
Submitted:
04 February 2025
Posted:
06 February 2025
You are already at the latest version
Abstract
Skin paraneoplastic syndromes (SPNS) are a group of disorders that arise as a consequence of cancer but are not directly related to the tumor mass itself. This review aims to provide a comprehensive overview of these syndromes, encompassing their pathophysiology, clinical features, diagnostic approaches, differential diagnosis, and management strategies. These syndromes, which include conditions such as Bazex syndrome, acanthosis nigricans, dermatomyositis, and necrolytic migratory erythema, often manifest prior to or concurrently with a cancer diagnosis, serving as potential early warning signs of underlying malignancies.
This review delves into the spectrum of SPNS and their associations with specific cancer types. Special emphasis is placed on the critical role of dermatologists and on-cologists in identifying these skin manifestations as potential markers of malignancy. By raising awareness of SPNS, this paper highlights the pivotal importance of prompt recognition and intervention in reducing cancer-related mortality.
Keywords:
cancer
; cutaneous paraneoplastic syndromes
; skin disorders
; differentiation
1. Introduction
Paraneoplastic skin syndromes (PNS) represent a fascinating and critical intersection between dermatology and oncology. These rare dermatological manifestations can be the first sign of an underlying malignancy, offering a unique opportunity for early cancer detection and intervention. Recognizing PNS is paramount, as these cutaneous indicators may precede the diagnosis of the primary tumor by months or even years, potentially altering the clinical course and prognosis of patients. However, the diagnostic process is fraught with challenges. The rarity and diverse presentation of PNS often lead to misdiagnosis or delayed recognition, complicating the clinical picture and delaying crucial oncological evaluations.
This article explores the significance of early identification of paraneoplastic skin syndromes in the context of cancer diagnosis and highlights the challenges healthcare providers face in recognizing these elusive conditions. Numerous skin lesions may be associated with tumors, often malignant. These lesions can present as direct metastases; however, there are also several syndromes that are not composed of the same cells as the primary tumor. Despite this, their occurrence is closely linked to specific types of malignancies.
There are some criteria which are helpful in making the diagnosis of paraneoplastic syndromes, which is Curth’s criteria [1,2]. At least one of them must be met to associate cutaneous lesion with underlying malignancy:
1.Coexstinence of dermatoses and cancer
2.Simultaneous development/resolution with the primary tumor. The reappearance of skin lesions indicates advancement of the cancer.
3.There is a distinct relationship between the type of tumor and the type of skin eruption. A particular malignancy is consistently linked with a specific skin condition
4.Reliable case-control studies demonstrate a significant statistical link between the type of cancer and the skin condition.
5.There is a genetic link between skin disorders and cancer
Among paraneoplastic syndromes we can distinguish those that do not pose diagnostic difficulties, but also those that may imitate other skin diseases in their appearance. In this situation histopathological examination may be conclusive. It is necessary to know that skin changes may by connected with internal malignancy, because this may lead to cancer being diagnosed sooner and starting treatment at an early stage which may provide to greater chance for recovery.
Paraneoplastic syndromes which can be problematic in recognition includes e.g., erythema gyratum repens, necrolytic migratory erythema, Bazex syndrome, Sweet syndrome, pyoderma gangrenosum and erythroderma. There is also some syndromes which lead to simple diagnosis and always require cancer’s screening, such as: paraneoplastic pemphigus, dermatomyositis, Laser-Trelat syndrome or acanthosis nigricans.
2. Paraneoplastic Syndromes That Pose Diagnostic Challenges
ERYTHEMA GYRATUM REPENS (EGR) – it is one of rare paraneoplastic syndrome which may cause diagnostic challenges. It is strongly associated with internal malignancy but there are also rare reports of this condition existing without tumors. [3] Clinically, it is a rapidly (usually 1 cm a day) spreading superficial erythema that creates an unusual pattern of intersecting coils and spirals. It resembles the rings of a cut tree, and it is sometimes referred to as “zebra skin.” [4]
Typical localization of skin lesions are trunk and proximal extremities. Rather it spares hands, feet and face. Desquamation may be observed at the edge of the erythema. It spreads centrifugally. Most patients experienced some degree of pruritus.
Histopathological examination is nonspecific. It may show mild to moderate hyperkeratosis, parakeratosis and spongiosis. The dermal vessels surrounded by lympho-histiocytic infiltrate with occasional eosinophils have been described. Mast cells may also be seen. [5] In some cases on direct immunofluorescence test (DIF), granular IgG and C3 deposition can be seen along the dermal-epidermal junction. [6,7]
While the precise etiology of EGR remains unclear, several immunological mechanisms have been proposed to explain its pathogenesis. One hypothesis suggests that the tumor may trigger the production of antibodies that cross-react with the skin’s basement membrane. Alternatively, it has been proposed that the tumor could secrete polypeptides that bind to skin antigens, thereby making them immunogenic. Another theory involves the deposition of tumor antigen-antibody complexes on the basement membrane, potentially leading to a reactive dermatitis. The presence of immunofluorescence patterns involving IgG, C3, and C4 at the basement membrane supports the likelihood of an underlying immunological mechanism. [3,8,9]
In nearly 82% of cases of EGR coexistence of internal malignancy have been reported. The most common are carcinoma of the lung, breasts, esophagus, cervix, stomach and pharynx. There is also some reports of prostate, bladder, intestinal, uterus and pancreas cancer. But some authors described also a patients with EGR without underlying malignancy. In that cases EGR was associated with bullous pemphigoid, CREST syndrome, tuberculosis and also pityriasis rubra pilaris [10,11]
The diagnosis of EGR is made mainly on the basis of the clinical appearance of the skin lesions. That is way it may provide to diagnostic difficulties. Sometimes the clinical pictures may resemble tinea corporis [12]; in these cases, we should perform a mycological examination of the lesion to exclude a fungal infection. Also a medical interview may be helpful if we are dealing with a farmer or an animal owner, mycosis is more likely. Some skin lesions may resemble psoriasis, then a positive family history of psoriasis may be decisive. Subacute cutaneous/discoid lupus erythematosus, bullous pemphigoid, erythema migrans and erythema annulare centrifugum should also be considered in the differential diagnosis.[13] There is nonspecific treatment for EGR. Since the course of the disease parallels that of the underlying disorder, therapy should be targeted at the tumor.
Various dermatologic and immunosuppressive therapies have been employed in the management of EGR, but their effectiveness remains limited. Systemic corticosteroids are often ineffective, and topical steroids, vitamin A, and azathioprine have also failed to alleviate the cutaneous symptoms. Improvement or resolution of EGR, along with its associated severe pruritus, largely depends on the identification and treatment of the underlying malignancy. In cases of widely metastatic disease, the response of EGR to chemotherapy is variable. In some patients, the rash may only resolve in the terminal stage of the disease, typically coinciding with profound immunosuppression. [3,14]
NECROLYTIC MIGRATORY ERYTHEMA (NME) – it is also one of the rare paraneoplastic syndrome. This disease most commonly coexists with cancers, with the highest number of cases described as prevalent in glucagonoma. However, a single case has been reported in the literature of necrolytic migratory erythema coexisting in a patient with lung adenocarcinoma treated with erlotinib [15]. There are also cases described of coexistence with zinc deficiency, liver disease, or pancreatitis, as well as two cases of NME induced in patients with non-small cell lung cancer treated with gefitinib [16,17].
In the pathogenesis of NME, amino acid deficiencies and deficiencies of other elements such as zinc are mentioned, which are directly related to elevated levels of glucagon in the blood. Since more than 90% of NME cases are associated with coexisting glucagonoma, patients often experience weight loss, diarrhea and general malaise. Typical skin lesions present as reddish-brown plaques that undergo superficial necrosis and crusting. Blistering reactions within the erythematous lesions and desquamation are often observed. The lesions can spread peripherally and typically affect the perioral region, trunk, extremities, and perineum. Nail and oral involvement is other associated manifestations. Patients often experience pruritus, pain, and a burning sensation at the site of the lesions. NME is one of the symptoms included in glucagonoma syndrome, in addition to glossitis, diabetes mellitus, angular cheilitis, venous thrombosis, weight loss, normochromic and normocytic anemia and neuropsychiatric symptoms. To help remember these classic characteristics, the acronym “4D” has been suggested, standing for dermatosis, diabetes, deep vein thrombosis, and depression. [18,19]
In histopathological examination some characteristic features for NME can be found. It is crucial to note that the histopathologic changes are confined entirely to the epidermis, especially the superficial epidermis. The hallmark of NME is the necrosis of the upper spinous layer of the epidermis. However, superficial epidermal necrolysis is relatively non-specific and may only be focally present, most likely at the edge of an active lesion. Other significant findings in NME include irregular acanthosis, and loss of the granular layer. Confluent parakeratosis overlying vacuolated superficial keratinocytes is quite characteristic of NME. Additionally, there may be mild perivascular lymphocytic or neutrophilic infiltrate and intraepidermal bullae. Multiple biopsies should be taken if NME is suspected. [20,21].
Dermatologic changes in NME may be misdiagnosed as they can suggest other dermatoses, such as e.g., acrodermatitis enteropatica. In this case erythema and erosions are limited to area around body orifices, but is responsive to zinc supplementation. Some cases of NME require differentiation from erythema multiforme (EM). However, EM is most often triggered by a specific factor (such as medications or herpes infection) and typically presents with a characteristic target or “bullseye” appearance.
These two conditions can also be distinguished by the good response of EM to removing the trigger and to systemic steroid therapy. Skin lesions in contact or atopic dermatitis can mimic those seen in NME. However, atopic dermatitis is often associated with a personal or family history of allergies, or there may be a specific trigger causing contact-related changes, like in contact dermatitis.
In the differential diagnosis, conditions such as erythrokeratoderma and psoriasis should also be considered. Also because of the ulceration in groin area Hailey-Hailey disease (HHD) must be taken into consideration during differentiation. To rule out HHD, a biopsy for histopathological examination should be taken. A family history of similar skin lesions can also be helpful in the differential diagnosis. [22]
Improvement in skin lesion is achieved with surgical resection of coexisting tumor, chemotherapy and long-acting somatostatin [23]
Medical therapy plays a pivotal role in managing patients with metastatic disease at presentation or those who are not suitable candidates for surgical intervention. Palliative treatment options include long-acting somatostatin analogs (e.g., octreotide, lanreotide) or interferon-alpha. Somatostatin analogs, particularly, have demonstrated significant efficacy in case reports by antagonizing glucagon activity, effectively mitigating both necrolytic migratory erythema (NME) and systemic symptoms associated with glucagonoma. Lanreotide, administered as a monthly depot injection, has been shown to prolong progression-free survival in patients with metastatic gastrointestinal neuroendocrine tumors.
While somatostatin analogs provide symptom relief and enhance survival, therapy must be maintained indefinitely, as discontinuation often leads to the rapid recurrence of NME and other glucagonoma-related symptoms.[24,25] Chemotherapy with agents like streptozotocin and 5-fluorouracil can be used for palliation in metastatic disease, though it is generally less preferred due to the tumor’s poor responsiveness to current chemotherapeutic regimens. Biologic agents, including sunitinib and everolimus, have shown promise in clinical trials for treating pancreatic neuroendocrine tumors (pNETs), including glucagonomas, and may be appropriate in specific clinical scenarios. [26]
Additionally, innovative treatments for liver metastases, such as liver resection, transplantation, percutaneous ablation, targeted radiotherapy, and chemoembolization, may also be considered to address metastatic disease in select cases.
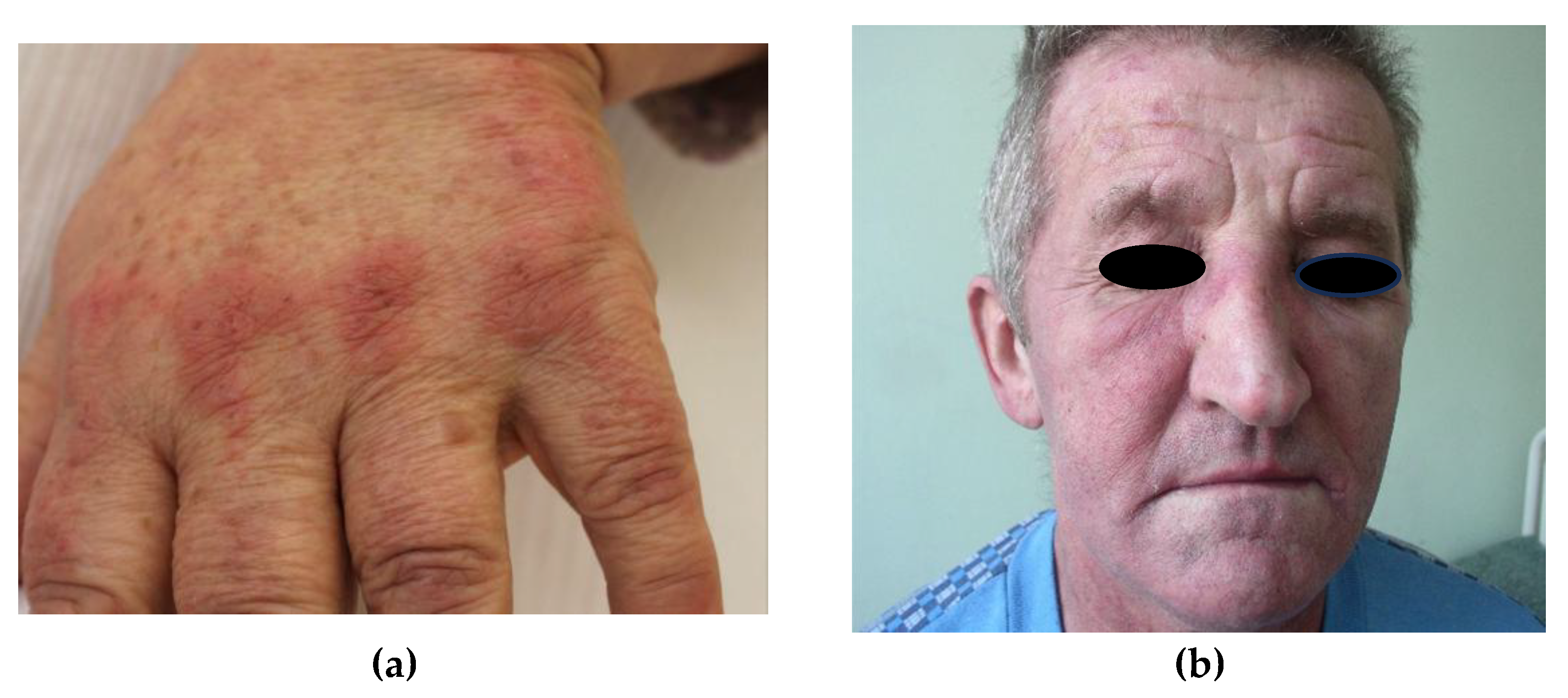
BAZEX SYNDROME - can mean either acrokeratosis or set of symptoms include: the genetic syndrome of basal cell carcinoma, hypotrichosis, disorders of sweating and follicular atrophoderma. Acrokeratosis paraneoplastica (APB) was first described in 1965 by Bazex, as a condition accompanying cancer of the pyriform fossa [27].
In Bazex syndrome, the most commonly affected areas for skin lesions are the hands, feet, ears, and nose. These skin eruptions are characterized by scaly, hyperkeratotic, and red to purple lesions. According to Bazex, the disorder progresses through three stages. The first stage involves a symmetrical erythemato-squamous eruption on the fingertips and toes, followed by the nose and typically the helices. The fingernails and toenails become abnormal, showing paronychia, subungual keratotic debris, and possibly onycholysis.
Figure 1.
Skin lesions in Bazex syndrome. (a)(b) The most commonly affected areas are the hands and feet. The lesions are erythematous with scaling and sometimes crack. (c) As the disease progresses, the lesions spread to other areas of the skin and may involve the facial skin.
Figure 1.
Skin lesions in Bazex syndrome. (a)(b) The most commonly affected areas are the hands and feet. The lesions are erythematous with scaling and sometimes crack. (c) As the disease progresses, the lesions spread to other areas of the skin and may involve the facial skin.
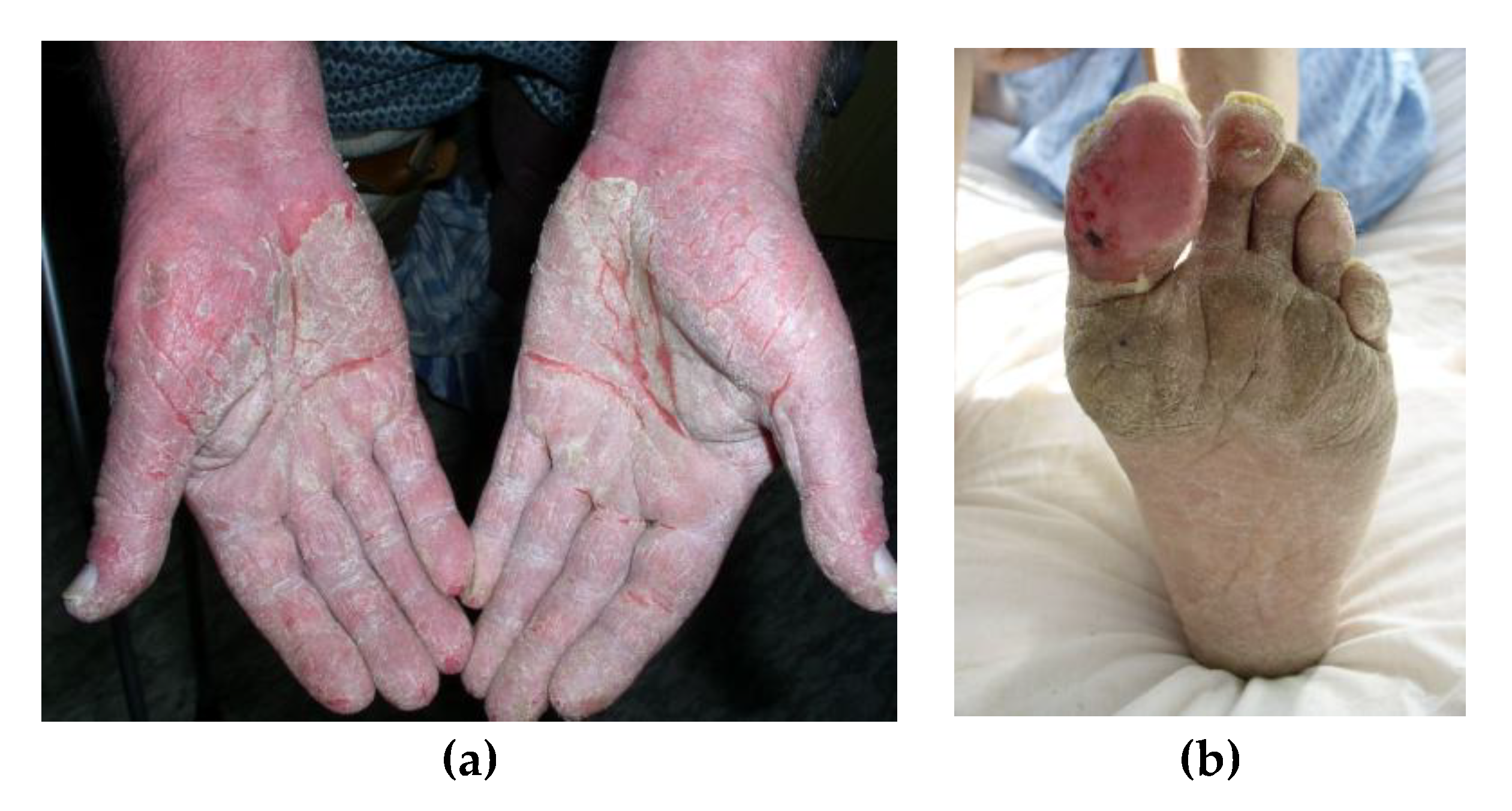
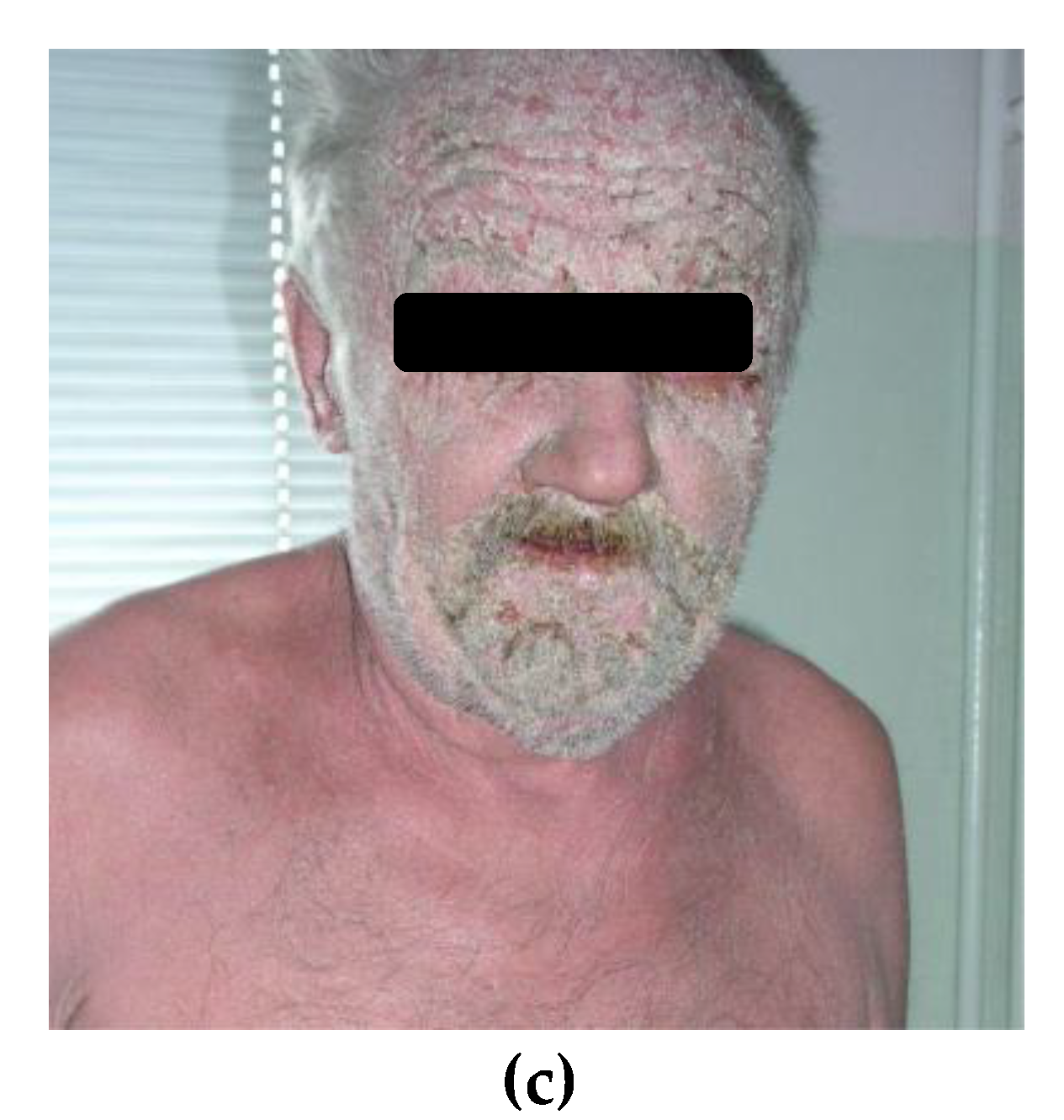
At this stage, an underlying tumor might have already metastasized to cervical or mediastinal lymph nodes, but the primary tumor is usually asymptomatic. In the second stage, tumor growth or cervical lymph node metastases usually become noticeable, and skin lesions spread inward. Areas such as the cheeks, forehead, elbows, thighs, and knees might be affected, with the borders of the skin lesions being poorly defined. Scraping a lesion may cause slight bleeding. In the third and final stage, the persistence of the underlying cancer leads to involvement of the extremities and trunk. The psoriasis-like skin lesions tend to develop fissures. Although itching is common, it is usually mild. [28,29,30].
The exact pathogenesis of Bazex syndrome remains unclear. Some researchers have proposed an immunological basis, supported by the detection of immunoglobulins (IgG, IgA, IgM) and complement components (C3) along the basement membrane of both affected and unaffected skin. An immune response to a shared antigen between the skin and the tumor has been suggested. Additionally, it has been hypothesized that the underlying malignancies may induce a shift towards a Th2-dominant immune response, which could, in turn, lead to increased expression of epidermal growth factor receptors (EGFR) in the affected keratinocytes. [31]
Histological features in Bazex syndrome are unspecific. Parakeratosis, hyperkeratosis, isolated necrosis of keratinocytes, acanthosis, and a perivascular lymphohistiocytic inflammatory infiltrate may occur. The immunofluorescence studies of lesional skin from patients with acrokeratosis paraneoplastica usually give negative results. [32].
In the review written by Shah and Ferazzano (2023), the cancers in which the coexistence of APB has been described so far were summarized. Among them, the most common were: lung cancer, oral cancer, hand and neck cancer, esophageal carcinoma and just a few cases of: hepatocellular, lip, pancreatic, cervical, gastric, colorectal and breast cancer, but also follicular lymphoma, T-cell lymphoma and acute myeloid leukemia. [33] The strong connection between APB and internal malignances, makes APB an essential indicator of hidden cancers.
Early detection is crucial for timely and effective treatment. The differential diagnosis primarily includes psoriasis. In this case, it is important to consider the recurrent nature of the lesions, the triggering factor preceding the appearance of skin changes (e.g., stress, infection, alcohol abuse), or family history, which may indicate a diagnosis of psoriasis. It can also be helpful to take skin scrapings for mycological examination or to perform PAS staining in a histopathological examination to rule out fungal infection. Hyperkeratotic skin lesions on the soles of the feet may also suggest a diagnosis of eczema. In such cases, patch tests can be useful to exclude potential allergic causes. A histopathological examination should be decisive in that case. There have been case reports of acrokeratosis paraneoplastica-like findings in patients with systemic lupus erythematosus. [34]
APB is characterized by resistance to treatment. Medications that have been tried and did not work include topical keratolytics, corticosteroids, tar, antifungals, and antibiotics.
Some successful treatments have been reported, such as oral etretinate and oral prednisone, but typically, removing the underlying cancer is necessary to clear the skin. [35]
The preferred approach to resolving the described skin lesions is effective treatment of the underlying tumor. Paraneoplastic skin lesions, such as those observed in Bazex syndrome, typically do not respond to standard dermatological therapies for inflammatory skin diseases. However, there have been reports of improvement with treatments like Vitamin D3, salicylic acid, and topical or systemic corticosteroids. Reported topical treatments include corticosteroids (e.g., clobetasol 0.05% or betamethasone 0.01%), salicylic acid 10% in vaseline, itraconazole, isosorbide dinitrate, fluconazole, cephalexin, keratolytic agents, neomycin, nystatin, zinc ointment, antibiotics, and emollients. Additionally, some authors have suggested the use of oral dexamethasone (10 mg/day), which has shown favorable outcomes in managing the cutaneous lesions.
In recent years, anecdotal evidence has highlighted the potential benefits of systemic and topical retinoids for these skin lesions. Early administration of acitretin has been reported to effectively improve acrokeratosis paraneoplastica in patients with incurable primary malignancies. [36] Retinoids may also be used in combination with oral corticosteroids. Furthermore, some studies have suggested that psoralen combined with ultraviolet light (PUVA) therapy could be beneficial for selected patients with localized skin lesions.
There are also reports indicating that zinc supplementation might help alleviate these skin manifestations.
ACUTE FEBRILE NEUTROPHILIC DERMATOSIS (SWEET SYNDROME) – for the first time described by Dr. Robert Douglas Sweet in 1964. [37] Neutrophilic dermatoses are a diverse group of inflammatory skin conditions, encompassing Sweet’s syndrome (SS), pyoderma gangrenosum, and subcorneal pustular dermatosis. Among these, Sweet’s syndrome is the most common. [38]
We can distinguish three clinical types of the disease, which is classic (or idiopathic) SS, drug-induced SS and malignancy-associated type. Typical eruptions consist of red, painful, well-demarcated plaques or nodules. The lesions are typically referred to as pseudovesicular or pseudopustular, though they can also present as fully pustular, bullous, or ulcerative. They may develop on face, neck, back, chest and also extremities. Mucosal changes are not specific for Sweet’s syndrome and occur in 3-30% of cases. In addition to skin lesions, rarely but may occur: arthritis, eye, kidney, CNS, lung, bones and liver involvement. [39]. Also pyrexia and general malaise may be present.
Distinctive criteria first described by Su and Liu [40] and modified by von den Driesch, are helpful in making the diagnosis of Sweet’s syndrome.
Two major criteria are identified: the rapid onset of tender, erythematous nodules or plaques, and the presence of neutrophilic infiltrates in histopathological examination that do not fulfill the criteria for leukocytoclastic vasculitis.
Minor criteria includes: fever over 38 degree; association with hematologic or visceral cancers, inflammatory conditions, or pregnancy, or occurring after an upper respiratory tract infection, gastrointestinal infection, or vaccination; rapid response to treatment with systemic corticosteroids or potassium iodide; deviations in laboratory tests (three of four): increased C-reactive protein level, level of leukocytes over 8000, with over 70% neutrophils; erythrocyte sedimentation rate >20mm/h.
To diagnose classical SS, both major and two minor criteria must be met.
In cases associated with malignancy, the condition should either precede, follow, or occur simultaneously with the diagnosis of the patient’s neoplasm, while also fulfilling the classical Sweet’s syndrome diagnostic criteria. [41].
Histological examination from the lesions should be helpful to make diagnosis. Distinguishing findings are: presence of a widespread neutrophilic infiltrate in the dermis, fragmentation and edema of neutrophil nuclei. The main cells constituting the infiltrate in the dermis of skin lesions in SS are mature neutrophils. Occasionally, eosinophils, lymphocytes or histiocytes may also be found in the inflammatory infiltrate. Neutrophils can also appear in the epidermis as neutrophilic spongiotic vesicles or subcorneal pustules, however if the infiltrate extend into the subcutaneous tissue and hypodermis, affecting adipocyte, it has recently been linked to myeloid disorders and it is called „subcutaneous Sweet’s syndrome” [42]
Many cases of coexistence of Sweet’s syndrome with cancer have been described in the literature, for the first time represented by Shapiro et al. [43] The most common are leukemia, particularly acute myeloid or myelomonocytic leukemia [44] SS may also been associated with chronic myeloid leukemia [45]. From the solid tumor the most frequently found is embryonal carcinoma of testis, ovarian carcinoma, gastric carcinoma and adenocarcinoma of breast, prostate, and rectum. [46] Unlike most other paraneoplastic diseases, this syndrome may occur at an early stage of development of these cancers that cure is possible. Even though there are specific criteria to help identify Sweet’s syndrome, it still poses diagnostic difficulties.
Sweet syndrome can also be associated with various inflammatory and autoimmune conditions, including inflammatory bowel diseases (ulcerative colitis or Crohn’s disease), rheumatoid arthritis, systemic lupus erythematosus, Sjögren’s syndrome, Hashimoto’s thyroiditis, Behçet’s disease, and dermatomyositis. [47] Post-infectious Sweet syndrome has been observed 1 to 3 weeks following upper respiratory or gastrointestinal infections. It has also been linked to other infections, such as human immunodeficiency virus (HIV), viral hepatitis, tuberculosis, and, in some cases, chlamydia infection.
Drug-induced Sweet syndrome is a well-recognized condition, with numerous medications implicated in its onset. Granulocyte colony-stimulating factor is the most commonly associated drug, but others include antibiotics (e.g., minocycline, nitrofurantoin, trimethoprim-sulfamethoxazole, norfloxacin, ofloxacin), antihypertensives (e.g., hydralazine, furosemide), nonsteroidal anti-inflammatory drugs (e.g., diclofenac, celecoxib), immunosuppressants (e.g., azathioprine) [48], antiepileptics (e.g., carbamazepine, diazepam), anticancer agents (e.g., bortezomib, imatinib mesylate, ipilimumab, lenalidomide, topotecan, vemurafenib), antipsychotics (e.g., clozapine), and antithyroid drugs (e.g., propylthiouracil). In these cases, Sweet syndrome typically develops after exposure or re-exposure to the offending drug and often resolves after drug withdrawal, with or without corticosteroid therapy.
Pregnancy is associated with the development of Sweet syndrome in approximately 2% of cases. The prognosis in pregnancy-related cases is favorable, with spontaneous resolution occurring after delivery and no reported maternal or infant morbidity or mortality. [49]
The most problematic in differentiation is erythema nodosum (EN), especially when skin lesions are located in lower extremities. In both cases we are dealing with painful, erythematous nodules, where histopathological examination is not always crucial. Accompanying symptoms in EN may also be similar (i.e., fever) or increased inflammatory markers. [41] Another disease which can be problematic in differentiation is toxic pustuloderma, which is a serious hypersensitivity reaction to medications like carbamazepine. Symptoms may include fever, an elevated white blood cell count, and involvement of the lymph nodes, liver, and kidneys. The histological appearance can resemble pustular SS; however, the presence of widespread, primarily isolated pustules, with or without association with hair follicles, helps to distinguish between the two conditions. When distinguishing it from SS, it’s important to also consider: periarteritis nodosa, granuloma faciale, leukocytoclastic vasculitis, erythema elevatum diutinum, and infectious disease such as erysipelas or impetigo contagiosum. All of this disorders can have similar histopathological features, but rather vary in another symptoms.
Paraneoplastic SS may develop more problems with diagnosis, because general malaise and pyrexia are not obligatory in this cases. In some patients with classical Sweet’s syndrome, lesions can persist for weeks to months without treatment but eventually heal on their own. While there are no established guidelines for treating Sweet’s syndrome, systemic corticosteroids are usually the first choice for treatment. For patients who cannot use corticosteroids, oral therapy with potassium iodide or colchicine often leads to a quick resolution of Sweet’s syndrome symptoms and lesions. [50] In cases of malignancy-associated Sweet’s syndrome, the skin condition sometimes resolves after the related cancer goes into remission.
The primary objective of pharmacotherapy in acute febrile neutrophilic dermatosis is to minimize morbidity and prevent complications. Systemic corticosteroids are considered the most effective first-line treatment, with topical corticosteroids being appropriate for localized lesions. In cases where corticosteroids are contraindicated, alternative options include anti-inflammatory or immunosuppressive agents.
PYODERMA GANGRENOSUM (PG) – is a rare, autoinflammatory neutrophilic dermatosis, that occurs with equal frequency in both sexes. This disease typically co-occurs with other autoimmune disorders, with the most common being arthritis, inflammatory bowel disease, and paraproteinemia. Pyoderma gangrenosum (PG) can also present as a paraneoplastic syndrome, most frequently associated with myeloproliferative neoplasms (acute myeloid leukemia, acute/chronic lymphatic leukemia, myelodysplastic syndrome, T-cell lymphoma), and less commonly with solid tumors such as breast, colon, bladder, or prostate cancer. [51]
Pyoderma gangrenosum comes in four main clinical types: ulcerative, bullous, pustular and vegetative (or superficial granulomatous). Skin lesions typically starts as one or more red pustules, nodules or blisters that can rapidly develop into painful, irregular ulcers with undermined edges. Ulceration is often covered with necrotic tissue. „Sieve sign” is also one of typical feature. Surrounding is covered with erythema. [52] In the case of PG, a pathognomonic sign known as pathergy is observed, meaning that new lesions can develop as a result of minor injuries. [53] Ulcers are located mainly on the lower limbs and trunk, but they can also develop in any part of the body.
Figure 2.
Pyoderma gangrenosum. (a) Ulcerations and erythema in surrounding area located on the face. (b) Ulceration covered with necrotic tissue located on the tibial area.
Figure 2.
Pyoderma gangrenosum. (a) Ulcerations and erythema in surrounding area located on the face. (b) Ulceration covered with necrotic tissue located on the tibial area.
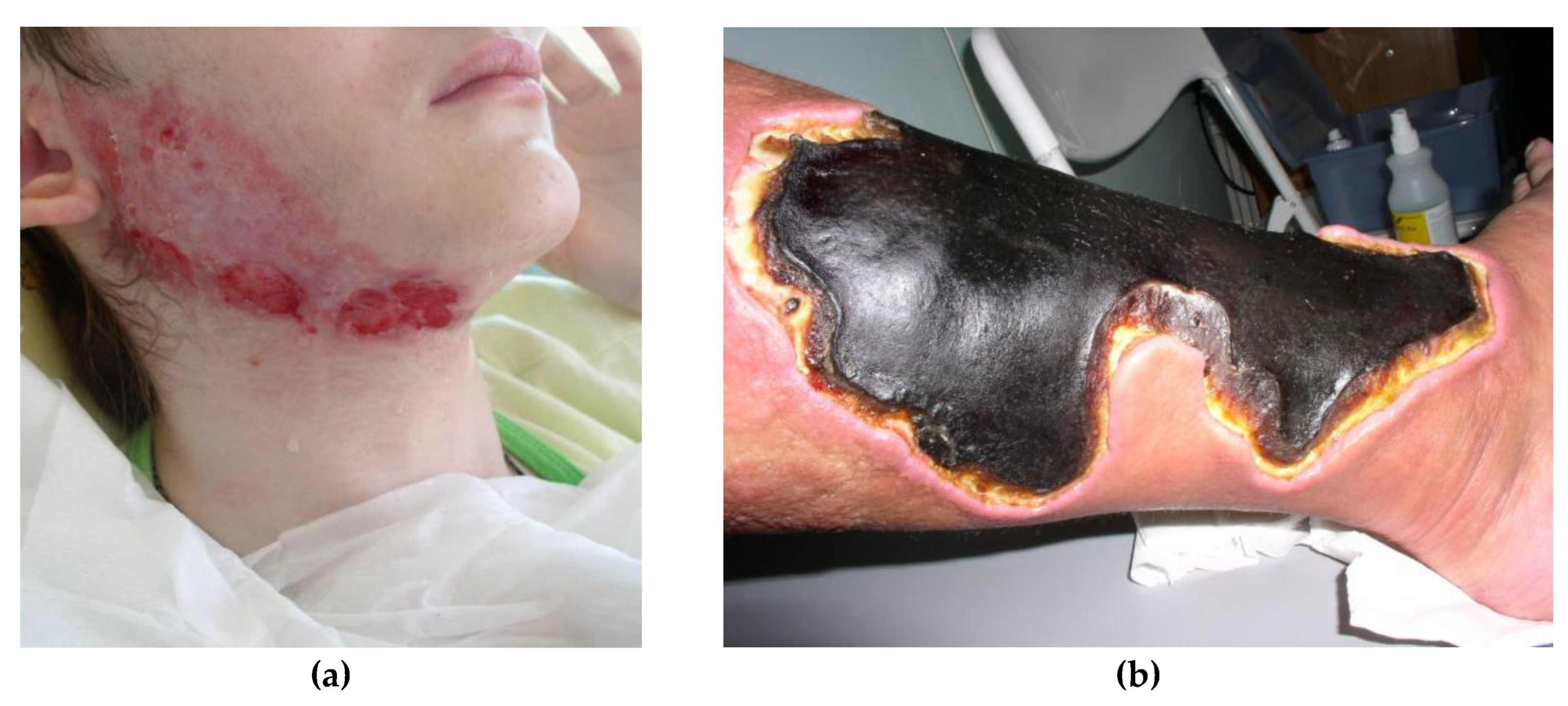
The findings in histopathological examination depending on the location and stage of the lesions. Biopsy samples from the ulcer’s edge typically reveal neutrophils and perivascular lymphocytic infiltrates along with dermal edema, while samples taken from the center of the ulcer predominantly show a neutrophilic infiltrate. It’s common to observe vascular damage characterized by fibrin deposition, thrombosis, and red blood cell extravasation. [54]
PG is typically diagnosed by ruling out other conditions, as there are no clear-cut laboratory tests or histopathological markers for it, which often results in it being misdiagnosed. [55]
The criteria established following a Delphi consensus exercise in 2018 replaced the Su criteria, introducing a single major criterion for diagnosing pyoderma gangrenosum: the presence of a neutrophilic infiltrate. When combined with four or more of the eight minor criteria, this approach achieves a sensitivity of 86% and a specificity of 90% for the condition. [56]
Major criterion
1.Histopathology of ulcer edge must show a neutrophilic infiltrate.
Minor criteria
1. Exclusion of infection
2. Pathergy
3. History of inflammatory bowel disease or inflammatory arthritis
4. History of papule, vesicle, or pustule ulcerating within four days
5. Peripheral erythema, undermining border, and tenderness at the ulcer site
6. Multiple ulcers, at least one on the anterior lower leg
7. Cribriform or wrinkled paper scars at the site of the healed ulcer
8. Decreased size of the ulcer within one month of initiating immunosuppressive
medication.
In the differential diagnosis, it is essential to consider other causes of chronic ulcers, such as venous or arterial insufficiency, Sweet’s syndrome, antiphospholipid syndrome, systemic lupus erythematosus, venous or arterial ulcers, tuberculosis, sporotrichosis, ulcerating skin tumors and lymphomas or vasculitis. [55] There are some features which can lead to PG suggestion, such as: repeated negative cultures results, failure to respond to antibiotics and neutrophilic infiltration in skin biopsy. [57]
In the treatment of the acute, initial phase of PG, systemic steroids are used. For chronic therapy, immunosuppressive drugs are chosen, most commonly cyclosporine A, dapsone, colchicine, and TNF-alpha inhibitors. It is important to perform surgical wound debridement carefully, as the pathergy phenomenon can cause the formation of new ulcers. There are also reported cases of spontaneous healing of PG-related ulcers following the successful treatment of an associated malignancy. [58]
EXFOLIATIVE DERMATITIS (ERYTHRODERMA) – is an inflammatory skin condition that affects more than 90% of the skin’s surface. It is not a disease in the strict sense, but symptom which can accompany other medical conditions. Erythroderma may result from the exacerbation of pre-existing dermatological conditions such as psoriasis, atopic dermatitis, pityriasis rubra pilaris and cutaneous lymphoma. [59] However, it can also be a symptom of a drug reaction or occur as a paraneoplastic syndrome in the course of internal malignancy.
The most common are hematologic malignancies apart from Sezary syndrome or mycosis fungoides. Also in publication we can’t find examples of carcinoma of the lung [60], thyroid and prostate, adenocarcinoma of liver, ovarian, rectal and mammary cancer, malignant melanoma and esophageal carcinoma. [61]
Typical symptoms of erythroderma include inflammation covering nearly 90% of the body, which may be accompanied by desquamation, excoriations and lichenification. Prolonged and severe erythroderma is often linked to diffuse alopecia, ectropion, nail deformities and keratoderma [62] Other clinical features that can be observed involved pyrexia, general malaise, and pruritus [63] Patients with erythroderma may experience tachycardia and high-output heart failure. Lymphadenopathy is typically related to this skin conditions and has been observed in the majority of erythrodermic patients. When lymphadenopathy occurs alongside organ enlargement, it may indicate drug hypersensitivity or malignancy. [64]
Establishing a definitive clinicopathologic correlation may require several skin biopsies taken repeatedly. In extensive studies of samples from erythrodermic patients, the histopathological findings are frequently nonspecific, typically displaying hyperkeratosis/parakeratosis, acanthosis, and chronic inflammatory infiltrates (with or without eosinophils). Even in cases with established dermatoses, the biopsies can still yield non diagnostic results. Although there are features that are often typical for cutaneous T-cell lymphoma (CTCL) like Pautrier’s microabscesses bandlike infiltrate with cerebriform nuclei and epidermotropism. Multiple biopsies may be necessary to clearly identify these characteristics. Sézary syndrome might show minimal epidermotropism, necessitating additional clinical information for diagnosis. [65] Other conditions, like drug-induced pseudolymphoma, can mimic CTCL histology, complicating differential diagnosis with lymphadenopathy and hepatosplenomegaly. Several cases of Hodgkin’s lymphoma associated with erythroderma have revealed Reed-Sternberg cells in skin biopsies, whereas certain leukemias exhibit deep monomorphous infiltrates along with mixed superficial infiltrates. Generally, erythroderma linked to malignancy presents nonspecific histology. To distinguish malignant from benign infiltrates, techniques like immunophenotyping, flow cytometry, and ultrastructural morphometric analysis have been assessed, though they aren’t completely reliable due to overlaps. [66]
Also a case where erythroderma evolved into erythema gyratum repens, an obligate paraneoplastic dermatosis, is also reported in the literature. [67] Paraneoplastic erythroderma can sometimes be distinguished from other forms of erythroderma by specific clinical features unique to the latter. If the condition is related to psoriasis, a family or personal history of psoriasis may indicate this background; additionally, typical nail changes (such as pitting, subungual keratosis, or oil spots) can also point toward this diagnosis. [68] If there are islands of healthy skin and the patient’s skin has a salmon hue, one should consider PRP [69]. Massive lymphadenopathy, ectropion, extensive skin infiltration, and recurrent erythroderma may suggest mycosis fungoides or Sézary syndrome. [70,71] The rapid onset of lesions and a history confirming the introduction of new medications, such as amoxicillin, allopurinol, carbamazepine, pregabalin, nifedipine or others, may suggest a drug-induced background for the changes. [72,73,74] Atopic history, a young patient age, and pre-existing lesions typical of atopic dermatitis may suggest an exacerbation caused by this condition.
Identifying a cause requires correlation between patient history, clinical presentation, biopsy and direct immune fluorescent test findings, and laboratory studies. The differential diagnosis is presented in Table 1.
Malignancy-associated erythroderma may be more progressive and its course dictated by the prognosis of the underlying malignancy and response to therapy, while idiopathic erythrodermic cases may be unpredictable with periods of relapse and remission.
The initial treatment for all forms of erythroderma is consistent, irrespective of the underlying cause. Focus is placed on ensuring proper nutrition and replacing fluids and electrolytes. Specifically, any potential drugs inducing erythroderma must be stopped. Treatment includes antihistamines to reduce itching, emollients, and medium-potency topical steroids. In severe cases, systemic medications are necessary, such as intramuscular or oral corticosteroids and immunosuppressive drugs, especially when the changes are due to chronic conditions like psoriasis or atopic dermatitis. However, it is important to consider the potential underlying cause of erythroderma as an internal malignancy, as the use of immunosuppressants may worsen the underlying condition. Typically, proper treatment of the cancers leads to an improvement in the erythroderma as well [75]
3. Paraneoplastic Syndromes That Are Easier to Recognize
PARANEOPLASTIC PEMPHIGUS (PNP) –is a distinct form of pemphigus characterized by clinical, histological, and immunological features that are pathognomonic. As the name suggests, it is usually associated with an internal neoplasm. Two-thirds of cases present with a known malignancy at the time the eruption begins.[76]
Recently, researchers have identified increased susceptibility to paraneoplastic pemphigus development in patients with HLA Class II Drb1*03 allele and HLA-Cw*14. These alleles are more common in Caucasian and Chinese populations, respectively. In contrast, HLA-DR4 and HLA-DR1-14 increase susceptibility to pemphigus vulgaris and pemphigus foliaceus, but have not shown an association with the development of PNP. [77,78]
PNP is most frequently linked to B-cell lymphoproliferative disorders. [79] Another most common malignancies associated with PNP include chronic lymphocytic leukemia, Hodgkin’s lymphoma thymomas, Waldenström’s macroglobulinemia, sarcomas and Castleman’s disease. [80]
PNP is characterized by polymorphous lesions that affect the skin and various mucous membranes. In almost all cases of PNP, oral lesions are seen, and are usually the first sign of the disease. [81] Mucosal lesions appear as widespread erosions and can involve the entire oral cavity, throat, or esophagus. Erosions in the lips are also frequently observed, where may be covered with crusts. It is also important to consider changes in the anogenital areas. Cutaneous lesions typically appear after the onset of mucosal lesions. [82] Skin lesions usually spare the face. They may present as tense or flaccid blisters with erosions and can coexist with lichenoid or psoriasis-like eruptions. Ocular manifestation in PNP is also not uncommon. Bilateral conjunctival erosions may occur, which can lead to pseudomembranous conjunctivitis, symblepharon, and even vision loss. [83] In contrast to other forms of pemphigus, which primarily affect squamous epithelium, PNP can involve other types of epithelium, including those of the gastrointestinal and respiratory tracts. They can manifest as obstructive lung disease, bronchiolitis obliterans and may even result in respiratory failure. [84]
Figure 3.
Paraneoplastic pemphigus. (a) Polymorphous skin lesions scattered on the trunk, including flaccid blisters and confluent erosions. (b) Lesions in the form of erosions covered with hemorrhagic and honey-yellow crusts, located on the skin and mucous membrane of the nasal cavity and lips.
Figure 3.
Paraneoplastic pemphigus. (a) Polymorphous skin lesions scattered on the trunk, including flaccid blisters and confluent erosions. (b) Lesions in the form of erosions covered with hemorrhagic and honey-yellow crusts, located on the skin and mucous membrane of the nasal cavity and lips.
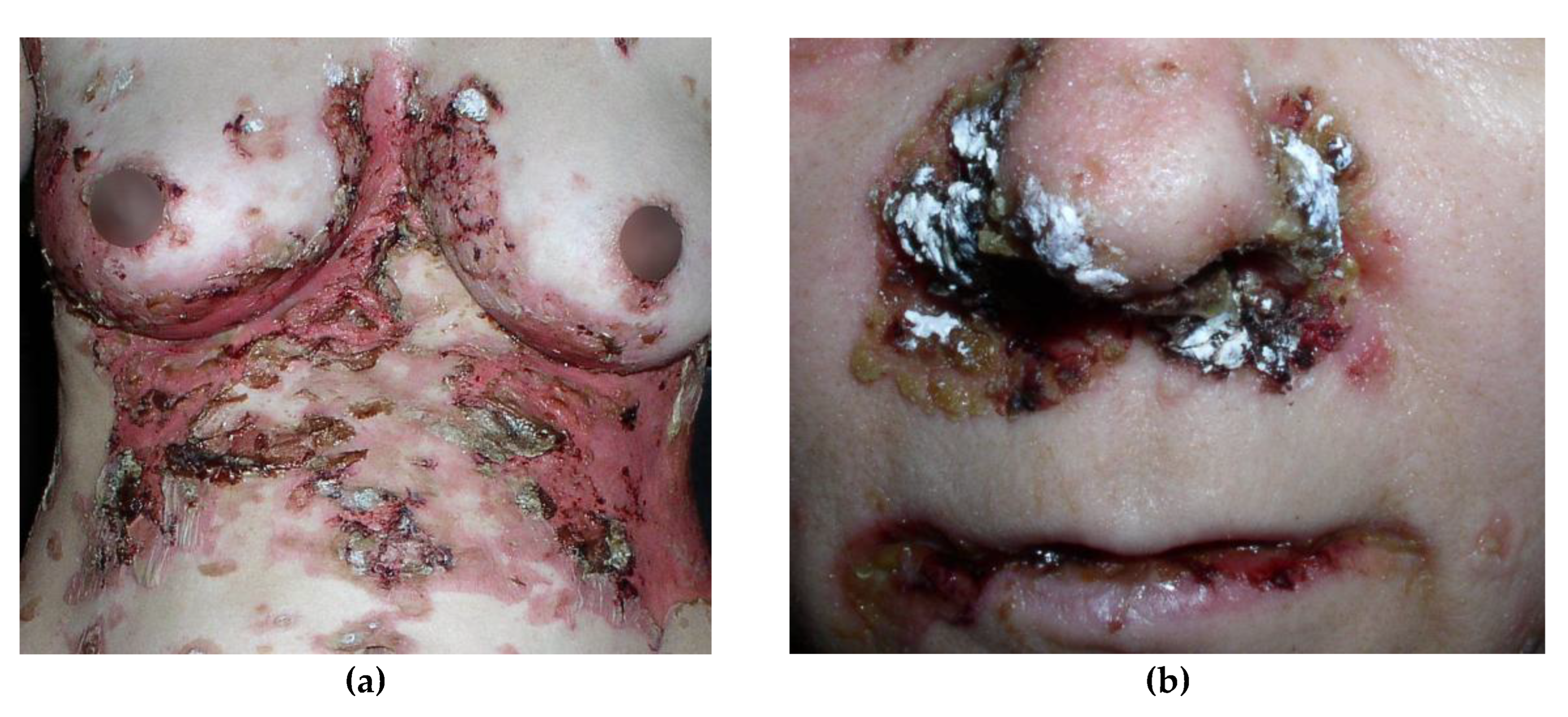
To diagnose paraneoplastic pemphigus (PNP), Helm and Camissa established major and minor criteria. The main criteria include a polymorphous skin lesions, the presence of an associated internal malignancy, and the detection of antibodies through a specific immunoprecipitation standard. The additional criteria consist of: intraepithelial acantholysis in histopathological examination, a linear pattern with IgG and C3 deposits along the basement membrane zone in direct immunofluorescence (DIF) examination and IIF using rat bladder epithelium as the substrate. For the diagnosis to be confirmed, either three major criteria or two major and one minor criterion must be met. [80]
Histopathological changes depend on the clinical features of the lesions. The most commonly observed characteristics include: suprabasal acantholysis, dyskeratotic keratinocytes, which may be present in all layers of the epidermis, a dense lichenoid infiltrate at the dermal-epidermal junction and migration of inflammatory cells into the epidermis. [85]
Direct immunofluorescence (DIF) on a perilesional biopsy can show intercellular IgG and C3 deposits. Additionally, linear IgG or C3 deposits at the basement membrane zone, caused by autoantibody binding to BPAG1, help distinguish PNP from other pemphigus types, where Ig deposits are limited to keratinocytes. [86] Positive DIF result is not required to confirm a diagnosis of PNP (Approximately 50% cases of PNP have negative DIF). Also false negative results are often, especially from oral mucosa, because of a lot necrotic tissue.
Indirect immunofluorescence using rat bladder is a key diagnostic tool for PNP, as the presence of autoantibodies to plakins is a hallmark feature. The most specific markers are autoantibodies against envoplakin and periplakin, followed by desmoplakin I and II. Immunoblotting and ELISA are alternative methods for detecting plakin autoantibodies in PNP. Immunoblotting can identify antibodies against desmoplakin I and II, periplakin, and envoplakin using cultured human keratinocyte extracts. Additionally, recombinant fragments of periplakin and envoplakin can also be utilized in both immunoblotting and ELISA for detection. [87] Proteins targeted by autoantibodies in paraneoplastic pemphigus are presented in Table 2. [82,88]
PNP can present a diagnostic challenge, especially when the skin lesions resemble other conditions. In differential diagnosis, it is important to consider other blistering diseases, including pemphigus vulgaris, pemphigus foliaceus, pemphigoid, as well as erythema multiforme, toxic epidermal necrolysis, graft-versus-host disease and lichen planus. [89,90,91] If extensive erosive lesions, including mucosal involvement, predominate, they may be mistaken for erythema multiforme or toxic epidermal necrolysis.
In such cases, a patient history indicating the use of a new medication can aid in differentiation. Additionally, direct and indirect immunofluorescence testing in PNP will confirm the presence of characteristic immunoglobulin deposits. Tense blisters, particularly on the lower legs, accompanied by mucosal erosions, may suggest bullous pemphigoid. In such cases, the patient’s age, associated conditions and symptoms, and the definitive role of direct immunofluorescence (DIF) testing are crucial for differentiation. [92] Targetoid and lichenoid lesions may also occur over the palms and soles in addition to blisters. When the lesions are papular, they may resemble lichen planus. Especially, after treatment and in chronic cases, the eruption is predominantly lichenoid in nature. In such cases, a medical history indicating recurrent lichen planus-like lesions can provide a clue for diagnosis. Histopathological examination of a biopsy taken from the lichenoid lesion area may be inconclusive. In these situations, the final diagnosis is established based on immunofluorescence studies. [93] When erosions and flaccid blisters predominate, PNP can be mistaken for pemphigus vulgaris or pemphigus foliaceus. In such cases, both DIF and IIF should be helpful in establishing the correct diagnosis. [94] For patients with paraneoplastic pemphigus (PNP) and no apparent malignancy, the diagnostic evaluation should be specifically directed towards identifying potential underlying cancers. The management of paraneoplastic pemphigus (PNP) is dual-focused. On one hand, treatment should target the associated malignancy; on the other hand, therapy must address the lesions caused by PNP. [95] Prognosis tends to be better in cases of less aggressive malignancies, such as thymoma or Castleman’s disease. Initial treatment typically involves high doses of systemic steroids. However, due to the adverse effects associated with long-term use of high-dose steroids, additional immunosuppressive agents, such as cyclosporine, azathioprine, or cyclophosphamide, should be included. In some cases, intravenous immunoglobulin infusions or plasmapheresis may be employed. In recent years, an increasing number of blistering disease cases have been treated with rituximab (RTX), a chimeric human-murine anti-CD20 monoclonal antibody. New studies have emerged confirming the effectiveness of RTX also in paraneoplastic pemphigus. [96] Cases have also been described in the literature where the treatment of the underlying disease, such as chronic lymphocytic leukemia with alemtuzumab, led to complete remission of lesions in coexisting paraneoplastic pemphigus. [97,98] Despite available treatments, PNP remains associated with high mortality. [99]
DERMATOMYOSITIS (DM) - is an infrequent autoimmune disease with numerous cutaneous and systemic symptoms. It primarily manifests with skeletal muscle weakness, multiple skin abnormalities and extramuscular symptoms such as esophageal motility disorder (EMD) and interstitial lung disease (ILD). [100]
Although, exact cause of this condition remains unclear several risk factors as genetic predisposition, exposure to UV radiation, geographic and environmental factors are believed to play a major role in its development.
Multiple studies have indicated that patients with particular human leukocyte antigen (HLA) types are at higher risk of dermatomyositis. High-risk haplotypes include HLA-A*68 in North American Whites, HLA-DRB1*0301 in African Americans, HLA-DQA1*0104 and HLA-DRB1*07 in Han Chinese, DQA1*05 and DQB1*02 in people from the UK. Also, DRB1*03-DQA1*05-DQB1*02 haplotype is strongly associated with the development of interstitial lung disease in dermatomyositis. [101,102]
Predominant age for DM is considered to be between 40 and 50 years old, and it occurs twice as often in women than in men. [103] Chronic nature of dermatomyositis is commonly characterized by periods of clinical remissions and exacerbations, which may also occur without noticeable changes in muscle function (Amyopathic dermatomyositis).
Diversified skin abnormalities can be present in the course of this disorder. Literature data distinguish pathognomonic symptom as Gottron papules which are characterized by presence of violaceous papules above the metacarpophalangeal and interphalangeal joints on hands. Other findings consist of: heliotrope rash (red or pink erythema of the eyelids accompanied by swelling of affected tissue), purple erythema on dorsal surfaces of the hands, elbows, knees and ankles (Gottron’s symptom), V-neck erythema and erythema of the neck and shoulders (scarf symptom), calcium deposits in the skin and other. In many cases cutaneous manifestations can be accompanied by photosensitivity and itching. [104] Symptoms of dermatomyositis are presented in Table 3.
Figure 4.
Pathognomic signs of dermatomyositis. (a) Erythematous papules located over the metacarpophalangeal joints. (b) Violaceous rash affecting upper eyelids with periorbital edema.
Figure 4.
Pathognomic signs of dermatomyositis. (a) Erythematous papules located over the metacarpophalangeal joints. (b) Violaceous rash affecting upper eyelids with periorbital edema.
In the diagnosis of DM, in addition to typical clinical symptoms, supportive tests also play a significant role. Elevated levels of muscle enzymes are observed in 95% of patients, especially aldolase and creatine kinase. Less specific enzymes, such as aspartate aminotransferase, alanine aminotransferase, and lactate dehydrogenase, may also be elevated. [105] Another diagnostic tool for detecting muscle inflammation in DM is electromyography (EMG). It is typically conducted on a proximal muscle, like the triceps. A positive EMG result largely confirms the presence of muscle inflammation in the course of DM. [106] Due to its non-invasive nature, MRI of the muscles is also gaining popularity and can be helpful in diagnosing muscle inflammation. [107]
However, a skin biopsy is not always diagnostic and may show findings similar to the histopathology of systemic lupus erythematosus (vacuolar changes of the basal layer, increased lymphocytic infiltrate, and increased mucin deposition in the dermis), muscle biopsy often shortens the diagnostic process and confirms diagnosis. Typical abnormalities observed in muscle histopathology include: perivascular and perimyosial inflammatory infiltrate, perifascicular atrophy and microangiopathy. [108]
- Perivascular and perimysial inflammatory infiltrate: The infiltrate in dermatomyositis is concentrated around the perivascular and interfascicular regions and consists of B cells, CD4+ T helper cells, macrophages, and plasmacytoid dendritic cells. In contrast to polymyositis, CD8+ T cells and NK cells are rarely present.
- Perifascicular atrophy: Atrophy of muscle fibers, especially around the periphery of fascicles, is a hallmark histopathological feature of dermatomyositis. Degenerating and regenerating muscle fibers may be observed in the perifascicular region.
- Microangiopathy: Injury to intramuscular blood vessels takes the form of immunoglobulin and complement (C5b-C9 membrane attack complex) deposits on endomysial capillaries. A reduced capillary density and endothelial hyperplasia may be observed.
Various studies shown that dermatomyositis has strong correlation with malignancies. [110,111] Over 30 % of adult patients with DM are at risk of developing cancer and it can be linked with the existence of specific autoantibodies. Many studies confirmed that patients with detectible Anti-TIF1-γ (transcription intermediary factor, anti-p155) and TIF1-α (anti p-140) antibodies were further diagnosed with cancer or other malignancy. [112] Immunological assessment of each DM patient can be an useful tool to forecast malignancy risk in the course of the disease. All adults confirmed with DM should undergo full screening to rule out possibility of malignant process. DM can occur before, concurrently or after diagnosis of cancer.
Malignancies related to DM include ovary, lung, pancreas, breast and gastrointestinal tract cancers. Non-Hodgkin’s lymphoma, testicular cancer and nasopharyngeal carcinoma were also described. [113] The most common malignancy associated with dermatomyositis is ovarian cancer. [114] The goal of treatment is to obtain improvement in patients comfort and daily activities, reduce muscle weakness and avoid systemic complications (including ILD, arthritis, cutaneous lesions and other).
Considering initial therapy, systemic corticosteroids are still believed to be the finest option yet still less effective in patients with underlying malignancy. Other considered options consists of with intravenous immunoglobulin (IVIG), rituximab, methotrexate, and hydroxycholoroquine. Conclusively, optimal and most favorable therapy in DM patients with concomitant malignancies is to coordinate oncological treatment along with immunosuppressants. [115]
LASER-TRELAT SYNDROME - is a rare paraneoplastic sign in which uncontrollable eruption of multiple seborrheic keratoses is associated with underlying malignancy. So far, no diagnostic criteria have been estimated and disease is confirmed on clinical data itself.
Leser-Trélat sign is oftentimes related to breast, gastrointestinal adenocarcinomas but renal, hepatic, and pancreatic malignancies were also reported in literature data. The definite cause of this phenomenon is not full elucidated, however, it is suspected that several growth factors (especially EGFR- epidermal growth factor) secreted from the tumor stimulates the uncontrolled growth of the seborrheic keratosis. [116]
The precise pathophysiology of the Leser-Trélat sign remains unclear. It is thought that the phenomenon may result from cytokines and growth factors released by the neoplasm, which stimulate the rapid proliferation of seborrheic keratoses. Overexpression of epidermal growth factor-α, transforming growth factor-α, and amphiregulin is believed to play a role in the eruptive growth of these lesions. Other growth factors potentially involved include human growth hormone, fibroblast growth factor receptor 3 (FGFR3), phosphatidylinositol 3-kinase catalytic subunit alpha (PIK3CA), and insulin-like growth factor. Seborrheic keratoses are common epidermal neoplasms that arise from the clonal expansion of immature keratinocytes. [117]
Seborrheic keratoses are benign epidermal neoplasms arising from clonal immature keratinocytes and have tendency to appear frequently, especially in in patients aged >40 years. Characteristics and frequency of those lesions induces skepticism regarding actual occurrence of this disease. Multiple seborrheic keratosis have been reported not only in cancerous patients, but also in the course of infectious diseases, such as HIV, COVID-19 infection or in post-transplant patients. The cause of this phenomenon is not fully understood. [118] Morphology of these benign neoplasm is manifested by presence of well demarcated macules, papules, and plaques with variety of colours. Pigmentation ranges from light to dark brown or black. In consideration of colour, they have a tendency to vary extensively amongst patients. The surface have a soft, greasy or scaly consistency, with diversity of size. Diagnosing seborrheic keratosis is usually straightforward. Dermoscopic examination reveals common findings as comedo-like (CL) openings, fissures and ridges (FR), and milia-like (ML) cysts. [119]
In doubtful or uncertain cases, histopathology is the procedure of choice. Regardless of the genesis of these lesions, histopathological examination presents identical features. [120] Literature data estimated that, the most prevalent histopathological type of seborrheic keratosis is acanthotic type, followed by mixed, hyperkeratotic, melanoacanthoma, clonal, irritated and adenoid types. Histological assessment is performed to rule out other skin lesions, including skin cancers.
Roh NK et al. proved that other pathologies that were mistaken for SK included: the common wart, flat wart, basal cell carcinoma, squamous cell carcinoma, dysplastic nevus, compound nevus, actinic keratosis, Bowen disease, melanoma and condyloma acuminatum. [121]
The therapy of concomitant cancer is of fundamental importance in the treatment of this disease Leser-Trélat syndrome. It is declared that approximately 50% of associated seborrheic keratosis resolve completely. Additionally, patient disturbing lesions may be managed with cryotherapy, curettage and electrodessication. [126]
ACANTHOSIS NIGRICANS (AN) - is a symptom that can be classified as benign or malignant – then is associated with malignant tumors. It appears as hyperpigmented skin with excessive keratinization, most commonly affecting symmetrically the armpits, groin, and the skin on the back of the neck. It can also localized on face, oral mucosa and anogenital area. Additionally, there could be hyperkeratosis of palms and soles, knuckles and fingers.
The distribution of lesions is often related to the underlying cause of AN. For example, facial lesions are most commonly observed in cases of metabolic syndrome, while involvement of the eyelids and mucous membranes may be a marker of malignancy. [127,128] Patients often complain of itching in the areas of skin lesions.
There are multiple factors involved in the development of acanthosis nigricans.
Increased circulating insulin activates keratinocyte insulin-like growth factor (ILGF) receptors, particularly IGF-1. At high concentrations, insulin may displace IGF-1 from IGF binding protein. Increased circulating IGF may lead to keratinocyte and dermal fibroblast proliferation. [129]
Hereditary variants are associated with fibroblast growth factor defects.
Increased transforming growth factor (TGF) appears to be the mechanism for malignancy-associated acanthosis nigricans. TGF acts on epidermal tissue via the epidermal growth factor receptor.
Mild forms of AN are most often associated with endocrine disorders, particularly those linked to metabolic syndrome and insulin resistance. It is also common for patients with diabetes to develop lesions typical of AN. [130] Moreover it can be triggered by specific medications, such as corticosteroids, nicotinic acid, and triazinate [20]. The link between acanthosis nigricans and internal malignancy was initially identified by Darier in 1893. [131]
“Malignant” acanthosis nigricans (MAN) is primarily diagnosed in individuals over the age of 40. The most commonly associated cancer is gastrointestinal adenocarcinoma. There have been reported cases where AN was found alongside tripe palms, mucosal papillomas, and the Leser-Trélat sign, especially when associated with gastric cancer. [132,133] Less commonly, AN may be a syndrome associated with carcinomas of the lung, kidney, and bladder, as well as mycosis fungoides and carcinomas of the ovary and pancreas. [134]
The correct diagnosis can be assisted by a histopathological analysis of the skin lesions. The features of acanthosis nigricans are distinctive. indeed pathognomonic. Typically involve significant papillomatosis with “finger-like” projections of the rete ridges. Acanthosis is generally mild and rather restricted to the valleys between the papillomatous formations. Hyperkeratosis is also observed, but it resembles a basket-weave pattern. [135] Although the changes in acanthosis nigricans (AN) are quite characteristic clinically, they can sometimes be mistaken for other conditions, especially in the early stages.
Conditions like hypothyroidism, Cushing’s syndrome, and polycystic ovary syndrome (PCOS) may also present with hyperpigmentation similar to AN. In such cases, hormonal and imaging tests are decisive. Chronic irritation or friction, especially in skin folds, can also lead to hyperpigmentation that might mimic AN. Due to their location in skin folds, the changes may initially be mistaken for fungal intertrigo. Under these circumstances, mycological examination can be helpful for differentiation.
After diagnosing acanthosis nigricans, it is essential to explore possible underlying causes. Especially if skin lesions appear in adulthood for the first time. When factors like family history, endocrine disorders, obesity or associated medications are not present, the emergence of this condition should prompt an evaluation for potential malignancy, with a focus on the gastrointestinal tract. [136] Once the tumor is removed, the condition may regress. This suggests that growth factors produced by the tumors could play a causal role in the development of AN.
Dermatologists sometimes prescribe keratolytics, such as topical retinoids (e.g., topical tretinoin 0.1% or a combination of tretinoin 0.05% and 12% ammonium lactate) [137,138] and podophyllin. Topical vitamin D analogs (e.g., calcipotriol (calcipotriene) 0.005%) act by decreasing keratinocyte proliferation and cause an improvement of the acanthosis nigricans lesions. [139] The success of these treatments is variable. Other agents that have been tried include metformin and etretinate.
4. Conclusion
This article has explored the various disease entities within the realm of paraneoplastic skin syndromes (PNS), highlighting their critical role as potential early indicators of underlying malignancies. The discussion emphasized the necessity for healthcare professionals, including dermatologists, oncologists, and primary care physicians, to maintain a high index of suspicion for PNS. Recognizing and differentiating these syndromes from other dermatological conditions is essential, despite the inherent challenges posed by their rarity and diverse clinical presentations. The article underscores the importance of continuous education and dialogue among medical practitioners to improve early detection and patient outcomes. By fostering awareness and knowledge sharing, the medical community can better navigate the complexities of PNS and enhance interdisciplinary collaboration in the diagnosis and management of associated malignancies.
Author Contributions
Conceptualization, A.R. and A.Ż.; methodology, A.R., K.T., W.L., A.K. and A.Ż.; software, A.R. A.Ż.; resources, A.R., K.T., W.L., A.K. A.Ż.; data curation, A.R. and A.Ż. writing—original draft preparation, A.R., A.K., K.T., W.L.; writing—review and editing, A.Ż.; visualization, A.R, A.Ż. A.K; supervision, A.Ż.; funding acquisition, A.Ż All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the research projects of Medical University of Lodz: No 503/1-152-01/503-11-002.
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Alter, M.; Mengoni, M.; Gaffal, E. Cutaneous manifestations of internal malignancy. J Dtsch Dermatol Ges. 2020, 18, 456–469. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.A.; Mesquita Kde, C.; Igreja, A.C.; Lucas, I.C.; Freitas, A.F.; Oliveira, S.M.; Costa, I.M.; Campbell, I.T. Paraneoplastic cutaneous manifestations: Concepts and updates. An Bras Dermatol. 2013, 88, 9–22. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Eubanks, L.E.; McBurney, E.; Reed, R. Erythema gyratum repens. Am J Med Sci. 2001, 321, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Rongioletti, F.; Fausti, V.; Parodi, A. Erythema gyratum repens is not an obligate paraneoplastic disease: A systematic review of the literature and personal experience. J Eur Acad Dermatol Venereol. 2014, 28, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Boyd, A.S.; Neldner, K.H.; Menter, A. Erythema gyratum repens: A paraneoplastic eruption. J Am Acad Dermatol. 1992, 26 Pt 1, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Holt, P.J.; Davies, M.G. Erythema gyratum repens--an immunologically mediated dermatosis? Br J Dermatol. 1977, 96, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Caux, F.; Lebbe, C.; Thomine, E.; Benyahia, B.; Flageul, B.; Joly, P.; Rybojad, M.; Morel, P. Erythema gyratum repens. A case studied with immunofluorescence, immunoelectron microscopy and immunohistochemistry. Br J Dermatol. 1994, 131, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Stone, S.P.; Buescher, L.S. Life-threatening paraneoplastic cutaneous syndromes. Clin Dermatol. 2005, 23, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Kleyn, C.E.; Lai-Cheong, J.E.; Bell, H.K. Cutaneous manifestations of internal malignancy. Am J Clin Dermatol. 2006, 7, 71–84. [Google Scholar] [CrossRef]
- Loucas, E.; Russo, G.; Millikan, L.E. Genetic and acquired cutaneous disorders associated with internal malignancy. Int J Dermatol. 1995, 34, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Boyd, A.S.; Neldner, K.H. Erythema gyratum repens without underlying disease. J Am Acad Dermatol. 1993, 28, 132. [Google Scholar] [CrossRef] [PubMed]
- Jablonska S, Blaszczyk M, Kozlowska A. Erythema gyratum repens-like psoriasis. Int J Dermatol. 2000 Sep;39(9):695-7. [CrossRef] [PubMed]
- Dermatol Rev/Przegl Dermatol 2017, 104, 439–445. [CrossRef]
- Skolnick, M.; Mainman, E.R. Erythema gyratum repens with metastatic adenocarcinoma. Arch Dermatol. 1975, 111, 227–229. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.H.; Li, C.Y. Necrolytic migratory erythema-like eruption associated with erlotinib in a patient with lung adenocarcinoma. Lung Cancer. 2020, 148, 173–174. [Google Scholar] [CrossRef] [PubMed]
- Trojan, A.; Jacky, E.; Follath, F.; Dummer, R. Necrolytic migratory erythema (glucagenoma)-like skin lesions induced by EGF-receptor inhibition. Swiss Med Wkly. 2003, 133, 22–23. [Google Scholar] [CrossRef] [PubMed]
- Seong, J.Y.; Kong, S.H.; Choi, Y.S.; Suh, H.S. Necrolytic Migratory Erythema–like Skin Lesion During Gefitinib Treatment: A Rare Cutaneous Adverse Reaction. JAMA Dermatol. 2016, 152, 947–948. [Google Scholar] [CrossRef]
- Tremblay, C.; Marcil, I. Necrolytic Migratory Erythema: A Forgotten Paraneoplastic Condition. J Cutan Med Surg. 2017, 21, 559–561. [Google Scholar] [CrossRef] [PubMed]
- Vinik, A.; Feliberti, E.; Perry, R. Glucagonoma syndrome. Endotext. South Dartmouth, MA: MDText.com; 2014. https://www.ncbi.nlm.nih.gov/books/NBK279041/.
- Wick, M.R.; Patterson, J.W. Cutaneous Paraneoplastic Syndromes. Semin. Diagn. Pathol. 2019. [Google Scholar] [CrossRef]
- Foss, M.G.; Hashmi, M.F.; Ferrer-Bruker, S.J. Necrolytic Migratory Erythema. 2023 Jul 25. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan–. [PubMed]
- Kovács, R.K.; Korom, I.; Dobozy, A.; Farkas, G.; Ormos, J.; Kemény, L. Necrolytic migratory erythema. J Cutan Pathol. 2006, 33, 242–245. [Google Scholar] [CrossRef] [PubMed]
- El Rassi, Z.; Partensky, C.; Valette, P.J.; Berger, F.; Chayvialle, J.A. Necrolytic migratory erythema, first symptom of a malignant glucagonoma: Treatment by long-acting somatostatin and surgical resection. Report of three cases. Eur J Surg Oncol. 1998, 24, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Al-Faouri, A.; Ajarma, K.; Alghazawi, S.; Al-Rawabdeh, S.; Zayadeen, A. Glucagonoma and Glucagonoma Syndrome: A Case Report with Review of Recent Advances in Management. Case Rep Surg. 2016, 2016, 1484089. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Caplin, M.E.; Pavel, M.; Ćwikła, J.B.; Phan, A.T.; Raderer, M.; Sedláčková, E.; Cadiot, G.; Wolin, E.M.; Capdevila, J.; Wall, L.; Rindi, G.; Langley, A.; Martinez, S.; Blumberg, J.; Ruszniewski, P.; CLARINET Investigators. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med. 2014, 371, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.C.; Shah, M.H.; Ito, T.; Bohas, C.L.; Wolin, E.M.; Van Cutsem, E.; Hobday, T.J.; Okusaka, T.; Capdevila, J.; de Vries, E.G.; Tomassetti, P.; Pavel, M.E.; Hoosen, S.; Haas, T.; Lincy, J.; Lebwohl, D.; Öberg, K.; RAD001 in Advanced Neuroendocrine Tumors, Third Trial (RADIANT-3) Study Group. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011, 364, 514–523. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Poole, S.; Fenske, N.A. Cutaneous markers of internal malignancy, I.I. Paraneoplastic dermatoses and environmental carcinogens. Journal of the American Academy of Dermatology 1993, 28, 147–164. [Google Scholar] [CrossRef]
- Sarkar, B.; Knecht, R.; Sarkar, C.; Weidauer, H. Bazex syndrome (acrokeratosis paraneoplastica). Eur Arch Otorhinolaryngol. 1998, 255, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Roy, B.; Lipner, S.R. A Review of Nail Changes in Acrokeratosis Paraneoplastica (Bazex Syndrome). Skin Appendage Disord. 2021, 7, 163–172. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Räßler, F.; Goetze, S.; Elsner, P. Acrokeratosis paraneoplastica (Bazex syndrome) - a systematic review on risk factors, diagnosis, prognosis and management. J Eur Acad Dermatol Venereol. 2017, 31, 1119–1136. [Google Scholar] [CrossRef] [PubMed]
- Amano, M.; Hanafusa, T.; Chikazawa, S.; Ueno, M.; Namiki, T.; Igawa, K.; Miura, K.; Yokozeki, H. Bazex Syndrome in Lung Squamous Cell Carcinoma: High Expression of Epidermal Growth Factor Receptor in Lesional Keratinocytes with Th2 Immune Shift. Case Rep Dermatol. 2016, 8, 358–362. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Valdivielso, M.; Longo, I.; Suárez, R.; Huerta, M.; Lázaro, P. Acrokeratosis paraneoplastica: Bazex syndrome. J Eur Acad Dermatol Venereol. 2005, 19, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.H.; Ferrazzano, C.; Karthikeyan, A.; Hejazi, H.; Bhattacharya, A.; Andrew Awuah, W.; Isik, A. Bazex Syndrome (Acrokeratosis Paraneoplastica): A Narrative Review of Pathogenesis, Clinical Manifestations, and Therapeutic Approaches. Cureus. 2023, 15, e45368. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Huilaja, L.; Soronen, M.; Karjalainen, A.; Tasanen, K. Acrokeratosis Paraneoplastica-like Findings as a Manifestation of Systemic Lupus Erythematosus. Acta Derm Venereol. 2019, 99, 333–334. [Google Scholar] [CrossRef] [PubMed]
- Bolognia, J.L. Bazex’ syndrome. Clin Dermatol. 1993, 11, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Vaynshtok, P.M.; Tian, F.; Kaffenberger, B.H. Acitretin amelioration of Acrokeratosis Paraneoplastica (Bazex Syndrome) in cases of incurable squamous cell carcinoma of the hypopharynx. Dermatol Online J. 2016, 22, 13030–qt8n96v45x. [Google Scholar] [CrossRef] [PubMed]
- Sweet, R.D. An acute febrile neutrophilic dermatosis. Br J Dermatol 1964, 76, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Fazili, T.; Duncan, D.; Wani, L. Sweet’s syndrome. Am J Med. 2010, 123, 694–696. [Google Scholar] [CrossRef] [PubMed]
- Burrall, B. Sweet’s syndrome (acute febrile neutrophilic dermatosis). Dermatol Online J. 1999, 5, 8. [Google Scholar] [PubMed]
- Su, W.P.; Liu, H.N. Diagnostic criteria for Sweet’s syndrome. Cutis. 1986, 37, 167–174. [Google Scholar] [PubMed]
- von den Driesch, P. Sweet’s syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. 1994, 31, 535–556, quiz 557-60. [Google Scholar] [CrossRef] [PubMed]
- Villarreal-Villarreal, C.D.; Ocampo-Candiani, J.; Villarreal-Martínez, A. Sweet Syndrome: A Review and Update. Actas Dermosifiliogr. 2016, 107, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, L.; Baraf, C.S.; Richheimer, L.L. Sweet’s syndrome (acute febrile neutrophilic dermatosis). Arch Dermatol 1971, 103, 81–84. [Google Scholar] [CrossRef]
- Pirard, C.; Delannoy, A. Sweet syndrome and acute myeloid leukemia. Ann Dermatol Venereol 1977, 104, 160–161. [Google Scholar]
- Cohen, P.R.; Kurzrock, R. Chronic myelogenous leukemia and Sweet syndrome. Am J Hematol 1989, 32, 134–137. [Google Scholar] [CrossRef] [PubMed]
- Hussein, K.; Nanda, A.; Al-Sabah, H.; Alsaleh, Q.A. Sweet’s syndrome (acute febrile neutrophilic dermatosis) associated with adenocarcinoma of prostate and transitional cell carcinoma of urinary bladder. J Eur Acad Dermatol Venereol 2005, 19, 597–599. [Google Scholar] [CrossRef] [PubMed]
- Adil, A.; Goyal, A.; Quint, J.M. Behcet Disease. 2023 Feb 22. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. [PubMed]
- Arun Kumar, A.U.; Elsayed, M.E.; Alghali, A.; Ali, A.A.; Mohamed, H.; Hussein, W.; Hackett, C.; Leonard, N.; Stack, A.G. Sweet syndrome: A rare feature of ANCA-associated vasculitis or unusual consequence of azathioprine-induced treatment. Allergy Asthma Clin Immunol. 2018, 14, 46. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Corbeddu, M.; Pilloni, L.; Pau, M.; Pinna, A.L.; Rongioletti, F.; Atzori, L. Treatment of Sweet’s syndrome in pregnancy. Dermatol Ther. 2018, 31, e12619. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.R.; Kurzrock, R. Sweet’s syndrome: A review of current treatment options. Am J Clin Dermatol. 2002, 3, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Kleyn, C.E.; Lai-Cheong, J.E.; Bell, H.K. Cutaneous Manifestations of Internal Malignancy. American Journal of Clinical Dermatology 2006, 7, 71–84. [Google Scholar] [CrossRef]
- Shah, M.; Sachdeva, M.; Gefri, A.; Jfri, A. Paraneoplastic pyoderma gangrenosum in solid organ malignancy: A literature review. Int J Dermatol. 2020, 59, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Duguid, C.M.; Powell, F.C. Pyoderma gangrenosum. Clin Dermatol. 1993, 11, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Maverakis, E.; Marzano, A.V.; Le, S.T.; Callen, J.P.; Brüggen, M.C.; Guenova, E.; Dissemond, J.; Shinkai, K.; Langan, S.M. Pyoderma gangrenosum. Nat Rev Dis Primers. 2020, 6, 81. [Google Scholar] [CrossRef] [PubMed]
- Alavi, A.; French, L.E.; Davis, M.D.; Brassard, A.; Kirsner, R.S. Pyoderma Gangrenosum: An Update on Pathophysiology, Diagnosis and Treatment. Am J Clin Dermatol. 2017, 18, 355–372. [Google Scholar] [CrossRef] [PubMed]
- Maverakis, E.; Ma, C.; Shinkai, K.; Fiorentino, D.; Callen, J.P.; Wollina, U.; Marzano, A.V.; Wallach, D.; Kim, K.; Schadt, C.; Ormerod, A.; Fung, M.A.; Steel, A.; Patel, F.; Qin, R.; Craig, F.; Williams, H.C.; Powell, F.; Merleev, A.; Cheng, M.Y. Diagnostic Criteria of Ulcerative Pyoderma Gangrenosum: A Delphi Consensus of International Experts. JAMA Dermatol. 2018, 154, 461–466. [Google Scholar] [CrossRef] [PubMed]
- George, C.; Deroide, F.; Rustin, M. Pyoderma gangrenosum - a guide to diagnosis and management . Clin Med 2019, 19, 224–228. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- You, H.R.; Ju, J.K.; Yun, S.J.; Lee, J.B.; Kim, S.J.; Won, Y.H.; Lee, S.C. Paraneoplastic Pyoderma Gangrenosum Associated with Rectal Adenocarcinoma. Ann Dermatol. 2018, 30, 79–82. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Karakayli, G.; Beckham, G.; Orengo, I.; Rosen, T. Exfoliative dermatitis. Am Fam Physician. 1999, 59, 625–630. [Google Scholar] [PubMed]
- Zervakis, S.G.; Spernovasilis, N.; Boutakoglou, E.; Panagiotakis, S.; Thomopoulou, K.; Samonis, G. Erythroderma as a paraneoplastic manifestation of small cell lung cancer. Curr Probl Cancer. 2020, 44, 100499. [Google Scholar] [CrossRef] [PubMed]
- Kerasia Maria Plachouri Paraneoplastic erythroderma: An insight on the existing data.
- Pal, S.; Haroon, T.S. Erythroderma: A clinico-etiologic study of 90 cases. Int J Dermatol 1998, 37104, 10.1046/j.1365-4362.1998.00228.x. [Google Scholar]
- Rothe, M.J.; Bialy, T.L.; Grant-Kels, J.M. Erythroderma. Dermatol Clin. 2000, 18, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, V.N. Srivastava G Exfoliative dermatitis: A prospective study of 80 patients. Dermatologica 1986, 173, 278. [Google Scholar] [CrossRef]
- Duangurai, K.; Piamphongsant, T.; Himmungnan, T. Sézary cell count in exfoliative dermatitis. Int J Dermatol. 1988, 27, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.C.; Jester, J.D.; King, L.E., Jr. Erythroderma and exfoliative dermatitis. Clin Dermatol. 1993, 11, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Muller CS, Lorenz MT, Tilgen W, € et al. Primary manifestation of erythema gyratum repens as a transient erythroderma in a patient with bronchial carcinoma. Int J Dermatol 2010, 49, 676–678. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Lee, K.M.; Ucmak, D.; Brodsky, M.; Atanelov, Z.; Farahnik, B.; Abrouk, M.; Nakamura, M.; Zhu, T.H.; Liao, W. Erythrodermic psoriasis: Pathophysiology and current treatment perspectives. Psoriasis 2016, 6, 93–104. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Roenneberg, S.; Biedermann, T. Pityriasis rubra pilaris: Algorithms for diagnosis and treatment. J Eur Acad Dermatol Venereol. 2018, 32, 889–898. [Google Scholar] [CrossRef] [PubMed]
- Jawed, S.I.; Myskowski, P.L.; Horwitz, S.; Moskowitz, A.; Querfeld, C. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): Part I. Diagnosis: Clinical and histopathologic features and new molecular and biologic markers. J Am Acad Dermatol. 2014, 70, 205.e1-16, quiz 221–222. [Google Scholar] [CrossRef] [PubMed]
- Roccuzzo, G.; Giordano, S.; Avallone, G.; Rubatto, M.; Canonico, S.; Funaro, A.; Ortolan, E.; Senetta, R.; Fava, P.; Fierro, M.T.; Ribero, S.; Quaglino, P. Sézary Syndrome: Different Erythroderma Morphological Features with Proposal for a Clinical Score System. Cells. 2022, 11, 333. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sharma, G.; Govil, D.C. Allopurinol induced erythroderma. Indian J Pharmacol. 2013, 45, 627–628. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Reynolds, N.J.; Jones, S.K.; Crossley, J.; Harman, R.R. Exfoliative dermatitis due to nifedipine. Br J Dermatol. 1989, 121, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Tietze, K.J.; Gaska, J.A. Cefoxitin-associated exfoliative dermatitis. Clin Pharm. 1983, 2, 582–584. [Google Scholar] [PubMed]
- Mistry, Nisha MD, FRCPC.; Gupta, Ambika; Alavi, Afsaneh MD, FRCPC.; Sibbald, R. Gary BSc, MD, MEd, FRCPC(Med)(Derm), FACP, FAAD, MAPWCA. A Review of the Diagnosis and Management of Erythroderma (Generalized Red Skin). Advances in Skin & Wound Care 28, p 228-236, May 2015. |. [CrossRef]
- Joly P, Richard C, Gilbert D, Courville P, Chosidow O, Roujeau JC; et al. Sensitivity and specificity of clinical, histologic, and immunologic features in the diagnosis of paraneoplastic pemphigus. J Am Acad Dermatol 2000, 43: 619-26. [CrossRef]
- Martel, P.; Loiseau, P.; Joly, P.; Busson, M.; Lepage, V.; Mouquet, H.; Courville, P.; Flageul, B.; Charron, D.; Musette, P.; Gilbert, D.; Tron, F. Paraneoplastic pemphigus is associated with the DRB1*03 allele. J Autoimmun. 2003, 20, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Bu, D.F.; Li, D.; Zhu, X.J. Genotyping of HLA-I and HLA-II alleles in Chinese patients with paraneoplastic pemphigus. Br J Dermatol. 2008, 158, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Sklavounou, A.; Laskaris, G. Paraneoplastic pemphigus: A review. Oral Oncol 1998, 34, 437–440. [Google Scholar] [CrossRef] [PubMed]
- Kimyai-Asadi, A.; Jih, M.H. Paraneoplastic pemphigus. Int J Dermatol. 2001, 40, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Edgin, W.A.; Pratt, T.C.; Grimwood, R.E. Pemphigus Vulgaris and Paraneoplastic Pemphigus. Oral Maxillofac Surg Clin North Am. 2008, 20, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Paolino, G.; Didona, D.; Magliulo, G.; Iannella, G.; Didona, B.; Mercuri, S.R.; Moliterni, E.; Donati, M.; Ciofalo, A.; Granata, G.; et al. Paraneoplastic Pemphigus: Insight into the Autoimmune Pathogenesis, Clinical Features and Therapy. Int. J. Mol. Sci. 2017, 18, 2532. [Google Scholar] [CrossRef]
- Meyers, S.J.; Varley, G.A.; Meisler, D.M.; Camisa, C.; Wander, A.H. Conjunctival involvement in paraneoplastic pemphigus. Am J Ophthalmol. 1992, 114, 621–624. [Google Scholar] [CrossRef] [PubMed]
- Nousari, H.C.; Deterding, R.; Wojtczack, H.; Aho, S.; Uitto, J.; Hashimoto, T.; Anhalt, G.J. The mechanism of respiratory failure in paraneoplastic pemphigus. N Engl J Med. 1999, 340, 1406–1410. [Google Scholar] [CrossRef] [PubMed]
- Nguyen VT, Ndoye A, Bassler KD et al. Classification, clinical manifestations, and immunopathological mechanisms of the epithelial variant of paraneoplastic autoimmune multiorgan syndrome: A reappraisal of paraneoplastic pemphigus. Arch. Dermatol. 2001; 137: 193–206.
- Maruta, C.W.; Miyamoto, D.; Aoki, V.; Carvalho, R.G.R.; Cunha, B.M.; Santi, C.G. Paraneoplastic pemphigus: A clinical, laboratorial, and therapeutic overview. An Bras Dermatol. 2019, 94, 388–398. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Probst C, Schlumberger W, Stöcker W et al. Development of ELISA for the specific determination of autoantibodies against envoplakin and periplakin in paraneoplastic pemphigus. Clin. Chim. Acta 2009; 410: 13–18. [CrossRef]
- Kartan, S.; Shi, V.Y.; Clark, A.K.; Chan, L.S. Paraneoplastic Pemphigus and Autoimmune Blistering Diseases Associated with Neoplasm: Characteristics, Diagnosis, Associated Neoplasms, Proposed Pathogenesis, Treatment. Am J Clin Dermatol. 2017, 18, 105–126. [Google Scholar] [CrossRef] [PubMed]
- Tsuchisaka, A.; Numata, S.; Teye, K.; Natsuaki, Y.; Kawakami, T.; Takeda, Y.; Wang, W.; Ishikawa, K.; Goto, M.; Koga, H.; Sogame, R.; Ishii, N.; Takamori, S.; Hoshino, T.; Brandt, O.; Pas, H.H.; Fujiwara, S.; Hashimoto, T. Epiplakin Is a Paraneoplastic Pemphigus Autoantigen and Related to Bronchiolitis Obliterans in Japanese Patients. J Invest Dermatol. 2016, 136, 399–408. [Google Scholar] [CrossRef]
- Didona, D.; Fania, L.; Didona, B.; Eming, R.; Hertl, M.; Di Zenzo, G. Paraneoplastic Dermatoses: A Brief General Review and an Extensive Analysis of Paraneoplastic Pemphigus and Paraneoplastic Dermatomyositis. Int J Mol Sci. 2020, 21, 2178. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yong, A.A.; Tey, H.L. Paraneoplastic pemphigus. Australas J Dermatol. 2013, 54, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Nam, K.H.; Lee, J.B.; Lee, J.Y.; Ihm, C.W.; Lee, S.E.; Oh, S.H.; Hashimoto, T.; Kim, S.C. Retrospective analysis of 12 Korean patients with paraneoplastic pemphigus. J Dermatol. 2012, 39, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Kershenovich, R.; Hodak, E.; Mimouni, D. Diagnosis and classification of pemphigus and bullous pemphigoid. Autoimmun Rev. 2014, 13, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Bowen, G.M.; Peters, N.T.; Fivenson, D.P.; Su, L.D.; Nousari, H.C.; Anhalt, G.J.; Cooper, K.D.; Stevens, S.R. Lichenoid dermatitis in paraneoplastic pemphigus: A pathogenic trigger of epitope spreading? Arch Dermatol. 2000, 136, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Wade, M.S.; Black, M.M. Paraneoplastic pemphigus: A brief update. Australas J Dermatol. 2005, 46, 1–8, quiz 9-10. [Google Scholar] [CrossRef] [PubMed]
- Murrell, D.F.; Peña, S.; Joly, P.; Marinovic, B.; Hashimoto, T.; Diaz, L.A.; Sinha, A.A.; Payne, A.S.; Daneshpazhooh, M.; Eming, R.; et al. Diagnosis and management of pemphigus: Recommendations of an international panel of experts. J Am Acad Dermatol. 2020, 82, 575–585.e1. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kikuchi, T.; Mori, T.; Shimizu, T.; Koda, Y.; Abe, R.; Kurihara, Y.; Funakoshi, T.; Yamagami, J.; Sato, H.; Tsunoda, K.; Amagai, M.; Okamoto, S. Successful treatment with bendamustine and rituximab for paraneoplastic pemphigus. Ann Hematol. 2017, 96, 1221–1222. [Google Scholar] [CrossRef]
- Bech, R.; Baumgartner-Nielsen, J.; Peterslund, N.A.; Steiniche, T.; Deleuran, M.; d’Amore, F. Alemtuzumab is effective against severe chronic lymphocytic leukaemia-associated paraneoplastic pemphigus. Br J Dermatol. 2013, 169, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Hohwy, T.; Bang, K.; Steiniche, T.; Peterslund, N.A.; d’Amore, F. Alemtuzumab-induced remission of both severe paraneoplastic pemphigus and leukaemic bone marrow infiltration in a case of treatment-resistant B-cell chronic lymphocytic leukaemia. Eur J Haematol. 2004, 73, 206–209. [Google Scholar] [CrossRef] [PubMed]
- Sevim, E.; Kobrin, D.; Casal-Dominguez, M.; Pinal-Fernandez, I. A comprehensive review of dermatomyositis treatments - from rediscovered classics to promising horizons. Expert Rev Clin Immunol. 2024, 20, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Kronzer, V.L.; Kimbrough, B.A.; Crowson, C.S.; Davis JM 3rd Holmqvist, M.; Ernste, F.C. Incidence, Prevalence, and Mortality of Dermatomyositis: A Population-Based Cohort Study. Arthritis Care Res 2023, 75, 348–355. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- O’Hanlon, T.P.; Rider, L.G.; Mamyrova, G.; Targoff, I.N.; Arnett, F.C.; Reveille, J.D.; Carrington, M.; Gao, X.; Oddis, C.V.; Morel, P.A.; Malley, J.D.; Malley, K.; Shamim, E.A.; Chanock, S.J.; Foster, C.B.; Bunch, T.; Reed, A.M.; Love, L.A.; Miller, F.W. HLA polymorphisms in African Americans with idiopathic inflammatory myopathy: Allelic profiles distinguish patients with different clinical phenotypes and myositis autoantibodies. Arthritis Rheum. 2006, 54, 3670–81. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Han, L.; Yuan, L.; Yang, Y.; Gou, G.; Sun, H.; Lu, L.; Bao, L. HLA class II alleles may influence susceptibility to adult dermatomyositis and polymyositis in a Han Chinese population. BMC Dermatol. 2014, 14, 9. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Okiyama, N.; Fujimoto, M. Cutaneous manifestations of dermatomyositis characterized by myositis-specific autoantibodies. F1000Res. 2019, 8, F1000. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Strowd, L.C.; Jorizzo, J.L. Review of dermatomyositis: Establishing the diagnosis and treatment algorithm. J Dermatolog Treat. 2013, 24, 418–421. [Google Scholar] [CrossRef] [PubMed]
- Pappu, R.; Seetharaman, M. Polymoysitis. In Medscape Reference. Available from http://emedicine.medscape.com/article/335925..accessed 8 June 2011.
- Adams, E.M.; Chow, C.K.; Premkumar, A.; Plotz, P.H. The idiopathic inflammatory myopathies: Spectrum of MR imaging findings. Radiographics. 1995, 15, 563–574. [Google Scholar] [CrossRef]
- Vattemi, G.; Mirabella, M.; Guglielmi, V.; Lucchini, M.; Tomelleri, G.; Ghirardello, A.; Doria, A. Muscle biopsy features of idiopathic inflammatory myopathies and differential diagnosis. Auto Immun Highlights. 2014, 5, 77–85. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sigurgeirsson, B.; Lindelöf, B.; Edhag, O.; Allander, E. Risk of cancer in patients with dermatomyositis or polymyositis. A population-based study. N Engl J Med. 1992, 326, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Emslie-Smith, A.M.; Engel, A.G. Microvascular changes in early and advanced dermatomyositis: A quantitative study. Ann Neurol. 1990, 27, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Maoz, C.R.; Langevitz, P.; Livneh, A.; Blumstein, Z.; Sadeh, M.; Bank, I.; Gur, H.; Ehrenfeld, M. High incidence of malignancies in patients with dermatomyositis and polymyositis: An 11-year analysis. Semin Arthritis Rheum. 1998, 27, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, D.; Casciola-Rosen, L. Autoantibodies to transcription intermediary factor 1 in dermatomyositis shed insight into the cancer-myositis connection. Arthritis Rheum. 2012, 64, 346–349. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ofori, E.; Ramai, D.; Ona, M.; Reddy, M. Paraneoplastic Dermatomyositis Syndrome Presenting as Dysphagia. Gastroenterology Res. 2017, 10, 251–254. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Callen, J.P.; Hyla, J.F.; Bole, G.G., Jr.; Kay, D.R. The relationship of dermatomyositis and polymyositis to internal malignancy. Arch Dermatol. 1980, 116, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Zhang, L.; Jia, H.; Nie, Z.; Zhang, L. Malignancy in dermatomyositis: A retrospective paired case-control study of 202 patients from Central China. Medicine (Baltimore). 2020, 99, e21733. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ponti, G.; Luppi, G.; Losi, L.; Giannetti, A.; Seidenari, S. Leser-Trélat syndrome in patients affected by six multiple metachronous primitive cancers. J Hematol Oncol. 2010, 3, 2. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Safa, G.; Darrieux, L. Leser-Trélat Sign without Internal Malignancy. Case Rep Oncol. 2011, 4, 175–177. [Hafner C, Vogt T. Seborrheic keratosis. J Dtsch Dermatol Ges. 2013, 6, 664–677. [Google Scholar] [CrossRef]
- Inamadar, A.C.; Palit, A. Eruptive seborrhoeic keratosis in human immunodeficiency virus infection: A coincidence or ’the sign of Leser-Trélat’? Br J Dermatol. 2003, 149, 435–436. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Salafranca, M.; Gómez-Martín, I.; Bañuls, J.; Serrano, P.; Medina, C.; Llambrich, A.; Pizarro, Á.; Ara, M.; Zaballos, P. Dermoscopy of inflamed seborrheic keratosis: A great mimic of malignancy. Australas J Dermatol. 2022, 63, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Myroshnychenko, M.S.; Moiseienko, T.M.; Torianyk, I.I.; Ivannik, V.Y.; Popova, N.G.; Mozhaiev, I.V.; Chastii, T.V.; Minukhin, V.V.; Leontiev, P.A.; Osolodchenko, T.P.; Parkhomenko, K.Y. Seborrheic Keratosis: Current State of the Problem. Wiad Lek. 2022, 75 Pt 2, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Roh, N.K.; Hahn, H.J.; Lee, Y.W.; Choe, Y.B.; Ahn, K.J. Clinical and Histopathological Investigation of Seborrheic Keratosis. Ann Dermatol. 2016, 28, 152–158. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gaduputi, V.; Chandrala, C.; Tariq, H.; Kanneganti, K. Sign of leser-trélat associated with esophageal squamous cell cancer. Case Rep Oncol Med. 2014, 2014, 825929. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Aouali, S.; Bensalem, S.; Saddouk, H.; Aissaoui, A.; Bennani, A.; Zizi, N.; Dikhaye, S. Leser-Trelat sign preceding male breast cancer. Ann Med Surg (Lond). 2021, 72, 103065. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Modi, K.; Chen, R.; Abubshait, L. Leser-Trélat Sign as a Marker for Underlying Pancreatic Cancer. Clin Pract Cases Emerg Med. 2023, 7, 202–204. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mathien, A.; Kuraitis, D. Leser-Trelat sign: Eruptive seborrheic keratoses and primary lung adenocarcinomas with an epidermal growth factor receptor mutation. JAAD Case Rep. 2023, 37, 38–40. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wollina, U. Recent advances in managing and understanding seborrheic keratosis. F1000Res. 2019, 8, F1000. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Valejo Coelho, M.M.; Prokop, J.; Fernandes, C. Acanthosis Nigricans Maligna. JAMA Dermatol. 2019, 155, 1069. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, R.; Liu, Y.; Zhang, S.; Qi, H. Malignant acanthosis nigricans: A case report. BMC Ophthalmol. 2020, 20, 446. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hermanns-Lê, T.; Scheen, A.; Piérard, G.E. Acanthosis nigricans associated with insulin resistance : Pathophysiology and management. Am J Clin Dermatol. 2004, 5, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Chung, V.Q.; Moschella, S.L.; Zembowicz, A.; Liu, V. Clinical and pathologic findings of paraneoplastic dermatoses. J Am Acad Dermatol. 2006, 54, 745–762, quiz 763-6. [Google Scholar] [CrossRef] [PubMed]
- Pollitzer, S. Acanthosis nigricans: A symptom of a disorder of the abdominal sympathetic. JAMA 1909, 53, 1369–1373. [Google Scholar] [CrossRef]
- Zhang, N.; Qian, Y.; Feng, A.P. Acanthosis nigricans, tripe palms, and sign of Leser-Trélat in a patient with gastric adenocarcinoma: Case report and literature review in China. Int J Dermatol. 2015, 54, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Yeh, J.S.; Munn, S.E.; Plunkett, T.A.; Harper, P.G.; Hopster, D.J.; du Vivier, A.W. Coexistence of acanthosis nigricans and the sign of Leser-Trélat in a patient with gastric adenocarcinoma: A case report and literature review. J Am Acad Dermatol. 2000, 42 Pt 2, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, W.J.; Goldin, H.M.; Bronson, D.M.; Brody, P.E. Acanthosis nigricans associated with mycosis fungoides. J Am Acad Dermatol. 1988, 19 Pt 2, 951–953. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Amador, V.; Esquivel-Pedraza, L.; Caballero-Mendoza, E.; Berumen-Campos, J.; Orozco-Topete, R.; Angeles-Angeles, A. Oral manifestations as a hallmark of malignant acanthosis nigricans. J Oral Pathol Med. 1999, 28, 278–281. [Google Scholar] [CrossRef] [PubMed]
- Rigel, D.S.; Jacobs, M.I. Malignant acanthosis nigricans: A review. J Dermatol Surg Oncol. 1980, 6, 923–927. [Google Scholar] [CrossRef] [PubMed]
- Darmstadt, G.L.; Yokel, B.K.; Horn, T.D. Treatment of acanthosis nigricans with tretinoin. Arch Dermatol. 1991, 127, 1139–1140. [Google Scholar] [CrossRef] [PubMed]
- Blobstein, S.H. Topical therapy with tretinoin and ammonium lactate for acanthosis nigricans associated with obesity. Cutis. 2003, 71, 33–34. [Google Scholar] [PubMed]
- Gregoriou, S.; Anyfandakis, V.; Kontoleon, P.; Christofidou, E.; Rigopoulos, D.; Kontochristopoulos, G. Acanthosis nigricans associated with primary hypogonadism: Successful treatment with topical calcipotriol. J Dermatolog Treat. 2008, 19, 373–375. [Google Scholar] [CrossRef] [PubMed]

Table 1.
Differential Diagnosis of Erythroderma.
| Category | Condition |
| Metabolic and Endocrine Disorders | Acanthosis nigricans |
| Granulomatous Diseases | Acute complications of sarcoidosis |
| Malignancies | Cutaneous T-cell lymphoma |
| Autoimmune Blistering Diseases | Bullous pemphigoid, Pemphigus foliaceus |
| Allergic Diseases | Irritant contact dermatitis, Pediatric atopic dermatitis, Allergic contact dermatitis |
| Inflammatory Skin Diseases | Plaque psoriasis, Lichen planus, Pityriasis rubra pilaris, Seborrheic dermatitis, Stasis dermatitis |
| Reumatologic Diseases | Reactive arthritis |
| Genodermatosis | Familial bening pemhigus (Hailey-Hailey disease) |
| Other diseases | Dermatologic manifestations of graft versus host disease |
Table 2.
Proteins targeted by autoantibodies in PNP.
| Protein name | Molecular Weight (kD) | Location |
|---|---|---|
| Desmoglein type 1 (Dsg1) Desmoglein type 3 (Dsg3) |
160 130 |
Desmosome, extracellular |
| Desmoplakin 1 Desmoplakin 2 |
250 210 |
Desmosome, intracellular |
| Bullous pemhigoid antigen 1 (BP230) | 230 | Hemidesmosome/lamina lucida |
| Envoplakin | 210 | Desmosome, intracellular |
| Epiplakin | >700 | Desmosome, intracellular |
| Periplakin | 190 | Desmosome, intracellular |
| Alpha-2-macroglobulin-like-1-antigen (A2ML1) | 170 | Protease inhibitor |
Table 3.
The symptoms of dermatomyositis.
| Skin finding | Description | |
| Pathognomic symptoms | Gottron papules | erythematous or violaceous papules with or without scaling or ulceration located over the interphalangeal or metacarpophalangeal joints |
| Heliotrope rash | violaceous, or an erythematous rash effecting upper eyelids with or without periorbital edema; may not be apparent in patients with dark skin | |
| Other symptoms | Gottron sign | erythematous macules or patches over the elbows or knees |
| Facial erythema | erythema over the cheeks and nasal bridge involving the nasolabial folds, may extend up to the forehead and ears | |
| Shawl sign | erythema over the posterior aspect of the neck, upper back and shoulders at times, extending to the upper arms | |
| V sign | ill-defined erythematous macules involving the anterior aspect of the neck and the upper chest | |
| Poikiloderma | atrophic skin with changes in pigmentation and teleangiectasia in photo-exposed or non-exposed areas | |
| Holster sign | poikilodermia involving the lateral aspects of the thighs | |
| Periungual involvement | teleangiectasis and cuticular overgrowth | |
| Mechanic’s hands | hyperkeratotic, cracked horizontal lines on the palmar and lateral aspects of the fingers | |
| Scalp involvement | diffuse poikilodermia, with scaling and pruritis | |
| Calcinosis cutis | calcium deposits in the skin |
Table 4.
Malignant etiologies of skin cancers.
| The most common causes | The rarer causes |
| Adenocarcinomas | Breast cancer |
|
Hematopoietic neoplasms Lung cancer Pancreatic malignancy |
|
Prostate malignancy Kidney malignancy |
| Laryngeal malignancy | |
| Ovarian and uterine malignancy | |
| Bladder malignancy | |
| Nasopharyngeal carcinoma | |
| Melanoma | |
| Mycosis fungoides | |
| Hepatocellular carcinoma | |
| Squamous cell carcinoma |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



