Submitted:
14 January 2025
Posted:
14 January 2025
You are already at the latest version
Abstract
Metabolic dysfunction-associated steatotic liver disease (MASLD) is a multifactorial disorder characterized by an excessive lipids accumulation in the liver which dysregulates this organ’s function. The key contributor to MASLD development seems to be insulin resistance (IR) which affects many organs (including adipose tissue, skeletal muscles and the liver), whereas the molecular background is associated with oxidative, nitrosative and carbonyl stress. Among molecules responsible for carbonyl stress effects, methylglyoxal (MGO) seems to play the major pathological function. MGO—a by-product of glycolysis, fructolysis and lipolysis (from glycerol and fatty acids-derived ketone bodies)—is implicated in hyperglycemia, hyperlipidemia, obesity, type 2 diabetes, hypertension and cardiovascular diseases. Its causative effect in the stimulation of prooxidative and proinflammatory pathways has been well documented. Since metabolic dysregulation leading to these pathologies underlies also MASLD, the role of MGO in MASLD is addressed in this review. Potential MGO participation in the mechanism of MASLD development is discussed in regard to its role in different signaling routes leading to pathological events accelerating the disorder. Moreover, treatment strategies including approved and potential therapies in MASLD are overviewed and discussed. Among them, medications aimed at attenuating MGO-induced pathological processes are addressed.
Keywords:
methylglyoxal
; advanced glycation end products
; AGEs
; AMPK
; MASLD
; MAFLD
; NAFLD
; steatohepatitis
; metformin
; silymarin
1. Introduction
This review comprises issues associated with the possible function of methylglyoxal in the development and progression of metabolic dysfunction-associated steatotic liver disease (MASLD). First, the description of MASLD with the mechanisms underlying this disorder are presented (Section 2). Subsequently, methylglyoxal metabolism and its detrimental effects in pathology are addressed (Section 3). The involvement of MGO in early and advanced MASLD stages is discussed in Section 4. Section 5 includes the discussion on possible MGO participation in fructose-mediated pathological routes leading to hepatic steatosis. Finally, the therapies in MASLD treatment are overviewed in Section 6. The detailed methodological approach associated with the literature search is described in Section 7. The review is complemented with the conclusions and the remarks on the future perspectives in regard to MGO impact on MASLD, as well as medicinal drugs which would inhibit pathological routes leading to MASLD development, including therapies aimed at the attenuation of MGO deleterious actions (Section 8).
2. Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD)
The disease entity “non-alcoholic fatty liver disease” (“NAFLD”) characterized mainly by steatosis was renamed in 2020 into “metabolic dysfunction-associated fatty liver disease” (“MAFLD”) [1], and now (according to the current Clinical Practice Guidelines) it is termed “metabolic dysfunction-associated steatotic liver disease” (“MASLD”) [2]. The change was proposed by an international panel of experts to underline the systemic metabolic dysregulation involved in the etiology of this type of liver dysfunction. Up to 5–20% of NAFLD cases develops from simple steatosis into non-alcoholic steatohepatitis (NASH) [3,4]. In around 20–30% of NASH cases fibrosis and cirrhosis are observed which further (in up to 2% of patients) can develop into hepatocellular carcinoma (HCC) [1,3,4,5].
Now, due to the change in the disorder term, also the subgroups of this liver dysfunction have been renamed. According to the present nomenclature: MASLD includes the condition concerning isolated liver steatosis called “metabolic dysfunction-associated steatotic liver” (“MASL”), whereas metabolic dysfunction reflected by steatohepatitis is termed “metabolic dysfunction-associated steatohepatitis” (“MASH”) [2]. Therefore, NAFLD has been renamed into MASLD, and NASH into MASH. Additionally, the new general definition of steatotic liver disease (SLD) has been coined, which encompasses liver dysfunctions associated with enhanced accumulation of lipids due to different etiologies. SLD comprises MASLD, MASLD with moderate (increased) alcohol intake (MetALD), alcohol-related liver disease (ALD), specific etiologies of SLD (e.g., drug induced, monogenic diseases) and cryptogenic SLD [2]. The diagnosis of MASLD requires documented (by imaging or biopsy) steatosis as well as the presence of at least one cardiometabolic risk factor which include: overweight or obesity, dysglycaemia or type 2 diabetes (T2DM), elevated plasma triacylglycerols (TAGs), decreased HDL-cholesterol (HDL-C) and elevated blood pressure. If additionally inflammation and hepatocytes ballooning is histologically detected, then MASH is diagnosed. Since the characteristics of NAFLD are aggregable with MASLD, these two terms can be used interchangeably [2].
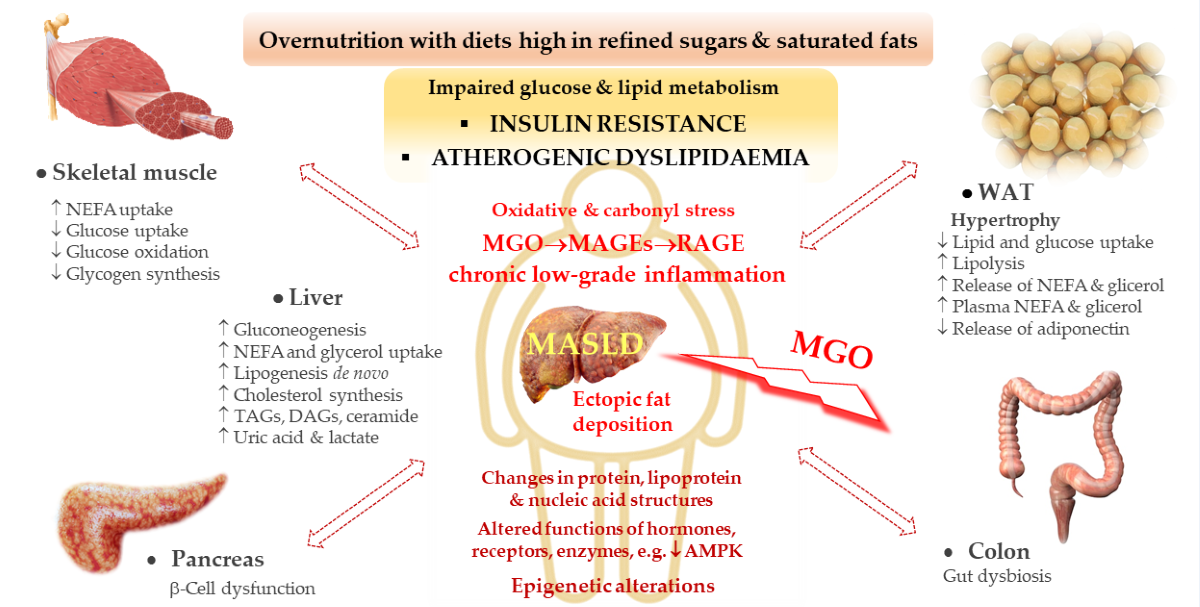
Among the theories explaining the development of NAFLD/MASLD, there have been “two-hit hypothesis” and “multiple-hit hypothesis” [3]. The first hypothesis assumes that the initial pathological event (first hit) is the accumulation of lipids in the liver due to the sedentary life style, high-fat diet, obesity and insulin resistance, which primes liver cells to the further pathological processes inducing inflammation and fibrogenesis—this is the second hit which promotes the progression to NASH/MASH and cirrhosis [3]. However, considering the complexity of the underlying events which induce and accelerate MASLD, both at the systemic and intracellular level, as well as the fact that also lean individuals suffer from this disorder, this is rather multiple-hit hypothesis which better addresses the mechanisms responsible for this type of liver dysfunction [6]. As reviewed by Buzzetti et al. [3], the leading driving force of NAFLD/MASLD is insulin resistance (IR) which impairs the adipose tissue metabolism promoting TAGs hydrolysis, thus enhancing non-esterified fatty acids (NEFA) release from the adipose tissue to the circulation. Additionally, IR downregulates glucose (Glc) transporters GLUT-4 in the skeletal muscles and adipose tissues which leads to hyperglycemia. These processes drive the uptake of both NEFA and Glc by other tissues including the liver, supplying this organ with substrates for de novo lipogenesis (DNL) and TAGs synthesis. Hence, both an excessive lipids accumulation in hepatocytes as well as their secretion from the liver to the circulation in the form of very low density lipoproteins (VLDL) are observed yielding hypertriglyceridemia. Additionally, obesity and IR induce proinflammatory processes in the adipose tissue conditioned by disturbances in adipokines release, which affects the liver and other organs [7,8]. Enhanced and prolonged influx of NEFA to the liver leads to lipotoxicity disturbing intracellular organelles and processes resulting in mitochondrial dysfunction which leads to oxidative stress development as well as endoplasmic reticulum (ER) stress [3]. Although the precise mechanism of MASLD has not been elucidated, some new findings in regard to intracellular organelle dysfunction, have been recently discussed by Li et al. [6]. The authors addressed the processes induced by lipotoxicity such as ER stress which due to its perseverance in liver cells is not regulated properly by unfolded protein response (UPR). In turn, instead of saving cells, signaling routes diverting cells into apoptosis are activated. Additionally, accumulation of lipids (such as saturated fatty acids) impairs mitochondria and lysosomes. For example, lipotoxicity-induced voltage-dependent anion channel (VDAC) increases the permeability of the outer mitochondrial membrane initiating cell death. Mitochondrial disturbances also lead to ROS/oxidative stress generation which fuels pathological processes. Moreover, lysosomal dysfunction in MASLD is probably associated with the impairment in lipophagy, which normally is responsible for the removal of lipid burdens. Finally, these organelles’ disturbances mediated by lipotoxicity-induced oxidative stress and inflammatory processes disrupt cellular homeostasis, which triggers cell death via different mechanisms (apoptosis, necroptosis, pyroptosis) [6]. Another type of liver cells death, which probably contributes to this organ injury in MASLD, is ferroptosis (described in Section 2.1).
Sedentary life-style and unhealthy diet are major contributors to MASLD. Especially high-lipid (rich in saturated fatty acids) and carbohydrate (glucose and fructose) food-stuffs are associated with the increased MASLD risk. In light of the growing MASLD prevalence in children and adolescents, paralleled by the rise in sweet beverages consumption, these are high-fructose corn syrup (HFCS)-enriched meals which are especially blamed for this disorder’s increasing rates [9]. Fructose and glucose contribute to the liver steatosis both delivering substrates for fatty acids (FAs) and TAGs synthesis (acetyl-CoA and glycerol molecules, respectively) and inducing transcription factors (SREBP-1c and ChREBP) which enhance lipid synthetic pathways [9]. Especially fructose (Fru) seems to augment hepatic lipogenesis due to the fact that, unlike glucose being metabolized in many tissues, Fru is mainly processed in the liver, where it additionally bypasses typical for Glc hormonal (e.g., insulin/glucagon-mediated) and nutritional regulation of the rate-limiting glycolytic enzyme (phosphofructo kinase-1) [9]. Additionally, Fru is uncontrollably phosphorylated which leads to the depletion of ATP (being converted into ADP/AMP whose levels increase) and rise in purine nucleotides degradation, ending with uric acid formation. Accumulation of uric acid as a consequence of fructose overload results in the generation of mitochondrial oxidative stress, the induction of ER stress and activation of SREBP-1c. These disturbances enhance lipogenesis (via acetyl-CoA carboxylase 1, fatty acid synthase, and stearoyl-CoA desaturase-1 activation) and, when persist for a long time, yield steatosis development [9]. Since fructose- and other simple sugars-rich diet can lead to addiction, as well as Fru metabolism can fuel similar pathological events as ethanol metabolism, the term “fructoholism” has been coined [10,11]. Fructoholism is understood as an excessive fructose consumption which leads to psychological and physical damage and fructoholic liver disease [11]. Similarly as enhanced ethanol metabolism in the liver, Fru yields intermediates which are involved in the stimulation of prooxidative (ROS generation) and proinflammatory routes (via JNK-1) as well as lipogenic factors (SREBP-1c), which in both cases results in hepatic insulin resistance and hepatic steatosis with possible further consequences (hepatitis, cirrhosis and CCA) [11,12]. Additionally, an excessive fructose metabolism in the intestine can impair its permeability leading to the release of endotoxins from gastrointestinal tract (GIT) to the circulation and liver [11]. The stimulatory effect of disturbances in the intestine, such as dysbiosis and increased intestine permeability, on pathological events in the liver has been well documented [13,14,15,16]. For example, an impaired microbiome can produce proinflammatory (lipopolysaccharide—LPS) and toxic (methylamines, alcohols) molecules which may be implicated in MASLD development [3,13,16].
All of these aforementioned factors would show more severe impact on liver dysfunction in individuals with unfavorable hereditary phenotypes predisposing to MASLD [17]. A variety of genetic polymorphisms/mutations are associated with the impairment of factors regulating lipid metabolism in the liver (de novo lipogenesis, β-oxidation and secretion of triacylglycerols) [17]. For example, a mutated variant of transmembrane 6 superfamily member 2 gene (TM6SF2) which impairs VLDL production, enhances hepatic steatosis and fibrosis [17,18,19]. Also, a common genetic variant (rs738409) coding for patatin-like phospholipase domain-containing3 (PNPLA3) is associated with fat accumulation in the liver and increases the risk of the steatosis, inflammation, fibrosis and HCC [17]. PNPLA3 encodes adiponutrin, which being attached to lipid droplets, enables TAGs hydrolysis by lipases, whereas its mutated counterpart inhibits this process leading to fat accumulation in the liver [9]. Moreover, some epigenetic factors seem to affect gene expression in the direction of steatotic liver development including microRNA oligonucleotides (miRNAs) [3]. Therefore, MASLD development with its possible further consequences in the form of fibrosis, cirrhosis and cancer is a multifaceted disorder both at the cellular and systemic level (Figure 1). Some mechanistic aspects underlying MASLD are addressed below.
2.1. Ferroptosis as a Possible Mechanism Contributing to Cell Death in MASLD
Ferroptosis is a type of a non-apoptotic regulated cell death stimulated by iron-dependent lipid peroxidation and characterized by cell swelling and plasma membrane rupture [20]. It is promoted in conditions of high concentration of free iron, reactive oxygen species (ROS) and membrane phospholipids containing polyunsaturated fatty acids (PUFAs). Free iron in the presence of superoxide radicals leads to the generation of hydroxyl radicals which stimulates lipid (PUFAs) peroxidation. This causes degradation of biological membranes and cell death. Therefore, among pro-ferroptotic factors are upregulated proteins involved in iron transport and generation (e.g., transferrin receptor or heme oxygenase—HO), as well as superoxide production (like NADPH oxidases—NOXs). Ferroptosis can be also promoted by selective autophagy such as ferritinophagy and lipophagy which are associated with the release of iron cations and fatty acids, respectively. In turn, the main mechanism restraining ferroptosis includes glutathione peroxidase 4 (GPX4)—a selenium-dependent antioxidative enzyme which reduces phospholipid hydroperoxides, as well as reduced glutathione—a co-substrate in GPX4 reaction. The sufficient level of GSH in the cell is conditioned by the influx of its precursor—cystine. Therefore, the transporter (xc- antiporter) involved in cystine transport to the cells plays an important function here. Another component protecting from ferroptosis is apoptosis inducing factor mitochondria associated 2A (AIFM2/FSP1) protein, which seems to act in two ways. Firstly, having an oxidoreductase activity, AIFM2 reduces coenzyme Q10 to its ubiquinol form, which generates a hydrophobic antioxidant. Secondly, it protects from membranes injury through activating the endosomal sorting complexes required for transport (ESCRT)-III–dependent membrane repair in the plasma membrane [20].
Except for other types of cell death such as apoptosis, necroptosis and pyroptosis, ferroptosis also seems to be responsible for liver damage in the course of MASLD [21,22,23,24]. Iron accumulation in MASLD patients’ hepatocytes as well as enhanced ferritin levels in their serum have been observed, whereas phlebotomy seems to improve the liver condition (ameliorating fibrosis, steatosis and hepatitis) [21,22]. Heightened iron level in MASLD patients may be associated with dysregulation of iron transport in the organism yielding an enhanced dietary iron absorption in the intestine (via upregulation of divalent metal transporter 1—DMT1) and its excessive uptake by the liver (through the upregulation of transferrin receptor 1—TfR1) [22]. Except for iron overload, MASLD patients are characterized by the increase in ferroptosis markers [23]. For instance, lipid peroxidation (LPO) breakdown product—malondialdehyde (MDA) level is increased especially in MASH patients, whereas oxidized phosphatidylcholine (which triggers ferroptosis) is detected in MASLD specimens [23]. Peleman et al. [23] have proposed the model of hepatic ferroptosis in which dying cells release damage-associated molecular patterns (DAMPs) which spread deleterious processes within liver tissue, propagating pathological events characteristic for MASLD. Therefore, along other cell death types, ferroptosis also seems to contribute to the liver damage in the course of this disease.
2.2. AMP-Activated Protein Kinase as the Major Signaling Node Impaired in MASLD
Among a variety of intracellular regulatory enzymes and signaling pathways, AMP-activated protein kinase (AMPK) is one of the most important in regard to its involvement in MASLD pathogenesis. AMPK is an energy sensor which regulates metabolism, switching the processes between catabolic and anabolic depending on the energetic status of the cell. AMPK requires threonine (Thr172) phosphorylation for its activation, which is achieved by three different kinases (liver kinase B1—LKB1, Ca21/calmodulin-dependent protein kinase kinase β—CaMKKβ, and TGFβ-activated kinase 1—TAK1) induced by various signals [25]. AMPK is sustained in this active phosphorylated form at low energy level by AMP (and ADP) binding, whereas higher ATP concentration inactivates the enzyme. Therefore, at low energy level reflected by high AMP/ATP ratio, AMPK is active and regulates specific target enzymes, increasing lipid oxidation and mitochondrial biogenesis, whereas the synthesis of lipids and glycogen is inhibited. In such a way, energy-consuming anabolic pathways are attenuated in favor of induced catabolic pathways aimed at the replenishment of energy. One of an important targets of AMPK is acetyl-CoA carboxylase (ACC) which produces malonyl-CoA. Malonyl-CoA is a substrate for palmitic acid synthesis, but also it is an inhibitor of carnitine palmitoyl-transferase 1 (CPT1)—an enzyme involved in transport of long-chain fatty acids to the mitochondrium for β-oxidation. AMPK phosphorylates and inhibits ACC which leads to the decrease in malonyl-CoA, thus attenuating palmitic acid synthesis. Simultaneously, a drop in malonyl-CoA level releases the inhibition of CPT1 which enables entry of FAs to the mitochondrium for β-oxidation. Consequently, the synthesis of fatty acids (DNL) in the liver decreases, whereas mitochondrial β-oxidation of FAs increases [25,26]. Additionally, AMPK phosphorylates and inhibits transcription factors (sterol regulatory element-binding proteins—SREBPs) responsible for the expression of enzymes involved in FAs, triacylglycerol and cholesterol synthesis. Thus, properly working AMPK attenuates hepatic steatosis. Besides its control over metabolism of lipids and carbohydrates, AMPK decreases the expression of proinflammatory mediators and attenuates inflammation. Namely, it inhibits signaling pathways leading through NF-κB and JNK routes, therefore suppressing the expression of monocyte chemoattractant protein 1 (MCP-1). Additionally, it reduces ROS generation implicated in the inflammatory processes, via the down-regulation of NOX genes and up-regulation of antioxidative enzymes (superoxide dismutase, catalase and thioredoxin—Trx) genes. Trx up-regulation is associated with the inhibition of inflammasome activation, pointing to another anti-inflammatory effect mediated by AMPK. AMPK activity is also necessary for the prevention of hepatocytes death via apoptosis followed by liver damage. It inhibits procaspase-6 through its phosphorylation, thus stopping the proapoptotic cascade [25,27]. In turn, AMPK signaling rather seems to protect liver cells from lipotoxicity diverting them into autophagy [28,29]. Finally, AMPK shows antifibrotic activity attenuating TGFβ-induced expression of fibrogenic genes in hepatic stellate cells (HSC), as well as diverting the routes from HSC proliferation and migration into the proapoptotic ones [25].
In metabolic disorders driven by overnutrition such as MASLD, AMPK is down-regulated by an excess of nutrients as well as low-grade inflammation (probably by TNFα), which leads to the worsening of the liver condition, whereas therapeutic effects are observed when AMPK activity is increased [25,26]. Therefore, up-regulation (restoring the proper activity level) of AMPK seems a promising therapeutic target in MASLD, especially in more advanced stages of the disorder [26,30].
2.3. Gut-Liver Axis—How Dysfunctional Gastrointestinal Tract (GIT) Affects MASLD and Vice Versa?
As shortly mentioned earlier, disturbances in the intestine due to e.g., poor diet or antibiotics overuse can deteriorate microbiota profile in the GIT and increase gut permeability thus stimulating prooxidative and proinflammatory processes in the course of MASLD. Considering a direct link between the intestines and the liver (via the hepatic portal vein) it is widely accepted that intestinal dysbiosis is a factor influencing energetic metabolism and exacerbating metabolic, liver as well as cardiovascular diseases (CVD) [15,31,32,33,34]. In MASLD microbiotic profile seems to be shifted towards microorganisms producing toxic/proinflammatory compounds (such as LPS, ethanol, phenylacetate and branched chain amino acids—BCAAs) at the cost of beneficial species generating short chain fatty acids (SCFAs) including butyrate. Pathogens and toxins are transported to the liver where they induce their respective receptors (pattern recognition receptors—PRR) expressed on Kupffer cells, macrophages and HSC thus triggering pro-oxidative and pro-inflammatory routes (via NF-κB) associated with the mobilization of T lymphocytes, neutrophils and monocytes. These processes lead to hepatocytes death (apoptosis and necrosis) and profibrotic events (HSC activation and proliferation coupled with TGF-β production) [35]. Microorganism-derived toxins, except for impairing intestinal environment (accelerating inflammatory processes and increasing permeability), also stimulate steatosis, hepatitis and fibrosis after being transported to the liver. For example, ethanol and especially its oxidized product—acetaldehyde—may be the culprit of a variety of deleterious processes characteristic for MASLD (and resembling ALD features) [16]. Also, such metabolites as phenylacetate and BCAAs are able to divert metabolism into excessive lipogenesis (in the first case) or impair mitochondrial function (BCAAs), enhancing pathological processes [35]. In turn, SCFAs seem to alleviate deleterious events both in the intestine (sealing intestinal mucosal barrier through the stimulation of mucus production and tight junction protein expression, as well as upregulating anti-inflammatory regulatory T cells) and in the liver (which accumulates less fat and shows lower inflammation upon supplementation with acetate, propionate or butyrate) [16,36]. These effects seem to be coupled with accelerating catabolic processes (via the induction of hepatic lipid oxidation), while attenuating lipogenic routes (via FAS downregulation) and proinflammatory signaling (via TNF decrease) [16]. Unfavorable microbiota profile patterns have been observed in MASLD patients, with most firmly substantiated decrease in butyrate-producing Ruminococcaceae [16]. However, the cause-and-effect relationships linking dysbiosis and MASLD are not clear. Nevertheless, the manipulation of the microbiota profile promoting the growth of beneficial species (e.g., these producing SCFAs) seems to be promising in alleviating MASLD. Another approach to the improvement of microbiota profile might be the design of a prebiotic mixture which would switch the energy source for bacteria from proteins (associated with toxic products generation) to indigestible carbohydrates (prebiotics) in the distal segment of the colon [16].
Apart from the impact of end products of gut microbiota-processed dietary compounds on the liver condition, there are also reports suggesting an interplay between hepatocytes-generated bile acids and their intestinal microbial-modified derivatives in regard to the MASLD course [37]. Primary bile acids produced in the liver are transported to the intestine where they are converted into the secondary bile acids by bacteria. Such a processing occurs many times a day during the rounds of enterohepatic circulation. Bile acids, except for their involvement in dietary lipids emulsification in digestion, have also many functions regulating metabolic pathways through the induction of a variety of receptors [37]. Therefore, acting as signaling molecules, they trigger many intracellular pathways through binding with both intracellular receptors (e.g., farnesoid X receptor—FXR) and plasma membrane receptors (G protein-coupled receptors). Their effects exerted through FXR receptors are associated with lipid and carbohydrate metabolism regulation, and are implicated in MASLD pathogenesis [16]. Hence, on the one hand unfavorable microbiotic profile would affect the processing of bile acids in the intestine, whereas on the other hand dysfunctional hepatocytes (during steatosis, hepatitis, fibrosis and cirrhosis development) would produce altered amounts of bile acids affecting intestinal microbiota. This would lead to the vicious circle of intestinal dysbiosis accelerating hepatic dysfunction and vice versa. As reviewed by Farooqui at al. [37], different alterations in bile acid levels in MASLD (especially fibrotic) patients have been observed.
2.4. Deleterious Effects of Advanced Glycation End Products Exerted Through the Induction of Their Receptors (Advanced Glycation End Products Receptors) in MASLD
Multiple pathological processes which lead to the development and exacerbation of MASLD are associated with enhanced accumulation of advanced glycation end products (AGEs) [38,39,40]. Most commonly, AGEs are formed between carbonyl/aldehyde or ketone functional groups of reducing sugars (mainly Glc and Fru) and their trioses derivatives (methylglyoxal—MGO, glyoxal—GO and glyceraldehyde—GA), and amino or guanidine residues of proteins and other compounds. Therefore, they modify their targets’ structures which leads to a variety of functional disturbances both intra- and extracellularly. This impairs the proper functioning of the cell, organ, and if widespread in the whole organism, deteriorates many disorders including MASLD. Except for endogenously produced AGEs, also highly-processed foodstuffs can load the organism with extra AGEs disturbing the homeostasis [38,41]. Generally, AGEs impact seems to be the resultant of their direct alteration of important macromolecules as well as their receptors-mediated signaling. Among AGEs receptors, probably the most studied (because implicated in many pathologies) are RAGEs whose induction triggers different signaling pathways, often leading to ROS generation and NF-κB activation. Such routes enhance oxidative stress and inflammatory response, being implicated in a variety of disorders. RAGE belonging to the receptor immunoglobulin superfamily, is a pattern recognition receptor which can be induced by different ligands. It is expressed on the surface of diverse cell types, including Kupffer cells (KCs—liver-resident macrophages) and hepatic stellate cells (HSCs) which are involved in the stimulation of inflammation and fibrosis—the phenomena responsible for MASLD development. Apart from being embedded in plasma membrane, RAGEs also undergo enzymatic exfoliation (yielding cRAGEs), as well as alternative splicing (releasing esRAGEs). This leads to the generation of soluble RAGE forms (sRAGEs) in the circulation, which competitively bind their respective ligands, thus attenuating the signaling effects mediated by their membranous counterparts [40]. A similar counteractive function, as compared with AGE/RAGE, is ascribed to AGE receptors 1 (AGEs-R1) which are involved in endocytosis and degradation of AGE-modified proteins, thus lowering their deleterious effects such as oxidative stress [42].
As discussed by Liu et al. [40], an accumulation of RAGE ligands (including AGEs) in the liver upregulates RAGE expression, thus accelerating oxidative stress and inflammation, and hence stimulating/exacerbating hepatic steatosis and fibrosis. For example, the induction of hepatic stellate cells’ RAGEs by (GA-derived) AGEs was shown to enhance human HSC (LI90 cell line) activation and proliferation. These effects were mediated by the upregulation of proinflammatory (MCP-1) and profibrotic (TGF-β1, α-SMA and collagen I) factors through the generation of ROS (produced by NADPH oxidase and electron transport chain) [43]. In turn, RAGE siRNA silencing in a rat model of (CCl4-induced) liver fibrosis ameliorated liver inflammation and fibrosis which was reflected by a decrease in liver damage markers (ALT, AST, ALP, bilirubin) as well as proinflammatory (IL-6 and TNFα) and profibrotic (hyaluronic acid, laminin and procollagen type III) factors in the animals serum. Additionally, RAGE downregulation resulted in the inhibition of hepatic NF-κB, α-SMA and collagen I expression [44]. Therefore, AGE/RAGE-induced NF-κB signaling seems to be involved in the development of liver inflammation and fibrosis. Accordingly, anti-RAGE antibodies improved liver condition (attenuating inflammation and fibrosis) and animals survival in a bile-duct ligation model of liver fibrosis [45]. Hence, either the reduction of AGEs and or the downregulation of RAGEs seems to be a promising approach to alleviate MASLD. Besides limiting endogenous AGEs generation (e.g., through the shift from high sugar/fat diet into the low-calories diet rich in fibers derived from vegetables and fruits), also highly-processed foods containing AGEs should be compromised. The reduction of dietary AGEs (dAGEs) seems reasonable in light of the experiments conducted by Leung et al. [38,41] who have reported further deterioration of liver condition in the MASLD rats which were fed a high dAGEs diet, in comparison with control MASLD animals. dAGEs feeding increased hepatic AGEs and TAGs levels, oxidative stress, steatosis, steatohepatitis (CD43, IL-6, TNF α) and fibrosis (α-SMA, CTGF, collagen I). Moreover, dAGEs enhanced the activation of both HSC and KC which was associated with their increased proliferation and oxidative stress. Additionally, HSC demonstrated proinflammatory and profibrotic characteristics which were inhibited by NOX downregulation. Since RAGE silencing reversed these effects, they seemed to be mediated by dietary AGEs stimulation of RAGEs on stellate and Kupffer cells [38,41]. Similar effects have been observed by Dehnad et al. [46] who reported upregulation of the RAGE and downregulation of AGER1 both in NASH/MASH patients and AGE-fed murine NASH/MASH model. Additionally, the authors analyzed signaling pathways which could lead from the AGEs induction of RAGE to AGER1 downregulation. In light of their findings, it seems that the accumulation of AGEs induces RAGE in hepatocytes which further triggers the signal transduction pathways through JNK, p38MAPK and TGFβ which induce SMAD3. Subsequently, SMAD3 stimulates NOX4 activity which further causes degradation of Nrf2. Finally, Nrf2-mediated protection against oxidative stress development is diminished, as well as AGER1 expression is inhibited. This leads to the acceleration of AGE/RAGE axis stimulating proinflammatory/profibrotic processes, and the attenuation of AGE/AGER1 axis responsible for the scavenging of AGEs from the circulation. Moreover, although NADPH oxidases in hepatic stellate cells (NOX2 and NOX4) and macrophages (NOX2) contributed to the development of oxidative stress, the authors reported more eminent paracrine effects of hepatocytic AGE/RAGE stimulation (mediated by NOX4) with respect to the activation of HSCs and macrophages [46]. Therefore, it seems that the detrimental processes associated with liver inflammation and fibrosis are the resultant of AGE/RAGE/NOX induction in HSCs, KCs as well as hepatocytes which, considering their prevalence in number in the liver, probably (at the certain advancement level) contribute most to these pathologies. Hence, the upregulation of extracellular AGEs’ scavenging receptors (soluble RAGE forms and AGER1) seems to be an encouraging therapeutic attitude in relieving MASLD symptoms [47].
3. Methylglyoxal (MGO)
Methylglyoxal (MGO) belonging to “reactive carbonyl species” (RCS) is mainly generated as a byproduct of glycolysis and fructolysis, which is further detoxified by glyoxalases system. Glyoxalase 1 (Glo1) catalyzes MGO transformation into lactoylglutathione—an intermediate being next converted into the final product; D-lactate by glyoxalase 2 (Glo2) [48,49,50,51]. MGO metabolism requires a reduced form of glutathione (GSH); a co-substrate in the first reaction, which in the second reaction is regenerated [51,52]. MGO is elevated in metabolic disturbances associated with hyperglycemia and hyperlipidemia such as metabolic syndrome (MetS), T2DM and CVD where it participates in MGO-AGEs (MAGEs) formation. This is associated with the destruction of macromolecules (proteins, lipoproteins and DNA) which impairs intracellular organelle (e.g., mitochondria) as well as extracellular matrix (ECM) functioning. Some of the deleterious effects are mediated by AGEs receptors (RAGEs) whose induction stimulates prooxidative and proinflammatory pathways underlying metabolic disturbances (recently reviewed in ref: [53])].
Except for glyoxalases, also DJ-1 might be implicated in protection from carbonyl stress decreasing MGO-induced glycation of macromolecules [54,55,56]. DJ-1 is a multifunctional protein which acts as a sensor of the cellular oxidative stress, in response to which it turns on protective mechanisms through several signaling pathways [57], as well as it controls the activity of mitochondria (being engaged in mitophagy) [58]. Additionally, DJ-1 seems to show glyoxalase and deglycase activities [54,55,56]. However, its function in MGO detoxification has been challenged in studies based on fruit flies and mammalian experimental models [59,60]. As observed by Mazza et al. [60], DJ-1 actually shows glyoxalase activity, but it is much weaker in comparison with Glo1, so its significance in vivo is questionable (but might be compensative e.g., during GSH depletion). DJ-1 deglycase activity is more controversial with some studies supporting it [55,56,61,62], whereas others showing contradictory results [59,60,63].
Apart from glucose and fructose entering glycolysis and fructolysis as the main MGO sources, and Glo1/2 system being the major MGO scavenger, in (physio)pathological processes also other molecules can contribute to MGO generation and additional enzymes can metabolize it. Minor MGO sources comprise amino acids, glycerol, ketone bodies, aldehydes and ketoaldehydes generated from lipid peroxidation, as well as glycated proteins [48,50,64,65,66]. These molecules can contribute more to MGO generation especially in metabolic disturbances, during which an excessive accumulation of MGO can result both from an accelerated glycolytic/fructolytic flux (fueled by pathologically increased Glc in T2DM or Fru in an excessive consumption of foods enriched in Fru), but also from lipid peroxidation or glycerol—augmented in hepatic steatosis—due to elevated fatty acids and glycerol flux into the liver (Figure 2). In turn, although MGO scavenging is mainly conducted by glyoxalases system (which metabolizes above 98% of this molecule) [48], also other enzymes can degrade it. They include NADPH-dependent aldehyde dehydrogenases—ALDHs—which convert MGO into pyruvate, and aldoketo reductases—AKRs—metabolizing MGO into hydroxyacetone [48]. These extra MGO-scavenging enzymes may play a compensatory function protecting from enhanced glycation especially in pathological processes coupled with Glo1/2 downregulation [51,67,68]. The main aspects of MGO metabolism are summarized in Figure 2.
The most common MGO-generated AGEs (MAGEs) are protein arginine (Arg) modifications resulting in hydroimidazolones generation. The major hydroimidazolone is MG-H1 (but MG-H2 and 3 isoforms also occur) [48,69,70,71]. Additionally, Arg can be converted into tetrahydropyrimidine (THP) or argpyrimidine (ArgP) by MGO [72,73]. Other amino acid residues modified by MGO include lysine (Lys) or cysteine (Cys) side chains [74]. Lys modification yields its carboxyethyl derivative (CEL) [48]. As discussed in a previous review paper [53], a variety of structurally and functionally important proteins can undergo MGO-glycation, which can lead to intra- and extracellular disturbances. They include albumin, hemoglobin, insulin, collagen, histones and mitochondrial proteins. For example, MAGEs can disrupt proteins structure to make them more resistant to degradation and/or impair proteasomal systems to limit proteolytic capacity of the cell [48,53]. Such phenomena might lead to the accumulation of misfolded proteins and activation of ER stress, which if exacerbated can cause cell death via different mechanisms. The implication of ER stress in MASLD has been recently reviewed [75,76]. MGO-modified insulin may attenuate an enhanced (by normal insulin) Glc uptake by muscles and adipose tissue, hence insulin-resistance (being involved in MASLD) can be stimulated. Collagen modification by MGO can impair extracellular matrix (ECM) integrity and enhance fibrosis in liver disease. MGO effect on mitochondrial proteins can lead to mitochondrial dysfunction stimulating oxidative stress (being one of the pathological processes observed in MASLD). Additionally, as proposed by Gugliucci et al. [77], MGO may modify AMPK, impairing this kinase activity, which would lead to the switching of metabolic pathways from catabolic into anabolic, enhancing FAs and TAGs synthesis in the liver (observed in MetS and MASLD). Some of MAGEs effects can lead through the alteration of gene expression, either directly by nucleic acids glycation (mainly forming CEdG and MG-dG derivatives of deoxyguanosine [48,51]), or via epigenetic regulation (through the modification of Arg and Lys residues on histones [54]). Therefore, a lot of MGO/MAGEs-induced routes leading to pathological processes are imaginable which accelerate the development of MASLD.
4. MGO in MASLD
Methylglyoxal has been associated with the development and progression of many pathological processes such as systemic insulin resistance which is a feature of metabolic syndrome and type 2 diabetes as has already been described in our previous paper [53]. The pathological changes leading to IR result either from aberrant insulin signaling pathways observed in this condition or from changes in the structure of insulin molecule itself that may be procured by MGO action. The background and factors accompanying MASLD and cardiovascular disturbances are interwoven with each other and these conditions often coexist. MGO involvement in MASLD-associated pathologies or comorbidities such as oxidative stress, low-grade inflammation, obesity, hypertension, (proatherogenic) dyslipidemia, hyperglycemia (prediabetes/T2DM), has been addressed in the earlier paper [53]. Therefore, in the present review we focused on the pathological events observed in the liver (and reflected in biological fluids) which may be mediated by MGO.
4.1. MGO in the Early MASLD
Among MASLD patients most of them show the features of simple steatosis, only in the minority of cases the disease develops into steatohepatitis and further may progress into cirrhosis and cancer [3,4]. Initial stages of MASLD are associated with the development of insulin-resistance, accumulation of FAs/TAGs in the liver and ballooning/low-grade inflammation. In an animal model of early MASLD (CCl4-treated rats showing some hepatic steatosis, apoptosis and ballooning, but no portal inflammation and fibrosis) [78], MGO concentration was elevated in the liver, however this change was not reflected by MGO level in the serum nor in the urine. Instead, hepatic MGO increase was paralleled by the rise in its detoxification product—D-lactate both in the liver and serum, as well as D-lactate urinary excretion was enhanced, in the fourth week of CCl4 treatment. Since the liver-damage markers were not enhanced in the serum, these findings suggest MGO implication in the early stages of MASLD, which precede more severe liver destruction [78] (Table 1). MGO involvement in the development of insulin resistance in hepatocytes has been postulated by Wei et al. [79]. In their experiments on rat hepatocytes, the authors observed that MGO disturbed insulin signaling components in an analogical way as Fru. Based on their findings, the authors concluded that detrimental actions of Fru excess were mediated by the increase in MGO in hepatocytes. These effects included the attenuation of insulin signaling through the inhibition of tyrosine phosphorylation on insulin receptor substrates (IRS-1 and 2), accompanied by the activation of serine307 phosphorylation on IRS-1 being a consequence of JNK activation [79,80]. Accordingly, MGO-mediated Fru influence on hepatocytes was shown to stimulate signaling routes associated with the activation of MKK7 and JNK kinases [79] (Table 1). Therefore, since MKK7 is an activator of JNK [81], it seems that Fru-high foodstuffs can induce the processes leading to IR in the liver through the elevation of MGO which in some way leads to the induction of signal transduction cascade activating MKK7 which subsequently stimulates JNK. Finally, the inhibition of insulin signaling via the change in phosphorylation pattern of IRS is observed (Figure 3).
To assess MGO and MAGEs implication in MASLD induction, an animal model with diet-induced obesity and methylglyoxal-induced glycation has been applied by Neves et al. [82]. MGO supplementation of rats fed high-fat diet (HFD) led to the initiation of the events characteristic for MASLD onset. Namely, MGO treatment caused the accumulation of its specific glycation products in the liver. This was associated with oxidative stress and infiltration of the liver with inflammatory cells (especially in portal regions), as well as the disturbances in lipid metabolism. MGO/MAGEs resulted in the increase in NEFA accompanied by the attenuation of (HFD-stimulated) TAGs generation in the liver, which leads to lipotoxicity. NEFA were elevated in the blood plasma of HFD/MGO-treated rats, whereas in their livers such a treatment lowered the level of glycerol esterification with FAs as well as altered the profile of saturated vs. unsaturated FAs in favor of saturated ones. Except for decreasing TAGs synthesis, MGO treatment seemed to enhance the synthesis of FAs through the upregulation of FAS and attenuation of ACC inhibition by HFD [82] (Table 1). Some of the observed effects could be mediated by MGO/MAGE-caused inhibition of AMPK signaling. Silencing of AMPK routes either directly (via MGO-modified functional Arg residues in AMPK [77]) or indirectly (by MGO-caused lowering adiponectin) would switch metabolic routes from catabolic into anabolic ones. As a consequence, lipogenesis instead of FA β-oxidation is accelerated. Additionally, insulin-resistance and dysglycemia features were observed upon MGO stimulation. They included hyperinsulinemia, increased HOMA values and enhanced Glc intolerance (AUC elevation), whereas raised by HFD adiponectin concentration dropped upon MGO treatment. Additionally, MGO/MAGEs probably inhibited insulin signaling and Glc uptake by hepatocytes diminishing the activation of insulin receptor and downregulating Glc transporters GLUT-2 whose expression rose upon HFD [82] (Table 1).
Peter at al. [83] studied non-pathological liver fat from liver biopsies performed due to the resection of solitary liver lesions. BMI of patients in the studied cohort was ranging from 24.6 to 29.8 kg/m2 (average 26.6 kg/m2). They observed that even at such an early stage, with upper range or slightly elevated BMI, that may, however, potentially proceed to MASLD development. Glo1 activity reflected body fat content, inversely correlating with TAGs. Glo1 activity also decreased alongside HOMA increase, reflecting changes in cells susceptibility to insulin already at this stage. However, what is of interest, they did not observe associations between Glo1 activity and dicarbonyl compounds (MGO, glyoxal) elevation or glycation indices. They concluded that Glo1 activity could be useful as a prognostic marker of MASLD. What may also have an impact on further studies and conclusions is the fact that they observed sex dimorphism in protein expression as well as activity of Glo1. Both of these factors were lower in females than males. At the same time there were no significant differences in MGO concentrations between sexes. Downregulation of Glo1 in females may imply that they may be more affected by effects of high levels of MGO and its AGEs than males.
Pathological features characteristic for early MASLD, such as oxidative stress and TAGs increase, have been associated with MGO accumulation in the liver, as observed in animal models (hereditary hypertriglyceridemic male rats—HHTg, and female rats with postmenopausal MetS) [84,85] (Table 1). In HHTg the application of salsalate (salicylate ester of salicylic acid) led to the increase in Glo1 which downregulated MGO and improved liver condition [84], however in the second model, MGO increase due to ovariectomy was not coupled with the change in Glo1 expression [85]. Nevertheless, the stimulation of Glo1 expression which would scavenge MGO and therefore attenuate its unfavorable actions, seems a promising strategy in ameliorating MASLD. Such an effect has been shown to result from genistein (soybean isoflavone) application in a murine model of metabolic syndrome, where upregulation of Glo1, Glo2 and aldose reductase in the liver and kidney was coupled with lowering MGO and AGEs levels in blood plasma. Additionally, genistein diminished AGEs generation in the liver and kidney. Except for the influence on MGO scavenging enzymes, the authors reported other mechanisms of MGO removal. Namely, the formation of adducts between genistein and MGO followed by their excretion from the organism in urine was reported. Moreover, as shown in this work downregulation of RAGE by genistein may also contribute to the attenuation of MGO deleterious effects exerted through these receptors induction [86] (Table 1). A different animal model of MASLD has been applied by Spanos et al. [87] who compared normal and apolipoprotein E knockout (ApoE−/−) mice fed normal (ND) or high fat (HFD) diet. After 12 and 16 weeks of HFD feeding ApoE−/− mice developed minimal to mild steatosis and showed significantly decreased expression of Glo1 in comparison with upregulated Glo1 in ApoE−/− mice on normal diet. Also, FAs treatment of hepatoma cells (HepG2), which led to lipids accumulation in the cells, downregulated Glo1 and elevated MGO levels [87]. The insight into the mechanism pointed towards Glo1 hyperacetylation followed by ubiquitination and finally proteasomal degradation of the enzymatic molecule [87]. Hence, due to lipotoxicity-induced insufficient scavenging capacity, this would lead to the accumulation of MGO in hepatocytes as well as its release to extracellular environment, with further consequences in the form of carbonyl stress and MAGEs formation in the liver. Disturbances in MGO metabolism in a murine MASH model have been also observed in the HFD fed LDLR−/− mice (C57BL/6J) [88]. The animals’ group which developed hepatosteatosis, hepatic damage, inflammation, oxidative stress and fibrosis, also showed lowered level of S-lactoylglutathione in the liver (an intermediate in MGO metabolism produced by Glo1), however neither Glo1 nor Glo2 mRNA levels were affected by diet [88].
In light of the findings presented above, MGO seems to be implicated in the development of MASLD, since its concentration in the liver increases in rodent models showing features of this disorder [78,84,85]. MGO accumulation in the liver parallels oxidative stress generation and lipogenesis. Additionally, rodent and cell line experiments (based on MGO or Fru supplementation) point to MGO as a factor contributing to insulin resistance development in the liver through the inhibitory effect on insulin receptor [82] and insulin receptor substrate (IRS) [79]—two main initial components in insulin-induced signaling pathway. MGO accumulation in the liver can be associated with its accelerated synthesis caused by the oversupply of lipid and carbohydrate substrates (Figure 2), but also with the impairment of MGO metabolism connected with the increased degradation of Glo1 stimulated by its (FAs-fueled) hyperacetylation [87]. TAGs accumulation is a major feature which leads to hepatosteatosis in MASLD, and it seems to accompany MGO increase [84,85]. However, MGO rather is involved in the stimulation of DNL and favoritism of saturated FAs generation, whereas its impact on TAGs synthesis was shown to be inhibitory [82]. Therefore, MGO accumulation is probably responsible for the generation of FAs and its toxic products which further contribute to detrimental effects on cell membranes, intracellular organelle and ECM components. MGO impact in early MASLD is summarized in Figure 4A.
4.2. MGO in Liver Cirrhosis
Liver tissue is composed of hepatocytes (parenchymal cells) and nonparenchymal cells which include Kupffer cells (KCs), hepatic stellate cells (HSCs) and liver sinusoidal endothelial cells (LSECs). Liver cells which are mostly involved in fibrosis and cirrhosis development are HSCs comprising 5–8% of normal liver cells [94,95]. Oxidative and inflammatory processes coupled with the production of TNF-α and IL-6 observed in the early stages of MASLD lead to the activation of KC which further activate HSC. Subsequently, HSCs transdifferentiate into myofibroblast-like cells (through TGF-β-dependent mechanisms) that produce the extracellular matrix (ECM) components including collagen [94,95]. These processes are mediated by signaling pathways leading to cell growth, proliferation, differentiation and contractility (via MAPKs and rho kinases), as well as proinflammatory routes activation (through NF-κB) [89,94]. Also LSECs are affected by the pathological events and further accelerate them, which leads to enhanced vasoconstriction and portal vein hypertension [94]. A commonly used marker reflecting HSC activation is alpha-smooth muscle actin (α-SMA) expression [95]. In liver cirrhosis two phases can be distinguished: compensated (where basic liver functions are still sustained, often proceeding asymptomatically) and decompensated (accompanied by ascites, gastrointestinal hemorrhages and hepatic encephalopathy). Inflammatory processes can lead to the transition from compensated to decompensated stage [93].
MGO and Glo1 have been implicated in the development of liver fibrosis and cirrhosis as shown in experiments on rats with induced cirrhosis as well as liver-derived cells in culture [89] (Table 1). Downregulation of Glo1 (at mRNA and protein levels) positively correlated with the severity of cirrhosis, and associated with the increase in MGO, was reported both in the whole livers and liver cells obtained from cirrhotic animals. Decreased Glo1 expression was noted in cirrhotic hepatocytes, LSECs as well as HSCs (in which additionally Glo1 activity was decreased). However, the inhibition of Glo1 activity with the application of its inhibitors ameliorated cirrhosis features in the animals livers, as well as reduced the activation of (LPS-induced) stellate cells in vitro. Similar attenuating effects were observed when LPS-activated HSCs were exposed to 0.1–10 mM MGO. Both Glo1 inhibition and MGO exposition resulted in the reduced release of inflammatory marker (TNF-α) and fibrotic markers (α-SMA and collagen-I) by HSCs [89] (Table 1). In humans serum MGO levels reflected the progression of liver cirrhosis being higher in decompensated than in compensated stage and correlated with liver dysfunction indices [93]. Especially high values were noted in patients with ascites. It has been also observed that the elevation of MGO was correlated with proinflammatory cytokines such as IL-6. The authors also demonstrated that the concentrations of a different dicarbonyl compound—glyoxal (GO) did not reflect the severity of the disease. Glyoxal is metabolized by a different route than MGO. Therefore, although no direct determinations were conducted concerning glyoxalase system in that study, the authors implied that the elevation of MGO observed alongside the progression of cirrhosis may be due to the downregulation of this system.
Hence, when it comes to the elucidation of Glo1 and MGO involvement in liver cirrhosis development, more research should be conducted, because from the cited papers’ results no ultimate conclusions can be drawn. On the one hand, it seems that Glo1 expression decrease during the development of cirrhosis leads to the elevation of MGO and further consequences yielding AGE formation and RAGE induction which triggers pro-oxidative and pro-inflammatory pathways through HSCs (and possibly Kupffer cells) activation. These routes further augment pro-fibrotic events such as an excessive generation of ECM components. On the other hand, attenuation of Glo1 activity/MGO elevation can compromise HSC activation—the phenomenon responsible for the acceleration of fibrotic events. It might be hypothesized that in the initial stages of MASLD at low oxidative stress and subtle inflammatory processes, MGO might work as a signaling molecule triggering healing processes, and only when its level overcomes certain threshold, then it accelerates pathological events. Such hormetic effects of MGO have been suggested earlier [48,96,97,98]. Nevertheless, due to a scarcity of the available data, such suggestions are only of a hypothetical value. The ambiguity of MGO participation in the liver cirrhosis in shown in Figure 4B.
4.3. MGO in Liver Cancer
MGO seems to show anticancer activity against hepatocellular carcinoma (HCC) compromising cancer cells adhesion, migration, and invasion [90] (Table 1). Such conclusions come from experiments on HCC cell lines exposed to MGO [90], as well as the observations on Glo1 expression in human liver cancer tissue which is upregulated in comparison with non-tumorous or cirrhotic tissue [91,92,99,100]. Proliferation of human HCC cell lines was inhibited by Glo1 silencing coupled with MGO accumulation [91] (Table 1). Therefore, the authors pointed to Glo1 inhibitors as potential medicines in HCC therapy [91]. Several compounds which inhibit Glo1 activity have actually shown encouraging properties ameliorating HCC [92,101] (Table 1). Michel et al. [92], except for supporting anti-proliferatory and anti-migratory effects of Glo1 inhibition in HCC cells, also reported downregulation of some signaling components involved in pathways promoting cancer growth and metastasis, and upregulation of Nrf2 transcription factor implicated in triggering protective mechanisms in oxidative stress conditions (Table 1).
Overexpression of Glo1 observed in HCC and other cancers is attributed to enhanced anaerobic glycolysis typical for cancer cells. An excessive glycolytic flux generates higher amounts of MGO (and D-lactate), therefore cancer cells produce more Glo1 to compromise MGO toxicity [102]. Hence, Glo1 inhibition followed by MGO accumulation would enhance MGO anti-cancer effects in HCC.
However, there are studies on other cancer types which show tumor promoting effects exerted by MGO. Such observations have been reported (on breast and glioblastoma cancer cells) by Nokin et al. [103] who found that low doses of MGO promoted cancer growth, whereas higher MGO levels caused the reduction of tumor volume. Therefore, as discussed by Bellier et al. [104] a dual impact of MGO on cancer is possible; lower MGO levels might be responsible for the adaptation of cancer cells to carbonyl stress increasing their survival rates (due to hormetic effect), whereas at higher concentrations MGO exerts toxic effects stimulating apoptotic death of cancer cells.
Therefore, although the mentioned above findings seem to point to MGO as a potential anticancer agent in HCC therapy, more research should be conducted, since the available data are mainly based on in vitro cell lines experiments. MGO effects on HCC cell lines are summarized in Figure 4C.
5. Contribution of Fructose-Derived MGO to MASLD Development
In light of the mounting body of evidence, Fru is placed in the center of factors contributing to MASLD [105]. An excess of Fru derived both from exogenous (HFCS and other refined sugars foodstuffs) and endogenous (Glc via polyol pathway) sources, promotes lipogenesis and inhibits FAs β-oxidation thus stimulating steatosis [105]. Additionally, Fru enhances prooxidative and proinflammatory processes which mostly seem to be mediated by uric acid actions [105]. Moreover, other multifaceted Fru effects at the systemic level contribute to the development of obesity, MetS, IR, T2DM, cardiovascular complications and hypertension [106,107]. These are disorders which coexist or increase the risk of MASLD development. Considering all these various Fru effects observed nowadays, which result from overnutrition due to excessive sugary diet, Johnson et al. [107] have put forward fructose survival hypothesis. They propose that during ages of evolution, human predecessors had adapted to the periods of famine, switching their metabolism in advance of crisis into the pathways promoting fat accumulation and lowering energy expenditure. Such organisms feeding on natural sources of Fru (fruits and honey) would accumulate energy which would later allow them to survive in scarcity times. This adaptation might have been favorable in times when the periods of sufficient food sources were interrupted by food depletions. However, presently due to the overconsumption of foodstuffs enriched with HFCS and sucrose, these evolutionarily conserved metabolic pathways get overstimulated and persevere contributing to many obesity-related dysfunctions. Supporting their theory, the authors summarize Fru effects which, although beneficial in the past, now fuel pathological routes. At the systemic level, Fru overload leads to the induction of leptin and insulin resistance. Leptin resistance attenuates satiety feeling, which stimulates the necessity for food and water intake. Insulin resistance downregulates glucose transporters in the skeletal muscle and adipose tissue (diminishing Glc uptake by these organs), thus saving blood plasma Glc for the brain. Additionally, Fru and IR promote lipogenesis and glycogen synthesis (simultaneously impairing FAs β-oxidation) in the liver. Generally, Fru lowers metabolic rate through the diminishing oxygen consumption (via the reduction of mitochondrial oxidative phosphorylation and stimulation of anaerobic glycolysis). These and other Fru-mediated effects (like blood pressure increase and the immune system activation) seem to have improved survival chances in critical conditions, whereas now they contribute to the development of metabolic disorders [107]. As a consequence of an excessive Fru phosphorylation by fructokinase C in the liver, ATP depletion coupled with uric acid generation is observed. Although normally being an antioxidant, when overproduced, uric acid mediates many of Fru deleterious events. However, also downstream reactions of fructolytic pathway are associated with the generation of intermediates involved in pathological events. These include trioses and their derivatives such as MGO and GA. According to the Brownlee and Giacco [108,109,110] hypothesis, enhanced Glc/Fru oxidative metabolism leads to ROS overproduction which (due to DNA damages) activates PARP—an enzyme which further inhibits GAPDH. Finally, glycolytic/fructolytic pathway gets obstructed at the trioses level resulting in the acceleration of upstream reactions. It will cause the accumulation of MGO, GA, DAGs and the induction of prooxidative and proinflammatory routes. Additionally, MGO has been shown to inhibit GAPDH (through the modification of the catalytic cysteine residues) [111], hence the accumulation of both reactive carbonyls and ROS seem to accelerate their own generation in a vicious cycle mode. GA has been demonstrated to exert similar effects as compared with MGO, modifying macromolecules (yielding GA-derived AGEs) and inducing pathological events thus stimulating MASLD [112]. In light of the Takeuchi et al. [112,113,114,115] and Sakasai-Sakai et al. [39,116] observations, (Fru/Glc-derived) GA seems to exert major cytotoxic impact mediated by GA-AGEs formation, in regard to MASLD and other lifestyle-related diseases onset and development. However, both MGO and GA can modify proteins in a similar mode; interacting with amino group of Lys and guanidino group of Arg, and yielding some common AGEs (e.g., MG-H1 and ArgP resulting from the action of both molecules) [117]. Therefore, analogical signaling pathways can be triggered by AGE/RAGE axis following the generation of either of these molecules. Except for the induction of ROS, NF-κB and other routes, also direct intracellular effects through functional proteins modifications seem to contribute to dysregulation of cellular homeostasis. An example is the proposed impact of MGO (and, according to Takeuchi findings, also GA) on AMPK Arg residues which would impair this enzyme’s susceptibility to regulation by the energy status. As hypothesized by Gugliucci [77,118], uncontrollable dietary Fru influx to the liver and its immediate phosphorylation should lead to the activation of AMPK by elevated AMP. However, the opposite phenomenon is observed as a consequence of an abundance of this sugar in the liver. Therefore, considering the involvement of three AMPK Arg residues in AMP allosteric regulation, as well as overgeneration of Fru-derived trioses being further converted into MGO, these might be MGO-derived Arg modifications which impair AMPK proper regulation. However, since both MGO and GA are produced from Fru (and other molecules, as presented in Figure 2), and both can modify Arg yielding hydroimidazolone AGEs (MG-H1), it might be suggested that the final effect is a resultant of both molecules’ actions.
When compared to alcoholic liver disease (ALD), MASLD shows analogical pathological stages which include steatosis, hepatitis, fibrosis, cirrhosis and HCC, conditioned by similar processes comprising lipotoxicity, mitochondrial dysfunction, oxidative stress, ER stress, inflammation, apoptotic cell death, as well as intestinal dysbiosis [112,119]. As mentioned earlier (see Section 2), both Fru and ethanol seem to trigger similar pathological routes in the liver [11]. In the major pathway of ethanol metabolism (catalyzed by alcohol and aldehyde dehydrogenases and yielding acetaldehyde and acetic acid, respectively) NADH is generated which leads to the increase in NADH/NAD+ and ATP/AMP ratios. This causes the inhibition of oxidative processes (TCA and FAs β-oxidation) and promotion of lipogenesis resulting in steatosis and TAGs export to the circulation (leading to hypertriglyceridemia) [120]. An important mechanism involved in these disturbances is the inhibition of AMPK due to AMP decline [120]. However, a decrease in AMPK activity observed in ALD might be also a consequence of this enzyme’s functional Arg residues modifications. Whereas in MASLD the glycating agents might be MGO and GA, in ALD it could be ethanol-derived acetaldehyde (AA) intermediate—a highly toxic (and accounted to carcinogens) molecule. Therefore, it might be supposed that these are highly reactive carbonyl intermediates in the metabolism of both Fru/Glc, lipids and ethanol which contribute to the dysregulation of metabolic homeostasis. Similarly as MGO and GA, also AA can interact with amino/guanidino groups of proteins thus forming its respective AGEs (AA-AGEs) which can impair structure and function of macromolecules as well as induce RAGE stimulating oxidative stress [112].
6. Approved and Potential Therapies in MASLD
Guidelines for the clinical practice, diagnosis, and treatment of patients with steatotic liver disease have been jointly developed by the European Association for the Study of the Liver (EASL), the European Association for the Study of Diabetes (EASD), and the European Association for the Study of Obesity (EASO) [2]. It states that the diagnosis of MASLD is established in an individual who has documented hepatic steatosis alongside at least one cardiometabolic risk factor that reflects the impact of abnormal carbohydrate and lipid metabolism. These factors include overweight or obesity (BMI), dysglycemia or T2DM (based on fasting plasma glucose levels and after 2 h in an OGTT, HbA1c), hypertriglyceridemia (fasting plasma TAGs), hypercholesterolemia (fasting plasma non-HDL cholesterol), and hypertension (Figure 5). They may also include peripheral insulin resistance (hyperinsulinemic-euglycemic clamp test, HOMA-IR based on insulin or C-peptide and fasting glucose levels), adipose tissue resistance to insulin (Adipo-IR index, may predict severity of liver fibrosis) [121] and hyperuricemia (fasting serum uric acid levels) [122,123]. IR has also been further identified as a distinct risk factor for cardiovascular events, even in non-diabetic patients [124]. Under conditions of insulin resistance, a primary contributor to TAGs and cholesterol accumulation appears to be the overproduction of VLDL, which are metabolized to VLDL remnants, intermediate-density lipoproteins (IDL), and low-density lipoproteins (LDL) [125].
Pharmacological therapies for MASLD are the subject of numerous preclinical and clinical studies. Several groups of therapeutic agents are currently under investigation, including antihyperglycemic agents that increase insulin sensitivity (biguanides, thiazolidinediones), stimulate insulin secretion (incretins, e.g., GLP-1 receptor agonists such as liraglutide and semaglutide; GIP receptor agonists, e.g., tirzepatide; DPP-4 inhibitors), and SGLT2 inhibitors (flozins, e.g., dapagliflozin), bile acid agonists (ursodeoxycholic acid), farnesoid X receptor/FXR agonists (obeticholic acid), peroxisome proliferator-activated receptor/PPAR agonists (PPAR-α/δ/γ lanifibranor, PPAR-α/δ elafibranor, PPAR-α/γ saroglitazar, PPAR-γ pioglitazone, PPAR-α pemafibrate), fibroblast growth factor 21/FGF21 analogs (efruxifermin), thyroid hormone receptor β/THR-β agonists (resmetirom), free fatty acid receptor 4/FFAR4 agonists (omega-3-acid ethyl esters, e.g., omacor), substances that restore AMPK activity (AMPK activators such as metformin), antioxidants (silymarin, vitamin E) [31,126,127,128], gut microbiota modulators (e.g., by promoting the production of selected SCFAs and regulating bile acid metabolism), and others [129]. However, it should be note that although there are antidiabetic agents that can improve IR, there are no approved medications specifically designed to treat IR in liver disease. To date, no specific medication has been developed to target MASLD (especially the simple steatosis stage). Nevertheless, there are several promising candidates in controlled clinical trials (RCTs) that improve insulin sensitivity, glucose, and lipid homeostasis and reduce inflammation and progressive liver fibrosis, such as obeticholic acid, elafibranor, cenicriviroc, selonsertib, and resmetirome [130]. Previous pharmacological studies have shown that these molecules have very different molecular mechanisms.
Obeticholic acid (6-ethylchenodeoxycholic acid) is a bile acid analogue, and FXR agonist indicated for the treatment of primary biliary cholangitis, also known as primary biliary cirrhosis (an autoimmune, inflammatory liver disease). The efficacy and safety of obeticho-lic acid in patients with MASH were evaluated in a meta-analysis of RCTs by Zhao et al. [131]. Elafibranor is a dual PPARα/δ agonist that improves glucose homeostasis, increases insulin metabolism, and reduces inflammation. On the other hand, cenicriviroc is a dual antagonist of chemokine receptors 2 and 5 (CCR2 and CCR5 promote the inflammatory response in liver injury) that has shown anti-fibrotic activity in preclinical models. Another candidate, selonsertib, is a selective inhibitor of apoptosis signal-regulating kinase 1 (ASK1, a member of the mitogen-activated protein kinase family), which is involved in the stress response. Activation of ASK1 by oxidative stress leads to liver inflammation, hepatocyte apoptosis, and fibrosis. Ongoing phase III clinical trials of the above molecules have evaluated their effect on improving liver histology, defined as the resolution of MASH without worsening fibrosis [130,132,133]. However, forty-eight weeks of selonsertib monotherapy did not show anti-fibrotic effects in patients with bridging fibrosis or compensated cirrhosis due to MASH [134]. The efficacy of cenicriviroc in treating histologically proven hepatic fibrosis in adults with MASH has also not been confirmed [135]. In contrast, elafibranor was approved by the U.S. Food and Drug Administration (FDA) in June 2024 for the treatment of primary biliary cholangitis in adults as monotherapy or in combination with ursodeoxycholic acid based on its confirmed ability to reduce alkaline phosphatase (ALP) levels. It has also received positive regulatory approval in the EU [136]. The only FDA-approved small molecule in clinical development for non-cirrhotic MASH with moderate to advanced liver fibrosis is resmetirom. Resmetirom (Figure 5) is a partial THR-β agonist that has shown beneficial effects on atherogenic lipid parameters in clinical trials. Compared to placebo, resmetirom significantly reduced LDL and non-HDL cholesterol, apolipoprotein B (ApoB), hepatic steatosis, and stiffness as measured by magnetic resonance imaging MRI-PDFF or FibroScan elastography. Resmetirome is also undergoing regulatory review by the European Medicines Agency (EMA) to treat MASH [137,138].
6.1. Recommended Therapies and Medications
It appears that the optimal treatment for reducing the risk of progression or reversing the course of MASLD should not target a single risk factor, but rather the often coexisting and causally related factors, as only then can they be mitigated or reversed. Given the multifactorial pathogenesis of MASLD, combination therapies aiming at multiple therapeutic targets may be a rational approach. Obesity often coexists with carbohydrate and lipid metabolism abnormalities and elevated blood pressure, but it is also a reversible cause of their development if treated appropriately. Increasing obesity and progressive metabolic disorders lead not only to the development of MASLD but also to other conditions, such as MetS and T2DM, which further increase the risk of CVD. For this reason, MASLD therapy should be personalized and include non-pharmacologic and pharmacologic management appropriate for comorbidities, and patients should remain under multispecialty care. Taking into account the above, the overall goal of treatment becomes gradual weight loss and restoration of glucose and lipid homeostasis, reduction of IR and dyslipidemia, as well as reduction of inflammation, achieving improvement or no worsening of histological features (improvement/stabilization of steatosis/fibrosis), which may consequently prevent disease progression and even reverse disease symptoms. Therefore, treatment approaches should be personalized and tailored to the individual needs of the patient.
MASLD develops slowly, is usually asymptomatic, and is primarily caused by metabolic factors. The disease is most commonly diagnosed in patients with obesity or T2DM, but it also affects lean patients. Steatotic liver disease occurs in patients who consume small amounts of alcohol (MASLD < 20–30 g/day), moderate amounts of alcohol (20–30 g/day < MetALD > 50–60 g/day), and in alcohol abusers (ALD > 50–60 g/day). Harmful alcohol consumption is known to accelerate the progression of liver disease in patients with MASLD and chronic hepatitis B and to contribute to the development of cirrhosis or HCC [2]. Obesity, metabolic syndrome, and diabetes also increase the risk of advanced liver disease in individuals who abuse alcohol. Looking at the medical recommendations for ALD, the most effective therapy to alleviate the clinical course of the disease and even reverse liver damage is long-term abstinence from alcohol [139]. Therefore, it seems justified that the MASLD approach of limiting excessive consumption of high-calorie products rich in refined sugars (fructose, HFCS, sucrose) and saturated fats, and excluding alcohol will produce relevant positive change [9,10,140]. Indeed, current medical recommendations [2,141,142] refer to changes in dietary habits (limiting the consumption of beverages and foods containing refined sugars and saturated fats, alcohol, and stimulants) and lifestyle in the broad sense, including increased physical activity. Thus, the introduction of a hypocaloric diet (caloric restriction, with or without increased physical activity) with adequate protein content and the counteraction of overweight and obesity by gradual weight reduction (by ≥5% to reduce hepatic steatosis, by 7–10% to improve hepatic inflammation, and by ≥10% to improve fibrosis) is the only recognized treatment strategy for MASLD, that positively affects all biochemical parameters, liver enzymes, steatosis, inflammation, and fibrosis, as well as IR, dyslipidemia, and comorbidities [2,141,142]. Weight loss improves glycemic control, lipid profile, and blood pressure and reduces the risk of T2DM and CVD. However, only a few patients are able to achieve and maintain weight loss. Therefore, behavioral or cognitive-behavioral therapy should be incorporated to overcome this problem, increase motivation for treating overweight or obesity, and improve adherence to dietary and physical activity guidelines [143]. In the case of patients with MASLD and regular body weight, however, there is no evidence of a beneficial effect of a hypocaloric diet on liver histology, fibrosis, and clinical liver-related outcomes. For these individuals, health benefits may be achieved by reducing the consumption of refined sugars, especially sweetened beverages, quitting smoking, and avoiding alcohol. Increasing physical activity and reducing visceral fat are also beneficial. Nevertheless, simple lifestyle changes are unlikely to cure advanced stages of MASH. Therefore, pharmacologic support is needed [2,31,126,144].
In addition to lifestyle interventions, there are established medications to reduce the risk of comorbidities associated with MASLD, such as obesity, MetS, T2DM, and CVD: anti-obesity agents (i.e., peripherally or centrally acting agents), lipid-modifying agents (statins, fibrates, bile acid sequestrants), diabetes medications (glucose-lowering agents), antihypertensives, diuretics, peripheral vasodilators, beta-blockers, calcium channel blockers, and others (Figure 5). Based on the available data, optimal management of comorbidities is recommended, including the use of incretin-based therapies (e.g., semaglutide, tirzepatide) in patients with T2DM or obesity when indicated [2,145,146]. In adults with non-cirrhotic MASH and significant liver fibrosis (stage ≥ 2), targeted therapy with resmetirom (an oral, liver-directed agent [137,138], which has shown efficacy in steatohepatitis and fibrosis with acceptable safety and tolerability) may be used. Unfortunately, there is no pharmacotherapy that would target MASH at the stage of cirrhosis [147,148]. There is also insufficient evidence to support the use of any other class of medications, including antihyperglycemic agents, for the treatment of steatohepatitis and liver fibrosis. Therefore, GLP-1 and GIP receptor agonists (liraglutide, semaglutide, tirzepatide), SGLT2 inhibitors (flozins), thiazolidinediones (pioglitazone), and metformin cannot be recommended in MASH-targeted therapy. These agents should be applied as indicated in patients with MASH and compensated cirrhosis, i.e., in co-existing obesity, T2DM, CVD and chronic kidney disease because they reduce cardiometabolic risk. They are also safe in MASLD. There is no definitive evidence that metformin can improve histology in MASH. However, data derived from observational studies in patients with T2DM and advanced fibrosis or cirrhosis associated with MASH indicate that metformin lowers ALT levels and sensitizes to insulin. Unfortunately, it does not significantly improve steatosis, inflammation, nor fibrosis in patients with MASLD. There is also evidence that metformin may have a protective effect against HCC. Therefore, metformin is recommended for patients with compensated cirrhosis and preserved renal function but not for those with decompensated cirrhosis or renal failure due to the risk of lactic acidosis [2,148].
According to the recommendations [2], statins may be used to reduce cardiovascular events in patients with chronic liver disease, including compensated cirrhosis. However, clinical, observational, and animal studies suggest that some statins (including atorvastatin, simvastatin, and rosuvastatin) may be associated with a small but statistically significant increase in the risk of diabetes, particularly in individuals with IR and prediabetes, despite lowering LDL cholesterol and improving endothelial dysfunction. This effect was confirmed in a retrospective cohort study in which statin use was associated with progression to diabetes, significant hyperglycemia, acute glycemic complications, and the need for glucose-lowering medications [149,150]. The exact mechanisms by which statins increase the risk of T2DM are not fully understood. However, evidence suggests that statins may contribute to peripheral insulin resistance and pancreatic β-cell dysfunction [150,151,152]. In an animal model, statin treatment was associated with worsening hepatic glycemic control [153]. Therefore, the effect of statins on glucose metabolism and the risk-benefit ratio should be considered in patients with diabetes [149]. Furthermore, other lipid-lowering agents may also be prescribed to control TAGs and cholesterol levels, which could help reduce lipid accumulation in the liver [127].
6.2. MGO, AGEs, and Gut Microbiota as Therapeutic Targets
The etiology of hepatic steatosis in different patients may be related to the co-occurrence of multiple overlapping pathogenic factors (“multiple-hit hypothesis”) resulting from dietary habits, addictions, physical activity levels, and genetic predisposition. Their impact on the risk of MASLD has not been definitively established. Lifestyle factors can be modified by eliminating unfavorable health behaviors, but it is not known how these modifications affect epigenetic changes related to energy metabolism disorders. In a word, it is unclear whether there is a chance to completely reverse the damage done and restore, as far as possible, regular carbohydrate and lipid metabolism in all crucial organs and tissues because of the induction of the metabolic memory phenomenon [156].
Potential therapeutic targets in the course of MASLD are located both in the liver tissue (hepatocytes, Kupffer cells, hepatic stellate cells) as well as in extrahepatic tissues in WAT (adipocytes) and skeletal muscles (myocytes) (Figure 1). However, much attention has been recently paid to the role of the microbiota and the gut-liver axis, as well as gut dysbiosis (an imbalance in the microbial community). The largest habitat of microbial communities in our body is the colon. Given the direct connection between the gut and the liver, gut dysbiosis is thought to be a factor that affects energy metabolism, which may contribute to the development of metabolic and cardiovascular diseases [15,31,32,33,34].
In individuals with MASLD and obesity, a low-calorie dietary intervention reduces body weight and intrahepatic lipid content [157]. Diet is also considered the most important factor shaping the microbiota ecosystem. However, due to the unique complexity of the interactions between dietary components and the gut microbiome profile and functioning, more data need to be gathered to better understand these relationships. Nevertheless, despite the fact that these mechanisms are not fully clear, specific dietary approaches are recommended to improve metabolic disturbances. For example, a low-carbohydrate diet significantly ameliorates the Firmicutes/Bacteroidetes ratio and metabolic markers of dyslipidemia [158]. Also a Mediterranean diet rich in fiber and phytochemicals is beneficial because it increases the production of SCFAs (propionate and butyrate) and stabilizes the integrity of the intestinal barrier [159]. A balanced plant-based diet (consisting of vegetables and fruits rich in fiber and phytochemicals) has also been shown to increase gut bacterial diversity and its metabolic activity, including SCFA levels [160,161], and provide significant cardiometabolic protection compared to a healthy omnivorous diet [162]. Introducing foods rich in fiber and carbohydrates (traditional foods) raises Firmicutes and Prevotella levels while increasing foods rich in fiber and animal protein increases Bacteroidetes levels [163]. Exercising also improves gut microbiome diversity. Similarly, protein intake is positively correlated with microbiome diversity [164]. Therefore, lifestyle changes, especially dietary modifications under the influence of personalized nutritional counseling, may restore the balance of the gut microbiota and be an effective therapeutic approach to reduce cardiometabolic risk (Figure 5) [33,34,35,165,166,167].
Many natural and synthetic substances (e.g., some pharmaceuticals) have been identified that are harmful to the liver. The development of ALD is induced by cytotoxic ethanol and its intermediate metabolite acetaldehyde, which is characterized by higher reactivity and harmfulness [119,168]. In the case of MASLD, these are probably RCS, the compounds with a similar chemical structure, of which MGO is the major contributor. Methylglyoxal is formed as a by-product of the metabolism of glucose, fructose, lipids, and some amino acids (threonine and glycine) (Figure 2) [65]. It has been found in some processed foods (root beer, apple juice, cola, brewed coffee, etc.) [169], although this source is probably of minor importance. A more relevant source of MGO seems to be bacteria inhabiting the gastrointestinal tract that metabolize carbohydrates non-absorbed in the upper part of the GIT [170,171]. Large amounts of MGO are produced by Proteus mirabilis, P. vulgaris, and Morganella morganii, which have the highest methylglyoxal synthase activity. Relatively high MGO production is also observed in Lactobacillus, Bifidobacterium, and Bacteroides. Methylglyoxal synthase activity and MGO production are significantly decreased in the presence of SCFAs (propionate, butyrate) and bile acids (cholic acid, deoxycholic acid), confirming that their concentration in the intestine may influence MGO production by colonizing bacteria [172]. Some bacteria have efficient enzymatic systems against MGO, such as GSH-dependent glyoxalase I and II in E. coli and bacillithiol/BSH-dependent glyoxalase A and B in Firmicutes, that convert MGO to D-lactate. Microorganisms with less efficient protective mechanisms are more sensitive to exogenous MGO, which may lead to quantitative and qualitative changes in the microbiota population under favorable conditions of excessive carbohydrate consumption (e.g., Fru, HFCS, sucrose) [173]. There is also evidence that the gut microbiota can be considered a source of AGEs. In particular, E. coli strains have been shown to release AGEs during growth [174].
Moreover, dietary AGEs (dAGEs) not absorbed in the upper GIT enter the colon, where gut microbiota can metabolize them. That may result in the production of low-molecular-weight compounds which cross the intestinal barrier. But dAGEs may also contribute to gut dysbiosis, chronic inflammation, and the progression of metabolic disorders by increasing AGE levels in plasma and viscera and altering microbiota-produced metabolites such as SCFAs (decreasing butyrate levels and increasing isobutyrate levels) [175,176,177]. In a recent cross-sectional study of the U.S. population a positive correlation between dAGEs, MG-H1, CML, and steatotic liver disease has been observed. This relationship was mainly found in obese individuals and those with increased waist circumference. Therefore, limiting the intake of dAGEs may be a priority for individuals with obesity to prevent liver disease. Moreover, restricting dAGE intake may improve central adiposity, inflammation, and IR in patients with MetS and T2DM [178].
Consequently, we suggest that the strategy targeting the MGO and AGEs (MAGEs, dAGEs) could be an additional pharmacological approach for the treatment of diseases associated with impaired carbohydrate and lipid metabolism, like steatotic liver disease, by reducing carbonyl stress, oxidative stress, nonenzymatic glycation, macromolecular cross-linking, and low-grade inflammation as a result of decreasing MGO and AGE production or scavenging/sequestering existing MGO and AGEs. Therapeutics with these properties may also improve microbiota diversity and dysbiosis.
6.3. MASLD Therapy with MGO Scavengers and Antiglycation Agents
The antiglycation and MGO scavenging properties are common for several therapeutic groups used in T2DM, MetS, CVD, and inflammation. These include biguanides (metformin), sulfonylureas (glibenclamide, gliclazide, glipizide), thiazolidinediones (pioglitazone), angiotensin II receptor antagonists, and angiotensin-converting enzyme inhibitors (valsartan, candesartan, olmesartan, irbesartan, losartan, telmisartan, captopril), calcium channel antagonists (lacidipine, semotiadil, amlodipine, nifedipine, diltiazem), hydrazinophthalazine derivatives (hydralazine), statins (atorvastatin, cerivastatin, pravastatin, rosuvastatin, simvastatin), purine derivatives (pentoxifylline), non-steroidal anti-inflammatory drugs (e.g., antithrombotic aspirin), bioflavonoids (e.g., rutin, hesperidin), and some B vitamins (e.g., B6 and benfotiamine) [53]. Metformin has been the most extensively studied in this field, and its ability to scavenge MGO may explain its pharmacological action beyond the inhibitory effect on hepatic gluconeogenesis. The antiglycation and anti-MGO properties of these therapeutics have been thoroughly discussed in our previous publication [53].
The ability to bind MGO is attributed to metformin [179,180] and many plant flavonoids [181,182,183], such as quercetin, taxifolin, luteolin, eriodictyol, hesperetin, phloretin (an aglycone of phloridzin or phlorizin, the first known SGLT1 and SGLT2 inhibitor), genistein, and others. The essential element of the flavonoid structure that enables MGO uptake is the phloroglucinol system in the A-ring [181], which is common to many compounds in this group, including quercetin, taxifolin, and silibinins (silybin) - the principal components of silymarin [154]. However, the structure of flavonoid adducts with MGO has not been fully elucidated. Based on spectroscopic studies, Bhuiyan et al. [183] proposed the structure of several monoMGO and diMGO-quercetin adducts. Figure 6 shows known MGO adducts formed with metformin [180] and quercetin [183].
Furthermore, flavonoids lower postprandial blood glucose levels by reducing carbohydrate digestion and absorption through the inhibition of α-amylase and α-glucosidase (e.g., quercetin, taxifolin, luteolin, eriodictyol, genistein, isoquercitrin, hyperoside, silybin, silymarin, citrus bioflavonoids) and monosaccharide transporters and cotransporters such as GLUT2, SGLT1 and SGLT2 (e.g., phloridzin/phlorizin, kurarinone, sophoraflavanone G) [184,185,186]. They can also inhibit DPP4 (e.g., quercetin, isoquercitrin, hyperoside, luteolin, isoliquiritigenin) [187]. For most of them, various research models have confirmed antioxidant, anti-inflammatory, and anti-atherogenic properties [183,188]. Although flavonoid aglycones (non-glycosylated forms) are known to have a higher MGO trapping potential, their glycosides (β-O-glucosides, β-O-galactosides, O-rutinosides, and others), which are stable in the upper gastrointestinal tract, are subject to degradation in the colon by microbiota, releasing the aglycones and sugar moieties that are an additional energy source in this environment. Therefore, flavonoid glycosides with other plant compounds, such as dietary fiber (including plant mucilage, β-glucan, galactomannans, pectins, lignin, etc.), are considered prebiotics that interact with the microbiota. This interaction leads to the production of bacterial metabolites from flavonoids, specifically low-molecular-weight phenols. Released metabolites exert local effects in the colon and help modulate the composition of the resident microorganisms. Once absorbed into the hepatic portal vein, these low-molecular phenols may contribute to liver effects alongside aglycones and their metabolites produced by the host [189]. For instance, DOPAC (3,4-dihydroxyphenylacetic acid), which is generated from quercetin glycosides by the gut microorganism Flavonifractor plautii, has been shown to protect cells from the cytotoxic effects of acetaldehyde [190].
The ability to trap MGO and reduce MAGEs in vitro is also attributed to eriocitrin (eriodictyol-7-O-rutinoside)—the citrus fruit flavonoid and the primary constituent of peppermint leaves [191]. Eriocitrin (200 mg/day) in a randomized controlled trial in prediabetic patients lowered glycemia, increased blood GLP-1 levels, moderately reduced the growth of microorganisms associated with gut dysbiosis, and increased the abundance of commensal bacteria (it decreased growth rate of Firmicutes and Lachnospiraceae associated with dysglycemia, Blautia associated with inflammation, and altered intestinal permeability, while it increased abundance of Ruminococcaceae linked with the production of SCFAs and anti-inflammatory cytokines). The microbiota changes promoted by eriocitrin have been linked with carbohydrate metabolism and increased GLP-1 production [192].
The best known hepatoprotective agent, which is used as adjunctive therapy in chronic liver diseases such as MASLD, MetALD, and ALD, is silymarin [193,194]. Silymarin is a complex of flavonolignans isolated from milk thistle fruits (Silybum mariani fructus, Silybum marianum (L.) Gaertner). Since the components of silymarin are derivatives of taxifolin, featuring a structure that enables covalent binding to MGO (the phloroglucinol motif in the A-ring), we hypothesize that the anti-MGO effect is crucial to its influence on liver steatosis. This action, combined with its anti-glycation, antioxidant, and anti-inflammatory properties, contributes significantly to the overall effect of silymarin. On the other hand, studies in an animal model of MASLD have shown that silymarin is potent in alleviating IR, primarily by reducing visceral adipose tissue, increasing lipolysis, and inhibiting gluconeogenesis [195]. Velussi et al. [196] have confirmed that silymarin (600 mg/day) directly increases insulin sensitivity in diabetic patients, further supporting its therapeutic potential in MASLD. The efficacy and safety of silymarin monotherapy or combined therapy (silymarin + vitamin E + phospholipids or silymarin + simvastatin) in MASLD have been completed by numerous RCTs and several meta-analyses [193,194,197]. In these studies, silymarin at the standard daily dose statistically and clinically significantly reduced aminotransferases (ALT, AST), fasting insulin, TAGs, and increased HDL-C levels. In contrast, there were no changes in BMI, and the effects on GGT, TC, LDL-C, fasting glucose, and HOMA-IR were uncertain. Furthermore, the meta-analysis by Li et al. [194] suggests that silymarin may reduce the fatty liver index (FLI) and improve the hepatic steatosis grade in MASLD patients. Unfortunately, only a few RCTs included pre- and post-treatment liver biopsy data. In a study by Kheong et al. [154], silymarin therapy also appeared to diminish liver fibrosis. However, in the RCT conducted by Navarro et al. [155], silymarin has not been shown to improve liver histology. Data on the underlying mechanisms of action, including interaction with the gut microbiota, were provided by Jin et al. [198]. In this RCT, silymarin at 103.2 mg/day significantly reduced liver stiffness (assessed by FibroScan) and ApoB levels compared to placebo but had no significant effect on other biochemical and non-invasive fibrosis indices. The authors suggested that silymarin may improve liver stiffness by modulating the gut microbiome. Silymarin promoted the growth of microorganisms that produce SCFAs and regulate bile acid metabolism, and microbiome analysis revealed an increase in species diversity and enrichment of Oscillospiraceae. Using a mouse model of MASLD, Yi et al. [199] also found a decrease in 7-keto-deoxycholic acid and an increase in taurodeoxycholic acid (a return to baseline) that restored the negative feedback of the FXR. These findings suggest that silymarin may modulate the gut microbiome and improve energy metabolism in MASLD patients.
Other studies suggest that various flavonoids, including those of dietary origin, can significantly improve dyslipidemia (lower TAGs, TC, and LDL-C) and liver steatosis. Li et al. [200] have indicated that flavonoids alleviate MASLD by beneficial effects on liver function, lipid profile, and inflammation. This systematic review and meta-analysis of RCTs showed that higher flavonoid intake was associated with a reduced risk of MASLD. The study reported the beneficial effect of flavonoid supplementation (≥ 500 mg daily) and a significant decrease in ALT (especially for dihydromyricetin), AST (especially for silymarin), GGT (especially for hesperidin), TAGs (especially for genistein), LDL-C (for hesperidin and dihydromyricetin), TC, steatosis scores, TNF-α, and NF-κB. On the other hand, there was no significant difference in HDL-C, high-sensitivity C-reactive protein (hs-CRP), fasting plasma glucose, and HOMA-IR.
Therefore, we can assume that silymarin, like quercetin and other flavonoids, exerts antiglycation and anti-AGE effects by scavenging and sequestering MGO. It can also modulate gut microbiota, improve energy metabolism, dyslipidemia, and IR, and reduce liver enzyme levels (in plasma). That supports the potential of silymarin as an adjunctive therapy in MASLD. However, further detailed studies are needed to confirm this hypothesized molecular mechanism.
7. Methodology
This review is based on the PRISMA 2020 statement [201] to provide a comprehensive, structured and transparent approach to collecting and analyzing relevant scientific literature on the current state of knowledge regarding the role of methylglyoxal (MGO) in the development and progression of metabolic dysfunction-associated steatotic liver disease (MASLD, formerly NAFLD) and therapeutic strategies for MASLD that address targets such as insulin resistance, energy metabolism, glucose and fructose metabolism, lipid metabolism, carbonyl stress, oxidative stress, nonenzymatic glycation and advanced glycation end products (AGEs), and chronic low-grade inflammation.
The research question for this review is: What is the role of methylglyoxal in the development and progression of steatotic liver disease associated with metabolic dysfunction, and are there any therapeutic strategies that target the metabolic pathways related to this molecule?
Literature search: A systematic literature search was conducted to identify relevant studies published in English between 1970 and 2024. The following electronic databases were screened: PubMed, Scopus, Web of Science, and Google Scholar. Keywords used included: “methylglyoxal”, “reactive carbonyl species (RCS)”, “carbonyl stress”, “oxidative stress”, “nonenzymatic glycation”, “advanced glycation endproducts (AGEs)”, “insulin resistance”, “steatotic liver disease”, “liver steatosis”, “hepatic steatosis”, “hepatosteatosis”, “fatty liver”, “MASLD”, “MAFLD”, “NAFLD”, “steatohepatitis” (“NASH”/”MASH”), “liver cirrhosis”, “liver cancer”, “liver cancer”, “hepatocellular carcinoma”, “energy/glucose/fructose/lipid metabolism in liver/liver diseases”, “steatotic/fatty liver pathology/etiology/pathogenesis”, and “steatotic/fatty liver therapy/treatment”, and combinations of these.
Selection of research: The article selection process was conducted in two stages—an initial selection of titles and abstracts, followed by full-text evaluation. Two independent reviewers analyzed the titles and abstracts of retrieved articles to identify potentially relevant reports. Pre-defined inclusion and exclusion criteria were used to retrieve the full text of potentially eligible articles.
Inclusion criteria:
Studies published in English between 1970 and 2024.
Studies on methylglyoxal and metabolic dysfunction-associated steatotic liver disease (MASLD/NAFLD).
Studies on carbonyl stress, oxidative stress, nonenzymatic glycation, AGEs, insulin resistance, chronic inflammation, gut dysbiosis, e.g., about their effects on energy/glucose/fructose/lipid metabolism and the etiology and pathogenesis of MASLD/NAFLD.
Exclusion criteria:
Studies published in languages other than English.
Studies not related to the etiology and pathogenesis of MASLD/NAFLD.
Studies not focused on methylglyoxal, reactive carbonyls, carbonyl stress, oxidative stress, nonenzymatic glycation, AGEs, insulin resistance, energy metabolism, glucose metabolism, fructose metabolism, lipid metabolism, the gut-liver axis, or liver disease.
Studies related to the involvement of methylglyoxal in metabolic dysfunctions associated with MASLD (such as obesity, metabolic syndrome, insulin resistance, diabetes, dyslipidemia, atherosclerosis, hypertension), which were covered in our previous work (see ref. [53]).
Data extraction: Two reviewers independently performed data extraction. The following information was extracted from each study: authors, year of publication, study objectives, study design, research models, statistical methods, areas of implementation, primary findings, and conclusions.
Data synthesis and analysis: The extracted data has been thematically organized, analyzed, and synthesized to provide a comprehensive overview of the current knowledge regarding the role of methylglyoxal (MGO) in the development and progression of liver steatosis associated with metabolic dysfunction, as well as therapeutic strategies targeting the involved metabolic pathways. The PRISMA flow diagram of the included studies/registers is presented in Figure 7.
8. Conclusions and Remarks for Future Research
An imbalance in calorie intake in favor of high-lipid and high-carbohydrate foods leads to metabolic disturbances mediated significantly by methylglyoxal (and other reactive carbonyl species). MGO triggers prooxidative/proinflammatory routes through RAGE induction. Furthermore, MGO may exert its effects via modification of important hormonal and enzymatic targets, such as insulin and its signaling components (causing insulin resistance), AMPK (amplifying anabolic processes), collagen (leading to fibrosis/cirrhosis through ECM dysfunction). Additionally, (M)AGE-associated damages to multiple proteins may be involved in ER stress due to an excessive accumulation of misfolded proteins. MGO possibly affects mitochondrial components contributing to this organellum impairment and oxidative stress. Finally, it may block glycolysis/fructolysis due to the inhibition of GAPDH [111] which favors the generation of by-products of these processes (Fru, trioses, DAGs, MGO, GA), thus accelerating detrimental pathways. Therefore, via multiple routes MGO can exacerbate MASLD where it seems to participate in insulin resistance, oxidative stress, inflammatory processes stimulation, and induction of lipogenesis. Since insulin resistance seems to be the central pathological knob in MASLD and its comorbidities, and MGO impact on systemic IR development has been well documented [202,203,204,205,206,207,208,209,210,211,212,213,214,215,216,217], the proposed MGO implication in IR is presented and discussed in Figure 8.
However, more research should be conducted to better elucidate MGO function in MASLD. On the one hand, its detrimental role in the generation of metabolic disturbances leading to the induction of MASLD is well substantiated (Figure 8). On the other hand, MGO involvement in the processes associated with the development of fibrosis and cirrhosis as well as liver cancer is not clear. There are only a few studies referring to these issues, and no consistent conclusions can be drawn. As discussed in Section 4.2, some data indicate profibrotic, and others antifibrotic actions of MGO. Experiments on MGO effects on hepatocellular carcinoma point to its anticancer actions. However, only several in vitro cell line studies have been performed in this area so far. Therefore, whereas MGO function in early MASLD is well evidenced, no ultimate conclusions can be drawn on its participation in the liver cirrhosis nor in the liver cancer. It can be only hypothesized that MGO effects are highly dependent on its concentration. Since MGO is constantly generated at low levels, mainly as a glycolytic by-product, it might be suggested that at low level it stimulates pro-survival processes in the cells. Such actions would activate protective mechanisms to prepare the cell against oxidative stress and inflammation. However, when the concentration of MGO crosses the threshold of the protective capacity of the cell, then its detrimental effects seem to prevail which triggers pathological routes. Such a dual role may be implied from studies on MGO involvement in liver cirrhosis development, as discussed in Section 4.2. A similar phenomenon may be observed in cancer cells which are characterized by increased anaerobic glycolysis associated with elevated MGO. Thus, at low levels MGO (and other glycolytic intermediates and by-products) may promote cancer growth, whereas only at higher concentration MGO gains anticancer activity [104] (as discussed in Section 4.3).
Despite the tremendous advances in our knowledge of liver steatosis, many important areas of MASLD require further research to improve its targeted therapy. Thus, there is an urgent need for continued studies in vitro and in vivo on the role of MGO and MAGEs in the etiology and pathogenesis of MASLD, considering the disease stage from simple steatosis to cirrhosis and HCC.
Several small molecules are currently being assessed in clinical trials to identify the most effective MASLD treatment. These agents differ in their mechanism of action, are directed against a variety of metabolic targets with the aim to extract a group of factors showing anti-inflammatory and anti-fibrotic effects. Unfortunately, except for resmetirome approved for non-cirrhotic MASLD therapy, phase III trials have not met expectations. Therefore, weight loss through a hypocaloric diet and lifestyle changes are the only accepted treatments for MASLD in its early stages. However, it appears that therapeutics that can capture and bind MGO, thereby reducing levels of cytotoxic methylglyoxal-derived AGEs (MAGEs), have beneficial effects on insulin resistance, dyslipidemia, aminotransferases levels, gut dysbiosis, and chronic low-grade inflammation. Although agents like metformin and silymarin exert modest effects on liver histology, their antiglycation, anti-MGO, antioxidant, anti-inflammatory, and gut microbiota-modulating effects could provide additional support for steatotic liver disease therapy. However, to conclusively prove their potential, especially concerning the mechanism targeting MGO and MAGEs, further well-designed studies in MASLD patients are required.
Author Contributions
Conceptualization, I.B. and I.F.; resources, I.B., I.F. and M.M.; writing—original draft preparation, I.B., I.F. and M.M.; writing—review and editing; I.B., I.F. and M.M.; visualization, I.F., I.B. and M.M.; supervision, I.B. and I.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ACC | acetyl-CoA carboxylase |
| AceCS | acetyl-CoA synthetase |
| AGEs | advanced glycation end products |
| AIFM2 | factor mitochondria associated 2A |
| ALP | alkaline phosphatase |
| ALT | alanine aminotransferase |
| AMPK | AMP-activated protein kinase |
| ArgP | Argpyrimidine |
| ApoE−/− | apolipoprotein E knockout |
| AST | aspartate aminotransferase |
| AUC | area under the curve |
| α-SMA | alpha-smooth muscle actin |
| BCAA | branched chain amino acids |
| CaMKKβ | Ca21/calmodulin-dependent protein kinase kinase β |
| CCl4 | carbon tetrachloride |
| CD43 | leukosialin (mucin-like protein expressed on the surface of most hematopoietic cells) |
| CEdG | N2-carboxyethyl-20–deoxyguanosine |
| CEL | Nε-(1-carboxyethyl)lysine = N6-(1-carboxyethyl)lysine |
| Chol | Cholesterol |
| ChREBP | carbohydrate-responsive element-binding protein |
| CRP | C-reactive protein |
| CTGF | connective tissue growth factor |
| CVD | cardiovascular diseases |
| DAGs | Diacylglycerols |
| DAMPs | damage-associated molecular patterns |
| DMT1 | divalent metal transporter 1 |
| DNL | de novo lipogenesis |
| ECM | extracellular matrix |
| FAs | fatty acids |
| FAS | fatty acid synthase |
| Fru | Fructose |
| FXR | farnesoid X receptor |
| GA | Glyceraldehyde |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| GGT | γ-glutamyl transpeptidase |
| GIT | gastrointestinal tract |
| Glc | Glucose |
| Glo1 | glyoxalase 1 |
| Glo2 | glyoxalase 2 |
| GLUT-4 | insulin-dependent glucose transporters in skeletal muscle and adipose tissue |
| GPx | glutathione peroxidase |
| GPX4 | glutathione peroxidase 4 |
| GSH | reduced glutathione |
| GSSG | oxidized glutathione |
| HCC | hepatocellular carcinoma |
| HDL-Chol | high-density lipoproteins cholesterol |
| Hep G2 | epithelial hepatoblastoma cell line |
| HFCS | high-fructose corn syrup |
| HFD | high-fat diet |
| HHTg | hereditary hypertriglyceridemic rats |
| HO | heme oxygenase |
| HOMA | homeostatic model assessment |
| HSCs | hepatic stellate cells |
| IR | insulin resistance |
| IRS-1,2 | insulin receptor substrate 1,2 |
| JNK | c-jun NH2-terminal kinase |
| KCs | Kupffer cells |
| LKB1 | liver kinase B1 |
| LPO | lipid peroxidation |
| LPS | lipopolysaccharide |
| LSECs | liver sinusoidal endothelial cells |
| MAGEs | MGO-derived advanced glycation end products |
| MAPKs | mitogen-activated protein kinases |
| MASLD | metabolic dysfunction-associated fatty liver disease |
| MCP-1 | monocyte chemoattractant protein 1 |
| MDA | malondialdehyde |
| MetS | metabolic syndrome |
| MG-dG | 3-(20–deoxyribosyl)-6,7-dihydro-6,7-dihydroxy-6/7-methylimidazo-[2,3-b]purin-9(8)one |
| MG-H1 | Nδ -(5-hydro-5-methyl-4-imidazolon-2-yl)-ornithine |
| MG-H2 | 2-amino-5-(2-amino-5-hydro-5-methyl-4- imidazolon-1-yl)-pentanoic acid |
| MG-H3 | 2-amino-5-(2-amino-4-hydro-4-methyl-5-imidazolon-1-yl)-pentanoic acid |
| MGO | methylglyoxal |
| MKK7 | mitogen-activated protein kinase kinase 7 |
| NEFA | non-esterified fatty acids |
| NF-κB | nuclear factor-kB |
| NOX | NADPH oxidase |
| Nrf2 | nuclear factor erythroid 2-related factor 2 |
| PARP | poly(ADP-ribose) polymerase |
| PRR | pattern recognition receptors |
| PUFAs | polyunsaturated fatty acids |
| p38 MAPK | p38 mitogen-activated protein kinase |
| RAGE | advanced glycation end products receptor |
| RCS | reactive carbonyl species |
| RCT | randomized controlled trial |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| SCFAs | short chain fatty acids |
| SMAD3 | a protein involved in TGFβ signal transduction |
| SOD | superoxide dismutase |
| SREBP | sterol regulatory element-binding protein |
| TAGs | triacylglycerols |
| TAK1 | TGFβ-activated kinase 1 |
| TBARS | thiobarbituric acid reactive substances |
| TC | total cholesterol |
| TCA | tricarboxylic acid cycle (Krebs cycle) |
| T2DM | type 2 diabetes mellitus |
| TfR1 | transferrin receptor 1 |
| TGFβ | transforming growth factor β |
| THP | tetrahydropyrimidine |
| TNFα | tumor necrosis factor alfa |
| Trx | thioredoxin |
| WR | Wistar rats |
References
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [CrossRef] [PubMed]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). European Association for the Study of the Liver (EASL). EASL-EASD-EASO Clinical Practice Guidelines on the management of metabolic dysfunction-associated steatotic liver disease (MASLD). J. Hepatol. 2024, 81, 492–542. [CrossRef] [PubMed]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [CrossRef] [PubMed]
- Loomba, R.; Friedman, S.L.; Shulman, G.I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 2021, 184, 2537–2564. [CrossRef] [PubMed]
- Gofton, C.; Upendran, Y.; Zheng, M.H.; George, J. MAFLD: How is it different from NAFLD? Clin. Mol. Hepatol. 2023, 29 (Suppl. S17–S31), 371. [CrossRef] [PubMed] [PubMed Central]
- Li, Y.; Yang, P.; Ye, J.; Xu, Q.; Wu, J.; Wang, Y. Updated mechanisms of MASLD pathogenesis. Lipids Health Dis. 2024, 23, 117. [CrossRef] [PubMed] [PubMed Central]
- Yao, J.; Wu, D.; Qiu, Y. Adipose tissue macrophage in obesity-associated metabolic diseases. Front. Immunol. 2022, 2; 977485. [CrossRef] [PubMed] [PubMed Central]
- Xu, L.; Yan, X.; Zhao, Y.; Wang, J.; Liu, B.; Yu, S.; Fu, J.; Liu, Y.; Su, J. Macrophage Polarization Mediated by Mitochondrial Dysfunction Induces Adipose Tissue Inflammation in Obesity. Int. J. Mol. Sci. 2022, 23, 9252. [CrossRef] [PubMed] [PubMed Central]
- Huneault, H.E.; Ramirez Tovar, A.; Sanchez-Torres, C.; Welsh, J.A.; Vos, M.B. The Impact and Burden of Dietary Sugars on the Liver. Hepatol. Commun. 2023, 7, e0297. [CrossRef] [PubMed] [PubMed Central]
- Salavrakos, M.; de Timary, P.; Ruiz Moreno, A.; Thissen, J.P.; Lanthier, N. Fructoholism in adults: The importance of personalised care in metabolic dysfunction-associated fatty liver disease. JHEP Rep. 2021, 4, 100396. [CrossRef] [PubMed] [PubMed Central]
- Ribeiro, A.; Igual-Perez, M.J.; Santos Silva, E.; Sokal, E.M. Childhood Fructoholism and Fructoholic Liver Disease. Hepatol. Commun. 2018, 3, 44-51. [CrossRef] [PubMed] [PubMed Central]
- Lustig, R.H. Fructose: Metabolic, hedonic, and societal parallels with ethanol. J. Am. Diet. Assoc. 2010, 110, 1307–1321. [CrossRef] [PubMed]
- Ji, J.; Wu, L.; Wei, J.; Wu, J.; Guo, C. The Gut Microbiome and Ferroptosis in MAFLD. J. Clin. Transl. Hepatol. 2023, 11, 174-187. [CrossRef] [PubMed] [PubMed Central]
- Jayachandran, M.; Qu, S. Non-alcoholic fatty liver disease and gut microbial dysbiosis- underlying mechanisms and gut microbiota mediated treatment strategies. Rev. Endocr. Metab. Disord. 2023, 24, 1189-1204. [CrossRef] [PubMed]
- Benedé-Ubieto, R.; Cubero, F.J.; Nevzorova, Y.A. Breaking the barriers: The role of gut homeostasis in Metabolic-Associated Steatotic Liver Disease (MASLD). Gut Microbes 2024, 16, 2331460. [CrossRef] [PubMed] [PubMed Central]
- Canfora, E.E.; Meex, R.C.R.; Venema, K.; Blaak, E.E. Gut microbial metabolites in obesity, NAFLD and T2DM. Nat. Rev. Endocrinol. 2019, 15, 261-273. [CrossRef] [PubMed]
- Lindén, D.; Romeo, S. Therapeutic opportunities for the treatment of NASH with genetically validated targets. J. Hepatol. 2023, 79, 1056-1064. [CrossRef] [PubMed]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjærg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [CrossRef] [PubMed] [PubMed Central]
- Liu, Y.L.; Reeves, H.L.; Burt, A.D.; Tiniakos, D.; McPherson, S.; Leathart, J.B.; Allison, M.E.; Alexander, G.J.; Piguet, A.C.; Anty, R.; et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5, 4309. [CrossRef] [PubMed] [PubMed Central]
- Chen, X.; Li, J.; Kang, R.; Klionsky, D.J.; Tang, D. Ferroptosis: Machinery and regulation. Autophagy 2021, 17, 2054-2081. [CrossRef] [PubMed] [PubMed Central]
- Shen, X.; Yu, Z.; Wei, C.; Hu, C.; Chen, J. Iron metabolism and ferroptosis in nonalcoholic fatty liver disease: What is our next step? Am. J. Physiol. Endocrinol. Metab. 2024, 326, E767–E775. [CrossRef] [PubMed]
- Zhu, B.; Wei, Y.; Zhang, M.; Yang, S.; Tong, R.; Li, W.; Long, E. Metabolic dysfunction-associated steatotic liver disease: Ferroptosis related mechanisms and potential drugs. Front. Pharmacol. 2023, 14, 1286449. [CrossRef] [PubMed] [PubMed Central]
- Peleman, C.; Francque, S.; Berghe, T.V. Emerging role of ferroptosis in metabolic dysfunction-associated steatotic liver disease: Revisiting hepatic lipid peroxidation. EBioMedicine 2024, 102, 105088. [CrossRef] [PubMed] [PubMed Central]
- Yao, C.; Lan, D.; Li, X.; Wang, Y.; Qi, S.; Liu, Y. Porphyromonas gingivalis is a risk factor for the development of nonalcoholic fatty liver disease via ferroptosis. Microbes Infect. 2023, 25, 105040. [CrossRef] [PubMed]
- Zhao, P.; Saltiel, A.R. From overnutrition to liver injury: AMP-activated protein kinase in nonalcoholic fatty liver diseases. J. Biol. Chem. 2020, 295, 12279-12289. [CrossRef] [PubMed] [PubMed Central]
- von Loeffelholz, C.; Coldewey, S.M.; Birkenfeld, A.L. A Narrative Review on the Role of AMPK on De Novo Lipogenesis in Non-Alcoholic Fatty Liver Disease: Evidence from Human Studies. Cells 2021, 10, 1822. [CrossRef] [PubMed] [PubMed Central]
- Zhao, P.; Sun, X.; Chaggan, C.; Liao, Z.; In Wong, K.; He, F.; Singh, S.; Loomba, R.; Karin, M.; Witztum, J.L.; et al. An AMPK-caspase-6 axis controls liver damage in nonalcoholic steatohepatitis. Science 2020, 367, 652-660. [CrossRef] [PubMed] [PubMed Central]
- Wu, P.; Zhao, J.; Guo, Y.; Yu, Y.; Wu, X.; Xiao, H. Ursodeoxycholic acid alleviates nonalcoholic fatty liver disease by inhibiting apoptosis and improving autophagy via activating AMPK. Biochem. Biophys. Res. Commun. 2020, 529, 834-838. [CrossRef] [PubMed]
- Lee, D.H.; Park, J.S.; Lee, Y.S.; Han, J.; Lee, D.K.; Kwon, S.W.; Han, D.H.; Lee, Y.H.; Bae, S.H. SQSTM1/p62 activates NFE2L2/NRF2 via ULK1-mediated autophagic KEAP1 degradation and protects mouse liver from lipotoxicity. Autophagy 2020, 16, 1949-1973. [CrossRef] [PubMed] [PubMed Central]
- Fang, C.; Pan, J.; Qu, N.; Lei, Y.; Han, J.; Zhang, J.; Han, D. The AMPK pathway in fatty liver disease. Front. Physiol. 2022, 13, 970292. [CrossRef] [PubMed] [PubMed Central]
- Zhu, S.; Wu, Z.; Wang, W.; Wei, L.; Zhou, H. A revisit of drugs and potential therapeutic targets against non-alcoholic fatty liver disease: Learning from clinical trials. J. Endocrinol. Investig. 2024, 47, 761-776. [CrossRef] [PubMed]
- Vallianou, N.G.; Kounatidis, D.; Psallida, S.; Vythoulkas-Biotis, N.; Adamou, A.; Zachariadou, T.; Kargioti, S.; Karampela, I.; Dalamaga, M. NAFLD/MASLD and the Gut-Liver Axis: From Pathogenesis to Treatment Options. Metabolites 2024, 14, 366. [CrossRef] [PubMed] [PubMed Central]
- Puetz, A.; Kappel, B.A. Gut Microbiome in Dyslipidemia and Atherosclerosis. In Gut Microbiome, Microbial Metabolites and Cardiometabolic Risk; Springer International Publishing: Cham, Switzerland, 2023; pp.1–29. [CrossRef]
- Al Samarraie, A.; Pichette, M.; Rousseau, G. Role of the Gut Microbiome in the Development of Atherosclerotic Cardiovascular Disease. Int. J. Mol. Sci. 2023, 24, 5420. [CrossRef] [PubMed] [PubMed Central]
- Bellucci, E.; Chiereghin, F.; Pacifici, F.; Donadel, G.; De Stefano, A.; Malatesta, G.; Valente, M.G.; Guadagni, F.; Infante, M.; Rovella, V.; et al. Novel therapeutic approaches based on the pathological role of gut dysbiosis on the link between nonalcoholic fatty liver disease and insulin resistance. Eur. Rev. Med. Pharmacol. Sci. 2023, 27, 1921-1944. [CrossRef] [PubMed]
- Mishra, S.P.; Karunakar, P.; Taraphder, S.; Yadav, H. Free Fatty Acid Receptors 2 and 3 as Microbial Metabolite Sensors to Shape Host Health: Pharmacophysiological View. Biomedicines 2020, 8, 154. [CrossRef] [PubMed] [PubMed Central]
- Farooqui, N.; Elhence, A.; Shalimar. A Current Understanding of Bile Acids in Chronic Liver Disease. J. Clin. Exp. Hepatol. 2022 12, 155-173. [CrossRef] [PubMed] [PubMed Central]
- Leung, C.; Herath, C.B.; Jia, Z.; Andrikopoulos, S.; Brown, B.E.; Davies, M.J.; Rivera, L.R.; Furness, J.B.; Forbes, J.M.; Angus, P.W. Dietary advanced glycation end-products aggravate non-alcoholic fatty liver disease. World J. Gastroenterol. 2016, 22, 8026–8040. [CrossRef] [PubMed] [PubMed Central]
- Sakasai-Sakai, A.; Takeda, K.; Takeuchi, M. Involvement of Intracellular TAGE and the TAGE-RAGE-ROS Axis in the Onset and Progression of NAFLD/NASH. Antioxidants 2023, 12, 748. [CrossRef] [PubMed] [PubMed Central]
- Liu, J.; Jin, Z.; Wang, X.; Jakoš, T.; Zhu, J.; Yuan, Y. RAGE pathways play an important role in regulation of organ fibrosis. Life Sci. 2023, 323, 121713. [CrossRef] [PubMed]
- Leung, C.; Herath, C.B.; Jia, Z.; Goodwin, M.; Mak, K.Y.; Watt, M.J.; Forbes, J.M.; Angus, P.W. Dietary glycotoxins exacerbate progression of experimental fatty liver disease. J. Hepatol. 2014, 60, 832–838. [CrossRef] [PubMed]
- Ott, C.; Jacobs, K.; Haucke, E.; Navarrete Santos, A.; Grune, T.; Simm, A. Role of advanced glycation end products in cellular signaling. Redox Biol. 2014, 2, 411–429. [CrossRef] [PubMed] [PubMed Central]
- Iwamoto, K.; Kanno, K.; Hyogo, H.; Yamagishi, S.; Takeuchi, M.; Tazuma, S.; Chayama, K. Advanced glycation end products enhance the proliferation and activation of hepatic stellate cells. J. Gastroenterol. 2008, 43, 298-304. [CrossRef] [PubMed]
- Cai, X.G.; Xia, J.R.; Li, W.D.; Lu, F.L.; Liu, J.; Lu, Q.; Zhi, H. Anti-fibrotic effects of specific-siRNA targeting of the receptor for advanced glycation end products in a rat model of experimental hepatic fibrosis. Mol. Med. Rep. 2014, 10, 306–314. [CrossRef] [PubMed]
- Xia, P.; Deng, Q.; Gao, J.; Yu, X.; Zhang, Y.; Li, J.; Guan, W.; Hu, J.; Tan, Q.; Zhou, L.; et al. Therapeutic effects of antigen affinity-purified polyclonal anti-receptor of advanced glycation end-product (RAGE) antibodies on cholestasis-induced liver injury in rats. Eur. J. Pharmacol. 2016, 779, 102–110. [CrossRef] [PubMed]
- Dehnad, A.; Fan, W.; Jiang, J.X.; Fish, S.R.; Li, Y.; Das, S.; Mozes, G.; Wong, K.A.; Olson, K.A.; Charville, G.W.; et al. AGER1 downregulation associates with fibrosis in nonalcoholic steatohepatitis and type 2 diabetes. J. Clin. Investig. 2020, 130, 4320-4330. [CrossRef] [PubMed] [PubMed Central]
- Asadipooya, K.; Lankarani, K.B.; Raj, R.; Kalantarhormozi, M. RAGE is a Potential Cause of Onset and Progression of Nonalcoholic Fatty Liver Disease. Int. J. Endocrinol. 2019, 2019, 2151302. [CrossRef] [PubMed] [PubMed Central]
- Schalkwijk, C.G.; Stehouwer, C.D.A. Methylglyoxal, a Highly Reactive Dicarbonyl Compound, in Diabetes, Its Vascular Complications, and Other Age-Related Diseases. Physiol. Rev. 2020, 100, 407–461. [CrossRef]
- Phillips, S.A.; Thornalley, P.J. The formation of methylglyoxal from triose phosphates. Investigation using a specific assay for methylglyoxal. Eur. J. Biochem. 1993, 212, 101–105. [CrossRef]
- Sousa Silva, M.; Gomes, R.A.; Ferreira, A.E.; Ponces Freire, A.; Cordeiro, C. The glyoxalase pathway: The first hundred years and beyond. Biochem. J. 2013, 453, 1–15. [CrossRef]
- Morgenstern, J.; Campos Campos, M.; Nawroth, P.; Fleming, T. The Glyoxalase System-New Insights into an Ancient Metabolism. Antioxidants 2020, 9, 939. [CrossRef]
- Phillips, S.A.; Mirrlees, D.; Thornalley, P.J. Modification of the glyoxalase system in streptozotocin-induced diabetic rats. Effect of the aldose reductase inhibitor Statil. Biochem. Pharmacol. 1993, 46, 805–811. [CrossRef]
- Berdowska, I.; Matusiewicz, M.; Fecka, I. Methylglyoxal in Cardiometabolic Disorders: Routes Leading to Pathology Counterbalanced by Treatment Strategies. Molecules 2023, 28, 7742. [CrossRef] [PubMed] [PubMed Central]
- Galligan, J.J.; Wepy, J.A.; Streeter, M.D.; Kingsley, P.J.; Mitchener, M.M.; Wauchope, O.R.; Beavers, W.N.; Rose, K.L.; Wang, T.; Spiegel, D.A.; et al. Methylglyoxal-derived posttranslational arginine modifications are abundant histone marks. Proc. Natl. Acad. Sci. USA. 2018, 115, 9228–9233. [CrossRef] [PubMed] [PubMed Central]
- Richarme, G.; Mihoub, M.; Dairou, J.; Bui, L.C.; Leger, T.; Lamouri, A. Parkinsonism-associated protein DJ-1/Park7 is a major protein deglycase that repairs methylglyoxal- and glyoxal-glycated cysteine, arginine, and lysine residues. J. Biol. Chem. 2015, 290, 1885–1897. [CrossRef] [PubMed] [PubMed Central]
- Richarme, G.; Liu, C.; Mihoub, M.; Abdallah, J.; Leger, T.; Joly, N.; Liebart, J.C.; Jurkunas, U.V.; Nadal, M.; Bouloc, P. et al. Guanine glycation repair by DJ-1/Park7 and its bacterial homologs. Science 2017, 357, 208–211. [CrossRef] [PubMed]
- Dolgacheva, L.P.; Berezhnov, A.V.; Fedotova, E.I.; Zinchenko, V.P.; Abramov, A.Y. Role of DJ-1 in the mechanism of pathogenesis of Parkinson’s disease. J. Bioenerg. Biomembr. 2019, 51, 175–188. [CrossRef] [PubMed] [PubMed Central]
- Smolders, S.; Van Broeckhoven, C. Genetic perspective on the synergistic connection between vesicular transport, lysosomal and mitochondrial pathways associated with Parkinson’s disease pathogenesis. Acta Neuropathol. Commun. 2020, 8, 63. [CrossRef] [PubMed] [PubMed Central]
- Pfaff, D.H.; Fleming, T.; Nawroth, P.; Teleman, A.A. Evidence Against a Role for the Parkinsonism-associated Protein DJ-1 in Methylglyoxal Detoxification. J. Biol. Chem. 2017, 292, 685–690. [CrossRef] [PubMed] [PubMed Central]
- Mazza, M.C.; Shuck, S.C.; Lin, J.; Moxley, M.A.; Termini, J.; Cookson, M.R.; Wilson, M.A. DJ-1 is not a deglycase and makes a modest contribution to cellular defense against methylglyoxal damage in neurons. J. Neurochem. 2022, 162, 245-261. [CrossRef] [PubMed] [PubMed Central]
- Richarme, G.; Abdallah, J.; Mathas, N.; Gautier, V.; Dairou, J. Further characterization of the Maillard deglycase DJ-1 and its prokaryotic homologs, deglycase 1/Hsp31, deglycase 2/YhbO, and deglycase 3/YajL. Biochem. Biophys. Res. Commun. 2018, 503, 703-709. [CrossRef] [PubMed]
- Richarme, G.; Dairou, J. Parkinsonism-associated protein DJ-1 is a bona fide deglycase. Biochem. Biophys. Res. Commun. 2017, 483, 387-391. [CrossRef] [PubMed]
- Andreeva, A.; Bekkhozhin, Z.; Omertassova, N.; Baizhumanov, T.; Yeltay, G.; Akhmetali, M.; Toibazar, D.; Utepbergenov, D. The apparent deglycase activity of DJ-1 results from the conversion of free methylglyoxal present in fast equilibrium with hemithioacetals and hemiaminals. J. Biol. Chem. 2019, 294, 18863-18872. [CrossRef] [PubMed] [PubMed Central]
- Rabbani, N.; Thornalley, P.J. Methylglyoxal, glyoxalase 1 and the dicarbonyl proteome. Amino Acids. 2012, 42, 1133–1142. [CrossRef] [PubMed]
- Hernandez-Castillo, C.; Shuck, S.C. Diet and Obesity-Induced Methylglyoxal Production and Links to Metabolic Disease. Chem. Res. Toxicol. 2021, 34, 2424–2440. [CrossRef] [PubMed]
- Kalapos, M.P. Where does plasma methylglyoxal originate from? Diabetes Res. Clin. Pract. 2013, 99, 260–271. [CrossRef] [PubMed]
- Schumacher, D.; Morgenstern, J.; Oguchi, Y.; Volk, N.; Kopf, S.; Groener, J.B.; Nawroth, P.P.; Fleming, T.; Freichel, M. Com-pensatory mechanisms for methylglyoxal detoxification in experimental & clinical diabetes. Mol. Metab. 2018, 18, 143–152. [CrossRef] [PubMed] [PubMed Central]
- Morgenstern, J.; Fleming, T.; Schumacher, D.; Eckstein, V.; Freichel, M.; Herzig, S.; Nawroth, P. Loss of Glyoxalase 1 Induces Compensatory Mechanism to Achieve Dicarbonyl Detoxification in Mammalian Schwann Cells. J. Biol. Chem. 2017, 292, 3224–3238. [CrossRef] [PubMed] [PubMed Central]
- Ahmed, N.; Argirov, O.K.; Minhas, H.S.; Cordeiro, C.A.; Thornalley, P.J. Assay of advanced glycation endproducts (AGEs): Surveying AGEs by chromatographic assay with derivatization by 6-aminoquinolyl-N-hydroxysuccinimidyl-carbamate and application to Nepsilon-carboxymethyl-lysine- and Nepsilon-(1-carboxyethyl)lysine-modified albumin. Biochem. J. 2002, 364, 1–14. [CrossRef]
- Klöpfer, A.; Spanneberg, R.; Glomb, M.A. Formation of arginine modifications in a model system of Nα-tert-butoxycarbonyl (Boc)-arginine with methylglyoxal. J. Agric. Food Chem. 2011, 59, 394–401. [CrossRef]
- Ahmed, N.; Dobler, D.; Dean, M.; Thornalley, P.J. Peptide mapping identifies hotspot site of modification in human serum albumin by methylglyoxal involved in ligand binding and esterase activity. J. Biol. Chem. 2005, 280, 5724–5732. [CrossRef]
- Oya, T.; Hattori, N.; Mizuno, Y.; Miyata, S.; Maeda, S.; Osawa, T.; Uchida, K. Methylglyoxal modification of protein. Chemical and immunochemical characterization of methylglyoxal-arginine adducts. J. Biol. Chem. 1999, 274, 18492–18502. [CrossRef]
- Lieuw-a-Fa, M.L.; Schalkwijk, C.G.; Engelse, M.; van Hinsbergh, V.W. Interaction of Nepsilon(carboxymethyl)lysine- and methylglyoxal-modified albumin with endothelial cells and macrophages. Splice variants of RAGE may limit the responsiveness of human endothelial cells to AGEs. Thromb. Haemost. 2006, 95, 320–328. [CrossRef]
- Lo, T.W.; Westwood, M.E.; McLellan, A.C.; Selwood, T.; Thornalley, P.J. Binding and modification of proteins by methylglyoxal under physiological conditions. A kinetic and mechanistic study with N alpha-acetylarginine, N alpha-acetylcysteine, and N alpha-acetyllysine, and bovine serum albumin. J. Biol. Chem. 1994, 269, 32299–32305.
- Zhang, J.; Guo, J.; Yang, N.; Huang, Y.; Hu, T.; Rao, C. Endoplasmic reticulum stress-mediated cell death in liver injury. Cell Death Dis. 2022, 13, 1051. [CrossRef] [PubMed] [PubMed Central]
- Ajoolabady, A.; Kaplowitz, N.; Lebeaupin, C.; Kroemer, G.; Kaufman, R.J.; Malhi, H.; Ren, J. Endoplasmic reticulum stress in liver diseases. Hepatology 2023, 77, 619-639. [CrossRef] [PubMed] [PubMed Central]
- Gugliucci, A. Fructose surges damage hepatic adenosyl-monophosphate-dependent kinase and lead to increased lipogenesis and] hepatic insulin resistance. Med. Hypotheses 2016, 93, 87–92. [CrossRef]
- Wang, W.C.; Chou, C.K.; Chuang, M.C.; Li, Y.C.; Lee, J.A. Elevated levels of liver methylglyoxal and d-lactate in early-stage hepatitis in rats. Biomed. Chromatogr. 2018, 32, e4039. [CrossRef] [PubMed]
- Wei, Y.; Wang, D.; Moran, G.; Estrada, A.; Pagliassotti, M.J. Fructose-induced stress signaling in the liver involves methylglyoxal. Nutr. Metab. 2013, 10, 32. [CrossRef] [PubMed] [PubMed Central]
- Wei, Y.; Pagliassotti, M.J. Hepatospecific effects of fructose on c-jun NH2-terminal kinase: Implications for hepatic insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E926–E933. [CrossRef] [PubMed]
- Tournier, C.; Whitmarsh, A.J.; Cavanagh, J.; Barrett, T.; Davis, R.J. Mitogen-activated protein kinase kinase 7 is an activator of the c-Jun NH2-terminal kinase. Proc. Natl. Acad. Sci. USA. 1997, 94, 7337–7342. [CrossRef] [PubMed] [PubMed Central]
- Neves, C.; Rodrigues, T.; Sereno, J.; Simões, C.; Castelhano, J.; Gonçalves, J.; Bento, G.; Gonçalves, S.; Seiça, R.; Domingues, M.R.; et al. Dietary Glycotoxins Impair Hepatic Lipidemic Profile in Diet-Induced Obese Rats Causing Hepatic Oxidative Stress and Insulin Resistance. Oxid. Med. Cell Longev. 2019, 2019, 6362910. [CrossRef] [PubMed] [PubMed Central]
- Peter, A.; Schleicher, E.; Kliemank, E.; Szendroedi, J.; Königsrainer, A.; Häring, H.U.; Nawroth, P.P.; Fleming, T. Accumulation of Non-Pathological Liver Fat Is Associated with the Loss of Glyoxalase I Activity in Humans. Metabolites 2024, 14, 209. [CrossRef] [PubMed] [PubMed Central]
- Hüttl, M.; Markova, I.; Miklánková, D.; Zapletalova, I.; Kujal, P.; Šilhavý, J.; Pravenec, M.; Malinska, H. Hypolipidemic and insulin sensitizing effects of salsalate beyond suppressing inflammation in a prediabetic rat model. Front. Pharmacol. 2023, 14, 1117683. [CrossRef] [PubMed] [PubMed Central]
- Malinská, H.; Hüttl, M.; Miklánková, D.; Trnovská, J.; Zapletalová, I.; Poruba, M.; Marková; I. Ovariectomy-Induced Hepatic Lipid and Cytochrome P450 Dysmetabolism Precedes Serum Dyslipidemia. Int. J. Mol. Sci. 2021, 22, 4527. [CrossRef] [PubMed] [PubMed Central]
- Zhao, Y.; Wang, P.; Sang, S. Dietary Genistein Inhibits Methylglyoxal-Induced Advanced Glycation End Product Formation in Mice Fed a High-Fat Diet. J. Nutr. 2019, 149, 776-787. [CrossRef] [PubMed]
- Spanos, C.; Maldonado, E.M.; Fisher, C.P.; Leenutaphong, P.; Oviedo-Orta, E.; Windridge, D.; Salguero, F.J.; Bermúdez-Fajardo, A.; Weeks, M.E.; Evans, C.; et al. Proteomic identification and characterization of hepatic glyoxalase 1 dysregulation in non-alcoholic fatty liver disease. Proteome Sci. 2018, 16, 4; Erratum in Proteome Sci. 2018, 16, 13. [CrossRef] [PubMed] [PubMed Central]
- Depner, C.M.; Traber, M.G.; Bobe, G.; Kensicki, E.; Bohren, K.M.; Milne, G.; Jump, D.B. A metabolomic analysis of omega-3 fatty acid-mediated attenuation of western diet-induced nonalcoholic steatohepatitis in LDLR-/-mice. PLoS ONE 2013, 8, e83756. [CrossRef] [PubMed] [PubMed Central]
- Hollenbach, M.; Thonig, A.; Pohl, S.; Ripoll, C.; Michel, M.; Zipprich, A. Expression of glyoxalase-I is reduced in cirrhotic livers: A possible mechanism in the development of cirrhosis. PLoS ONE 2017, 12, e0171260. [CrossRef] [PubMed] [PubMed Central]
- Loarca, L.; Sassi-Gaha, S.; Artlett, C.M. Two α-dicarbonyls downregulate migration, invasion, and adhesion of liver cancer cells in a p53-dependent manner. Dig. Liver Dis. 2013, 45, 938–946. [CrossRef] [PubMed]
- Hu, X.; Yang, X.; He, Q.; Chen, Q.; Yu, L. Glyoxalase 1 is up-regulated in hepatocellular carcinoma and is essential for HCC cell proliferation. Biotechnol. Lett. 2014, 36, 257–263. [CrossRef] [PubMed]
- Michel, M.; Hollenbach, M.; Pohl, S.; Ripoll, C.; Zipprich, A. Inhibition of Glyoxalase-I Leads to Reduced Proliferation, Migration and Colony Formation, and Enhanced Susceptibility to Sorafenib in Hepatocellular Carcinoma. Front. Oncol. 2019, 9, 785. [CrossRef] [PubMed] [PubMed Central]
- Michel, M.; Hess, C.; Kaps, L.; Kremer, W.M.; Hilscher, M.; Galle, P.R.; Moehler, M.; Schattenberg, J.M.; Wörns, M.A.; Labenz, C.; et al. Elevated serum levels of methylglyoxal are associated with impaired liver function in patients with liver cirrhosis. Sci. Rep. 2021, 11, 20506. [CrossRef] [PubMed] [PubMed Central]
- Hollenbach, M. The Role of Glyoxalase-I (Glo-I), Advanced Glycation Endproducts (AGEs), and Their Receptor (RAGE) in Chronic Liver Disease and Hepatocellular Carcinoma (HCC). Int. J. Mol. Sci. 2017, 18, 2466. [CrossRef] [PubMed] [PubMed Central]
- Fernando, D.H.; Forbes, J.M.; Angus, P.W.; Herath, C.B. Development and Progression of Non-Alcoholic Fatty Liver Disease: The Role of Advanced Glycation End Products. Int. J. Mol. Sci. 2019, 20, 5037. [CrossRef] [PubMed] [PubMed Central]
- Nigro, C.; Leone, A.; Fiory, F.; Prevenzano, I.; Nicolò, A.; Mirra, P.; Beguinot, F.; Miele, C. Dicarbonyl Stress at the Crossroads of Healthy and Unhealthy Aging. Cells 2019, 8, 749. [CrossRef]
- Zemva, J.; Fink, C.A.; Fleming, T.H.; Schmidt, L.; Loft, A.; Herzig, S.; Knieß, R.A.; Mayer, M.; Bukau, B.; Nawroth, P.P. et al. Hormesis enables cells to handle accumulating toxic metabolites during increased energy flux. Redox Biol. 2017, 13, 674–686. [CrossRef]
- Ravichandran, M.; Priebe, S.; Grigolon, G.; Rozanov, L.; Groth, M.; Laube, B.; Guthke, R.; Platzer, M.; Zarse, K.; Ristow, M. Impairing L-Threonine Catabolism Promotes Healthspan through Methylglyoxal-Mediated Proteohormesis. Cell Metab. 2018, 27, 914–925.e5. [CrossRef]
- Zhang, S.; Liang, X.; Zheng, X.; Huang, H.; Chen, X.; Wu, K.; Wang, B.; Ma, S. Glo1 genetic amplification as a potential therapeutic target in hepatocellular carcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 2079–2090. [PubMed] [PubMed Central]
- Liu, Y.; Zhu, X.; Zhu, J.; Liao, S.; Tang, Q.; Liu, K.; Guan, X.; Zhang, J.; Feng, Z. Identification of differential expression of genes in hepatocellular carcinoma by suppression subtractive hybridization combined cDNA microarray. Oncol. Rep. 2007, 18, 943–951. [PubMed]
- Taïbi, N.; Al-Balas, Q.A.; Bekari, N.; Talhi, O.; Al Jabal, G.A.; Benali, Y.; Ameraoui, R.; Hadjadj, M.; Taïbi, A.; Boutaiba, Z.M.; et al. Molecular docking and dynamic studies of a potential therapeutic target inhibiting glyoxalase system: Metabolic action of the 3, 3 ‘-[3-(5-chloro-2-hydroxyphenyl)-3-oxopropane-1, 1-diyl]-Bis-4-hydroxycoumarin leads overexpression of the intracellular level of methylglyoxal and induction of a pro-apoptotic phenomenon in a hepatocellular carcinoma model. Chem. Biol. Interact. 2021, 345, 109511. [CrossRef] [PubMed]
- Thornalley, P.J.; Rabbani, N. Glyoxalase in tumourigenesis and multidrug resistance. Semin. Cell Dev. Biol. 2011, 22, 318–325. [CrossRef] [PubMed]
- Nokin, M.J.; Durieux, F.; Bellier, J.; Peulen, O.; Uchida, K.; Spiegel, D.A.; Cochrane, J.R.; Hutton, C.A.; Castronovo, V.; Bellahcène, A. Hormetic potential of methylglyoxal, a side-product of glycolysis, in switching tumours from growth to death. Sci. Rep. 2017, 7, 11722. [CrossRef] [PubMed] [PubMed Central]
- Bellier, J.; Nokin, M.J.; Lardé, E.; Karoyan, P.; Peulen, O.; Castronovo, V.; Bellahcène, A. Methylglyoxal, a potent inducer of AGEs, connects between diabetes and cancer. Diabetes Res. Clin. Pract. 2019, 148, 200–211. [CrossRef] [PubMed]
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.H.; et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 1063–1075. [CrossRef] [PubMed] [PubMed Central]
- Mortera, R.R.; Bains, Y.; Gugliucci, A. Fructose at the crossroads of the metabolic syndrome and obesity epidemics. Front. Biosci.-Landmark 2019, 24, 186–211. [CrossRef] [PubMed]
- Johnson, R.J.; Lanaspa, M.A.; Sanchez-Lozada, L.G.; Tolan, D.; Nakagawa, T.; Ishimoto, T.; Andres-Hernando, A.; Rodriguez-Iturbe, B.; Stenvinkel, P. The fructose survival hypothesis for obesity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2023, 378, 20220230. [CrossRef] [PubMed] [PubMed Central]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [CrossRef]
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [CrossRef]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [CrossRef]
- Barinova, K.V.; Serebryakova, M.V.; Melnikova, A.K.; Medvedeva, M.V.; Muronetz, V.I.; Schmalhausen, E.V. Mechanism of inactivation of glyceraldehyde-3-phosphate dehydrogenase in the presence of methylglyoxal. Arch. Biochem. Biophys. 2023, 733, 109485. [CrossRef] [PubMed]
- Takeuchi, M.; Takino, J.I.; Sakasai-Sakai, A.; Takata, T.; Tsutsumi, M. Toxic AGE (TAGE) Theory for the Pathophysiology of the Onset/Progression of NAFLD and ALD. Nutrients 2017, 9, 634. [CrossRef] [PubMed] [PubMed Central]
- Takeuchi, M.; Suzuki, H.; Takeda, K.; Sakai-Sakasai, A. (2024). Toxic advanced glycation end-products (TAGE) are major structures of cytotoxic AGEs derived from glyceraldehyde. Med. Hypotheses 2024, 183, 111248. [CrossRef]
- Takeuchi, M.; Sakasai-Sakai, A.; Takata, T.; Takino, J.I.; Koriyama, Y.; Kikuchi, C.; Furukawa, A.; Nagamine, K.; Hori, T.; Matsunaga, T. Intracellular Toxic AGEs (TAGE) Triggers Numerous Types of Cell Damage. Biomolecules 2021, 11, 387. [CrossRef] [PubMed] [PubMed Central]
- Takeuchi, M. Toxic AGEs (TAGE) theory: A new concept for preventing the development of diseases related to lifestyle. Diabetol. Metab. Syndr. 2020, 12, 105. [CrossRef] [PubMed] [PubMed Central]
- Sakasai-Sakai, A.; Takata, T.; Takino, J.I.; Takeuchi, M. Impact of intracellular glyceraldehyde-derived advanced glycation end-products on human hepatocyte cell death. Sci. Rep. 2017, 7, 14282. [CrossRef] [PubMed] [PubMed Central]
- Sakai-Sakasai, A.; Takeda, K.; Suzuki, H.; Takeuchi, M. Structures of Toxic Advanced Glycation End-Products Derived from Glyceraldehyde, A Sugar Metabolite. Biomolecules 2024, 14, 202. [CrossRef] [PubMed] [PubMed Central]
- Gugliucci, A. Formation of Fructose-Mediated Advanced Glycation End Products and Their Roles in Metabolic and Inflammatory Diseases. Adv. Nutr. 2017, 8, 54–62. [CrossRef] [PubMed] [PubMed Central]
- Petagine, L.; Zariwala, M.G.; Patel, V.B. Alcoholic liver disease: Current insights into cellular mechanisms. World J. Biol. Chem. 2021, 12, 87–103. [CrossRef] [PubMed] [PubMed Central]
- Wilson, D.F.; Matschinsky, F.M. Ethanol metabolism: The good, the bad, and the ugly. Med. Hypotheses. 2020, 140, 109638. [CrossRef] [PubMed]
- Kalavalapalli, S.; Leiva, E.G.; Lomonaco, R.; Chi, X.; Shrestha, S.; Dillard, R.; Budd, J.; Romero, J.P.; Li, C.; Bril, F.; et al. Adipose Tissue Insulin Resistance Predicts the Severity of Liver Fibrosis in Patients With Type 2 Diabetes and NAFLD. J. Clin. Endocrinol. Metab. 2023, 108, 1192–1201. [CrossRef] [PubMed]
- He, L.; Qiu, K.; Zheng, W.; Kong, W.; Zeng, T. Uric acid may serve as the sixth cardiometabolic criterion for defining MASLD. J. Hepatol. 2024, 80, e152–e153. [CrossRef] [PubMed]
- Choi, Y.; Yang, H.; Jeon, S.; Cho, K.W.; Kim, S.J.; Kim, S.; Lee, M.; Suh, J.; Chae, H.W.; Kim, H.S.; et al. Prediction of insulin resistance and elevated liver transaminases using serum uric acid and derived markers in children and adolescents. Eur. J. Clin. Nutr. 2024, 78, 864–871. [CrossRef] [PubMed]
- Kosmas, C.E.; Bousvarou, M.D.; Kostara, C.E.; Papakonstantinou, E.J.; Salamou, E.; Guzman, E. Insulin resistance and cardiovascular disease. J. Int. Med. Res. 2023, 51, 3000605231164548. [CrossRef] [PubMed] [PubMed Central]
- Wang, S.; Zhang, Q.; Qin, B. Association between remnant cholesterol and insulin resistance levels in patients with metabolic-associated fatty liver disease. Sci. Rep. 2024, 14, 4596. [CrossRef] [PubMed] [PubMed Central]
- Genua, I.; Cusi, K. Pharmacological Approaches to Nonalcoholic Fatty Liver Disease: Current and Future Therapies. Diabetes Spectr. 2024, 37, 48–58. [CrossRef] [PubMed] [PubMed Central]
- Paulino, P.J.I.V.; Cuthrell, K.M.; Tzenios, N. Non Alcoholic Fatty Liver Disease; Disease Burden, Management, and Future Perspectives. Int. Res. J. Gastroenterol. Hepatol. 2024, 7, 1–13. http://archive.pcbmb.org/id/eprint/1800.
- Shinozaki, S.; Tahara, T.; Miura, K.; Lefor, A.K.; Yamamoto, H. Pemafibrate therapy for non-alcoholic fatty liver disease is more effective in lean patients than obese patients. Clin. Exp. Hepatol. 2022, 8, 278–283. [CrossRef] [PubMed] [PubMed Central]
- Zhang, S.; Zhao, J.; Xie, F.; He, H.; Johnston, L.J.; Dai, X.; Wu, C.; Ma, X. Dietary fiber-derived short-chain fatty acids: A potential therapeutic target to alleviate obesity-related nonalcoholic fatty liver disease. Obes. Rev. 2021, 22, e13316. [CrossRef] [PubMed]
- Alkhouri, N.; Scott, A. An Update on the Pharmacological Treatment of Nonalcoholic Fatty Liver Disease: Beyond Lifestyle Modifications. Clin. Liver Dis. 2018, 11, 82–86. [CrossRef] [PubMed] [PubMed Central]
- Zhao, J.; Li, B.; Zhang, K.; Zhu, Z. The effect and safety of obeticholic acid for patients with nonalcoholic steatohepatitis: A systematic review and meta-analysis of randomized controlled trials. Medicine 2024, 103, e37271.
- Harrison, S.A.; Bedossa, P.; Guy, C.D.; Schattenberg, J.M.; Loomba, R.; Taub, R.; Labriola, D.; Moussa, S.E.; Neff, G.W.; Rinella, M.E.; et al. MAESTRO-NASH Investigators. A Phase 3, Randomized, Controlled Trial of Resmetirom in NASH with Liver Fibrosis. N. Engl. J. Med. 2024, 390, 497–509. [CrossRef] [PubMed]
- Harrison, S.A.; Taub, R.; Neff, G.W.; Lucas, K.J.; Labriola, D.; Moussa, S.E.; Alkhouri, N.; Bashir, M.R. Resmetirom for nonalcoholic fatty liver disease: A randomized, double-blind, placebo-controlled phase 3 trial. Nat. Med. 2023, 29, 2919–2928. [CrossRef] [PubMed] [PubMed Central]
- Harrison, S.A.; Wong, V.W.; Okanoue, T.; Bzowej, N.; Vuppalanchi, R.; Younes, Z.; Kohli, A.; Sarin, S.; Caldwell, S.H.; Alkhouri, N.; et al. Selonsertib for patients with bridging fibrosis or compensated cirrhosis due to NASH: Results from randomized phase III STELLAR trials. J. Hepatol. 2020, 73, 26–39. [CrossRef] [PubMed]
- Anstee, Q.M.; Neuschwander-Tetri, B.A.; Wai-Sun Wong, V.; Abdelmalek, M.F.; Rodriguez-Araujo, G.; Landgren, H.; Park, G.S.; Bedossa, P.; Alkhouri, N.; Tacke, F.; et al. Cenicriviroc Lacked Efficacy to Treat Liver Fibrosis in Nonalcoholic Steatohepatitis: AURORA Phase III Randomized Study. Clin. Gastroenterol. Hepatol. 2024, 22, 124–134.e1. [CrossRef] [PubMed]
- Blair, H.A. Elafibranor: First Approval. Drugs 2024, 84, 1143–1148; Erratum in Drugs 2024, 84, 1165. [CrossRef] [PubMed]
- Keam, S.J. Resmetirom: First Approval. Drugs 2024, 84, 729–735. [CrossRef] [PubMed]
- Petta, S.; Targher, G.; Romeo, S.; Pajvani, U.B.; Zheng, M.H.; Aghemo, A.; Valenti, L.V.C. The first MASH drug therapy on the horizon: Current perspectives of resmetirom. Liver Int. 2024, 44, 1526–1536. [CrossRef] [PubMed]
- Jophlin, L.L.; Singal, A.K.; Bataller, R.; Wong, R.J.; Sauer, B.G.; Terrault, N.A.; Shah, V.H. ACG Clinical Guideline: Alcohol-Associated Liver Disease. Am. J. Gastroenterol. 2024, 119, 30–54. [CrossRef] [PubMed] [PubMed Central]
- Yki-Järvinen, H.; Luukkonen, P.K.; Hodson, L.; Moore, J.B. Dietary carbohydrates and fats in nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 770–786. [CrossRef] [PubMed]
- Chan, W.K.; Chuah, K.H.; Rajaram, R.B.; Lim, L.L.; Ratnasingam, J.; Vethakkan, S.R. Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD): A State-of-the-Art Review. J. Obes. Metab. Syndr. 2023, 32, 197–213. [CrossRef] [PubMed] [PubMed Central]
- Hansen, C.D.; Gram-Kampmann, E.M.; Hansen, J.K.; Hugger, M.B.; Madsen, B.S.; Jensen, J.M.; Olesen, S.; Torp, N.; Rasmussen, D.N.; Kjærgaard, M.; et al. Effect of Calorie-Unrestricted Low-Carbohydrate, High-Fat Diet Versus High-Carbohydrate, Low-Fat Diet on Type 2 Diabetes and Nonalcoholic Fatty Liver Disease: A Randomized Controlled Trial. Ann. Intern. Med. 2023, 176, 10–21. [CrossRef] [PubMed]
- Cha, J.Y.; Kim, S.Y.; Lim, Y.W.; Choi, K.H.; Shin, I.S. Comparative Effectiveness of Cognitive Behavioral Therapy and Behavioral Therapy in Obesity: A Systematic Review and Network Meta-Analysis. J. Clin. Psychol. Med. Settings. 2024. [CrossRef] [PubMed]
- Hegazi, O.E.; Alalalmeh, S.O.; Shahwan, M.; Jairoun, A.A.; Alourfi, M.M.; Bokhari, G.A.; Alkhattabi, A.; Alsharif, S.; Aljehani, M.A.; Alsabban, A.M.; et al. Exploring Promising Therapies for Non-Alcoholic Fatty Liver Disease: A ClinicalTrials.gov Analysis. Diabetes Metab. Syndr. Obes. 2024, 17, 545–561. [CrossRef] [PubMed] [PubMed Central]
- Gastaldelli A, Cusi K, Fernández Landó L, Bray R, Brouwers B, Rodríguez, Á. Effect of tirzepatide versus insulin degludec on liver fat content and abdominal adipose tissue in people with type 2 diabetes (SURPASS-3 MRI): A substudy of the randomised, open-label, parallel-group, phase 3 SURPASS-3 trial. Lancet Diabetes Endocrinol. 2022, 10, 393–406. [CrossRef] [PubMed]
- Gastaldelli, A.; Cusi, K.; Landó, L.F.; Bray, R.; Brouwers, B.; Rodríguez, Á.; Menzen, M. Effect of Tirzepatide Versus Insulin Degludec on Liver Fat Content and Abdominal Adipose Tissue in Patients with Type 2 Diabetes (SURPASS-3 MRI). Diabetol. Und Stoffwechs. 2023, 18 (Supp. S15–S16), P-004. [CrossRef]
- Koh, B.; Xiao, J.; Ng, C.H.; Law, M.; Gunalan, S.Z.; Danpanichkul, P.; Ramadoss, V.; Sim, B.K.L.; Tan, E.Y.; Teo, C.B.; et al. Comparative efficacy of pharmacologic therapies for MASH in reducing liver fat content: Systematic review and network meta-analysis. Hepatology 2024. [CrossRef] [PubMed]
- Huang, Y.; Wang, X.; Yan, C.; Li, C.; Zhang, L.; Zhang, L.; Liang, E.; Liu, T.; Mao, J. Effect of metformin on nonalcoholic fatty liver based on meta-analysis and network pharmacology. Medicine 2022, 101, e31437. [CrossRef] [PubMed] [PubMed Central]
- Mansi, I.A.; Chansard, M.; Lingvay, I.; Zhang, S.; Halm, E.A.; Alvarez, C.A. Association of Statin Therapy Initiation with Diabetes Progression: A Retrospective Matched-Cohort Study. JAMA Intern. Med. 2021, 181, 1562–1574. [CrossRef] [PubMed] [PubMed Central]
- Mansi, I.A.; Sumithran, P.; Kinaan, M. Risk of diabetes with statins. BMJ 2023, 381, e071727. [CrossRef] [PubMed]
- Abbasi, F.; Lamendola, C.; Harris, C.S.; Harris, V.; Tsai, M.S.; Tripathi, P.; Abbas, F.; Reaven, G.M.; Reaven, P.D.; Snyder, M.P.; et al. Statins Are Associated with Increased Insulin Resistance and Secretion. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2786–2797. [CrossRef] [PubMed] [PubMed Central]
- Galicia-Garcia, U.; Jebari, S.; Larrea-Sebal, A.; Uribe, K.B.; Siddiqi, H.; Ostolaza, H.; Benito-Vicente, A.; Martín, C. Statin Treatment-Induced Development of Type 2 Diabetes: From Clinical Evidence to Mechanistic Insights. Int. J. Mol. Sci. 2020, 21, 4725. [CrossRef] [PubMed] [PubMed Central]
- Ling, Z.; Shu, N.; Xu, P.; Wang, F.; Zhong, Z.; Sun, B.; Li, F.; Zhang, M.; Zhao, K.; Tang, X.; et al. Involvement of pregnane X receptor in the impaired glucose utilization induced by atorvastatin in hepatocytes. Biochem. Pharmacol. 2016, 100, 98–111. [CrossRef] [PubMed]
- Wah Kheong, C.; Nik Mustapha, N.R.; Mahadeva, S. A Randomized Trial of Silymarin for the Treatment of Nonalcoholic Steatohepatitis. Clin. Gastroenterol. Hepatol. 2017, 15, 1940–1949.e8. [CrossRef] [PubMed]
- Navarro, V.J.; Belle, S.H.; D’Amato, M.; Adfhal, N.; Brunt, E.M.; Fried, M.W.; Reddy, K.R.; Wahed, A.S.; Harrison, S. Silymarin in NASH and C Hepatitis (SyNCH) Study Group. Silymarin in non-cirrhotics with non-alcoholic steatohepatitis: A randomized, double-blind, placebo controlled trial. PLoS ONE 2019, 14, e0221683; Erratum in PLoS ONE 2019, 14, e0223915. [CrossRef] [PubMed] [PubMed Central]
- Ceriello, A. Hypothesis: The “metabolic memory”, the new challenge of diabetes. Diabetes Res. Clin. Pract. 2009, 86 (Suppl. S1), S2–S6. [CrossRef] [PubMed]
- Dobbie, L.J.; Burgess, J.; Hamid, A.; Nevitt, S.J.; Hydes, T.J.; Alam, U.; Cuthbertson, D.J. Effect of a Low-Calorie Dietary Intervention on Liver Health and Body Weight in Adults with Metabolic-Dysfunction Associated Steatotic Liver Disease (MASLD) and Overweight/Obesity: A Systematic Review and Meta-Analysis. Nutrients 2024, 16, 1030. [CrossRef] [PubMed] [PubMed Central]
- Ma, Y.; Sun, Y.; Sun, L.; Liu, X.; Zeng, R.; Lin, X.; Li, Y. Effects of gut microbiota and fatty acid metabolism on dyslipidemia following weight-loss diets in women: Results from a randomized controlled trial. Clin. Nutr. 2021, 40, 5511–5520. [CrossRef] [PubMed]
- Seethaler, B.; Nguyen, N.K.; Basrai, M.; Kiechle, M.; Walter, J.; Delzenne, N.M.; Bischoff, S.C. Short-chain fatty acids are key mediators of the favorable effects of the Mediterranean diet on intestinal barrier integrity: Data from the randomized controlled LIBRE trial. Am. J. Clin. Nutr. 2022, 116, 928–942. [CrossRef] [PubMed]
- Losasso, C.; Eckert, E.M.; Mastrorilli, E.; Villiger, J.; Mancin, M.; Patuzzi, I.; Di Cesare, A.; Cibin, V.; Barrucci, F.; Pernthaler, J.; et al. Assessing the Influence of Vegan, Vegetarian and Omnivore Oriented Westernized Dietary Styles on Human Gut Microbiota: A Cross Sectional Study. Front. Microbiol. 2018, 9, 317. [CrossRef] [PubMed] [PubMed Central]
- De Filippis, F.; Pellegrini, N.; Vannini, L.; Jeffery, I.B.; La Storia, A.; Laghi, L.; Serrazanetti, D.I.; Di Cagno, R.; Ferrocino, I.; Lazzi, C.; et al. High-level adherence to a Mediterranean diet beneficially impacts the gut microbiota and associated metabolome. Gut 2016, 65, 1812–1821. [CrossRef] [PubMed]
- Landry, M.J.; Ward, C.P.; Cunanan, K.M.; Durand, L.R.; Perelman, D.; Robinson, J.L.; Hennings, T.; Koh, L.; Dant, C.; Zeitlin, A.; et al. Cardiometabolic Effects of Omnivorous vs. Vegan Diets in Identical Twins: A Randomized Clinical Trial. JAMA Netw. Open 2023, 6, e2344457; Erratum in JAMA Netw. Open 2023, 6, e2350422. [CrossRef] [PubMed] [PubMed Central]
- Tanaka, M.; Nakayama, J. Development of the gut microbiota in infancy and its impact on health in later life. Allergol. Int. 2017, 66, 515–522. [CrossRef] [PubMed]
- Monda, V.; Villano, I.; Messina, A.; Valenzano, A.; Esposito, T.; Moscatelli, F.; Viggiano, A.; Cibelli, G.; Chieffi, S.; Monda, M.; et al. Exercise Modifies the Gut Microbiota with Positive Health Effects. Oxid. Med. Cell Longev. 2017, 2017, 3831972. [CrossRef] [PubMed] [PubMed Central]
- Sun, C.; Li, A.; Xu, C.; Ma, J.; Wang, H.; Jiang, Z.; Hou, J. Comparative Analysis of Fecal Microbiota in Vegetarians and Omnivores. Nutrients 2023, 15, 2358. [CrossRef] [PubMed] [PubMed Central]
- Prochazkova, M.; Budinska, E.; Kuzma, M.; Pelantova, H.; Hradecky, J.; Heczkova, M.; Daskova, N.; Bratova, M.; Modos, I.; Videnska, P.; et al. Vegan Diet Is Associated with Favorable Effects on the Metabolic Performance of Intestinal Microbiota: A Cross-Sectional Multi-Omics Study. Front. Nutr. 2022, 8, 783302. [CrossRef] [PubMed] [PubMed Central]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14. [CrossRef] [PubMed] [PubMed Central]
- Takeuchi, M.; Suzuki, H.; Sakai-Sakasai, A. Major generation route of cytotoxic protein adducts derived from acetaldehyde, a metabolite of alcohol. Med. Hypotheses 2024, 189, 111385. [CrossRef]
- Hayashi, T.; Shibamoto, T. Analysis of methyl glyoxal in foods and beverages. J. Agric. Food Chem. 1985, 33, 1090–1093. https://pubs.acs.org/doi/pdf/10.1021/jf00066a018.
- Hopper, D.J.; Cooper, R.A. The regulation of Escherichia coli methylglyoxal synthase; a new control site in glycolysis? FEBS Lett. 1971, 13, 213–216. [CrossRef] [PubMed]
- Booth, I.R.; Ferguson, G.P.; Miller, S.; Li, C.; Gunasekera, B.; Kinghorn, S. Bacterial production of methylglyoxal: A survival strategy or death by misadventure? Biochem. Soc. Trans. 2003, 31 Pt 6,1406–1408. [CrossRef] [PubMed]
- Baskaran, S.; Rajan, D.P.; Balasubramanian, K.A. Formation of methylglyoxal by bacteria isolated from human faeces. J. Med. Microbiol. 1989, 28, 211–215. [CrossRef] [PubMed]
- Chandrangsu, P.; Dusi, R.; Hamilton, C.J.; Helmann, J.D. Methylglyoxal resistance in Bacillus subtilis: Contributions of bacillithiol-dependent and independent pathways. Mol. Microbiol. 2014, 91, 706–715. [CrossRef] [PubMed] [PubMed Central]
- Srebreva, L.N.; Stoynev, G.A.; Ivanov, I.G. Evidence for excretion of glycation agents from E. coli cells during growth. Biotechnol. Biotechnol. Equip. 2009, 23, 1068–1071. [CrossRef]
- Chen, Y.; Guo, T.L. Dietary advanced glycation end-products elicit toxicological effects by disrupting gut microbiome and immune homeostasis. J. Immunotoxicol. 2021, 18, 93–104. [CrossRef] [PubMed] [PubMed Central]
- Yuan, X.; Liu, J.; Nie, C.; Ma, Q.; Wang, C.; Liu, H.; Chen, Z.; Zhang, M.; Li, J. Comparative Study of the Effects of Dietary-Free and -Bound Nε-Carboxymethyllysine on Gut Microbiota and Intestinal Barrier. J. Agric. Food Chem. 2024, 72, 5014–5025. [CrossRef] [PubMed]
- Aschner, M.; Skalny, A.V.; Gritsenko, V.A.; Kartashova, O.L.; Santamaria, A.; Rocha, J.B.T.; Spandidos, D.A.; Zaitseva, I.P.; Tsatsakis, A.; Tinkov, A.A. Role of gut microbiota in the modulation of the health effects of advanced glycation end-products (Review). Int. J. Mol. Med. 2023, 51, 44. [CrossRef] [PubMed] [PubMed Central]
- Xie, F.; Zhao, J.; Liu, D.; Wan, Z.; Sun, K.; Wang, Y. Associations of dietary advanced glycation end products with liver steatosis via vibration controlled transient elastography in the United States: A nationwide cross-sectional study. Eur. J. Nutr. 2024, 63, 173–183. [CrossRef] [PubMed]
- Beisswenger, P.J.; Howell, S.K.; Touchette, A.D.; Lal, S.; Szwergold, B.S. (1999). Metformin reduces systemic methylglyoxal levels in type 2 diabetes. Diabetes 1999, 48, 198–202. [CrossRef]
- Kinsky, O.R.; Hargraves, T.L.; Anumol, T.; Jacobsen, N.E.; Dai, J.; Snyder, S.A.; Monks, T.J.; Lau, S.S. Metformin Scavenges Methylglyoxal to form a Novel Imidazolinone Metabolite in Humans. Chem. Res. Toxicol. 2016, 29, 227–234. [CrossRef] [PubMed] [PubMed Central]
- Shao, X.; Chen, H.; Zhu, Y.; Sedighi, R.; Ho, C.T.; Sang, S. Essential Structural Requirements and Additive Effects for Flavonoids to Scavenge Methylglyoxal. J. Agric. Food Chem. 2014, 62, 3202–3210. [CrossRef] [PubMed]
- Zhang, Y.; Zhan, L.; Wen, Q.; Feng, Y.; Luo, Y.; Tan, T. Trapping Methylglyoxal by Taxifolin and Its Metabolites in Mice. J. Agric. Food Chem. 2022, 70, 5026–5038. [CrossRef] [PubMed]
- Bhuiyan, M. N. I., Mitsuhashi, S., Sigetomi, K., & Ubukata, M. (2017). Quercetin inhibits advanced glycation end product formation via chelating metal ions, trapping methylglyoxal, and trapping reactive oxygen species. Bioscience, biotechnology, and biochemistry, 81(5), 882-890. [CrossRef]
- Zhu, J.; Chen, C.; Zhang, B.; Huang, Q. The inhibitory effects of flavonoids on α-amylase and α-glucosidase. Crit. Rev. Food Sci. Nutr. 2020, 60, 695–708. [CrossRef]
- Proença, C.; Ribeiro, D.; Freitas, M.; Fernandes, E. Flavonoids as potential agents in the management of type 2 diabetes through the modulation of α-amylase and α-glucosidase activity: A review. Crit. Rev. Food Sci. Nutr. 2022, 62, 3137–3207. [CrossRef] [PubMed]
- Lim, J.; Ferruzzi, M.G.; Hamaker, B.R. Structural requirements of flavonoids for the selective inhibition of α-amylase versus α-glucosidase. Food Chem. 2022, 370, 130981. [CrossRef] [PubMed]
- Pan, J.; Zhang, Q.; Zhang, C.; Yang, W.; Liu, H.; Lv, Z.; Liu, J.; Jiao, Z. Inhibition of Dipeptidyl Peptidase-4 by Flavonoids: Structure-Activity Relationship, Kinetics and Interaction Mechanism. Front. Nutr. 2022, 9, 892426. [CrossRef] [PubMed] [PubMed Central]
- Chen, S.; Wang, X.; Cheng, Y.; Gao, H.; Chen, X. A Review of Classification, Biosynthesis, Biological Activities and Potential Applications of Flavonoids. Molecules 2023, 28, 4982. [CrossRef] [PubMed] [PubMed Central]
- Wang, M.; Lu, Y.; Wu, Q.; Chen, G.; Zhao, H.; Ho, C.T.; Li, S. Biotransformation and Gut Microbiota-Mediated Bioactivity of Flavonols. J. Agric. Food Chem. 2023, 71, 8317–8331. [CrossRef] [PubMed]
- Liu, Y.; Kurita, A.; Nakashima, S.; Zhu, B.; Munemasa, S.; Nakamura, T.; Murata, Y.; Nakamura, Y. 3,4-Dihydroxyphenylacetic acid is a potential aldehyde dehydrogenase inducer in murine hepatoma Hepa1c1c7 cells. Biosci. Biotechnol. Biochem. 2017, 81, 1978–1983. [CrossRef] [PubMed]
- Fecka, I.; Bednarska, K.; Kowalczyk, A. In Vitro Antiglycation and Methylglyoxal Trapping Effect of Peppermint Leaf (Mentha × piperita L.) and Its Polyphenols. Molecules 2023, 28, 2865. [CrossRef] [PubMed] [PubMed Central]
- Ramos, F.M.M.; Ribeiro, C.B.; Cesar, T.B.; Milenkovic, D.; Cabral, L.; Noronha, M.F.; Sivieri, K. Lemon flavonoids nutraceutical (Eriomin®) attenuates prediabetes intestinal dysbiosis: A double-blind randomized controlled trial. Food Sci. Nutr. 2023, 11, 7283–7295. [CrossRef] [PubMed] [PubMed Central]
- Malik, A.; Malik, M.; Qureshi, S. Effects of silymarin use on liver enzymes and metabolic factors in metabolic dysfunction-associated steatotic liver disease: A systematic review and meta-analysis. Can. Liver J. 2024, 7, 40–53. [CrossRef] [PubMed] [PubMed Central]
- Li, S.; Duan, F.; Li, S.; Lu, B. Administration of silymarin in NAFLD/NASH: A systematic review and meta-analysis. Ann. Hepatol. 2024, 29, 101174. [CrossRef] [PubMed]
- Yao, J.; Zhi, M.; Gao, X.; Hu, P.; Li, C.; Yang, X. Effect and the probable mechanisms of silibinin in regulating insulin resistance in the liver of rats with non-alcoholic fatty liver. Braz. J. Med. Biol. Res. 2013, 46, 270–277. [CrossRef] [PubMed] [PubMed Central]
- Velussi, M.; Cernigoi, A.M.; De Monte, A.; Dapas, F.; Caffau, C.; Zilli, M. Long-term (12 months) treatment with an anti-oxidant drug (silymarin) is effective on hyperinsulinemia, exogenous insulin need and malondialdehyde levels in cirrhotic diabetic patients. J. Hepatol. 1997, 26, 871–879. [CrossRef] [PubMed]
- Zhong, S.; Fan, Y.; Yan, Q.; Fan, X.; Wu, B.; Han, Y.; Zhang, Y.; Chen, Y.; Zhang, H.; Niu, J. The therapeutic effect of silymarin in the treatment of nonalcoholic fatty disease: A meta-analysis (PRISMA) of randomized control trials. Medicine 2017, 96, e9061. [CrossRef] [PubMed] [PubMed Central]
- Jin, Y.; Wang, X.; Chen, K.; Chen, Y.; Zhou, L.; Zeng, Y.; Zhou, Y.; Pan, Z.; Wang, D.; Li, Z.; et al. Silymarin decreases liver stiffness associated with gut microbiota in patients with metabolic dysfunction-associated steatotic liver disease: A randomized, double-blind, placebo-controlled trial. Lipids Health Dis. 2024, 23, 239. [CrossRef] [PubMed] [PubMed Central]
- Yi, M.; Manzoor, M.; Yang, M.; Zhang, H.; Wang, L.; Zhao, L.; Xiang, L.; Qi, J. Silymarin targets the FXR protein through microbial metabolite 7-keto-deoxycholic acid to treat MASLD in obese mice. Phytomedicine 2024, 133, 155947. [CrossRef] [PubMed]
- Li, L.; Ji, K.; Du, F.; Jin, N.; Boesch, C.; Farag, M.A.; Li, H.; Liu, X.; Xiao, J. Does Flavonoid Supplementation Alleviate Non-Alcoholic Fatty Liver Disease? A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Mol. Nutr. Food Res. 2023, 67, e2300480. [CrossRef] [PubMed]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. Syst. Rev. 2021, 10, 89. [CrossRef]
- Guo, Q.; Mori, T.; Jiang, Y.; Hu, C.; Osaki, Y.; Yoneki, Y.; Sun, Y.; Hosoya, T.; Kawamata, A.; Ogawa, S.; et al. Methylglyoxal contributes to the development of insulin resistance and salt sensitivity in Sprague-Dawley rats. J. Hypertens. 2009, 27, 1664–1671. [CrossRef]
- Dhar, A.; Desai, K.M.; Wu, L. Alagebrium attenuates acute methylglyoxal-induced glucose intolerance in Sprague-Dawley rats. Br. J. Pharmacol. 2010, 159, 166–175. [CrossRef]
- Nigro, C.; Raciti, G.A.; Leone, A.; Fleming, T.H.; Longo, M.; Prevenzano, I.; Fiory, F.; Mirra, P.; D’Esposito, V.; Ulianich, L.; et al. Methylglyoxal impairs endothelial insulin sensitivity both in vitro and in vivo. Diabetologia 2014, 57, 1485–1494. [CrossRef]
- Nigro, C.; Leone, A.; Raciti, G.A.; Longo, M.; Mirra, P.; Formisano, P.; Beguinot, F.; Miele, C. Methylglyoxal-Glyoxalase 1 Balance: The Root of Vascular Damage. Int. J. Mol. Sci. 2017, 18, 188. [CrossRef]
- Shamsaldeen, Y.A.; Mackenzie, L.S.; Lione, L.A.; Benham, C.D. Methylglyoxal, A Metabolite Increased in Diabetes is Associated with Insulin Resistance, Vascular Dysfunction and Neuropathies. Curr. Drug Metab. 2016, 17, 359–367. [CrossRef]
- Mey, J.T.; Haus, J.M. Dicarbonyl Stress and Glyoxalase-1 in Skeletal Muscle: Implications for Insulin Resistance and Type 2 Diabetes. Front. Cardiovasc. Med. 2018, 5, 117. [CrossRef]
- Riboulet-Chavey, A.; Pierron, A.; Durand, I.; Murdaca, J.; Giudicelli, J.; Van Obberghen, E. Methylglyoxal impairs the insulin signaling pathways independently of the formation of intracellular reactive oxygen species. Diabetes 2006, 55, 1289–1299. [CrossRef]
- Engelbrecht, B.; Mattern, Y.; Scheibler, S.; Tschoepe, D.; Gawlowski, T.; Stratmann, B. Methylglyoxal impairs GLUT4 trafficking and leads to increased glucose uptake in L6 myoblasts. Horm. Metab. Res. 2014, 46, 77–84. [CrossRef]
- Jia, X.; Wu, L. Accumulation of endogenous methylglyoxal impaired insulin signaling in adipose tissue of fructose-fed rats. Mol. Cell Biochem. 2007, 306, 133–139. [CrossRef]
- Dhar, A.; Dhar, I.; Jiang, B.; Desai, K.M.; Wu, L. Chronic methylglyoxal infusion by minipump causes pancreatic beta-cell dysfunction and induces type 2 diabetes in Sprague-Dawley rats. Diabetes 2011, 60, 899–908. [CrossRef]
- Matafome, P.; Santos-Silva, D.; Crisóstomo, J.; Rodrigues, T.; Rodrigues, L.; Sena, C.M.; Pereira, P.; Seiça, R. Methylglyoxal causes structural and functional alterations in adipose tissue independently of obesity. Arch. Physiol. Biochem. 2012, 118, 58–68. [CrossRef]
- Hüttl, M.; Markova, I.; Miklankova, D.; Makovicky, P.; Pelikanova, T.; Šeda, O.; Šedová, L.; Malinska, H. Adverse Effects of Methylglyoxal on Transcriptome and Metabolic Changes in Visceral Adipose Tissue in a Prediabetic Rat Model. Antioxidants 2020, 9, 803. [CrossRef]
- Rodrigues, T.; Matafome, P.; Sereno, J.; Almeida, J.; Castelhano, J.; Gamas, L.; Neves, C.; Gonçalves, S.; Carvalho, C.; Arslanagic, A.; et al. Methylglyoxal-induced glycation changes adipose tissue vascular architecture, flow and expansion, leading to insulin resistance. Sci. Rep. 2017, 7, 1698. [CrossRef]
- Jia, X.; Olson, D.J.; Ross, A.R.; Wu, L. Structural and functional changes in human insulin induced by methylglyoxal. FASEB J. 2006, 20, 1555–1557. [CrossRef]
- Fiory, F.; Lombardi, A.; Miele, C.; Giudicelli, J.; Beguinot, F.; Van Obberghen, E. Methylglyoxal impairs insulin signalling and insulin action on glucose-induced insulin secretion in the pancreatic beta cell line INS-1E. Diabetologia 2011, 54, 2941–2952. Erratum in: Diabetologia 2012, 55, 272. [CrossRef]
- Bo, J.; Xie, S.; Guo, Y.; Zhang, C.; Guan, Y.; Li, C.; Lu, J.; Meng, Q.H. Methylglyoxal Impairs Insulin Secretion of Pancreatic β-Cells through Increased Production of ROS and Mitochondrial Dysfunction Mediated by Upregulation of UCP2 and MAPKs. J. Diabetes Res. 2016, 2016, 2029854. [CrossRef]
Figure 1.
Multifactorial routes leading to MASLD development. Poor life style associated with unhealthy diet and low mobility leads to insulin resistance which impairs energy metabolism in adipose tissue, skeletal muscles and the liver. This stimulates pro-oxidative and pro-inflammatory processes which disturb intracellular organelles in these organs. Pathological events are mediated by MGO originated from impaired carbohydrate/lipid metabolism. MGO, either through the direct damage to macromolecules (e.g., AMPK), or the induction of advanced glycation end products receptor (RAGE) signaling pathways accelerates pro-oxidative/pro-inflammatory routes as well as diverts metabolic pathways from catabolic into anabolic reactions. These pathological routes impair intracellular homeostasis (damage endoplasmic reticulum, lysosomes and mitochondria), thus inducing cell death (via apoptosis, necroptosis, pyroptosis and ferroptosis). Finally, the liver (and other organs) dysfunction develops leading to steatosis, steatohepatitis and cirrhosis. Due to the similar pathological background, MASLD is coupled with the co-existence of other disorders such as obesity, metabolic syndrome (MetS), type 2 diabetes mellitus (T2DM), cardiovascular diseases (CVD) and cancer. These pathological events are exacerbated by dysbiosis and increased gut permeability which are associated with an influx of toxins to the liver. Therefore, except for the main MGO pool generated in the liver from an excessive supply of fructose, glucose, glycerol and fatty acids, also MGO derived from diet or gut microbiota metabolism may additionally contribute to detrimental effects exerted by this molecule in the liver.
Figure 1.
Multifactorial routes leading to MASLD development. Poor life style associated with unhealthy diet and low mobility leads to insulin resistance which impairs energy metabolism in adipose tissue, skeletal muscles and the liver. This stimulates pro-oxidative and pro-inflammatory processes which disturb intracellular organelles in these organs. Pathological events are mediated by MGO originated from impaired carbohydrate/lipid metabolism. MGO, either through the direct damage to macromolecules (e.g., AMPK), or the induction of advanced glycation end products receptor (RAGE) signaling pathways accelerates pro-oxidative/pro-inflammatory routes as well as diverts metabolic pathways from catabolic into anabolic reactions. These pathological routes impair intracellular homeostasis (damage endoplasmic reticulum, lysosomes and mitochondria), thus inducing cell death (via apoptosis, necroptosis, pyroptosis and ferroptosis). Finally, the liver (and other organs) dysfunction develops leading to steatosis, steatohepatitis and cirrhosis. Due to the similar pathological background, MASLD is coupled with the co-existence of other disorders such as obesity, metabolic syndrome (MetS), type 2 diabetes mellitus (T2DM), cardiovascular diseases (CVD) and cancer. These pathological events are exacerbated by dysbiosis and increased gut permeability which are associated with an influx of toxins to the liver. Therefore, except for the main MGO pool generated in the liver from an excessive supply of fructose, glucose, glycerol and fatty acids, also MGO derived from diet or gut microbiota metabolism may additionally contribute to detrimental effects exerted by this molecule in the liver.
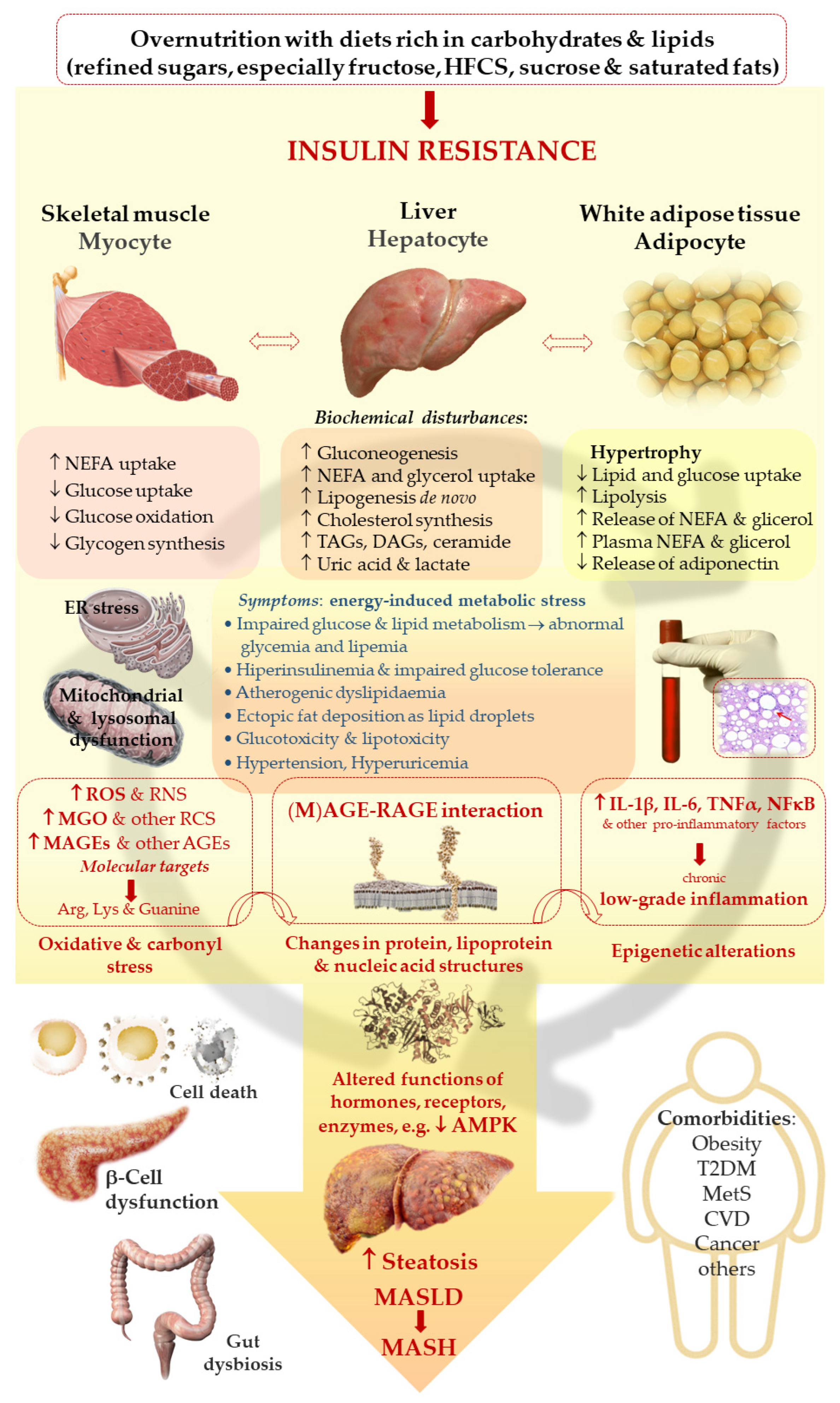
Figure 2.
The sources and fates of methylglyoxal (MGO). The main route of MGO generation and metabolism is shown in the center, visualized by the widest arrows. Glucose and fructose metabolites are the major MGO source, and glyoxalases 1 and 2 (Glo 1&2) comprise the main MGO scavenging system. However, in metabolic disturbances initiated by insulin resistance, lipid sources probably contribute significantly to the hepatic MGO generation. They include glycerol (excessively entering the liver due to enhanced lipolysis in white adipose tissue—WAT), as well as (keto)aldehydes—products of lipid peroxidation which increases due to oxidative stress. Also, the activity of glyoxalases, sometimes impaired in pathological conditions, might be compensated for by NADPH-dependent dehydrogenases (NADPH-dependent DHs), as well as aldoketo reductases (AKRs). Substrates and routes leading to MGO synthesis are shown in purple. Enzymatic systems involved in MGO scavenging and the final products of MGO metabolism are depicted in green.
Figure 2.
The sources and fates of methylglyoxal (MGO). The main route of MGO generation and metabolism is shown in the center, visualized by the widest arrows. Glucose and fructose metabolites are the major MGO source, and glyoxalases 1 and 2 (Glo 1&2) comprise the main MGO scavenging system. However, in metabolic disturbances initiated by insulin resistance, lipid sources probably contribute significantly to the hepatic MGO generation. They include glycerol (excessively entering the liver due to enhanced lipolysis in white adipose tissue—WAT), as well as (keto)aldehydes—products of lipid peroxidation which increases due to oxidative stress. Also, the activity of glyoxalases, sometimes impaired in pathological conditions, might be compensated for by NADPH-dependent dehydrogenases (NADPH-dependent DHs), as well as aldoketo reductases (AKRs). Substrates and routes leading to MGO synthesis are shown in purple. Enzymatic systems involved in MGO scavenging and the final products of MGO metabolism are depicted in green.
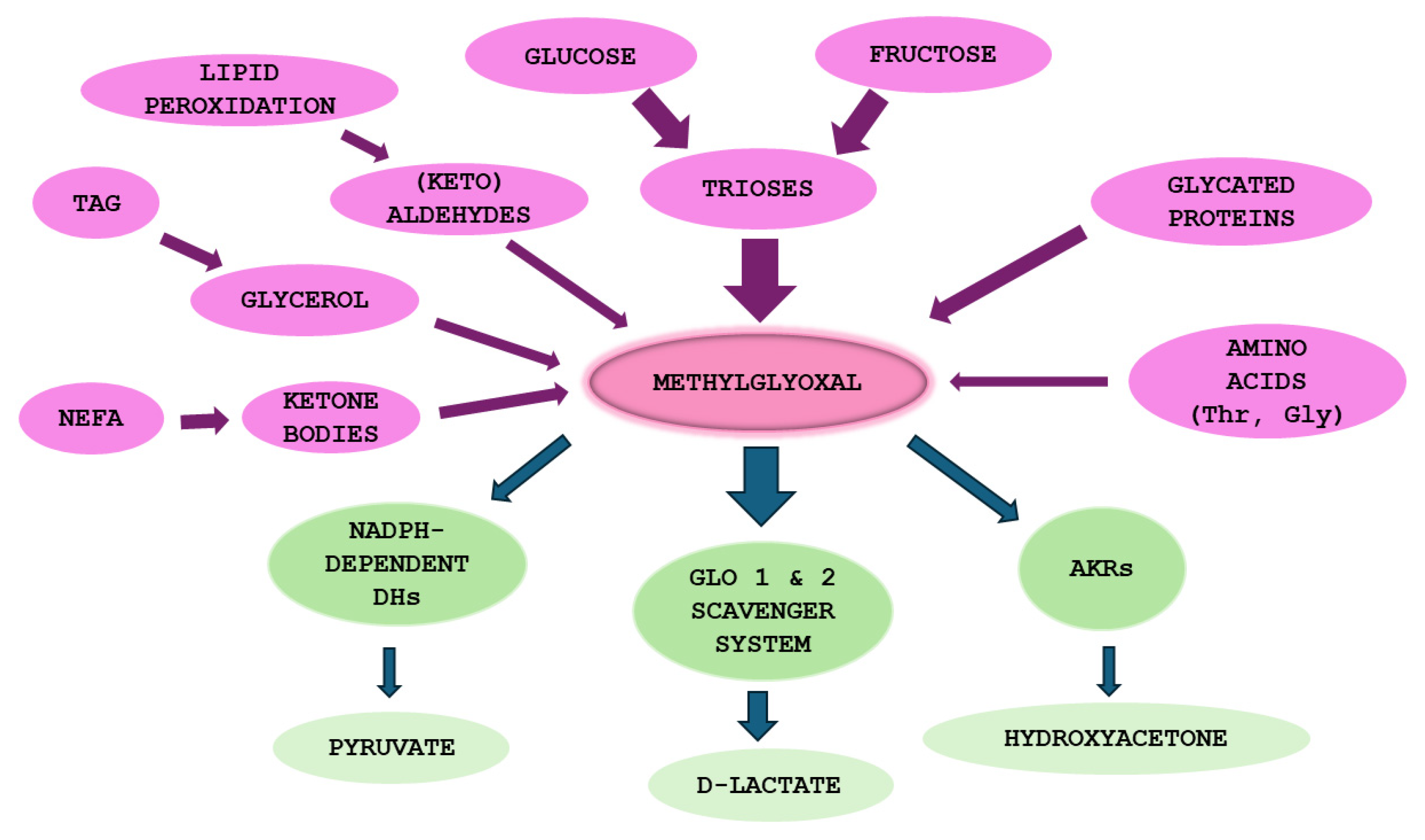
Figure 3.
A possible pathway of insulin resistance induction by Fru-derived MGO in the liver [79]. MGO accumulation in hepatocytes causes activation of kinases cascade which leads to the inhibition of tyrosine phosphorylation on insulin receptor substrate paralleled by enhanced serine phosphorylation. This impairs insulin-triggered signaling cascade which results in insulin resistance development. MKK7, mitogen-activated protein kinase kinase 7; JNK, c-jun NH2-terminal kinase; IRS-TyrP, insulin receptor substrate phosphorylated at tyrosine; IRS-SerP, insulin receptor substrate phosphorylated at serine; IR, insulin resistance.
Figure 3.
A possible pathway of insulin resistance induction by Fru-derived MGO in the liver [79]. MGO accumulation in hepatocytes causes activation of kinases cascade which leads to the inhibition of tyrosine phosphorylation on insulin receptor substrate paralleled by enhanced serine phosphorylation. This impairs insulin-triggered signaling cascade which results in insulin resistance development. MKK7, mitogen-activated protein kinase kinase 7; JNK, c-jun NH2-terminal kinase; IRS-TyrP, insulin receptor substrate phosphorylated at tyrosine; IRS-SerP, insulin receptor substrate phosphorylated at serine; IR, insulin resistance.
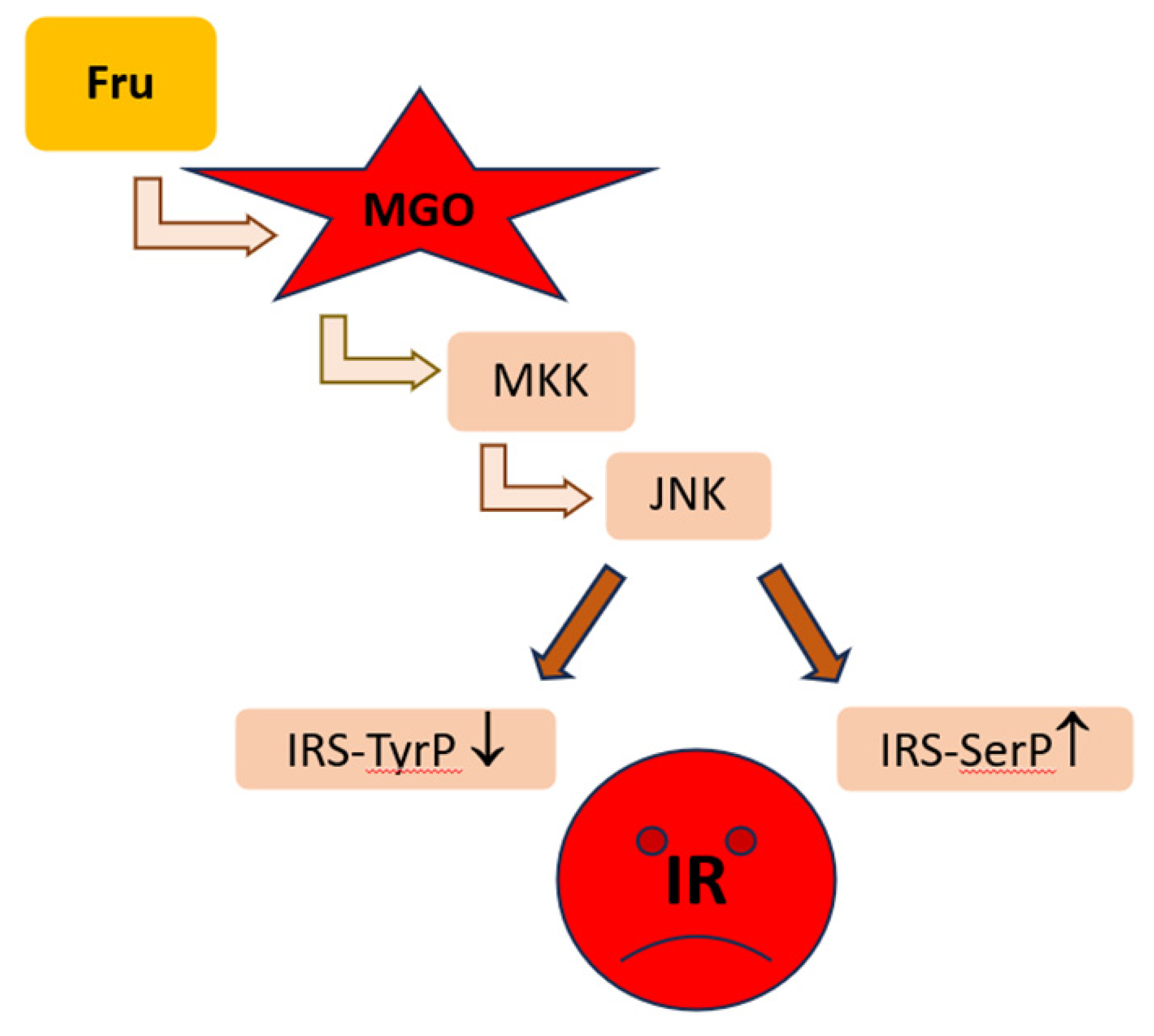
Figure 4.
Effects associated with MGO involvement in early MASLD [78,79,82,84,85], liver cirrhosis [89,93] and liver cancer [90,91,92]. Whereas a detrimental MGO impact in early MASLD is well documented, its function in liver cirrhosis and liver cancer is not clear due to the scarcity of available data.
Figure 4.
Effects associated with MGO involvement in early MASLD [78,79,82,84,85], liver cirrhosis [89,93] and liver cancer [90,91,92]. Whereas a detrimental MGO impact in early MASLD is well documented, its function in liver cirrhosis and liver cancer is not clear due to the scarcity of available data.
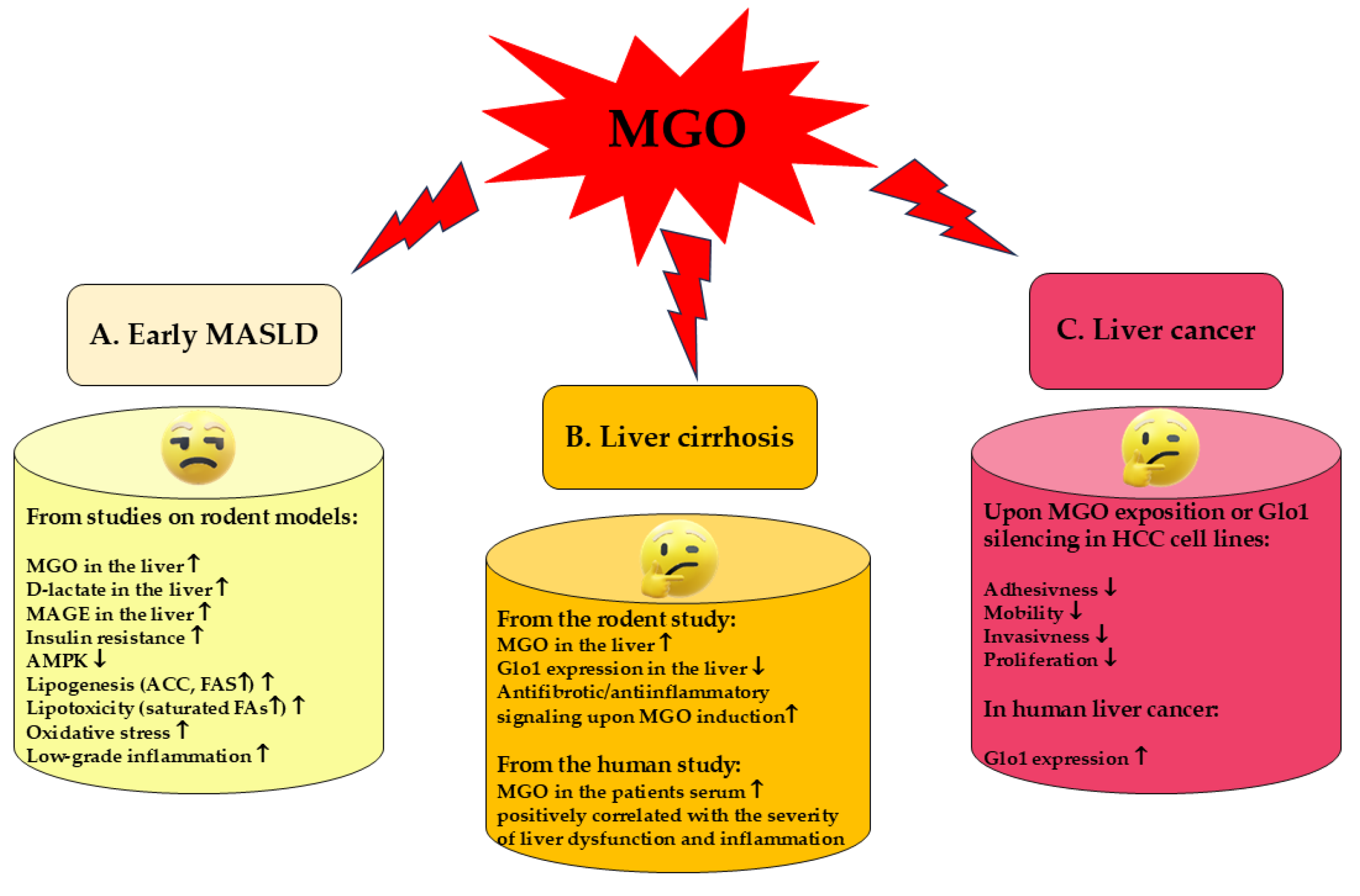
Figure 5.
General information on diagnosis and treatment of MASLD; Abbreviations: Adipo-IR, adipose tissue insulin resistance; BMI, body mass index; DPP-4, dipeptidyl peptidase-4; GIP, glucose-dependent insulinotropic polypeptide; GLP-1, glucagon-like peptide-1; HbA1c, glycated hemoglobin; HDL, high-density lipoprotein; HFCS, high-fructose corn syrup; HMG CoA reductase, 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase; HOMA-IR, homeostatic model assessment of insulin resistance; IR, insulin resistance; LDL, low-density lipoprotein; MetS, metabolic syndrome; NEFA, non-esterified fatty acids; OGTT, oral glucose tolerance test; SGLT2, sodium-glucose co-transporter 2; TAGs, triacylglycerols; T2DM, type 2 diabetes mellitus.
Figure 5.
General information on diagnosis and treatment of MASLD; Abbreviations: Adipo-IR, adipose tissue insulin resistance; BMI, body mass index; DPP-4, dipeptidyl peptidase-4; GIP, glucose-dependent insulinotropic polypeptide; GLP-1, glucagon-like peptide-1; HbA1c, glycated hemoglobin; HDL, high-density lipoprotein; HFCS, high-fructose corn syrup; HMG CoA reductase, 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase; HOMA-IR, homeostatic model assessment of insulin resistance; IR, insulin resistance; LDL, low-density lipoprotein; MetS, metabolic syndrome; NEFA, non-esterified fatty acids; OGTT, oral glucose tolerance test; SGLT2, sodium-glucose co-transporter 2; TAGs, triacylglycerols; T2DM, type 2 diabetes mellitus.

Figure 6.
Potential MGO adducts with metformin and quercetin; M, molecular mass.
Figure 7.
PRISMA flowchart of included studies and registries.
Figure 8.
Hypothetical route starting from an excessive production and accumulation of MGO in the liver, and leading to the development of insulin resistance, which further accelerates MGO generation. The pathological processes characteristic for MASLD and its comorbidities and partially mediated by MGO include: glycation of structurally and functionally vital macromolecules, which disturbs mitochondria, lysosomes and endoplasmic reticulum. Subsequently, oxidative stress, pro-inflammatory and pro-fibrotic (?) processes are enhanced, which leads to cell death and liver tissue damage. MGO contribution to IR development may be exerted through the modification of insulin, insulin receptor and insulin receptor substrate (as well as other down-stream signaling components). Furthermore, other signal transduction pathways can be impaired due to MGO-modification of their components. A possible intracellular signaling cascade disturbed by MGO can be AMPK. AMPK inhibition leads to the shift from catabolic into anabolic reactions which stimulates lipogenesis and inhibits FAs oxidation. This causes lipid accumulation contributing to hepatosteatosis. Apart from a direct effect of MGO on macromolecules, it also stimulates pro-oxidative and pro-inflammatory pathways through the induction of RAGE.
Figure 8.
Hypothetical route starting from an excessive production and accumulation of MGO in the liver, and leading to the development of insulin resistance, which further accelerates MGO generation. The pathological processes characteristic for MASLD and its comorbidities and partially mediated by MGO include: glycation of structurally and functionally vital macromolecules, which disturbs mitochondria, lysosomes and endoplasmic reticulum. Subsequently, oxidative stress, pro-inflammatory and pro-fibrotic (?) processes are enhanced, which leads to cell death and liver tissue damage. MGO contribution to IR development may be exerted through the modification of insulin, insulin receptor and insulin receptor substrate (as well as other down-stream signaling components). Furthermore, other signal transduction pathways can be impaired due to MGO-modification of their components. A possible intracellular signaling cascade disturbed by MGO can be AMPK. AMPK inhibition leads to the shift from catabolic into anabolic reactions which stimulates lipogenesis and inhibits FAs oxidation. This causes lipid accumulation contributing to hepatosteatosis. Apart from a direct effect of MGO on macromolecules, it also stimulates pro-oxidative and pro-inflammatory pathways through the induction of RAGE.
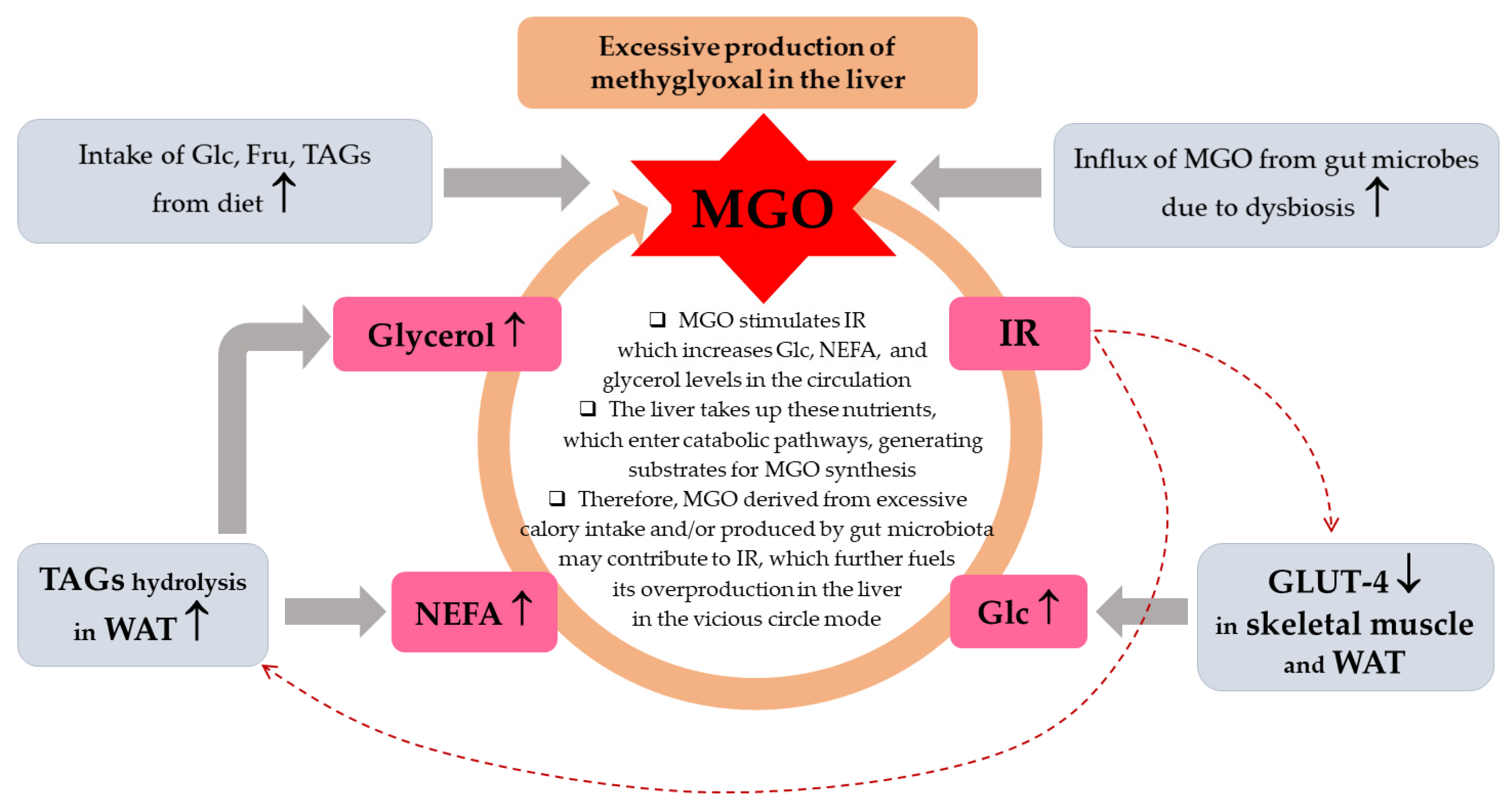

Table 1.
Methylglyoxal and glyoxalase 1 in MASLD.
| Experimental Model | Detailed Observations | Major Findingsin the Liver Tissue/Cells | Ref./Year |
|---|---|---|---|
| Early MASLD | |||
| Seven-week-old male Wistar rats (WR) divided into 2 groups: (1) WR injected with (0.3 mL/kg/week of 40%) CCl4 (in soy bean oil) for 4 weeks. (2) WR injected with the same volume of soybean oil (control group) |
In the serum (as compared with control group): increase in D-lactate; no change in AST, ALT and MGO concentration. In the liver: increase in MGO level and D-lactate. In the urine: increase in D-lactate; no change in MGO concentration. |
In the model of early MASLD: MGO and D-lactate in the liver ↑ |
[78]/2018 |
| (1) 6-month-old male hereditary hypertriglyceridemic rats (HHTg) as the non-obese prediabetic model treated or not-treated with salsalate (2) WR as the control group |
In HHTg rats (in comparison with WR and attenuated by salsalate); In the liver: increase in MGO, TAGs, Chol; increase in oxidative stress (TBARS ↑, GSH/GSSG ↓, SOD ↓). Upon salsalate treatment in HHTg: increased expression of Glo1 gene associated with MGO decrease. |
In the model of hypertriglyceridemia/prediabetes: MGO, lipids and oxidative stress in the liver ↑ |
[84]/2023 |
| Female Wistar rats (WR) divided into 2 groups: (1) Ovariectomized WR used as a model of postmenopausal MetS (W-OVX); (2) Sham-operated WR as a control (W-sham) |
In W-OVX rats (in comparison with W-sham rats); In the serum: increase in leptin, FAs, HDL-Chol, MCP-1; no change in TAGs and Chol. In the liver: increase in MGO and TAG; no change in Glo1 (mRNA and activity) and Chol; increase in oxidative stress (TBARS ↑, GSH/GSSG ↓, GPx ↓); In the muscle: increase in TAGs. |
In the model of postmenopausal MetS: MGO, TAGs and oxidative stress in the liver ↑ |
[85]/2021 |
| Male WR divided into four groups: (1) control (Ct) with standard diet A03 (5% triglycerides and 45% carbohydrates) (2) methylglyoxal group (MG) with standard diet and MGO administration (rats fed 75 mg MGO kg-1 daily for 18 weeks) (3) high-fat diet-fed group (HFD) (40% triglycerides and 10% carbohydrates) (4) high-fat diet group with MGO supplementation (rats fed 75 mg MGO kg-1 daily for 18 weeks) (HFDMG) |
Effect of MGO supplementation (HFDMG group compared to control and/or MG or HFD rats); In blood plasma: increase in NEFA; decrease in albumin; decrease in adiponectin (as compared to raised adiponectin in HFD). In the liver: increase in inflammatory cells (F4/80 ↑—a marker of macrophages/Kupffer cells); increase in MAGEs (MG-H1 ↑, CEL ↑, but ArgP=); decrease in insulin receptor phosphorylation at Tyr1163; decrease in phosphorylation of ACC (ACC activity ↑);decrease in phosphorylation of AMPK (AMPK activity ↓) decrease in cardiolipin 70:2; increase in expression of FAS and AceCS; no change in membrane RAGE; no change in Glo1 expression (but Glo1 activity ↑ in MG; Glo1 activity ↓ in HFDMG) |
Upon MGO supplementation in the liver: inflammation, MAGE, IR, ACC ↑ AMPK ↓ IR ↑ |
[82]/2019 |
| Male C57BL/6J mice divided into 8 groups: Study 1 (mice fed for 16 weeks with): (1) low-fat diet (10% fat energy) (LF) (2) very-high-fat diet (60% fat energy) (VHF) (3) very-high-fat diet with 0.25% genistein (VHF-G). Study 2 (mice fed for 18 weeks with): (4) low-fat diet (10% fat energy) (LF) (5) moderately high-fat diet (HF) (6) moderately high-fat diet with MGO (110–145 mg/kg/day) (HFM) (7) moderately high-fat diet with MGO and 0.067% genistein (HFM-GL) (8) moderately high-fat diet with MGO and 0.2% genistein (HFM-GH) |
Genistein effect (VHF-G vs. VHF and HFM-GH vs. HFM); In blood plasma: decrease in MGO, AGEs, Glc, Chol, ALT, AST. In the liver and kidney: decrease in AGEs; increase in Glo1/2 and aldose reductase expression; decrease in RAGE expression. In the liver: decrease in TAGs level. |
Upon genistein supplementation in the liver Glo1/2, TAGs, RAGE and AGEs ↓ |
[86]/2019 |
| (1) Primary rat hepatocytes (isolated from WR) (PRH) incubated with Glc (8 mM) and inulin (0.12%) with or without inulinase in the absence or presence of insulin for up to 4 h. (2) PRH incubated with Glc (8 mM) and inulin (0.12%) and MGO (20 µM) in the absence or presence of insulin for 4 h. |
Effects of Fru delivery in PRH (in comparison with PRH exposed only to Glc): around 2-fold increase in MGO in hepatocytes. Effects of Fru delivery or MGO exposition in PRH: increase in phosphorylation of MKK7, JNK and serine307 of IRS-1 (in the absence and presence of insulin); decrease in insulin-stimulated tyrosine phosphorylation of IRS-1 and IRS-2. |
Upon Fru treatment in rat hepatocytes: MGO ↑ Upon Fru/MGO treatment in rat hepatocytes: IR ↑ |
[79]/2013 |
| Liver cirrhosis | |||
| (1) Male WR treated with CCl4 and phenobarbital for 8 weeks (early cirrhosis without ascites) or 12–14 weeks (advanced cirrhosis with ascites) (2) Male WR treated with CCl4 for 12–14 weeks, and Glo1 inhibitor (ethyl pyruvate—EP) starting from week 8. Primary rat hepatocytes (pHEP), primary hepatic stellate cells (pHSC) and primary liver sinusoidal endothelial cells (pLSEC) isolated from control and cirrhotic WR Normal hepatic stellate cells (HSZ-B-S1) |
In comparison with pHEP: decreased Glo1 expression in pHSC and pLSEC derived from control WR. In the whole liver, and pHEP, pHSC and pLSEC in cirrhosis (in comparison with healthy WR): decreased Glo1 expression (and lower in advanced cirrhosis as compared to early cirrhosis). In pHSC and pLSEC in cirrhosis (in comparison with healthy WR): decreased Glo1 activity. In the whole liver and pHEP in cirrhosis (in comparison with healthy WR): increased Glo1 activity In the whole liver in cirrhosis (in comparison with healthy WR): increased MGO level (and higher increase in advanced cirrhosis as compared to early cirrhosis) Upon LPS induction of HSZ-B-S1: increase in Glo1 activity. Upon EP or MGO treatment of LPS-induced HSZ-B-S1: decrease in TNF-α, collagen-I and α-SMA. Upon EP treatment of LPS-induced HSZ-B-S1: decrease in LPS-induced NF-κB stimulation, inhibition of LPS-induced reduction of Nrf2, reduction of LPS-induced pERK, no effect on ERK expression. Effect of EP treatment on cirrhotic WR (compared to cirrhotic livers without EP treatment): reduction of fibrotic tissue, reduction of α-SMA, TGF-β, NF-κB expression, increase in Nrf2 expression. |
In the model of cirrhosis: in the liver MGO ↑ in the liver and liver cells Glo1 expression ↓ in the liver and hepatocytes Glo1 activity ↑ |
[89]/2017 |
| Hepatocellular carcinoma | |||
| Human HCC cell lines: Huh-7, HepG2 and Hep3B. |
Effect of 1 µM MGO on Huh-7 and HepG2 cells (but not Hep3B): inhibition of cells adhesion to collagen, and invasion through Matrigel (via promoting p53 localization in the nucleus by MGO). |
Upon MGO treatment: attenuation of cancer cells invasiveness |
[90]/2013 |
| Human HCC cell lines: Hep3B, SK-HEP-1 and SMMC-7721 |
Effect of Glo1 knock-down in Hep3B, SK-HEP-1 and SMMC-7721 cell lines: inhibition of the cells proliferation; Effect of Glo1 over-expression in Hep3B, SK-HEP-1 and SMMC-7721 cell lines: no effect on the cells growth. |
Upon Glo1 silencing: inhibition of cancer cells proliferation |
[91]/2014 |
| Human HCC cell lines: Huh-7 and HepG2 Murine hepatocyte cell line AML12 |
In comparison with normal AML12 cells: Glo1 up-regulation in Huh-7 cells (at mRNA, protein and activity levels); Glo1 up-regulation in HepG2 cells (only at mRNA level); Effects of Glo1 inhibition in Huh-7 cells (by 1–20 mM ethyl pyruvate or 1–10 µM BrBzGSHCp2): reduction of proliferation, migration and colony formation; decreased expression of PDGFR-β, VEGFR2, VEGF, pERK/ERK, NF-κB; increased expression of Nrf2 Effects of 2.5–10 µM sorafenib (a multi-tyrosine kinase inhibitor approved for the therapy of advanced HCC): increase in Glo1 and MGO. |
Upon Glo1 silencing: inhibition of cancer cells proliferation, migration and invasiveness |
[92]/2019 |
| Human HCC cell line HepG2 incubated with palmitic or oleic acids for 24 h. | Glo1 expression: decrease in oleic acid treated HepG2 cells. MGO concentration: increase in both palmitic and oleic acids treated HepG2 and their culture media. |
Upon FAs treatment of hepatoma cells: MGO ↑ Glo1 ↓ |
[87]/2018 |
“↑”—increased levels; “↓”—decreased levels; “=”—no change.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



