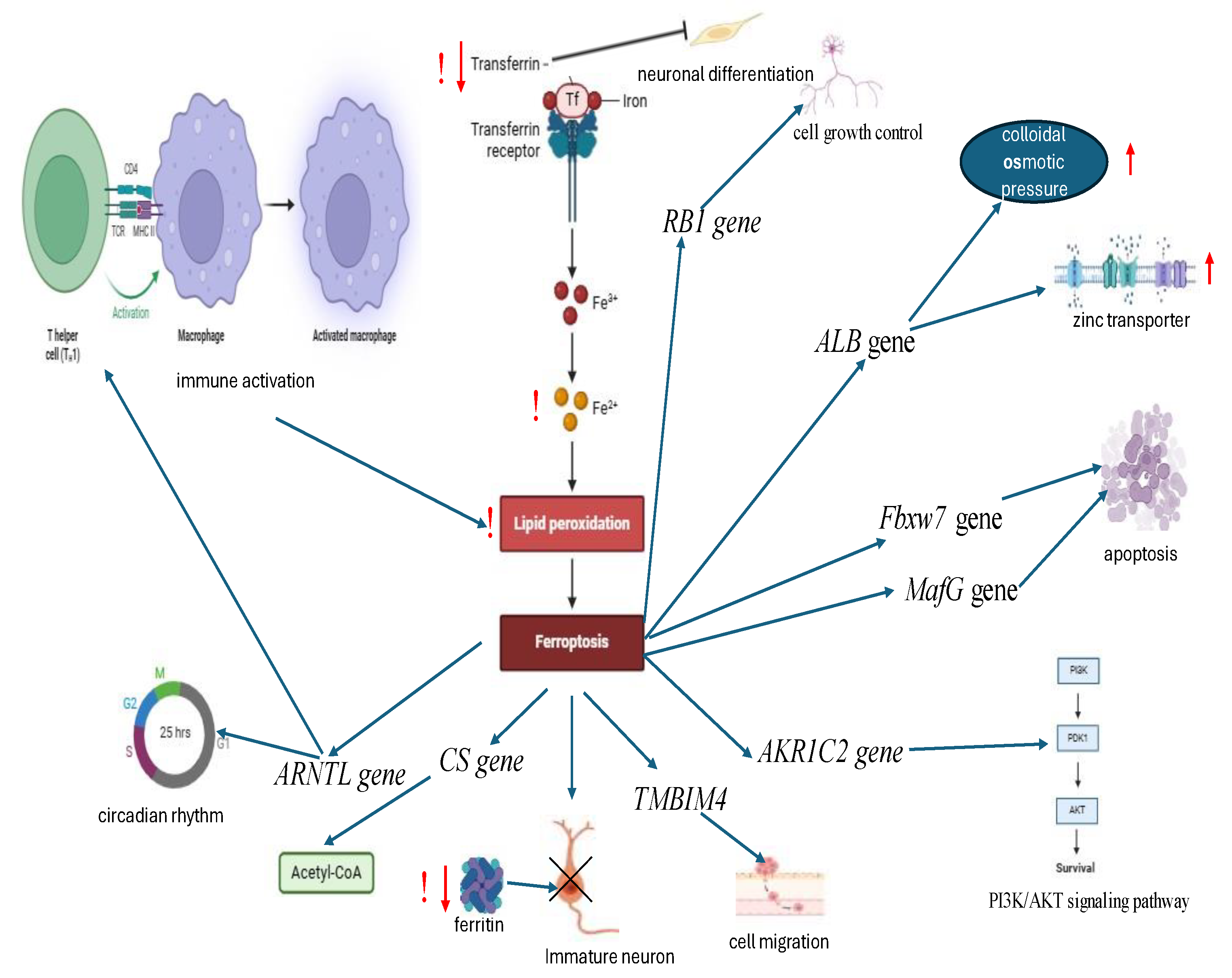
Figure 4.
Dysregulation of serotonin pathways in ASD. In ASD the variations in the serotonin transporter gene (SLC6A4) have been demonstrated to result in impaired neuronal metabolism, decreased excitability and reduced brain serotonin clearance. Consequently, the changings in serotonin level can give rise to a state of dysregulation of 5HT2A receptors, located in the frontal and parietal cortices, in relation to inhibition of dopamine and neurohypophyseal hormones secretion in the hippocampus, and inhibition of 5HT1A receptors located in the striatum. A decrease in tryptophan levels, a precursor of serotonin, has been demonstrated to result in diminished expression of genes within the tryptophan pathway. This phenomenon has been shown to exert an influence on the enzymes involved in serotonin clearance, neuroprotection, and N-methyl-D-aspartate (NMDA) receptor activation. The observed changes are concomitant with the elevation of serum BDNF levels. The following provides a synopsis of signs that are associated with ASD. Impaired SERT activation could trigger a cascade of serotonin pathway dysregulation, causing a vicious circle and the progression of ASD symptoms. Potential biomarkers marked with “!”.
Figure 4.
Dysregulation of serotonin pathways in ASD. In ASD the variations in the serotonin transporter gene (SLC6A4) have been demonstrated to result in impaired neuronal metabolism, decreased excitability and reduced brain serotonin clearance. Consequently, the changings in serotonin level can give rise to a state of dysregulation of 5HT2A receptors, located in the frontal and parietal cortices, in relation to inhibition of dopamine and neurohypophyseal hormones secretion in the hippocampus, and inhibition of 5HT1A receptors located in the striatum. A decrease in tryptophan levels, a precursor of serotonin, has been demonstrated to result in diminished expression of genes within the tryptophan pathway. This phenomenon has been shown to exert an influence on the enzymes involved in serotonin clearance, neuroprotection, and N-methyl-D-aspartate (NMDA) receptor activation. The observed changes are concomitant with the elevation of serum BDNF levels. The following provides a synopsis of signs that are associated with ASD. Impaired SERT activation could trigger a cascade of serotonin pathway dysregulation, causing a vicious circle and the progression of ASD symptoms. Potential biomarkers marked with “!”.
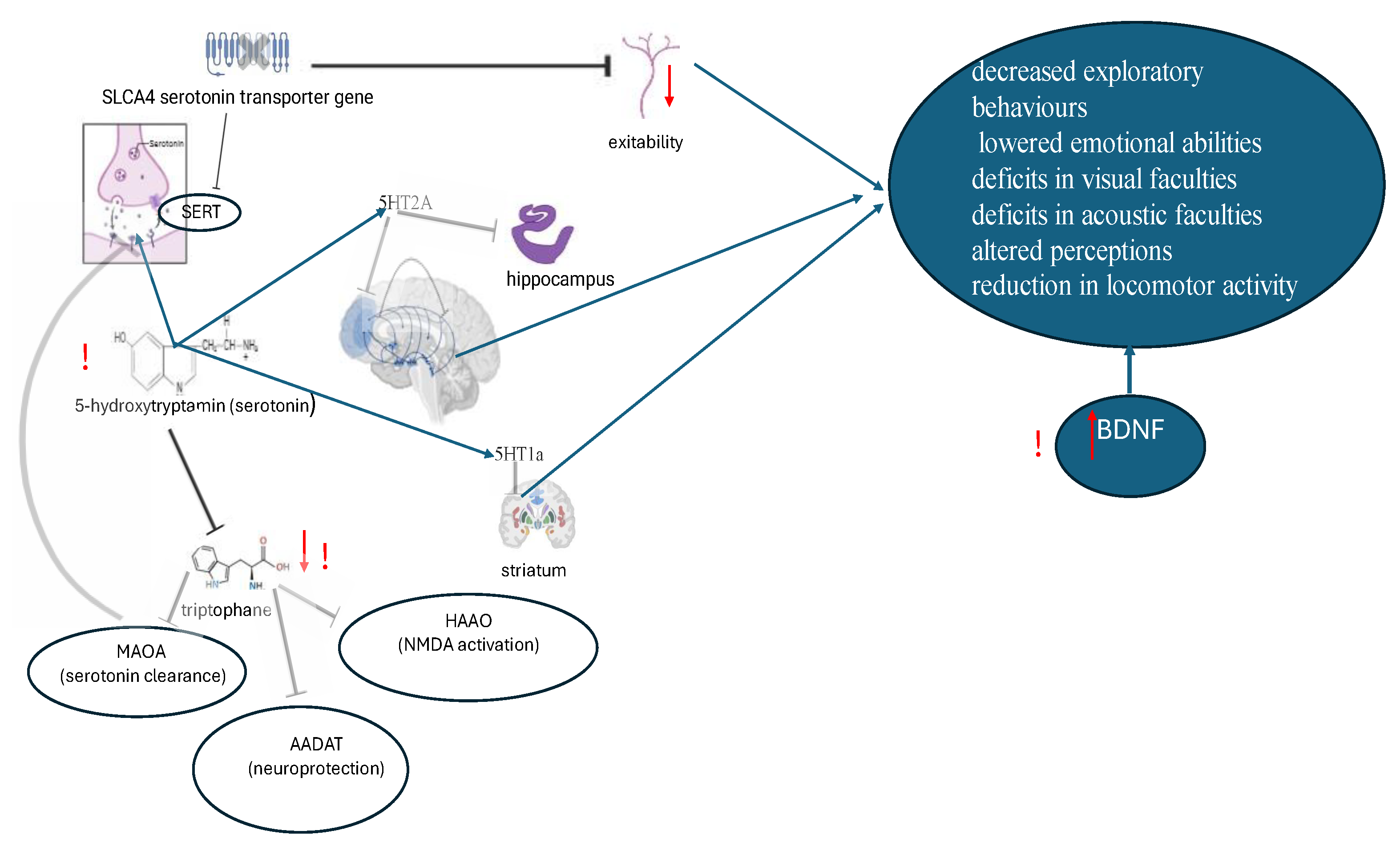
Figure 5.
Oxytocin pathways alterations in ASD. The hypothesis (denoted with "?") suggests that folic acid cycle gene mutations may result in impaired methylation and alterations to the OXTR- oxytocin pathway, as well as folate deficiency affects serotonin and dopamine production. Methylation of the OXTR gene has been observed to result in reduced expression and heightened binding levels of the OXTR in the nucleus basalis with diminished expression levels in the ventral pallidum. Diminished serotonin has the potential to modify the expression of both serotonin and dopamine, thereby inducing. This could result in induced disorganization within the domains of visual attention and motivated behaviors processing, a phenomenon that has been observed in individuals diagnosed with ASD. Potential biomarkers marked with “!”.
Figure 5.
Oxytocin pathways alterations in ASD. The hypothesis (denoted with "?") suggests that folic acid cycle gene mutations may result in impaired methylation and alterations to the OXTR- oxytocin pathway, as well as folate deficiency affects serotonin and dopamine production. Methylation of the OXTR gene has been observed to result in reduced expression and heightened binding levels of the OXTR in the nucleus basalis with diminished expression levels in the ventral pallidum. Diminished serotonin has the potential to modify the expression of both serotonin and dopamine, thereby inducing. This could result in induced disorganization within the domains of visual attention and motivated behaviors processing, a phenomenon that has been observed in individuals diagnosed with ASD. Potential biomarkers marked with “!”.
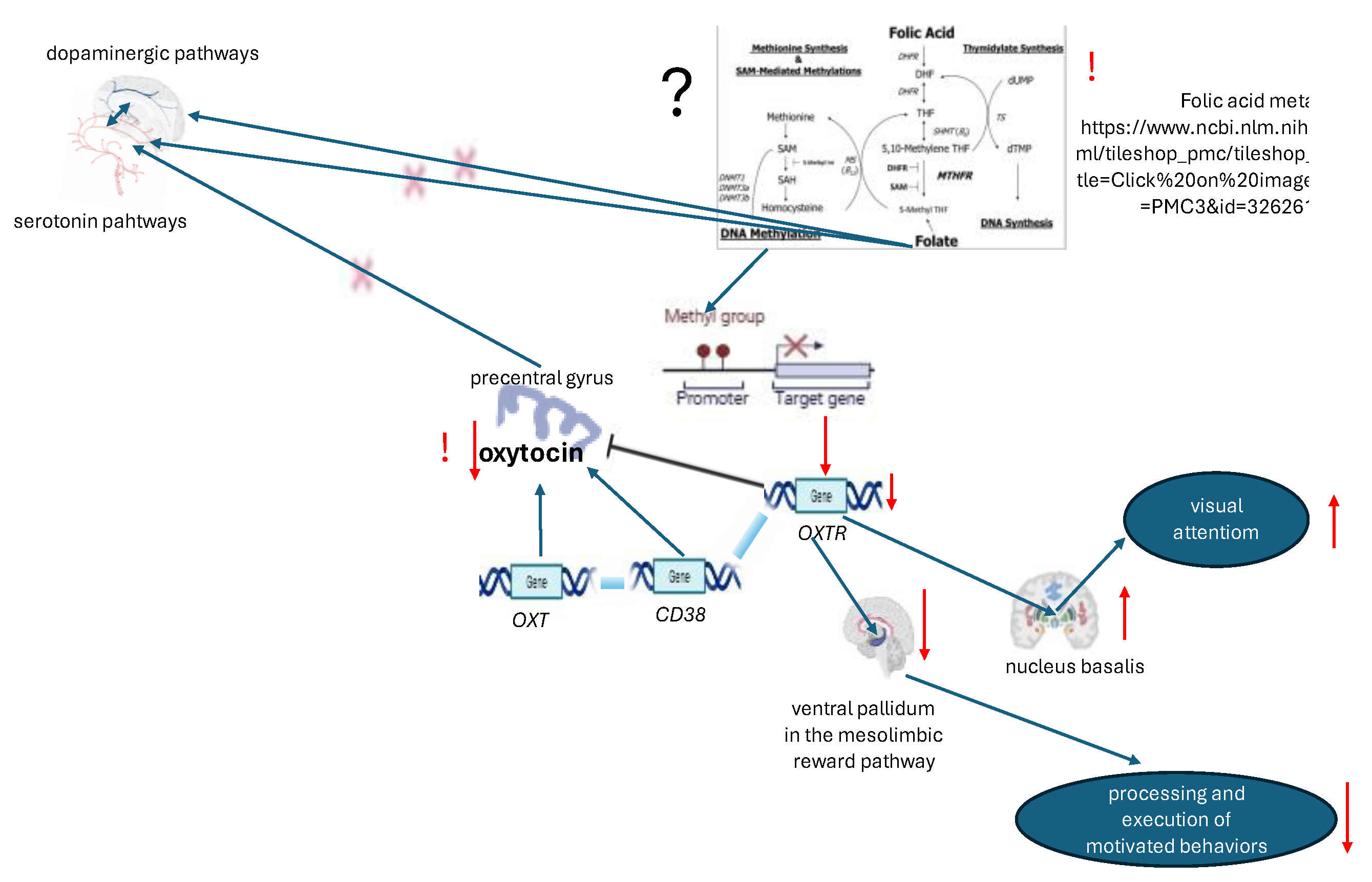

Table 1.
Prevailed ASD signs, suggested biomarkers and signs associated combined modulatory substances.
Table 1.
Prevailed ASD signs, suggested biomarkers and signs associated combined modulatory substances.
| Prevailed ASD signs | Suggested biomarkers | Suggested combined modulation |
| History of prenatal viral exposure, DSM -5 relevant signs of DA deficit (unusual mood or emotional reactions, anxiety, stress, or excessive worry) | Retinoid status, dopamine, IgG antibodies to DA1 and DA2 receptors, BDNF, IgF-1, NSE, oxidative stress markers (malonic dialdehyde) | Folinic acid (leucovorin or L-methylfolate) or all-trans retinoic acid, antioxidants (glutation +Vit C + NAC), IgF-1 derived neurotrophins (mecasermine, trofinetid) |
| Parasomnias, restless legs syndrome, delayed language skills, DA deficit | Ferritin, transferrin, dopamine, IgG antibodies to DA1 and DA2 receptors, oxidative stress markers, Vit B 6 levels, NSE, HSP | DA agonists, antioxidants, Vit B6 |
| Negative DSM -5 relevant signs (avoids or does not keep eye contact, does not respond to name by 9 months of age, does not show facial expressions such as happy, sad, angry, and surprised by 9 months of age) | Serotonin, BDNF, IgF-1, tryptophan, folates, Vit B 6 levels | SSRI, folinic acid, VitB6 |
| DSM-5 relevant repetitive signs (focusing on parts of objects, obsessive interests, extreme interest or knowledge of specific, narrow topics, strong attachment to a certain object) | Oxytocin, folic circle genes, folates, vit B 6 levels. | Intranasal oxytocin, folates, vit B6, neurotrophins. |
| DSM-5 relevant cognitive signs (delayed cognitive or learning skills, hyperactive, impulsive, and/or inattentive behavior) | Plasma glutamate, magnesium, zinc, NSE, BDNF, IgF-1. | NMDARs antagonists, glycine, magnesium-containing drugs, zinc-containing drug |
Notes: SSRI- selective serotonin reuptake inhibitors.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



