
Submitted:
13 February 2025
Posted:
13 February 2025
You are already at the latest version
Abstract
Despite enormous progress in the development of therapeutic agents for persons with hemophilia A and B (HA, HB), several unmet needs persist. These are disease- and treatment-related. Prophylaxis with clotting factor replacement is the gold standard but not feasible in HA and HB with inhibitors. Whereas persons with HA with inhibitors can receive prophylaxis with a factor-mimicking agent, emicizumab, there is no recommendation on the agents to use as prophylaxis in persons with HB with inhibitors, as there are no available molecules. Concizumab is a novel, subcutaneous prophylaxis option in persons with HA or HB with inhibitors that can potentially improve long-term outcomes. Here, we review the available data on concizumab and discuss its possible positioning in the armamentarium to treat hemophilia with inhibitors.
Keywords:
concizumab
; hemophilia A
; hemophilia B
; inhibitors
; non-replacement therapy
; rebalancing agents
; tissue factor pathway inhibitor
; unmet needs
1. Introduction
Hemophilia A and B (HA, HB) are rare, congenital recessive chromosome X-linked bleeding disorders in which clotting factor VIII (FVIII) and IX (FIX), respectively, are lacking or deficient [1]. Both factors are part of the intrinsic coagulation pathway involved in the amplification phase of clotting [2]. FVIII and FIX deficiency compromises factor X (FX) activation, leading to suboptimal thrombin generation and inadequate strength of early clot [3]. Thus, especially in the absence of treatment, persons with severe forms of HA and HB suffer from spontaneous bleeds into major joints and intramuscular bleeds and possible intracranial hemorrhages with consequent comorbidities, whereas persons with moderate or mild disease may experience post-traumatic or surgery-related bleeds [1]. Joint bleeds are a serious complication of hemophilia leading to disability [4].
Replacement of the deficient clotting factor continues to be the gold standard of therapy for hemophilia [1,5,6]. Replacement therapy can be delivered through prophylaxis or ‘on-demand’. Prophylaxis aims at raising background levels of circulating coagulation factors to prevent spontaneous bleeds and results in better outcomes than treatment on-demand [6]. Treatment-related unmet needs remain. Recently, there has been growing interest in developing treatment strategies that do not involve deficient clotting factors. Accordingly, several non-replacement approaches are being explored. These include factor mimetics or rebalancing agents, such as antithrombin inhibitors, activated protein C inhibitors, and tissue factor pathway inhibitor (TFPI) inhibitors[7].
Concizumab is a monoclonal antibody that inhibits TFPI, and was the first drug of its class to be approved by the regulatory agency in Canada [8]. Late in 2023, concizumab made it onto the list of “antibodies to watch in 2024”, an annual summary of therapeutic monoclonal antibodies in late stages of clinical development, or regulatory review or those for which a first approval was recently granted [9].
This narrative review aims to summarize the available data on concizumab and map its place in the context of the unmet needs of persons with HA and HB with inhibitors and the existing treatments for the two bleeding disorders.
2. Unmet Needs
The primary unmet need in hemophilia, regardless of whether HA or HB, is the possibility of living a normal life without the constraints of the disease and its treatments [10]. None of the currently available therapies can ensure a fully normal life for persons with hemophilia. Although prophylaxis with factor replacement therapy is the standard of care, it has its limitations. Replacement therapies have been linked to an important treatment burden due to the frequency of intravenous infusions needed (partially overcome by the development of extended half-life products), the need for intravenous access, treatment-related pain, lengthy infusions, fridge storage, and the logistics of organizing everyday life and work around the injections, to name just a few issues [5,11,12]. When treated with replacement factor concentrates, some patients develop neutralizing antibodies (inhibitors) against exogenous clotting factors. Such inhibitor development occurs in up to 30% of persons with HA and up to 10% of persons with HB [1,6,13,14,15,16,17], and results in inactivation of infused factors, difficult management of bleeding episodes, and a greater disease burden [6,18,19,20,21].
Persons with HA and HB with inhibitors are the two populations for which few therapeutic options are available. For persons with HA with inhibitors, there is an unmet need for treatment options that prevent bleeds to be used concomitantly with on-demand treatments without additional safety concerns. Activated prothrombin complex concentrate (aPCC) and emicizumab are the only approved drugs for prophylaxis in the setting of HA with inhibitors [6,22,23]. However, the greatest and most urgent need for novel treatments is for persons with HB with inhibitors [24]. Despite extensive evidence on the general benefits of prophylaxis, the World Federation of Hemophilia has no recommendations for persons with HB with inhibitors [6]. Immune tolerance induction is possible but difficult and costly, and it fails in up to one-third of individuals [17,25,26]. In fact, persons with HB with inhibitors may have anaphylactic reactions to products containing FIX [6]. The development of inhibitors is also an exclusion criterion for gene therapy eligibility [27,28]. Gene therapy has the potential to transform treatment, but it is not available for all persons with hemophilia. Besides persons with HA and HB with inhibitors, individuals with pre-existing immunity against viral vectors used for gene delivery are also currently ineligible for gene therapy [29]. Moreover, there are uncertainties concerning: the duration of transgene expression, with factor expression declining over time [28,30], and the long-term safety of such approaches, particularly regarding liver toxicity and genotoxicity [28,31].
The limitations of traditional factor replacement therapy led to research into non-replacement treatments, taking advantage of varied mechanisms of action [32]. Emicizumab was the first non-replacement treatment approved by regulatory agencies worldwide for routine prophylaxis in persons with HA with inhibitors and persons with moderate-to-severe HA without inhibitors [33,34]. Emicizumab is a bispecific monoclonal antibody that bridges FIXa and FXa to restore hemostatic function. Its development filled the gap in therapy for persons with HA with inhibitors, but emicizumab cannot be used in persons with HB with inhibitors. Although long-term study outcomes confirmed low annualized bleeding rates (ABRs) and no new safety signals, real-world data reported a variable incidence of bleeding episodes, some severe, in persons with HA on emicizumab prevention [35,36,37,38,39]. Although emicizumab has minimal immunogenicity, several cases of neutralizing anti-emicizumab antibodies in persons with HA without FVIII inhibitors have been described [40]. Taken together, these data show that there are still unmet needs in persons with hemophilia with inhibitors. Hemostatic rebalancing agents may be able to meet these needs.
3. Rationale for Inhibiting TFPI
Hemostasis is a complex process that ensures blood flow and prevents losses after injury [41,42]. For correct functioning of the hemostatic system, there must be a balance between natural procoagulants and anticoagulants; any imbalance can lead to either pathologic bleeding or thrombosis [43]. Natural procoagulants are the clotting factors, whereas anticoagulants include antithrombin, TFPI, heparin cofactor II, protease nexin 1, Z-dependent protease inhibitor, and activated protein C [3,43]. Synergistic modulation of the tendency to bleed by anticoagulant and fibrinolytic factors was described in persons with hemophilia, in whom deficiency of those factors was associated with a milder bleeding phenotype [44]. Inhibition of natural anticoagulants can restore hemostasis in patients with bleeding disorders [45].
Among natural anticoagulants, TFPI, a multivalent Kunitz-type proteinase with inhibitor, prevents unrestricted amplification of the clotting cascade. It inhibits not only FXa, but also activated factor VII (FVIIa), thereby avoiding FXa generation by the extrinsic clotting pathway [46]. TFPI inhibition allows for sustained thrombin generation, despite FVIII or FIX deficiency in persons with HA/HB [3]. Thrombin generation is crucial for blood coagulation: it converts fibrinogen to fibrin to form a clot. Thrombin is generated by FXa, which, in turn, is activated either by the tissue factor/FVIIa complex or by the complex composed of activated factors FVIII/FIX, the latter being two factors missing or deficient in persons with HA/HB [3]. Without the amplification phase, the attenuation of TFPI inhibition can restore thrombin generation [47]. The idea of deploying TFPI inhibition in the treatment of HA and HB dates back to 1991 [48].
4. Concizumab: Mechanism of Action
Concizumab is a humanized monoclonal immunoglobulin G4 antibody against TFPI that binds to the Kunitz-2 domain of TFPI and prevents TFPI from directly binding to FXa and from indirectly binding to the tissue factor/FVIIa complex (Figure 1); the result is increased thrombin generation through the extrinsic pathway [49]. Concizumab binds to both soluble and membrane-bound TFPI with high affinity, contributing to non-linear pharmacokinetics (i.e., target-mediated drug disposition [TMDD]) [50]. TMDD, which can be considered a consequence of pharmacodynamics affecting pharmacokinetics [51], describes a situation in which a large proportion of an administered drug dose binds with high affinity to its target, contributing to significant drug elimination until saturation of target binding. At low drug concentrations, the administration of increasing doses is associated with an apparent decrease in steady-state volume of distribution until the target is saturated [48]. The impact of TMDD on the pharmacokinetics of concizumab was studied in Cynomolgus monkeys and showed that terminal half-life depended on plasma concentration [52]. Phase 1 studies confirmed that concizumab half-life ranged from 31.1 to 74.2 h and depended on plasma concentration and route of administration [50]. At steady state, following multiple subcutaneous injections, the estimated half-life is approximately 38 h [53]. Such a short half-life is an advantage when discontinuation of concizumab administration is needed, as a quick washout is attained.
The hemostatic effect of concizumab was first confirmed in vitro and in a rabbit model of hemophilia [49], and later, in the first human trial [50]. In all trials, concizumab plasma concentration correlated with increased thrombin generation and elevated levels of fibrin D-dimer and prothrombin fragments 1+2. Given that fibrin D-dimer and prothrombin fragments 1+2 are biomarkers of coagulation activation [54,55], their elevated levels proved the hemostatic effect of concizumab [50,56,57].
Mechanistically, concizumab does not affect downstream regulation of coagulation and can be combined with treatments for breakthrough bleeds [58,59]. Such bleeds in persons with HA or HB on concizumab prophylaxis can be treated with bypassing agents (e.g., aPCC, recombinant FVIIa [rFVIIa]), or with recombinant FVIII (rFVIII) or FIX (rFIX) depending on each patient’s inhibitor status. In the presence of concizumab, rFVIIa, aPCC, rFVIII, and rFIX enhance plasma thrombin generation potential, and available in vitro data support their use to treat mild and moderate breakthrough bleeds in patients on concizumab. Dosing during on-demand treatment needs to be adjusted to ensure patient safety (i.e., the lowest approved dose, as per label, should be administered) [59]. Concizumab has no antidote, but it has the advantage of a quick washout [60].
The product characteristics of concizumab are summarized in Table 1.
5. Efficacy and Safety of Concizumab in Persons with HA or HB with Inhibitors
Concizumab efficacy in persons with HA or HB with inhibitors was evaluated in the explorer4 and 7 trials [56,57,62]. The explorer4 trial was a successful, phase 2, proof-of-concept trial in persons with HA or HB with inhibitors [48,62]. The primary objective of explorer4 was to assess the efficacy of once-daily concizumab in preventing bleeding episodes in persons with HA or HB with inhibitors; efficacy was evaluated as the number of bleeding episodes during at least 24 weeks from treatment initiation. Starting with a maintenance dose of 0.15 mg/kg, the dose was escalated to 0.20 mg/kg and then to 0.25 mg/kg in patients who had ≥3 treatment-requiring spontaneous bleeding episodes within the 12 weeks before concizumab treatment, during both the main and extension study parts [62]. The estimated ABR during the main and extension study parts at the last dose level was 4.8 (95% confidence interval [CI]: 3.2–7.2; median ABR 3.6), and for spontaneous bleeds was 1.8 (95% CI: 1.2–2.6). Importantly, switching from no prophylaxis in the main study part to prophylaxis with concizumab in the extension part decreased the estimated ABR from 18.6 (95% CI: 12.9–26.9) to 4.9 (95% CI: 2.2–10.6) [57].
explorer7 was designed as a phase 3 safety and efficacy trial in persons with HA or HB with inhibitors [56]. The primary endpoint compared the number of treated spontaneous and traumatic bleeding episodes in group 1 (no prophylaxis for at least 24 weeks) with the number in group 2 (prophylaxis with concizumab for at least 32 weeks) [56]. The estimated mean ABR ratio for treated spontaneous and traumatic bleeding episodes between group 1 and group 2 was 0.14 (95% CI: 0.07–0.29) confirming the superiority of concizumab prophylaxis over no prophylaxis. The median ABR was 9.8 (interquartile range [IQR] 6.5–20.2) episodes in group 1 (i.e., estimated mean ABR 11.8; 95% CI: 7.0–19.9), and 0.0 (IQR 0.0–3.3) episodes in group 2 (i.e., estimated mean ABR 1.7; 95% CI: 1.0–2.9). The overall median ABR for patients receiving concizumab (groups 2, 3, and 4) was 0.0 (IQR 0.0–3.3) episodes [56].
Joint health outcomes are an important aspect of any treatment modality used in persons with hemophilia. The explorer4 and 7 trials demonstrated good protection against joint bleeds for persons with HA or HB with inhibitors [56,57,62]. In explorer4, a joint ABR of 2.7 (95% CI: 1.6–4.6) was observed. Moreover, the estimated mean joint ABR decreased from 13.8 (95% CI: 9.6–19.9) to 2.9 (95% CI: 1.1–7.7) in patients switched from no prophylaxis to prophylaxis with concizumab [57]. explorer7 further confirmed that concizumab prophylaxis helped joint health, as the median ABR for joint bleeding episodes was higher in group 1 than group 2 (6.5 [IQR 3.2–13.1] vs. 0.0 [IQR 0.0–2.6]); the median ABR for target joint bleeding episodes was 0.0 (IQR 0.0–2.2) in group 1 versus 0.0 (IQR 0.0–0.0) in group 2 [56]. Also, concizumab prophylaxis resolved 91.8% of target joints (i.e., joints with recurrent bleeding) in persons with HA or HB with inhibitors, usually within 12 months. The median ABR for treated spontaneous and traumatic target joint bleeding episodes at 56 weeks in persons with HA or HB with inhibitors was 0.0 [63].
Equally important is the performance of bleeding prophylaxis in persons with hemophilia undergoing scheduled surgery. Results from phase 3 trials showed that minor surgeries could be performed in patients receiving concizumab prophylaxis. A total of 11% of patients (30/278) in those trials had minor surgical, mostly dental, procedures. Surgery- related bleeding occurred in 24 patients: most bleeding episodes were mild or moderate, and 17 episodes were treated in 15 patients [64].
Safety was the primary endpoint in the explorer1, 2, and 3 trials,, during which adverse event (AE) analysis confirmed the safety of concizumab: all reported AEs were mild, and there were no serious AEs [50,65,66]. In the proof-of-concept trials, during both the main and extension parts, most AEs were mild, with no deaths, no events leading to withdrawal, and no thromboembolic events [57,62]. There was, however, a serious safety issue that led to a pause of the explorer7 study. Three patients on concizumab, including one patient from the explorer7 trial, and all with thrombotic risk factors at baseline, experienced nonfatal thromboembolic events, which included a renal infarct in one patient. All three patients were receiving concomitant hemostatic medication before or on the day of their thromboembolic event and two patients were at the higher end of the concizumab exposure range in phase 3 trials [67]. Following careful analysis of the available data from phase 2 and 3 trials, a risk-mitigation strategy was implemented and consisted of guidelines on the concomitant use of hemostatic agents in the treatment of bleeding episodes while on concizumab prophylaxis, and updated concizumab dosing (i.e., the daily maintenance dose was lowered from 0.25 to 0.20 mg/kg, with dose adjustment based on concizumab plasma concentrations within the initial 5–8 weeks of treatment). The explorer7 study was later resumed with an amended protocol, and no thromboembolic events were observed after implementation of the risk-mitigation strategy [56]. Post-authorization pharmacovigilance is also very important in hemophilia research [68]. Such efforts should be made for all available treatments, and for products in late-stage clinical development, as in future, these efforts will help associate patient profiles with the most appropriate treatments.
A summary of results from the explorer clinical development program, with a focus on persons with HA or HB with inhibitors, is shown in Table 2.
6. Adherence to Treatment
Despite the undeniable benefits of prophylaxis with replacement factors, adherence to this treatment modality has been a significant problem [6]. The barriers to adherence include factors that are patient-related, condition-related, and treatment-related (including intravenous administration), and socioeconomic and healthcare-system factors. Strategies to improve adherence involve education, monitoring and reminder systems, and incentives [69]. It is expected that less burdensome non-replacement treatments will improve adherence [6].
Concizumab prophylaxis is delivered as a daily subcutaneous injection. In a Canadian study of time trade-off utilities, prophylaxis delivered subcutaneously was associated with higher utility values than intravenous prophylaxis or on-demand treatment [70]. In chronic diseases, once-daily treatment was linked to higher adherence rates than treatment schemes requiring more frequent medicine administration [71]. Infrequent bleeds constitute an important barrier to adherence; in fact, medication adherence may diminish over time when patients become asymptomatic [69]. Daily injections may help the development of a routine. Providing patients with device training and concurrent teaching/development of routines (i.e., anchoring the when, where, and how of the injection) or rituals (e.g., dimming lights, using the same chair) may be useful to reduce anxiety and improve adherence to injection therapy, as shown by a study on self-injections of biologic therapy [72].
Therapy with concizumab is delivered through a pre-filled multidose pen, much like the pens used by persons with diabetes using insulin, to be self-administered or injected by a caregiver [8]. Although unlikely erroneous intramuscular administration is a possibility, especially in thin individuals, with adequate training, patients or their carers easily learn how to perform the injections [53]. In a study on patient and caregiver preference for the pre-filled concizumab pen over current systems to deliver prophylaxis (current systems included intravenous factor replacement in 41 individuals and subcutaneous emicizumab prophylaxis in 39), 98% of participants considered the pre-filled pen ‘easy’ or ‘very easy’ to use. The pen gave participants high confidence in a full dose being delivered correctly. Overall, 88% of participants preferred the pre-filled pen to the system they had been using [73]. Subcutaneous drug administration was preferred to intravenous delivery in a systematic review that included different drugs and patients. Such preference was determined by time saving and the possibility of home treatment [74]. Pre-filled pens were preferred over pre-filled syringes by patients with ulcerative colitis and by parents of children with juvenile idiopathic arthritis [75,76]. In addition, the precise weight-based dosing of concizumab, with its specialized pen, will avoid the product wastage encountered with emicizumab [77].
Choosing the optimal treatment should be patient-oriented and based on shared decision making between patients and physicians. Such an approach has a good chance of improving adherence [78]. In the explorer7 trial, 93% of patients (77/83) who responded to the Hemophilia-Patient Preference Questionnaire preferred concizumab over their previous treatment, 6% (5/77) expressed no preference, and 1% (1/77) had a preference for the previous treatment [56].
7. Outcome Measures
The development of novel therapies has markedly reduced the burden of hemophilia and its treatment. However, novel outcome measures are needed, and potential outcome measures in hemophilia have been reviewed [79]. To evaluate the outcomes of treatment in persons with a near-normal life expectancy, and the lower disease burden (such as bleeding rates approaching zero) and lower treatment burden (resulting in improved quality of life), objective clinical methodologies should be combined with patient-reported outcomes [79].
Bleeding phenotype, joint health status, physical activity expectations, drug half-life and properties, and capacity to adhere to treatment should guide the optimization of prophylaxis in hemophilia to secure the best outcomes [10,78]. However, joint health assessments with adequate sensitivity and specificity continue to be an unmet need [80]. Incidentally, adults with hemophilia with inhibitors have more severe and rapidly progressing hemophilic arthropathy than those without inhibitors [81]. This underlines the importance of effective joint health assessment in this patient category. Point-of-care ultrasound for joints could help in the early detection of arthropathy and monitor its progression, together with assessing treatment efficacy [80].
Moreover, health outcomes that are important for persons with HA or HB should not be neglected [79]. Knowing what matters to patients is essential for delivering patient-centered care. Patients’ perceptions of joint damage, pain, and the impact of hemophilia and its treatment on daily living can best be gauged using patient-reported outcome measures. One new tool for assessing disease and treatment burden from patients’ perspectives is the Hem-TEM tool [82]. Obtaining good health-related quality of life in hemophilia should be considered a new therapeutic goal [83]. Patient-reported outcomes in the explorer7 study showed improvements favoring concizumab in the health-related quality-of-life domains of ‘feeling’, ‘treatment’, ‘view of yourself’, and ‘sport and leisure’. Improvements were also perceived regarding treatment burden, and patients’ treatment preferences in persons with HA or HB with inhibitors during concizumab prophylaxis [84].
8. Discussion
There has been enormous progress over the years in the management of HA and HB [28]. Persons with HA or HB who were once dying of bleeding, or subsequently of AIDS after transfusion of blood-derived factors, now have near-normal life expectancy [85]. Traditionally, there were limited options for persons with HB with inhibitors [24]. However, with the advent of rebalancing agents, such as concizumab, this gap is now being filled.
Advances like the development of concizumab also provide exciting opportunities to improve the management of persons with HA with inhibitors. Indeed, even though non-factor replacement therapies have significantly improved clinical outcomes in this group, concizumab may represent a further therapeutic option for personalized care, especially in patients unable to attain adequate hemostasis and who still experience bleeding episodes during treatment with currently approved modalities.
Concizumab is effective (improved ABRs and patient-reported outcomes) and delivered in way that patients prefer, which will probably enhance adherence to treatment [56,73]. Furthermore, the explorer clinical development program showed that accurate data analysis and appropriate risk-mitigation and safety measures can ward off the potential for thrombosis seen in earlier trials [56,67].
Additional real-world evidence is now being acquired for concizumab, which is already available in five countries, and it will be extremely important to consider the inclusion of concizumab, together with other innovative therapies, in national and international guidelines on hemophilia management as soon as possible.
Hemophilia is expensive to treat, and action is required to make treatments more widely available. This is particularly true for prophylaxis that is superior to on-demand treatment but also more expensive [6].
In this review, we have discussed the patient-centered approaches that are becoming everyday practice. People affected by hemophilia were once called ‘hemophiliacs’, but are now referred to as ‘persons with HA or HB’. Hemophilia is not a disease but a genetic health condition. Patients’ preferences must be heard and incorporated into clinical decision making.
Where will we be in 5 or 10 years’ time? One could ask whether to concentrate all research efforts in hemophilia management on gene therapy that is hoped to provide an ultimate cure. At present, gene therapy is not available in persons with inhibitors, although some studies showed waning inhibitor titers following liver-directed gene therapy in animal models [86]. There are also ethical implications of gene therapy: e.g., the available products may not yet provide a permanent cure, or the inaccessibility of gene therapy to most persons with hemophilia because of cost [87]. Thus, there is a need (continuing into the foreseeable future) for all available treatments, including concizumab.
Clearly, the management of hemophilia will have to become personalized. As our understanding of genetic and phenotypic variations in hemophilia deepens, treatments will increasingly be tailored to individual patient profiles, needs, and expectations [88]. The ultimate goals in the field are to improve clinical and patient-reported outcomes for persons living with hemophilia, and to provide normal and fulfilling lives for persons with hemophilia worldwide.
9. Conclusions
Because of its unique mechanism of action, concizumab is the first prophylactic treatment approved for persons with HB with inhibitors. It represents a promising therapeutic option for persons with HA with inhibitors who continue to experience bleeds during prophylaxis with emicizumab. Since 2023, concizumab has been approved in Canada, Australia, Japan, and Switzerland, and in France through early-access authorization [53,89,90,91,92]. The development of a non-replacement treatment modality (e.g., concizumab), with a convenient mode of subcutaneous administration using a multi-dose pen, offers a chance at normal life to persons with HA or HB with inhibitors, who have traditionally had a long history of compromised health-related quality of life; moreover, the short half-life and quick washout of concizumab make this therapy a highly adaptable treatment option.
Funding
Editorial assistance was funded by Novo Nordisk S.p.A.
Acknowledgments
The outline and the first and second drafts of this review were written by Alicja M. Gruszka, an independent medical writer on behalf of Springer Healthcare Italia. Medical writing was funded by Novo Nordisk.
Conflicts of Interest
GC received fees for lectures, presentations, speakers’ bureau, or educational events from Bioviiix, Biomarin, CSL Behring, Sanofi, Novo Nordisk, LFB, Roche, SOBI, and Takeda. Participation in Data Safety Monitoring Boards or Advisory Boards for Bayer, CSL Behring, Biomarin, Sanofi, Novo Nordisk, Takeda, LFB, and Roche. VJ-Y has received reimbursement for attending symposia/congresses and/or honoraria for speaking and/or honoraria for consulting, and/or funds for research from Takeda, Bayer, BioMarin, CSL Behring, Grifols, Novo Nordisk, Sobi, Roche, Octapharma, and Pfizer. JM has received research grant/research support from Biomarin, CSL, Novo Nordisk, Pfizer, Roche, Sanofi, Spark, and Vega, and has been a consultant/scientific board member for Biomarin, CSL Behring, Novo Nordisk, Roche, Takeda, Sanofi, SPA Pharma, and Vega; and is a member of the speaker bureau of Novo Nordisk, Pfizer, Roche, Sanofi, and Takeda.
Abbreviations
The following abbreviations are used in this manuscript:
| ABR | Annualized bleeding rate |
| AE | Adverse event |
| aPCC | Activated prothrombin complex concentrate |
| CI | Confidence interval |
| ELISA | Enzyme-linked immunosorbent assay |
| FIX | Factor IX |
| FVIIa | Activated factor VII |
| FVIII | Factor VIII |
| FX | Factor X |
| HA | Hemophilia A |
| HB | Hemophilia B |
| IQR | Interquartile range |
| rFIX | Recombinant factor IX |
| rFVIII | Recombinant factor VIII |
| TF | Tissue factor |
| TFPI | Tissue factor pathway inhibitor |
| TMDD | Target-mediated drug disposition |
References
- Berntorp, E.; Fischer, K.; Hart, D.P.; Mancuso, M.E.; Stephensen, D.; Shapiro, A.D.; Blanchette, V. Haemophilia. Nat Rev Dis Primers 2021, 7, 45. [CrossRef]
- Troisi, R.; Balasco, N.; Autiero, I.; Sica, F.; Vitagliano, L. New insight into the traditional model of the coagulation cascade and its regulation: illustrated review of a three-dimensional view. Res. Pr. Thromb. Haemost. 2023, 7, 102160. [CrossRef]
- van den Berg, H.M.; Srivastava, A. Hemostasis – a balancing act. N Engl J Med 2023, 389, 853-856. [CrossRef]
- Leuci, A.; Dargaud, Y. Blood-Induced Arthropathy: A Major Disabling Complication of Haemophilia. J. Clin. Med. 2023, 13, 225. [CrossRef]
- Nogami, K.; Shima, M. Current and future therapies for haemophilia—Beyond factor replacement therapies. Br. J. Haematol. 2022, 200, 23–34. [CrossRef]
- Srivastava, A.; Santagostino, E.; Dougall, A.; Kitchen, S.; Sutherland, M.; Pipe, S.W.; Carcao, M.; Mahlangu, J.; Ragni, M.V.; Windyga, J.; et al. WFH Guidelines for the Management of Hemophilia, 3rd edition. Haemophilia 2020, 26 (Suppl. 6), 1–158. [CrossRef]
- Swan, D.; Mahlangu, J.; Thachil, J. Non-factor therapies for bleeding disorders: A primer for the general haematologist. eJHaem 2022, 3, 584–595. [CrossRef]
- Keam, S.J. Concizumab: First Approval. Drugs 2023, 83, 1053–1059. [CrossRef]
- Crescioli, S.; Kaplon, H.; Chenoweth, A.; Wang, L.; Visweswaraiah, J.; Reichert, J.M. Antibodies to watch in 2024. mAbs 2024, 16, 2297450. [CrossRef]
- Núñez, R.; Álvarez-Román, M.T.; Bonanad, S.; González-Porras, J.R.; De La Corte-Rodriguez, H.; Berrueco, R.; Jiménez-Yuste, V. The Limitations and Unmet Needs of the Five Cornerstones to Guarantee Lifelong Optimization of Prophylaxis in Hemophilia Patients. TH Open 2022, 06, e365–e377. [CrossRef]
- Ozelo, M.C.; Yamaguti-Hayakawa, G.G. Impact of novel hemophilia therapies around the world. Res. Pr. Thromb. Haemost. 2022, 6, e12695. [CrossRef]
- Tischer, B.; Marino, R.; Napolitano, M. Patient preferences in the treatment of hemophilia A: impact of storage conditions on product choice. Patient Preference Adherence 2018, ume 12, 431–441. [CrossRef]
- Giangrande, P.L.F.; Hermans, C.; O'Mahony, B.; de Kleijn, P.; Bedford, M.; Batorova, A.; Blatny, J.; Jansone, K.; European Haemophilia Consortium (EHC); the European Association for Haemophilia and Allied Disorders. European principles of inhibitor management in patients with haemophilia. Orphanet J Rare Dis 2018, 13, 66. [CrossRef]
- Male, C.; Andersson, N.G.; Rafowicz, A.; Liesner, R.; Kurnik, K.; Fischer, K.; Platokouki, H.; Santagostino, E.; Chambost, H.; Nolan, B.; et al. Inhibitor incidence in an unselected cohort of previously untreated patients with severe haemophilia B: a PedNet study. Haematologica 2020, 106, 123–129. [CrossRef]
- Puetz, J.; Soucie, J.M.; Kempton, C.L.; Monahan, P.E.; Hemophilia Treatment Center Network (HTCN) Investigators Prevalent inhibitors in haemophilia B subjects enrolled in the Universal Data Collection database. Haemophilia 2013, 20, 25–31. [CrossRef]
- Berg, H.M.v.D.; Fischer, K.; Carcao, M.; Chambost, H.; Kenet, G.; Kurnik, K.; Königs, C.; Male, C.; Santagostino, E.; Ljung, R. Timing of inhibitor development in more than 1000 previously untreated patients with severe hemophilia A. Blood 2019, 134, 317–320. [CrossRef]
- Wight, J.; Paisley, S.; Knight, C. Immune tolerance induction in patients with haemophilia A with inhibitors: a systematic review. Haemophilia 2003, 9, 436–463. [CrossRef]
- D'Angiolella, L.S.; Cortesi, P.A.; Rocino, A.; Coppola, A.; Hassan, H.J.; Giampaolo, A.; Solimeno, L.P.; Lafranconi, A.; Micale, M.; Mangano, S.; et al. The socioeconomic burden of patients affected by hemophilia with inhibitors. Eur. J. Haematol. 2018, 101, 435–456. [CrossRef]
- Dolan, G. Partnering to change the world for people with haemophilia: 7th Haemophilia Global Summit, Madrid, Spain 22–24 September 2016. Eur. J. Haematol. 2017, 99, 3–9. [CrossRef]
- Oladapo, A.O.; Lu, M.; Walsh, S.; O’hara, J.; Kauf, T.L. Inhibitor clinical burden of disease: a comparative analysis of the CHESS data. Orphanet J. Rare Dis. 2018, 13, 198. [CrossRef]
- Walsh, C.E.; Soucie, J.M.; Miller, C.H.; United States Hemophilia Treatment Center Network. Impact of inhibitors on hemophilia a mortality in the United States. Am. J. Hematol. 2015, 90, 400–405. [CrossRef]
- Berntorp, E.; Hermans, C.; Solms, A.; Poulsen, L.; Mancuso, M.E. Optimising prophylaxis in haemophilia A: The ups and downs of treatment. Blood Rev. 2021, 50, 100852. [CrossRef]
- Brackmann, H.H.; Schramm, W.; Oldenburg, J.; Cano, V.; Turecek, P.L.; Négrier, C. Origins, Development, Current Challenges and Future Directions with Activated Prothrombin Complex Concentrate for the Treatment of Patients with Congenital Haemophilia with Inhibitors. Hamostaseologie 2020, 40, 606–620. [CrossRef]
- Hermans, C.; Giangrande, P.L.F.; O'Mahony, B.; de Kleijn, P.; Bedford, M.; Batorova, A.; Blatny, J.; Jansone, K.; European Haemophilia, C.; the European Association for, H., et al. European principles of inhibitor management in patients with haemophilia: implications of new treatment options. Orphanet J Rare Dis 2020, 15, 219. [CrossRef]
- Hay, C.R.M.; DiMichele, D.M.; International Immune Tolerance, S. The principal results of the International Immune Tolerance Study: a randomized dose comparison. Blood 2012, 119, 1335–1344. [CrossRef]
- Ljung, R.; Auerswald, G.; Benson, G.; Dolan, G.; Duffy, A.; Hermans, C.; Jimenez-Yuste, V.; Lambert, T.; Morfini, M.; Zupancic-Salek, S., et al. Inhibitors in haemophilia A and B: Management of bleeds, inhibitor eradication and strategies for difficult-to-treat patients. Eur J Haematol 2019, 102, 111-122. [CrossRef]
- Doshi, B.S.; Arruda, V.R. Gene therapy for hemophilia: what does the future hold? Ther Adv Hematol 2018, 9, 273-293. [CrossRef]
- Mancuso, M.E.; Mahlangu, J.N.; Pipe, S.W. The changing treatment landscape in haemophilia: from standard half-life clotting factor concentrates to gene editing. Lancet 2021, 397, 630–640. [CrossRef]
- Miesbach, W.; Klamroth, R.; Oldenburg, J.; Tiede, A. Gene therapy for hemophilia – opportunities and risks. Dtsch Arztebl Int 2022, 119, 887-894. [CrossRef]
- Miesbach, W.; O’mahony, B.; Key, N.S.; Makris, M. How to discuss gene therapy for haemophilia? A patient and physician perspective. Haemophilia 2019, 25, 545–557. [CrossRef]
- Nowrouzi, A.; Penaud-Budloo, M.; Kaeppel, C.; Appelt, U.; Le Guiner, C.; Moullier, P.; von Kalle, C.; O Snyder, R.; Schmidt, M. Integration Frequency and Intermolecular Recombination of rAAV Vectors in Non-human Primate Skeletal Muscle and Liver. Mol. Ther. 2012, 20, 1177–1186. [CrossRef]
- Jiménez-Yuste, V.; Auerswald, G.; Benson, G.; Dolan, G.; Hermans, C.; Lambert, T.; Ljung, R.; Morfini, M.; Santagostino, E.; Šalek, S.Z. Practical considerations for nonfactor-replacement therapies in the treatment of haemophilia with inhibitors. Haemophilia 2021, 27, 340–350. [CrossRef]
- Mahlangu, J.; Iorio, A.; Kenet, G. Emicizumab state-of-the-art update. Haemophilia 2022, 28 Suppl 4, 103-110. [CrossRef]
- European Medicines Agency. Hemlibra. Summary of product characteristics. 2018. Availabe online: https://www.ema.europa.eu/en/documents/product-information/hemlibra-epar-product-information_en.pdf (accessed on 23 May 2024).
- Abbattista, M.; Ciavarella, A.; Noone, D.; Peyvandi, F. Hemorrhagic and thrombotic adverse events associated with emicizumab and extended half-life factor VIII replacement drugs: EudraVigilance data of 2021. J. Thromb. Haemost. 2023, 21, 546–552. [CrossRef]
- Arcudi, S.; Gualtierotti, R.; Scalambrino, E.; Clerici, M.; Hassan, S.; Begnozzi, V.; Boccalandro, E.A.; Novembrino, C.; Valsecchi, C.; Palla, R.; et al. Predictive parameters for spontaneous joint bleeding during emicizumab prophylaxis. Blood Adv. 2024, 8, 2901–2907. [CrossRef]
- Batsuli, G.; Wheeler, A.P.; Weyand, A.C.; Sidonio, R.F., Jr.; Young, G. Severe muscle bleeds in children and young adults with hemophilia A on emicizumab prophylaxis: Real-world retrospective multi-institutional cohort. Am. J. Hematol. 2023, 98, E285–E287. [CrossRef]
- Levy-Mendelovich, S.; Brutman-Barazani, T.; Budnik, I.; Avishai, E.; Barg, A.A.; Levy, T.; Misgav, M.; Livnat, T.; Kenet, G. Real-World Data on Bleeding Patterns of Hemophilia A Patients Treated with Emicizumab. J. Clin. Med. 2021, 10, 4303. [CrossRef]
- Warren, B.B.; Chan, A.; Manco-Johnson, M.; Branchford, B.R.; Buckner, T.W.; Moyer, G.; Gibson, E.; Thornhill, D.; Wang, M.; Ng, C.J. Emicizumab initiation and bleeding outcomes in people with hemophilia A with and without inhibitors: A single-center report. Res. Pract. Thromb. Haemost. 2021, 5, e12571. [CrossRef]
- Kizilocak, H.; Guerrera, M.F.; Young, G. Neutralizing antidrug antibody to emicizumab in patients with severe hemophilia A: Case report of a first noninhibitor patient and review of the literature. Res. Pract. Thromb. Haemost. 2023, 7, 102194. [CrossRef]
- Chaudhry, R.; Usama, S.M.; Babiker, H.M. Physiology, coagulation pathways. In StatPearls, Treasure Island (FL), 2023.
- Zaidi, A.; Green, L. Physiology of haemostasis. Anaesth Intensive Care Med 2022, 23, 111-117.
- Mehic, D.; Colling, M.; Pabinger, I.; Gebhart, J. Natural anticoagulants: A missing link in mild to moderate bleeding tendencies. Haemophilia 2021, 27, 701–709. [CrossRef]
- Shetty, S.; Vora, S.; Kulkarni, B.; Mota, L.; Vijapurkar, M.; Quadros, L.; Ghosh, K. Contribution of natural anticoagulant and fibrinolytic factors in modulating the clinical severity of haemophilia patients. Br. J. Haematol. 2007, 138, 541–544. [CrossRef]
- Lane, D.A. Correcting the hemophilic imbalance. Blood 2017, 129, 10–11. [CrossRef]
- Kato, H. Tissue factor pathway inhibitor; its structure, function and clinical significance. Pol J Pharmacol. 1996, 48, 67–72.
- Chowdary, P. Anti-tissue factor pathway inhibitor (TFPI) therapy: a novel approach to the treatment of haemophilia. Int. J. Hematol. 2018, 111, 42–50. [CrossRef]
- Chowdary, P. Inhibition of Tissue Factor Pathway Inhibitor (TFPI) as a Treatment for Haemophilia: Rationale with Focus on Concizumab. Drugs 2018, 78, 881–890. [CrossRef]
- Hilden, I.; Lauritzen, B.; Sørensen, B.B.; Clausen, J.T.; Jespersgaard, C.; Krogh, B.O.; Bowler, A.N.; Breinholt, J.; Gruhler, A.; Svensson, L.A.; et al. Hemostatic effect of a monoclonal antibody mAb 2021 blocking the interaction between FXa and TFPI in a rabbit hemophilia model. Blood 2012, 119, 5871–5878. [CrossRef]
- Chowdary, P.; Lethagen, S.; Friedrich, U.; Brand, B.; Hay, C.; Karim, F.A.; Klamroth, R.; Knoebl, P.; Laffan, M.; Mahlangu, J.; et al. Safety and pharmacokinetics of anti-TFPI antibody (concizumab) in healthy volunteers and patients with hemophilia: a randomized first human dose trial. J. Thromb. Haemost. 2015, 13, 743–754. [CrossRef]
- An, G. Concept of pharmacologic target-mediated drug disposition in large-molecule and small-molecule compounds. J Clin Pharmacol 2020, 60, 149-163. [CrossRef]
- Agersø, H.; Overgaard, R.V.; Petersen, M.B.; Hansen, L.; Hermit, M.B.; Sørensen, M.H.; Petersen, L.C.; Hilden, I. Pharmacokinetics of an anti-TFPI monoclonal antibody (concizumab) blocking the TFPI interaction with the active site of FXa in Cynomolgus monkeys after iv and sc administration. Eur. J. Pharm. Sci. 2014, 56, 65–69. [CrossRef]
- Novo Nordisk Canada. AlhemoTM (concizumab injection): Product Monograph. 2023. Availabe online: https://www.novonordisk.ca/content/dam/nncorp/ca/en/products/alhemo-en-product-monograph.pdf (accessed on 23 May 2024).
- Favresse, J.; Lippi, G.; Roy, P.M.; Chatelain, B.; Jacqmin, H.; Ten Cate, H.; Mullier, F. D-dimer: Preanalytical, analytical, postanalytical variables, and clinical applications. Crit Rev Clin Lab Sci 2018, 55, 548-577.
- Capecchi, M.; Scalambrino, E.; Griffini, S.; Grovetti, E.; Clerici, M.; Merati, G.; Chantarangkul, V.; Cugno, M.; Peyvandi, F.; Tripodi, A. Relationship between thrombin generation parameters and prothrombin fragment 1+2 plasma levels. Int. J. Lab. Hematol. 2021, 43, E248–E251. [CrossRef]
- Matsushita, T.; Shapiro, A.; Abraham, A.; Angchaisuksiri, P.; Castaman, G.; Cepo, K.; D’oiron, R.; Frei-Jones, M.; Goh, A.-S.; Haaning, J.; et al. Phase 3 Trial of Concizumab in Hemophilia with Inhibitors. New Engl. J. Med. 2023, 389, 783–794. [CrossRef]
- Shapiro, A.D.; Angchaisuksiri, P.; Astermark, J.; Benson, G.; Castaman, G.; Eichler, H.; Jiménez-Yuste, V.; Kavakli, K.; Matsushita, T.; Poulsen, L.H.; et al. Long-term efficacy and safety of subcutaneous concizumab prophylaxis in hemophilia A and hemophilia A/B with inhibitors. Blood Adv. 2022, 6, 3422–3432. [CrossRef]
- Shapiro, A.D. Concizumab: a novel anti-TFPI therapeutic for hemophilia. Blood Adv. 2021, 5, 279–279. [CrossRef]
- Kjalke, M.; Kjelgaard-Hansen, M.; Andersen, S.; Hilden, I. Thrombin generation potential in the presence of concizumab and rFVIIa, APCC, rFVIII, or rFIX: In vitro and ex vivo analyses. J. Thromb. Haemost. 2021, 19, 1687–1696. [CrossRef]
- Young, G. Nonfactor Therapies for Hemophilia. HemaSphere 2023, 7, e911. [CrossRef]
- Anandani, G.; Patel, T.; Parmar, R. The Implication of New Developments in Hemophilia Treatment on Its Laboratory Evaluation. Cureus 2022, 14, e30212. [CrossRef]
- Shapiro, A.D.; Angchaisuksiri, P.; Astermark, J.; Benson, G.; Castaman, G.; Chowdary, P.; Eichler, H.; Jiménez-Yuste, V.; Kavakli, K.; Matsushita, T.; et al. Subcutaneous concizumab prophylaxis in hemophilia A and hemophilia A/B with inhibitors: phase 2 trial results. Blood 2019, 134, 1973–1982. [CrossRef]
- Castaman, G.; Abraham, A.; Angchaisuksiri, P.; Martinez, L.V.; Nogami, K.; Sathar, J.; Shen, C.; Zaw, J.J.T.; Young, G. The Effect of Concizumab Prophylaxis on Target Joints, Resolution and Joint Bleeds in Patients With Hemophilia A or B With or Without Inhibitors in Phase 3 Clinical Trials. Blood 2023, 142, 284–284. [CrossRef]
- Chan, A.K.; Barnes, C.; Mathias, M.; Linari, S.; Jaime, F.J.L.; Poulsen, L.H.; Bovet, J.; Odgaard-Jensen, J.; Matsushita, T. Surgical Procedures and Hemostatic Outcome in Patients with Hemophilia Receiving Concizumab Prophylaxis during the Phase 3 explorer7 and explorer8 Trials. Blood 2023, 142, 30–30. [CrossRef]
- Eichler, H.; Angchaisuksiri, P.; Kavakli, K.; Knoebl, P.; Windyga, J.; Jiménez-Yuste, V.; Hyseni, A.; Friedrich, U.; Chowdary, P. A randomized trial of safety, pharmacokinetics and pharmacodynamics of concizumab in people with hemophilia A. J. Thromb. Haemost. 2018, 16, 2184–2195. [CrossRef]
- Waters, E.K.; Sigh, J.; Friedrich, U.; Hilden, I.; Sørensen, B.B. Concizumab, an anti-tissue factor pathway inhibitor antibody, induces increased thrombin generation in plasma from haemophilia patients and healthy subjects measured by the thrombin generation assay. Haemophilia 2017, 23, 769–776. [CrossRef]
- Seremetis, S.V.; Cepo, K.; Rasmussen, J.S.; Rose, T.H.; Tamer, S.; Porstmann, T.; Haaning, J. Risk Mitigation Strategy for Concizumab Clinical Trials after Pause Due to Non-Fatal Thrombotic Events. Blood 2020, 136, 40–40. [CrossRef]
- Peyvandi, F.; Garagiola, I.; Mannucci, P.M. Post-authorization pharmacovigilance for hemophilia in Europe and the USA: Independence and transparency are keys. Blood Rev. 2021, 49, 100828. [CrossRef]
- Thornburg, C.D.; Duncan, N. Treatment adherence in hemophilia. Patient Preference Adherence 2017, ume 11, 1677–1686. [CrossRef]
- Johnston, K.; Stoffman, /.J.M.; Mickle, A.T.; Klaassen, R.J.; Diles, D.; Olatunde, S.; Eliasson, L.; Bahar, R. Preferences and Health-Related Quality-of-Life Related to Disease and Treatment Features for Patients with Hemophilia A in a Canadian General Population Sample. Patient Preference Adherence 2021, ume 15, 1407–1417. [CrossRef]
- Coleman, C.I.; Limone, B.; Sobieraj, D.M.; Lee, S.; Roberts, M.S.; Kaur, R.; Alam, T. Dosing Frequency and Medication Adherence in Chronic Disease. J. Manag. Care Pharm. 2012, 18, 527–539. [CrossRef]
- Coyne, M.; Rinaldi, A.; Brigham, K.; Hawthorne, J.; Katsaros, D.; Perich, M.; Carrara, N.; Pericaud, F.; Franzese, C.; Jones, G. Impact of Routines and Rituals on Burden of Treatment, Patient Training, Cognitive Load, and Anxiety in Self-Injected Biologic Therapy. Patient Preference Adherence 2022, ume 16, 2593–2607. [CrossRef]
- Rasmussen, N.K.; Berg, B.; Christiansen, A.S.L.; Neergaard, J.S.; Ter-Borch, G.; Hildebrand, E.; Gonczi, M.; Sparre, T. The Concizumab Pen-Injector is Easy to Use and Preferred by Hemophilia Patients and Caregivers: A Usability Study Assessing Pen-Injector Handling and Preference. Patient Preference Adherence 2024, ume 18, 1713–1727. [CrossRef]
- Stoner, K.L.; Harder, H.; Fallowfield, L.J.; Jenkins, V.A. Intravenous versus Subcutaneous Drug Administration. Which Do Patients Prefer? A Systematic Review. Patient - Patient-Centered Outcomes Res. 2014, 8, 145–153. [CrossRef]
- Roszkiewicz, J.; Swacha, Z.; Smolewska, E. Prefilled pen versus prefilled syringe: a pilot study evaluating two different methods of methotrexate subcutaneous injection in patients with JIA. Pediatr Rheumatol Online J 2020, 18, 1–8. [CrossRef]
- Vermeire, S.; D'Heygere, F.; Nakad, A.; Franchimont, D.; Fontaine, F.; Louis, E.; Van Hootegem, P.; Dewit, O.; Lambrecht, G.; Strubbe, B.; et al. Preference for a prefilled syringe or an auto-injection device for delivering golimumab in patients with moderate-to-severe ulcerative colitis: a randomized crossover study. Patient Preference Adherence 2018, ume 12, 1193–1202. [CrossRef]
- Young, G. The dosing conundrum of emicizumab: to waste product or not? Res Pract Thromb Haemost 2023, 7, 100087. [CrossRef]
- Hermans, C.; Noone, D.; Benson, G.; Dolan, G.; Eichler, H.; Jiménez-Yuste, V.; Königs, C.; Lobet, S.; Pollard, D.; Zupančić-Šalek, S.; et al. Hemophilia treatment in 2021: Choosing the”optimal” treatment using an integrative, patient-oriented approach to shared decision-making between patients and clinicians. Blood Rev. 2021, 52, 100890. [CrossRef]
- Castaman, G.; Jimenez-Yuste, V.; Gouw, S.; D'Oiron, R. Outcomes and outcome measures. Haemophilia 2024, 30, 112–119. [CrossRef]
- Di Minno, M.N.D.; Martinoli, C.; Pasta, G.; la Corte-Rodriguez, H.; Samy, I.; Stephensen, D.; Timmer, M.A.; Winburn, I. How to assess, detect, and manage joint involvement in the era of transformational therapies: role of point-of-care ultrasound. Haemophilia 2023, 29, 1-10. [CrossRef]
- Dutreil, S. Physical and psychosocial challenges in adult hemophilia patients with inhibitors. J. Blood Med. 2014, 5, 115–122. [CrossRef]
- Brod, M.; Bushnell, D.M.; Neergaard, J.S.; Waldman, L.T.; Busk, A.K. Understanding treatment burden in hemophilia: development and validation of the Hemophilia Treatment Experience Measure (Hemo-TEM). J. Patient-Reported Outcomes 2023, 7, 1–23. [CrossRef]
- Thachil, J.; Connors, J.M.; Mahlangu, J.; Sholzberg, M. Reclassifying hemophilia to include the definition of outcomes and phenotype as new targets. J. Thromb. Haemost. 2023, 21, 1737–1740. [CrossRef]
- Tran, H.; von Mackensen, S.; Abraham, A.; Castaman, G.; Hampton, K.; Knoebl, P.; Linari, S.; Odgaard-Jensen, J.; Neergaard, J.S.; Stasyshyn, O.; et al. Concizumab prophylaxis in persons with hemophilia A or B with inhibitors: patient-reported outcome results from the phase 3 explorer7 study. Res. Pr. Thromb. Haemost. 2024, 8, 102476. [CrossRef]
- Mannucci, P.M. Hemophilia treatment innovation: 50 years of progress and more to come. J. Thromb. Haemost. 2023, 21, 403–412. [CrossRef]
- Arruda, V.R.; Samelson-Jones, B.J. Gene therapy for immune tolerance induction in hemophilia with inhibitors. J. Thromb. Haemost. 2016, 14, 1121–1134. [CrossRef]
- Baas, L.; van der Graaf, R.; van Hoorn, E.S.; Bredenoord, A.L.; Meijer, K.; consortium, S. The ethics of gene therapy for hemophilia: a narrative review. J. Thromb. Haemost. 2023, 21, 413–420. [CrossRef]
- Spadarella, G.; Di Minno, A.; Milan, G.; Franco, N.; Polimeno, M.; Castaldo, F.; Di Minno, G. Paradigm shift for the treatment of hereditary haemophilia: Towards precision medicine. Blood Rev. 2020, 39, 100618. [CrossRef]
- Haute Autorite de Sante. ALHEMO (concizumab) - hemophilia A and B with inhibitors - early access decision. Posted on 6 Oct 2023 [in French]. 2023. Availabe online: https://www.has-sante.fr/jcms/p_3466237/fr/alhemo-concizumab-hemophilie-a-et-b-avec-inhibiteurs (accessed on 2 Oct 2024).
- Pharmaceutical Evaluation Division Pharmaceutical Safety and Environmental Health Bureau. Report on the deliberation results - Alhemo. 2023. Availabe online: https://www.pmda.go.jp/files/000268789.pdf (accessed on 2 Oct 2024).
- Swissmedic. Alhemo® (active substance: concizumab). 2023. Availabe online: https://www.swissmedic.ch/swissmedic/en/home/about-us/publications/public-summary-swiss-par/public-summary-swiss-par-alhemo.html (accessed on 2 Oct 2024).
- Therapeutic Goods Administration. Alhemo. 2023. Availabe online: https://www.tga.gov.au/resources/auspmd/alhemo (accessed on 2 Oct 2024).
Figure 1.
Mechanism of action of tissue factor pathway inhibitors (TFPIs), such as concizumab (reproduced from [48]). A Tissue factor-based initiation of coagulation and generation of activated factor X (Xa) by the extrinsic tenase complex. B Inhibition of Xa and activated factor VIIa (VIIa) by TFPI. C Binding of the different Kunitz (K) domains by the various anti-TFPI antibodies. X, factor X.
Figure 1.
Mechanism of action of tissue factor pathway inhibitors (TFPIs), such as concizumab (reproduced from [48]). A Tissue factor-based initiation of coagulation and generation of activated factor X (Xa) by the extrinsic tenase complex. B Inhibition of Xa and activated factor VIIa (VIIa) by TFPI. C Binding of the different Kunitz (K) domains by the various anti-TFPI antibodies. X, factor X.
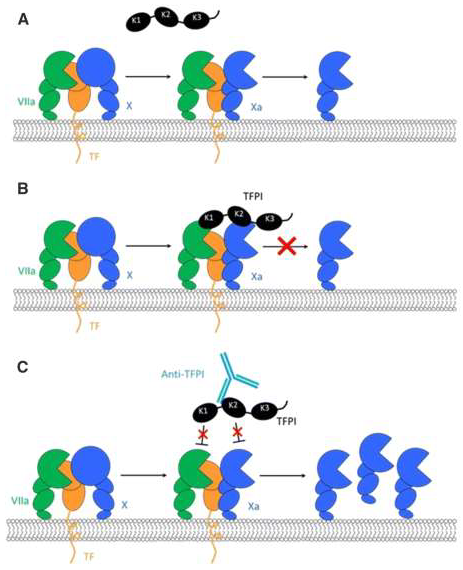

Table 1.
Summary of product characteristics of concizumab.
| Characteristic | Description |
|---|---|
| Mechanism of action [48] | Concizumab binds to the Kunitz-2 domain of the TFPI protein and prevents TFPI from binding to FXa and to the TF/FVIIa complex; the inhibition of TFPI increases thrombin generation |
| Administration [53] | Subcutaneous using a prefilled multidose pen |
| Half-life [53] | 38 h |
| Frequency of administration [53] | Once daily |
| Dose calculation [53] | Patient bodyweight (kg) × dose (1.00, 0.15, 0.20 or 0.25 mg/kg) = total amount (mg) of concizumab to be administered in a single daily injection |
| Antidote [60] | None, but quick washout |
| Laboratory monitoringa [61] |
Monitoring drug concentration: Measurement of TFPI levels using ELISA Measurement of residual TFPI activity using specific activity assays, e.g., diluted PT-based assay or TF-dependent chromogenic assays Monitoring drug efficacy: Thrombin generation, thromboelastography, clot waveform analysis before and after treatment commencement |
| Breakthrough bleed treatment | No concizumab dose adjustment needed Bypassing agents (rFVIIa, aPCC, plasma-derived FVIIa/FX), factor concentrates |
| Laboratory monitoring during concomitant treatment with concizumab and bypassing agents | Thrombin generation |
| Treatment management during surgery |
Minor surgery: No concizumab dose adjustment needed Major surgery: Concizumab should be paused 4 days prior to surgery and resumed at the normal daily maintenance dose (either 0.15, 0.20, or 0.25 mg/kg) 10–14 days after surgery, considering each patient’s overall clinical pictureb |
| Immunogenicity [57] | In the explorer4 and 5 trials [57], 25% of patients developed mostly low-titer and transient neutralizing anti-concizumab antibodies |
| AEs (frequency in the explorer7 trial) [56] |
Common AEs (occurring in ≥5% of patients): injection-site reactions (22.8%), arthralgia (11.4%), upper respiratory tract infections (7.0%), headache (5.3%), pyrexia (5.3%) Less common AEs: hypersensitivity (2.6%), thromboembolic events (0.9%), pruritus (0.9%) |
aIn the explorer7 trial [56], concizumab concentration, free TFPI concentration, and thrombin peak were measured. bPatients with planned surgery were excluded from the concizumab studies. AE, adverse event; aPCC, activated prothrombin complex concentrate; ELISA, enzyme-linked immunosorbent assay; FVIIIa, activated factor VIII; FX, factor X; FXa, activated factor X; PT, prothrombin time; rFVIIIa, recombinant activated factor VIII; TF, tissue factor; TFPI, tissue factor pathway inhibitor.
Table 2.
Summary of the explorer trials.
| Trial ID | Study type | Intervention | Number of participants | Findings |
|---|---|---|---|---|
| explorer1 (NCT01228669) Phase 1 |
A multicenter, randomized, double-blind, placebo-controlled, single-dose, dose-escalation trial investigating safety, PK and PD of NNC 0172-0000-2021 administered intravenously and SC to healthy male subjects and persons with HA or HB | Concizumab or placebo | 52 (28 healthy volunteers, 24 persons with HA or HB) | Primary endpoint: safety 76 AEs (75% mild) |
| explorer2 (NCT01631942) Phase 1 |
A multicenter, open-label, multiple-dosing trial investigating safety, PK and PD of NNC 0172-2021 administered SC to healthy male subjects and persons with HA or HB | Low, medium, or high dose of concizumab | 22 (4 healthy volunteers, 18 persons with HA or HB) | Primary endpoint: safety No severe or unexpected AEs Increased thrombin generation with concizumab in thrombin generation assay ex vivo and in vivo |
| explorer3 (NCT02490787) Phase 1b |
A multicenter, randomized, placebo-controlled, double-blind, multiple-dose trial investigating safety, PK and PD of concizumab administered SC to persons with HA | Placebo, or five escalating doses of concizumab | 24 | 56 AEs in 19 persons (54 mild and 2 moderate); 91 bleeds (almost all mild) |
| explorer4 (NCT03196284) Phase 2 |
A multicenter, randomized, open-label, controlled trial evaluating the efficacy and safety of prophylactic administration of concizumab in persons with HA or HB with inhibitors | Concizumab (main and extension phases), with eptacog alfa administered on-demand during bleeding episodes | 26 | Estimated ABR 4.5 (95% CI: 3.2–6.4) in the concizumab arm vs. 20.4 (95% CI: 14.4–29.1) in the rFVIIa on-demand arm Low AE rates, no severe AEs reported, no AE-related withdrawals, no thromboembolic events, and no deaths |
| explorer6 (NCT03741881) Phase 3 |
A prospective, multinational, non-interventional study in persons with HA or HB with or without inhibitors treated according to routine clinical practice | No treatment given | 231* | No published results |
| explorer7 (NCT04083781) Phase 3 |
Efficacy and safety of concizumab prophylaxis in persons with HA or HB with inhibitors | No prophylaxis for ≥24 weeks (group 1), or prophylaxis with concizumab for ≥32 weeks (group 2), or nonrandomly assigned to prophylaxis with concizumab for ≥24 weeks (groups 3 and 4) | 133(19 in group 1; 33 in group 2; 21 in group; and 60 in group 4) | Median ABR was 9.8 (IQR 6.5–20.2) in group 1 vs. 0.0 (IQR 0.0–3.3) in group 2 Overall median ABR in the concizumab groups was 0.0 No thromboembolic events after resuming the therapy |
*Enrolment as of November 2015. ABR, annualized bleeding rate; AE, adverse event; CI, confidence interval; HA, hemophilia A; HB, hemophilia B; IQR, interquartile range; PD, pharmacodynamics; PK, pharmacokinetics; rFVIIa, recombinant activated factor VII; SC, subcutaneously.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



