Submitted:
24 March 2025
Posted:
25 March 2025
You are already at the latest version
Abstract
The Reverse Transcriptase of the Human Immunodeficiency Virus (HIV) is distinguished for its high rate of homologous recombination. A less-studied consequence of this phenomenon is the increased occurrence of non-homologous recombination, which results in length polymorphism. While most of these genome-wide variations are sporadic, some provide a replication advantage to variant strains, such as those in the Long Terminal Repeat (LTR) and p6-Gag regions. By analyzing sequences from these two regions in the HIV-1 databases, we categorize all non-homologous recombination into four groups based on the presence or absence of two molecular features. Additionally, drawing on established models of homologous recombination, we propose a model that describes the process of sequence duplication. This model can also be applied to explain non-homologous recombination in different types in HIV and other viruses.
Keywords:
HIV
; reverse transcriptase
; sequence duplication
; non-homologous recombination
1. Introduction
Human Immunodeficiency Virus (HIV) is distinguished by its ability to generate variants at a remarkably high rate. This ability is primarily driven by three molecular features – first, the low fidelity of the RT, which is due to the lack of proofreading activity [1,2] second, the pseudo-diploid nature of the viral genome, allowing the enzyme to switch between two viral RNA strands to generate recombinants; and third, the rapid rate of replication of the virus, enabling the production of multiple variant strains [2,3]. These unique qualities collectively enable the virus to exist as a quasispecies within the host. As a result, over 95% of viral genomes in a host are defective due to large internal deletions, hypermutations (catalysed by the antiviral activity of the APOBEC family of enzymes; [3,4], premature stop codons, and/or frameshift mutations.
While significant focus has been placed on genetic variations of different types within the viral genome, relatively limited attention has been given to sequence duplications, which are present in approximately 17% of HIV-1B and HIV-1C sequences in the Los Alamos Sequence database. Sequence duplications, though mainly sporadic, can occur at several locations of the viral genome. These variant viral strains are often overlooked due to the sporadic nature of the duplications, their relatively low frequency, and the absence of apparent positive selection and expansion [5,6,7]. For instance, sequence duplications have been reported at the Sp1 sites in the Long Terminal Repeat (LTR, [5,8]), the Trans-Activation Response (TAR) element [9], RT [10,11], the env [12], and the nef genes [13]. However, evaluating the biological significance of a sequence motif duplication can be challenging when only a few additional amino acid residues are added to a reading frame of a viral protein without causing a frameshift mutation. For example, the significance of adding only three additional amino acid residues to the PTAP motif of the p6-Gag of HIV-1B is not well understood, although this modification represents the most common sequence duplication in this subtype [14]. Approximately 4.7% of HIV-1B sequences in the databases contain the partial duplication of the PTAP motif [14].
Against this backdrop, we highlight a class of sequence motif duplications in the HIV-1 genome that occur at a much higher frequency in natural infection and, importantly, appear to confer a significant replication advantage on the variant viral strains. In HIV-1C, we identified and characterized two such hotspots of sequence motif duplications in the LTR and p6-Gag. For example, variant viral strains harbouring an additional copy of the PTAP motif in p6-Gag dominate the canonical viral strains in both the plasma and the latent viral reservoir compartments [14]. Similarly, we previously demonstrated that the presence of an additional copy of the NF-κB motif in the HIV-1 LTR enhances transcriptional strength, allowing the variant viral strains to dominate the canonical strain in several experimental models and, importantly, in natural infection [15].
Of note, we recently demonstrated the emergence of several LTR-variant viral strains in HIV-1C that contain additional copies of existing transcription factor binding sites (TFBS) created by sequence duplication [16]. These viral strains are characterized by variations in the TFBS copy number, sequences of the duplicated motifs, and the relative positions of the duplicated motifs. The overall profile of TFBS in the LTR appears to influence the transcriptional activity and latent reservoir properties of the variant viral strains. For instance, the addition of a second copy of the RBEIII motif renders the variant viral strains significantly resistant to latency reversal [17], whereas the co-duplication of NF-κB motif offsets this effect (Bhange, unpublished).
A careful observation of the sequence motif duplication in these two hotspots of the HIV-1 genome, p6-Gag and the LTR, confirms that the phenomenon is highly diverse despite a common underlying framework, indicating potentially diverse mechanisms contributing to the duplication of each motif independently. Although sequence duplications at these two hotspots are shared among all HIV-1 subtypes, subtype-specific traits are evident, especially in HIV-1C. For example, NF-κB motif duplication in the viral enhancer, which can enhance the transcriptional strength of the LTR, is unique for HIV-1C and not seen in other HIV-1 genetic families [15]. The creation of the fourth copy of the NF-κB motif in HIV-1C LTR is genetically distinct yet highly conserved, regardless of geographical and temporal differences.
The canonical HIV-1C LTR contains three copies of the NF-κB motif, two of an identical sequence motif (5’- GGGACTTTCC – 3’, labelled the H-κB site) and one with a sequence variation (5’- GGGGAGTTTCC – 3’, called the C-κB site). Given the order of these three NF-κB motifs from the 5’ end to the 3’ end, the viral promoter is designated as HHC-LTR. In a variant viral strain containing four copies of the NF-κB motif (designated FHHC, ‘F’ representing the fourth motif, 5’-GGGACTTTCT-3’), a sequence of 22 base pairs (5’-GCTGACACAGAAGGGACTTTCT-3’), comprising the upstream H-κB motif and the sequences immediately upstream is copied. Notably, the ultimate base at position 10 of the F-κB motif is invariably changed to a ‘T’, thus distinguishing the F-κB motif from the canonical H-κB motif through a 'C-to-T' substitution at position 10.
In contrast to the highly faithful duplication of the F-κB motif of precisely 22 base pairs, the duplication of the RBEIII motif (designated as ‘R’) in the modulatory region of the LTR exhibits significant length, ranging from 10 to 33 residues, and genetic variations in the inserted sequences [16]. Further, while the RBEIII motif core sequence of eight bases (5’ – ACTGCTGA – 3’) is fully conserved following sequence duplication, the co-duplicated flanking sequence motifs, which serve as binding sites for other transcription factors, harbour subtype-specific variations possibly modulating latency kinetics among different HIV-1 subtypes. Furthermore, sequence motif duplication in HIV-1C becomes more complicated with permutations and combinations of several TFBS, even blurring the distinction between the enhancer and modulatory regions of the viral promoter and leading to the generation of several LTR-variant viral strains.
A similar degree of sequence length variation is evident among HIV-1 subtypes in the duplication of the PTAP motif in p6-Gag. In HIV-1B, only a partial PTAP motif is duplicated, the biological significance of which is not fully established, although sporadic reports implicate a compensatory role in drug resistance [18]. In contrast, in HIV-1C, not only is the motif duplicated in its entirety at a significantly higher frequency, but the length of the duplicated motif is also highly variable, ranging from 3 to 14 amino acids, although the core motif sequence (PTAPP) is highly conserved [14,19].
Thus, given the complex nature of sequence motif duplications in HIV-1C, comprising copy number, sequence, combinatorial, and flanking sequence variations, a single molecular model cannot satisfactorily explain the phenomenon, warranting a broader classification schematic. RT-mediated recombination during reverse transcription between the two viral genome copies co-packaged into the viral particles is the basis for sequence motif duplications in the viral genome. Sequence duplications arise due to non-homologous, rather than homologous, recombination events mediated by HIV-1 RT.
Several studies have characterized homologous recombination in HIV-1, which is estimated to occur at a frequency of three to five template switches between the two copies of the viral genome per round of viral replication [1]. In homologous recombination, an absolute or significant complementarity must exist between the nascent DNA and the template RNA. The most widely accepted model for homologous recombination in retroviruses is the dynamic-copy-choice model [20]. This model posits that a template switch is primarily driven by the disruption of the delicate equilibrium between the polymerase and RNase H activities of RT. Variations affecting either of these enzyme activities or the presence of RNA secondary structures can influence the rate of recombination.
A related model, the dock-and-lock model, suggests that acceptor strand invasion catalyzes the hybridization of the nascent DNA to the acceptor RNA, slowing the RT and thus facilitating strand switching [21,22,23]. In this model, the acceptor RNA progressively hybridizes with the nascent complementary DNA (cDNA), eventually displacing the donor RNA molecule and prompting the enzyme to switch to the acceptor RNA-cDNA hybrid for continued polymerization [24,25,26].
In contrast, non-homologous recombination, essential for sequence motif duplication, is expected to operate in the absence of perfect sequence complementarity, is much rarer, and has been less frequently studied. The frequency of non-homologous recombination is estimated to be at least a thousand times lower than that of homologous recombination. Therefore, while the dynamic-copy-choice paradigm and dock-and-lock model offer insights into viral recombination in HIV-1, a more comprehensive framework is needed to address the complexities associated with sequence motif duplications.
Here, using data from existing sequence databases and our own observations, we provide a scaffold for classifying sequence motif duplications into four distinct subgroups. Additionally, we propose a model to explain the mechanism by which HIV-1 RT generates these sequence duplications. Given that duplications are more frequent and varied in HIV-1C [14,15,16], we will use this subtype as the point of reference while delineating the salient features of the phenomenon.
2. A Classification of Sequence Motif Duplications in HIV-1
Two distinct molecular traits – the presence of a triplet motif flanking the sequence to copy and the presence of a mismatched base pair at the 3’ end of the nascent DNA - permit the classification of the sequence motif duplications in the HIV-1 genome into four groups as outlined below. The triplet motif flanking the duplicated motif lacks a defined label. It has been called variously as a short-direct repeat [27], short regions of homology [28], or short regions of sequence identity [29]. A search of HIV-1 sequences at the LANL database identified sequences with or without the flanking repeat sequence motifs (see below). Furthermore, we observed the presence of a mismatched base at the 3’ terminal end following a strand switch (see sections 2.2, 2.4), leading to the creation of additional variation in the duplicated sequence.
Based on these two molecular features, the presence or absence of the triplet motif flanking the duplicated sequence and the presence or absence of a mismatched base at the growing end of the nascent DNA following strand switch, we classify the non-homologous recombination of HIV-1 into four categories (Figure 1). Importantly, we propose a label for the three-base motif as the ‘micro-homology domain’ (MHD) since such a sequence of only three bases or even smaller can permit non-homologous recombination (See section 3.0). The proposed classification system is further corroborated by the observation that these sequence duplications occur at an increased frequency when either of these molecular features, i.e., the MHD or a matched-terminal base pair (Figure 1) are present in the duplicated sequence [14,15]. Further, the four models of recombination we describe here can explain every kind of sequence motif duplication identified in the two hotspots of the HIV-1 genome and possibly elsewhere. These models, especially the MR-ME model, can be applied to explain sequence motif duplications observed in other viruses, including Influenza [30], SARS-Cov2, Hepatitis E [32], and the Japanese Encephalitis Virus (Zhang et. al, 2021). The relatively low recombination rates mediated by the RNA polymerases in these viruses make the frequency of these events rarer compared to that in HIV-1 [1].
Since these sequence duplications appear at the highest frequency in HIV-1C, we will use this subtype as a reference in our classification. Additionally, while naming the variant viral strains, we will follow the nomenclature established previously [17,18,19].
2.1. The MR–ME Model of Sequence Duplication
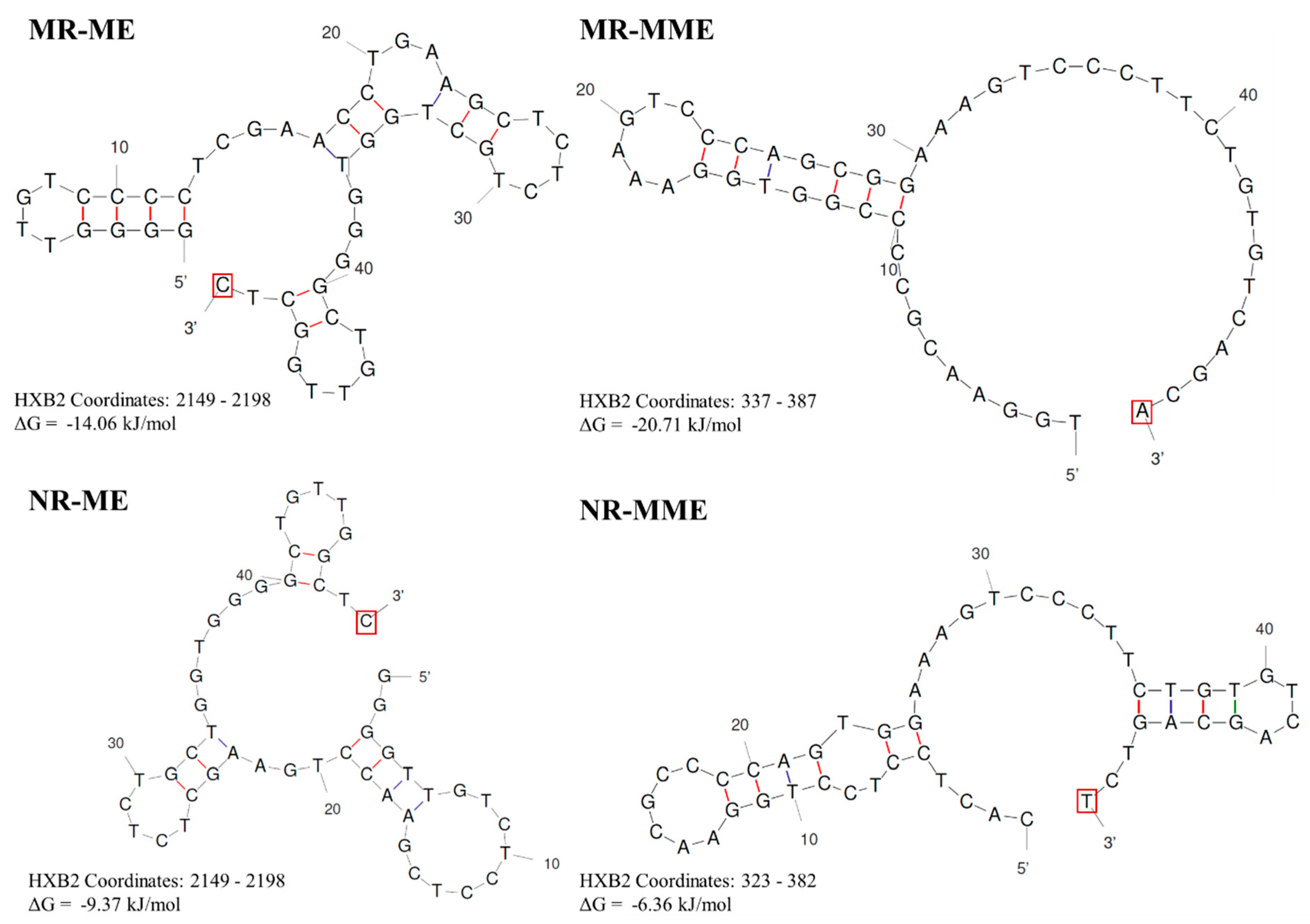
In this model, a perfectly conserved triplet MHD flanks the sequence to be duplicated, and the base present at the 3’ end on the cDNA is complementary to the base on the acceptor RNA. PTAP motif duplications of three different lengths (7, 12, and 14 aa) use this strategy for the motif duplication (Figure 1). To illustrate the model, we consider the example of the seven amino acid core PTAP motif (EPTAPPA) duplication of HIV-1C [14]. A 21-base pair sequence (5 – GAG CCA ACA GCC CCA CCA GAA – 3’) encoding the core PTAP motif is duplicated and inserted immediately downstream of the original motif as the RT continues to polymerize. Analysis of the nucleic acid sequence identifies a unique triplet ‘GAG’ flanking both motifs. When a single PTAP motif is encoded by the RNA sequence, two copies of the GAG triplet are present at either end of the sequence to be duplicated. In contrast, three copies of the GAG triplet are found flanking the two copies of the PTAP motif core sequence following duplication (Figure 2A), suggesting that the GAG triplet functions as the MHD for the PTAP sequence duplication of 21 base pairs. The three MHD motifs are designated as MHDs for the motif ‘stalling’ the RT polymerization, MHDa for the motif ‘accepting’ the cDNA at template switching, and MHDc for the motif created as an additional ‘copy’.
Based on this observation, we propose the following schema for the 7 amino acid PTAP motif duplication. The RT stalls after copying the 5’ G-residue of the GAG triplet, the MHDs motif, on the donor RNA, prompting the nascent cDNA to switch strands from the donor RNA to the acceptor RNA molecule. When the RT switch is faithfully aligned with the complementary sequence on the acceptor RNA, which is expected to be the most common event, the polymerization will resume without the sequence motif duplication. However, in a rare event, the ‘CTC’ triplet present at the 3’ end of the cDNA may misalign with the GAG triplet, the MHDa motif, present at the 3’ end of the sequence to be copied on the acceptor RNA template (Figure 2B). Notably, there is no mismatch at the growing end of the cDNA in this scenario as the ‘CTC’ triplet of the cDNA is a perfect match to the ‘GAG’ triplet on the acceptor RNA molecule. As the RT resumes polymerization, from these misaligned complementary sequences, the 21 bases encoding the seven amino acid sequences of the PTAP motif are copied again, creating an additional copy of the PTAP motif, including the GAG triplet, the MHDc motif. We designate this model as the MHD-dependent Recombination and Matched-base Extension (MR-ME).
2.2. The MR–MME Model of Sequence Duplication
The creation of a fourth copy of the NF-κB motif in HIV-1C LTR, which confers a significant replication fitness advantage on the variant viral strains [15], serves as an excellent example when the RT uses the MHD-dependent recombination strategy leading to the sequence motif duplication. However, unlike in the MR-ME model described above, the RT here encounters a mismatched base at the growing end of the cDNA that is non-complementary to the acceptor RNA template. The new κB-motif added to the viral enhancer, called the F-κB motif, demonstrates distinct molecular features, which must be satisfactorily explained by the MR-MME model of recombination. First, although a typical NF-κB binding site consists of only 10 base pairs, a sequence of 22 bp is copied faithfully. The 22 bp duplicated sequence (5’-GCTGACACAGAAGGGACTTTCT-3’) encompasses a five-base partial binding site of RBEIII motif (5’-GCTGA-3’), followed by an overlapping seven-base binding site for TFII-I (5’-TGACACA-3’), finally followed by the 10 bases of the F-κB binding site (Figure 3A). The 22 bp duplicated sequence is highly conserved without an evident sequence variation regardless of the geographical and temporal origin of the viral strains. Second, the F-κB-motif, comprising the 10 base pairs (italicized) at the 3’ end of the 22 base sequence duplicated, is genetically distinct from the canonical H-κB motif (5’-GGGACTTTCC-3’). The F-κB motif contains a C-to-T base substitution at position 10 of the element (5’-GGGACTTTCT-3’), a molecular trait highly consistent without exception. Thus, a recombination model must satisfactorily explain how the 22 bases of the motif-cluster sequence are duplicated along with a mechanism that accounts for the ‘C-to-T’ variation at position 10 of the κB-motif.
The MHD-dependent Recombination and MisMatched-base extension (MR-MME) model posits that a series of successive processes lead to the sequence motif duplication: the RT stalling at the MHDs triplet, the non-specific addition of an ‘A’ residue to the growing end of the cDNA by the extendase activity of the RT, the RT strand-switching to the MHDa motif on the donor RNA template, and a potential mismatch extension by the RT [33]. Considering the sequences with and without the 22-base duplication, we suggest that the RT pauses at the 5’ ‘G’ residue of the GCU motif serving as the MHDs, following the reverse transcription of the H-κB motif, the TFII-I motif, and a partial RBEIII core motif (Figure 3B). The paused RT adds a nucleotide to the growing end of the DNA in the RNA:DNA hybrid in a template-independent manner, typically with a strong preference for an ‘A’ residue [34]. Subsequently, the nascent DNA and donor RNA hybrid dissociates, and the nascent DNA anneals with the acceptor RNA template. When the nascent DNA hybridizes with a cognate complementary sequence on the acceptor RNA, only homologous recombination occurs without sequence duplication. A new RT molecule recognizes the resulting ‘nascent DNA-acceptor RNA’ complex and resumes polymerization that should not introduce variation as the non-specifically added ‘A’ can pair with the natural ‘U’ present on the acceptor template RNA at this position. Alternatively, and rarely, the nascent DNA may misalign with the acceptor RNA template at the MHDa ‘GCU’ triplet, serving as the acceptor for hybridization (Figure 3B). However, this hybridization causes the misalignment of non-complementary bases (C Vs. A) at the growing end of the cDNA. The RT then proceeds to extend the mismatched base at the growing end of the nascent DNA, efficiently resuming polymerization, resulting in the ‘C to T’ transition of the F-κB element at position 10 of the motif.In summary, the duplication of the 22-base sequence motif in HIV-1C depends not only on where the RT pauses in the viral modulator region but also on the RT's ability to non-specifically add an ‘A’ residue to the growing end of the nascent DNA molecule using the extendase activity and to extend the mismatched base pair at the growing end of the cDNA while resuming polymerization [33].
A second pathway to the creation of the F-κB motif may also exist, where the RT stalls at the 5’ U, immediately upstream of the MHDs GCU sequence. In this case, the RT would add the corresponding ‘A’ nucleotide on the cDNA before transitioning to the MHDa on the acceptor strand, bypassing the addition of the non-templated ‘A’. The subsequent steps of misalignment on the acceptor RNA and extension of the mismatched bases would be identical to the previous pathway described above. Additional examples of sequence motif duplications caused by the MR-MME model are enlisted (Figure 1).
2.3. The NR–ME Model of Sequence Duplication
An examination of several other events of sequence duplications in HIV-1C did not reveal the presence of a triplet that could serve as an MHD. Nevertheless, we identified a single base within the triplet at the growing end of the cDNA being complementary to the acceptor RNA sequence. Of note, several studies previously illustrated recombination events mediated by single-base pair complementarity [28,35,36]. Thus, depending on whether the single base at the terminal position of the cDNA serves as a complementarity residue, the NR model of recombination may be classified into two categories. We provide here examples of some of these recombination events from the duplication hotspots of HIV-1C.
The six amino-acid long PTAP motif (EPTAPP) duplication in HIV-1C p6-Gag serves as a notable example of the NR-NE recombination model. As mentioned previously, the six amino acid PTAP motif duplication is the second most event commonly duplicated in the gag sequences of HIV-1C (23.7%) and HIV-1B (21.7%) available in the extant databases. According to this model, the RT is expected to stall at the 5’ G residue of the ‘GAG’ codon, encoding the Aspartic Acid residue of the ‘EPTAPP’ motif on the donor RNA. Subsequently, the RT switches to the acceptor RNA and mis-pairs with the ‘GCA’ codon encoding the c-terminal Alanine residue of the ‘EPTAPP’ motif on the donor RNA molecule. The pairing adjacent to the ‘CCA’ codon of the cDNA and the ‘GCA’ codon of the acceptor RNA molecule leads to a perfect match at the terminal base located at the growing end, which is recognized by an RT molecule to initiate polymerization. Thus, despite the absence of an evident triplet MHD motif, the misaligned sequence allows the terminal ‘C’ of the cDNA to pair with the ‘G’ of the ‘GCA’ codon, resulting in a Non-MHD-dependent Recombination and Matched-base Extension (NR-ME) duplication (Figure 4). Additional examples of sequence motif duplications using the NR-ME model have been illustrated (Figure 1).
2.4. The NR–MME Model of Sequence Duplication
Previously, we reported the presence of several LTR-variant viral strains of HIV-1 in India [16]. An example of LRhR-HHC LTR, one variant LTR, serves aptly to explain the NR-MME model of sequence duplication; additional examples are enlisted (Figure 1). The sequence motif duplication creates an additional copy of the RBEIII binding site (R), and an additional copy of the κB-like motif (‘h’, 5’-GGGACTTTCA-3’, deviates from the canonical ‘H-κB motif at position 10, difference underlined), whereas the ‘L’ represents the TCFα/LEF-1 binding site. HHC represents two H-κB and one C-κB motifs in the enhancer. The generation of the LRhR-HHC variant entails the duplication of a 26 bp sequence (5’ – AGACTGCTGACACAGAAGGGACTTTC – 3’) accommodating binding sites for three TF families – RBEIII (bolded), TFII-I (underlined), and the κB-like element (italicized).
Analysis of the variant LTR sequence fails to find a triplet MHD motif flanking the duplicated sequence to serve as a catalyst to promote a misaligned recombination event. Sequence analysis suggests that the RT pauses at the 'A' residue immediately upstream of the RBEIII motif on the donor RNA template (Figure 5). Subsequently, the RT switches the template strands and aligns with the 'CGC' triplet on the acceptor RNA immediately downstream of the 5’ H-κB site, thus creating a base pair mismatch between the growing end of the cDNA and the acceptor RNA. The RT commences reverse transcription, disregarding the 'C-to-T' mismatch at the growing end of the cDNA, leading to the duplication of the
26 bp sequence and fixing of the 'C-to-A' variation at position 10 of the newly formed NF-κB motif, and resulting in a Non-MHD-dependent Recombination and Mis-Matched-base Extension (NR-MME)
3. A Model Elucidating the Structural Constraints the RT Encounters While Duplicating Sequence Motifs
The RT encounters two or three structural and mechanistic challenges while attempting to accomplish the task of duplicating a sequence motif. Misalignment of the cDNA growing end to the MHDa causes the formation of a single-stranded DNA loop structure, which should be accommodated within the confines of the RT interior during the process of polymerization. The additional DNA loop occupying the limited space within the active zone of the RT may create mechanistic barriers that may prevent the polymerization process.
We propose here a schematic model integrating the fundamental principles of retroviral recombination with insights gleaned from the literature and our own theoretical analysis to elucidate how the RT overcomes these structural constraints to complete the process of polymerization successfully, leading to the creation of a duplicated sequence.
To depict this model, we will illustrate the earlier example of the 22 base pair sequence duplication in the LTR using the MR-MME mechanism, leading to the creation of an additional NF-κB motif, the F-κB motif (Figure 3). During minus strand synthesis, the RT after pausing at the ‘GCU’ MHDs triplet, adds an ‘A’ residue to the growing end of the cDNA by the template-independent process (Figure 3A and 6). At this time, 24 base pairs of the RNA-DNA hybrid are enclosed within the RT [37,38].
The downstream sequence of the donor RNA, having served as the template for reverse transcription, is hydrolyzed by the RNase H activity of the RT. Consistent with the dock-and-lock model, the complementary single-stranded DNA hybridizes with the acceptor RNA molecule in this region of the viral genome. Consequently, the nascent DNA is hybridized with both the donor and the acceptor RNA molecules in different regions, thus generating a trimolecular complex (Figure 6A). Subsequently, when the RT dissociates from the donor RNA, the cDNA aligns with the cognate complementary sequence on the acceptor RNA molecule, and the polymerization process resumes efficiently.
Alternatively, in rare cases, when the RT dissociates from this trimolecular complex, the free bases on the growing end of the cDNA form an intramolecular secondary structure, such as a loop, causing the cDNA to misalign with the MHDa triplet on the acceptor RNA molecule. The secondary structure on the cDNA could be a contributory factor to the RT favouring such a misalignment. Importantly, the formation of a looped secondary structure of the intervening bases of the cDNA represents an inevitable outcome, since these bases lack a complementary region to hybridize with.
The unhybridized sequences, therefore, must extend outward from the central DNA binding groove of the RT (Figure 6A). The intervening DNA sequences forming such a loop structure could be as long as 48 base pairs, based on sequence evidence available from the extant databases. The RT then extends the cDNA despite the base pair mismatch at the growing end due to the propensity of the HIV-1 RT to read through such mismatches [39,40,41,42,43], (Figure 3B and Figure 6B).
Importantly, the DNA loop protruding from the RT complex would be a significant impediment when polymerization resumes. Analyzing the crystal structure of HIV-1B RT archived in the Protein Data Bank (PDB ID: 5J2M), we determined that the width of the DNA binding groove ranges from 16.3 Å at its narrowest point between the R78 and L289 residues of the p66 chain to 46.1 Å at its widest between the G15 and D471 residues of the p51 and p66 chains, respectively (Figure 6B). The space available within the confines of the RT central cleft is adequate for the DNA loop to extend outward, away from the RT.
Notably, during polymerization, each time a fresh base is added to the growing end of the cDNA, the RT must undergo a rotation of approximately 20 - 30° relative to the axis of polymerization [44]. The DNA loop also must rotate concomitantly along with the RT as a fresh base is added to the cDNA. However, due to the orientation of the DNA loop within the groove, a free unhindered rotation is permitted only up to 111° (as measured across the G359, K395, and G15 residues). Therefore, the RT can add a maximum of three to five nucleotides before the DNA loop encounters the p51 subunit, which impedes further reverse transcription. At this juncture, the RT complex must disassemble, freeing the DNA-RNA complex, and polymerization must resume with a different RT molecule, reassembling the polymerization complex. The process of the addition of up to five nucleotides, the complex dissociation, and reassembly must repeat for several rounds until the DNA loop resolves and exits the RT complex, allowing the RT to resume normal polymerization. Our model predicts that incorporating the initial 3-6 base pairs poses the greatest challenge for the enzyme due to the proximity of the loop to the active site. As the loop moves away from the active site, the rate of polymerization accelerates until the process is normalized. This prediction aligns with the observation from the databases that the error rate is highest in bases within three to six positions from the junction between the original and duplicated sequences in the absence of an MHD (Panchapakesan et. al, unpublished data).
Our model incorporates the widely accepted tenets of retroviral recombination, representing the dynamic copy choice model and the strand invasion theory to explain how sequence duplications are generated. Importantly, building on the earlier proposition that the cDNA must loop to generate a sequence duplication [27], our model posits that this looped DNA secondary structure is the central driving force underlying a sequence duplication. The formation of this secondary structure is supported by DNA secondary structure predictions using the Unafold Webserver with the 50 terminal bases of representative sequences from all four types of sequence duplications described above. Importantly, in all the secondary structure predictions shown, the terminal base is free for extension by the RT (Figure 7). Further, our model proposes that the events leading up to the RT switching between the two templates are identical between homologous and non-homologous recombination processes. The difference lies in the nonavailability of the looped bases for hybridisation with the acceptor RNA template. Therefore, the cDNA is forced to misalign and duplicate the bases copied already.
While the focus of this manuscript is on sequence duplication, the same principles could be extrapolated to sequence deletion. Typically, sequence deletions are far more frequent than sequence motif duplications [45,46], although both events share a common mechanism. For a sequence deletion to occur, the looped secondary structure must form on the template RNA, unlike in sequence duplication, where the cDNA forms the secondary structure. While sequence duplications typically result from inter-molecular switching of the RT enforced by the degradation of the donor RNA template by the RNase H activity of the enzyme, in contrast, deletions can occur due to both inter- and intra-molecular switching of RT [27]. Furthermore, RNA secondary structures are far more numerous than those of DNA, partially explaining the significantly higher proportion of deletions compared to duplications [46].
4. Despite Being Rare, Sequence Motif Duplications Are Positively Selected in HIV-1
Nearly 95-98% of all HIV-1 proviruses present in the latent reservoir are impaired by sequence deletions, frameshift mutations, G to A hypermutations, and other defects [45,47,48]. Consequently, substantial efforts have been dedicated to assessing the impact of such genetic defects on latent reservoir properties. In contrast, sequence duplication remains a relatively unexplored area in HIV-1 research, even though sequence duplications of variable length dot the viral genome in many viral strains. However, such sequence duplications are sporadic and lack evident evolutionary patterns. The nature of sequence motif duplications described here in two hotspots, the LTR and p6-Gag, of the viral genome, especially in HIV-1C, deviate by conferring a significant replication fitness advantage on the variant viral strains.
Non-homologous recombination between two template RNA molecules is a precondition for the emergence of sequence motif duplication. However, the frequency of non-homologous recombination is expected to be much lower than that of homologous recombination. For instance, the RT typically switches strands three to five times per replication round, with a recombination probability estimated at 0.03% - 0.05% per base. Non-homologous recombination, which is responsible for duplication, occurs 100 to 1,000 times less frequently than homologous recombination [27,35]. Since only a fraction of these events result in duplications, the mean duplication rate at a specific genome location is approximately 0.000015% - 0.0025%.
Importantly, although the frequency of the sequence motif duplication is expected to be extremely low, the variant viral strains are subjected to strong Darwinian selection. Despite their rarity, databases contain a notable presence of variant viral strains with motif duplications. Two factors contribute to this phenomenon. Firstly, the high replication rate of HIV-1 produces approximately 10¹⁰ to 10¹² new virions [49] daily in an infected individual, partially compensating for the infrequent appearance of sequence duplications, despite the less defined numbers of infected cells and reverse transcription events. Secondly, certain variant strains containing motif duplications exhibit a replication advantage over wild-type strains, experiencing a strong positive selection. We have previously shown that in competitions between viral variants discordant for a PTAP duplication, double-PTAP variants consistently outperformed single-PTAP strains in various conditions, including natural infection [14]. Consequently, intense positive selection ensures the continued propagation of these variants despite their low frequencies. The domination of these variant viral strains over their canonical counterparts may also influence their transmission potential. In summary, the delicate balance between the rarity of sequence duplications and subsequent strong positive selection ensures the replication fitness of these variant viral strains.
4.1. Sequence Motif Duplications Represent an Important Evolutionary Mechanism
Sequence duplications that create an additional copy of a biologically significant motif can confer a replication fitness advantage on variant viral strains through quantitative and/or qualitative gains of function. For instance, the viral enhancer of HIV-1B, which contains two tandemly arranged and genetically identical copies of the canonical H-κB motif (5’-GGGACTTTCC-3’), enhances the transcriptional strength of the viral promoter by recruiting the same host factor complexes, such as the p50-p65 heteroduplex, at both sites. In contrast, the canonical enhancer of HIV-1C, which harbors three tandem copies of the NF-κB motif that genetically represent two variable binding sites (two copies of the H-κB motif and one copy of the C-κB motif, 5’-GGGGCGTTCC-3’, differences underlined), may benefit from an additional advantage of recruiting diverse NF-κB dimers to the enhancer. Similarly, the creation of the fourth NF-κB motif in the HIV-1C enhancer by the duplication of 22 bases (Figure 3) introduces additional genetic diversity and proportionately increases transcriptional strength (Bachu M, 2012). Thus, viruses like HIV appear to prefer the duplication of short-length sequences, given the packaging restrictions. Consequently, they tend to add copies of sequence motifs of biological significance, conferring an immediate gain of function.
The duplication of shorter and biologically important motifs appears to play a crucial evolutionary role in viruses. The addition of short-length sequences does not significantly alter the genome size, thus avoiding packaging problems. While the original copy of the motif ensures the functional integrity of the biological property, the acquired and genetically variable copies of the motif may benefit variant viral strains by broadening their biological function [14,15,16,17,18]. These variations, whether in open reading frames or regulatory elements, are subjected to strong selection processes.
Taken collectively, the non-homologous recombination models presented here can explain the various observations associated with sequence duplications in HIV-1C. These include genetic variations within the duplicated regions, the high mutational rate at the junctions, the presence or absence of the MHD in certain sequences, and the relatively higher frequencies of some duplications over others. Furthermore, unlike a few published reports [27,50], our model emphasizes the importance cDNA secondary structure in hindering its correct base pairing with the acceptor RNA molecule, thereby serving as the primary driving force for sequence motif duplication. Additionally, our models account for the differences in sequence duplications observed. Empirical validation of the recombination models proposed here could prove challenging, given the extremely low frequencies of non-homologous recombination.
Author Contributions
AP and UR jointly conceptualized the ideas and prepared the manuscript.
Funding
We acknowledge the financial support from the Core Research Grant CRG/2019/000820 provided by the Science and Engineering Research Board, Government of India, to UR and the Corporate Social Responsibility funds from Gennova Biopharmaceuticals Ltd, Maharashtra, India.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank Dr. Vinayaka Prasad, Albert Einstein College of Medicine, for the critical reading of the manuscript and Chhavi Saini for her help in the preparation of the figures.
Conflicts of Interest
The authors declare no competing interests.
List of Abbreviations
HIV – Human Immunodeficiency Virus
RT – Reverse Transcriptase
APOBEC - Apolipoprotein B mRNA Editing Catalytic Polypeptide-like
LTR – Long Terminal Repeat
TAR – Trans-Activation Response
TFBS – Transcription Factor Binding Site
MHD – Micro-Homology Domain
MR-ME – MHD-dependent Recombination and Matched-base Extension
MR-MME – MHD-dependent Recombination and Mis-Matched-base Extension
NR-ME - Non-MHD-dependent recombination and Matched-base Extension
NR-MME - Non-MHD-dependent recombination and Mis-Matched-base Extension
References
- Onafuwa-Nuga, A.; Telesnitsky, A. The remarkable frequency of human immunodeficiency virus type 1 genetic recombination. Microbiol. Mol. Biol. Rev. 2009, 73, 451–480. [Google Scholar] [CrossRef] [PubMed]
- Hu WS, Hughes SH. HIV-1 reverse transcription. Cold Spring Harbor perspectives in medicine. 2012 Oct 1;2(10):a006882.
- Smyth RP, Davenport MP, Mak, J. The origin of genetic diversity in HIV-1. Virus research. 2012 Nov 1;169(2):415-29.
- Cuevas JM, Geller R, Garijo R, López-Aldeguer, J., Sanjuán, R. Extremely high mutation rate of HIV-1 in vivo. PLoS biology. 2015 Sep 16;13(9):e1002251.
- Michael NL, d'Arcy L, Ehrenberg PK, Redfield RR. Naturally occurring genotypes of the human immunodeficiency virus type 1 long terminal repeat display a wide range of basal and Tat-induced transcriptional activities. Journal of virology. 1994 May;68(5):3163-74.
- Alexander L, Aquino-DeJesus MJ, Chan, M., Andiman WA. Inhibition of human immunodeficiency virus type 1 (HIV-1) replication by a two-amino-acid insertion in HIV-1 Vif from a nonprogressing mother and child. Journal of virology. 2002 Oct 15;76(20):10533-9.
- Naghavi MH, Salminen MO, Sonnerborg, A., Vahlne, A. DNA sequence of the long terminal repeat of human immunodeficiency virus type 1 subtype A through G. AIDS research and human retroviruses. 1999 Mar 20;15(5):485-8.
- Berkhout B, Verhoef K, van Wamel JL, Back NK. Genetic instability of live, attenuated human immunodeficiency virus type 1 vaccine strains. Journal of virology. 1999 Feb 1;73(2):1138-45.
- Emiliani S, Coudronnière N, Delsert, C., Devaux, C. Structural and functional properties of HIV-1 (GER) TAR sequences. Journal of Biomedical Science. 1996 Aug 11;3(1):31-40.
- Sato H, Tomita Y, Ebisawa K, Hachiya A, Shibamura K, Shiino T, Yang R, Tatsumi M, Gushi, K., Umeyama, H., Oka, S. Augmentation of human immunodeficiency virus type 1 subtype E (CRF01_AE) multiple-drug resistance by insertion of a foreign 11-amino-acid fragment into the reverse transcriptase. Journal of Virology. 2001 Jun 15;75(12):5604-13.
- Harrigan PR, Mo T, Hirsch J, Brumme ZL, McKenna P, Bacheler, L. A 21-base pair insertion/duplication at codon 69 of the HIV type 1 reverse transcriptase in a patient undergoing multiple nucleoside therapy. AIDS research and human retroviruses. 2007 Jul 1;23(7):895-9.
- Guglietta S, Pantaleo G, Graziosi, C. Long sequence duplications, repeats, and palindromes in HIV-1 gp120: Length variation in V4 as the product of misalignment mechanism. Virology. 2010 Mar 30;399(1):167-75.
- Hiebenthal-Millow, K., Kirchhoff, F. The most frequent naturally occurring length polymorphism in the HIV-1 LTR has little effect on proviral transcription and viral replication. Virology. 2002 Jan 5;292(1):169-75.
- Sharma S, Arunachalam PS, Menon M, Ragupathy V, Satya RV, Jebaraj J, Aralaguppe SG, Rao C, Pal S, Saravanan S, Murugavel KG. PTAP motif duplication in the p6 Gag protein confers a replication advantage on HIV-1 subtype C. Journal of Biological Chemistry. 2018 Jul 27;293(30):11687-708.
- Bachu M, Yalla S, Asokan M, Verma A, Neogi U, Sharma S, Murali RV, Mukthey AB, Bhatt R, Chatterjee S, Rajan RE. Multiple NF-κB sites in HIV-1 subtype C long terminal repeat confer superior magnitude of transcription and thereby the enhanced viral predominance. Journal of Biological Chemistry. 2012 Dec 28;287(53):44714-35.
- Bhange D, Prasad N, Singh S, Prajapati HK, Maurya SP, Gopalan BP, Nadig S, Chaturbhuj D, Jayaseelan B, Dinesha TR, Ahamed SF. The evolution of regulatory elements in the emerging promoter-variant strains of HIV-1 subtype C. Frontiers in microbiology. 2021 Nov 16;12:779472.
- Ali H, Bhange D, Mehta K, Gohil Y, Prajapati HK, Byrareddy SN, Buch, S., Ranga, U. An emerging and variant viral promoter of HIV-1 subtype C exhibits low-level gene expression noise. Retrovirology. 2021 Dec;18:1-2.
- Martins AN, Waheed AA, Ablan SD, Huang W, Newton A, Petropoulos CJ, Brindeiro RD, Freed EO. Elucidation of the molecular mechanism driving duplication of the HIV-1 PTAP late domain. Journal of virology. 2016 Jan 15;90(2):768-79.
- Sharma S, Aralaguppe SG, Abrahams MR, Williamson C, Gray C, Balakrishnan P, Saravanan S, Murugavel KG, Solomon, S., Ranga, U. The PTAP sequence duplication in HIV-1 subtype C Gag p6 in drug-naive subjects of India and South Africa. BMC infectious diseases. 2017 Dec;17:1-9.
- Hwang CK, Svarovskaia ES, Pathak VK. Dynamic copy choice: Steady state between murine leukemia virus polymerase and polymerase-dependent RNase H activity determines frequency of in vivo template switching. Proceedings of the National Academy of Sciences. 2001 Oct 9;98(21):12209-14.
- Negroni, M., Buc, H. Copy-choice recombination by reverse transcriptases: Reshuffling of genetic markers mediated by RNA chaperones. Proceedings of the National Academy of Sciences. 2000 Jun 6;97(12):6385-90.
- Roda RH, Balakrishnan M, Hanson MN, Wöhrl BM, Le Grice SF, Roques BP, Gorelick RJ, Bambara RA. Role of the reverse transcriptase, nucleocapsid protein, and template structure in the two-step transfer mechanism in retroviral recombination. Journal of Biological Chemistry. 2003 Aug 22;278(34):31536-46.
- Roda RH, Balakrishnan M, Kim JK, Roques BP, Fay PJ, Bambara RA. Strand transfer occurs in retroviruses by a pause-initiated two-step mechanism. Journal of Biological Chemistry. 2002 Dec 6;277(49):46900-11.
- Negroni, M., Buc, H. Retroviral recombination: What drives the switch? Nature Reviews Molecular Cell Biology. 2001 Feb 1;2(2):151-5.
- Balakrishnan M, Roques BP, Fay PJ, Bambara RA. Template dimerization promotes an acceptor invasion-induced transfer mechanism during human immunodeficiency virus type 1 minus-strand synthesis. Journal of virology. 2003 Apr 15;77(8):4710-21.
- Delviks-Frankenberry K, Galli A, Nikolaitchik O, Mens H, Pathak VK, Hu WS. Mechanisms and factors that influence high frequency retroviral recombination. Viruses. 2011 Sep 9;3(9):1650-80.
- Pathak VK, Hu WS. “Might as well jump!” Template switching by retroviral reverse transcriptase, defective genome formation, and recombination. InSeminars in virology 1997 Jan 1 (Vol. 8, No. 2, pp. 141–150). Academic Press.
- Yin PD, Pathak VK, Rowan AE, Teufel 2nd RJ, Hu WS. Utilization of nonhomologous minus-strand DNA transfer to generate recombinant retroviruses. Journal of virology. 1997 Mar;71(3):2487-94.
- Temin, H.M. Retrovirus variation and reverse transcription: Abnormal strand transfers result in retrovirus genetic variation. Proceedings of the National Academy of Sciences. 1993 Aug 1;90(15):6900-3.
- Perdue ML, Garcı́a M., Senne, D., Fraire, M. Virulence-associated sequence duplication at the hemagglutinin cleavage site of avian influenza viruses. Virus research. 1997 Jun 1;49(2):173-86.
- Garushyants SK, Rogozin IB, Koonin EV. Template switching and duplications in SARS-CoV-2 genomes give rise to insertion variants that merit monitoring. Communications biology. 2021 Nov 30;4(1):1343.
- Lhomme S, Nicot F, Jeanne N, Dimeglio C, Roulet A, Lefebvre C, Carcenac R, Manno M, Dubois, M., Peron JM, Alric, L. Insertions and duplications in the polyproline region of the hepatitis E virus. Frontiers in microbiology. 2020 Jan 31;11:1.
- Golinelli MP, Hughes SH. Nontemplated nucleotide addition by HIV-1 reverse transcriptase. Biochemistry. 2002 May 7;41(18):5894-906.
- Wu W, Blumberg BM, Fay PJ, Bambara RA. Strand transfer mediated by human immunodeficiency virus reverse transcriptase in vitro is promoted by pausing and results in misincorporation. Journal of Biological Chemistry. 1995 Jan 6;270(1):325-32.
- Zhang, J., Temin HM. Rate and mechanism of nonhomologous recombination during a single cycle of retroviral replication. Science. 1993 Jan 8;259(5092):234-8.
- Zhang QY, Liu SQ, Li XD, Li JQ, Zhang YN, Deng CL, Zhang HL, Li XF, Fang CX, Yang FX, Zhang, B. Sequence duplication in 3′ UTR modulates virus replication and virulence of Japanese encephalitis virus. Emerging Microbes & Infections. 2022 Dec 31;11(1):123-35.
- Huang H, Chopra R, Verdine GL, Harrison SC. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: Implications for drug resistance. Science. 1998 Nov 27;282(5394):1669-75.
- Salie ZL, Kirby KA, Michailidis E, Marchand B, Singh K, Rohan LC, Kodama EN, Mitsuya H, Parniak MA, Sarafianos SG. Structural basis of HIV inhibition by translocation-defective RT inhibitor 4′-ethynyl-2-fluoro-2′-deoxyadenosine (EFdA). Proceedings of the National Academy of Sciences. 2016 Aug 16;113(33):9274-9.
- Preston BD, Poiesz BJ, Loeb LA. Fidelity of HIV-1 reverse transcriptase. Science. 1988 Nov 25;242(4882):1168-71.
- Bebenek K, Abbotts J, Roberts JD, Wilson SH, Kunkel TA. Specificity and mechanism of error-prone replication by human immunodeficiency virus-1 reverse transcriptase. Journal of Biological Chemistry. 1989 Oct 5;264(28):16948-56.
- Ji, J., Loeb LA. Fidelity of HIV-1 reverse transcriptase copying a hypervariable region of the HIV-1 env gene. Virology. 1994 Mar 1;199(2):323-30.
- Daddacha W, Noble E, Nguyen LA, Kennedy EM, Kim, B. Effect of ribonucleotides embedded in a DNA template on HIV-1 reverse transcription kinetics and fidelity. Journal of Biological Chemistry. 2013 May 3;288(18):12522-32.
- Kharytonchyk S, King SR, Ndongmo CB, Stilger KL, An, W., Telesnitsky, A. Resolution of specific nucleotide mismatches by wild-type and AZT-resistant reverse transcriptases during HIV-1 replication. Journal of molecular biology. 2016 Jun 5;428(11):2275-88.
- Lapkouski M, Tian L, Miller JT, Le Grice SF, Yang, W. Complexes of HIV-1 RT, NNRTI and RNA/DNA hybrid reveal a structure compatible with RNA degradation. Nature structural & molecular biology. 2013 Feb;20(2):230-6.
- Katz RA, Skalka AM. Generation of diversity in retroviruses. Annual review of genetics. 1990 Jan 1;24:409-45.
- Bruner KM, Murray AJ, Pollack RA, Soliman MG, Laskey SB, Capoferri AA, Lai J, Strain MC, Lada SM, Hoh, R., Ho YC. Defective proviruses rapidly accumulate during acute HIV-1 infection. Nature medicine. 2016 Sep;22(9):1043-9.
- Ho YC, Shan L, Hosmane NN, Wang J, Laskey SB, Rosenbloom DI, Lai J, Blankson JN, Siliciano JD, Siliciano RF. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell. 2013 Oct 24;155(3):540-51.
- Kuniholm J, Coote C, Henderson AJ. Defective HIV-1 genomes and their potential impact on HIV pathogenesis. Retrovirology. 2022 Jun 28;19(1):13.
- Perelson AS, Neumann AU, Markowitz, M., Leonard JM, Ho DD. HIV-1 dynamics in vivo: Virion clearance rate, infected cell life-span, and viral generation time. Science. 1996 Mar 15;271(5255):1582-6.
- Parthasarathi S, Varela-EchavarríA A, Ron Y, Preston BD, Dougherty JP. Genetic rearrangements occurring during a single cycle of murine leukemia virus vector replication: Characterization and implications. Journal of virology. 1995 Dec;69(12):7991-8000.
Figure 1.
Classification of Sequence Duplications. A schematic depiction of a classification system categorizing the non-homologous recombination events in HIV-1 leading to sequence motif duplication of four types. MR-ME: MHD-dependent recombination and matched-base extension. MR-MME: MHD-dependent recombination and mismatched-base extension. NR-ME: Non-MHD-dependent recombination and matched-base extension. NR-MME: Non-MHD-dependent recombination and mismatched-base extension.
Figure 1.
Classification of Sequence Duplications. A schematic depiction of a classification system categorizing the non-homologous recombination events in HIV-1 leading to sequence motif duplication of four types. MR-ME: MHD-dependent recombination and matched-base extension. MR-MME: MHD-dependent recombination and mismatched-base extension. NR-ME: Non-MHD-dependent recombination and matched-base extension. NR-MME: Non-MHD-dependent recombination and mismatched-base extension.
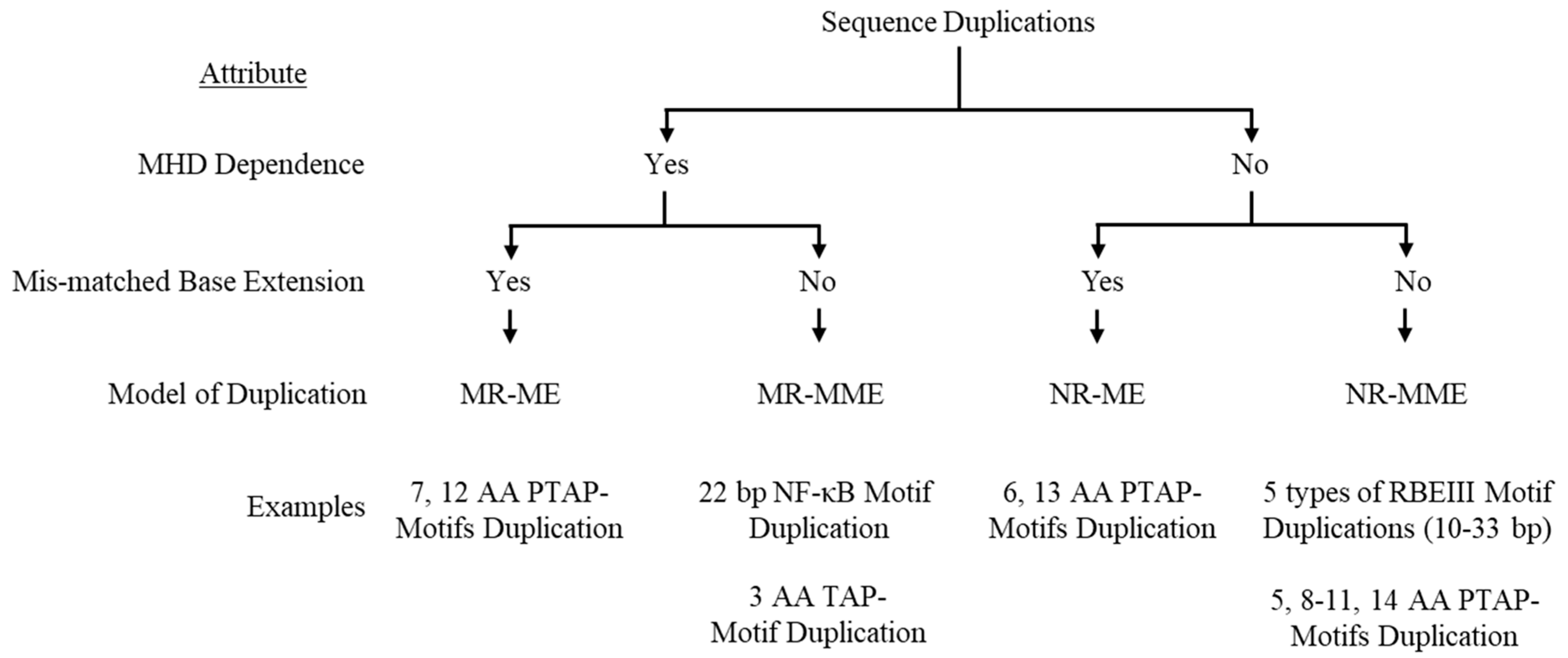
Figure 2.
A schematic model portraying the seven amino acid PTAP motif duplication in HIV-1C by the MR-ME model. (A) Two copies of the PTAP motif of seven amino acid length (EPTAPPA) and the corresponding nucleic acid sequence are presented. Solid and dashed arrows represent the original and the duplicated sequences, respectively. The MHDs, MHDa, and MHDc triplets are underlined. The 21-base sequence in p6-Gag corresponds to 2145-2169 coordinates in HXB2. (B) The donor and acceptor RNAs are shown using blue and red lines, respectively, and the black line represents the nascent cDNA. Dashed lines represent the template RNA degraded by the RNase H activity of the RT. The MHD motifs are highlighted in gray.
Figure 2.
A schematic model portraying the seven amino acid PTAP motif duplication in HIV-1C by the MR-ME model. (A) Two copies of the PTAP motif of seven amino acid length (EPTAPPA) and the corresponding nucleic acid sequence are presented. Solid and dashed arrows represent the original and the duplicated sequences, respectively. The MHDs, MHDa, and MHDc triplets are underlined. The 21-base sequence in p6-Gag corresponds to 2145-2169 coordinates in HXB2. (B) The donor and acceptor RNAs are shown using blue and red lines, respectively, and the black line represents the nascent cDNA. Dashed lines represent the template RNA degraded by the RNase H activity of the RT. The MHD motifs are highlighted in gray.
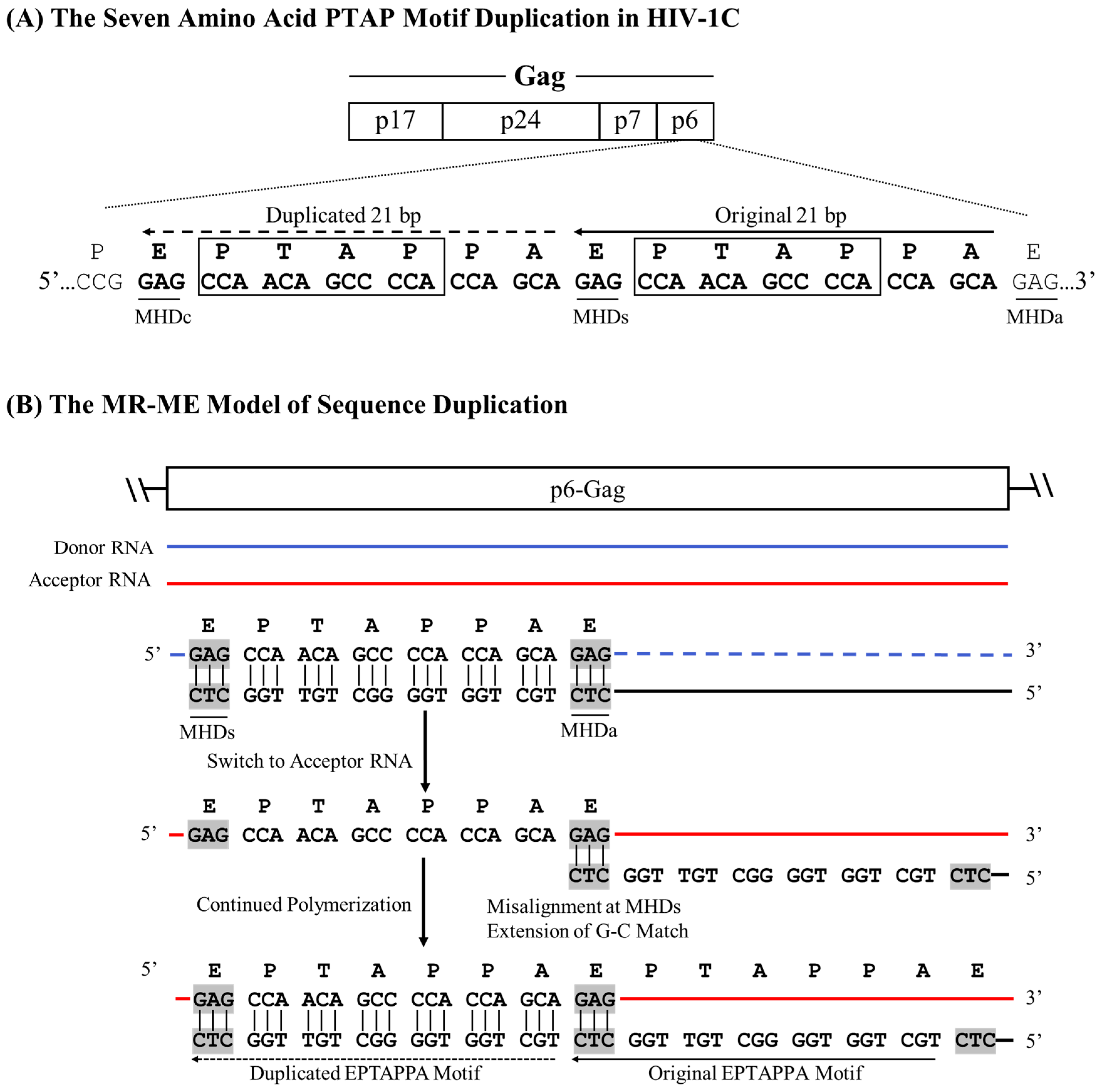
Figure 3.
A schematic model portraying the 22 bp NF-κB motif duplication in HIV-1C by the MR-MME model. (A) The architecture of the HIV-1C LTR depicting the relative positions of the four NF-κB and other important TFBS, along with their nucleic acid sequences. Solid and dashed arrows represent the original and the duplicated sequences, respectively. MHDs, MHDa, and MHDc triplets are underlined. The asterisks represent the C-to-T variation between the H- and F-κB motifs. The 22-base pair sequence in the LTR corresponds to 325-352 coordinates in HXB2. (B) The donor and acceptor RNAs are shown using blue and red lines, respectively, and the black line represents the nascent cDNA. Dashed lines represent the template RNA degraded by the RNase H activity of the RT. The MHD motifs are highlighted in gray.
Figure 3.
A schematic model portraying the 22 bp NF-κB motif duplication in HIV-1C by the MR-MME model. (A) The architecture of the HIV-1C LTR depicting the relative positions of the four NF-κB and other important TFBS, along with their nucleic acid sequences. Solid and dashed arrows represent the original and the duplicated sequences, respectively. MHDs, MHDa, and MHDc triplets are underlined. The asterisks represent the C-to-T variation between the H- and F-κB motifs. The 22-base pair sequence in the LTR corresponds to 325-352 coordinates in HXB2. (B) The donor and acceptor RNAs are shown using blue and red lines, respectively, and the black line represents the nascent cDNA. Dashed lines represent the template RNA degraded by the RNase H activity of the RT. The MHD motifs are highlighted in gray.
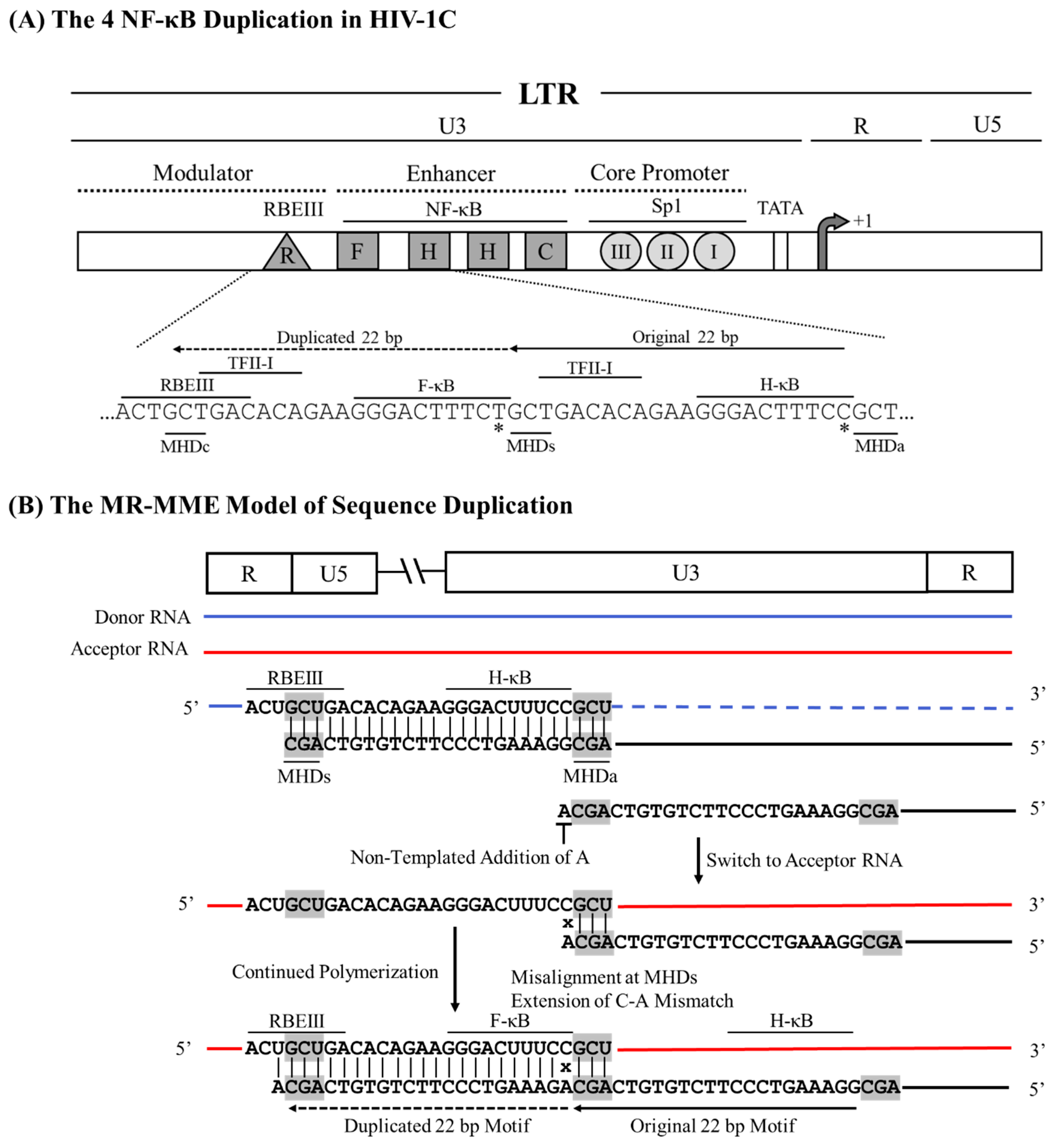
Figure 4.
A schematic model portraying the six amino acid PTAP motif duplication in HIV-1C by the NR-ME model. (A) Two copies of the PTAP motif of six amino acid length (EPTAPP) and the corresponding nucleic acid sequence are presented. Solid and dashed arrows represent the original and the duplicated sequences, respectively. The 18-base sequence in p6-Gag corresponds to 2145-2166 coordinates in HXB2. (B) The donor and acceptor RNAs are shown using blue and red lines, respectively, and the black line represents the nascent cDNA. The triplets highlighted by square boxes show the alleged MHD that do not exist in this strategy, and the mismatched bases are represented with an x.
Figure 4.
A schematic model portraying the six amino acid PTAP motif duplication in HIV-1C by the NR-ME model. (A) Two copies of the PTAP motif of six amino acid length (EPTAPP) and the corresponding nucleic acid sequence are presented. Solid and dashed arrows represent the original and the duplicated sequences, respectively. The 18-base sequence in p6-Gag corresponds to 2145-2166 coordinates in HXB2. (B) The donor and acceptor RNAs are shown using blue and red lines, respectively, and the black line represents the nascent cDNA. The triplets highlighted by square boxes show the alleged MHD that do not exist in this strategy, and the mismatched bases are represented with an x.
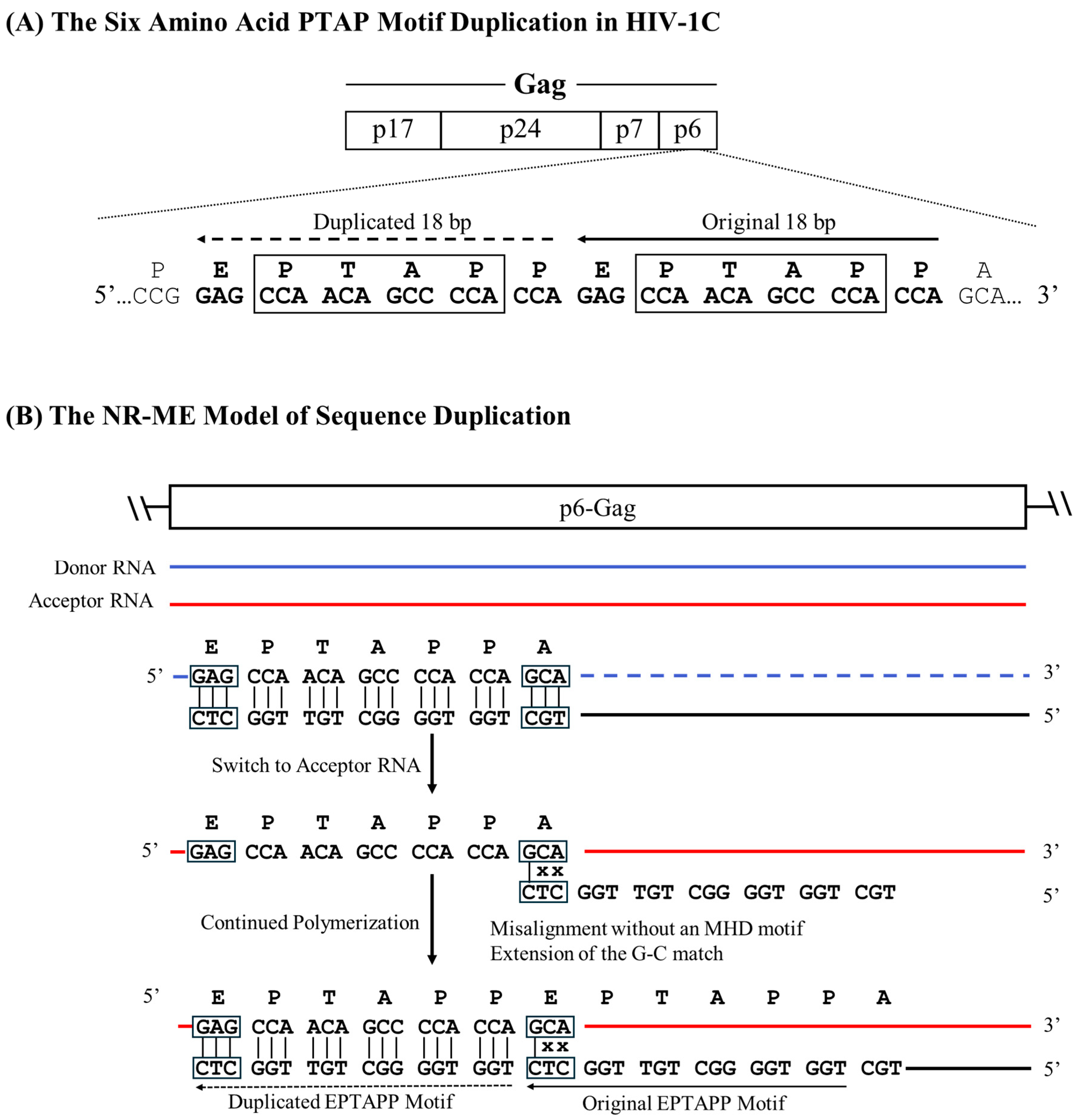
Figure 5.
A schematic model portraying the 26 bp RBEIII cluster duplication in HIV-1C by the NR-MME model. (A) The organization of the HIV-1C LTR presents the RBEIII cluster duplication, the important TFBS, and their corresponding nucleic acid sequences. Solid and dashed arrows represent the original and the duplicated sequences, respectively. The 26-base pair sequence in the LTR corresponds to 320-352 coordinates in HXB2. (B) The donor and acceptor RNAs are shown using blue and red lines, respectively, and the black line represents the nascent cDNA. The absence of the MHD is highlighted using black boxes, and the mismatched bases are represented with an x.
Figure 5.
A schematic model portraying the 26 bp RBEIII cluster duplication in HIV-1C by the NR-MME model. (A) The organization of the HIV-1C LTR presents the RBEIII cluster duplication, the important TFBS, and their corresponding nucleic acid sequences. Solid and dashed arrows represent the original and the duplicated sequences, respectively. The 26-base pair sequence in the LTR corresponds to 320-352 coordinates in HXB2. (B) The donor and acceptor RNAs are shown using blue and red lines, respectively, and the black line represents the nascent cDNA. The absence of the MHD is highlighted using black boxes, and the mismatched bases are represented with an x.
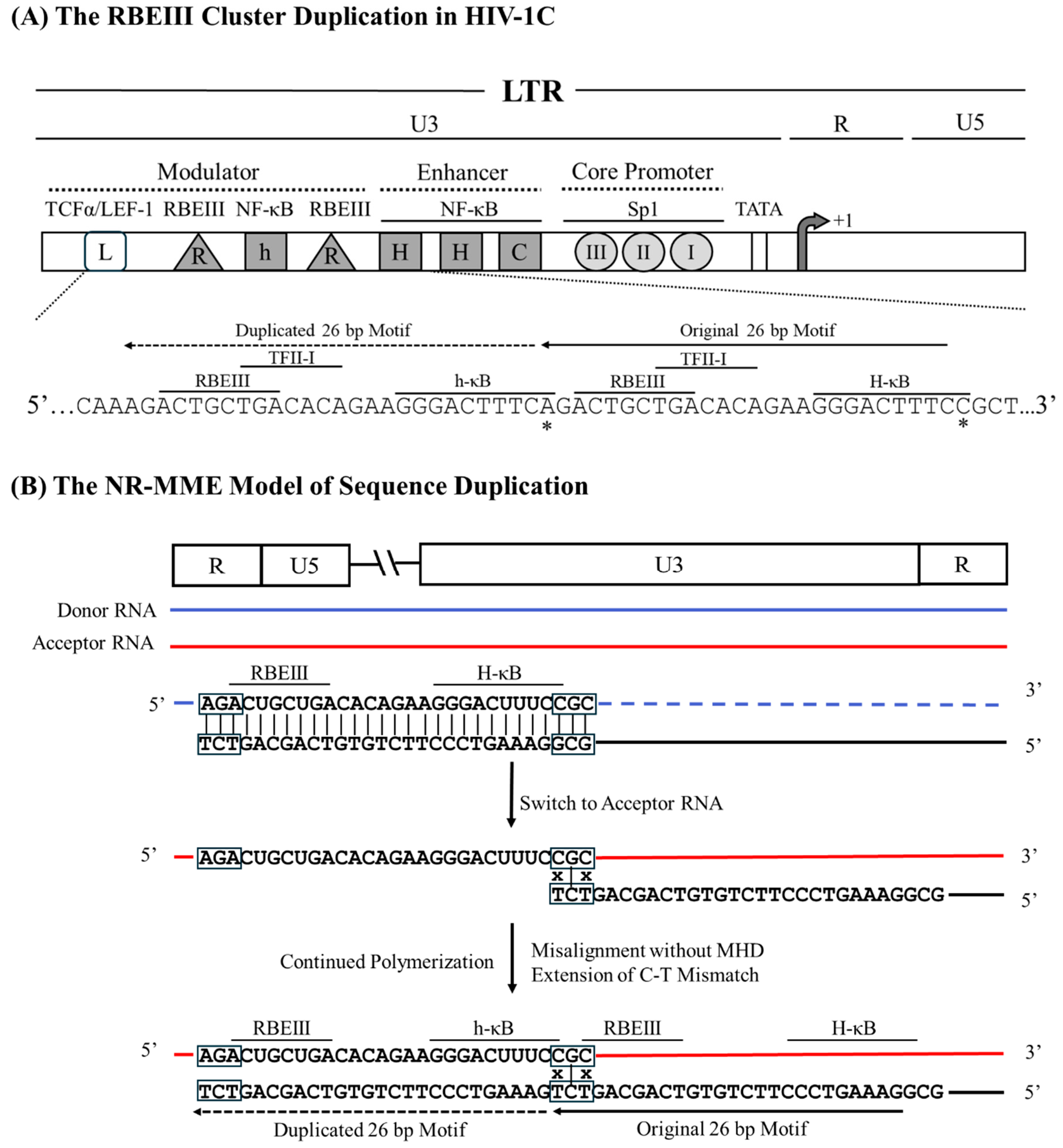
Figure 6.
A generalized model for sequence duplication. The model portrays the duplication of an approximately 22 bp sequence motif. The blue and red horizontal lines represent the two template RNA molecules, donor and acceptor, respectively, and the black line the nascent cDNA being polymerized by the RT. The dashed lines represent template RNA hydrolysed by the RNase H activity of the RT. Three dots in dark and light purple color represent the triplet micro-homology domains MHDa, and the MHDs, respectively. The terminal base that RT extends during sequence duplication is highlighted in green. Different RT domains are depicted in different colors, as shown. (A) The depiction of the RT stalling at the MHD and the acceptor RNA invasion from the 3’ end of the nascent DNA. The two possible outcomes at this event are shown using the bifurcated arrows. For clarity, the RT complex is rotated 90° from the top panel. To aid the visualization of the polymerase active site in this view, the finger domain is rendered transparent, with the blue dotted lines depicting its outline. The first outcome of the strand-switch, resulting in homologous recombination, is shown in the central left panel, where the alignment of the cDNA and the acceptor RNA template is perfect. The central right panel displays the misalignment of the cDNA at the MHDs, leading to the loop formation. The base on the template which is paired incorrectly is shown using a green dot on the DNA. (B) The maximum angle available for the rotation of the RT-nucleic acid hybrids in the central space of the RT is depicted after rotating the complex by a further 90° from the central panel, as shown. This view represents the RT as it is seen by the cDNA when it enters the cleft. The direction of the polymerization is perpendicular to the page surface. The available angle is measured across the G15, K 395, and G359 residues, as shown. Dashed lines are used to display the sections of the angle that are obstructed by the domains of the RT.
Figure 6.
A generalized model for sequence duplication. The model portrays the duplication of an approximately 22 bp sequence motif. The blue and red horizontal lines represent the two template RNA molecules, donor and acceptor, respectively, and the black line the nascent cDNA being polymerized by the RT. The dashed lines represent template RNA hydrolysed by the RNase H activity of the RT. Three dots in dark and light purple color represent the triplet micro-homology domains MHDa, and the MHDs, respectively. The terminal base that RT extends during sequence duplication is highlighted in green. Different RT domains are depicted in different colors, as shown. (A) The depiction of the RT stalling at the MHD and the acceptor RNA invasion from the 3’ end of the nascent DNA. The two possible outcomes at this event are shown using the bifurcated arrows. For clarity, the RT complex is rotated 90° from the top panel. To aid the visualization of the polymerase active site in this view, the finger domain is rendered transparent, with the blue dotted lines depicting its outline. The first outcome of the strand-switch, resulting in homologous recombination, is shown in the central left panel, where the alignment of the cDNA and the acceptor RNA template is perfect. The central right panel displays the misalignment of the cDNA at the MHDs, leading to the loop formation. The base on the template which is paired incorrectly is shown using a green dot on the DNA. (B) The maximum angle available for the rotation of the RT-nucleic acid hybrids in the central space of the RT is depicted after rotating the complex by a further 90° from the central panel, as shown. This view represents the RT as it is seen by the cDNA when it enters the cleft. The direction of the polymerization is perpendicular to the page surface. The available angle is measured across the G15, K 395, and G359 residues, as shown. Dashed lines are used to display the sections of the angle that are obstructed by the domains of the RT.
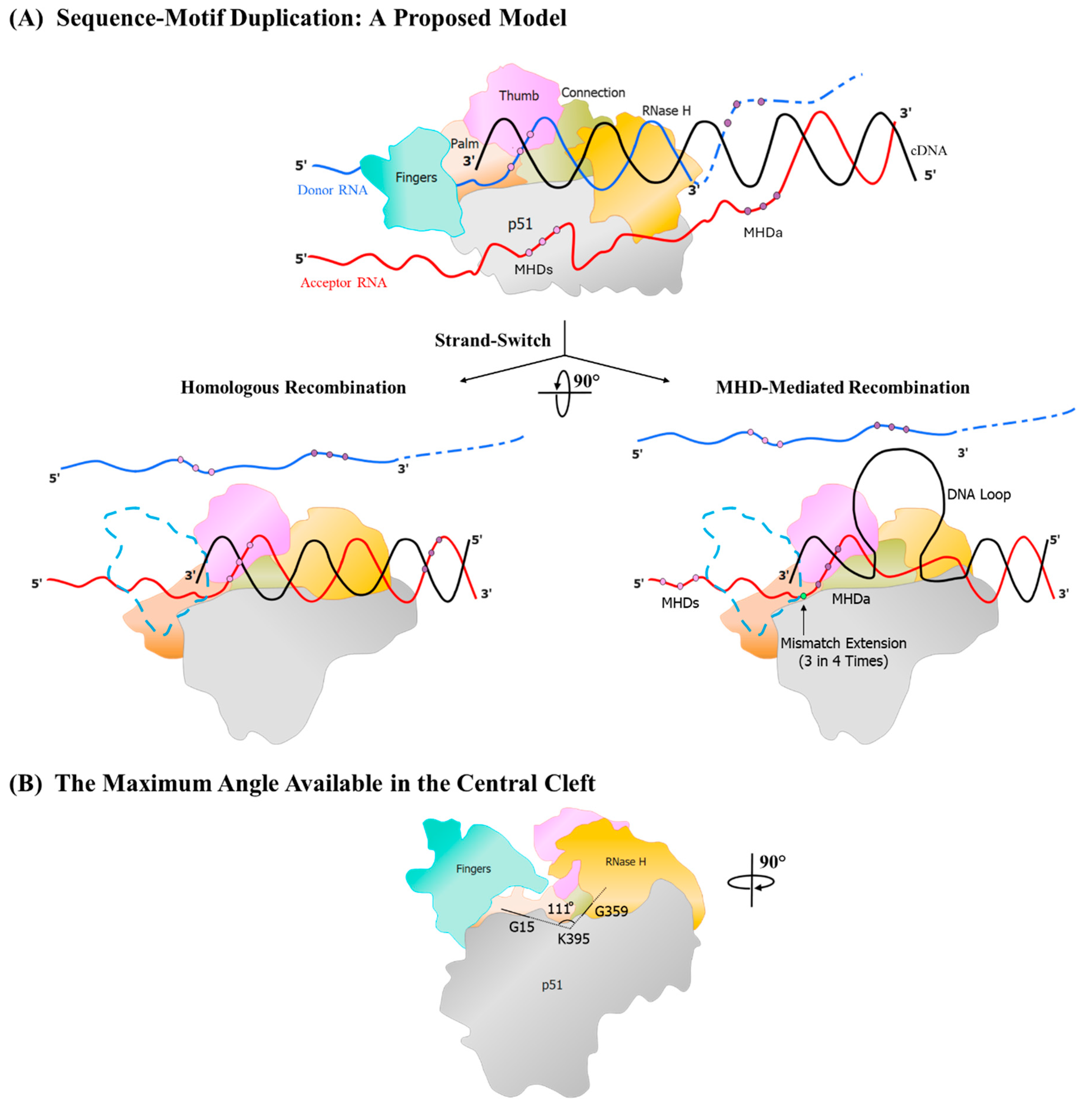
Figure 7.
DNA secondary structures in four types of sequence duplications: A representative minus strand cDNA sequence from each of the four proposed sequence duplication categories was used as input for the Unafold DNA secondary structure prediction software available at http://www.unafold.org/. The conditions for the structure prediction software were set at physiological concentrations of 14 mM and 0.5 mM for Na+ and Mg++ ions, respectively. The predictions characterized by the least ΔG values are presented. The terminal base at the 3’ end that the RT that must be extended in each case is highlighted using a red square box.
Figure 7.
DNA secondary structures in four types of sequence duplications: A representative minus strand cDNA sequence from each of the four proposed sequence duplication categories was used as input for the Unafold DNA secondary structure prediction software available at http://www.unafold.org/. The conditions for the structure prediction software were set at physiological concentrations of 14 mM and 0.5 mM for Na+ and Mg++ ions, respectively. The predictions characterized by the least ΔG values are presented. The terminal base at the 3’ end that the RT that must be extended in each case is highlighted using a red square box.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



