Submitted:
28 March 2025
Posted:
31 March 2025
You are already at the latest version
Abstract
Phytoplasmas of the X-disease group (16SrIII) are economically significant pathogens in South America, causing severe crop losses. Traditional classification based on the 16S rRNA gene has limitations in resolving closely related strains, prompting the exploration of alternative markers. This study focuses on the immunodominant membrane proteins imp and idpA, which exhibit high variability and play crucial roles in host-pathogen interactions. Through molecular characterization of imp and idpA genes in 16SrIII subgroups, we identified significant genetic diversity and distinct evolutionary pressures. The imp gene, under positive selection, showed high variability in its hydrophilic extracellular domain, suggesting adaptation to host immune responses. In contrast, idpA exhibited strong negative selection, indicating functional conservation. Phylogenetic analyses revealed that imp and idpA provide higher resolution than the 16S rRNA gene, enabling finer differentiation within subgroups. These findings highlight the potential of imp and idpA as complementary markers for phytoplasma classification and diagnostics.
Keywords:
membrane proteins
; X-disease
; phylogeny
; selection
; imp
; idpA
1. Introduction
Phytoplasmas are cell wall-less bacteria with an obligate parasitic lifestyle, associated with diseases in hundreds of plant species [1,2]. These pathogens are transmitted between plants by sap-sucking insect vectors, including leafhoppers, planthoppers, and psyllids [3]. The classification of phytoplasmas has traditionally relied on sequence analysis of the 16S rRNA gene, with a sequence identity threshold of 98.65% used to delineate species [4]. Established in the 1990s, this system further employs restriction fragment length polymorphism (RFLP) analysis of a 1.25 kb fragment of the 16S rRNA gene to define ribosomal (16Sr) groups and subgroups, resulting in the classification of more than 37 groups and over 150 subgroups to date [2,5,6]. Despite its widespread adoption, the 16Sr classification system has several limitations. These include its reliance on variability at restriction sites rather than the full sequence, intragenomic heterogeneity of 16S rRNA genes, and insufficient resolution for inferring phylogenetic relationships [7,8,9]. To address these challenges, supplementary analyses using protein-coding genes and multilocus sequence typing (MLST) have been introduced as more robust alternatives. MLST, which leverages multiple single-copy markers, provides enhanced resolution for distinguishing closely related strains. For example, studies have successfully differentiated phytoplasma strains using genes such as secY, rp, and leuS in combination with the 16S rRNA gene [10,11,12]. The development of genome-enabled multilocus sequence analysis (MLSA) markers has further advanced phytoplasma classification. By leveraging available genome sequences, researchers can design primers and evaluate marker performance through genome-scale phylogenies [13]. This approach has been particularly effective in well-studied groups such as 16SrI, which benefit from extensive genomic data [14]. As sequencing technology continues to advance, a transition toward whole genome-based taxonomy is anticipated. Metrics such as average nucleotide identity (ANI) are expected to play a key role in defining species boundaries, offering a more reliable alternative to the traditional 16S rRNA gene-based approach [15].
Phytoplasmas of the 16SrIII group are agriculturally significant in South America, infecting a wide range of plants, including weeds, vegetables, fruit trees, and staple crops [16,17,18]. The 16SrIII-J subgroup is particularly relevant in South America, associated with X-disease in Argentina, Southern Brazil, and Chile, causing significant crop losses [16,19,20,21,22,23]. Another important subgroup is 16SrIII-B, found in a variety of hosts such as China tree, peach, plum, tomato, and cassava, in Argentina, Brazil, and Paraguay [24,25,26]. Other subgroups within 16SrIII include 16SrIII-W, associated with Heterothalamus alienus causing leaf reduction, and 16SrIII-X, found in Erigeron bonariensis and lettuce, showing witch's broom and malformed flowers [16,27]. Additionally, 16SrIII-L has been linked to frogskin disease in cassava, where symptoms primarily affect tubers [28]. The diversity of these subgroups, including 16SrIII-B, 16SrIII-J, and 16SrIII-X, highlights their adaptability to various hosts and environments, posing ongoing challenges for disease management. Their widespread distribution and economic impact in South America emphasize the need for continued research and improved diagnostic and management strategies. Immunodominant membrane proteins (IDPs) of phytoplasmas, such as Imp, IdpA, and Amp, are highly abundant proteins located on the outer membrane, interacting directly with plant and insect hosts. These proteins play a key role in host-pathogen interactions, including adhesion to insect vectors and plant colonization [29]. IDPs exhibit high sequence variability due to positive selection, reflecting their adaptation to evading host immune responses and optimizing transmission [30,31]. This variability, combined with their membrane localization and abundance, makes them ideal candidates for molecular markers of diversity. Re-combinant expression of IDPs can generate specific antibodies, enabling their detection and characterization across phytoplasma strains [31,32]. Therefore, IDPs are essential for understanding phytoplasma biology and are valuable tools for strain differentiation and epidemiological studies. Given the agricultural significance of the X-disease group and the need for more precise molecular markers, this work aims to molecularly characterize these membrane proteins in phytoplasmas of the X-disease group (subgroups 16SrIII-J, 16SrIII-B, and 16SrIII-X) in Argentina and their association with various plant hosts.
2. Materials and Methods
2.1. Plant samples
Total DNA from eleven isolates previously reported in Argentina and belonging to the X-disease group (subgroups 16SrIII-J, 16SrIII-B, and 16SrIII-X) were used for molecular assays (Table 1). DNA was isolated from leaf midribs according to the Doyle and Doyle [33] CTAB protocol. Detection and classification of phytoplasmas was performed by direct and nested PCR amplification of 16S rRNA using the primer pairs P1/P7 [34] and R16F2n/R16R2 [35], respectively. RFLP profiles of nested amplicons were analyzed as described in previous works [16,22].
2.2. Identification of imp and idpA homologues
The coding sequences for the imp and idpA genes were identified in published phytoplasma genomes belonging to the X-disease group (16SrIII) available in the NCBI database (NCBI: txid85623) (Table 2). To identify these genes, BLASTp searches were conducted using the imp (AP314487.1) and idpA (AP31480.1) sequences from the WX phytoplasma as queries. For phytoplasmas lacking annotations in the NCBI database, functional annotations were performed using Prokka [36]. Subsequently, BLASTp searches were carried out using Geneious R.10 software (Biomatters Ltd., Auckland, New Zealand) to identify homologous sequences.
2.3. Cloning and PCR amplification of imp and idpA genes
Specific PCR primers were designed to clone the complete coding sequences of the imp and IdpA proteins. The genomic context of each gene (imp and idpA) was manually examined, and conserved regions across all X-disease genomes were identified for primer design. The final primer sets were validated using Primer3 2.3.7 within Geneious R.10 software. PCR amplification of genomic fragments was performed in a 40 µL reaction volume containing 100 ng of DNA, 0.4 mM of each primer, 200 µM of each dNTP, 1 U of GoTaq® DNA polymerase, 1X polymerase buffer (Promega, USA), and sterile water. The thermal cycling conditions for imp and idpA genes were as follows: an initial denaturation at 94 °C for 3 minutes, followed by 35 cycles of 94 °C for 1 minute, 54 or 58 °C for 1 minute (for imp or idpA, respectively), and 72 °C for 1 minute 30 seconds, with a final extension at 72 °C for 8 minutes. PCR amplicons were visualized by electrophoresis on 1% agarose gels stained with GelRed® and imaged under UV light.
2.4. Sequencing of imp and idpA genes
PCR amplicons were purified using commercial columns (PBL, Argentina) and cloned into pGEM®-T Easy Vector System I (Promega, USA) following the manufacturer’s recommendations. E. coli DH5α competent cells were used for transformation. For each sample, three clones were bidirectionally sequenced (3X minimum coverage per base) using an automated Sanger DNA sequencer service (Macrogen, Korea). Consensus sequences were assembled using Geneious R.10 software and deposited in the NCBI GenBank. Open reading frames were estimated using ORF Finder and annotated using BLASTp (nr, BLOSUM62, word size 6). The deduced amino acid sequences of Imp and IdpA proteins were analyzed using SignalP 5.0 [37] for prediction of signal peptide sequences. Transmembrane helix domains were predicted using TMHMM v2.0 [38].
2.5. Genetic diversity
The nucleotide and amino acid identity of the sequences were calculated using MAFFT v7.505 [39]. The number of polymorphic sites and nucleotide diversity (Pi, Jukes and Cantor) were evaluated using DnaSP v6.0 [40]. For the target genes (imp and idpA), synonymous (dS) and non-synonymous (dN) nucleotide substitution values were calculated using the Nei-Gojobori (Jukes-Cantor) method implemented in MEGA v7.0 [41]. Codon-based Z-Test of Neutrality was used to reject the null hypothesis of strict neutrality (dN = dS). For positive selection, dN-dS values >1 and p-value < 0.05 were considered significant [42]. Maximum Likelihood analysis of natural selection codon-by-codon was conducted using the HyPhy software package [43] implemented in MEGA v7.0.
2.6. Phylogenetic analysis of X-disease phytoplasmas
Phylogenetic relationships among X-disease phytoplasmas were evaluated using the nucleotide sequences of the imp, idpA, and 16S rRNA genes from nine X-disease phytoplasma genomes available on NCBI (Table 2), along with six isolates sequenced in this study. Multiple sequence alignments were performed using MAFFT (L-ins-I; k=2). Phylogenetic trees were constructed with IQ-TREE [44] using an automatic substitution model and an ultrafast bootstrap analysis with 1000 replicates.
3. Results
3.1. Identification of imp and idpA ORFs in X-disease genomes
In this analysis, the sequences of nine genomes described in Table 2 were used. Of the nine phytoplasma genomes analyzed, two belong to subgroup 16SrIII-J, two to subgroup 16SrIII-B, two to subgroup 16SrIII-F, and three to subgroup 16SrIII-A (Ca. Phytoplasma pruni). The complete sequences of the imp and idpA genes were identified in all isolates.
3.2. PCR amplifications and sequencing
For the imp gene, a primer pair (imp-Fw: 5'-ATCTCGTCCTCTTAAACCGCATCC-3'; imp-Rv: 5'-AGACTCTTAACTGGCAACG-3') was designed to amplify a ~1.0 kb fragment covering the complete ORF of the imp protein. Similarly, for the idpA gene, a primer pair (idpA-Fw: 5'-CCCTTCTGCTCCGCCAATTA -3'; idpA-Rv: 5'- TTGCCGAGCAAAAGAGCAAT -3') was designed to amplify a ~1.4 kb fragment encompassing the entire ORF of the idpA protein. PCR amplification of 1.0 kb (imp gene) and 1.4 kb (idpA gene) was successfully achieved in eight out of eleven strains, respectively, as shown in Table 1. No amplification was observed in the negative controls (control mix and healthy samples). The complete coding sequences for the imp and idpA proteins were obtained from the FooBeetWY, GDIII, BellVir, CicWB (subgroup 16SrIII-J), CaesLL (subgroup 16SrIII-B), BidPhy and LWB (subgroup 16SrIII-X) strains. For ChTDIII strain (subgroup 16SrIII-B) isolate, although amplicons were obtained for both genes, the sequences obtained from the genome were used for analysis. The size of the coding region of the imp and idpA genes analyzed in this work falls within the expected range for these genes according to previous reports [45]. For the imp gene, we found sizes ranged from 522 bp (ChTDIII, 16SrIII-B) to 546 bp (BidPhy/LWB, both from 16SrIII-X), while for the idpA gene, sizes varied from 522 bp (ChTDIII, 16SrIII-B) to 546 bp (BidPhy/LWB, both from 16SrIII-X).
3.3. Sequences homology and predicted protein structure
The analysis of amino acid sequences from all strains sequenced in this study, as well as those retrieved from NCBI, reveals a conserved domain structure consistent with previous findings for the imp [31,46] and IdpA protein [47,48]. In the case of imp, a typical hydrophilic C-terminal domain was observed in all sequences, while the N-terminal region encoded a hydrophobic domain (putative transmembrane helix) of approximately 40 amino acids, which likely serve as an anchor to the phytoplasma cell membrane (Figure 1). We did not infer the presence of a signal peptide or putative cleavage motif in any of the analyzed sequences. Regarding similarity, we found 100% identity values among all isolates of subgroup 16SrIII-J, except for the Vc33 isolate, which showed a value of 97.73% compared to the other members of the subgroup. On the other hand, isolates from subgroup 16SrIII-A exhibited amino acid identity values ranging from 97.73% to 91.57%, while those from 16SrIII-F had an identity of 90.29%. In 16SrIII-B, the phytoplasmas ChTDIII and CaesLL exhibited identity values of 71.84% with each other. However, when compared to the MA1 isolate, the identity value dropped to 51.98% for ChTDIII and 55.93% for CaesLL. For the sequences of 16SrIII-X, the identity values ranged from 54.4% compared to the VAC isolate to 44.75% compared to the CaesLL isolate (Supplementary Figure S1). The overall homology was lower in the exposed hydrophilic region, with a pairwise % identity value of 61.50%, compared to the overall sequence with a value of 66.40%.
For all analyzed strains, the structure of IdpA protein showed a large central hydrophilic region flanked by two hydrophobic regions (putative transmembrane helices) near the C- and N-terminus (Figure 2). A signal peptide (35 aa) was inferred in all sequences, while no putative cleavage motif was identified. Regarding similarity, we found that the identity values among all isolates of 16SrIII-J varied between 100% and 87.04% (Vc33 vs all). In subgroup 16SrIII-A, IdpA identity values ranged from 100% (JR-1 vs PR2021) to 63.83% (PR2021 vs CX), while within 16SrIII-B, these values ranged from 86.77% (ChTDIII vs GDIII) to 67.48% (CaesLL vs MA-1). For 16SrIII-F, the identity was 81.71% (MW-1 vs MA-1), and for 16SrIII-X, it was 100% (BidPhy vs LWB) (Supplementary Figure S1). Like the findings for Imp, the global homology was lower in the exposed hydrophilic region, with a pairwise % identity value of 68.69%, compared to the overall sequence with a value of 73.20%.
3.3. Genetic diversity
In this study, sixteen sequences of the imp protein coding genes were used for the selection pressure analysis. Multiple alignments of 471 positions (157 codons) were evaluated and dN-dS was calculated for each codon. Eighty codons showed dN-dS>0 values, indicating that the protein would be under positive selection pressure (dN-dS general= 3.474 p=0.01) , of which seventy were found to encode for amino acids in the hydrophilic region exposed. The same analysis was performed with sixteen sequences of the gene encoding for the idpA protein, where two hundred fifty-three codons (253) one hundred and fifteen (115) showed dN-dS>0 values (dN-dS general= -3.090 p=0.002) , demonstrating that this protein would be under negative selection pressure. Within these two hundred fifty-three codons (253), one hundred and four (104) encode for amino acids in the hydrophilic region exposed. The results of these analyses determined that the highest number of codons with dN-dS values > 0 occurred in the extracellular region [Table 3], indicating that in both proteins this domain (hydrophilic) is the most variable.
3.4. Phylogeny based on 16S rRNA, idpA and imp genes
The phylogenetic analysis based on 16S rRNA, idpA, and imp genes provides complementary insights into the evolutionary relationships among 16SrIII phytoplasma subgroups. The 16S rRNA tree establishes a well-defined taxonomic framework, with 16SrIII-J isolates (BellVir, CiWB, Vc33, FdWY, GDIII) forming a strongly supported monophyletic group (bootstrap = 94-100%) , clearly distinct from 16SrIII-B (ChTDIII, CaesLL, MA1). Similarly, 16SrIII-A/S (CX, JR1, PR2021/WX) clusters with strong support (97%), while 16SrIII-F (MW1, VAC1) and 16SrIII-X (BidPhy, LWB) are more phylogenetically distinct (Figure 3). However, while the 16S rRNA gene is highly conserved and effective for broad classification, it lacks the resolution to detect functional differentiation or evolutionary adaptation [9]. Notably, the 16SrIII-B and 16SrIII-F subgroups show a degree of association, but their placement remains separate.
The idpA gene phylogeny introduces additional differentiation within subgroups. While 16SrIII-J members remain clustered (bootstrap = 95-100%), Vc33 is more divergent, suggesting increased variation in idpA compared to the highly conserved 16S sequences (Figure 4.A). The 16SrIII-B subgroup is less cohesive in idpA, with MA-1 appearing more distantly related to ChTDIII and CaesLL. Additionally, 16SrIII-F (MW-1, VAC-1) now clusters more closely with 16SrIII-B (MA-1), reinforcing a pattern not evident in the 16S-based tree. The 16SrIII-A/s sungroups retains its structure (bootstrap = 100%) but shows an increased association with 16SrIII-F. The 16SrIII-X isolates (BidPhy and LWB) remain the most divergent. Overall, idpA phylogeny provides higher resolution than 16S rRNA, revealing subgroup differentiation and evolutionary interactions. The imp gene phylogeny exhibits the highest degree of sequence divergence, indicating distinct evolutionary pressures acting on this locus (Figure 4.B). 16SrIII-J members remain partially clustered but show a looser relationship compared to the previous trees, with Vc33 displaying significant divergence. The 16SrIII-B subgroup (ChTDIII, CaesLL, MA-1) is even more fragmented, with CaesLL and ChTDIII forming a strongly supported clade (bootstrap = 100%), yet at a considerable evolutionary distance from other subgroups. Interestingly, MW-1 and VAC-1 (16SrIII-F) remain associated but are now positioned closer to 16SrIII-B, reinforcing a trend seen in idpA. The 16SrIII-A/S subgroup remains well-defined (bootstrap = 100%) but appears more closely linked to 16SrIII-F, suggesting possible functional adaptation. Finally, BidPhy and LWB (16SrIII-X) exhibits extreme divergence, consistent with its distinct evolutionary trajectory. Overall, the imp phylogeny highlights strong functional divergence across subgroups, suggesting it is more influenced by selective pressures than either idpA or 16S rRNA. Together, these three markers provide a comprehensive view of phytoplasma evolution, with 16S rRNA clarifying taxonomy, idpA refining subgroup differentiation, and imp revealing a puative adaptive divergence.
4. Discussion
Phytoplasmas belonging to the X-disease group (16SrIII) exhibit remarkable genetic diversity and are widely distributed across South America, affecting multiple plant species [16,17,18,22]. In Argentina, several subgroups within this group have been reported, including 16SrIII-B, J, X, and W [16,22]. Traditionally, diversity within this group has been assessed using the 16S rRNA gene; however, this approach has limitations such as incomplete sequence coverage, low phylogenetic resolution, and intragenic variability [9]. To overcome these constraints, alternative phylogenetic gene markers like tuf, secA, and secY have been proposed [49,50]. In this context, genes encoding immunodominant membrane proteins, such as idpA (Immunodominant Protein A) and imp (Immunodominant Membrane Protein), offer promising alternatives for resolving phytoplasma diversity at a finer scale.
In the present study, we analyzed the genetic variability of the imp and idpA genes in phytoplasmas belonging to the 16SrIII group. Homologous sequences retrieved from the NCBI database facilitated the design of primers for the successful amplification of genomic fragments. Diversity analysis revealed that imp and idpA provide higher phylogenetic resolution than the 16S rRNA gene, with imp enabling a better classification at the subgroup level, while idpA discriminates isolates within the same subgroup. This suggests that these genes could serve as complementary markers for phytoplasma classification.
Selection pressure analyses indicated that the imp gene is under positive selection, with a general dN/dS value of 3.474 (p=0.01) [42], suggesting adaptive evolution. Most positively selected sites were in the hydrophilic C-terminal region, which is exposed to the host environment and may be involved in host-pathogen interactions. This aligns with previous findings indicating that imp is highly variable among phytoplasmas, a characteristic often linked to its role in host adaptation and immune evasion [31]. Structural studies further support this hypothesis, showing that Imp functions as an F-actin-binding protein, potentially influencing host cytoskeletal dynamics and facilitating infection [51]. The selective pressure on its extracellular domain reinforces the idea that imp plays a role in host recognition and transmission efficiency. Given its high variability, exposure to the host immune system, and functional relevance, imp emerges not only as a valuable phylogenetic marker [52] but also as a key determinant of phytoplasma pathogenicity.
In contrast, the idpA gene exhibited strong negative selection pressure (dN-dS = -3.090, p=0.002) [42], with 115 out of 253 codons showing dN-dS>0 values, predominantly in the hydrophilic extracellular region. This suggests that idpA is highly conserved, likely due to functional constraints essential for phytoplasma survival. Unlike imp, which undergoes strong positive selection and exhibits high variability, idpA appears to be under purifying selection, preserving its structure and function across different strains. Previous studies have shown that idpA expression levels vary across phytoplasma species. For example, in Western X-disease phytoplasma (Ca. Phytoplasma pruni, subgroup III-S), IdpA has been identified as the major immunodominant membrane protein [47], whereas in PoiBI Phytoplasma (Ca. Phytoplasma pruni, subgroup III-A), imp is more abundantly expressed, with idpA only detectable through immunohistochemistry but not by Western blot, likely due to lower expression levels [48]. These findings reinforce that imp and idpA are not homologous genes and that their relative expression differs among phytoplasma species, possibly reflecting distinct functional roles in host adaptation and transmission [31].
Phylogenetic analyses revealed that the 16S rRNA gene, though useful for broad classification, lacks resolution for distinguishing closely related phytoplasma strains [53]. In contrast, imp and idpA genes provided enhanced differentiation at both subgroup and intra-subgroup levels. For example, while 16SrIII-J isolates formed a monophyletic cluster based on 16S rRNA sequences, imp and idpA genes uncovered finer genetic differences, demonstrating their value for resolving intra-subgroup diversity. These findings are further supported by whole-genome analyses, which have shown that isolates from the 16SrIII-J subgroup, such as Cicuta witches' broom phytoplasma (CicWB) and Vc33, form a distinct clade separate from other 16SrIII subgroups. This separation is strongly supported by genomic metrics such as Average Nucleotide Identity (ANI) and digital DNA-DNA hybridization (dDDH), which exceed 97% and 70%, respectively, within the 16SrIII-J subgroup [54]. These metrics, now integral to modern classification schemes for Candidatus Phytoplasma species [2,4], provide robust evidence for the distinct evolutionary trajectory of the 16SrIII-J subgroup. The correlation between gene-based phylogenies (imp, idpA) and whole-genome analyses underscores the importance of integrating multiple approaches to achieve a more accurate and comprehensive classification of phytoplasmas, particularly within highly diverse groups like the X-disease phytoplasmas (16SrIII). These findings highlight the potential of combining genomic and gene-specific markers to refine our understanding of phytoplasma diversity and evolution.
The imp gene, beyond its role as a marker of genetic diversity and phylogenetic classification, has significant potential for the development of diagnostic tools and management strategies for phytoplasma-associated diseases. Its high intra- and interspecies variability makes it an excellent candidate for population and evolutionary studies, as demonstrated by [52], who identified 17 imp genotypes in 'Ca. Phytoplasma pyri', revealing its utility in distinguishing strains and understanding host adaptation. This variability also opens the door to the development of specific antisera or monoclonal antibodies targeting imp, which could be used for serological detection. For instance, [55] successfully developed an anti-Imp ELISA assay for detecting "flavescence dorée" phytoplasmas in grapevine, insect vectors, and host plants, demonstrating the feasibility of using imp-based serological tools for field diagnostics. Furthermore, the use of anti-Imp antibodies has been shown to improve phytoplasma genome sequencing efforts. [56] employed immunoprecipitation with anti-Imp antibodies to enrich phytoplasma DNA, enabling the assembly of high-quality genomes, such as that of 'Ca. P. aurantifolia' NCHU2014. In addition, the study by [45] highlights the utility of imp gene as a target for recombinase polymerase amplification (RPA) assays, which demonstrated comparable sensitivity to PCR for detecting 'Ca. Phytoplasma pruni' in sweet cherry tissues. Their findings revealed that imp is highly expressed in infected plants, with RNA transcript levels significantly higher than those of idpA, suggesting that imp may be the major immunodominant protein in this phytoplasma subgroup.
In summary, the study of imp and idpA genes has provided valuable insights into the diversity, evolution, and pathogenicity of phytoplasmas within the 16SrIII group. These findings not only enhance our understanding of phytoplasma biology but also pave the way for the development of more accurate diagnostic tools and effective management strategies.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure Supplementary 1. Pairwise sequence identity matrix (%) comparing two proteins, imp (lower diagonal) and idpA (upper diagonal), across the same bacterial isolates. Values represent percentage identity, with colors (in the original heatmap) ranging from cool tones (low identity, e.g., 40-60%) to warm tones (high identity, e.g., 80-100%). The diagonal (100.0) serves as a self-identity reference.
Author Contributions
Conceptualization, F.D.F and F.I.A; methodology, F.D.F and F.I.A.; formal analysis, F.I.A, F.D.F, V.A.B., L.R.C.; writing—original draft preparation, F.D.F and F.I.A; writing—review and editing, F.I.A, F.D.F, L.R.C.; supervision, F.D.F., L.R.C., C.M; project administration, L.R.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by INTA (PD I085, I081 and I090) and FONCyT (PICT-2016-0862, PICT2017-3068 and PICT 2020-0230). Alessio FI holds a doctoral fellow of FONCyT (PICT-2016-0862).
Data Availability Statement
Nucleotide and Aminoacidic sequence from imp and idpA membrane proteins obtained in this paper were deposited in the NCBI repository under the accession numbers PQ429233 to PQ429243 and PQ871563.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Maejima, K.; Oshima, K.; Namba, S. Exploring the phytoplasmas, plant pathogenic bacteria. J. Gen. Plant Pathol. 2014, 80, 210-221. [CrossRef]
- Wei, W.; Zhao, Y. Phytoplasma Taxonomy: Nomenclature, Classification, and Identification. Biology 2022, 11, 1119. [CrossRef]
- Weintraub, P.G.; Beanland, L. Insect vectors of phytoplasmas. Annu. Rev. Entomol. 2006, 51, 91-111. [CrossRef]
- Bertaccini, A.; Arocha-Rosete, Y.; Contaldo, N.; Duduk, B.; Fiore, N.; Montano, H. G.; Kube, M.; Kuo, C.H.; Martini, M.; Oshima, K.; Qualigno, F.; Schenider, B.; Wei, W.; Zamorano, A. Revision of the ‘Candidatus Phytoplasma’ species description guidelines. Int. J. Syst. Bacteriol. 2022, 72(4). [CrossRef]
- Gundersen, D.E.; Lee, I.M.; Schaff, D.A.; Harrison, N.A.; Chang, C.J.; Davis, R.E.; Kingsbury, D.T. Genomic diversity and differentiation among phytoplasma strains in 16S rRNA groups I (aster yellows and related phytoplasmas) and III (X-disease and related phytoplasmas). Int. J. Syst. Evol. Microbiol. 1996, 46, 64-75. [CrossRef]
- Zhao, Y.; Wei, W.; Lee, I.M.; Shao, J.; Suo, X.; Davis, R.E. Construction of an interactive online phytoplasma classification tool, iPhyClassifier, and its application in analysis of the peach X-disease phytoplasma group (16SrIII). Int. J. Syst. Evol. Microbiol. 2009, 59, 2582-2593. [CrossRef]
- Liefting, L.W.; Andersen, M.T.; Beever, R.E.; Gardner, R.C.; Forster, R.L. Sequence heterogeneity in the two 16S rRNA genes of Phormium yellow leaf phytoplasma. Appl. Environ. Microbiol. 1996, 62, 3133-3139. [CrossRef]
- Zhao, Y.; Davis, R.E. Criteria for phytoplasma 16Sr group/subgroup delineation and the need of a platform for proper registration of new groups and subgroups. Int. J. Syst. Evol. Microbiol. 2016, 66, 2121–2123. [CrossRef]
- Cho, S.T.; Kung, H.J.; Huang, W.; Hogenhout, S.A.; Kuo, C.H. Species boundaries and molecular markers for the classification of 16SrI phytoplasmas inferred by genome analysis. Front. Microbiol. 2020, 11, 1531. [CrossRef]
- Arnaud, G.; Malembic-Maher, S.; Salar, P.; Bonnet, P.; Maixner, M.; Marcone, C.; Boudon-Padieu, E.; Foissac, X. Multilocus sequence typing confirms the close genetic interrelatedness of three distinct flavescence dorée phytoplasma strain clusters and group 16SrV phytoplasmas infecting grapevine and alder in Europe. Appl. Environ. Microbiol. 2007, 73, 4001–4010. [CrossRef]
- Davis, R.E.; Zhao, Y.; Dally, E.L.; Lee, I.M.; Jomantiene, R.; Douglas, S.M. 'Candidatus Phytoplasma pruni', a novel taxon associated with X-disease of stone fruits, Prunus spp.: multilocus characterization based on 16S rRNA, secY, and ribosomal protein genes. Int. J. Syst. Evol. Microbiol. 2013, 63, 766-776. [CrossRef]
- Abeysinghe, S.; Abeysinghe, P.D.; Kanatiwela-de Silva, C.; Udagama, P.; Warawichanee, K.; Aljafar, N.; Dickinson, M. Refinement of the taxonomic structure of 16SrXI and 16SrXIV phytoplasmas of gramineous plants using multilocus sequence typing. Plant Dis. 2016, 100, 2001–2010. [CrossRef]
- Kakizawa, S.; Kamagata, Y. A multiplex-PCR method for strain identification and detailed phylogenetic analysis of AY-group phytoplasmas. Plant Dis. 2014, 98, 299–305. [CrossRef]
- Toth, R.; Ilic, A.M.; Huettel, B.; Duduk, B.; Kube, M. Divergence within the Taxon 'Candidatus Phytoplasma asteris' Confirmed by Comparative Genome Analysis of Carrot Strains. Microorganisms 2024, 12, 1016. [CrossRef]
- Hugenholtz, P.; Chuvochina, M.; Oren, A.; Parks, D.H.; Soo, R.M. Prokaryotic taxonomy and nomenclature in the age of big sequence data. ISME J. 2021, 15, 1879–1892. [CrossRef]
- Galdeano, E.; Guzmán, F.; Fernández, F.; Conci, L. Genetic diversity of 16SrIII group phytoplasmas in Argentina. Predominance of subgroups 16SrIII-J and B and two new subgroups 16SrIII-W and X. Eur. J. Plant Pathol. 2013, 137, 753–764. [CrossRef]
- Perez-Lopez, E.; Luna-Rodríguez, M.; Olivier, C.Y.; Dumonceaux, T.J. The underestimated diversity of phytoplasmas in Latin America. Int. J. Syst. Evol. Microbiol. 2016, 66, 492–513. [CrossRef]
- Montano, H.G.; Bertaccini, A.; Fiore, N. Phytoplasma-Associated Diseases in South America: Thirty Years of Research. Microorganisms 2024, 12, 1311. [CrossRef]
- Amaral Mello, A.P.O.; Eckstein, B.; Flôres, D.; Kreyci, P.F.; Bedendo, I.P. Identification by computer-simulated RFLP of phytoplasmas associated with eggplant giant calyx representative of two subgroups, a lineage of 16SrIII-J and the new subgroup 16SrIII-U. Int. J. Syst. Evol. Microbiol. 2011, 61, 1454–1461 . [CrossRef]
- Gonzalez, F.; Zamorano, A.; Pino, A.M.; Paltrinieri, S.; Bertaccini, A.; Fiore, N. Identification of phytoplasma belonging to X-disease group in cherry in Chile. Bull. Insectol. 2011, 64 (Suppl.), S235–S236.
- Rappussi, M.C.C.; Eckstein, B.; Flôres, D.; Haas, I.C.R.; Amorim, L.; Bedendo, I.P. Cauliflower stunt associated with a phytoplasma of subgroup 16SrIII-J and the spatial pattern of disease. Eur. J. Plant Pathol. 2012, 133, 829–840. [CrossRef]
- Fernández, F.; Uset, A.; Baumgratz, G.; Conci, L. Detection and identification of a 16SrIII-J phytoplasma affecting cassava (Manihot esculenta Crantz) in Argentina. Australas. Plant Dis. Notes 2018, 13, 1–5. [CrossRef]
- Fernández, F.D.; Guzmán, F.A.; Baffoni, P.; Reinoso, L.; Kiehr, M.; Delhey, R.; Conci, L.R. Phytoplasmas of subgroup 16SrIII-J associated with Beta vulgaris in Argentina. Trop. Plant Pathol. 2020, 45, 143–147. [CrossRef]
- Arneodo, J.D.; Galdeano, E.; Orrego, A.; Stauffer, A.; Nome, S.F.; Conci, L.R. Identification of two phytoplasmas detected in China-trees with decline symptoms in Paraguay. Australas. Plant Pathol. 2005, 34, 583–585. [CrossRef]
- Flôres, D.; Haas, I.C.; Canale, M.C.; Bedendo, I.P. Molecular identification of a 16SrIII-B phytoplasma associated with cassava witches’ broom disease. Eur. J. Plant Pathol. 2013, 137, 237–242. [CrossRef]
- Galdeano, E.; Torres, L.E.; Meneguzzi, N.; Guzmán, F.; Gomez, G.G.; Docampo, D.M.; Conci, L.R. Molecular characterization of 16S ribosomal DNA and phylogenetic analysis of two X-disease group phytoplasmas affecting China-tree (Melia azedarach L.) and garlic (Allium sativum L.) in Argentina. J. Phytopathol. 2004, 152, 174–181. [CrossRef]
- Fernandez, F.D.; Carloni, E.; Alessio, F.; Bongiorno, V.; Conci, L.R. First report of a 16SrIII-X phytoplasma associated with Lactuca sativa witches’ broom in Argentina. New Dis. Rep. 2022, 46, e12103. [CrossRef]
- Alvarez, E.; Mejía, J.F.; Llano, G.; Loke, J.; Calari, A.; Duduk, B.; Bertaccini, A. Characterization of a phytoplasma associated with frogskin disease in cassava. Plant Dis. 2009, 93, 1139–1145. [CrossRef]
- Konnerth, A., Krczal, G., & Boonrod, K. (2016). Immunodominant membrane proteins of phytoplasmas. Microbiol., 162(8), 1267-1273. [CrossRef]
- Kakizawa, S.; Oshima, K.; Jung, H.Y.; Suzuki, S.; Nishigawa, H.; Arashida, R.; Lee, J.T.; Miyata, S.; Ugaki, M.; Namba, S. Positive selection acting on a surface membrane protein of the plant-pathogenic phytoplasmas. J. Bacteriol. 2006, 188, 3424–3428. [CrossRef]
- Kakizawa, S.; Oshima, K.; Ishii, Y.; Hoshi, A.; Maejima, K.; Jung, H.Y.; Yamaji, Y.; Namba, S. Cloning of immunodominant membrane protein genes of phytoplasmas and their in planta expression. FEMS Microbiol. Lett. 2009, 293, 92–101. [CrossRef]
- Galetto, L.; Siampour, M.; Marzachì, C. Preparation of phytoplasma membrane recombinant proteins. Methods Mol. Biol. 2013, 938, 351–369. [CrossRef]
- Doyle, J.J.; Doyle, J.L. Isolation of plant DNA from fresh tissue. Focus 1990, 12, 13–15.
- Deng, S.; Hiruki, C. Amplification of 16S rRNA genes from culturable and unculturable Mollicutes. J. Microbiol. Methods 1991, 14, 53–61. [CrossRef]
- Lee, I.M.; Hammond, R.W.; Davis, R.E.; Gundersen, D.E. Universal amplification and analysis of pathogen 16S rDNA for classification and identification of MLOs. Phytopathology 1993, 83, 834–842. [CrossRef]
- Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [CrossRef]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420-423. [CrossRef]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [CrossRef]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: Oxford, UK, 2000; p. 174.
- Pond, S.L.K.; Frost, S.D.W.; Muse, S.V. HyPhy: hypothesis testing using phylogenies. Bioinformatics 2005, 21, 676–679. [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [CrossRef]
- Villamor, D.E.V.; Eastwell, K.C. Multilocus characterization, gene expression analysis of putative immunodominant protein coding regions, and development of recombinase polymerase amplification assay for detection of 'Candidatus Phytoplasma pruni' in Prunus avium. Phytopathology 2019, 109, 983–992. [CrossRef]
- Siampour, M.; Izadpanah, K.; Galetto, L.; Salehi, M.; Marzachi, C. Molecular characterization, phylogenetic comparison and serological relationship of the Imp protein of several 'Candidatus Phytoplasma aurantifolia' strains. Plant Pathol. 2013, 62, 452–45. [CrossRef]
- Blomquist, C.L.; Barbara, D.J.; Davies, D.L.; Clark, M.F.; Kirkpatrick, B.C. An immunodominant membrane protein gene from the Western X-disease phytoplasma is distinct from those of other phytoplasmas. Microbiology 2001, 147, 571–580. [CrossRef]
- Neriya, Y.; Sugawara, K.; Maejima, K.; Hashimoto, M.; Komatsu, K.; Minato, N.; Miura, C.; Kakizawa, S.; Yamaji, Y.; Oshima, K.; Namba, S. Cloning, expression analysis, and sequence diversity of genes encoding two different immunodominant membrane proteins in poinsettia branch-inducing phytoplasma (PoiBI). FEMS Microbiol. Lett. 2011, 324, 38–47. [CrossRef]
- Martini, M.; Quaglino, F.; Bertaccini, A. Multilocus genetic characterization of phytoplasmas. In Phytoplasmas: Plant Pathogenic Bacteria-III: Genomics, Host Pathogen Interactions and Diagnosis; Bertaccini, A., Oshima, K., Kube, M., Rao, G.P., Eds.; Springer: Singapore, 2019; pp. 161–200. [CrossRef]
- Zhang, R.Y.; Wang, X.Y.; Shan, H.L.; Li, J.; Li, Y.H.; Li, W.F.; Huang, Y.K. Multilocus sequence typing reveals two distinct populations of "Candidatus Phytoplasma sacchari" in China. Trop. Plant Pathol. 2023, 48, 199–206. [CrossRef]
- Liu, C.Y.; Cheng, H.P.; Lin, C.P.; Liao, Y.T.; Ko, T.P.; Lin, S.J.; Wang, H.C. Structural insights into the molecular mechanism of phytoplasma immunodominant membrane protein. IUCrJ 2024, 11, 384–394. [CrossRef]
- Bohunická, M.; Valentová, L.; Suchá, J.; Nečas, T.; Eichmeier, A.; Kiss, T.; Cmejla, R. Identification of 17 'Candidatus Phytoplasma pyri' genotypes based on the diversity of the imp gene sequence. Plant Pathol. 2018, 67, 971–977. [CrossRef]
- Pusz-Bochenska, K.; Perez-Lopez, E.; Wist, T.J.; Bennypaul, H.; Sanderson, D.; Green, M.; Dumonceaux, T.J. Multilocus sequence typing of diverse phytoplasmas using hybridization probe-based sequence capture provides high resolution strain differentiation. Front. Microbiol. 2022, 13, 959562. 10.3389/fmicb.2022.959562.
- Fernández, F.D.; Guzmán, F.A.; Conci, L.R. Draft genome sequence of Cicuta witches' broom phytoplasma, subgroup 16SrIII-J: A subgroup with phytopathological relevance in South America. Trop. Plant Pathol. 2024, 49, 558–565. [CrossRef]
- Filippin, L.; Trivellone, V.; Galetto, L.; Marzachì, C.; Elicio, V.; Angelini, E. Development of an anti-Imp serological assay for the detection of "flavescence dorée" phytoplasmas in grapevine, insect vectors and host plants. Phytopathog. Mollicutes 2019, 9, 75–76. 10.5958/2249-4677.2019.00038.0.
- Tan, C.M.; Lin, Y.C.; Li, J.R.; Chien, Y.Y.; Wang, C.J.; Chou, L.; Yang, J.Y. Accelerating complete phytoplasma genome assembly by immunoprecipitation-based enrichment and MinION-based DNA sequencing for comparative analyses. Front. Microbiol. 2021, 12, 766221. 10.3389/fmicb.2021.766221.
Figure 1.
Multiple sequence alignment of imp proteins from representative phytoplasma strains. Conserved residues are indicated by a gradient of blue intensity. A putative transmembrane helix domain (grey box) and a putative extracellular domain (orange box) are highlighted.
Figure 1.
Multiple sequence alignment of imp proteins from representative phytoplasma strains. Conserved residues are indicated by a gradient of blue intensity. A putative transmembrane helix domain (grey box) and a putative extracellular domain (orange box) are highlighted.
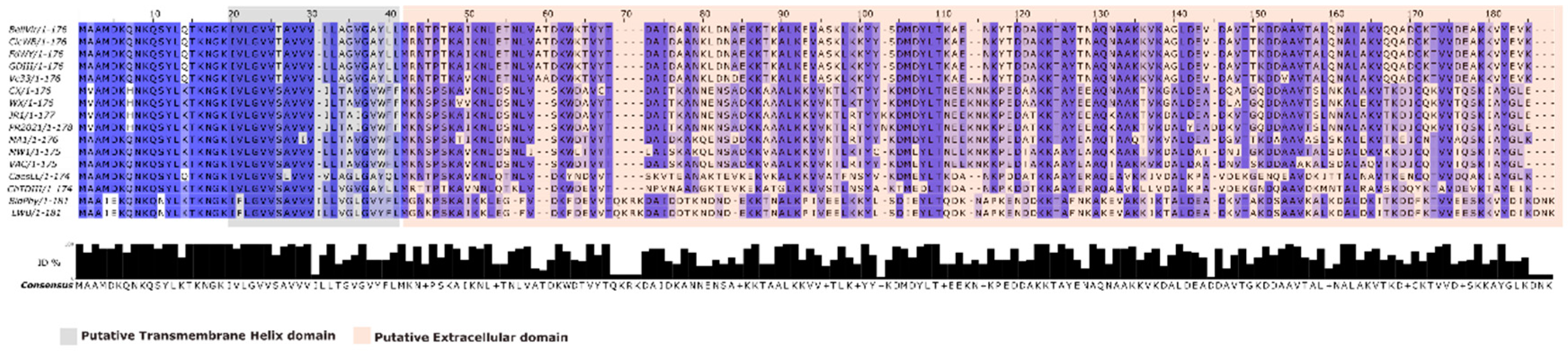
Figure 2.
Multiple sequence alignment of idpA proteins from representative phytoplasma strains. Conserved residues are indicated by a gradient of blue intensity. A putative transmembrane helix domain (grey box) and a putative extracellular domain (orange box) are highlighted.
Figure 2.
Multiple sequence alignment of idpA proteins from representative phytoplasma strains. Conserved residues are indicated by a gradient of blue intensity. A putative transmembrane helix domain (grey box) and a putative extracellular domain (orange box) are highlighted.
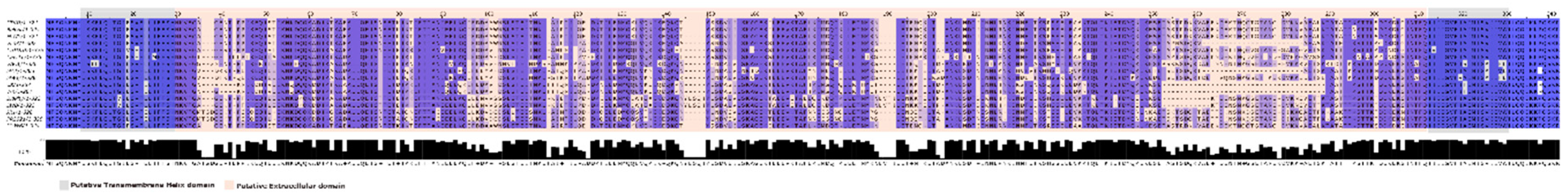
Figure 3.
Phylogenetic tree and heatmap based on the 16S rRNA gene sequences of X-disease group phytoplasma strains. The dendrogram was inferred using the maximum-likelihood method, and bootstrap values (>50%) are indicated at the nodes. The accompanying heatmap shows pairwise nucleotide identity percentages among the strains. The color gradient reflects nucleotide identity ranging from 98.0% (light yellow) to 100.0% (dark green).
Figure 3.
Phylogenetic tree and heatmap based on the 16S rRNA gene sequences of X-disease group phytoplasma strains. The dendrogram was inferred using the maximum-likelihood method, and bootstrap values (>50%) are indicated at the nodes. The accompanying heatmap shows pairwise nucleotide identity percentages among the strains. The color gradient reflects nucleotide identity ranging from 98.0% (light yellow) to 100.0% (dark green).
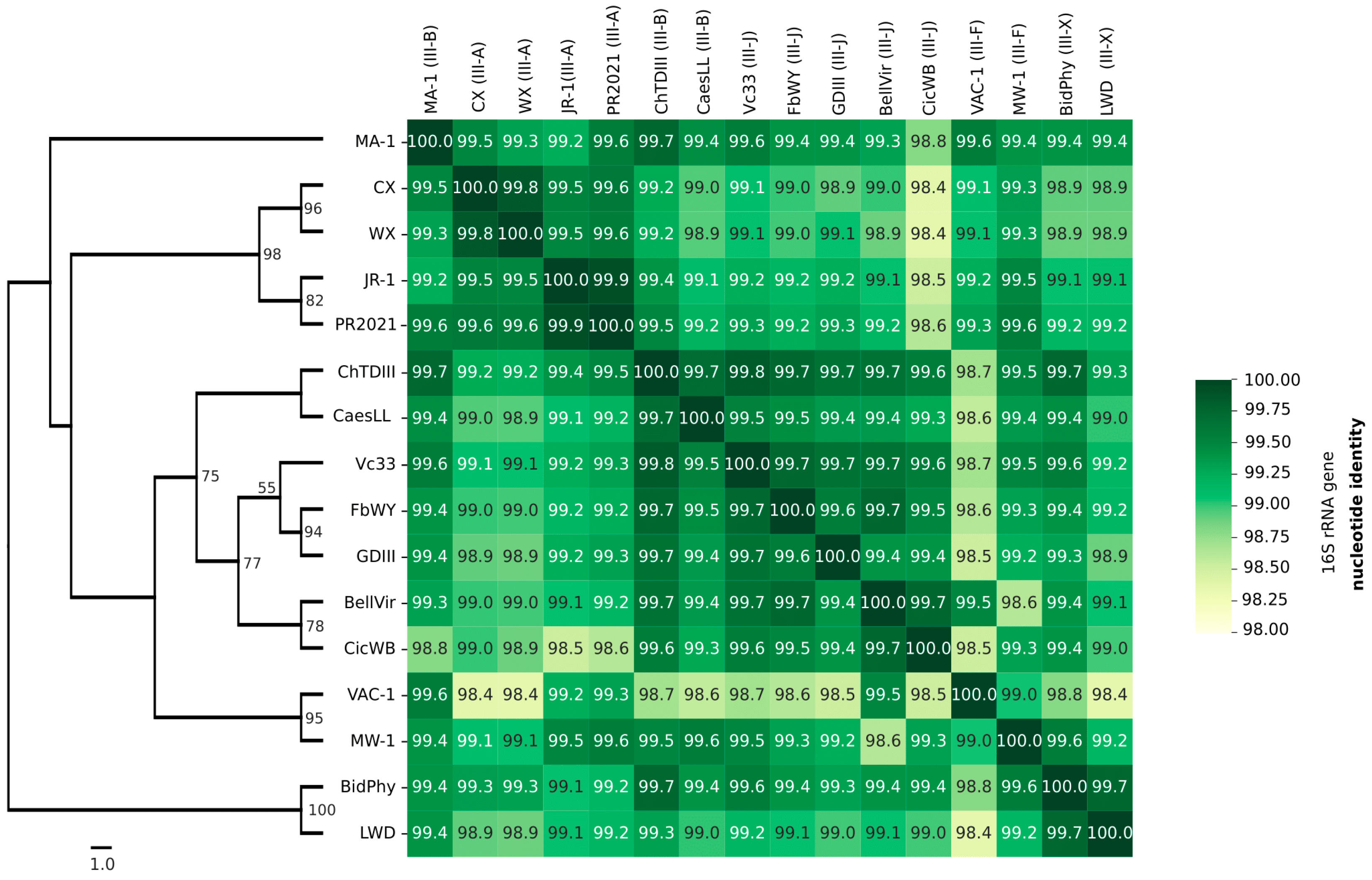
Figure 4.
Phylogenetic trees and pairwise nucleotide identity matrices based on the imp (A) and idpA (B) gene sequences of X-disease group phytoplasmas. The heatmaps show the pairwise percentage of nucleotide identity, while the dendrograms represent the phylogenetic relationships inferred from the multiple sequence alignments of each gene using the ML method. Color gradients indicate identity values ranging from 63% (light yellow) to 100% (dark green). In bold sequence obtained in this work.
Figure 4.
Phylogenetic trees and pairwise nucleotide identity matrices based on the imp (A) and idpA (B) gene sequences of X-disease group phytoplasmas. The heatmaps show the pairwise percentage of nucleotide identity, while the dendrograms represent the phylogenetic relationships inferred from the multiple sequence alignments of each gene using the ML method. Color gradients indicate identity values ranging from 63% (light yellow) to 100% (dark green). In bold sequence obtained in this work.
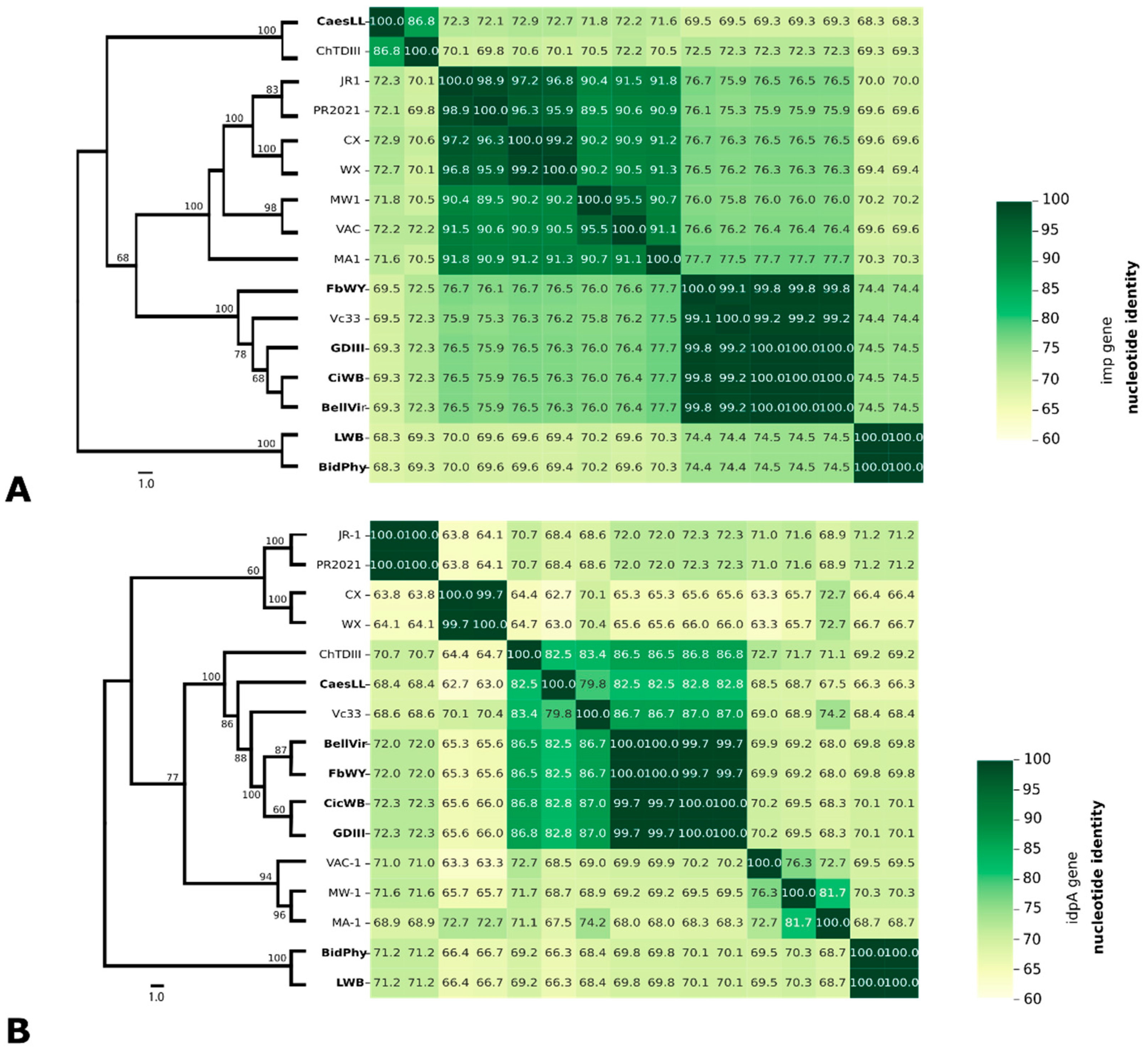

Table 1.
List of strain used in PCR reactions to amplify imp and idpA ORFs. * 16SrIII-subgroup based in 16S rRNA (1.2kb) RFLP profiles, imp PCR/ idpA PCR (number of samples tested/ number of samples PCR positives); (-): non amplification.
Table 1.
List of strain used in PCR reactions to amplify imp and idpA ORFs. * 16SrIII-subgroup based in 16S rRNA (1.2kb) RFLP profiles, imp PCR/ idpA PCR (number of samples tested/ number of samples PCR positives); (-): non amplification.
| Phytoplasma strain | 16SrIII* | Host |
imp PCR (+) |
idpA PCR (+) |
#Accession (imp/idpA) |
| Bellis virescence (BellVir) | III-J | Bellis perennis | 2/2 | 2/2 | MG435348.1/MG435349.1 |
| Garlic Decline (GDIII) | III-J | Allium sativum | 2/2 | 2/2 | PQ429243.1/PQ429237.1 |
| Fodder Beet Wilting- Yellowing (FbWY) | III-J | Beta vulgaris var. rapacea | 2/2 | 2/2 | PQ429242.1/PQ429236.1 |
| Sugar Beet Wilting-Yellowing (SugBeetWY) | III-J | Beta vulgaris var. altissima | 0/3 | 0/3 | - |
| Cicuta Witches Broom (CicWB) | III-J | Conium maculatum | 2/2 | 2/2 | PQ429241.1/PQ429238.1 |
| China tree decline (ChTDIII) | III-B | Melia azedarach | 3/3 | 3/3 | NWN45603.1/NWN45596.1 |
| Caesalpinia little leaf (CaesLL) | III-B | Caesalpinia gilliesii | 2/2 | 2/2 | PQ429239.1/PQ429233.1 |
| Argentinean Peach Yellows (ArPY) | III-B | Prunus persica | 0/3 | 0/3 | - |
| Lettuce Witches’ Broom (LWB) | III-X | Lactuca sativa | 2/2 | 2/2 | PQ871563/ PQ429235.1 |
| Bidens Phyllody (BidPhy) | III-X | Bidens subalternans | 1/3 | 1/3 | PQ429240.1/PQ429234.1 |
| Heterosperma Phyllody (HetPhy) | III-X | Heterosperma ovatifolium | 0/3 | 0/3 | - |
Table 2.
List of reference genomes used in this study. * 16S rRNA subgroup based on analysis in iPhyclassifier (https://plantpathology.ba.ars.usda.gov/cgi-bin/resource/iphyclassifier.cgi).
Table 2.
List of reference genomes used in this study. * 16S rRNA subgroup based on analysis in iPhyclassifier (https://plantpathology.ba.ars.usda.gov/cgi-bin/resource/iphyclassifier.cgi).
|
Phytoplasma [strain] |
16SrIII* | Host | Location | GenBank accession |
|
Ca. Phytoplasma pruni [WX] |
III-S | Prunus avium | USA | AF533231.1 |
|
Ca. Phytoplasma pruni [CX] |
III-A | Prunus domestica | USA | LHCF00000000.1 |
|
Ca. Phytoplasma pruni [PR2021] |
III-A | Euphorbia pulcherrima | Taiwan | CP119306.1 |
| Poinsettia branch-inducing [JR1] |
III-A | Euphorbia pulcherrima | USA | AKIK00000000.1 |
| Clover Phyllody [MA1] |
III-B | Chrysanthemum leuchantemum | Italy | AKIM00000000.1 |
| Vaccinium Witches’ Broom [VAC1] |
III-F | Vaccinium myrtillus | Italy | AKIN00000000.1 |
| Milkweed Yellows [MW1] |
III-F | Asclepias syriaca | USA | AKIL00000000.1 |
|
Ca. Phytoplasma sp [Vc33] |
III-J | Catharanthus roseus | Chile | LLKK00000000.1 |
| Chinaberry tree decline [ChTDIII] |
III-B | Melia azedarach | Argentina | JABUOH000000000.1 |
Table 3.
Selection pressure analysis in imp and ipdA proteins. Nº: number of sequences, S: segregating sites, P: nucleotic diversity, dN-dS: statistic test, dS and dN are the numbers of synonymous and nonsynonymous substitutions per site, respectively, p-value: the probability of rejecting the null hypothesis of strict-neutrality (dN = dS), TM, HD: numbers of codons with normalized dN-dS value >0 in Transmembrane or hydrophilic domain. %: proportion of normalized codons with dN-dS>0/ total codons, #codons: total numbers of codon.
Table 3.
Selection pressure analysis in imp and ipdA proteins. Nº: number of sequences, S: segregating sites, P: nucleotic diversity, dN-dS: statistic test, dS and dN are the numbers of synonymous and nonsynonymous substitutions per site, respectively, p-value: the probability of rejecting the null hypothesis of strict-neutrality (dN = dS), TM, HD: numbers of codons with normalized dN-dS value >0 in Transmembrane or hydrophilic domain. %: proportion of normalized codons with dN-dS>0/ total codons, #codons: total numbers of codon.
| Normalized dN-dS >0 | |||||||||
| Dataset | Nº | S | P | dN-dS | p-value | TM | HD | #codons | % |
| imp | 16 | 258 | 0.18272 | 3,474 | 0.01 | 10 | 70 | 157 | 50.955 |
| idpA | 16 | 268 | 0.10407 | -3,090 | 0.002 | 11 | 104 | 253 | 45.454 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



