
Submitted:
26 March 2025
Posted:
26 March 2025
You are already at the latest version
Abstract
Alzheimer’s disease (AD) is the most common neurodegenerative disease which has rather complex pathophysiology. During its course several neurotransmitter neuronal systems get affected as acetylcholinergic, glutamatergic, gamma-aminobutyric acid (GABA)ergic system and etc. Such complex physiology requires sophisticated approach of pharmaceutical management. Therefore, multi-target drugs seem to be an appealing solution.
In the present study we designed and synthesized a hybrid molecule - N-sinapoylamide of memantine, whose parent molecules memantine (MEM) and sinapic acid has been shown in vivo to impact glutamatergic and acetylcholinergic and GABA-ergic systems respectively.
In silico comparative testing of these molecules was performed and their patterns of interaction with the target enzymes or molecular complexes were analyzed and some of the mechanisms of action were proposed. Consequently, in vivo testing was performed on scopolamine mice model of AD and the results overly confirm part of the in silico findings. Therefore the hybrid molecule (N-Sinapoyl-memantine) seems to be a potent candidate for further evaluation in the management of AD.
Keywords:
Alzheimer’s disease
; memantine
; sinapic acid
; in silico
1. Introduction
Alzheimer’s disease (AD) is the most common neurodegenerative disease worldwide [Zhang et al., 2016]. Among its key disturbances is the neuronal loss in the acetylcholinergic system, disregulation of its nicotinic and muscarinic receptors, reduced availability of its neurotransmitter – acetylcholine [Hampel, et al., 2018]. As a result, many of the researchers have focused on the development of acetylcholinesterase (AChE) inhibitors as a means to combat some of these disturbances. However, the officially approved AChE inhibitors as galantamine etc. are still scarce and do not stop the disease progression in the long run [Hampel, et al., 2018]. Hereto and hopefully better AChE inhibitors are wanted and developed. Another major neurotransmitter system significantly affected by the AD is the glutamatergic one. The glutamatergic N-methyl-D-aspartate receptors (NMDARs) ion channels conduct monovalent ions and Ca2+ which elicit not only electric signal but also can trigger Ca2+ dependent intracellular signaling processes therefore their dysfunction of the in AD leads to impairments of memory and learning, increased oxidative stress and impaired Ca2+ homeostasis and etc. ultimately causing neuronal death [Zhang et al., 2016; Liu et al., 2019]. Therefore, the glutamatergic NMDA receptors are another target for the drug designers involved in the research on the management of the AD. Currently, the number of the approved drugs for these receptors as memantine is rather limited [Zhang et al., 2016, Limapichat et al., 2013]. The difficulty for good design of such drugs arises from the fact that the NMDA receptors are of pivotal importance and their full inhibition leads to is similarly undesired as their over-excitation [Limapichat et al., 2013].
Additionally, growing evidence suggests that the main inhibitory neurotransmitter system in the mammalian CNS is also disturbed in the course of the AD. Besides some studies showed that GABA-ergic system dysfunctions are related to the Aβ depositions and pathology in AD patients – one of the hallmarks for AD [Garcia-Marin, et al., 2009]. So the GABA- receptors and the GABA-ergic system and the related symptoms in AD, also need to be addressed by the therapeutic interventions and management of AD. One of the ways probably can be by using modulators of the GABAA receptors as the results from a recent study may suggest [Petrache, et al., 2019].
Sinapic acid (3,5-dimethoxy-4-hydroxycinnamic acid; SINA) belongs to one of the fundamental classes of phenolic acids, namely hydroxycinnamic acids (ferulic-, coumaric-, caffeic acids), which are widely distributed in the plant kingdom [Nićiforović & Abramovič, 2014; Hameed, et al., 2016]. Besides its free form of existence, this secondary plant metabolite may be also found as diverse natural or synthetic conjugates (esters, amides and other).
It has been reported that SINA is active against diverse pathological conditions such as infections [Maddox et al., 2010], oxidative stress [Kikuzaki et al., 2002; Shahid et al., 2022], inflammation [Zou, et al., 2002; Yun et al., 2008], cancer [Hudson et al., 2000], diabetes [Kanchana et al., 2011], neurodegeneration [Kim et al., 2010; Chen, 2016], and so on. Generally, the pharmaceutical potential of sinapic acid (or other hydroxycinnamic acids) can be attributed to its potent antioxidant ability against various radicals [Georgiev et al., 2012; Rice-Evans et al., 1996; 1997; Nenadis & Tsimidou, 2002; Hotta, et al., 2002]. However, when the imbalance between antioxidants and free radicals occurs, the physiological state has been known as “oxidative stress”. The latter is suspected to be associated with the development of various diseases, including neurodegenerative, cancer, cardiovascular diorders, and so on. In particular, NO and superoxide being considerable cytotoxic molecules exist at high concentrations during inflammation. Moreover, they are also implicated in the pathology of various brain neurodegenerative disorders, ischemic stroke, aging of the brain and etc. [Brown & Borutaite, 2006]. Thus, the peroxynitrite can be easily formed by NO and superoxide are rather reactive and can provoke protein modifications leading to the progress of various diseases such as AD and Parkinson disease [Torreilles et al., 1999; Klotz et al., 2002].
Several previous in vitro studies suggest that SINA has neuroprotective effect through GABA receptor agonistic property [Yoon et al., 2007; Kim et al., 2010] and peroxynitrite scavenging activity in neurodegenerative diseases. Moreover, the neuroprotective effect of sinapic acid has also been found in a mouse model of amyloid β1–42 protein-induced Alzheimer's disease (AD) [Lee et al., 2012].
Considering the involvement of oxidative stress hypothesis in the complex multifactorial nature of AD, the antioxidant molecules could play a special role in prevention and therapy of AD.
Currently, memantine (МЕМ) is well known as the only N-methyl-D-aspartate (NMDA) receptor channel blocker, used for the treatment of moderate-to-severe forms of the AD disease [Xia et al., 2010; Song et al., 2018]. However, besides the other available anti-AD drugs, MEM offers only a symptomatic relief to patients and is not able to stop the AD – progression.
In our earlier study by Chochkova et al., (2021), we developed a series of antioxidant hydroxycinnamic hybrids based on МЕМ scaffold. Among all memantine hybrids, N-sinapoylamide of memantine (N-Sinapoyl-memantine; SINA-MEM) was found to be highly effective antioxidant in all tested in vitro radical scavenging tests. Moreover, the single crystal X-ray analysis of SINA-MEM defines that amide crystallizes in a non-centrosymmetric manner in trigonal crystal system, space group R3. Further it was found that the aromatic ring system present in the sinapoyl moiety is substantially planar. As for the other parent moiety – the memantine one – it was found that its geometry was highly preserved. The neighbor intermolecular groups and bonds (·NH·; ·CO·; ·CH=CH·) also seems to stabilize the 3D structure of the new hybrid molecule according to our data and structure analysis [Chochkova et al., 2021]. In the testing of anti-Alzheimer effects in vitro (against Aβ toxicity, excitotoxicity, oxidative stress damage, hypoxia injury and neuroinflammation), the memantine hybrids displayed neuroprotection in the moderate levels. Despite the tested memantine hybrids were slightly weaker than the positive control MEM, they may be able to enhance the efficacy of AD therapy in vivo [Chochkova et al., 2021; Chochkova et al., 2022].
Following our line of research based on a particularly attractive approach for multi-target ligands for AD treatment, herein we further explore in silico the interaction with couple host target molecules of interest and consequently we check in vivo some of the possible resulting neuroprotective activities of sinapic acid hybrid with memantine from such modelled interactions.
2. Results and Discussion
2.1. Compound Synthesis
In our previous papers we reported the synthesis of SINA-MEM and other hydroxycinnamoyl hybrids by EDC/HOBt method following the synthetic route (see Figure 1) [Chochkova et al., 2021; M. Spasova et al., 2006].
Based on the manifested in vitro antioxidant and neuroprotection investigations, SINA-MEM was selected in order to study its ability to stop the disease progression in mice.
2.2. Physico-Chemical Analysis of the Compounds
The comparison of the data from the X-ray diffraction between the parental and the new hybrid compounds (see Table 1 and Figure 2) showed that the lengths of all bonds are changed. Least pronounced are the changes of the C-H bonds which change in the range of -0.5% - +0.5%, with exception of the ones of the –OCH3 group where the change can go up to 0.15%. Next least affected type of bonds are the C-C ones from the aromatic rings which change in the range -1.2% - +1.2%. More affected are the C-C bonds from the aliphatic chain which change by 1.9% and -2.48% respectively. From the C-O bonds least affected the ones in the carbonyl group with 0.95% reduction. The C-O bonds with carbon atoms from the aromatic ring are increased by slightly more than 1%. More pronounced are the changes of the C-O bonds in the –OCH3 groups which length can change by up to 2.8%. The most pronounced change is found in the OH bond of the hydroxyl group at para position of the aromatic ring where length change of over 7% is observed.
The comparison of the data from the X-ray diffraction between the parental MEM and the MEM-moiety of the new hybrid compound showed that the lengths of the C-C bonds are changed within the range -0.3% to + 0.5%, greater changes where observed in the bonds with the methyl groups – approximately 0.8%. The change of the bond length with the nitrogen atom is reduced by 0.6%. And the maximal change in the whole MEM-moiety is observed at the N-H bond with length reduction of over 5.5%.
Comparing the spatial arrangement of the atoms in the new hybrid molecule with the parental SINA is found that the planarity is preserved in the respective part of the molecule.
Analyzing the acquired attenuated total reflectance infrared spectra (ATR-IR) showed that many of the more pronounced peaks are shifted in the new hybrid molecule in compare to the parents (see Figure 3 and Table 2). Comparing the parent compound SINA with the corresponding moiety of the new hybrid was determined that most of the peaks of the bonds in the aromatic ring decreased with 1-2 cm-1 in the hybrid molecule. On the other side the wavenumber of the bonds at the aliphatic chain connecting with MEM-moiety increased their absolute value by 2-5 cm-1. However, the biggest observed change was at the most remote group from the MEM-moiety, namely the hydroxyl one at the para position of the aromatic ring which shifted by almost 9 cm-1.
The comparison of the data from the ATR-IR between the parental MEM and the MEM-moiety of the new hybrid compound were hindered mostly due to the low intensity, overlapping and masking of some the characteristic peaks thus turning it less informative.
2.3. QSAR and Docking Results
The QSAR evaluation of the SINA-MEM compound compared to its parent molecules shows that it has no expected mutagenicity and carcinogenicity (see Table 4). Also, it has moderate probability for hepatotoxicity (0.5750) and oral toxicity (0.4847), which is higher than the one of its parents and on pair with the other. It has high probability of intestinal absorption (0.9434) as its parents but lower oral bioavailability (0.5571). The compound has high probability to cross the blood brain barrier (0.9426) as its parents. Its subcellular localization is expected to be the mitochondria.
The in silico experiments with molecular docking revealed that all of the tested substances can interact with the active site of one of the key target enzymes in the management of AD – acetylcholinesterase (AChE).
The active site of AChE is unusual one and is deeply recessed at the bottom of a relatively long gorge (approximately 20A) [Deb, et al., 2012]. It can be divided to several subsites each with different amino acid residues namely: a). catalytic triad called also esteratic subsite, encompassing the residues Ser203, His447, Glu334; b). acyl binding pocket (ABP) - Trp236, Phe295, Phe297, Phe338; c). peripheral anionic subsite (PAS) - Asp74, Tyr124, Ser125, Trp286, Tyr337, Tyr341; d). anionic subsite - Trp86, Tyr133, Glu202, Gly448, Ile451; e). oxyanion hole - Gly121, Gly122, Ala204, and other residues of the omega loop (Thr83, Asn87, Pro88) (see Figure 5). The omega loop is a disulphide-linked loop (Cys69 - Cys96) that covers the active site of AChE, which is buried at the bottom the gorge.
In order to compare the docking results parameters and how the studied substances interact and probably can inhibit the AChE enzyme we used one of the established and clinically approved AChE inhibitors – galantamine – as referent molecule [Atanasova, et al., 2015].
The hybrid molecule has relatively low binding energy of -8.61 kcal/mol and forms several hydrogen bonds with few amino acid residues which are part of the active sites of the enzyme. All of these hydrogen bonds are formed with participation of the oxygen atoms from the sinapoyl moiety. Two of these bonds are with amino acid residues His447 and Ser203 part of the catalytic triad, the other hydrogen bond is with Ser125 (PAS).
The molecule also engages in several hydrophobic interactions with the following AChE amino acid residues (some of them are part of its active sites): Trp286 (PAS), Leu289, Ile294, Arg296, Phe297 (ABP), Phe338 (ABP), Tyr341 (PAS).
The SINA has binding energy of -5.36 kcal/mol which is higher than the one of the hybrid molecule. However it forms more hydrogen bonds with the surrounding amino acid residues at the AChE catalytic center (see Figure 6). These amino acids are Tyr124 (PAS), Ser293, Phe295 (ABP), Arg296, Tyr337 (PAS). It engages in hydrophobic interactions with the following amino acid residues: Phe297 (ABP), Phe338 (ABP), Tyr341 (PAS).
MEM has relatively low binding energy of -8.02 kcal/mol. It forms two hydrogen bonds with Ser293 and Tyr341 (PAS) through its only nitrogen atom (see Figure 7). It also engages in hydrophobic interactions with Trp286 (PAS), Ile294, Phe297 (ABP), Phe338 (ABP), Tyr341 (PAS).
Galantamine has binding energy of -8.79 kcal/mol which is the lowest among the tested molecules. It forms 3 hydrogen bonds (but longer than 3,19A, therefore probably are not counted by the 2D-plotting software). These bonds are with the amino acid moieties Tyr72, Tyr124 (PAS), Tyr337 (PAS) (see Figure 8). It engages in hydrophobic interactions with Phe338 (ABP), Tyr341 (PAS). However, unlike the other molecules it also interacts through pi-stacking.
As we can see all of the molecules regardless their similarities interact differently with the active site of AChE. SINA-MEM forms hydrogen bonds with residues from CT and PAS, while SINA forms such bonds with residues from ABP and PAS, and MEM only with residues from PAS. The pattern of hydrophobic interactions also differs. The common residues for SINA-MEM, MEM and SINA are Phe297 (ABP), Phe338 (ABP), Tyr341 (PAS). SINA-MEM interacts with 4 more residues than SINA and with 2 more than Mem.
As for the Galantamine additionally it differs from the rest by the pi-stacking interaction. All of the tested molecules have hydrophobic interactions with Phe338 (ABP) and Tyr341 (PAS). And none of them interacts with the oxyanion hole, anionic subsite and omega loop. Only SINA-MEM interacts with the catalytic triad.
The detailed results of the re-docking experiments generated similar profile of the interaction of the amino-acid residues (see Figure 9).
Most of the tested compounds have relatively low binding energy below -8.0 kcal/mol. Only SINA has higher value of -5.36 kcal/mol. In comparison the acquired conformations of ACH have binding energy in the range above -4.80 kcal/mol (see Table 5).
According to Silman and Sussman [Silman & Sussman, 2008] there is narrowing in the middle of the active site gorge, produced by the juxtaposition of Phe330 and Gly121, whose cross-section they estimate to be ∼5 Å. So probably bigger than acetylcholine molecules can pass through such obstacle but only if their 3D structure is planar and/or sufficiently flexible. MEM and its synthesized analogue SINA-MEM are the bulkiest molecules among the tested batch. The approximate distance between the carbon atoms of its methyl moieties is 5 A, similar to the one between the nitrogen in the amino group and the carbons from the methyl groups.
So regardless that the MEM has rather low value of the binding energy of the interaction with the active site of AChE in reality we cannot benefit from such affinity simply because the molecule is spatially obscured with the access to this site.
However, the situation with the other bulky molecule - the MEM hybrid SINA-MEM is different. This molecule comprises of two bulky moieties connected by long carbon chain. Howbeit one of these bulky moieties – the aromatic ring from the sinapic acid – has mostly planar structure and is less likely to be obscured by the gorge narrowing. In fact in all of the found docking conformations for this molecule that bind to the amino acid residues in the AChE active site this moiety is the one inserted in it, while the MEM one remains outside or at its entrance. As it can be seen from the 2D-plotting diagram the sinapic acid moiety is behind the Gly121 amino acid residue, while the MEM one remains outside the active center.
Docking simulations were also performed with another possible molecular target in the brain – the GABAA receptors. These receptors are part of the GLIC-family and form ion channels in the neuronal membranes. Since this type of protein complexes are notoriously hard for study until recently there was no 3D protein structure of them available for the scientific community. Recently the 3D structure of human GABAA receptor was published [Miller & Aricescu, 2014; Zhu et al., 2018], followed by chimera 3D assembly for mouse [Laverty et al., 2017].
The docking with the classic benzodiazepine flumazenil revealed that the ligand binds to the benzodiazepine binding site of GABAA receptors with binding energy of -7.55 kcal/mol.
Flumazenil forms with three hydrogen bonds with both protein chains (α and γ) with the following amino acid residues: Thr142γ, Ser205α and Ser206α. It also has 7 hydrophobic interactions with Tyr58γ, Phe77γ, Tyr160α and Tyr210α (see Figure 10).
MEM has slightly higher binding energy of -7.37 kcal/mol than flumazenil. It forms fewer hydrogen bonds amino acids from both protein chains namely: Asp56γ and Ser206α (see Figure 11). It has 4 hydrophobic interactions with amino acids from γ protein chain: Tyr58, Phe77 and Ala79.
SINA has the lowest binding energy among the tested substances of -5.76 kcal/mol. It forms more hydrogen bonds with amino acids but only from α-protein chain namely: His102, Ser159, Ala161 and Ser205 (see Figure 12). It has fewest hydrophobic interactions with amino acids only from γ protein chain: Phe77. And unlike the previous compounds it interacts through π-stacking with Tyr210α.
The derivative SINA-MEM scores second by binding energy after flumazenil with value of -7.5 kcal/mol. It forms three hydrogen bonds with amino acids only from α-protein chain namely: Lys156 and Ser205 (see Figure 13). It has 4 hydrophobic interactions with amino acids from both protein chains: Phe77γ, Tyr160α and Tyr210α.
All of the tested compounds form hydrogen bonds with one of the polar serine residues at α-chain (Ser205 or Ser206). And all of them have hydrophobic interaction with Phe77γ, which is an important amino acid residue in the benzodiazepine binding pocket [Zhu, et al., 2018]. Among them SINA is the compound with the weakest inhibitory potential, while the rest have estimated inhibitory constants Ki in the range 2.9 - 4.0 (see Table 6).
The detailed results of the re-docking experiments generated similar profile of the interaction of the amino-acid residues (see Figure 14).
Since MEM is among the very few approved drugs for management of AD as NMDA receptor antagonist and is part of the hybrid molecule, it was decided to be performed docking simulation with the active site of the NMDA receptor.
MEM has relatively weak to moderate binding energy of -6.13 kcal/mol. It forms two hydrogen bonds amino acids from both protein chains namely: Asn599d and Asn606a (see Figure 15). It has six hydrophobic interactions with amino acids from two protein chains: Asn598d, Asn599d, Ala622d, Val623d, Leu626d and Asn606a.
SINA has relatively weak binding energy of -4.59 kcal/mol. It forms four hydrogen bonds amino acids from both protein chains namely: Leu595d, Asn598d, Asn599d and Leu604a (see Figure 16). It has only one hydrophobic interaction with amino acids from protein chain d: Val623d.
The memantine hybrid molecule has the lowest binding energy among the tested compounds with value -8.91 kcal/mol. It forms two hydrogen bonds amino acids from both protein chains namely: Asn599d and Leu605a (see Figure 17). It has six hydrophobic interactions with amino acids from one protein chain: Leu595d, Asn598d, Asn599d, Ala622d, Val623d, Leu626d.
It seems that MEM and SINA-MEM rely more on the hydrophobic interactions for binding to the ion channel protein chains and they share most of the amino acid residues they interact with. While the SINA relies more on forming hydrogen bonds with the surrounding amino acid residues. All of the compounds form hydrogen bonds with the important for the ion channel blockage Asn residues or/and they have hydrophobic interactions with them (Asn598d, Asn599d, Asn606a).
The detailed results of the re-docking experiments generated similar profile of the interaction of the amino-acid residues (see Figure 18).
Sinapic acid is the weakest ion channel inhibitor with highest binding energy (-4.59 kcal/mol) and estimated inhibitory constant (428.54 µM). While the hybrid molecule SINA-MEM is the most potent inhibitor with lowest binding energy (-8.91 kcal/mol) and estimated inhibitory constant (294.18 nM) (see Table 7).
It is noteworthy to mention that both MEM and SINA-MEM show preference for binding at the ion channel opening vicinity, while the SINA shows no such trend.
2.4. Biochemical Testing Results
The results of the AChE activity assay show that the treatment with SINA has the strongest impact among the tested compounds, followed by GNT, an then SINA-MEM in reducing the AChE activity (see Figure 19) which is in concordance with the results from a study of AD related pathological conditions as the induced brain hypoxia [Bais et al., 2017]. Another in vitro study shows that sinapic acid is potent AChE inhibitor [Szwajgier & Borowiec, 2012], which may explain our results, however our study further shows that the N-Sinapoyl-memantine has weaker impact upon the AChE activity which probably can be accounted to the additional memantine group. Since memantine being NMDA receptor antagonist has no inhibitory effects upon acetylcholinesterase activity as our study confirms plus it is rather bulky moiety which probably may hinder or obstruct the inhibitory action of the sinapic acid moiety in the synthesized molecule.
Part of these results superficially may seem to contradict some of the findings from the molecular docking. For example, SINA showed the most pronounced effect upon AChE acivity in our in vitro testing, but has the weakest score in the docking results among the tested compounds, which is comparable with ACH itself. However, when ACH enters in the AChE active site compartment it rather quickly is catalyzed to smaller products some of which are removed either through the backdoor in the active site bottom as Silman and Sussman [Silman & Sussman, 2008] suggest or they diffuse through the AChE gorge entrance. The situation with the SINA is different. When it enters the AChE active site compartment trough the narrow opening it actually gets trapped there. Additionally, it has three oxygen atoms that can induce strong polarization, which is predisposition for various relatively strong non-covalent interactions with the amino acid residues. We presented here in detail only the best conformation of interaction of the ligand with the protein. However, our molecular docking results show that SINA is capable to interact with many different residues in the AChE active site vicinity. So, once it gets detached from one conformation it is very possible to bind to another part of the active site instead to leave the active site completely.
The hybrid compound SINA-MEM from another side has rather low binding energy but does not perform so well in the in vitro assay (see Figure 19). This to some extend can be attributed to the fact that less than half of the molecule enters the AChE active site compartment while the bigger part of it remains outside the narrowing of the gorge. Here we should take into account that in reality the enzyme actually “breathes” as Silman and Sussman [Silman & Sussman, 2008] suggest. With such long bulky tail it is easier for the enzyme to exhale the inhibiting molecule at some point regardless its good binding capacities. And also probably it is more difficult for such long and bulky molecule to enter the gorge and pass through the mentioned narrowing.
2.5. Behavioral Testing Results
The animals with AD model show higher anxiety than the control group, while the tested compounds reduce significantly this parameter at P<0,001, with best results with sinapic acid, which nearly restores it to the level of the control group (see Figure 20). This corresponds to the results from previous studies with oral treatment with sinapic acid in mice [Yoon et al., 2007]. This effect probably is mediated with action upon the GABA receptors, which are known to be involved in the anxiety related states [Yoon et al., 2007]. The - hybrid molecule shows less pronounced anxiolytic effects, but still higher than to the other parent molecule – memantine which is also known to have some anxiolytic effects in rodents and to be NMDA receptor antagonist [Minkeviciene et al., 2008]. Therefore, the anxiolytic action of the hybrid molecule probably can be attributed to its impact upon GABA and NMDA receptors.
The performance on the passive avoidance test has some mixed effects (see Figure 21). There was improvement in all of the tested compounds which is with concordance with previous studies with the parental molecules in AD animal models [Karakida et al., 2007; Abdel-Aal et al., 2011], however only the synthesized molecule had statistically significant (at P<0.05) improvement of the learning and memory performance in the Sco AD model with levels comparable to the control group and the already approved AChE inhibitor GNT. Such less pronounced effects in the case with the parental molecules may be attributed to the relatively low doses in this particular model of AD, since another study showed a dose dependence in the observed cognitive ameliorative effects [Abdel-Aal et al., 2011].
Additionally, these in vivo results only partly confirm the in silico findings showing the SINA-MEM compound as the most potent NMDA ion channel inhibitor among the tested ones. Such subpar results can be attributed to the sophisticated involvement of the NMDAR in the pathophysiology in AD and it's complex molecular mechanisms of regulation as shown in recent molecular studies of the inhibition and control of these channels with MEM and other agents [Chou, et al., 2022; Song et al., 2018].
3. Discussion
The results from the NMR and ATR-IR spectroscopy and X-ray diffraction analysis revealed that the new hybrid molecule inherits the planarity of the SINA parent and to significant extent the 3D structure of the MEM parent. However the addition of the MEM moiety to SINA has some more pronounced far reaching impact due to the resonant properties of the formed conjugated quantum electronic system. Thus the most pronounced changes were observed besides the ones near the –C-N-C- bonds, but also at the most distant from them – the hydroxyl group at the aromatic ring, which shortens the overall length of the molecule. This means that the charge distribution, polarization pattern and dipole moment are tweaked to various extent. And this is predisposition for different pharmacological behavior in the delicate fine tuned micro environment of the active centers and other functional sites of the target biomolecules as enzymes and receptors. Such modifications of the electronic shield are probably to be blamed for the differences in the interaction profiles since the overall spatial changes of the atoms in the new hybrid molecule are often less than 0.01 A.
With its polarized aromatic ring paired with specific aliphatic chain that further extends the conjugated quantum system enables the new hybrid molecule to pair with other aromatic amino acid moieties trough π- stacking and pairing and other weak interactions as the one like Tyrosine significantly present in the vicinity of the active sites of AChE and the GABAA receptor. On the other side the presence of strongly electronegative atoms as oxygen facilitates the Van der Waals interactions and H-bond formation with the respective polarized amino acid moieties which are also present within and around the active sites. Thus the new hybrid compound can interact with most of the important amino acid moieties in the active sites and partly mimics the interaction of the established pharmacological agents. These observations are in concordance with the in vivo and biochemical results were it scored admirably being on par with GNT on the in vivo testing.
As for the effects upon the important NMDAR receptors the results are less straightforward. Although the new hybrid molecule has best binding energy according to the docking results it performed slightly worse than the established pharmacological agent in the management of these receptors – MEM. Among the possible reasons for these discrepancies in the results are that the NMDAR receptors have relatively complex and dynamic mechanisms of their regulation as recent studies with molecular dynamics and other biophysical methods revealed [Chou, et al., 2022; Song et al., 2018]. And especially for their usage in Alzheimer's disease the agent for NMDAR receptors is the one that only partly inhibits their function and not in a static fashion but in dynamic one. And that is among the reasons why MEM performs so well there – because it has low polarizability but due to its specific cyclic geometric shape it has specific polarized charge distribution pattern that facilitates weak interactions in dynamic way which is necessary to control the micro currents in the vicinity of the NMDAR ion channel. Therefore although the new hybrid molecule has the MEM moiety with relatively preserved geometry and charge distribution, it can have slightly lesser in vivo effect. Because for this particular receptor the ideal pharmacological agent must be able to interact dynamically in the channel core – not too strongly, but not too weekly either. MEM performs relatively well in this regard, while its child SINA-MEM performs slightly subpar probably due to its slightly stronger interaction than the optimal level for this pathological physiological condition.
Overall the QSAR analysis and the in vivo studies show that the new hybrid molecule with its low toxicity and low to medium penetration and bioavailability has significant pharmacological potential in the management of the Alzheimer's disease.
4. Conclusions
The synthesized N-Sinapoyl-memantine molecule showed better ameliorative effects in the key AD symptom as the cognitive decline than the parental molecules. Additionally, it has good anxiolytic properties and excellent anti-oxidant potency, which is also important since it, is known that AD is oxidative stress related disease. Therefore, the synthesized compound deserves further exploration and testing as prospective AD drug.
5. Materials and Methods
5.1. Reagents
(E)-3,5-dimethoxy-4-hydroxycinnamic (sinapic acid, SINA), 1-[3-(dimethylamino)propyl]-3-ethyl carbodiimide hydrochloride (EDC), HOBt (N-hydroxybenzotriazole), NMM (N-methylmorpholine were purchased from Sigma-Aldrich. All solvents were reagent grade and used without further purification.
5.2. Chemical and Structural Analysis
5.2.1. X-Ray Diffraction Analysis
To get single crystals of sufficient grade, the compound dry powder was dissolved in acetone at 290 K. Slow evaporation produced large crystals (0.3 × 0.2 × 0.15 mm3) appropriate for single crystal X-ray experiments in 2-3 days.
From each compound a single crystal was put on a glass capillary, and all data were acquired at 290 K using an Oxford diffraction Supernova diffractometer utilizing Mo-K α radiation (λ = 0.71013 ˚A) from a micro-focus source. CrysalisPro [Agilent Technologies, 2011] was used to determine unit cell characteristics, integrate data, scale, and make absorption adjustments. The structures were solved using direct techniques (ShelxS-2018 [Sheldrick, 2008]). The structures were refined over numerous cycles using full-matrix least-squares on F 2 with the ShelxL-2018 package [Sheldrick, 2008]. The N hydrogen atoms were positioned using the difference Fourier map, whereas all other hydrogen atoms were placed in idealized places. The non-hydrogen atoms were refined anisotropically, whereas hydrogen atoms were refined using the riding model. ORTEP [Farrugia, 2012] was used to create visual representations of the molecules in the asymmetric unit, and Mercury [Macrae, et al., 2008] was used to depict 3D packing and hydrogen bonding interactions. The Crystallographic Information File's data (coordinates, structure factors, etc.) were checked by IUCr checkCIF/ PLATON [Spek, 2009]. Crystallographic data (excluding structure factors) for the structural investigation for the novel hybrid compound were deposited with the Cambridge Crystallographic Data Centre, CCDC No. Deposition Number 1,957,027.
Further analysis of the data was done with software packages as Vesta and Biovia Studio [Momma, & Izumi, 2011; Biovia, 2019].
5.2.2. ATR-IR Spectroscopy
Measurements for the attenuated total reflectance infrared spectroscopy (ATR-IR) were made using the Thermo Scientific Nicolet iS10 FT-IR equipment with ID5 ATR accessory (diamond crystal) at temperature at 293 K. The NMR spectra were captured using a Bruker Avance II + spectrometer (14.09 T magnet) with a 5 mm BBO probe and a z-gradient coil, running at 600.01 MHz 1H frequencies. Using a Bruker B-VT 30 0 0 temperature unit with an airflow of 535 L/h, the temperature is kept at 293 K. Every chemical shift is expressed in parts per million (ppm), using tetramethylsilane (TMS, 0.00 ppm) or the residual solvent signal of 2.5 ppm for DMSO as a reference.
5.2.3. NMR Spectroscopy
The sample was prepared as follows: a stock solution of trimethoxybenzene (TMB) in DMSO-d6 at a concentration of 8.44 mM (7.1 mg per 5 ml) was produced. Weighted quantities of the compounds were dissolved in 0.6 ml stock solution, and 1H spectra were obtained.
The Bruker Avance II + spectrometer (14.09 T magnet) with a 5 mm BBO probe and z-gradient coil was used to record all NMR spectra. It operated at 600.01 MHz 1H frequencies. A Bruker B-VT 30 0 0 temperature unit with an airflow of 535 L/h is used to keep the temperature at 293 K. Tetramethylsilane (TMS, 0.00 ppm) or the residual solvent signal of 2.5 ppm for DMSO are used as references for all chemical shifts, which are expressed in parts per million (ppm).
The Bruker pulse sequences cosygpmfqf and hsqcedetgpsp.3 were used to capture two-dimensional (2D) 1 H/ 1 H COZY and (2D) 1 H/ 13 C HSQC spectra.
5.2.4. Animals
The experimental male IRC mice (18-22 g) were maintained in a controlled environment (20 ± 2°C, 50 ± 10% relative humidity, 12-h light/dark cycle) housed in plastic cages where free access to food and water were provided. Each of the experimental groups consisted of 8-10 animals. Compliance of the experiments with the guidelines and practice established by the Ethical Committees of the Institute of Neurobiology, Sofia, Bulgaria and in conformance with the European Convention on Animal Protection and Guidelines on Research Animal Use was strictly followed.
5.2.5. Experimental Protocol of the AD Model
The model of AD was induced in the experimental animals through daily treatment with single i.p. injection of 1 mg/kg scopolamine for duration of 11 days [Tancheva et al., 2018]. The model was verified by using cognitive and biochemical assays and markers, as learning and memory behavioral tasks and acetylcholine esterase (AChE) activity in brain.
5.2.6. Drug Treatment and Experimental Design
The mice were separated into the following groups: control group; SCO group; MEM group; SINA group and SINA-MEM group. The control group was treated daily with single saline injections (saline 0.1ml/10g, i.p).The other groups received daily injections of scopolamine (scopolamine solution 1mg/kg, i.p) plus the specific substance for each group with doses as follows: SCO group (scopolamine solution 1mg/kg, i.p); MEM group (memantine 20 mg/kg, i.p) + (scopolamine 1mg/kg, i.p); SINA group (sinapic acid 10 mg/kg, i.p) + (scopolamine 1mg/kg, i.p); SINA-MEM group (SINA-MEM 20 mg/kg, i.p) + (scopolamine 1mg/kg, i.p). The daily treatment with the respective substances for all groups continued for 11 days.
The selected doses of the used compounds were chosen on the basis of our previous preliminary studies (data not published) and data from the literature [Li et al., 2013; de Moura et al., 2015].
5.3. Behavioral Tests
5.3.1. Step Trough Passive Avoidance Learning Test
The passive avoidance learning test (Step through test) was utilized to evaluate the learning and memory performance in the mice [Jarvik and Kopp, 1967]. The utilized apparatus in this test consisted of two compartments: the first compartment was illuminated and the other one was kept dark. The floor of the later compartment was built of stainless steel rods through which it was delivered an electrical shock to the animals. These compartments were kept separated by a wall with guillotine door. During the acquisition phase every animal was placed in the lightened compartment. Shortly after the animal entered into the dark compartment they received electrical foot shock (0.5 mA, 1 s). Therefore, during this phase the rodent learned that the entrance into the dark compartment is related with negative consequences. In this phase it was recorded the initial latency (IL, acquisition latency time) of the entrance into the dark chamber, and mice with ILs > 60 s were excluded from the study. The test phase was done 1 hour after the acquisition phase and also on the 6th and 10th day, during which each animal was placed in the lightened chamber for evaluation of passive avoidance. The measured parameter (named as step through latency (STL)) was the duration between the placement in the illuminated compartment and the entry into the dark compartment. The behavioural tests were carried out between 9 a.m. to 12 a.m.
After the completion of the planned behavioral tests the animals were euthanized with CO2 followed by quick removal of their brains further biochemical analyses.
5.3.2. Hole Board Test
For the test it was used a special apparatus comprising of perforated floorboard which was made of plastic (45 cm × 45 cm × 25 cm) and it was drilled with 16 evenly spaced holes (3 cm in diameter); the board was delimited by a square box having 50 cm in height made of plexiglass [Aguirre-Hernández et al., 2007]. The registered parameter for each animal was the number of head-dips during a 3 min trial.
The testing was done 1 hour and also on the 5th and 10th day after the treatment with scopolamine/saline of the animals.
5.3.3. Molecular Docking and Screening
For the in silico molecular docking experiments were used two software packages AutoDock Tools 4.2.6 and iGemdock, which use different scoring algorithms for cross validation purposes according to the protocols by Shlyahtun et al. and Azad, et al. [Shlyahtun et al. 2021; Azad, et al., 2021]. Briefly with custom scripts were performed screening of the active sites of the target molecular complexes with box of 20A. The most prospective conformations were selected and further evaluated at ligand - amino acid residue level. The target molecular complexes were acquired from the publicly available protein data banks. Microsoft Excel and Origin Pro packages were used for the data processing.
Funding
This work was funded by the Bulgarian National Science Fund (BNSF), grant number KP-06-Russia/7-2019.
Institutional Review Board Statement
The animal study protocol was approved by the Commission of Bioethics (CBE) at the Institute of Neurobiology, Bulgarian Academy of Sciences—BAS/CBE/018/2020.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Abdel-Aal, R. A., Assi, A. A. A., & Kostandy, B. B. (2011). Memantine prevents aluminum-induced cognitive deficit in rats. Behavioural brain research, 225(1), 31-38. [CrossRef]
- Agilent Technologies (2011). P. CrysAlis , Version 171.36. 20, Agilent Technologies UK Ltd, Yarnton, Oxfordshire, UK.
- Aguirre-Hernández, E., Martínez, A. L., González-Trujano, M. E., Moreno, J., Vibrans, H., & Soto-Hernández, M. (2007). Pharmacological evaluation of the anxiolytic and sedative effects of Tilia americana L. var. mexicana in mice. Journal of ethnopharmacology, 109(1), 140-145. [CrossRef]
- Atanasova, M., Yordanov, N., Dimitrov, I., Berkov, S., & Doytchinova, I. (2015). Molecular docking study on galantamine derivatives as cholinesterase inhibitors. Molecular informatics, 34(6-7), 394-403. [CrossRef]
- Azad, I., Khan, T., Maurya, A. K., Irfan Azad, M., Mishra, N., & Alanazi, A. M. (2021). Identification of severe acute respiratory syndrome coronavirus-2 inhibitors through in silico structure-based virtual screening and molecular interaction studies. Journal of Molecular Recognition, 34(10), e2918. [CrossRef]
- Bais, S., Kumari, R., & Prashar, Y. (2017). Therapeutic effect of Sinapic acid in aluminium chloride induced dementia of Alzheimer's type in rats. Journal of Acute Disease, 6(4), 154-162. [CrossRef]
- Biovia, D.S. (2019) Biovia Studio. Dassault Systèmes, San Diego, USA.
- Brown, G. C., & Borutaite, V. (2006). Interactions between nitric oxide, oxygen, reactive oxygen species and reactive nitrogen species. Biochemical Society transactions, 34(Pt 5), 953–956. https://doi.org/10.1042/BST0340953. [CrossRef]
- Chen, C. (2016). Sinapic acid and its derivatives as medicine in oxidative stress-induced diseases and aging. Oxidative medicine and cellular longevity, 2016(1), 3571614. [CrossRef]
- Chochkova, M., Georgieva, A., Ilieva, T., Andreeva, M., Pramatarov, G., Petek, N., ... & Svete, J. (2022). Hybridization of Aminoadamantanes with cinnamic acid analogues and elucidation of their antioxidant profile. Journal of Chemistry, 2022(1), 7582587. [CrossRef]
- Chochkova, M., Jiang, H., Kyoseva, R., Stoykova, B., Tsvetanova, E., Alexandrova, A., ... & Shivachev, B. (2021). Cinnamoyl-memantine hybrids: Synthesis, X-ray crystallography and biological activities. Journal of Molecular Structure, 1234, 130147. [CrossRef]
- Chou, T. H., Epstein, M., Michalski, K., Fine, E., Biggin, P. C., & Furukawa, H. (2022). Structural insights into binding of therapeutic channel blockers in NMDA receptors. Nature structural & molecular biology, 29(6), 507-518. [CrossRef]
- de Moura, F. C., Godinho, W. D., Vieira, L. L., da Silva, C., Martins, P. M. S., & de Castro Brito, G. A. (2015). Current Research On Therapy In Alzheimer’s Disease Experimental Model: Beta-Amyloid1-42 Induction. Journal of Advance Research in Pharmacy & Biological Science ISSN, 2208, 2360.
- Deb, P. K., Sharma, A., Piplani, P., & Akkinepally, R. R. (2012). Molecular docking and receptor-specific 3D-QSAR studies of acetylcholinesterase inhibitors. Molecular Diversity, 16, 803-823. [CrossRef]
- Farrugia, L. J. (2012). WinGX and ORTEP for Windows: an update. Applied Crystallography, 45(4), 849-854.
- Garcia-Marin, V., Blazquez-Llorca, L., Rodriguez, J. R., Boluda, S., Muntane, G., Ferrer, I., & DeFelipe, J. (2009). Diminished perisomatic GABAergic terminals on cortical neurons adjacent to amyloid plaques. Frontiers in neuroanatomy, 3, 1102. [CrossRef]
- Georgiev, L., Chochkova, M., Totseva, I., Seizova, K., Marinova, E., Ivanova, G., ... & Milkova, T. (2013). Anti-tyrosinase, antioxidant and antimicrobial activities of hydroxycinnamoylamides. Medicinal Chemistry Research, 22, 4173-4182.
- Hameed, H., Aydin, S., & Başaran, N. (2016). Sinapic acid: is it safe for humans. FABAD Journal of Pharmaceutical Sciences, 41(1), 39.
- Hampel, H., Mesulam, M. M., Cuello, A. C., Farlow, M. R., Giacobini, E., Grossberg, G. T., ... & Khachaturian, Z. S. (2018). The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain, 141(7), 1917-1933. [CrossRef]
- Hotta, H., Nagano, S., Ueda, M., Tsujino, Y., Koyama, J., & Osakai, T. (2002). Higher radical scavenging activities of polyphenolic antioxidants can be ascribed to chemical reactions following their oxidation. Biochimica et Biophysica Acta (BBA)-General Subjects, 1572(1), 123-132. [CrossRef]
- Hudson, E. A., Dinh, P. A., Kokubun, T., Simmonds, M. S., & Gescher, A. (2000). Characterization of potentially chemopreventive phenols in extracts of brown rice that inhibit the growth of human breast and colon cancer cells. Cancer Epidemiology Biomarkers & Prevention, 9(11), 1163-1170.
- Jarvik, M. E., & Kopp, R. (1967). An improved one-trial passive avoidance learning situation. Psychological reports, 21(1), 221-224. [CrossRef]
- Kanchana, G., Shyni, W. J., Rajadurai, M., & Periasamy, R. (2011). Evaluation of antihyperglycemic effect of sinapic acid in normal and streptozotocin-induced diabetes in albino rats. Global Journal of Pharmacology, 5(1), 33-39.
- Karakida, F., Ikeya, Y., Tsunakawa, M., Yamaguchi, T., Ikarashi, Y., Takeda, S., & Aburada, M. (2007). Cerebral protective and cognition-improving effects of sinapic acid in rodents. Biological and Pharmaceutical Bulletin, 30(3), 514-519. [CrossRef]
- Kikuzaki, H., Hisamoto, M., Hirose, K., Akiyama, K., & Taniguchi, H. (2002). Antioxidant properties of ferulic acid and its related compounds. Journal of agricultural and food chemistry, 50(7), 2161-2168. [CrossRef]
- Kim, D. H., Yoon, B. H., Jung, W. Y., Kim, J. M., Park, S. J., Park, D. H., ... & Ryu, J. H. (2010). Sinapic acid attenuates kainic acid-induced hippocampal neuronal damage in mice. Neuropharmacology, 59(1-2), 20-30. [CrossRef]
- Klotz, L. O., Schroeder, P., & Sies, H. (2002). Peroxynitrite signaling: receptor tyrosine kinases and activation of stress-responsive pathways. Free Radical Biology and Medicine, 33(6), 737-743. [CrossRef]
- Laverty, D., Thomas, P., Field, M., Andersen, O. J., Gold, M. G., Biggin, P. C., ... & Smart, T. G. (2017). Crystal structures of a GABAA-receptor chimera reveal new endogenous neurosteroid-binding sites. Nature structural & molecular biology, 24(11), 977-985. [CrossRef]
- Lee, H. E., Kim, D. H., Park, S. J., Kim, J. M., Lee, Y. W., Jung, J. M., ... & Ryu, J. H. (2012). Neuroprotective effect of sinapic acid in a mouse model of amyloid β1–42 protein-induced Alzheimer's disease. Pharmacology Biochemistry and Behavior, 103(2), 260-266. [CrossRef]
- Li, F., Chen, X., Wang, F., Xu, S., Chang, L., Anwyl, R., & Wang, Q. (2013). Chronic pre-treatment with memantine prevents amyloid-beta protein-mediated long-term potentiation disruption. Neural regeneration research, 8(1), 49-55. [CrossRef]
- Limapichat, W., Yu, W. Y., Branigan, E., Lester, H. A., & Dougherty, D. A. (2013). Key binding interactions for memantine in the NMDA receptor. ACS chemical neuroscience, 4(2), 255-260. [CrossRef]
- Liu, J., Chang, L., Song, Y., Li, H., & Wu, Y. (2019). The role of NMDA receptors in Alzheimer’s disease. Frontiers in neuroscience, 13, 43. [CrossRef]
- Macrae, C. F., Bruno, I. J., Chisholm, J. A., Edgington, P. R., McCabe, P., Pidcock, E., ... & Wood, P. A. (2008). Mercury CSD 2.0–new features for the visualization and investigation of crystal structures. Applied Crystallography, 41(2), 466-470.
- Maddox, C. E., Laur, L. M., & Tian, L. (2010). Antibacterial activity of phenolic compounds against the phytopathogen Xylella fastidiosa. Current microbiology, 60, 53-58. [CrossRef]
- Miller, P. S., & Aricescu, A. R. (2014). Crystal structure of a human GABAA receptor. Nature, 512(7514), 270-275. [CrossRef]
- Minkeviciene, R., Banerjee, P., & Tanila, H. (2008). Cognition-enhancing and anxiolytic effects of memantine. Neuropharmacology, 54(7), 1079-1085. [CrossRef]
- Momma, K., & Izumi, F. (2011). VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. Applied Crystallography, 44(6), 1272-1276.
- Nenadis, N., & Tsimidou, M. (2002). Observations on the estimation of scavenging activity of phenolic compounds using rapid 1, 1-diphenyl-2-picrylhydrazyl (DPPH•) tests. Journal of the American Oil Chemists' Society, 79, 1191-1195. [CrossRef]
- Nićiforović, N., & Abramovič, H. (2014). Sinapic acid and its derivatives: natural sources and bioactivity. Comprehensive reviews in food science and food safety, 13(1), 34-51. [CrossRef]
- Petrache, A. L., Rajulawalla, A., Shi, A., Wetzel, A., Saito, T., Saido, T. C., ... & Ali, A. B. (2019). Aberrant excitatory–inhibitory synaptic mechanisms in entorhinal cortex microcircuits during the pathogenesis of Alzheimer’s disease. Cerebral Cortex, 29(4), 1834-1850. [CrossRef]
- Rice-Evans, C. A., Miller, N. J., & Paganga, G. (1996). Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free radical biology and medicine, 20(7), 933-956. [CrossRef]
- Rice-Evans, C., Miller, N., & Paganga, G. (1997). Antioxidant properties of phenolic compounds. Trends in plant science, 2(4), 152-159.
- Shahid, M., Raish, M., Ahmad, A., Bin Jardan, Y. A., Ansari, M. A., Ahad, A., ... & Al-Jenoobi, F. I. (2022). Sinapic acid ameliorates acetic acid-induced ulcerative colitis in rats by suppressing inflammation, oxidative stress, and apoptosis. Molecules, 27(13), 4139. [CrossRef]
- Sheldrick, G. M. (2008). A short history of SHELX. Foundations of crystallography, 64(1), 112-122.
- Shlyahtun A. G., Raduta E. F., Sutko I. P., Kasper E. V., Bogdevich E. V., Sevko A. A. (2021). Screening of molecular targets that determine the hypoglycemic effect of lupane triterpenoids. In Proceedings of Int. Scientific Conference "Physicochemical biology as the basis of modern medicine", Minsk, Belarus, May 21, 2021. Edited by V. V. Khrustalev, A. D. Taganovich, T. A. Khrustaleva.. BGMU Press, Minsk, Belarus, 2021. p. 362-363.
- Silman, I., & Sussman, J. L. (2008). Acetylcholinesterase: how is structure related to function?. Chemico-biological interactions, 175(1-3), 3-10.
- Song, X., Jensen, M. Ø., Jogini, V., Stein, R. A., Lee, C. H., Mchaourab, H. S., ... & Gouaux, E. (2018). Mechanism of NMDA receptor channel block by MK-801 and memantine. Biophysical Journal, 114(3), 24a-25a. [CrossRef]
- Spasova, M., Kortenska-Kancheva, V., Totseva, I., Ivanova, G., Georgiev, L., & Milkova, T. (2006). Synthesis of cinnamoyl and hydroxycinnamoyl amino acid conjugates and evaluation of their antioxidant activity. Journal of Peptide Science: An Official Publication of the European Peptide Society, 12(5), 369-375. [CrossRef]
- Spek, A. L. (2009). Structure validation in chemical crystallography. Biological crystallography, 65(2), 148-155. [CrossRef]
- Szwajgier, D., & Borowiec, K. (2012). Phenolic acids from malt are efficient acetylcholinesterase and butyrylcholinesterase inhibitors. Journal of the Institute of Brewing, 118(1), 40-48. [CrossRef]
- Tancheva, L. P., Popatanasov, A. B., Dragomanova, S. T., Tzvetanova, E. R., Aleksandrova, S. M., Alova, L. G., ... & Kalfin, R. E. (2018). New mechanisms in preventive effect of ellagic acid on cognition in mice with Alzheimer’s disease type dementia. Bulg. Chem. Commun, 50, 20-24.
- Torreilles, F., Salman-Tabcheh, S., Guérin, M. C., & Torreilles, J. (1999). Neurodegenerative disorders: the role of peroxynitrite. Brain Research Reviews, 30(2), 153-163. [CrossRef]
- Xia, P., Chen, H. S. V., Zhang, D., & Lipton, S. A. (2010). Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autapses. Journal of Neuroscience, 30(33), 11246-11250. [CrossRef]
- Yoon, B. H., Jung, J. W., Lee, J. J., Cho, Y. W., Jang, C. G., Jin, C., ... & Ryu, J. H. (2007). Anxiolytic-like effects of sinapic acid in mice. Life Sciences, 81(3), 234-240. [CrossRef]
- Yun, K. J., Koh, D. J., Kim, S. H., Park, S. J., Ryu, J. H., Kim, D. G., ... & Lee, K. T. (2008). Anti-inflammatory effects of sinapic acid through the suppression of inducible nitric oxide synthase, cyclooxygase-2, and proinflammatory cytokines expressions via nuclear factor-κB inactivation. Journal of agricultural and food chemistry, 56(21), 10265-10272. [CrossRef]
- Zhang, J., Gao, Y., Gao, Y., Munsell, B. C., & Shen, D. (2016). Detecting anatomical landmarks for fast Alzheimer’s disease diagnosis. IEEE transactions on medical imaging, 35(12), 2524-2533. [CrossRef]
- Zhang, Y., Li, P., Feng, J., & Wu, M. (2016). Dysfunction of NMDA receptors in Alzheimer’s disease. Neurological Sciences, 37, 1039-1047. [CrossRef]
- Zhu, S., Noviello, C. M., Teng, J., Walsh Jr, R. M., Kim, J. J., & Hibbs, R. E. (2018). Structure of a human synaptic GABAA receptor. Nature, 559(7712), 67-72.
- Zhu, S., Noviello, C. M., Teng, J., Walsh Jr, R. M., Kim, J. J., & Hibbs, R. E. (2018). Structure of a human synaptic GABAA receptor. Nature, 559(7712), 67-72.
- Zou, Y., Kim, A. R., Kim, J. E., Choi, J. S., & Chung, H. Y. (2002). Peroxynitrite scavenging activity of sinapic acid (3, 5-dimethoxy-4-hydroxycinnamic acid) isolated from Brassica juncea. Journal of Agricultural and Food Chemistry, 50(21), 5884-5890.
Figure 1.
Synthesis of memantine-based multitarget-directed ligand: (i) EDC/HOBt, NMM, CH2Cl2/ DMF, 1h at 0⁰C, then 23h at rt.
Figure 1.
Synthesis of memantine-based multitarget-directed ligand: (i) EDC/HOBt, NMM, CH2Cl2/ DMF, 1h at 0⁰C, then 23h at rt.
Figure 2.
3D structures of the compounds with labeled atoms. A – SINA; B. – SINA-MEM.
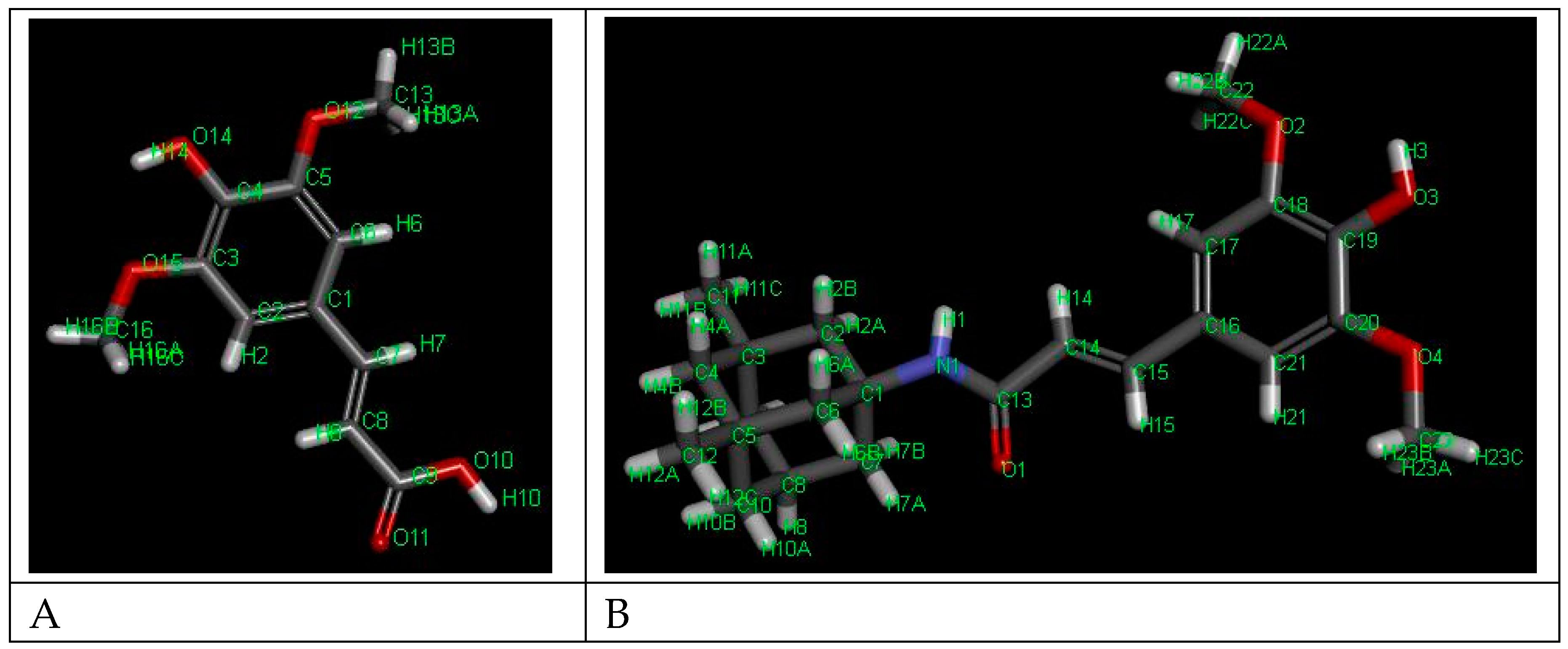
Figure 3.
Comparison of the ATR-IR spectra of the parental and the new hybrid compounds. X axis – wavenumbers; Y axis – transmittance intensity.
Figure 3.
Comparison of the ATR-IR spectra of the parental and the new hybrid compounds. X axis – wavenumbers; Y axis – transmittance intensity.
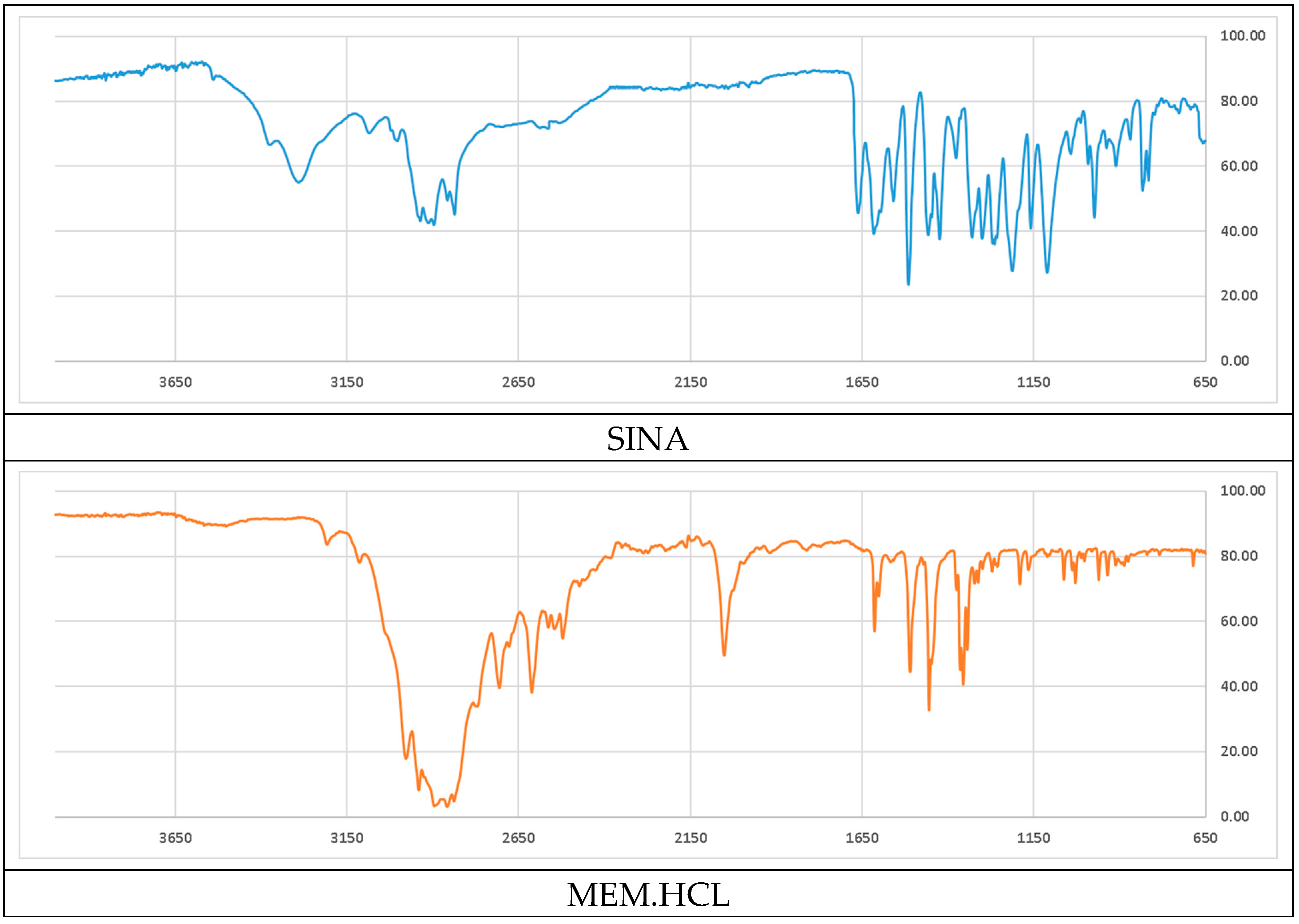
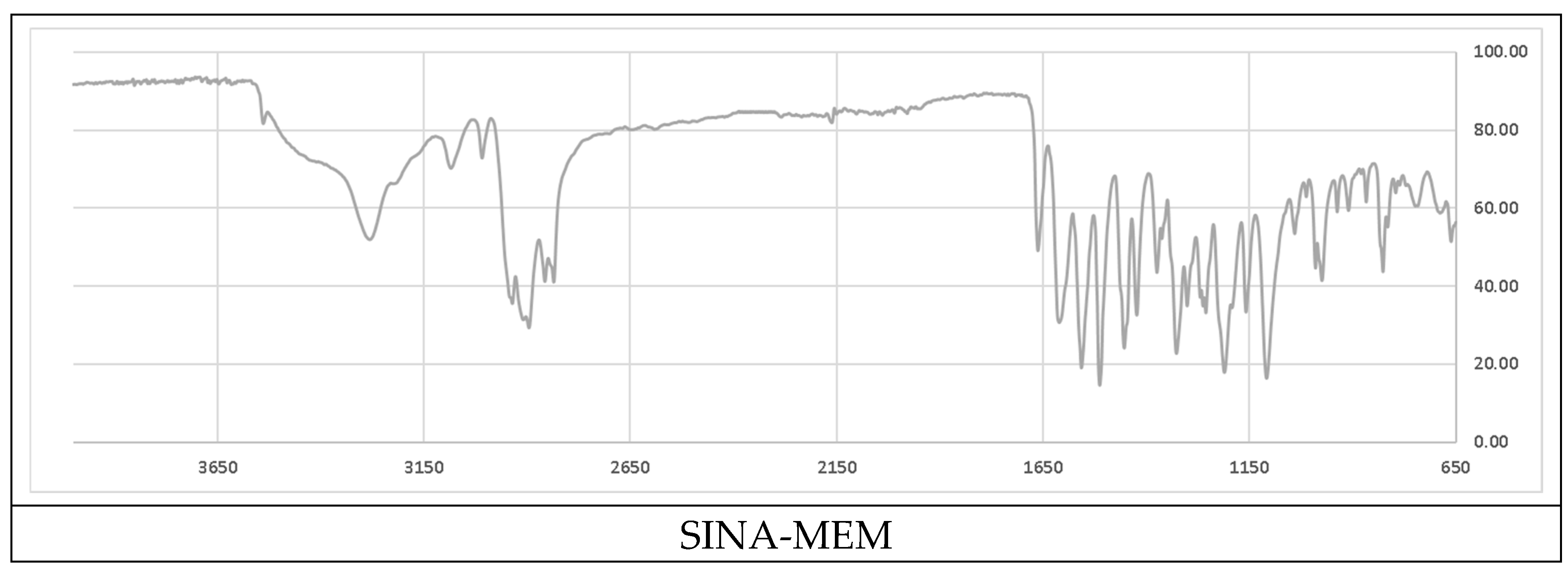
Figure 4.
NMR spectra and correlation analysis of the new hybrid compound.
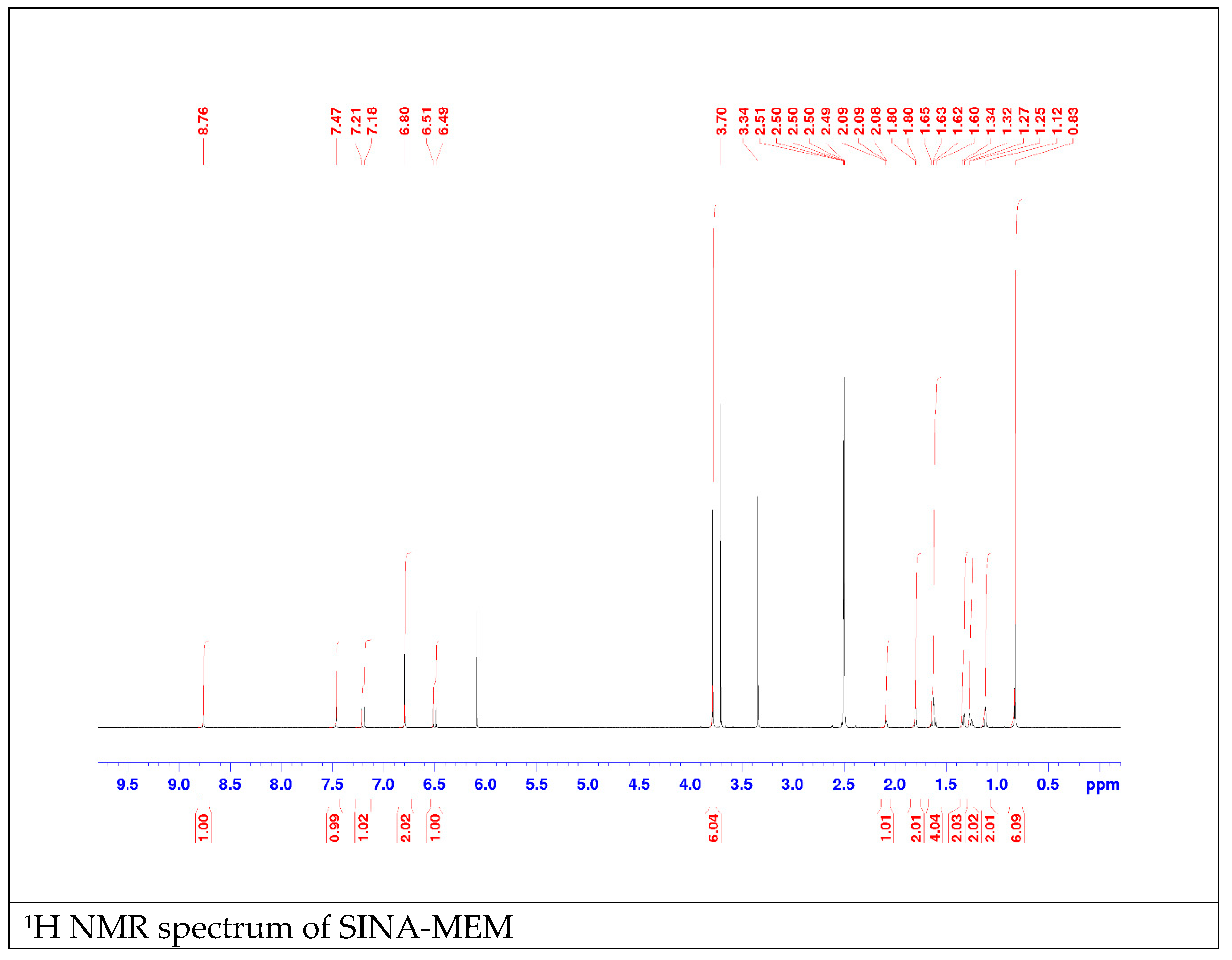
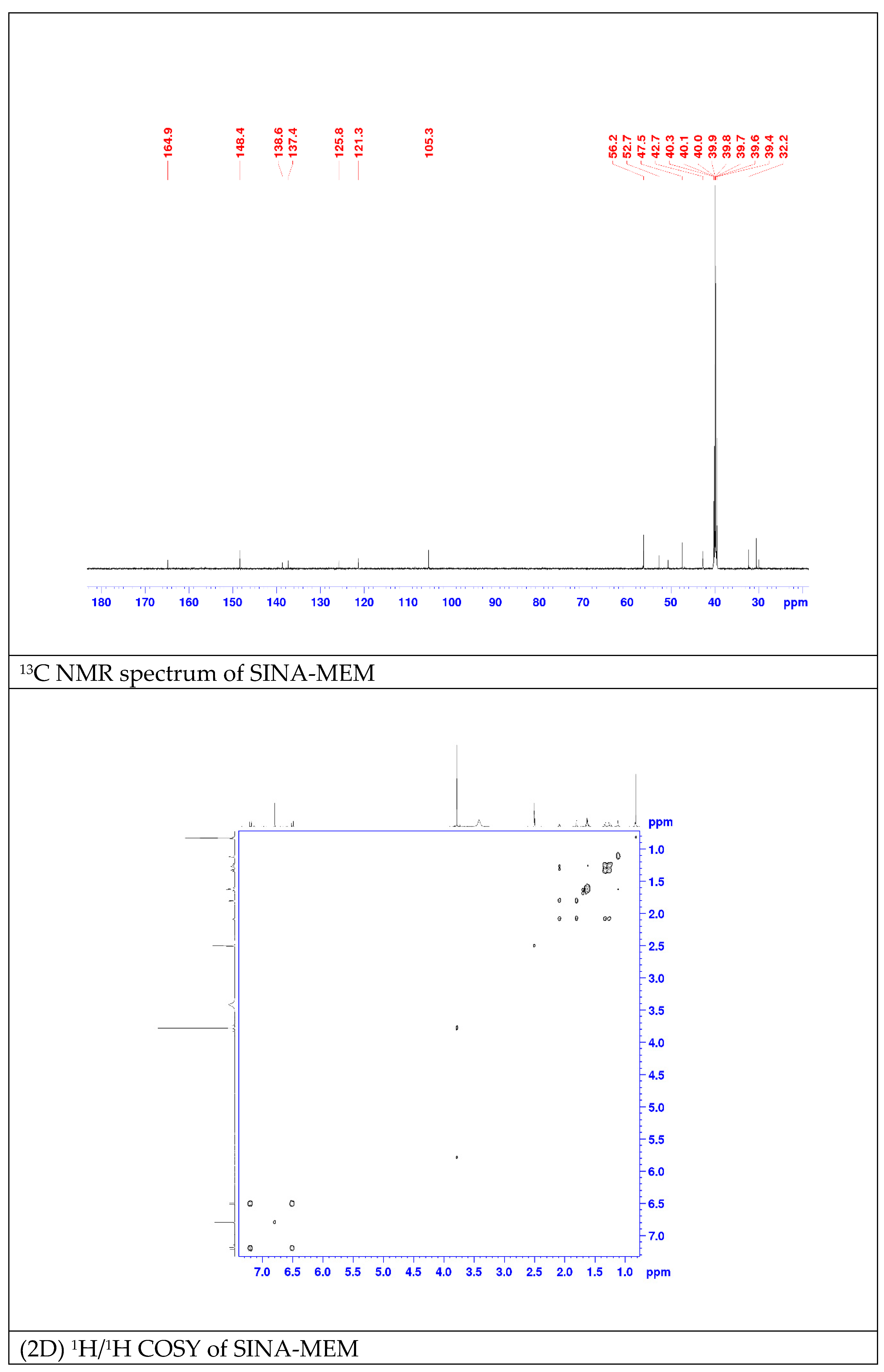
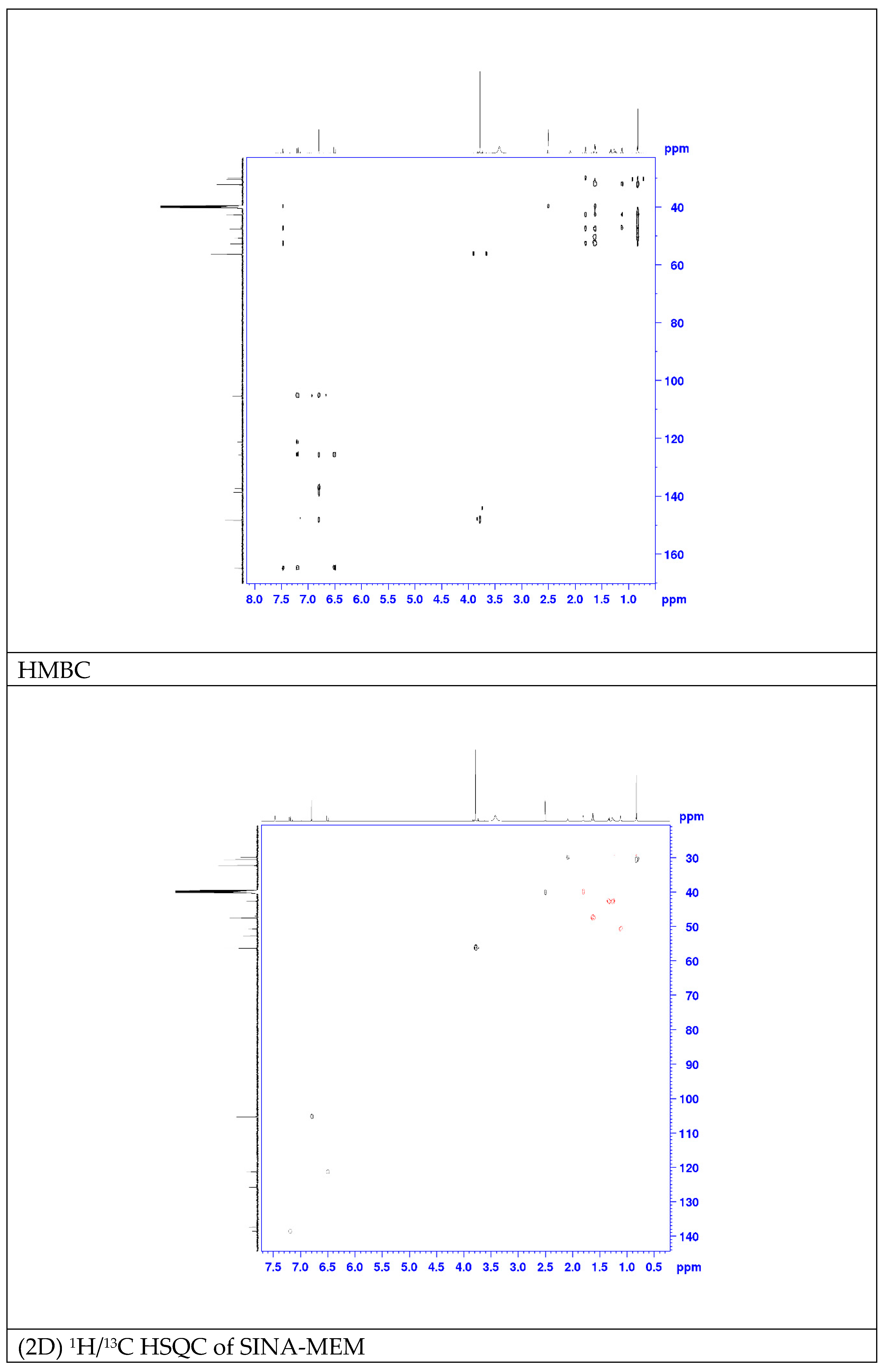
Figure 5.
2D- (A) and 3D- (B) diagrams of the interactions of SINA-MEM with AChE active site.
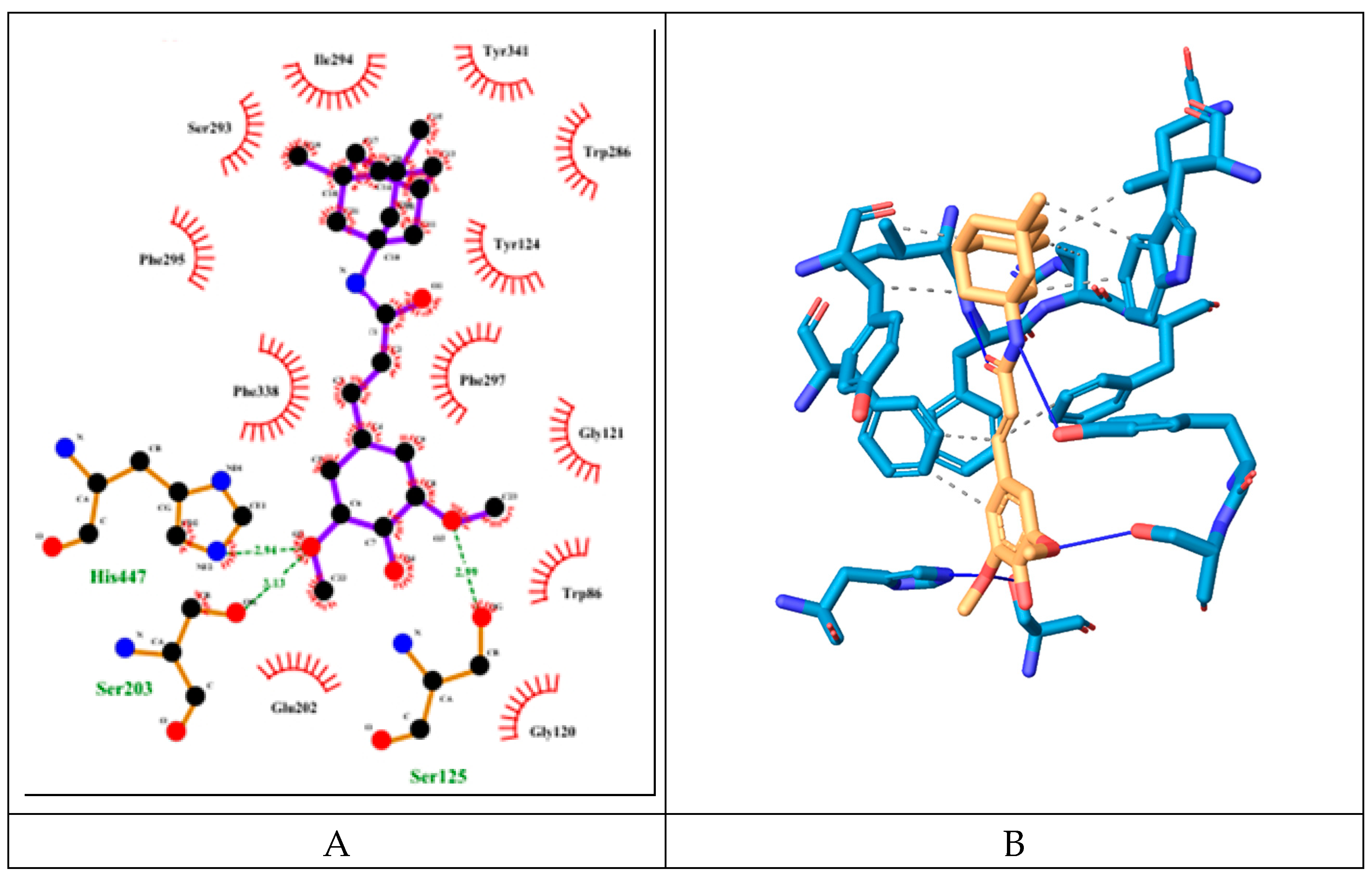
Figure 6.
2D- (A) and 3D- (B) diagrams of the interactions of SINA with AChE active site.
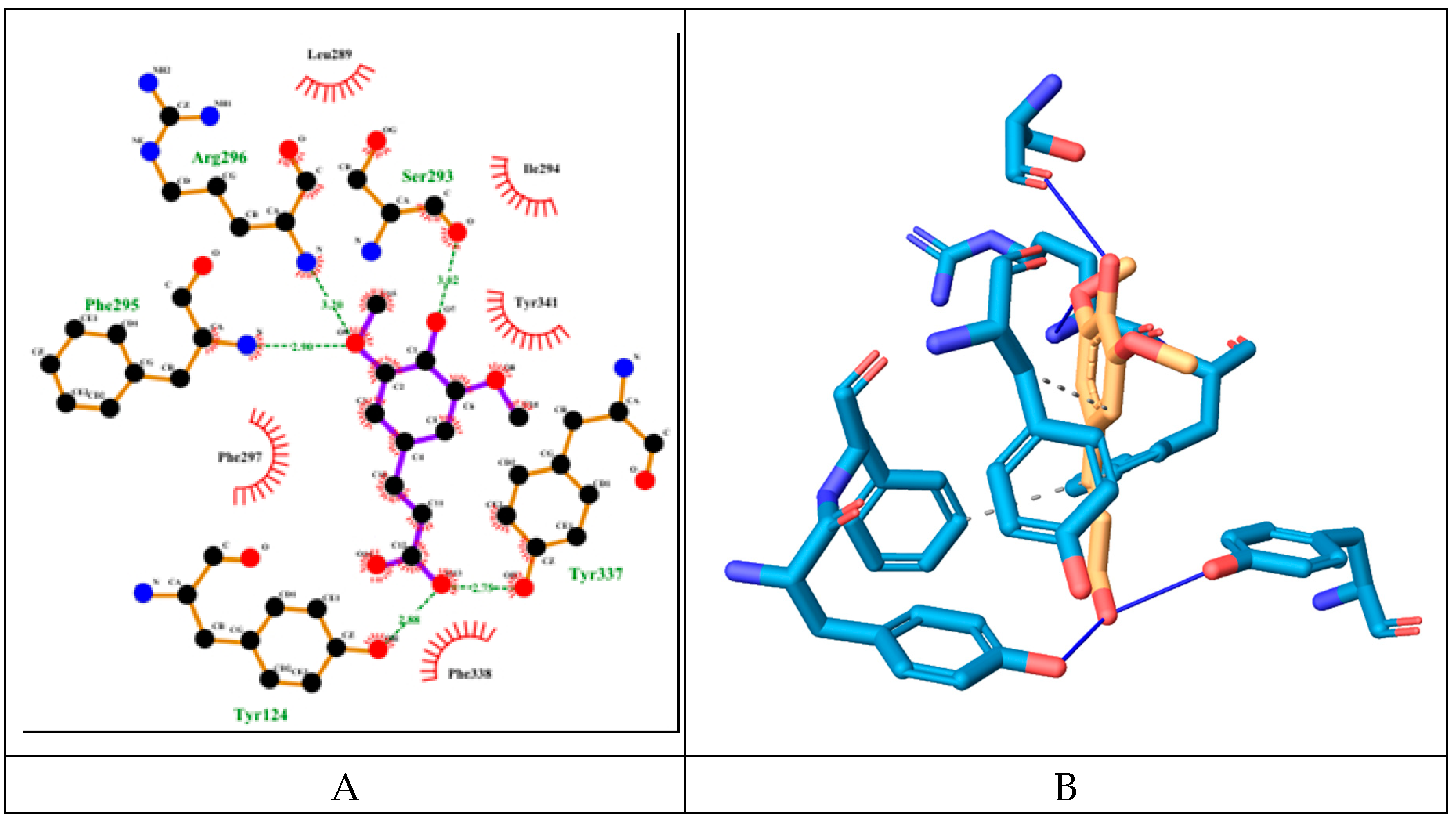
Figure 7.
2D- (A) and 3D- (B) diagrams of the interactions of MEM with AChE active site.
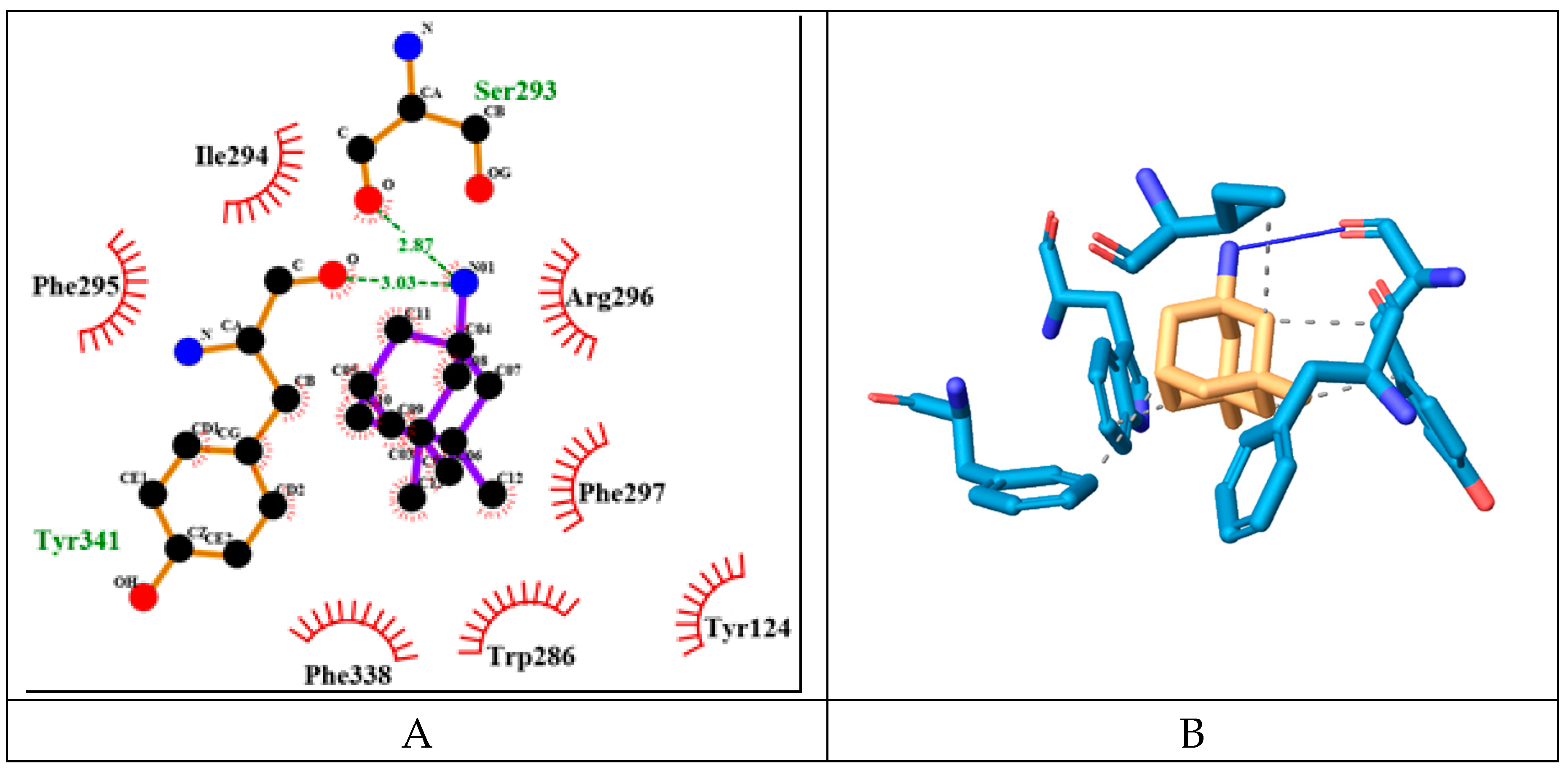
Figure 8.
2D- (A) and 3D- (B) diagrams of the interactions of GNT with AChE active site.
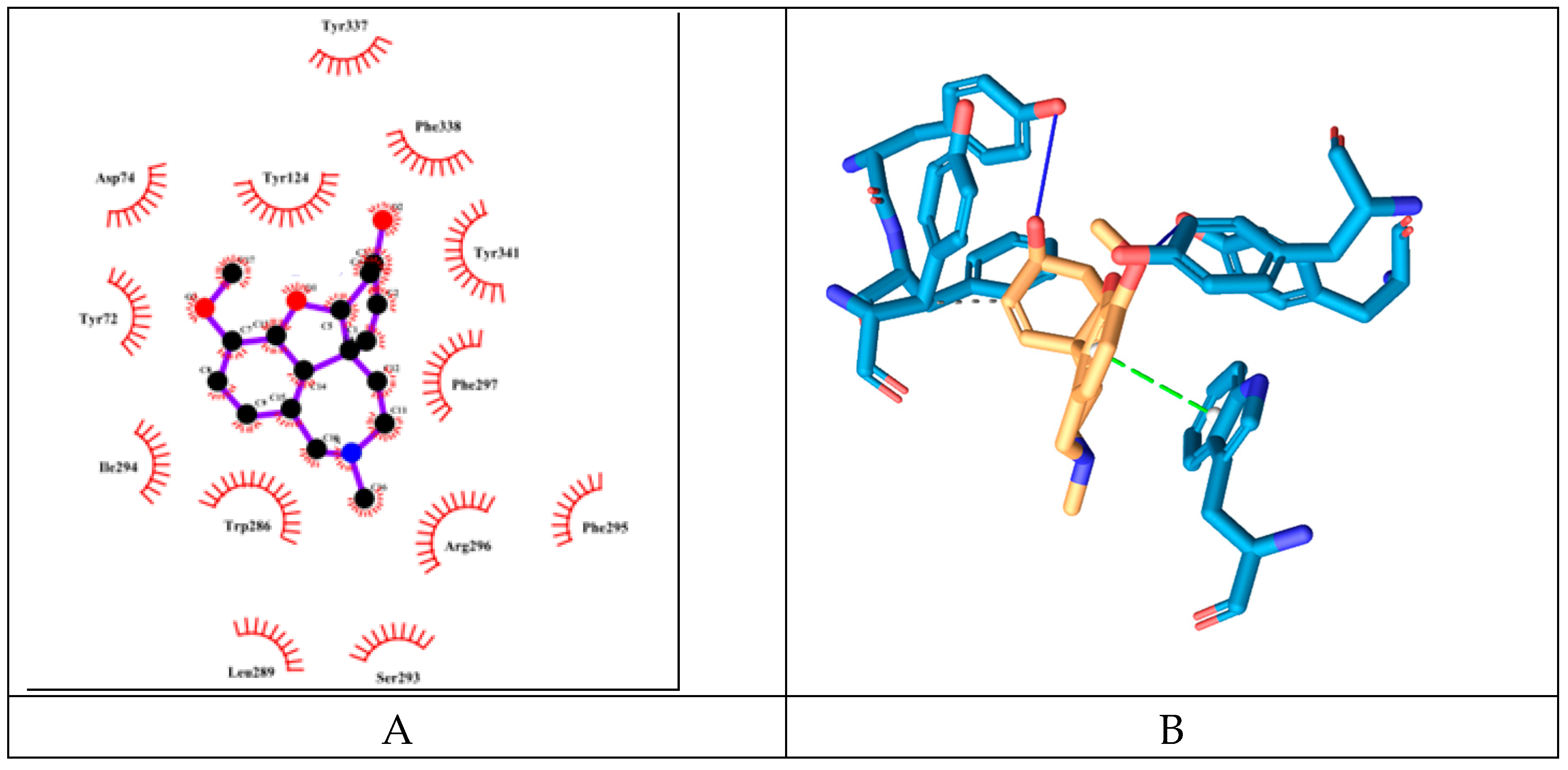
Figure 9.
Amino acid interactions profiles of the compounds with AChE active site derived from the re-docking experiments (V – Van der Waals interaction; H – hydrogen bond; S – strong; M – medium). Only interactions with value bellow -2.5 are shown.
Figure 9.
Amino acid interactions profiles of the compounds with AChE active site derived from the re-docking experiments (V – Van der Waals interaction; H – hydrogen bond; S – strong; M – medium). Only interactions with value bellow -2.5 are shown.
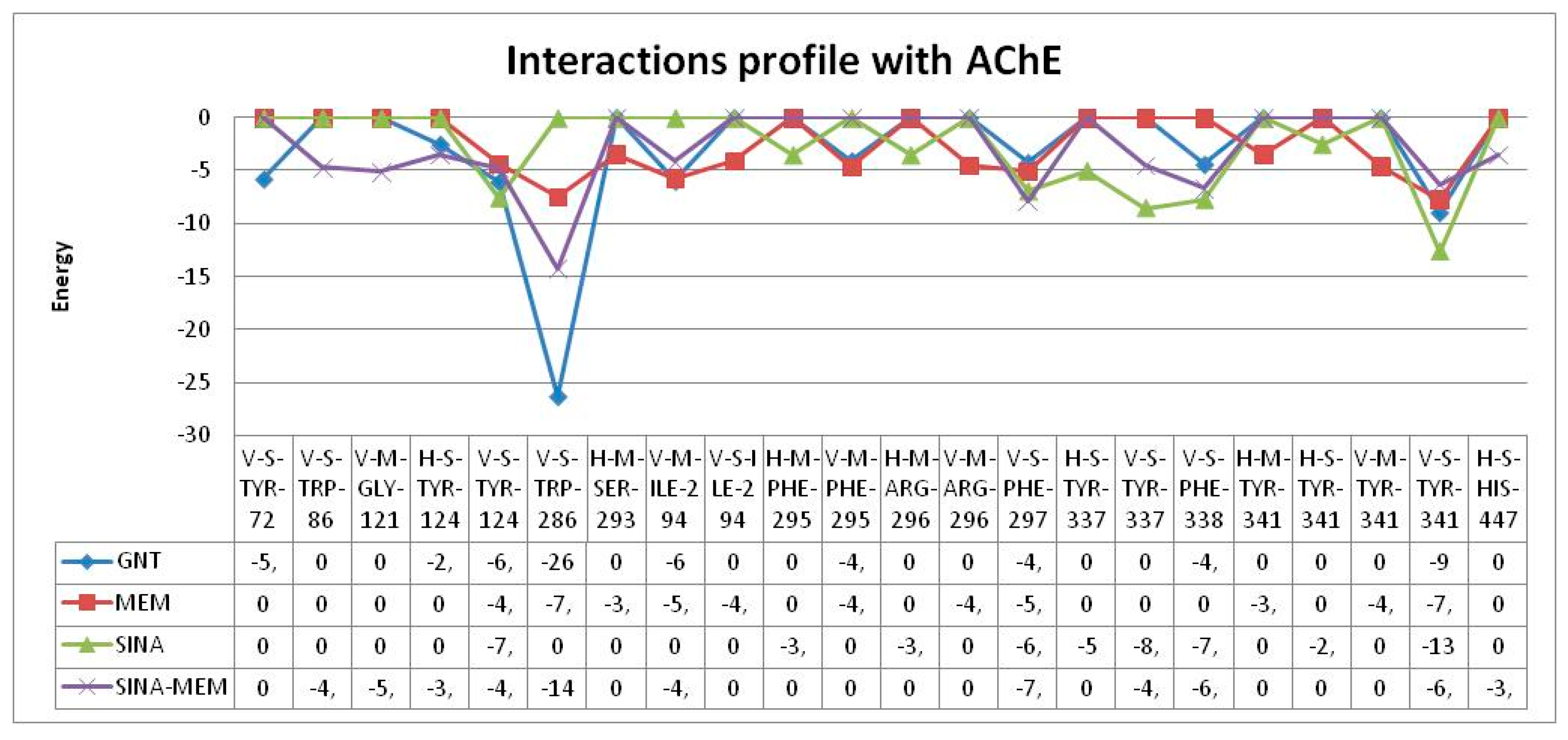
Figure 10.
2D- (A) and 3D- (B) diagrams of the interactions of Flumazenil with GABA A receptor’s benzodiazepine active site.
Figure 10.
2D- (A) and 3D- (B) diagrams of the interactions of Flumazenil with GABA A receptor’s benzodiazepine active site.
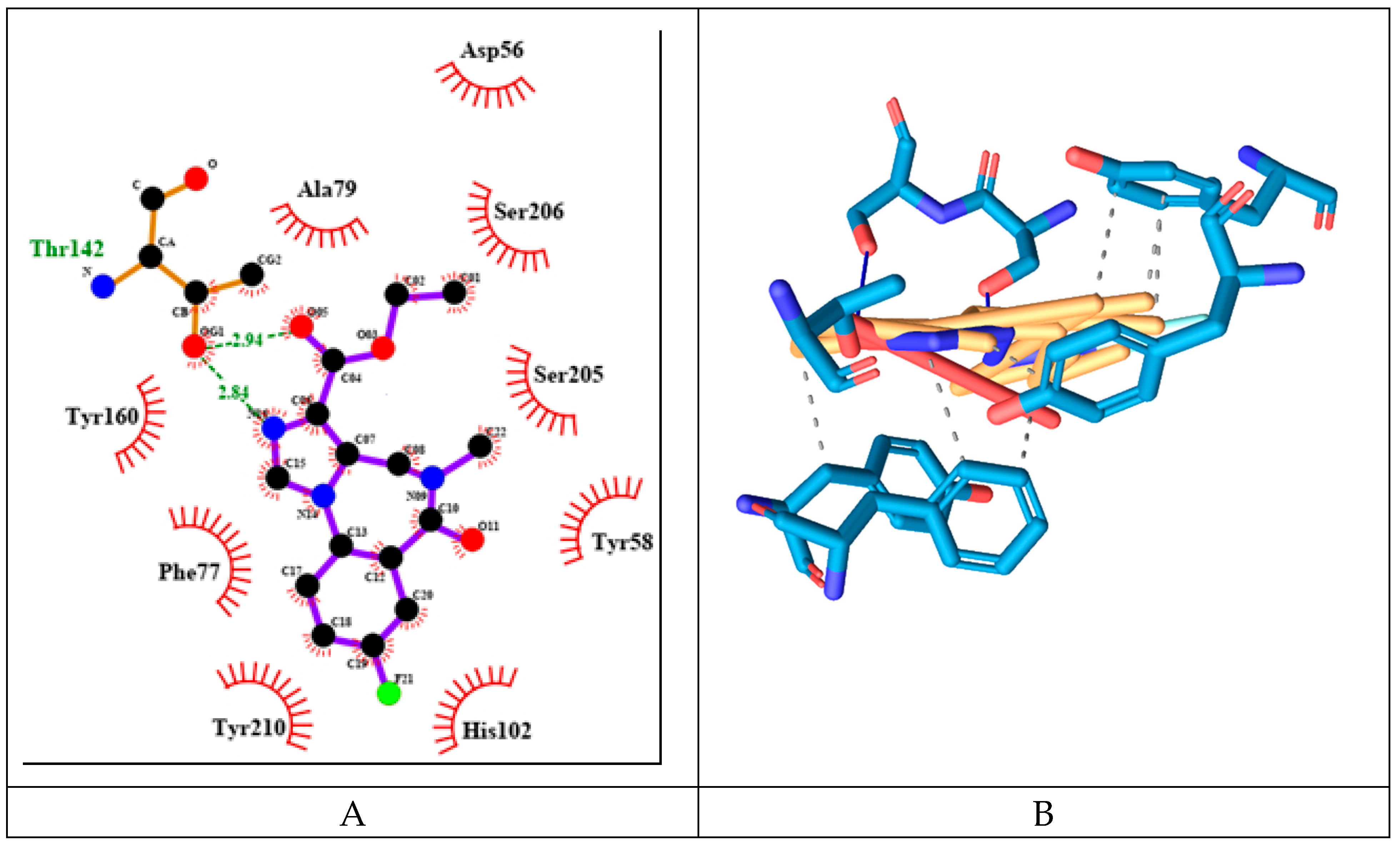
Figure 11.
2D- (A) and 3D- (B) diagrams of the interactions of MEM with GABA A receptor’s benzodiazepine active site.
Figure 11.
2D- (A) and 3D- (B) diagrams of the interactions of MEM with GABA A receptor’s benzodiazepine active site.
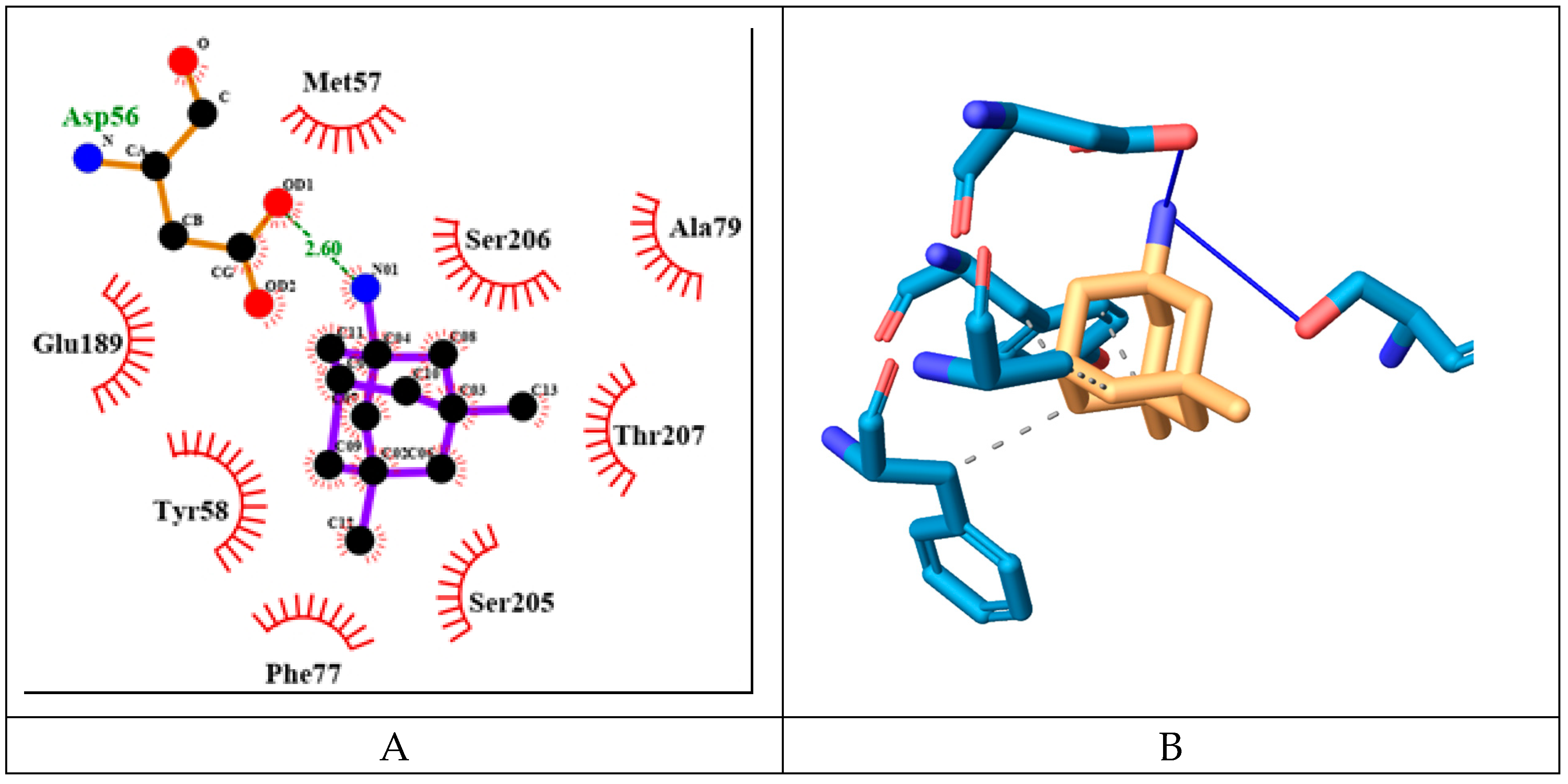
Figure 12.
2D- (A) and 3D- (B) diagrams of the interactions of SINA with GABA A receptor’s benzodiazepine active site.
Figure 12.
2D- (A) and 3D- (B) diagrams of the interactions of SINA with GABA A receptor’s benzodiazepine active site.
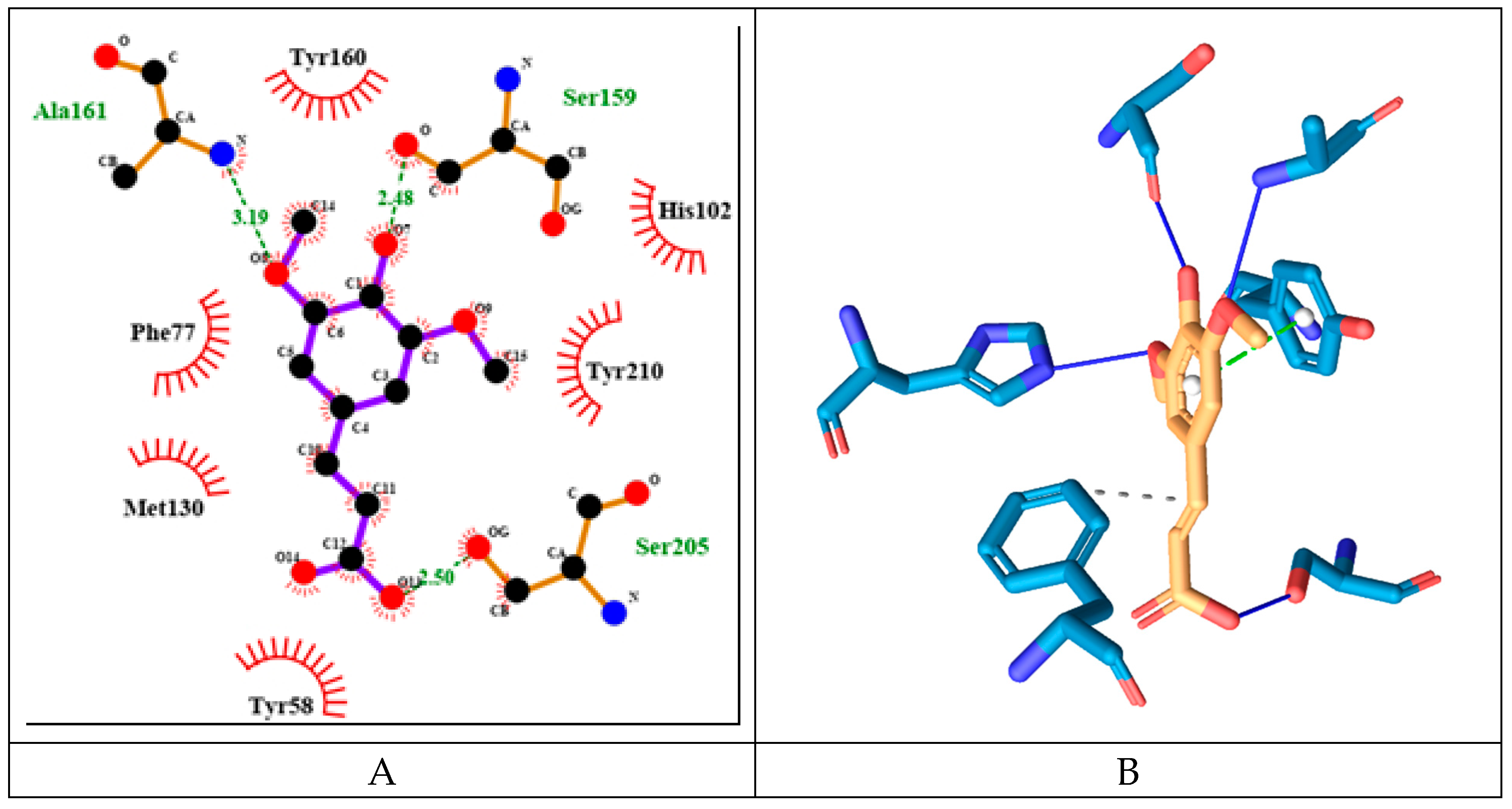
Figure 13.
2D- (A) and 3D- (B) diagrams of the interactions of SINA-MEM with GABA A receptor benzodiazepine active site.
Figure 13.
2D- (A) and 3D- (B) diagrams of the interactions of SINA-MEM with GABA A receptor benzodiazepine active site.
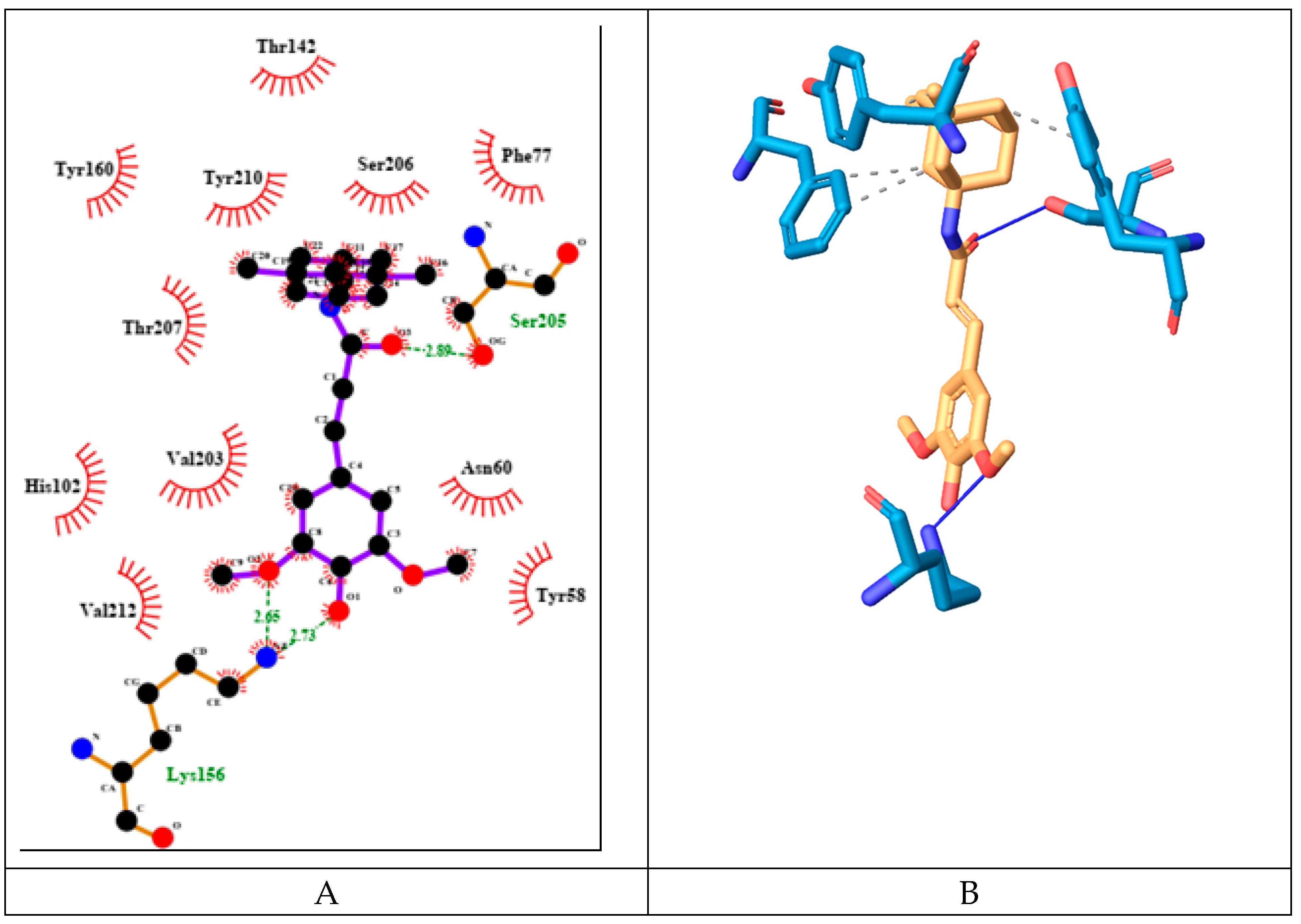
Figure 14.
Amino acid interactions profiles of the compounds with GABA A receptor’s benzodiazepine active site derived from the re-docking experiments (V – Van der Waals interaction; H – hydrogen bond; S – strong; M – medium). Only interactions with value bellow -2.5 are shown.
Figure 14.
Amino acid interactions profiles of the compounds with GABA A receptor’s benzodiazepine active site derived from the re-docking experiments (V – Van der Waals interaction; H – hydrogen bond; S – strong; M – medium). Only interactions with value bellow -2.5 are shown.
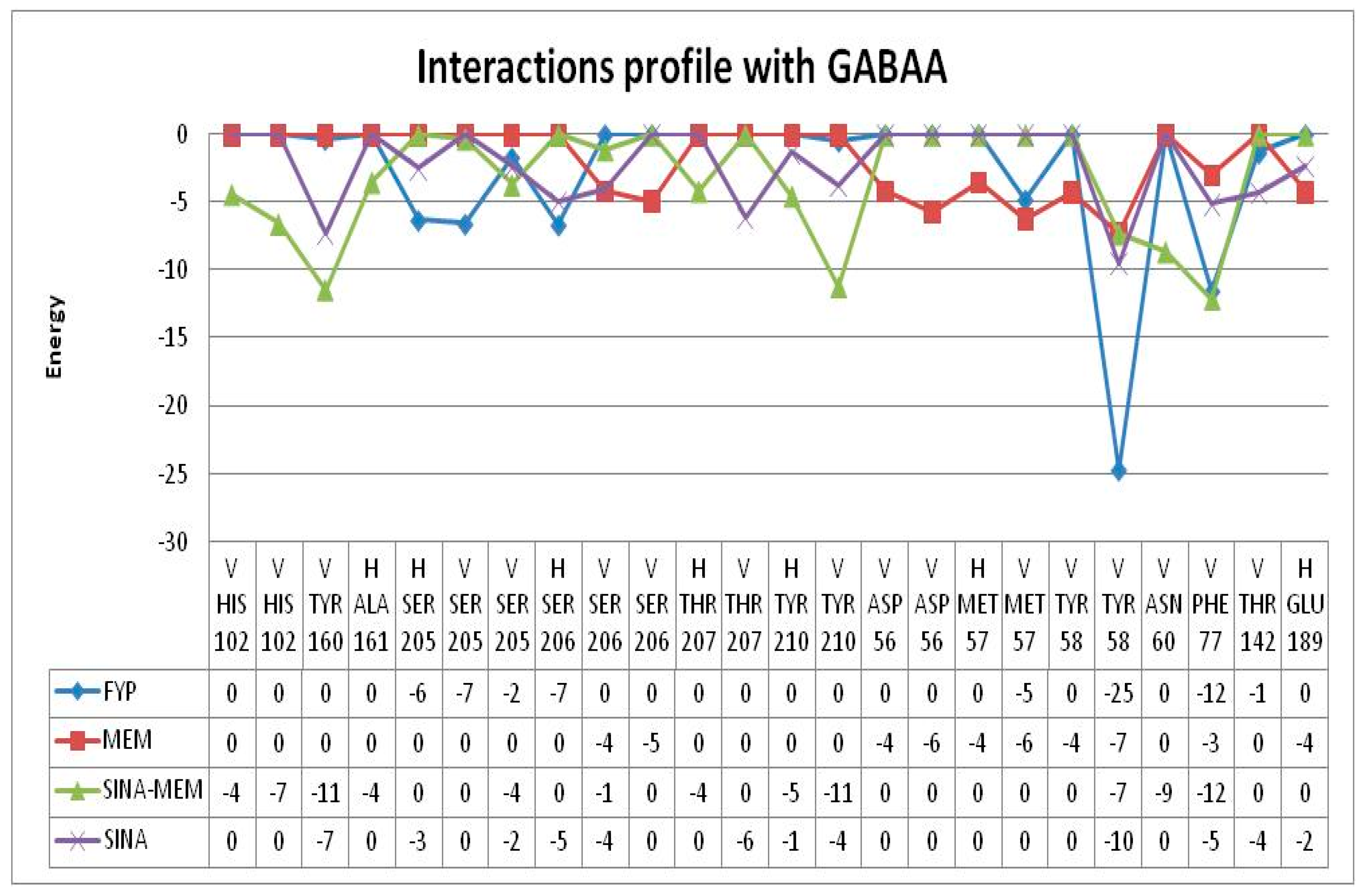
Figure 15.
2D- (A) and 3D- (B) diagrams of the interactions of MEM with NMDA receptor ion channel gate.
Figure 15.
2D- (A) and 3D- (B) diagrams of the interactions of MEM with NMDA receptor ion channel gate.
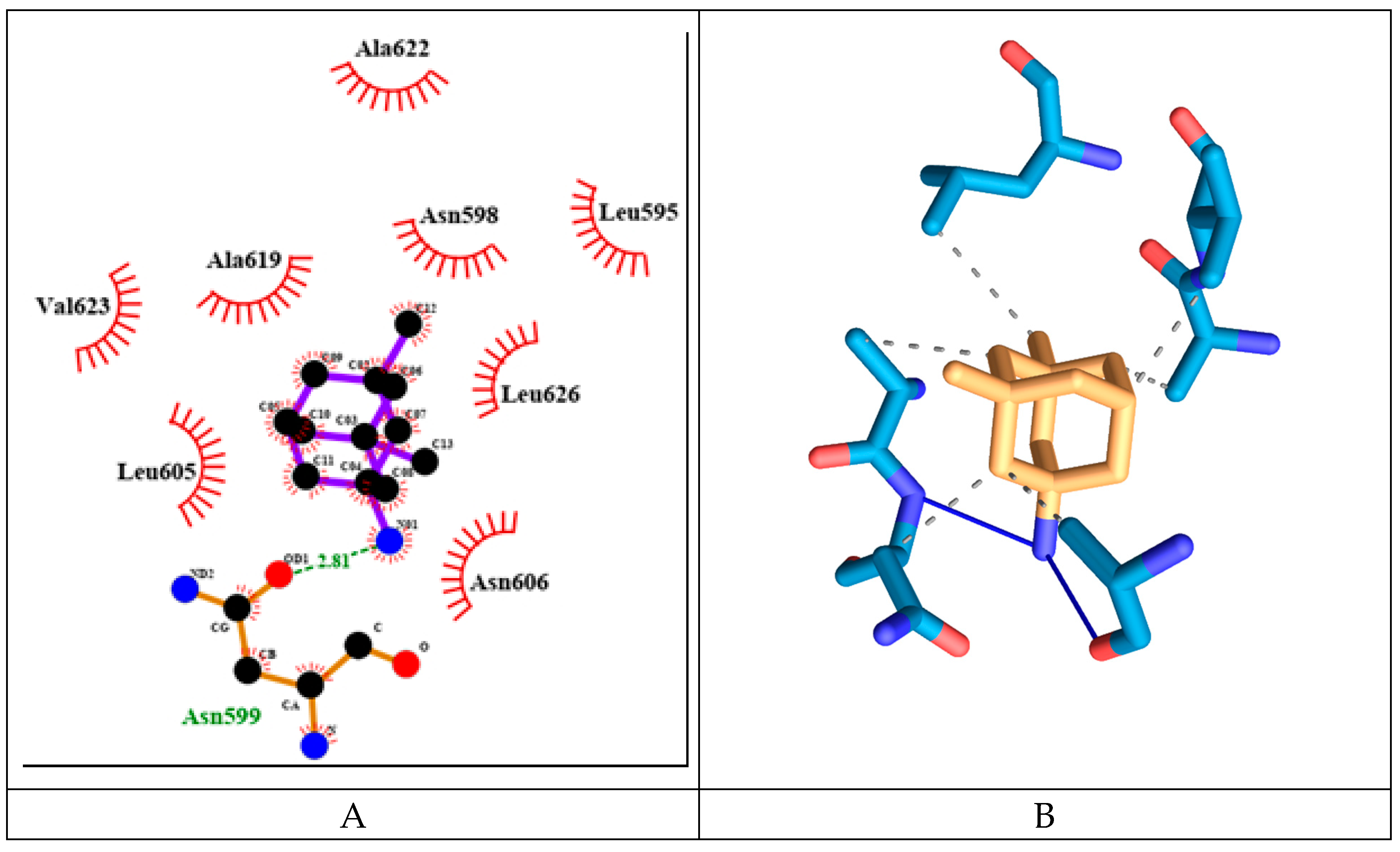
Figure 16.
2D- (A) and 3D- (B) diagrams of the interactions of SINA with NMDA receptor ion channel gate.
Figure 16.
2D- (A) and 3D- (B) diagrams of the interactions of SINA with NMDA receptor ion channel gate.
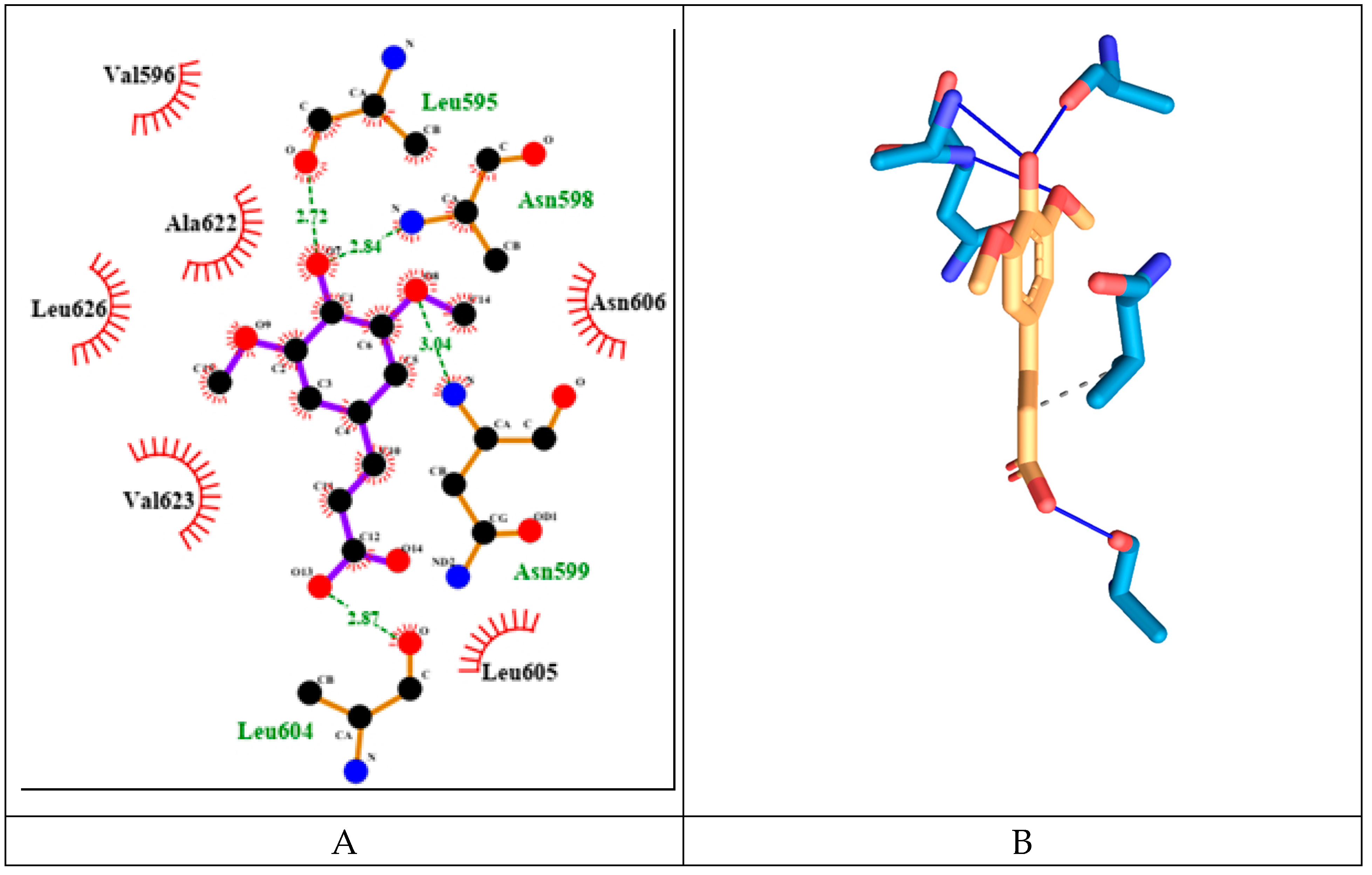
Figure 17.
2D- (A) and 3D- (B) diagrams of the interactions of SINA-MEM with NMDA receptor ion channel gate.
Figure 17.
2D- (A) and 3D- (B) diagrams of the interactions of SINA-MEM with NMDA receptor ion channel gate.
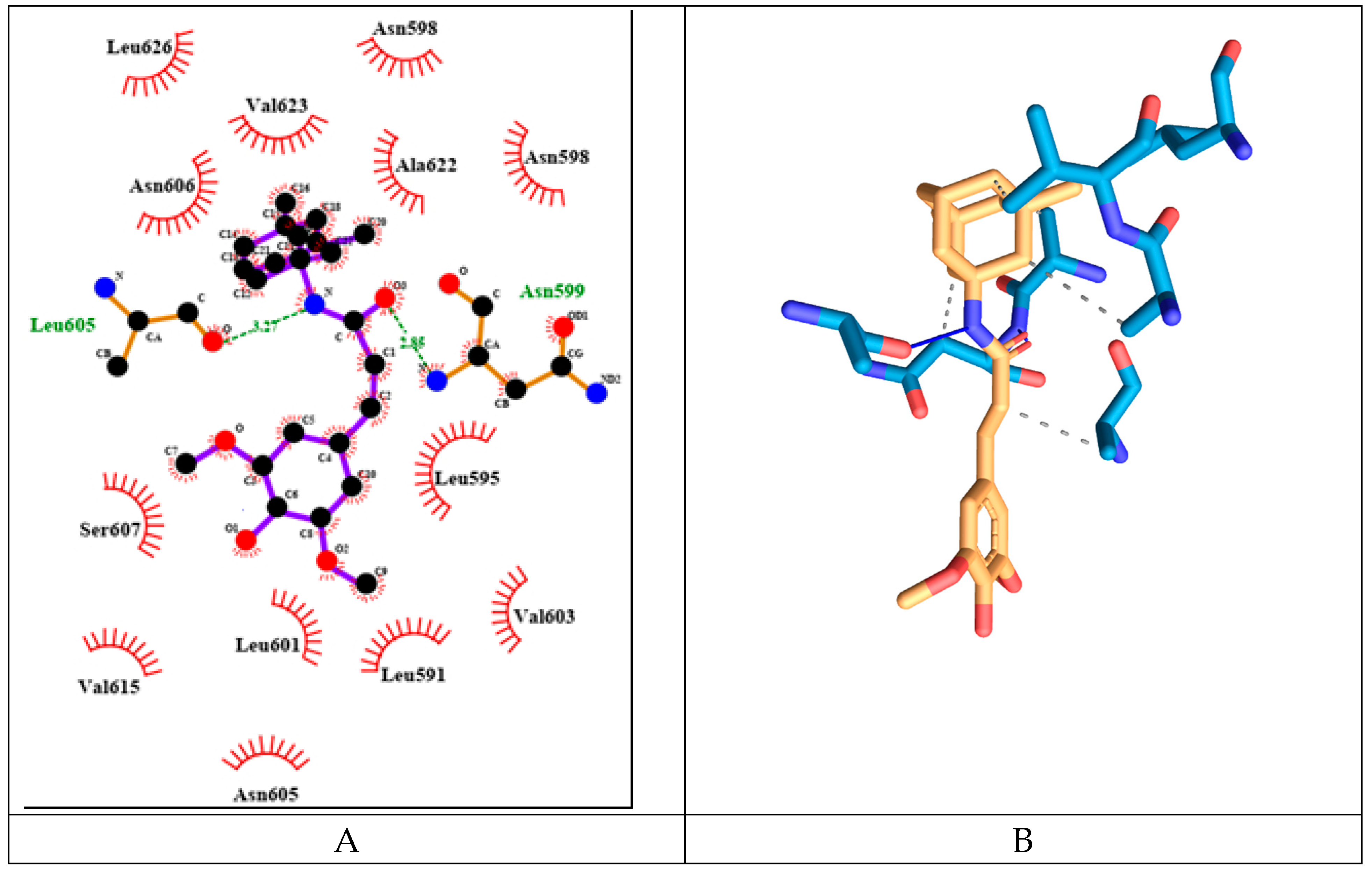
Figure 18.
Amino acid interactions profiles of the compounds with NMDA receptor ion channel gate derived from the re-docking experiments (V – Van der Waals interaction; H – hydrogen bond; S – strong; M – medium). Only interactions with value bellow -2.5 are shown.
Figure 18.
Amino acid interactions profiles of the compounds with NMDA receptor ion channel gate derived from the re-docking experiments (V – Van der Waals interaction; H – hydrogen bond; S – strong; M – medium). Only interactions with value bellow -2.5 are shown.
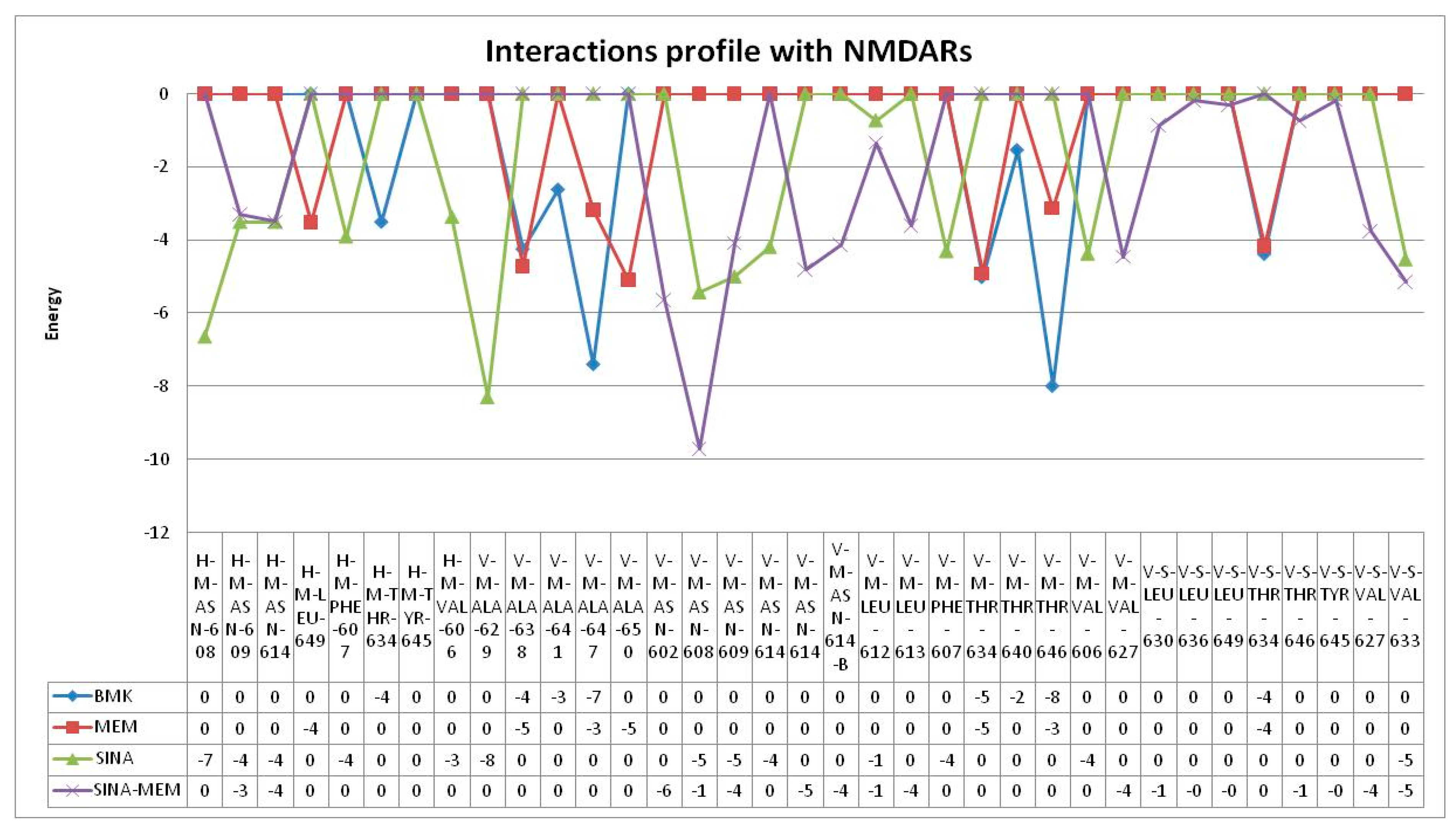
Figure 19.
Impact upon the AChE activity of the tested compounds.
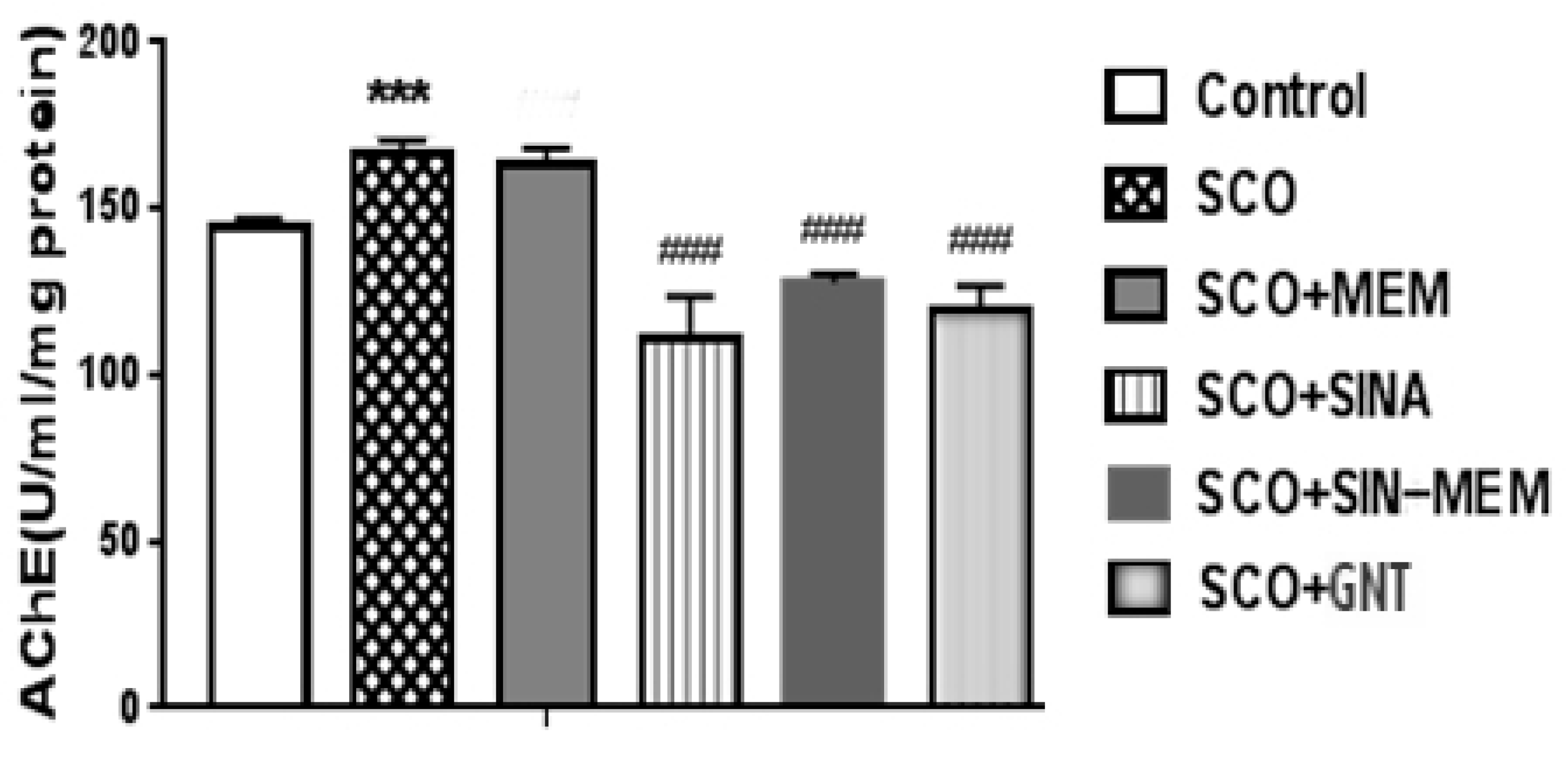
Figure 20.
Effects of the tested compounds on the performance of the animals in the Hole board test.
Figure 20.
Effects of the tested compounds on the performance of the animals in the Hole board test.
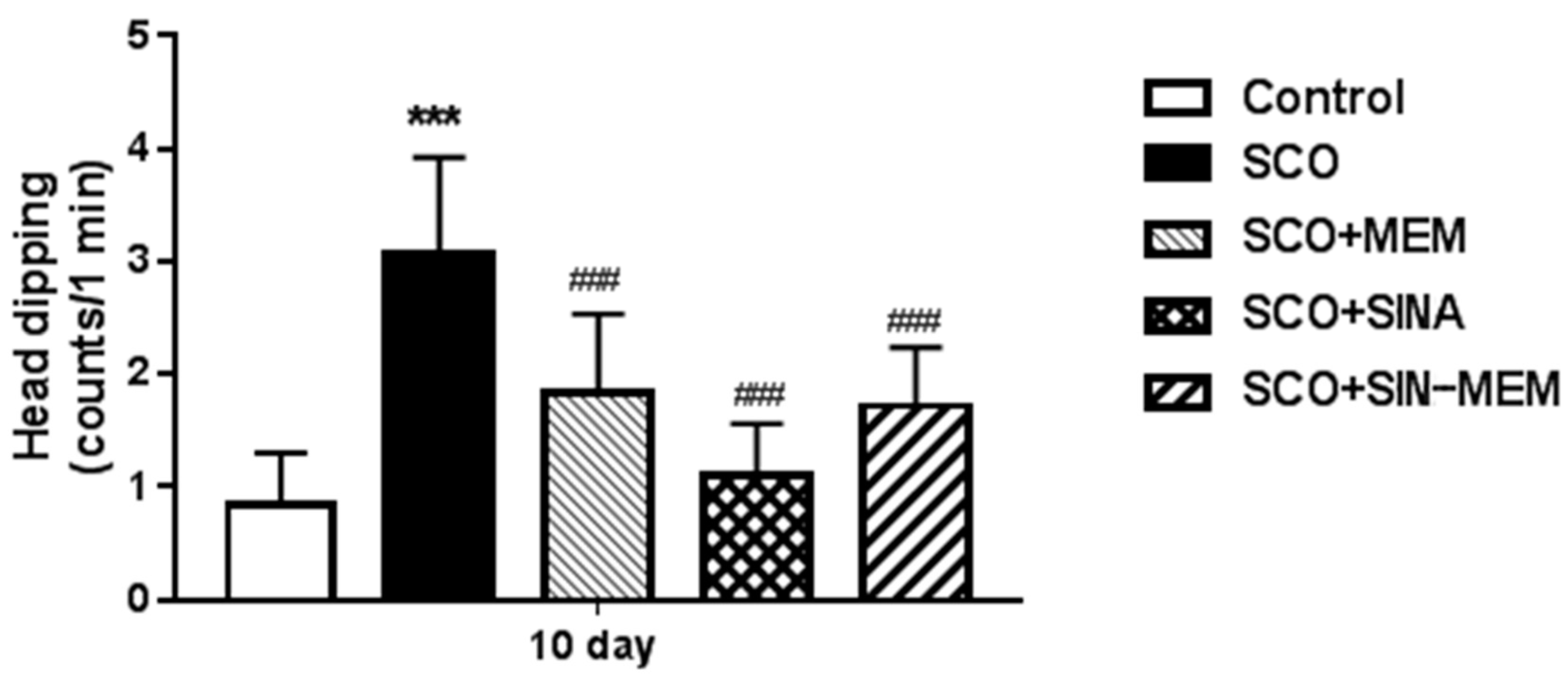
Figure 21.
Effects of the tested compounds on the performance of the animals in the Step-through test.
Figure 21.
Effects of the tested compounds on the performance of the animals in the Step-through test.

Table 1.
Comparison of the bond lengths as percentage difference between SINA and SINA-MEM as determined by the X-ray diffraction.
Table 1.
Comparison of the bond lengths as percentage difference between SINA and SINA-MEM as determined by the X-ray diffraction.
| Compound 1 | Bond 1 | Length diff. % | Bond 2 | Compound 2 | Order | Type |
| SINA | C1 - C2 | 0.890 | C16 - C17 | SINA-MEM | 2 | Double |
| SINA | C1 - C6 | 0.840 | C16 - C21 | SINA-MEM | 1 | Single |
| SINA | C1 - C7 | -0.880 | C16 - C15 | SINA-MEM | 1 | Single |
| SINA | C2 - H2 | -0.015 | C17 - H17 | SINA-MEM | 1 | Single |
| SINA | C2 - C3 | -1.041 | C17 - C18 | SINA-MEM | 1 | Single |
| SINA | C3 - O15 | -1.022 | O2 - C18 | SINA-MEM | 1 | Single |
| SINA | C3 - C4 | 1.272 | C19 - C18 | SINA-MEM | 2 | Double |
| SINA | C4 - C5 | -1.079 | C20 - C19 | SINA-MEM | 1 | Single |
| SINA | C4 - O14 | -0.851 | O3 - C19 | SINA-MEM | 1 | Single |
| SINA | C5 - O12 | 1.177 | O4 - C20 | SINA-MEM | 1 | Single |
| SINA | C5 - C6 | 0.476 | C20 - C21 | SINA-MEM | 2 | Double |
| SINA | C6 - H6 | 0.039 | C15 - H15 | SINA-MEM | 1 | Single |
| SINA | C7 - H7 | -0.021 | C14 - H14 | SINA-MEM | 1 | Single |
| SINA | C7 - C8 | 1.900 | C14 - C15 | SINA-MEM | 2 | Double |
| SINA | C8 - C9 | -2.483 | C13 - C14 | SINA-MEM | 1 | Single |
| SINA | C8 - H8 | -0.003 | C21 - H21 | SINA-MEM | 1 | Single |
| SINA | C9 - O11 | 0.953 | O1 - C13 | SINA-MEM | 2 | Double |
| SINA | O12 - C13 | 2.789 | O2 - C22 | SINA-MEM | 1 | Single |
| SINA | C13 - H13B | 0.047 | C22 - H22A | SINA-MEM | 1 | Single |
| SINA | C13 - H13A | 0.079 | C22 - H22B | SINA-MEM | 1 | Single |
| SINA | C13 - H13C | 0.020 | C22 - H22C | SINA-MEM | 1 | Single |
| SINA | O14 - H14 | 7.100 | O3 - H3 | SINA-MEM | 1 | Single |
| SINA | O15 - C16 | -1.160 | O4 - C23 | SINA-MEM | 1 | Single |
| SINA | C16 - H16C | 0.159 | C23 - H23A | SINA-MEM | 1 | Single |
| SINA | C16 - H16A | 0.013 | C23 - H23B | SINA-MEM | 1 | Single |
| SINA | C16 - H16B | -0.035 | C23 - H23C | SINA-MEM | 1 | Single |
Table 2.
Comparison of the bond wavenumbers of some of the characteristic peaks between SINA and the corresponding moiety of SINA-MEM as determined by the ATR-IR.
Table 2.
Comparison of the bond wavenumbers of some of the characteristic peaks between SINA and the corresponding moiety of SINA-MEM as determined by the ATR-IR.
| O-H (Phenol) | C=O (Carboxilyc) | C=C (trans) | C=C (aromatic) | C-O-C_1 | C-O-C_2 | -OCH3 | |
| SINA | 3290.5 | 1660.83 | 1615.22 | 1558.02 | 1328.84 | 1041.45 | 2836.12 |
| SINA-MEM | 3281.25 | 1662.42 | 1610.81 | 1557.07 | 1326.88 | 1040.33 | 2835.49 |
Table 3.
Analysis of NMR spectra data of the new hybrid compound.
| Atom number | 1H chemical shift (multiplicity, J in Hz) | 13C chemical shift |
| 1 | - | 52.7 |
| 2,9 | 1.64 (d, 12.1) | |
| 1.61 (d, 12.0) | 47.5 | |
| 3, 5 | - | 32.2 |
| 4 | 1.12 (m) | 50.6 |
| 6, 10 | 1.34 (d, 12.0) | |
| 1.26 (d, 11.9) | 42.7 | |
| 7 | 2.09 (m) | 29.9 |
| 8 | 1.81 (m) | 39.9 |
| 11, 12 | 0.83 (s) | 30.5 |
| 1‘ | - | 164.9 |
| 2‘ | 6.51 (d, 15.6) | 121.3 |
| 3‘ | 7.20 (d, 15.6) | 138.6 |
| 4‘ | - | 125.8 |
| 5‘ | 6.8 (s) | 105.3 |
| 6‘ | - | 148.4 |
| 7‘ | - | 137.4 |
| 6’-ОМе | 3.78 (s) | 56.2 |
| NH | 7.47 (s) | |
| Numeration of the carbon atoms in the new hybrid compound | 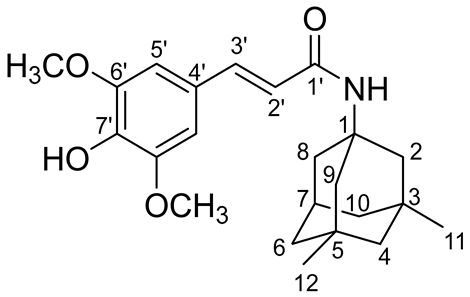 |
|
Table 4.
QSAR predicted pharmacological values of the tested compounds.
| FACTORS | SINA-MEM VALUE | SINA-MEM PROBABILITY | MEM VALUE | MEM PROBABILITY | SINA VALUE | SINA PROBABILITY |
|---|---|---|---|---|---|---|
| Human Intestinal Absorption | + | 0.9434 | + | 0.9759 | + | 0.9774 |
| Caco-2 | - | 0.6593 | + | 0.7723 | + | 0.5291 |
| Blood Brain Barrier | + | 0.9426 | + | 0.9968 | + | 0.8958 |
| Human oral bioavailability | + | 0.5571 | + | 0.8857 | + | 0.6286 |
| Subcellular localzation | Mitochondria | 0.4927 | Lysosomes | 0.9686 | Mitochondria | 0.8118 |
| OATP2B1 inhibitior | - | 1.0000 | - | 0.8649 | - | 1.0000 |
| OATP1B1 inhibitior | + | 0.8969 | + | 0.9698 | + | 0.8888 |
| OATP1B3 inhibitior | + | 0.9596 | + | 0.9500 | + | 0.9763 |
| MATE1 inhibitior | - | 0.8600 | - | 0.5400 | - | 0.9200 |
| OCT2 inhibitior | - | 0.9500 | - | 0.6500 | - | 0.9750 |
| BSEP inhibitior | + | 0.6056 | - | 0.8326 | - | 0.7876 |
| P-glycoprotein inhibitior | - | 0.7762 | - | 0.9625 | - | 0.9628 |
| P-glycoprotein substrate | - | 0.7512 | - | 0.8811 | - | 0.9400 |
| CYP3A4 substrate | + | 0.5868 | - | 0.6095 | - | 0.6637 |
| CYP2C9 substrate | - | 0.7936 | - | 1.0000 | - | 0.6110 |
| CYP2D6 substrate | - | 0.8276 | + | 0.3579 | - | 0.8502 |
| CYP3A4 inhibition | - | 0.8143 | - | 0.8309 | - | 0.8748 |
| CYP2C9 inhibition | - | 0.8227 | - | 0.9281 | - | 0.8380 |
| CYP2C19 inhibition | - | 0.7947 | - | 0.9025 | - | 0.7182 |
| CYP2D6 inhibition | - | 0.8631 | - | 0.8720 | - | 0.9297 |
| CYP1A2 inhibition | - | 0.5936 | - | 0.9327 | - | 0.8445 |
| CYP inhibitory promiscuity | - | 0.5429 | - | 0.6795 | - | 0.7608 |
| UGT catelyzed | + | 0.8000 | - | 0.0000 | + | 0.6000 |
| Carcinogenicity (binary) | - | 0.6581 | - | 0.7286 | - | 0.8025 |
| Carcinogenicity (trinary) | Non-required | 0.4507 | Non-required | 0.5777 | Non-required | 0.6699 |
| Ames mutagenesis | - | 0.5400 | - | 0.7000 | - | 0.8200 |
| Human either-a-go-go inhibition | - | 0.5375 | - | 0.7219 | - | 0.8521 |
| micronuclear | + | 0.8000 | - | 0.7400 | + | 0.7000 |
| Hepatotoxicity | + | 0.5750 | - | 0.9250 | - | 0.5750 |
| Acute Oral Toxicity (c) | III | 0.4847 | III | 0.7138 | III | 0.4500 |
| Estrogen receptor binding | + | 0.8350 | - | 0.8414 | - | 0.5000 |
| Androgen receptor binding | + | 0.7126 | - | 0.5614 | + | 0.5248 |
| Thyroid receptor binding | + | 0.8121 | - | 0.6810 | - | 0.5940 |
| Glucocorticoid receptor binding | + | 0.6864 | - | 0.8488 | - | 0.7463 |
| Aromatase binding | + | 0.8041 | - | 0.7437 | - | 0.8089 |
| PPAR gamma | + | 0.7385 | - | 0.7060 | - | 0.6295 |
Table 5.
Calculated energies and inhibitory constant from the molecular docking of the tested compounds.
Table 5.
Calculated energies and inhibitory constant from the molecular docking of the tested compounds.
| Mem | SINA-MEM | SINA | GNT | |
| Estimated Free Energy of Binding [kcal/mol] | -8.02 | -8.61 | -5.36 | -8.79 |
| Estimated Inhibition Constant, Ki [T = 298.15 K] | 1.32 µM | 488.86 nM | 118.51 µM | 360.71 nM |
| Final Intermolecular Energy [kcal/mol] | -8.32 | -10.40 | -7.15 | -9.39 |
| Electrostatic Energy [kcal/mol] | -0.87 | -0.19 | +0.06 | -0.52 |
| Final Total Internal Energy [kcal/mol] | +0.07 | -0.66 | -1.11 | -0.85 |
| Torsional Free Energy [kcal/mol] | +0.30 | +1.79 | +1.79 | +0.60 |
| Unbound System's Energy [=(2)] [kcal/mol] | +0.07 | -0.66 | -1.11 | -0.85 |
Table 6.
Summary of the parameters of the compound interactions with GABA A receptor’s benzodiazepine active site.
Table 6.
Summary of the parameters of the compound interactions with GABA A receptor’s benzodiazepine active site.
| MEM | SINA | SINA-MEM | FYP | |
| Estimated Free Energy of Binding [kcal/mol] | -7.37 | -5.76 | -7.52 | -7.55 |
| Estimated Inhibition Constant, Ki [T = 298.15 K] | 3.99 µM | 59.96 µM | 3.08 µM | 2.94 µM |
| Final Intermolecular Energy [kcal/mol] | -7.66 | -7.55 | -9.31 | -8.44 |
| Electrostatic Energy [kcal/mol] | -1.92 | -0.42 | -0.51 | +0.03 |
| Final Total Internal Energy [kcal/mol] | +0.07 | -1.17 | -1.50 | -0.56 |
| Torsional Free Energy [kcal/mol] | +0.30 | +1.79 | +1.79 | +0.89 |
| Unbound System's Energy [=(2)] [kcal/mol] | +0.07 | -1.17 | -1.50 | -0.56 |
Table 7.
Summary of the parameters of the compound interactions with NMDAR ion channel.
| MEM | SINA-MEM | SINA | |
| Estimated Free Energy of Binding [kcal/mol] | -6.13 | -8.91 | -4.59 |
| Estimated Inhibition Constant, Ki [T = 298.15 K] | 32.34 µM | 294.18 nM | 428.54 µM |
| Final Intermolecular Energy [kcal/mol] | -6.42 | -10.70 | -6.38 |
| Electrostatic Energy [kcal/mol] | -0.32 | -0.08 | +0.20 |
| Final Total Internal Energy [kcal/mol] | +0.07 | -1.48 | -1.17 |
| Torsional Free Energy [kcal/mol] | +0.30 | +1.79 | +1.79 |
| Unbound System's Energy [=(2)] [kcal/mol] | +0.07 | -1.48 | -1.17 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



