Submitted:
14 January 2025
Posted:
15 January 2025
You are already at the latest version
Abstract
Primary pulmonary hypertension (PPH), now known as pulmonary arterial hypertension (PAH), has envisioned significant treatment breakthroughs in the past decade. Treatment has focused on improving patient survival and quality of life and delaying disease progression. Current therapies are categorized based on targeting different pathways known to contribute to PAH, including endothelin receptor antagonists (ERAs), phosphodiesterase-5 inhibitors (PDE-5 inhibitors), prostacyclin analogs, soluble guanylate cyclase stimulators, activin signaling inhibitors as Sotatercept. The latest addition to treatment options is soluble guanylate cyclase stimulators, such as Riociguat, which directly stimulates the nitric oxide pathway, facilitating vasodilation. Looking to the future, advancements in PAH treatment focus on precision medicine involving the sub-stratification of patients through a deep characterization of altered Transforming Growth Factor- β(TGF-β) signaling and molecular therapies. Gene therapy, targeting specific genetic mutations linked to PAH, and cell-based therapies, such as mesenchymal stem cells, are under investigation. Novel pharmacologic targets are also developing, including growth factors and inflammation-modulating pathways. Ongoing clinical trials aim to refine these emerging treatments and better understand their long-term benefits and safety profiles. This review will discuss the current therapies that help delay the progression and improve survival and future PAH treatments that hold potential for curative approaches targeting underlying disease mechanisms.
Keywords:
Pulmonary arterial hypertension
; endothelin receptor antagonists
; phosphodiesterase-5 inhibitors
; prostacyclin analogs
; soluble guanylate cyclase stimulators
; activin signaling inhibitors
Introduction
Pulmonary Hypertension (PH) is a chronic debilitating condition affecting the pulmonary vasculature, characterized by increased pressure of pulmonary circulation. Hemodynamically, it is defined as the mean Pulmonary Artery Pressure (mPAP) at rest of more than or equal to 20 mmHg[1,2].
PH is not a single clinical entity. Instead, it is a cluster of different conditions. Based on the similarity of pathophysiology, hemodynamic parameters, and management options, PH is classified into five groups as follows[1,2]:
- Group 1 PH: Pulmonary Arterial Hypertension (PAH).
- Group 2 PH: PH due to Left Heart Disease.
- Group 3 PH: PH due to lung diseases and/or hypoxia.
- Group 4 PH: PH due to Pulmonary artery obstructions.
- Group 5 PH: PH due to multifactorial mechanisms.
This article discusses the current management strategies and prospects for Group 1 PH.
Pulmonary Arterial Hypertension (PAH) is a progressive disease, incurable to date, that ultimately leads to Right Heart failure and death. PAH is a precapillary PH with its hemodynamic features, as shown in Table 1[2].
A population-based study done in Canada showed PAH accounting for about 15% of cases with PH, affecting more females (59.7%) as compared to males. The mean age of diagnosis for PAH was 55.4±28 years. In children less than 16 years of age diagnosed with PH, PAH accounted for approximately 65% of cases[3].
Before the advent of PAH-specific therapy in the late 1900s, the median survival of PAH patients was 2.8 years. Only about 68% of patients diagnosed with PAH were able to survive the first year after diagnosis. The survival rates at 3 and 5 years were only 48% and 34%, respectively[4]. Whereas, after about two decades of the modern treatment era, the situation has improved a lot. In 2012, it was found that the median survival of PAH patients was 7 years, with survival rates from diagnosis at 1 year, 3 years, 5 years, and 7 years being 85%, 68%, 57%, and 49%, respectively[5].
Among the subclassifications, the most common form is Idiopathic PAH, accounting for 39.2% of total cases of PAH, followed by PAH associated with Connective tissue disease (15.3%). Other forms like PAH associated with congenital heart disease (11.3%), PAH associated with portal Hypertension (10.4%), PAH associated with anorexigenic (9.5%), PAH associated with HIV (6.2%), and Heritable PAH (3.9%) are less common [6].
Histologically, PAH is characterized by diffuse medial hypertrophy and intimal/adventitial thickening. Such lesions suggest excessive production of vasoconstrictors like Endothelin or loss of vasorelaxants like Nitric Oxide (NO) and Prostacyclin[7]. These findings form the basis for the use of PAH-specific therapy, namely, Endothelin Receptor Antagonists (ERAs), Phosphodiesterase 5 inhibitors, guanylate cyclase stimulators, Prostacyclin analogs, and prostacyclin receptor agonists.
The pathophysiological overview and site of action of the drugs are shown in Figure 1.
Endothelin Receptor Antagonists (ERAs):
Endothelin is a potent vasoconstrictor with 4 isoforms. Endothelin-1 (ET-1) is the most common isoform produced by endothelial cells, which acts upon vascular smooth muscle cells and endothelial cells in an autocrine or paracrine fashion[8,9]. It leads to vascular remodeling, resulting in medial hypertrophy and plexiform lesions in patients with PAH[7]. The actions of Endothelin are mediated via Endothelin Receptors (ER). ETA is the isoform of the endothelin receptor abundantly present in vascular smooth muscles and contributes to vasoconstriction and cellular proliferation. In contrast, ETB is primarily present in endothelial cells and contributes to Endothelin clearance and vasodilation[8,9].
ERAs act by blocking the actions of Endothelin at Endothelin receptors, mainly ETA. Bosentan and Macitentan are dual endothelin receptor antagonists with blocking effects at both ETA and ETB, whereas Ambrisentan only antagonizes ETA[10,11,12].
Bosentan is the pioneer drug in the group, approved by the FDA in 2001 based on the results of the BREATHE-1 trial in patients with idiopathic PAH and PAH associated with connective tissue diseases. It showed improvement in the 6-minute walking test (6 MWT), World Health Organization (WHO) functional class, and time to clinical worsening. The efficacy of Bosentan was similar in both the dosage forms used in the trial (125 mg twice daily or 250 mg twice daily). In contrast, the most common side effect, i.e., abnormal hepatic function, was dose-dependent.
About 10% of patients suffered from an increase in hepatic enzymes, whereas 7% of it was seen in patients taking Bosentan 250 mg twice daily[13]. Thus, the recommended maximum dosage for Bosentan for the treatment of PAH is 125mg twice daily[1].
Ambrisentan, the second drug in the group, was approved by the FDA in 2007 in patients with Idiopathic PAH, Heritable PAH, and PAH associated with connective tissue diseases. In contrast to Bosentan, it has once daily dosing[14]. ARIES-1 and ARIES-2, the concurrent randomized controlled trial, showed improvement in the 6-minute walk test (6 MWT) after 12 weeks of treatment with different dosages of Ambrisentan, the maximum dose being 10 mg once daily. The average change from baseline in 6-minute walk distance was 40 m (95% Confidence Interval, 33 to 48 m) in week 12 and 39 m (95% CI, 29 to 49 m) at week 48, thus suggesting sustained improvement in primary endpoint by the continued use of Ambrisentan. The most worrying adverse effects of Bosentan, i.e., raised liver enzymes, were not seen with Ambrisentan. Peripheral edema, Headache, and Nasal congestion were observed in patients treated with Ambrisentan due to systemic vasodilation. Nasal congestion was the only side effect found to have a dose-response relationship. ARIES study showed that 5 mg and 10 mg once daily dosages showed favorable efficacy to safety profile. Improvement of exercise capacity (6 MWT) showed a positive dose-response trend, which supports the clinical decision to increase the dose from initial 5 mg once daily to 10 mg once daily if the drug is well tolerated[15].
Similarly, Macitentan, the latest addition to the group in 2013, is an oral formulation with once daily dosing that antagonizes both the receptor isoforms. The SERAPHIN trial showed that long-term treatment with Macitentan reduces morbidity and mortality among patients with PAH. Unlike previous short-term trials of 12-16 weeks considering improvement of exercise capacity as the primary endpoint, SERAPHIN trial used the time from the initiation of treatment to the first event related to PAH (worsening of PAH, initiation of treatment with intravenous or subcutaneous prostanoids, lung transplantation, or atrial septostomy) or death from any cause up to the end of treatment as their primary endpoint. A total of 742 patients were enrolled in this multicenter trial suffering from Idiopathic PAH, Heritable PAH, Drug/Toxin-induced PAH, PAH associated with Connective Tissue Diseases, PAH with repaired congenital left to right shunts, and PAH with HIV infection. Over the median period of 115 weeks, about half of patients receiving a placebo had events related to PAH or death due to any cause. The percentage was lesser with patients receiving Macitentan (38.0% and 31.4% in patients receiving 3 mg and 10 mg, respectively), demonstrating a significant reduction in morbidity compared to placebo. The trial also showed improvement in exercise capacity and WHO Functional class as in previous trials. The most worrying adverse effects of hepatic transaminitis didn’t appear to be significant in this trial, as both placebo and drug groups showed similar increments in the level of liver enzymes. Systemic vasodilation causing headache and nasopharyngitis were of concern. Anemia is one of the significant adverse effects of Macitentan, and 13.2% and 8.8% of patients treated with 10 mg and 3 mg of Macitentan suffered from anemia, in contrast to 3.2% of those receiving placebo. Unfortunately, the trial was not powered enough to show any mortality benefits[16].
A meta-analysis for the safety of ERAs in the treatment of PAH studied the adverse effects of 3 ERAs. Table 3 shows the Relative Risks (RR) of 3 different ERAs concerning their most common adverse effects.
It demonstrated that Bosentan 125 mg twice daily had the highest risk of abnormal liver function; Ambrisentan 10 mg once daily had the highest risk of peripheral edema, and Macitentan 10 mg had the highest risk of anemia[17].
ERAs can be considered as initial monotherapy in patients having PAH with cardiac comorbidities, and in patients with PAH without cardiac comorbidities, ERAs are part of initial combination therapy[1].
Nitric Oxide-cyclic Guanosine Monophosphate (cGMP) Stimulators:
Nitric Oxide (NO) is synthesized from L-Arginine with the help of the enzyme NO-Synthase (NOS). There are 3 isoforms of NOS, namely, neuronal NOS (nNOS), inducible NOS (iNOS), and endothelial NOS (eNOS). Endothelial NOS is expressed by most endothelial cells and is responsible for producing NO, which has vasodilatory, anti-thrombotic (anti-platelets aggregatory), and anti-proliferative effects. NO acts via its second messenger, cGMP, produced by the action of soluble Guanylate Cyclase (sGC) upon GTP. This cGMP is reduced to GMP with no residual functions of cGMP by the action of the enzyme named Phosphodiesterase (PDE), as shown in Figure 1[18]. PDE-5 is the isoform of PDE mostly expressed in Pulmonary Artery smooth muscle cells. It is responsible for hydrolysis of about 80% of cGMP produced in circulation. PDE-5 is overexpressed in pulmonary circulation smooth muscle cells in patients with PAH and forms the basis for treating PAH with PDE-5 inhibitors (PDE-5is)[19].
Phosphodiesterase-5 Inhibitors (PDE-5i):
PDE-5i was initially approved for erectile dysfunction. Sildenafil and Tadalafil are the commonly used PDE-5is that are later used for PAH based on different clinical trials.
Sildenafil, a PDE-5 inhibitor, was the second oral drug to be studied for PAH therapy after Bosentan. SUPER-1, a 12-week, multicentric, double-blinded, placebo-controlled trial, was designed to study the efficacy and safety of Sildenafil for PAH therapy, which included 278 patients with Idiopathic PAH, PAH associated with Connective Tissue Diseases and PAH with repaired congenital systemic to pulmonary shunts. All patients were randomized into 4 groups, i.e., placebo and Sildenafil, with dosages of 20 mg three times daily, 40 mg three times daily, and 80 mg three times daily.
Improvement in 6-minute walk distance (6 MWD) was considered the primary endpoint. Sildenafil demonstrated improvement in 6 MWT irrespective of the dosage used. The mean placebo-corrected improvements were 45m, 46m, and 50m with 20 mg, 40 mg, and 80 mg of Sildenafil, respectively. Sildenafil showed statistically significant improvements in hemodynamic parameters, i.e., a decrease in mPAP and pulmonary vascular resistance (PVR). However, there was no delay in time to clinical worsening with Sildenafil as compared to placebo, while improvement in WHO- Functional class was seen[17]. The extension period in the SUPER-1 trial showed the mean change in 6 MWD to be 51m after 12 months of monotherapy, suggesting its sustained effect. SUPER-2 trial, an open-label uncontrolled extension of SUPER-1, demonstrated that 46% of patients had increased 6 MWD after 3 years of treatment and 29% of patients had improved their WHO FC while 31% were able to remain in the same WHO-FC without further clinical deterioration with monotherapy. Patients with baseline 6 MWD <325m in the SUPER-1 trial and those who showed no improvement in 6 MWD even after 12 weeks of treatment with Sildenafil had poor survival. Thus, close monitoring and early aggressive treatment options could benefit such cases[20].
Mostly mild to moderate side effects related to PDE-5is were observed during the trial, like headache, dyspepsia, diarrhea, blurred vision, etc. Left Ventricular Dysfunction and postural hypotension were serious adverse effects observed in one patient during the first 12 weeks of the study period. Other perceived serious adverse effects were seizures, drug hypersensitivity, angioedema, and subcapsular cataracts. Though the drugs in different dosages were well tolerated, no dose-response relationship was evident regarding efficacy outcomes, likely due to the complete inhibition of PDE-5 with the lowest dose. Thus, 20 mg three times daily is the only approved dose of Sildenafil based on the statistical benefit at the 12-week trial[20,21].
Like Sildenafil, Tadalafil’s safety and efficacy were studied with different dosage forms (2.5mg, 10 mg, 20 mg, and 40 mg once daily). PHIRST trial included 405 patients with Idiopathic PAH, Heritable PAH, Drug and toxin-induced PAH, PAH associated with Connective tissue disease, HIV, and congenital left to right shunt. Tadalafil showed improvement in 6-minute walk distance (6 MWD) with different dosage forms except 2.5 mg once daily compared to placebo. Although the improvement was clinically significant, only 40 mg OD achieved a pre-specified statistical significance value. Unlike Sildenafil, Tadalafil demonstrated a delay in time to clinical worsening, whereas no improvement in the WHO Functional class was seen. Improvement in hemodynamic parameters like mPAP and PVR were observed, as with Sildenafil. Subgroup analysis in patients taking Tadalafil 40 mg revealed that the placebo-adjusted change in 6-MWD was 44m in treatment naïve patients. In contrast, it was only 23m in patients with background Bosentan therapy. Though the exact mechanism is unclear, it is thought that the pharmacokinetic interaction between Bosentan and Tadalafil, mediated via Cytochrome P450, is responsible for the decreased efficacy of Tadalafil in combination therapy [22,23].
The long-term extension (PHIRST-2) revealed sustained improvement in the exercise capacity of patients taking Tadalafil regularly[24]. The most common side effects observed during the 16-week trial were Headache, Myalgia, and flushing with mild to moderate intensity. Serious side effects like nausea, vomiting, retinal artery occlusion, dyspnea, priapism, esophageal varices hemorrhage, hypotension, gastritis, menorrhagia, histiocytosis hematophagic syndrome, and drug hypersensitivity were rare. The prevalence of headaches was lower in long-term extensions than in the initial 16-week trial, suggesting that the side effects waned with time[22,24]. As the improvement in exercise capacity was dose-dependent, whereas the side effects were similar in all dosage forms, 40 mg once daily was approved as the dosage form of Tadalafil for PAH therapy[22,24].
A meta-analysis of PAH patients treated with PDE-5is has shown that the treatment with PDE-5is will likely improve WHO FC and exercise capacity of patients measured in terms of 6MWD. About 22% of cases are less likely to die within 14 weeks of treatment with PDE-5i as compared to placebo. It showed that the treatment's most common adverse effects are headache, GI upset, flushing, muscle aches, and joint pains[25]. Like ERAs, PDE-5is are also considered first-line monotherapy for patients having PAH with Cardiac comorbidities. They are considered for combination therapy with ERAs in the case of low to intermediate-risk patients and combinations with ERAs and Prostacyclin analogs in high-risk PAH cases[1].
Soluble Guanylate Cyclase (sGC) stimulator
Riociguat is the sole drug in the group that stimulates the production of cGMP and promotes the action of nitric oxide (NO). Its phase II trial demonstrated promising efficacy and safety for use in both Group 1 and Group 4 PH. It became one of the drugs to be approved for treatment in two different groups of PH[26]. In the PATENT trial, 443 patients were enrolled with Idiopathic PAH, Heritable PAH, Drug or toxin-induced PAH, PAH associated with Connective tissue disease, congenital heart disease, and Portal Hypertension with cirrhosis. 50% of the participants were treatment naïve, whereas 44% received ERA and 6% received Iloprost as background therapy. Riociguat was used in different individualized dosage forms, the maximum dose being 2.5 mg three times daily. It showed improvement in 6-minute walk distance (6 MWD) at 12 weeks in both subgroups of patients, whether it be treatment naïve or under background therapy[27]. The 6 MWD was improved during long-term extension by 51m at 1 year and 47m at 2 years, suggesting its sustained effect[28,29]. Though Riociguat is equally effective in either treatment naïve patients or patients with background therapy, it is not recommended to combine Rioiciguat with PDE-5is due to the increased risk for hypotension[30].
Improvement in hemodynamic parameters (mPAP and PVR), NTproBNP levels, WHO-FC, and time to clinical worsening were also seen in the 12-week trial of Riociguat[27]. There was an improvement in WHO-FC of about 33% of patients, and 61% of patients’ WHO-FC was maintained at the end of 1 year of treatment with the drug[28]. Syncope, worsening PH, chest pain, and Right Ventricular failure were common adverse effects seen. The worrisome adverse effects were hemoptysis and pulmonary hemorrhage. In the PATENT-2 study, 10 patients (3%) suffered from hemoptysis and pulmonary hemorrhage, among which, in 7 (2%) cases, it was resolved without drug withdrawal or dose reduction, whereas 1 patient (0.3%) died of it, 1 patient (0.3%) underwent drug withdrawal and death later and in 1 (0.3%) it didn’t resolve till study period. Though the exposure-adjusted rate per 100 patient years for pulmonary hemorrhage was 1.8 in PATENT-2 as compared to 4.3 in PATENT-1, the potential risk of bleeding with the drug is a concern[28].
The survival rate for 1 year was estimated to be 97%, and the incidences of clinical worsening and hospitalization due to PAH were relatively low (21% and 10%, respectively). These results are in favor of the long-term efficacy of Riociguat in patients with PAH[28].
Prostacyclin analogues and receptor agonists:
Prostacyclin (PGI2) and its analogues are potent vasodilators with antiproliferative, anti-thrombotic, anti-inflammatory, and anti-platelet aggregatory nature. They mediate their actions via Prostacyclin (IP) receptor-stimulating G protein-coupled increase in cAMP[30]. In PAH patients, Prostacyclin Synthase (PGI2-S), responsible for the synthesis of PGI2, decreased in the pulmonary circulation, which could be the reason for thrombosis and inflammatory changes in vessels[31]. Currently, 3 prostacyclin analogs are used for the treatment of PAH patients, viz. Epoprostenol, Treprostinil and Iloprost.
Epoprostenol is a synthetic analog of prostacyclin with a very short half-life of <3-5 minutes (like that of endogenous prostacyclin), requiring intravenous (IV) administration for its action. As it irritates the vein, the peripheral IV route is not preferred; rather, administration as a continuous IV infusion via central venous catheter is preferred [32]. It is the first drug to be used for the treatment of PAH and is a significant therapy to date. The first trial with Epoprostenol was conducted in 1990, and it was randomized for the first 8 weeks, followed by an unrandomized extension of 18 months. It showed improvement in hemodynamic parameters like PVR and mPAP, which was sustained for 18 months but required dose adjustment[33]. Later, 81 patients with PAH in WHO-FC III/IV were enrolled for a 12-week trial where Epoprostenol combined with conventional therapy (anticoagulants, vasodilators, diuretics, digoxin) versus traditional therapy alone was studied. Patients in the treatment group showed statistically significant improvement in exercise capacity. The mean 6 MWD improved by 32m in the treatment group versus a decrease of 15m in a conventional therapy group. 40% of patients showed improvement in their WHO-FC, and 48% could maintain WHO-FC without deterioration. Hemodynamic parameters like mPAP, PVR, and Cardiac Index (CI) significantly improved. Eight patients died during the 12-week trial, where all deaths were from the conventional therapy group. The 6 MWD at baseline was significantly lower in the dead patients when compared to that of survivors, thus indicating 6 MWD as an independent predictor of survival in PAH patients[34]. The survival benefit of Epoprostenol found in short-term studies was not limited. Survival with Epoprostenol therapy at 1, 2, and 3 years was 87.8%, 76.3%, and 62.8%, respectively, and was significantly better than the expected survival of 58.9%, 46.3%, and 35.4% based on historical data[35]. Patients with a history of Right Heart failure, absence of significant hemodynamic improvement after 3 months of Epoprostenol therapy, and persistence of WHO-FC after 3 months of treatment were linked with poor survival. These could be suitable candidates to be considered for lung transplantation[36]. The common side effects of Epoprostenol use were jaw pain, diarrhea, flushing, headache, nausea, vomiting, etc., mainly related to its vasodilatory effects. The serious side effects to be considered were catheter-related sepsis, catheter-related thrombotic events, infection at the catheter site, and bleeding or pain at the catheter site. Interruption in continuous IV Epoprostenol has been shown to increase symptoms in patients, leading to clinical worsening[34]. The main limitation of Epoprostenol therapy is the difficulty in drug handling and its delivery process.
Unlike Epoprostenol, with molecular instability and a short half-life, Treprostinil is a stable compound with a half-life of about 4.5 hours. It is available in four forms for administration, i.e., Subcutaneous (SC), Intravenous (IV), Inhalation, and oral. Subcutaneous Treprostinil was approved for the treatment of PAH patients with WHO-FC II-IV in 2002[32]. A 12-week, double-blind, placebo-controlled, multicentric trial included 470 patients suffering from Idiopathic PAH, PAH associated with connective tissue disease and congenital heart disease between November 1998 to October 1999. The initiation dose for continuous subcutaneous infusion of Treprostinil was 1.25ng/kg/min, and the maximum allowable dose in 12 weeks was 22.5ng/kg/min. The treatment group showed only modest improvement in 6 MWD (the difference in median distance walked was 16m), but it was statistically significant. Interestingly, the improvement in exercise capacity measured by 6 MWD was observed to be significantly improved in those with poor capacity at baseline. The inclusion of relatively stable patients in the trial compared to those of Epoprostenol might be the reason for modest improvement in exercise capacity. Moreover, the most common adverse effect, i.e., infusion site pain, didn’t allow for an adequate increase in the dosage of Treprostinil in some patients[37]. This trial showed improvement in multiple other hemodynamic parameters and clinical signs and symptoms of patients. Improvements in patient outcomes were dose-dependent, with maximum improvement seen in patients receiving dosages of >13.8 ng/kg/min. Though this trial did not demonstrate the mortality benefit, it showed significant improvement and could act as an alternative to the tiresome administration of Epoprostenol with multiple catheter-related complications. The most common adverse effect of SC Treprostinil was Infusion site pain. 85% of patients had this side effect, and 8% of cases discontinued the treatment. It didn’t appear to be dose-related but instead correlated with the rate of dose increment. Other significant side effects were irritation, bleeding, and bruising at the infusion site and side effects related to vasodilatory effects of the drug include jaw pain, flushing, and edema. Unlike Epoprostenol therapy, interruption in drug delivery due to infusion system malfunction didn’t lead to any serious adverse effects in patients with SC Treprostinil. Though 3 episodes of GI bleeding were noted, it was uneventful and was not considered to be due to Treprostinil. Instead, it was thought to be due to other conventional therapies[37]. Long-term therapy with the drug demonstrates continuous improvement in exercise capacity and clinical symptoms in patients with PAH. Event-free survival rates, i.e., survival without events like hospitalization for clinical worsening, transition to IV Epoprostenol, and need for combination therapy or atrial septostomy, were 83.2% and 69% at 1 year and 3 years, respectively, which appear similar to that with IV Epoprostenol[38].
Among different routes of administration of Treprostinil, FDA has approved SC, IV and Inhalation routes for the treatment of PAH. A 12-week, open-label, multicentric study including 16 patients with Idiopathic PAH, PAH associated with Connective Tissue Disease, and PAH associated with congenital heart disease in WHO-FC III and IV demonstrated improvement in exercise capacity, dyspnea, and hemodynamic parameters with IV Treprostinil. The mean increment in 6MWD was 82m, only 16m in the treatment group with SC Treprostinil. There was no deterioration in WHO-FC during the study, and no patients were in WHO-FC IV by the end of the treatment period. Improvement in dyspnea was more significant with more rapid dose escalations, and higher dosages were given to the patients than in the previous trial of the SC route[39]. IV administration of Treprostinil is advantageous over IV Epoprostenol because of its longer half-life and stability at room temperature. The chance of a life-threatening crisis due to an inadvertent infusion interruption is less as compared to IV Epoprostenol because of its longer half-life. Besides, the storage and preparation of infusions are less tiresome and can enhance patient convenience. The transition from Epoprostenol to Treprostinil is also found to be safe and effective[39,40]. The most worrisome adverse effects of IV Treprostinil were extremity pain and jaw pain, reported by 69% of participants. Other side effects like nausea, headache, flushing, and diarrhea were minor ones. The main issue with SC Treprostinil, i.e., infusion site pain, is not seen with the IV route, thus improving patient compliance[39].
Inhaled Treprostinil was also found to be an effective adjunct therapy in patients who remained symptomatic despite treatment with ERAs or PDE-5is. A 12-week trial showed the placebo-corrected median change from baseline in 6 MWD of 20 m[41]. Along with Inhalation via Nebulization, a dry powder inhalation (DPI) form has been FDA-approved for use in PAH treatment. Transition from Nebulization to DPI form was demonstrated to be safe and convenient in BREEZE trial[42]. Compared to SC and IV administration of prostacyclin, a more straightforward route like inhalation can aid in initiating prostacyclin earlier in the treatment plan. Inhaled Treprostinil has also been approved by the FDA for the treatment of Group 3 PH [43,44].
Iloprost is an FDA-approved inhalation therapy for PAH. A 12-week, multicentric, placebo-controlled trial with patients suffering from Severe PAH and chronic thromboembolic PH (CTEPH) showed significant improvement in exercise capacity and functional classification. This study used a combined primary endpoint of at least 10% improvement in 6MWD and improvement in WHO-FC (then termed as NYHA FC). 16.8% of participants met the primary endpoint, whereas in about 40% of patients, the improvement in 6 MWD was more than 10%. The mean increase in 6MWD was 36.4m but when Primary Pulmonary Hypertension was only taken into account, it was 58.8m. There was no significant improvement in hemodynamic parameters, but no further deterioration was seen. The mean dosage for inhalation was 0.37ng/kg/min, which is comparatively lesser, and it is assumed that due to the direct action of the inhalation route over pulmonary circulation, its potency is higher as compared to other forms of administration[45]. The disadvantage of the inhalation route is repeated inhalation of the drug about 6-9 times per day via nebulization. Apart from side effects related to prostacyclin, syncopal attack was considered severe in treatment with Iloprost. It occurred in patients usually after 2-9 hours of last therapy and could be due to loss of pharmacological effect of Iloprost[45]. Iloprost has shown efficacy and safety not only as an add-on to conventional treatment but also as efficacious and safe while adding it on with Bosentan [45,46].
Selexipag, an oral prostacyclin (IP) receptor agonist, is chemically different from prostacyclins yet is considered in the group because of its similar pharmacological action via the prostacyclin receptor. In PAH patients with stable treatment with ERA and/or PDE-5i, the addition of Selexipag showed a statistically significant improvement of 30.3% in PVR after 17 weeks [47]. The GRIPHON trial, an event-driven, phase III RCT, studied Selexipag in patients with Idiopathic PAH, Heritable PAH, Drug and toxin induced PAH, and PAH associated with Connective Tissue Disease, HIV, and congenital heart disease. The primary endpoint for the study was a composite of death from any cause or complication related to PAH up to the end of treatment. 41.6% of patients in the placebo group and 27% in the treatment group met the primary endpoint. Disease progression and hospitalization accounted for 81.9% of events. No significant difference in mortality was seen in two groups. The dosage of Selexipag was individualized and variable in the study. The initial dose was 200 micrograms twice daily, increasing until adverse effects were observed, and the maximum tolerated dose was calculated. The maximum dose in this trial was 1600 micrograms twice daily. Efficacy was similar among patients receiving low, medium, or high dose. Similarly, the drug was found to have a similar effect whether the patient was not under any treatment or stable background therapy with ERA, PDE-5i, or both. Adverse effects were related to the vasodilatory effects of the drug. Headache, Diarrhea, and nausea were significant side effects that even led to discontinuation of the therapy[48].
2022 European Society of Cardiology (ESC)/European Respiratory Society (ERS) guidelines for diagnosing and treating Pulmonary Hypertension have recommended using combination therapy of ERA and PDE-5i for patients with low or intermediate risk and without cardiac complications[1].
Combination with Bosentan has been limited due to drug interactions, as discussed earlier, whereas the combination of Tadalafil with Ambrisentan and Macitentan showed promising results. In an event-driven, randomized controlled trial, it was observed that in a treatment naïve patient, initial combination therapy with Ambrisentan and Tadalafil resulted in a significantly lower risk (50% lower risk) of clinical failure events than the risk with Ambrisentan or Tadalafil monotherapy. Thus, targeting multiple pathways with early combination therapy could benefit patients in PAH [49].
Fixed-Dose Combination Drug (Macitentan/Tadalafil)
A DUE study, a multicentric, double-blind, adaptive phase III trial, studied the efficacy and safety of combination therapy in the form of Fixed Dose Combination over 16 weeks. Patients suffering from PAH in WHO FC II and III were included in the trial. They were randomized into 3 groups receiving Macitentan 10 mg/Tadalafil 40 mg Fixed-Dose Combination (M/T FDC), Macitentan 10 mg monotherapy, and Tadalafil 40 mg monotherapy. There was significant improvement in PVR in subjects treated with M/T FDC as compared to monotherapy. Changes in PVR from Baseline to week 16 are shown in Table 4[50].
There was an improvement in exercise capacity measured in terms of 6 MWD in patients of all groups, but no statistically significant differences were observed when compared between the groups. The improvements in symptoms and WHO-FC were also not significantly different among the groups[50].
Headache and peripheral edema were the most common treatment-emergent adverse effects. Anemia, hypotension, and edema were more common in the M/T FDC group than others. No new or unexpected adverse effects were observed during the trial, and the drug was well tolerated. Owing to the reduction in pill burden with the benefit of combination therapy, the study supported the use of single-tablet combination therapy for initial management in patients with PAH[50].
Activin signaling Inhibitor:
Activin and Bone Morphogenetic protein (BMP) are the Transforming Growth Factor β (TGF-β) family members[51]. Activin A is thought to increase proliferation and induce gene expression of Endothelin-1 and Plasminogen activator inhibitor-1 in pulmonary artery smooth muscle cells, ultimately contributing to vascular remodeling [52]. On the other hand, BMP 2 is antiproliferative and can inhibit smooth muscle cell proliferation without stimulating extracellular matrix synthesis[53]. In normal individuals, expression of BMP Receptor (BMPR2) is prominent on vascular endothelium, whereas in PAH cases, especially those with heterozygous BMPR2 mutations, its expression is markedly reduced in peripheral lung tissue, and expression of Activin A is found to be increased[54,55]. Activin A mediates its action by binding at its Activin Receptor Type II A [ActRIIA)[51,55].
The pathophysiologic overview and action of Sotatercept is shown in Figure 2.
Sotatercept, a decoy Activin receptor, is a fusion protein (ActRIIA-Fc) composed of the extracellular domain of human Activin Receptor Type II A fused with the Fc portion of IgG1[55]. It prevents the binding of Activin A to its receptor and prevents vascular remodeling in the case of PAH. It is an FDA-approved drug that has shown promising results in Phase II and III trials. Those trials included patients with PAH (Idiopathic PAH, Heritable PAH, Drug/Toxin-induced PAH, PAH associated with Connective Tissue Disease and congenital heart disease) in WHO-FC II and III who were under stable background therapy for at least 3 months with PAH-specific drugs[56,57]. Sotatercept was given subcutaneously every 3 weeks, and its efficacy and safety were measured. It showed significant improvement in PVR and WHO functional classes. The difference between the treatment group and placebo in terms of change in 6 MWD from baseline at 24 weeks was 40.8 m[56,57,58]. The risk of death or nonfatal clinical worsening events, assessed up to the end of the trial, was 84% lower with sotatercept than with placebo[57]. Thrombocytopenia was the most common adverse effect of interest during the trial, yet no thrombocytopenia-induced bleeding occurred or platelet transfusion was required. However, thrombocytopenia resulted in the discontinuation of Sotatercept in one patient, and another withdrew the consent. Erythrocytosis was another effect of concern; three patients had to withdraw from the trial as their Hemoglobin level raised above 18 mg/dl, and they underwent phlebotomy[56]. Thus, Sotatercept had a favorable benefit-to-risk ratio combined with stable background PAH-specific therapy. It could be helpful add-on therapy for patient’s refractory to PAH treatment.
Treatment Algorithm
As discussed, there are multiple pharmacological options for the treatment of PAH. The decision regarding the treatment plan is based on pretreatment assessment for baseline risk. Three strata models segregating the patients into low, intermediate and high-risk groups are used in the initial treatment plan. It was suggested by the 2015 ESC/ERS Guidelines for the diagnosis and treatment of PH. It is a comprehensive model and includes different parameters like clinical features of right heart failure, progression of symptoms, syncope, 6 MWD, WHO-FC, Biomarkers (BNP/ NTproBNP), different parameters of Echocardiography, cardiac MRI, cardiopulmonary exercise testing (CPET) and Hemodynamic parameters.
A few abbreviated approaches of the three strata risk stratification tool have been validated using the Swedish Pulmonary Arterial Hypertension Registry (SPAHR), the Comparative, Prospective Registry of Newly Initiated Therapies for PH (COMPERA), and the French PH Registry (FPHR). The United States Registry has developed other risk-stratification tools to Evaluate Early and Long-term PAH disease management (US REVEAL), including the REVEAL2.0 risk score calculator and REVEAL Lite 2[1]The objective of all these stratification models is to determine whether the patient is at high risk, which is essential for making decisions regarding treatment initiation with prostacyclin or other medications.
Surgical Strategies in Severe Pulmonary Arterial Hypertension (PAH):
Risk Assessment and Decision-Making for Treatment:
Pulmonary arterial hypertension (PAH) represents a challenging condition requiring a multidisciplinary approach. Severe PAH (WHO functional class III/IV) is associated with poor survival despite advanced medical therapies. Surgical options become crucial in these patients, aiming to palliate symptoms, improve hemodynamics, and enhance quality of life. Decision-making for surgical intervention involves complex considerations, including risk assessment, timing of intervention, and patient-specific factors[1,60].
Effective risk stratification is critical for identifying surgical candidates in severe PAH. Key components include[1]:
Hemodynamic Parameters: Right atrial pressure (RAP), cardiac index (CI), and pulmonary vascular resistance (PVR) are pivotal hemodynamic predictors. RAP >15 mmHg and CI <2 L/min/m² indicate high-risk patients unsuitable for extensive surgeries.
Biomarkers: Elevated NT-proBNP and troponin levels correlate with right ventricular dysfunction and adverse outcomes.
Functional Capacity: The 6-minute walk test (6MWT) and cardiopulmonary exercise testing (CPET) are standard assessments to evaluate functional reserve and surgical candidacy.
Surgical Strategies:
Right to Left Shunting:
Right to left shunting increases systemic blood flow due to increased left ventricle preload from right ventricle decompression. This procedure involves Atrial Septostomy and Potts Shunt[60].
Atrial Septostomy
Atrial septostomy creates a right-to-left shunt, reducing right atrial pressure and increasing cardiac output. While primarily a palliative intervention, it is effective in relieving symptoms of refractory PAH in carefully selected patients. Hemodynamic stability during the procedure is critical for success[60,61,62].
Potts Shunt
The Potts shunt creates an anastomosis between the left pulmonary artery and the descending aorta, leading to reduced oxygen saturation in the lower extremities. It had improved the systolic function of right ventricle, exercise tolerance, and functional status in children with end stage IPAH[60,63].
Pulmonary artery denervation (PADN):
Thoracic Organ Transplantation:
Thoracic organ transplantation can be applicable to patients with refractory PAH. It includes lung transplantation and heart-lung transplantation.
Lung Transplantation
Heart-Lung Transplantation (HLT)
PAH with irreversible heart failure can be considered for heart-lung transplantation. However, at present days HLT has been reduced due to significant improvement in PAH patients following lung transplant[60].
Decision-Making for Surgical Treatment[1,60,61]:
Early referral for surgical evaluation improves outcomes, particularly in patients with rapidly deteriorating hemodynamics or refractory symptoms despite optimized medical therapy. Collaboration among pulmonologists, cardiologists, and thoracic surgeons ensures comprehensive evaluation of risks and benefits. Advanced imaging and hemodynamic monitoring guide decision-making. Risk stratification tools such as the REVEAL risk calculator and ESC/ERS guidelines provide frameworks for quantifying surgical risks.
Emerging Surgical Strategies:
Hybrid Approaches
Hybrid procedures, combining atrial septostomy with medical therapies or mechanical support, are gaining traction for patients with severe hemodynamic compromise.
Mechanical Support Devices
Precision Medicine in Surgical PAH Management
Genetic profiling and molecular biomarkers are being integrated into risk assessment and post-surgical prognostication[67,68].
Surgical management in severe PAH is limited by high perioperative mortality, especially in patients with advanced right ventricular dysfunction. Future advancements focus on refining patient selection criteria, improving surgical techniques, and incorporating novel perioperative care strategies.
Surgical strategies play a critical role in the management of severe PAH when medical therapies fail. Comprehensive risk assessment, multidisciplinary collaboration, and timely intervention are essential for optimizing outcomes. Emerging approaches, including hybrid procedures and mechanical support, hold promises for improving the survival and quality of life in this high-risk population.
Palliative care: An overlooked extra panel of support for PAH therapy:
Palliative care is a specialized medical care designed to provide ease from symptoms and stress resulting from chronic illness such as PAH, cancer. PAH undoubtedly affects physical, social and emotional functioning, contributing to deterioration of patient’s quality of life. Furthermore, adverse effects of PAH specific therapies add on to the worsening standard of living. Palliative care, being classified as an invasive and non-invasive therapy, overlaps with other treatment modalities (specific treatment and surgical intervention) as shown in Table 5.
What is on the Horizon?
Regenerative Medicine: A potential curative approach for a patient with PAH
As discussed, the management of PAH demands a holistic approach that encompasses both supportive and specific therapies. Current therapeutic modalities mainly revolve around the Endothelin pathway, NO-sGC-cGMP (Nitric Oxide-soluble guanylate cyclase-cyclic guanosine monophosphate), and Prostacyclin pathway. Despite improved survival rates following recent therapies, introducing novel curative advanced therapy seems crucial. Modern therapeutic advancements (Regenerative Medicine) in PAH comprising stem cell therapies, gene therapies, and epigenetic medicines have shown promising results in preclinical studies, thus appearing as a potential cure with serious consideration[71,72,73,74,75]. Regenerative Medicine mainly emphasizes repairing, replacing, or regenerating cells, tissues, or organs to restore their impaired function[72,73].
Stem Cell Therapy
Stem cells are undifferentiated cells that can self-renew and convert to specialized cells under exceptional circumstances. Depending upon differentiation potential, it can be classified as unipotent, oligopotent, multipotent, totipotent, and pluripotent stem cells[74,76,77,78]. It can be transplanted directly to the target site of the body or into the patient as stem cells derived from mature cells following in vitro processing[79]. As these stem cells can differentiate into vascular cell lineages, they can potentially cure PAH[76,79,80]. Adult stem cell therapy is one of the trending issues in managing PAH due to its competency in restoring normal endothelial function and preventing the proliferation of smooth muscle cells in the pulmonary artery. There are three significant varieties of stem cell therapy: Endothelial progenitor cell therapy, Mesenchymal stem cell therapy, and Induced pluripotent stem cell therapy[75,78,81].
Figure 4.
Mechanism of Pulmonary Arterial Hypertension and role of stem cell transplant in its treatment[78,82].
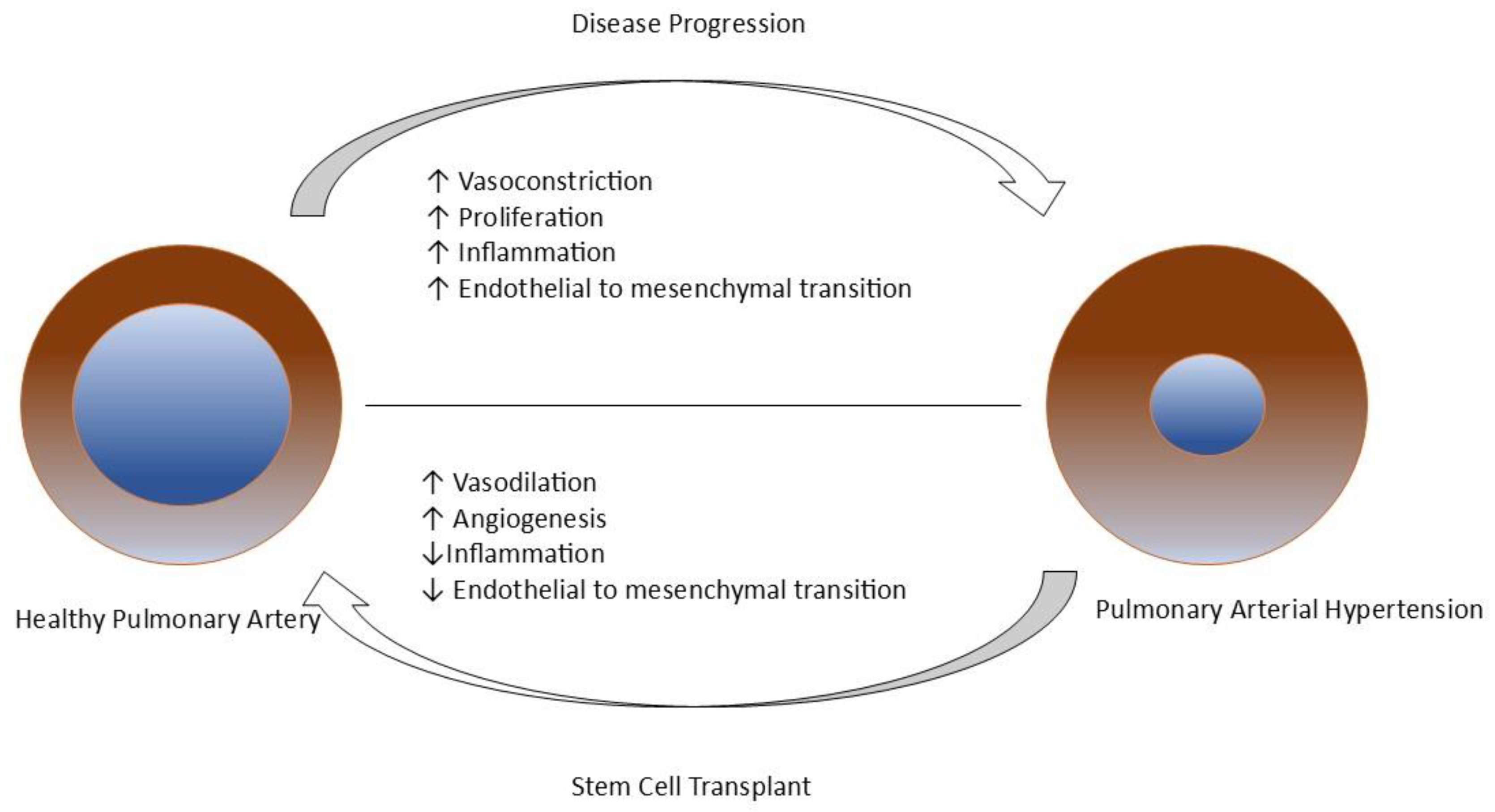
Endothelial progenitor cells (EPCs) are oligopotent stem cells predominantly found in bone marrow with a tendency for endothelial cell differentiation. PAH is associated with a change in the number of circulating EPCs and the destruction of its migratory and adhesive capacity, leading to impaired vascular network formations[75,83]. EPC therapy is expected to improve PAH by differentiating into mature endothelial cells, repairing tissue, and restoring endothelial function. Furthermore, it can be diagnostically helpful as circulating biomarkers to predict the risk of the disease[78,80,82,83]. During preclinical studies, EPC therapy had improved hemodynamic parameters such as mean pulmonary arterial pressure (mPAP), Right Ventricular (RV) systolic pressure & pulmonary vascular resistance (PVR), and survival [84,85]. The PHACeT trial (Pulmonary Hypertension and Angiogenic Cell therapy) demonstrated short-term hemodynamic improvement and long-term improvement in functional and quality of life assessments, with two adverse effects being sepsis and death[86]. Recent clinical studies have also shown improvements in exercise capacity and hemodynamics[83,87]. These findings suggest EPC therapy has potential scope in the future.
Mesenchymal stem cells (MSCs) are non-hematopoietic multipotent stem cells with the potency to differentiate into various cell types, with their specialty in releasing paracrine factors that contribute to the recovery of injured tissue. It can be easily isolated from bone marrow, adipose tissue, umbilical cord, or lung[75,81,82,88]. MSC therapy is beneficial in a patient with PAH by repairing vascular function, inhibiting endothelial-to-mesenchymal transition, and inhibiting pro-inflammatory and pro-apoptotic factors in lung tissues. The principal mechanism to achieve the treatment goal in PAH occurs due to paracrine effects[78,88,89]. Studies of MSC therapy in animal models have demonstrated reversal of vascular remodeling, increase in mPAP, pulmonary inflammation, and right ventricular hypertrophy[75,81,82,89]. Numerous advantages of this therapy make it a desirable alternative.
Induced pluripotent stem cells (iPSCs) are artificial pluripotent stem cells that are genetically reprogrammed from adult somatic cells to embryonic stem cell states using various transcription factors[75,78,80]. Huang et al. illustrated significant improvement in right ventricular systolic pressure and hypertrophy of right ventricle in monocrotaline (MCT) rats[90]. As the limitations are apparent, such as tumorigenicity, genetic and epigenetic abnormalities, and long-term safety and efficacy, iPSCs are often used as cell models for investigating different phenotypes, drug screening, RNA sequencing, and the influence of gene mutation[74,78,80,91].
Gene Therapy
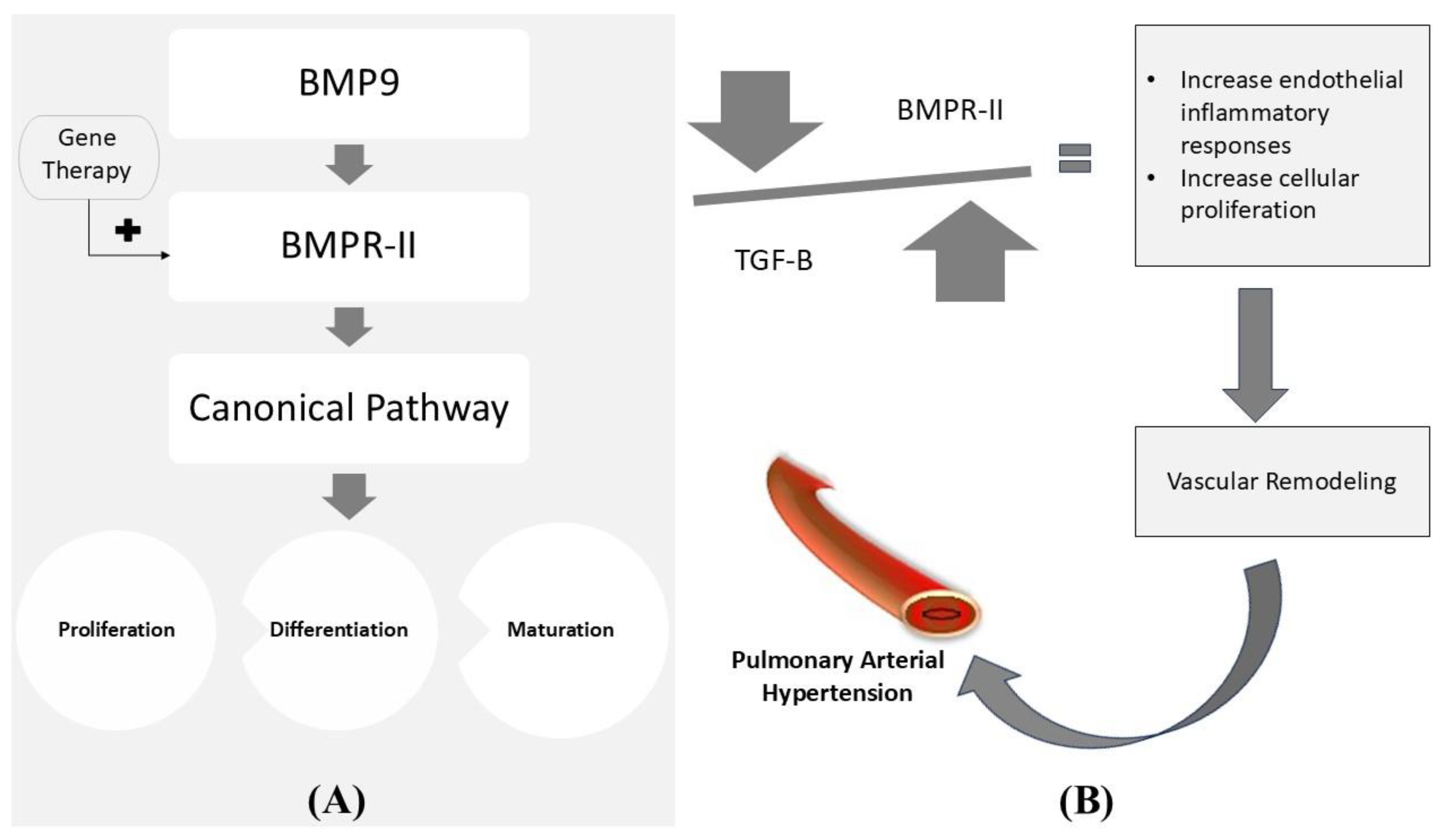
The ability to correct the mutated genes or site-specific modifications via genetics and bioengineering to achieve therapeutic goals is known as gene therapy[92]. Genetic factors play a significant role in PAH's development, primarily Heritable PAH (HPAH). Among numerous genetic mutations, BMPR-II (Bone Morphogenic Protein Receptor Type II), which is expressed predominantly in pulmonary endothelium, having complex interactions with the TGF-β (Transforming Growth Factor β) signaling pathway, is one of the remarkable factors responsible for causing PAH[75,93,94,95]. BMPR-II gene therapy prevented increment in TGF-β and reduced right ventricular hypertrophy, pulmonary vascular resistance and remodeling[75,95,96]. These results reflect the demand of gene therapy for the management of PAH.
Figure 5.
(A) It depicts the normal mechanism of stimulation of the canonical signaling pathway via the BMPR-II receptor, which ultimately controls the proliferation, differentiation, and maturation of the cell. (B) It depicts the imbalance in BMPR-II and TGF-β leading to PAH. The above diagram illustrates gene therapy's possibility in maintaining the normal canonical pathway. BMP9: Bone Morphogenic Protein-9; BMPR-II: Bone Morphogenic Protein Receptor Type II; TGF-β: Transforming Growth Factor-β[75,97,98].
Figure 5.
(A) It depicts the normal mechanism of stimulation of the canonical signaling pathway via the BMPR-II receptor, which ultimately controls the proliferation, differentiation, and maturation of the cell. (B) It depicts the imbalance in BMPR-II and TGF-β leading to PAH. The above diagram illustrates gene therapy's possibility in maintaining the normal canonical pathway. BMP9: Bone Morphogenic Protein-9; BMPR-II: Bone Morphogenic Protein Receptor Type II; TGF-β: Transforming Growth Factor-β[75,97,98].
Epigenetic Medicines
The heritable changes in the genome due to changes in gene expression without affecting the DNA sequence, such as DNA & RNA methylation, histone modifications, and non-coding RNA modification, is known as epigenetics. PAH-induced vascular cell changes occur at pathological and biochemical levels by affecting epigenetic-sensitive pathways. The advantage of epigenetic medicine, particularly DNA methylation, is that it is pharmacologically reversible and allows effective primary prevention[75,99,100,101].
Regenerative medicine collectively is expected to have reversible changes in PAH treatment. Thus making it an attractive treatment strategy in the future.
Conclusions
Initiation of combination therapy early in the treatment plan is highly recommended for the management of this progressive and incurable disease. Long-term trials with morbidity and mortality benefits as outcome measures are needed to further understand the safety and effectiveness of treatment options.
Palliative care with different invasive and non-invasive measures could help promote patients’ comfort and alleviate distress. Regenerative medicine as a group including stem cell therapy, gene therapy, and epigenetic therapy marks the new horizon for the cure of PAH which needs further exploration.
Author Contributions
Conceptualization by M.S., drafting of Manuscript done by all authors (M.S., V.P., S.K.S, R.T, N.K, S.S.), supervision done by M.S., S.S.
Conflicts of Interest
Authors declare no conflict of interest.
References
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.M.F.; Brida, M.; Carlsen, J.; Coats, A.J.; Escribano-Subias, P.; Ferrari, P.; et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Respir. J. 2023, 61, 2200879. [Google Scholar] [CrossRef]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef] [PubMed]
- Wijeratne, D.T.; Lajkosz, K.; Brogly, S.B.; Lougheed, M.D.; Jiang, L.; Housin, A.; Barber, D.; Johnson, A.; Doliszny, K.M.; Archer, S.L. Increasing Incidence and Prevalence of World Health Organization Groups 1 to 4 Pulmonary Hypertension: A Population-Based Cohort Study in Ontario, Canada. Circ. Cardiovasc. Qual Outcomes 2018, 11, e003973. [Google Scholar] [CrossRef] [PubMed]
- D'Alonzo, G.E.; Barst, R.J.; Ayres, S.M.; Bergofsky, E.H.; Brundage, B.H.; Detre, K.M.; Fishman, A.P.; Goldring, R.M.; Groves, B.M.; Kernis, J.T.; et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann. Intern. Med. 1991, 115, 343–349. [Google Scholar] [CrossRef] [PubMed]
- McGoon, M.D.; Miller, D.P. REVEAL: A contemporary US pulmonary arterial hypertension registry. Eur. Respir. Rev. 2012, 21, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Sitbon, O.; Chaouat, A.; Bertocchi, M.; Habib, G.; Gressin, V.; Yaici, A.; Weitzenblum, E.; Cordier, J.F.; Chabot, F.; et al. Pulmonary arterial hypertension in France: Results from a national registry. Am. J. Respir. Crit. Care Med. 2006, 173, 1023–1030. [Google Scholar] [CrossRef]
- Pietra, G.G.; Capron, F.; Stewart, S.; Leone, O.; Humbert, M.; Robbins, I.M.; Reid, L.M.; Tuder, R. Pathologic assessment of vasculopathies in pulmonary hypertension. J Am Coll Cardiol. 2004, 43, S25–S32. [Google Scholar] [CrossRef] [PubMed]
- Lüscher, T.F.; Barton, M. Endothelins and Endothelin Receptor Antagonists: Therapeutic Considerations for a Novel Class of Cardiovascular Drugs. Circulation 2000, 102, 2434–2440. [Google Scholar] [CrossRef] [PubMed]
- Hynynen, M.M.; Khalil, R.A. The Vascular Endothelin System in Hypertension - Recent Patents and Discoveries. Recent Patents Cardiovasc. Drug Discov. 2006, 1, 95–108. [Google Scholar] [CrossRef]
- Clozel, M.; Breu, V.; A Gray, G.; Kalina, B.; Löffler, B.M.; Burri, K.; Cassal, J.M.; Hirth, G.; Müller, M.; Neidhart, W. Pharmacological characterization of bosentan, a new potent orally active nonpeptide endothelin receptor antagonist. J. Pharmacol. Exp. Ther. 1994, 270, 228–235. [Google Scholar] [CrossRef]
- Kingman, M.; Ruggiero, R.; Torres, F. Ambrisentan, an endothelin receptor type A-selective endothelin receptor antagonist, for the treatment of pulmonary arterial hypertension. Expert Opin. Pharmacother. 2009, 10, 1847–1858. [Google Scholar] [CrossRef]
- Bolli, M.H.; Boss, C.; Binkert, C.; Buchmann, S.; Bur, D.; Hess, P.; et al. The Discovery of N -[5-(4-Bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]- N ′-propylsulfamide (Macitentan), an Orally Active, Potent Dual Endothelin Receptor Antagonist. J Med Chem. 2012, 55, 7849–7861. [Google Scholar] [CrossRef]
- Rubin, L.J.; Badesch, D.B.; Barst, R.J.; Galiè, N.; Black, C.M.; Keogh, A.; Pulido, T.; Frost, A.; Roux, S.; Leconte, I.; et al. Bosentan Therapy for Pulmonary Arterial Hypertension. N. Engl. J. Med. 2002, 346, 896–903. [Google Scholar] [CrossRef]
- Rivera-Lebron, B.N.; Risbano, M.G. Ambrisentan: A review of its use in pulmonary arterial hypertension. Ther. Adv. Respir. Dis. 2017, 11, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Galiè, N.; Olschewski, H.; Oudiz, R.J.; Torres, F.; Frost, A.; Ghofrani, H.A.; et al. Ambrisentan for the treatment of pulmonary arterial hypertension: Results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation 2008, 117, 3010–3019. [Google Scholar] [CrossRef] [PubMed]
- Pulido, T.; Adzerikho, I.; Channick, R.N.; Delcroix, M.; Galiè, N.; Ghofrani, A.; Jansa, P.; Jing, Z.-C.; Le Brun, F.-O.; Mehta, S.; et al. Macitentan and Morbidity and Mortality in Pulmonary Arterial Hypertension. N. Engl. J. Med. 2013, 369, 809–818. [Google Scholar] [CrossRef]
- Zhang, Y.-J.; Wang, N.; Gu, Z.-C.; Wei, A.-H.; Cheng, A.-N.; Fang, S.-S.; Du, H.-L.; Wang, L.-Z.; Zhang, G.-Q. A network meta-analysis for safety of endothelin receptor antagonists in pulmonary arterial hypertension. Cardiovasc. Diagn. Ther. 2019, 9, 239–249. [Google Scholar] [CrossRef]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef]
- Wharton, J.; Strange, J.W.; Møller, G.M.O.; Growcott, E.J.; Ren, X.; Franklyn, A.P.; Phillips, S.C.; Wilkins, M.R. Antiproliferative Effects of Phosphodiesterase Type 5 Inhibition in Human Pulmonary Artery Cells. Am. J. Respir. Crit. Care Med. 2005, 172, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Galiè, N.; Ghofrani, H.A.; Torbicki, A.; Barst, R.J.; Rubin, L.J.; Badesch, D.; Fleming, T.; Parpia, T.; Burgess, G.; Branzi, A.; et al. Sildenafil Citrate Therapy for Pulmonary Arterial Hypertension. N. Engl. J. Med. 2005, 353, 2148–2157. [Google Scholar] [CrossRef]
- Rubin, L.J.; Badesch, D.B.; Fleming, T.R.; Galiè, N.; Simonneau, G.; Ghofrani, H.A.; et al. Long-term treatment with sildenafil citrate in pulmonary arterial hypertension: The SUPER-2 study. Chest 2011, 140, 1274–1283. [Google Scholar] [CrossRef]
- Galie, N.; Brundage, B.H.; Ghofrani, H.A.; Oudiz, R.J.; Simonneau, G.; Safdar, Z.; Shapiro, S.; White, R.J.; Chan, M.; Beardsworth, A.; et al. Tadalafil Therapy for Pulmonary Arterial Hypertension. Circulation 2009, 119, 2894–2903. [Google Scholar] [CrossRef]
- Wrishko, R.E.; Dingemanse, J.; Yu, A.; Darstein, C.; Phillips, D.L.; Mitchell, M.I. Pharmacokinetic interaction between tadalafil and bosentan in healthy male subjects. J. Clin. Pharmacol. 2008, 48, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Oudiz, R.J.; Brundage, B.H.; Galiè, N.; Ghofrani, H.A.; Simonneau, G.; Botros, F.T.; Chan, M.; Beardsworth, A.; Barst, R.J. Tadalafil for the Treatment of Pulmonary Arterial Hypertension. Journal of the American College of Cardiology 2012, 60, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Barnes, H.; Brown, Z.; Burns, A.; Williams, T. Phosphodiesterase 5 inhibitors for pulmonary hypertension. Cochrane Database Syst Rev. 2019, 1, CD012621. [Google Scholar] [CrossRef] [PubMed]
- Ghofrani, H.; Hoeper, M.; Halank, M.; Meyer, F.; Staehler, G.; Behr, J.; Ewert, R.; Weimann, G.; Grimminger, F. Riociguat for chronic thromboembolic pulmonary hypertension and pulmonary arterial hypertension: A phase II study. Eur. Respir. J. 2010, 36, 792–799. [Google Scholar] [CrossRef] [PubMed]
- Ghofrani, H.-A.; Galiè, N.; Grimminger, F.; Grünig, E.; Humbert, M.; Jing, Z.-C.; Keogh, A.M.; Langleben, D.; Kilama, M.O.; Fritsch, A.; et al. Riociguat for the Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2013, 369, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Rubin, L.J.; Galiè, N.; Grimminger, F.; Grünig, E.; Humbert, M.; Jing, Z.-C.; Keogh, A.; Langleben, D.; Fritsch, A.; Menezes, F.; et al. Riociguat for the treatment of pulmonary arterial hypertension: A long-term extension study (PATENT-2). Eur. Respir. J. 2015, 45, 1303–1313. [Google Scholar] [CrossRef] [PubMed]
- Ghofrani, H.-A.; Grimminger, F.; Grünig, E.; Huang, Y.; Jansa, P.; Jing, Z.-C.; Kilpatrick, D.; Langleben, D.; Rosenkranz, S.; Menezes, F.; et al. Predictors of long-term outcomes in patients treated with riociguat for pulmonary arterial hypertension: Data from the PATENT-2 open-label, randomised, long-term extension trial. Lancet Respir. Med. 2016, 4, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Majed, B.H.; Khalil, R.A. Molecular Mechanisms Regulating the Vascular Prostacyclin Pathways and Their Adaptation during Pregnancy and in the Newborn. Pharmacol. Rev. 2012, 64, 540–582. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Cool, C.D.; Geraci, M.W.; Wang, J.; Abman, S.H.; Wright, L.; Badesch, D.; Voelkel, N.F. Prostacyclin Synthase Expression Is Decreased in Lungs from Patients with Severe Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 1999, 159, 1925–1932. [Google Scholar] [CrossRef]
- Gomberg-Maitland, M.; Olschewski, H. Prostacyclin therapies for the treatment of pulmonary arterial hypertension. Eur. Respir. J. 2008, 31, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Rubin, L.J. Treatment of Primary Pulmonary Hypertension with Continuous Intravenous Prostacyclin (Epoprostenol): Results of a Randomized Trial. Ann Intern Med. 1990, 112, 485. [Google Scholar] [CrossRef] [PubMed]
- Barst, R.J.; Rubin, L.J.; Long, W.A.; McGoon, M.D.; Rich, S.; Badesch, D.B.; Groves, B.M.; Tapson, V.F.; Bourge, R.C.; Brundage, B.H.; et al. A Comparison of Continuous Intravenous Epoprostenol (Prostacyclin) with Conventional Therapy for Primary Pulmonary Hypertension. N. Engl. J. Med. 1996, 334, 296–301. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, V.V.; Shillington, A.; Rich, S. Survival in primary pulmonary hypertension: The impact of epoprostenol therapy. Circulation. 2002, 106, 1477–1482. [Google Scholar] [CrossRef]
- Sitbon, O.; Humbert, M.; Nunes, H.; Parent, F.; Garcia, G.; Hervé, P.; et al. Long-term intravenous epoprostenol infusion in primary pulmonary hypertension: Prognostic factors and survival. J Am Coll Cardiol. 2002, 40, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Barst, R.J.; Galie, N.; Naeije, R.; Rich, S.; Bourge, R.C.; et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: A double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med. 2002, 165, 800–804. [Google Scholar] [CrossRef] [PubMed]
- Lang, I.; Gomez-Sanchez, M.; Kneussl, M.; Naeije, R.; Escribano, P.; Skoro-Sajer, N.; Vachiery, J.-L. Efficacy of Long-term Subcutaneous Treprostinil Sodium Therapy in Pulmonary Hypertension. Chest 2006, 129, 1636–1643. [Google Scholar] [CrossRef] [PubMed]
- Tapson, V.F.; Gomberg-Maitland, M.; McLaughlin, V.V.; Benza, R.L.; Widlitz, A.C.; Krichman, A.; et al. Safety and efficacy of IV treprostinil for pulmonary arterial hypertension: A prospective, multicenter, open-label, 12-week trial. Chest 2006, 129, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Gomberg-Maitland, M.; Tapson, V.F.; Benza, R.L.; McLaughlin, V.V.; Krichman, A.; Widlitz, A.C.; Barst, R.J. Transition from Intravenous Epoprostenol to Intravenous Treprostinil in Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2005, 172, 1586–1589. [Google Scholar] [CrossRef]
- McLaughlin, V.V.; Benza, R.L.; Rubin, L.J.; Channick, R.N.; Voswinckel, R.; Tapson, V.F.; et al. Addition of inhaled treprostinil to oral therapy for pulmonary arterial hypertension: A randomized controlled clinical trial. J Am Coll Cardiol. 2010, 55, 1915–1922. [Google Scholar] [CrossRef] [PubMed]
- Spikes, L.A.; Bajwa, A.A.; Burger, C.D.; Desai, S.V.; Eggert, M.S.; El-Kersh, K.A.; et al. BREEZE: Open-label clinical study to evaluate the safety and tolerability of treprostinil inhalation powder as Tyvaso DPITM in patients with pulmonary arterial hypertension. Pulm circ. 2022, 12, e12063. [Google Scholar] [CrossRef]
- West, N.; Smoot, K.; Patzlaff, N.; Miceli, M.; Waxman, A. Plain Language Summary of the INCREASE Study: Inhaled Treprostinil (Tyvaso) for the Treatment of Pulmonary Hypertension Due to Interstitial Lung Disease. Futur. Cardiol. 2023, 19, 229–239. [Google Scholar] [CrossRef]
- Arslan, A.; Smith, J.; Qureshi, M.R.; Uysal, A.; Patel, K.K.; Herazo-Maya, J.D.; et al. Evolution of pulmonary hypertension in interstitial lung disease: A journey through past, present, and future. Front Med (Lausanne) 2023, 10, 1306032. [Google Scholar] [CrossRef]
- Olschewski, H.; Simonneau, G.; Galiè, N.; Higenbottam, T.; Naeije, R.; Rubin, L.J.; et al. Inhaled Iloprost for Severe Pulmonary Hypertension. N Engl J Med. 2002, 347, 322–329. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, V.V.; Oudiz, R.J.; Frost, A.; Tapson, V.F.; Murali, S.; Channick, R.N.; et al. Randomized study of adding inhaled iloprost to existing bosentan in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2006, 174, 1257–1263. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Torbicki, A.; Hoeper, M.M.; Delcroix, M.; Karlócai, K.; Galiè, N.; Degano, B.; Bonderman, D.; Kurzyna, M.; Efficace, M.; et al. Selexipag: An oral, selective prostacyclin receptor agonist for the treatment of pulmonary arterial hypertension. Eur. Respir. J. 2012, 40, 874–880. [Google Scholar] [CrossRef] [PubMed]
- Sitbon, O.; Channick, R.; Chin, K.M.; Frey, A.; Gaine, S.; Galiè, N.; Ghofrani, H.-A.; Hoeper, M.M.; Lang, I.M.; Preiss, R.; et al. Selexipag for the Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2015, 373, 2522–2533. [Google Scholar] [CrossRef]
- Galiè, N.; Barberà, J.A.; Frost, A.E.; Ghofrani, H.-A.; Hoeper, M.M.; McLaughlin, V.V.; Peacock, A.J.; Simonneau, G.; Vachiery, J.-L.; Grünig, E.; et al. Initial Use of Ambrisentan plus Tadalafil in Pulmonary Arterial Hypertension. N. Engl. J. Med. 2015, 373, 834–844. [Google Scholar] [CrossRef]
- Grünig, E.; Jansa, P.; Fan, F.; Hauser, J.A.; Pannaux, M.; Morganti, A.; Rofael, H.; Chin, K.M. Randomized Trial of Macitentan/Tadalafil Single-Tablet Combination Therapy for Pulmonary Arterial Hypertension. J Am Coll Cardiol. 2024, 83, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGF-beta signal transduction. Annu Rev Biochem. 1998, 67, 753–791. [Google Scholar] [CrossRef] [PubMed]
- Yndestad, A.; Larsen, K.-O.; Øie, E.; Ueland, T.; Smith, C.; Halvorsen, B.; Sjaastad, I.; Skjønsberg, O.H.; Pedersen, T.M.; Anfinsen, O.-G.; et al. Elevated levels of activin A in clinical and experimental pulmonary hypertension. J. Appl. Physiol. 2009, 106, 1356–1364. [Google Scholar] [CrossRef]
- Nakaoka, T.; Gonda, K.; Ogita, T.; Otawara-Hamamoto, Y.; Okabe, F.; Kira, Y.; Harii, K.; Miyazono, K.; Takuwa, Y.; Fujita, T. Inhibition of rat vascular smooth muscle proliferation in vitro and in vivo by bone morphogenetic protein-2. J. Clin. Investig. 1997, 100, 2824–2832. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, C.; Stewart, S.; Upton, P.D.; Machado, R.; Thomson, J.R.; Trembath, R.C.; Morrell, N.W. Primary Pulmonary Hypertension Is Associated With Reduced Pulmonary Vascular Expression of Type II Bone Morphogenetic Protein Receptor. Circulation 2002, 105, 1672–1678. [Google Scholar] [CrossRef] [PubMed]
- Yung, L.-M.; Yang, P.; Joshi, S.; Augur, Z.M.; Kim, S.S.J.; Bocobo, G.A.; Dinter, T.; Troncone, L.; Chen, P.-S.; McNeil, M.E.; et al. ACTRIIA-Fc rebalances activin/GDF versus BMP signaling in pulmonary hypertension. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Humbert, M.; McLaughlin, V.; Gibbs, J.S.R.; Gomberg-Maitland, M.; Hoeper, M.M.; Preston, I.R.; et al. Sotatercept for the Treatment of Pulmonary Arterial Hypertension. N Engl J Med. 2021, 384, 1204–1215. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Badesch, D.B.; Ghofrani, H.A.; Gibbs, J.S.R.; Gomberg-Maitland, M.; McLaughlin, V.V.; Preston, I.R.; Souza, R.; Waxman, A.B.; Grünig, E.; et al. Phase 3 Trial of Sotatercept for Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2023, 388, 1478–1490. [Google Scholar] [CrossRef] [PubMed]
- Nasrollahizadeh, A.; Soleimani, H.; Nasrollahizadeh, A.; Hashemi, S.M.; Hosseini, K. Navigating the Sotatercept landscape: A meta-analysis of clinical outcomes. Clin Cardiol. 2024, 47, e24173. [Google Scholar] [CrossRef]
- Chin, K.M.; Gaine, S.P.; Gerges, C.; Jing, Z.-C.; Mathai, S.C.; Tamura, Y.; McLaughlin, V.V.; Sitbon, O. Treatment algorithm for pulmonary arterial hypertension. Eur. Respir. J. 2024, 64, 2401325. [Google Scholar] [CrossRef]
- Stącel, T.; Latos, M.; Urlik, M.; Nęcki, M.; Antończyk, R.; Hrapkowicz, T.; Kurzyna, M.; Ochman, M. Interventional and Surgical Treatments for Pulmonary Arterial Hypertension. J. Clin. Med. 2021, 10, 3326. [Google Scholar] [CrossRef] [PubMed]
- Keogh, A.M.; Mayer, E.; Benza, R.L.; Corris, P.; Dartevelle, P.G.; Frost, A.E.; Kim, N.H.; Lang, I.M.; Pepke-Zaba, J.; Sandoval, J. Interventional and Surgical Modalities of Treatment in Pulmonary Hypertension. J Am Coll Cardiol. 2009, 54, S67–S77. [Google Scholar] [CrossRef]
- Zebadua, R.; Zayas, N.; Lopez, J.; Zorrilla, L.; Pozas, M.; Villalobos, M.; et al. Survival of patients diagnosed with Pulmonary Hypertension undergoing Atrial Septostomy. In: 1301 - Pulmonary hypertension [Internet]. European Respiratory Society; 2022 [cited 2025 Jan 8]. p. 4186. Available online: http://publications.ersnet.org/lookup/doi/10.1183/13993003.congress-2022.4186.
- Baruteau, A.-E.; Serraf, A.; Lévy, M.; Petit, J.; Bonnet, D.; Jais, X.; Vouhé, P.; Simonneau, G.; Belli, E.; Humbert, M. Potts Shunt in Children With Idiopathic Pulmonary Arterial Hypertension: Long-Term Results. Ann. Thorac. Surg. 2012, 94, 817–824. [Google Scholar] [CrossRef]
- Zhang, H.; Wei, Y.; Zhang, C.; Yang, Z.; Kan, J.; Gu, H.; et al. Pulmonary Artery Denervation for Pulmonary Arterial Hypertension: A Sham-Controlled Randomized PADN-CFDA Trial. JACC Cardiovasc Interv. 2022, 15, 2412–2423. [Google Scholar] [CrossRef] [PubMed]
- Otto, M.; McGiffin, D.; Whitford, H.; Kure, C.; Snell, G.; Diehl, A.; Orosz, J.; Burrell, A.J. Survival and left ventricular dysfunction post lung transplantation for pulmonary arterial hypertension. J. Crit. Care 2022, 72, 154120. [Google Scholar] [CrossRef]
- Hecker, F.; Keller, H.; Vasa-Nicotera, M.; Iken, S.; Holubec, T. Percutaneous dual-outflow extracorporeal membrane oxygenation support in secondary right ventricular failure. JTCVS Tech. 2022, 13, 125–127. [Google Scholar] [CrossRef]
- Leopold, J.A.; Maron, B.A. Precision Medicine in Pulmonary Arterial Hypertension. Circ. Res. 2019, 124, 832–833. [Google Scholar] [CrossRef] [PubMed]
- Benincasa, G.; DeMeo, D.L.; Glass, K.; Silverman, E.K.; Napoli, C. Epigenetics and pulmonary diseases in the horizon of precision medicine: A review. Eur Respir J. 2021, 57, 2003406. [Google Scholar] [CrossRef] [PubMed]
- Khirfan, G.; Tonelli, A.R.; Ramsey, J.; Sahay, S. Palliative care in pulmonary arterial hypertension: An underutilised treatment. Eur. Respir. Rev. 2018, 27, 180069. [Google Scholar] [CrossRef]
- Christiansen, D.; Porter, S.; Hurlburt, L.; Weiss, A.; Granton, J.; Wentlandt, K. Pulmonary Arterial Hypertension: A Palliative Medicine Review of the Disease, Its Therapies, and Drug Interactions. J Pain Symptom Manage. 2020, 59, 932–943. [Google Scholar] [CrossRef]
- Loisel, F.; Provost, B.; Haddad, F.; Guihaire, J.; Amsallem, M.; Vrtovec, B.; et al. Stem cell therapy targeting the right ventricle in pulmonary arterial hypertension: Is it a potential avenue of therapy? Pulm Circ. 2018, 8, 2045893218755979. [Google Scholar] [CrossRef]
- Brouwer, K.M.; Hoogenkamp, H.R.; Daamen, W.F.; van Kuppevelt, T.H. Regenerative medicine for the respiratory system: Distant future or tomorrow’s treatment? Am J Respir Crit Care Med. 2013, 187, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Mason, C.; Dunnill, P. A Brief Definition of Regenerative Medicine. Regen. Med. 2008, 3, 1–5. [Google Scholar] [CrossRef]
- Weissman, I.L. Stem cells: Units of development, units of regeneration, and units in evolution. Cell 2000, 100, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Bisserier, M.; Pradhan, N.; Hadri, L. Current and emerging therapeutic approaches to pulmonary hypertension. Rev. Cardiovasc. Med. 2020, 21, 163–179. [Google Scholar] [CrossRef] [PubMed]
- Jaenisch, R.; Young, R. Stem Cells, the Molecular Circuitry of Pluripotency and Nuclear Reprogramming. Cell 2008, 132, 567–582. [Google Scholar] [CrossRef] [PubMed]
- Dulak, J.; Szade, K.; Szade, A.; Nowak, W.; Józkowicz, A. Adult stem cells: Hopes and hypes of regenerative medicine. Acta Biochim. Pol. 2015, 62, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Zheng, R.; Xu, T.; Wang, X.; Yang, L.; Wang, J.; Huang, X. Stem cell therapy in pulmonary hypertension: Current practice and future opportunities. Eur. Respir. Rev. 2023, 32, 230112. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Jung, J.-H.; Ahn, K.-J.; Jang, A.Y.; Byun, K.; Yang, P.C.; Chung, W.-J. Stem Cell and Exosome Therapy in Pulmonary Hypertension. Korean Circ. J. 2022, 52, 110–122. [Google Scholar] [CrossRef]
- Pu, X.; Du, L.; Hu, Y.; Fan, Y.; Xu, Q. Stem/Progenitor Cells and Pulmonary Arterial Hypertension. Arter. Thromb. Vasc. Biol. 2021, 41, 167–178. [Google Scholar] [CrossRef]
- Sun, Q.W.; Sun, Z. Stem Cell Therapy for Pulmonary Arterial Hypertension: An Update. J. Hear. Lung Transplant. 2022. [Google Scholar] [CrossRef]
- Dierick, F.; Solinc, J.; Bignard, J.; Soubrier, F.; Nadaud, S. Progenitor/Stem Cells in Vascular Remodeling during Pulmonary Arterial Hypertension. Cells 2021, 10, 1338. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-X.; Pan, Y.-Y.; Zhao, Y.-Y.; Wang, X.-X. Endothelial Progenitor Cell-Based Therapy for Pulmonary Arterial Hypertension. Cell Transplant. 2013, 22, 1325–1336. [Google Scholar] [CrossRef]
- Nagaya, N.; Kangawa, K.; Kanda, M.; Uematsu, M.; Horio, T.; Fukuyama, N.; Hino, J.; Harada-Shiba, M.; Okumura, H.; Tabata, Y.; et al. Hybrid Cell–Gene Therapy for Pulmonary Hypertension Based on Phagocytosing Action of Endothelial Progenitor Cells. Circulation 2003, 108, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.D.; Courtman, D.W.; Deng, Y.; Kugathasan, L.; Zhang, Q.; Stewart, D.J. Rescue of monocrotaline-induced pulmonary arterial hypertension using bone marrow-derived endothelial-like progenitor cells: Efficacy of combined cell and eNOS gene therapy in established disease. Circ Res. 2005, 96, 442–450. [Google Scholar] [CrossRef]
- Granton, J.; Langleben, D.; Kutryk, M.B.; Camack, N.; Galipeau, J.; Courtman, D.W.; et al. Endothelial NO-Synthase Gene-Enhanced Progenitor Cell Therapy for Pulmonary Arterial Hypertension: The PHACeT Trial. Circulation Research. 2015, 117, 645–654. [Google Scholar] [CrossRef]
- Wang, X.X.; Zhang, F.R.; Shang, Y.P.; Zhu, J.H.; Xie, X.D.; Tao, Q.M.; et al. Transplantation of Autologous Endothelial Progenitor Cells May Be Beneficial in Patients With Idiopathic Pulmonary Arterial Hypertension. Journal of the American College of Cardiology. 2007, 49, 1566–1571. [Google Scholar] [CrossRef] [PubMed]
- Farkas, L.; Kolb, M. Vascular Repair and Regeneration as a Therapeutic Target for Pulmonary Arterial Hypertension. Respiration 2013, 85, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Fukumitsu, M.; Suzuki, K. Mesenchymal stem/stromal cell therapy for pulmonary arterial hypertension: Comprehensive review of preclinical studies. J. Cardiol. 2019, 74, 304–312. [Google Scholar] [CrossRef]
- Huang, W.-C.; Ke, M.-W.; Cheng, C.-C.; Chiou, S.-H.; Wann, S.-R.; Shu, C.-W.; Chiou, K.-R.; Tseng, C.-J.; Pan, H.-W.; Mar, G.-Y.; et al. Therapeutic Benefits of Induced Pluripotent Stem Cells in Monocrotaline-Induced Pulmonary Arterial Hypertension. PLoS ONE 2016, 11, e0142476. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, M.W.; Ting, C.Y.; Chan, D.Z.H.; Cheng, Y.C.; Lee, Y.C.; Hsu, C.C.; et al. Utility of iPSC-Derived Cells for Disease Modeling, Drug Development, and Cell Therapy. Cells 2022, 11, 1853. [Google Scholar] [CrossRef]
- Gonçalves, G.A.R.; Paiva R de, M.A. Gene therapy: Advances, challenges and perspectives. Einstein (Sao Paulo). 2017, 15, 369–375. [Google Scholar] [CrossRef]
- Wang, M.-T.; Charng, M.-J.; Chi, P.-L.; Cheng, C.-C.; Hung, C.C.; Huang, W.-C. Gene Mutation Annotation and Pedigree for Pulmonary Arterial Hypertension Patients in Han Chinese Patients. Glob. Hear. 2021, 16. [Google Scholar] [CrossRef] [PubMed]
- Fazal, S.; Bisserier, M.; Hadri, L. Molecular and Genetic Profiling for Precision Medicines in Pulmonary Arterial Hypertension. Cells 2021, 10, 638. [Google Scholar] [CrossRef]
- Reynolds, A.M.; Holmes, M.D.; Danilov, S.M.; Reynolds, P.N. Targeted gene delivery of BMPR2 attenuates pulmonary hypertension. Eur. Respir. J. 2012, 39, 329–343. [Google Scholar] [CrossRef]
- Harper, R.L.; Reynolds, A.M.; Bonder, C.S.; Reynolds, P.N. BMPR2 gene therapy for PAH acts via Smad and non-Smad signalling. Respirology 2016, 21, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Theilmann, A.L.; Hawke, L.G.; Hilton, L.R.; Whitford, M.K.; Cole, D.V.; Mackeil, J.L.; Dunham-Snary, K.J.; Mewburn, J.; James, P.D.; Maurice, D.H.; et al. Endothelial BMPR2 Loss Drives a Proliferative Response to BMP (Bone Morphogenetic Protein) 9 via Prolonged Canonical Signaling. Arter. Thromb. Vasc. Biol. 2020, 40, 2605–2618. [Google Scholar] [CrossRef]
- Xu, B.; Xu, G.; Yu, Y.; Lin, J. The role of TGF-β or BMPR2 signaling pathway-related miRNA in pulmonary arterial hypertension and systemic sclerosis. Arthritis Res. Ther. 2021, 23, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Benincasa, G.; DeMeo, D.L.; Glass, K.; Silverman, E.K.; Napoli, C. Epigenetics and pulmonary diseases in the horizon of precision medicine: A review. Eur. Respir. J. 2021, 57, 2003406. [Google Scholar] [CrossRef]
- Dave, J.; Jagana, V.; Janostiak, R.; Bisserier, M. Unraveling the epigenetic landscape of pulmonary arterial hypertension: Implications for personalized medicine development. J. Transl. Med. 2023, 21, 1–18. [Google Scholar] [CrossRef]
- Napoli, C.; Benincasa, G.; Loscalzo, J. Epigenetic Inheritance Underlying Pulmonary Arterial Hypertension. Arter. Thromb. Vasc. Biol. 2019, 39, 653–664. [Google Scholar] [CrossRef]
- Clinical trials.gov [Internet].
Figure 1.
Schematic representation of pathophysiology and pharmacotherapy of Pulmonary Arterial Hypertension.Endothelin (ET) pathway: Endothelin-1 (ET-1) mediates its action via Endothelin Receptor (ER). Endothelin Receptor Antagonists (ERAs) inhibit (-) the action of Endothelin -1 on the Endothelin Receptor by blocking it.Nitric Oxide (NO) pathway: Nitric Oxide acts upon smooth muscles via its second messenger cyclic Guanosine Monophosphate (cGMP). Soluble Guanylate Cyclase (sGC) stimulator, Riociguat stimulates (+) soluble Guanylate Cyclase (sGC) and increases the conversion of Guanosine Triphosphate (GTP) into cyclic Guanosine Monophosphate (cGMP) whereas, Phosphodiesterase 5 inhibitors (PDE-5is) inhibit (-) the action of Phosphodiesterase 5(PDE5) and prevent conversion of cGMP into Guanosine Monophosphate (GMP). Prostacyclin (PGI2) pathway: Prostacyclin mediates its action via the Prostacyclin receptor (IP). Prostacyclin Analogues (PCAs) and Prostacyclin Receptor Agonists (PRAs) act upon the prostacyclin receptor and stimulate the production of cyclic Adenosine Monophosphate(cAMP). AC: Adenylyl cyclase, ATP: Adenosine Triphosphate.
Figure 1.
Schematic representation of pathophysiology and pharmacotherapy of Pulmonary Arterial Hypertension.Endothelin (ET) pathway: Endothelin-1 (ET-1) mediates its action via Endothelin Receptor (ER). Endothelin Receptor Antagonists (ERAs) inhibit (-) the action of Endothelin -1 on the Endothelin Receptor by blocking it.Nitric Oxide (NO) pathway: Nitric Oxide acts upon smooth muscles via its second messenger cyclic Guanosine Monophosphate (cGMP). Soluble Guanylate Cyclase (sGC) stimulator, Riociguat stimulates (+) soluble Guanylate Cyclase (sGC) and increases the conversion of Guanosine Triphosphate (GTP) into cyclic Guanosine Monophosphate (cGMP) whereas, Phosphodiesterase 5 inhibitors (PDE-5is) inhibit (-) the action of Phosphodiesterase 5(PDE5) and prevent conversion of cGMP into Guanosine Monophosphate (GMP). Prostacyclin (PGI2) pathway: Prostacyclin mediates its action via the Prostacyclin receptor (IP). Prostacyclin Analogues (PCAs) and Prostacyclin Receptor Agonists (PRAs) act upon the prostacyclin receptor and stimulate the production of cyclic Adenosine Monophosphate(cAMP). AC: Adenylyl cyclase, ATP: Adenosine Triphosphate.
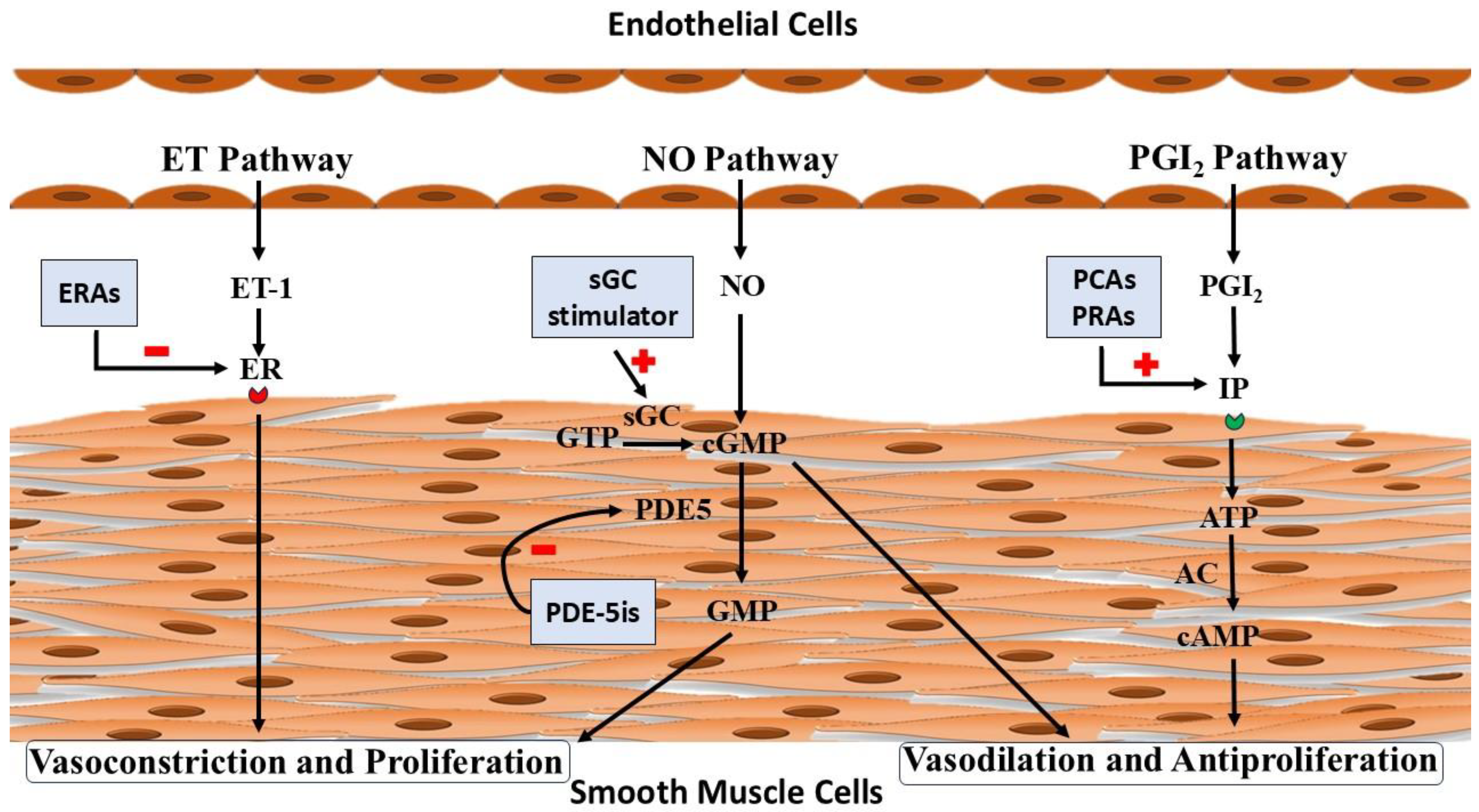
Figure 2.
(A) Unbalanced action of Activin due to Bone Morphogenetic Protein Receptor II (BMPR- II) mutation leads to increased proliferation. (B) Sotatercept binding with Activin counteracts the proliferative effect of Activin. BMP: Bone Morphogenetic Protein.
Figure 2.
(A) Unbalanced action of Activin due to Bone Morphogenetic Protein Receptor II (BMPR- II) mutation leads to increased proliferation. (B) Sotatercept binding with Activin counteracts the proliferative effect of Activin. BMP: Bone Morphogenetic Protein.
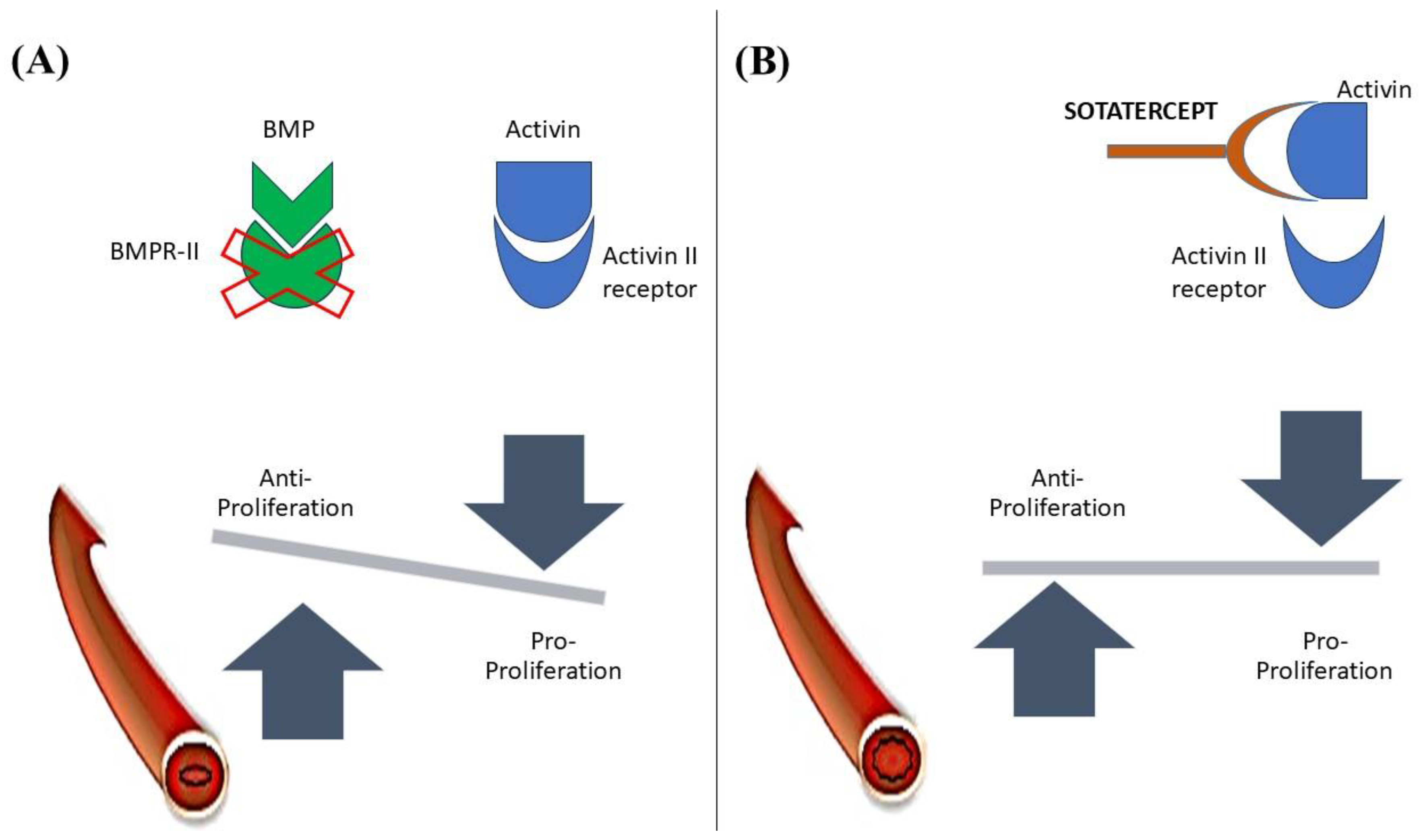
Figure 3.
Treatment Algorithm for PAH patients. ERA: Endothelin Receptor Antagonist, IV: Intravenous, PDE-5i: Phosphodiesterase 5 inhibitor, SC: subcutaneous.
Figure 3.
Treatment Algorithm for PAH patients. ERA: Endothelin Receptor Antagonist, IV: Intravenous, PDE-5i: Phosphodiesterase 5 inhibitor, SC: subcutaneous.
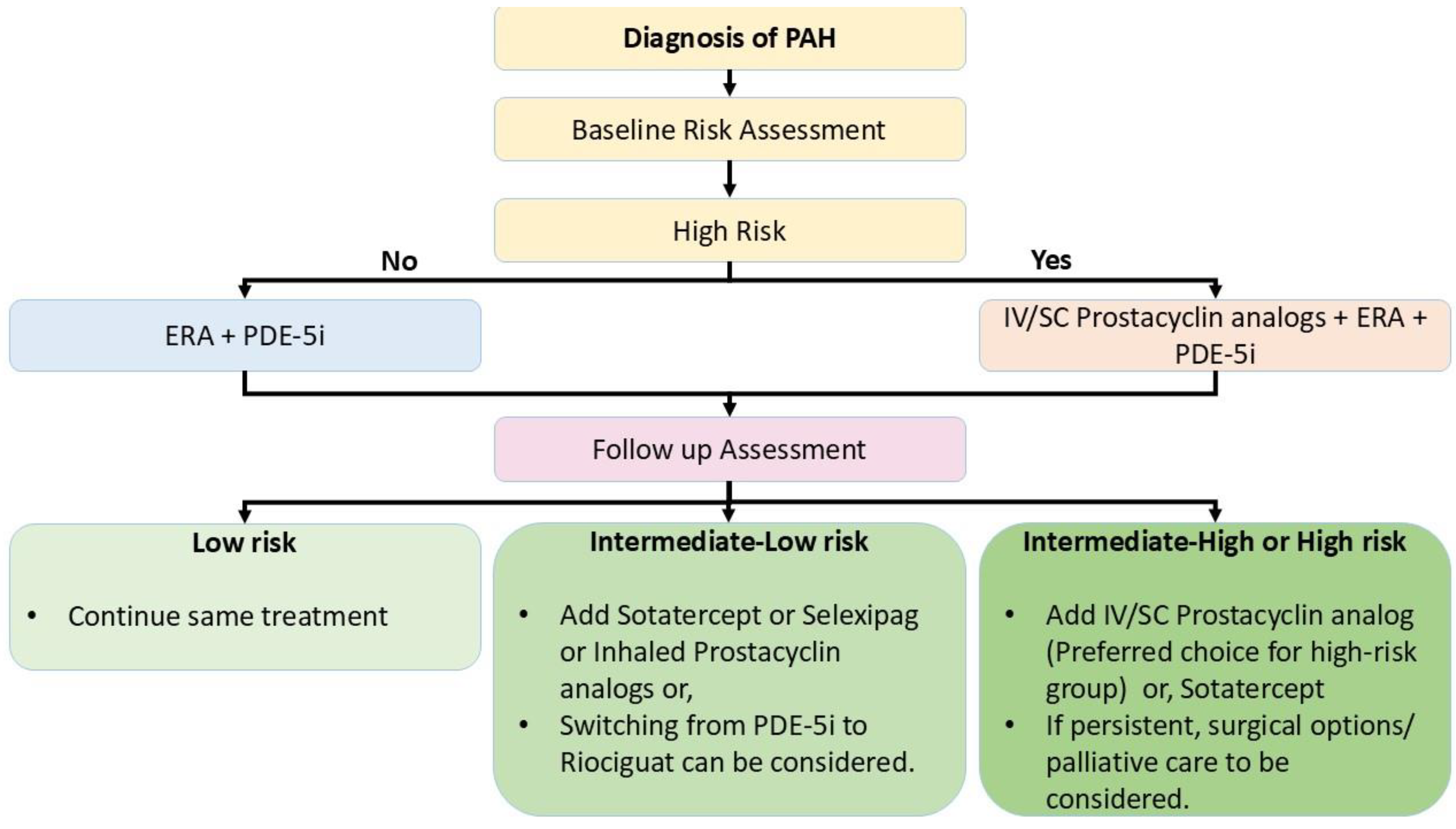

Table 1.
Hemodynamic Definitions of Pulmonary Arterial Hypertension (PAH).
| Mean Pulmonary Artery Pressure (mPAP) | ≥ 20 mmHg |
| Pulmonary Arterial Wedge Pressure (PAWP) | ≤ 15 mmHg |
| Pulmonary Vascular Resistance (PVR) | ≥ 2 WU (Wood units) |
Table 2.
Classification of PAH.
|
Idiopathic PAH (IPAH) Heritable PAH (HPAH) Drug and toxin induced PAH (DT-PAH) PAH associated with: Connective tissue disease (CTD) HIV infection Portal Hypertension Congenital Heart Disease (CHD) Schistosomiasis PAH long term responders to Calcium channel blockers PAH with overt features of venous/capillaries (PVOD/PCH) involvement. Persistent PH of the newborn syndrome. |
Table 3.
Relative Risk (RR) of adverse effects of Endothelin Receptor Antagonists (ERAs).
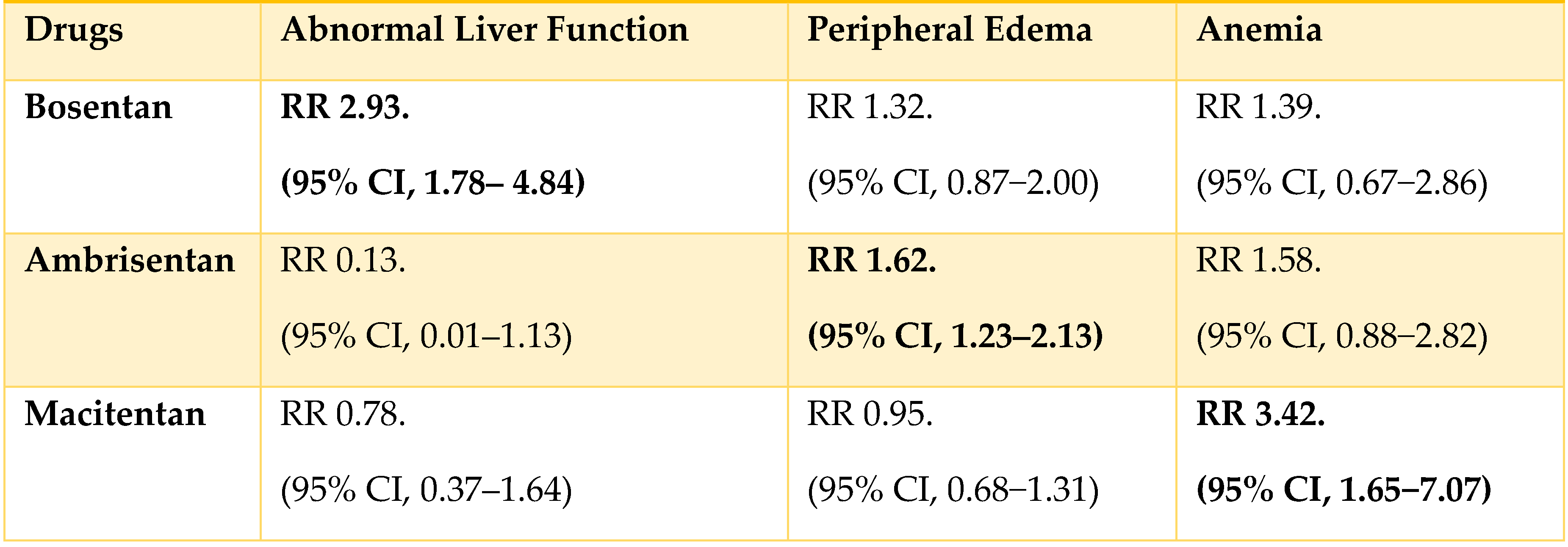
Table 4.
Primary End Point (Change in PVR from baseline to week 16).
| M/T FDC | Macitentan | Treatment Effect | |
|---|---|---|---|
| Reduction in PVR | 45% | 23% | 29% reduction |
| Geometric mean ratio of change in PVR | 0.55 (95% CI= 0.50-0.60) | 0.77 (95% CI= 0.69-0.87) | 0.71 (95% CI=0.61-0.82) |
| M/T FDC | Tadalafil | Treatment Effect | |
| Reduction in PVR | 44% | 22% | 28% reduction |
| Geometric mean ratio of change in PVR | 0.56(95% CI= 0.52-0.60) | 0.78 (95% CI= 0.72-0.84) | 0.72 (95% CI= 0.64-0.80) |
Table 5.
Palliative care in End-stage PAH patient AS: Atrial Septostomy; ECMO: Extracorporeal Membrane Oxygenation; RVAD: Right Ventricle Assist Device [60,69].
| Invasive therapy | Right to left shunting (AS, Potts shunt) Pulmonary artery denervation Others: RVAD, Right Ventricular Pacing, ECMO |
| Non-invasive therapy | Pain management Symptomatic treatment Treatment of underlying psychiatric disorders Specific therapy |
| Others | Counselling Financial assistance |
Table 6.
Some of the upcoming trials in PAH.
| Title | Primary Outcome Measures | Time Frame |
|---|---|---|
| Positioning Imatinib for Pulmonary Arterial Hypertension (PIPAH) | Identifying the highest tolerated doseChange in pulmonary vascular resistance (PVR) | 12 months24 months |
| Clinical Trial of 2- hydroxbenzylamine (2-HOBA) in Pulmonary Arterial Hypertension | Change in acetylated Superoxide Dismutase 2 (SOD2) and Long-chain acyl-CoA dehydrogenase (LCAD) in plasma. | Baseline and 12-weeks |
| Apabetalone for Pulmonary Arterial Hypertension (APPROACH-2) | Placebo-corrected change from baseline in PVR at week 24 | Baseline, and 24 weeks |
| Metabolic Remodeling in Pulmonary Arterial Hypertension (PAH) | Change in ratio of oxidative metabolism to glycolysis | Baseline and 6 months |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



