Submitted:
10 March 2025
Posted:
11 March 2025
You are already at the latest version
Abstract
Leigh syndrome (LS) is a severe neurodegenerative condition with an early onset, typically during early childhood or infancy. The disorder exhibits substantial clinical and genetic diversity. From a clinical standpoint, Leigh syndrome showcases a broad range of irregularities, ranging from severe neurological issues to minimal or no discernible abnormalities. The central nervous system is most affected, resulting in psychomotor retardation, seizures, nystagmus, ophthalmoparesis, optic atrophy, ataxia, dystonia, or respiratory failure. Some patients also experience involvement of the peripheral nervous system, such as polyneuropathy or myopathy, as well as non-neurological anomalies like diabetes, short stature, hypertrichosis, cardiomyopathy, anemia, renal failure, vomiting, or diarrhea (Leigh-like syndrome). Mutations associated with Leigh syndrome impact genes in both the mitochondrial and nuclear genomes. Presently, LS remains without a cure and shows limited response to various treatments, although certain case reports suggest potential improvement with supplements. Ongoing preclinical studies are actively exploring new treatment approaches. This review comprehensively outlines the genetic underpinnings of LS, its current treatment methods, and preclinical investigations, with a particular focus on treatment.
Keywords:
Leigh syndrome
; Genetics
; Neurology
; Therapy
; Preclinical Research
1. Introduction
1.1. Brief History
Leigh syndrome (LS) is a rare incurable mitochondrial disease with an early onset, typically during early childhood or infancy. Prof. Denis Leigh gave a very clear definition of subacute necrotizing encephalomyelopathy in 1951. He described a neuropathological entity characterized by early-onset development of necrotizing lesions in the grey nuclei of the brainstem, subthalamic region, and basal ganglia, including often the deep structures of the cerebellum and descending up to the rostral segment of the spinal cord. These lesions are usually symmetric, with neuronal loss, initial inflammation followed by gliosis, and maintenance of capillary hyperproliferation. Any Leigh syndrome or Leigh-like syndrome (whose definition is much more uncertain) must contain these essential neuropathological features[1].
1.2. Most Common Clinical Presentation
The symptoms are various: when the long tracts (e.g., the mesencephalic anterior descending tracts) are also affected, a motor impairment with axial hypotonia and spastic tetraparesis can occur. In all cases, movement disorders, due to lesions in the basal ganglia and subthalamus, are present, as well as respiratory irregularities (alterations of the respiratory centers), eye movement disorders (due to medial longitudinal fasciculus and oculomotor nuclei involvement), ataxia (cerebellum and spinocerebellar tracts), sometimes hypoacusia, sometimes optic atrophy (retinitis pigmentosa is rather rare and more often detected in the m.8993T>G mutation[2]). A general psychomotor regression and cognitive delay or stagnation may occur. Characteristically, there are no consistent dysmorphic or malformation features, whereas is typical the presence of "disease-free" windows lasting variably from 0 to up to 6 months. The first or second infection of the child usually precipitates the situation revealing the general neurological impairment. The heart is usually unaffected; myopathy is not prominent and typically ragged red fibers are usually absent. Peripheral neuropathy may be present but it is not the main problem. Other organs are marginally or not affected at all, although a DeToni-Debré-Fanconi proximal tubular impairment may occasionally be seen[3]. Usually, serum lactate is relatively high, and sometimes, especially during infection, metabolic acidosis may occur. The natural history is that of progressive, rapid disease, rarely do these patients live beyond the first decade. Adult or late-onset conditions are rare and incompletely understood, but are certainly documented[4]. Biochemically, most of the cases are associated with a mitochondrial bioenergetic disorder, either for mutations in nuclear DNA, nDNA, or mitochondrial DNA, mtDNA (e.g., in the high heteroplasmic levels of the 8993T>G mutation of ATPase6 in mitochondrial inherited Leigh syndrome[5]). In several cases, Complex I (CI) mutations, either in structural subunits or assembly factors, are associated with Leigh syndrome, but the single most frequent gene causing nDNA Leigh syndrome is loss of function mutations in SURF1, an assembly factor of cytochrome c oxidase IV[4].
Leigh Syndrome is defined by the Online Mendelian Inheritance in Man Database (ONIM 2014) as follows (1) a neurodegenerative disease with variable symptoms (2) caused by mitochondrial dysfunction from a hereditary genetic defect and (3) accompanied by bilateral central nervous system (CNS) lesions. These criteria are not always met, and, in those cases, the term “Leigh-like syndrome” is preferred[6,7]. Leigh and Leigh-like syndromes have defects in the respiratory chain, coenzyme Q, or pyruvate dehydrogenase complex[8]. The absence of well-defined criteria for LS results from the broadest genetic heterogeneity, in fact, nonmitochondrial disorders also may present as LS[9]. LS often shows a progressive decline of CNS because of focal necrotizing lesions of the basal ganglia, cerebellum, diencephalon, or brainstem as already mentioned[10]. The estimated incidence is 1 per 40000 live births[11]. Nonetheless, the precise prevalence of Leigh syndrome is not known, as many cases are wrongly diagnosed[12]. LS is a pan-ethnic disorder, yet some genetic subtypes have a high prevalence in certain geographical regions, as discussed in the following sections.
1.3. Faroe Islands Variant
A Faroe variant has been described in the Faroe Islands where the incidence is higher due to a founder effect and a carrier frequency of 1 in 33[11]. The term Faroe variant of mitochondrial disease has historically been used to describe a specific form of SUCLA2 deficiency, a genetic disorder affecting succinate-CoA ligase, an essential enzyme in the tricarboxylic acid (TCA) cycle. The name derives from the increased prevalence of this condition in the Faroe Islands, attributed to a founder effect. However, the term "Faroe variant" is misleading, as SUCLA2 mutations have been identified in multiple populations worldwide. This genetic variation is associated with the early onset of muscle weakness (hypotonia), muscle atrophy, compromised motor skills, hearing problems, seizures, and neuroimaging findings that point to Leigh syndrome[13]. Individuals with this variation often experience recurring respiratory tract infections. Many of these patients exhibit abnormal muscle tone (dystonia) or excessive involuntary movements (hyperkinesia), which may manifest as athetoid or choreiform movements[13]. Scoliosis is a common occurrence in most of these patients, often requiring a supportive corset[13].
1.4. French-Canadian Variant
The clinical profile of individuals affected by the French-Canadian variant, also known as the Saguenay variant, includes developmental delay, moderate muscle weakness (hypotonia), limited facial and limb movements, unsteady movement of the trunk (truncal ataxia), a cautious wide-based gait, and intention tremor. These patients do not display cardiomyopathy, glycosuria, and aminoaciduria which are frequently present in LS[14]. The majority of patients with this variant present severe lactacidosis, hepatic steatosis, or a neurodegenerative disorder[14]. Unfortunately, most of these individuals succumb to a rapid and severe acidic crisis before reaching five years of age[15]. This condition is predominantly observed in the Saguenay–Lac-Saint-Jean region of Quebec, Canada, due to a founder effect in the population. It is caused by mutations in the LRPPRC (Leucine-rich pentatricopeptide repeat-containing protein) gene, which encodes a protein critical for the transcriptional and translational regulation of mitochondrial DNA[16]. This dysfunction leads to severe mitochondrial impairment, particularly affecting the brain, liver, and other high-energy-demanding tissues. The LRPPRC plays a pivotal role in mitochondrial gene expression. It functions in stabilizing mitochondrial mRNA, regulating oxidative phosphorylation (OXPHOS), and maintaining mitochondrial homeostasis[17,18].
1.5. Objective
The objective of this review is to provide a clinical presentation of the different Leigh and Leigh-like syndromes with a particular focus on current treatment and future directions.
2. Search Strategy
We performed a comprehensive search in the online database PubMed. We did not exert any limitation in terms of language or time of publication. We used search terms such as Leigh syndrome, Leigh disease, Subacute Necrotizing Encephalomyelopathies, and Juvenile Leigh Disease in combination with treatment. Reference lists of retrieved articles and relevant reviews were also manually searched. More than 1331 results were included in the literature research, conducted until 15 February 2025. Retrieved studies were revised and included based on the objective of the review. Unpublished data was not included.
3. Clinical Presentation
The original case described by Dr. Leigh in 1951 was that of a 7-month-old boy who developed rapid progressive neurological regression with subsequent death[19]. The child had a normal birth and development until he suddenly developed lethargy with feeding difficulty at 5 months, after exposure to a viral illness. Post-mortem examination demonstrated bilateral symmetrical lesions of necrotic nature in the brainstem, thalami, and spinal cord[19]. Initial characterization of the disease was thought to be limited to the central nervous system (CNS) and peripheral nervous system (PNS) involvement, it is now known that non-neurologic manifestations also happen in LS, especially in Leigh-like syndrome. Age onset is infancy or early childhood, but it can also occur rarely in adolescents and adults[20]. 75% of patients show signs and symptoms of the disease by 1 month of age[21]. Presenting symptoms of LS include sudden death, developmental delays, and regression of neurological abilities[22,23]. LS is considered a mitochondrial disease because it impairs mitochondrial oxidative phosphorylation, this results in reduced ATP production and consequent increase in glycolysis[24]. This can influence cells during development and may be the reason why developmental delay is the most common feature of LS as shown in a meta-analysis of clinical manifestation of LS, where developmental delay was reported as the most common clinical sign in 57% of patients[25]. Developmental delay was followed by respiratory dysfunction (34%), epileptic seizures ( 33%), poor feeding (29%), and weakness (27%)[25]. A hallmark of LS is the occurrence of acute neurological and, at times, systemic events called “decompensation,” which are linked to psychomotor delay or regression, resulting in the loss of previously acquired skills[26].
Figure 1 provides a summary of neurological and non-neurological clinical manifestations of LS.
3.1. Neurological Manifestations
Most of the patients present with neurological manifestations of both central and peripheral nervous system, without other organ involvements[27]. Central nervous system manifestations include psychomotor retardation, optic atrophy, ataxia, nystagmus, ophthalmoparesis, dysphagia, cranial nerve palsies, general weakness, hypotonia, dystonia, deafness, and retinitis pigmentosa[28,29]. Respiratory manifestations can be classified as neurological manifestations too since they are often driven by brainstem involvement and/or severe myopathy. Acute respiratory failure is a frequent feature of Leigh syndrome, and it may happen without prodromal manifestations. When prodromal manifestations are present, they include irregular breathing, hyperventilation, hiccups, and lethargy[13,30,31].
Epileptic seizures are frequently observed in LS, with a reported prevalence between 40% and 79%. The types of seizures can vary, including generalized tonic-clonic, myoclonic, and focal seizures. Seizure occurrence is often associated with episodes of metabolic decompensation[7,32]. Focal seizures seem to be the most common type in a work from South Korea[33].
Ophthalmologic abnormalities associated with LS include optic atrophy, retinitis pigmentosa, strabismus, ptosis, nystagmus, and ophthalmoparesis. Pigmentary retinopathy was identified in patients with MT-ATP6 variant[34].
Movement disorders are commonly encountered in LS and include dystonia (resulting in involuntary muscle contractions, abnormal postures, and movements) and ataxia (resulting in gait instability and difficulties with fine motor tasks), the first being the most prevalent one[35]. Choreiform movements have been observed in Leigh syndrome, particularly in children with ATPase 6 mutations[36].
Peripheral nervous system manifestations include neuropathy and myopathy, although more commonly in Leigh-like syndromes.
Myopathy in LS is characterized by muscle weakness, hypotonia, and exercise intolerance due to impaired oxidative phosphorylation within skeletal muscle fibers. Mutations in mitochondrial genes such as MT-ND5 have been associated with myopathy[37].
3.2. Non-Neurological Manifestations
Non-neurological manifestations of LS may include dysmorphic features although very rarely, cardiac abnormalities (hypertrophic/dilated cardiomyopathy) although less frequently reported, endocrine features (hypertrichosis, short height), gastrointestinal symptoms[8,38], diabetes mellitus, anemia, sleep disturbances, renal failure, hearing loss, scoliosis. The majority of patients die before 3 years of age from sudden respiratory failure[39].
Cardiac manifestations, such as hypertrophic or dilated cardiomyopathy and conduction defects, have been reported in approximately 15% of individuals with LS[40]. Cardiomyopathy has been observed in up to 70% of individuals with an MT-ND5 variant[41].
Gastrointestinal symptoms were even more prevalent in patients with COX-deficient Leigh syndrome caused by mutations in the SURF1 gene[42]. Infants and children with Leigh syndrome often experience poor feeding, leading to failure to thrive. This can be due to central nervous system involvement, hypotonia, and poor suck-swallow coordination[42]. Gastroesophageal reflux, gastroparesis, and delayed gastric emptying are commonly observed and probably due to neuromuscular and mitochondrial dysfunction[43]. Hepatic manifestations, such as elevated liver transaminases, hepatomegaly, or liver failure, have been observed in about 10% of individuals with LS[44,45].
3.3. Leigh-Like Syndrome
Leigh-like syndrome has unusual neurological or radiological characteristics that share similarities with LS[51]. Individuals with this syndrome typically lack respiratory issues linked to brainstem involvement[52]. However, they may experience delayed onset of muscle weakness (myopathy) and minimal to no problems with eye movements[52]. Leigh-like syndrome may present itself with seizures (such as infantile spasms or hypsarrhythmia), alterations in behavior, decreased muscle tone (hypotonia), involuntary muscle contractions (dystonia), nerve-related problems (neuropathy), or abnormalities in the retina (retinopathy)[28]. Many experts believe Leigh syndrome and Leigh-like syndrome to be part of a disease continuum driven by shared or partially overlapping pathophysiological mechanisms and genetic mutations[50].
3.4. Presentation in Youth and Adults
A small subset of individuals diagnosed with either LS or Leigh-like syndrome manage to surpass the age of 10 years. Adolescent and adult-onset LS may arise in those with congenital LS who survive into adulthood, or when the syndrome first presents in late childhood or early adolescence[53]. An observational longitudinal cohort study, enrolled seventy-two children with LS over 2.6 years, who completed the Newcastle Pediatric Mitochondrial Disease Scale (NPMDS), a scale used to quantify the disease burden and the rate of progression of LS[43]. The scale is composed of three sections and each item has 4 responses: normal (0), mild (1), moderate (2), and severe (3) impairment. The total score from all 3 sections can be categorized into mild (0–14), moderate (15–25), and severe (>25). The median NPMDS score improved from 18 at baseline to 24 at follow-up. At the same time, the number of children needing gastrostomy or nasogastric tubes doubled, rising from 22.2% to 45.8%. The percentage of children experiencing epileptic seizures also increased, going from 29.2% to 37.5%[43]. During this time, twelve children died, and predictors of poor outcomes were SURF1 gene variants and symmetrical hyperintensities on caudate, globus pallidus, and putamen[43]. When LS manifests in adults, it is often associated with minimal neurological abnormalities[54] or with typical features consistent with LS or a mitochondrial disorder[53]. Patients with a primary coenzyme-Q deficiency may exhibit symptoms consistent with adult LS[55]. Such individuals commonly display symptoms like encephalopathy, growth retardation, ataxia, and deafness[55]. Notably, neuropathological investigations conducted postmortem on a patient who had experienced a gradual progression of sensorimotor neuropathy, along with deafness, retinitis pigmentosa, and ataxia, showed characteristic LS lesions, undetectable on previous cerebral magnetic resonance imaging, despite their demise at age 37[56].
4. Genetics
Leigh syndrome has been reported to be caused by defects of 16 mitochondrial genes and almost 100 nuclear genes[12]. MtDNA mutations seem to account for 21.5 to 47% of cases in many cohorts of genetically confirmed Leigh syndrome[47,57]. LS caused by mtDNA mutations can be maternally inherited or sporadic. Maternally inherited cases usually happen in a clinically unaffected mother. Inheritance of nuclear-encoded LS is typically autosomal recessive and rarely X-linked[12].
4.1. Complex I Deficiency
Complex I (CI) is the primary and largest complex, weighing approximately 1 MDa, whose main function involves the transfer of electrons from NADH to coenzyme Q10 while also transporting H+ ions. It consists of 44 subunits, including 14 core subunits crucial for its catalytic function, with seven subunits encoded by mtDNA and the remaining core subunits encoded by nDNA. Mutations in 24 CI subunit-encoding genes and several assembly factors have been linked to Leigh syndrome, highlighting the importance of CI in mitochondrial function. Notably, mutations in the nDNA-encoded NDUFS4 gene, encoding subunit four of CI, induce 'mitochondrial complex I deficiency, nuclear type 1' (MC1DN1), and Leigh syndrome in pediatric patients[58]. The lack of NDUFS4 in various mouse tissues leads to reduced activity and stability of complex I. This instability causes a greater disconnect between electron influx from the NADH dehydrogenase module and the whole complex[58-60]. A dystonic later-onset form of NDUFS4 was described with an onset of toes walking, dysarthric speech, nystagmus, and mental deterioration[61].
NADH ubiquinone oxidoreductase core subunit S8 (NDUFS8) is another crucial core component of the iron-sulfur (FeS) fragment in mitochondrial complex I, playing a direct role in electron transfer and energy metabolism. Pathogenic variants of NDUFS8 are associated with Leigh syndrome, as well as cancer and diabetes mellitus[61,62].
Several mtDNA mutations associated with Leigh syndrome are concentrated in MTND ‘hotspots,’ particularly in MTND1, MTND3, and MTND5[63]. Isolated complex I deficiency is the most common oxidative phosphorylation enzyme defect, leading to a diverse clinical presentation that includes Leigh syndrome. The m.10191T>C mutation in the MTND3 subunit is relatively frequent and can be either maternally inherited or a recurrent sporadic mutation. This mutation should be considered when evaluating individuals with complex I deficiency and Leigh syndrome features[64]. Defects of complex I are usually associated with visual disturbance, nystagmus, optic atrophy, and epilepsy[12].
4.2. Complex II-V Deficiency
Complex II subunits are all nuclear-encoded: four structural subunits (SDHA, SDHB, SDHC, and SDHD) and two known assembly factor genes (SDHAF1 and SDHAF2). Complex II is distinct in that it serves as a component of both the respiratory chain and the Krebs cycle[65]. Mitochondrial diseases linked to isolated complex II deficiency are uncommon, they can result in Leigh syndrome or familial pheochromocytomas and paragangliomas[66]. Interestingly in some cases of complex II deficiency magnetic resonance spectroscopy of the brain can reveal a succinate peak[67].
Complex III defect involves proteins like BCS1L, TTC19, and in mouse models PARL. Mutations in TTC19 have been found to impair the function of Complex III, leading to a diverse clinical presentation that includes Leigh syndrome[68]. Clinical presentation of TTC19 disease is variable and may consist of spinocerebellar ataxia and psychiatric manifestations, with the observation that hypertrophic olivary degeneration in the brain MRI may be of diagnostic value[69]. PARL deficiency, as shown in mouse models, leads to forms of Leigh-like syndrome[70]. A single variant in one subunit (UQCRQ) has been reported in more than 20 affected individuals of consanguineous Israeli family[71].
Complex IV comprises three mtDNA subunits (COX I to III encoded by MT-CO1 to 3) and three nuclear-encoded subunits (COX4I1, COX8A, and NDUFA4). NDUFA4 encodes a subunit of the respiratory chain Complex IV. NDUFA4 dysfunction is recognized as the underlying cause of mitochondrial Complex IV deficiency nuclear type 21 (MC4DN21, OMIM 619065), a relatively mild presentation of Leigh syndrome[72]. SURF1 is another involved protein in Complex IV deficiency and is one of the most common causes of LS. To date, over sixty distinct SURF1 mutations have been identified as causes of SURF1-associated Leigh syndrome[73]. The majority of SURF1-associated Leigh syndrome cases follow a typical course, leading to early mortality before the age of ten. However, approximately 10% of cases exhibit an atypical progression with milder symptoms and a longer life expectancy[73]. Onset is usually in infancy with poor feeding and later on developmental regression. Ataxia, neuropathy ophthalmoplegia, and hypertrichosis are the most important manifestations[74]. In a murine model, SURF1−/− mice exhibited lower birth weights but eventually reached body weights similar to their siblings; however, they demonstrated a mild motor delay[75]. Other COX assembly factor defects associated with LS are deficiencies of COX10, COX15, SCO2, PET100, PET117, and TACO1[12].
Mitochondrial Complex V plays a crucial role in oxidative phosphorylation by facilitating ATP production. One of the first recognized genetic causes of LS was a recurrent single nucleotide mtDNA variant in the MT-ATP6 gene, with a clinical spectrum varying according to the variable mutation load[76]. ATP5PO, which encodes the oligomycin sensitivity-conferring protein, is one of the proteins whose role has been recognized in Leigh syndrome presentation[77].
4.3. Other Mutations Associated with Leigh and Leigh-Like Syndrome
LS is present in about one-third of cases of pyruvate dehydrogenase complex (PDHc) deficiency[78]. Five genetic defects of PDH have been linked to LS such as PDHA1, PDHB, PDHX, DLT, and DLD mutations. The most frequently encountered is the PDHA1 X-linked mutations[79]. These disorders do not always meet the clinical and pathological features for LS diagnosis.
Disorders of vitamins and cofactor metabolism have also been associated with LS, such as biotinidase deficiency which leads to biotin-dependent enzyme impairment[80]. Another treatable cause of LS is SLC19A13 mutation which leads to Wernicke-like encephalopathy presentation, when not fatal[81,82].
Disorders of mitochondrial DNA maintenance associated with LS include biallelic pathogenetic variants of SUCLA2, SUCLG1, POLG, and RNASEH1. The most relevant ones are SUCL2 and SUCLG1 mutations, which encode two subunits of succinyl-CoA ligase, a Krebs cycle enzyme that also helps maintenance of mtDNA. Patients with SUCL2 variants usually have sensorineural hearing loss and dystonia, while SUCLG1 patients have systemic involvement, especially in the liver and heart[83]. Many experts believe POLG mutations never meet the diagnostic criteria for LS as they often present themselves with a clinically different spectrum such as Alpers-Huttenlocher syndrome.
Many other disorders of mitochondrial proteins have been linked to LS, among these mutations in the SERAC1 gene lead to 3-methylglutaconic aciduria, deafness, and encephalopathy, Leigh-like (MEGDEL) syndrome, which is a disorder of lipid remodeling[84].
4.4. Coenzyme-Q Deficiency
Coenzyme-Q (CoQ) deficiency can have different manifestations and can resemble LS[46]. For instance, a patient with coenzyme-Q deficiency may exhibit congenital muscle weakness (hypotonia), seizures starting at age 3 months that are difficult to control and spread throughout the body, progressive muscle weakness, challenges in feeding, intermittent vomiting since age 7 months, the need for tube feeding, swelling due to low protein levels linked to poor feeding and kidney-related issues (nephrotic syndrome), ultimately resulting in death at 8 months[46]. In another case involving two sisters, primary coenzyme-Q deficiency presented as an adult-onset syndrome characterized by brain dysfunction, slowed growth, lack of muscle coordination (ataxia), and hearing impairment[55]. Both individuals significantly improved with coenzyme-Q replacement[55]. CoQ10 deficiency primarily affects the brain, muscles, and kidneys due to their high energy demand. Symptoms range from severe in infancy to mild in later life. It can cause ataxia, nephrotic syndrome, and hypertrophic cardiomyopathy. This entity should not be included in a Leigh-like syndrome per se.
5. Diagnosis
5.1. Laboratory Findings
Commonly, resting levels of lactate or pyruvate in the blood are elevated[8,27]. Elevated lactate levels (hyperlactatemia) may not be initially observed at the onset of symptoms but may develop as the disease progresses[85]. Moreover, in cases where skeletal muscles are affected, levels of creatine kinase are elevated[86]. Anemia has also been reported in isolated cases[85]. Patients with the Faroe variant typically display elevated levels of methylmalonic acid in their urine[11]. In cases of mitochondrial Leigh syndrome, there may be increased excretion of Krebs cycle intermediates in the urine. Lactate levels may be elevated in specific patients[87]. In patients with the Faroe variant, methylmalonic acid levels in the urine are typically high[11]. Patients with non-mitochondrial LS may display increased urinary excretion of 3-methylglutaconic acid or 3-methylglutaric acid[9,88]. In most cases, the levels of lactate, pyruvate, or the lactate/pyruvate ratio are elevated in the cerebrospinal fluid[8,89,90]. In a case involving a patient with Leigh syndrome resulting from a deficiency in coenzyme-Q, there was a significant reduction in coenzyme-Q levels[55].
5.2. Electrophysiological Assessments
Needle electromyography can detect irregular spontaneous activity[91]. Nerve-conduction studies have provided insights into potential polyneuropathy[91]. Auditory-evoked brainstem potentials may exhibit extended latencies even before clinical symptoms manifest[90,92,93]. Assessing visually evoked potentials may reveal elongation or reduction in amplitude of the P100 component[89], or in some cases, its complete absence[90]. Finally, an electroencephalogram may demonstrate irregular baseline activity and focal epileptic indicators, potentially leading to secondary generalization and hypsarrhythmia[86].
5.3. Neuroimaging
When clinical evaluations and laboratory tests trigger suspicion of LS, a cerebral MRI should be obtained. Distinctive observations in LS patients typically include the presence of bilateral and symmetrical hyperintensities evident in T2-weighted images. These hyperintensities are primarily located in specific brain regions, notably the basal ganglia (especially the putamen) and various parts of the brainstem, such as the substantia nigra, nucleus ruber, and medulla oblongata[94,95]. Other findings include microcephaly, ventricular enlargement, intracranial pseudo-cysts, and white matter abnormalities[50]. Lesions in LS usually evolve over time; nonetheless, some case series reported partial or complete regression of lesions in follow-up MRI[39,96]. The reversibility of Leigh lesions underscores the importance of ongoing efforts to develop treatments that prevent brain damage[96]. Moreover, the absence of visible lesions does not rule out a diagnosis of Leigh syndrome, as patients may develop them later in the disease course[96]. It is also recommended to complement conventional MRI with proton magnetic resonance spectroscopy (MRS). The identification of a lactate peak in either brain parenchyma or cerebrospinal fluid (CSF) is considered a hallmark of mitochondrial disease.
Notably, there are correlations between MRI findings and the underlying genetic mutations in LS. For instance, in cases with complex I deficiency, brain MRIs often reveal bilateral symmetric brainstem lesions, at least one striatal anomaly, and an observable lactate peak in MRS[90]. Rarely, Leigh syndrome mimics tectal glioma, with symmetrical lesions in the midbrain and the pons, described as “giant panda” or “double panda sign” [97]. Furthermore, some patients with POLG and SURF1 mutations have been reported to exhibit hypertrophic olivary degeneration, which may manifest clinically as tremors[15]. POLG mutations are associated with a variety of manifestations, in children in particular by Alpers-Huttenlocher disease or, later, by Spinocerebellar ataxia and epilepsy. Therefore these mutations are usually not included in the Leigh-like syndrome spectrum. Bindu and colleagues described the presence of bilateral hypertrophic olivary degeneration on MRI in a cohort of 10 children diagnosed with Leigh and Leigh-like syndrome[98]. Finally, the progression of LS over time is not always followed by a progression of lesions on MRI. Regression of lesions was demonstrated in 5 of 12 patients[39]. Moreover, in another study three patients had complete resolution of their lesions, demonstrating that LS lesions can be reversible and that the lack of lesions cannot rule out a diagnosis of LS[95].
5.4. Muscle Biopsies and Cultured Fibroblasts
Muscle tissue is frequently impacted in mitochondrial diseases due to its substantial energy requirements. Confirmation or exclusion of an OXPHOS disorder can be solely achieved through the analysis of a muscle biopsy[14]. An essential aspect of OXPHOS deficiency diagnosis involves the biochemical assessment of muscle biopsies extracted from either the quadriceps femoris muscle or soleus muscle. To comprehensively assess the entire OXPHOS system, immediate processing of a freshly obtained biopsy is imperative. Notably, the necessity of a muscle biopsy in the diagnostic procedure is not universal. In cases where the clinical and biochemical phenotype strongly points to a specific mutation, it is advisable to commence a genetic analysis. Another valuable diagnostic tool is the examination of cultured fibroblasts derived from skin biopsies. This approach becomes pertinent when muscle tissue accessibility is limited or when validation and clarification of muscle biopsy results are required. However, it's worth noting that OXPHOS disorders in muscle tissue may exhibit less marked symptoms or even be absent in cultured fibroblasts.
5.6. Genetic Diagnosis
Mitochondrial disorders occur with various clinical and biochemical features, implicating hundreds of genes present in both mtDNA and nDNA. When clinical features lack specificity or a multitude of candidate genes exist, a broader approach is necessary.A primary step in genetic diagnosis involves full sequence analysis of mtDNA, often extracted from affected tissue, primarily muscle. This approach is particularly useful for identifying mutations with high heteroplasmy levels. In cases of mtDNA depletion without detectable mutations, consideration of POLG gene mutations, encoding polymerase gamma essential for mtDNA replication and repair, becomes crucial. If mtDNA mutations are ruled out, the focus shifts to sequencing candidate nuclear genes based on the patient's clinical and biochemical features. The conventional strategy involves sequencing the most frequently mutated genes among the selected candidates. However, nowadays next-generation sequencing (NGS) technologies enable the sequencing of multiple candidate genes or even entire exomes.
5.7. Genetic Counseling
Genetic counseling is crucial for parents with children affected by LS who want to elucidate the underlying genetic factors. Categorizing mitochondrial disorders based on the identified mutations—whether in nuclear-encoded or mtDNA-encoded genes—provides a framework for risk assessment and future family planning.
Table 1 provides information on the known mtDNA and nDNA genes responsible for LS.
Table adapted from Rahman S, Thorbur D.Nuclear Gene-Encoded Leigh Syndrome Spectrum Overview. 2015 Oct 1 [Updated 2020 Jul 16]. In: Adam MP, Mirzaa GM, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023.
5.8 Differential DiagnosisSeveral disorders share clinical features with LS, necessitating a meticulous medical history and targeted laboratory investigations. Key differential diagnoses include neonatal asphyxia, Wernicke encephalopathy, various metabolic disorders, and toxic-induced basal ganglia lesions.
6. Treatment
To date, no definitive treatment has been identified for LS. Current therapies include supplements (coenzyme Q10 and its derivatives), vitamins, pyruvate, dichloroacetate, and a ketogenic diet. Table 2 provides information on the current drugs and therapies used for LS and preclinical in vivo studies. However, recent systematic reviews have highlighted additional therapeutic interventions under investigation, including experimental treatments and clinical trials[99].
6.1. Coenzyme Q10 and EPI-743 (Vatiquinone)
Coenzyme Q10, also referred to as ubiquinone, plays a vital role in shuttling electrons between complex II and complex III. In a specific case study, a patient with LS and an m.10197 G>A mutation experienced notable improvement after receiving CoQ10 supplementation for three months[100]. Furthermore, an MRI scan conducted during a one-year follow-up showed the complete disappearance of the previously observed lesion. It is worth noting, however, that CoQ10 supplementation does not consistently produce positive results in cases where there is a CoQ10 deficiency[46]. Six studies evaluated CoQ10 as a standalone treatment, with four reporting clinical improvement in patients harboring mutations in m.10197G>A, COQ2, and COQ4[99]. However, it was observed that patients in advanced stages of the disease showed clinical deterioration despite CoQ10 initiation, underscoring the importance of early intervention[99]. Improvements in renal function were noted in some cases following CoQ10 supplementation[101]. Conversely, Scalais et al. reported a case of a child who developed severe proteinuria despite receiving CoQ10 therapy from infancy[102].
Some LS patients have shown positive responses to alternative treatments such as EPI-743 and idebenone[103]. EPI-743 is a para-benzoquinone that repletes intracellular glutathione more potently than coenzyme Q10 or idebenone. It has been tested in open-label clinical trials for mitochondrial diseases, including LS. Enns et al. found that 11 out of 12 participants showed both clinical and radiological improvements[104]. Additionally, Martinelli et al. observed significant improvements in 10 children with genetically confirmed LS treated with EPI-743[105]. While EPI-743 appears to have broad efficacy across genetic variations, and other drugs are currently undergoing clinical trials, not all outcomes have been satisfactory[46].
6.2. Sodium Dichloroacetate and Sodium Pyruvate
Another compound, Dichloroacetate (DCA), enhances pyruvate dehydrogenase activity by reducing lactate buildup. DCA treatment has shown effectiveness in LS patients with the T8993C mutation in ATPase 6, a subunit of complex V[106], as well as PDHc and CI deficiency[107]. However, studies have reported mixed clinical outcomes with DCA treatment. While it has shown potential benefits, DCA is also associated with the risk of worsening peripheral neuropathy, even when thiamine is administered prophylactically to mitigate side effects[108]. For instance, Fujii et al. documented successful improvement in midbrain hyperintensities in a patient with the MT-ATP6 m.8993T>C mutation following DCA and thiamine therapy[106]. In contrast, Koga et al. described a case of a child with a PDHE1 mutation who experienced clinical deterioration with DCA but subsequently responded positively to pyruvate therapy[109].
It is important also to mention Pyruvate, which plays a crucial role in remedying the malfunction of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) by supplying NAD, thus restoring the NADH/NAD+ ratio that becomes disrupted in disorders marked by OXPHOS deficiency[110]. In a specific case study, the oral administration of sodium pyruvate over one year resulted in a significant enhancement of exercise tolerance, ventricular ejection fraction, and the normalization of an abnormal echocardiographic presentation[111]. The majority of studies focused on biochemical pyruvate dehydrogenase deficiencies, which were not confirmed radiologically or genetically as Leigh syndrome (LS). Only two papers met all inclusion and exclusion criteria. The highest level of evidence came from a case series by Fujii et al., which included two genetically confirmed LS patients with the variants m.8993T>G and m.9176T>C. Both patients, who were bedbound, showed improvements in the Newcastle Pediatric Mitochondrial Disease Scale (NPMDS) following pyruvate treatment[112].
6.3. KH176
KH176 is a promising redox-modulating agent under investigation for mitochondrial diseases, including Leigh syndrome (LS). A Phase 1 study (NCT02544217) evaluating KH176 in patients with various mitochondrial disorders, including LS, demonstrated that the agent was generally well-tolerated. However, at higher doses, it was associated with QTc prolongation and T-wave morphological changes, necessitating careful dose optimization and monitoring[113]. Building on these findings, randomized, multi-center Phase 2 trials are currently recruiting participants with a range of mitochondrial disorders, such as MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes), MIDD (maternally inherited diabetes and deafness), LS, mitochondrial myopathies, and mitochondrial encephalopathies. These studies aim to further evaluate the safety and efficacy of KH176 across different mitochondrial disease phenotypes. Notably, a safety and efficacy trial focusing on m.3243A>G-associated mitochondrial disease has already been completed, providing valuable insights into the therapeutic potential of KH176 in this specific genetic context[114].
6.4. Vitamin Supplementation
Vitamin supplementation is another approach to ameliorate the neurological symptoms of LS. High doses of riboflavin enhance muscle strength and alleviate lactic acidosis in the context of complex I deficiency due to mutation in ACAD9 mutation, which is a crucial factor for the operation of the OXPHOS system and ATP production[115,116]. Nonetheless, ACAD9 mutation seems to show a different pathology from the one observed in LS, ACAD9 mutations are associated with a non-Leigh complex encephalopathy and often cardiomyopathy in most of the cases. Riboflavin was utilized in 18 studies, though it was predominantly administered as part of a "mitochondrial cocktail" rather than as a standalone treatment. Only three studies specifically evaluated riboflavin monotherapy. Among these, two studies demonstrated clinical improvement in patients with ACAD9 mutations[116,117]. Notably, Gerards et al. reported the restoration of complex I activity in a family with a homozygous ACAD9 (c.1594C>T) mutation following riboflavin monotherapy[116].
Moreover, early supplementation with both thiamine and biotin in patients with LS caused by a mutation in SLC19A3, a gene important to thiamine transporter-2 deficiency, has been found to have an immediate therapeutic effect[118,119]. The SLC19A3 gene encodes a thiamine transporter, and mutations in this gene are associated with Biotin-Responsive Basal Ganglia Disease (BBGD). A review of 22 and 57 studies revealed that biotin and thiamine supplementation respectively, are commonly used as part of a "mitochondrial cocktail" for treating mitochondrial disorders. However, only two case series have directly compared biotin or thiamine monotherapy versus combination therapy. Debs et al. reported clinical and radiological improvement in two siblings with SLC19A3 missense mutations, both with biotin monotherapy and combination therapy[120]. In contrast, Tabarki et al. found no significant difference in the Burke-Fahn-Marsden Dystonia Rating Scale (BFMDRS) scores between thiamine monotherapy and combination therapy in an open-label prospective study involving 20 children with the SLC19A3 c.1264A>G (p.Thr422Ala) mutation. However, combination therapy was associated with a significantly faster recovery from acute crises (2 days versus 3 days) compared to monotherapy[121].
6.5. N-Acetylcysteine and Carnitine
N-acetylcysteine (NAC) is a compound that serves as a precursor to glutathione, a powerful antioxidant that helps neutralize toxic sulfides[122]. In one study, Viscomi et al. reported positive outcomes in five children with homozygous 505 + 1G>T splice-site mutations in the ETHE1 gene, which is responsible for a syndrome that shows a different pathology but shares many similarities with LS. These children experienced a reduction in seizure frequency, neurological improvements, and a decrease in symptoms such as acrocyanosis (bluish discoloration of extremities) and petechiae (small red or purple spots on the skin). Additionally, their episodes of diarrhea improved, which was likely enhanced by the combined use of Metronidazole, a bactericidal and prokinetic agent[123]. Another case, described by Shayota et al., documented developmental improvements in a patient with an ECHS1 mutation following NAC treatment. However, the patient was also on a valine-restricted diet, making it difficult to determine the exact contribution of NAC to the observed progress[124]. While these findings are encouraging, further studies are needed to clarify NAC’s role in managing mitochondrial disorders and to determine its effectiveness as a standalone treatment.
Carnitine plays a crucial role in fatty acid oxidation and is commonly included in mitochondrial cocktail therapies with combinations of nutraceuticals, co-factors, and antioxidants aimed at supporting mitochondrial function and bypassing defects in the electron transport chain[125]. However, its effectiveness remains uncertain, as many case series and reports have documented its use with little to no noticeable benefit[99]. One notable case, described by Toth et al., involved a patient with the m.8993T>C mutation who experienced sudden loss of mobility following an upper respiratory tract infection. The patient’s condition later improved after receiving carnitine supplementation, but it is unclear whether this was due to the treatment itself or simply the natural resolution of the infection. The authors noted that the patient had low plasma and muscle carnitine levels before treatment. Later, at age 13, the patient experienced worsening ataxia and muscle weakness, which improved after the carnitine dose was adjusted based on body weight[126]. While carnitine remains a widely used supplement in mitochondrial disease management, further studies are needed to determine its true therapeutic value, particularly regarding disease progression and symptomatic relief.
6.6. Ketogenic Diet
KD seems to extend longevity and ameliorate mental problems[127]. Moreover, it seems to reverse the oculomotor palsy of LS caused by the pathogenic NDUFV1 variant. Ketogenic diets are believed to enhance fatty acid β-oxidation, providing an alternative energy pathway when oxidative phosphorylation is impaired[128]. These diets have shown effectiveness in controlling epilepsy, improving eye movements, and supporting mental development in patients with mutations in genes such as ECHS1, POLG, TMEM126B, and PDHA 1, which causes pyruvate dehydrogenase deficiency[99]. Additionally, protein and valine-restricted diets have been reported to benefit patients with mutations affecting valine degradation pathways, such as those in HIBCH and ECHS1[128]. These dietary interventions are thought to prevent the buildup of toxic metabolites like methacrylyl-CoA and acryloyl-CoA[124]. However, ketogenic diets can also pose risks, such as inducing metabolic acidosis and potentially worsening clinical symptoms in some LS patients[129].
7. Preventive Approach
7.1. Spindle Nuclear Transfer
An innovative technology called spindle nuclear transfer (SNT) for females with LS prevents vertical transmission of mutated mtDNA. SNT consists of transferring the nucleus to another egg from a healthy donor with normal mitochondria but stripped of its nucleus. This remarkable technique holds great promise in mitochondrial disorders, although ethical concerns have been raised[130], and long-term follow-up of the child's longitudinal development remains to be seen[131]. The spindle transfer is a preventative approach which has been prohibited in the US, whereas the UK and Australia have adopted the pronuclear transfer but so far no positive publication about the clinical efficacy of this approach has been released.
8. Preclinical Research
8.1. Gene Therapy
Gene therapy holds great potential for treating orphan diseases, particularly with the advent of precision medicine. Administering adenovirus-associated virus (AAV) has been shown to alleviate respiratory abnormalities and lead to mild clinical improvements in NDUFS4 KO mice[132]. Adeno-associated virus (AAV) recombinant vectors have emerged as a promising gene therapy approach for LS. AAV vectors are particularly advantageous due to their ability to transduce post-mitotic cells, including neurons, with long-term gene expression and minimal immunogenicity. In the context of Leigh syndrome, AAV-mediated gene therapy aims to deliver functional copies of nuclear-encoded mitochondrial genes or introduce therapeutic molecules that enhance mitochondrial function. Studies exploring AAV-based interventions have shown potential in preclinical models, particularly in targeting defects associated with complex I deficiencies, the most common cause of Leigh syndrome. However, challenges remain, including optimizing tissue-specific targeting, overcoming the blood-brain barrier, and ensuring sustained expression without adverse immune responses. As research advances, AAV vectors hold promise for the development of targeted therapies, and as of now represent the only possibility for a cure as they convey the missing gene to the brain[133].
Interestingly, findings from a recent study indicate that new approaches involving mitochondrial transfer hold potential as a therapeutic strategy for treating mitochondrial diseases, including Leigh syndrome[134].
8.2. Disease Models of Leigh Syndrome
A variety of disease models of LS have been developed, leading to important findings into LS pathophysiology, with the most prominent model of LS being the NDUFS4 knockout mouse model[135].
NDUFS4 is a nuclear gene encoding the NADH-dehydrogenase subunit S4 of the OXPHOS complex I, its mutation leads to mitochondrial dysfunction. Mitochondrial dysfunction in neurons can lead to the buildup of lipid droplets (LD) in glial cells and may occur before LS symptoms appear. Mouse models of NDUFS4 mutations developed bilateral spongiform lesions, and systemic inflammation which seems to result from LD accumulation in glial cells[60]. In both mouse and Drosophila models, LD accumulation was associated with increased reactive oxygen species (ROS) levels, and both effects were alleviated by antioxidants. This indicates that oxidative stress can drive lipid droplet accumulation during mitochondrial dysfunction. Consequently, ROS-induced lipid peroxidation and subsequent neuroinflammation may contribute to the underlying disease mechanism[58]. These mouse models show how mTOR inhibition and antioxidants could be a promising approach to mitigate the severe phenotype in patients with Leigh syndrome. Treating NDUFS4 mice with AD4 helped reduce LD levels, slightly delaying the onset of neurodegenerative signs and improving motor function[60]. Repletion of nicotinamide adenine dinucleotide (NAD+) granted extended lifespan in murine models of NDUFS4[136]. Compounds such as rapamycin and doxycycline improved and doubled the lifespan of the NDUFS4 knockout mouse model[137-139]. Mouse models by the Mootha lab have also demonstrated that one of the most effective interventions in NDUFS4 models is prolonged, continuous exposure to mild hypoxia, resulting in a significant extension of lifespan and both pathological and radiological improvement[140-143].
Yeast models have also been developed, among these the S. cerevisiae model of deficiency by editing the SURF1 homolog Shy1 helped confirm the importance of this assembly factor of the complex IV[144]. Zebrafish SURF1 models provided evidence for cysteamine bitartrate and N-Acetylcysteine in ameliorating ROS in SURF1-associated LS[145].
Non-mammalian animal models have also been used in LS. Burman et al generated a Drosophila model carrying a deletion in the MT-ND2 gene[146]. This knockout model revealed complex I reduced activity with features of mitochondrial disease. This model was used to test rapamycin as a potential treatment, with evidence of fat storage defect rescue[147]. In Drosophila, mutations in the ND23 subunit of mitochondrial complex I—analogous to the mammalian NDUFS8—mimic key features of LS. Mutations in the mitochondrial complex I subunit ND23 enhance susceptibility to isoflurane-induced toxicity and oxidative stress in Drosophila. Asymptomatic flies carrying ND23 and MT-ND2 mutations become increasingly vulnerable to isoflurane and halothane toxicity due to aging and genetic background[148,149]. This model helped gain an understanding of the danger related to anesthetic usage in LS patients.
Also, patient fibroblast models carrying mtDNA mutations in MT-ND1,3 and 5 were helpful in showing possible therapeutic compounds such as hispidin, a natural fungal compound, which helped inhibit the pro-oxidative pathway mediated by p66Shc protein[150]. Another LS disease model came after the discovery of a method for reprogramming fibroblast cultures into induced pluripotent stem cells (iPSCs)[151]. An iPSC model for MT-ATP6 mutation was able to show some benefit in helping mitochondrial membrane potential restoration with the PDE5 inhibitor avanafil[152]. An iPSCs model of NDUFS4 helped provide evidence for nicotinamide riboside (NR) as a potential treatment strategy for LS[153]. NR, a vitamin B3 analogue and precursor of nicotinamide adenine dinucleotide (NAD+), supports beneficial effects in LS through a mechanism dependent on acetylation. Targeted metabolomic analysis of heart and brain samples from LS mice, both under basal conditions and after NR supplementation, revealed disruptions in NAD+-dependent metabolic enzymes. These disruptions were restored following NR treatment. As NAD+ serves as a co-substrate for Sirtuin deacetylases, its depletion leads to reduced Sirtuin 1 (SIRT1) activity, resulting in increased protein acetylation[154]. Possible therapies to ameliorate SIRT1 activity are discussed in the following section.
8.3. Sirtuins
Sirtuins (SIRTs) are a class of histone deacetylases that rely on nicotinamide adenine dinucleotide (NAD+) as a cofactor. They play crucial roles in regulating vital signaling pathways in both prokaryotes and eukaryotes, contributing to various biological processes. Currently, there are seven mammalian homologs of yeast Sir2, known as SIRT1 to SIRT7. These proteins are involved in a range of essential cellular processes, including inflammation, metabolism, oxidative stress, and apoptosis. Consequently, they are considered promising therapeutic targets for a wide array of pathologies, including cancer, cardiovascular disease, respiratory issues, and other medical conditions. SIRT1, for example, regulates mitochondrial energy metabolism and is known to boost mitochondrial biogenesis by activating PGC-1α[155,156]. One emerging strategy in LS involves activating SIRT1, a key regulator of cellular metabolism and mitochondrial function. SIRT1 activation has been explored using NAD+ precursors and poly(ADP-ribose) polymerase (PARP) inhibitors, which enhance mitochondrial biogenesis and function. Enhancing SIRT1 activity may counteract mitochondrial dysfunction in LS by promoting energy homeostasis, reducing oxidative stress, and increasing cellular resilience[157]. NAD+ is a crucial coenzyme in mitochondrial metabolism and a direct modulator of SIRT1 activity. NAD+ precursors, such as nicotinamide riboside and nicotinamide mononucleotide, have been shown to boost NAD+ levels, thereby enhancing SIRT1 function. Studies in mitochondrial disease models indicate that NAD+ supplementation improves mitochondrial respiration and reduces neurodegeneration[158]. Ongoing clinical trials are assessing the benefits of NAD+ augmentation in mitochondrial diseases, including Leigh syndrome, with promising preliminary results. Another strategy to preserve NAD+ levels involves the inhibition of PARP. PARP enzymes play a critical role in DNA repair but excessively consume NAD+, leading to energy depletion in mitochondrial diseases. PARP inhibitors (PARPis) such as olaparib and veliparib have been investigated for their ability to prevent NAD+ depletion and enhance mitochondrial function. PARP inhibition has been reported to improve cellular energy balance and mitochondrial dynamics in neurodegenerative conditions models[157]. By preserving NAD+ levels, PARP inhibitors may sustain SIRT1 activity and improve mitochondrial resilience in LS patients. Therapies targeting SIRT1 activation, including NAD+ precursors and PARP inhibitors, offer a promising avenue for mitigating mitochondrial dysfunction in Leigh syndrome. By enhancing mitochondrial biogenesis, reducing oxidative stress, and preserving cellular energy balance, these approaches hold potential for improving patient outcomes.
8.4. Rapamycin
Rapamycin, also recognized as sirolimus, is commonly administered to prevent immunological rejection after kidney transplantation. Rapamycin, a macrolide antibiotic and potent inhibitor of the mechanistic target of rapamycin (mTOR), has shown therapeutic potential in preclinical models of mitochondrial dysfunction[159]. One of the most significant mechanisms through which rapamycin exerts neuroprotective effects is by enhancing autophagy and mitophagy. In mitochondrial diseases, dysfunctional mitochondria accumulate and contribute to oxidative damage and cellular dysfunction. Rapamycin-induced autophagy facilitates the degradation and recycling of damaged mitochondria, thereby improving cellular homeostasis. Increased mitophagy helps to remove defective mitochondria, reducing the burden of oxidative stress and improving mitochondrial network function[159]. Rapamycin mitigates oxidative stress by upregulating antioxidant defense mechanisms, such as increased expression of superoxide dismutase (SOD) and nuclear factor erythroid 2-related factor 2 (NRF2). Additionally, rapamycin modulates inflammatory pathways by reducing the activation of pro-inflammatory cytokines and microglial activation, thereby limiting neuroinflammation, which is a key contributor to neurodegeneration in complex I deficiency[160]. Rapamycin has also been shown to influence the integrated stress response (ISR), a cellular pathway activated in response to mitochondrial dysfunction. By modulating ISR signaling, rapamycin promotes cell survival and adaptation to mitochondrial stress, improving neuronal resilience in complex I-defective models[160]. In a mouse model designed to simulate LS, rapamycin showed promising results by improving neurological symptoms and extending the lifespan to approximately 110 days, which is twice as long as the untreated control group, whose lifespan was around 50 days[161]. Rapamycin also emulates the effects of caloric restriction (CR), a practice known to extend lifespan and reduce the risk of age-related health issues in yeast[162]. However, it's essential to acknowledge that there are prevalent side effects associated with rapamycin usage, including hyperlipidemia, immunosuppression, and impaired wound healing. These side effects can present challenges, particularly when considering their long-term use[163].
8.5. From Preclinical Model to New Interventions
Preclinical models have helped gain insight into new possible therapeutic options. Treatment of LS usually involves vitamins, supplementation, and a ketogenic diet[135]. Experimental models provided preclinical evidence for human trials for the mTOR inhibitors, such as the nab-sirolimus trial (NCT03747328) recently withdrawn, and sirolimus (NCT06843811) currently enrolling. Another EPI-743 trial was recently completed (NCT02352896) and results are waiting to be posted. The European Medical Agency (EMA) issued an orphan drug designation for Cannabidiol (EU/3/23/2800) and Sildenafil (EU/3/23/2831), both showing some efficacy in mouse models[152,164]. Gene replacement therapy using the AAV9/hSURF1 vector in LS patients with SURF1 mutations has obtained an orphan drug designation by EMA (EU/3/21/2531), after proving successful in SURF1 knockout mice[165].
9. Prognosis and Conclusions
Despite notable progress in medical treatments, the outlook for individuals with Leigh syndrome remains a complex and challenging one. Regrettably, a significant portion of cases result in fatality, often occurring before the age of five. Nevertheless, by implementing vigilant monitoring of patients who exhibit respiratory problems and employing a range of diagnostic assessments to assess brainstem function, including magnetic resonance imaging, auditory-evoked brainstem potentials, somatosensory-evoked potentials, blink reflex, or polysomnography, there is the potential to prevent sudden and early fatalities in individuals with early-onset Leigh syndrome. However, most of the interventions are palliative and not radical treatments. Among current treatments, AAV vectors hold promise for the development of targeted therapies, and as of now represent the only possibility for a cure as they convey the missing gene to the brain. Further clinical studies are needed to evaluate the safety and efficacy of gene therapy.
Author Contributions
GM wrote the first draft of the manuscript and was responsible for the first literature search. GM, VL, and FT helped with the PubMed research and reviewed the manuscript. All authors had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Funding
There was no funding source for this study.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki. Ethical review and approval were waived for this study, due to the nature of the study.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We all thank our family and loved ones, for their continuous support.
Conflicts of Interest
The author declares no conflict of interest.
References
- Schubert Baldo, M.; Vilarinho, L. Molecular basis of Leigh syndrome: a current look. Orphanet J Rare Dis 2020, 15, 31. [CrossRef]
- Balasubramaniam, S.; Lewis, B.; Mock, D.M.; Said, H.M.; Tarailo-Graovac, M.; Mattman, A.; van Karnebeek, C.D.; Thorburn, D.R.; Rodenburg, R.J.; Christodoulou, J. Leigh-Like Syndrome Due to Homoplasmic m.8993T>G Variant with Hypocitrullinemia and Unusual Biochemical Features Suggestive of Multiple Carboxylase Deficiency (MCD). JIMD Rep 2017, 33, 99-107. [CrossRef]
- Ogier, H.; Lombes, A.; Scholte, H.R.; Poll-The, B.T.; Fardeau, M.; Alcardi, J.; Vignes, B.; Niaudet, P.; Saudubray, J.M. de Toni-Fanconi-Debré syndrome with Leigh syndrome revealing severe muscle cytochrome c oxidase deficiency. J Pediatr 1988, 112, 734-739. [CrossRef]
- Lake, N.J.; Compton, A.G.; Rahman, S.; Thorburn, D.R. Leigh syndrome: One disorder, more than 75 monogenic causes. Ann Neurol 2016, 79, 190-203. [CrossRef]
- Kucharczyk, R.; Rak, M.; di Rago, J.P. Biochemical consequences in yeast of the human mitochondrial DNA 8993T>C mutation in the ATPase6 gene found in NARP/MILS patients. Biochim Biophys Acta 2009, 1793, 817-824. [CrossRef]
- Baertling, F.; Rodenburg, R.J.; Schaper, J.; Smeitink, J.A.; Koopman, W.J.; Mayatepek, E.; Morava, E.; Distelmaier, F. A guide to diagnosis and treatment of Leigh syndrome. J Neurol Neurosurg Psychiatry 2014, 85, 257-265. [CrossRef]
- Finsterer, J. Leigh and Leigh-like syndrome in children and adults. Pediatr Neurol 2008, 39, 223-235. [CrossRef]
- Chol, M.; Lebon, S.; Bénit, P.; Chretien, D.; de Lonlay, P.; Goldenberg, A.; Odent, S.; Hertz-Pannier, L.; Vincent-Delorme, C.; Cormier-Daire, V.; et al. The mitochondrial DNA G13513A MELAS mutation in the NADH dehydrogenase 5 gene is a frequent cause of Leigh-like syndrome with isolated complex I deficiency. J Med Genet 2003, 40, 188-191. [CrossRef]
- Di Rocco, M.; Caruso, U.; Moroni, I.; Lupino, S.; Lamantea, E.; Fantasia, A.R.; Borrone, C.; Gibson, K.M. 3-Methylglutaconic aciduria and hypermethioninaemia in a child with clinical and neuroradiological findings of Leigh disease. J Inherit Metab Dis 1999, 22, 593-598. [CrossRef]
- Cooper, M.P.; Qu, L.; Rohas, L.M.; Lin, J.; Yang, W.; Erdjument-Bromage, H.; Tempst, P.; Spiegelman, B.M. Defects in energy homeostasis in Leigh syndrome French Canadian variant through PGC-1alpha/LRP130 complex. Genes Dev 2006, 20, 2996-3009. [CrossRef]
- Rahman, S.; Blok, R.B.; Dahl, H.H.; Danks, D.M.; Kirby, D.M.; Chow, C.W.; Christodoulou, J.; Thorburn, D.R. Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann Neurol 1996, 39, 343-351. [CrossRef]
- Rahman, S. Chapter 4 - Leigh syndrome. In Handbook of Clinical Neurology, Horvath, R., Hirano, M., Chinnery, P.F., Eds.; Elsevier: 2023; Volume 194, pp. 43-63.
- Ostergaard, E.; Hansen, F.J.; Sorensen, N.; Duno, M.; Vissing, J.; Larsen, P.L.; Faeroe, O.; Thorgrimsson, S.; Wibrand, F.; Christensen, E.; et al. Mitochondrial encephalomyopathy with elevated methylmalonic acid is caused by SUCLA2 mutations. Brain 2007, 130, 853-861. [CrossRef]
- Morin, C.; Mitchell, G.; Larochelle, J.; Lambert, M.; Ogier, H.; Robinson, B.H.; De Braekeleer, M. Clinical, metabolic, and genetic aspects of cytochrome C oxidase deficiency in Saguenay-Lac-Saint-Jean. Am J Hum Genet 1993, 53, 488-496.
- Merante, F.; Petrova-Benedict, R.; MacKay, N.; Mitchell, G.; Lambert, M.; Morin, C.; De Braekeleer, M.; Laframboise, R.; Gagné, R.; Robinson, B.H. A biochemically distinct form of cytochrome oxidase (COX) deficiency in the Saguenay-Lac-Saint-Jean region of Quebec. Am J Hum Genet 1993, 53, 481-487.
- Oláhová, M.; Hardy, S.A.; Hall, J.; Yarham, J.W.; Haack, T.B.; Wilson, W.C.; Alston, C.L.; He, L.; Aznauryan, E.; Brown, R.M.; et al. LRPPRC mutations cause early-onset multisystem mitochondrial disease outside of the French-Canadian population. Brain 2015, 138, 3503-3519. [CrossRef]
- Hong, C.-M.; Na, J.-H.; Park, S.; Lee, Y.-M. Clinical Characteristics of Early-Onset and Late-Onset Leigh Syndrome. Frontiers in Neurology 2020, 11. [CrossRef]
- Debray, F.G.; Morin, C.; Janvier, A.; Villeneuve, J.; Maranda, B.; Laframboise, R.; Lacroix, J.; Decarie, J.C.; Robitaille, Y.; Lambert, M.; et al. LRPPRC mutations cause a phenotypically distinct form of Leigh syndrome with cytochrome c oxidase deficiency. J Med Genet 2011, 48, 183-189. [CrossRef]
- Leigh, D. Subacute necrotizing encephalomyelopathy in an infant. Journal of neurology, neurosurgery, and psychiatry 1951, 14, 216-221. [CrossRef]
- Arii, J.; Tanabe, Y. Leigh syndrome: serial MR imaging and clinical follow-up. AJNR Am J Neuroradiol 2000, 21, 1502-1509.
- Piao, Y.S.; Tang, G.C.; Yang, H.; Lu, D.H. Clinico-neuropathological study of a Chinese case of familial adult Leigh syndrome. Neuropathology 2006, 26, 218-221. [CrossRef]
- Koenig, M.K. Presentation and diagnosis of mitochondrial disorders in children. Pediatr Neurol 2008, 38, 305-313. [CrossRef]
- Lee, J.S.; Yoo, T.; Lee, M.; Lee, Y.; Jeon, E.; Kim, S.Y.; Lim, B.C.; Kim, K.J.; Choi, M.; Chae, J.H. Genetic heterogeneity in Leigh syndrome: Highlighting treatable and novel genetic causes. Clin Genet 2020, 97, 586-594. [CrossRef]
- Chen, L.; Cui, Y.; Jiang, D.; Ma, C.Y.; Tse, H.-F.; Hwu, W.-L.; Lian, Q. Management of Leigh syndrome: Current status and new insights. Clinical Genetics 2018, 93, 1131-1140. [CrossRef]
- Chang, X.; Wu, Y.; Zhou, J.; Meng, H.; Zhang, W.; Guo, J. A meta-analysis and systematic review of Leigh syndrome: clinical manifestations, respiratory chain enzyme complex deficiency, and gene mutations. Medicine (Baltimore) 2020, 99, e18634. [CrossRef]
- Ball, M.; Thorburn, D.R.; Rahman, S. Mitochondrial DNA-Associated Leigh Syndrome Spectrum. In GeneReviews(®), Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington, Seattle.
- Copyright © 1993-2025, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved.: Seattle (WA), 1993.
- Yang, Y.L.; Sun, F.; Zhang, Y.; Qian, N.; Yuan, Y.; Wang, Z.X.; Qi, Y.; Xiao, J.X.; Wang, X.Y.; Qi, Z.Y.; et al. Clinical and laboratory survey of 65 Chinese patients with Leigh syndrome. Chin Med J (Engl) 2006, 119, 373-377.
- Bugiani, M.; Tiranti, V.; Farina, L.; Uziel, G.; Zeviani, M. Novel mutations in COX15 in a long surviving Leigh syndrome patient with cytochrome c oxidase deficiency. J Med Genet 2005, 42, e28. [CrossRef]
- Desguerre, I.; Pinton, F.; Nabbout, R.; Moutard, M.L.; N'Guyen, S.; Marsac, C.; Ponsot, G.; Dulac, O. Infantile spasms with basal ganglia MRI hypersignal may reveal mitochondrial disorder due to T8993G MT DNA mutation. Neuropediatrics 2003, 34, 265-269. [CrossRef]
- Pequignot, M.O.; Desguerre, I.; Dey, R.; Tartari, M.; Zeviani, M.; Agostino, A.; Benelli, C.; Fouque, F.; Prip-Buus, C.; Marchant, D.; et al. New splicing-site mutations in the SURF1 gene in Leigh syndrome patients. J Biol Chem 2001, 276, 15326-15329. [CrossRef]
- Mak, S.C.; Chi, C.S.; Tsai, C.R. Mitochondrial DNA 8993 T > C mutation presenting as juvenile Leigh syndrome with respiratory failure. J Child Neurol 1998, 13, 349-351. [CrossRef]
- Sofou, K.; De Coo, I.F.M.; Isohanni, P.; Ostergaard, E.; Naess, K.; De Meirleir, L.; Tzoulis, C.; Uusimaa, J.; De Angst, I.B.; Lönnqvist, T.; et al. A multicenter study on Leigh syndrome: disease course and predictors of survival. Orphanet Journal of Rare Diseases 2014, 9, 52. [CrossRef]
- Lee, S.; Na, J.H.; Lee, Y.M. Epilepsy in Leigh Syndrome With Mitochondrial DNA Mutations. Front Neurol 2019, 10, 496. [CrossRef]
- Ng, Y.S.; Martikainen, M.H.; Gorman, G.S.; Blain, A.; Bugiardini, E.; Bunting, A.; Schaefer, A.M.; Alston, C.L.; Blakely, E.L.; Sharma, S.; et al. Pathogenic variants in MT-ATP6: A United Kingdom-based mitochondrial disease cohort study. Ann Neurol 2019, 86, 310-315. [CrossRef]
- Macaya, A.; Munell, F.; Burke, R.E.; De Vivo, D.C. Disorders of movement in Leigh syndrome. Neuropediatrics 1993, 24, 60-67. [CrossRef]
- Tranchant, C.; Anheim, M. Movement disorders in mitochondrial diseases. Revue Neurologique 2016, 172, 524-529. [CrossRef]
- Alston, C.L.; Morak, M.; Reid, C.; Hargreaves, I.P.; Pope, S.A.S.; Land, J.M.; Heales, S.J.; Horvath, R.; Mundy, H.; Taylor, R.W. A novel mitochondrial MTND5 frameshift mutation causing isolated complex I deficiency, renal failure and myopathy. Neuromuscular Disorders 2010, 20, 131-135. [CrossRef]
- Farina, L.; Chiapparini, L.; Uziel, G.; Bugiani, M.; Zeviani, M.; Savoiardo, M. MR findings in Leigh syndrome with COX deficiency and SURF-1 mutations. AJNR Am J Neuroradiol 2002, 23, 1095-1100.
- Sofou, K.; Steneryd, K.; Wiklund, L.M.; Tulinius, M.; Darin, N. MRI of the brain in childhood-onset mitochondrial disorders with central nervous system involvement. Mitochondrion 2013, 13, 364-371. [CrossRef]
- Stenton, S.L.; Zou, Y.; Cheng, H.; Liu, Z.; Wang, J.; Shen, D.; Jin, H.; Ding, C.; Tang, X.; Sun, S.; et al. Leigh Syndrome: A Study of 209 Patients at the Beijing Children's Hospital. Ann Neurol 2022, 91, 466-482. [CrossRef]
- Kistol, D.; Tsygankova, P.; Krylova, T.; Bychkov, I.; Itkis, Y.; Nikolaeva, E.; Mikhailova, S.; Sumina, M.; Pechatnikova, N.; Kurbatov, S.; et al. Leigh Syndrome: Spectrum of Molecular Defects and Clinical Features in Russia. Int J Mol Sci 2023, 24. [CrossRef]
- Rahman, S. Gastrointestinal and hepatic manifestations of mitochondrial disorders. Journal of Inherited Metabolic Disease 2013, 36, 659-673. [CrossRef]
- Lim, A.Z.; Ng, Y.S.; Blain, A.; Jiminez-Moreno, C.; Alston, C.L.; Nesbitt, V.; Simmons, L.; Santra, S.; Wassmer, E.; Blakely, E.L.; et al. Natural History of Leigh Syndrome: A Study of Disease Burden and Progression. Ann Neurol 2022, 91, 117-130. [CrossRef]
- Naess, K.; Freyer, C.; Bruhn, H.; Wibom, R.; Malm, G.; Nennesmo, I.; von Döbeln, U.; Larsson, N.G. MtDNA mutations are a common cause of severe disease phenotypes in children with Leigh syndrome. Biochim Biophys Acta 2009, 1787, 484-490. [CrossRef]
- Van Hove, J.L.; Saenz, M.S.; Thomas, J.A.; Gallagher, R.C.; Lovell, M.A.; Fenton, L.Z.; Shanske, S.; Myers, S.M.; Wanders, R.J.; Ruiter, J.; et al. Succinyl-CoA ligase deficiency: a mitochondrial hepatoencephalomyopathy. Pediatr Res 2010, 68, 159-164. [CrossRef]
- López, L.C.; Schuelke, M.; Quinzii, C.M.; Kanki, T.; Rodenburg, R.J.; Naini, A.; Dimauro, S.; Hirano, M. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am J Hum Genet 2006, 79, 1125-1129. [CrossRef]
- Sofou, K.; de Coo, I.F.M.; Ostergaard, E.; Isohanni, P.; Naess, K.; De Meirleir, L.; Tzoulis, C.; Uusimaa, J.; Lönnqvist, T.; Bindoff, L.A.; et al. Phenotype-genotype correlations in Leigh syndrome: new insights from a multicentre study of 96 patients. J Med Genet 2018, 55, 21-27. [CrossRef]
- Sonam, K.; Khan, N.A.; Bindu, P.S.; Taly, A.B.; Gayathri, N.; Bharath, M.M.S.; Govindaraju, C.; Arvinda, H.R.; Nagappa, M.; Sinha, S.; et al. Clinical and magnetic resonance imaging findings in patients with Leigh syndrome and SURF1 mutations. Brain and Development 2014, 36, 807-812. [CrossRef]
- Østergaard, E.; Bradinova, I.; Ravn, S.H.; Hansen, F.J.; Simeonov, E.; Christensen, E.; Wibrand, F.; Schwartz, M. Hypertrichosis in patients with SURF1 mutations. American Journal of Medical Genetics Part A 2005, 138A, 384-388. [CrossRef]
- Gerards, M.; Sallevelt, S.C.E.H.; Smeets, H.J.M. Leigh syndrome: Resolving the clinical and genetic heterogeneity paves the way for treatment options. Molecular Genetics and Metabolism 2016, 117, 300-312. [CrossRef]
- Zhang, Y.; Yang, Y.L.; Sun, F.; Cai, X.; Qian, N.; Yuan, Y.; Wang, Z.X.; Qi, Y.; Xiao, J.X.; Wang, X.Y.; et al. Clinical and molecular survey in 124 Chinese patients with Leigh or Leigh-like syndrome. J Inherit Metab Dis 2007, 30, 265. [CrossRef]
- Munaro M, T.V., Sandonà D, et al. . A single cell complementation class is common to several cases of cytochrome c oxidase- defective Leigh’s syndrome. Hum Mol Genet 1997, 6, 221-228.
- Nagashima, T.; Mori, M.; Katayama, K.; Nunomura, M.; Nishihara, H.; Hiraga, H.; Tanaka, S.; Goto, Y.; Nagashima, K. Adult Leigh syndrome with mitochondrial DNA mutation at 8993. Acta Neuropathol 1999, 97, 416-422. [CrossRef]
- Debray, F.G.; Lambert, M.; Chevalier, I.; Robitaille, Y.; Decarie, J.C.; Shoubridge, E.A.; Robinson, B.H.; Mitchell, G.A. Long-term outcome and clinical spectrum of 73 pediatric patients with mitochondrial diseases. Pediatrics 2007, 119, 722-733. [CrossRef]
- Van Maldergem, L.; Trijbels, F.; DiMauro, S.; Sindelar, P.J.; Musumeci, O.; Janssen, A.; Delberghe, X.; Martin, J.J.; Gillerot, Y. Coenzyme Q-responsive Leigh's encephalopathy in two sisters. Ann Neurol 2002, 52, 750-754. [CrossRef]
- Malandrini, A.; Palmeri, S.; Fabrizi, G.M.; Villanova, M.; Berti, G.; Salvadori, C.; Gardini, G.; Motti, L.; Solimé, F.; Guazzi, G.C. Juvenile Leigh syndrome with protracted course presenting as chronic sensory motor neuropathy, ataxia, deafness and retinitis pigmentosa: a clinicopathological report. J Neurol Sci 1998, 155, 218-221. [CrossRef]
- Alves, C.A.P.F.; Teixeira, S.R.; Martin-Saavedra, J.S.; Guimarães Gonçalves, F.; Lo Russo, F.; Muraresku, C.; McCormick, E.M.; Falk, M.J.; Zolkipli-Cunningham, Z.; Ganetzky, R.; et al. Pediatric Leigh Syndrome: Neuroimaging Features and Genetic Correlations. Annals of Neurology 2020, 88, 218-232. [CrossRef]
- van de Wal, M.A.E.; Adjobo-Hermans, M.J.W.; Keijer, J.; Schirris, T.J.J.; Homberg, J.R.; Wieckowski, M.R.; Grefte, S.; van Schothorst, E.M.; van Karnebeek, C.; Quintana, A.; et al. Ndufs4 knockout mouse models of Leigh syndrome: pathophysiology and intervention. Brain 2022, 145, 45-63. [CrossRef]
- Calvaruso, M.A.; Willems, P.; van den Brand, M.; Valsecchi, F.; Kruse, S.; Palmiter, R.; Smeitink, J.; Nijtmans, L. Mitochondrial complex III stabilizes complex I in the absence of NDUFS4 to provide partial activity. Hum Mol Genet 2012, 21, 115-120. [CrossRef]
- Liu, L.; Zhang, K.; Sandoval, H.; Yamamoto, S.; Jaiswal, M.; Sanz, E.; Li, Z.; Hui, J.; Graham, B.H.; Quintana, A.; et al. Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell 2015, 160, 177-190. [CrossRef]
- Procaccio, V.; Wallace, D.C. Late-onset Leigh syndrome in a patient with mitochondrial complex I NDUFS8 mutations. Neurology 2004, 62, 1899-1901. [CrossRef]
- Wang, S.; Kang, Y.; Wang, R.; Deng, J.; Yu, Y.; Yu, J.; Wang, J. Emerging Roles of NDUFS8 Located in Mitochondrial Complex I in Different Diseases. Molecules 2022, 27. [CrossRef]
- Kirby, D.M.; Kahler, S.G.; Freckmann, M.L.; Reddihough, D.; Thorburn, D.R. Leigh disease caused by the mitochondrial DNA G14459A mutation in unrelated families. Ann Neurol 2000, 48, 102-104.
- Nesbitt, V.; Morrison, P.J.; Crushell, E.; Donnelly, D.E.; Alston, C.L.; He, L.; McFarland, R.; Taylor, R.W. The clinical spectrum of the m.10191T>C mutation in complex I-deficient Leigh syndrome. Dev Med Child Neurol 2012, 54, 500-506. [CrossRef]
- Rustin, P.; Rötig, A. Inborn errors of complex II--unusual human mitochondrial diseases. Biochim Biophys Acta 2002, 1553, 117-122. [CrossRef]
- Alston, C.L.; Davison, J.E.; Meloni, F.; van der Westhuizen, F.H.; He, L.; Hornig-Do, H.T.; Peet, A.C.; Gissen, P.; Goffrini, P.; Ferrero, I.; et al. Recessive germline SDHA and SDHB mutations causing leukodystrophy and isolated mitochondrial complex II deficiency. J Med Genet 2012, 49, 569-577. [CrossRef]
- Brockmann, K.; Bjornstad, A.; Dechent, P.; Korenke, C.G.; Smeitink, J.; Trijbels, J.M.F.; Athanassopoulos, S.; Villagran, R.; Skjeldal, O.H.; Wilichowski, E.; et al. Succinate in dystrophic white matter: A proton magnetic resonance spectroscopy finding characteristic for complex II deficiency. Annals of Neurology 2002, 52, 38-46. [CrossRef]
- Atwal, P.S. Mutations in the Complex III Assembly Factor Tetratricopeptide 19 Gene TTC19 Are a Rare Cause of Leigh Syndrome. JIMD Rep 2014, 14, 43-45. [CrossRef]
- Koch, J.; Feichtinger, R.G.; Freisinger, P.; Pies, M.; Schrödl, F.; Iuso, A.; Sperl, W.; Mayr, J.A.; Prokisch, H.; Haack, T.B. Disturbed mitochondrial and peroxisomal dynamics due to loss of MFF causes Leigh-like encephalopathy, optic atrophy and peripheral neuropathy. Journal of Medical Genetics 2016, 53, 270-278. [CrossRef]
- Spinazzi, M.; Radaelli, E.; Horré, K.; Arranz, A.M.; Gounko, N.V.; Agostinis, P.; Maia, T.M.; Impens, F.; Morais, V.A.; Lopez-Lluch, G.; et al. PARL deficiency in mouse causes Complex III defects, coenzyme Q depletion, and Leigh-like syndrome. Proc Natl Acad Sci U S A 2019, 116, 277-286. [CrossRef]
- Barel, O.; Shorer, Z.; Flusser, H.; Ofir, R.; Narkis, G.; Finer, G.; Shalev, H.; Nasasra, A.; Saada, A.; Birk, O.S. Mitochondrial Complex III Deficiency Associated with a Homozygous Mutation in UQCRQ. The American Journal of Human Genetics 2008, 82, 1211-1216. [CrossRef]
- Misceo, D.; Strømme, P.; Bitarafan, F.; Chawla, M.S.; Sheng, Y.; Bach de Courtade, S.M.; Eide, L.; Frengen, E. Biallelic NDUFA4 Deletion Causes Mitochondrial Complex IV Deficiency in a Patient with Leigh Syndrome. Genes (Basel) 2024, 15. [CrossRef]
- Lee, I.C.; Chiang, K.L. Clinical Diagnosis and Treatment of Leigh Syndrome Based on SURF1: Genotype and Phenotype. Antioxidants (Basel) 2021, 10. [CrossRef]
- Wedatilake, Y.; Brown, R.M.; McFarland, R.; Yaplito-Lee, J.; Morris, A.A.; Champion, M.; Jardine, P.E.; Clarke, A.; Thorburn, D.R.; Taylor, R.W.; et al. SURF1 deficiency: a multi-centre natural history study. Orphanet J Rare Dis 2013, 8, 96. [CrossRef]
- Bartke, A. New findings in gene knockout, mutant and transgenic mice. Exp Gerontol 2008, 43, 11-14. [CrossRef]
- Thorburn, D.R.; Rahman, J.; Rahman, S. Mitochondrial DNA-Associated Leigh Syndrome and NARP; University of Washington, Seattle, Seattle (WA): 1993.
- Ganapathi, M.; Friocourt, G.; Gueguen, N.; Friederich, M.W.; Le Gac, G.; Okur, V.; Loaëc, N.; Ludwig, T.; Ka, C.; Tanji, K.; et al. A homozygous splice variant in ATP5PO, disrupts mitochondrial complex V function and causes Leigh syndrome in two unrelated families. J Inherit Metab Dis 2022, 45, 996-1012. [CrossRef]
- Patel, K.P.; O'Brien, T.W.; Subramony, S.H.; Shuster, J.; Stacpoole, P.W. The spectrum of pyruvate dehydrogenase complex deficiency: Clinical, biochemical and genetic features in 371 patients. Molecular Genetics and Metabolism 2012, 106, 385-394. [CrossRef]
- Quinonez, S.C.; Thoene, J.G. Dihydrolipoamide Dehydrogenase Deficiency; University of Washington, Seattle, Seattle (WA): 1993.
- Mitchell, G.; Ogier, H.; Munnich, A.; Saudubray, J.M.; Shirrer, J.; Charpentier, C.; Rocchiccioli, F. Neurological Deterioration and Lactic Acidemia in Biotinidase Deficiency. A Treatable Condition Mimicking Leigh's Disease 1986, 17, 129-131. [CrossRef]
- Gerards, M.; Kamps, R.; van Oevelen, J.; Boesten, I.; Jongen, E.; de Koning, B.; Scholte, H.R.; de Angst, I.; Schoonderwoerd, K.; Sefiani, A.; et al. Exome sequencing reveals a novel Moroccan founder mutation in SLC19A3 as a new cause of early-childhood fatal Leigh syndrome. Brain 2013, 136, 882-890. [CrossRef]
- Fassone, E.; Rahman, S. Complex I deficiency: clinical features, biochemistry and molecular genetics. Journal of Medical Genetics 2012, 49, 578-590. [CrossRef]
- Carrozzo, R.; Verrigni, D.; Rasmussen, M.; de Coo, R.; Amartino, H.; Bianchi, M.; Buhas, D.; Mesli, S.; Naess, K.; Born, A.P.; et al. Succinate-CoA ligase deficiency due to mutations in SUCLA2 and SUCLG1: phenotype and genotype correlations in 71 patients. Journal of Inherited Metabolic Disease 2016, 39, 243-252. [CrossRef]
- Wortmann, S.B.; van Hasselt, P.M.; Barić, I.; Burlina, A.; Darin, N.; Hörster, F.; Coker, M.; Ucar, S.K.; Krumina, Z.; Naess, K.; et al. Eyes on MEGDEL: distinctive basal ganglia involvement in dystonia deafness syndrome. Neuropediatrics 2015, 46, 98-103. [CrossRef]
- Bénit, P.; Chretien, D.; Kadhom, N.; de Lonlay-Debeney, P.; Cormier-Daire, V.; Cabral, A.; Peudenier, S.; Rustin, P.; Munnich, A.; Rötig, A. Large-scale deletion and point mutations of the nuclear NDUFV1 and NDUFS1 genes in mitochondrial complex I deficiency. Am J Hum Genet 2001, 68, 1344-1352. [CrossRef]
- Horváth, R.; Abicht, A.; Holinski-Feder, E.; Laner, A.; Gempel, K.; Prokisch, H.; Lochmüller, H.; Klopstock, T.; Jaksch, M. Leigh syndrome caused by mutations in the flavoprotein (Fp) subunit of succinate dehydrogenase (SDHA). J Neurol Neurosurg Psychiatry 2006, 77, 74-76. [CrossRef]
- Lebon, S.; Chol, M.; Benit, P.; Mugnier, C.; Chretien, D.; Giurgea, I.; Kern, I.; Girardin, E.; Hertz-Pannier, L.; de Lonlay, P.; et al. Recurrent de novo mitochondrial DNA mutations in respiratory chain deficiency. J Med Genet 2003, 40, 896-899. [CrossRef]
- Wortmann, S.; Rodenburg, R.J.; Huizing, M.; Loupatty, F.J.; de Koning, T.; Kluijtmans, L.A.; Engelke, U.; Wevers, R.; Smeitink, J.A.; Morava, E. Association of 3-methylglutaconic aciduria with sensori-neural deafness, encephalopathy, and Leigh-like syndrome (MEGDEL association) in four patients with a disorder of the oxidative phosphorylation. Mol Genet Metab 2006, 88, 47-52. [CrossRef]
- Martín, M.A.; Blázquez, A.; Gutierrez-Solana, L.G.; Fernández-Moreira, D.; Briones, P.; Andreu, A.L.; Garesse, R.; Campos, Y.; Arenas, J. Leigh syndrome associated with mitochondrial complex I deficiency due to a novel mutation in the NDUFS1 gene. Arch Neurol 2005, 62, 659-661. [CrossRef]
- Malfatti, E.; Bugiani, M.; Invernizzi, F.; de Souza, C.F.; Farina, L.; Carrara, F.; Lamantea, E.; Antozzi, C.; Confalonieri, P.; Sanseverino, M.T.; et al. Novel mutations of ND genes in complex I deficiency associated with mitochondrial encephalopathy. Brain 2007, 130, 1894-1904. [CrossRef]
- Rossi, A.; Biancheri, R.; Bruno, C.; Di Rocco, M.; Calvi, A.; Pessagno, A.; Tortori-Donati, P. Leigh Syndrome with COX deficiency and SURF1 gene mutations: MR imaging findings. AJNR Am J Neuroradiol 2003, 24, 1188-1191.
- Loeffen, J.; Smeitink, J.; Triepels, R.; Smeets, R.; Schuelke, M.; Sengers, R.; Trijbels, F.; Hamel, B.; Mullaart, R.; van den Heuvel, L. The first nuclear-encoded complex I mutation in a patient with Leigh syndrome. Am J Hum Genet 1998, 63, 1598-1608. [CrossRef]
- Araki, S.; Hayashi, M.; Yasaka, A.; Maruki, K. Electrophysiological brainstem dysfunction in a child with Leigh disease. Pediatr Neurol 1997, 16, 329-333. [CrossRef]
- Martin, E.; Burger, R.; Wiestler, O.D.; Caduff, R.; Boltshauser, E.; Boesch, C. Brainstem lesion revealed by MRI in a case of Leigh's disease with respiratory failure. Pediatr Radiol 1990, 20, 349-350. [CrossRef]
- Bonfante, E.; Koenig, M.K.; Adejumo, R.B.; Perinjelil, V.; Riascos, R.F. The neuroimaging of Leigh syndrome: case series and review of the literature. Pediatr Radiol 2016, 46, 443-451. [CrossRef]
- Bonfante, E.; Koenig, M.K.; Adejumo, R.B.; Perinjelil, V.; Riascos, R.F. The neuroimaging of Leigh syndrome: case series and review of the literature. Pediatric Radiology 2016, 46, 443-451. [CrossRef]
- Rio, M.; Lebre, A.S.; de Lonlay, P.; Valayannopoulos, V.; Desguerre, I.; Dufier, J.L.; Grévent, D.; Zilbovicius, M.; Tréguier, C.; Brunelle, F.; et al. Mitochondrial ND5 mutations mimicking brainstem tectal glioma. Neurology 2010, 75, 93. [CrossRef]
- Bindu, P.S.; Taly, A.B.; Sonam, K.; Govindaraju, C.; Arvinda, H.R.; Gayathri, N.; Bharath, M.M.; Ranjith, D.; Nagappa, M.; Sinha, S.; et al. Bilateral hypertrophic olivary nucleus degeneration on magnetic resonance imaging in children with Leigh and Leigh-like syndrome. Br J Radiol 2014, 87, 20130478. [CrossRef]
- Tiet, M.Y.; Lin, Z.; Gao, F.; Jennings, M.J.; Horvath, R. Targeted Therapies for Leigh Syndrome: Systematic Review and Steps Towards a 'Treatabolome'. J Neuromuscul Dis 2021, 8, 885-897. [CrossRef]
- Chen, Z.; Zhao, Z.; Ye, Q.; Chen, Y.; Pan, X.; Sun, B.; Huang, H.; Zheng, A. Mild clinical manifestation and unusual recovery upon coenzyme Q₁₀ treatment in the first Chinese Leigh syndrome pedigree with mutation m.10197 G>A. Mol Med Rep 2015, 11, 1956-1962. [CrossRef]
- Montini, G.; Malaventura, C.; Salviati, L. Early coenzyme Q10 supplementation in primary coenzyme Q10 deficiency. N Engl J Med 2008, 358, 2849-2850. [CrossRef]
- Scalais, E.; Chafai, R.; Van Coster, R.; Bindl, L.; Nuttin, C.; Panagiotaraki, C.; Seneca, S.; Lissens, W.; Ribes, A.; Geers, C.; et al. Early myoclonic epilepsy, hypertrophic cardiomyopathy and subsequently a nephrotic syndrome in a patient with CoQ10 deficiency caused by mutations in para-hydroxybenzoate-polyprenyl transferase (COQ2). Eur J Paediatr Neurol 2013, 17, 625-630. [CrossRef]
- Haginoya, K.; Miyabayashi, S.; Kikuchi, M.; Kojima, A.; Yamamoto, K.; Omura, K.; Uematsu, M.; Hino-Fukuyo, N.; Tanaka, S.; Tsuchiya, S. Efficacy of idebenone for respiratory failure in a patient with Leigh syndrome: a long-term follow-up study. J Neurol Sci 2009, 278, 112-114. [CrossRef]
- Enns, G.M.; Kinsman, S.L.; Perlman, S.L.; Spicer, K.M.; Abdenur, J.E.; Cohen, B.H.; Amagata, A.; Barnes, A.; Kheifets, V.; Shrader, W.D.; et al. Initial experience in the treatment of inherited mitochondrial disease with EPI-743. Mol Genet Metab 2012, 105, 91-102. [CrossRef]
- Martinelli, D.; Catteruccia, M.; Piemonte, F.; Pastore, A.; Tozzi, G.; Dionisi-Vici, C.; Pontrelli, G.; Corsetti, T.; Livadiotti, S.; Kheifets, V.; et al. EPI-743 reverses the progression of the pediatric mitochondrial disease--genetically defined Leigh Syndrome. Mol Genet Metab 2012, 107, 383-388. [CrossRef]
- Fujii, T.; Ito, M.; Miyajima, T.; Okuno, T. Dichloroacetate therapy in Leigh syndrome with a mitochondrial T8993C mutation. Pediatr Neurol 2002, 27, 58-61. [CrossRef]
- Kimura, S.; Osaka, H.; Saitou, K.; Ohtuki, N.; Kobayashi, T.; Nezu, A. Improvement of lesions shown on MRI and CT scan by administration of dichloroacetate in patients with Leigh syndrome. J Neurol Sci 1995, 134, 103-107. [CrossRef]
- Spruijt, L.; Naviaux, R.K.; McGowan, K.A.; Nyhan, W.L.; Sheean, G.; Haas, R.H.; Barshop, B.A. Nerve conduction changes in patients with mitochondrial diseases treated with dichloroacetate. Muscle Nerve 2001, 24, 916-924. [CrossRef]
- Koga, Y.; Povalko, N.; Katayama, K.; Kakimoto, N.; Matsuishi, T.; Naito, E.; Tanaka, M. Beneficial effect of pyruvate therapy on Leigh syndrome due to a novel mutation in PDH E1α gene. Brain Dev 2012, 34, 87-91. [CrossRef]
- Tanaka, M.; Nishigaki, Y.; Fuku, N.; Ibi, T.; Sahashi, K.; Koga, Y. Therapeutic potential of pyruvate therapy for mitochondrial diseases. Mitochondrion 2007, 7, 399-401. [CrossRef]
- Komaki, H.; Nishigaki, Y.; Fuku, N.; Hosoya, H.; Murayama, K.; Ohtake, A.; Goto, Y.; Wakamoto, H.; Koga, Y.; Tanaka, M. Pyruvate therapy for Leigh syndrome due to cytochrome c oxidase deficiency. Biochim Biophys Acta 2010, 1800, 313-315. [CrossRef]
- Fujii, T.; Nozaki, F.; Saito, K.; Hayashi, A.; Nishigaki, Y.; Murayama, K.; Tanaka, M.; Koga, Y.; Hiejima, I.; Kumada, T. Efficacy of pyruvate therapy in patients with mitochondrial disease: a semi-quantitative clinical evaluation study. Mol Genet Metab 2014, 112, 133-138. [CrossRef]
- Koene, S.; Spaans, E.; Van Bortel, L.; Van Lancker, G.; Delafontaine, B.; Badilini, F.; Beyrath, J.; Smeitink, J. KH176 under development for rare mitochondrial disease: a first in man randomized controlled clinical trial in healthy male volunteers. Orphanet J Rare Dis 2017, 12, 163. [CrossRef]
- Janssen, M.C.H.; Koene, S.; de Laat, P.; Hemelaar, P.; Pickkers, P.; Spaans, E.; Beukema, R.; Beyrath, J.; Groothuis, J.; Verhaak, C.; et al. The KHENERGY Study: Safety and Efficacy of KH176 in Mitochondrial m.3243A>G Spectrum Disorders. Clin Pharmacol Ther 2019, 105, 101-111. [CrossRef]
- Garone, C.; Donati, M.A.; Sacchini, M.; Garcia-Diaz, B.; Bruno, C.; Calvo, S.; Mootha, V.K.; Dimauro, S. Mitochondrial encephalomyopathy due to a novel mutation in ACAD9. JAMA Neurol 2013, 70, 1177-1179. [CrossRef]
- Gerards, M.; van den Bosch, B.J.; Danhauser, K.; Serre, V.; van Weeghel, M.; Wanders, R.J.; Nicolaes, G.A.; Sluiter, W.; Schoonderwoerd, K.; Scholte, H.R.; et al. Riboflavin-responsive oxidative phosphorylation complex I deficiency caused by defective ACAD9: new function for an old gene. Brain 2011, 134, 210-219. [CrossRef]
- Garone, C.; Donati, M.A.; Sacchini, M.; Garcia-Diaz, B.; Bruno, C.; Calvo, S.; Mootha, V.K.; DiMauro, S. Mitochondrial Encephalomyopathy Due to a Novel Mutation in ACAD9. JAMA Neurology 2013, 70, 1177-1179. [CrossRef]
- Ortigoza-Escobar, J.D.; Molero-Luis, M.; Arias, A.; Oyarzabal, A.; Darín, N.; Serrano, M.; Garcia-Cazorla, A.; Tondo, M.; Hernández, M.; Garcia-Villoria, J.; et al. Free-thiamine is a potential biomarker of thiamine transporter-2 deficiency: a treatable cause of Leigh syndrome. Brain 2016, 139, 31-38. [CrossRef]
- Haack, T.B.; Klee, D.; Strom, T.M.; Mayatepek, E.; Meitinger, T.; Prokisch, H.; Distelmaier, F. Infantile Leigh-like syndrome caused by SLC19A3 mutations is a treatable disease. Brain 2014, 137, e295. [CrossRef]
- Debs, R.; Depienne, C.; Rastetter, A.; Bellanger, A.; Degos, B.; Galanaud, D.; Keren, B.; Lyon-Caen, O.; Brice, A.; Sedel, F. Biotin-responsive basal ganglia disease in ethnic Europeans with novel SLC19A3 mutations. Arch Neurol 2010, 67, 126-130. [CrossRef]
- Tabarki, B.; Alfadhel, M.; AlShahwan, S.; Hundallah, K.; AlShafi, S.; AlHashem, A. Treatment of biotin-responsive basal ganglia disease: Open comparative study between the combination of biotin plus thiamine versus thiamine alone. Eur J Paediatr Neurol 2015, 19, 547-552. [CrossRef]
- Bottani, E.; Lamperti, C.; Prigione, A.; Tiranti, V.; Persico, N.; Brunetti, D. Therapeutic Approaches to Treat Mitochondrial Diseases: “One-Size-Fits-All” and “Precision Medicine” Strategies. Pharmaceutics 2020, 12. [CrossRef]
- Viscomi, C.; Burlina, A.B.; Dweikat, I.; Savoiardo, M.; Lamperti, C.; Hildebrandt, T.; Tiranti, V.; Zeviani, M. Combined treatment with oral metronidazole and N-acetylcysteine is effective in ethylmalonic encephalopathy. Nat Med 2010, 16, 869-871. [CrossRef]
- Shayota, B.J.; Soler-Alfonso, C.; Bekheirnia, M.R.; Mizerik, E.; Boyer, S.W.; Xiao, R.; Yang, Y.; Elsea, S.H.; Scaglia, F. Case report and novel treatment of an autosomal recessive Leigh syndrome caused by short-chain enoyl-CoA hydratase deficiency. Am J Med Genet A 2019, 179, 803-807. [CrossRef]
- Tarnopolsky, M.A. The mitochondrial cocktail: rationale for combined nutraceutical therapy in mitochondrial cytopathies. Adv Drug Deliv Rev 2008, 60, 1561-1567. [CrossRef]
- Tóth, G.; Morava, E.; Bene, J.; Selhorst, J.J.; Overmars, H.; Vreken, P.; Molnár, J.; Farkas, V.; Melegh, B. Carnitine-responsive carnitine insufficiency in a case of mtDNA 8993T>C mutation associated Leigh syndrome. J Inherit Metab Dis 2001, 24, 421-422. [CrossRef]
- Wexler, I.D.; Hemalatha, S.G.; McConnell, J.; Buist, N.R.; Dahl, H.H.; Berry, S.A.; Cederbaum, S.D.; Patel, M.S.; Kerr, D.S. Outcome of pyruvate dehydrogenase deficiency treated with ketogenic diets. Studies in patients with identical mutations. Neurology 1997, 49, 1655-1661. [CrossRef]
- Laugel, V.; This-Bernd, V.; Cormier-Daire, V.; Speeg-Schatz, C.; de Saint-Martin, A.; Fischbach, M. Early-onset ophthalmoplegia in Leigh-like syndrome due to NDUFV1 mutations. Pediatr Neurol 2007, 36, 54-57. [CrossRef]
- Banka, S.; de Goede, C.; Yue, W.W.; Morris, A.A.; von Bremen, B.; Chandler, K.E.; Feichtinger, R.G.; Hart, C.; Khan, N.; Lunzer, V.; et al. Expanding the clinical and molecular spectrum of thiamine pyrophosphokinase deficiency: a treatable neurological disorder caused by TPK1 mutations. Mol Genet Metab 2014, 113, 301-306. [CrossRef]
- R., S. Three-parent baby' claim raises hopes and ethical concerns.
- Available online: http://www.nature.com/news/three-parent-baby-claim-raises-hopes-and-ethical-concerns-1. (accessed on 1st October 2023).
- Zhang, J.; Liu, H.; Luo, S.; Lu, Z.; Chávez-Badiola, A.; Liu, Z.; Yang, M.; Merhi, Z.; Silber, S.J.; Munné, S.; et al. Live birth derived from oocyte spindle transfer to prevent mitochondrial disease. Reprod Biomed Online 2017, 34, 361-368. [CrossRef]
- Quintana, A.; Zanella, S.; Koch, H.; Kruse, S.E.; Lee, D.; Ramirez, J.M.; Palmiter, R.D. Fatal breathing dysfunction in a mouse model of Leigh syndrome. J Clin Invest 2012, 122, 2359-2368. [CrossRef]
- Hanaford, A.R.; Cho, Y.J.; Nakai, H. AAV-vector based gene therapy for mitochondrial disease: progress and future perspectives. Orphanet J Rare Dis 2022, 17, 217. [CrossRef]
- Nakai, R.; Varnum, S.; Field, R.L.; Shi, H.; Giwa, R.; Jia, W.; Krysa, S.J.; Cohen, E.F.; Borcherding, N.; Saneto, R.P.; et al. Mitochondria transfer-based therapies reduce the morbidity and mortality of Leigh syndrome. Nat Metab 2024, 6, 1886-1896. [CrossRef]
- Henke, M.T.; Prigione, A.; Schuelke, M. Disease models of Leigh syndrome: From yeast to organoids. J Inherit Metab Dis 2024, 47, 1292-1321. [CrossRef]
- Lee, C.F.; Caudal, A.; Abell, L.; Nagana Gowda, G.A.; Tian, R. Targeting NAD+ Metabolism as Interventions for Mitochondrial Disease. Scientific Reports 2019, 9, 3073. [CrossRef]
- Johnson, S.C.; Yanos, M.E.; Kayser, E.-B.; Quintana, A.; Sangesland, M.; Castanza, A.; Uhde, L.; Hui, J.; Wall, V.Z.; Gagnidze, A.; et al. mTOR Inhibition Alleviates Mitochondrial Disease in a Mouse Model of Leigh Syndrome. Science 2013, 342, 1524-1528, doi:doi:10.1126/science.1244360.
- Felici, R.; Cavone, L.; Lapucci, A.; Guasti, D.; Bani, D.; Chiarugi, A. PARP Inhibition Delays Progression of Mitochondrial Encephalopathy in Mice. Neurotherapeutics 2014, 11, 651-664. [CrossRef]
- Martin-Perez, M.; Grillo, A.S.; Ito, T.K.; Valente, A.S.; Han, J.; Entwisle, S.W.; Huang, H.Z.; Kim, D.; Yajima, M.; Kaeberlein, M.; et al. PKC downregulation upon rapamycin treatment attenuates mitochondrial disease. Nature Metabolism 2020, 2, 1472-1481. [CrossRef]
- Jain, I.H.; Zazzeron, L.; Goli, R.; Alexa, K.; Schatzman-Bone, S.; Dhillon, H.; Goldberger, O.; Peng, J.; Shalem, O.; Sanjana, N.E.; et al. Hypoxia as a therapy for mitochondrial disease. Science 2016, 352, 54-61, doi:doi:10.1126/science.aad9642.
- Ferrari, M.; Jain, I.H.; Goldberger, O.; Rezoagli, E.; Thoonen, R.; Cheng, K.-H.; Sosnovik, D.E.; Scherrer-Crosbie, M.; Mootha, V.K.; Zapol, W.M. Hypoxia treatment reverses neurodegenerative disease in a mouse model of Leigh syndrome. Proceedings of the National Academy of Sciences 2017, 114, E4241-E4250, doi:doi:10.1073/pnas.1621511114.
- Walker, M.A.; Miranda, M.; Allred, A.; Mootha, V.K. On the dynamic and even reversible nature of Leigh syndrome: Lessons from human imaging and mouse models. Current Opinion in Neurobiology 2022, 72, 80-90. [CrossRef]
- Jain, I.H.; Zazzeron, L.; Goldberger, O.; Marutani, E.; Wojtkiewicz, G.R.; Ast, T.; Wang, H.; Schleifer, G.; Stepanova, A.; Brepoels, K.; et al. Leigh Syndrome Mouse Model Can Be Rescued by Interventions that Normalize Brain Hyperoxia, but Not HIF Activation. Cell Metabolism 2019, 30, 824-832.e823. [CrossRef]
- Barrientos, A.; Korr, D.; Tzagoloff, A. Shy1p is necessary for full expression of mitochondrial COX1 in the yeast model of Leigh's syndrome. Embo j 2002, 21, 43-52. [CrossRef]
- Haroon, S.; Yoon, H.; Seiler, C.; Osei-Frimpong, B.; He, J.; Nair, R.M.; Mathew, N.D.; Burg, L.; Kose, M.; Venkata, C.R.M.; et al. N-acetylcysteine and cysteamine bitartrate prevent azide-induced neuromuscular decompensation by restoring glutathione balance in two novel surf1-/- zebrafish deletion models of Leigh syndrome. Hum Mol Genet 2023, 32, 1988-2004. [CrossRef]
- Burman, J.L.; Itsara, L.S.; Kayser, E.B.; Suthammarak, W.; Wang, A.M.; Kaeberlein, M.; Sedensky, M.M.; Morgan, P.G.; Pallanck, L.J. A Drosophila model of mitochondrial disease caused by a complex I mutation that uncouples proton pumping from electron transfer. Dis Model Mech 2014, 7, 1165-1174. [CrossRef]
- Wang, A.; Mouser, J.; Pitt, J.; Promislow, D.; Kaeberlein, M. Rapamycin enhances survival in a Drosophila model of mitochondrial disease. Oncotarget 2016, 7, 80131-80139. [CrossRef]
- Olufs, Z.P.G.; Ganetzky, B.; Wassarman, D.A.; Perouansky, M. Mitochondrial Complex I Mutations Predispose Drosophila to Isoflurane Neurotoxicity. Anesthesiology 2020, 133, 839-851. [CrossRef]
- Borchardt, L.A.; Scharenbrock, A.R.; Olufs, Z.P.G.; Wassarman, D.A.; Perouansky, M. Mutations in Complex I of the Mitochondrial Electron-Transport Chain Sensitize the Fruit Fly (Drosophila melanogaster) to Ether and Non-Ether Volatile Anesthetics. Int J Mol Sci 2023, 24. [CrossRef]
- Wojtala, A.; Karkucinska-Wieckowska, A.; Sardao, V.A.; Szczepanowska, J.; Kowalski, P.; Pronicki, M.; Duszynski, J.; Wieckowski, M.R. Modulation of mitochondrial dysfunction-related oxidative stress in fibroblasts of patients with Leigh syndrome by inhibition of prooxidative p66Shc pathway. Mitochondrion 2017, 37, 62-79. [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663-676. [CrossRef]
- Lorenz, C.; Lesimple, P.; Bukowiecki, R.; Zink, A.; Inak, G.; Mlody, B.; Singh, M.; Semtner, M.; Mah, N.; Auré, K.; et al. Human iPSC-Derived Neural Progenitors Are an Effective Drug Discovery Model for Neurological mtDNA Disorders. Cell Stem Cell 2017, 20, 659-674.e659. [CrossRef]
- Prigione, A.; Viscomi, C. How to mitigate neurological and cardiac decompensation in Leigh syndrome: Can nicotinamide riboside be an answer? Clinical and Translational Discovery 2022, 2, e124. [CrossRef]
- Bieganowski, P.; Brenner, C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell 2004, 117, 495-502. [CrossRef]
- Price, N.L.; Gomes, A.P.; Ling, A.J.; Duarte, F.V.; Martin-Montalvo, A.; North, B.J.; Agarwal, B.; Ye, L.; Ramadori, G.; Teodoro, J.S.; et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab 2012, 15, 675-690. [CrossRef]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 2005, 434, 113-118. [CrossRef]
- Bai, P.; Cantó, C.; Oudart, H.; Brunyánszki, A.; Cen, Y.; Thomas, C.; Yamamoto, H.; Huber, A.; Kiss, B.; Houtkooper, R.H.; et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab 2011, 13, 461-468. [CrossRef]
- Cantó, C.; Menzies, K.J.; Auwerx, J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab 2015, 22, 31-53. [CrossRef]
- Martin-Perez, M.; Grillo, A.S.; Ito, T.K.; Valente, A.S.; Han, J.; Entwisle, S.W.; Huang, H.Z.; Kim, D.; Yajima, M.; Kaeberlein, M.; et al. PKC downregulation upon rapamycin treatment attenuates mitochondrial disease. Nat Metab 2020, 2, 1472-1481. [CrossRef]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: one drug, many effects. Cell Metab 2014, 19, 373-379. [CrossRef]
- Johnson, S.C.; Yanos, M.E.; Kayser, E.B.; Quintana, A.; Sangesland, M.; Castanza, A.; Uhde, L.; Hui, J.; Wall, V.Z.; Gagnidze, A.; et al. mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science 2013, 342, 1524-1528. [CrossRef]
- Johnson, S.C. Translational Medicine. A target for pharmacological intervention in an untreatable human disease. Science 2014, 346, 1192. [CrossRef]
- Geach, T. Neurometabolic disease: Treating mitochondrial diseases with mTOR inhibitors--a potential treatment for Leigh syndrome? Nat Rev Neurol 2014, 10, 2. [CrossRef]
- Puighermanal, E.; Luna-Sánchez, M.; Gella, A.; van der Walt, G.; Urpi, A.; Royo, M.; Tena-Morraja, P.; Appiah, I.; de Donato, M.H.; Menardy, F.; et al. Cannabidiol ameliorates mitochondrial disease via PPARγ activation in preclinical models. Nature Communications 2024, 15, 7730. [CrossRef]
- Ling, Q.; Rioux, M.; Hu, Y.; Lee, M.; Gray, S.J. Adeno-associated viral vector serotype 9-based gene replacement therapy for SURF1-related Leigh syndrome. Mol Ther Methods Clin Dev 2021, 23, 158-168. [CrossRef]
Figure 1.
Clinical characteristics of Leigh syndrome. Predominantly impacting the brain, muscles, and eyes, the most common clinical features are highlighted. Additionally, abnormalities extend to the cardiovascular, gastrointestinal, renal, and hematological systems. The origin of Leigh syndrome lies in pathogenic mutations found in either nuclear DNA (nDNA) or mitochondrial DNA (mtDNA), leading to disruptions in the oxidative phosphorylation (OXPHOS) capabilities of the mitochondria.
Figure 1.
Clinical characteristics of Leigh syndrome. Predominantly impacting the brain, muscles, and eyes, the most common clinical features are highlighted. Additionally, abnormalities extend to the cardiovascular, gastrointestinal, renal, and hematological systems. The origin of Leigh syndrome lies in pathogenic mutations found in either nuclear DNA (nDNA) or mitochondrial DNA (mtDNA), leading to disruptions in the oxidative phosphorylation (OXPHOS) capabilities of the mitochondria.
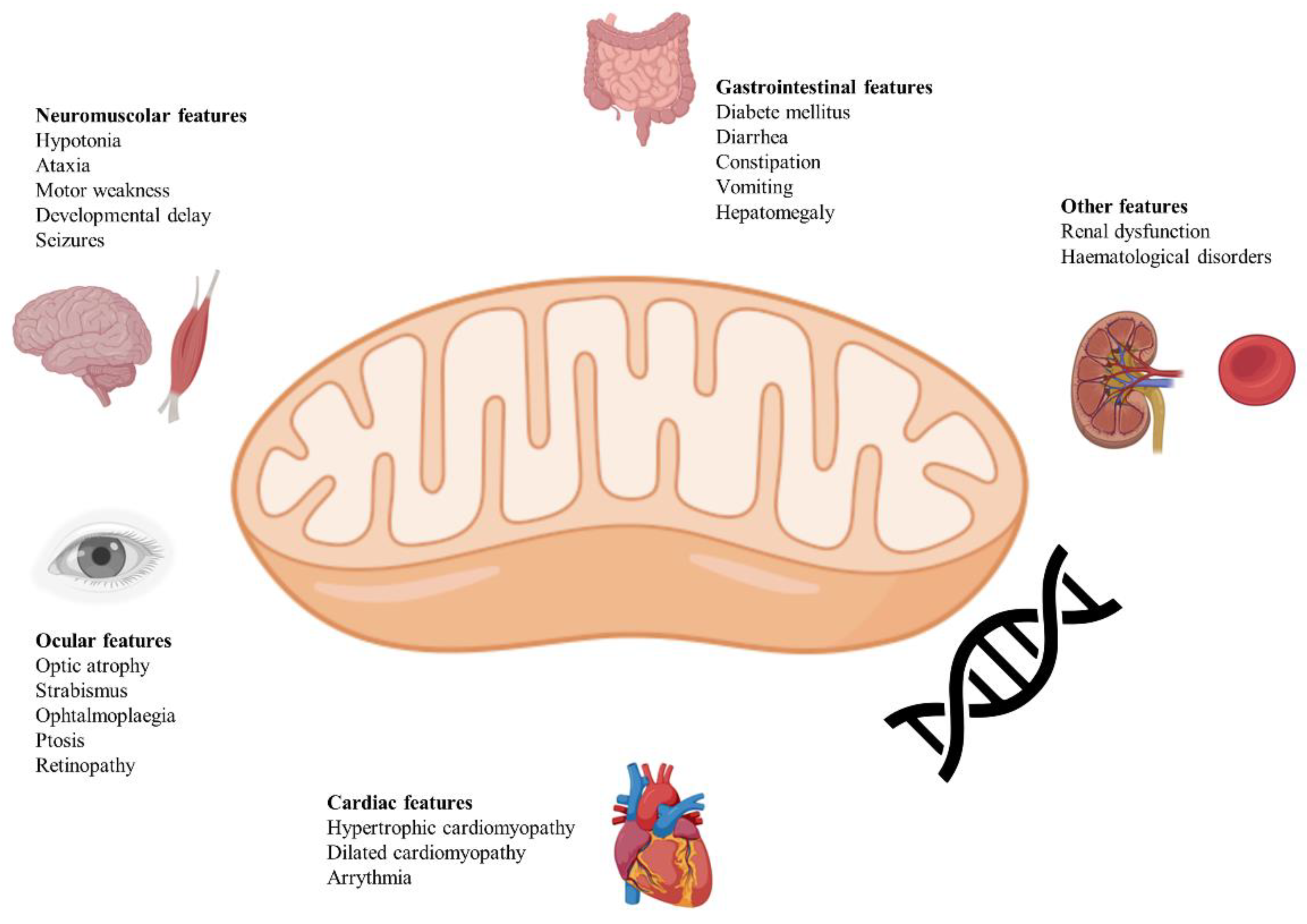

Table 1.
Summary of genes involved in the Leigh Syndrome (LS).
| Inheritance | ||||
| Autosomal Recessive/(Maternal/sporadic) | Genetic mutations | |||
| Complex I | NDUFS1, NDUFS2, NDUFS3, NDUFS4, NDUFS7, NDUFS8, NDUFV1, NDUFV2, NDUFA2, NDUFA9, NDUFA10, NDUFA12, NDUFA13, NDUFAF2, NDUFAF4, NDUFAF5, NDUFAF6, FOXRED1, NUBPL, NDUFAF8, TIMMDC1, NDUFB8, NDUFC2(MT-ND1, MT-ND2, MT-ND3, MT-ND4, MT-ND5,MT-ND6) | |||
| Complex II | SDHA, SDHAF1* | |||
| Complex III | UQCRQ, TTC19, BCS1L | |||
| Complex IV | SURF1, NDUFA4, COX4I1, COX8A, COX10, COX15, SCO2, LRPPRC, TACO1, PET100, PET117(MT-CO1, MT-CO2, MT-CO3) | |||
| Complex V | ATP5MD (MT-ATP6) | |||
| mitochondrial DNA maintenance | POLG*, SUCLA2, SUCLG1, FBXL4, SLC25A4, SSBP1, RNASEH1, GTPBP3 | |||
| mitochondrial gene expression | TRMU, GTPBP3, MTFMT, EARS2, FARS2, IARS2, NARS2, PTCD3, MRPS34, GFM1, GFM2, TSFM, MTRFR, PNPT1, C12ORF65, TARS2(MT-TI, MT-TK, MT-TL1, MT-TL2, MT-TV, MT-TW) | |||
| mitochondrial cofactor | PDSS2, COQ9, LIAS, LIPT1, MECR | |||
| mitochondrial membrane | SERAC1, MFF, SLC25A46, CLPB | |||
| mitochondrial toxicity | HIBCH, ECHS1, ETHE1*, SQOR, SLC39A8, NAXE | |||
| mitochondrial (other) | LONP1, VPS13D, OPA1 | |||
| pyruvate dehydrogenase complex | PDHB, DLAT, DLD, PDHX | |||
| B vitamin transport and metabolism | SLC25A19, TPK1, BTD, SLC19A3 | |||
| miscellaneous | HPDL, ADAR, NUP62, RANBP2, MORC2 | |||
| Autosomal dominant | DNM1L | |||
| X-linked | PDHA1*, NDUFA1, AIFM1* | |||
*These mutations include syndromes that may resemble Leigh syndrome but show a different pathology. Ethylmalonic encephalopathy (due to mutations of ETHE1) has a different, complex neuropathology evidenced by both autoptic and MRI examination. POLG mutations are associated with a variety of manifestations, in children in particular by Alpers-Huttenlocher disease or, later, by Spinocerebellar ataxia and epilepsy. Therefore these mutations are not usually included in the Leigh-like syndrome spectrum. Defects in PDHA can also appear as Leigh syndrome at MRI, but usually, they are associated with other lesions, for instance micropore accumulation, especially in the frontal region.
Table 2.
Proposed therapies and target mutation/pathways. *These mutations include syndromes that may resemble Leigh syndrome but show a different pathology: ACAD9 mutations are associated with a non-Leigh complex encephalopathy and often cardiomyopathy.
Table 2.
Proposed therapies and target mutation/pathways. *These mutations include syndromes that may resemble Leigh syndrome but show a different pathology: ACAD9 mutations are associated with a non-Leigh complex encephalopathy and often cardiomyopathy.
| Treatment | Specific mutations or deficiencies reported |
|---|---|
| Coenzyme Q10 | ND3-m.10197 G>A, Succinate: cytochrome c oxidoreductase deficiency, m.9185 T > C, m.10191 T > C, PDSS2/CoQ10 deficiency |
| EPI-743 | ND1-G3697A, SUCLA2, ETHE1*, ND5-G13513A, EARS2, SURF1, ND1-G3697A, ND6-T14487C |
| Idebenone | Unknown |
| KH176 | Mitochondrial m.3243A>G Spectrum Disorders |
| Riboflavin | ACAD9*- c.1240C>T |
| Thiamine | SLC19A3/Thiamine transporter-2 deficiency |
| Thiamine+Biotin+ CoenzymeQ10 +Vitamin E+ Vitamin C+Carnitine | SLC19A3/Thiamine transporter-2 deficiency |
| Sodium Pyruvate | Unknown, m.8993 T>G, m.9176 T>C |
| Sodium Dichloroacetate | ATP6-m.8993 T>C, ATP6-m.8993 T>G |
| N-acetylcysteine | ETHE1* |
| Carnitine | m.8993T>C mutation |
| Ketogenic diet | PDHc deficiency, NDUFV1 |
| Plasmapheresis + IVIG | ATP6-m.9176 T>C |
| Preclinical studies | |
| Targeted mechanism | |
| Rapamycin | Unknown, mTOR pathway |
| Hypoxia | Oxygen restriction |
| AVV gene therapy | Insertion of a target gene |
| Olaparib, veliparib | PARP inhibitors (PARPis) |
| Nicotinamide riboside and nicotinamide mononucleotide | (NAD+ precursors) enhancing SIRT1 function |
| AD4 | Lipid metabolism |
| Hispidin | MTND |
| Avanafil | MT-APT6 mutation, membrane potential |
Adapted from: Chen L, Cui Y, Jiang D, Ma CY, Tse HF, Hwu WL, Lian Q. Management of Leigh syndrome: Current status and new insights. Clin Genet. 2018 Jun;93(6):1131-1140.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



