Submitted:
03 February 2025
Posted:
04 February 2025
You are already at the latest version
Abstract
The development of the human immune system starts already during the fetal period in a largely, but probably not completely, sterile environment. During and after birth the immune system is exposed to an increasingly complex microbiota. The first microbiota encountered during passage through the birth canal colonize the infant gut and induce tolerance of the immune system. Transplacental derived maternal IgG as well as IgA from breast milk protect the infant from in-fections during the first 100 days, during which the immune system further matures, and im-munological memory is formed. Weaning and introduction of solid food expose the immune system to novel (food) antigens and allow for other microbiota to colonize. The cells and mole-cules involved in the mutual, intricate interaction between microbiota and developing immune system are now beginning to be recognized. These include bacterial components such as poly-saccharide A from Bacteroides fragilis, as well as bacterial metabolites such as the short chain fatty acid butyrate, indole-3-aldehyde, and indole-3-propionic acid. All these, and probably more, bacterial metabolites have specific immunoregulatory functions which shape the development of the human immune system during the first 1000 days of life.
Keywords:
Gut microbiota
; Neonatal immune system
; Bacterial colonization
; Mode of delivery
; Prenatal delivery
; Short-chain fatty acids (SCFAs)
1. Introduction
Directly after birth, the human newborn enters a world teeming with micro-organisms. In order to protect itself against pathogenic micro-organisms, innate and adaptive immune systems have developed during fetal life. During postnatal life, the intestines, as well as all other body surfaces exposed to the outside world become colonized with micro-organisms. While pathogenic micro-organisms should be recognized and eliminated by the immune system, commensal micro-organisms contribute to human health and well-being and therefore should not be attacked. Philippe Sansonetti has termed this situation as war and peace on mucosal surfaces [1]. In this review we describe the development of the human immune system during the first 1000 days of life, a period during which the immune system further matures and diversifies, and the mutual interactions with the microbiota, especially those residing in the gastrointestinal (GI) tract [2]. The cellular and molecular details of these interactions will be discussed and put in context.
2. Pre- and Postnatal Development of the Immune System
2.1. Prenatal Development of the Immune System
The first steps in the development of the immune system have already been taken during early embryonic development. Thanks to large-scale, single-cell genomics imaging, the concurrent development of the hematopoietic and immune systems has been mapped throughout early prenatal life. In the 1st post conception week (PCW), the primary human yolk sac first appears and is closely associated with the development of the first fetal blood cells (Figure 1) [3]. Up to 3 weeks after fertilization, trophoblast cells arise from the blastocyst, initiating implantation into the uterine wall and early placental development [4]. Around the same time, the main site of hematopoiesis is initiated in the yolk sac around 2 PCW before eventually transitioning to the aorta-gonad-mesonephros (AGM) region. By 4 to 8 PCW, hematopoietic activity gradually shifts to the fetal liver and is concentrated here until 20 PCW, when hematopoiesis shifts once more to predominantly occur in the fetal bone marrow and spleen, producing all types of blood cells, including the cells of the immune system, continuing after birth, throughout adulthood [2,5].
2.1.1. Development of Innate Immune System
At 4 PCW, the yolk sac contains definitive hematopoietic progenitors, macrophages, mast cells, innate lymphoid cells, monocytes as well as megakaryocytes [6,7,8]. Neutrophils, basophils and eosinophils, on the other hand, remain absent until hematopoiesis in the bone marrow is established [6,9]. During the 6th PCW, the embryonic pancreas, brain, and placenta are populated with precursors of antigen presenting cells such as macrophages, microglia, and Hofbauer cells (fetal placental macrophages), respectively. These cells appear before blood cell formation starts in the fetal liver, indicating that macrophages of erythromyeloid progeny from the yolk sac or AGM are able to migrate to populate other tissues [6,10,11,12]. These macrophages are in an activated state and can contribute to immune regulation and phagocytosis halfway through gestation. They begin expressing proinflammatory genes (e.g., NKFB1, FOS, JUN) as early as 8 to 10 PCW, and can exhibit scavenger functions by 12 to 16 PCW [13,14].
While yolk sac-derived macrophages persist in certain tissues, they are gradually phased out and replaced in other locations, such as the lung, heart, and gut, by monocytes derived from hematopoietic stem cells [15,16]. Monocytes account for approximately 3% of cells in the fetal blood, with their levels increasing linearly with gestational age [13]. These cells are initially detected in the fetal liver but later shift their production site to the bone marrow [5,17]. Although a distinct mast cell population is phenotypically developing in the fetal skin around 12 to 14 PCW, these cells express minimal levels of detectable granules, and a low but maturing expression of the IgE receptor on their surfaces [18,19]. This demonstrates a minimal role in allergic responses but a potential contribution to mucosal immunity and angiogenesis.
Innate lymphoid tissue inducers are cells that aid in forming secondary lymphoid organs [2,6,20], specifically embryonic lymph nodes, by interacting with lymphoid tissue organizer cells [6,21], to generate conditions that facilitate innate lymphocytes to develop very early in the human embryo. At 18-23 PCW, mucosal-associated invariant T cells (MAIT) can be found in the fetal small intestine, liver and lung [22].
2.1.2. Development of the Humoral Immune System
At 7 PCW, the earliest signs of the B cell lineage appear, with mature B cells observable by 9 P[6,7]. Although intestinal fetal B cells initially derive from follicular and transitional B cell types, by mid-gestation, the bone marrow becomes the primary source of B cells. By 9 to 12 PCW, lymphoid progenitor cells differentiate, with some maturing in the thymus and others developing into B cells and natural killer (NK) cells. Moreover, maturing B cells have also been confirmed to exist outside blood vessels in the submucosa of the gut and the dura mater of the meninges [13,23,24].
By 16 PCW, B cells begin producing immunoglobulins [25], while NK cells become active in innate immune responses. The adaptive immune system becomes more robust as gestation progresses, with B cells increasing immunoglobulin production to strengthen humoral immunity. While fetal B cells attain a progressive increase of B cell receptor diversity from early stages [6,21,25], the formation of germinal centers and hence the specialization of B cells are limited until antigen exposure after birth [6,7]. NK cells play a role in the innate immune response and immune surveillance when mature. A high concentration of fetal NK cells is present in the liver, lungs, and spleen, whereas lower levels are found in the fetal bone marrow and mesenteric lymph nodes [26]. Despite fetal NK cells from the liver and spleen being functionally immature and less active, compared to adult NK cells, they exhibit signs of cytotoxic activity. They are abundant within the infant intestines and show higher degranulation levels than their adult NK cell counterparts [26,27,28]. In the final stages of fetal development, the immune system undergoes fine-tuning and maturation to prepare for exposure to the extrauterine and external environments.
2.1.3. Development of the Cellular Immune System
At 8 weeks, early lymphoid progenitor cells migrate from the fetal liver to the thymus, where they are able to develop, giving rise to naïve T cells (Figure 1) [29]. Prenatally, innate lymphoid cells exist in subtypes. These include cells expressing EOMES and TBX21 (type 1), cells expressing RORC and CCR6 (type 3) (similar to T helper 17 cells and NKT-like cells), and PDCD1-expressing CD8aa cells (similar to CD8aa+ T cells). Postnatally, the levels of PDCD1-expressing CD8aa cells have been shown to increase, reaching their peak during childhood [13,30]. Distinct cell types in the thymic microenvironment such as the stromal cells: thymic epithelial cells, mesenchymal cells, endothelial cells and other non T lineage immune cells interact extensively, facilitating the continuous progression from early thymic progenitors to various mature T cell types, promoting T lymphopoiesis [31,32,33]. Following this interaction, by 12 to 14 PCW, circulating T cells can be observed in the periphery, indicating that the thymus has now developed the ability to produce functional, responsive T cells [34,35]. Studies on neonatal adaptive immunity suggests that the early-life T cell populations are primed to generate tolerogenic responses, characterized by effector T cells with innate-like functions and an increase in peripherally induced regulatory T cells (pTregs) [36,37,38,39,40].
Although dendritic cell (DC) production is initiated and first observed in the fetal liver around 7 PCW, the potential production of plasmacytoid DCs from both myeloid and lymphoid origins in the yolk sac have been discovered [7]. By 13 PCW, DC subsets can also be found in the fetal spleen, skin, thymus, and lung [41]. As pregnancy progresses between 17 and 20 PCW, splenic DCs mature and gradually take on roles as antigen presenting cells for T cells [41,42]. Fetal DCs contribute to immune regulation. They induce the differentiation of regulatory T cells (T-regs) and promote T cell IL-4 production, supporting a Th2 response. They also inhibit T cell TNF-α by increasing arginase 2 expression in conventional DCs within the fetal spleen to further aid in the suppression of inflammation [41].
2.1.4. Development of Immunological Memory
Analysis of umbilical cord shows that T cells and B cells in the peripheral blood of the human newborn are all virtually in the naïve state (Figure 2). This makes sense because the intrauterine environment is virtually devoid of antigenic stimuli (see also below). Yet, T cells with a memory phenotype have been identified in the fetal intestine, implying the possibility of antigen recognition [6,43,44,45,46]. In studying CD4+ T cells in the intestines, memory T cells and T-regs showed signs of proliferation and activation [6,45,46]. During the prenatal stage, ingestion of amniotic fluid in the womb could potentially present microbial antigens (see below), which could lead to the activation of the mucosal immune system of the gut.
2.2. Postnatal Development of the Immune System
Directly after birth the infant immune system is exposed to an overwhelming number of antigenic stimuli, both of microbial nature and otherwise. While their immune system is perfectly capable of responding to these stimuli (with one exception, discussed below), every immune response is a primary response, taking up to 10 days to reach its peak and plateau. Neonates, in the first period after birth are protected by transplacental-obtained IgG from mother [47]. Because of the 21 days half-life of IgG, by 100 days of age, most of the maternal IgG has gone, and from that moment onwards the infant depends on the functionality of its own immune system [48].
During the first 1-2 years of life, children experience many respiratory and GI infections, commonly referred to as childhood diseases. In the Dutch language, the term “childhood diseases” is also used in a non-medical context, namely for inevitable start-up problems, which are overcome by themselves. In English, teething troubles is the expression used for these events. The frequency of childhood diseases decreases over time, which reflects the building up of immunological memory, conferring clinical protection against pathogens that have been encountered before.
The exceptions to neonatal immune capabilities/capacities are infections with encapsulated bacteria, such as Streptococcus pneumoniae and Salmonella typhi. The B-lymphocyte responses to the capsular polysaccharides of these bacteria are absent at birth and cannot be demonstrated until 1.5-2 years of age. Bacterial polysaccharides are so-called T-cell independent antigens, and in order to achieve full B-lymphocyte activation, co-stimulation via complement receptor type 2 (CD21) is necessary [49,50]. B-lymphocytes with high expression of CD21 reside in the marginal zone of the spleen (Figure 3, panel b) and it is at this site where polysaccharide antigens are encountered, and a response initiated. In infants and young children, marginal zone B-lymphocytes lack CD21 expression and thus are unable to respond to polysaccharides [51]. Conjugation of bacterial polysaccharides with carrier proteins (so called conjugate vaccines) changes the nature of the polysaccharide antigens and overcomes neonatal unresponsiveness. Conjugate vaccines against S. pneumoniae, Haemophilus influenza type b, and Neisseria meningitidis have been successfully implemented in childhood vaccination programs [52].
The intimate interaction between gut microbiota and the developing immune system is also reflected in the occurrence of anti-A and anti-B blood group antibodies. A person with blood group A has anti-B antibodies, blood group B has anti-A antibodies. Anti-blood group antibodies generally are not detectable before one year of age. Although, from the work of Karl Landsteiner, this phenomenon is already known for almost a century [53], the underlying mechanism still has not been proven in detail. The current view is that gut microbiota carrying oligosaccharides with strong antigenic similarity with blood group antigens can induce an antibody response. While microbiota with blood group A or B will be present in every individual, the immune system is tolerant for self-antigens and thus will not generate an immune response to their own blood group antigen. Indeed, persons with the blood group AB do not generate blood group antibodies [54]. The microbiota which carry the oligosaccharides similar to human blood group antigens have not been specified yet. Apparently they are not highly pathogenic, because the infection frequency of persons with blood group AB is not different from others. It should be noted that blood group A and AB persons may have been more susceptible to smallpox [55].
During the first 500 days of life, T- and B cell numbers gradually increase and obtain a mature phenotype [56,57]. Afterwards, gradual changes take place until adulthood. After the physiological dip at 100 days of age, serum IgG levels increase and reach adult levels at 500-1000 days. IgM and IgA levels increase at a slower rate and reach adult levels later [58,59].
3. Development of the Infant Gut Microbiome
3.1. Maternal Microbiota
As discussed above, the development of the immune system takes place within the sterile environment of the womb. This implies that the immune system of the newborn has not yet been in contact with microbial or viral antigens from the external environment. Thus, the immune system of the newborn can be considered to be immunologically naïve. This does not necessarily mean that the developing immune system has not been in contact with microbial antigens, especially as derived from the maternal microbiota.
3.1.1. The Role of Maternal Microbiota During Fetal Development
The earliest interaction between the immune system of the developing embryo and external microbiota already takes place with maternal microbial antigens. The maternal microbiome has been shown to be vital in producing essential vitamins, amino acids, and other metabolites needed to facilitate the growth and development of the fetal immune system [60]. This finding is supported by the discovery that the maternal microbiome undergoes a dramatic change during pregnancy, establishing a particularly stable bacterial population in the vaginal tract (characterized by a surge in L. crispatus, L. gasseri, L. jensenii, Limosilactobacillus vaginalis populations), and an embryo-nurturing population in the maternal GI tract [61,62]. Additionally, short-chain fatty acids (SCFAs), vital bacterial products in healthy development are synthesized via fermentation in the maternal gut (from a surge in SCFA-producing bacteria) and play a central role in the establishment of a healthy immune system and metabolic profile within the developing embryo [62,63,64]. This change indicates that bacterial abundance promotes the healthy growth of the developing embryo through metabolite production. Although it is outside the scope of this review, it is interesting to note that maternal microbiota play a role in the fetal neurodevelopment, and that Th17 cells are involved in this interaction [65,66,67].The resident microbiota of the maternal gut and vaginal tract may also play a role in placental colonization. This will be explored further in subsequent sections. For now, Aagaard et al. characterized a unique placental microbiome composed of symbiotic species which were compositionally similar to the microbiome of the oral cavity and the upper GI tract [68].
In summary, the unique composition of the maternal microbiome could play a vital role in the healthy development of the fetus through providing a stable input of essential metabolites and preventing the colonization with potentially Th17-mediated inflammation-inducing bacteria. The stability of the maternal vaginal microbiome also plays a role in preventing colonization with pathogenic species which may play a role in adverse pregnancy outcomes (APOs).
3.1.2. The Composition of Maternal Microbiota Can Be Changed Through External Factors
Despite the observed stability of the maternal microbiome, alterations in diet, mental state, and medication can result in microbiota alterations, which could have an effect on the developing fetus. The primary modulator of the maternal gut microbiome (and thus the healthy development of the fetus) is maternal diet which may be altered during pregnancy. Dramatic changes in maternal diet have been shown to shift the composition of the maternal gut microbiome, with lower levels of Bifidobacteria and Bacteroides subspecies, and higher levels of Firmicutes subspecies being associated with a diet rich in saturated fatty acids and carbohydrates. The secondary modulator of maternal microbiota is maternal stress. A study by Jašerevič et al. demonstrated that maternal stress during prenatal development modulated the dynamics and composition of the gut and vaginal microbiome. The observed change included a shift toward facultative anaerobe species in the gut (Mucispirillum and Desulfovibrionaceae) and a disruption in the dynamics of vaginal microbiota diversity. This change in maternal microbiota then reflected the composition of the neonatal microbiome vertical transmission and may have altered fetal development through the formation of inflammation-associated metabolites such as hydrogen sulfide [69]. Finally, the administration of antibiotic treatment during pregnancy has been shown to disrupt the maternal gut microbiome and affect fetal development [70]. This provides further evidence for the link between the maternal microbiome and prenatal development, as antibiotic use has been associated with multiple APOs [70]. An additional link between the maternal microbiome and the developing fetus can be seen in the case of infection-based APOs. Intrauterine infections are the most common causes of preterm delivery, accounting for 20% of preterm births and resulting in 450 preventable neonatal deaths per hour worldwide [71,72]. This showcases the strong relation between the composition and dynamics of the maternal microbiome and prenatal development.
3.2. Prenatal Bacterial Colonization
There is clear evidence that the maternal gut microbiome influences the development of the fetus; however, there is an ongoing debate on the extent to which the maternal microbiome would lead to bacterial colonization of the fetus before birth. This debate is between the sterile womb hypothesis, whose supporters pose that primary bacterial colonization takes place during and after birth, and the in-utero hypothesis which proposes that the fetus is exposed to microbiota prenatally through the amniotic fluid, placenta, and umbilical cord. The evidence for both hypotheses is based on bacterial culturing and 16S ribosomal sequencing of both the meconium and the composition of the womb.
3.2.1. The Sterile Womb Hypothesis
The sterile womb hypothesis proposes a view of the womb which is devoid of possible bacterial colonization [73]. This older view became prevalent because of the failure to culture bacterial colonies from the infant meconium [74]. The argument has been strengthened by the outcome of molecular techniques, such as 16S rRNA sequencing, indicating that the meconium does not have a distinct microbiome [75]. Kennedy et al. investigated the fetal meconium through rectal swabs during elective caesarean section before the application of antibiotics and labor induction. These samples were then compared to fecal samples of infant’s hours, and days after birth through 16S rRNA sequencing. The results of this study indicate that no microbial signal was detected in most of the fetal meconium samples. The samples with a detectable biomass were most likely skin contaminants as their profile consisted mostly of Staphylococcus epidermidis [76]. Further evidence for the sterile womb hypothesis comes from taxonomic analysis of gut microbiota in relation to different delivery methods. The microbial composition of neonates delivered vaginally reflect the composition of the maternal vaginal microbiome, with a more homogeneous multi-microbial composition, while neonates delivered through caesarean section were inoculated with the maternal skin microbiome which is less homogeneous and dominated by particular taxa including B. licheniformis [77]. Despite the evidence proposed by this hypothesis, studies into the biomass of the placenta, amniotic fluid, and fetus have provided interesting insights into a possible alternative.
3.2.2. The Sterility of the Placenta, Amniotic Fluid, and Fetus
The primary evidence for a non-sterile womb is the finding of a substantial microbiome in the placenta. Under sterile conditions, Aagaard et al. examined the microbiological composition of extracted placental specimens from 320 subjects. These specimens were then compared to the compositions of other human body site niches including the oral cavity, skin, nasal cavity, and gut from non-pregnant controls. The results of this ribosomal analysis showed that the placenta harbors a unique microbiome composed of multiple phyla including Firmicutes, Tenericutes, Proteobacteria, Bacteroidetes, and Fusobacteria. Additionally, the comparative analysis demonstrated that the microbiome of the placenta is most akin to the oral microbiome of non-pregnant controls in composition. Finally, this study showed that the microbiome in question was associated with a history of antenatal infections [68]. Additional evidence for the colonization of the womb comes from 16s rRNA sequencing of amniotic fluid samples which have been found to house a low biomass characterized by low richness and diversity, mostly consisting of Proteobacteria [78]. Additionally, past studies have shown that the volume of the amniotic fluid is not constant throughout pregnancy and may be altered by fetal ingestion [79]. When taken together, these ideas may contribute to the conclusion that fetal colonization may occur through amniotic ingestion and placental exposure. The sterility of the fetus is particularly disputed and will be explored below.
3.2.3. The In-Utero Hypothesis
The in-utero hypothesis is the antithesis to the sterile womb hypothesis and poses the idea that fetal colonization takes place before inoculation with vaginal or dermal microbiota during birth. This hypothesis responds to the methodological shortcomings of the sterile womb hypothesis. A particular argument was made that negative culture data do not rule out low-level colonization.
16s rRNA sequencing of the umbilical cord blood of healthy infants born by elective caesarean section has shown DNA from commensal bacteria of the Enterococcus, Streptococcus, Staphylococcus, or Propionibacterium genus [80]. This suggests a possible mother-to-child axis of microbial colonization. In their research on rodent models, Jiménez et al. isolated a strain of E. faecium and labelled it before maternal oral inoculation. The presence of this labelled strain could be detected in the meconium of the neonatal mice obtained by caesarean section one day before scheduled delivery [81].
Further analysis of the meconium of neonates with DGGE and HITChip methods provided additional evidence of bacterial presence. Specifically, the meconium was found to contain Bacilli and other Firmicutes bacterial species while fecal samples of the same infants days later were dominated by Proteobacteria. Additional culturing techniques showed that the meconium may be colonized with Staphylococcus strains while fecal samples of older infants contained Enterococcus, Escherichia coli, Escherichia fergusonii, Klebsiella pneumoniae and Serratia marcescens [82].
Despite these findings of possible bacterial colonization in-utero, methodological issues remain. Environmental contamination of samples is a great risk. A study by Dos Santos et al. demonstrated the ease of false positivity in the analysis of meconium samples which were not above background signals. Additionally, the bacterial contamination that was confirmed was particularly uncommon and was consistent with post-natal skin colonization [75]. Finally, due to the low biomass of possible microbiota, 16s rRNA sequencing is the prevalent technique for bacterial recognition. However, this sequencing technique is prone to false positives at the hand of dead bacteria whose contents may be recognized. The role of the placenta is particularly debatable in this regard as the presence of bacterial biomass may not be a marker of fetal colonization, but instead of placental function in waste management [74]. These criticisms are exemplified in a study by Hansen et al. where the genetic sequencing was able to recognize bacterial colonization in two thirds of meconium samples analyzed, at levels too low to be reliably confirmed by PCR [83]. Therefore, although there may be evidence for an in-utero origin to bacterial colonization, the methodology behind these findings may be limited.
3.3. Bacterial Colonization of Premature Children
Despite the inconsistencies in evidence surrounding fetal exposure to microbiota in utero, the maternal and fetal exposure to pathogenic bacteria has been a persistent and observed cause of APOs. Globally, severe infection and preterm birth account for a combined 54% of neonatal deaths [71]. Both factors are associated with pathogenic bacterial exposure and are exceedingly prevalent in low-income countries [71]. In the case of neonatal death, exposure to bacterial biomass or metabolites becomes a critical step in pathogenesis, however; neonates born prematurely because of non-pathogen related causes are also distinct in their microbial colonization.
3.3.1. The Development of the Gut Microbiome Is Specific to Gestational Age
Gestational age (GA) is a determining factor in the colonization and development of the neonatal gut microbiome. Preterm infants have been shown to contain a functionally different gut microbiome compared to full-term infants, with preferential colonization by facultative anaerobe species [84,85]. Studies into differences of initial microbial composition of preterm infants compared to full-term infants showed predominant Proteobacteria colonization, as well as high levels of Staphylococcus and Haemophilus which decreased after delivery in preterm infants [86]. Additional analysis of very preterm infants delivered at GA ≤ 30 weeks showed drastic differences in bacterial strains, with Staphylococcus species composing the majority of fecal biomass. Additionally, very few infants exhibited colonization by Bifidobacterium, Bacteroides, and Atopobium species at birth and during the early periods of development [87]. Thus, preterm infants also differ from full-term infants in their exposure to bacterial metabolites. Concentrations of SCFAs are particularly low in preterm infants due to differences in bacterial colonization, possibly shaping the development of the neonatal immune system [88].
Due to the instability of the preterm gut microbiome, administration of antibiotics shortly after birth can be particularly damaging. The deleterious effect of broad-spectrum pre-partum antibiotics in premature infants has been modelled by observing increased colonization by Enterobacteriaceae species after birth. It is important to note that preterm infants are often subject to pathogenic colonization (group B Streptococci) as a result of possible bacterial infection leading to APOs. In these cases, antibiotics are a standard part of treatment. However, these results raise doubts on the need of broad-spectrum antibiotic use in patients with no observable need for antibiotic treatment as it may threaten the colonization process and subsequently alter infant development [89,90].
Finally, despite these differences in bacterial colonization of full-term infants, preterm infants have been shown to be able to catch up to full-term infants through nutritional and probiotic interventions [91]. Optimization of infant nutrition allows for a catch-up period projected to allow preterm infants to achieve the growth and development parameters of full-term infants [92,93].
3.4. Neonatal Bacterial Colonization
Full-term infants have a specific path towards bacterial colonization distinct from that of preterm infants which may be influenced by in utero colonization. The main determining factor in infant gut microbiota composition is the method of delivery.
3.4.1. Delivery Method
Using 16s rRNA sequencing of the neonatal skin, oral mucosa, nasopharyngeal aspirate and meconium Dominguez-Bello et al. were able to compare the microbiome of the neonate with the dermal, oral, and vaginal microbiome of the mother in relation to delivery method. The results of this study indicate that the method of delivery determines the contents of vertical transmission with neonates delivered vaginally taking on vaginal microbiota dominated by Lactobacillus, Prevotella, or Sneathia species and neonates delivered via C-section resembling the microbiome of the maternal dermis, dominated by Staphylococcus, Corynebacterium, and Propionibacterium species [94,95]. Put together, these observations support a variation of the sterile womb hypothesis: the bacterial baptism hypothesis. The assumption of this hypothesis is that bacterial colonization occurs during birth as a result of exposure to the bacterial composition of the vaginal canal or the dermis. Additional observation that supports this hypothesis is the finding that infants delivered through C-section are more prone to pathogenic bacterial infection then infants delivered vaginally as the baptism in pathogenic dermal bacteria leads to increased concentrations of Klebsiella, Enterococcus, and Clostridium species [96].
Despite the popularity of this hypothesis, researchers disagree about the importance of environmental factors when considering infant primary inoculation with bacteria. A twin study by Palmer et al. observed compositional differences of microbial profiles between dizygotic twins over the first six months of life, before stabilizing into an adult-like profile by the end of the first year of life [97]. These results suggest an environmentally dependent microbial composition after birth. This does not necessary counter the bacterial baptism hypothesis but calls into question the importance of initial inoculation.
Further complications arise when considering the importance of vertical transmission in caesarean delivery. Studies into the causal factor behind the discrepancy in bacterial colonization seen in infants born by C-section disagree. The main culprit may be considered the procedure itself which seems to compromise the natural vertical transmission of bacterial mass through vaginal colonization and potential fecal exposure [98]. However, some research proposes that the procedure plays a less active role in colonization. Instead, a shift to assessing the effect of intrapartum antibiotic administration, absence of labor, differences in breastfeeding behaviors, maternal obesity, and gestational age has provided evidence that a lack of vaginal exposure may not be the main reason for a different microbial phenotype [99]. Antibiotic use has been particularly studied in this regard as it has been linked to long term disruptions of the natural microbiome, leading to developmental disruptions [98]. The results of these studies indicate a need for reevaluating the overall use of antibiotics in full-term deliveries when antibiotic use is not needed. Paired with the importance of environmental factors, these results propose a nurture-based view of microbial colonization, affected by drug exposure, nutrition, and post-natal care to establish the basis of an adult-like microbial phenotype.
3.4.2. Long-Term Development of Gut Microbiota
The maturation of the infant gut microbiome occurs during the first years of life and is impacted by several environmental factors in reaching an adult-like composition. In their longitudinal study on the development of the gut microbiome in the first 2 years of life, Wernroth et al. conclude three broad findings about the change. Firstly, the infant gut microbiome is low in diversity and compositionally different between individuals. Secondly, through exposure to environmental factors such as maternal breastfeeding and nutrition, the perinatal factors of colonization, such as mode of delivery, can be overcome in preference for a mature microbiome. Finally, neonatal oral and gut microbiota share similarities in the early months after birth but become distinct by the two-year mark [100].Additional findings highlighting the importance of environmental factors come from a study by Barker-Tejeda et al. whose comparative analysis between the microbial composition of the infant gut and the microbiome of three generations of their ancestors showed that the microbiome of the infant is low in diversity and metabolite concentration, suggesting a strong environmental pressure for the development of a mature microbiome [101].
The progression of microbial stabilization within the infant gut takes place over the early years of life. The neonatal microbiome is particularly rich in Proteobacteria species whose population gradually dwindles over the first year of life, reaching a concentration of < 5% of overall gut biomass around the two-year mark, reflecting the composition of the adult gut microbiome [100]. The dominant anaerobic bacteria are discussed further below.
3.4.2.1. Bifidobacterium
Bifidobacteria belonging to the phylum Actinomycetes, are gram positive anaerobic bacilli. Their concentrations within the infant gut oscillate during the first months of life, before finally reaching their stable concentrations at around 2 years of age [100]. Early peaks in Bifidobacterial concentrations can be explained by their central role in the processing and metabolism of prebiotic human milk oligosaccharides (HMOs) which directs the development of the infant gut microbiome during breastfeeding [102]. Additional evidence for their importance in healthy gut development comes from Bifidobacterium supplementation in preterm infants, leading to a stabilization in bacterial composition [103].
3.4.2.2. Lactobacillacea
Lactobacilli, belonging to the phylum Firmicutes, experience a universally significant increase in concentrations during early development before acquiring stable concentrations at 24 to 30 months [100]. As mentioned previously, Lactobacilli are a dominant species of the vaginal microbiome and are vertically transmitted during vaginal delivery onto the neonate. This is notable as infants delivered through caesarean section have a distinct lack of Lactobacilli colonization. An additional source of Lactobacillacea colonization is maternal milk during breastfeeding, which accounts for the continual increase in concentration from birth [104].
3.4.2.3. Clostridium
Clostridia also belong to the phylum Firmicutes, however; unlike Lactobacilli, they are mostly pathogenic. Despite this, the infant gut is particularly prone to colonization by this genus and presenting asymptomatically. The exact colonization pattern and importance of the genus is not fully understood; however, one study indicates that the colonization prevalence of this genus in infants can be as high as 27.2%, and even higher in infants < 6 months of age [105]. The function of this colonization is not agreed upon but may be linked to pathogenic resistance as the administration of Clostridium increases the colonization resistance of the infant gut and promotes homeostasis [106].
3.4.2.4. Bacteroides
As opposed to other phyla, Bacteroides experience a low change in diversity during infant development and seem rather to be implanted at a stable concentration during delivery [100]. Method of delivery is particularly important in this case, as infants delivered vaginally have an overall higher concentration in Bacteroide species after birth [107]. The function of this genus in the infant gut includes fermentation of HMOs and synthetizing SCFAs for the promotion of healthy maturation. Additionally, some Bacteroide species, such as B. fragilis, have been found to be pathogenic and may play a role in the onset of allergies as an increase in their concentration is seen in allergic infants [108].
3.4.3. First 1000 Days of Life and Nutritional Weaning
During the early months of infant life, the primary source of nutrition is human breastmilk which provides an opportunity for further colonization and exposure to prebiotic agents such as HMOs. Overall, the infant gut experiences a selective pressure which decreases the concentrations of facultative anaerobes present shortly after birth (Enterococcus, Staphylococcus, Streptococcus, Enterobacteriaceae) as the levels of free oxygen in the gut decrease. In the absence of oxygen, anaerobic species such as Bifidobacteria, Lactobacilli, and Bacteroides take hold [109].
Additional selection pressure comes from breast milk, containing secretory IgA as well as prebiotics such as HMOs. HMOs can be fermented by specialized bacterial strains, allowing for the diversity of Bifidobacteria, Lactobacilli and Clostridium species to increase. The role of HMOs is particularly important in this regard as their fermentation requires a particular enzymatic profile capable of degrading HMOs into monosaccharides. The prevalence of these oligosaccharides in the nutrition of the infant promotes the growth of species capable of their fermentation and the addition of further metabolic pathways for the benefit of the host. Additionally, the increased concentrations of these bacteria in the gut provides a protective mechanism against colonization with potential pathogenic bacteria at this vital point in infant development [110].
Usually around 6 to 12 months after birth, the diet of the child shifts from exclusive breast milk to a solid diet, with dramatic changes in the infant gut microbiota as a consequence. During this transition (weaning), the overall concentrations of HMO related bacteria such as Bifidobacterium, Lactobacillus, and Enterobacter decreases while Bacteroides, Akkermansia, and Ruminococcaceae species expand as their enzymatic profiles capable of processing broader classes of macromolecules become beneficial and environmentally dictated [109].
At this point in infant development, the particular nutritional choices of the parent(s) become the dominant factor in directing microbial composition. A study on the development of the natural metabolic profile of infants has found that the composition of the first complimentary food may be influential, as first nutrition lacking in iron has been associated with inflammation and disruptions to the microbial profile of the infant [111].
The weaning from breast milk and introduction of solid food thus has dramatic consequences for the composition of the gut microbiota. During weaning, the overall concentrations of HMO related bacteria such as Bifidobacterium, Lactobacillus, and Enterobacter decreases while Bacteroides, Akkermansia, and Ruminococcaceae species increase. In mouse models it has been shown that weaning causes a temporary peak in TNF-α and γ-IFN. A delay in the introduction of solid food of less than 1 week (day 21 to day 27 post-partum) abrogates this spike in TNF-α and γ-IFN. The relevance of this weaning reaction only becomes evident in adult life of the mouse: the delayed weaned mice are more susceptible to immune mediated diseases such as colitis [112]. Comparable data for humans are unknown, so the optimal timepoint of weaning from breastfeeding and introduction of solid food, as well as potential changes in microbiota composition and functionality will have to be determined. A multicenter study into the impact of introduction of solid food and weaning on the colonization pattern of the infant gut is ongoing [113].
4. Cellular and Molecular Interactions Between Microbiota and the Immune System
As indicated and discussed above, the gut is the major interface between the microbiota and the developing immune system. It is estimated that 70-80% of immune cells reside in and around the gut, where they maintain a fine balance between anti- and pro-inflammatory responses [114]. The maintenance of intestinal homeostasis is highly dependent on the regulation of the immune system. As indicated above, microbe-host interactions during the neonatal period determine the immune mechanisms that maintain homeostasis and tolerance [115,116,117]. The GI tract and the mucosal immune system are highly interconnected, and it is important to understand the cellular and molecular interactions involved.
4.1. Gastrointestinal Microbe Activity
The GI tract is composed of a mucosal layer, exposed to the lumen, a single layer of epithelial cells and the lamina propria [118]. The mucous layer represents a physical barrier between the intestinal epithelial and the microbiota which reside in the lumen [115,119]. Below the mucosal layer is the epithelial layer composed of tightly packed epithelial cells whose major function is the absorption of nutrients such as sugars, vitamins, and amino acids [120]. Interspersed between the epithelial cells are immune cells such as T-cells [115,118]. Below the epithelium is the lamina propria, a layer of connective tissue which supports the epithelium above and is rich in immune and other cells [115,118]. Finally, located within the lamina propria are Peyer’s patches; areas of lymphoid tissues which help monitor for, recognize, and process antigens [115,121].
Gut microbiota metabolizes proteins and carbohydrates thereby producing metabolites, such as vitamins, signaling molecules, and SCFAs, most notably butyrate, propionate, and acetate [116,122,123,124,125]. SCFAs act on many different cell types, through diverse mechanisms, to regulate important processes. They help with cell growth regulation, injury recovery, maintaining barrier integrity, and initiating, enhancing, limiting, and terminating immune responses [116,125,126]. They have also been shown to be involved in shaping certain structures of the immune system, such as Peyer’s patches, regulating amounts of immune cell types, and influencing the development of immune cells [114,127,128,129]. Commensal microbiota also adopt many mechanisms to prevent infection and the resulting spread and damage of pathogens. For instance, in a process termed ‘colonization resistance’ commensal microbiota make the microbiome inhospitable to pathogens by altering pH and competing for nutrients, adhesion sites, and receptors [114,130]. They also secrete certain antimicrobial peptides (AMPs) such as α-defensins, RegIII, and lysozymes [115,131]. Additionally, bacteria self-regulate via quorum sensing, which is the ability to use secreted signaling molecules to monitor their environment and change gene expression in response to received signals, causing changes in motility, adherence, and population density [119,121,132]. Commensal bacteria use quorum sensing to maintain homeostasis, while pathogens can use it to limit immune response among other activities. The interaction of microbiota, both commensal and pathogenic, with the GI tract results in continuous immune signaling, which is important for proper immune stimulation and development.
The neonatal immune system is immature and both innate and adaptive immune responses are not fully functional (see above section 2). Microbial interaction with the neonatal gut likely helps direct immune system maturation, playing a key role in developing a balanced innate and adaptive immune system [115,118,130]. Certain pioneer microbiota such as, Bifidobacteria, Firmicutes, and Bacteroidetes, are likely responsible for early education of the neonatal immune system [130,133,134,135,136]. Proteobacteria are important for early priming of innate and adaptive systems [117]. As mentioned previously, these anaerobic bacteria are also some of the most common and important colonizers in the neonatal gut. These bacteria contribute to the creation of a more desirable environment for beneficial microbial activity through mechanisms such as creating an anaerobic environment and colonization resistance [130,134,137]. Additionally, they produce and present specific compounds including microbe-associated molecular patterns (MAMPs), which help train immune tolerance [114,130]. The most important immune consequences of these bacteria involve the activation of T-cells, B-cells and immunoglobulins, and dendritic cells (DCs). These immune components, as well as the function of the mucosal and epithelial layers, will be discussed in further detail below.
4.1.1. Interactions at Mucosal Surfaces
The mucosal immune system comes into direct contact with many external threats, especially from pathogenic microbes, so it has many structures and mechanisms to maintain immunological homeostasis and tolerance to environmental encounters. The mucosal layer is primarily composed of highly structured glycoproteins known as mucins, which provide an important source of nutrients for commensal bacteria [118,128,138]. Up to this point 21 different mucin genes (Muc) have been identified [139]. Muc2 is the most expressed mucin in mucus [138,139]. It can bind and inhibit microbial translocation and is crucial for the proper functioning of the mucosal barrier [118]. The structure of the mucin network contributes to the viscoelasticity of mucus [140,141]. Other components such as proteins and DNA also contribute to the viscosity of mucus [138]. The mucus also acts as a storage for antimicrobial molecules, such as secretory immunoglobulin A (sIgA), defensins, and AMPs [138,142]. The mucus is produced and maintained by goblet cells, located in the epithelium [118,138]. The mucus layer is a dynamic barrier and is able to change thickness and coverage in response to immune interactions [118].
The mucus barrier is still immature at birth. The protective function of the neonatal mucus may be limited, as neonates have a thinner mucus layer (relative to adults) covering the epithelial layer, due to reduced expression of major mucins Muc2, Muc3, and Muc5ac [128]. The reduced thickness and amount of mucus is associated with increased permeability, which is then also associated with an increased potential for bacterial translocation [143]. Rat studies have shown increased susceptibility to certain infections in younger pups in comparison to older pups, the change in susceptibility was shown to be associated with the maturation and thickening of the mucosal layer, as well as the increase of mucin, DNA, and IgA amounts which impact viscosity [118,143]. One proposed explanation for reduced mucus production in early neonates is reduced goblet cell density, this is supported by findings from pig studies [144]. Additionally, trefoil factor (peptides secreted by goblet cells) levels are significantly lower in preterm over term infants, giving further support to the theory [118,145]. Trefoil factors are important for barrier restoration after injury, especially from damage caused by inflammation [146,147]. Therefore, it is possible that the immaturity of the neonatal mucosal barrier not only increases susceptibility to infections, but also reduces the ability to recover from possible harm.
The mucus layer is one of the main GI areas impacted by microbiota [115]. It continuously comes into contact with microbes and is responsible for providing protection against pathogens while maintaining tolerance of commensal microbiota. It also allows for the segregation of microbiota and host immune cells, due to its selective permeability, which helps to prevent abnormal immune responses [115]. Secretion and degradation of mucus are regulated by the host’s recognition of bacterial metabolites and MAMPs [114,148]. Mucus production and composition can also be altered by changes in inflammation and microbiota composition [130,149]. For example, SCFAs, which are the main metabolites produced by microbiota, can increase the secretion of mucus and AMPs from IECs [114,150]. The mucosal layer is the primary location of interaction between the microbiota and the immune system and serves an essential function in neonatal immunity.
4.1.2. Epithelium
Despite the need for defense against potential harm from foreign substances, the gut epithelium consists of a single layer of cells only, connected by tight-junctions [114,118,151]. Because of its location directly below the mucosal layer, it is exposed to the contents of the lumen. The epithelial barrier matures during gestation and is fully developed in term infants [128]. The development of the epithelial structure is a closely regulated process. The intestinal crypts and villi begin to develop in utero from intestinal stem cells (ISCs) between 8-24 weeks gestation [152,153]. During this period, the crypts deepen, the villi lengthen, and the ISCs proliferate and migrate to the crypt bases [152]. The ISCs also differentiate into many IEC subtypes, such as goblet cells and Paneth cells [153,154,155]. Differentiated cells migrate from the crypts up to the villi, however Paneth cells and undifferentiated ISCs remain in the crypt bases [154].
The integrity of the epithelium is crucial for limiting foreign material, such as microorganisms and toxins, from entering the body. Interaction between gut microbiota and IECs can impact the permeability and function of the epithelial barrier [116,118,126,149,156]. For example, some pathogenic microbiota can release toxic metabolites that disrupt the formation of the tight-junctions between IECs, reducing epithelial integrity and increasing permeability [151]. On the other hand, commensal SCFA-producing microbes improve barrier integrity by reducing inflammation and regulating cell turnover [116,130]. The continuous exposure of microbes in the gut causes adaptations in the neonatal gut tissue, however this process is not yet fully understood [130]. The immature immune system of a neonate most likely plays a role in this, as it is characterized by dampened pro-inflammatory cytokine responses, resulting in greater tolerance of microbiota establishment [130,157]. Tolerance is the main difference in function between neonatal and adult immune cells [133].
IECs can passively and actively recognize and take-up SCFAs through passive diffusion and carrier-mediated absorption through solute transporters [125]. Transporters allow for more efficient transport of SCFAs from the lumen of the gut into IECs, the lamina propria, and eventually the blood circulation. The primary transporters for SCFAs are SLC5a8 (sodium-coupled monocarboxylate transporter 1) and SLC16a1 (mono-carboxylate transporter 1) [125,158,159,160,161]. SCFAs are the most abundant microbial metabolite in the lumen [116,125]. They act as a significant source of energy to support cells when metabolized, as they can readily be converted into acetyl-CoA, fueling the tricarboxylic acid (TCA) cycle for ATP production [125,130,162]. They are also an important carbon source for IECs [116]. Additionally, SCFAs induce proliferation of IECs and innate lymphoid cells, such as ILC3, which in turn produce IL-22, a cytokine which induces an anti-inflammatory response [125,150]. The metabolism of butyrate, a major SCFA, by IECs helps establish an anti-inflammatory environment [116,126]. Additionally, SCFAs can also help IECs produce AMPs, which help to limit epithelium and pathogen interaction [150]. Other metabolites besides SCFAs also have immunoregulatory properties. Bacterial tryptophan metabolites, such as iodine-3-propionic acid (IPA), impact the metabolism of tryptophan, an important amino acid that has been shown to improve epithelial integrity and help maintain homeostasis of the mucosal immune system.
The regulation of immune responses is crucial for the maintenance of gut homeostasis. IECs direct appropriate responses to commensal and pathogenic microbiota by producing immune regulatory signals. For instance, Paneth cells, located in the crypts of Lieberkühn in the intestinal epithelium, are secretory cells that produce AMPs including α-defensins and lysozyme [115,118,138]. These cells first appear at about 12 weeks of gestation but do not fully mature until the postnatal period [163]. Preterm infants may have reduced AMP secretion due to reduced levels of Paneth cells compared to term infants [118]. Microbial metabolites help mediate communication between the epithelium and immune cells. For instance, commensal microbiota use signaling molecules to communicate between each other, as well as the epithelium [114]. Much like the mucosal layer, the epithelium provides a physical and chemical barrier which is essential to maintaining homeostasis and normal functioning in the body.
4.1.3. Introduction of Breast Milk and Its Impact on the Gut Microbiota’s Immune System
By week 12, the fetal gut begins to form villi and crypts, and around this time, the fetus starts to ingest amniotic fluid through the mouth, coming into contact with external stimuli for the first time [164]. The fetal gut is believed to have a stable immune landscape from the second trimester until birth, though as discussed above, it remains unclear if a microbiome is present before birth. Necrotizing enterocolitis in fetuses has also been raised as an argument for the presence of bacteria in the fetal gut, as these bacteria could inflame mucosal cells, leading to apoptosis and necrotizing enterocolitis. This condition, characterized by a necrotizing bowel, can rapidly progress to severe illness and death [165].
Human milk oligosaccharides (HMOs) have been shown to promote healthy gut bacteria, counteracting harmful microbes in the fetal gut. The weaning period is marked by a "weaning reaction," which triggers the increased expression of genes involved in antimicrobial immunity, including Reg3 defensins, mucins, and CC and CXC chemokines in dams [36,112,166]. This reaction supports the "hygiene hypothesis," and posits that exposure to diverse, colonizing microbiota in the infant's GI tract is crucial for the development of a transient microbiome in addition to proper immune system priming in infants to influence the onset of allergy and asthma later in life [112,167]. While there is strong evidence for this in mice, it is more challenging to confirm in humans. However, longitudinal stool sampling and in-depth immune profiling have revealed innate and adaptive immune responses during early and mid-infancy, likely induced by intestinal microbes settling in after exposure to breast milk and the mother's skin, similar to the weaning reaction observed in mice [36,168,169,170].
Dysbiosis of the fetal microbiome has been shown to have long-term negative effects on an infant's immune system, regardless of later colonization [171]. There is a window of opportunity for microbiome education and exposure to environmental cues, although the exact timing of this window remains unclear [134,172]. It is believed, however, that food-derived antigens play a key role in establishing a threshold for immune responses and future host-microbe interactions, directly and indirectly shaping gut microbiota [36,172,173,174,175].
Nutrition plays an important role in shaping neonatal mucosal immunity, as it introduces antigens to the infant for the first time, helping the immune system learn to build tolerance to non-self, foreign entities on the gut's mucosal surface. Breast milk contains HMOs, which promote the growth of beneficial bacteria such as Bifidobacterium, creating a distinct microbiome in breastfed infants compared to those fed only with formula [36,176]. Infants exposed to breast milk have been shown to have increased microbial diversity and enhanced stabilization of the gut microbiome. This is believed to lay the foundation for a more robust and resilient immune system, influencing acquisition of atopic diseases, obesity, and type 1 diabetes as they transition into adulthood [36,177,178].
4.1.4. Introduction of Solid Food and Its Impact on the Gut Microbiota and the Immune System
The introduction of solid food into the diet of an infant sees a change in the rapid decline in Bifidobacterium species and an increase of Bacteroides, Bilophila, Roseburia, Clostridium, and Anaerostipes in the infant gut microbiome [36,137,179]. The composition of the microbiota (and hence, the production of SCFAs) shifts with the introduction of solid food, leading to an increased level of pro-inflammatory cytokines (TNF-α and IFN-γ) and promotion the differentiation of RORγt+ T-regs in the intestine [112].
Indirect interaction through maternal microbial metabolites can also influence the development of a fetus's immune system, even before exposure to solid food, as studies have shown that limited exposure to E. coli during pregnancy can lead to changes in the fetal immune system, specifically affecting certain immune cells in the gut, like type III innate lymphoid cells (ILC3s) and mononuclear cells, if the fetus was exposed to them indirectly, prenatally [180,181].
4.2. Interaction Between Microbiota and Immune Cells and Molecules
Beyond the structural components of the epithelial and mucosal layer there are also the complex interactions of the many immune signals produced by the microbiota, IECs, and the immune cells which reside in the epithelial layer and lamina propria.
4.2.1. MAIT-Cells
Mucosal-associated invariant T cells (MAIT) are induced early during postnatal life by vitamin B-2 synthesizing commensal bacteria [182]. MAIT cells are derived from CD4+CD8+ thymocytes and are IL-23 dependent [182]. These cells respond to riboflavin metabolites produced by commensals and presented by MR1 MHC molecules. The interaction between MAIT cells and microbiota is essential for their development and function in tissue repair during adult life.
4.2.1.1. Anti-Inflammatory Effects of Bacterial Metabolites
Within the gut microbiome, the immune environment is primarily anti-inflammatory. This is especially important for neonates, who ingest an incredible amount of unfamiliar foreign material, including microbiota as well as food particles. Signaling by commensal bacteria promotes and maintains immune tolerance. For instance, bacterial metabolites such as the SCFAs butyrate, propionate, and acetate, suppress inflammatory cytokine production in Th cells. They can increase IL-10 and decrease IL-6 secretion, promoting differentiation of naïve T-cells into T-regs and inhibiting differentiation into Th17 [116]. Butyrate also induces CTLs to produce granzymes and inhibits the activity of pro-inflammatory cytokine IL-17 [125,183]. Furthermore, SCFA signaling can also enhance the function of T-regs, as butyrate promotes the release of anti-inflammatory cytokine IL-10 from T-regs [115,116,130]. Additionally, butyrate and, to a lesser degree, propionate inhibit histone deacetylase (HDAC) activity, resulting in epigenetic gene alterations and increasing T-reg interaction with dendritic cells (DCs) [116,122,125,184]. Therefore, butyrate and other SCFA signaling increases expression of the transcription factor Foxp3 which is associated with extrathymic T-reg production [116,126,130,184].
Because the repertoire of the antigen receptors on B- and T-lymphocytes is considered to be complete, the immune system has the capacity for specific recognition of all possible antigens. By definition this would then include self-antigens. In order to avoid an auto-immune attack of auto-reactive B- and T-lymphocytes, these cells should be eliminated or at least inactivated during development. For auto-reactive precursor T-lymphocytes which differentiate in the thymus, auto-antigens are presented by medullary thymic epithelial cells (by virtue of AIRE, the autoimmune regulator) This gene plays a role in the promotion of self-tolerance by initiating destruction of autoreactive T cells that respond to self-antigens. Thetis cells are a class of RORγt+ antigen-presenting cells present in the intestinal lymph nodes during a critical window in early life, coinciding with the wave of peripheral regulatory T cell differentiation [185]. Based on the expression of unique transcription factors, including Spi-B and PU.1, Thetis cells resemble medullary thymic epithelial cells. This suggest that they play a role in the induction of immunological tolerance (39). This aspect will be further discussed below.
Regulatory T-cells are immunosuppressive T-cells which can directly and indirectly cause anti-inflammatory responses through a variety of mechanisms. Naïve T-cells in fetuses are more likely to differentiate into T-regs than pro-inflammatory T-cells [128,130]. Gut microbiota such as Bacteroides, Clostridium, Lactobacillus, Bifidobacterium, and Streptococcus, impact T-reg proliferation and activity [130]. For instance, their SCFA metabolites promote T-reg acquisition [129,184,186,187]. They can prime T-reg production [114,128]. Bacteroides fragilis downregulates pro-inflammatory cytokine IL-8 and interferon gamma (IFNγ), supporting the anti-inflammatory function of T-regs [130]. B. fragilis may also activate and enhance T-reg function by activating toll-like receptor 2 (TLR2) with their lipopolysaccharide surface polysaccharide A (PSA) [115,130].
Bacterial polysaccharides can have immunoregulatory effects and benefits and also impact T-regs. In particular, polysaccharide A (PSA), a capsular carbohydrate with anti-inflammatory effects produced by Bacteroides fragilis, has been studied extensively in this respect [115,127,130,188]. In mouse models, B. fragilis has significant beneficial effects on the immune system, in particular in inflammatory bowel diseases [127,188,189]. These immunologic benefits result from its production of PSA 19-11. PSA helps prevent inflammatory disease by inducing IL-10 producing CD4+ T-cell proliferation, including T-regs, shown with mice and in vitro studies [127]. These cells have immunosuppressive activities. B. fragilis and PSA can also suppress expression of certain pro-inflammatory cytokines [115,188]. For example, PSA-producing B. fragilis has been shown to inhibit IL-23 release [127]. However, when B. fragilis is genetically modified to prevent synthesis of PSA this is not observed, indicating the immunosuppressive impacts derived from PSA [127]. PSA can inhibit the production of TNF-α and IL-17 by intestinal immune cells [127,190]. PSA is a zwitterionic polysaccharide (ZPS), a class of bacterial carbohydrates [188]. All identified ZPSs have immunoregulatory properties [191,192,193,194].
Indole-3-propionic acid (IPA) is another metabolite of tryptophan which significantly impacts immune function, especially related to T-cells. IPA is a metabolite of tryptophan, a SCFA produced by many microbiota [195]. Tryptophan is converted into indole-3-lactic acid (ILA) and then into IPA with the help of microbes, such as Lactobacillus johnsonii and Clostridium sporogenes, see Figure 5. IPA regulates the stemness of CD8+ T-cells [195]. IPA also promotes the proliferation of progenitor exhausted CD8+ T-cells by acetylation modification of H3K27 [195]. Furthermore, IPA can inhibit inflammatory factors, including TNF-α, IL-1β, and IL-6 [196].
4.2.1.2. Pro-Inflammatory Effects of Microbiota
While signaling molecules from microbiota within the gut are primarily anti-inflammatory, they can still be recognized as antigens and trigger an inflammatory immune response. Most notably, intraepithelial cells, like TCRαβ+ and TCRγδ+ T-cells, help protect IECs from injury and trigger immune responses by expressing cytokines [115,197,198,199]. Th cells secrete pro-inflammatory cytokines IL-22 and IL-17 in the lamina propria [114,115]. Pathogenic microbiota, including segmented filamentous bacteria, can promote pro-inflammatory Th17 development [65,115,128,184]. In addition to promotion of Th activation, signaling molecules can also activate and prime CTLs [115,126,195]. Butyrate inhibits HDAC in CTLs which induces CTL differentiation into cytotoxic memory T-cells and SCFA signals also prime CTLs [126].
4.2.2. B-Cells and Immunoglobulin A
Peyer’s patches along the GI tract consist largely of B-cells. The production and activation of B-cells causes the generation of plasma cells which can express and produce serum IgA (sIgA) [115]. Germ-free mice have been shown to have reduced IgA-secreting plasma cells [200]. This indicates that gut microbiota drive IgA production. The release of sIgA is a major defensive mechanism of the mucosal immune system. It is responsible for blocking microbe adhesion to epithelial cells without causing inflammation [130,201]. In neonates and young children, a significant part of IgA is derived from breast milk [118,128].
Microbiota induce sIgA production when their antigens are taken up by microfold cells (M cells) found in the epithelial layer, causing B-cell activation and the eventual increased production of IgA [115,118]. In addition, IgA may influence the microbiome by regulating the expansion of microbes by controlling their gene-expression, thereby impacting microbiome diversity. While part of adaptive immunity, IgAs have been shown to be polyreactive and attach to a range of certain microbiota, indicating possible innate recognition.
4.2.3. Dendritic Cells Connecting Gut Microbiota to Immune Systems
Dendritic cells (DCs) are important immune cells that link the innate and adaptive systems by expressing pattern recognition receptors (PRRs) and antigens [6]. Host-microbe interaction relies on the recognition of MAMPs using PRRs [114]. Bacteria are sensed differently depending on the type of PRR that interacts with their MAMPs [202,203]. Commensal microbiota are recognized by the mucosal immune system through MAMPs, which is important to induce tolerance [114,130]. This is also important for the healthy colonization of microbes. Fetal DCs contribute to immune regulation. They induce the differentiation of regulatory T cells (T-regs) and promote T cell IL-4 production, supporting a Th2 response. They also inhibit T cell TNF-α by increasing arginase 2 expression in conventional DCs within the fetal spleen to further aid in the suppression of inflammation [41]. Therefore, DCs are vital components of the mucosal immune system and are crucial for the initiation and control of immune responses.
These immune cells are considered immature at birth. Healthy neonates appear to express adult-level Toll-like receptors (TLRs) on DCs, an important PRR [115,130,204]. However, the production of innate inflammatory mediators is reduced in early life [130]. The response of TLRs to ligands expressed by microbes causes the release of chemokines and regulatory cytokines. Neonatal TLR signaling also promotes tolerogenic phenotypes of DCs, which results in immune responses to greatly involve T-regs [130]. Neonatal DCs promote differentiation of Foxp3+ T-regs and limit TNF-α production by inducing proliferation of allogeneic T-cells [41]. Further induction of T-regs occurs due to C-type lectin receptors on DCs, which recognize and bind to certain microbiota [130,205]. Furthermore, DCs also secrete anti-inflammatory cytokines. IFNβ DCs, which release IFN-I, can be induced by the glycolipids released by B. fragilis [206].
5. Conclusions and Interpretative View
The intimate and intricate interaction between gut microbiota and neonatal immune system is crucial for the regulated development of immunity: efficient and adequate defense against pathogens while maintaining tolerance for commensal microbiota. Homo sapiens and its microbiota function as a superorganism with mutual benefits: the intestines offer safe niches for a great diversity of micro-organisms, which in turn contribute to the health and well-being of their host by supporting the immune system, as well as providing vitamins and other metabolites. When Homo sapiens and its microbiota function as one, the immune system must consider the (antigens expressed by) commensal microbiota as self. Induction of self-tolerance takes place in the thymus and bone marrow during maturation of T-cells and B-cells, respectively. Microbial antigens are not expressed at those sites, so tolerance induction necessarily has to take place at other sites, of which mucosal surfaces are the most likely. As discussed in paragraph 2.4., during the intrauterine period, the fetus can swallow amniotic fluid, and this fluid contains microbial antigens from the mother [41,148,204]. Via such a mechanism, the fetal immune system could become tolerant for maternal antigens, including maternal microbiota. The second, and most dramatic, wave of exposure to the outside microbial world takes place during and immediately after birth (in the timeline of Figure 5, day 0). During passage through the birth canal, the neonate comes into direct and intense contact with maternal microbiota (paragraph 3.4.). Activation of pattern recognition receptors, including the ones expressed on buccal cells, initially triggers a strong inflammatory response, but within days this response wanes [207]. In the first weeks after birth, a rapid succession of bacterial species in the infant gut takes place, with some species appearing and taking hold, while others can be detected for just one or several days and then disappear [208]. A third wave of exposure of the immature mucosal immune system to microbial antigens takes place during introduction of solid food. At least in mice, this is accompanied with big changes in composition of gut microbiota, as well as exposure of the immune system to a wide array of food antigens. At the same period, Thetis cells accumulate in the lamina propria [147]. Thetis cells, which have features of both dendritic cells as well as ILCs, induce peripheral Treg cells. The current concept is that Thetis cells in such a way fulfill a role in induction of tolerance for non-self antigens, in particular commensal bacteria and food antigens. Little is known yet about Thetis cells in humans, including the timing of their appearance and exact role in the development of tolerance. Still, it is tempting to postulate that these cells play an important role in the establishment of tolerance of the immune system for commensal bacteria. After introduction of solid food in the diet, and weaning from breast feeding, the diversity of gut microbiota progresses at a slower rate and by day 1000 of life reaches a stable plateau. Apart from major and permanent changes in diet and antibiotics, the composition of gut microbiota remains stable well into adulthood.
A consequence of the above integration of microbiological and immunological data is that the functionality of the immune system determines the composition and diversity of gut microbiota. Because the fine-specificity of the immune system is individually determined (being the consequence of rearrangements of immunoglobulin- and T cell receptor gene segments), the composition of gut microbiota is also individually determined. Another implication would be that a permanent change in gut microbiota composition would not be possible, apart from drastic measures such as permanent dietary changes. Finally, a deeper understanding of the cellular and molecular interactions between microbiota and the immune system would allow for targeted interventions in cases when the immune system would function super- or sub-optimally.
Author Contributions
Conceptualization, G.R, F.v.O., S.L.; resources, S.V.; writing—original draft preparation, M.T., I.L., S.V., F.v.O., G.R.; writing—review and editing, M.T., I.L., S.V., F.v.O., G.R.; visualization, S.V.; supervision, G.R.; project administration, G.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data was created or used for this review.
Acknowledgments
We acknowledge the artistic contribution of Stella Simons, BSc (University College Roosevelt, Middelburg, The Netherlands) in designing Figure 1 and the Graphical Abstract. Furthermore, we are grateful to Professor Wim Timens, University Medical Center Groningen, The Netherlands for allowing us to use Figure 3 (CD21 expression).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Sansonetti, P.J. War and peace at mucosal surfaces. Nat Rev Immunol. 2004, 4, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Cupedo, T.; Crellin, N.K.; Papazian, N.; Rombouts, E.J.; Weijer, K.; Grogan, J.L.; Fibbe, W.E.; Cornelissen, J.J.; Spits, H. Human fetal lymphoid tissue-inducer cells are interleukin 17-producing precursors to RORC+ CD127+ natural killer-like cells. Nat Immunol. 2009, 10, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.; Boroviak, T.E. Origin and function of the yolk sac in primate embryogenesis. Nat Commun. 2020, 11, 3760. [Google Scholar] [CrossRef] [PubMed]
- Enders, A.C. Implantation in the macaque: expansion of the implantation site during the first week of implantation. Placenta. 2007, 28, 794–802. [Google Scholar] [CrossRef]
- Xie, X.; Gou, F.; Zheng, Z.; Zhang, Y.; Zhang, Y.; Dong, F.; Cheng, T.; Cheng, H. Decoding human bone marrow hematopoietic stem and progenitor cells from fetal to birth. iScience. 2024, 27, 110445. [Google Scholar] [CrossRef]
- Park, J.E.; Jardine, L.; Gottgens, B.; Teichmann, S.A.; Haniffa, M. Prenatal development of human immunity. Science. 2020, 368, 600–603. [Google Scholar] [CrossRef]
- Popescu, D.M.; Botting, R.A.; Stephenson, E.; Green, K.; Webb, S.; Jardine, L.; Calderbank, E.F.; Polanski, K.; Goh, I.; Efremova, M.; et al. Decoding human fetal liver haematopoiesis. Nature. 2019, 574, 365–371. [Google Scholar] [CrossRef]
- Goh, I.; Botting, R.A.; Rose, A.; Webb, S.; Engelbert, J.; Gitton, Y.; Stephenson, E.; Quiroga Londoño, M.; Mather, M.; Mende, N.; et al. Yolk sac cell atlas reveals multiorgan functions during human early development. Science. 2023, 381, eadd7564. [Google Scholar] [CrossRef]
- Slayton, W.B.; Li, Y.; Calhoun, D.A.; Juul, S.E.; Iturraspe, J.; Braylan, R.C.; Christensen, R.D. The first-appearance of neutrophils in the human fetal bone marrow cavity. Early Hum Dev. 1998, 53, 129–144. [Google Scholar] [CrossRef]
- Vento-Tormo, R.; Efremova, M.; Botting, R.A.; Turco, M.Y.; Vento-Tormo, M.; Meyer, K.B.; Park, J.E.; Stephenson, E.; Polański, K.; Goncalves, A.; et al. Single-cell reconstruction of the early maternal-fetal interface in humans. Nature. 2018, 563, 347–353. [Google Scholar] [CrossRef]
- Banaei-Bouchareb, L.; Peuchmaur, M.; Czernichow, P.; Polak, M. A transient microenvironment loaded mainly with macrophages in the early developing human pancreas. J Endocrinol. 2006, 188, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Menassa, D.A.; Gomez-Nicola, D. Microglial dynamics during human brain development. Front Immunol. 2018, 9:1014.. eCollection 02018. [CrossRef]
- Jardine, L.; Schim van der Loeff, I.; Haq, I.J.; Sproat, T.D.R. Gestational development of the human immune system. Immunol Allergy Clin North Am. 2023, 43, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Alsinet, C.; Primo, M.N.; Lorenzi, V.; Bello, E.; Kelava, I.; Jones, C.P.; Vilarrasa-Blasi, R.; Sancho-Serra, C.; Knights, A.J.; Park, J.E.; et al. Robust temporal map of human in vitro myelopoiesis using single-cell genomics. Nat Commun. 2022, 13, 2885. [Google Scholar] [CrossRef]
- Hoeffel, G.; Ginhoux, F. Fetal monocytes and the origins of tissue-resident macrophages. Cell Immunol. 2018, 330:5-15.. Epub 2018 Jan 1012. [CrossRef]
- Ginhoux, F.; Guilliams, M. Tissue-resident macrophage ontogeny and homeostasis. Immunity. 2016, 44, 439–449. [Google Scholar] [CrossRef]
- Jardine, L.; Webb, S.; Goh, I.; Quiroga Londoño, M.; Reynolds, G.; Mather, M.; Olabi, B.; Stephenson, E.; Botting, R.A.; Horsfall, D.; et al. Blood and immune development in human fetal bone marrow and Down syndrome. Nature. 2021, 598, 327–331. [Google Scholar] [CrossRef]
- Schuster, C.; Vaculik, C.; Prior, M.; Fiala, C.; Mildner, M.; Eppel, W.; Stingl, G.; Elbe-Bürger, A. Phenotypic characterization of leukocytes in prenatal human dermis. J Invest Dermatol. 2012, 132, 2581–2592. [Google Scholar] [CrossRef]
- Msallam, R.; Balla, J.; Rathore, A.P.S.; Kared, H.; Malleret, B.; Saron, W.A.A.; Liu, Z.; Hang, J.W.; Dutertre, C.A.; Larbi, A.; et al. Fetal mast cells mediate postnatal allergic responses dependent on maternal IgE. Science. 2020, 370, 941–950. [Google Scholar] [CrossRef]
- Hoorweg, K.; Narang, P.; Li, Z.; Thuery, A.; Papazian, N.; Withers, D.R.; Coles, M.C.; Cupedo, T. A stromal cell niche for human and mouse type 3 innate lymphoid cells. J Immunol. 2015, 195, 4257–4263. [Google Scholar] [CrossRef]
- van de Pavert, S.A.; Mebius, R.E. New insights into the development of lymphoid tissues. Nat Rev Immunol. 2010, 10, 664–674. [Google Scholar] [CrossRef]
- Leeansyah, E.; Loh, L.; Nixon, D.F.; Sandberg, J.K. Acquisition of innate-like microbial reactivity in mucosal tissues during human fetal MAIT-cell development. Nat Commun 2014, 5:3143. [CrossRef]
- He, P.; Lim, K.; Sun, D.; Pett, J.P.; Jeng, Q.; Polanski, K.; Dong, Z.; Bolt, L.; Richardson, L.; Mamanova, L.; et al. A human fetal lung cell atlas uncovers proximal-distal gradients of differentiation and key regulators of epithelial fates. Cell. 2022, 185, 4841–4860.e4825. [Google Scholar] [CrossRef]
- Schafflick, D.; Wolbert, J.; Heming, M.; Thomas, C.; Hartlehnert, M.; Börsch, A.L.; Ricci, A.; Martín-Salamanca, S.; Li, X.; Lu, I.N.; et al. Single-cell profiling of CNS border compartment leukocytes reveals that B cells and their progenitors reside in non-diseased meninges. Nat Neurosci. 2021, 24, 1225–1234. [Google Scholar] [CrossRef] [PubMed]
- Rechavi, E.; Lev, A.; Lee, Y.N.; Simon, A.J.; Yinon, Y.; Lipitz, S.; Amariglio, N.; Weisz, B.; Notarangelo, L.D.; Somech, R. Timely and spatially regulated maturation of B and T cell repertoire during human fetal development. Sci Transl Med. 2015, 7, 276ra225. [Google Scholar] [CrossRef] [PubMed]
- Ivarsson, M.A.; Loh, L.; Marquardt, N.; Kekäläinen, E.; Berglin, L.; Björkström, N.K.; Westgren, M.; Nixon, D.F.; Michaëlsson, J. Differentiation and functional regulation of human fetal NK cells. J Clin Invest. 2013, 123, 3889–3901. [Google Scholar] [CrossRef] [PubMed]
- Sagebiel, A.F.; Steinert, F.; Lunemann, S.; Körner, C.; Schreurs, R.; Altfeld, M.; Perez, D.; Reinshagen, K.; Bunders, M.J. Tissue-resident Eomes(+) NK cells are the major innate lymphoid cell population in human infant intestine. Nat Commun. 2019, 10, 975. [Google Scholar] [CrossRef] [PubMed]
- Angelo, L.S.; Bimler, L.H.; Nikzad, R.; Aviles-Padilla, K.; Paust, S. CXCR6(+) NK cells in human fetal liver and spleen possess unique phenotypic and functional capabilities. Front Immunol. 2019, 10:469.. eCollection 02019. [CrossRef]
- Haynes, B.F.; Heinly, C.S. Early human T cell development: analysis of the human thymus at the time of initial entry of hematopoietic stem cells into the fetal thymic microenvironment. J Exp Med. 1995, 181, 1445–1458. [Google Scholar] [CrossRef]
- Suo, C.; Dann, E.; Goh, I.; Jardine, L.; Kleshchevnikov, V.; Park, J.E.; Botting, R.A.; Stephenson, E.; Engelbert, J.; Tuong, Z.K.; et al. Mapping the developing human immune system across organs. Science. 2022, 376, eabo0510. [Google Scholar] [CrossRef]
- Wang, H.; Zúñiga-Pflücker, J.C. ThymicmMicroenvironment: Interactions between innate immune cells and developing thymocytes. Front Immunol. 2022, 13:885280.. eCollection 882022. [CrossRef]
- Park, J.E.; Botting, R.A.; Domínguez Conde, C.; Popescu, D.M.; Lavaert, M.; Kunz, D.J.; Goh, I.; Stephenson, E.; Ragazzini, R.; Tuck, E.; et al. A cell atlas of human thymic development defines T cell repertoire formation. Science. 2020, 367, eaay3224. [Google Scholar] [CrossRef]
- Zeng, Y.; Liu, C.; Gong, Y.; Bai, Z.; Hou, S.; He, J.; Bian, Z.; Li, Z.; Ni, Y.; Yan, J.; et al. Single-cell RNA sequencing resolves spatiotemporal development of pre-thymic lymphoid progenitors and thymus organogenesis in human embryos. Immunity. 2019, 51, 930–948.e936. [Google Scholar] [CrossRef]
- Haynes, B.F.; Martin, M.E.; Kay, H.H.; Kurtzberg, J. Early events in human T cell ontogeny. Phenotypic characterization and immunohistologic localization of T cell precursors in early human fetal tissues. J Exp Med. 1988, 168, 1061–1080. [Google Scholar] [CrossRef]
- Rackaityte, E.; Halkias, J. Mechanisms of fetal T cell tolerance and immune regulation. Front Immunol. 2020, 11:588.. eCollection 02020. [CrossRef]
- Ignacio, A.; Czyz, S.; McCoy, K.D. Early life microbiome influences on development of the mucosal innate immune system. Semin Immunol. 2024, 73:101885. [CrossRef]
- Mold, J.E.; Venkatasubrahmanyam, S.; Burt, T.D.; Michaëlsson, J.; Rivera, J.M.; Galkina, S.A.; Weinberg, K.; Stoddart, C.A.; McCune, J.M. Fetal and adult hematopoietic stem cells give rise to distinct T cell lineages in humans. Science. 2010, 330, 1695–1699. [Google Scholar] [CrossRef]
- Hayakawa, S.; Ohno, N.; Okada, S.; Kobayashi, M. Significant augmentation of regulatory T cell numbers occurs during the early neonatal period. Clin Exp Immunol. 2017, 190, 268–279. [Google Scholar] [CrossRef] [PubMed]
- McDavid, A.; Laniewski, N.; Grier, A.; Gill, A.L.; Kessler, H.A.; Huyck, H.; Carbonell, E.; Holden-Wiltse, J.; Bandyopadhyay, S.; Carnahan, J.; et al. Aberrant newborn T cell and microbiota developmental trajectories predict respiratory compromise during infancy. iScience. 2022, 25, 104007. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.L.; Patel, R.K.; Reynaldi, A.; Grenier, J.K.; Wang, J.; Watson, N.B.; Nzingha, K.; Yee Mon, K.J.; Peng, S.A.; Grimson, A.; et al. Developmental origin governs CD8(+) T cell fate decisions during infection. Cell. 2018, 174, 117–130.e114. [Google Scholar] [CrossRef]
- McGovern, N.; Shin, A.; Low, G.; Low, D.; Duan, K.; Yao, L.J.; Msallam, R.; Low, I.; Shadan, N.B.; Sumatoh, H.R.; et al. Human fetal dendritic cells promote prenatal T-cell immune suppression through arginase-2. Nature. 2017, 546, 662–666. [Google Scholar] [CrossRef]
- Feyaerts, D.; Urbschat, C.; Gaudillière, B.; Stelzer, I.A. Establishment of tissue-resident immune populations in the fetus. Semin Immunopathol. 2022, 44, 747–766. [Google Scholar] [CrossRef]
- Schreurs, R.R.C.E.; Baumdick, M.E.; Sagebiel, A.F.; Kaufmann, M.; Mokry, M.; Klarenbeek, P.L.; Schaltenberg, N.; Steinert, F.L.; van Rijn, J.M.; Drewniak, A.; et al. Human fetal TNF-α-cytokine-producing CD4(+) effector memory T cells promote intestinal development and mediate inflammation early in life. Immunity. 2019, 50, 462–476.e468. [Google Scholar] [CrossRef]
- Zhang, X.; Mozeleski, B.; Lemoine, S.; Dériaud, E.; Lim, A.; Zhivaki, D.; Azria, E.; Le Ray, C.; Roguet, G.; Launay, O.; et al. CD4 T cells with effector memory phenotype and function develop in the sterile environment of the fetus. Sci Transl Med. 2014, 6, 238ra272. [Google Scholar] [CrossRef]
- Li, N.; van Unen, V.; Guo, N.; Abdelaal, T.; Somarakis, A.; Eggermont, J.; Mahfouz, A.; Chuva de Sousa Lopes, S.M.; Lelieveldt, B.P.F.; Koning, F. Early-life compartmentalization of immune cells in human fetal tissues revealed by high-dimensional mass cytometry. Front Immunol. 2019, 10:1932.. eCollection 02019. [CrossRef]
- Stras, S.F.; Werner, L.; Toothaker, J.M.; Olaloye, O.O.; Oldham, A.L.; McCourt, C.C.; Lee, Y.N.; Rechavi, E.; Shouval, D.S.; Konnikova, L. Maturation of the human intestinal immune system occurs early in fetal development. Dev Cell. 2019, 51, 357–373.e355. [Google Scholar] [CrossRef]
- van den Berg, J.P.; Westerbeek, E.A.; van der Klis, F.R.; Berbers, G.A.; van Elburg, R.M. Transplacental transport of IgG antibodies to preterm infants: a review of the literature. Early Hum Dev. 2011, 87, 67–72. [Google Scholar] [CrossRef]
- Hong, R. Transient hypogammaglobulinemia of infancy. J Pediatr. 1978, 92, 521–522. [Google Scholar] [CrossRef]
- Griffioen, A.W.; Toebes, E.A.; Zegers, B.J.; Rijkers, G.T. Role of CR2 in the human adult and neonatal in vitro antibody response to type 4 pneumococcal polysaccharide. Cell Immunol. 1992, 143, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Griffioen, A.W.; Franklin, S.W.; Zegers, B.J.; Rijkers, G.T. Expression and functional characteristics of the complement receptor type 2 on adult and neonatal B lymphocytes. Clin Immunol Immunopathol. 1993, 69, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Timens, W.; Boes, A.; Rozeboom-Uiterwijk, T.; Poppema, S. Immaturity of the human splenic marginal zone in infancy. Possible contribution to the deficient infant immune response. J Immunol. 1989, 143, 3200–3206. [Google Scholar] [CrossRef] [PubMed]
- Rijkers, G.; Dekker, K.; Berbers, G. Oswald Avery: Pioneer of bacterial vaccines and the first to discover the function of DNA. Cureus. 2024, 16, e71465. [Google Scholar] [CrossRef]
- Goldman, A.S.; Schmalstieg, F.C. Karl Otto Landsteiner (1868-1943). Physician-biochemist-immunologist. J Med Biogr. 2019, 27, 67–75. [Google Scholar] [CrossRef]
- Zorn, E. ABO antibody development, a new look at a very old question. Transplantation. 2023, 107, 2311–2312. [Google Scholar] [CrossRef]
- Adalsteinsson, S. Possible changes in the frequency of the human ABO blood groups in Iceland due to smallpox epidemics selection. Ann Hum Genet. 1985, 49, 275–281. [Google Scholar] [CrossRef]
- Comans-Bitter, W.M.; de Groot, R.; van den Beemd, R.; Neijens, H.J.; Hop, W.C.; Groeneveld, K.; Hooijkaas, H.; van Dongen, J.J. Immunophenotyping of blood lymphocytes in childhood. Reference values for lymphocyte subpopulations. J Pediatr. 1997, 130, 388–393. [Google Scholar] [CrossRef]
- Blanco, E.; Pérez-Andrés, M.; Arriba-Méndez, S.; Contreras-Sanfeliciano, T.; Criado, I.; Pelak, O.; Serra-Caetano, A.; Romero, A.; Puig, N.; Remesal, A.; et al. Age-associated distribution of normal B-cell and plasma cell subsets in peripheral blood. J Allergy Clin Immunol. 2018, 141, 2208–2219.e2216. [Google Scholar] [CrossRef]
- Irjala, K.; Koskinen, P.; Icen, A.; Palosuo, T. Reference intervals for immunoglobulins IgA, IgG and IgM in serum in adults and in children aged 6 months to 14 years. Scand J Clin Lab Invest. 1990, 50, 573–577. [Google Scholar] [CrossRef]
- Grunewald, O.; Lopez, B.; Brabant, S.; Rogeau, S.; Deschildre, A.; Phrommavanh, V.; Lefort, M.; Moitrot, E.; Gyselinckx, D.; Deleplancque, A.S.; et al. Immunoglobulin G (IgG) and IgG subclass reference intervals in children, using Optilite® reagents. Clin Chem Lab Med. 2018, 56, 1319–1327. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, J.G.; Milani, C.; de Giori, G.S.; Sesma, F.; van Sinderen, D.; Ventura, M. Bacteria as vitamin suppliers to their host: a gut microbiota perspective. Curr Opin Biotechnol. 2013, 24, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Romero, R.; Hassan, S.S.; Gajer, P.; Tarca, A.L.; Fadrosh, D.W.; Nikita, L.; Galuppi, M.; Lamont, R.F.; Chaemsaithong, P.; Miranda, J.; et al. The composition and stability of the vaginal microbiota of normal pregnant women is different from that of non-pregnant women. Microbiome. 2014, 2, 4. [Google Scholar] [CrossRef]
- Altemani, F.; Barrett, H.L.; Gomez-Arango, L.; Josh, P.; David McIntyre, H.; Callaway, L.K.; Morrison, M.; Tyson, G.W.; Dekker Nitert, M. Pregnant women who develop preeclampsia have lower abundance of the butyrate-producer Coprococcus in their gut microbiota. Pregnancy Hypertens. 2021, 23:211-219.. Epub 2021 Jan 1026. [CrossRef]
- Yao, Y.; Cai, X.; Fei, W.; Ye, Y.; Zhao, M.; Zheng, C. The role of short-chain fatty acids in immunity, inflammation and metabolism. Crit Rev Food Sci Nutr 2022, 62, 1–12. [Google Scholar] [CrossRef]
- Koren, O.; Goodrich, J.K.; Cullender, T.C.; Spor, A.; Laitinen, K.; Bäckhed, H.K.; Gonzalez, A.; Werner, J.J.; Angenent, L.T.; Knight, R.; et al. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell. 2012, 150, 470–480. [Google Scholar] [CrossRef]
- Ivanov, II; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009, 139, 485-498. [CrossRef]
- Kim, S.; Kim, H.; Yim, Y.S.; Ha, S.; Atarashi, K.; Tan, T.G.; Longman, R.S.; Honda, K.; Littman, D.R.; Choi, G.B.; et al. Maternal gut bacteria promote neurodevelopmental abnormalities in mouse offspring. Nature. 2017, 549, 528–532. [Google Scholar] [CrossRef]
- Choi, G.B.; Yim, Y.S.; Wong, H.; Kim, S.; Kim, H.; Kim, S.V.; Hoeffer, C.A.; Littman, D.R.; Huh, J.R. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science. 2016, 351, 933–939. [Google Scholar] [CrossRef]
- Aagaard, K.; Ma, J.; Antony, K.M.; Ganu, R.; Petrosino, J.; Versalovic, J. The placenta harbors a unique microbiome. Sci Transl Med. 2014, 6, 237ra265. [Google Scholar] [CrossRef]
- Jašarević, E.; Howard, C.D.; Misic, A.M.; Beiting, D.P.; Bale, T.L. Stress during pregnancy alters temporal and spatial dynamics of the maternal and offspring microbiome in a sex-specific manner. Sci Rep. 2017, 7:44182. [CrossRef]
- Zhou, P.; Zhou, Y.; Liu, B.; Jin, Z.; Zhuang, X.; Dai, W.; Yang, Z.; Feng, X.; Zhou, Q.; Liu, Y.; et al. Perinatal antibiotic exposure affects the transmission between maternal and neonatal microbiota and is associated with early-onset sepsis. mSphere. 2020, 5, e00984–00919. [Google Scholar] [CrossRef]
- Lawn, J.E.; Cousens, S.; Zupan, J. 4 million neonatal deaths: When? Where? Why? Lancet. 2005, 365, 891–900. [Google Scholar] [CrossRef]
- Goldenberg, R.L.; Hauth, J.C.; Andrews, W.W. Intrauterine infection and preterm delivery. N Engl J Med. 2000, 342, 1500–1507. [Google Scholar] [CrossRef] [PubMed]
- Perez-Muñoz, M.E.; Arrieta, M.C.; Ramer-Tait, A.E.; Walter, J. A critical assessment of the "sterile womb" and "in utero colonization" hypotheses: implications for research on the pioneer infant microbiome. Microbiome. 2017, 5, 48. [Google Scholar] [CrossRef] [PubMed]
- Milani, C.; Duranti, S.; Bottacini, F.; Casey, E.; Turroni, F.; Mahony, J.; Belzer, C.; Delgado Palacio, S.; Arboleya Montes, S.; Mancabelli, L.; et al. The first microbial colonizers of the human gut: Composition, activities, and health implications of the infant gut microbiota. Microbiol Mol Biol Rev. 2017, 81, e00036–00017. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, S.J.; Pakzad, Z.; Elwood, C.N.; Albert, A.Y.K.; Gantt, S.; Manges, A.R.; Dumonceaux, T.J.; Maan, E.J.; Hill, J.E.; Money, D.M. Early neonatal meconium does not have a demonstrable microbiota determined through use of robust negative controls with cpn60-based microbiome profiling. Microbiol Spectr. 2021, 9, e0006721. [Google Scholar] [CrossRef]
- Kennedy, K.M.; Gerlach, M.J.; Adam, T.; Heimesaat, M.M.; Rossi, L.; Surette, M.G.; Sloboda, D.M.; Braun, T. Fetal meconium does not have a detectable microbiota before birth. Nat Microbiol. 2021, 6, 865–873. [Google Scholar] [CrossRef]
- Shi, Y.C.; Guo, H.; Chen, J.; Sun, G.; Ren, R.R.; Guo, M.Z.; Peng, L.H.; Yang, Y.S. Initial meconium microbiome in Chinese neonates delivered naturally or by cesarean section. Sci Rep. 2018, 8, 3255. [Google Scholar] [CrossRef]
- Collado, M.C.; Rautava, S.; Aakko, J.; Isolauri, E.; Salminen, S. Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Sci Rep. 2016, 6:23129. [CrossRef]
- Gilbert, W.M.; Brace, R.A. Amniotic fluid volume and normal flows to and from the amniotic cavity. Semin Perinatol. 1993, 17, 150–157. [Google Scholar]
- Jiménez, E.; Fernández, L.; Marín, M.L.; Martín, R.; Odriozola, J.M.; Nueno-Palop, C.; Narbad, A.; Olivares, M.; Xaus, J.; Rodríguez, J.M. Isolation of commensal bacteria from umbilical cord blood of healthy neonates born by cesarean section. Curr Microbiol. 2005, 51, 270–274. [Google Scholar] [CrossRef]
- Jiménez, E.; Marín, M.L.; Martín, R.; Odriozola, J.M.; Olivares, M.; Xaus, J.; Fernández, L.; Rodríguez, J.M. Is meconium from healthy newborns actually sterile? Res Microbiol. 2008, 159, 187–193. [Google Scholar] [CrossRef]
- Moles, L.; Gómez, M.; Heilig, H.; Bustos, G.; Fuentes, S.; de Vos, W.; Fernández, L.; Rodríguez, J.M.; Jiménez, E. Bacterial diversity in meconium of preterm neonates and evolution of their fecal microbiota during the first month of life. PLoS One. 2013, 8, e66986. [Google Scholar] [CrossRef]
- Hansen, R.; Scott, K.P.; Khan, S.; Martin, J.C.; Berry, S.H.; Stevenson, M.; Okpapi, A.; Munro, M.J.; Hold, G.L. First-pass meconium samples from healthy term vaginally-delivered neonates: An analysis of the microbiota. PLoS One. 2015, 10, e0133320. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.J.; Lynch, D.B.; Murphy, K.; Ulaszewska, M.; Jeffery, I.B.; O'Shea, C.A.; Watkins, C.; Dempsey, E.; Mattivi, F.; Tuohy, K.; et al. Evolution of gut microbiota composition from birth to 24 weeks in the INFANTMET Cohort. Microbiome. 2017, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Rougé, C.; Goldenberg, O.; Ferraris, L.; Berger, B.; Rochat, F.; Legrand, A.; Göbel, U.B.; Vodovar, M.; Voyer, M.; Rozé, J.C.; et al. Investigation of the intestinal microbiota in preterm infants using different methods. Anaerobe. 2010, 16, 362–370. [Google Scholar] [CrossRef]
- Cong, X.; Xu, W.; Janton, S.; Henderson, W.A.; Matson, A.; McGrath, J.M.; Maas, K.; Graf, J. Gut microbiome developmental patterns in early life of preterm infants: Impacts of feeding and gender. PLoS One. 2016, 11, e0152751. [Google Scholar] [CrossRef] [PubMed]
- Jacquot, A.; Neveu, D.; Aujoulat, F.; Mercier, G.; Marchandin, H.; Jumas-Bilak, E.; Picaud, J.C. Dynamics and clinical evolution of bacterial gut microflora in extremely premature patients. J Pediatr. 2011, 158, 390–396. [Google Scholar] [CrossRef]
- Arboleya, S.; Binetti, A.; Salazar, N.; Fernández, N.; Solís, G.; Hernández-Barranco, A.; Margolles, A.; de Los Reyes-Gavilán, C.G.; Gueimonde, M. Establishment and development of intestinal microbiota in preterm neonates. FEMS Microbiol Ecol. 2012, 79, 763–772. [Google Scholar] [CrossRef]
- Arboleya, S.; Sánchez, B.; Milani, C.; Duranti, S.; Solís, G.; Fernández, N.; de los Reyes-Gavilán, C.G.; Ventura, M.; Margolles, A.; Gueimonde, M. Intestinal microbiota development in preterm neonates and effect of perinatal antibiotics. J Pediatr. 2015, 166, 538–544. [Google Scholar] [CrossRef]
- Rutten, N.B.; Rijkers, G.T.; Meijssen, C.B.; Crijns, C.E.; Oudshoorn, J.H.; van der Ent, C.K.; Vlieger, A.M. Intestinal microbiota composition after antibiotic treatment in early life: the INCA study. BMC Pediatr. 2015, 15:204. [CrossRef]
- Cooke, R.J. Improving growth in preterm infants during initial hospital stay: principles into practice. Arch Dis Child Fetal Neonatal Ed. 2016, 101, F366–370. [Google Scholar] [CrossRef]
- Tadros, J.S.; Llerena, A.; Sarkar, A.; Johnson, R.; Miller, E.M.; Gray, H.L.; Ho, T.T.B. Postnatal growth and gut microbiota development influenced early childhood growth in preterm infants. Front Pediatr. 2022, 10:850629.. eCollection 852022. [CrossRef]
- van den Akker, C.H.P.; van Goudoever, J.B.; Shamir, R.; Domellöf, M.; Embleton, N.D.; Hojsak, I.; Lapillonne, A.; Mihatsch, W.A.; Berni Canani, R.; Bronsky, J.; et al. Probiotics and preterm infants: A position paper by the European Society for Paediatric Gastroenterology Hepatology and Nutrition Committee on Nutrition and the European Society for Paediatric Gastroenterology Hepatology and Nutrition Working Group for Probiotics and Prebiotics. J Pediatr Gastroenterol Nutr. 2020, 70, 664–680. [Google Scholar] [CrossRef]
- Dominguez-Bello, M.G.; Costello, E.K.; Contreras, M.; Magris, M.; Hidalgo, G.; Fierer, N.; Knight, R. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A. 2010, 107, 11971–11975. [Google Scholar] [CrossRef]
- Yang, R.; Gao, R.; Cui, S.; Zhong, H.; Zhang, X.; Chen, Y.; Wang, J.; Qin, H. Dynamic signatures of gut microbiota and influences of delivery and feeding modes during the first 6 months of life. Physiol Genomics. 2019, 51, 368–378. [Google Scholar] [CrossRef]
- Montoya-Williams, D.; Lemas, D.J.; Spiryda, L.; Patel, K.; Carney, O.O.; Neu, J.; Carson, T.L. The neonatal microbiome and its partial role in mediating the association between birth by Cesarean section and adverse pediatric outcomes. Neonatology 2018, 114, 103–111. [Google Scholar] [CrossRef]
- Palmer, C.; Bik, E.M.; DiGiulio, D.B.; Relman, D.A.; Brown, P.O. Development of the human infant intestinal microbiota. PLoS Biol. 2007, 5, e177. [Google Scholar] [CrossRef]
- Korpela, K.; de Vos, W.M. Early life colonization of the human gut: microbes matter everywhere. Curr Opin Microbiol. 2018, 44:70-78.. Epub 2018 Aug 1014. [CrossRef]
- Stinson, L.F.; Payne, M.S.; Keelan, J.A. A critical review of the bacterial baptism hypothesis and the impact of Cesarean delivery on the infant microbiome. Front Med (Lausanne). 2018, 5:135.. eCollection 02018. [CrossRef]
- Wernroth, M.L.; Peura, S.; Hedman, A.M.; Hetty, S.; Vicenzi, S.; Kennedy, B.; Fall, K.; Svennblad, B.; Andolf, E.; Pershagen, G.; et al. Development of gut microbiota during the first 2 years of life. Sci Rep. 2022, 12, 9080. [Google Scholar] [CrossRef]
- Barker-Tejeda, T.C.; Zubeldia-Varela, E.; Macías-Camero, A.; Alonso, L.; Martín-Antoniano, I.A.; Rey-Stolle, M.F.; Mera-Berriatua, L.; Bazire, R.; Cabrera-Freitag, P.; Shanmuganathan, M.; et al. Comparative characterization of the infant gut microbiome and their maternal lineage by a multi-omics approach. Nat Commun. 2024, 15, 3004. [Google Scholar] [CrossRef]
- Sakanaka, M.; Gotoh, A.; Yoshida, K.; Odamaki, T.; Koguchi, H.; Xiao, J.Z.; Kitaoka, M.; Katayama, T. Varied pathways of infant gut-associated Bifidobacterium to assimilate human milk oligosaccharides: Prevalence of the gene set and its correlation with Bifidobacteria-rich microbiota formation. Nutrients. 2019, 12, 71. [Google Scholar] [CrossRef]
- Alcon-Giner, C.; Dalby, M.J.; Caim, S.; Ketskemety, J.; Shaw, A.; Sim, K.; Lawson, M.A.E.; Kiu, R.; Leclaire, C.; Chalklen, L.; et al. Microbiota supplementation with Bifidobacterium and Lactobacillus modifies the preterm infant gut microbiota and metabolome: An observational study. Cell Rep Med. 2020, 1, 100077. [Google Scholar] [CrossRef]
- Jamyuang, C.; Phoonlapdacha, P.; Chongviriyaphan, N.; Chanput, W.; Nitisinprasert, S.; Nakphaichit, M. Characterization and probiotic properties of Lactobacilli from human breast milk. 3 Biotech. 2019, 9, 398. [Google Scholar] [CrossRef]
- Al Radaideh, A.J.; Badran, E.F.; Shehabi, A.A. Diversity of toxin genotypes and antimicrobial susceptibility of Clostridium perfringens isolates from feces of infants. Germs. 2019, 9, 28–34. [Google Scholar] [CrossRef]
- Kim, Y.G.; Sakamoto, K.; Seo, S.U.; Pickard, J.M.; Gillilland, M.G., 3rd; Pudlo, N.A.; Hoostal, M.; Li, X.; Wang, T.D.; Feehley, T.; et al. Neonatal acquisition of Clostridia species protects against colonization by bacterial pathogens. Science. 2017, 356, 315–319. [Google Scholar] [CrossRef]
- Gregory, K.E.; LaPlante, R.D.; Shan, G.; Kumar, D.V.; Gregas, M. Mode of birth influences preterm infant intestinal colonization with bacteroides over the early neonatal period. Adv Neonatal Care. 2015, 15, 386–393. [Google Scholar] [CrossRef]
- Suzuki, S.; Shimojo, N.; Tajiri, Y.; Kumemura, M.; Kohno, Y. A quantitative and relative increase in intestinal bacteroides in allergic infants in rural Japan. Asian Pac J Allergy Immunol. 2008, 26, 113–119. [Google Scholar]
- Biagioli, V.; Volpedo, G.; Riva, A.; Mainardi, P.; Striano, P. From birth to weaning: A window of opportunity for microbiota. Nutrients. 2024, 16, 272. [Google Scholar] [CrossRef]
- Schwab, C. The development of human gut microbiota fermentation capacity during the first year of life. Microb Biotechnol. 2022, 15, 2865–2874. [Google Scholar] [CrossRef]
- Qasem, W.; Azad, M.B.; Hossain, Z.; Azad, E.; Jorgensen, S.; Castillo San Juan, S.; Cai, C.; Khafipour, E.; Beta, T.; Roberts, L.J., 2nd; et al. Assessment of complementary feeding of Canadian infants: effects on microbiome & oxidative stress, a randomized controlled trial. BMC Pediatr. 2017, 17, 54. [Google Scholar] [CrossRef]
- Al Nabhani, Z.; Dulauroy, S.; Marques, R.; Cousu, C.; Al Bounny, S.; Déjardin, F.; Sparwasser, T.; Bérard, M.; Cerf-Bensussan, N.; Eberl, G. A weaning reaction to microbiota is required for resistance to immunopathologies in the adult. Immunity. 2019, 50, 1276–1288.e1275. [Google Scholar] [CrossRef]
- Dizzell, S.; Stearns, J.C.; Li, J.; van Best, N.; Bervoets, L.; Mommers, M.; Penders, J.; Morrison, K.M.; Hutton, E.K. Investigating colonization patterns of the infant gut microbiome during the introduction of solid food and weaning from breastmilk: A cohort study protocol. PLoS One. 2021, 16, e0248924. [Google Scholar] [CrossRef]
- Wiertsema, S.P.; van Bergenhenegouwen, J.; Garssen, J.; Knippels, L.M.J. The interplay between the gut microbiome and the immune system in the context of infectious diseases throughout life and the role of nutrition in optimizing treatment strategies. Nutrients. 2021, 13, 886. [Google Scholar] [CrossRef]
- Shi, N.; Li, N.; Duan, X.; Niu, H. Interaction between the gut microbiome and mucosal immune system. Mil Med Res. 2017, 4:14.. eCollection 42017. [CrossRef]
- Yoo, J.Y.; Groer, M.; Dutra, S.V.O.; Sarkar, A.; McSkimming, D.I. Gut microbiota and immune system interactions. Microorganisms. 2020, 8, 1587. [Google Scholar] [CrossRef]
- Mulligan, C.M.; Friedman, J.E. Maternal modifiers of the infant gut microbiota: metabolic consequences. J Endocrinol. 2017, 235, R1–R12. [Google Scholar] [CrossRef]
- Golubkova, A.; Hunter, C.J. Development of the neonatal intestinal barrier, microbiome, and susceptibility to NEC. Microorganisms. 2023, 11, 1247. [Google Scholar] [CrossRef]
- Iacob, S.; Iacob, D.G.; Luminos, L.M. Intestinal microbiota as a host defense mechanism to infectious threats. Front Microbiol. 2018, 9:3328.. eCollection 02018. [CrossRef]
- Kong, S.; Zhang, Y.H.; Zhang, W. Regulation of Intestinal Epithelial Cells Properties and Functions by Amino Acids. Biomed Res Int. 2018, 2018:2819154.. eCollection 2812018. [CrossRef]
- Lazar, V.; Ditu, L.M.; Pircalabioru, G.G.; Gheorghe, I.; Curutiu, C.; Holban, A.M.; Picu, A.; Petcu, L.; Chifiriuc, M.C. Aspects of gut microbiota and immune system interactions in infectious diseases, immunopathology, and cancer. Front Immunol. 2018, 9:1830.. eCollection 02018. [CrossRef]
- Chen, Y.; Chen, Y.X. Microbiota-associated metabolites and related immunoregulation in colorectal cancer. Cancers (Basel). 2021, 13, 4054. [Google Scholar] [CrossRef]
- Macfarlane, G.T.; Macfarlane, S. Human colonic microbiota: ecology, physiology and metabolic potential of intestinal bacteria. Scand J Gastroenterol Suppl 1997, 222:3-9. [CrossRef]
- Flint, H.J.; Scott, K.P.; Duncan, S.H.; Louis, P.; Forano, E. Microbial degradation of complex carbohydrates in the gut. Gut Microbes. 2012, 3, 289–306. [Google Scholar] [CrossRef]
- Kim, C.H. Control of lymphocyte functions by gut microbiota-derived short-chain fatty acids. Cell Mol Immunol. 2021, 18, 1161–1171. [Google Scholar] [CrossRef]
- Verhoef, J.I.; Klont, E.; van Overveld, F.J.; Rijkers, G.T. The long and winding road of faecal microbiota transplants to targeted intervention for improvement of immune checkpoint inhibition therapy. Expert Rev Anticancer Ther. 2023, 23, 1179–1191. [Google Scholar] [CrossRef]
- Mazmanian, S.K.; Round, J.L.; Kasper, D.L. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature. 2008, 453, 620–625. [Google Scholar] [CrossRef]
- Torow, N.; Marsland, B.J.; Hornef, M.W.; Gollwitzer, E.S. Neonatal mucosal immunology. Mucosal Immunol. 2017, 10, 5–17. [Google Scholar] [CrossRef]
- Ahern, P.P.; Maloy, K.J. Understanding immune-microbiota interactions in the intestine. Immunology. 2020, 159, 4–14. [Google Scholar] [CrossRef]
- Dzidic, M.; Boix-Amorós, A.; Selma-Royo, M.; Mira, A.; Collado, M.C. Gut microbiota and mucosal immunity in the neonate. Med Sci (Basel). 2018, 6, 56. [Google Scholar] [CrossRef]
- van Es, J.H.; Jay, P.; Gregorieff, A.; van Gijn, M.E.; Jonkheer, S.; Hatzis, P.; Thiele, A.; van den Born, M.; Begthel, H.; Brabletz, T.; et al. Wnt signalling induces maturation of Paneth cells in intestinal crypts. Nat Cell Biol. 2005, 7, 381–386. [Google Scholar] [CrossRef]
- Thompson, J.A.; Oliveira, R.A.; Xavier, K.B. Chemical conversations in the gut microbiota. Gut Microbes 2016, 7, 163–170. [Google Scholar] [CrossRef]
- Wopereis, H.; Oozeer, R.; Knipping, K.; Belzer, C.; Knol, J. The first thousand days - intestinal microbiology of early life: establishing a symbiosis. Pediatr Allergy Immunol. 2014, 25, 428–438. [Google Scholar] [CrossRef]
- Gensollen, T.; Iyer, S.S.; Kasper, D.L.; Blumberg, R.S. How colonization by microbiota in early life shapes the immune system. Science. 2016, 352, 539–544. [Google Scholar] [CrossRef]
- Jost, T.; Lacroix, C.; Braegger, C.P.; Chassard, C. New insights in gut microbiota establishment in healthy breast fed neonates. PLoS One 2012, 7, e44595. [Google Scholar] [CrossRef]
- Houghteling, P.D.; Walker, W.A. Why is initial bacterial colonization of the intestine important to infants' and children's health? J Pediatr Gastroenterol Nutr. 2015, 60, 294–307. [Google Scholar] [CrossRef]
- Bäckhed, F.; Roswall, J.; Peng, Y.; Feng, Q.; Jia, H.; Kovatcheva-Datchary, P.; Li, Y.; Xia, Y.; Xie, H.; Zhong, H.; et al. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe. 2015, 17, 690–703. [Google Scholar] [CrossRef]
- Managlia, E.; Yan, X.; De Plaen, I.G. Intestinal epithelial barrier function and necrotizing enterocolitis. Newborn (Clarksville). 2022, 1, 32–43. [Google Scholar] [CrossRef]
- Yang, S.; Yu, M. Role of goblet cells in intestinal barrier and mucosal immunity. J Inflamm Res. 2021, 14:3171-3183.. eCollection 312021. [CrossRef]
- Godl, K.; Johansson, M.E.; Lidell, M.E.; Mörgelin, M.; Karlsson, H.; Olson, F.J.; Gum, J.R., Jr.; Kim, Y.S.; Hansson, G.C. The N terminus of the MUC2 mucin forms trimers that are held together within a trypsin-resistant core fragment. J Biol Chem. 2002, 277, 47248–47256. [Google Scholar] [CrossRef]
- McGuckin, M.A.; Lindén, S.K.; Sutton, P.; Florin, T.H. Mucin dynamics and enteric pathogens. Nat Rev Microbiol. 2011, 9, 265–278. [Google Scholar] [CrossRef]
- Chairatana, P.; Nolan, E.M. Defensins, lectins, mucins, and secretory immunoglobulin A: microbe-binding biomolecules that contribute to mucosal immunity in the human gut. Crit Rev Biochem Mol Biol. 2017, 52, 45–56. [Google Scholar] [CrossRef]
- Birchenough, G.M.; Dalgakiran, F.; Witcomb, L.A.; Johansson, M.E.; McCarthy, A.J.; Hansson, G.C.; Taylor, P.W. Postnatal development of the small intestinal mucosa drives age-dependent, regio-selective susceptibility to Escherichia coli K1 infection. Sci Rep. 2017, 7, 83. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Hui, Y.; Obelitz-Ryom, K.; Brandt, A.B.; Kot, W.; Nielsen, D.S.; Thymann, T.; Sangild, P.T.; Nguyen, D.N. Neonatal gut and immune maturation is determined more by postnatal age than by postconceptional age in moderately preterm pigs. Am J Physiol Gastrointest Liver Physiol. 2018, 315, G855–G867. [Google Scholar] [CrossRef] [PubMed]
- Henderickx, J.G.E.; Zwittink, R.D.; Renes, I.B.; van Lingen, R.A.; van Zoeren-Grobben, D.; Jebbink, L.J.G.; Boeren, S.; van Elburg, R.M.; Knol, J.; Belzer, C. Maturation of the preterm gastrointestinal tract can be defined by host and microbial markers for digestion and barrier defense. Sci Rep. 2021, 11, 12808. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, W. Trefoil factors TFF (trefoil factor family) peptide-triggered signals promoting mucosal restitution. Cell Mol Life Sci. 2005, 62, 2932–2938. [Google Scholar] [CrossRef] [PubMed]
- Bossenmeyer-Pourié, C.; Kannan, R.; Ribieras, S.; Wendling, C.; Stoll, I.; Thim, L.; Tomasetto, C.; Rio, M.C. The trefoil factor 1 participates in gastrointestinal cell differentiation by delaying G1-S phase transition and reducing apoptosis. J Cell Biol. 2002, 157, 761–770. [Google Scholar] [CrossRef]
- Johansson, M.E.; Jakobsson, H.E.; Holmén-Larsson, J.; Schütte, A.; Ermund, A.; Rodríguez-Piñeiro, A.M.; Arike, L.; Wising, C.; Svensson, F.; Bäckhed, F.; et al. Normalization of host intestinal mucus layers requires long-term microbial colonization. Cell Host Microbe. 2015, 18, 582–592. [Google Scholar] [CrossRef]
- Berg, D.; Clemente, J.C.; Colombel, J.F. Can inflammatory bowel disease be permanently treated with short-term interventions on the microbiome? Expert Rev Gastroenterol Hepatol. 2015, 9, 781–795. [Google Scholar] [CrossRef]
- Schnupf, P.; Gaboriau-Routhiau, V.; Cerf-Bensussan, N. Modulation of the gut microbiota to improve innate resistance. Curr Opin Immunol. 2018, 54:137-144.. Epub 2018 Sep 1018. [CrossRef]
- Libertucci, J.; Young, V.B. The role of the microbiota in infectious diseases. Nat Microbiol. 2019, 4, 35–45. [Google Scholar] [CrossRef]
- Frazer, L.C.; Good, M. Intestinal epithelium in early life. Mucosal Immunol. 2022, 15, 1181–1187. [Google Scholar] [CrossRef]
- Condino, A.A.; Barleycorn, A.A.; Lu, W.; Maheshwari, A.; Christensen, R.D.; Calhoun, D.A. Abnormal intestinal histology in neonates with congenital anomalies of the gastrointestinal tract. Biol Neonate 2004, 85, 145–150. [Google Scholar] [CrossRef]
- Barker, N.; van Es, J.H.; Kuipers, J.; Kujala, P.; van den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007, 449, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009, 459, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Kayama, H.; Okumura, R.; Takeda, K. Interaction between the microbiota, epithelia, and immune cells in the intestine. Annu Rev Immunol. 2020, 38:23-48. [CrossRef]
- Kamada, N.; Chen, G.Y.; Inohara, N.; Núñez, G. Control of pathogens and pathobionts by the gut microbiota. Nat Immunol. 2013, 14, 685–690. [Google Scholar] [CrossRef]
- Li, H.; Myeroff, L.; Smiraglia, D.; Romero, M.F.; Pretlow, T.P.; Kasturi, L.; Lutterbaugh, J.; Rerko, R.M.; Casey, G.; Issa, J.P.; et al. SLC5A8, a sodium transporter, is a tumor suppressor gene silenced by methylation in human colon aberrant crypt foci and cancers. Proc Natl Acad Sci U S A. 2003, 100, 8412–8417. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, S.; Gopal, E.; Fei, Y.J.; Ganapathy, V. Functional identification of SLC5A8, a tumor suppressor down-regulated in colon cancer, as a Na(+)-coupled transporter for short-chain fatty acids. J Biol Chem. 2004, 279, 13293–13296. [Google Scholar] [CrossRef]
- Yanase, H.; Takebe, K.; Nio-Kobayashi, J.; Takahashi-Iwanaga, H.; Iwanaga, T. Cellular expression of a sodium-dependent monocarboxylate transporter (Slc5a8) and the MCT family in the mouse kidney. Histochem Cell Biol. 2008, 130, 957–966. [Google Scholar] [CrossRef]
- Suzuki, T.; Yoshida, S.; Hara, H. Physiological concentrations of short-chain fatty acids immediately suppress colonic epithelial permeability. Br J Nutr. 2008, 100, 297–305. [Google Scholar] [CrossRef]
- Kim, M.; Qie, Y.; Park, J.; Kim, C.H. Gut microbial metabolites fuel host antibody responses. Cell Host Microbe. 2016, 20, 202–214. [Google Scholar] [CrossRef]
- Barreto, E.B.L.; Rattes, I.C.; da Costa, A.V.; Gama, P. Paneth cells and their multiple functions. Cell Biol Int. 2022, 46, 701–710. [Google Scholar] [CrossRef]
- Grassi, R.; Farina, R.; Floriani, I.; Amodio, F.; Romano, S. Assessment of fetal swallowing with gray-scale and color Doppler sonography. AJR Am J Roentgenol. 2005, 185, 1322–1327. [Google Scholar] [CrossRef]
- Battersby, C.; Santhalingam, T.; Costeloe, K.; Modi, N. Incidence of neonatal necrotising enterocolitis in high-income countries: a systematic review. Arch Dis Child Fetal Neonatal Ed. 2018, 103, F182–F189. [Google Scholar] [CrossRef] [PubMed]
- Rakoff-Nahoum, S.; Kong, Y.; Kleinstein, S.H.; Subramanian, S.; Ahern, P.P.; Gordon, J.I.; Medzhitov, R. Analysis of gene-environment interactions in postnatal development of the mammalian intestine. Proc Natl Acad Sci U S A. 2015, 112, 1929–1936. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.H. Revisiting the hygiene hypothesis for allergy and asthma. J Allergy Clin Immunol. 2015, 136, 860–865. [Google Scholar] [CrossRef]
- Henrick, B.M.; Rodriguez, L.; Lakshmikanth, T.; Pou, C.; Henckel, E.; Arzoomand, A.; Olin, A.; Wang, J.; Mikes, J.; Tan, Z.; et al. Bifidobacteria-mediated immune system imprinting early in life. Cell. 2021, 184, 3884–3898.e3811. [Google Scholar] [CrossRef]
- Stewart, C.J.; Ajami, N.J.; O'Brien, J.L.; Hutchinson, D.S.; Smith, D.P.; Wong, M.C.; Ross, M.C.; Lloyd, R.E.; Doddapaneni, H.; Metcalf, G.A.; et al. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature. 2018, 562, 583–588. [Google Scholar] [CrossRef]
- Sanidad, K.Z.; Zeng, M.Y. Neonatal gut microbiome and immunity. Curr Opin Microbiol. 2020, 56:30-37.. Epub 2020 Jul 1014. [CrossRef]
- Tejesvi, M.V.; Turunen, J.; Salmi, S.; Reunanen, J.; Paalanne, N.; Tapiainen, T. Delivery mode and perinatal antibiotics influence the infant gut bacteriome and mycobiome: A network analysis. J Fungi (Basel). 2023, 9, 718. [Google Scholar] [CrossRef]
- Torow, N.; Hornef, M.W. The neonatal window of opportunity: Setting the stage for life-long host-microbial interaction and immune homeostasis. J Immunol. 2017, 198, 557–563. [Google Scholar] [CrossRef]
- Vatanen, T.; Kostic, A.D.; d'Hennezel, E.; Siljander, H.; Franzosa, E.A.; Yassour, M.; Kolde, R.; Vlamakis, H.; Arthur, T.D.; Hämäläinen, A.M.; et al. Variation in microbiome LPS immunogenicity contributes to autoimmunity in humans. Cell. 2016, 165, 842–853. [Google Scholar] [CrossRef]
- Braun-Fahrländer, C.; Riedler, J.; Herz, U.; Eder, W.; Waser, M.; Grize, L.; Maisch, S.; Carr, D.; Gerlach, F.; Bufe, A.; et al. Environmental exposure to endotoxin and its relation to asthma in school-age children. N Engl J Med. 2002, 347, 869–877. [Google Scholar] [CrossRef]
- Verhasselt, V. A newborn's perspective on immune responses to food. Immunol Rev. 2024, 326, 117–129. [Google Scholar] [CrossRef]
- Lawson, M.A.E.; O'Neill, I.J.; Kujawska, M.; Gowrinadh Javvadi, S.; Wijeyesekera, A.; Flegg, Z.; Chalklen, L.; Hall, L.J. Breast milk-derived human milk oligosaccharides promote Bifidobacterium interactions within a single ecosystem. ISME J. 2020, 14, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Fassarella, M.; Blaak, E.E.; Penders, J.; Nauta, A.; Smidt, H.; Zoetendal, E.G. Gut microbiome stability and resilience: elucidating the response to perturbations in order to modulate gut health. Gut. 2021, 70, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.Y.; Tan, L.T.; Law, J.W.; Hong, K.W.; Ratnasingam, V.; Ab Mutalib, N.S.; Lee, L.H.; Letchumanan, V. Exploring the potential of human milk and formula milk on infants' gut and health. Nutrients. 2022, 14, 3554. [Google Scholar] [CrossRef]
- Davis, M.Y.; Zhang, H.; Brannan, L.E.; Carman, R.J.; Boone, J.H. Rapid change of fecal microbiome and disappearance of Clostridium difficile in a colonized infant after transition from breast milk to cow milk. Microbiome. 2016, 4, 53. [Google Scholar] [CrossRef] [PubMed]
- Hussain, T.; Murtaza, G.; Kalhoro, D.H.; Kalhoro, M.S.; Yin, Y.; Chughtai, M.I.; Tan, B.; Yaseen, A.; Rehman, Z.U. Understanding the immune system in fetal protection and maternal infections during pregnancy. J Immunol Res. 2022, 2022:7567708.. eCollection 7562022. [CrossRef]
- Gomez de Agüero, M.; Ganal-Vonarburg, S.C.; Fuhrer, T.; Rupp, S.; Uchimura, Y.; Li, H.; Steinert, A.; Heikenwalder, M.; Hapfelmeier, S.; Sauer, U.; et al. The maternal microbiota drives early postnatal innate immune development. Science. 2016, 351, 1296–1302. [Google Scholar] [CrossRef]
- Constantinides, M.G.; Link, V.M.; Tamoutounour, S.; Wong, A.C.; Perez-Chaparro, P.J.; Han, S.J.; Chen, Y.E.; Li, K.; Farhat, S.; Weckel, A.; et al. MAIT cells are imprinted by the microbiota in early life and promote tissue repair. Science. 2019, 366, eaax6624. [Google Scholar] [CrossRef]
- Luu, M.; Weigand, K.; Wedi, F.; Breidenbend, C.; Leister, H.; Pautz, S.; Adhikary, T.; Visekruna, A. Regulation of the effector function of CD8(+) T cells by gut microbiota-derived metabolite butyrate. Sci Rep. 2018, 8, 14430. [Google Scholar] [CrossRef]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; van der Veeken, J.; deRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 2013, 504, 451–455. [Google Scholar] [CrossRef]
- Akagbosu, B.; Tayyebi, Z.; Shibu, G.; Paucar Iza, Y.A.; Deep, D.; Parisotto, Y.F.; Fisher, L.; Pasolli, H.A.; Thevin, V.; Elmentaite, R.; et al. Novel antigen-presenting cell imparts T(reg)-dependent tolerance to gut microbiota. Nature. 2022, 610, 752–760. [Google Scholar] [CrossRef]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013, 504, 446–450. [Google Scholar] [CrossRef]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly, Y.M.; Glickman, J.N.; Garrett, W.S. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013, 341, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Troy, E.B.; Kasper, D.L. Beneficial effects of Bacteroides fragilis polysaccharides on the immune system. Front Biosci (Landmark Ed). 2010, 15, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Mazmanian, S.K.; Liu, C.H.; Tzianabos, A.O.; Kasper, D.L. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell. 2005, 122, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.H.; Lee, S.M.; Vanlare, J.M.; Kasper, D.L.; Mazmanian, S.K. Regulation of surface architecture by symbiotic bacteria mediates host colonization. Proc Natl Acad Sci U S A. 2008, 105, 3951–3956. [Google Scholar] [CrossRef]
- Tzianabos, A.O.; Onderdonk, A.B.; Rosner, B.; Cisneros, R.L.; Kasper, D.L. Structural features of polysaccharides that induce intra-abdominal abscesses. Science. 1993, 262, 416–419. [Google Scholar] [CrossRef]
- Tzianabos, A.O.; Wang, J.Y.; Lee, J.C. Structural rationale for the modulation of abscess formation by Staphylococcus aureus capsular polysaccharides. Proc Natl Acad Sci U S A. 2001, 98, 9365–9370. [Google Scholar] [CrossRef]
- Tzianabos, A.O.; Finberg, R.W.; Wang, Y.; Chan, M.; Onderdonk, A.B.; Jennings, H.J.; Kasper, D.L. T cells activated by zwitterionic molecules prevent abscesses induced by pathogenic bacteria. J Biol Chem. 2000, 275, 6733–6740. [Google Scholar] [CrossRef]
- McLoughlin, R.M.K., D. L. Immunomodelation by zwitterionic polysaccharides. In Microbial glycobiology. Structure, relevance and applications., Holst, O.B., P. J.; von Itzstein, M., Ed.; Elsevier: 2009; pp. 957-980. [CrossRef]
- Jia, D.; Wang, Q.; Qi, Y.; Jiang, Y.; He, J.; Lin, Y.; Sun, Y.; Xu, J.; Chen, W.; Fan, L.; et al. Microbial metabolite enhances immunotherapy efficacy by modulating T cell stemness in pan-cancer. Cell. 2024, 187, 1651–1665.e1621. [Google Scholar] [CrossRef]
- Zhang, B.; Jiang, M.; Zhao, J.; Song, Y.; Du, W.; Shi, J. The mechanism underlying the influence of indole-3-propionic acid: A relevance to metabolic disorders. Front Endocrinol (Lausanne). 2022, 13:841703.. eCollection 842022. [CrossRef]
- Wang, H.C.; Zhou, Q.; Dragoo, J.; Klein, J.R. Most murine CD8+ intestinal intraepithelial lymphocytes are partially but not fully activated T cells. J Immunol. 2002, 169, 4717–4722. [Google Scholar] [CrossRef]
- Moretto, M.; Weiss, L.M.; Khan, I.A. Induction of a rapid and strong antigen-specific intraepithelial lymphocyte response during oral Encephalitozoon cuniculi infection. J Immunol. 2004, 172, 4402–4409. [Google Scholar] [CrossRef]
- Boismenu, R.; Havran, W.L. Modulation of epithelial cell growth by intraepithelial gamma delta T cells. Science. 1994, 266, 1253–1255. [Google Scholar] [CrossRef] [PubMed]
- Crabbé, P.A.; Bazin, H.; Eyssen, H.; Heremans, J.F. The normal microbial flora as a major stimulus for proliferation of plasma cells synthesizing IgA in the gut. The germ-free intestinal tract. Int Arch Allergy Appl Immunol 1968, 34, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Macpherson, A.J.; Yilmaz, B.; Limenitakis, J.P.; Ganal-Vonarburg, S.C. IgA function in relation to the intestinal microbiota. Annu Rev Immunol. 2018, 36:359-381.. Epub 042018 Jan 042626. [CrossRef]
- Swiatczak, B.; Rescigno, M. How the interplay between antigen presenting cells and microbiota tunes host immune responses in the gut. Semin Immunol. 2012, 24, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Konieczna, P.; Groeger, D.; Ziegler, M.; Frei, R.; Ferstl, R.; Shanahan, F.; Quigley, E.M.; Kiely, B.; Akdis, C.A.; O'Mahony, L. Bifidobacterium infantis 35624 administration induces Foxp3 T regulatory cells in human peripheral blood: potential role for myeloid and plasmacytoid dendritic cells. Gut. 2012, 61, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Kollmann, T.R.; Levy, O.; Montgomery, R.R.; Goriely, S. Innate immune function by Toll-like receptors: distinct responses in newborns and the elderly. Immunity. 2012, 37, 771–783. [Google Scholar] [CrossRef]
- Figdor, C.G.; van Kooyk, Y.; Adema, G.J. C-type lectin receptors on dendritic cells and Langerhans cells. Nat Rev Immunol. 2002, 2, 77–84. [Google Scholar] [CrossRef]
- Stefan, K.L.; Kim, M.V.; Iwasaki, A.; Kasper, D.L. Commensal microbiota modulation of natural resistance to virus infection. Cell. 2020, 183, 1312–1324.e1310. [Google Scholar] [CrossRef]
- Menckeberg, C.L.; Hol, J.; Simons-Oosterhuis, Y.; Raatgeep, H.R.; de Ruiter, L.F.; Lindenbergh-Kortleve, D.J.; Korteland-van Male, A.M.; El Aidy, S.; van Lierop, P.P.; Kleerebezem, M.; et al. Human buccal epithelium acquires microbial hyporesponsiveness at birth, a role for secretory leukocyte protease inhibitor. Gut. 2015, 64, 884–893. [Google Scholar] [CrossRef]
- Favier, C.F.; de Vos, W.M.; Akkermans, A.D. Development of bacterial and bifidobacterial communities in feces of newborn babies. Anaerobe. 2003, 9, 219–229. [Google Scholar] [CrossRef]
Figure 1.
Ontogeny of the fetal immune system during prenatal development. Key steps in development of cells and organs of the innate and adaptive immune system are indicated. See text for further details.
Figure 1.
Ontogeny of the fetal immune system during prenatal development. Key steps in development of cells and organs of the innate and adaptive immune system are indicated. See text for further details.
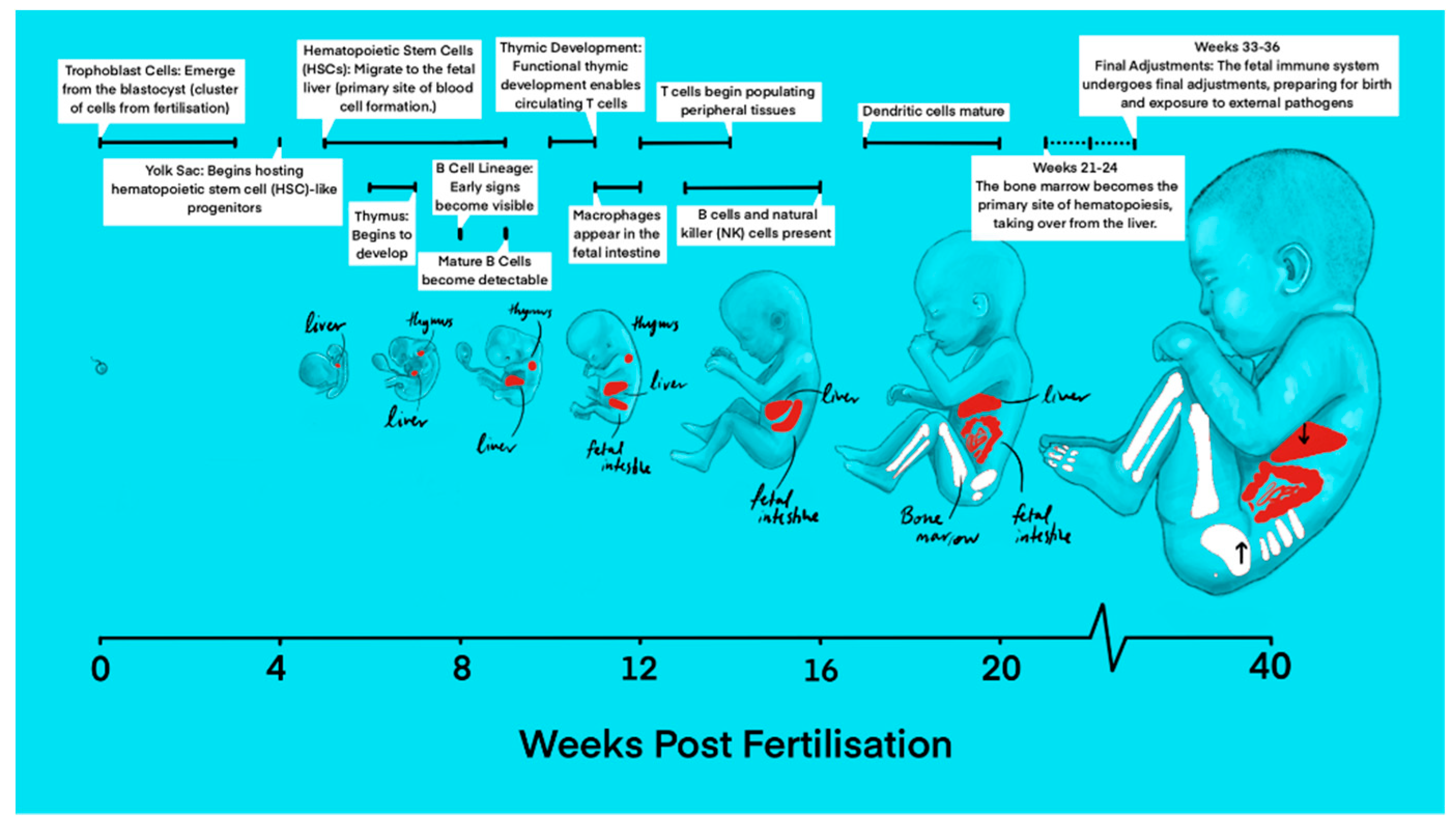
Figure 2.
Naïve B- and T-lymphocytes in umbilical cord blood. In the upper panels, blood mononuclear cells were stained with CD19, IgD, and CD27 antibodies. B-lymphocytes were gated on CD19, and expression of surface IgD was plotted against CD27. In adult peripheral blood (panel a), memory B-lymphocytes, characterized by the phenotype surface IgD- CD27+, are enclosed in the red circle. Memory B-lymphocytes in cord blood are virtually absent (panel b). In the lower panels, mononuclear cells were stained with CD3, CD45RA, and CD45RO. T-lymphocytes were gated on CD3, and memory T-lymphocytes, characterized by the phenotype CD45RA- CD45RO+, are abundant in adult blood (panel c) and almost absent in cord blood (panel d). Data from GT Rijkers, unpublished.
Figure 2.
Naïve B- and T-lymphocytes in umbilical cord blood. In the upper panels, blood mononuclear cells were stained with CD19, IgD, and CD27 antibodies. B-lymphocytes were gated on CD19, and expression of surface IgD was plotted against CD27. In adult peripheral blood (panel a), memory B-lymphocytes, characterized by the phenotype surface IgD- CD27+, are enclosed in the red circle. Memory B-lymphocytes in cord blood are virtually absent (panel b). In the lower panels, mononuclear cells were stained with CD3, CD45RA, and CD45RO. T-lymphocytes were gated on CD3, and memory T-lymphocytes, characterized by the phenotype CD45RA- CD45RO+, are abundant in adult blood (panel c) and almost absent in cord blood (panel d). Data from GT Rijkers, unpublished.
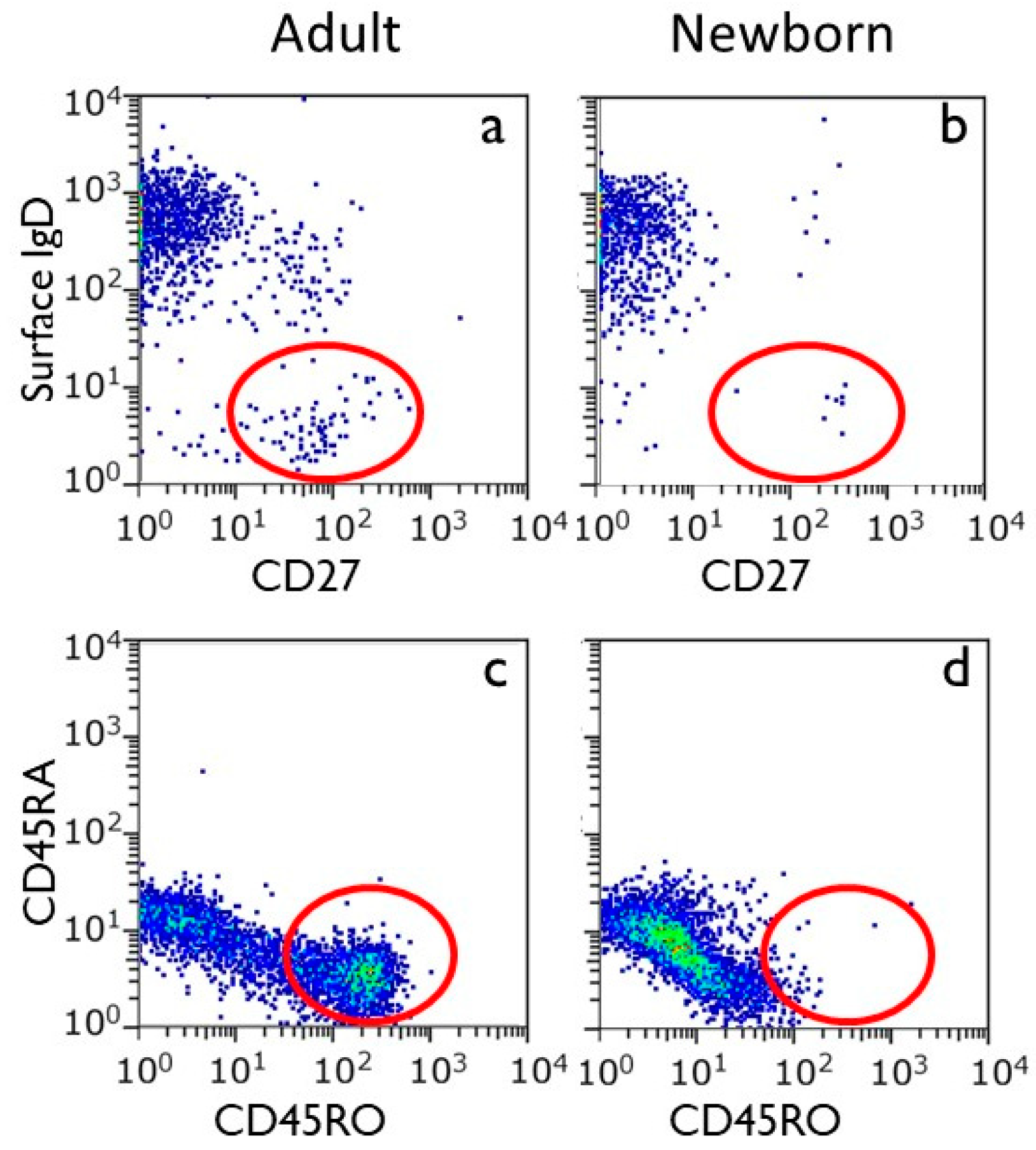
Figure 3.
Expression of CD21 on human marginal zone B cells. Immunohistochemistry of the HB5 (anti-CD21) antibody on sections of a spleen of a 12 months old infant (panel A)) and a 13 years old child (panel B)). In panel B) the marginal zone (MZ) B cells show strong expression of CD21, but the infant MZ B cells (panel A) are lacking CD21 expression. GC, germinal center; C, corona; R, red pulp. Courtesy of Prof. W. Timens. The photograph in panel B) has been published before [51].
Figure 3.
Expression of CD21 on human marginal zone B cells. Immunohistochemistry of the HB5 (anti-CD21) antibody on sections of a spleen of a 12 months old infant (panel A)) and a 13 years old child (panel B)). In panel B) the marginal zone (MZ) B cells show strong expression of CD21, but the infant MZ B cells (panel A) are lacking CD21 expression. GC, germinal center; C, corona; R, red pulp. Courtesy of Prof. W. Timens. The photograph in panel B) has been published before [51].
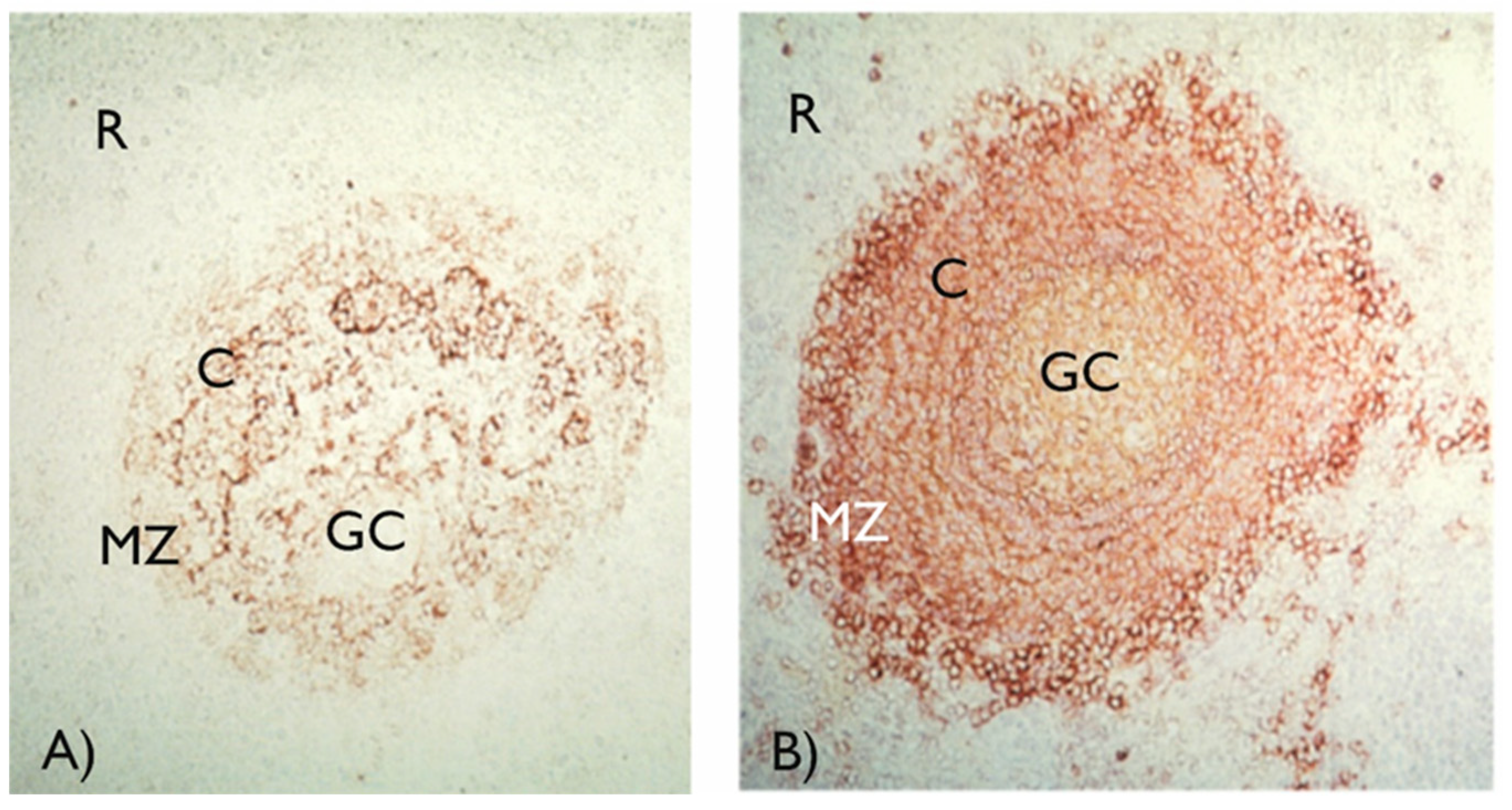
Figure 4.
Major developmental factors in microbial colonization. The progressive colonization of the infant gut is shown under the categorizations of natal status. From left to right, the figure depicts the progressive colonization of the gut with commensal (gray), probiotic (green), or pathogenic (red) bacteria. The progression of tight junction maturation is also presented (purple).
Figure 4.
Major developmental factors in microbial colonization. The progressive colonization of the infant gut is shown under the categorizations of natal status. From left to right, the figure depicts the progressive colonization of the gut with commensal (gray), probiotic (green), or pathogenic (red) bacteria. The progression of tight junction maturation is also presented (purple).
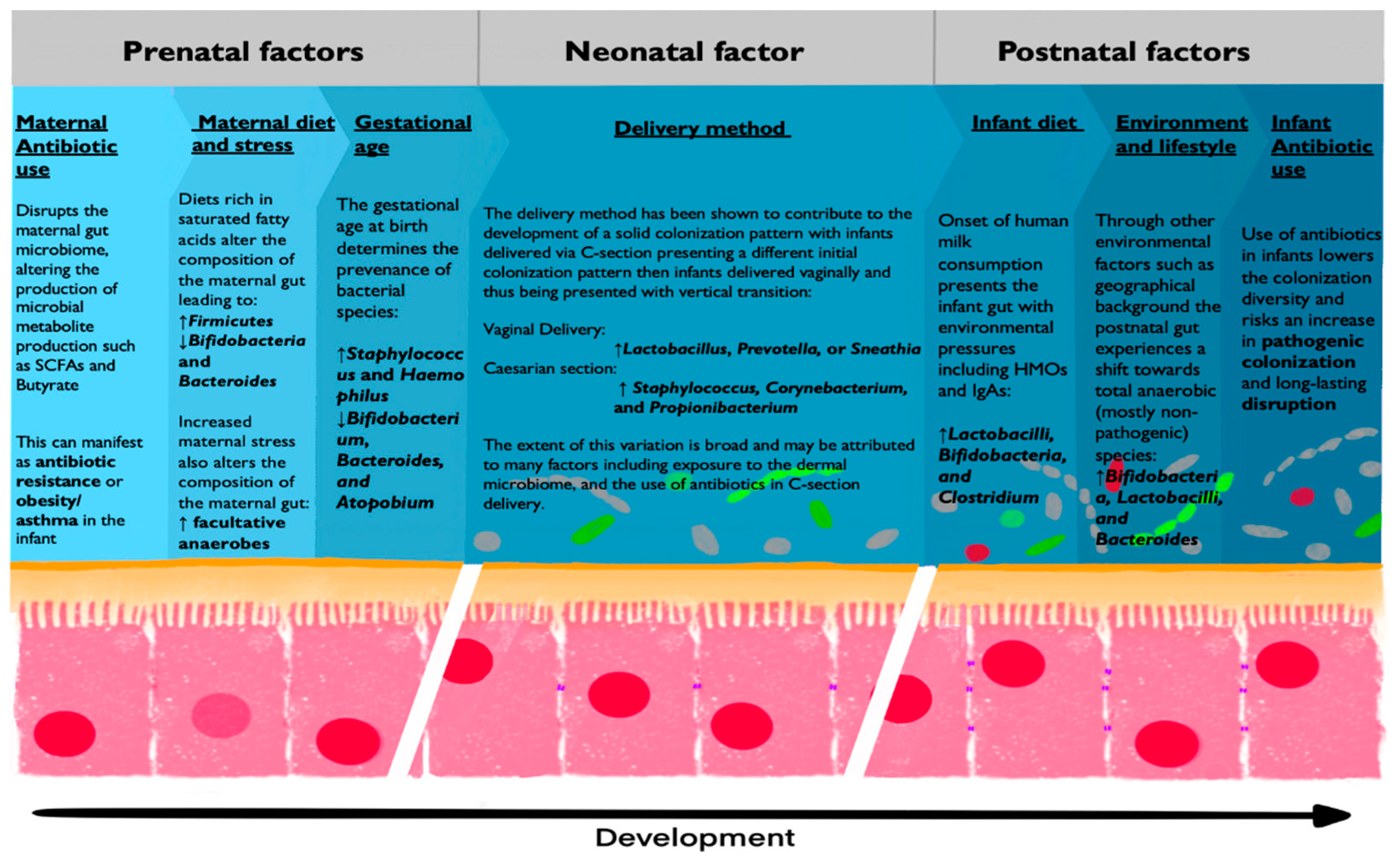
Figure 5.
Microbiota metabolites with an impact on the functionality of the immune system. Panel a shows that various bacteria are able to produce the short chain fatty acid butyrate as a byproduct of the fermentation of indigestible carbohydrates. Butyrate inhibits histone deacetylase (HDAC) which leads to activation of FOXP3 in regulatory T cells. Butyrate also activates CD8+ cytotoxic T cells which produce γ−interferon and granzymes. Dietary tryptophan can be metabolized by L. reuteri into indole-3-aldehyde (I3A) (panel b) or by L. johnsonii and subsequently C. sporogenes into indole-3-propionic acid (IPA) (panel c). I3A, via unknown mechanisms, activates CD8+ cytotoxic T cells, IPA leads to activation of the Tcf7 transcription factor, resulting in expansion of progenitor exhausted CD8+ cells (Tpex). See text for further explanation.
Figure 5.
Microbiota metabolites with an impact on the functionality of the immune system. Panel a shows that various bacteria are able to produce the short chain fatty acid butyrate as a byproduct of the fermentation of indigestible carbohydrates. Butyrate inhibits histone deacetylase (HDAC) which leads to activation of FOXP3 in regulatory T cells. Butyrate also activates CD8+ cytotoxic T cells which produce γ−interferon and granzymes. Dietary tryptophan can be metabolized by L. reuteri into indole-3-aldehyde (I3A) (panel b) or by L. johnsonii and subsequently C. sporogenes into indole-3-propionic acid (IPA) (panel c). I3A, via unknown mechanisms, activates CD8+ cytotoxic T cells, IPA leads to activation of the Tcf7 transcription factor, resulting in expansion of progenitor exhausted CD8+ cells (Tpex). See text for further explanation.
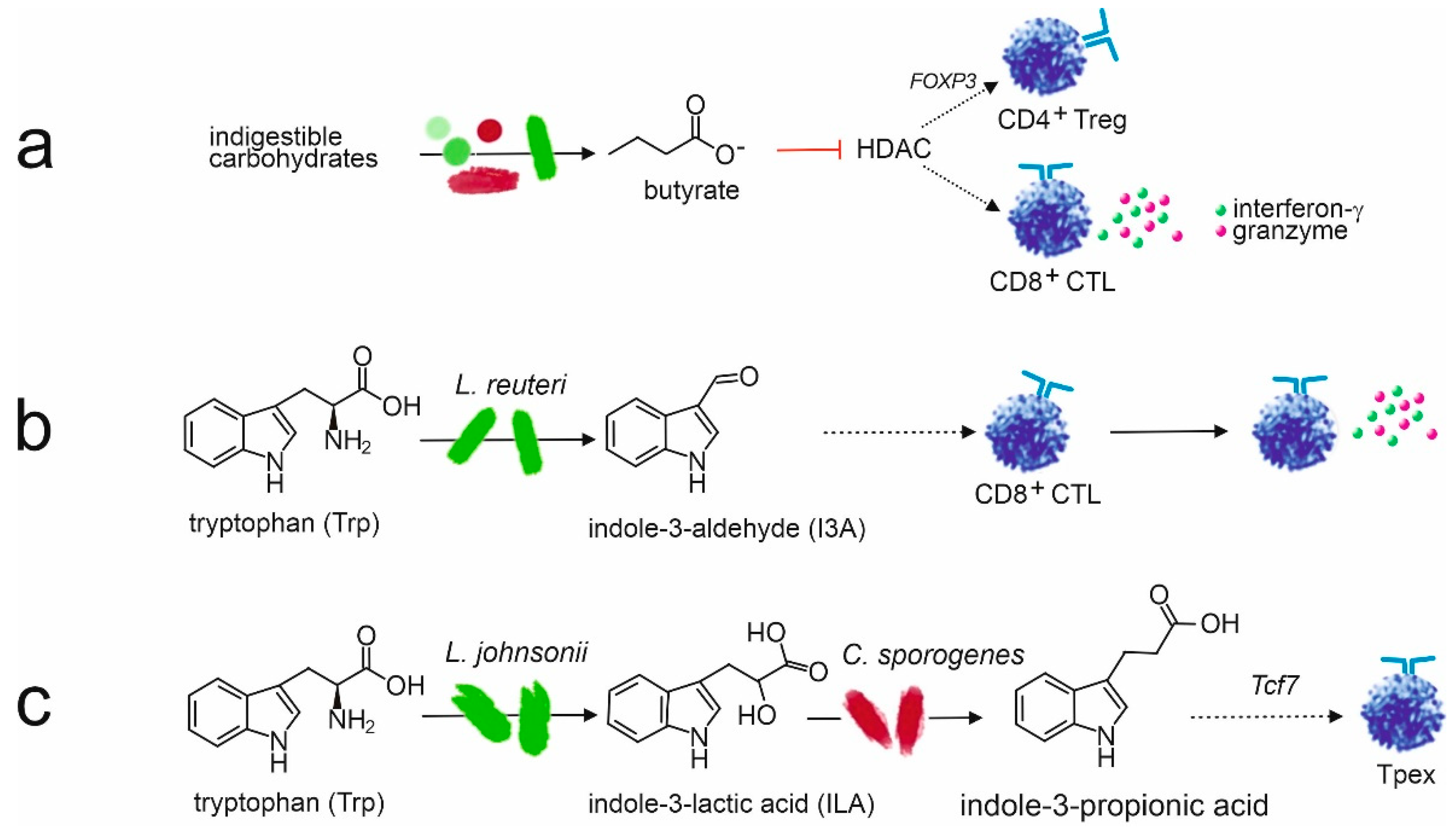
Figure 6.
Immunoglobulin Levels and Gut Microbiome Development in Early Human Life: IgG, IgM, and IgA levels are shown across fetal development, birth, and the first 1000 days postnatally. During the fetal period, IgG levels rise due to maternal transfer via the placenta. Postnatally, IgG levels initially decline as maternal antibodies are metabolized, but they gradually increase as the infant's immune system matures and begins endogenous IgG production. IgM and IgA levels are present in smaller amounts, with both rising at a slower rate compared to IgG. The line representing bacterial diversity and abundance in the infant gut microbiome demonstrates a rapid postnatal expansion. This increase begins with early colonization and continues through the first 1000 days.
Figure 6.
Immunoglobulin Levels and Gut Microbiome Development in Early Human Life: IgG, IgM, and IgA levels are shown across fetal development, birth, and the first 1000 days postnatally. During the fetal period, IgG levels rise due to maternal transfer via the placenta. Postnatally, IgG levels initially decline as maternal antibodies are metabolized, but they gradually increase as the infant's immune system matures and begins endogenous IgG production. IgM and IgA levels are present in smaller amounts, with both rising at a slower rate compared to IgG. The line representing bacterial diversity and abundance in the infant gut microbiome demonstrates a rapid postnatal expansion. This increase begins with early colonization and continues through the first 1000 days.
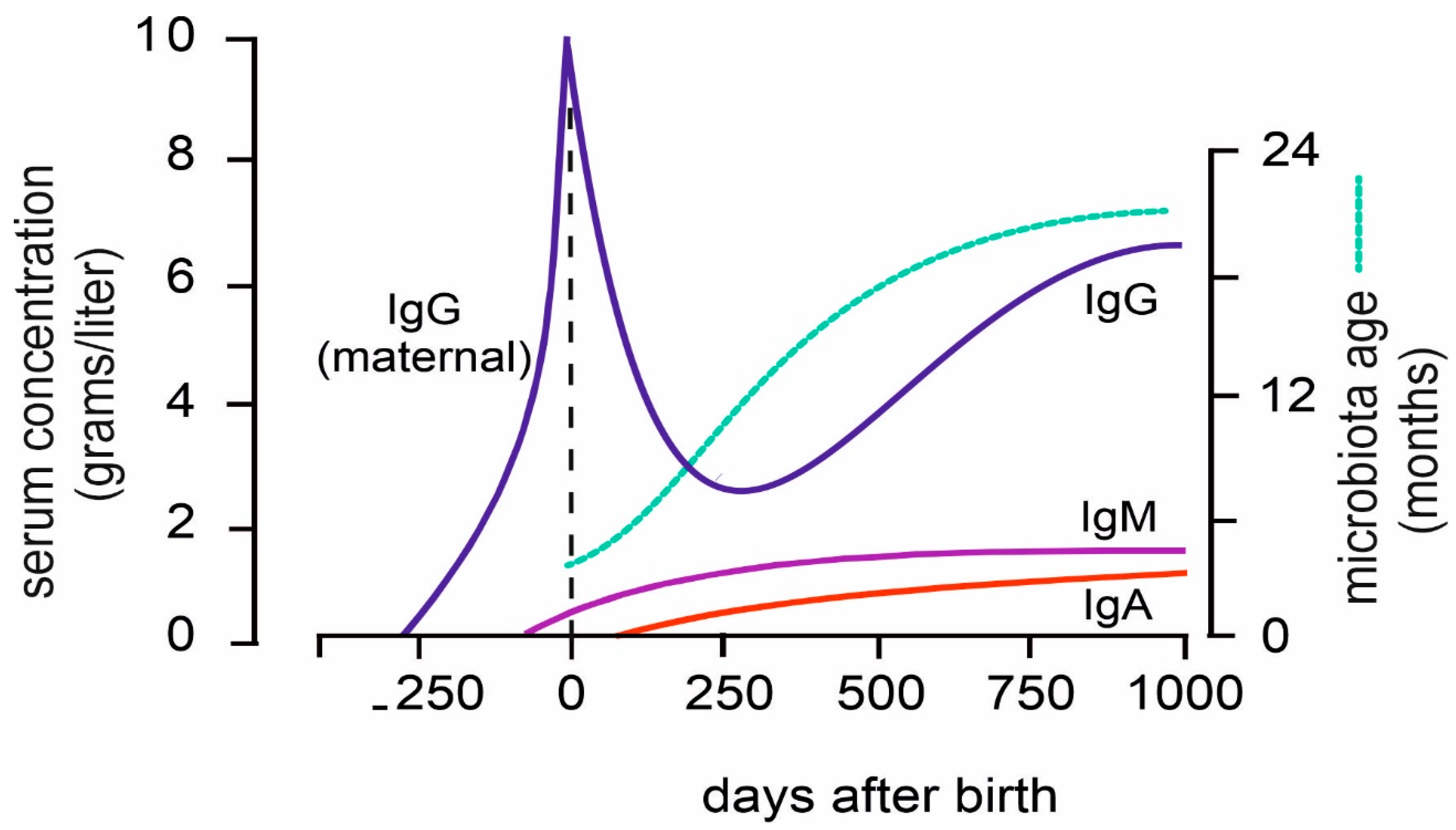
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



