Submitted:
20 March 2025
Posted:
21 March 2025
You are already at the latest version
Abstract
Drink spiking is a significant public safety issue, often linked to crimes such as theft and sexual assault. The detection of drugs used in these incidents is challenging due to the low concentrations (<ng) and complex matrices involved. This review explores the application of gas chromatography (GC) and gas chromatography-mass spectrometry (GC-MS) for identifying drugs in spiked beverages. GC-MS offers high sensitivity and specificity, capable of detecting drugs at ng/mL levels and distinguishing between compounds with similar structures. The review highlights the advantages of GC-MS, including its ability to analyze multiple substances simultaneously and provide detailed molecular information. Various methods for detecting gamma-hydroxybutyrate (GHB), benzodiazepines, and other drugs in beverages are discussed, emphasizing the importance of derivatization to enhance volatility and chromatographic performance. The paper also addresses the challenges of analyzing complex beverage matrices and the need for continuous improvement in detection techniques to keep pace with the evolving drug market. Overall, GC and GC-MS are powerful tools for forensic analysis in drink spiking cases, offering reliable and accurate results essential for legal and investigative processes.
Keywords:
Gas Chromatography (GC)
; Mass Spectrometry (MS)
; Drink Spiking
; Forensic Analysis
; Gamma-Hydroxybutyrate (GHB)
; Benzodiazepines
; ketamine
; Drug-Facilitated Sexual Assault (DFSA)
1. Introduction
Drink spiking, or spiking, is the illicit addition of drugs or alcohol to a beverage belonging to a victim without their knowledge, leading to the victim experiencing drowsiness, confusion, nausea, and, potentially, memory loss. In more severe cases, victims may be rendered unconscious and experience harmful long-term psychological effects. Spiking events most often occur in bars, nightclubs, or house parties or other situation involving crowds of people such as music festivals. A person might be spiked to make them vulnerable for various motives, such as a prank or to enable a theft or sexual assault to be committed. It is believed that drug-facilitated sexual assault (DFSA) is underreported, but evidence suggests it is becoming an increasingly common offence [1,2]. Countries vary in how they address DFSA, with some focusing on punitive measures while others emphasize prevention and education. In the United Kingdom [3], spiking offences could lead to a perpetrator being prosecuted under several laws, including the Offences Against the Person Act 1861 [4] and Criminal Justice Act 1988 [5], and if an individual has attempted a DFSA against a victim after they may have become disorientated or incapacitated, the Sexual Offences Act 2003 [6]. Depending on the severity of an offence, an individual prosecuted under these laws may be required to pay substantial fines and could face a sentence ranging from several years to life imprisonment. In the recent King’s Speech, the UK government at the time proposed to introduce new legislation to make drink spiking a specific criminal offence with strict liability. This would mean that intent would not need to be proven for a conviction, potentially increasing conviction rates due to the challenges in proving intent. Identifying spiked drinks can be difficult as many drugs used for spiking are chosen for their ability to dissolve without noticeable visual changes in the drink. This makes it hard for unsuspecting victims to notice the addition of a drug to their drink and to identify the perpetrator. Such factors lead to a general underreporting of incidents complicating legal actions and victim support. Efforts to combat drink spiking include awareness campaigns, improved venue security, and the development of detection tools like drink-testing coasters [7], and wearable sensors [8]; these measures, however have not yet been widely implemented and can often be circumvented. Overall, drink spiking remains a significant public safety issue, requiring continued efforts in education, prevention, and support for victims. Detection of drugs used in DFSA is challenging due to the short biological half-lives [9] of these substances and the frequent delays in reporting by victims. Similarly, the analysis of small sample volumes with potentially degraded contents is required. Training responders to gather comprehensive data and encourage early sample collection is essential. Continuous improvement of detection methods is necessary to keep up with the evolving drug market.
The focus of this review is on the detection of drugs used in drink-spiking-related DFSA. There have recently been reports of DFSA resulting from what has been termed needle spiking, also known as injection spiking. Here, individuals are surreptitiously injected with drugs in crowded environments like nightclubs. Victims are reported to feel a sudden pinprick sensation, followed by symptoms such as dizziness, confusion, and blackouts. Despite numerous reports, there is skepticism among experts about the feasibility of such injections being administered unnoticed in crowded settings. Nevertheless, methods for the simple sample preparation of syringe residues for subsequent gas chromatography-mass spectrometry (GC-MS) analysis has been given [10]. Here, fresh portions of methanol are repeatedly drawn into the syringe, expelled, and collected in a test tube. This is then concentrated under nitrogen for reconstitution in a known solvent volume and introduction to the GC-MS.
Costa et al. [11] in 2020 reviewed the drugs commonly reported in DFSA. They highlighted that ethanol, benzodiazepines, ketamine, and gamma-hydroxybutyrate were the most commonly reported. However, a more recent review by Burrell et al. [12] revealed that other than ethanol, these drugs are rarely detected. For instance, they highlighted that Scott-Ham and Burton [13] found no covert use of the commonly DFSA associated benzodiazepine, Rohypnol, and Caballero et al. in 2017 [14] reported commonly perceived 'date rape drugs' in only 3 out of 152 DFSA cases. More recently, Orts et al. [15] have highlighted the emergence of the so-called designer benzodiazepines, such as; clonazolam and flubromazepam. In other regions of the world, such as Iran, opioids, such as dextromethorphan, the antihistamines; promethazine and cyproheptadine, and the antiemetics, metoclopramide are the most commonly reported [16]. Many other substances, including alcohol, over-the-counter medications, and prescription drugs, can also be used to commit these crimes [9]. These substances can have stronger effects when combined with alcohol and are often easier to obtain. Such findings highlight the need for analytical techniques capable of dealing with this complexity and changing demands.
Gas chromatography and GC-MS offer several key advantages for the determination of drug spiking cases. They can detect very low concentrations of drugs, often in the ng/mL range and in the case of GC-MS be used to identify unknown sample components. The chromatographic element allows for the analysis of beverages that may contain various ingredients and additives, and to simultaneously determine multiple different drugs and their metabolites in a single sample. This is important as drink spiking can involve a variety of substances. The mass spectrometry component provides detailed molecular information, in the form of mass spectra allowing for precise identification of compounds. This helps in distinguishing between substances of with similar structures, reducing false positives. Table 1 summarizes the GC applications given in this review.
1.1. Gas Chromatography
Gas chromatography, as with all chromatographic separations is based on partitioning of compounds between two phases. Unlike high-performance liquid chromatography (HPLC), the mobile phase is an inert gas and pays less of an involvement chromatographically, serving as a carrier to move components though the GC column where they can interact with the column stationary phase. For the sample to be carried effectively by the gas, it needs to be vaporized and converted into a gaseous state. For this, a small sample volume (typically 0.1 to 1.0 µL) is introduced into the injection port using a micro-syringe either manually or via an autosampler. The injection port is heated to a temperature higher than the boiling points of the analytes to ensure rapid vaporization. The carrier gas sweeps the vaporized analytes into the analytical column. Initial chromatographic focusing of the sample can then be achieved by either cold trapping or solvent focusing [17]. In cold trapping, the initial section of the column is kept at a low temperature to condense and focus the analytes into a narrow band. This is achieved by programming the oven temperature or using a cryogenic cooling system. In solvent focusing, the solvent vaporizes first and condenses at the front of the column, carrying the analytes with it. The column oven is then programmed to follow a temperature gradient. Initially, the temperature is kept low to allow for the chromatographic focusing of the analytes. It is then slowly increased, allowing for the separation of analytes based on their volatilities and interactions with the column stationary phase.
Components with low affinity for the stationary phase exit the column quickly, while those with higher affinity take longer. The separation is influenced by the analyte’s vapour pressures and their interactions with the stationary phase. Early GC methods used a single temperature (isothermal), but modern techniques employ temperature programming, starting at a low temperature and gradually increasing it to separate components more effectively. This approach achieves high chromatographic efficiencies, with modern capillary columns offering theoretical plate values exceeding 100,000 [18], notably higher than that obtained by other commonly applied separation techniques such as; high performance liquid chromatography (8000 to 12,000 plates) [19].
1.2. Gas Chromatography-Flame Ionisation Detection
As the separated components exit the column, they pass through to a detector, which generates a signal proportional to the amount of each component. The detector's signal is recorded as a chromatogram, a plot of detector response versus time. Each peak on the chromatogram corresponds to a different component of the sample, allowing for qualitative and quantitative analysis. There are a number of different detectors including the flame ionization detector (FID), electron capture detector (ECD) [20], flame photometric detector (FPD), and the nitrogen-phosphorus detector (NPD) [21] that have been used with GC. Mass spectrometry has become a preferred detector for GC over traditional detectors for several reasons. The MS can determine a wide range of compounds, including those that are not easily detected by other detector formats. It provides both quantitative and qualitative data; allowing for both quantification of substance and also its identification and the possibility of identifying unknowns. Nevertheless, FID remains popular for GC due to its cost-effectiveness, robustness, and sensitivity to hydrocarbons coupled to a wide linear dynamic range.
The FID [22], works by burning compounds eluting from the GC column in a small hydrogen and air-based flame. The high temperature of the flame causes organic compounds to ionize giving positively charged ions, and electrons. A potential difference is placed across the flame and the formation of these ions and electrons results in the generation of a small current directly proportional to the amount of analyte in the flame. The current formed is then converted into a voltage signal and is recorded as a peak on the chromatogram. The FID is highly sensitive to organic compounds, making it ideal for detecting a wide range of hydrocarbon-based drugs and other carbon-containing substances. It is relatively simple and robust, with a relatively large linear range and a stable baseline. However, it is this selective response to most organic compounds that makes it non-specific when used against biological and beverage-based samples. It is also not suitable for detecting inorganic compounds or compounds without carbon atoms.
1.3. Gas Chromatography-Mass Spectrometry
Most modern laboratories also utilize GC with mass spectrometry-based detectors which offer a number of advantages. Modern GC-MS systems can be used in two modes: full scan and selected ion monitoring (SIM). In full scan mode, the system scans a range of mass values, usually from m/z 50 to m/z 500, depending on the expected sample components. This mode gives a complete picture of the sample, detecting any ionizable compounds that produce fragments within the scanned range. The full scan mode allows for the mass spectrum of each compound to be obtained and to be matched with library spectra to identify unknowns. In full scan GC-MS, the mass spectrometer collects data across a wide range of m/z for each point in the chromatogram. Single or multiple m/z values can be also be “extracted” from this, and displayed in an ion extracted chromatogram (EIC) [23]. Post run, specific ions of interest corresponding to the compounds of interest can be selected from the full scan data. The software extracts the signal for these specific ions creating an EIC. The EIC can be used to highlight specific compounds within a complex mixture. Selected ion monitoring (SIM) mode is more targeted, and sensitive mode focusing only on specific fragments (m/z values). This approach is more sensitive as it scans a smaller range, increasing the number of scans per unit of time [23]. It also reduces interference from other ions and improves signal-to-noise ratios, enhancing detection limits.
1.4. Gas Chromatography Tandem Mass Spectrometry
Various detectors are available in single-dimension, classical GC–MS, such as ion trap and time-of-flight, but quadrupole-based systems are the most common due to their simplicity and cost-effectiveness. Quadrupole systems are also prevalent in gas chromatography tandem mass spectrometry (GC–MS/MS). The primary difference [23] between GC-MS/MS and conventional GC-MS systems is that GC-MS/MS has three quadrupole mass filters in series, whereas GC-MS has only one. In GC-MS/MS [23,24], the first quadrupole (Q1) functions like the single quadrupole in traditional GC–MS, selecting ions for the electron multiplier detector in full-scan or selected ion monitoring modes. The second quadrupole (Q2) induces collision fragmentation of ions from Q1, creating new fragments. The third quadrupole (Q3) then selects and analyses these fragments. GC-MS/MS can be operated in three modes. It can function like a single quadrupole system, with Q2 and Q3 passing ions to the detector without further alteration. In a product ion scan, Q1 performs selected ion monitoring, Q2 induces fragmentation, and Q3 analyses the new fragments, aiding in structural confirmation. In a precursor ion scan, Q1 operates in full scan, passing all fragments to Q2 for re-ionization, with Q3 monitoring a single fragment. The most sensitive and selective mode for quantitative GC-MS/MS is multiple reaction monitoring (MRM), where Q1 and Q3 analyze single ions, enhancing selectivity and reducing noise. GC–MS/MS is particularly useful in targeted analysis of a few analytes, especially when extreme sensitivity or low detection limits are needed, or when dealing with complex sample matrices. MRM can resolve issues with unresolved chromatographic peaks. However, MS/MS detectors are expensive, require special training, and need more maintenance. Their high sensitivity necessitates clean carrier gases and meticulous sample preparation, as errors and contamination are amplified.
1.5. Derivatisation
Gas chromatography requires analytes to be volatile and thermally stable, in the case of illicit substances with higher molecular weights versus small metabolites derivatization is often required to improve their volatility, and chromatographic performance. Common derivatisation methods include the formation of trimethylsilyl derivatives. Trimethylsilylation involves introducing a trimethylsilyl (TMS) group (–Si(CH₃)₃) into a molecule. This is usually undertaken using reagents such as N,O-bis(trimethylsilyl)acetamide (BSA), N-methyl-N-(trimethylsilyl)trifluoroacetamide (MSTFA), or trimethylsilyl chloride (TMSCl). The TMS group reacts with functional groups such as hydroxyl (–OH), carboxyl (–COOH), amino (–NH₂), or thiol (–SH) groups in the compound [25]. Trimethylsilylation significantly increases the volatility and thermal stability of the compound making them more amenable to GC analysis. Water and oxygen can cause significant issues during TMS derivatizations. Trace amounts of water can lead to partial or complete desilylation [26], resulting in unexpected peaks in the mass spectrum or the formation of the original compound and trimethylsilanol via hydrolysis. This is particularly challenging when attempting to isolate illicit substances from spiked beverages. A number of different derivatization methods have also been developed, such as those based on the formation of methyl esters via transesterification using methanol and a mineral acid catalyst or via the application of diazomethane [27]. However, these are beyond the remit of this review.
2. Methods
To compile this paper our primary search engine was Google Scholar; however, we also used Web of Science, and the University of the West of England Library. Only papers with full access and English as the published language were included. Since the nature of the paper is to discuss the role of GC/GC-MS in the analysis of beverages sample, we did not set an exclusion date range to allow for a broad overview. The search terms began with GC or GC-MS with the AND Boolean operator followed by one or more of: drugs AND beverages or drink.
3. Results
3.1. Ethanol
Ethanol is commonly determined by gas chromatography for investigations of both product quality [28] and alcohol consumption [29]. The forensic application of headspace gas chromatography flame ionization detector (GC-FID) of ethanol in blood has recently been described [30]. However, little in the literature exists on the application of GC or GC-MS for the determination of ethanol in beverages associated with DFSA. Recently, the presence of ethanol and other alcohols such as the more toxic methanol [31] in beverages has been determined using headspace GC-FID. The study reported methanol levels of between 32.0% to 58.3% v/v in samples of illicit alcoholic beverages taken from various parts of Rwanda. Evidently, the determination of both the type and concentration of the alcohol present in beverages is highly important.
3.2. Gamma-Hydroxybutyric Acid and Gamma-Butyrolactone
The structures of both gamma-hydroxybutyric acid (GHB) and its lactone, gamma-butyrolactone (GBL), are shown in Figure 1. These are central nervous system depressants that have been misused for their euphoric and relaxing effects. They are also used in DFSA due to their sedative and amnesic properties at higher doses [32]. Their increased abuse and involvement in criminal cases have led forensic laboratories to develop methods to detect these substances in various samples. Restrictions on the availability of GHB [33,34] led to consumers turning to its pro-drug, gamma-butyrolactone (GBL) [35,36], which is easier to obtain as it is commercially available as an industrial solvent [37]. Gamma-butyrolactone can be readily converted to GHB through simple chemical reactions or following consumption, enzymatically in the body [38]. More recently in the UK the 2022 regulations [39] have placed GBL and the related 1,4-butanediol (1,4-BD) under stricter control, requiring industrial users to obtain a controlled drugs license. From the 13th April 2022 GHB, has been reclassified as a Class B drug along with GBL and 1,4-BD [40].
Gas chromatography has been commonly used to detect GHB and the related compounds; GBL and 1,4-BD in beverages and biological fluids. Methods generally involve the derivatization of GHB to its trimethylsilyl derivative, which is then analyzed by GC-MS. This approach enhances the volatility and so the chromatographic behaviour of GHB. Alternatively, by acidifying the sample, GHB can be converted to its less polar, more volatile from, GBL. However, such approaches have been questioned as they can affect the equilibrium levels of GHB and GBL that may be present in the sample. Beverages, such as beer, wine, rum, Tequila, fruit juice and tonic water naturally contain ng/mL levels of GHB [41,42], which can serve to complicate understanding of the GHB levels detected. However, GHB levels commonly reported in drink spiking and sexual assaults are generally much greater, in the g/L region.
Tucci et al. [41] have reported a method based on ion-exchange solid phase extraction (SPE) of GHB followed by SIM GC-MS. One millilitre of degassed beverage sample was adjusted with 1 mL 0.025 M HEPES buffer pH 8.2 and 100 µL of the internal standard, GHB-d6 1 mg/mL. The resulting solution was then extracted with previously conditioned ion-exchange solid SPE cartridges. The SPE was washed with 6 mL MilliQ water and 2 mL acetone. Elution was then performed with 2 mL methanol containing 0.05 M glacial acetic acid under vacuum. The eluent was collected, evaporated to dryness under nitrogen, and 100 µL ethyl acetate and 50 µL 1% N,O-bis(trimethylsilyl)trifluoroacetamide (BSTFA)–trimethylchlorosilane (TMCS) were then added to derivatise the dried extract. After incubation at 65⁰C for 20 minutes, the derivatised extracts were cooled and introduced to the GC-MS. The developed method was reported to be linear over the range 20—1000 ng/mL; with a lowest limit of quantification of 6.7 ng/mL and a limit of detection of 4.5 ng/mL being reported. Both alcoholic and non-alcoholic beverages were investigated. The concentrations of GHB was shown in tonic water and lemon tonic water (130—180 ng/mL), rum (100—150 ng/mL), vodka (160—190 ng/mL), tequila (210—270 ng/mL) and fruit juice (380—500 ng/mL) beer (330—430 ng/mL) and in red (9300—12,000 ng/mL) and white wine (2500—3200 ng/mL).
Elliott and Fais [42] have investigated the concentrations of naturally occurring GHB in various non-alcoholic beverages including tonic water and lemon-flavored tonic water, were purchased from local stores to provide further evidence of its endogenous presence. Each beverage was sampled at 0, 24, and 96 hours after opening. Samples were degassed in an ultrasonic bath for 15 minutes before extraction. The analysis was performed using gas chromatography coupled to tandem mass spectrometry (GC–MS/MS) on an Agilent 6890/7000C Triple Quadrupole. The method involved liquid-liquid extraction with acidified ethyl acetate and MSTFA derivatization. The sensitivity of the method was determined with a limit of quantitation (LOQ) of 2.5 ng/mL and a limit of detection (LOD) of 1.3 ng/mL. Calibration curves and quality controls were used to ensure accuracy. GHB was detected in all beverage samples at very low concentrations, ranging from 89 to 145 ng/mL (0.089–0.145 mg/L). There was no significant variation in GHB concentration over the 96-hour period. The study confirmed that the detected GHB levels were far below those required to produce any pharmacological effect. GHB was detected in all beverage samples at very low concentrations, ranging from 89 to 145 ng/mL (0.089–0.145 mg/L). The study confirmed that the detected GHB levels were far below those required to produce any pharmacological effect.
The levels of GHB reported by Elliott and Fais are lower than that previously reported by Elliott and Burgess [43]. They reported that naturally occurring GHB and GBL were detected in those beverages involving the fermentation of white and particularly red grapes. No GHB or GBL was detected in other drinks such as beer, juice, spirits or liqueurs. Levels of GHB/GBL was detected in red wine vermouth (8.2 mg/L), sherry (9.7 mg/L), port (GBL), red wine (4.1–21.4 mg/L) and white wine (<3–9.6 mg/L). Their study utilized GC–FID for the detection of GHB and GBL as ‘‘total GBL’’ as it involved complete conversion to GBL prior analysis. This was undertaken by acidification with 6 M sulfuric acid and extraction with chloroform, using hexanoic acid as an internal standard. Levels of GHB was determined by full scan mode GC–MS following deviantization with TMS, using GHB-D6 as an internal standard.
Meyers and Almirall [44] developed a method using solid-phase microextraction (SPME) to extract GHB from water and beverage samples followed by on-fibre derivatization and analysis via GC/MS. The method demonstrated linearity from 0.01 mg/mL to 0.25 mg/mL for the detection of GHB in aqueous samples, without the need for sample manipulation that could possibly lead to the interconversion of GHB and its lactone, GBL. The method was successfully applied to detect GHB in spiked water and beverage samples. The SPME fibre was immersed in a solution of aqueous of GHB for 15 minutes with stirring for 15 minutes. The exposed fibre was then exposed to the atmosphere for 1 minute before being exposed to the headspace of 50 µL of BSTFA/TMCS (99:1) at 60⁰C for 40 minutes. The fibre was then again exposed to the atmosphere for 1 minute and then placed in the GC injection port set at set to 220⁰C to desorb for 12 minutes. Figure 2 shows the resulting mass spectrum obtained. Under the conditions used for analysis, an ionization voltage of 70 eV, no molecular ion was detected. This is common for TMS ethers as they tend to fragment easily under electron ionization (EI) conditions. A common fragmentation pathway involves the loss of a methyl group, resulting in a significant loss of m/z 15, rather than the molecular ion itself. The molecular ions of a TMS ethers can be unstable and may decompose before detection. This instability can be exacerbated by the presence of trace amounts of water or oxygen, which can lead to partial or complete desilylation. Even when molecular ions are formed, they are often of low abundance compared to other fragment ions. This makes them harder to detect and less prominent in the mass spectrum. Therefore, the authors utilized the peak at m/z 233 ([M- 15]+ peak) to show the presence of the derivatized GHB. Another ion indicative of derivatized GHB is m/z 159, resulting from the further loss of a TMS group. However, the base peak, m/z 147 ion, is not indicative of GHB, but is commonly observed in mass spectrometry of trimethylsilyl (TMS) derivatives in general [45]. Similarly, the ion m/z 73 in TMS derivatisation corresponds to the fragment (CH3)3Si+, which is the trimethylsilyl cation. This ion is formed during the EI process when a TMS group is cleaved from the molecule. The stability of the trimethylsilyl cation makes it a common and prominent fragment in the mass spectra of TMS derivatives.
Total vaporisation solid-phase microextraction (TV-SPME) involves heating the sample to completely vaporise the analytes, which are then adsorbed onto a solid-phase microextraction fibre. The fibre is coated with a sorbent material that selectively captures the analytes. This offers greater sensitivity compared to traditional liquid injection and SPME methods, making it suitable for detecting low-concentration analytes. Davies et al. [46] have used a TV-SPME based GC-MS approach to determine both GHB and GBL in water, beer, wine, liquor, and mixed drinks. The SPME fibre was initially exposed to the BSTFA +1% TMCS derivatizing agent. Once the fibre was saturated, it was transferred to the sample vial, where the sample was vaporized by heating to 60°C. After a 10-minute extraction period, the fibre was moved to the GC inlet for desorption at 250°C in splitless mode. The oven temperature started at 60°C and was held for 1 minute, then increased to 250°C at a rate of 15°C/minute, and maintained at the final temperature for 1 minute. The mass range scanned was from m/z 40 to m/z 550. Total ion chromatograms were generated, and extracted ion profiles were used to identify the analyte of interest in each sample. All compounds were identified using the SWGDRUG and/or NIST libraries.
Meng et al. [47] have reported a method based on dispersive liquid-liquid microextraction (DLLME) followed by GC–MS/MS for the determination of GHB in beverages and hair. For beverage samples, a 1 mL aliquot was placed into a 2-mL centrifuge tube, containing the internal standard, GHB-d6 at a concentration of 50 ng/mL. The pH of this was adjusted to 4.3 by adding ammonium dihydrogen phosphate to saturation. Afterwards, the 180 μL ethyl acetate as extractant was added into the solution. The centrifuge tube was sealed and sonicated in an ultrasonic bath for 3 minutes to form a cloudy suspension, facilitating mass transfer of target analytes into the ethyl acetate extraction solvent. The tube was then centrifuged and 30 µL of the upper ethyl acetate layer was withdrawn and derivatised with 30 µL BSTFA. This was then heated and following cooling, introduced to the GC–MS/MS. The instrument was operated in the scan mode for qualitative analysis in the range from m/z 40 to 500 and MRM mode for quantitative analysis. Monitored mass transitions were m/z 233/147 and 233/73 for GHB-TMS and m/z 239/147 and 239/73 for GHB-d6-TMS. The monitored mass transitions of m/z 233/147 and 239/147 were used for quantification.
The possible advantages of GC-MS compared to HPLC for the determination of GHB and gamma-hydroxyvalerate (GHV) (Figure 1) have been investigated by Mercer et al. [48]. Gamma-hydroxyvaleric, a 4-methyl analogue of GHB, which has also been abused and marketed as a dietary supplement. Studies by Carter et al. [49] aimed to compare the pharmacological and behavioural profiles of GHV and GHB. However, GHV showed lower affinity and did not significantly affect GABAB receptors and did not mimic the discriminative effects of GHB or that of the muscle relaxant drug, baclofen. However, GHV did share other effects like sedation, catalepsy, and ataxia, at higher doses.
Using a GC-MS based approach, following derivatization with N,O-bis(trimethylsilyl)trifluoroacetamide–trimethylchlorosilane (BSTFA + TMCS) a LOD of 1 pg on column was reported. The possibility of determining GHB in a variety of beverages was investigated including: water, Tropicanas cranberry juice cocktail with Barton s vodka (1.5 oz vodka and 10.5 oz cranberry juice), Coca Colas, Guinness Stouts beer, Coors Lights beer, and Willi Haags Riesling. A percentage recovery of 97% was also reported using an internal standard was 1,5-pentanediol and a surrogate standard of 1,2-hexanediol. The study also investigated the possibility of using HPLC with UV detection at 254 nm using a C-18 column and a mobile phase of 20:80% v/v methanol: dibasic phosphoric buffer (10 mM, pH 3) at a flow rate of 1.0 mL/min. This approach was found to be less sensitive (LOD 0.05 µg on column) for direct analysis of aqueous samples. In attempt to improve on this, and the presence of number of interferences observed from the beverage sample, an LLE was tested prior to HPLC analysis, but this resulted in a notable decreased in sensitivity (LOD 100 µg on column) and inconsistent peak profiles. Taken together both GC, and GC-MS have been successful multiple times in the detection of GHB and GBL at concentrations that are forensically relevant.
3.3. Xylazine
The utilization of xylazine (Figure 3), a veterinary drug, had recently been reported in DFSA. Sadiq et al. [50] have developed a GC-MS and GC-FID based method for the determination of xylazine in liquid, droplet, and dry beverage samples. The presence of xylazine was identified by GC-MS using the full scan mode (m/z 41 to m/z 500). The mass spectrum of xylazine was characterised by a base peak of m/z 205.1 and a molecular ion of m/z 220.1 (70%). Quantification was then undertaken by GC-FID. Samples of water, an energy drink, a carbonated drink, and a fruit-based drink were investigated. A sample aliquot of 1.0 mL of beverage sample was transferred into a vial and the pH adjusted ca. pH 11 using 13% sodium hydroxide solution. This was then extracted with two 0.5 mL portions of dichloromethane (DCM). The organic phases were then combined, passed through anhydrous sodium sulphate, and blown down under nitrogen to dryness. The resulting residue was reconstituted in 0.1 mL of 10 µg/mL internal standard solution (2,2,2-triphenylacetophenone). Three different drink sample types were investigated: Liquid drink itself, droplets of drink residue and dry drink residue. For the extraction of liquid drink; 1.0 mL of the sample was taken and subjected to liquid-liquid extraction (LLE). Droplets of drink residue and dry drink residue were recovered by rinsing with 1.0 mL of distilled water, and the resulting sample extract then subjected to LLE. Higher recoveries of xylazine were achieved from beverage samples in liquid form (77.2-97.3%) compared to droplets (50.8% – 80.0%) and dry samples (39.8% – 66.9%). A limit of detection (LOD) and limit of quantitation (LOQ) were reported at 0.08 µg/mL and 0.26 µg/mL, respectively. Despite the decreased recovery in the droplets and dry samples there is sufficient recovery such as to still be forensically relevant. The chromatographic separation of xylazine was shown to be possible from a number of drugs including paracetamol, caffeine, ketamine, codeine, morphine, 6-monoacetylmorphine and heroin.
3.4. Benzodiazepines
1,4-benzodiazepines are commonly used as tranquilizers and antidepressants in clinical practice. However, their easy availability and synergistic effects with alcohol make them appealing for criminal misuse. Milk-based alcoholic drinks, such as whiskey creams, are a popular beverage, but represent a complex samples matrix; containing proteins and fatty acids, that several of which can interfere with extraction and quantification. Famiglini et al. [51] have investigated the possibility of determining the eight benzodiazepines; diazepam (Valium), chlordiazepoxide (Librium), clobazam, flunitrazepam (Rohypnol), bromazepam, flurazepam, nitrazepam (Mogadon), and clonazepam (Figure 4) in milk-based alcoholic drinks (whiskey creams) using a QuEChERS based extraction method prior to quantification by GC–MS. To simulate realistic crime scene scenario conditions, 0.5 mL of whiskey cream (the typical residue volume found at the bottom of a glass) was diluted with Millipore water to a final volume of 10 mL. An aliquot of 10 mL of acetonitrile was then added, and the resulting mixture manually shaken for one minute before extraction and placed in the extraction tube. This was stirred for one minute, and then centrifuged. The supernatant (10 mL) was collected and concentrated to 1 mL under a nitrogen flow. The final volume was transferred to the purification tube, manually shaken and centrifuge. The supernatant (0.5 mL) was collected, and the final volume was adjusted to 0.5 mL with acetonitrile. A 5.0 µL aliquot of internal standard (Medazepam) was added to give a final concentration of 0.5 µg/mL. One microlitre of the resulting solution was then introduced to the GC-MS. The mass spectrometer was operated in SIM mode, and Limits of detection and limits of quantitation were in the range of 0.02–0.1 and 0.1–0.5 µg/mL, respectively. Whiskey cream beverages fortified with commercial drugs at 20 µg/mL gave percentage recoveries of between 47% to 64% with RSD between 11 % and 14 % for all eight benzodiazepines.
The possibility determining the benzodiazepines; in beer and peach juice by full scan GC-MS has been reported [52]. The compounds were extracted from spiked drinks by LLE with chloroform: isopropanol 1:1(v/v). Benzodiazepine pills containing flunitrazepam, clonazepam, alprazolam, diazepam and ketamine (Figure 5) were used to fortify samples of peach juice and beer. Samples were fortified at concentrations reported to approximate doses that represent peak response and amnesia. An aliquot of 1.0 mL of the fortified beverage was then extracted with 1 mL of a 1:1 (v/v) solution of chloroform : isopropanol. The resulting organic extract was then and evaporated under nitrogen and the resulting residue was reconstituted in 0.5 mL internal standard (medazepam) solution and introduced to the GC-MS using a scan range of m/z 50–600. Extracted ion chromatograms were used to determine the analytes and internal standard peaks from the total ion chromatograms.
The collection of blood and urine samples is commonly undertaken in cases of DFSA. However, a number of these drugs have short half-lives [9]. Accordingly, there is interest in alternative samples such as the possibly spiked beverages and their residues. Consequently, there is interest in understanding the stability of drugs in various beverages. Gautam et al. [53] have investigated the behaviour of three benzodiazepines (diazepam, flunitrazepam and temazepam) in five drinks, an alcopop (flavoured alcoholic drink), a beer, a white wine, a spirit, and a fruit based non-alcoholic drink (J2O). Stability of the benzodiazepines under two different storage conditions, uncontrolled room temperature and refrigerator (4 ⁰C) over a 25-day period. Reportedly, all drugs could be detected in all beverages investigated over this time period. Diazepam was found to be stable in all of the beverages, except the J2O, under both storage conditions. The stability flunitrazepam changed after being stored in both wine and J2O. This was concluded to be due to the pH or alcohol content, as both beverages have a pH of 3.2 and the wine investigated had an ethanol concentration of 12.5%, while J2O was alcohol-free. Temazepam also degraded in all drinks apart from in beer stored at 4°C. The stability of benzodiazepines varied by beverage type, with J2O showing significant changes for all substances investigated. Factors like pH, alcohol content, extraction, and analysis might affect stability, but this investigation was unable to show any clear patterns.
Solid-phase extraction (SPE) combined with dispersive liquid–liquid microextraction was used to extract trace amounts of diazepam, midazolam, and alprazolam from water, tap water, fruit juices, and urine [54]. The analytes were adsorbed onto octadecyl silica SPE columns from 60 mL sample volumes. After elution with acetone, the eluent was injected into water, extracted, and centrifuged. The sedimented phase was analyzed using by GC-FID. The method showed low detection limits (0.02-0.05 µg/L), with a linear range (0.1-100 µg/L).
Jain et al. [55] have explored the possibility of using fabric phase sorptive extraction (FPSE) for the determination of sedative-hypnotic drugs in food and drink samples combined with GC-MS. Fabric phase sorptive extraction is a sample preparation technique where the extraction medium is a fabric substrate coated with a sol-gel sorbent, a thin layer of an inorganic or organically modified inorganic polymer, enhancing the ability of the fabric to interact with and retain analytes. The FPSE membrane is immersed in the sample and stirred to enhance interaction between the analytes and the FPSE membrane. Following extraction, the membrane is then back extracted in a small volume of solvent, and the resulting solution is then introduced to the GC-MS. Beverage samples of flavored milk, juice, water, tea and beer along with food samples of chocolate, cream, and cake were investigated. Sol-gel coated FPSE membranes, were coated with Carbowax 20M (CW-20M) and used for the extraction of food and drink samples fortified with diazepam, chlordiazepoxide, and ketamine and diluted with ultrapure water adjusted to pH 12. Extraction was undertaken following dilution with ultrapure water and adjustment to pH 12. The FPSE membranes were then immersed in the prepared sample with stirring for 45 minutes. The exposed membrane was then transferred to an Eppendorf tube containing 0.5 mL of methanol for 10 minutes to desorb the analytes. A suitable aliquot of this was then examined by GC-MS. Chromatograms were obtained using full scan GC-MS with subsequent processing to give ion-extracted chromatograms. The method was reported to be linear over the range of 0.3–10 µg/mL with a R² values ranging from 0.996 to 0.999. Detection limits of between 0.020–0.069 µg/mL for liquid samples and 0.056–0.090 µg/g for solid samples were reported. The FPSE based GC-MS method was successfully applied to real forensic food samples involved in drug-facilitated crimes, demonstrating its effectiveness in detecting diazepam, chlordiazepoxide, and ketamine residues.
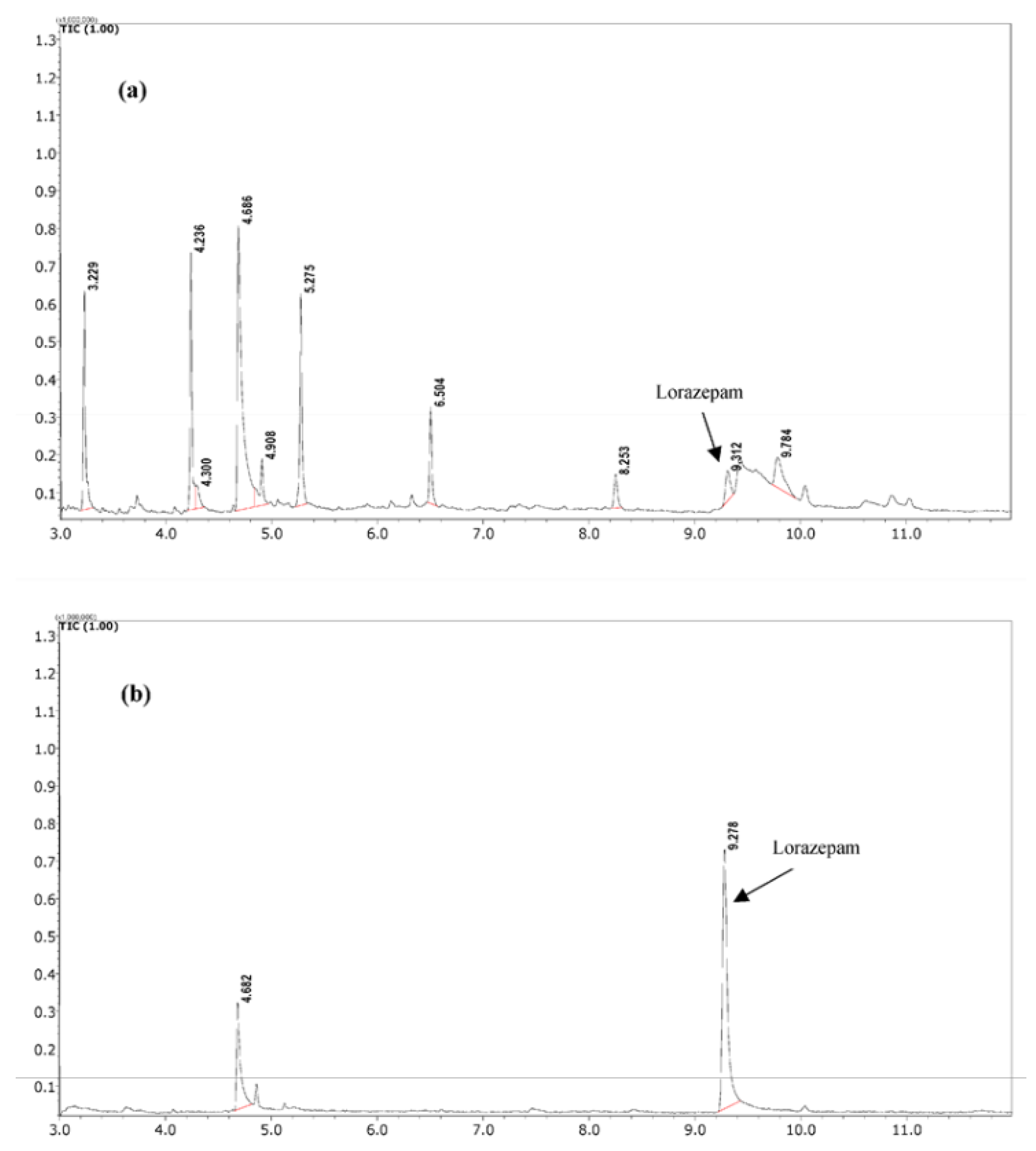
Jain et al. have also investigated the related technique of cellulose paper sorptive extraction (CPSE) [56] for determination of lorazepam residues in food and tea samples using GC-MS. Samples of tea are diluted and adjusted to pH 12. Pieces of cellulose paper (1.5 cm x 1.5 cm) were introduced into the diluted food matrices and stirred on a rotary shaker at 200 rpm for 30 minutes. The cellulose papers were then dried and the adsorbed lorazepam back-extracted into 2 mL of methanol. The methanol extract was investigated by GC-MS in the SIM mode using m/z 275, 303, 239. The method was reported to be linear over the range of 0.2–10 µg/mL with a R² values ranging from 0.996 to 0.998. A LOD of 0.054 µg/g for cream biscuits and 0.05 µg/mL for tea samples with LOQ of 0.18 µg/g for cream biscuits and 0.16 µg/mL for tea samples were reported. The method was applied to real forensic food samples involved in drug-facilitated crimes, demonstrating its effectiveness in the extraction and giving cleaner extracts for the determination of lorazepam residues.
Figure 6.
Comparison of sample clean-up capacity of (a) LLE and (b) CPSE in a real forensic sample (tea sample from case 1) in TIC mode. Peak identification: Caffeine at 4.68 min; Lorazepam at 9.29 min. form reference [56].
Figure 6.
Comparison of sample clean-up capacity of (a) LLE and (b) CPSE in a real forensic sample (tea sample from case 1) in TIC mode. Peak identification: Caffeine at 4.68 min; Lorazepam at 9.29 min. form reference [56].
3.5. Other Drugs
As well the problems associated with drinks being spiked with drugs reports have also shown issues relating to the deliberate use of drugs in the manufacture and formulation of the beverage itself. The composition and drugs used to formulate these beverages are broad and the resulting names these are known by are equally as broad. One notable example is that commonly sold as “Dirty Sprite” [57]; a preparation prepared from mixing soft drink with cough medicines containing codeine and promethazine. The GC-MS determination of both codeine and promethazine in such beverages has been recently reported by Rosenberger et al. [57]. Samples were extracted following a modification of the liquid/liquid extraction method described by Meatherall [58]. Following adjustment to pH ≥ 9 with ammonium hydroxide solution, a sample aliquot of 1.0 mL was extracted with an equal volume of 1-chlorobutane. The resulting extract was then blown down to dryness and reconstituted in 0.1 mL of methanol and introduced to the GC-MS. Screening was undertaken by GC–MS and was reported to revealed the presence of promethazine, dihydrocodeine, codeine and cocaine (Figure 7). Quantification was undertaken using SIM, and concentrations of between 68 to 75 mg/L promethazine and 130 mg/L codeine were recorded. The presence of other drugs such as cocaine at 3.4 mg/L, and dihydrocodeine at 91 mg/L was also reported. Further screening by LC-MS/MS did not reveal the presence of any other drugs.
Phonchai et al. [59] have investigated what is known colloquially as a “lean cocktail”; an improvised drink made from mixing cough or prescription medicines with a beverage. It is also sometimes referred to as a “dirty sprite” amongst other names. The Phonchai et al. [59] investigation focused on the determination of tramadol and diphenhydramine, codeine, promethazine (Figure 8) and caffeine in “Lean Cocktails” obtained from five high schools located in the Songkhla, Satun, and Yala provinces of Southern Thailand. The authors developed a “dilute and shoot” GC-FID based method for nine “Lean cocktail” samples. Aliquots of 1.0 mL of sample were filtered, and then diluted with 1:5 in methanol and a suitable aliquot directly introduced to the GC-FID. Three of the samples were reported to contained just tramadol at levels between 195 to 859 mg/L and another three only promethazine between 71 to 87 mg/L. Both tramadol and promethazine were detected in a further three of samples at levels between 179 to 576 mg/L and 7 to 47 mg/L, respectively. None of the samples were reported to contained caffeine, codeine, or diphenhydramine. The injector was held at a temperature of 260⁰C with the initial GC column temperature set at 120⁰C for 1.0 minute before being ramped to 240⁰C at a rate of 10⁰C/minute, and held isothermal for 4.0 minutes. The temperature was then increased to 280⁰C at 20⁰C/minute and then held at this temperature for 4.0 minutes.
The fabrication of SPME fibres modified with a graphene oxide, and a metal-organic framework (MOF) zeolitic Imidazolate Framework-8 (GO@ZIF-8 MOF) combined with molecularly imprinted polymers (MIP) has been recently reported [60]. The SPME was used for the extraction of the amphetamines; dextroamphetamine, methamphetamine, methylphenidate, and modafinil (figure 8). The levels of these was then determined by GC-MS in the SIM mode in a number of beverages and snacks. The GO@ZIF-8 MOF has properties such as; high surface area, porosity, and chemical and thermal stability, making them a good candidate for modification with MIPs and application as SPMEs. Following optimization of the GO@ZIF-8 MOF MIP SPME, a linear range between 0.1 and 400 μg/L with a R2 >0.9976 was reported for the compounds investigated. Detection limits for amphetamine derivatives were found to be between 0.023 and 0.033 μg/L. Samples include cappuccino, espresso, Nescafe coffee, breakfast cereal, dark chocolate, energy drinks, Gummi candies, ginseng drinks, truffles, marshmallows and toffee. Ten grams of sample was homogenised and a 500 mg aliquot dissolve in 200 mL of water with the aid of ultrasound. The solution was then further diluted with deionized water and following adjustment to pH 3.5, extracted using GO@ZIF-8 MOF/MIP-SPME. Before extraction, monolithic fibre bundles were loaded into a 5 mL Teflon syringe and inserted through the vial septum. The extraction was performed by immersing 1 cm of the fibre into the sample for 35 minutes with stirring. The fibres were then transferred into 1-octanol for desorption, with the aid of ultrasonication. The solvent was then evaporated to dryness under nitrogen, and the residue was re-dissolved in 20 μL of methanol for GC-MS analysis. Between samples, the fibres were reconditioned by immersing them in methanol and ultrapure water. The GC injection port was set at 260⁰C in split less pulse mode. An HP-5MS fused silica capillary column was used for separation, with a temperature program that included an initial temperature of 67⁰C and a programmed increase to finally 270⁰C.
Vortex-assisted dispersive liquid–liquid microextraction-gas chromatography (VADLLME-GC) has been investigated for the determination of ketamine, nimetazepam, and xylazine [61]. The study focused on the application of a novel extraction method for these drugs in various forms of spiked mineral water, carbonated drink, tea, beer, and orange juice for quantification by GC FID. Beverages were spiked with known quantities of the drugs and prepared in three forms: liquid, droplet, and dry residues. The extraction involved using dichloromethane as the extraction solvent and ethanol as the dispersive solvent. The study optimised the extraction parameters of the VADLLME procedure, including the choice and volume of solvents, vortex agitation time, centrifugation rate and time, and pH adjustment. The samples were vortexed, centrifuged, and the organic phase was collected and evaporated before introduction to the GC. The method was reported to give a LOD of 0.08 μg/mL for ketamine and xylazine, and 0.16 μg/mL for nimetazepam. Higher recoveries were achieved for liquid samples (51%–97%) compared to droplet (48%–96%) and dry samples (44%–93%). Intra-day and inter-day precision (%RSD) below 7.2% and accuracy (% recovery) between 92.8% and 103.5%.

Table 1.
Determination of drugs in beverage samples by gas chromatography and gas chromatography-mass spectrometry.
Table 1.
Determination of drugs in beverage samples by gas chromatography and gas chromatography-mass spectrometry.
| Analytes | Sample Matrix | Derivatisation | Sample Pre-Treatment | Type of GC | LOD/LOQ (mg/L) | Comments | Ref |
|---|---|---|---|---|---|---|---|
| GHB | Beer, wine, rum, Tequila, fruit juice and tonic water | BSTFA 1% TMCS | Ion-exchange solid phase extraction. | SIM GC-MS. HP-5MS (30 m × 0.25 mm I.D, 0.25 µm film | LOQ 0.0067 and LOD 0.0045. | Internal standard GHB-d6. Naturally occurring levels of GHB determined. | [41] |
| GHB | Tonic water and lemon-flavored tonic water | MSTFA | liquid-liquid extraction with acidified ethyl acetate | MRM GC–MS/MS. HP-5MS column 30 m x 0.25 mm x 0.025 µm). | LOQ 0.0025 and LOD 0.0013 | Internal standard GHB-d6. Naturally occurring levels of GHB determined. | [42] |
| GHB and GBL | Beer, juice, spirits, liqueurs, sherry, port, white and red wine | TMS for GHB and conversion via acidification to give total GBL. | LLE with chloroform. | Full scan GC-MS and GC-FID. DB5 MS capillary column, 30 m x 0.25 mm, 0.25 μm film thickness. | GHB LOD 3.0 | Internal standard GHB-d6. Naturally occurring levels of GHB and GBL determined. | [43] |
| GHB | Water, Coca Cola, beer, lemonade. | On-fibre derivatisation with BSTFA/TMCS (99:1). | SPME. | GC-MS. A 30-m HP5-MS column with a 0.25 µm film thickness and 0.25 mm | LOQ 1.5. | Internal standard GHB-d6. | [44] |
| GHB and GBL | Water, beer, wine, liquor, coca cola and mixed drinks. | On-fibre derivatisation with BSTFA/TMCS (99:1). | Total vaporisation SPME. | Full scan GC-MS. Extracted ion profiles were used to identify the analyte. | LOD 1.0. | [46] | |
| GHB | Beverage samples and hair. | BSTFA 1% | Dispersive liquid-liquid microextraction with ethyl acetate following adjustment to pH 4.3 with ammonium dihydrogen phosphate. | GC-MS/MS. DB-5MS capillary column (30m × 0.32mm ID, 0.25 μm film. | LOD 0.0005. | Internal standard GHB-d6. | [47] |
| GHB | Water, Tropicanas cranberry juice cocktail with Barton s vodka, Coca Cola, Guinness Stouts beer, Coors Lights beer, and Willi Haags Riesling. | BSTFA + TMCS | Dilution in internal standard solution. | GHB was quantitated using the peak area of the ion at m/z 233 and derivatized GHV was quantitated using the peak area of the ion at m/z 117. HP-5, 30 m x 0.25 mm i.d. x 0.25 µm film. | 1.0 pg on column. | Internal standard 1,5-pentanediol. Reverse phase HPLC also investigated. | [48] |
| Xylazine | Energy drink, a carbonated drink, and a fruit-based drink. | None | LLE with dichloromethane following adjustment to pH 11 using 13% sodium hydroxide solution. | Full-scan GC-MS and GC-FID. 5%-phenyl)-methylpolysiloxane (HP5) capillary column (30 m × 0.32 μm i.d., 0.25 µm film thickness, | LOD and LOQ were reported at 0.08 and 0.26 respectively by GC-FID. | Internal standard 2,2,2-triphenylacetophenone | [50] |
| Diazepam, chlordiazepoxide, clobazam, flunitrazepam, bromazepam, flurazepam, nitrazepam, and clonazepam. | Milk-based alcoholic drinks (whiskey creams). | None. | QuEChERS based extraction. | SIM GC-MS. HP-5MS (30 m 0.25 mm i.d., 0.25 µm film thickness | LOD and LOQ were in the range of 0.02–0.1 and 0.1–0.5, respectively. | Internal standard medazepam. | [51] |
| Flunitrazepam, clonazepam, alprazolam, diazepam and Ketamine | Peach juice and beer. | None. | LLE with chloroform: isopropanol 1:1 (v/v). | Full scan GC-MS (m/z 50–600). Ion extracted chromatograms used. HP-5MS 30 m x 250 µm i.d. x 0.25 µm film. | LOD between 1.3 and 34.2. LOQ 3.9 and 103.8. | Internal standard medazepam. | [52] |
| Diazepam, midazolam, and alprazolam | Tap water, fruit juices, and urine | None. | SPE combined with dispersive liquid–liquid microextraction | GC-FID. HP-5 30 m x 0.32 mm and 0.25 µm film thickness | LODs of between 0.00002 - 0.00005 | [54] | |
| Diazepam, chlordiazepoxide, and ketamine | Flavoured milk, juice, water, tea and beer | None. | Fabric phase sorptive extraction (FPSE) | Full scan GC-MS with subsequent processing to give ion-extracted chromatograms | LODs of between 0.020–0.069 | Food samples of chocolate, cream, and cake also investigated. | [55] |
| Lorazepam | Tea | None. | Cellulose paper sorptive extraction (CPSE) | GC-MS in the SIM mode using m/z 275, 303, 239. Txi-5Sil MS capillary column (30 m length x 0.25 mm internal diameter x 0.25 µm film thickness) with a stationary phase of 95% dimethylpolysiloxane and 5% phenyl. | LOD 0.05 | Cream biscuits also investigated. | [56] |
| Codeine and promethazine | “Dirty Sprite” beverage formulated from a mixture of soft drink, and cough medicine. | None. | LLE with 1-chlorobutane following adjustment to pH ≥9 with ammonium hydroxide solution. | Full scan and SIM GC-MS. DB-5 ms, 30 m, i.d. 0.25 mm, film thickness 0.25 μm | LOD/LOQ were 0.3/0.9 for cocaine, 0.3/1.0 for promethazine, 0.3/1.0 for dihydrocodeine, and 0.3/1.0 for codeine, respectively. | Dihydrocodeine, and cocaine also determined. Internal standards: promethazine-D3, codeine-D3, cocaine-D3 and dihydrocodeine-D6. | [57] |
| Tramadol, caffeine, diphenhydramine, codeine, and promethazine | “Lean Cocktail” An improvised drink containing prescription drugs. | None. | Beverage filtered and diluted five times in methanol and introduced to the GC. | GC-FID. DB-5 capillary analytical column (30 m x 0.25 mm i.d. x 0.25 µm film thickness. | LOD 1.25 for tramadol and codeine and 2.5 for caffeine, diphenhydramine, and promethazine. LOQ 2.5 for tramadol and 5.0 for the other analytes. | “Dilute and shoot” method employed. | [59] |
| Dextroamphetamine, methamphetamine, methylphenidate, and modafinil. | Cappuccino, espresso, Nescafe coffee, energy drinks, ginseng drinks. | None. | GO@ZIF-8 MOF MIP SPME | GC-MS in the SIM mode. Column 25 m × 0.32 mm film thickness of 0.5 μm. | LOD between s0.000023 and 0.000033 | Breakfast cereal, dark chocolate, gummi candies, truffles, marshmallows and toffee also investigated. | [60] |
| ketamine, nimetazepam, and xylazine | Mineral water, carbonated drink, tea, beer, and orange juice | None. | Optimised VADLLME procedure | 5%-phenyl)-methylpolysiloxane (HP-5) capillary column (30 m × 0.32 μm i.d., 0.25 μm film thickness). | LOD, nimetazepam 0.16; ketamine and xylazine, 0.08. LOQ ranged from 0.26 to 0.53. | Internal standard 2,2,2-triphenylacetophenone. | [61] |
BSTFA, N,O-bis(trimethylsilyl)trifluoroacetamide; GC×GC, two-dimensional gas chromatography; GO@ZIF-8 MOF MIP SPME, graphene oxide modified zeolitic imidazolate framework molecularly imprinted polymer solid-phase microextraction; HPLC, high performance liquid chromatography; LLE, liquid-liquid extraction; LOD, limit of detection; LOQ, limit of quantification; MIP, molecular imprinted polymer; MRM, multiple reaction monitoring; QuEChERS, quick, easy, cheap, effective, rugged, and safe; SIM, select ion monitoring; SPME, solid-phase microextraction; TMCS, trimethylchlorosilane; TMS, trimethylsilyl chloride; ZIF-8, zeolitic imidazolate framework; VADLLME, Vortex-assisted dispersive liquid–liquid microextraction.
4. Discussion
Gas chromatography is a powerful analytical technique. However, it has some limitations for the determination of drugs in beverages. It is best suited for the determination of volatile compounds and many drugs and their metabolites are not sufficiently volatile without derivatization making them unsuitable for direct GC analysis. Some drugs may also decompose at the temperatures required for GC analysis. A notable number of applications have focused on the determination of GHB. This is believed to reflect the general changes in analytical laboratories to techniques based on liquid chromatography-mass spectrometry (LC-MS). Liquid chromatography-mass spectrometry can often be preferred for analyzing drugs in beverages because they can handle non-volatile and thermally labile compounds more effectively. However, this is generally only true for compounds that can give ions or distinct transitions that are large enough to demonstrate some degree of uniqueness; a problem when attempting the determination of GHB by LC-MS. Different methods of criminal spiking other than drink spiking should be investigated, for example, injection needle spiking and the illegal additions made to food.
The evolving drug market requires ongoing updates to analytical methods to keep pace with new substances. The technique of gas chromatography is capable of potentially detecting these compounds especially when coupled with sophisticated detectors such as mass spectrometry.
Future developments in the application of gas chromatography (GC) for determining drugs in drinks and beverages are focusing on several key areas. There is a growing emphasis on incorporating green chemistry principles into GC methods. This includes reducing the use of harmful solvents and minimizing waste, making the process more environmentally friendly. Advances in detector technology, such as the development of more sensitive mass spectrometers, are improving the ability to detect and quantify trace amounts of drugs in complex beverage matrices. The trend towards automation and miniaturization of GC systems is making the analysis faster and more efficient. Portable GC systems [62] are being developed for on-site testing, which could be particularly useful in forensic and regulatory settings. The use of comprehensive two-dimensional gas chromatography (GC×GC) is expanding [63]. This technique offers enhanced separation capabilities, allowing for better resolution of complex mixtures and more accurate identification of compounds. Innovations in sample preparation, such as solid-phase microextraction (SPME) and dispersive liquid-liquid microextraction (DLLME), are being integrated with GC to enhance the extraction and concentration of drugs from beverages. The application of advanced data analysis techniques, including machine learning, is helping to interpret complex GC data more effectively. This can lead to more accurate identification and quantification of drugs in beverages.
5. Conclusions
Gas chromatography and gas chromatography-mass spectrometry are powerful analytical techniques for detecting drugs in beverages, particularly in cases of drink spiking. These methods offer high sensitivity and specificity, capable of identifying drugs at trace levels and distinguishing between compounds with similar structures. The review highlights the effectiveness of GC and GC-MS in analyzing complex beverage matrices and its ability to simultaneously detect multiple substances, providing detailed molecular information essential for forensic investigations.
Despite the advantages, GC and GC-MS face challenges such as the need for derivatization to enhance volatility and the potential thermal instability of some drugs. The evolving drug market necessitates continuous improvement in detection techniques to keep pace with new substances. Future developments in GC technology, including green chemistry integration, enhanced detector sensitivity, automation, and advanced data analysis, promise to further improve the efficiency and accuracy of drug detection in beverages.
Overall, GC and GC-MS remain indispensable tools in forensic science, offering reliable and accurate results crucial for legal and investigative processes. Continued advancements in these techniques will enhance their application in combating drug-facilitated crimes and ensuring public safety.
Author Contributions
Conceptualization, K.C.H.; methodology, K.C.H.; validation, O.G., K.C.H. and H.K.; investigation, K.C.H; resources, K.C.H.; data curation, K.C.H.; writing—original draft preparation, K.C.H, O.G. and H.K.; writing—review and editing, O.G., K.C.H and H.K.; visualization, K.C.H; supervision, K.C.H.; project administration, K.C.H.; funding acquisition, K.C.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| 1,4-BD | 1,4-butanediol |
| BSA | N,O-bis(trimethylsilyl)acetamide |
| BSTFA | N,O-bis(trimethylsilyl)trifluoroacetamide |
| CAF | Caffeine |
| COD | Codeine |
| CPSE | Cellulose paper sportive extraction |
| DCM | Dichloromethane |
| DLLME | Dispersive liquid-liquid microextraction |
| DPH | Diphenhydramine |
| ECD | Electron capture detector |
| EI | Electron ionization |
| EIC | Ion extracted chromatogram |
| FID | Flame ionization |
| FPD | Flame photometric detector |
| FPSE | Fabric phase sorptive extraction |
| GBL | Gamma-butyrolactone |
| GC | Gas chromatography |
| GC-FID | Gas chromatography flame ionization detector |
| GC-MS | Gas chromatography mass spectrometry |
| GC-MS/MS DFSA |
Gas chromatograph tandem mass spectrometry. Drug-facilitated sexual assault |
| GC×GC | Comprehensive two-dimensional gas chromatography |
| GHB | Gamma-hydroxybutyrate |
| GHV | Gamma-hydroxyvalerate |
| GO@ZIG-8 MOF | Zeolitic Imidazolate Framework-8 |
| HPLC | High-performance liquid chromatography |
| LC-MS | Liquid chromatography-mass spectrometry |
| LC-MS/MS | Liquid chromatography tandem mass spectrometry |
| LLE | Liquid-liquid extraction |
| LOD | Limit of detection |
| LOQ | Limit of quantitation |
| MIP | Molecular imprinted polymers |
| MOF | Metal-organic framework |
| MRM | Multiple reaction monitoring |
| MSTFA | N-methyl-N-(trimethylsilyl)trifluoroacetamide |
| PRO | Promethazine |
| SIM | Selected ion monitoring |
| SPE | Solid phase extraction |
| SPME | Solid-phase microextraction |
| TMCS | Trimethylchlorosilane |
| TMS | Trimethylsilyl |
| TMSCI | Trimethylsilyl chloride |
| TRA | Tramadol |
| TV-SPME | Total vaporization solid-phase microextraction |
| VADLLME-GC | Vortex-assisted dispersive liquid-liquid microextraction-gas chromatography |
References
- Forsberg, C.; Gray, C. Girls Just Want to be Safe: An Analysis of Drugged Drinking and Prevention Amongst Students at the University of South Carolina. Senior Theses 2023, 640. Available online: https://scholarcommons.sc.edu/senior_theses/640 (accessed on 5 February 2025).
- Swan, S.C.; Lasky, N.V.; Fisher, B.S.; Woodbrown, V.D.; Bonsu, J.E.; Schramm, A.T.; Warren, P.R.; Coker, A.L.; Williams, C.M. Just a Dare or Unaware? Outcomes and Motives of Drugging (“Drink Spiking”) Among Students at Three College Campuses. Psychol. Violence 2017, 7, 253-264. [CrossRef]
- House of Commons Home Affairs Committee. Spiking Ninth Report of Session 2021–22; Report, together with formal minutes relating to the report. Ordered by the House of Commons to be printed 20 April 2022. Available online: https://committees.parliament.uk/publications/21969/documents/165662/default/ (accessed on 22 February 2025).
- Offences against the Person Act 1861. Chapter 100. Legislation.gov.uk. Available online: https://www.legislation.gov.uk/ukpga/Vict/24-25/100/section/23 (accessed on 31 January 2025).
- Criminal Justice Act 1988. Chapter 33. Legislation.gov.uk. Available online: https://www.legislation.gov.uk/ukpga/1988/33/section/39 (accessed on 31 January 2025).
- Sexual Assaults Act 2003. Chapter 42. Legislation.gov.uk. Available online: https://www.legislation.gov.uk/ukpga/2003/42/section/61/notes#:~:text=The%20offence%20applies%20both%20where,into%20B's%20drink%20than%20A (accessed on 31 January 2025).
- Germain, M.; Desharnais, B.; Motard, J.; Doyon, A.; Bouchard, C.; Marcoux, T.; Audette, E.; Muehlethaler, C.; Mireault, P. On-site drug detection coasters: an inadequate tool to screen for GHB and ketamine in beverages. Forensic Sci. Int. 2023, 352, 111817. [CrossRef]
- Stojanović, G.M.; Milić, L.; Endro, A.A.; Simić, M.; Nikolić, I.; Savić, M.; Savić, S. Textile-based wearable device for detection of date rape drugs in drinks. Text. Res. J. 2024, 94, 763–776. [CrossRef]
- United Nations Office on Drugs and Crime. Guidelines for the Forensic Analysis of Drugs Facilitating Sexual Assault and Other Criminal Acts. United Nations. Available online: www.unodc.org/unodc/en/scientists/guidelines-for-the-forensic-analysis-of-drugs-facilitating-sexual-assault-and-other-criminal-acts_new.html (accessed on 5 January 2025).
- Brettell, T.A.; Lum, B.J. Analysis of drugs of abuse by gas chromatography–mass spectrometry (GC-MS). Analysis of Drugs of Abuse 2018, pp. 29–42.
- Costa, Y.R.S.; Lavorato, S.N.; Baldin, J.J.C.M.C. Violence against women and drug-facilitated sexual assault (DFSA): A review of the main drugs. J. Forensic Leg. Med. 2020, 74, 102020. [CrossRef]
- Burrell, A.; Woodhams, J.; Gregory, P.; Robinson, E. Spiking Prevalence and Motivation. 2023. Available online: https://www.researchgate.net/profile/Amy-Burrell/publication/373895390_Spiking_Prevalence_and_Motivation/links/6501dadd9763a22fa3df74e0/Spiking-Prevalence-and-Motivation.pdf (accessed on 2 January 2025).
- Scott-Ham, M.; Burton, F.C. Toxicological findings in cases of alleged drug-facilitated sexual assault in the United Kingdom over a 3-year period. J. Clin. Forensic Med. 2005, 12, 175–186. [CrossRef]
- Caballero, C.G.; Jorge, O.Q.; Landeira, A.C. Alleged drug-facilitated sexual assault in a Spanish population sample. Forensic Chem. 2017, 4, 61-66. [CrossRef]
- Orts, M.P.; van Asten, A.; Kohler, I. The Evolution Toward Designer Benzodiazepines in Drug-Facilitated Sexual Assault Cases. J. Anal. Toxicol. 2023, 47, 1–25. [CrossRef]
- Gharedaghi, F.; Hassanian-Moghaddam, H.; Akhgari, M.; Zamani, N.; Taghadosinejad, F. Drug-Facilitated Crime Caused by Drinks or Foods. Egypt. J. Forensic Sci. 2018, 8, 1-7. [CrossRef]
- Hyver, K.J.; Sandra, P. High Resolution Gas Chromatography, 1989, 3rd edition, Hewlett Packard, 3-28 – 3 29.
- Stafford, D.T.; Brettell, T.A. Forensic Gas Chromatography; CRC Press: Boca Raton, FL, USA, 2019; ISBN 9780203016411/0203016416.
- Phillips, D.J.; Caparella, M.; El Fallah, Z.; Neue, U.D. Small particle columns for faster high performance liquid chromatography. Waters Column 1996, 6, 1–7. Available online: https://www.waters.com/webassets/cms/library/docs/wc6-2-1.pdf (accessed on 20 August 2023).
- Pellizzari, E.D. Electron capture detection in gas chromatography. J. Chromatogr. A 1974, 98, 323–361. [CrossRef]
- Burgett, C.A.; Smith, D.H.; Bente, H.B. The nitrogen-phosphorus detector and its applications in gas chromatography. J. Chromatogr. A 1977, 134, 57–64. [CrossRef]
- Hyver, K.J.; Sandra, P. High Resolution Gas Chromatography; 3rd ed.; Hewlett Packard: 1989; pp. 48–416.
- Snow, N. Flying High with Sensitivity and Selectivity: GC–MS to GC–MS/MS. LCGC North America 2021, 39, 61-67.
- de Hoffmann, E. Tandem mass spectrometry: a primer. J. Mass Spectrom. 1996, 31, 129-137.
- Harvey, D.J.; Vouros, P. Mass spectrometric fragmentation of trimethylsilyl and related alkylsilyl derivatives. Mass Spectrom. Rev. 2020, 39(1-2), 105–211. [CrossRef]
- Rontani, J.-F.; Aubert, C. Unexpected peaks in electron ionisation mass spectra of trimethylsilyl derivatives resulting from the presence of trace amounts of water and oxygen in GC-QTOF systems. Rapid Commun. Mass Spectrom. 2019, 33, 741–743.
- Farajzadeh, M.A.; Nouri, N.; Khorram, P. Derivatization and microextraction methods for determination of organic compounds by gas chromatography. TrAC Trends Anal. Chem. 2014, 55, 14-23. [CrossRef]
- Wiśniewska, P.; Śliwińska, M.; Dymerski, T.; Wardencki, W.; Namieśnik, J. Application of Gas Chromatography to Analysis of Spirit-Based Alcoholic Beverages. Crit. Rev. Anal. Chem. 2015, 45, 201–225. [CrossRef]
- Musshoff, F., Chromatographic methods for the determination of markers of chronic and acute alcohol consumption, J. Chromatogr. B Biomed. Appl. 2002, 781, 457-480. [CrossRef]
- Cerioni, A.; Mietti, G.; Cippitelli, M.; Ricchezze, G.; Buratti, E.; Froldi, R.; Cingolani, M.; Scendoni, R. Validation of a Headspace Gas Chromatography with Flame Ionization Detection Method to Quantify Blood Alcohol Concentration (BAC) for Forensic Practice. Chemosensors 2024, 12, 133. [CrossRef]
- Ndikumana, E.; Niyonizera, E.; Kabera, J.N.; Pandey, A. Analysis of Methanol and Ethanol Content in Illegal Alcoholic Beverages using Headspace Gas Chromatography: Case Studies at Rwanda Forensic Institute. Braz. J. Anal. Chem. 2024, 11, 18–33. [CrossRef]
- Dufayet, L.; Bargel, S.; Bonnet, A.; Boukerma, A.K.; Chevallier, C.; Evrard, M.; Guillotin, S.; Loeuillet, E.; Paradis, C.; Pouget, A.M.; Reynoard, J.; Vaucel, J.-A. Gamma-Hydroxybutyrate (GHB), 1,4-Butanediol (1,4BD), and Gamma-Butyrolactone (GBL) Intoxication: A State-of-the-Art Review. Regul. Toxicol. Pharmacol. 2023, 142, 105435. [CrossRef]
- The Misuse of Drugs Act 1971 (Amendment) Order. 2009. Available online: http://www.opsi.gov.uk/si/si2009/draft/ukdsi_9780111486610_en_1 (accessed on 22 March 2022).
- Savino, J.O.; Turvey, B.E. Rape Investigation Handbook; Academic Press: Waltham, MA, USA, 2011; p. 335.
- Mason, P.E.; Kerns, W.P. II. Gamma hydroxybutyric acid (GHB) intoxication. Acad. Emerg. Med. 2002, 9, 730–739.
- Weng, T.-I.; Chen, L.-Y.; Chen, J.-Y.; Chen, G.-y.; Mou, C.-W.; Chao, Y.-L.; Fang, C.-C. Characteristics of patients with analytically confirmed γ-hydroxybutyric acid/γ-butyrolactone (GHB/GBL)-related emergency department visits in Taiwan. J. Formos. Med. Assoc. 2021, 120, 1914–1920. [CrossRef]
- WHO. Gamma-butyrolactone (GBL): Critical Review Report, Agenda item 4.3. Expert Committee on Drug Dependence, World Health Organisation, 2014. Available online: https://www.who.int/medicines/areas/quality_safety/4_3_Review.pdf (accessed on 24 December 2021).
- Ciolino, L.A.; Mesmer, M.Z.; Satzger, R.D.; Machal, A.C.; McCauley, H.A.; Mohrhaus, A.S. The chemical interconversion of GHB and GBL: Forensic issues and implications. J. Forensic Sci. 2001, 46, 1315–1323. [CrossRef]
- UK Home Office, Circular 004/2022: Gamma-Butyrolactone (GBL) and 1,4-Butanediol (1,4-BD): revocation of rescheduling. https://www.gov.uk/government/publications/circular-0042022-gamma-butyrolactone-gbl-and-14-butanediol-14-bd-revocation-of-rescheduling/circular-0042022-gamma-butyrolactone-gbl-and-14-butanediol-14-bd-revocation-of-rescheduling Accessed 29/12/2024.
- Home Office. Harsher sentences introduced for ‘spiking’ drugs. 13 April 2022. Available online: https://www.gov.uk/government/news/harsher-sentences-introduced-for-spiking-drugs (accessed on 3 January 2025).
- Tucci, M.; Stocchero, G.; Pertile, R.; Favretto, D. Detection of GHB at low levels in non-spiked beverages using solid phase extraction and gas chromatography—mass spectrometry. Toxicol. Anal. Clin. 2017, 29, 225–233. [CrossRef]
- Elliott, S.P.; Fais, P. Further Evidence for GHB Naturally Occurring in Common Non-Alcoholic Beverages. Forensic Sci. Int. 2017, 277, e36–e38. [CrossRef]
- Elliott, S.; Burgess, V. The presence of gamma-hydroxybutyric acid (GHB) and gamma-butyrolactone (GBL) in alcoholic and non-alcoholic beverages. Forensic Sci. Int. 2005, 151, 289–292. [CrossRef]
- Meyers, J.; Almirall, J. Analysis of Gamma-Hydroxybutyric Acid (GHB) in Spiked Water and Beverage Samples Using Solid Phase Microextraction (SPME) on Fiber Derivatization/Gas Chromatography-Mass Spectrometry (GC/MS). J. Forensic Sci. 2005, 50, 31–36. [CrossRef]
- Diekman, J.; Thomson, J.B.; Djerassi, C. Mass spectrometry in structural and stereochemical problems. CLV. Electron impact induced fragmentations and rearrangements of some trimethylsilyl ethers of aliphatic glycols, and related compounds. J. Org. Chem. 1968, 33, 2271-2284. [CrossRef]
- Davis, K.E.; Hickey, L.D.; Goodpaster, J.V. Detection of ɣ-hydroxybutyric acid (GHB) and ɣ-butyrolactone (GBL) in alcoholic beverages via total vaporization solid-phase microextraction (TV-SPME) and gas chromatography–mass spectrometry. J. Forensic Sci. 2021, 66, 846–853.
- Meng, L.; Chen, S.; Zhu, B.; Zhang, J.; Mei, Y.; Cao, J.; Zheng, K. Application of dispersive liquid-liquid microextraction and GC–MS/MS for the determination of GHB in beverages and hair. J. Chromatogr. B Biomed. Appl. 2020, 1144, 122058. [CrossRef]
- Mercer, J.W.; Oldfield, L.S.; Hoffman, K.N.; Shakleya, D.M.; Bell, S.C. Comparative Analysis of Gamma-Hydroxybutyrate and Gamma-Hydroxyvalerate Using GC/MS and HPLC. J. Forensic Sci. 2007, 52, 383–388. [CrossRef]
- Carter, L.P.; Chen, W.; Wu, H.; Mehta, A.K.; Hernandez, R.J.; Ticku, M.K.; Coop, A.; Koek, W.; France, C.P. Comparison of the Behavioral Effects of Gamma-Hydroxybutyric Acid (GHB) and Its 4-Methyl-Substituted Analog, Gamma-Hydroxyvaleric Acid (GHV). Drug Alcohol Depend. 2005, 78, 91–99. [CrossRef]
- Sadiq, N.S.M.; Teoh, W.K.; Saisahas, K.; Phoncai, A.; Kunalan, V.; Muslim, N.Z.M.; Limbut, W.; Chang, K.H.; Abdullah, A.F.L. Determination of Residual Xylazine by Gas Chromatography in Drug-Spiked Beverages for Forensic Investigation. Malays. J. Anal. Sci. 2022, 26, 774–787.
- Famiglini, G.; Capriotti, F.; Palma, P.; Termopoli, V.; Cappiello, A. The Rapid Measurement of Benzodiazepines in a Milk-Based Alcoholic Beverage Using QuEChERS Extraction and GC–MS Analysis. J. Anal. Toxicol. 2015, 39, 306–312. [CrossRef]
- Acikkol, M.; Mercan, S.; Karadayi, S. Simultaneous determination of benzodiazepines and ketamine from alcoholic and nonalcoholic beverages by GC-MS in drug facilitated crimes. Chromatographia 2009, 70, 1295-1298.
- Gautam, L.; Sharratt, S.D.; Cole, M.D. Drug facilitated sexual assault: detection and stability of benzodiazepines in spiked drinks using gas chromatography-mass spectrometry. PLoS ONE 2014, 9, e89031. [CrossRef]
- Ghobadi, M.; Yamini, Y.; Ebrahimpour, B. SPE coupled with dispersive liquid–liquid microextraction followed by GC with flame ionization detection for the determination of ultra-trace amounts of benzodiazepines. J. Sep. Sci. 2014, 37, 287-294. [CrossRef]
- Jain, B.; Jain, R.; Kabir, A.; Zughaibi, T.; Bajaj, A.; Sharma, S. Exploiting the potential of fabric phase sorptive extraction for forensic food safety: Analysis of food samples in cases of drug facilitated crimes. Food Chem. 2024, 432, 137191. [CrossRef]
- Jain, B.; Jain, R.; Kabir, A.; Ghosh, A.; Zughaibi, T.; Chauhan, V.; Koundal, S.; Sharma, S. Cellulose Paper Sorptive Extraction (CPSE) Combined with Gas Chromatography–Mass Spectrometry (GC–MS) for Facile Determination of Lorazepam Residues in Food Samples Involved in Drug Facilitated Crimes. Separations 2023, 10, 281. [CrossRef]
- Rosenberger, W.; Teske, J.; Klintschar, M.; Dziadosz, M. Detection of Pharmaceuticals in “Dirty Sprite” Using Gas Chromatography and Mass Spectrometry. Drug Test. Anal. 2022, 14, 539-544. [CrossRef]
- Meatherall R. GC-MS quantitation of codeine, morphine, 6-acetylmorphine, hydrocodone, hydromorphone, oxycodone, and oxymorphone in blood. J. Anal. Toxicol. 2005, 29, 301-308. [CrossRef]
- Phonchai, A.; Pinsrithong, S.; Janchawee, B.; Prutipanlai, S.; Botpiboon, O.; Keawpradub, N. Simultaneous Determination of Abused Prescription Drugs by Simple Dilute-and-Shoot Gas Chromatography – Flame Ionization Detection (GC-FID). Anal. Lett. 2021, 54, 716-728. [CrossRef]
- Kardani, F.; Khezeli, T.; Rashedinia, M.; Hashemi, M.; Zarei Jelyani, A.; Shariati, S.; Mahdavinia, M.; Noori, S.M.A. Application of GO@ZIF-8 MOF/molecularly imprinted polymer composite for multiple monolithic fiber solid phase microextraction: Determination of illegally added amphetamines in snacks and beverages and quantitation by GC-MS. J. Food Compos. Anal. 2025, 137, 106977. [CrossRef]
- Teoh, W.K.; Mohamed Sadiq, N.S.; Saisahas, K.; Phoncai, A.; Kunalan, V.; Md Muslim, N.Z.; Limbut, W.; Chang, K.H.; Abdullah, A.F.L. Vortex-Assisted Dispersive Liquid–Liquid Microextraction-Gas Chromatography (VADLLME-GC) Determination of Residual Ketamine, Nimetazepam, and Xylazine from Drug-Spiked Beverages Appearing in Liquid, Droplet, and Dry Forms. J. Forensic Sci. 2022, 67, 1836–1845.
- Chen, J.; Kuang, Y.; Feng, X.; Mao, C.; Zheng, J.; Ouyang, G. Solid Phase Microextraction Coupled with Portable Gas Chromatography-Mass Spectrometry for On-Site Analysis. Green Anal. Chem. 2024, 11, 100164. [CrossRef]
- Mondello, L.; Cordero, C.; Janssen, H.-G.; Synovec, R.E.; Zoccali, M.; Tranchida, P.Q. Comprehensive two-dimensional gas chromatography–mass spectrometry. Nat. Rev. Methods Primers 2025, 5, 7.
Figure 1.
Structures of gamma-hydroxybutyric acid (GHB), gamma-butyrolactone (GBL) and gamma-hydroxyvalerate (GHV).
Figure 1.
Structures of gamma-hydroxybutyric acid (GHB), gamma-butyrolactone (GBL) and gamma-hydroxyvalerate (GHV).
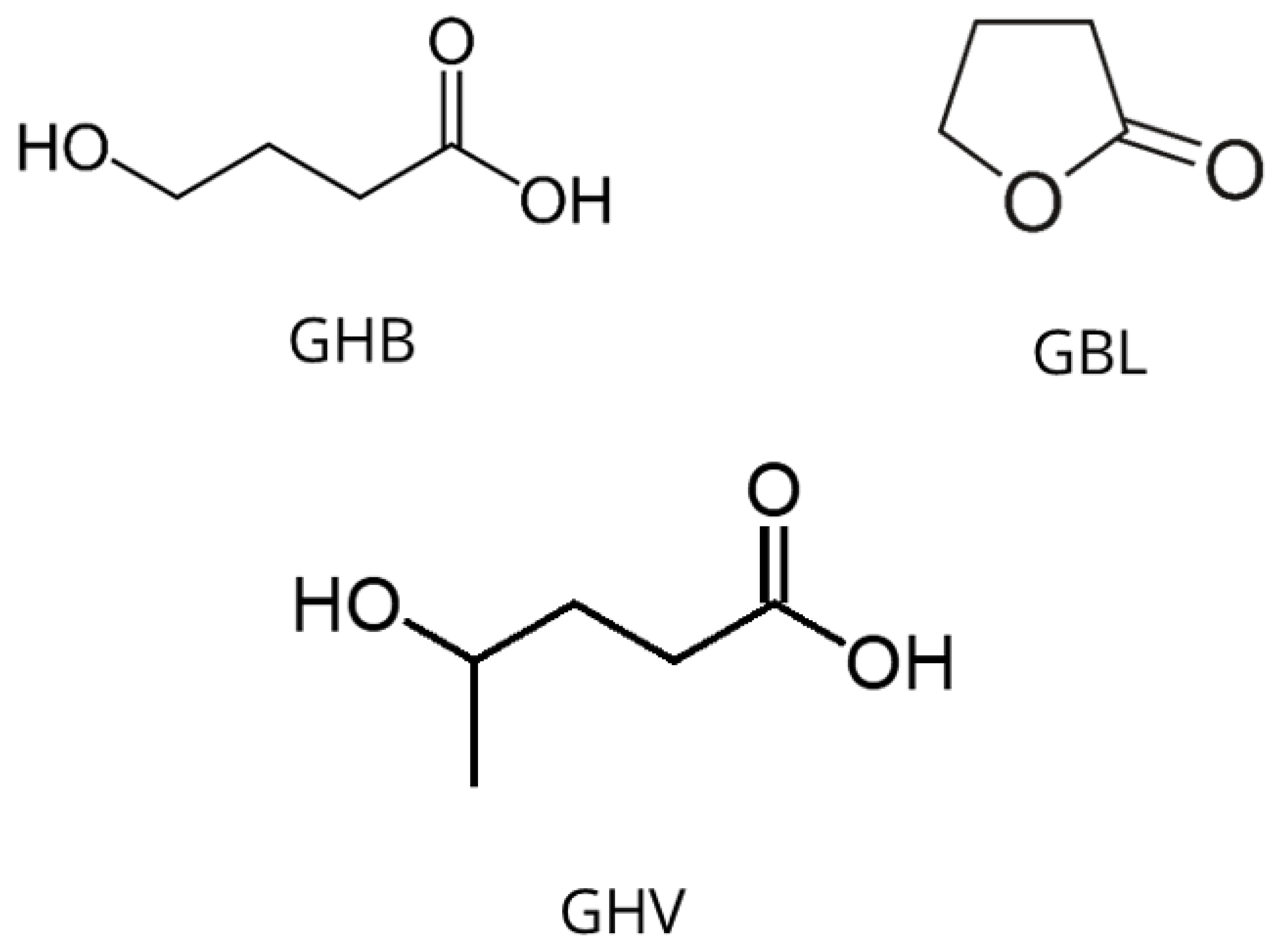
Figure 2.
Mass spectrum of derivatized GHB after Meyers and Almirall [44].
Figure 2.
Mass spectrum of derivatized GHB after Meyers and Almirall [44].
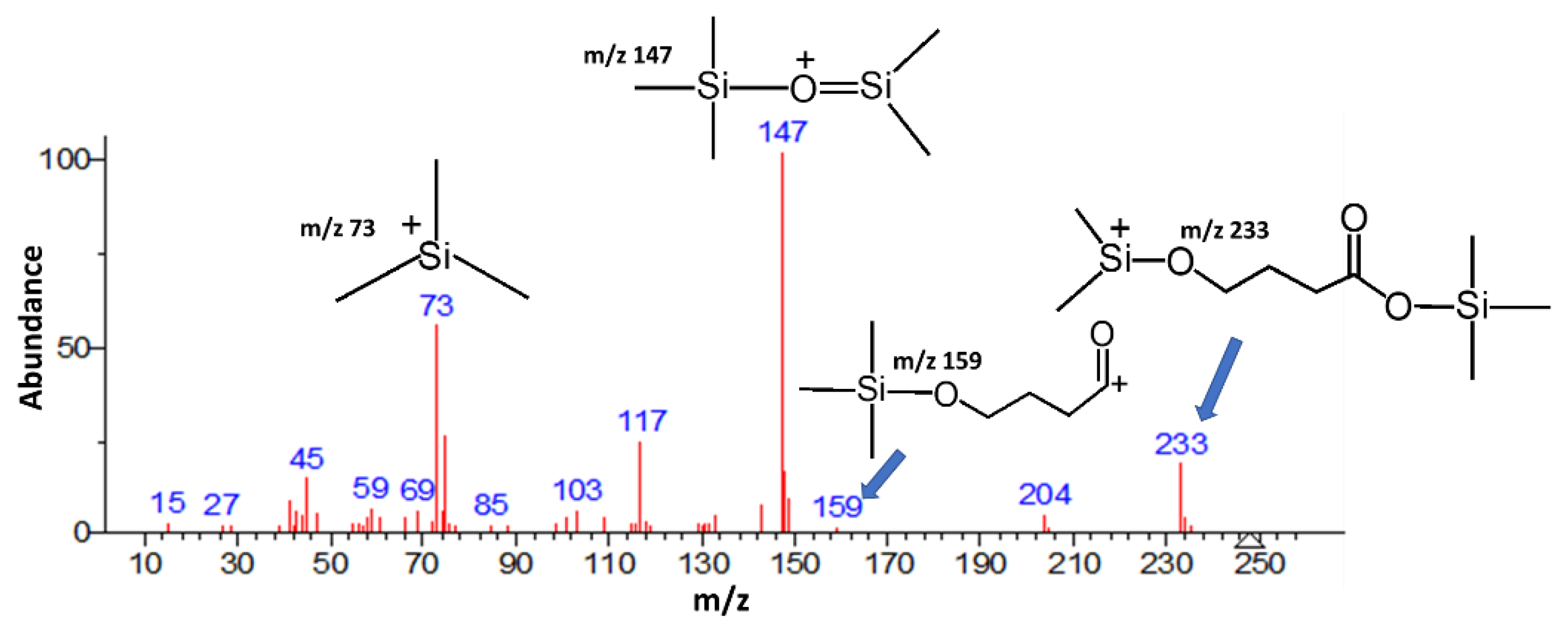
Figure 3.
Structure of xylazine.
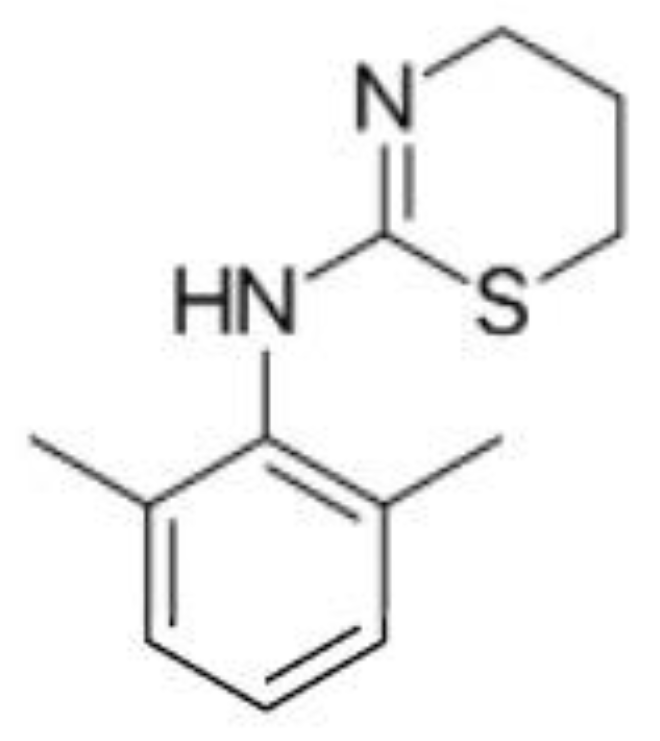
Figure 4.
Structures of benzodiazepines studied by Famiglini et al. [51] in milk-based alcoholic drinks, such as whiskey creams.
Figure 4.
Structures of benzodiazepines studied by Famiglini et al. [51] in milk-based alcoholic drinks, such as whiskey creams.
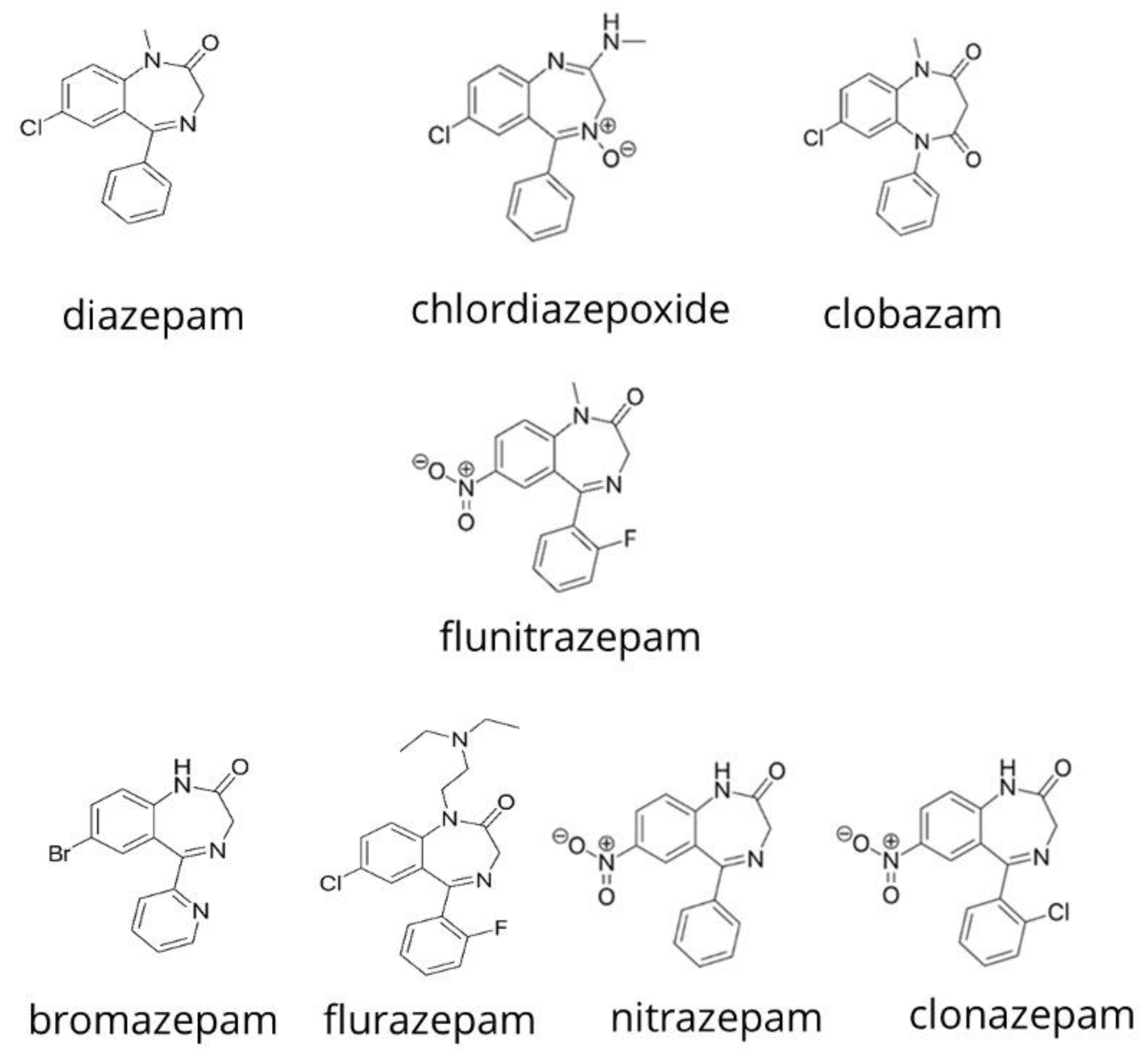
Figure 5.
Structures of ketamine and the benzodiazepines studied by Acikkol et al. [52] in peach juice and beer.
Figure 5.
Structures of ketamine and the benzodiazepines studied by Acikkol et al. [52] in peach juice and beer.
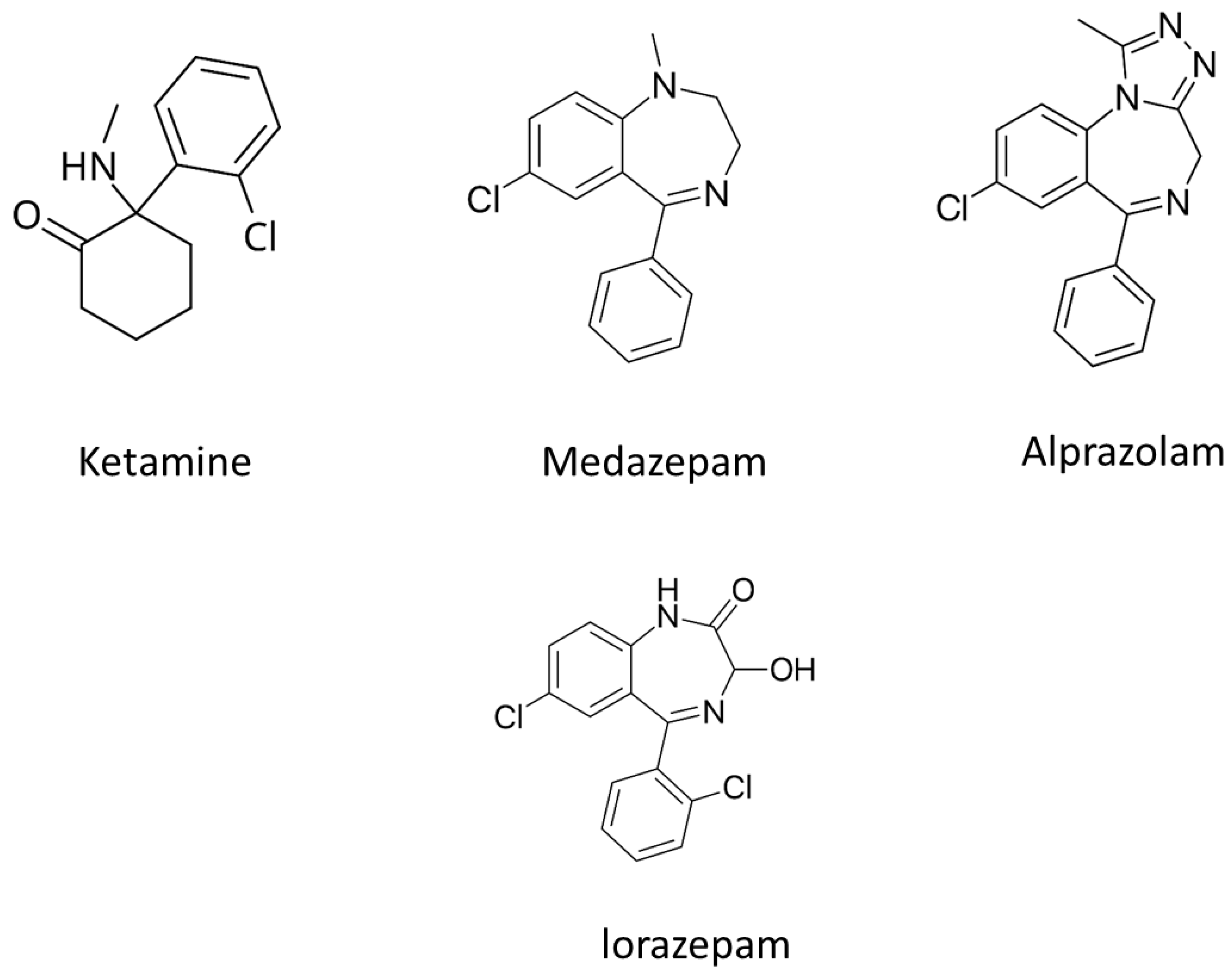
Figure 7.
Structures of promethazine, codeine, dihydrocodeine and cocaine.
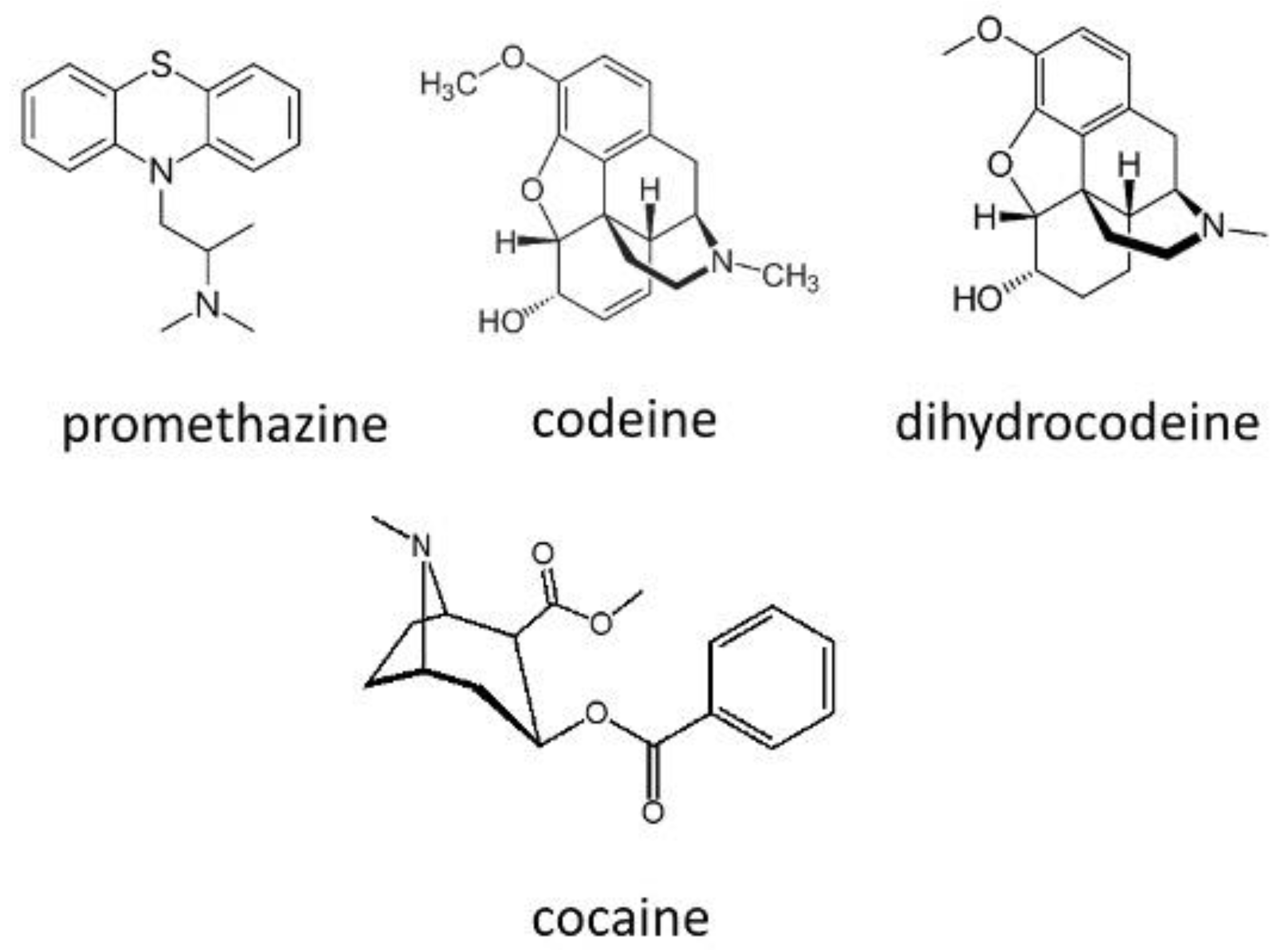
Figure 8.
Structures of tramadol, diphenhydramine, dextroamphetamine, methamphetamine, methylphenidate, modafinil and nimetazepam.
Figure 8.
Structures of tramadol, diphenhydramine, dextroamphetamine, methamphetamine, methylphenidate, modafinil and nimetazepam.
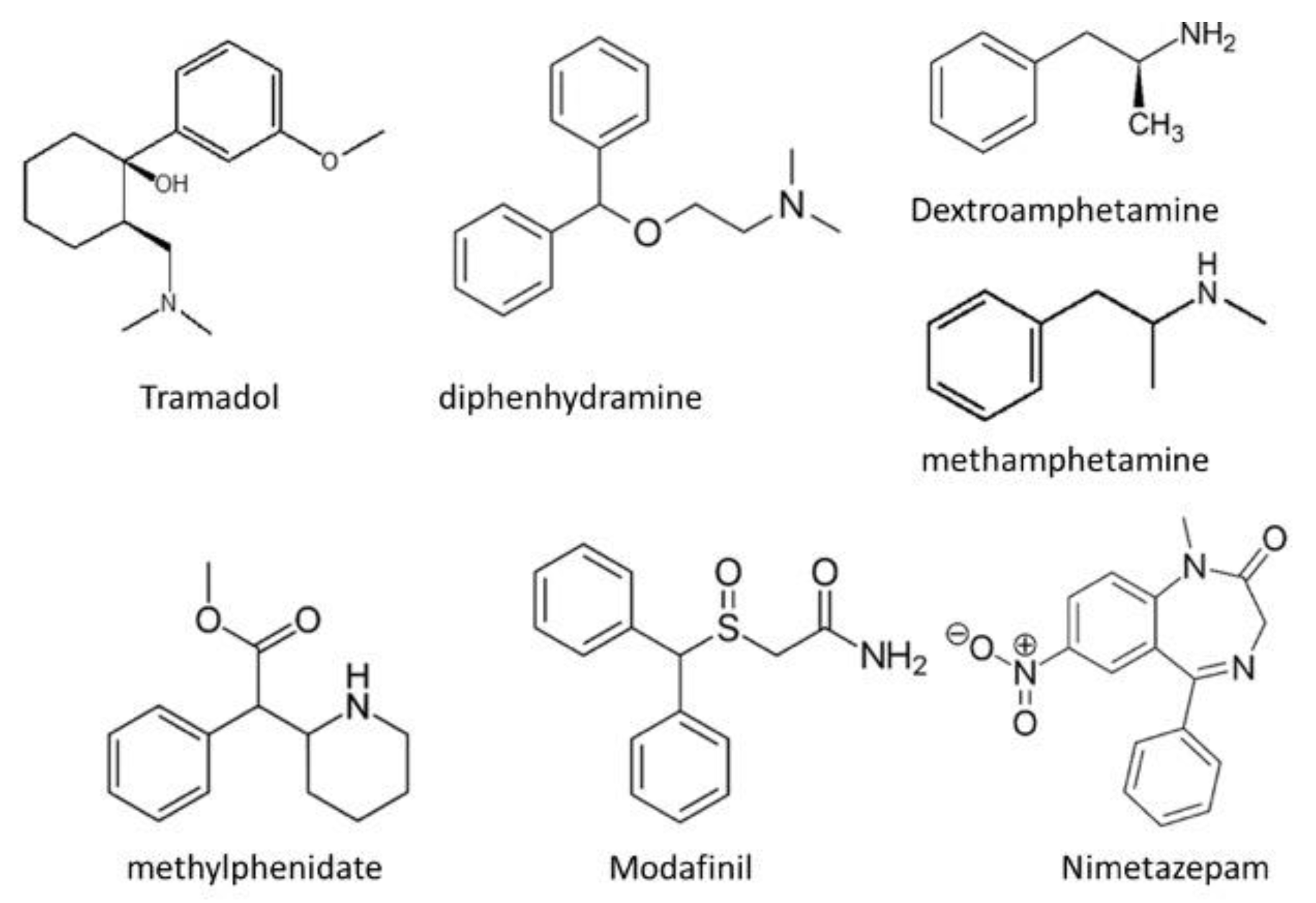
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



