Submitted:
23 January 2025
Posted:
24 January 2025
You are already at the latest version
Abstract
This study introduces an optimized microwave-assisted extraction (MAE) method for the recovery of fatty acids from wheat bran, strategically implemented to maximize efficiency while fully adhering to sustainability practices. For an environmentally friendly approach, ethanol was selected as solvent, used alongside potassium hydroxide (KOH) to facilitate the extraction. The effect of water content on extraction yields was investigated, with results confirming that water negatively affects ethanolysis. The method was systematically refined to reduce solvent consumption and streamline the process, resulting in consistent fatty acid recovery yields and demonstrating the reproducibility of the approach. To ensure the efficiency of the implemented extraction protocol, nonadecanoic acid (C19) was used as an internal standard, thus further validating the method’s precision. The recovered fatty acids will serve as a hydrophobic component in the synthesis of biodegradable surfactants when conjugated with inulin, a renewable polysaccharide. Overall, this optimized MAE method offers a sustainable and reproducible approach for the recovery of fatty acids from agrifood waste, contributing to the circular economy. agri-food byproducts, microwave, extraction, fatty acids, GC-MS.
Keywords:
agri-food byproducts
; microwave
; extraction
; fatty acids
; GC-MS
1. Introduction
In the context of growing emphasis on sustainability, crop recycling gained prominence as a solution to minimize waste and enhancing resource efficiency. Agrifood sectors, especially widely cultivated crop such as wheat and corn, play a pivotal role in this transition since various and significant by-products are generated. Specifically, husks, bran and germ are often underutilized or discarded. Within the circular economy framework, the agricultural by-products can be integrated in the production cycle, thus providing high value products for other processes and closing the system’s loop. By shifting towards a more sustainable food system, staple crops could provide a significant contribution to support both economic and environmental goals.
The vegetable oil extraction industry commonly uses large amounts of organic solvents and time-consuming extraction processes. To overcome the risks associated with solvent emissions, recent advancements have been made, developing and optimizing greener extraction methods in the food industry.
Among the mostly consumed solvents is hexane. Despite it is permitted by food safety authorities like the FDA and the European Commission, it is deemed hazardous compared to green solvents such as ethanol and water. [1] The use of greener options indeed, enhances the efficiency of the process leading to shorter extraction times, decreased solvent consumption, and lower energy requirements. [2]
The necessity to overcome the disadvantages associated to conventional extraction methods prompted the incorporation of eco-friendly solvents and effective techniques to assess their performance with various oil sources. In this context, microwave-assisted irradiation has emerged as a promising approach for vegetal samples treatment. Microwave-assisted extraction (MAE) relies on the use of electromagnetic waves that directly heat the solvent, resulting in rapid and uniform energy transfer leading to faster procedure with enhanced yields. Among the numerous advantages, temperature control represents a crucial benefit of MAE as it allows to preserve the integrity of heat-sensitive compounds. [3]
By optimizing energy use and minimizing solvent waste, MAE provides an ideal method for the sustainable extraction of bioactive compounds from agrifood by-products, making the process both cost-effective and environmentally friendly, thus aligning with green chemistry principles. [4]
Since microwave radiation is primarily absorbed by substances characterized by high dielectric constants, to achieve optimal extractability, careful solvent selection is crucial since it must effectively dissolve the target compounds and absorb heat efficiently. [5]
In this regard, polar solvents as methanol, ethanol and water provide a suitable choice. [6] Indeed, the direct interaction of microwaves with polar molecules within the solvent and the sample matrix accelerates molecular movement thus causing rapid heating. As a result, cell walls rupture is induced, enhancing the release of bioactive compounds. [7]
Another key aspect of the extraction process involves the use of homogeneous catalysts, such as sodium hydroxide (NaOH) or potassium hydroxide (KOH), which favours the breaking down of triglycerides into free fatty acids, simplifying the recovery. [8] This approach enables the simultaneous extraction and transesterification of fatty acids through conversion of triacylglycerols, thus drastically shortening the synthesis time, reducing the usage of solvents and limiting the number of steps.
In the present work, MAE was employed in combination with a variety of organic solvents to achieve the extraction of fatty acids from wheat bran, aiming to refine and to enhance the process while aligning with environmentally friendly practices. Preliminary experiments were carried out on corn kernels due to their substantial oil content, the associated economic and nutritional benefits and the abundant availability.
Throughout the evaluated approaches for the fatty acids recovery from wheat bran, ethanolysis in the presence of KOH was systematically investigated. Additionally, to ensure accurate quantification and process efficiency, nonadecanoic acid was utilized as an internal standard to assess recovery yields and validate the effectiveness of the optimized processes.
Within the framework of circular economy, this research aims advancing the production of biodegradable surfactants. In this context, fatty acids recovered from wheat bran serve as the hydrophobic component, while inulin, a polysaccharide derived from radice amara di Soncino, acts as the hydrophilic part. Inulin needs to be conjugated to the fatty acid to form amphiphilic surfactant molecules, resulting in the conversion of secondary products with low economic value into useful ones. This work thus highlights potential of integrating renewable resources and innovative extraction techniques to design eco-friendly surfactants entirely derived from vegetable sources.
2. Results
2.1. Determination of Moisture Content in Corn Kernels and Wheat Bran
Aiming to determine the moisture content, the corn and wheat samples were heated at 100°C, in a oven, up to constant mass. Thus, the water content was subtracted from the original mass throughout the subsequent investigations. Water content estimation was thus essential to guarantee accuracy. The results are summarized in Table 1:
2.2. Oil Extraction Yield
Corn is known as a valuable resource for oil extraction; notably, corn oil composition exhibits a favourable fatty acids profile, with high levels of linoleic (52%) and oleic (30%) acids; [9] moreover, this higher-quality vegetable oils offers significant amounts of polyunsaturated and monounsaturated fats that are beneficial for health. Aiming to optimize the fatty acids recovery, preliminary experiments with microwave-assisted extraction (MAE) were firstly carried out on easely available corn kernels.
For the purpose, a variety of organic solvents were explored including hexane, DCM, isopropanol, and ethanol. Owing to their different properties, the process efficiency was evaluated relying on solvent polarity and their ability to interact with fatty acids.
In this context, non-polar solvents, like n-hexane, are particularly indicated for hydrophobic fatty acids extraction, compared to slightly more polar solvents as DCM. DCM indeed provides an effective alternative to extract a broader spectrum of fatty acids, promoting faster and higher microwave absorption due to its enhanced dielectric properties; additionally, the DCM low boiling point is suitable for easy solvent recovery. Isopropanol was selected for its higher polarity, due to its ability to disrupt cell membranes enhancing the subsequent lipid extraction from the matrix. [10] Ultimately, in the context of sustainability, ethanol was taken into account as it is considered a green solvent, [11] thus offering environmental benefits. The oil extraction yields obtained for each corn sample are summarized in Table 2.
As expected from the outline, the data collected revealed that the highest efficiency belongs to the most polar solvents, isopropanol and ethanol, with isopropanol (12.85%) slightly outperforming ethanol (11.45%). The combination of MAE with suitable polar solvents like ethanol or isopropanol indeed, further improves the process performance, owing to a more effective solvent penetration. [12] Overall, DCM (9.33%) and n-hexane (7.41%) were found to be less effective under the tested conditions.
Following the preliminary extractions, the direct hydrolysis with NaOH (1M) under microwave conditions was attempted, given that water is the most valuable solvent in the context of ecology. Alkali hydrolysis can indeed directly break down triglycerides into free fatty acids and glycerol, accelerating the extraction process by promoting saponification.
Nevertheless, the applied method exhibited significant drawbacks, particularly the high viscosity of the media hindered the work-up process, leading to a very low oil recovery yield (2.59%). Additionally, the TLC analysis of the alkaline crude extract showed several side products and impurities, compared to the previously described approaches with organic solvents, thus further complicating the purification.
The challenges faced with both corn kernels and aqueous media, prompted a shift toward ethanolysis; thus, we investigated the potential of MAE combined with the use of ethanol as solvent and KOH as a catalyst. This way allows a lower environmental impact, compared to conventional organic solvents like hexane and DCM, since ethanol is a renewable solvent. This new approach was directly applied to wheat bran germ, the subject of interest of the present study and chromatography purification was implemented.
Microwave-assisted ethanolysis relies on the conversion of triglycerides into ethyl esters and glycerol using potassium hydroxide (KOH) as a catalyst. The solid-liquid extraction concomitant to the transesterification was followed by acidification with hydrochloric acid (HCl) to a pH of 1. This step jump started the hydrolysis of ethyl esters back into free fatty acids. The free fatty acids were then isolated through extraction with hexane.
For the purpose, two different extraction media distinguished by the water content, SA and SB, whose preparation is described in paragraph 4.8, were tested.
In this context, a transition from measuring solvent volume in millilitres (mL) to mass in grams (g) was performed. Mass measurement indeed, provides greater precision not being affected by the typical volume measurements limitations such as the errors from temperature fluctuations and meniscus reading. Furthermore, using mass eliminates issues of volume contraction that occur when preparing solutions with strong bases. This allows for more consistent control over solute concentration, regardless of density variations or temperature effects. Throughout the modifications introduced, the solid-liquid mixture from the MAE was subjected to centrifugation instead of filtration through a sintered glass funnel to separate the solid residue from the supernatant, aiming to minimize sample loss. The experimental conditions and the yields are reported in Table 3.
The data collected demonstrated that a single 30 min extraction with 18g of SB, thus minimizing the amount of water in the reaction mixture, provided higher yields (3.10%) compared to the sample treated with SA (2.47%). Conversely, multiple extraction cycles using smaller quantities of solvent, freshly replaced at each step, yielded consistent results, ranging between 2.86% and 2.99%.
Based on the results, it was evident that the presence of water significantly impacts the extraction process. Specifically, the yields obtained with solvent system SA, which contains additional water, are consistently lower compared to those achieved with media lacking added water. This corroborates the observation that water adversely affects the efficiency of ethanolysis, as it appears to negatively influence the yield of ethyl esters formation. [13]
In line with the principles of circular economy, further optimizations were performed by reducing the volumes of water and hexane during the washing steps post MAE extraction. The revised procedure, conducted with SB media and a single 30-min MAE cycle, was repeated twice to ensure data consistency and the results highlighted variable yields of 2.81% and 2.29%, respectively. To improve the control over the total procedure and to reduce yield variability, the pre-washing steps before acidification were eliminated, as chromatography was ultimately applied for purification. The experiments were repeated three times and the results are summarized in Table 4.
The data collected exhibited consistent yields across the three replicates, resulting in a mean value of 4.62% with a standard deviation of 0.20%.
In summary, the results validated the effectiveness and reproducibility of the optimized protocol, achieving high oil recovery with reduced solvent usage. The process evolved systematically from the use of traditional solvent extraction methods including hexane and DCM, to NaOH hydrolysis, and finally to ethanolysis with KOH. The outlined adjustments demonstrated that the protocol not only improved the extraction performance but provides an environmentally friendly approach for extracting fatty acids from wheat.
2.3. Wheat Oil Composition
The GC-MS chromatographic method successfully separated and profiled FAME mixtures within an 86-minute analysis. Experimental data concerning corn kernels, not reported since not relevant in this study, are in agreement with literature data. [14] The relative abundances of each component identified in the wheat germ samples, along with the corresponding m/z values and retention times are summarized in Table 5.
The wheat oil profiling revealed the presence of a variety of fatty acids, including lauric acid (C12:0) and myristic acid (C14:0), both present in relatively low amounts, 1.12% and 0.39%, respectively. Pentadecanoic acid (C15:0) was detected at 0.31%, while palmitoleic acid (C16:1) appeared in slightly higher abundance at 0.83%. Palmitic acid (C16:0), comprising 17.53% of the total, and linoleic acid (C18:2), making up 53.15%, represented the most abundant components alongside the significant contribution from the 10-trans,12-cis-linoleic isomer, which accounted for 20.35%.
The observed presence of 10-trans,12-cis-linoleic acid directly results from the microwave-assisted extraction with ethanol and KOH. This method indeed leads to mass and heat transfer enhancement, thus promoting a more effective isomerization of linoleic acid. [15]
Stearic acid (C18:0), gondoic acid (C20:1), and arachidic acid (C20:0) were present in lower amounts, being 2.28%, 1.60%, and 0.42%, respectively. Other minor components included erucic acid (C22:1), behenic acid (C22:0), tricosanoic acid (C23:0), nervonic acid (C24:1), and lignoceric acid (C24:0), with relative abundances ranging from 0.14% to 0.66%.
Notably, the relative abundances are consistent with previously reported data in the literature, [16,17] confirming the reliability of the results.
A further characterization of the fatty acid mixture was achieved employing 1H-NMR spectroscopy, which provided detailed insights into the chemical structure of the molecular species. The spectra revealed distinct chemical shifts: terminal methyl groups (CH₃) appeared at 0.8-1.0 ppm, methylene groups (CH₂) within the fatty acid chains were observed at 1.2-1.6 ppm, whereas β and allylic protons were detected at 1.6-1.9 ppm and 1.9-2.0 ppm respectively. Alpha protons were identified at 2.2-2.5 ppm. The peak at 2.5-3.0 ppm are indicative of the presence of unsaturated fatty acids, further supported by the vinylic protons (CH=CH) signal observed at 5.3-5.5 ppm. The 1H-NMR spectrum (Figure 1) corroborates the findings from the GC-MS analysis, confirming the presence of both saturated and unsaturated fatty acid components in the mixture.
2.4. C19 Standard Calibration Curve
The calibration curve for methyl decanoate (Figure 2) was built by analysing solutions with concentrations ranging from 1 to 50 µg/mL using GC-MS. The data showed a strong linear relationship, with an R² value of 0.9974. The equation of the curve is y = 281636x - 438250.
The injection of the reference molecule within the same concentration range used for the calibration curve allowed to assess the repeatability, which was expressed as the relative standard deviation (RSD). For the purpose, each sample was injected three times. The results demonstrated reliable precision for peak areas, with RSD values ranging between 0.3% and 2.5%, with an average RSD value of 1.3%.
2.5. Internal Standard Recovery
In the extraction process, nonadecanoic acid (C19) was employed as an internal standard to evaluate the efficiency of the implemented microwave-assisted extraction (MAE) methodology. The internal standard was added to the extraction medium and treated as described in detail in paragraph 4.13. The quantification of the recovered C19 was performed integrating the area of the peak at 52.40 min (Figure 3) and relying on the calibration curve. The experiment was conducted in triplicate to assess the reproducibility. The average recovery of the internal standard throughout the extraction was determined to be 83.2%, with a relative standard deviation (RSD) of 5.5%. Given the complexity of matrix, of the procedure and the number of steps involved that influence the overall recovery yield, the optimized extraction process was found to be efficient, resulting in limited losses of the molecules of interest. Furthermore, the RSD reflects a moderate variability between replicates, indicating a good level of precision. This suggests that the method is robust and reproducible, despite the inherent challenges posed by the extraction process.
3. Discussion
The present study demonstrates the successful application of microwave-assisted extraction (MAE) combined to ethanolysis for effective recovery of fatty acids from wheat bran germ. Wheat bran germ was selected as a more promising substrate due to its relevance to the study’s objective of valorizing sustainable agricultural by-products. The choice of ethanol together with potassium hydroxide (KOH) as a catalyst enhanced sustainability and efficiency of the processes. Known to be renewable and less harmful to the environment, ethanol is seen as a safer and better alternative to traditional solvents like hexane or dichloromethane (DCM). The process optimization demonstrated that the absence of additional water (SB medium) led to higher yields, indicating that water negatively impacts ethanolysis. This effect can be attributed to water's propensity to promote saponification and emulsion formation, which interfere with the efficient extraction of free fatty acids. The study also explored the effects of different extraction cycles and solvent-to-sample ratios. The single-cycle extraction with SB medium consistently provided higher yields compared to multiple cycles with smaller solvent volumes. This finding suggests that a prolonged single exposure to microwave irradiation allows for more effective disruption of the cell matrix and enhanced release of fatty acids, while minimizing solvent consumption. Furthermore, the analysis of the extracted fatty acids revealed a composition rich in linoleic acid (C18:2) and palmitic acid (C16:0), alongside other beneficial unsaturated fatty acids. The presence of the 10-trans,12-cis-linoleic acid isomer, which is known to arise from isomerization processes, indicates that the MAE method may induce structural modifications. This isomerization highlights the potential for tailored extraction processes to enhance specific fatty acid profiles. The reproducibility of the extraction protocol was demonstrated through multiple experimental replicates, yielding a mean recovery rate of 4.62% with a low standard deviation (0.20%). The use of nonadecanoic acid (C19) as an internal standard provided a reliable means of quantifying extraction efficiency and method precision, with an average recovery of 83.2% and a relative standard deviation (RSD) of 5.5%. The optimized MAE protocol thus offers a sustainable and reproducible method for extracting fatty acids from wheat bran, contributing to the circular economy by transforming low-value agricultural waste into valuable bio-based products. The study’s findings pave the way for further exploration of MAE’s application in extracting bioactive compounds from various substrates, with potential implications for the production of eco-friendly surfactants.
4. Materials and Methods
4.1. Materials
Reagents and solvents of the purest grade available for GC-MS, hydrochloric acid, trifluoroacetic acid, thionyl chloride, dry methanol, MeONa in dry MeOH (25% solution), sodium hydroxide and Amberlite IR120 were supplied by Sigma Aldrich. Silica gel chromatography was purchased from Merck (Supelco, high-purity grade, pore size 60 Å, 230-400 mesh particle size). Absolute ethanol, diisopropyl ether and hexane were purchased from Carlo Erba, potassium hydroxide was acquired from Panreac and anhydrous sodium sulphate (Na₂SO₄) from Alfa Aesar GmbH (Karlsruhe, Germany).
Deionized water was obtained using a Milli-Q Water purification system (Millipore Corporation, Bedford, MA). were purchased from Sigma-Aldrich.
Dry milled corn and wheat bran where kindly provided by Mulino Sobrino, La Morra (CN), Italy and stored in a freezer at -20°C.
4.2. Instruments
Centrifugation was performed with a 5804-R (Eppendorf s.r.l., Milan, Italy) centrifuge. Microwave (MW) assisted solid-liquid extraction was conducted with a Biotage initiator+ instrument (Uppsala, Sweden) with a maximum power of irradiation of 400W (2.45 GHz).
GC/MS analyses were carried out on a Thermo Scientific DSQII single quadrupole GC/MS system (TraceDSQII mass spectrometer, Trace GC Ultra gas chromatograph, TriPlus autosampler - ThermoFisher Scientific, Waltham, MA, USA).
The characterization of purified FFA mixtures were performed recording the NMR spectra on a Bruker Advance III spectrometer operating at 400 MHz (Bruker Corporation, Billerica, MA, USA) setting standard pulse sequences provided in the Bruker Topspin 3.6 software. Chemical shifts of 1H NMR spectra are reported in parts per million (ppm) and the coupling constants are measured in Hertz (Hz).
4.3. Microwave-Assisted Extraction of Fatty Acids from Wheat and Corn
Dry milled corn was initially used to carry out preliminary extractions combining microwave irradiation with the use of organic solvents. For the purpose, a variety of organic solvents have been tested. Specifically, 1g of milled corn was suspended in 10 mL of either n-hexane, dichloromethane, ethanol or isopropanol and subjected to microwave extraction. The procedure was carried out for 10 min, setting the temperature at the boiling point of the solvent: n-hexane (70°C), dichloromethane (40°C), isopropanol (82°C) and ethanol (78°C). The suspension was then filtered over a sintered glass filter and the solvent evaporated in vacuo. The thus obtained oily residue was transesterified with MeONa in dry MeOH (25% solution) under inert atmosphere. The reaction was monitored through TLC and quenched with Amberlite IR120 upon complete consumption of the starting material. The acidic resin was removed through filtration and the solvent dried under reduced pressure. The fatty acids methyl esters (FAME) were thus characterized by GC-MS without further purification. The extraction procedure afforded the fatty acids mixture as FAME, but for subsequent conjugation with hydrophilic substances, pure free fatty acids (FFAs) were required. Due to the formation of by-products during the hydrolysis of FAME, the conditions were modified to directly obtain FFAs, bypassing the intermediate steps.
Thus, the extraction and hydrolysis were conducted simultaneously under the following conditions: dry milled corn (1 g) was suspended in 10 mL of 1M NaOH solution and heated in a microwave at 100°C for 10 minutes, followed by filtration through a Büchner funnel. The aqueous solution was extracted with n-hexane (3 x 50 mL) to remove the unsaponifiable fraction. The organic layers were collected, washed with water, and allowed to equilibrate before separation. The combined aqueous fractions were acidified with 6N HCl to pH 1 to release the free fatty acids, which were then extracted with n-hexane (3 x 100 mL). The organic phases were washed with water, with NaCl added to clear any remaining emulsion, and allowed to settle before separation. After drying with anhydrous Na2SO4, the solvent was filtered and evaporated under reduced pressure at 35°C. The crude product was purified by flash chromatography using n-hexane and diisopropyl ether (60/40 v/v) with the addition of 0.2% trifluoroacetic acid. After solvent removal, the oily residue was dried in vacuo to constant weight.
The focus was then directed to wheat bran, which offers significant potential for contributing to circular economy efforts. The aforementioned procedure was thus repeated on wheat bran and to overcome issues of viscosity of the media, which significantly hindered handling, the procedure was modified accordingly. This involved changing the media composition and utilizing a centrifuge instead of a Büchner funnel, enhancing manageability and efficiency.
In addition to these modifications, we switched from measuring the solvent volume from millilitres (mL) to grams (g) for improved accuracy.
Thus, a suspension of 1g of wheat bran in absolute ethanol was prepared and an aqueous solution of 20M KOH was added. The heterogeneous mixture was then heated in a microwave reactor at 78°C with magnetic stirring. Two extraction media were tested, differing in water content: SA, which included an additional 5% water, and SB, which contained no extra water. Specifically, SA was prepared by mixing absolute ethanol, 20M KOH, and water in the ratios of 15.3g ethanol, 1.8g KOH, and 0.9g water for a total of 18g. For smaller batches, 5.1g ethanol, 0.6g KOH, and 0.3g water were combined to yield 6g of SA. SB was prepared using 16.2g of ethanol and 1.8g of 20M KOH for 18g of solution, while the 6g batch consisted of 5.4g ethanol and 0.6g KOH.
The total quantity of extraction media was determined based on the number of cycles. A single 30-minute extraction was performed using 18g of the ethanolic solution, while for a three-cycle process, 6g of solvent was used for each 10-minute extraction. After each cycle, the solvent was replaced with fresh 6g aliquots, reaching a total of 18g over three cycles. Additionally, a validation experiment was carried out using two consecutive 15-minute extraction cycles.
Once the solid-liquid extraction was completed, the ethanolic solution was collected, diluted in 20 mL of deionized water and centrifuged (4500g) for 20 min at room temperature (25°C) to allow the wheat residue to settle. The solvent was thus removed and replaced with fresh water (20 mL) before another centrifuge cycle. Overall, the process was repeated 4 times. The aqueous solution was then subjected to hexane washes (3 x 50 mL) in order to remove the lipophile impurities. Hexane was then collected and extracted with water (2 x 100 mL). The aqueous fractions were thus gathered all together and acidified to pH=1 with HCl 6N to induce the protonation of the extracted fatty acids. Subsequently, hexane extractions were performed (3 x 100 mL) and the collected organic phase was then washed with water (3 x 100 mL). Hexane was thus treated with dry Na2SO4, filtered and evaporated under reduced pressure. The oily residue was let dry in vacuo to constant weight and the crude product purified through silica gel chromatography (60:40, hexane: diisopropyl ether (v/v) + 0.2% trifluoroacetic acid). A schematic representation of the extraction of the vegetable oil from wheat bran is provided in Figure 4.
4.4. Optimization of Liquid-Liquid Extraction
In the framework of circular economy principles, the primary objective was to minimize the use of organic solvents. Therefore, the liquid-liquid extraction following microwave-assisted extraction was optimized. To achieve this, the experiment involving the suspension of 1g of wheat bran in SB for a single 30-minute extraction cycle was replicated using the revised methodology. This procedure was repeated three times to ensure data comparability.
Initially, the volume of water used to dilute the ethanolic solution and wash the wheat bran residue upon centrifugation was reduced from 80 mL to 40 mL: three centrifugations were performed using 20 mL at first and 10 mL for the last two rounds (1 x 20 mL + 2 x 10 mL). Similarly, the volume of hexane for subsequent washes was decreased from 50 mL to 30 mL (3 x 30 mL), and the final water wash volume was reduced from 100 mL to 50 mL (3 x 50 mL). After acidifying the aqueous phase with HCl (4 mL), the extraction was carried out using 50 mL of hexane (3 x 50 mL), while water was reduced to 50 mL (3 x 50 mL). The crude product was purified through silica gel chromatography using a mixture of hexane and diisopropyl ether (60:40, hexane:diisopropyl ether (v/v)) with the addition of 0.2% trifluoroacetic acid and the oily residue was then let dry in vacuo to constant weight.
Moreover, in order to further reduce solvent consume and streamline the process, a final column chromatography replaced the pre-washes before acidification, making the workup less time-consuming without compromising the quality of the extraction.
4.5. Esterification of the Free Fatty Acids Mixture
Aiming to determine the composition of the isolated products, the free fatty acids were treated with thionyl chloride for the conversion into methyl esters fatty acids (FAME). The overall reaction can be summarized as follows:
R-COOH + SOCl2 + CH3OH→R-COOCH3 + SO2 + CO2 + HCl
Referring to the literature [18], the oily residue was dissolved in dry MeOH in a round bottomed flask filled with nitrogen and placed in an ice-bath at 0°C. Thionyl chloride (1.5 eq.) was then added in a dropwise manner under stirring. In the end, the flask was sealed and the mixture let react for 1h. The progression of the reaction was monitored through TLC. Upon completion, the solvent was evaporated in vacuo, and the product used without any further purification.
4.6. Determination of Corn Kernels and Wheat Bran Moisture Content
Dry milled corn kernels and wheat bran (500 mg) were placed in an oven heated at 100°C till constant weight, aiming to determine their humidity content.
Given the initial weight of the samples (Wi) and their weight after the thermal treatment (Wf), the moisture content was calculated as follows:
The yield of each extraction process was determined taking into account the moisture content of the sample to ensure accurate determination of the dry matter extracted. The assays were performed in triplicate and the results are expressed as the mean ± standard deviation.
4.7. Oil Extraction Yield
The oil extraction yield for the overall process was calculated as the ratio of the grams of oil obtained to the weight of corn or wheat suspended in the solvent, expressed as a percentage:
Specifically, for the preliminary extractions with corn the amount of oil in the numerator refers to the product obtained after the liquid-liquid extraction, since the mixture was analysed without further purification. In contrast, the yield for wheat was calculated after the silica gel chromatography purification and taking into account the moisture content.
4.8. Solutions Preparation
- SA and SB solutions
In the context of the optimization of extraction procedures, the preparation of two different extraction media, SA and SB, was undertaken. These media differed in their water content to assess their efficiency in extracting free fatty acids from wheat bran.
For the preparation of SA, the mixture included absolute ethanol, a 20M KOH solution, and an additional 5% of water. Specifically, to prepare 18 grams of SA, 15.3 grams of absolute ethanol were combined with 1.8 grams of 20M KOH and 0.9 grams of water. For the samples subjected to three MW extraction, 6 grams of SA were prepared for each cycle by mixing 5.1 grams of absolute ethanol, 0.6 grams of 20M KOH, and 0.3 grams of water.
Conversely, the preparation of SB did not include any additional water, relying solely on absolute ethanol and the 20M KOH solution. To prepare 18 grams of SB, 16.2 grams of absolute ethanol were mixed with 1.8 grams of 20M KOH. Similarly, for 6 grams, SB was prepared using 5.4 grams of absolute ethanol and 0.6 grams of 20M KOH.
The control experiment was then conducted preparing 6 grams of the extraction media and using it for two 15-minute extraction cycles.
SA and SB were freshly prepared for each use and utilized immediately thereafter.
- KOH solution
20M KOH was freshly prepared in a volumetric flask daily in 5 mL aliquots to minimize waste and was utilized throughout the day. If there was a delay between preparations, the solution was stored in the refrigerator (4°C) for a few hours to maintain stability.
- Standard stock and working solutions
The calibration curve of nonadecanoic acid (C19) was built through GC-MS. In order to detect the standard, it was at first esterified as described in paragraph 4.5.
A stock solution of methyl nonadecanoate (1 mg/mL) was prepared by dissolving the standard in analytical-grade DCM and stored at -20°C. For the linearity study, the stock solution was appropriately diluted with DCM to achieve concentrations ranging from 1 to 50 µg/mL (3.3-167.5 µM).
4.9. GC-MS for Fatty Acids Analysis
The esterified fatty acids were dissolved in analytical-grade dichloromethane (DCM) and appropriately diluted to ensure optimal conditions for GC-MS analysis. Chromatography was performed on a Rxi-5Sil MS capillary column (30 m length x 0.25 mm ID x 0.25 µm film thickness, Restek, Milan, Italy) with helium (>99.99%) as the carrier gas at a constant flow rate of 1.0 mL/min. An injection volume of 1 µL was employed, with the injector temperature set at 250°C and operated in split mode, with a split flow of 10 mL/min. The oven temperature program started at 45°C (isothermal for 4 min), then increased to 175°C at a rate of 13°C/min, and finally to 215°C (isothermal for 35 min) at a rate of 4°C/min. The mass transfer line temperature was set at 250°C and the total GC runtime was 86 minutes. Mass spectra were acquired using an electron ionization system (EI, Electron Impact mode) with ionization energy of 70 eV, a source temperature of 250°C and a spectral acquisition in Full Scan mode, positive polarity, over a mass range of 35-650 Da with a scan rate of 804 amu/s.
Chromatogram acquisition, mass spectral peak detection, and waveform processing were carried out using Xcalibur MS Software Version 2.1 (Thermo Scientific Inc.). Chemical structures were assigned to chromatographic peaks by comparing the mass spectra with the databases from the NIST Mass Spectral Library (NIST 08) and the Wiley Registry of Mass Spectral Data (8th Edition).
FAMEs were identified by extracting their molecular ions from the total ion current, and the peaks at their corresponding retention times were integrated.
4.10. Wheat Oil Composition
The composition of wheat oil extracts was analysed using GC-MS to determine the area percentage and relative concentration of each fatty acid component. The relative abundance of each fatty acid methyl ester (FAME) was calculated by integrating the area under each FAME peak in the GC chromatogram. Specifically, the relative abundance was determined by dividing the area of each FAME peak by the total area of all FAME peaks and then multiplying by 100, yielding the percentage of each fatty acid relative to the overall mixture.
4.11. C19 Standard Calibration Curve
Solutions of methyl nonadecanoate acid with concentrations ranging from 1 to 50 µg/mL (3.3-167.5 µM) were prepared and injected in the gas chromatography system. Linearity was evaluated across five concentration levels, with three replicates run for each concentration. Calibration curves were generated using linear regression analysis.
4.12. Precision
The precision of the method for methyl nonadecanoate was evaluated by examining the repeatability of the data. Five standard solutions, each representing a concentration within the range of the calibration curve, were analyzed in triplicate. The relative standard deviations (RSDs) of the peak areas at each concentration level were calculated, along with the overall average RSD, to assess precision. The results are expressed as the mean ± standard deviation (SD).
4.13. Internal Standard Recovery
The optimized MAE conditions were applied to monitor the overall efficiency of the extraction process. For the purpose, nonadecanoic acid (C19) (4 mg) was employed to spike the extraction medium in which 1g of wheat bran was suspended in 16.2g of absolute ethanol and 1.8g of 20M KOH solution. The sealed flask was subjected to microwave extraction for 10 minutes at 78°C, followed by a simplified work-up, omitting the pre-washing steps before acidification as described in paragraph 4.4. The mixture of free fatty acids (FFAs) was subsequently purified, esterified, and analysed via GC-MS. All experiments were performed in triplicate to ensure repeatability and accuracy.
The internal standard signal was identified by extracting its ion current and integrating the corresponding peak. The peak area for each batch was determined and applied to the calibration curve equation to quantify the reference compound. Recovery (%) was calculated using the following formula:
The data collected are expressed as the mean recovery (%) with the corresponding RSD.
5. Conclusions
This study focused on the development and optimization of a microwave-assisted extraction (MAE) process for extracting fatty acids from wheat bran, with a particular emphasis on sustainability practices. The use of traditional solvents, such as hexane or dichloromethane, was replaced by the use of ethanol in combination with KOH, being the most efficient extraction medium. Ethanol was selected due to its ecofriendly and renewable properties, thus providing an optimal solvent to align to sustainability principles.
A crucial point in the optimization procedure included the evaluation of water content effect over the fatty acids extraction yields. In this context, two media, SA and SB with and without 5% of additional water respectively, were investigated under identical experimental conditions. The results displayed higher extractions yields for SB, thus indicating that water has a negative influence on ethanolysis. The data corroborates the influence of water in transesterification reactions, where water excess can lead to reduced reaction efficiency due to saponification and emulsion formation.
After the identification of the optimal solvent, the microwave extraction protocol was further refined in order to reduce the number of steps and the solvent consumption. Triplicate experiments were carried out giving rise to an overall yield of 4.62% with a relative standard deviation of 0.20%; these results assessed the protocol consistency, ensuring reliable results.
Then, nonadecanoic acid (C19) was added in the extraction media, as an internal standard, to evaluate the method robustness. The validation allowed a precise quantification of recovery rates, with an average recovery of 83.2% and a relative standard deviation (RSD) of 5.5%, indicating a high level of reproducibility and precision.
GC-MS analysis confirmed the composition of the extracted fatty acids mixture, showing a profile rich in linoleic acid (C18:2) and palmitic acid (C16:0), alongside a variety of beneficial unsaturated fatty acids. The presence of the 10-trans12-cis-linoleic acid isomer as a result of the microwave extraction, underscored the method’s capability to enhance isomerization reactions.
In conclusion, this MAE protocol achieves high yields, reduces environmental impact and offers a reproducible approach in the extraction of fatty acids from wheat bran. In addition, the overarching aim of combining fatty acids from wheat bran and inulin from radice amara di Soncino, enhances the value of this work in the production of sustainable and biodegradable surfactants, contributing to waste reduction in alignment with circular economy principles.
Author Contributions
Conceptualization, G.B., M.P.A., D.M. and E.C.; methodology, G.D.S. and G.B.; investigation, G.D.S., G.B, G.M. and F.R.; resources, D.M. and E.C.; data curation, G.D.S. and G.M.; writing—original draft preparation, G.D.S.; writing—review and editing, G.D.S., G.B., F.R. and E.C.; supervision, G.B. and E.C.; project administration, D.M. and E.C.; funding acquisition, D.M. and E.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by European Union – NextGenerationEU within the PRIN: PROGETTI DI RICERCA DI RILEVANTE INTERESSE NAZIONALE – Bando 2022 PNRR Prot. P2022WKBH7. Research project title: AGRIECOSURF – AGRIcultural by-products recycling for healthcare ECOlogic SURFactants production.
Data Availability Statement
The original contributions presented in this study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author(s).
Acknowledgments
In this section, you can acknowledge any support given which is not covered by the author contribution or funding sections. This may include administrative and technical support, or donations in kind (e.g., materials used for experiments).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- European Parliament, Council of the European Union (2008). Directive of the European and of the Council on the Approximation of the Laws of the Member States on Extraction Solvents Used in the Production of Foodstuffs and Food Ingredients.
- Chemat, F.; Rombaut, N.; Meullemiestre, A.; Turk, M.; Perino, S.; Fabiano-Tixier, A.S.; Abert-Vian, M. Review of Green Food Processing Techniques. Preservation, Transformation, and Extraction. Innov. Food Sci. Emerg. Technol. 2017, 41, 357–377. [Google Scholar] [CrossRef]
- Alara, O.R.; Abdurahman, N.H.; Ukaegbu, C.I. Extraction of Phenolic Compounds: A Review. Curr Res Food Sci 2021, 4, 200–214. [Google Scholar] [CrossRef] [PubMed]
- Biesaga, M. Influence of Extraction Methods on Stability of Flavonoids. J Chromatogr A 2011, 1218, 2505–2512. [Google Scholar] [CrossRef]
- Zhang, Q.-W.; Lin, Li.-G.; Ye, W.-C. Techniques for Extraction and Isolation of Natural Products: A Comprehensive Review. Chin. Med. 2018, 13, 1–26. [Google Scholar] [CrossRef]
- Dudley, G.B.; Richert, R.; Stiegman, A.E. On the Existence of and Mechanism for Microwave-Specific Reaction Rate Enhancement. Chem Sci 2015, 6, 2144–2152. [Google Scholar] [CrossRef]
- Kratchanova, M.; Pavlova, E.; Panchev, I. The Effect of Microwave Heating of Fresh Orange Peels on the Fruit Tissue and Quality of Extracted Pectin. Carbohydr Polym 2004, 56, 181–185. [Google Scholar] [CrossRef]
- Vicente, G.; Martínez, M.; Aracil, J. Integrated Biodiesel Production: A Comparison of Different Homogeneous Catalysts Systems. Bioresour Technol 2004, 92, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Oswell, N.J.; Gunstone, F.D.; Pegg, R.B. Vegetable Oils. In Bailey’s Industrial Oil and Fat Products; Wiley, 2020; pp. 1–30.
- Halim, R.; Gladman, B.; Danquah, M.K.; Webley, P.A. Oil Extraction from Microalgae for Biodiesel Production. Bioresour Technol 2011, 102, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Rincón-Cervera, M.Á.; Galleguillos-Fernández, R.; González-Barriga, V.; Valenzuela, R.; Speisky, H.; Fuentes, J.; Valenzuela, A. Fatty Acid Profile and Bioactive Compound Extraction in Purple Viper’s Bugloss Seed Oil Extracted with Green Solvents. JAOCS, Journal of the American Oil Chemists’ Society 2020, 97, 319–327. [Google Scholar] [CrossRef]
- Moretto, J.A.; Ossanes de Souza, A.; Berneira, L.M.; Brigagao, L.G.G.; Pereira de Pereira, C.M.; Converti, A.; Pinto, E. Microwave-Assisted Extraction of Fatty Acids from Cultured and Commercial Phytoplankton Species. Appl. Sci. 2022, 12, 2407–2415. [Google Scholar] [CrossRef]
- Fiilières, R.; Bouchra, B.-M.; Delmas, M. Ethanolysis of Rapeseed Oil: Quantitation of Ethyl Esters, Mono-, Di-, and Triglycerides and Glycerol by High-Performance Size-Exclusion Chromatography. J. Am. Oil. Chem. Soc. 1995, 72, 427–432. [Google Scholar] [CrossRef]
- Barrera-Arellano, D.; Badan-Ribeiro, A.P.; Serna-Saldivar, S.O. Corn Oil: Composition, Processing, and Utilization. In Corn: Chemistry and Technology, 3rd Edition; Elsevier, 2018; pp. 593–613.
- San, K.N.E.; Fan, Y.; Fang, Y.; Xia, Y.; Ma, L. A Soap-Free Strategy for One-Pot Synthesizing Conjugated Linoleic Acid: Alkali Isomerization of Linoleic Acid through Preferential Esterification Pathway. J. Am. Oil Chem. Soc. 2020, 97, 1021–1028. [Google Scholar] [CrossRef]
- Nemzer, B.; Al-Taher, F. Analysis of Fatty Acid Composition in Sprouted Grains. Foods 2023, 12, 1853–1869. [Google Scholar] [CrossRef]
- Slama, A.; Cherif, A.; Boukhchina, S. Importance of New Edible Oil Extracted from Seeds of Seven Cereals Species. J. Food Qual. 2021, 2021, 5531414–55311421. [Google Scholar] [CrossRef]
- Tran, H.-L.; Ryu, Y.-J.; Ho, D.S.; Lim, S.-M.; Lee, C.-G. An Effective Acid Catalyst for Biodisel Production from Impure Raw Feedstocks. Biotechnol. Bioprocess Eng. 2013, 18, 242–247. [Google Scholar] [CrossRef]
Figure 1.
1H-NMR spectrum (400 MHz, CDCl3) of the free fatty acids mixture obtained from wheat bran after purification.
Figure 1.
1H-NMR spectrum (400 MHz, CDCl3) of the free fatty acids mixture obtained from wheat bran after purification.
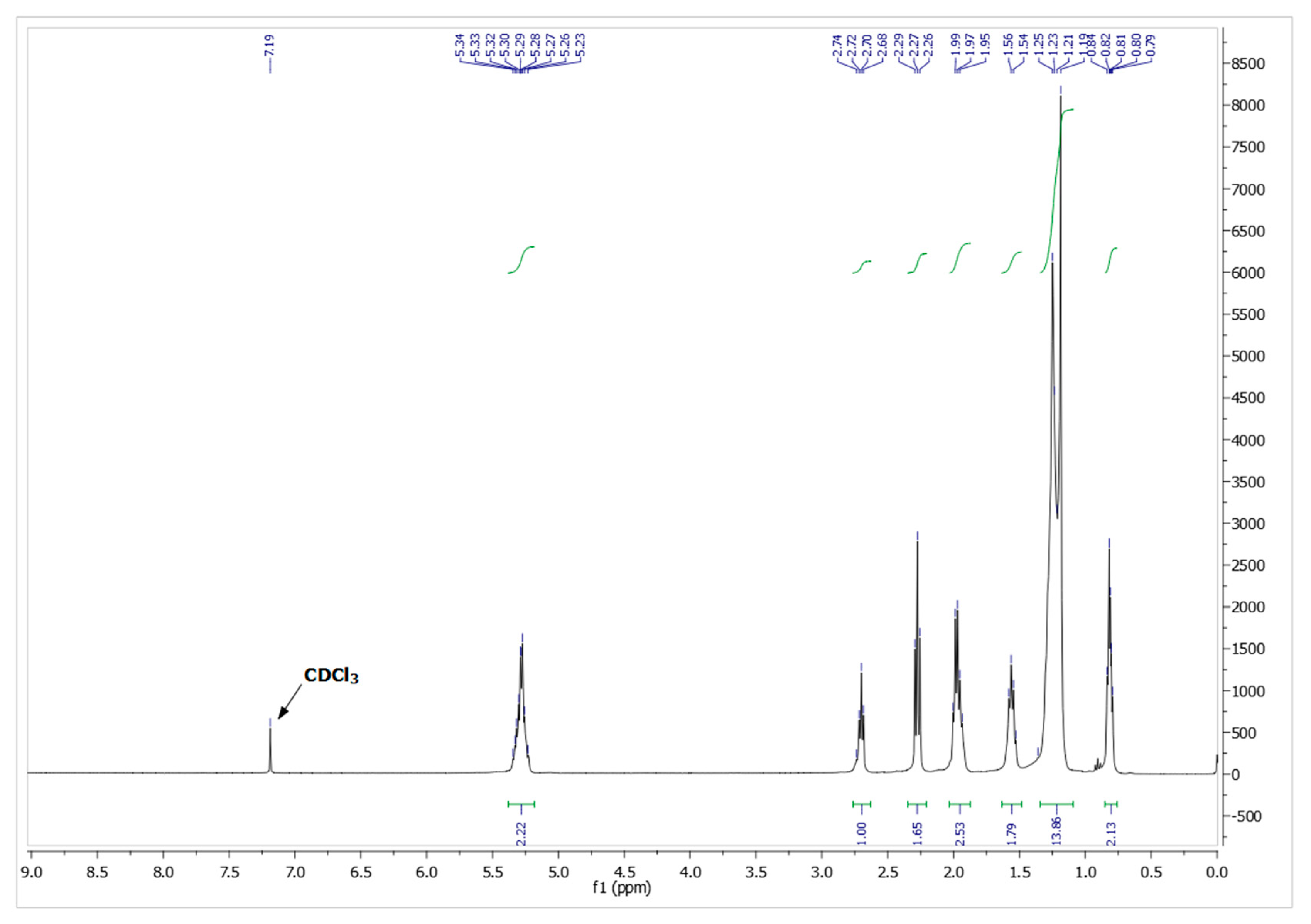
Figure 2.
GC-MS calibration curve of methyl decanoate in the 1 to 50 µg/mL range.
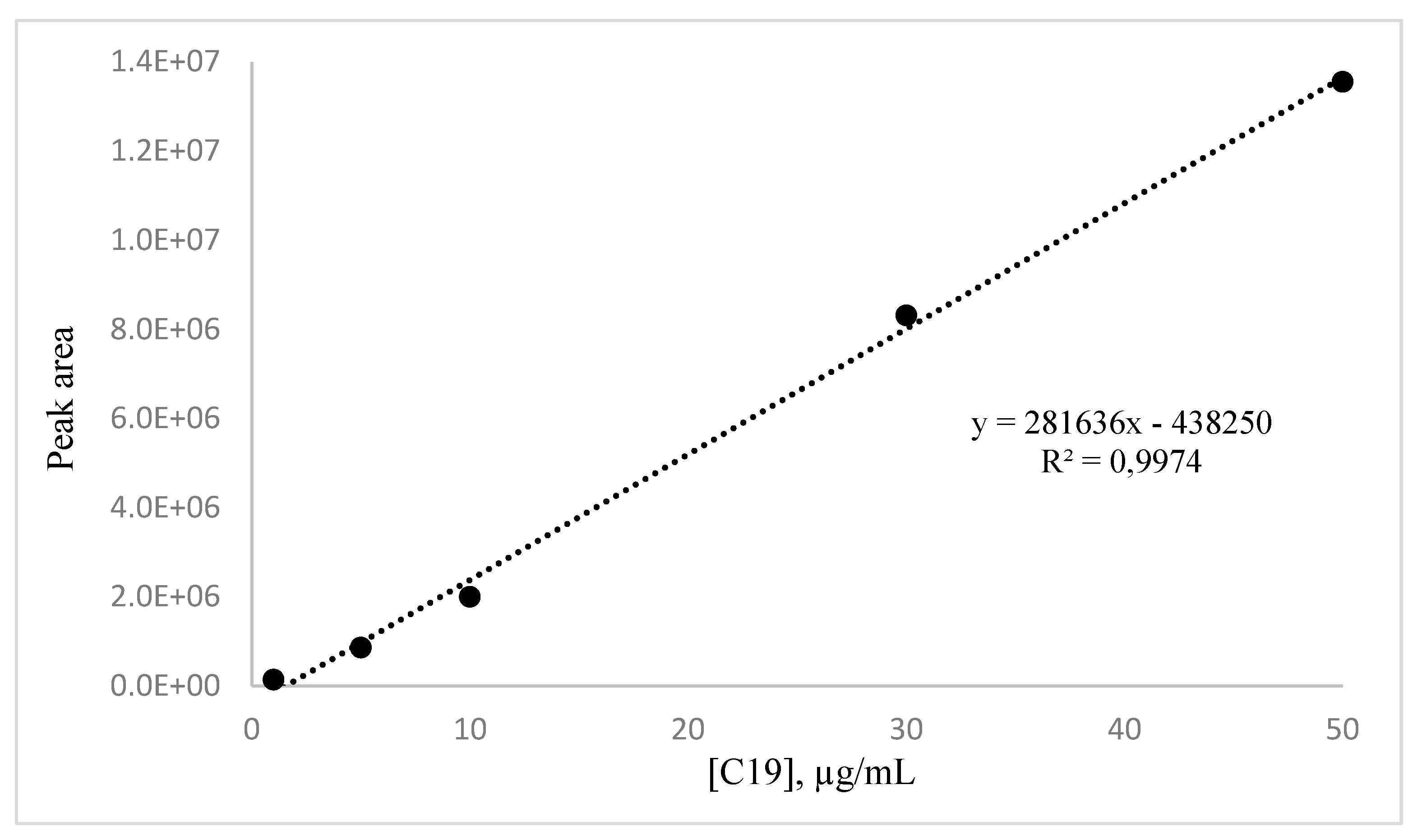
Figure 3.
Chromatographic trace of a) the extracted fatty acids mixture enriched with C19 standard and of b) C19 internal standard.
Figure 3.
Chromatographic trace of a) the extracted fatty acids mixture enriched with C19 standard and of b) C19 internal standard.
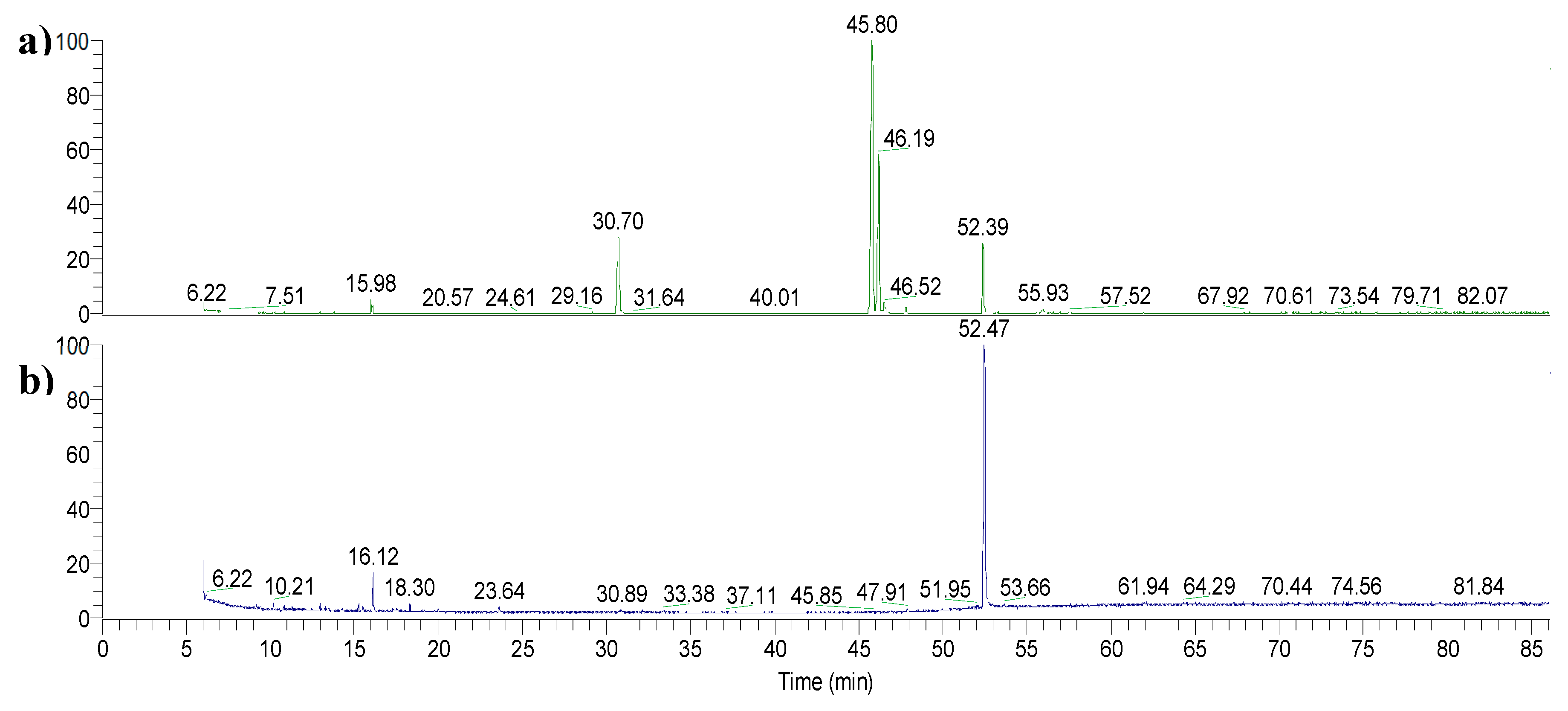
Figure 4.
Illustration of the extraction process steps.
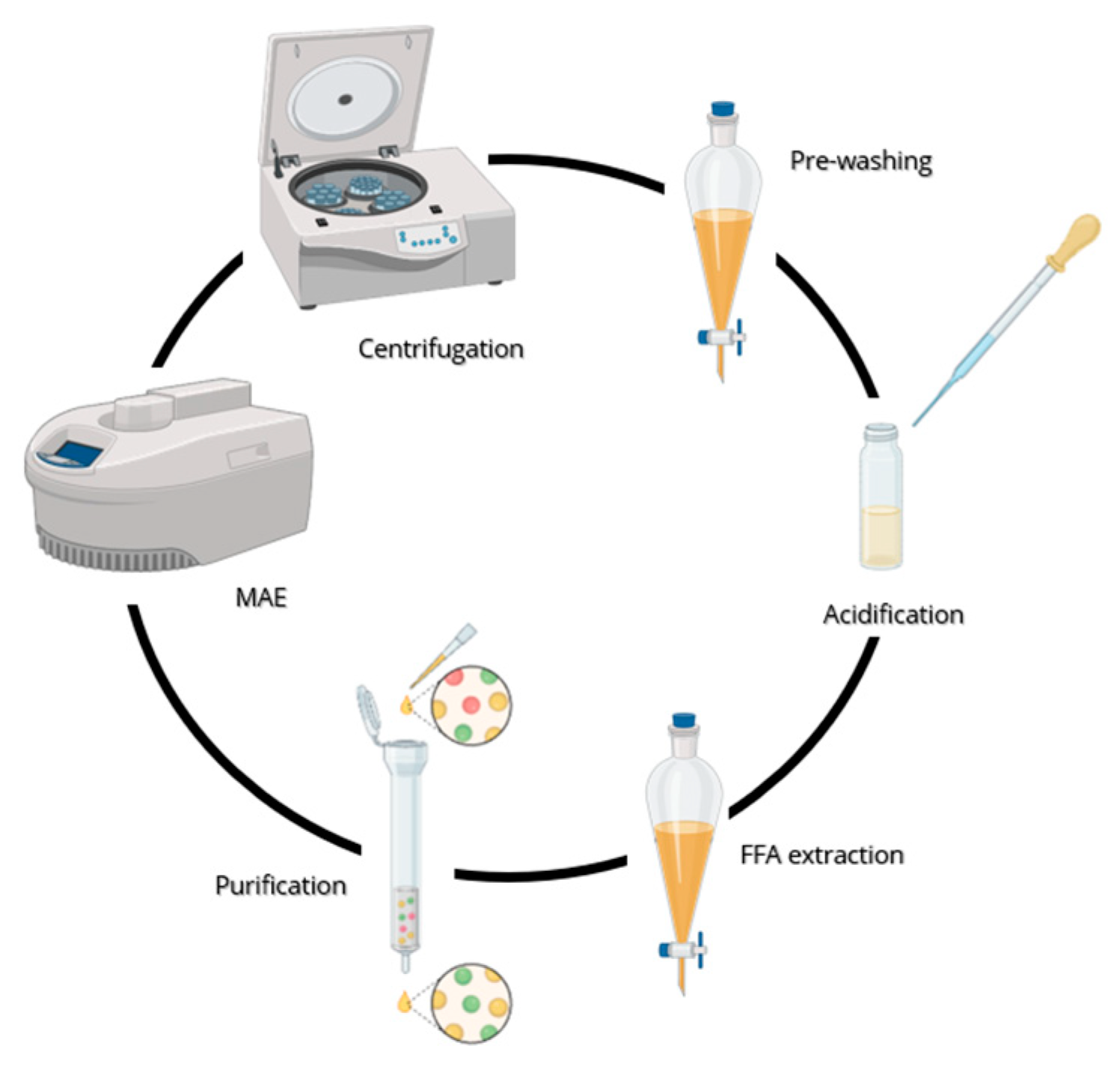

Table 1.
Moisture content (%) data obtained upon thermal treatment for corn and wheat.
| Matrix | Moisture content (%) |
|---|---|
| Corn | 6.1 ± 0.1 |
| Wheat | 6.3 ± 0.8 |
Table 2.
Vegetable oil yields from corn using n-hexane, DCM, isopropanol and EtOH with the corresponding temperatures and extraction times.
Table 2.
Vegetable oil yields from corn using n-hexane, DCM, isopropanol and EtOH with the corresponding temperatures and extraction times.
| Solvent | Temperature (°C) | Extraction time (min) | Yield (%) |
|---|---|---|---|
| n-hexane | 70 | 10 | 7.41 |
| DCM | 40 | 10 | 9.33 |
| Isopropanol | 82 | 10 | 12.85 |
| EtOH | 78 | 10 | 11.45 |
Table 3.
Experimental conditions set for the extraction of fatty acids from wheat bran with the corresponding yields.
Table 3.
Experimental conditions set for the extraction of fatty acids from wheat bran with the corresponding yields.
| Experiment # | Solvent | Solvent weight (g) | MW cycles |
|---|---|---|---|
| 1 2 3 |
SA | 18 | 1 |
| SB | 18 | 1 | |
| SA | 6 | 3 | |
| 4 5 |
SB | 6 | 3 |
| SB | 6 | 2 |
Table 4.
Wheat bran extraction yields upon replication of the optimized procedure using SB as MAE media.
Table 4.
Wheat bran extraction yields upon replication of the optimized procedure using SB as MAE media.
| Experiment # | Solvent | Solvent weight (g) | MW cycles | Cycle time (min) | Yield (%) |
|---|---|---|---|---|---|
| 1 2 3 |
SB | 18 | 1 | 30 | 4.45 |
| SB | 18 | 1 | 30 | 4.84 | |
| SB | 18 | 1 | 30 | 4.56 |
Table 5.
Fatty acid profile of wheat bran oil listed with the corresponding FAME, their mass-to-charge ratios (m/z), retention times (tr) and relative abundances.
Table 5.
Fatty acid profile of wheat bran oil listed with the corresponding FAME, their mass-to-charge ratios (m/z), retention times (tr) and relative abundances.
| Fatty acid | FAME (m/z) | tr (min) | Relative abundance (%) | |
|---|---|---|---|---|
| C12:0 C14:0 C15:0 |
Lauric | 214.34 | 14.77 | 1.12 |
| Myristic | 242.40 | 18.14 | 0.39 | |
| Pentadecanoic | 256.42 | 20.95 | 0.31 | |
| C16:1 C16:0 |
Palmitoleic | 268.40 | 24.10 | 0.83 |
| Palmitic | 270.45 | 25.47 | 17.53 | |
| C18:2 | Linoleic | 294.47 | 38.46 | 53.15 |
| C18:2 | 10-trans,12-cis-linoleic | 294.47 | 38.97 | 20.35 |
| C18:0 | Stearic | 298.50 | 41.10 | 2.28 |
| C20:1 | Gondoic | 324.54 | 51.10 | 1.60 |
| C20:0 | Arachidic | 326.56 | 52.27 | 0.42 |
| C22:1 | Erucic | 352.61 | 61.40 | 0.57 |
| C22:0 | Behenic | 354.61 | 63.37 | 0.49 |
| C23:0 | Tricosanoic | 368.64 | 71.83 | 0.14 |
| C24:1 | Nervonic | 380.65 | 79.67 | 0.16 |
| C24:0 | Lignoceric | 382.66 | 83.26 | 0.66 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



