
Submitted:
11 February 2025
Posted:
12 February 2025
You are already at the latest version
Abstract
Disease eponyms could be confusing, difficult to remember, scientifically non-robust; lacking implications on and relationship with cell lineage, histogenesis and pathogenesis. This review is geared for revisiting hematolymphoid diseases with eponyms in the light of recent advances in technology and science, by searching the literature using Scopus and Google Scholar with the keywords “eponyms, hematolymphoid, diseases, lymphoma, benign, malignant, lymph node, spleen, liver, bone marrow, leukemia” for the past fifty years. With advances in science and technology, there is accumulation of information on the morphologic nuances, immunologic, immunophenotypic and genetic features of various hematolymphoid eponymic diseases; thus shedding light on important issues of etiology and pathogenesis with implications on therapy in various non-neoplastic (Castleman, Kikuchi-Fujimoto, IgG4-related diseases) and neoplastic (Hodgkin, Burkitt, NK/T-cell lymphomas, dendritic/histiocytic neoplasms and Sezary syndrome) diseases. This contributes to modern nomenclature, classification, subtyping, prognostication and discoveries on new treatment strategies of hematolymphoid eponymic diseases.
Keywords:
Eponyms
; hematolymphoid diseases
; new facets
1. Introduction
In the old days, due to the lack of clinical investigative methods and technology, many diseases were named after persons, usually physicians who first identified the diseases. Less commonly, they were named after patients who first had the diseases, a fictional character depicted with the diseases or places where the diseases were prevalent.
A meeting on classification and nomenclature of morphologic defects was held at the National Institute of Health, USA; with the proposal published in the Lancet in 1974 that eponyms be used only in the absence of a reasonable descriptive designation, be limited to one proper name and until the disease basis is recognized [1]. Though eponyms are memorable of the credited persons or places, they are scientifically non-robust with no relevance or implications to cell lineage, etiology, histogenesis, pathogenesis, immunophenotypic or genetic features. This compromises further investigations and discovery of important prognostic and therapeutic attributes of the diseases. Furthermore, the same eponym may be used for different diseases (e.g. Murphy’s sign for acute cholecystitis and acute appendicitis); thus potentially causing confusion.
Eponyms have been used for many diseases in a plethora of organ systems. This review aims to comprehensively look into eponyms used in hematolymphoid neoplastic and non-neoplastic diseases. A literature research with Google Scholar and Scopus for the past 50 years using the keywords “eponyms, hematolymphoid, diseases, lymphoma, benign, malignant, lymph node, spleen, liver, bone marrow, leukemia” was performed. The various relevant eponymic hematolymphoid diseases were then reviewed in the light of impactful advances in science and technology.
2. Hematolymphoid Non-Neoplastic Eponymic Diseases
Storage diseases represent a major disease group designated with eponyms. They are inborn errors of metabolism with specific enzyme deficiencies leading to abnormal accumulation of substances ranging from glycogen to lipids. Patients often present with hepatosplenomegaly or blood cytopenias, thus masquerading as hematolymphoid diseases. Differential diagnosis from other hematolymphoid diseases is often not difficult due to early presentation in life, hereditary basis, clinical picture, characteristic histology and electron microscopy features, with confirmation by enzyme studies. Due to the distinctive clinical and investigative features, they are not included in this review.
2.1. Castleman Disease (CD)
CD was first described by Benjamin Castleman in 1956 as localized enlarged mediastinal lymph nodes which he and colleagues in 1972 described pathologically as the hyaline vascular (HV) type with the plasma cell (PC) type [2,3]. As more CD cases were reported, it is increasingly recognized that HV and PC types often occur together as a “mixed” type, albeit the HV pattern is more common in unicentric (UC) and the PC pattern more common in multicentric (MC) CD [4,5].
2.1.1. Epidemiology and Pathogenesis
This is poorly understood, mostly because CD is heterogeneous encompassing several lymphoproliferative disorders with distinct clinicopathologic entities [6,7]. CD is classified under “tumour-like lesions with B-cell predominance” in the 5th edition of the WHO Classification of Haematolymphoid Tumours [7]. In UCCD, a clonal neoplastic proliferation of stromal and possibly follicular dendritic cells with some harboring platelet-derived growth factor receptor beta mutations has been demonstrated [5,7,8,9,10,11]. IL-6 is a critical driver in many idiopathic MCCD (iMCCD) as supported by the clinical effectiveness of using IL-6 inhibitors [4,5,7,12,13,14]. However, IL-6 is not universally elevated in iMCCD, and increased T-cell activation, elevated serum vascular endothelial growth factor (VEGF), immunoglobulin (Ig) gene rearrangement, chromatin remodeling gene mutations, PIK3/AKT/mTOR, MAK, JAK-STAT, and IL signaling pathways alterations may also be at play [5,7]. There is a key role of B cells in pathogenesis of Kaposi sarcoma-associated herpesvirus/human herpesvirus 8 (KSHV/HHV8)+ MCCD [6,7,15] as demonstrated by efficacy of rituximab treatment. IL-6 (including viral IL-6 and secreted human IL-6) is also important in driving KSHV/HHV8 MCC. Other KSHV/HHV8 gene products (vFLIP,vIRF3/LANA2, viral microRNA) are important in affecting infected cell survival and host/cytokine responses [7]. The role of autoimmunity has also been considered as autoantibodies are present in one-third of iMCCD [5,16]. Monoclonal plasma cells also play a role in polyneuropathy, organomegaly, endocrinopathy, monoclonal plasma cell disorder, skin changes (POEMS) syndrome-associated MCCD [5,6].
2.1.2. Classification of CD
Due to heterogeneity and lack of a unifying concept with regard to etiology or pathogenesis, classification of CD is based on its centricity and known etiologic or syndromic associations [5,6,7].
- (1)
- Unicentric CD (UCCD), when the disease is localized to one lymph node or a single lymph node station. This accounts for 50-70% CD.
- (2)
- Multicentric CD (MCCD), when the disease affects multiple lymph nodes or lymph node stations, including:
- (a)
- POEMS (polyneuropathy, organomegaly, endocrinopathy, M-protein and skin changes) -associated CD
- (b)
- iMCCD
- (i)
- iMCCD-TAFRO (thrombocytopenia, ascites, reticulin fibrosis, renal dysfunction, organomegaly). This is a severe form of MCCD.
- (ii)
- iMCCD, not otherwise specified (NOS).
- (c)
- KSHV/HHV8 associated MCCD
- (i)
- Human immunodeficiency virus (HIV) negative
- (ii)
- HIV positive
2.1.3. Pathology
Excised lymph nodes are required for histopathologic diagnosis as small biopsies may not contain full diagnostic features. There are 3 histopathologic patterns [4,5,6,7].
- (a) Hyaline vascular (HV) pattern.
This is most common in UCCD featuring atretic follicles with regressed germinal centers depleted of lymphoid cells, with follicular dendritic cell retention, frequent radial traversion by hyalinized sclerotic blood vessels to impart a “lollipop” appearance and concentric rings of mantle or marginal lymphocytes to portray the characteristic “onion-skin” picture. Twining of 2 or more atretic follicles by rings of mantle lymphocytes is often present. The interfollicular region shows marked vascular proliferation with plump endothelial cells and inconspicuous or absent lymph node sinuses (Figure 1 A-D). This pattern may also be present in the plasma cell type CD, where the interfollicular lymph node sinuses are patent. The follicular dendritic cells of the germinal centers and interfollicular region may show dysplastic changes.
- (b) Plasma cell (PC) pattern
This is characterized by hyperplastic lymphoid follicles and sheets of PC in the interfollicular region. In KSHV/HHV8 associated MCCD, some follicles and mantle zones may contain clusters of large immunoblasts or plasmablasts, often expressing monotypic IgM-λ, though polyclonality is demonstrated on molecular analysis.
- (c) Mixed pattern
This shows both HV and PC patterns in varying proportions, and may occur in UCCD and MCCD.
- (d) Histologic variants
This occurs in the HV pattern and mostly in UCCD. The interfollicular region is expanded with stromal, angiomyoid or follicular dendritic cell proliferation. The latter may progress to follicular dendritic cell sarcoma [17,18,19,20]. Occurrence of Kikuchi lymphadenitis-like change in UCCD has also been reported [21].
2.1.4. Diagnosis and Differential Diagnosis
Diagnosis of CD requires correlation of histology, clinical findings and exclusion of a myriad of reactive and neoplastic mimickers [5,6].
UCCD - usually present as single site lymph node enlargement with or without local compression symptoms and no systemic symptoms. Histology is often adequate for diagnosis.
MCCD - characterized by multicentric lymphadenopathy, systemic clinical features and laboratory abnormalities in 80% cases (cytopenias, liver and kidney dysfunction). In addition, the following types of MCCD are diagnosed by specific tests and clinical/syndromic features.
- (i)
- KSHV/HHV8 associated MCCD - positive HHV8 testing
- (ii)
- iMCCD-TAFRO - associated TAFRO signs and symptoms (thrombocytopenia, anarsaca, fever, renal insufficiency, organomegaly) and negative for KSHV/HHV8 and HIV.
- (iii)
- POEMS-MCCD - associated POEM syndrome and monoclonal gammopathy, and negative for KSHV/HHV8 and HIV.
2.1.5. Treatment and Outcome
UCCD - Complete surgical excision usually yields excellent outcome. Radiotherapy, immunochemotherapy or embolization may be used for unresectable disease or compressing symptoms. Anti-IL6, immunotherapy may be required in patients with inflammatory symptoms.
MCCD - Appropriate treatment is varied due to unclear etiology.
- iMCCD : anti-IL-6 directed therapy, with about one third of patients responding. Steroids, chemotherapy, rituximab, immunomodulators, intravenous immunoglobulins and thalidomide applied singly or in combinations may be required in anti-IL6 non-responders and very ill patients.
- POEM-MCCD : when no bone lesions; iMCCD-like therapy
: when bone lesions present; myeloma type therapy
- HHV8+MCCD : anti-retrovirus therapy, rituximab
2.1.6. Secondary Malignancies
2.2. Kikuchi/Kikuchi-Fujimoto Disease (KFD)
KFD was described in 1972 independently by Kikuchi [22] and Fujimoto [23]. The greatest dimension of the involved lymph node could be large and up to 7cm [24]. Rarely, extranodal involvement of skin, liver, spleen, central nervous system, kidney and heart has been reported [24]. KFD affects mostly Asians with female predominance, though non-Asians are also affected [25,26,27]. Patients present with fever, leukopenia, with self-limiting disease duration of up to 6 months. Recurrence may occur up to 2 years after the initial presentation [24,28]. The involved lymph nodes characteristically show cortical and paracortical variably-sized discrete or confluent pale nodular areas constituted by many histiocytes which are phagocytic or non-phagocytic with special morphologic types including crescentic histiocytes, signet ring histiocytes and plasmacytoid dendritic cells (pDC). There are admixed variable quantities of immunoblasts. karyorrhectic and eosinophilic debris, and sometimes coagulative necrosis and focal fibrovascular organization. Neutrophils are conspicuously absent or very rare (Figure 1 E-H). Kuo [24] recognized 3 histologic patterns: proliferative, necrotizing and xanthomatous, which are not patterns of temporal progression but histologic variations related to individual host immune response. Immunohistochemically, the histiocytic cells are CD163+, CD68+, CD4+ and myeloperoxidase+. The T-cells are mostly CD8+, and with rarely CD4+ T-cells. The pDC are positive for CD123, CD4 and CD68. CD20+ B cells are practically absent. DNA ploidy shows diploidy, supporting reactive nature [24]. KFD is classified under “tumour-like lesion with T-cell predominance” in the 5th edition WHO Classification of Haematolymphoid Tumours, in line with the important role of exuberant T-cell mediated response in the disease [7].
Fine needle aspiration (FNA) cytology yields the characteristic histiocytes, lack of neutrophils, karyorrhectic debris and necrosis corresponding to the histologic features. In the proper clinical context, FNA is diagnostic [29]. With regard to pathogenesis, recent studies demonstrated involvement of cytokine and chemokine pathways of interferon (IF) γ, IL-18, MIG(CXCL9), IFγ - induced protein 10 and IF-1 [30]. The most important differential diagnosis is from lymphoma and systemic lupus erythematosus [21,24,28,31]. Etiology is unknown, autoimmune or infection mediated mechanisms have not been substantiated [30]. Due to the frequent presence of two histologic features, histiocytes and necrosis; the term histiocytic necrotizing lymphadenitis has been recommended. However, in the 5th edition WHO Classification of Haematolymphoid Tumours, Kikuch-Fujimoto disease is classified under ”tumour-like lesions with T-cell predominance” and involves an exuberant T-cell response with CD-8+ cytotoxic cell induced apoptosis in genetically predisposed subjects [7]. The disease is self-limiting, usually not requiring treatment.
2.3. Kuttner’s-KT, Mikulicz’s Disease-MD, Ormond’s Disease-OD, Riedel’s Thyroiditis-RT
KT, MD, OD and RT are chronic fibroinflammatory diseases affecting salivary glands,lacrimal glands, retroperitoneum and thyroid gland. MD was initially described by Mikulicz in 1888 being characterized by symmetrical bilateral lacrimal, parotid and submandibular glands enlargement. However, MD was later described by Napp to be related to leukemia, lymphoma, sarcoidosis and tuberculosis by Napp [32]. These entities are now recognized as IgG4-related diseases(IgG4RD) with local manifestations [33]. IgG4RD, was initially recognized in early 2000 as sclerosing pancreatitis or autoimmune pancreatitis associated with raised serum IgG4 or tissue IgG4-positive plasma cells [34,35]. With increased recognition, there is a proliferation of reports on IgG4RD [36], culminating in the consensus statement on the pathology of IgG4RD in 2012 [37]. In the latter, there are 3 possible characteristic histological features of IgG4RD. (1) Dense lymphoplasmacytic infiltrate, (2) fibrosis, usually storiform in character, and (3) obliterative phlebitis. The IgG4+ PC count ranges from 10 to 200 cells/HPF, depending on the involved organ. An elevated IgG4+/IgG+ PC ratio of >40% (>50% for aorta specimens) is also necessary. Accordingly, there are 3 diagnostic categories. (1) highly suggestive of IgG4RD, when 2 of the 3 characteristic histologic features with fulfilled IgG4+ PC count are observed. (2) probable IgG4RD when only one characteristic histologic feature and the required IgG4+ PC count are fulfilled. (3) insufficient histopathological evidence of IgG4RD, when neither categories (1) or (2) are met (Figure 1 I-M).
IgG4RD involves multiple organs and tissues, including superficial or deep, with synchronous or metachronous involvement of 3 organs on average. The affected deep sites include pancreas, hepatobiliary system, liver, retroperitoneum, mesentery, mediastinum, aorta, lung, pleura, kidney and urinary tract, central nervous system, thyroid, prostate, seminal vesicles, maxillary sinus, nasal septum, paranasal sinus and pericardium. Lymph nodes are involved in 30-60% cases manifesting 5 histologic patterns, with the pseudotumor-like pattern more specific for IgG4RD [36]. The involved superficial sites include orbit, lacrimal gland, salivary gland, skin and breast [36,38]. In the pancreas, pancreatectomy and Whipple’s operation is not infrequently performed for tumor-like lesions caused by IgG4RD [39,40]. However, true malignancies including lymphoma, pancreatic ductal adenocarcinoma, salivary duct carcinoma, pulmonary adenocarcinoma, gastrointestinal clear cell sarcoma have been described in a backdrop of IgG4RD [36]. The patients are in good general condition with symptoms attributable to local mass lesions. Serum IgG4 level correlates with disease activity [37]. Treatment does not require radical surgery and steroids are usually effective [36,38].
Pathogenesis and Etiology
3. Hematolymphoid Neoplastic Eponymic Diseases
3.1. Histiocytic/Dendritic Cell Neoplasm
The eponymic diseases encompassed under this category include Hashimoto-Pritzker disease (HPD), Hand-Schuller-Christian disease (HSCD), Letterer-Sieve disease (LSD), Langerhans cell histiocytosis (LCH), Langerhans cell sarcoma (LCS), Erdheim-Chester disease (ECD) and Rosai-Dorfman disease (RDD).
3.1.1. Langerhans Cell Histiocytosis (LCH-Including HPD, HSCD and LSD)
Langerhans cells (LC) were named after Paul Langerhans, who discovered the LC as part of the peripheral nervous system in the epidermis [43]. The LC is however later confirmed to be a hematopoietic cell [44]. There are 2 forms of LCH. The primarily cutaneous HPD (also known as congenital self-healing Langerhans cell histiocytosis – CSHLCH), which is mostly self-healing and causes vesicular papulonodular skin lesions in neonates, infants and very young children [45,46]. CSHLCH (HPD) does not harbor BRAF mutations, in contrast to systemic LCH [45,46]. Epidermal LCs develop from yolk sac-derived primitive myeloid progenitors [47]. Mutated epidermal LCs are unable to to replicate [46], which may be related to the self-healing behavior of CSHLCH. Systemic LCH is mostly a clonal neoplastic disease of bone marrow-derived immature myeloid dendritic cells and not from epidermal LCs [7,48,49]. There are recurrent BRAF V600E and MAP2K1 gene mutations that activate the mitogen-activated protein kinase (MAPK) pathway causing phosphorylated MEK and ERK proteins [7,48,49,50,51,52,53,54,55]. Mutations in BRAF V600E and MAP2K1 are mutually exclusive [52,54]. Additional hematologic malignancy co-existing with LCH has been reported [44]. In the 5th edition of the WHO Classification of Haematolymphoid Tumours, LCH is classified as a histiocytic/dendritic cell neoplasm and a close relationship of LCH with bone marrow-derived myeloid dendritic cells is described [7].
LCH is classified as single system (SS) or multisystem (MS) disease with either unifocal (UF) or multifocal (MF) involvement in SS. HSCD is MFSS-LCH and LSD is MS-LCH. SS-LCH commonly affects skin, pituitary or bone while in MS-LCH, bone, skin, liver, spleen, lymph node, lungs and bone marrow are involved. Involvement of Bone marrow, spleen or liver involvement is high risk LCH. SS-LCH affects older children and adults while MS-LCH neonates and infants with systemic symptoms, cytopenias, disturbed liver functions hepatosplenomegaly, endocrine dysfunction, diabetes insipidus and central nervous system involvement, with grave prognosis [7,54]. LCH is a rare disease with an annual incidence of 5-9 new cases per million pediatric and 1-2 new cases per million in the adult population. There is a slight male predominance affecting more commonly Caucasians [7,54].
Pathology
There is characteristic accumulation of large oval to round histiocytic cells with irregular nuclei, often with nuclear grooves, abundant pale cytoplasm and minimal nuclear atypia in a backdrop of giant cells, eosinophils, lymphocytes and multinucleated histiocytes. LCH cells express CD1a, S100, CD207 and CD68 (Figure 2 A-E). CD207 is a surrogate for ultrastructural Birbeck granules which are tennis-racket or zipper-like structures. Staining for downstream markers of MAPK activation; cyclin D1 and p-ERK, is positive [7,54].
Treatment and Prognosis
This is related to stage, SS or MS involvement, and whether high risk organ involvement is present. SS-LCH usually requires only local control with a 90% survival rate. MS-LCH requires systemic therapy with variable clinical course. About 60% achieve no evidence of disease after LCH-III therapy for 1 year and about 37% relapse rate 1 year after vinblastine and prednisone. Targeted therapy may be useful [7,53,55].
3.1.2. Langerhans Cell Sarcoma (LCS)
LCS is a very rare highly aggressive neoplasm with high grade LC morphology. The incidence is 0.02 per million. It is a multisystem disease affecting skin, lung, bone, soft tissue, liver, spleen and lymph nodes. Forty-five percent is widespread disease and isolated lymph node involvement occurs in 20% of cases. LCS is a clonal neoplasm harboring mutations, including KRAS and BRAF V600E, in the MAPK pathway. Other implicated genes include CDKN2A, TP53 and PTEN. Secondary hematolymphoid malignancies (follicular lymphoma, chronic lymphocytic leukemia, B-lymphoblastic leukemia and hairy cell leukemia) with shared clonality/molecular aberrations are reported, possibly by transdifferentiation from LCS [7,54].
Pathology
Treatment and Prognosis
This is a highly aggressive disease. Localized disease fares better. Targeted therapy or chemotherapy has produced responses in some cases [7].
3.1.3. Erdheim-Chester Disease (ECD)
ECD is a rare non-LC histiocytic neoplasm affecting adults between the 5th and 7th decades. It is a multisystem disease with the commonly involved sites including bone, central nervous system, cardiovascular system, lungs, kidneys and skin. Lymph nodes, liver and spleen, however, are generally spared. There is BRAF V600E mutation in 50% patients. Patients may be asymptomatic but more often show systemic symptoms, bone pain, exophthalmos, xanthelasma and diabetes insipidus. 3-15% ECD may be associated with myeloid neoplasms including acute myeloblastic leukemia, chronic myelomonocytic leukemia; and multiple myeloma [7,49]. There is a gain of function somatic alteration in BRAFV600E in 50-60%cases, activating the MAPK signaling pathway. Other implicated mutations include BRAF, ARAF, NRAS, KRAS, MAPZK1 and PIK3CA. Relationship with bone marrow progenitors is proposed [7,49].
Pathology
There is infiltration by foamy, lipid-laden small histiocytic cells in a backdrop of Touton giant cells, plasma cells, lymphocytes and neutrophils, but not eosinophils, mimicking xanthogranuloma. Fibrosis may be prominent. There may be co-existing LCH and Rosar-Dorfuman disease (RDD). The tumor cells are positive for CD163, CD68, CD14 and CD4 but negative for CD1a and CD207. Expression of pERK is frequent and diffuse strong cytoplasmic staining with VE1 may reflect BRAF V600E mutation [7,56].
Treatment and Prognosis
3.1.4. Rosai-Dorfman Disease (RDD)
RDD is classified as a histiocytic/macrophage neoplasm in the 5th edition (2024) WHO Classification of Hematolymphoid Tumors [7] and is a clonal histiocytic disease with nodal or extranodal accumulation of large S-100 - positive histiocytes, commonly exhibiting emperipolesis [7,57,58]. Some RDD are familial and may be related to the inherited autoimmune lymphoproliferative syndrome with disordered immune functions and defects in Fas-mediated apoptosis with germline mutations in the TNFRSF6 gene encoding Fas [59]. Classic sporadic RDD affects children and young adults with an average age of 21 years, more common in African subjects, with slight male predominance, massive bilateral painless cervical lymphadenopathy, fever, weight loss and night sweats. Inguinal, retroperitoneal and mediastinal lymph nodes; and skin, nasal cavity, and central nervous system involvement may occur. Cutaneous RDD exhibits higher female predominance and old age (44 years), more Asian and white subjects, with papulonodular skin lesions [7,57].
Pathology
There is extensive lymph nodal sinusoidal expansion or effacement of nodal architecture (advanced cases) by histiocytes with hyperchromatic nuclei, distinct nucleoli, pale crispy cytoplasm characteristically featuring hematolymphoid cells within vacuoles; a feature known as emperipolesis (Figure 2 F-I). The cortex comprises B cells, plasma cells and some follicles, which with the pale histiocytic cells, constitute alternating dark and light zonal appearance. Many IgG4-positive plasma cells may be present which may be confused with IgG4RD [52]. Marked fibrosis, sometimes storiform, may occur. Extranodal disease is similar, usually with more lymphoid follicles and less emperipolesis. There may be coexisting lymphoma, LCH and ECD. The histiocytic cells are S-100+, CD68+, CD163+ and CD1a-, CD207 [7,57].
Pathogenesis and Etiology
Familial RDD may show the Fas gene TNFRSF6 germline mutations [59]. Approximately 50% sporadic carry mutations in the MAPK/ERK pathway, including KRAS, NRAS, MAP2K1, ARAF, CSF1R, BRAF V600E. Others include SNX24, CIC, INTS2, SFR1, BRD4, PHOX2B, PD55A, MUC4, ERCC2, LATS2, BRCA1, ATM and USP25 [7,57,58], thus supporting the neoplastic nature of at least a proportion of RDD.
Classification
RDD may be sporadic or familial. Sporadic RDD is most common, including nodal, extranodal, neoplasia-associated and immune disease-associated. Familial RDD includes H syndrome and autoimmune lymphoproliferative syndrome-associated RDD [57,59,60]. Sporadic RDD is classified under “R group” and cutaneous RDD under “C group” of histiocytosis by the Histiocyte Society [57,60].
Prognosis
3.2. Hodgkin Lymphoma (HL)
Described by Thomas Hodgkin in 1832 [61], subsequent microscopic studies on reported cases that were diagnosed by macroscopy showed that the originally described HL may have included other lymphomas, tuberculosis or syphilis [62]. HL is subsequently discovered to be a GC B-cell lymphoma [7,63,64,65,66], after uncovering high load of somatic mutations in the rearranged immunoglobulin heavy chain variable region (IGHV) gene. It is categorized in the 5th edition WHO Classification of Haematolymphoid Tumours under B-cell lymphoid proliferations and lymphomas. There are 2 types of Hodgkin lymphoma: classic HL and nodular lymphocyte predominant HL [7].
3.2.1. Classic HL (CHL)
The characteristic and diagnostic Hodgkin and Reed-Sternberg (HRS) cells are the minority neoplastic cells in a reactive cell milieu which accounts for a major component of the disease. CHL is primarily a lymph node disease with supradiaphragmatic predilection (cervical, supraclavicular and mediastinal nodes). Less than 5% CHL is infradiaphragmatic. The nodes are painless and spread is typically predictable in a contiguous fashion. Primary extranodal involvement is extremely rare and when present (spleen, lungs, liver, bone marrow) usually represent Stage IV disease. Incidence is higher in socioeconomically developed countries (2.63 per 100,000 persons-year in USA) with slight male predominance (except nodular sclerosis subtype) [7].
Pathogenesis and Etiology
Derived from crippled pre-apoptotic GC B cells, HRS cells lose B-lineage specific gene expression program and B-cell identity [7,63,64,65,66]. Recurrent gene mutations of NF-KB, JAK/STAT, MAPK/ERK, AP1, PIK3/AKT and NOTCH1 cause activation of the respective signaling pathways. CHL may be EBV+ (latency II) or EBV-. The reactive cell milieu and HRS cells are symbiotic [7,63,64,65,66].
Pathology
There are 4 subtypes: nodular sclerosis (NSCHL), mixed cellularity (MCCHL), lymphocyte rich (LRCHL) and lymphocyte depleted CHL (LDCHL, rare, <2% CHL). NSHL is characterized by capsular fibrosis, sclerotic bands and neoplastic lacunar cells (Figure 3 A-F). Sheets of neoplastic cells constitute the syncytial variant. MCCLH shows architectural effacement and more frequent HRS cells (Figure 3 G-I). LRCHL is nodular (less commonly diffuse) and lymphocyte rich. LDCHL exhibits diffuse infiltration of pleomorphic HRS cells with strong EBV association. The reactive milieu is exuberant in MCCHL, rare in eosinophils in LRCHL and lymphocyte sparse in LDCHL. HRS and lacunar cells are CD30+(almost all cases), CD15+(majority of cases), PAX5+, CD20+ (some cases) and CD45-, J chain-, IG heavy and light chains-, Oct2-, BOB1-, PU1- and BCL6-. Aberrant T-cell marker expression may occur. EBV staining is variable [7].
Prognosis and Treatment
More than 80% CHL can be cured with modern polychemotherapy. Prognosis is related to stage, B symptoms and prognostic score [7].
3.2.2. Nodular Lymphocyte Predominant CHL (NLPHL)
NLPHL is a GC B-cell lymphoma [7,67,68] characterized by LP cells with multilobulated nuclei (popcorn appearance, thus nicknamed “popcorn cells”) arranged in nodules. This is mostly a nodal disease, with less supradiaphragmatic, bulky and Stage IV disease compared to CHL. This is rare (0.11 per 100,000 person-years) with male predominance and wide age distribution (childhood to >80 years of age) [7].
Pathogenesis and Etiology
Etiology is unknown and there is no association with EBV. Genetic predisposition [69,70] and primary immunodeficiency may be contributory [71]. The neoplastic cells are clonal GC B cells with ongoing somatic hypermutation (PIM1, RhoH, PAX5, MYC, SOCS1, JUNB, DUSP2, SGK1), intraclonal diversity in rearranged IG genes [7,67,68] and BCL6 translocations or amplifications [72]. Preceding bacterial infection may be a triggering event [73].
Pathology
Nodular, sometimes with diffuse areas, containing characteristic LP (popcorn) cells featuring multilobulated nuclei, variable nucleoli and pale cytoplasm occurring singly or in small clusters. Reactive B, T and histiocytic cells but no eosinophils or neutrophils constitute the backdrop microenvironment. Granulomas are sometimes present. LP cells are positive for B-cell markers (CD20, CD79a, OCT2, BOB1, PU1), GC markers (BCL6, LMO2, HGAL), rarely for CD15; and negative for CD30. NLPHL can transform to large B-cell lymphoma, with infradiaphragmatic and splenic involvement and clonal IG rearrangement (at diagnosis) being risk factors [7,74,75].
Prognosis and Treatment
3.3. Burkitt Lymphoma (BL)
Described by Dennis Burkitt in 1958 [76], BL is an aggressive mature B-cell lymphoma with GC phenotype, high proliferation and tumor burden [7], involving mostly extranodal tissues; and classified under “mature B-cell lymphoma” in the 5th edition of the WHO Classification of Haematolymphoid Tumours [7]. BL affects mostly extranodal sites. There are three epidemiologic types: endemic (equatorial Africa and Papua New Guinea), common in children causing facial bones and abdominal disease and associated with EBV in 90% cases; sporadic in the USA and Western Europe, affecting children and adults causing commonly abdominal disease with EBV in 20% cases; and immunodeficiency (IDF)-associated, mostly associated with HIV infection (affecting those with higher CD4 count) than other causes of immunosuppression and commonly causes lymph node disease. Leukemic picture and bone marrow involvement occurs in 20% cases and is more common in IDF-associated BL. CNS involvement occurs in any of these types, with a frequency of 10-30% [77] and portends worse prognosis among high stage BL patients [78].
EBV (latency 1) is implicated in almost 100% endemic and 25-40% IDF associated BL; being involved in early pathogenesis by apoptosis evasion [79]. The EBNA1 protein is the most consistently detected transforming EBV protein in EBV-positive BL [7]. Therefore, EBV may be the defining feature of BL types, and EBV-positive and EBV-negative BL differ in their underlying biology and pathogenesis [80,81], despite conventional subtying of BL into endemic, sporadic and immunodeficiency-associated.
Malaria (particularly mixed P falciparum infection) is also pathogenetically related, probably causing chronic B cell stimulation and T cell suppression, and induction of activation-induced cytidine deaminase which is associated with MYC translocation [82]. In 70% BL, there is IG::MYC translocation (involving genes of heavy t(8;14) or light chains t(8;22); t(2;8)). The MYC translocation is not sufficient for oncogenesis and other genetic changes including TCF3, ID3, CCND3, TP53 alterations; resulting in BCR, PIK3, SWI/SNF and GPCR signaling activation are also at play [7,83].
3.3.1. Pathology
Diffuse infiltrate of monomorphic medium-sized lymphoid cells with squared cell borders, round nuclei, basophilic paracentric nucleoli, basophilic cytoplasm, brisk mitoses and frequent apoptosis; punctuated by tingible body macrophages portraying “starry sky” appearance. Focal granulomatous reactions may sometimes be present. The tumor cells are positive for B-cell markers (CD19, CD20, CD79a, CD22, PAX5), GC markers (CD10, BCL6, CD38, HGAL, MEF2B), IgM, Ki67>95%; and negative for BCL2 and TdT (Figure 4 A-F) [7].
3.3.2. Prognosis and Treatment
3.4. Stewart Granuloma (SG)
Stewart granuloma was first reported in 1897 by McBride as “Case of rapid destruction of the nose and face” [85] and later in 1928 by Stewart [86], whose name became the disease eponym. This disease is characterized by midfacial disfiguring, extirpative and ulcerative disease of the nose, paranasal sinuses, nasopharynx, tonsils, palate, hypopharynx and larynx, often accompanied by distant metastasis at presentation [7,85,86,87,88]. It has therefore attracted many descriptive names including lethal midline granuloma, malignant/mutilating granuloma of the nose, granuloma ganrenescens, reticulo-endothelial sarcoma and later malignant histiocytosis or lymphoma [85,86,88]. The latter was a misnomer because the prominent presence of histiocytes represents reactive hemophagocytic lymphohistiocytosis [7,87,88]. The application of modern immunohistochemistry and molecular analysis unraveled the true nature of the disease, which was not possible in the past due the need for functional studies to confirm natural killer cell lineage [88]. SG has been described as a T-cell lymphoma which is confirmed by the expression of surface CD3 and T-cell receptor (TCR) and TCR gene rearrangement. However, the cases that are surface CD3-, TCR- and lack TCR rearrangement, and express NK cell markers (often CD56) are of NK-cell lineage [7,88,89]. With this modern development, SG is now reclassified as extranodal NK/T-cell lymphoma, nasal type (ENNKTL). Most ENNKTL (60-90%) are of NK-lineage, with the remaining 10-40% being of T-cell lineage.
3.4.1. Etiology and Pathogenesis
There is prevalence in eastern Asia, Central and South America. Prevalence in haplotypes HLA-DPB1, HLA-DRB1 and IL18RAP in eastern Asia predisposes these populations to the disease. There is strong association with EBV which occurs in clonal episomal forms in the tumor cells. EBV type A is usually involved, with type II latency [7,88,90,91,92,93]. Immunodeficiency may also be a predisposing factor [7]. Complex genetic changes have been documented, which involve genes of the JAK/STAT and NF-KB and DPGFR signaling pathways, epigenetic modifiers, tumor suppressor genes, RNA helicase gene DDX3X and non-coding RNAs (microRNAs and long intervening/intergenic non-coding RNA) [7,88].
3.4.2. Pathology
There is infiltration by atypical lymphoid cells which range from small, medium to large size, usually occurring in mixture. Typically, epithelial invasion leading to lymphoepithelial lesions, angiocentricity, angioinvasion and extensive geographical necrosis are evident. The tumor cells are immunophenotyically surface CD3 and TCR+ in T-cell type and surface CD3- and TCR-, with CD56 positivity in the NK-cell type ENNKTL (Figure 4 G-K). TCR rearrangement is present in the former and negative in the latter. Cytoplasmic CD3 may be present in the NK-cell type. Typically, both types express cytotoxic proteins. ENNKTL is positive for EBV, which is required for definitive diagnosis [7,88].
3.4.3. Treatment and Prognosis
Much advancement has been made in treating ENNKTL. Radiotherapy is the mainstay of treatment. For local disease, concurrent chemoradiotherapy and sequential chemoradiotherapy is used. The most recent protocol for advanced disease is combined radiotherapy and chemotherapy administered synchronously or metachronously. Chemotherapeutic agents include L-asparaginase, Aspa-Met-Dex, SMILE, DeVIC, ESHAP, DEP, VIPD, VIDL, DDGP, MESA, GELOX, PGEMOX, and pegaspargase is used in patients allergic to Escherichia coli L-asparaginase [88,94,95,96,97,98].
3.5. Sezary Syndrome (SS)
Named after Albert Sezary [102], SS is A T-cell neoplasm featuring the triad erythroderma, generalized lymphadenopathy and clonally proliferated T cells featuring cerebriform nuclei. SS is rare, and affects adults aged >60 years, with male predominance, presenting as leukemia, skin and lymph node involvement. Almost the whole skin (>80%)is affected, with extensive erythroderma. In advanced stages, lymph nodes and various viscera, more commonly oropharynx, lungs and CNS, are affected. This is a rare disease with an incidence of 0.36/100,000 person-years and accounts for 2-3% of cutaneous T-cell lymphomas [7].
3.5.1. Etiology and Pathogenesis
Two genetic mutational signatures are identified. Signature 1 indicative of age-related deamination that is prevalent among T-cell lymphomas and signature 7 characteristic of UV exposure prevalent in cutaneous T-cell lymphomas [103]. Environmental UV exposure is causal in transforming T cells in the skin. A high tumor mutation burden reflective of ultraviolet induced mutations, gene mutations affecting T-cell signaling and upregulation of NF-KB activity (PLCG1, CARD11, CD28, CARMIL2), mutations in the DNA damage response pathway (TP53, POT1, ATM) and mutations affecting JAK/STAT signaling (STAT5B, JAK3), chromatin modifiers (ARID1A, TRRAP, DMNT3A, TET2) are detected [7,103,104,105].
3.5.2. Pathology
CD3+, CD4+, CD8-, PD1 (CD279)+, CLA (cutaneous lymphocyte antigen)+, CCR4 (skin homing receptor)+ neoplastic medium-sized T cells with cerebriform nuclei infiltrate involved tissues and circulate in the peripheral blood. In the skin, epidermotropism may be absent. In lymph nodes, partial or complete infiltration may occur. In bone marrow, there may be interstitial or sparse infiltration6. The major histologic differential diagnosis is Mycosis fungoides, which is distinguishable by being an indolent disease primarily affecting the skin with consistent epidermotropism [7,102].
3.5.3. Prognosis
4. Conclusions
Eponyms have fulfilled a meritorious service - assembling one or more similar/related diseases with comparable manifestations into disease entities for easier recognition and management. With scientific and technological advances in the past decades, the nature of many have been elucidated with regard to cell lineage, histogenesis, morphologic nuances, immunophenotypic and genetic characteristics. New nomenclature, classification, prognostic predictions and treatment strategies have thus been developed for these eponymic diseases. Eye-opening discoveries have uncovered GC B-cell lineage of Hodgkin lymphoma, GC B-cell phenotype in Burkitt lymphoma, NK/T-cell lineage of Stewart granuloma which is reclassified as NK/T-cell lymphoma, nasal type. Classic Hodgkin lymphoma is further found to originate from crippled GC B cells which have mostly lost their B-cell identity, thus explaining non-expression of many B-cell markers in HL. NK/T-cell lymphomas are discovered to be etiologically and pathogenetically EBV-related, and UV light exposure is causally related to cutaneous T-cell transformation in Sezary syndrome. Langerhans cell histiocytosis, Erdheim-Chester disease and Rosai-Dorfman disease have also been discovered to be neoplasms of histiocytic/dendritic cell lineage. A classification system has been formulated for Castleman disease based on etiologic (KSHV/HHV8) and clinical syndromic (POEMS. TAFRO) findings. A number of fibroinflammatory diseases of various sites are reclassified as IgG4-related disease. Kikuchi-Fujimoto disease is discovered to be the sequel of a prominent T-cell response to some unknown antigens. Recent discoveries on hematolymphoid eponymic diseases are summarized in Table 1. While best avoided, many eponyms of these diseases are here to stay and it remains to be seen if they could be renamed or reclassified as more findings in state of art technology unfold.
Author Contributions
Ng CS- Conception, conceptualization, literature review, curation of data, drafting manuscript, finalization and approval of manuscript. Qin JL- Retrieval and review of cases, image preparation of figures, review and approval of final manuscript.
Funding
The authors declare that no funding was obtained for this work.
Acknowledgments
The authors thank Yvonne Chan for preparing the manuscript.
References
- [No authors listed] Classification and nomenclature of morphological defects. The Lancet 1975;305:513. [CrossRef]
- Castleman B, Ivenon L, Menendez VP. Localized mediastinal lymph node hyperplasia resembling thymoma. Cancer 9(1956), pp822-830.
- Keller AR, Hochholzer L, Castleman B. Hyaline vascular and plasma-cell types of giant lymph node hyperplasia of the mediastinum and other locations. Cancer 29(1972), pp670-683.
- van Rhee F, Stone K, Szmania S, Barlogie B, Singh Z. Castleman disease in the 21st century: an update on diagnosis, assessment, and therapy. Clin Adv Hematol Oncol 8(2010), pp486-498. [PubMed]
- Dispenzieri A, Fajgenbaum D. Overview of Castleman disease. Blood 135(2020),pp1353-1364. [CrossRef]
- Wu D, Lim MS, Jaffe ES. Pathology of Castleman disease. Hematol Oncol Clin N Am 32(2018), pp37-52. [CrossRef]
- WHO Classification of Tumours Editorial Board. WHO Classification of Tumours. Haematolymphoid Tumours. 5th ed. Vol 11. Beta version ahead of print. Lyon: International Agency for Research on Cancer; 2024. https://tumourclassification.iarc.who.
- Chang KC, Wang YC, Hung LY et al. Monoclonality and cytogenetic abnormality in hyaline vascular Castleman disease. Mod Pathol 27(2014), pp823-831.
- Li Z, Lan X, Li C, Zhang Y, Wang Y, Xue W et al. Recurrent PDGFRB mutations in unicentric Castleman disease. Leukemia 33(2019), pp:1035-1038. [CrossRef]
- Butzmann A, Kumar J, Sridhar K et al. A review of genetic abnormalities in unicentric and multicentric Castleman disease. Biology 10(2021),251. [CrossRef]
- Goodman AM, Jeong A, Philips A et al. Novel somatic alterations in unicentric and idiopathic multicentric Castleman disease. Eur J Haematol 107(2021), pp642-649. [CrossRef]
- Nishimoto N, Karakura Y, Aozasa K, Johkoh T, Nakamura M, Nakano S et al. Humanized anti-interleukin 6 receptor antibody treatment of multicentric Castleman disease. Blood 106(2005), pp2627-2632. [CrossRef]
- van Rhee F, Fayad L, Voorhees P et. Situximab, a novel anti-interleukin 6 monoclonal antibody for Castleman disease. J Clin Oncol 28(2010), pp3701-3708.
- van Rhee F, Oksenhendler E, Sokalovic G et al. International evidence-based consensus diagnostic and treatment guidelines for unicentric Castleman disease. Blood Adv 4(2020), pp6039-6050. [CrossRef]
- Suda T, Katano H, Delso G, Kakiuchi C, Nakamura T, Shiota M et al. HHV-8 infection status of AIDS-unrelated and AIDS-associated multicentric Castleman disease. Pathol Int 51(2001), pp671-679. [CrossRef]
- Liu AY, Nabel CS, Finkelman BS, Ruth JR, Kurzrock R, van Rhee F et al. Idiopathic multicentric Castleman disease: a systematic literature review. Lancet Haematol 3(2016), e163-e175. [CrossRef]
- Lin O, Frizzera G. Angiomyoid and follicular dendritic cell proliferative lesions in Castleman disease of hyaline-vascular type: a study of 10 cases. Am J Surg Pathol 21(1997), pp1295-1306.
- IzumiM, Mochizuki M, Kuroda M, Iwaya K, Mukai K. Angiomyoid proliferative lesion: an unusual stroma-rich variant of Castleman disease of hyaline-vascular type. Virchow Arch 441(2002), pp400-405.
- Walsh-Jahake R, Cui W, Zhang D. Late recurrence of Castleman’s disease with mixed angiomyoid, histiocytic reticulum cell, follicular dendritic cell stroma-rich proliferations: a case report and review of the literature. J Hematopathol 8(2015), pp43-47.
- Chan JKC, Tsang WY, Ng CS. Follicular dendritic cell tumor and vascular neoplasm complicating hyaline-vascular Castleman disease. Am J Surg Pathol 18(1994), pp517-525. [CrossRef]
- Chan JKC, Luk SC, Ho PL. Stroma-rich Castleman’s disease with superimposed Kikuchi’s lymphadenitis-like changes. Int J Surg Pathol 4(1997), pp197-202. doi.org/10.1177/106689699700400401.
- Kikuchi, M. Lymphadenitis showing focal reticulum cell hyperplasia with nuclear debris and phagocytosis/ Acta Hematol Jpn 35(1972), pp379-380.
- Fujimoto Y, Koyima Y, Yamaguchi K. Cervical subacute necrotizing lymphadenitis. Naika 30(1972), pp920-927.
- Kuo, TT. Kikuchi’s disease (Histiocytic necrotizing lymphadenitis). A clinicopathologic study of 79 cases with an analysis of histologic subtypes, immunohistology and DNA ploidy. Am J Surg Pathol 19(1995), pp798-809. [CrossRef]
- Pileri S, Kikuchi M, Helbron D, Lennert K. Histiocytic necrotizing lymphadenitis without granulocytic infiltration. Virchow Arch A Pathol Anat 395(1982), pp257-271.
- Papadimitriou CS, Rapacharalampous NX. Histiocytic necrotizing lymphadenitis without granulocytic infiltration. Arch Pathol Lab Med 109(1985), pp107-108.
- Dorfman RF, Berry GJ. Kikuchi’s histiocytic necrotizing lymphadenitis: an analysis of 108 cases with emphasis on differential diagnosis. Sem Diagn Pathol 5(1988), pp329-345.
- Tsang WYW, Chan JKC, Ng CS. Kikuchi’s lymphadenitis. A morphologic analysis of 75 cases with special reference to unusual features. Am J Surg Pathol 18(1994), pp219-231. [PubMed]
- Tsang WYW, Chan JKC. Fine-needle aspiration cytologic diagnosis of Kikuchi’s lymphadenitis: a report of 27 cases. Am J Clin Pathol 102(1994), pp454-458. doi.org/10.1093/ajcp/102.4.454.
- Deaven D, Horna P, Cualing H, Soloi L. Pathogenesis, diagnosis and management of Kikuchi-Fujimoto disease. Cancer Control 21(2014), pp313-321. [CrossRef]
- Xu F, Ba X, Yang H et al. Kikuchi disease with an exuberant proliferation of large T-cells: a study of 25 cases that can mimic T-cell lymphoma. Histopathology 82(2023), pp340-353. Doi.org/10.1111/his.14821.
- Yamamoto M, Takahashi H, Ohara M, Suzuki C, Naishiro Y, Yamamoto H et al. A new conceptualization for Mikulicz’s diseases as an IgG4-related plasmacytic disease. Mod Rheumatol 16(2006), pp335-340. [CrossRef]
- Mahajan VS, Mattoo H, Deshpande V, Pillai SS, Stone JH. IgG4-related disease. Ann Rev Pathol 9(2014), pp315-347. [CrossRef]
- Hamano H, Kawa S, Horiuchi A, Unno H, Furuya N, Akamatsu T et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Eng J Med 344(2001), pp732-738. [CrossRef]
- Kamisawa T, Funata N, Hayashi Y et al. A new clinicopathological entity of IgG4-related autoimmune disease. J Gastroenterol 38(2003), pp982-984.
- Cheuk W, Chan JKC. IgG4-related sclerosing disease. A critical appraisal of an evolving clinicopathologic entity. Adv Anat Pathol 17(2010), pp303-332. [CrossRef]
- Deshpande V, Zea Y, Chan JKC,Yi EE, Sato Y, Yoshino Y et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol 25(2012), pp1181-1192. [CrossRef]
- Wang X, Ng CS, Yin W. A comparative study of Kimura’s disease and IgG4-related disease: similarities, differences and overlapping features. Histopathology 2021;79(5):801-809. [CrossRef]
- Nehring P, Przybytkowski A. Think twice before operating on a pancreatic mass: could it be IgG4-related disease? Lancet 395(2020), 816.doi org/10.1016/S0140-6736(20)30169-0.
- Saavedra-Perez D, Vaquero EC, Ayuso JR, Fernandez-Cruz L. Autoimmune pancreatitis: a surgical dilemma. Cir Esp (English ed) 92(2014), pp645-653. [CrossRef]
- Munemura R, Maehara T, Murakami Y, Koga R, Aoyagi R, Kaneko N et al..Distinct disease-specific Tfh cell populations in 2 different fibrotic diseases: IgG4-related disease and Kimura disease. J Allergy Clin Immunol 150(2022), pp440-155. Doi.org/10.1016/j.jaci.2022.03.034.
- Jarrell J, Baker MC, Perugino CA, Liu H, Bloom MS, Maehara T et al. Neutralizing anti-IL-1 receptor antagonist autoantibodies include inflammatory and fibrotic mediators in IgG4-related disease. J Allergy Clin Immunol 149(2022), pp358-368. doi.org/10.1016/jaci 2021.05.002.
- Langerhans, P. Langerhans P. Ueber die Nerven der menschlichen Ilaut. Arch Anat Physiol Kin Med. 44(1868), pp325-338. [CrossRef]
- Rowden G, Lewis MG, Sullivan AK. Ia antigen expression on human epidermal Langerhans cells. Nature 268(1977), pp192-193. [CrossRef]
- Kapur P, Reickson C, Rakheja D, Carden KR, Hoang MP. Congenital self-healing reticulohistiocytosis (Hashimoto-Pritzker disease): ten-year experience at Dallas Children’s Medical Center. J Am Acad Dermatol 56(2007), pp290-294. [CrossRef]
- Takayama S, Matubayashi T, Koizumi M. BRAF mutation analysis in two cases of congenital self-healing Langerhans cell histiocytosis. Cureus 14(2022), e32497. [CrossRef]
- Hoeffel G, Wang Y, Greter M, See P, Teo P, Malleret B et al. Adult Langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac-derived macrophages. J Exp Med 209(2012), pp1167-1181. [CrossRef]
- Allen CE, Li L, Peters TL, Leung HCE, Yu A, Man TK et al. Cell-specific gene expression in Langerhans cell histiocytosis lesions reveal a distinct profile compared with epidermal Langerhans cells. J Immunol 184(2010), pp4557-4567. [CrossRef]
- Kemps PG, Hebeda KM, Pals ST, Verdijk RM, Lam KH, Bruggink AH et al. Spectrum of histiocytic neoplasms associated with diverse haematological malignancies bearing the same oncogenic mutation. J Pathol Clin Res 7(2021), pp10-26. [CrossRef]
- Badalian-Veoy G, Verogilo J-A, Degar BA et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood 116(2010), pp1919-1923. [CrossRef]
- Yousem SA, Dacic S, Nikiforov YE, Nikiforov M. pulmonary Langerhans cell histiocytosis: profiling of multifocal tumors using next-generation sequencing identifies concordant occurrence of BRAFV600E mutations. Chest 143(2013), pp1679-1684.
- Brown NA, Furtado LV, Betz BL, Kiel MJ, Weigelin HC, Lim MS et al. High prevalence of somatic MAP2k1 mutations in BRAF V600E-negative Langerhans cell histiocytosis. Blood 124(2014), pp1655-1658. [CrossRef]
- Allen CE, Ladisch S, McClain KL. How I treat Langerhans cell histiocytosis. Blood 126(2015), pp26-35.
- Harmon CM, Brown N. Langerhans cell histiocytosis. A clinicopathologic review and molecular pathogenetic update. Arc Pathol Lab Med 139(2015), pp1211-1214. [CrossRef]
- Hutter C, Minkoy M. Insights into the pathogenesis of Langerhans cell histiocytosis: the development of targeted therapies. Immunotargets Ther 5(2016), pp81-91. [CrossRef]
- Mazor RD, Manerish-Mazor M, Shoenfield Y. Erdheim-Chester disease: a comprehensive review of the literature. Orphanet J Rare DS 137(2013). doi.org/10.1186/1750-1172-8-137.
- Bruce-Brand C, Schneider JW, Schubert P. Rosai-Dorfman disease: an overview. J Clin Pathol 73(2020), pp697-705. [CrossRef]
- Garces S, Medeiros LJ, Patel KP, Li S, Pina-oviedo S, Li J et al. Mutually exclusive recurrent KRAS and MAP2K1 mutations in Rosai-Dorfman disease. Mod Pathol 30(2017). Pp1367-1377. [CrossRef]
- Maoic I, Pittaluga S, Dale JK, Niemela JE, Delsol G, Diment J et al. Histologic features of sinus histiocytosis with massive lymphadenopathy in patients with autoimmune lymphoproliferative syndrome. Am j Surg Pathol 29(2005), pp903-911.
- Emile J-F, Abla O, Fraitag S et al. Revised classification of histiocytosis and neoplasms of the macrophage-dendritic cell lineages. Blood 127(2016), pp2672-2681.
- Hodgkin, T. On some morbid appearance of the absorbent glands and spleen. Med Chir Trans 17(1832), 0068-114.
- Lakhtakia R, Burney I. A historical tale of two lymphomas. Sultan Quaboos Uni Med J, 15(2015), ppe202-206.
- Kanzler H, Kuppers R, Hansmann ML, Rajewshy K. Hodgkin and Reed-Sternberg cells in Hodgkin’s disease represent the outgrowth of a dominant tumor clone derived from (crippled) germinal center B cells. J Exp Med 184(1996), pp1495-1505. [CrossRef]
- Kuppers R, Schwering I, Brauninger A, Rajewsky K, Hansmann M-L. Biology of Hodgkin’s lymphoma. Ann Oncol 13(2002), pp11-18.
- Marafiot T, Hummel M, Foss HD, Laumen H, Korbjuhn P, Anagnostopoulos I et al. Hodgkin and reed-sternberg cells represent an expansion of a single clone originating from a germinal center B-cell with functional immunoglobulin gene rearrangements but defective immunoglobulin transcription. Blood 95(2000), pp1443-1450. [PubMed]
- Schwering I, Brauninger A, Klein U, Jungnickel B, Tinguely M, Diehl V et al.. Loss of the B-lineage –specific gene expression program in Hodgkin and Reed-Sternberg cells of Hodgkin lymphoma. Blood 101(2003), pp1505-1512. [CrossRef]
- Marafiot T, Hummel M, Anagnostopoulos I, Foss HD, Falini B, Delsol G et al.. Origin of nodular lymphocyt-predominant Hodgkin’s disease from a clonal expansion of highly mutated germinal center B cells. N Engl J Med 33(1997), pp453-458. [CrossRef]
- Braeuninger A, Kuppers R, Strickler JG, wacker HH, Rajewsky K, Hansmann ML. Hodgkin and Reed-Sternberg cells in predominant Hodgkin disease represent clonal populations of germinal center-derived tumor B cells. Proc Natl Acad Sci USA 94(1997), pp9337-9342. [CrossRef]
- Saarinen S, Aavikko M, Aittomaki K et al. Exome sequencing reveals germline NPAT mutation as a candidate risk factor for Hodgkin lymphoma. Blood 118(2011), pp493-498.
- Strobbe L, Valke LLFG, Diets IJ, van den Brand M, Aben K, Raemaekers JMM et al. A 20-year population-based study on the epidemiology, clinical features, treatment, and outcome of nodular lymphocyte predominant Hodgkin lymphoma. Ann Hematol 95(2016), pp417-423. [CrossRef]
- Van den Berg A, Maggio E, Diepstra A et al. Germline FAS gene mutation in a case of ALPS and NLP Hodgkin lymphoma. Blood 15(2002), pp1492-1494.
- Elodarska I, Nooyen P, Maes B et al. Frequent occurrence of BCL6 rearrangements in nodular lymphocyte predominant Hodgkin lymphoma but not in classical Hodgkin lymphoma. Blood 101(2003), pp706-710. [CrossRef]
- Thurner L, Hartmann S, Fadle N et al. Lymphocyte predominant cells detect Moraxella catarrhalis-derived antigens in nodular lymphocyte predominant Hodgkin lymphoma. Nat Commun 11(2020), 2465. [CrossRef]
- Al-Mansour M, Connors JM, Gasciyne RD, Skinner B, Savage KJ. Transformation to aggressive lymphoma in nodular lymphocyte-predominant Hodgkin lymphoma. J Clin Oncol 28(2010), pp793-799. [CrossRef]
- Paschold L, Willscher E, Bein J et al. Evolutionary clonal trajectories in nodular lymphcyte-predominant Hodglin lymphoma with high risk transformation. Haematologica 106(2021), pp2654-2666. [CrossRef]
- Burkitt, D. A sarcoma involving the jaws in African childen. Br J Surg 46(1958), pp218-223.
- Roschewski M, Dunleavy K, Abramson JS et l. Multicenter study of risk-adapted therapy with dose-adjusted EPOCH-R in adults with intreated Burkitt lymphoma, J Clin Oncol 38(2020), pp2519-2529. [CrossRef]
- Salzburg J, Burkhardt B, Zimmermann M, Wachowski O, Woessmann W, Oschlies I et al. Prevalence, clinical pattern and outcome of CNS involvement in childhood and adolescent non-Hodgkin's lymphoma differ by non-Hodgkin’s lymphoma subtype: a Berlin-Frankfurt-Munster Group Report. J Clin Oncol 25(2007), pp3915-3922. [CrossRef]
- Fitzsimmons L, Boyce AJ, Wei W et al. Coordinated repression of BIM and PUMA by Epstein-Barr virus latent genes maintains the survival of Burkitt lymphoma cells. Cell Death Differ 25(2018), pp241-254. [CrossRef]
- Bellan C, Lazzi S, Hummel M et al. Immunoglobulin gene analysis reveals 2 distinct cells of origin for EBV-positive and EBV-negative Burkitt lymphoma. Blood 106(2005), pp1031-1036. [CrossRef]
- Grande BM, Gerhard DS, Jiang A et al. Genome-wide discovery of somatic coding and noncoding mutations in pediatric endemic and sporadic Burkitt lymphoma. Blood 133(2019), pp1313-1324. [CrossRef]
- Robbiani D, Deroubaix S, Feldhahn N, Oliveira TY, Callen E, Wang Q et al. Plasmodium infection promotes genomic instability and AID-dependent B cell lymphoma. Cell 162(2015), pp727-737. [CrossRef]
- Schmitz R, Young RM, Ceribelli M. Jhavar S, Xiao W, Zhang M et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 490(2012), pp116-120. [CrossRef]
- Saleh K, Michot JM, Camara-Clayette V, Vassetsky Y, Ribrag V. Burkitt and Burkitt-like lymphomas: a systematic review. Currr Oncol Rep22(2020), 33. Doi”:10.1007/s11912-020-0898-8.
- McBride, P. Case of rapid destruction of the Nose and Face. J Laryngol Otol xii( 1897), 64.
- Stewart, JP. The Hisopathology of Mastoiditis. J Laryngol Otol xlii( 1928), 689.
- Ng CS, Chan JKC, Cheng PNM et al. Nasal T cell lymphoma associated with hemophagocytic sundrome. Cncer 58(1986), pp67-71.
- Ng, CS. From the midfacial destructive drama to the unfolding EBV story: a short history of EBV-positive NK-cell and T-cell lymphoproliferative diseases. Pathology 56(2024), pp773-785.
- Ng CS, Chan JKC, Lo STH. Expression of natural killer cell markers in non-Hodgkin’s lymphoma. Hum Pathol 18(1987), pp1257-1262.
- Tse E, Kwong YL. The diagnosis and management of NK/T-cell lymphomas. J Hematol Oncol 10(2017), 85.
- Harabuchi Y, Imai S, Wakashima J et al. Nasal T-cell lymphoma causally associated with Epstein-Barr virus: clinicopathologic, phenotypic and genotypic studies. Cancer 77(1996), pp2137-2149.
- Yoon TY, Lee HT, Chang SH. Nasal type T/natural killer cell angiocentric lymphoma. Epstein Barr virus associated and showing clonal T-cell receptor gamma gene rearrangement. Br J Dermatol 140(1999), pp505-508.
- Nagata H, Konno A, Kimura N et al. Characterization of novel natural killer (NK) –cell and gammadelta T-cell lines established from primary lesions of nasal T/NK-cell lymphomas associated with Epstein-Barr Virus. Blood 97(2001), pp708-713.
- Wang H, Fu BB, Gale RP et al. NK/T-cell lymphomas. Leukemia 35(2021), pp2460-2468.
- Qi S-N, Yang Y, Zhang Y-J et al. Risk-based, response-adapted therapy for early stage extranodal nasal-type NK/T-cell lymphoma in the modern chemotherapy era: a China Lymphoma Collaborative Group (CLCG) study. Am J Hematol 95(2020), pp1046-1057.
- Kim SJ, Yang DH, Kim JS et al. Concurrent chemoraiotherapyfollowed by L-asparaginase-containing chemotherapy, VIDL, for localised nasal extranodal NK/T cell lymphoma . [CrossRef]
- Huang Y, Yang J, Liu P et al. Intensity-modulated radiation therapy followed by GDP chemotherapy for newly diagnosed Stage I/II extranodal natural killer/T cell lymphoma, naal type. Ann Hematol 96(2017), pp1477-1483.
- Kim HJ, Ock CY, Kim TM et al. Comparison of native Escheria Coli L-asparaginase versus pegylated asparaginase in combination with ifosfamide, methotrexate, ectoposide and prednisolone (IMEP), I extranodal NK/T cell lymphoma, nasal type (NTCL). Cancer Res Treat 50(2018), pp670-680.
- Suzuki, R. Pathogenesis and treatment of extranodal natural killer/T-cell lymphoma. Semin Hematol 51( 2014), pp42–51. [CrossRef] [PubMed]
- Kwong YL, Chan TSY, Tan D et al. PD1 blockade with pembrolizumab is highly effective in relapsed or refractory NK/T-cell lymphoma failing L-asparaginase. Blood 129(2017), PP2437-2442.
- Cho S-G, Kim N, Sohn H-J et al. Long-term outcome of extranodal NK/T cell lymphoma patients treated with postremission therapy using EBV LMP1 and LMP2a-specific CTLs. Mod Ther 23(2015), pp1501-1509.
- Steffen, C. the man behind the eponym dermatology in historical perspective: Albert Sezary and the Sezary syndrome. Am j Dermatopathol 28(2006), pp357-367. [CrossRef]
- Jones L, Degasperi A, Grandi V, Amarante TD, Genomics England Research Consortium, Mitchell TJ et al. Spectrum of mutational signatures in T-cell lymphoma reveals a key role for UV radiation in cutaneous T-cell lymphoma. Sci Rep 11(2021), 3962. [CrossRef]
- Park J, Daniels J, Wartewig T et al. Integrated genomic analysis of cutaneous T-cell lymphomas reveal the molecular basis for disease heterogeneity. Blood 138(2021), pp1225-1236. [CrossRef]
- Da Silva Almeida AC, Abate F, Khiabanian H et al. The mutational landscape of cutaneous T cell lymphoma and sezart syndrome. Nat Genet 47( 2015), pp1465–1476. [CrossRef]
- Williamze R, Jafe ES, Burg G et al. WHO-EORTC classification for cutaneous lymphomas. Blood 105(2005), pp3768-3785. [CrossRef]
Figure 1.
(A) – (D) Unicentric Castleman disease. F/47, 10 cm mediastinal mass. (A) Hyperplastic follicles with prominent germinal centers and interfollicular lymphoplasmacytic cells. H&E x100 (B) Hyperplastic follicles with radial vessels (lollipop appearance) and concentric rings of mantle lymphocytes (onion skinning) H&E x400. (C) & (D) “Twining” of follicles. (C) CD21, (D) CD20 x100. (E) to (H) Kikuchi-Fujimoto disease. M/13, left cervical lymph node. (E) Large pale “necrotic” area involving paracortex and cortex. H&E x100. (F) Nuclear debris in “necrotic” area. H&E x200. (G) Characteristic histiocytes in “necrotic area. H&E x400. (H) CD68 x100. (I) to (M) IgG4-related disease. M/85, left orbital mass. (I) Extensive fibrosis in lymphoplasmacytic infiltrate. x100. (J) Storiform fibrosis. H&E x200. (K) Obliterative phlebitis. H&Ex400. (L) Obliterated vein. H&Ex200. (M) IgG4-positive plasma cells x200.
Figure 1.
(A) – (D) Unicentric Castleman disease. F/47, 10 cm mediastinal mass. (A) Hyperplastic follicles with prominent germinal centers and interfollicular lymphoplasmacytic cells. H&E x100 (B) Hyperplastic follicles with radial vessels (lollipop appearance) and concentric rings of mantle lymphocytes (onion skinning) H&E x400. (C) & (D) “Twining” of follicles. (C) CD21, (D) CD20 x100. (E) to (H) Kikuchi-Fujimoto disease. M/13, left cervical lymph node. (E) Large pale “necrotic” area involving paracortex and cortex. H&E x100. (F) Nuclear debris in “necrotic” area. H&E x200. (G) Characteristic histiocytes in “necrotic area. H&E x400. (H) CD68 x100. (I) to (M) IgG4-related disease. M/85, left orbital mass. (I) Extensive fibrosis in lymphoplasmacytic infiltrate. x100. (J) Storiform fibrosis. H&E x200. (K) Obliterative phlebitis. H&Ex400. (L) Obliterated vein. H&Ex200. (M) IgG4-positive plasma cells x200.
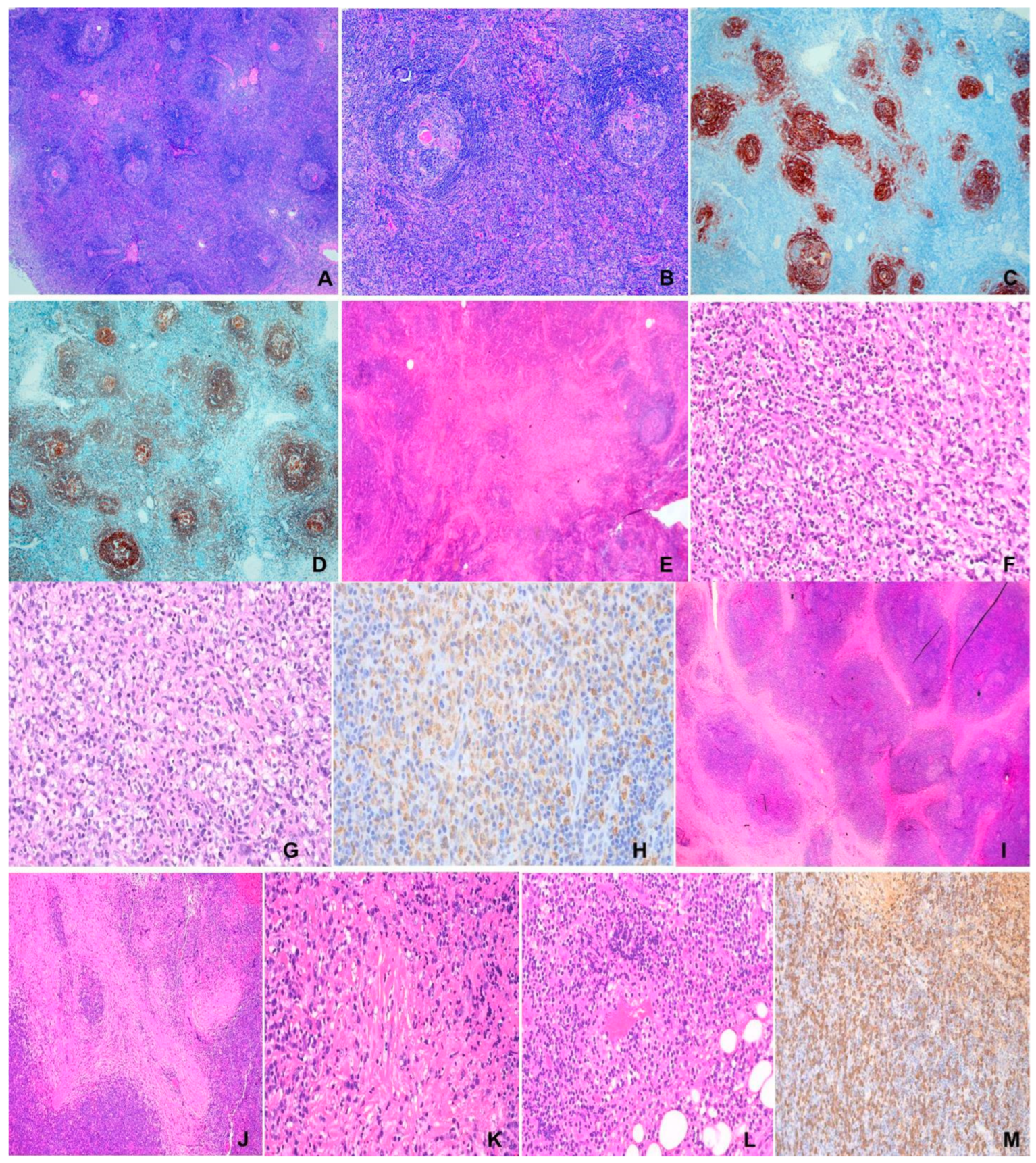
Figure 2.
(A) to (E) Langerhans cell histiocytosis. F/1 Right orbital mass. (A) Infiltrates of eosinophils among histiocytes and giant cells. H&E x200. (B) Characteristic irregular or grooved nuclei of histiocytes and giant cells. H&E x400. (C) S100 X200. (D) CD1a. x200. (E) CD68 X200. (F) to (I) Rosai Dorfman disease. F/31, 1.5 m lung mass. (F) “Sinusoidal” accumulation of large pale histiocytes in a backdrop of plasma cells and lymphocytes. H&E x100. (G) Large pale histiocytes exhibiting emperipolesis. H&E x 400. (H) CD163 x200. (I) Cyclin D1 x200.
Figure 2.
(A) to (E) Langerhans cell histiocytosis. F/1 Right orbital mass. (A) Infiltrates of eosinophils among histiocytes and giant cells. H&E x200. (B) Characteristic irregular or grooved nuclei of histiocytes and giant cells. H&E x400. (C) S100 X200. (D) CD1a. x200. (E) CD68 X200. (F) to (I) Rosai Dorfman disease. F/31, 1.5 m lung mass. (F) “Sinusoidal” accumulation of large pale histiocytes in a backdrop of plasma cells and lymphocytes. H&E x100. (G) Large pale histiocytes exhibiting emperipolesis. H&E x 400. (H) CD163 x200. (I) Cyclin D1 x200.
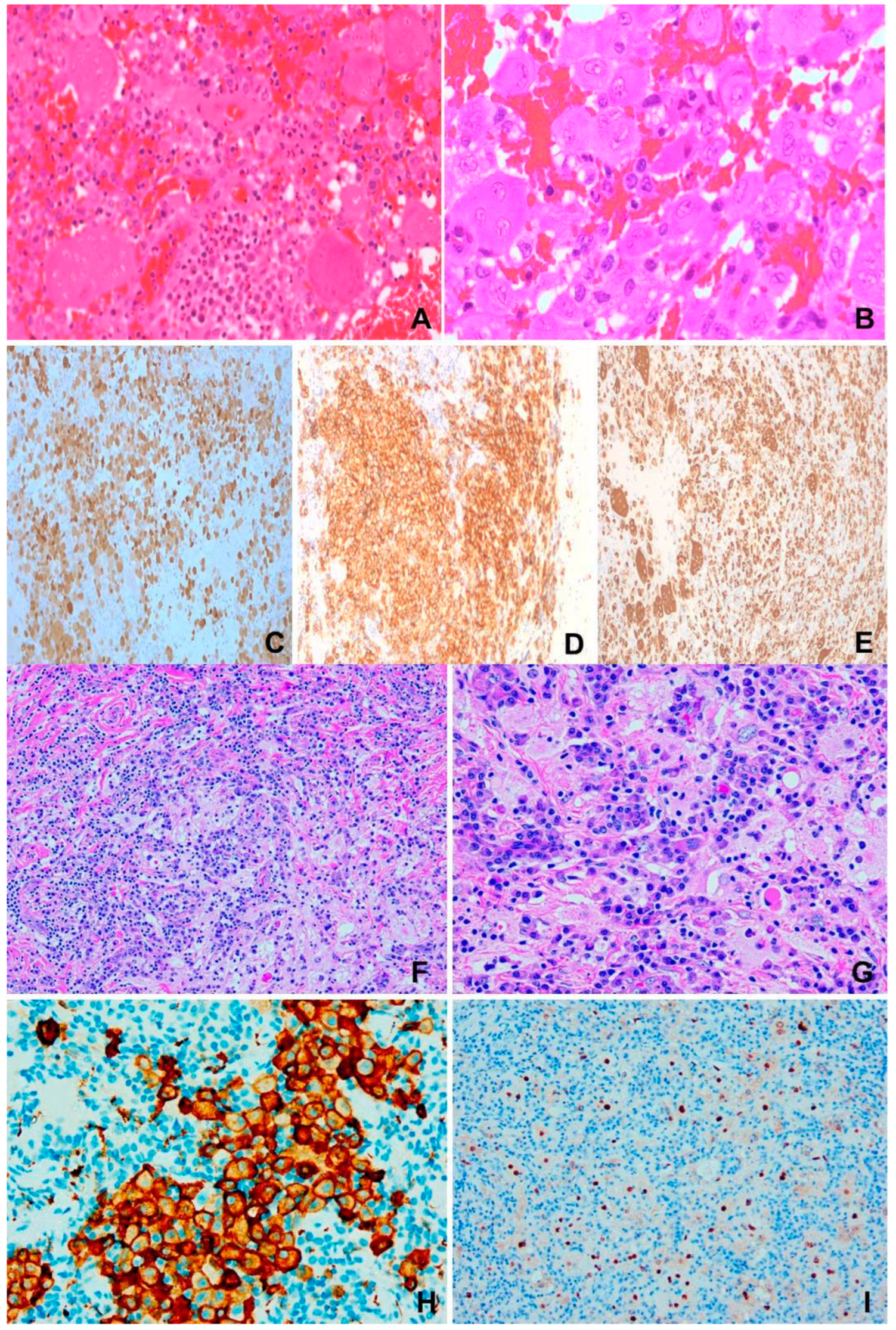
Figure 3.
Hodgkin lymphoma. (A) to (F) Nodular sclerosing type. M/14, neck mass. (A) Extensive fibrosis dissecting cellular islands of lymphoma tissue. H&E x100. (B) Aggregates of “lacunar cells” in a reactive cellular milieu. H&E x200. (C) Large “lacunar cells”. H&E x400. (D) CD30. X200. (E) CD15 x200. (F) CD20 showing rare positively stained “lacunar cells” x100. (G) to (I) Mixed cellularity type. (G) & (H) Reed-Sternberg and Hodgkin cells in mixed cellular milieu. H&E x200 (G), x400 (H). (I) CD30 x400.
Figure 3.
Hodgkin lymphoma. (A) to (F) Nodular sclerosing type. M/14, neck mass. (A) Extensive fibrosis dissecting cellular islands of lymphoma tissue. H&E x100. (B) Aggregates of “lacunar cells” in a reactive cellular milieu. H&E x200. (C) Large “lacunar cells”. H&E x400. (D) CD30. X200. (E) CD15 x200. (F) CD20 showing rare positively stained “lacunar cells” x100. (G) to (I) Mixed cellularity type. (G) & (H) Reed-Sternberg and Hodgkin cells in mixed cellular milieu. H&E x200 (G), x400 (H). (I) CD30 x400.
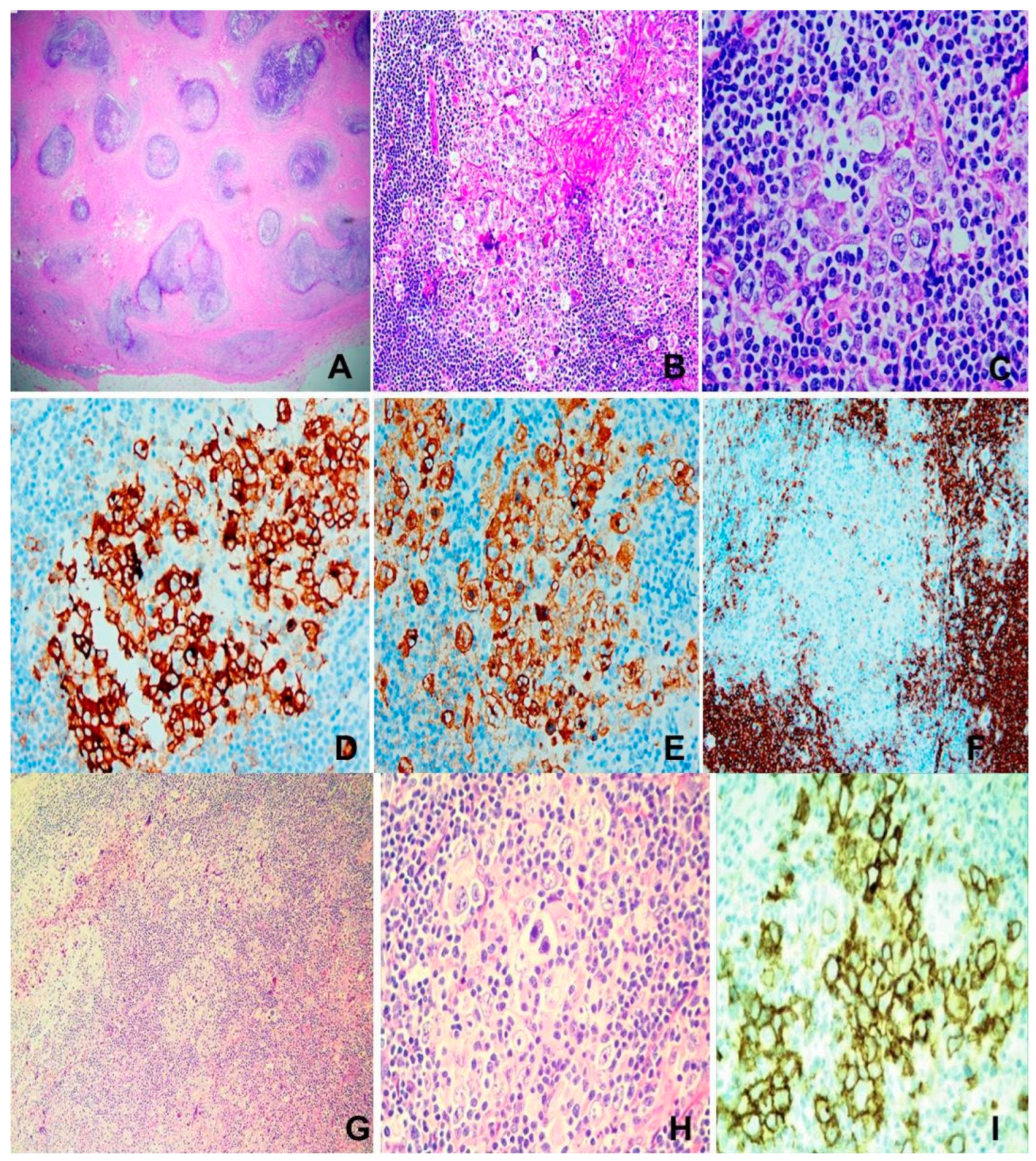
Figure 4.
(A) to (F) Burkitt lymphoma. M/74, small intestinal tumor. (A) Intestinal wall mural invasion. H&Ex20. (B) & (C) “Starry sky” appearance. Pale histiocytes in a “dark” backdrop of tumor cells. H&Ex100 & 200. (D) Medium-sized tumor cells with squaring and brisk mitosis. H&Ex400. (E) Necrosis of tumor. H&Ex200. (F) CD20 immunostaining x200. (G) to (K) NK/T-cell lymphoma, nasal type. M/47, nasal mass. (G) Polymorphic lymphoid infiltrate. H&E x200, (H) Epithelial invasion. H&E x 400, (I) Angioinvasion and necrosis. H&E x 400, (J) CD56 x200, (K) EBER x 200.
Figure 4.
(A) to (F) Burkitt lymphoma. M/74, small intestinal tumor. (A) Intestinal wall mural invasion. H&Ex20. (B) & (C) “Starry sky” appearance. Pale histiocytes in a “dark” backdrop of tumor cells. H&Ex100 & 200. (D) Medium-sized tumor cells with squaring and brisk mitosis. H&Ex400. (E) Necrosis of tumor. H&Ex200. (F) CD20 immunostaining x200. (G) to (K) NK/T-cell lymphoma, nasal type. M/47, nasal mass. (G) Polymorphic lymphoid infiltrate. H&E x200, (H) Epithelial invasion. H&E x 400, (I) Angioinvasion and necrosis. H&E x 400, (J) CD56 x200, (K) EBER x 200.
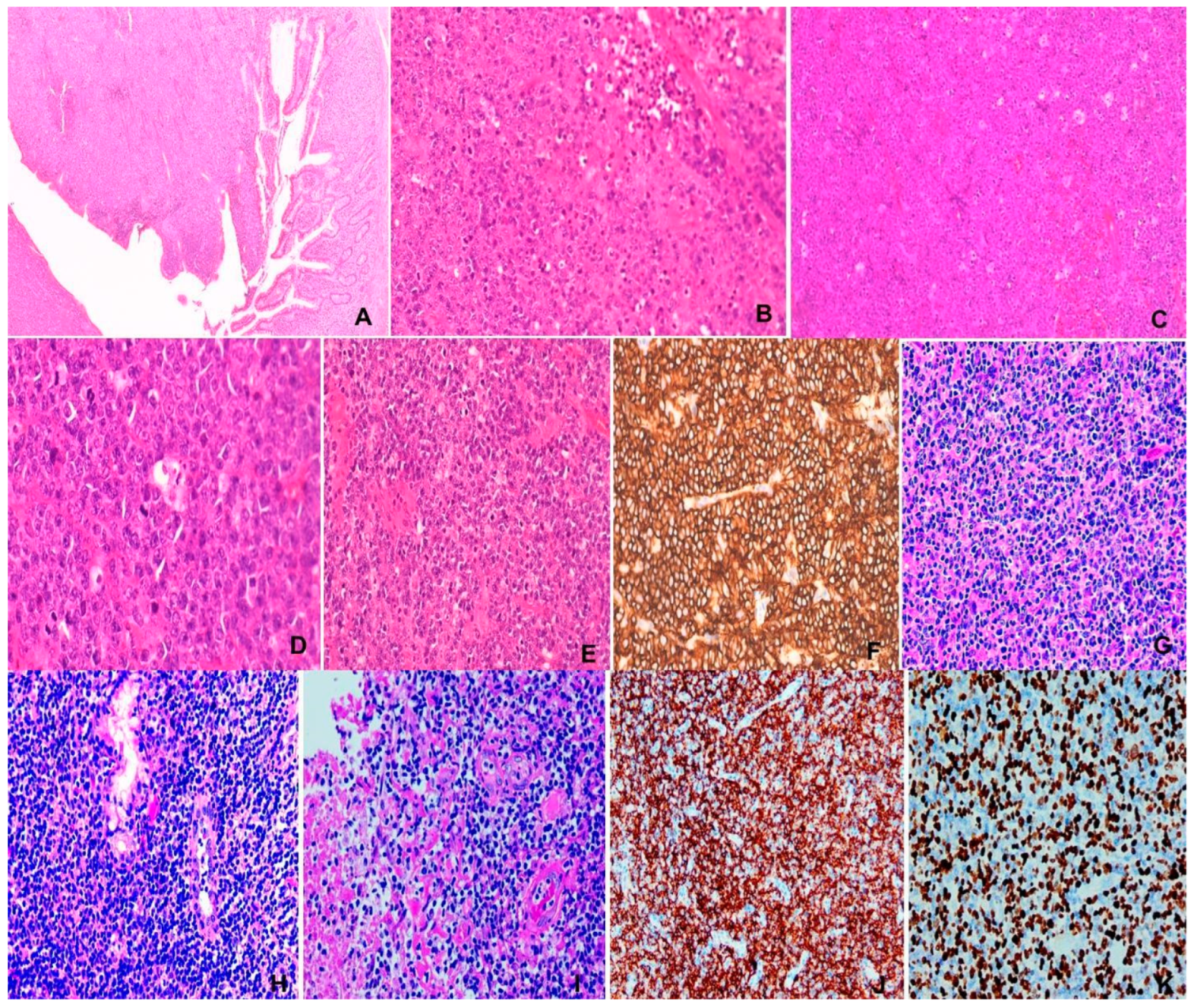

Table 1.
Recent discoveries on hematolymphoid eponymic diseases.
| Eponymic disease | Cell lineage/histogenesis | Pathogenesis | Classification/nomenclature/subtyping |
| Castleman disease |
|
|
|
| Kikuchi-Fujimoto disease |
|
|
|
| Knutter’s disease, Mikulicz’s disease, Ormond disease, Riedel’s thyroiditis. |
|
|
|
| LC/LS/ECD/RDD |
|
|
|
| Hodgkin lymphoma (HL) |
|
|
|
| Burkitt lymphoma |
|
|
|
| Stewart granuloma |
|
|
|
| Sezary Syndrome |
|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



