Submitted:
11 February 2025
Posted:
12 February 2025
You are already at the latest version
Abstract
Background/Objectives: Chronic obstructive pulmonary disease (COPD) is a heterogeneous inflammatory condition characterized by progressive airflow limitation which may be caused by genetic and environmental factors. Beside, epigenetic mechanisms could provide valuable insights into the complex interactions between environment and genes and subsequent development of the disease. The aim of this study is to provide a systematic review of the latest knowledge on epigenetic modifications that characterize COPD summarizing epigenetic factors that could serve as potential novel biomarkers and therapeutic targets for the treatment of COPD patients.
Methods: We queried the PubMed and Scopus electronic databases with specific keywords, in May 2024, according to the PRISMA guidelines, and articles were included if they met all the inclusion criteria and survived a quality assessment
Results: We identified 5414 publications in our systematic search. Among them, only 51 articles met the criteria of COPD-associated epigenetic modifications in human patients compared to control group. 8 studies described DNA methylation, 1 study histone modifications, and 42 studies non-coding RNAs.
Conclusion: Apoptosis and inflammatory pathways have been found to be the main mechanisms regulated by epigenetic elements in COPD patients. In addition, non-coding RNAs may be useful as biomarkers or therapeutic targets of pulmonary disease. Future studies will be needed to confirm the role of epigenetic elements associated with COPD.
Keywords:
COPD
; epigenetic mechanisms
; DNA methylation
; histone modification
; non-coding RNA
1. Introduction
Chronic Obstructive Pulmonary Disease (COPD) is a heterogeneous inflammatory condition that results in largely irreversible and progressive airflow limitation [1]. Before the COVID-19 pandemic, COPD was described as the third leading cause of death worldwide, with approximately 3.3 million deaths, between 1990 and 2019 [2]. The Global Initiative for Chronic Obstructive Lung Disease (GOLD) report 2023 defined the COPD as a: “heterogeneous lung condition characterized by chronic respiratory symptoms (dyspnea, cough, expectoration and/or exacerbations) due to abnormalities of the airways (bronchitis, bronchiolitis) and/or alveoli (emphysema) that cause persistent, often progressive, airflow obstruction” and established the criteria for categorizes airflow limitation into 4 stages (GOLD classification): mild (GOLD1), moderate (GOLD2), severe (GOLD3) and very-severe (GOLD4) COPD [3].
Cigarette smoking has long been considered as the main risk factor for the COPD [4]. The complex mixture found in tobacco smoke, including liquid droplets, volatile and semi-volatile compounds, and gases can induce DNA damage, hinder the inflammatory response, and increase the production of reactive oxidative substances, leading to various levels of lung impairment [5]. However, the development of COPD also in non-smokers patients suggested that other elements in addition to environmental factors (cigarette smoking and air pollution) can contribute to onset of disease. The genetic contribution to COPD is still being learned. The first Genome-Wide Association Study (GWAS) provided evidence of hedgehog interacting protein (HHIP), SERPINA, family with sequence similarity 13 member A (FAM13A), advanced glycosylation end-product specific receptor (AGER), cholinergic receptor nicotinic alpha 5 subunit (CHRNA5), and interleukin 27 (IL27) association with COPD [6,7]. However, GWAS studies using larger sample sizes have failed to confirm these results, finding other gene variants associated to COPD, including genomic regions near Ras and Rab interactor 3 (RIN3), cytochrome P450 family 2 subfamily A member 6 (CYP2A6), and desmoplakin (DSP) [6,8]. To date, GWAS analysis has identified COPD genetic loci that could only explain ~10% of European disease cases [7,8]. Moreover, smoking and genetic variants considered as individual risk factors for lung disease, showed a moderate impact on COPD. Therefore, it has been suggested that COPD could be recognized as a progressive illness resulting from a combination of environmental, genetic, and epigenetic factors [4].
Epigenetic regulation can occur at various levels, including DNA methylation, histone modifications, and noncoding RNA (ncRNA) modulation [9,10]. Emerging evidence indicates that epigenetic regulation plays a crucial role in COPD development and progression [9,10,11]. The reversibility of epigenetic alterations renders them valuable therapeutic targets for treating COPD patients [9,10]. Moreover, the possibility of associating epigenetic alterations with disease progression could suggest the use of epigenetic elements as diagnostic and prognostic COPD biomarkers. This study seeks to offer an objective and comprehensive overview of the latest insights into the epigenetic modifications post COVID-19 associated with COPD, and aims to summarize the epigenetic factors that could serve as potential new biomarkers and therapeutic targets for the treatment of patients with COPD.
2. Materials and Methods
2.1. Study Design and Search Strategy
We conducted this systematic review following the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines [12,13]. PRISMA comprises a 27-item checklist to ensure and promote the quality of systematic reviews; this checklist is reported in Supplementary Tables S1 and S3 (for abstract checklist). The protocol employed in the current systematic review has been submitted for registration to Open Science Framework platform (available online at https://osf.io/4ja25/; accessed on 2025-01-31). The retrieval process consisted of three phases.
2.1.1. Systematic Search Phases
1. Preliminary research and definition of keywords
During the first phase, started in May 2024, we carried out a preliminary analysis of the literature in the absence of specific keywords to define the scientific databases to be analyzed, the keywords to be used, and the inclusion/exclusion criteria to be applied. Based on the research question and the exploratory research of literature, the strategy was to define keywords referring to the four macro areas of interest: (1) keywords related to COPD; (2) keywords related to molecules can be used to establish the presence, development or treatment of COPD; (3) keywords related to epigenetic mechanisms; (4) keywords related to pathophysiological mechanisms underlying COPD. The keywords belonging to each category were summarized in Table 1.

Table 1.
Keywords.
| KEY WORDS |
|---|
| Chronic obstructive pulmonary disease, COPD |
| Biomarker*, therapeutic target* |
| Epigenome-Wide Association*, sequencing, epigenetic*, DNA methylation*, long noncoding RNA*, circRNA*, miRNA*, histone* deacetylation*, histone* protein*, HDAC |
| Molecular mechanism*, extracellular vesicle* |
- 2. Systematic Search and Definition of PICOS
The second phase, conducted in May 2024, consisted of a systematic search among titles, abstracts, and keywords of scientific papers, using the electronic databases PubMed and Scopus, based on the selected keywords, properly combined through the Boolean operators “AND” and “OR”. We limited our search to articles, English, published from 01-01-2020 to the date of the search (09-05-2024) to include only the most recent papers in our systematic review and to avoid the massive inclusion of papers without dated analysis techniques. The search strategy, including all keywords used and the number of studies found from each database, was analytically reported in Table 2.
Table 2.
Search Strategy.
| Database | Steps | Query | Research in | Items found |
|---|---|---|---|---|
| PubMed | #1 | (((((((((((((Biomarker*[Title/Abstract]) OR ("therapeutic target*"[Title/Abstract])) OR ("Epigenome-Wide Association*"[Title/Abstract])) OR (sequencing[Title/Abstract])) OR (epigenetic*[Title/Abstract])) OR ("DNA methylation*"[Title/Abstract])) OR ("long noncoding RNA*"[Title/Abstract])) OR (circRNA*[Title/Abstract])) OR (miRNA*[Title/Abstract])) OR ("histone* deacetylation*"[Title/Abstract])) OR ("histone* protein*"[Title/Abstract])) OR (HDAC[Title/Abstract])) OR ("Molecular mechanism*"[Title/Abstract])) OR ("extracellular vesicle*"[Title/Abstract]) | Title/Abstract | 1,460,188 |
| #2 | "COPD"[Title/Abstract] OR "Chronic obstructive pulmonary disease"[Title/Abstract] | Title/Abstract | 83,977 | |
| #3 | Combine #1 AND #2 | 5,834 | ||
| #4 | Limit to (English) | 5,663 | ||
| #5 | Limit after 2020 | 2,509 | ||
| Scopus | #1 | TITLE-ABS-KEY ("Biomarker*" OR "therapeutic target*" OR "Epigenome-Wide Association*" OR "sequencing" OR "epigenetic*" OR " DNA methylation*" OR "long noncoding RNA*" OR "circRNA*" OR "miRNA*" OR "histone* deacetylation*" OR "histone* protein*" OR "HDAC" OR "molecular mechanism*" OR "extracellular vesicle*" ) | Title/Abstract/Keywords | 1,989,075 |
| #2 | TITLE-ABS-KEY (“COPD” OR "Chronic Obstructive Pulmonary Disease") | Title/Abstract/Keywords | 102,296 | |
| #3 | Combine #1 AND #2 | 7,433 | ||
| #4 | Limit to (English) and (Italian) | 7,078 | ||
| #5 | Limit after 2020 | 2,906 |
The final selection of papers for inclusion was carried out according to the Population, Intervention, Comparison, Outcomes, and Study Design (PICOS) worksheet [12,13], summarized in Table 3. We defined the following PICOS criteria: Population, we chose “Studies in Humans” and “Studies including COPD patients” as the inclusion criteria and “In vitro and in vivo studies” and “Participants with other malignancies” as exclusion criteria because we aimed to avoid including humans with COPD but with any malignancies that might influence the study results, thus preventing the introduction of bias; in addition, we excluded all studies conducted exclusively in vivo and in vitro; Interventions, we chose as inclusion criteria “Assessment of DNA methylation, Hystone modification, and ncRNA expression” to including only work that studied these epigenetic mechanisms to the exclusion of everything else; Comparisons, we choose as inclusion criterion "Control group (between or within design)" to have a comparison between two populations to identify any differences in epigenetic mechanisms between healthy and COPD groups or during the evolution of the disease within patients group; Results, we chose as inclusion criteria "provide an unbiased and comprehensive overview of the current knowledge on epigenetic modifications associated with COPD" and "summarize epigenetic modifications translated into clinical therapeutic interventions and biomarkers for COPD", to have a complete picture of the impact of epigenetic mechanisms in the disease as well as the possibility of using epigenetic elements for the management of patients with COPD. Study Design, we chose “Review, Scoping Review, Narrative Review, Systematic Review, Meta-Analysis, Editorial, Book, Case Report, Conference Review, and Conference Paper” as exclusion criteria following the guidelines to carry out a Systematic Review.
Table 3.
Population, intervention, comparison, outcomes and study design (PICOS) worksheet.
| Parameters | Inclusion Criteria | Exclusion Criteria |
|---|---|---|
| Participants | Studies in humans Studies including COPD patients |
In vitro and in vivo studies Participants with other malignancies |
| Interventions | Assessment of DNA methylation, Hystone modification, and ncRNA expression | Others |
| Comparisons | Control Group | Others |
| Outcomes | 1) to provide unbiased and exhaustive overview of the current knowledge on the epigenetic modification associated COPD; 2) to summarize the epigenetic modifications translated into clinical therapeutic interventions and biomarkers for COPD. |
Others |
| Study Design | Original studies in English | Review, Scoping Review, Narrative Review, Systematic Review, Meta-Analysis, Editorial, Book, Case Report, Conference Review, and Conference Paper |
- 3. Application of PICOS Study Design Exclusion Criteria
The final phase consisted of a first step in which, following PICOS criteria related to the Study Design section, we excluded all reviews, both narrative and systematic ones, meta-analyses, and conference papers, to substantially reduce the number of included studies.
2.1.2. Title and Abstract Selection
By reading the titles and abstract we excluded all studies that did not match with the research question.
2.1.3. Full-Text Selection According to PICOS Criteria
Finally, we included in the systematic review only clinical studies that investigated the epigenetic mechanisms in COPD. The included papers were read thoroughly to obtain the data of our interest.
2.1.4. Synthesis Method
The included papers were clustered according to epigenetic mechanisms involved in COPD (molecular and cellular processes, as biomarker or therapeutic target). Table 4, Table 5, Table 6, Table 7, Table 8 and Table 9 described the extracted information, including: Study = name of first author et al., year; Country (Region) = where the study took place; Number of participants = sample size; Type of sample = biological sample employed; Gene affected = gene or group of genes whose expression can be "regulated" by epigenetic mechanisms; Epigenetic alteration = type of epigenetic alteration observed in the presence of disease; Activity in COPD = involvement of epigenetic elements in different molecular and cellular mechanisms associated with COPD; Role of epigenetic mechanisms = epigenetic modifications that can be used to explain the pathophysiology of COPD or as biomarkers and therapeutic targets.
Table 4.
DNA methylation in COPD.
| Study | Country | Number of participants | Type of sample | Gene affected | Epigenetic alteration | Activity in COPD | Role of epigenetic mechanisms |
|---|---|---|---|---|---|---|---|
| Kachroo P, et al 2021 [16] | Boston (USA) | N=78 fetal N=160 adult COPD |
Lung tissue | Transcription factors, oxido-reductase, VEGFA-VEGFR2 | Hyper-/hypo-methylation | Air flow limitation, inflammation activation, lung remodeling | Fetal origin of COPD |
| Kachroo P, et al 2020 [17] | Boston (USA) | N=78 fetal N=160 adult COPD |
Lung tissue | Co-methylation: Wnt, Pi3K/AKT, MAPK, Hippo | DNA methylation imbalance | Low lung function | Fetal origin of COPD |
| Schwartz U, et al 2023 [18] | Heidelberg and Munich (Germany) Huston (USA) |
N=3 control N=3 COPD I N=5 COPD II-IV |
Parenchymal fibroblasts (lung tissue) |
3 cluster of genes involved in cell proliferation, DNA repair and extracellular matrix organization | Hyper-/hypo-methylation | Low lung function | Kinetics of DNA methylation in COPD |
| Strom JE, et al 2022 [19] |
Northern Sweden | N=15 control N=18 COPD |
Macrophage from Broncho alveolar lavage (BAL) | DMPs co-localized with COPD-associated SNPs | DNA methylation imbalance | --- | Pathophysiology of COPD |
| Cordero AIH, et al, 2022 [10] | Vancouver (Canada) | N=27 control N=15 COPD |
Small airway epithelial brushings and buffy coat blood | DNAmGrimAge | DNA methylation imbalance | Biomarker for assessing accelerated aging in the airways of individuals with COPD | Biomarker |
| Morrow JD, et al 2020 [20] | Boston (USA) | N=336 control N=331 COPD |
Blood samples | Pi3KCD cg03971555 cg12033075 |
Hyper methylation | Predictive biomarker | Biomarker |
| Zhang Z, et al 2021 [21] | Wuxi (China) |
N=18/17 control N=8/16 COPD |
Lung tissue/bronchoscopies (bronco epithelial cells) | Nfr2 | Hyper methylation | Increased oxidative stress and cell death | Therapeutic target |
| Chen Q, et al 2022 [22] |
Groningen (Netherlands) |
N=966/8 control N=595/14 COPD |
whole blood/airway epithelial cells | AHRR cg05575921 cg21161138 |
Hypo methylation | Airway epithelial cell proliferation, dysregulate mitochondrial function, and reduce apoptotic processes | Therapeutic target |
Table 5.
Histone modification in COPD.
| Study | Country | Number of participants | Type of sample | Gene affected | Epigenetic alteration | Activity in COPD | Role of epigenetic mechanisms |
|---|---|---|---|---|---|---|---|
| Günes GG, et al, 2022 [25] | Ghent (Belgium) |
N=40 control N=111 COPD |
Monocytes/lung tissue | PRMT7 | Histone methylation | Chronic inflammation | Pathophysiology of COPD /Therapeutic target |
Table 6.
circular RNA in COPD.
| Study | Country | Number of participants | Type of sample | Gene affected | Epigenetic alteration | Activity in COPD | Role of epigenetic mechanisms |
|---|---|---|---|---|---|---|---|
| Duan R, et al, 2020 [28] | Beijing (China) |
N= 21 control N= 21 COPD |
Peripheral blood mononuclear cells | Gene involved in natural killer T cell activation and T-helper cell differentiation | Differential expression in COPD compared to control | Immune balance alteration | Pathophysiology of COPD /Therapeutic target |
| Xie T, et al, 2024 [29] | Hainan (China) |
N= 5 control N= 10 (acute and stable) COPD |
Peripheral blood mononuclear cells | Caspase 1, IL-18, IL-1b | UP-regulation hsa-circ_0008833-57aa |
Pyroptosis | Pathophysiology of COPD |
| Liu P, et al 2022 [27] | Anhui (China) |
N= 3 control N= 3 COPD |
Blood samples | miR-1273h-3p; miR-411-5p; miR-122-5p; miR-615-5p; miR-519d-3p; miR-485-3p; miR-3646; miR-4714-5p; miR-203b-5p; miR-193a-5p; miR-1261; miR-4690-5p; miR-939-5p; miR-9-5p; miR-2113; miR-7977 | UP-regulation circFCHO2; circMBOAT2, circPTPN22; circTBC1D22A; circACADM; circCKAP5 |
---- | Pathophysiology of COPD |
| Zhang C, et al, 20220[30] | Jiangsu (China) |
N= 17 COPD non-smoker N=23 COPD smoker |
Lung tissue | miR-24/PHPPL2 axis | Down regulation Circ_0006892 | Inflammatory injury | Pathophysiology of COPD |
| Wang Z, et al,2021 [31] | Hebei (China) |
N= 27 control N=21COPD |
Lung tissue | miR-145-5p/BRD4 axis | UP-regulation Circ_ANKRDII |
Inflammation, apoptosis and oxidative stress | Pathophysiology of COPD |
| Tang S, et al,2023 [32] | Hefei (China) |
N=30 control N=30 COPD |
Plasma samples | ---- | Differential expression circ_0008882; circ_00089763; circ_00062683; circ_00077607 | Immune balance alteration | Biomarkers |
| Shen X, et al, 2024 [33] | Jiangsu (China) |
N=29 control N=41 COPD |
Peripheral blood mononuclear cells | ---- | Differential expression circ_0049875 and circ_0042590 |
Acute exacerbation of COPD | Biomarkers |
Table 7.
Long non-coding RNA in COPD.
| Study | Country | Number of participants | Type of sample | Gene affected | Epigenetic alteration | Activity in COPD | Role of epigenetic mechanisms |
|---|---|---|---|---|---|---|---|
| Wu S, et al, 2020 [37] | Taipei(Taiwan) | N=35 control N=64 COPD |
Peripheral blood mononuclear cells | IL-8, VCAM1, E-SEL | Down regulation lncRNA-IL7R | Inflammatory processes | Pathophysiology of COPD |
| Zhou Ai, et al, 2020 [38] | Xiangya (China) | N=3 control N=7COPD |
Lung tissue | Notch1 | Down regulation lncRNA- HOXA-AS2 | Cell viability and Inflammatory processes | Pathophysiology of COPD |
| Wang Y, et al,2020 [39] | Wuhan (China) |
N=80 control N=80 stable COPD N=80 AECOPD |
Peripheral blood mononuclear cells | Mir-146a/TNF, IL-6, IL-8, IL-1IL-17 | Upregulation LncRNA-PVT1 |
Inflammatory processes | Prognostic biomarker |
| Liu S, et al, 2020 [42] | Wuhan (China) |
N=120control N=120 stable COPD N=120 AECOPD |
Blood samples | miR-125b, miR-133, miR-146a, miR-203/TNF, IL-6, IL-8, IL-1IL-17, IL-23 | Up regulation LncRNA-MALAT1 |
Inflammatory processes | Prognostic biomarker |
| Liu P, et al, 2021 [43] | Shanghai (China) |
N=90 control N=50 COPD |
Blood samples | Mir-18a-5p/TNF, IL-6, IL-8, IL-1 | Down regulation lncRNA-CASC2 | Inflammatory processes | Diagnostic biomarker |
| Zhao S, et al,2021 [44] | Jiangsu (China) |
N=150 control N=70 COPD |
Blood samples | Mir-181-5p/Wnt/b-catenin axis | Upregulation LncRNA-LUCAT1 |
Apoptotic/Inflammatory processes | Biomarker/therapeutic target |
| Dai Z, et al, 2022 [45] | Xiangya (China) | N=8 control N=5COPD |
Lung tissue | Bcl-2 | Up regulation lncRNA-HOTAIR | Apoptotic processes | Therapeutic target |
| Zong D, et al, 2022 [46] | Xiangya (China) | N=10 control N=10 COPD |
Lung tissue | Mir-152-3p/ERK | Up regulation lncRNA-CCT1 | Inflammatory processes | Therapeutic target |
Table 8.
Micro RNA in COPD.
| Study | Country | Number of participants | Type of sample | Gene affected | Epigenetic alteration | Activity in COPD | Role of epigenetic mechanisms |
|---|---|---|---|---|---|---|---|
| Wang L, et al 2022 [48] | Xiangya (China) |
N= 12 control N=12 COPD |
Peripheral blood mononuclear cells | IL-8 signaling; *********************** iCOS-iCOSL signaling |
Aberrant expression: miR-4453; miR-4736; miR-3118; miR-6967-5p; miR-132-3p; miR-96-5p; miR-4497 ***************** miR-16-5p; miR-1964-5p; miR-29b-3p; miR-2355-3p; miR-18a-5p; miR-1234-3p; miR-148-3p; miR-21-5p; miR-1184; miR-140-5p; miR-19b-3p; miR-223-3p; miR-1246; miR-130a-3p |
Inflammatory processes | Pathophysiology of COPD |
| Hu J, et al 2022 [49] | Wuhan (China) |
N= 3/9 control N=3/9 COPD |
BALF/ blood samples |
MAPK, RAS, FOXO | miR-129-5p; miR-3529-3p; miR-365b-3p; miR-6503-5p; miR-26-3p; miR-34b-5p; miR-4748; miR-491-5p; miR-158-3p |
Oxidative/inflammatory process | Pathophysiology of COPD |
| Zhang J, et al 2020 [50] | Xuzhou (China) |
N= 14/75 control N=36/53 COPD |
Alveolar macrophages/ Peripheral blood mononuclear cells | HAT1/TNF-IL-6-IL-8 | Up regulation miR-486-5p |
Inflammatory processes | Pathophysiology of COPD |
| Yang H, et al 2021 [52] | Xuzhou (China) |
N= 27 control N=21COPD |
Lung tissue | CDKN1B | Down regulation miR-221-3p |
Apoptotic and Inflammatory processes | Pathophysiology of COPD |
| Chang C, et al 2024 [54] | Beijing (China) |
N= 19 control N= 13 COPD |
Lung tissue | RAGE | Down regulation miR-23a-5p |
Oxidative/inflammatory process | Pathophysiology of COPD |
| Kim R, et al 2021 [55] | Newcastle, New South Wales (Australia) |
N= 5 control N= 10 COPD |
Lung tissue | SATB1/S100A9/NF-kB | Up regulation miR-21 |
Inflammatory processes | Pathophysiology of COPD |
| De Smet E, et al 2020 [56] | Ghent (Belgium) | N= 44 control N= 48 COPD |
Lung tissue | MMP12, ADAM19 | Up regulation miR-155 |
Inflammatory processes | Pathophysiology of COPD |
| Zhu Y, et al 2022 [57] | Brigham (USA) |
N= 17control N=8COPD |
Alveolar macrophages | LDLR | Down regulation miR-103a |
Oxidative/inflammatory process | Pathophysiology of COPD |
| Yang Z, et al 2023 [59] | Suzhou (China) |
N= 14 control N=14 COPD |
Lung tissue | NOS1 | Up regulation miR-4640-5p |
Pulmonary hypertension | Pathophysiology of COPD |
| Tasena H, et al 2022 [63] | Groningen (Netherlands) | N=6 control N=3 COPD |
Lung tissue | MICS5AC, COL4A1, COL5A1 | Up regulation miR-708-5p, let-7a-5p, miR-31-5p, miR-146a-5p |
Epithelial differentiation, chronic mucus hypersecretion (CMH) |
Pathophysiology of COPD |
| Singh P, et al 2024 [60] | Birmingham (USA) |
N= 9 control N=13 COPD |
Lung tissue | ANO1 | Down regulation miR-381 |
Mucus production and secretion | Pathophysiology /therapeutic target |
| Zheng L, et al 2021 [61] | Hefei (China) | N= 400 control N=400 COPD N=50 control+COPD |
Blood samples/ Lung tissue |
E-cadherin, α-SMA,Vimentin,N-cadherin | Down regulation miR-30 |
Epithelial-mesenchymal transition | Pathophysiology of COPD |
| Shi X, et al 2022 [51] | Qinghai (China) |
N= 40 control N=40 COPD |
Blood samples | ------ | Up regulation miR-486-5p; miR-106b-5p |
Hypoxia/Pulmonary Hypertension | Biomarkers |
| Shen Y, et al 2021[ 53] | Nanjing (China) |
N=77 control N=155 COPD |
Blood samples | TNF, IL-6, IL-8, IL-1 | Up regulation miR-221-3p; miR-92a-3p |
Inflammatory processes | Biomarkers |
| Zhang L, et al 2022 [64] | Guizhou (China) |
N= 6 control N=6 COPD |
Blood samples | SLC17A9 | Down regulation miR-548ar-3p |
------ | Biomarkers |
| Nadi E, et al 2022 [65] | Hamadan (Iran) |
N=60 control N=60 COPD |
Blood samples | ------ | Down regulation miR-146a;miR-216 |
Oxidative/inflammatory process | Biomarkers |
| Ding Y, et al 2023 [66] | Hefei (China) |
N=26 control N=59 COPD |
Blood samples | ------ | Down regulation miR-150-5p |
Inflammatory processes | Biomarkers |
| Zhang X, et al 2022 [67] | ChongQing (China) |
N=33 control N=36 COPD |
Blood samples | ------ | Down regulation miR-423-5p |
------ | Biomarkers |
| Tao S, et al 2024 [68] | Xiangya (China) |
N= 23 control N= 240 COPD |
Blood samples | ------ ***** PIK3R2 |
Down regulation miR-1290; miR-1246 ****** Up regulation miR-4433a-5p |
------ ***** Apoptotic and Inflammatory processes |
Biomarkers |
| Cazola-Rivero S, et al 2020 [69] | Tenerife (Spain) |
N=13 control N=24 COPD |
Blood samples | MAPK, chemokines, Wnt | Down regulation miR-1246 |
Emphysema development | Biomarkers |
| Wang C, et al 2021 [70] | Jiaozuo (China) |
N=70 control N=140 COPD |
Blood samples | TNF, IL-1, IL-6 | Up regulation miR-126 |
Inflammatory processes | Biomarkers |
| Huang H, et al 2020 [71] | Kunshan (China) |
N=80 control N=160 COPD |
Blood samples | ------ | Up regulation miR-210 |
Pulmonary hypertension | Biomarkers |
| Burke H, et al 2022 [72] | Southampton (United Kingdom) |
N=20 control N=24 COPD |
BALF/Extra vesicles | ------ | Down regulation miR-338-3p; miR-204-5p ****** Up regulation miR-223-3p; miR-182-5p; miR-2110 |
Inflammatory patterns | Biomarkers |
| Wang F, et al 2023 [73] | Peking (China) |
N=42 control N=111COPD |
Blood samples/Exosome | ------ | Up regulation miR-1258 |
Inflammatory processes | Biomarkers |
| Shen H, et al 2021 [74] | Binzhou (China) |
N=20 control N=20 COPD |
Blood samples | ARHGEF12, BCAT1 |
Up regulation miR-196-5p Down regulation miR-361-5p |
Epithelial hyperplasia | Therapeutic target |
Table 9.
Competing endogenous RNA in COPD.
| Study | Country | Number of participants | Type of sample | Gene affected | Epigenetic alteration | Activity in COPD | Role of epigenetic mechanisms |
|---|---|---|---|---|---|---|---|
| Li B, et al 2023 [76] | Ningxia (China) |
N=6 control N=14 COPD |
Lung tissue | 18 hub genes | ceRNA aberrant expression | Immune cells infiltration/differentiation; cell proliferation | Pathophysiology of COPD |
| Feng X, et al 2023 [77] | Ningxia (China) |
N=6 control N=7COPD |
Lung tissue | TNF/NF-kb; IL-6/JAK/STAT3 | ceRNA aberrant expression | Inflammatory processes | Pathophysiology of COPD |
2.1.5. Study Risk of Bias Assessment
We assessed the risk of bias for each study in Supplementary Table S2 by compiling the AXIS tool [1][].
3. Results
3.1. Flow Diagram
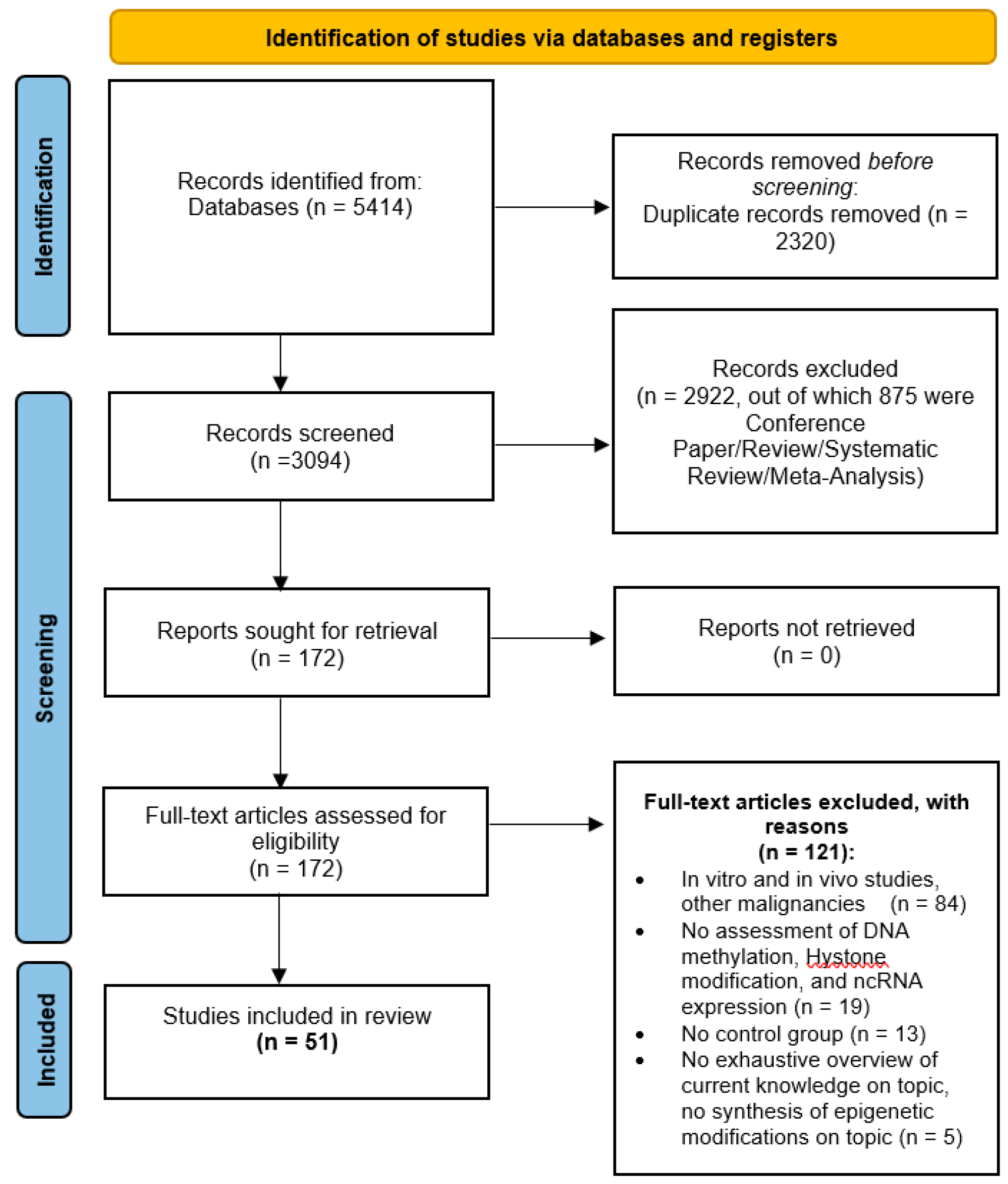
Figure 1 shows all the phases in the systematic review process.
The research carried out on PubMed and Scopus yielded 2509 and 2905 studies, respectively. These 5414 papers were merged in a non-redundant database and 3094 papers remained. Then, by eliminating all studies not related to our research question and all studies that did not match PICOS criteria related to Study Design (exclusion for all types of reviews, meta-analysis, and conference papers), the number of studies was reduced to 172. Finally, by applying all the PICOS criteria, we obtained 51 studies to be included in the systematic review.
Figure 1.
Flow Diagram.
3.2. Study Selection and Characteristics
Two independent reviewers (R.R. and P.B.) checked the pool of 3094 abstracts collected from PubMed and Scopus search engine outputs; any disagreement was discussed with C.C. as the arbiter. Titles and abstracts were screened, and 2922 were removed through a semi-automatic script created via MATLAB (The MathWorks, Inc., Natick, MA, USA), according to PICOS criteria related to Study Design (875 papers) and irrelevance to the research question (2047 papers). The remaining 172 papers were checked for eligibility according to the remaining PICOS criteria, all in vitro and in vivo studies and studies with participant with other malignancies (84), all studies without assessment of DNA methylation, Histone modification or ncRNA expression (19), all studies with no control group (13) and lastly, all studies that did not provide a comprehensive overview of current knowledge on the topic or did not have a summary of epigenetic changes on the topic (5) were excluded. Finally, 51 articles were included, summarized in Table 4, Table 5, Table 6, Table 7, Table 8 and Table 9, which shows the main information of each, divided according to epigenetic mechanisms in COPD patients.
3.3. Synthesized Findings
In this subsection, for each study, we reported the main findings of pertinence for this systematic review grouped according to epigenetic mechanisms in COPD patients.
Our search criteria identified 51 recent studies focusing on epigenetic mechanisms: DNA methylation (8 studies), histone modifications/chromatin remodeling (1 study), circRNAs (7 studies), lncRNAs (8 studies), miRNAs (25 studies), and competing RNAs (3 studies). All studies were conducted using human samples such as lung tissue, bronchoalveolar lavage (BAL) fluid, and blood. Moreover, several studies employed also in vitro and in vivo models to explain the effects of the epigenetic changes identified in clinical samples and to verify their use as potential therapeutic targets.
3.4. DNA Methylation
DNA methylation, the most extensively studied epigenetic mechanism, is able to inhibit gene expression by adding a methyl group to cytosine and by recruiting proteins involved in gene transcription repression (Table 4) [15]. This epigenetic mechanism affects mainly specific sites called CpG islands [15]. The imbalance in DNA methylation status can disrupt lung development, accelerating cellular aging and interfering with the inflammatory response to injuries [16].
Kachroo and colleagues investigated the relationship between fetal DNA methylation perturbations in fetal lung tissue exposed to maternal smoking, and DNA methylation in COPD development using an original network (pathways)-CpG approach that focused on few gene clusters relevant in COPD [16,17]. Genome-wide DNA methylation assay from fetal lung samples (n=42 tissue exposed to maternal smoking and n=36 unexposed) were compared with that from adult-lung tissue (n=160, 114 with COPD) [16,17]. No significant differences were found among the Genome-wide DNA methylation assay of fetal lung tissue exposed to maternal smoking compared to unexposed [16,17], and CpGs in fetal lung tissue exposed to maternal smoking overlapped with DNA methylation profile in adult COPD patients [17]. In particular, the co-methylation in “gene modules” involved in cell growth and development as Wnt, phosphatidylinositol 3-kinase (Pi3K)/ serine/threonine kinase (AKT), mitogen-activated protein kinase (MAPK) and serine/threonine-protein kinase (Hippo) in fetal lung tissues exposed to maternal smoking matched with the same co-methylation perturbations found in adult COPD [17]. These studies, also using a “gene promoter analysis” approach to validate COPD-related DNA methylation changes associated with age [16,17], suggested that, in adult subjects, specific CpGs associated with disrupted fetal development could be used to predict the chronological age (epigenetic clock) of the individual and to establish what subjects have the greatest risk for age-associated lung diseases [16,17].
Schwartz and colleagues examined the entire epigenomic landscape including gene promoter and enhancers to identify DNA methylation changes associated with genes and pathways essential in COPD pathophysiology and disease progression [18]. Parenchymal fibroblasts were isolated from lung tissues of ex-smoker control subjects (no COPD), patients with mild COPD (stage I, according with GOLD classification) and established COPD patients (stage II-IV). Tagmentation-based whole-genome bisulfite sequencing (T-WGBS) and RNA sequencing were performed to identify the differentially methylated regions (DMRs) profile and to evaluate gene expression changes in COPD patients and control groups [18]. DMRs profile and changes in expression of genes involved in cell proliferation, DNA repair and extracellular matrix organization were already found in COPD I patients compared to control [18]. Moreover, the changes in DNA methylation considering gene promoter and enhancer region suggested that all the epigenetic landscape should be take into account in the analysis of molecular mechanisms associated to COPD pathophysiology [18].
Eriksson Ström and colleague conducted epigenome-wide association studies (EWAS) on BAL cells from COPD and control smoker subjects to assess DNA methylation patterns and to verify a potential relationship among age and disease manifestation or progression [19]. The main cell type in BAL was macrophages, and 1155 differential methylation positions (DMPs) were found in COPD patients [19]. Moreover, 638 of 1155 DMPs showed higher mean methylation in COPD samples compared to control [19]. The most significant impact of COPD-DNA methylation alterations was observed in genes such as zinc finger DHHC-type palmitoyltransferase 14 (ZDHHC14), negative regulator of ubiquitin-like proteins 1 (NUB1), NLR family pyrin domain containing 3 (NLRP3), and zinc finger protein 322 (ZNF322) [19]. A strong correlation among DNA methylation and chronological age was found in COPD patients, but no association among epigenetic changes and progression of illness was observed. Interestingly, 38.7% of DMPs were in proximity to COPD-associated SNPs, helping to explain why some smokers are prone to develop COPD while others are not[19].
DNA methylation clocks evaluated in peripheral blood could be used as biomarkers to stratify patients with COPD according to their risk for lung complications. Cordero and colleagues calculated epigenetic age in COPD and control subject using six DNA methylation clocks [DNAmAge (pan-tissue), DNAmAgeHannum (blood-specific), DNAmAgeSkinBlood (fibroblast- and blood-specific), DNAmPhenoAge DNAmGrimAge and DNAmTelomereLength (DNAmTL)] 10. Blood and airway epithelial samples were tested and the DNAmGrimAge pattern showed a strong correlation in epigenetic age between the blood and the airways [10]. Moreover, DNAmGrimAge was suggested to have a role as a blood epigenetic age biomarker for assessing accelerated aging in the airways of individuals with COPD and in the control group [10].
A differentially methylation of two CpG sites of Pi3K catalytic subunit delta (Pi3KCD) (cg03971555 and cg12033075) was analyzed in blood collected from COPD patients and in the control group 20. The CpG sites were hypermethylated in COPD patients compared to control and were significantly associated with COPD mortality. Although the physio-pathological significance of these change in DNA methylation has yet to be clarified, the measured Pi3KCD hypermethylation could be used as a prognostic biomarker of COPD mortality [20].
The recent studies on changes in DNA methylation in COPD patients increased the understanding of the disease's molecular mechanisms suggesting potential treatment targets. The nuclear factor E2-related factor 2 (Nfr2), a key antioxidant element, regulates the expression of glutathione peroxidase, heme oxygenase-1 and NAD(P)H quinone oxidoreductase, protecting the cells from oxidative damage [21]. Zhang and colleagues found aberrant hyper methylation in CpG sites of Nrf2 promoter both in lung tissue and primary bronchial epithelial cells obtained from COPD patients compared to control group [21]. The Nrf2 downregulation leading to increased oxidative stress, cell death, and the progression of COPD [21]. As a result, targeting the methylation of CpG sites in the Nrf2 promoter was suggested as a potential treatment approach for COPD progression [21]. Another potential therapeutic target proposed for the treatment of COPD is the aryl hydrocarbon receptor repressor (AHRR), a repressor of aryl hydrocarbon receptor (AHR), that regulated the xenobiotic metabolism and the apoptotic processes [22,23]. The methylation of AHRR (CpG sites) gene promoter was lower in whole blood and airway epithelial cells from COPD patients compared to control, but only in active smokers22. The increased levels of AHRR could modify epithelial cell proliferation, dysregulate mitochondrial function, and reduce apoptotic processes upon cigarette smoke exposure, favoring worsening of lung function and COPD progression [22]. Regulation of AHRR CpGs methylation could, therefore, be considered as a therapeutic strategy in the treatment of COPD [22].
3.5. Histone Modifications
Histone modifications are epigenetic mechanisms involved in regulation of gene transcription, genomic instability, premature aging, inflammatory activation and DNA damage/repair (Table 5) [24]. In the most recent study exploring the relationship between histone modifications and COPD pathophysiology was conducted in 2022, the high expression levels of protein arginine methyltransferase 7 (PRMT7) were detected in subjects with COPD when compared to smoke controls [25]. In monocytes, PRMT7 was able to methylate histones on the arginine residues of the Ras-related protein 1A (Rap1a) enhancer, increasing the adhesion and migration capacity of monocytes onto lung tissue and negatively influencing the outcome of COPD patients [25]. These data were confirmed by in vivo study, where mice Prmt7+/- exposed to cigarette smoke (CSE) showed less monocytes/macrophage infiltration in lungs and were protected from chronic lung disease [25], thus suggesting the use of PRMT7 inhibitors as a possible therapeutic strategy to treat COPD.
3.6. Non-Coding RNA
3.6.1. Circular RNA
Circular RNAs (circRNAs) are a type of non-coding RNAs that form closed loops because of the lack of free 3′ and 5′ ends (Table 6). Some circRNAs have multiple miRNA-binding sites and can prevent miRNA-mRNA targets from binding, a phenomenon known as the "sponge effect." Apart from acting as miRNA sponges, circRNAs can also act as protein sponges, protein decoys, and scaffolds for forming protein complexes. Additionally, a few circRNAs have been found to regulate gene expression [26]. Aberrant expression of circRNAs is involved in the pathogenesis of several diseases including cardiovascular and pulmonary disorders, such as lung cancer, pulmonary fibrosis, tuberculosis and acute lung injury [26]. However, the association between circRNA and COPD development is poorly explored. circRNAs (N=81) were found to be differentially expressed between COPD and healthy subjects [27]. Out of these 81 circRNAs, Liu and colleagues focused on circ_FCHO2, circ_MBOAT2, circ_PTPN22, circ_TBC1D22A, circ_ACADM and circ_CKAP5 [27]. Bioinformatics tools demonstrated that these six circRNAs act as sponges of 18 miRNAs that were also differentially expressed between COPD patients and healthy subjects [27].
Two recent studies examined the expression profiles of circRNAs in peripheral blood mononuclear cells (PBMCs) of COPD patients and compared them with those of control subjects [28,29]. In the first study, 2132 circRNAs were found differentially expressed in patients with COPD compared to normal controls [28]. The mRNA targeted from dysregulated circRNA obtained by gene ontology (GO) enrichment analysis indicated that various signaling pathways, including cell death and apoptosis, as well as MAPK activation, may play a role in molecular and cellular processes associated with the COPD development [28]. Moreover, the Kyoto Encyclopedia of Genes and Genomes (KEGG) molecular pathway enrichment analysis revealed that the differentially expressed circRNAs in COPD could influence the disease development by affecting the NOD-like receptor signaling, the Toll-like receptor signaling, and Type 1 T helper (Th1) /Th2 cell differentiation 28. In the second study, the analysis of circRNAs profile in PBMCs showed high expression levels of hsa-circ_0008833 in COPD patients compared to controls [29]. The functional investigation analysis suggested that circ_0008833 could promote COPD progression by inducing a distinct form of inflammatory cell death (pyroptosis) in bronchial epithelial cells [29]. The hsa-circ_0008833 derived from the SMAD3 gene and resulted able to encode to a small peptide (57aa). Study in vitro using 16HBE bronchial epithelial cells confirmed the ability of hsa-circ-0008833-57aa pathway to increase the mRNA of Caspase 1, IL-18, IL-1, inducing pyroptosis [29].
In an attempt to explain the role of circRNA in COPD pathogenesis, Zhang and colleagues examined the effect of circ_0006892 in bronchial epithelial cell injuries [30]. Circ_0006892 acts as a sponge for miR-24, reducing the bronchial epithelial cell inflammatory injury [30], confirming that the circ_0006892 expression was downregulated in the lung tissue of COPD smokers compared to non-smoker COPD [30]. Moreover, in COPD patients, the disequilibrium in circ_0006892-miR-24 reduced the expression of anti-apoptotic and inflammatory elements, thus promoting the exacerbation of COPD processes through low levels of PH domain and leucine-rich repeat protein phosphatase 2 (PHLPP2) [30]. Differently from circ_0006892, the expression of circ_ANKRDII was found to be up-regulated in the lung tissue collected from COPD smoker patients when compared to the smoker and non-smoker subjects without COPD [31]. The authors demonstrated that circ_ANKRDII binds to miR-145-5p, neutralizing its protective effects and contributing to the molecular mechanisms underlying COPD pathogenesis, such as inflammation, oxidative stress, and apoptosis [31].
CircRNAs, owing to their distinctive structural conformation that makes them particularly stable in circulation, have been suggested as potential biomarkers. In a recent study, 119 circular RNAs were identified to be differentially expressed in plasma samples from individuals with very severe COPD compared to the control group (90 upregulated and 29 downregulated circular RNAs) [32]. After validation by real-time PCR, only the 4 circRNAs confirmed to be differentially expressed between the two groups were considered as potential prognostic biomarkers of severe COPD [32]. Moreover, GO and KEGG enrichment analysis indicated that circ_0008882, circ_00089763, circ_00062683, and circ_00077607 may be involved in the onset and progression of COPD by influencing immune cell development and activity [32]. Additional study suggested a role for circ_0049875 and circ_0042590 as diagnostic biomarkers of COPD [33]. Shen and colleagues observed that circ_0042590 was up regulated while circ_0049875 was down regulated in PBMCs collected from COPD compared to control [33]. Moreover, a strong correlation among circ_0049875 and circ_0042590 levels and acute exacerbation of COPD was verified [33].
3.6.2. Long Non-Coding RNA
Long non-coding RNAs (lncRNAs) are a type of non-coding transcripts longer than 200 nucleotides and characterized by secondary or tertiary structures which resemble proteins (Table 7) [34]. Most lncRNAs are found in the nucleus, but can also be detected in cytoplasm, serum and extracellular vesicles [34]. LncRNAs play a role in regulating gene expression through epigenetic, transcriptional, and post-transcriptional mechanisms [34].
Several studies before 2020 already suggested that among lncRNAs, the lncTUG1 and ANRIL may be involved in inflammatory process affecting airway epithelial cells of COPD patients [35,36]. Studies since 2020 have confirmed the close relationship between inflammation/lncRNAs/COPD. Wu and colleagues explored the role of lncRNA-IL7 receptor (lncRNA-IL7R) in COPD pathophysiology [37]. The lncRNA-IL7R is an anti-inflammatory lncRNA-s induced by toll like receptor (TLR)2 and 4 activation [37]. In PBMCs, lncRNA-IL7R is located in the nucleus and can regulated the expression of IL-8 by the deacetylation of lysine 9 on histone H3 (H3K9) and the tri-methylation of lysine 9 on histone H3 (H3K9me3) and histone H3 lysine 27 (H3K27me3) [37]. The low expression levels of lncRNA-IL7R were correlated with inflammation and acute exacerbation phenotype of COPD [37].
Long non-coding RNA homeobox A cluster antisense RNA 2 (lncRNA- HOXA-AS2) is a lncRNA located between the human HOXA3 and HOXA4 gene [38]. The role of lncRNA- HOXA-AS2 in cellular and molecular mechanisms associated to COPD were explain by in vitro study using Human Pulmonary Microvascular Endothelial Cells (HPMEC) under CSE [38]. Low levels of lncRNA- HOXA-AS2 in HPMEC were linked to reduced cell proliferation and viability, contributing to endothelial dysfunction through the activation of inflammatory responses [38]. The lncRNA- HOXA-AS2 resulted down regulated in lung tissue samples collected from COPD patients compared to control [38]. Moreover, the authors suggested that the effects of lncRNA- HOXA-AS2 were mediated by notch receptor 1 (Notch1) (receptor capable of monitoring proliferation, differentiation, apoptosis, and stem cell maintenance) [38].
Besides their role in the pathogenesis of chronic pulmonary disease, lncRNAs are also proposed as novel biomarkers for the progression and exacerbation of COPD. Wang and colleagues investigated the ability of lncRNA- plasmacytoma variant traslocation 1(lncRNA-PVT1) as predictor of COPD exacerbation [39]. The expression levels of lncRNA-PVT1 were evaluated in PBMCs collected from healthy subjects, stable COPD and acute exacerbation COPD (AECOPD) patients [39]. Data indicated that lncRNA-PVT1 levels were significantly elevated in COPD patients compared to controls. Additionally, lncRNA-PVT1 levels were able to distinguish AECOPD patients from stable COPD [39]. Previous research indicated that lncRNA-PVT1 plays a pro-inflammatory role in other diseases by reducing the function of miR-146a [40,41]. LncRNA-PVT1 levels were positively correlated with expression of inflammatory mediators such as IL-6, TNF, IL-8 and IL-17 in both stable COPD and AECOPD patient [39]. Furthermore, the expression of anti-inflammatory miR-146a showed a negative correlation with lncRNA-PVT1 levels and the progression of COPD [39]. These findings suggest that the pro-inflammatory pathway of lncRNA-PVT1 relied on the inhibition of its specific target, miR-146a [39]. Similarly, also lncRNA-metastasis associated lung adenocarcinoma transcript1 (lncRNA-MALT1) was proposed as potential biomarker for predicting acute exacerbation risk and disease progression of COPD [42]. Circulating lncRNA-MALAT1 levels were positively associated to GOLD stage and inflammatory mediator (TNF, IL-6, IL-8, IL-1IL-17 and IL-23) levels and negatively related to expression levels of miRNA potentially involved in the reduction of inflammatory processes (miR-133, miR-125b, miR-146a and miR-203) [42]. Moreover, blood levels of lncRNA-MALAT1 were described as good tool for distinguish AECOPD from COPD patients [42]. A recently study proposed lncRNA-cancer susceptibility candidate 2 (lncRNA-CASC2) as a diagnostic biomarker of COPD [43]. In a vitro study using human bronchial epithelial cell line, it was observed that under normal conditions, lncRNA-CASC2 acts as a sponge for miR-18a-5p, reducing the expression of pro-inflammatory cytokines [43]. Circulating levels of lncRNA-CASC2 were found to be significantly decreased in COPD patients compared to smoking control subjects and were able to distinguish severe COPD to mild and moderate cases [43]. Moreover, the levels of lncRNA-CASC2 were negatively correlated with increased miR-18a-5p and inflammatory cytokines, thus confirming its role in regulating inflammation [43].
High circulating levels of lncRNA-LUCAT1 were detected in the blood samples collected form patients with liver, pulmonary, gastric and breast cancer [44]. Zhao and colleagues investigated the expression of lncRNA-LUCAT1 in COPD and its potential role as a biomarker [44]. The data indicated that serum levels of lncRNA-LUCAT1 could effectively distinguish COPD patients from healthy individuals [44]. Additionally, lncRNA-LUCAT1 levels were found to be higher in smokers with COPD compared to non-smokers with COPD [44]. Mechanistically, it was suggested that lncRNA-LUCAT1 impaired the anti-inflammatory and anti-apoptotic effects of miR-181-5p in bronchial epithelial cells, thereby contributing to the progression of COPD [44].
Recent studies have highlighted the possibility of using lncRNAs also as COPD therapeutic targets [45,46].The lncRNA-HOX transcript antisense (lncRNA-HOTAIR) is involved in various carcinogenic processes, and, specifically, was up-regulated in COPD lung tissue compared to healthy tissue [45]. In human pulmonary endothelial cells treated with CSE the level of lncRNA-HOTAIR increased, thus contributing to the apoptosis process by hypermethylation of Bcl-2 gene promoter [45] and to the increased levels of IL-1b and IL-6. The expression levels of lncRNA-colon cancer associated transcript 1 (lncRNA-CCT1), similarly to lncRNA-HOTAIR, was found to be up-regulated in lung tissue collected from COPD patients compared to control group [46]. Mechanistically, it was suggested that lncRNA-CCT1, through the inhibition of the anti-inflammatory miR-152-3p, could promote the inflammatory processes by activating ERK signaling [46]. Given the role of lncRNA-HOTAIR and lncRNA-CCT1 in the molecular mechanisms linked to COPD, these findings indicated that these lncRNAs may serve as novel therapeutic targets for COPD prevention [45,46].
3.6.3. Micro RNA
MicroRNAs (miRNAs), a family of single-stranded, non-coding small RNAs, approximately 22 nucleotides in length (Table 8) 47, play a crucial role in regulating various physiological and pathological processes by interacting in a sequence-specific manner with mRNAs in the cytoplasm [47]. miRNAs directly influence gene expression by recruiting different protein complexes to the promoter or enhancer regions of target genes, which can lead to either the activation or repression of transcription [47]. Several studies explored the role of abnormal miRNAs expression in COPD patients, indicating their significant function in the pathophysiology of disease and their potential use as biomarkers and therapeutic targets of COPD.
The analysis of PBMCs transcriptome and miRNome profiles in severe COPD patients and control group were assessed to identify the mRNAs and miRNAs associated with disease [48]. The application of bioinformatics tools to identify altered pathways in PBMCs from COPD patients suggested that the IL-8 and inducible T cell costimulator (iCOS)-iCOS ligand (iCOSL) signaling were primarily involved in disease progression [48]. Among differentially expressed miRNAs, 7 miRNAs were found to be able to target genes involved in IL-8 signaling, while 12 miRNAs were associated with iCOS-iCOSL signaling regulation, suggesting a role for these miRNAs in inflammatory processes [48]. In addition to PBMC, miRNome was carried out also on BALF and plasma samples collected from COPD patients and compared with control group [49]. A total of 6 miRNAs-BALF were found differentially expressed between COPD and healthy subjects, while only 3 miRNAs were significantly down regulated in COPD plasma samples compared to control [49]. The GO enrichment analysis and KEGG analysis indicated that the differentially expressed miRNAs in both BALF and blood contributed to the oxidative and inflammatory processes underlying COPD through the regulation of the MAPK, RAS, and FOXO signaling pathways [49].
In addition to exploratory studies, other research has examined the specific role of miRNAs in COPD. The impact of miR-486-5p on COPD using alveolar macrophages and peripheral monocytes was studied [50]. Expression levels of miR-486-5p were significantly up regulated in monocytes/macrophages collected from COPD patients compared to control group [50]. Moreover, the increased levels of miR-486-5p were associated to TLR4 and pro-inflammatory cytokines expression [50]. In vitro study, on a CSE/lung alveolar macrophage cell line, confirmed that miR-486-5p contributed to the overexpression of TLR4 and cytokines by inhibiting the expression of histone acetyltransferase (HAT1) [50]. Moreover, high circulating levels of miR-486-5p together with miR-106b-5p were observed in blood samples from COPD patients when compared to control group, suggesting the possible use of these ncRNAs as diagnostic biomarkers of hypoxia and pulmonary hypertension (PH) [51].
Still in the context of the role of inflammation in COPD, contrasting results were obtained studying the expression levels of miR-221-3p in COPD patients [52,53]. The miR-221-3p expression was down regulated in COPD lung tissue compared to control [52] and the low levels of miR-221-3p were negatively related with expression of cyclin dependent kinase inhibitor 1B (CDKN1B), pro-inflammatory and apoptotic elements [52]. In contrast to the lung expression, in the serum samples collected from AECOPD and stable COPD patients the circulating levels of miR-221-3p were significantly higher compared to control [53]. The 16HBEC treated with CSE expressed higher miR-221-3p levels compared to untreated cells. Moreover, the increased levels of miR-221-3p were positively correlated with the mRNA of IL-6, IL-1b, TNFα, Collagen IV, Fibronectin, and a-SMA [53]. Finally, Shen and colleagues suggested that circulating miR-221-3p in combination with miR-92a-3p could be used as a diagnostic biomarker of COPD, able to distinguish also COPD exacerbation from stable COPD [53].
The expression of Receptor for Advanced Glycation End-products (RAGE), a multi-ligand receptor of the immunoglobulin superfamily able to activate ROS signaling and MAPK pathway, was higher in lung tissues collected from COPD patients compared to control [54]. In vivo and in vitro studies showed that increasing miR-23a-5p levels could reduce the inflammatory and oxidative processes involved in COPD, specifically by targeting RAGE [54]. The anti-inflammatory effect of miR-23a-5p may be absent in COPD, considering the low expression levels of miRNA in patients compared to a control group 54 . Other inflammatory miRNAs linked to the pathogenesis of COPD are miR-21 and miR-155 [55,56]. In bronchial biopsies taken from COPD patients, the expression levels of miR-21 and miR-155 were found to be up-regulated compared to those in healthy subjects [55,56]. Through appropriate in vitro and in vivo studies was demonstrated that miR-21, by inhibiting the expression of the anti-inflammatory factor special AT-rich binding protein 1 (SATB1), would induce the increase in the expression of a chemotactic factor such as calgranulins S100 calcium binding proteinA9 (S100A9) and the activity of the “inflammatory transcription factor” NF-kB [55]. In patients with COPD, elevated lung levels of miR-155 could contribute to inflammation by inducing the expression of MMP12 and ADAM19, which are proteases that cleave extracellular matrix proteins and release cytokines, such as TNFα [56].
Zhu and colleagues investigated the role of miR-103a in COPD pathophysiology and discovered a connection between the miRNA and the impaired function of alveolar macrophages in COPD patients [57]. Surfactant proteins are produced from Type II alveolar cells to preserve the surface tension. CSE increased the oxidation of surfactant proteins. Under normal conditions, alveolar macrophages should remove oxidized proteins protecting alveolar compliance [57]. In alveolar COPD patients the increase in oxidized proteins was associated with the formation of lipid-laden macrophages or foam cells [57]. The down regulation of miR-103a in alveolar macrophages collected by bronco alveolar lavage from COPD patients could explain the dysfunction of these inflammatory cells [57] Both in vitro and in vivo studies confirmed that CSE induces a reduction the expression of miR-103a by increased surfactant oxidative protein, leading to an increased in its mRNA target LDLR and resulting in lipid and oxidized lipid accumulation in macrophages [57].
One of the most common complications of COPD is PH, characterized by the remodeling of the pulmonary vessels, leading to thickening and stiffening of the vessels [58]. Yang and colleagues explored the miRNAs potentially involved in COPD-PH severity and progression [59]. miR-4640-5p was up regulated in lung tissues collected from COPD-PH compared to normal lung [59] and high levels of miR-4640-5p were able, in pulmonary vascular smooth muscle cells, to induce a decrease of NOS1 expression and an increase of cell proliferation [59], thus causing thickening of the vessel wall and progression of the disease [59]. In addition to PH, the higher production and accumulation of mucus in airways was observed in COPD patients. Anoctamin 1 (ANO1), a calcium-activated chloride channel present in the airway epithelium, is involved in mucus secretion and regulation of vascular contraction. Singh and colleagues verified that in the lung tissue of COPD smokers, the expression levels of ANO1 were higher compared to the control [60]. Moreover, the exposition to low doses of cadmium, a toxic substance released by burning tobacco, increased the expression of ANO1 by downregulating miR-381[ 60]. In vitro studies confirmed that miR-381 directly regulates ANO1 mRNA, suggesting that miR-381 could be a potential target of treatment for patients with COPD [60]. In addition to mucus secretion, in COPD patients, the cadmium exposition and accumulation in the blood can contribute to epithelial-mesenchymal transition (EMT) and COPD progression [61]. Elevated levels of cadmium in plasma correlated with a decrease in the pulmonary expression of epithelial markers, such as E-cadherin, and with an increase in mesenchymal biomarkers, such as N-cadherin, Vimentin, and α-SMA [61]. Zheng and colleagues found that circulating levels of miR-30 negatively correlated with plasma cadmium concentration in COPD patients [61]. Furthermore, in vitro experiments confirmed that exposure to cadmium promoted EMT in lung epithelial cells by reducing miR-30 expression [61].
Other miRNAs associated with chronic mucus hypersecretion (CMH) in COPD patients were observed [62]. Tasena and colleagues investigated the role of some CMH-associated miRNAs in aberrant fibroblast-epithelial cell crosstalk in COPD patients [63]. Using a co-culture of primary bronchial epithelial cells (PBECs) and pulmonary airway fibroblasts (PAFs) collected from COPD-CMH patients, an increase in the expression levels of miR-708-5p was observed in the PBECs. In contrast, let-7a-5p and miR-31-5p showed higher expression levels in the PAFs after their interaction with the epithelial cells [63]. Tasena and colleagues observed that in physiological condition the expression levels of miR-708-5p decreased in PBECs during mucociliary differentiation and that an increased in miR-708-5p suppressed MUCS5AC secretion, as demonstrated after miRNA-mimic transfection study [63]. Considering that in miR-708-5p expression increased in PBECs collected from COPD patients, it was suggested that the regulatory mechanisms of this miRNA on MUCS5AC were altered by other factors [63]. The changes in the expression levels of let-7a-5p and miR-31-5p in co-cultured PAFs were linked to a decrease in the expression of COL4A1 and COL5A1. This decrease may contribute to abnormalities in the basement membrane, which could, in turn, affect epithelial differentiation and promote CMH [63].
The necessity of identifying diagnostic and prognostic biomarkers for the early detection of COPD onset and progression has prompted numerous studies to explore the potential use of miRNAs as markers in patients suffering from chronic lung disease. In this context, several studies reported that circulating miR-548ar-3p, miR-146a, miR-218, miR-150-5p, miR-423-5p, miR-1290 and miR-1246 were under-expressed in COPD patients compared to control group [64,65,66,67,68]. miR-146a and miR-218 involved in inflammatory response and oxidative processes were able to discriminate smokers COPD from non-smokers COPD [65], while the miR-150-5p, miR-423-5p, miR-1290 and miR-1246 could be used as markers of disease worsening considering that the circulating levels of miRNAs were relatively lower in patients with severe lung function decline compared to stable COPD [66,67,68]. Moreover, the changes in the circulating levels of miR-1246 during 10 years of follow-up in relation to COPD progression were studied [69]. Cazola-Rivero and colleagues suggested that circulating levels of miR-1246 do not effectively differentiate between COPD patients and healthy individuals [69] . Furthermore, no significant changes in miR-1246 levels were observed in COPD patients at follow-up compared to baseline measurements [69]. However, it was confirmed that miR-1246 could distinguish between COPD patients and those with both COPD/emphysema, suggesting the use of miRNA as biomarker of disease severity [69].
Conversely, circulating levels of miR-4433a-5p, miR-126 and miR-210 were elevated in COPD patients compared to controls [68,70,71]. The expression of miR-4433a-5p gradually increased in blood samples of COPD patients with the progression of disease [68]. miR-4433a-5p targeting PIK3R2 gene could promote the apoptosis and inflammatory processes in COPD patients [68]. The miR-126 is a marker of inflammation and can contribute directly to processes regulating the synthesis of pro-inflammatory cytokines [70]. Wang and colleagues found that circulating levels of miR-126 were elevated in COPD patients compared to a control group. This miRNA was also able to differentiate patients with acute exacerbations of the disease from those with stable COPD [70]. Finally, miR-210 was described as early biomarker of PH in COPD patients [71]. No differences in blood miR-210 levels were found when stable COPD were compared with healthy subjects [71]. In patients with COPD-induced pulmonary hypertension, miR-210 expression was significantly higher compared to patients with stable COPD [71].
In addition to free miRNAs in various biological fluids, it is possible to identify miRNAs encapsulated within extracellular vesicles (EVs), which could also be used as biomarkers. Very recently, the profile of EV-associated miRNAs in BALF from COPD patients was compared to that of a control group [72]. The number of EVs in the control groups was lower than in COPD patients, and the combination of EVs- miR-2110, miR-223-3p, and miR-182-5p was able to distinguish between healthy patients and patients with lung disease [72]. Moreover, miR-2110 and miR-182-5p were associated with neutrophil expression [72]. In contrast, EVs-miRNAs like miR-223-3p, miR-338-3p, and miR-204-5p were linked to eosinophil expression. This suggests that these specific EVs-miRNAs could be used to identify the inflammatory patterns underlying COPD [72]. Among the EVs, exosomes, the small nanovesicles released in extracellular space, were studied as carriers of miRNAs in different diseases. In serum, the levels of free or exosom-miR-1258 were found significantly higher in COPD patients compared to control 73. Moreover, exosome-miR-1258 proved to be a more accurate marker for distinguishing COPD patients experiencing acute exacerbations of the disease from those with stable COPD [73].
In a recent work, miRNAs were proposed as therapeutic targets in treating patients with COPD [74]. It is well known that chronic inflammation disrupts the balance between apoptosis and cellular proliferation, leading to one of the adverse effects of COPD progression: airway endothelial cell hyperplasia [74]. miR-196-5p and miR-361-5p were differentially expressed in blood samples collected from COPD patients compared to control [74]. These miRnas were able to promote hyperplasia in endothelial cells reducing the mRNA-ARHGEF12 (miR-196-5p) and increasing the expression levels of mRNA-BCAT1(miR-361-5p) [74]. In an in vitro study, gamma-sterol (GS) was shown to reduce epithelial cell hyperplasia by decreasing the expression of miR-196-5p and increasing the levels of miR-361-5p suggesting its possible use in the treatment of COPD patients [74].
3.6.4. Competing Endogenus RNA
In the recent years, a new theory called "competing endogenous RNA" (ceRNA) has been explored to study various diseases [75]. The ceRNA theory suggests that mRNA, lncRNA, and circRNA could function as miRNA sponges, thereby regulating the expression of specific mRNAs and biological networks [75]. In relation to the development of COPD, two works have attempted to identify the ceRNA networks (Table 9) [76,77]. The mRNA/lncRNA/circRNA/miRNA profile were analyzed in lung tissue collected from COPD patients and healthy subjects [76,77], and 1289 mRNAs, 69 miRNAs, 32 circRNAs and 433 lncRNAs were found differentially expressed between two groups [76,77]. After an appropriate bioinformatics analysis, the authors conclude that these ceRNAs differentially expressed between healthy and COPD patients could be involved in the regulation of 18 gene hubs that including TGF signaling and Wnt/-catenin signaling, immune cell infiltration, M2 macrophage differentiation, and NK cell activation [76,77].
4. Discussion
This systematic review summarized the available evidence regarding epigenetic alterations in COPD patients after the pandemic emergency due to SARS-CoV-2. Current evidence provides detailed description of epigenetic elements and their related pathways involved in COPD onset, which could be used for the development of the diagnostic and therapeutic strategies for the lung disease. A key aspect highlighted in many publications is that, at the molecular level, the epigenetic elements associated with COPD play a role in regulating the expression of factors involved in inflammation, oxidative stress, apoptosis, and cell proliferation. In certain studies, it was observed that COPD patients exhibited elevated levels of circ_0008833, miR-486-5p, and miR-21, while the levels of lncR-17R were decreased in PBMC and/or lung tissue when compared to a control group [29,37,50,55]. These changes contributed to an increased expression of pro-inflammatory cytokines and chemokines and an impaired lung function. On the other hands, aberrant expression levels of circANKRDII, lncR-HOXA-AS2, miR-221-3p and miR-23a-5p resulted in dysregulation of apoptosis/cell proliferation and oxidative stress [31,38,52,54]. Moreover, some miRNAs have shown the ability to interfere also with processes essential for disease progression, such as mucus production, and mechanisms related to both epithelial-mesenchymal transition and PH [59,60,63].
The epigenetic studies also have evidenced a great potential to identify probable biomarkers of COPD. Seventeen out of the 51 studies in this systematic review discuss the use of epigenetic factors as potential biomarkers. Once again, a lengthy list of circRNAs, lncRNAs, and miRNAs related to an inflammatory process has been identified as able to indicate the presence of a disease. Considering the studies involving a larger number of COPD subjects and normal subjects, ncRNAs, including circ_00089763, circ_0008882, lncR-PVt1, lncR-MALAT1, miR-126, miR-221-3p, and miR-92a-3p, have been described as excellent inflammatory biomarkers able to establishing the presence of COPD [32,39,42,53,70]. Studies on changes in DNA methylation related to COPD reported that alterations in CpG methylation linked to disrupted fetal development are similar to the DNA methylation patterns observed in adult COPD patients [16,17]. These specific DNA methylation regions could potentially help in predicting which individuals are at a higher risk for developing age-related lung diseases.
The most interesting aspect of epigenetic modifications is their reversibility, which positions them as promising candidates for pharmacological treatments. This review reports the most recent studies on the treatment of COPD that target epigenetic factors. Among them, Chen and colleagues suggest that restoring AHRR gene methylation (which reduces AHRR mRNA and protein levels) may be an effective therapeutic strategy. This approach may improve mitochondrial function and help reduce programmed cell death in COPD patients exposed to cigarette smoke [22]. Another promising therapeutic target for preventing COPD has been identified in Lnc-HOTAIR. Reducing Lnc-HOTAIR expression may lead to decreased apoptosis by enhancing Bcl-2 production in the lung [45]. These studies will require further verification, also considering that the dysregulation of multiple epigenetic factors occur during the onset of COPD, and therefore re-modulating only the methylation of a gene or the expression of an ncRNA may not have the desired therapeutic effect.
What emerged in this systematic review is that the primary criticism of epigenetic studies concerning COPD is the variability in sample size, type, and methods used for evaluating epigenetic changes. Moreover, while most DNA methylation and histone modification studies were conducted on European and American populations, the non-coding RNA studies were carried out in China.
Finally, the above data support that epigenetic regulation affects the pathophysiology of COPD disorders with potential diagnostic or therapeutic utility. Factors like ethnicity and environmental influences may act as confounding variables in the study of epigenetic mechanisms related to COPD. Therefore, it is essential to approach the topic of epigenetics in COPD by considering multiethnic studies. This approach could help to reduce these confounding factors and reveal the fundamental mechanisms that regulate the onset of the disease.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Supplementary Table S1: PRISMA checklist; Supplementary Table S2: AXIS tool; Supplementary Table S3: PRISMA abstract checklist.
Author Contributions
Conceptualization, R.R. and C.C.; methodology, R.R and P.B; validation, A.T.,M.L and C.C; investigation, R.R, P.B. AND C.C; writing—original draft preparation, R.R. and P.B.; writing—review and editing, A.T., M.L. and C.C.; supervision, C.C. Funding Acquisition, A.T, M.L.
Funding
Horizon Europe - TOLIFE - HORIZON-HLTH-2021-DISEASE-04, Project number 101057103.
Data Availability Statement
Registration to Open Science Framework platform (available online at https://osf.io/4ja25/; accessed on 2025-01-31) and supplementary tables.
Acknowledgments
This project has received funding from the European Union’s Horizon Europe Research and Innovation Programme under grant agreement No. 101057103 – project TOLIFE.
Conflicts of Interest
The authors declare no conflict of interest.
Disclaimer
Funded by the European Union. Views and opinions expressed are however those of the authors only and do not necessarily reflect those of the European Union or the European Health and Digital Executive Agency. Neither the European Union nor the granting authority can be held responsible for them.
References
- Barnes, P.J. Inflammatory Mechanisms in Patients with Chronic Obstructive Pulmonary Disease. Journal of Allergy and Clinical Immunology, Mosby Inc. July 1, 2016, pp 16–27. [CrossRef]
- WHO. Https://Www.Who.Int/News-Room/Fact-Sheets/Detail/Chronic-Obstructive-Pulmonary-Disease-(Copd); 2023.
- Agustí, A.; Celli, B.R.; Criner, G.J.; Halpin, D.; Anzueto, A.; Barnes, P.; Bourbeau, J.; Han, M.L.K.; Martinez, F.J.; de Oca, M.M.; et al. Global Initiative for Chronic Obstructive Lung Disease 2023 Report: GOLD Executive Summary. European Respiratory Journal 2023, 61. [Google Scholar] [CrossRef] [PubMed]
- Safiri, S.; Carson-Chahhoud, K.; Noori, M.; Nejadghaderi, S.A.; Sullman, M.J.M.; Ahmadian Heris, J.; Ansarin, K.; Mansournia, M.A.; Collins, G.S.; Kolahi, A.A.; et al. Burden of Chronic Obstructive Pulmonary Disease and Its Attributable Risk Factors in 204 Countries and Territories, 1990-2019: Results from the Global Burden of Disease Study 2019. The BMJ 2022. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Aarsand, R.; Schotte, K.; Han, J.; Lebedeva, E.; Tsoy, E.; Maglakelidze, N.; Soriano, J.B.; Bill, W.; Halpin, D.M.G.; et al. Tobacco and COPD: Presenting the World Health Organization (WHO) Tobacco Knowledge Summary. Respiratory Research, BioMed Central Ltd December 1, 2024. [CrossRef]
- Lee, Y.J.; Choi, S.; Kwon, S.Y.; Lee, Y.; Lee, J.K.; Heo, E.Y.; Chung, H.S.; Kim, D.K. A Genome-Wide Association Study in Early Copd: Identification of One Major Susceptibility Loci. International Journal of COPD 2020, 15, 2967–2975. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Bakshi, A.; Zhu, Z.; Hemani, G.; Vinkhuyzen, A.A.E.; Lee, S.H.; Robinson, M.R.; Perry, J.R.B.; Nolte, I.M.; Van Vliet-Ostaptchouk, J.V.; et al. Genetic Variance Estimation with Imputed Variants Finds Negligible Missing Heritability for Human Height and Body Mass Index. Nat Genet 2015, 47, 1114–1120. [Google Scholar] [CrossRef]
- Cho, M.H.; Hobbs, B.D.; Silverman, E.K. Genetics of Chronic Obstructive Pulmonary Disease: Understanding the Pathobiology and Heterogeneity of a Complex Disorder. The Lancet Respiratory Medicine, Elsevier Ltd May 1, 2022, pp 485–496. [CrossRef]
- Feng, X.; Dong, H.; Li, B.; Yu, L.; Zhu, J.; Lou, C.; Zhang, J. Integrative Analysis of the Expression Profiles of Whole Coding and Non-Coding RNA Transcriptomes and Construction of the Competing Endogenous RNA Networks for Chronic Obstructive Pulmonary Disease. Front Genet 2023, 14. [Google Scholar] [CrossRef]
- Hernandez Cordero, A.I.; Yang, C.X.; Li, X.; Yang, J.; Shaipanich, T.; MacIsaac, J.L.; Lin, D.T.S.; Kobor, M.S.; Horvath, S.; Man, S.F.P.; et al. The Blood DNA Methylation Clock GrimAge Is a Robust Surrogate for Airway Epithelia Aging. Biomedicines 2022, 10. [Google Scholar] [CrossRef]
- Li, B.; Zhang, J.; Dong, H.; Feng, X.; Yu, L.; Zhu, J.; Zhang, J. Systematic Analysis of Various RNA Transcripts and Construction of Biological Regulatory Networks at the Post-Transcriptional Level for Chronic Obstructive Pulmonary Disease. J Transl Med 2023, 21. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 Statement: An Updated Guideline for Reporting Systematic Reviews. Syst Rev 2021, 10. [Google Scholar] [CrossRef]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; Antes, G.; Atkins, D.; Barbour, V.; Barrowman, N.; Berlin, J.A.; Clark, J.; et al. Preferred Reporting Items for Systematic Reviews and Meta-Analyses: The PRISMA Statement. PLoS Medicine, Public Library of Science July 1, 2009. [CrossRef]
- Downes, M.J.; Brennan, M.L.; Williams, H.C.; Dean, R.S. Development of a Critical Appraisal Tool to Assess the Quality of Cross-Sectional Studies (AXIS).
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology, January 2013, pp 23–38. [CrossRef]
- Kachroo, P. Priyadarshini Kachroo 2021.
- Kachroo, P.; Morrow, J.D.; Kho, A.T.; Vyhlidal, C.A.; Silverman, E.K.; Weiss, S.T.; Tantisira, K.G.; DeMeo, D.L. Co-Methylation Analysis in Lung Tissue Identifies Pathways for Fetal Origins of COPD. European Respiratory Journal 2020, 56. [Google Scholar] [CrossRef]
- Schwartz, U.; Llamazares Prada, M.; Pohl, S.T.; Richter, M.; Tamas, R.; Schuler, M.; Keller, C.; Mijosek, V.; Muley, T.; Schneider, M.A.; et al. High-resolution Transcriptomic and Epigenetic Profiling Identifies Novel Regulators of COPD. EMBO J 2023, 42. [Google Scholar] [CrossRef]
- Strom, J.E.; Merid, S.K.; Pourazar, J.; Blomberg, A.; Lindberg, A.; Ringh, M.V.; Hagemann-Jensen, M.; Ekstrom, T.J.; Behndig, A.F.; Melen, E. Chronic Obstructive Pulmonary Disease Is Associated with Epigenome-Wide Differential Methylation in BAL Lung Cells. Am J Respir Cell Mol Biol 2022, 66, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Morrow, J.D.; Make, B.; Regan, E.; Han, M.L.; Hersh, C.P.; Tal-Singer, R.; Quackenbush, J.; Choi, A.M.K.; Silverman, E.K.; DeMeo, D.L. DNA Methylation Is Predictive of Mortality in Current and Former Smokers. Am J Respir Crit Care Med 2020, 201, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Fu, C.; Liu, J.; Sai, X.; Qin, C.; Di, T.; Yang, Y.; Wu, Y.; Bian, T. Hypermethylation of the Nrf2 Promoter Induces Ferroptosis by Inhibiting the Nrf2-GPX4 Axis in COPD. International Journal of COPD 2021, 16, 3347–3362. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Nwozor, K.O.; van den Berge, M.; Slebos, D.J.; Faiz, A.; Jonker, M.R.; Boezen, H.M.; Heijink, I.H.; de Vries, M. From Differential DNA Methylation in COPD to Mitochondria: Regulation of AHRR Expression Affects Airway Epithelial Response to Cigarette Smoke. Cells 2022, 11. [Google Scholar] [CrossRef]
- Kodal, J.B.; Kobylecki, C.J.; Vedel-Krogh, S.; Nordestgaard, B.G.; Bojesen, S.E. AHRR Hypomethylation, Lung Function, Lung Function Decline and Respiratory Symptoms. European Respiratory Journal 2018, 51. [Google Scholar] [CrossRef]
- Yao, H.; Rahman, I. Role of Histone Deacetylase 2 in Epigenetics and Cellular Senescence: Implications in Lung Inflammaging and COPD. Am J Physiol Lung Cell Mol Physiol 2012, 303, 557–566. [Google Scholar] [CrossRef]
- Günes Günsel, G.; Conlon, T.M.; Jeridi, A.; Kim, R.; Ertüz, Z.; Lang, N.J.; Ansari, M.; Novikova, M.; Jiang, D.; Strunz, M.; et al. The Arginine Methyltransferase PRMT7 Promotes Extravasation of Monocytes Resulting in Tissue Injury in COPD. Nat Commun 2022, 13. [Google Scholar] [CrossRef]
- Santer, L.; Bär, C.; Thum, T. Circular RNAs: A Novel Class of Functional RNA Molecules with a Therapeutic Perspective. Molecular Therapy 2019, 27, 1350–1363. [Google Scholar] [CrossRef]
- Liu, P.; Wang, Y.; Zhang, N.; Zhao, X.; Li, R.; Wang, Y.; Chen, C.; Wang, D.; Zhang, X.; Chen, L.; et al. Comprehensive Identification of RNA Transcripts and Construction of RNA Network in Chronic Obstructive Pulmonary Disease. Respir Res 2022, 23. [Google Scholar] [CrossRef]
- Duan, R.; Niu, H.; Yu, T.; Cui, H.; Yang, T.; Hao, K.; Wang, C. Identification and Bioinformatic Analysis of Circular RNA Expression in Peripheral Blood Mononuclear Cells from Patients with Chronic Obstructive Pulmonary Disease. International Journal of COPD 2020, 15, 1391–1401. [Google Scholar] [CrossRef]
- Xie, T.; Yang, Z.; Xian, S.; Lin, Q.; Huang, L.; Ding, Y. Hsa_circ_0008833 Promotes COPD Progression via Inducing Pyroptosis in Bronchial Epithelial Cells. Exp Lung Res 2024, 50, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Gu, S.; Kang, X. CircRNA Circ_0006892 Regulates MiR-24/PHLPP2 Axis to Mitigate Cigarette Smoke Extract-Induced Bronchial Epithelial Cell Injury. Biotechnol Appl Biochem 2022, 69, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zuo, Y.; Gao, Z. CircANKRD11 Knockdown Protects HPMECs from Cigarette Smoke Extract-Induced Injury by Regulating MiR-145-5p/BRD4 Axis. International Journal of COPD 2021, 16, 887–899. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Ding, Y.; Zhou, Z.; Yang, W. Identification and Bioinformatic Analysis of CircRNAs in the Plasma of Patients with Very Severe Chronic Obstructive Pulmonary Disease. BMC Pulm Med 2023, 23. [Google Scholar] [CrossRef]
- Shen, X.R.; Liu, Y.Y.; Qian, R.Q.; Zhang, W.Y.; Huang, J.A.; Zhang, X.Q.; Zeng, D.X. Circular RNA Expression of Peripheral Blood Mononuclear Cells Associated with Risk of Acute Exacerbation in Smoking Chronic Obstructive Pulmonary Disease. International Journal of COPD 2024, 19, 789–797. [Google Scholar] [CrossRef]
- Mattick, J.S.; Amaral, P.P.; Carninci, P.; Carpenter, S.; Chang, H.Y.; Chen, L.L.; Chen, R.; Dean, C.; Dinger, M.E.; Fitzgerald, K.A.; et al. Long Non-Coding RNAs: Definitions, Functions, Challenges and Recommendations. Nat Rev Mol Cell Biol 2023, 24, 430–447. [Google Scholar] [CrossRef]
- Tang, W.; Shen, Z.; Guo, J.; Sun, S. Screening of Long Non-Coding RNA and TUG1 Inhibits Proliferation with TGF-β Induction in Patients with COPD. International Journal of COPD 2016, 11, 2951–2964. [Google Scholar] [CrossRef]
- Devadoss, D.; Long, C.; Langley, R.J.; Manevski, M.; Nair, M.; Campos, M.A.; Borchert, G.; Rahman, I.; Chand, H.S. Long Noncoding Transcriptome in Chronic Obstructive Pulmonary Disease. American Journal of Respiratory Cell and Molecular Biology, American Thoracic Society 2019, pp 678–688. [CrossRef]
- Wu, S.M.; Feng, P.H.; Chuang, H.C.; Ho, S.C.; Fan Chung, K.; Chen, K.Y.; Wu, G.S.; Chen, T.T.; Tseng, C.H.; Liu, W.T.; et al. Impaired Lnc-IL7R Modulatory Mechanism of Toll-like Receptors Is Associated with an Exacerbator Phenotype of Chronic Obstructive Pulmonary Disease. FASEB Journal 2020, 34, 13317–13332. [Google Scholar] [CrossRef]
- Zhou, A.Y.; Zhao, Y.Y.; Zhou, Z.J.; Duan, J.X.; Zhu, Y.Z.; Cai, S.; Chen, P. Microarray Analysis of Long Non-Coding RNAs in Lung Tissues of Patients with Copd and Hoxa-As2 Promotes Hpmecs Proliferation via Notch1. International Journal of COPD 2020, 15, 2449–2460. [Google Scholar] [CrossRef]
- Wang, Y.; Lyu, X.; Wu, X.; Yu, L.; Hu, K. Long Non-Coding RNA PVT1, a Novel Biomarker for Chronic Obstructive Pulmonary Disease Progression Surveillance and Acute Exacerbation Prediction Potentially through Interaction with MicroRNA-146a. J Clin Lab Anal 2020, 34. [Google Scholar] [CrossRef]
- Feng, F.; Qi, Y.; Dong, C.; Yang, C. Pvt1 Regulates Inflammation and Cardiac Functiovia the Mapk/Nf-Κb Pathway in a Sepsis Model. Exp Ther Med 2018, 16, 4471–4478. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zeng, Z.; Shen, X.; Wu, Z.; Dong, Y.; Cheng, J.C.H. MicroRNA-146a-5p Negatively Regulates pro-Inflammatory Cytokine Secretion and Cell Activation in Lipopolysaccharide Stimulated Human Hepatic Stellate Cells through Inhibition of Toll-like Receptor 4 Signaling Pathways. Int J Mol Sci 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, M.; Dong, L. The Clinical Value of LncRNA MALAT1 and Its Targets MiR-125b, MiR-133, MiR-146a, and MiR-203 for Predicting Disease Progression in Chronic Obstructive Pulmonary Disease Patients. J Clin Lab Anal 2020, 34. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Zhang, H.; Zeng, H.; Meng, Y.; Gao, H.; Zhang, M.; Zhao, L. LncRNA CASC2 Is Involved in the Development of Chronic Obstructive Pulmonary Disease via Targeting MiR-18a-5p/IGF1 Axis. Ther Adv Respir Dis 2021, 15. [Google Scholar] [CrossRef]
- Zhao, S.; Lin, C.; Yang, T.; Qian, X.; Lu, J.; Cheng, J. Expression of Long Non-Coding RNA LUCAT1 in Patients with Chronic Obstructive Pulmonary Disease and Its Potential Functions in Regulating Cigarette Smoke Extract-Induced 16HBE Cell Proliferation and Apoptosis. J Clin Lab Anal 2021, 35. [Google Scholar] [CrossRef]
- Dai, Z.; Liu, X.; Zeng, H.; Chen, Y. Long Noncoding RNA HOTAIR Facilitates Pulmonary Vascular Endothelial Cell Apoptosis via DNMT1 Mediated Hypermethylation of Bcl-2 Promoter in COPD. Respir Res 2022, 23. [Google Scholar] [CrossRef]
- Zong, D.; Liu, X.; Li, J.; Long, Y.; Ouyang, R.; Chen, Y. LncRNA-CCAT1/MiR-152-3p Is Involved in CSE-Induced Inflammation in HBE Cells via Regulating ERK Signaling Pathway. Int Immunopharmacol 2022, 109. [Google Scholar] [CrossRef]
- Billi, M.; De Marinis, E.; Gentile, M.; Nervi, C.; Grignani, F. Nuclear MiRNAs: Gene Regulation Activities. International Journal of Molecular Sciences, Multidisciplinary Digital Publishing Institute (MDPI) June 1, 2024. [CrossRef]
- Wang, L.; Zhao, H.; Raman, I.; Yan, M.; Chen, Q.; Li, Q.Z. Peripheral Blood Mononuclear Cell Gene Expression in Chronic Obstructive Pulmonary Disease: MiRNA and MRNA Regulation. J Inflamm Res 2022, 15, 2167–2180. [Google Scholar] [CrossRef]
- Hu, J.; Wang, W.; Lu, Q.; Du, L.; Qin, T. Differential Expression of MiRNAs in Bronchoalveolar Lavage Fluid and Plasma from Patients with Chronic Obstructive Pulmonary Disease. Medicine (United States) 2022, 101. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, Z.; Kong, L.; Gao, H.; Zhang, Y.; Zheng, Y.; Wan, Y. Mirna-486-5p Promotes Copd Progression by Targeting Hat1 to Regulate the Tlr4-Triggered Inflammatory Response of Alveolar Macrophages. International Journal of COPD 2020, 15, 2991–3001. [Google Scholar] [CrossRef]
- Shi, X. feng; He, X.; Sun, Z. rui; Wang, J. xiang; Gu, Y. hai; Xie, Y. bang; Duo, J. Different Expression of Circulating MicroRNA Profile and Plasma SP-D in Tibetan COPD Patients. Sci Rep 2022, 12. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhang, L.; Wang, Q. MicroRNA-221-3p Alleviates Cell Apoptosis and Inflammatory Response by Targeting Cyclin Dependent Kinase Inhibitor 1B in Chronic Obstructive Pulmonary Disease. Bioengineered 2021, 12, 5705–5715. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Lu, H.; Song, G. MiR-221-3p and MiR-92a-3p Enhances Smoking-Induced Inflammation in COPD. J Clin Lab Anal 2021, 35. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Huang, K.; Xu, X.; Duan, R.; Yu, T.; Chu, X.; Chen, C.; Li, B.; Yang, T. MiR-23a-5p Alleviates Chronic Obstructive Pulmonary Disease through Targeted Regulation of RAGE-ROS Pathway. Respir Res 2024, 25. [Google Scholar] [CrossRef]
- Kim, R.Y.; Sunkara, K.P.; Bracke, K.R.; Jarnicki, A.G.; Donovan, C.; Hsu, A.C.; Ieni, A.; Beckett, E.L.; Galvão, I.; Wijnant, S.; et al. A MicroRNA-21-Mediated SATB1/S100A9/NF-κB Axis Promotes Chronic Obstructive Pulmonary Disease Pathogenesis; 2021; Vol. 13. https://www.science.org.
- De Smet, E.G.; Van Eeckhoutte, H.P.; Avila Cobos, F.; Blomme, E.; Verhamme, F.M.; Provoost, S.; Verleden, S.E.; Venken, K.; Maes, T.; Joos, G.F.; et al. The Role of MiR-155 in Cigarette Smoke-Induced Pulmonary Inflammation and COPD. Mucosal Immunol 2020, 13, 423–436. [Google Scholar] [CrossRef]
- Zhu, Y.; Han, Y.; Almuntashiri, S.; Dutta, S.; Wang, X.; Owen, C.A.; Zhang, D. Dysregulation of MiR-103a Mediates Cigarette Smoking–Induced Lipid-Laden Macrophage Formation. Am J Respir Cell Mol Biol 2022, 67, 695–707. [Google Scholar] [CrossRef]
- Arif, R.; Pandey, A.; Zhao, Y.; Arsenault-Mehta, K.; Khoujah, D.; Mehta, S. Treatment of Pulmonary Hypertension Associated with COPD: A Systematic Review. ERJ Open Research, European Respiratory Society January 1, 2022. [CrossRef]
- Yang, Z.; Li, P.; Yuan, Q.; Wang, X.; Ma, H.H.; Zhuan, B. Inhibition of MiR-4640-5p Alleviates Pulmonary Hypertension in Chronic Obstructive Pulmonary Disease Patients by Regulating Nitric Oxide Synthase 1. Respir Res 2023, 24. [Google Scholar] [CrossRef]
- Singh, P.; Li, F.J.; Dsouza, K.; Stephens, C.T.; Zheng, H.; Kumar, A.; Dransfield, M.T.; Antony, V.B. Low Dose Cadmium Exposure Regulates MiR-381–ANO1 Interaction in Airway Epithelial Cells. Sci Rep 2024, 14. [Google Scholar] [CrossRef]
- Zheng, L.; Jiang, Y.L.; Fei, J.; Cao, P.; Zhang, C.; Xie, G.F.; Wang, L.X.; Cao, W.; Fu, L.; Zhao, H. Circulatory Cadmium Positively Correlates with Epithelial-Mesenchymal Transition in Patients with Chronic Obstructive Pulmonary Disease. Ecotoxicol Environ Saf 2021, 215. [Google Scholar] [CrossRef]
- Tasena, H.; Faiz, A.; Timens, W.; Noordhoek, J.; Hylkema, M.N.; Gosens, R.; Hiemstra, P.S.; Spira, A.; Postma, D.S.; Tew, G.W.; et al. MicroRNA–MRNA Regulatory Networks Underlying Chronic Mucus Hypersecretion in COPD. European Respiratory Journal 2018, 52. [Google Scholar] [CrossRef]
- Tasena, H.; Timens, W.; Van Den Berge, M.; Van Broekhuizen, J.; Kennedy, B.K.; Hylkema, M.N.; Brandsma, C.A.; Heijink, I.H. MicroRNAs Associated with Chronic Mucus Hypersecretion in COPD Are Involved in Fibroblast–Epithelium Crosstalk. Cells 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, X.; Zheng, Y.; Du, F.; He, G. MiR-548ar-3p Increases Cigarette Smoke Extract-Induced Chronic Obstructive Pulmonary Disease (COPD) Injury through Solute Carrier Family 17 Member 9 (SLC17A9). Arch Biol Sci 2022, 74, 97–105. [Google Scholar] [CrossRef]
- Nadi, E.; Geramirad, G.; Kahramfar, Z.; Rasouli-Saravani, A.; Solgi, G. Peripheral Blood Expressions of MicroRNA-146a and MicroRNA-218 in Chronic Obstructive Pulmonary Disease with/without Cigarette Smoke Exposure. Iran J Allergy Asthma Immunol 2022, 21, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Tang, S.; Zhou, Z.; Wei, H.; Yang, W. Plasma MiR-150-5p as a Biomarker for Chronic Obstructive Pulmonary Disease. International Journal of COPD 2023, 18, 399–406. [Google Scholar] [CrossRef]
- Zhang, X.; Shi, Q.; Xiong, L.; Shi, S.; Li, Y.; Wang, Y.; Zhang, M. Clinical Relevance of MiR-423-5p Levels in Chronic Obstructive Pulmonary Disease Patients. Clinics 2022, 77. [Google Scholar] [CrossRef]
- Tao, S.; Liao, C.; Wang, Y.; Xu, D.; Li, Z.; Li, F. Differential MiRNA Profiling Reveals MiR-4433a-5p as a Key Regulator of Chronic Obstructive Pulmonary Disease Progression via PIK3R2- Mediated Phenotypic Modulation. Comb Chem High Throughput Screen 2023, 27, 2323–2334. [Google Scholar] [CrossRef]
- Cazorla-Rivero, S.; Mura-Escorche, G.; Gonzalvo-Hernández, F.; Mayato, D.; Córdoba-Lanús, E.; Casanova, C. Circulating Mir-1246 in the Progression of Chronic Obstructive Pulmonary Disease (Copd) in Patients from the Bode Cohort. International Journal of COPD 2020, 15, 2727–2737. [Google Scholar] [CrossRef]
- Wang, C.; Feng, D.; Dong, S.; He, R.; Fan, B. Dysregulated Circulating MicroRNA-126 in Chronic Obstructive Pulmonary Disease: Linkage with Acute Exacerbation Risk, Severity Degree, and Inflammatory Cytokines. J Clin Lab Anal 2022, 36. [Google Scholar] [CrossRef]
- Huang, H.; Wu, F.; Yang, J.; Li, H.; Cai, M.; Shan, K.; Shi, S. Increased Plasma Level of MiR-210 as a Potential Diagnostic Marker for Chronic Obstructive Pulmonary Disease Induced Pulmonary Hypertension. Clin Lab 2020, 66, 971–977. [Google Scholar] [CrossRef]
- Burke, H.; Cellura, D.; Freeman, A.; Hicks, A.; Ostridge, K.; Watson, A.; Williams, N.P.; Spalluto, C.M.; Staples, K.J.; Wilkinson, T.M.A. Pulmonary EV MiRNA Profiles Identify Disease and Distinct Inflammatory Endotypes in COPD. Front Med (Lausanne) 2022, 9. [Google Scholar] [CrossRef]
- Wang, F.; Yang, B.; Qiao, J.; Bai, L.; Li, Z.; Sun, W.; Liu, Q.; Yang, S.; Cui, L. Serum Exosomal MicroRNA-1258 May as a Novel Biomarker for the Diagnosis of Acute Exacerbations of Chronic Obstructive Pulmonary Disease. Sci Rep 2023, 13. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.F.; Liu, Y.; Qu, P.P.; Tang, Y.; Li, B.B.; Cheng, G.L. Mir-361-5p/Abca1 and Mir-196-5p/Arhgef12 Axis Involved in γ-Sitosterol Inducing Dual Anti-Proliferative Effects on Bronchial Epithelial Cells of Chronic Obstructive Pulmonary Disease. International Journal of COPD 2021, 16, 2741–2753. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A CeRNA Hypothesis: The Rosetta Stone of a Hidden RNA Language? Cell, Elsevier B.V. August 5, 2011, pp 353–358. [CrossRef]
- Li, B.; Zhang, J.; Dong, H.; Feng, X.; Yu, L.; Zhu, J.; Zhang, J. Systematic Analysis of Various RNA Transcripts and Construction of Biological Regulatory Networks at the Post-Transcriptional Level for Chronic Obstructive Pulmonary Disease. J Transl Med 2023, 21. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Dong, H.; Li, B.; Yu, L.; Zhu, J.; Lou, C.; Zhang, J. Integrative Analysis of the Expression Profiles of Whole Coding and Non-Coding RNA Transcriptomes and Construction of the Competing Endogenous RNA Networks for Chronic Obstructive Pulmonary Disease. Front Genet 2023, 14. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



