
Submitted:
03 February 2025
Posted:
05 February 2025
You are already at the latest version
Abstract
Multiple sequence alignment trimming can help improve phylogenetic signal and reduce computational load. ClipKIT trims multiple sequence alignments by retaining phylogenetically informative sites and removing all others. Here, we present a web browser application for ClipKIT, which supports DNA, protein, and codon data types in FASTA format. The web browser application can process one or many multiple sequence alignment files, which users can subsequently download. Users can also view the trimmed multiple sequence alignment using a web-based multiple sequence alignment viewer. ClipKIT is available at https://clipkit.genomelybio.com, is free and open to all users, and there is no login requirement. ClipKIT in the browser aims to broaden the accessibility of web-based tools for phylogenetics research.
Keywords:
Phylogenetics
; Genomics
; Phylogenomics
; Multiple Sequence Alignment
; Filtering
; Phylogenetic Signal
; Parsimony-Informative Site
; Orthology
; Homology
; Molecular Phylogenetics
GRAPHICAL ABSTRACT
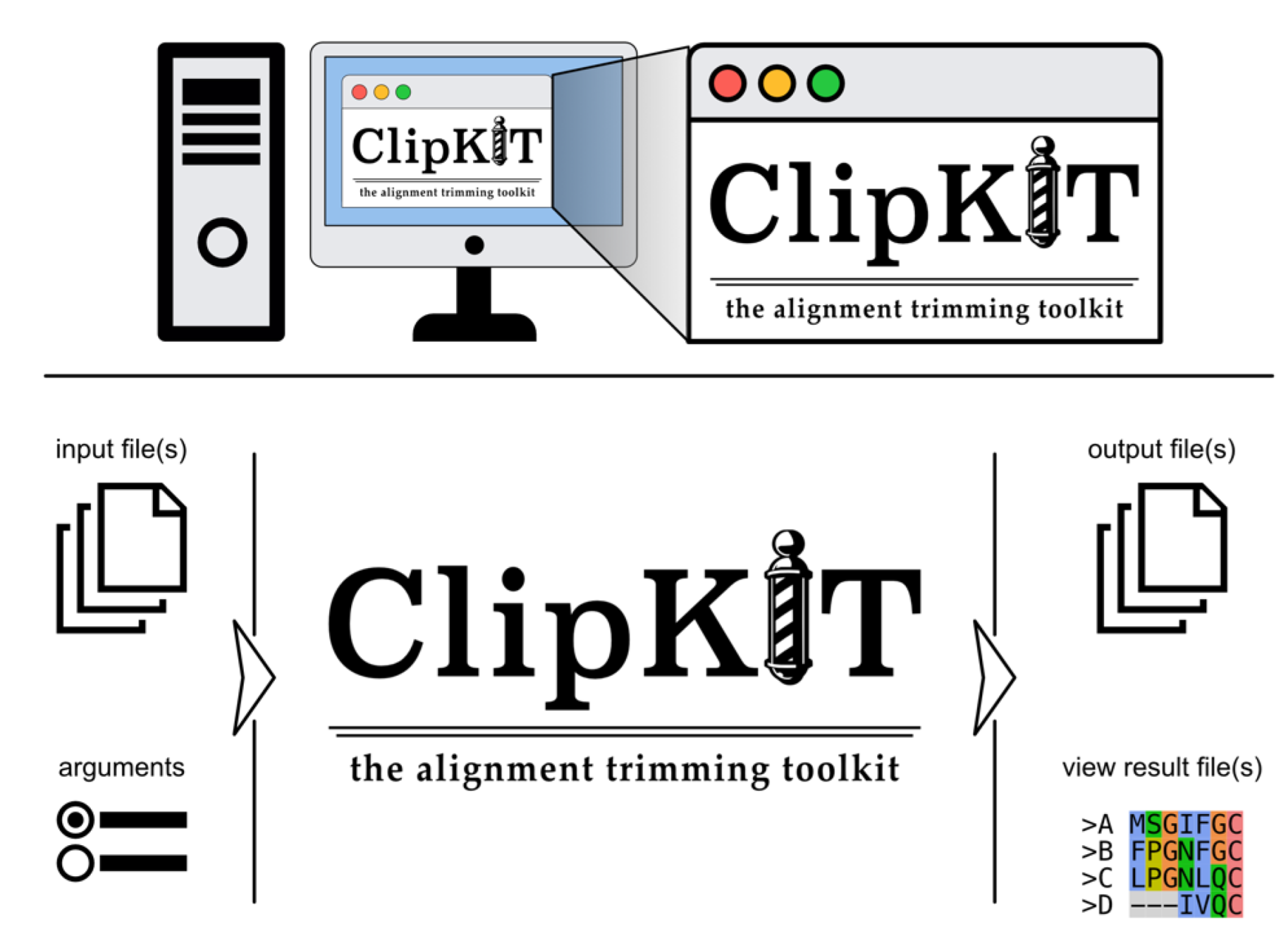
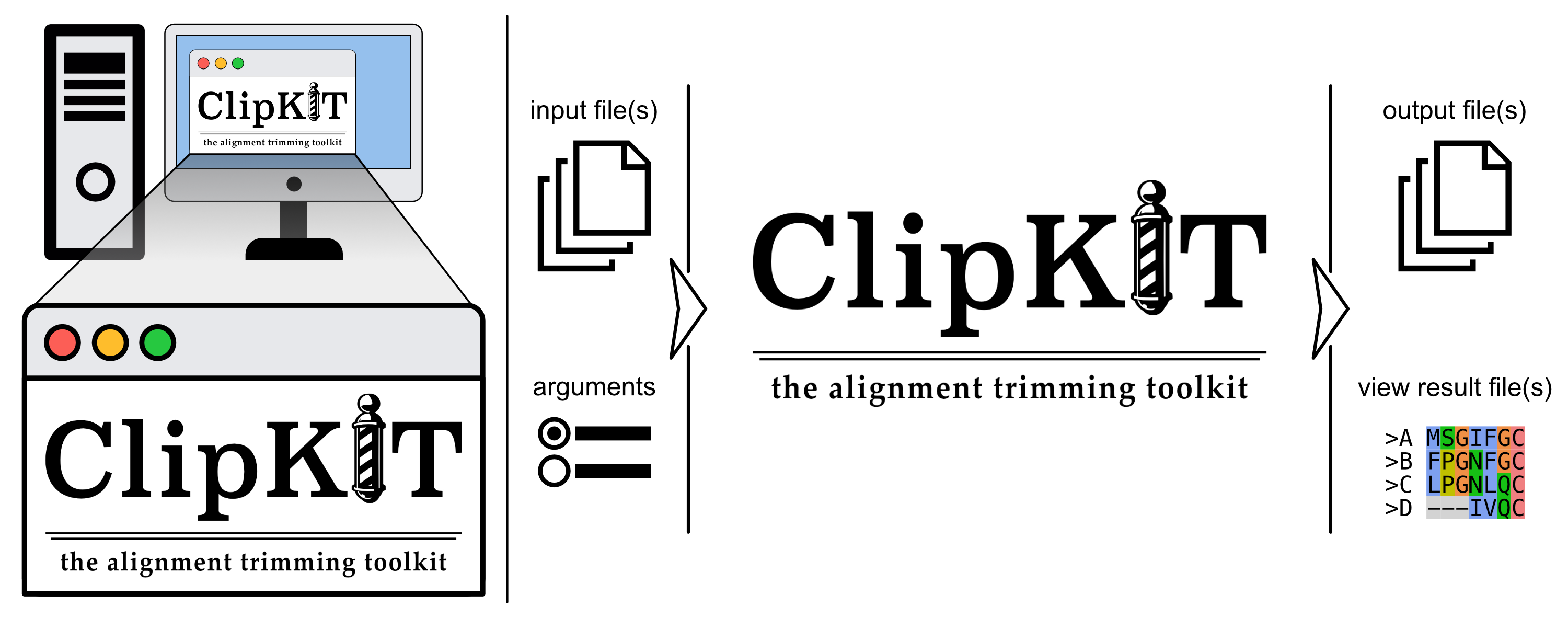
ClipKIT, a multiple sequence alignment trimming toolkit, is available in the browser. FASTA file(s) and arguments are input and processed in the cloud. Output files can be downloaded and viewed.
Introduction
ClipKIT is an efficient software that conducts multiple sequence alignment trimming for phylogenomics [1]. While most algorithms aim to identify and remove highly divergent sites in multiple sequence alignments [2], ClipKIT identifies phylogenetic informative sites and removes all others. Benchmarking revealed that ClipKIT outperformed other multiple sequence alignment trimming tools, such as Gblocks [2], BMGE [3], trimAl [4], and Noisy [5]. ClipKIT is flexible, featuring numerous modes for multiple sequence alignment trimming.
Although ClipKIT has been adopted by numerous researchers, ClipKIT is only available as a command-line tool and is, therefore, difficult to use for non-expert bioinformaticians. Moreover, there is a dearth of tools that enable multiple sequence alignment trimming in the browser [6], underscoring the broad inaccessibility of trimming multiple sequence alignments to non-experts.
Here, we present ClipKIT in the browser, a user-friendly application for multiple sequence alignment trimming using cloud-based resources. Currently, ClipKIT runs using resources from Amazon Web Services (https://aws.amazon.com/). Since first launch, ClipKIT in the browser has processed about 250 files per month.
CLIPKIT WEB-APPLICATION
ClipKIT in the browser works on all web browsers. The web interface provides a ‘Help’ section, which includes exemplary files, a tutorial, and other helpful information for using the toolkit (Figure 1a). Minimally, users upload a multiple sequence alignment file. Then, the input file and default argument specifications are sent and processed by cloud resources, alleviating the user from providing any computational resources. Elements of the web interface are discussed below.
Input Data and Arguments
ClipKIT in the browser takes one or more FASTA files as input (Figure 1b). The user can then specify the trimming mode used, sequence type (default is to auto-detect sequence type), and whether the multiple sequence alignment is a codon-based multiple sequence alignment (Figure 1c). The various ClilpKIT modes are kpic (keep only parsimony informative and constant sites), kpi (keep only parsimony informative sites), gappy (keep sites with few gap characters based on a hard threshold), and smart-gap (dynamic determination of gappyness threshold); combinations of kpic/kpi and gappy/smart-gap (such as kpic-gappy) can also be used. ClipKIT also supports codon-based trimming; if one site in a codon is trimmed, the whole codon will be removed. ClipKIT also has a c3 mode for trimming, eliminating the third codon position from an alignment. Thereafter, users can catalyze file processing (Figure 1d). After doing so, the ‘Trim FASTA(s)’ button will update with a loading spinner and the text will read as ‘Trimming’, indicating to the user that processing is underway.
Results and Output Information
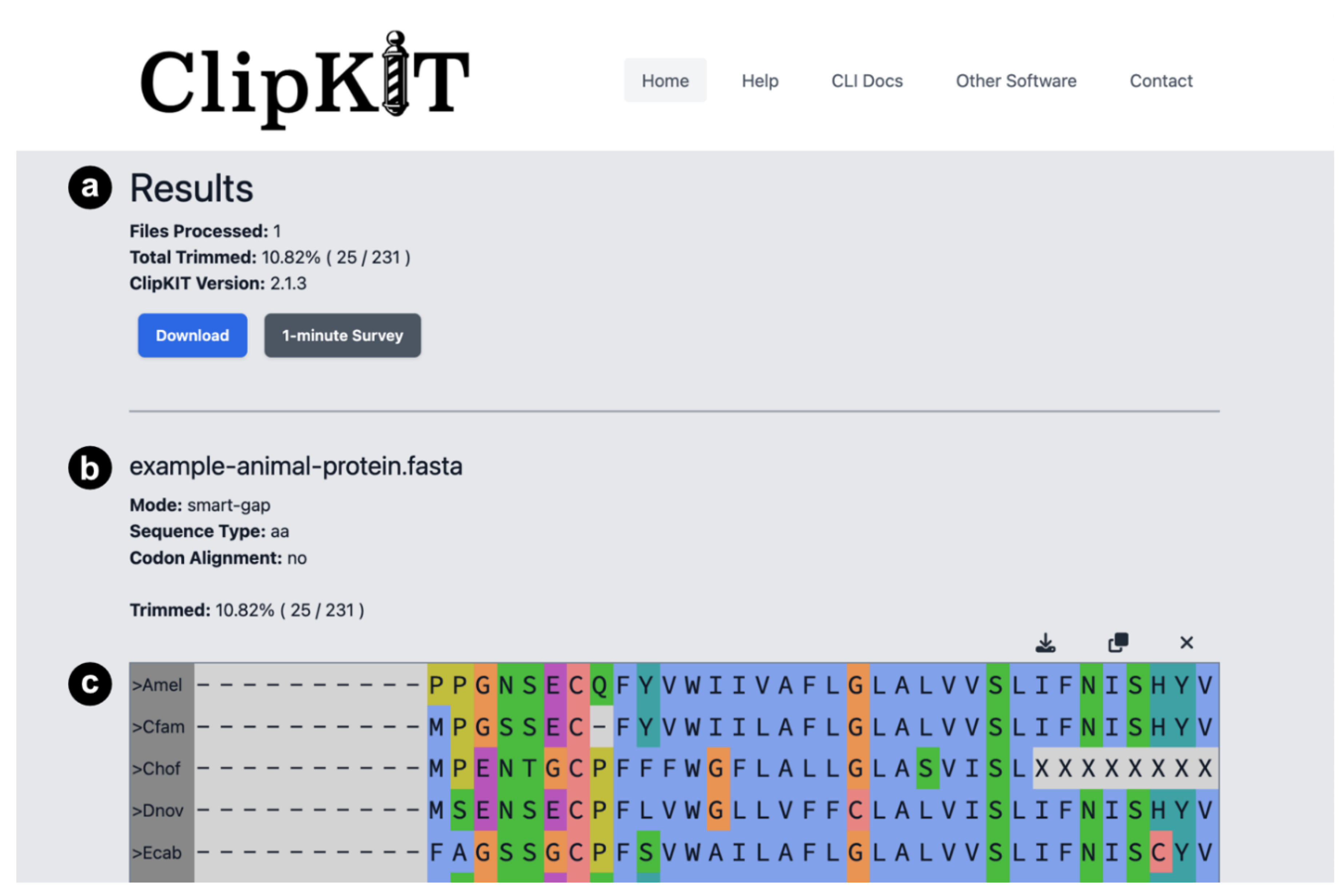
After processing the file(s) using cloud resources, the browser will automatically update with the results and descriptive statistics. Specifically, there are summary statistics about the number of files processed and the total percentage and number of trimmed sites (Figure 2a). Users can also download all results files at this time.
Thereafter, information regarding individually processed files is presented (Figure 2b). This includes what trimming mode was used, the sequence type, if the alignment is a codon, and the percentage of the alignment trimmed, including the number of sites trimmed out of the total number of possible sites. A multiple sequence alignment viewer also displays the trimmed alignment (Figure 2c), enabling users to quickly inspect the resulting output file. Above the multiple sequence alignment viewer to the right side are buttons to download, copy results to the clipboard, or close an individually processed file.
Funding
JLS is a Howard Hughes Medical Institute Awardee of the Life Sciences Research Foundation.
Data Availability Statement
ClipKIT is freely accessible in the browser at https://clipkit.genomelybio.com. The user interface was developed using Vue and JavaScript. The backend was written in Python. Tutorials and documentation are available on the ‘Help’ page https://clipkit.genomelybio.com/#/help.
Acknowledgments
We thank the King lab for the helpful discussion and comments.
Conflicts of Interest
JLS is an advisor to ForensisGroup Inc. JLS is a scientific consultant to FutureHouse Inc. JLS is a Bioinformatics Visiting Scholar at MantleBio Inc.
References
- Steenwyk, J.L.; Buida, T.J.; Li, Y.; Shen, X.-X.; Rokas, A. ClipKIT: A multiple sequence alignment trimming software for accurate phylogenomic inference. PLOS Biol. 2020, 18, e3001007. [Google Scholar] [CrossRef] [PubMed]
- Talavera, G.; Castresana, J. Improvement of Phylogenies after Removing Divergent and Ambiguously Aligned Blocks from Protein Sequence Alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Criscuolo, A.; Gribaldo, S. BMGE (Block Mapping and Gathering with Entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol. Biol. 2010, 10, 210. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Dress, A.W.; Flamm, C.; Fritzsch, G.; Grünewald, S.; Kruspe, M.; Prohaska, S.J.; Stadler, P.F. Noisy: Identification of problematic columns in multiple sequence alignments. Algorithms Mol. Biol. 2008, 3, 7. [Google Scholar] [CrossRef] [PubMed]
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.-F.; Guindon, S.; Lefort, V.; Lescot, M.; et al. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008, 36, W465–W469. [Google Scholar] [CrossRef] [PubMed]
- Steenwyk, J.L.; Buida, T.J.; Labella, A.L.; Li, Y.; Shen, X.-X.; Rokas, A. PhyKIT: a broadly applicable UNIX shell toolkit for processing and analyzing phylogenomic data. Bioinformatics 2021, 37, 2325–2331. [Google Scholar] [CrossRef] [PubMed]
- Steenwyk, J.L.; Martínez-Redondo, G.I.; Buida, T.J.; Gluck-Thaler, E.; Shen, X.; Gabaldón, T.; Rokas, A.; Fernández, R. PhyKIT: A Multitool for Phylogenomics. Curr. Protoc. 2024, 4, e70016. [Google Scholar] [CrossRef] [PubMed]
- Steenwyk, J.L.; Buida, T.J.; Gonçalves, C.; Goltz, D.C.; Morales, G.; Mead, M.E.; LaBella, A.L.; Chavez, C.M.; Schmitz, J.E.; Hadjifrangiskou, M.; et al. BioKIT: a versatile toolkit for processing and analyzing diverse types of sequence data. Genetics 2022, 221, iyac079. [Google Scholar] [CrossRef] [PubMed]
- Steenwyk, J.L.; Buida, T.J.; Rokas, A.; King, N. OrthoHMM: Improved Inference of Ortholog Groups using Hidden Markov Models 2024. [CrossRef]
- Steenwyk, J.L.; Goltz, D.C.; Buida, T.J.; Li, Y.; Shen, X.-X.; Rokas, A. OrthoSNAP: A tree splitting and pruning algorithm for retrieving single-copy orthologs from gene family trees. PLOS Biol. 2022, 20, e3001827. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
ClipKIT in the browser landing page. The landing page directly takes user’s to where files are uploaded. (a) The header bar provides key links, including the ‘Home’ page (depicted here), the ‘Help’ page that contains additional information about using ClipKIT in the browser and exemplary files to test running the software. Other tabs include documentation for the command-line interface (CLI) tool, other software our team has developed — such as PhyKIT [7,8], BioKIT [9], OrthoHMM [10], OrthoSNAP [11], and other algorithms — that may be of interest to users, and, lastly, contact information in case users have feature requests, comments, or questions. (c) Users can specify what trimming mode to use, and sequence type (amino acid, nucleotide, or the default, auto-detect). Users can also specify if the input data is a codon alignment; if so, sequence type is ignored and assumed to be nucleotides. (d) The ‘Trim FASTA(s)’ button catalyzes the file processing using cloud-based computing resources.
Figure 1.
ClipKIT in the browser landing page. The landing page directly takes user’s to where files are uploaded. (a) The header bar provides key links, including the ‘Home’ page (depicted here), the ‘Help’ page that contains additional information about using ClipKIT in the browser and exemplary files to test running the software. Other tabs include documentation for the command-line interface (CLI) tool, other software our team has developed — such as PhyKIT [7,8], BioKIT [9], OrthoHMM [10], OrthoSNAP [11], and other algorithms — that may be of interest to users, and, lastly, contact information in case users have feature requests, comments, or questions. (c) Users can specify what trimming mode to use, and sequence type (amino acid, nucleotide, or the default, auto-detect). Users can also specify if the input data is a codon alignment; if so, sequence type is ignored and assumed to be nucleotides. (d) The ‘Trim FASTA(s)’ button catalyzes the file processing using cloud-based computing resources.
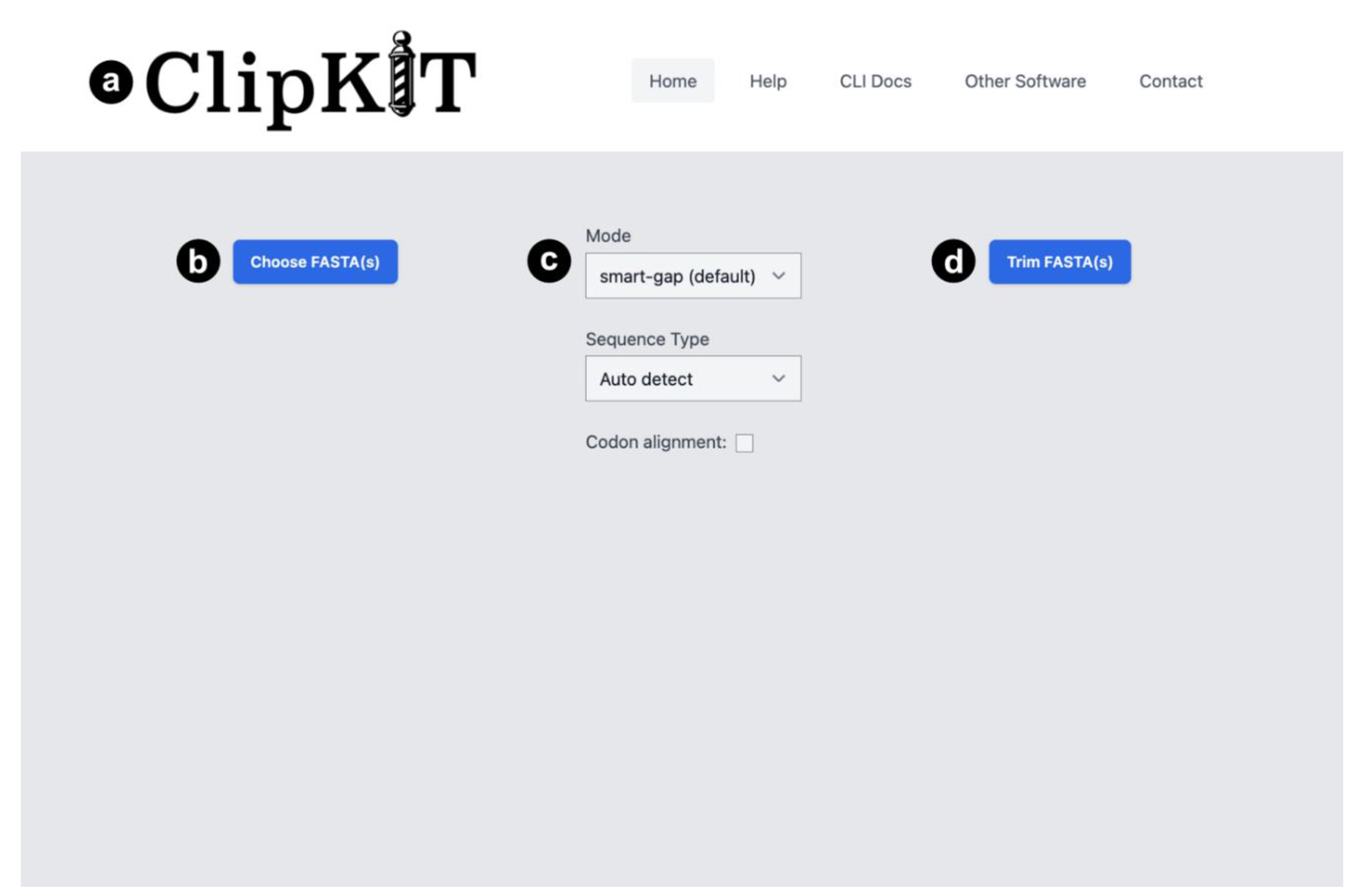
Figure 2.
ClipKIT in the browser results and output. (a) Summary information about all processed files is provided. This includes how many files were processed and the total percentage and number of trimmed sites as well as the total number of sites examined. The version of ClipKIT is also specified. (b) Thereafter, information about individually processed files is provided, including the trimming mode used, the sequence type, whether the file represents a codon alignment, and the percentage of sites trimmed. (c) A multiple sequence alignment viewer enables users to easily examine the resulting trimmed multiple sequence alignment file. Buttons are also included above the viewer to the right for downloading, copying the results to the clipboard, and closing the file information.
Figure 2.
ClipKIT in the browser results and output. (a) Summary information about all processed files is provided. This includes how many files were processed and the total percentage and number of trimmed sites as well as the total number of sites examined. The version of ClipKIT is also specified. (b) Thereafter, information about individually processed files is provided, including the trimming mode used, the sequence type, whether the file represents a codon alignment, and the percentage of sites trimmed. (c) A multiple sequence alignment viewer enables users to easily examine the resulting trimmed multiple sequence alignment file. Buttons are also included above the viewer to the right for downloading, copying the results to the clipboard, and closing the file information.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



