Submitted:
13 January 2025
Posted:
14 January 2025
You are already at the latest version
Abstract
Structural virology has emerged as the foundation for the development of effective antiviral therapeutics. It is pivotal in providing crucial insights into the three-dimensional frame of viruses and viral proteins at atomic or near atomic-level resolution. Structure-based assessment of viral components, including capsids, envelope proteins, replication machinery, and host interaction interfaces, are instrumental in unravelling the multiplex mechanisms of viral infection, replication, and pathogenesis. The structural elucidation of viral enzymes, including proteases, polymerases, and integrases, has been essential in combating viruses like HIV, SARS-CoV-2, and influenza. Techniques including X-ray crystallography, Nuclear Magnetic Resonance spectroscopy, Cryo-electron Microscopy, and Cryo-electron Tomography have revolutionized the field of virology and significantly aided in the discovery of antiviral therapeutics. The ubiquity of chronic viral infections, along with the emergence and re-emergence of new viral threats necessitate the development of novel antiviral strategies and agents, while the extensive structural diversity of viruses and their high mutation rates further underscore the critical need for structural analysis of viral proteins to aid antiviral development. This review highlights the significance of structure-based investigations for bridging the gap between structure and function, thus facilitating the development of effective antiviral therapeutics, vaccines, and antibodies for tackling emerging viral threats.
Keywords:
Structural virology
; antiviral therapeutics
; viral proteins
; viral replication enzymes
; X-ray crystallography
; nuclear magnetic resonance (NMR)
; cryo-EM
; emerging and re-emerging viruses
; rational drug design
; bioinformatics
1. Introduction
Viruses constitute a diverse group of sub-microscopic infectious agents that are reliant on the host cells' metabolism to replicate. They lack cellular structures and possess a genome composed of deoxyribonucleic Acid (DNA) or ribonucleic acid (RNA), which can be single-stranded or double-stranded. The genome can range from 3000 to over 1,000,000 nucleotides, and virus size can vary from 10 to 1000 nm [1] (Figure 1). According to the International Committee on Taxonomy of Viruses (ICTV), there are 314 families of viruses as of 2024 [2]. Viruses replicate via a series of complex steps, including initial attachment to the host cell, followed by entry, subsequent uncoating, genome replication, protein synthesis, virion assembly, and release of viral particles. Viral infection can thereby hijack the cellular machinery and disrupt cellular metabolism. These perturbations can manifest as a broad range of pathological outcomes for the host organism and even death [1]. Owing to the wide variety of viruses and the diversity of hosts, including bacteria, blue-green algae, fungi, plants, insects, and vertebrates, viruses pose a threat of infection across the three cellular domains of life- Archaea, Bacteria, and Eukarya.
Archaeal viruses can be broadly divided into archaea-specific viruses and cosmopolitan archaeal viruses, classified into 12 and 5 families, respectively. Most known archaeal viruses have been isolated from extreme environments, from hyperthermophiles or hyperhalophiles. These viruses are known to encode anti-clustered regularly interspaced short palindromic repeats (CRISPR) proteins. It is suggested that the viruses play a significant role in ocean biogeochemical cycling [3,4]. Bacteriophages infect bacteria and exhibit ubiquitous distribution in the environment and diverse genomes. They exhibit lytic or lysogenic life cycles and can facilitate horizontal gene transfer, playing a crucial role in microbial ecology and evolutionary dynamics. Temperate phages can form a mutually beneficial relationship with their host. Phages have played a significant role in developing several molecular biology techniques, including CRISPR-Cas (CRISPR-associated protein) system for genome editing. They have recently been utilized in phage display technology and as phage therapy to combat antimicrobial resistance [5,6]. Amongst eukaryotes, protists, including amoebae, ciliates, and flagellates, can be infected by protist-infecting viruses. Giant viruses (GVs) such as Mimivirus belonging to phylum Nucleocytoviricota have genome and particle sizes comparable to prokaryotes and small eukaryotes [7,8]. Algal viruses, such as chloroviruses and phaeoviruses infecting Chlorella and brown algae, respectively, influence host evolution via predator-prey selection and genetic exchange, thereby affecting host fitness and microbial community composition. The infections can lead to aquatic "viral shunt," i.e., alteration of organic matter composition and distribution [9]. Viruses that infect fungi are known as mycoviruses and are classified into 23 families and the genus Botybirnavirus. Mycoviruses infecting plant pathogenic fungi are the primary research focus due to their potential to act as biocontrol agents against the host fungi. They are reliant on hyphal anastomosis for intracellular spread and lack an extracellular transmission mechanism, limiting cross-strain spread [10]. Plant viruses pose a significant threat to agriculture and food security and can potentially cause pandemics and epidemics globally [11,12]. They are predominantly RNA viruses transmitted via vectors such as aphids, nematodes, whiteflies, fungi, or mechanical injury. Inside the plant host, they employ plasmodesmata and vasculature to spread internally, exhibiting symptoms such as mosaics, chlorosis, stunting, and wilting. Notable examples include Tobacco mosaic virus (TMV), Potato virus Y (PVY), and Cucumber mosaic virus (CMV) [11,12].
Animal viruses infect an extensive range of hosts, including vertebrates and invertebrates such as insects [1]. They display significant structural and genetic diversity and are categorized into several families. Insect viruses include families such as Baculoviridae and Iridoviridae, which are known to infect lepidopteran larvae and other insects, respectively [13]. These viruses can be vital for pest management and agriculture. Moreover, arthropods serve as vectors for the transmission of arboviruses, such as members of Flaviviridae, Togaviridae, and Nairoviridae, to other animals, including humans [14,15,16]. Infection with animal viruses can manifest into numerous disease pathologies, including localized and systemic infection in wild and domestic animals. Members of Orthomyxoviridae (e.g., influenza viruses) [17,18] and Rhabdoviridae (e.g., rabies virus- RABV) [19] have caused substantial disease burden and mortality. A subset of animal viruses is represented by human viruses, which include notable pathogens, such as members of Coronaviridae (e.g., Severe Acute Respiratory Syndrome Coronavirus 2- SARS-CoV-2) [20,21], Orthomyxoviridae (e.g., influenza viruses) [17,18], Herpesviridae (e.g., herpes simplex virus- HSV) [22,23,24,25], Papillomaviridae (e.g., human papillomavirus- HPV) [26,27,28], Retroviridae (e.g., human immunodeficiency virus- HIV) [29,30], and Picornaviridae (e.g., poliovirus) families [31,32]. Seven identified human oncoviruses, including Epstein-Barr virus (EBV), human T-cell leukemia virus type 1 (HTLV-1), hepatitis B virus (HBV), HPV, hepatitis C virus (HCV), Kaposi's sarcoma-associated herpesvirus (KSHV or HHV-8), and Merkel cell polyomavirus (MCV or MCPyV) account for causing an estimated 12-15% of cancers globally [33]. Additionally, the zoonotic spillover from animals to humans or reverse zoonosis from humans to animals, potentially facilitated by wildlife farming and trade, is of great concern [34,35]. Examples of zoonoses include rabies and avian influenza. All known coronaviruses (HCoVs) are believed to have originated in animals, with five of the seven HCoVs originating in bats [36]. Most recently, the Coronavirus disease 2019 (COVID-19) outbreak is believed to have transmitted from bats to humans. Therefore, adopting a One Health approach, considering human, animal and environmental health, for disease prevention and control is imperative.
Throughout history, viral infections have periodically emerged as epidemics and pandemics, resulting in significant loss of life. It has been estimated that there have been at least 14 influenza pandemics since 1500, including the Russian flu (1889-1893), Spanish flu (1918-1920), Asian flu (1957-1959), Hong Kong flu (1968-1970), and the first influenza pandemic of the 21st century, Swine flu (2009-2010) [37,38]. The ongoing HIV/ AIDS pandemic (1981-present) has claimed millions of lives [39,40]. Severe acute respiratory syndrome coronavirus (SARS-CoV) [41], Middle East respiratory syndrome coronavirus (MERS-CoV) [42], and SARS-CoV-2 [20,21] are distinct coronaviruses that emerged in 2002, 2012, and 2019, respectively. On 11 March 2020, the World Health Organization (WHO) declared COVID-19, caused by SARS-CoV-2, a pandemic, which raised an alarming situation and caused ~7 million fatalities worldwide [43]. Other significant outbreaks include Smallpox epidemics in the 17th century [44], polio epidemics in the 20th century [45], frequent outbreaks of Ebola [46], Dengue fever [47], yellow fever [48], Zika [49], Measles [50], Chikungunya [51], Japanese encephalitis [52], West Nile fever [53] and rabies [19,54]. As many RNA viruses are emerging and re-emerging viruses, they can evolve and reappear in the future with mutations [55,56]. The frequent viral outbreaks and lack of effective treatment and vaccination strategies underscore the urgent need to identify and develop antiviral therapeutics and advanced drug discovery for preparedness against future viral pandemics.
The study of three-dimensional (3D) structures of proteins has been recognized as crucial to expediting drug discovery. It offers insights into the shape of targets, hydrophobic and hydrophilic behaviours of macromolecules, and their interactions with substrates. Structural biology techniques are employed to study the key components of viruses, including structural proteins, replication proteins, and host interaction sites, thereby bridging the gap between viral structure and function, playing a pivotal role in shaping the development of antiviral therapies. The present review provides a comprehensive summary of structure-based investigations in the field of virology that lead to the identification and development of antiviral therapeutics and advanced drug discovery and explores the potential of structural virology in addressing emerging viral threats.
2. Exploring the 3D Protein Landscape: Structural Biology Techniques
Structural biology aims to understand the 3D structure of biological macromolecules, including proteins. These techniques have been employed in the study of viruses for close to a century. It not only furthers our understanding of life's molecular machinery but also enhances our ability to design targeted therapeutic interventions against disease. Researchers can gain valuable insights into biochemical activities and mechanisms by solving protein complex structures, which can be instrumental in drug design, and biotechnology. These methods have helped us visualize the molecular world and expose dynamic and transient stages of proteins [57]. The evolution of structural biology from basic chemical analysis to advanced imaging techniques mirrors scientific inquiry and technology. Chemical degradation and conventional optical microscopy initially provided limited information regarding molecular composition and structure. However, the discovery of X-ray crystallography by the pioneering work of Max von Laue and William Henry Bragg in the early 20th century provided atomic-level resolution and paved the way for structure-guided molecular biology [58,59].
2.1. X-ray Crystallography
X-ray crystallography has been instrumental in antiviral research by enabling the analysis of high-resolution atomic details of crystallized proteins and complexes by interpreting the diffraction patterns. [60,61,62]. TMV was the first virus to be crystallized by Wendell Stanley in 1935. It was demonstrated that the infectivity of the virus was retained in crystalline form. He was awarded the Nobel Prize in Chemistry in 1946 [63,64]. The virus structure was described in detail for the first time by Bernal and Fankuchen, who examined TMV suspension via X-ray diffraction [65]. The first atomic-resolution structure of a virus was provided in a pioneering study by Harrison et al., revealing an icosahedral arrangement of 180 capsid protein subunits of the Tomato bushy stunt virus (TBSV) at 2.9 Å resolution [66]. Parallelly, Aaron Klug and colleagues determined the structure of the TMV protein disk at a resolution of 2.8 Å [67] and revealed the structure of nucleosome core particle at 7 Å resolution [68]. Aaron Klug was awarded the Nobel Prize in Chemistry in 1982 for his development of crystallographic electron microscopy and his structural elucidation of biologically important nucleic acid-protein complexes [64]. With the development of therapeutics against HIV and HCV, X-ray crystallography became pivotal in antiviral and vaccine research in the late 20th century. Protein crystallization faces challenges such as limited solubility, unresolved protein dynamics, and chemical heterogeneity, complicating structural determination and drug discovery. Additionally, X-ray crystallography offers only static snapshots of molecules, potentially missing important dynamic interactions. The limitations of X-ray crystallography, especially its reliance on crystalline crystals, spurred further advancements [69,70]. Despite these limitations, the impact of X-ray crystallography on fields such as biochemistry, pharmacology, and virology has been profound. The primary advantage of the technique over others is its ability to provide highly detailed atomic resolution structures, essential for understanding the precise molecular mechanics of biological processes. It has enabled the detailed mapping of the interaction sites for drug molecules, providing a foundation for rational drug design and a deeper understanding of fundamental biological processes. Overall, X-ray crystallography remains a vital method in structural biology, complemented by newer techniques that provide insights into the structures of non-crystallizable molecules [71].
2.2. Nuclear Magnetic Resonance (NMR)
NMR spectroscopy was developed as a complement to study chemicals, including biomolecules in solution, revealing insights into their conformational flexibility. NMR spectroscopy has evolved from a chemical analysis tool to a fundamental technique in structural biology since the mid-20th century. Advances in technology, higher magnetic field strengths, and computational methods during the 1970s and 1980s allowed NMR to determine the structures of proteins in solution, providing dynamic molecular insights and study of dynamics and interactions within hosts [72]. The technique's ability to reveal protein dynamics and atomic interactions opened new avenues for the study of protein folding, enzyme activity and ligand interaction critical to viral pathogenesis and lifecycle, and eventually identifying antivirals to target the viral antigen proteins. NMR serves as a distinctive investigative tool for obtaining atom-resolved information regarding the structural and dynamic characteristics of highly flexible and disordered proteins, such as intrinsically disordered proteins (IDPs). In contrast to the more compact structures of globular protein domains, IDPs significantly influence NMR observables, necessitating the customization of NMR experiments for their study. In this context, 13C direct detection NMR has emerged as a valuable instrument for the characterization of IDPs/IDRs at an atomic resolution [73]. NMR is, therefore, uniquely suited for examining physiological states and complex biological processes. However, it is limited by its applicability mainly to smaller proteins (up to about 35 kDa), the requirement of large sample amounts, and extensive time [73]. Its spectral complexity demands high expertise for data interpretation, presenting challenges in high-throughput environments. Despite its limitations, NMR has profoundly impacted structural biology. The conjugation of X-ray crystallography and NMR aids in a deeper understanding of the protein structures [70]. NMR spectroscopy aids antiviral drug discovery by identifying ligand-protein interactions, optimizing drug properties, detecting false positives, and supporting multidisciplinary approaches. NMR spectroscopy can help accelerate the design of antiviral drugs. It has been instrumental in studying the HCV non-structural proteins, including protease, helicase, and polymerase, optimizing drug properties, and validating hits from screening and identifying peptidomimetics against HCV non-structural protein (NS3) serine protease [74].
2.3. Transmission electron microscopy (TEM)
Electron microscopy is used to visualize the ultrastructure of specimens using focused electron beams. Helmut Ruska made significant contributions to the field of virology by visualizing viruses in the 1930s. In the next decade, he detailed the sub-microscopic structures of various viruses, including poxviruses, TMV, varicella-zoster virus (VZV), and bacteriophages primarily employing TEM [75]. During the 1940s, TEM was employed for the diagnosis of smallpox and chicken pox [76]. In 1959, a negative staining method for high-resolution electron microscopy of viruses was developed [77]. TEM has since been used to understand viral structures, virus-host interaction studies, vaccine development, mutation monitoring, nanomedicine imaging, and diagnostics [78]. The challenges of TEM, such as uneven specimen staining and staining-induced distortions, were overcome when the first successful implementation of cryo-electron microscopy (cryo-EM) was reported.
2.4. Cryo-electron microscopy (cryo-EM)
Cryo-EM methodology acquires images of specimens cooled at cryogenic temperatures, aiding in the visualization of proteins, viruses, and complexes in their native state [79]. Its advent revolutionized the field of structural biology as it allowed the study of large protein complexes and fleeting protein states that are hard to crystallize. The technique gained popularity as it facilitates the visualization of biomolecules in their native, hydrated conformations and can achieve near-atomic resolutions comparable to X-ray crystallography without crystallization. With more sensitive detectors and better image processing tools, cryo-EM has become the standard for structural studies of viruses and components such as viral capsids, membrane proteins, and protein complexes [80]. This has provided invaluable insights into viral assembly, infection mechanisms, and interactions with host cells, directly impacting the development of antiviral drugs and vaccines [81]. One of the key advantages of Cryo-EM over other techniques is its ability to study complex and large biomolecular assemblies at near-atomic resolutions, enabling the study of membrane proteins, large protein complexes, and viruses. Additionally, Cryo-EM can capture snapshots of multiple conformational states of a molecule, providing a dynamic perspective on the functional mechanisms. For instance, the structure of RNA polymerase determined using Cryo-EM has advanced the understanding of the dynamic behaviour of the enzyme [82]. The technique is not without challenges, including the need for expensive, high-maintenance equipment and the requirement for significant computational resources to process large datasets. Moreover, achieving the highest resolutions often necessitates many images and averaging to obtain the 3D structures, which can be time-consuming to collect and analyze. Nevertheless, it enabled unprecedented molecular insights, facilitating structure-guided therapeutic design and driving continual advancements in deciphering intricate biological processes [83].
2.5. Small Angle X-ray Scattering (SAXS)
Building upon the principles of X-ray crystallography, SAXS offers a complementary approach by allowing the study of macromolecules in solution, providing insights into their size, shape, and conformational changes. Unlike X-ray crystallography, which requires the formation of crystals and primarily gives high-resolution static structures, SAXS can analyze samples that are difficult to crystallize and provides low-resolution data on flexible and dynamic assemblies in near-native conditions [84]. SAXS is particularly advantageous for examining large complexes and conducting rapid screenings of samples under various conditions, making it a valuable tool in cases where X-ray crystallography is not feasible. SAXS has been essential in studying IDPs, revealing 3D structures of aggregates and identifying different stages of protein aggregation due to their flexible domains and smaller size [85]. Conversely, for detailed atomic resolution structures necessary for precise molecular interactions, X-ray crystallography remains the superior technique [86].
2.6. Cryo-electron tomography (Cryo-ET)
Leaning on the capabilities of cryo-EM, cryo-ET helps study structural biology further by providing detailed 3D visualizations of cells and viruses in their native environment. While cryo-EM offers revolutionary insights into individual proteins and complexes, cryo-ET extends this by allowing scientists to examine the spatial organization and interactions within entire cells or tissues at near-atomic resolutions. In the early 2000s, cryo-ET provided the first visualization of HIV-1 envelope glycoproteins (Env) on the virion surface, revealing their unique tripod-like structure [87]. Subsequent studies have further elucidated Env's conformational dynamics, aiding in the design of broadly neutralizing antibodies and vaccines [88]. This makes Cryo-ET a superior technique for understanding complex viral infection mechanisms and cellular architecture dynamics, as it captures biological processes in situ without the need for sample sectioning or markers, providing a more comprehensive and realistic view of molecular biology [89].
2.7. Emerging Techniques
Together, these techniques complement each other, and ongoing and future advances in technologies such as X-ray Free Electron Laser (XFEL) imaging are broadening the horizons of structural biology as these may disclose new biomolecular behaviour, especially in cells and in reaction to inhibitors [90]. Advancements in computational biology have enabled simulations that predict protein folding and dynamics based on known sequences. Techniques such as molecular dynamics (MD) simulations complement experimental data by providing insights into conformational changes over time. AlphaFold 3 is the latest iteration of Google DeepMind's artificial intelligence (AI) tool that offers unparalleled accuracy in predicting 3D protein structures and complex biomolecular assemblies, including protein-nucleic acid and protein-small molecule complexes. In 2024, the Nobel Prize for Chemistry was awarded to David Baker for computational protein design, and to Demis Hassabis and John M. Jumper (Google DeepMind) for protein structure prediction [91].
5. Host-targeted antivirals:
Protein-protein interactions (PPIs) are fundamental to any virus infection. Detailed understanding of protein interactions is essential for understanding viral pathogenesis. Many host proteins act as receptors for viruses, mediating attachment and entry (Figure 2). C-C chemokine receptor type 5 (CCR5) and C-X-C chemokine receptor type 4 (CXCR4) act as co-receptors for the entry of HIV-1. Antagonists of these co-receptors are being utilized as antiviral strategies. Drug candidates like Aplaviroc, Cenicriviroc and Vicriviroc have shown efficacy in inhibiting HIV-1 replication. Available structural data for CCR5 and CXCR4 can prove to be valuable for improving inhibitor design and therapeutic potential [282,283,284]. Structure of the CCR5 chemokine receptor with FDA approved inhibitor, Maraviroc has been reported [285]. Ibalizumab, a CD4-directed post-attachment inhibitor was the first monoclonal antibody to be approved for the treatment of HIV-1 infection [286,287].
Additionally, glycosylation, autophagy, actin polymerization, fatty acid biosynthesis, programmed ribosomal frameshifting (PRF) and proteolytic cleavage are some of the host-mediated metabolic processes that can be targeted for antiviral development. Key pathways that can be targeted include host lipid pathway, host glycolytic pathways, host ubiquitination pathways, polyamine metabolic pathway, host nucleoside synthesis pathway, cytokine signalling and inflammatory pathways, and stress granule (SGs) machinery (Figure 3). [288,289]. SGs are assemblies of stalled mRNA and proteins that form in response to cellular stress, such as viral infection. Viral proteins like N-protein of SARS-CoV-2 and nsP3 of CHIKV in virus-infected cells recruit SG proteins G3BP1 (Ras GTPase-activating protein SH3-domain-binding protein 1) and G3BP2. This interaction suppresses SG formation, enhancing virus replication and assembly of new virions. Therefore, antivirals targeting the host protein G3BP can aid in SG-mediated antiviral response [290,291]. Viruses establish virus–host protein interactions to exploit cellular machinery essential for critical stages such as entry, genome replication, translation, assembly and release [292,293,294]. Increased resistance to antiviral agents and the need for broad-spectrum antiviral therapeutics have steered efforts to targeting proviral host factors and cellular mechanisms [288,295,296,297,298].
Viral infection can lead to the activation of various innate immune responses, with interferons (IFNs) playing a central role. IFNs, particularly IFN-α, have been utilized in antiviral therapies for infections like hepatitis B and C, with ongoing efforts to enhance therapeutic efficacy. Pegylated interferon afla 2b (PegIFNα-2b), interferon alfacon 1 (CIFN), pegylated interferon alfa 2b + ribavirin (PegIFNα-2b+RBV), pegylated interferon afla 2a (PegIFN-α2a) are some of the interferon therapeutics that are approved against HCV infections. Podofilox (PDX) is an antimitotic drug that interrupts cell division, approved for HPV-related diseases. Imiquimod (IQM) stimulates cytokines and sinecatechins (SINE) is an immunomodulatory drug approved against HPV-related diseases [61].
Cyclophilins (Cyps), key cellular factors playing role in transcription regulation, immune response, protein secretion, and mitochondrial function. Cyclophilin A (CypA), a mediator of cyclosporin A’s (CsA) immunosuppressive effects, also supports the replication of multiple viruses, including HIV-1, HCV, and influenza. Interaction of CypA with viral proteins, facilitates virus replication, as seen with HIV-1, HCV, influenza virus, HCV, VSV, vaccinia virus, SARS-CoV, rotavirus (RV), and HPV. Cyclophilin inhibitors, such as alisporivir and NIM811, are reported to display potent antiviral activity against HIV and HCV [270].
Figure 2.
Interactions Between Viral Families and Host Receptors with Structural Insights. The figure highlights key interactions between viral families and host receptors critical for viral attachment and entry. For adenoviruses, the Coxsackievirus and Adenovirus Receptor (CAR) (PDB: 1KAC), heparan sulfate (PDB: 5UF6), and sialylated glycoproteins (PDB: 6QU6) are key receptors. Coronavirus interactions include ACE-2 (PDB: 7VXM), integrins, heparan sulfate (PDB: 8XUT), sialic acid (PDB: 6NK), and the DC-SIGN receptor (PDB: 7NL6) [299,300]. Alphaviruses engage with heparan sulfate (PDB: 6ODF), C-type lectin receptors (PDB: 1K9I), and MXRA8 (PDB: 6JO8) [301]. Similarly, flaviviruses interact with heparan sulfate, DC-SIGN (PDB: 2B6B), and C-type lectin receptors [302,303]. Poxviruses target heparan sulfate (PDB: 3VOP) and C-type lectin receptors (PDB: 3K7B)[304]. PDB identifiers highlight viral protein-host receptor complexes, offering molecular-level insights into receptor diversity and viral entry mechanisms, supported by recent structural and experimental studies.
Figure 2.
Interactions Between Viral Families and Host Receptors with Structural Insights. The figure highlights key interactions between viral families and host receptors critical for viral attachment and entry. For adenoviruses, the Coxsackievirus and Adenovirus Receptor (CAR) (PDB: 1KAC), heparan sulfate (PDB: 5UF6), and sialylated glycoproteins (PDB: 6QU6) are key receptors. Coronavirus interactions include ACE-2 (PDB: 7VXM), integrins, heparan sulfate (PDB: 8XUT), sialic acid (PDB: 6NK), and the DC-SIGN receptor (PDB: 7NL6) [299,300]. Alphaviruses engage with heparan sulfate (PDB: 6ODF), C-type lectin receptors (PDB: 1K9I), and MXRA8 (PDB: 6JO8) [301]. Similarly, flaviviruses interact with heparan sulfate, DC-SIGN (PDB: 2B6B), and C-type lectin receptors [302,303]. Poxviruses target heparan sulfate (PDB: 3VOP) and C-type lectin receptors (PDB: 3K7B)[304]. PDB identifiers highlight viral protein-host receptor complexes, offering molecular-level insights into receptor diversity and viral entry mechanisms, supported by recent structural and experimental studies.
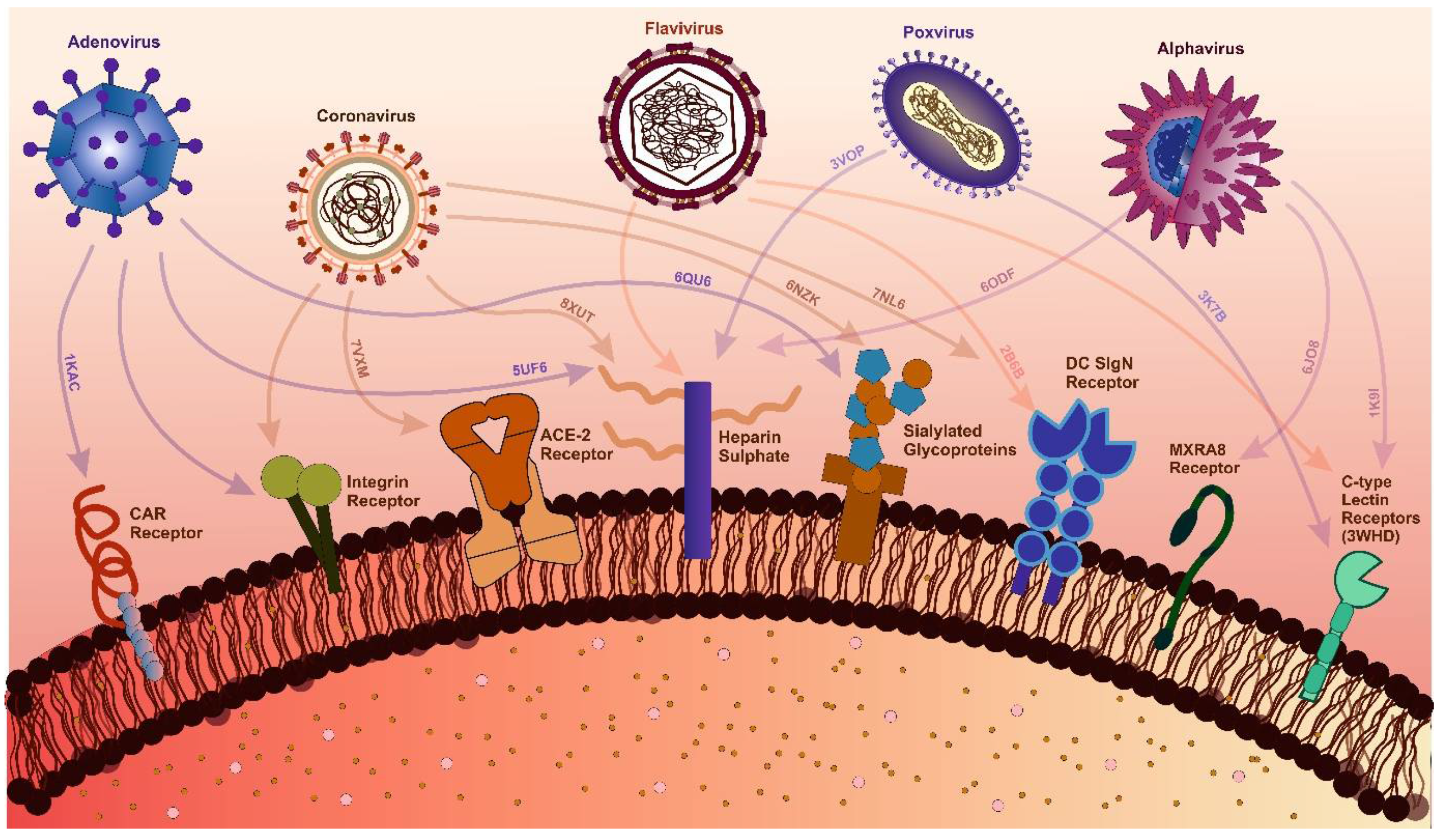
Figure 3.
Stress granule formation in response to viral infection. Viruses hijack host cell replication machinery, but antiviral molecules counteract this by promoting stress granule formation, inducing activation of Interferon-Stimulated Genes (ISGs), and producing Type I Interferons, directly inhibiting viral replication, thereby creating an antiviral environment that restricts viral survival and propagation.
Figure 3.
Stress granule formation in response to viral infection. Viruses hijack host cell replication machinery, but antiviral molecules counteract this by promoting stress granule formation, inducing activation of Interferon-Stimulated Genes (ISGs), and producing Type I Interferons, directly inhibiting viral replication, thereby creating an antiviral environment that restricts viral survival and propagation.

6. Rational Drug Design
Viral outbreaks, such as COVID-19 and Monkeypox, highlight the challenges posed by viral mutations that enhance immune escape and virulence, potentially leading to pandemics. Predicting the next mutation remains difficult [305]. Therefore, global preparedness is essential to counter future pandemics [306]. The pursuit of advanced, structure-guided treatments through AI-driven technologies is crucial to address viral mutations that could reduce therapeutic efficacy [307]. For instance, after the COVID-19 outbreak, it became evident that a strategic pipeline is required to identify antigenic sites, design non-cytotoxic ligands, and enable mass production and distribution of drugs [308]. Similarly, the Monkeypox outbreak reinforced the need to identify viral targets and develop specific drugs against them [309].
Visualization forms the foundation of rational drug design, enabling researchers to analyze viral structures [310]. This structural understanding facilitates precise drug design based on viral protein architecture, as demonstrated during the COVID-19 pandemic [311]. Unlike traditional methods, which relied on screening drugs for antiviral activity without prior molecular insight, rational drug design employs high-resolution structural techniques—X-ray crystallography, NMR spectroscopy, and cryo-EM—to study viral proteins and processes essential for replication [312]. The process begins by identifying a target viral protein critical to replication or host entry. Structural analyses then reveal active sites and binding pockets where small molecules can inhibit protein function [313]. Using this data, scientists design molecules tailored to fit these pockets, thereby blocking protein activity. This targeted strategy improves efficacy and minimizes side effects by avoiding interactions with non-target proteins.
Rational drug design combines structural and biochemical insights to create highly specific therapies, transforming antiviral treatment approaches. For example, the small molecule Nirmatrelvir targets SARS-CoV-2's main protease, showing high antiviral efficacy [314]. Similarly, analogues like spirolactam, derived from zamnair, improve antiviral potency, while peptides such as VIR250 selectively inhibit the papain-like protease of SARS-CoV-2 [315]. Nanoparticles, like the ICO-RBD nanovaccine, mimic virus-like structures to boost immunogenicity [316]. Future developments in rational drug design promise faster and more precise solutions for emerging pathogens, leveraging computational tools and structural biology advancements.
Rational drug design has transformed structural biology by providing a direct pathway from understanding a virus's atomic structure to developing effective antiviral therapies [317]. This approach has led to the creation of antiviral drugs for diseases such as HIV/AIDS and influenza, where structural studies of viral enzymes and surface proteins play a key role in designing inhibitors. Additionally, this method is vital for quickly developing treatments against emerging viral threats, as demonstrated by the rapid creation of SARS-CoV-2 spike protein inhibitors during the COVID-19 pandemic. AI has further accelerated this process by reducing screening times for antivirals, improving predictive models for binding affinity, and enabling data mapping to track trends in viral mutations and forecast future changes [318,319]. AI technologies have enhanced drug discovery by optimizing, generating, and identifying molecules with drug-like properties [320]. The need for such advancements in rational drug design is clear, as they support the rapid development of therapeutics to combat future pandemics. Ultimately, rational drug design not only deepens our understanding of drug mechanisms but also advances public health by enabling the swift creation of targeted treatments for both existing and emerging viral diseases [321].
6.1. De novo designing - a targeted approach with improved features
De novo protein design has emerged as a promising strategy to create molecules from scratch, contributing significantly to combating viral infections [322]. Computational pipelines can be developed and deployed for the streamlined development of antibody-based therapeutic interventions against emerging pathogens [323]. Applications include an in silico affinity maturation pipeline developed and employed to successfully bioengineer nanobodies with enhanced affinity [324]. With the integration of generative AI, it is now possible to engineer proteins targeting specific protein structures. This advancement also enables personalized therapeutic research tailored to individual patients [325,326]. Large language models, such as PALM-H3, further enhance these capabilities by generating antibodies using pre-trained models, allowing for the de novo synthesis of antibodies [327]. Beyond drug design, vaccines targeting viral proteins can also be developed to present surface glycoproteins and elicit immunization responses. Modelling tools such as SabPred assist further in modelling antibodies and in silico validation of structures [328]. For instance, HIV vaccines displaying envelope glycoproteins have demonstrated strong neutralizing titers, emphasizing the effectiveness of nanoparticle-based strategies [329]. Moreover, generative AI can be utilized to produce antibodies specific to target antigens, enhancing therapeutic applications [330].
Breakthroughs such as AlphaFold have revolutionized protein modeling, enabling the accurate prediction of protein structures [331]. These tools facilitate the identification of protein functions and their interactions with other molecules [332]. Further advancements, like RFdiffusion, simplify the design of protein binders that target specific antigen sites, eliciting neutralizing responses against viruses [333]. Various approaches taken for designing these antivirals are mentioned in Table 3. Rational approach has not only been evident in designing antivirals but antibody and vaccines designing too.
7. Identifying the Threat of Future – a structural approach:
Viruses pose a significant global health threat due to their potential to cause epidemics and pandemics. The WHO R&D Blueprint for Epidemics prioritizes identifying high-risk viruses for early detection, targeted research, efficient resource allocation, and global collaboration (WHO Pathogen Prioritization Report, 2024) [350] (Table 4). “Pathogen X”, an unknown future threat, underscores the need for preparedness. Identifying new pathogens involves robust surveillance, genomic sequencing, and epidemiological studies. Targeting Pathogen X requires focusing on pathogen families, studying prototype pathogens, fostering international collaboration, and investing in R&D [351]. Prioritizing high-risk viruses and preparing for Pathogen X enhances global response capabilities, ultimately saving lives and protecting public health (WHO Pathogen Prioritization Report, 2024) [350]. Having the knowledge of the threat, knowing their structures and with rational approach to designing therapeutics, there is hope to fight the upcoming pandemics with advanced knowledge of structural virology.
8. Conclusion and future direction
Structural virology has played an instrumental role in shaping our understanding of viral mechanisms and enabling the development of targeted therapeutics. Key techniques like X-ray crystallography, cryo-EM, and NMR spectroscopy have yielded high-resolution images of viral structures, revealing key molecular targets on enzymes, receptors, and structural proteins. With the ever-increasing data and research on virus-encoded proteins, structural biology has proven to be an indispensable tool for rational design and optimization of antiviral drugs. Additionally, computational methods have become integral to the early stages of drug discovery and development. The review delves into existing and new technological advancements to further the depth of structural understanding of viral proteins, interactions, and the strategies for antiviral therapeutic development.
Although notable strides have been made in the field, the structural data of many of the key proteins are yet to be elucidated. Therefore, it is imperative to develop strategies for obtaining structural data and establishing a link between structure and function. Covid-19 demonstrated the significance of pandemic preparedness to the world. Based on historical precedents of pandemics and epidemics that have plagued the world, debilitated global healthcare systems, and caused substantial mortality and lasting health impact. Covid-19 is unlikely to be the final pandemic. Therefore, continuous efforts from the scientific community to develop antiviral therapeutics against emerging pathogens and pathogens with pandemic potential is the need of the hour. Further investigations are crucial to identify druggable host factors for the development of broad-spectrum therapeutics.
Due to rapid rate of mutation, many viruses are emerging and reemerging pathogens. Mutation prediction, leveraging sequence and structural data along with surveillance data, are now critical in forecasting evolution, aiding to guide the design of adaptive therapeutic strategies that can keep up with the rapid rate of viral evolution. As witnessed during the SARS-CoV-2 pandemic, available and ongoing research to elucidate the protein structures expedited vaccine development, whereas the rapid identification of variants underscored the importance of anticipating viral evolution in real time. In the future, real-time structural surveillance, coupled with AI-powered tools, will be pivotal in swiftly identifying mutations and new pathogens. It will be essential to assess the impact of viral mutations on transmissibility and immune escape.
Author Contributions
Conceptualization, T.H., A.S., S.T.; writing—original draft preparation T.H., A.S, and S.T.; writing—review and editing, T.H., A.S., S.T., A.N., E.R., P.K., and J.S. All authors have read and agreed to the published version of the manuscript.
Acknowledgement
The authors PK and ST duly acknowledge the financial support from the Scheme for Transformational and Advanced Research in Sciences (STARS), Ministry of Education (MoE) (project reference no. STARS2/2023-0209) for supporting this study. A.S. thanks Prime Minister's Research Fellows (PMRF) scheme, MOE for research fellowship. T.H. acknowledges MHRD for research fellowship.
Conflicts of Interest
The authors declare that they have no competing interest concerning the publication of this review.
References
- Payne, S. Introduction to Animal Viruses. In Viruses; Elsevier, 2017; pp. 1–11.
- Current ICTV Taxonomy Release | ICTV Available online:. Available online: https://ictv.global/taxonomy (accessed on 27 December 2024).
- Wirth, J.; Young, M. The Intriguing World of Archaeal Viruses. PLoS Pathog 2020, 16. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Cvirkaite-Krupovic, V.; Iranzo, J.; Prangishvili, D.; Koonin, E. V. Viruses of Archaea: Structural, Functional, Environmental and Evolutionary Genomics. Virus Res 2018, 244, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Mahler, M.; Costa, A.R.; van Beljouw, S.P.B.; Fineran, P.C.; Brouns, S.J.J. Approaches for Bacteriophage Genome Engineering. Trends Biotechnol 2023, 41, 669–685. [Google Scholar] [CrossRef] [PubMed]
- Gordillo Altamirano, F.L.; Barr, J.J. Phage Therapy in the Postantibiotic Era. Clin Microbiol Rev 2019, 32. [Google Scholar] [CrossRef]
- Queiroz, V.F.; Tatara, J.M.; Botelho, B.B.; Rodrigues, R.A.L.; Almeida, G.M. de F.; Abrahao, J.S. The Consequences of Viral Infection on Protists. Communications Biology 2024 7:1 2024, 7, 1–15. [Google Scholar] [CrossRef]
- Mimiviruses: Giant Viruses with Novel and Intriguing Features (Review). Available online: https://www.spandidos-publications.com/10.3892/mmr.2022.12723 (accessed on 11 January 2025).
- Coy, S.R.; Gann, E.R.; Pound, H.L.; Short, S.M.; Wilhelm, S.W. Viruses of Eukaryotic Algae: Diversity, Methods for Detection, and Future Directions. Viruses 2018, 10, 487. [Google Scholar] [CrossRef]
- Hough, B.; Steenkamp, E.; Wingfield, B.; Read, D. Fungal Viruses Unveiled: A Comprehensive Review of Mycoviruses. Viruses 2023, 15, 1202. [Google Scholar] [CrossRef]
- Tatineni, S.; Hein, G.L. Plant Viruses of Agricultural Importance: Current and Future Perspectives of Virus Disease Management Strategies. Phytopathology 2023, 113, 117–141. [Google Scholar] [CrossRef]
- Jones, R.A.C.; Janssen, D. Global Plant Virus Disease Pandemics and Epidemics. Plants 2021, 10, 233. [Google Scholar] [CrossRef]
- Bertola, M.; Mutinelli, F. A Systematic Review on Viruses in Mass-reared Edible Insect Species. Viruses 2021, 13, 2280. [Google Scholar] [CrossRef]
- Garrison, A.R.; Alkhovsky, S. V.; Avšič-Županc, T.; Bente, D.A.; Bergeron, É.; Burt, F.; Paola, N. Di; Ergünay, K.; Hewson, R.; Kuhn, J.H.; et al. ICTV Virus Taxonomy Profile: Nairoviridae. Journal of General Virology 2020, 101, 798–799. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Becher, P.; Bukh, J.; Gould, E.A.; Meyers, G.; Monath, T.; Muerhoff, S.; Pletnev, A.; Rico-Hesse, R.; Smith, D.B.; et al. ICTV Virus Taxonomy Profile: Flaviviridae. Journal of General Virology 2017, 98, 2–3. [Google Scholar] [CrossRef] [PubMed]
- Bolling, B.G.; Weaver, S.C.; Tesh, R.B.; Vasilakis, N. Insect-Specific Virus Discovery: Significance for the Arbovirus Community. Viruses 2015, 7, 4911–4928. [Google Scholar] [CrossRef] [PubMed]
- Javanian, M.; Barary, M.; Ghebrehewet, S.; Koppolu, V.; Vasigala, V.K.R.; Ebrahimpour, S. A Brief Review of Influenza Virus Infection. J Med Virol 2021, 93, 4638–4646. [Google Scholar] [CrossRef]
- Hutchinson, E.C. Influenza Virus. 2018. [CrossRef]
- Brunker, K.; Mollentze, N. Rabies Virus. Trends Microbiol 2018, 26, 886–887. [Google Scholar] [CrossRef]
- Li, G.; Hilgenfeld, R.; Whitley, R.; De Clercq, E. Therapeutic Strategies for COVID-19: Progress and Lessons Learned. Nature Reviews Drug Discovery 2023, 22, 449–475. [Google Scholar] [CrossRef]
- Ciotti, M.; Ciccozzi, M.; Terrinoni, A.; Jiang, W.C.; Wang, C. Bin; Bernardini, S. The COVID-19 Pandemic. Crit Rev Clin Lab Sci 2020, 365–388. [Google Scholar] [CrossRef]
- Crimi, S.; Fiorillo, L.; Bianchi, A.; D’amico, C.; Amoroso, G.; Gorassini, F.; Mastroieni, R.; Marino, S.; Scoglio, C.; Catalano, F.; et al. Herpes Virus, Oral Clinical Signs and QoL: Systematic Review of Recent Data. Viruses 2019, Vol. 11, Page 463 2019, 11, 463. [Google Scholar] [CrossRef]
- Rechenchoski, D.Z.; Faccin-Galhardi, L.C.; Linhares, R.E.C.; Nozawa, C. Herpesvirus: An Underestimated Virus. Folia Microbiol (Praha) 2017, 62, 151–156. [Google Scholar] [CrossRef]
- Connolly, S.A.; Jardetzky, T.S.; Longnecker, R. The Structural Basis of Herpesvirus Entry. Nature Reviews Microbiology 2020 19:2 2020, 19, 110–121. [Google Scholar] [CrossRef]
- Agut, H.; Bonnafous, P.; Gautheret-Dejean, A. Laboratory and Clinical Aspects of Human Herpesvirus 6 Infections. Clin Microbiol Rev 2015, 28, 313–335. [Google Scholar] [CrossRef] [PubMed]
- de Sanjosé, S.; Brotons, M.; Pavón, M.A. The Natural History of Human Papillomavirus Infection. Best Pract Res Clin Obstet Gynaecol 2018, 47, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Burd, E.M.; Dean, C.L. Human Papillomavirus. Diagnostic Microbiology of the Immunocompromised Host 2016, 177–195. [Google Scholar] [CrossRef]
- Schiffman, M.; Doorbar, J.; Wentzensen, N.; De Sanjosé, S.; Fakhry, C.; Monk, B.J.; Stanley, M.A.; Franceschi, S. Carcinogenic Human Papillomavirus Infection. Nature Reviews Disease Primers 2016 2:1 2016, 2, 1–20. [Google Scholar] [CrossRef]
- Yoshimura, K. Current Status of HIV/AIDS in the ART Era. Journal of Infection and Chemotherapy 2017, 23, 12–16. [Google Scholar] [CrossRef]
- Bekker, L.G.; Beyrer, C.; Mgodi, N.; Lewin, S.R.; Delany-Moretlwe, S.; Taiwo, B.; Masters, M.C.; Lazarus, J. V. HIV Infection. Nature Reviews Disease Primers 2023 9:1 2023, 9, 1–21. [Google Scholar] [CrossRef]
- Lévêque, N.; Semler, B.L. A 21st Century Perspective of Poliovirus Replication. PLoS Pathog 2015, 11, e1004825. [Google Scholar] [CrossRef]
- Marzi, A.; Blanco, J.R.; Gibellini, D.; Mbani, C.J.; Pandoua Nekoua, M.; Moukassa, D.; Hober, D. The Fight against Poliovirus Is Not Over. Microorganisms 2023, Vol. 11, Page 1323 2023, 11, 1323. [Google Scholar] [CrossRef]
- Cao, J.; Li, D. Searching for Human Oncoviruses: Histories, Challenges, and Opportunities. J Cell Biochem 2018, 119, 4897–4906. [Google Scholar] [CrossRef]
- Noguera Z., L. P.; Charypkhan, D.; Hartnack, S.; Torgerson, P.R.; Rüegg, S.R. The Dual Burden of Animal and Human Zoonoses: A Systematic Review. PLoS Negl Trop Dis 2022, 16, e0010540. [Google Scholar] [CrossRef]
- Zeller, M.A.; Carnevale de Almeida Moraes, D.; Ciacci Zanella, G.; Souza, C.K.; Anderson, T.K.; Baker, A.L.; Gauger, P.C. Reverse Zoonosis of the 2022–2023 Human Seasonal H3N2 Detected in Swine. npj Viruses 2024 2:1 2024, 2, 1–12. [Google Scholar] [CrossRef]
- Lv, J.X.; Liu, X.; Pei, Y.Y.; Song, Z.G.; Chen, X.; Hu, S.J.; She, J.L.; Liu, Y.; Chen, Y.M.; Zhang, Y.Z. Evolutionary Trajectory of Diverse SARS-CoV-2 Variants at the Beginning of COVID-19 Outbreak. Virus Evol 2024, 10. [Google Scholar] [CrossRef] [PubMed]
- Piret, J.; Boivin, G. Pandemics Throughout History. Front Microbiol 2021, 11, 631736. [Google Scholar] [CrossRef] [PubMed]
- Taubenberger, J.K.; Morens, D.M. Influenza: The Once and Future Pandemic. http://dx.doi.org/10.1177/00333549101250S305 2010, 125, 15–26. [CrossRef]
- Eisinger, R.W.; Fauci, A.S. Ending the HIV/AIDS Pandemic. Emerg Infect Dis 2018, 24, 413. [Google Scholar] [CrossRef]
- Reflections on 40 Years of AIDS. 2021, 231–245.
- Chan-Yeung, epidemiologyM; Xu, R.; Chan-Yeung, M.; Chan-yeung, M. SARS: Epidemiology. Respirology 2003, 8, S9–S14. [CrossRef]
- Raj, V.S.; Osterhaus, A.D.M.E.; Fouchier, R.A.M.; Haagmans, B.L. MERS: Emergence of a Novel Human Coronavirus. Curr Opin Virol 2014, 5, 58–62. [Google Scholar] [CrossRef]
- Listings of WHO’s Response to COVID-19 Available online:. Available online: https://www.who.int/news/item/29-06-2020-covidtimeline (accessed on 27 December 2024).
- Duggan, A.T.; Perdomo, M.F.; Piombino-Mascali, D.; Marciniak, S.; Poinar, D.; Emery, M. V.; Buchmann, J.P.; Duchêne, S.; Jankauskas, R.; Humphreys, M.; et al. 17th Century Variola Virus Reveals the Recent History of Smallpox. Current Biology 2016, 26, 3407–3412. [Google Scholar] [CrossRef]
- Bandyopadhyay, A.S.; Garon, J.; Seib, K.; Orenstein, W.A. Polio Vaccination: Past, Present and Future. Future Microbiol 2015, 10, 791–808. [Google Scholar] [CrossRef]
- Jacob, S.T.; Crozier, I.; Fischer, W.A.; Hewlett, A.; Kraft, C.S.; Vega, M.A. de La; Soka, M.J.; Wahl, V.; Griffiths, A.; Bollinger, L.; et al. Ebola Virus Disease. Nature Reviews Disease Primers 2020 6:1 2020, 6, 1–31. [Google Scholar] [CrossRef]
- Guo, C.; Zhou, Z.; Wen, Z.; Liu, Y.; Zeng, C.; Xiao, D.; Ou, M.; Han, Y.; Huang, S.; Liu, D.; et al. Global Epidemiology of Dengue Outbreaks in 1990–2015: A Systematic Review and Meta-Analysis. Front Cell Infect Microbiol 2017, 7, 275966. [Google Scholar] [CrossRef]
- Douam, F.; Ploss, A. Yellow Fever Virus: Knowledge Gaps Impeding the Fight Against an Old Foe. Trends Microbiol 2018, 26, 913–928. [Google Scholar] [CrossRef]
- Baud, D.; Gubler, D.J.; Schaub, B.; Lanteri, M.C.; Musso, D. An Update on Zika Virus Infection. The Lancet 2017, 390, 2099–2109. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.T.; Halsey, N.A. The Clinical Significance of Measles: A Review. J Infect Dis 2004, 189, S4–S16. [Google Scholar] [CrossRef] [PubMed]
- Weibel Galluzzo, C.; Kaiser, L.; Chappuis, F. Reemergence of Chikungunya Virus. Rev Med Suisse 2015, 11, 1012–1016. [Google Scholar] [CrossRef] [PubMed]
- Turtle, L.; Neurology, T.S.-N.R.; 2018, undefined Japanese Encephalitis—the Prospects for New Treatments. nature.comL Turtle, T SolomonNature Reviews Neurology, 2018•nature.com.
- Campbell, G.L.; Marfin, A.A.; Lanciotti, R.S.; Gubler, D.J. West Nile Virus. Lancet Infectious Diseases 2002, 2, 519–529. [Google Scholar] [CrossRef]
- Baer, G.M. History of Rabies and Global Aspects. 2017, 1–24.
- Sperk, M.; Van Domselaar, R.; Rodriguez, J.E.; Mikaeloff, F.; Sá Vinhas, B.; Saccon, E.; Sönnerborg, A.; Singh, K.; Gupta, S.; Végvári, Á.; et al. Utility of Proteomics in Emerging and Re-Emerging Infectious Diseases Caused by RNA Viruses. J Proteome Res 2020, 19, 4259–4274. [Google Scholar] [CrossRef]
- Çelik, İ.; Saatçi, E.; Eyüboğlu, F.Ö. Emerging and Reemerging Respiratory Viral Infections up to Covid-19. Turk J Med Sci 2020, 50, 557–562. [Google Scholar] [CrossRef]
- Curry, S. Structural Biology: A Century-Long Journey into an Unseen World. Interdisciplinary Science Reviews 2015, 40, 308–328. [Google Scholar] [CrossRef]
- Brooks-Bartlett, J.C.; Garman, E.F. The Nobel Science: One Hundred Years of Crystallography. Interdisciplinary Science Reviews 2015, 40, 244–264. [Google Scholar] [CrossRef]
- Thomas, J.M. Centenary: The Birth of X-Ray Crystallography. Nature 2012, 491, 186–187. [Google Scholar] [CrossRef]
- Shi, Y. A Glimpse of Structural Biology through X-Ray Crystallography. Cell 2014, 159, 995–1014. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E.; Li, G. Approved Antiviral Drugs over the Past 50 Years. Clin Microbiol Rev 2016, 29, 695–747. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Handing, K.B.; Zimmerman, M.D.; Shabalin, I.G.; Almo, S.C.; Minor, W. X-Ray Crystallography over the Past Decade for Novel Drug Discovery – Where Are We Heading Next? Expert Opin Drug Discov 2015, 10, 975–989. [Google Scholar] [CrossRef] [PubMed]
- Stanley, W.M. ISOLATION OF A CRYSTALLINE PROTEIN POSSESSING THE PROPERTIES OF TOBACCO-MOSAIC VIRUS. Science 1935, 81, 644–645. [Google Scholar] [CrossRef]
- Norrby, E. Nobel Prizes and the Emerging Virus Concept. Arch Virol 2008, 153, 1109–1123. [Google Scholar] [CrossRef]
- Bernal, J.D.; Fankuchen, I. X-RAY AND CRYSTALLOGRAPHIC STUDIES OF PLANT VIRUS PREPARATIONS : I. INTRODUCTION AND PREPARATION OF SPECIMENS II. MODES OF AGGREGATION OF THE VIRUS PARTICLES. J Gen Physiol 1941, 25, 111–146. [Google Scholar] [CrossRef]
- Harrison, S.C.; Olson, A.J.; Schutt, C.E.; Winkler, F.K.; Bricogne, G. Tomato Bushy Stunt Virus at 2.9 A Resolution. Nature 1978, 276, 368–373. [Google Scholar] [CrossRef]
- Bloomer, A.C.; Champness, J.N.; Bricogne, G.; Staden, R.; Klug, A. Protein Disk of Tobacco Mosaic Virus at 2.8 A Resolution Showing the Interactions within and between Subunits. Nature 1978, 276, 362–368. [Google Scholar] [CrossRef]
- Richmond, T.J.; Finch, J.T.; Rushton, B.; Rhodes, D.; Klug, A. Structure of the Nucleosome Core Particle at 7 A Resolution. Nature 1984, 311, 532–537. [Google Scholar] [CrossRef]
- Zheng, H.; Handing, K.B.; Zimmerman, M.D.; Shabalin, I.G.; Almo, S.C.; Minor, W. X-Ray Crystallography over the Past Decade for Novel Drug Discovery -Where Are We Heading Next? Expert Opin Drug Discov 2015, 10, 975–989. [Google Scholar] [CrossRef]
- Schirò, A.; Carlon, A.; Parigi, G.; Murshudov, G.; Calderone, V.; Ravera, E.; Luchinat, C. On the Complementarity of X-Ray and NMR Data. J Struct Biol X 2020, 4. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Handing, K.B.; Zimmerman, M.D.; Shabalin, I.G.; Almo, S.C.; Minor, W. X-Ray Crystallography over the Past Decade for Novel Drug Discovery -Where Are We Heading Next? Expert Opin Drug Discov 2015, 10, 975–989. [Google Scholar] [CrossRef] [PubMed]
- Vincenzi, M.; Leone, M. The Fight against Human Viruses: How NMR Can Help? Curr Med Chem 2021, 28, 4380–4453. [Google Scholar] [CrossRef]
- Yu, H. Extending the Size Limit of Protein Nuclear Magnetic Resonance. Proc Natl Acad Sci U S A 1999, 96, 332–334. [Google Scholar] [CrossRef]
- LaPlante, S.R.; Coric, P.; Bouaziz, S.; França, T.C.C. NMR Spectroscopy Can Help Accelerate Antiviral Drug Discovery Programs. Microbes Infect 2024, 26, 105297. [Google Scholar] [CrossRef]
- Kruger, D.H.; Schneck, P.; Gelderblom, H.R. Helmut Ruska and the Visualisation of Viruses. Lancet 2000, 355, 1713–1717. [Google Scholar] [CrossRef]
- Nagler, F.P.; Rake, G. The Use of the Electron Microscope in Diagnosis of Variola, Vaccinia, and Varicella. J Bacteriol 1948, 55, 45–51. [Google Scholar] [CrossRef]
- Brenner, S.; Horne, R.W. A Negative Staining Method for High Resolution Electron Microscopy of Viruses. Biochim Biophys Acta 1959, 34, 103–110. [Google Scholar] [CrossRef]
- Tyrrell, D.A.J.; Almeida, J.D. Direct Electron-Microscopy of Organ Cultures for the Detection and Characterization of Viruses. Arch Gesamte Virusforsch 1967, 22, 417–425. [Google Scholar] [CrossRef]
- Adrian, M.; Dubochet, J.; Lepault, J.; McDowall, A.W. Cryo-Electron Microscopy of Viruses. Nature 1984 308:5954 1984, 308, 32–36. [Google Scholar] [CrossRef]
- Schoehn, G.; Chenavier, F.; Crépin, T. Advances in Structural Virology via Cryo-EM in 2022. Viruses 2023, Vol. 15, Page 1315 2023, 15, 1315. [Google Scholar] [CrossRef] [PubMed]
- Dutta, M.; Acharya, P. Cryo-Electron Microscopy in the Study of Virus Entry and Infection. Front Mol Biosci 2024, 11, 1429180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, S.; Zhang, K. Cryo-EM: A Window into the Dynamic World of RNA Molecules. Curr Opin Struct Biol 2024, 88, 102916. [Google Scholar] [CrossRef] [PubMed]
- Renaud, J.P.; Chari, A.; Ciferri, C.; Liu, W.T.; Rémigy, H.W.; Stark, H.; Wiesmann, C. Cryo-EM in Drug Discovery: Achievements, Limitations and Prospects. Nature Reviews Drug Discovery 2018 17:7 2018, 17, 471–492. [Google Scholar] [CrossRef]
- Boldon, L.; Laliberte, F.; Liu, L. Review of the Fundamental Theories behind Small Angle X-Ray Scattering, Molecular Dynamics Simulations, and Relevant Integrated Application. Nano Rev 2015, 6, 25661. [Google Scholar] [CrossRef]
- Handa, T.; Kundu, D.; Dubey, V.K. Perspectives on Evolutionary and Functional Importance of Intrinsically Disordered Proteins. Int J Biol Macromol 2023, 224, 243–255. [Google Scholar] [CrossRef]
- Barradas-Bautista, D.; Rosell, M.; Pallara, C.; Fernández-Recio, J. Structural Prediction of Protein–Protein Interactions by Docking: Application to Biomedical Problems. Adv Protein Chem Struct Biol 2018, 110, 203–249. [Google Scholar] [CrossRef]
- Zhu, P.; Winkler, H.; Chertova, E.; Taylor, K.A.; Roux, K.H. Cryoelectron Tomography of HIV-1 Envelope Spikes: Further Evidence for Tripod-like Legs. PLoS Pathog 2008, 4. [Google Scholar] [CrossRef]
- Dutta, M.; Acharya, P. Cryo-Electron Microscopy in the Study of Virus Entry and Infection. Front Mol Biosci 2024, 11, 1429180. [Google Scholar] [CrossRef]
- Baumeister, W. Cryo-Electron Tomography: The Power of Seeing the Whole Picture. Biochem Biophys Res Commun 2022, 633, 26–28. [Google Scholar] [CrossRef]
- Meents, A.; Wiedorn, M.O. Virus Structures by X-Ray Free-Electron Lasers. Annu Rev Virol 2019, 6, 161–176. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, K. AlphaFold Gets an Upgrade (and a Nobel). Nature Medicine 2024 30:12 2024, 30, 3393–3393. [Google Scholar] [CrossRef] [PubMed]
- FENNER, F.; BACHMANN, P.A.; GIBBS, E.P.J.; MURPHY, F.A.; STUDDERT, M.J.; WHITE, D.O. Structure and Composition of Viruses. In Veterinary Virology; Elsevier, 1987; pp. 3–19.
- Zheng, B.; Duan, M.; Huang, Y.; Wang, S.; Qiu, J.; Lu, Z.; Liu, L.; Tang, G.; Cheng, L.; Zheng, P. Discovery of a Heparan Sulfate Binding Domain in Monkeypox Virus H3 as an Anti-Poxviral Drug Target Combining AI and MD Simulations. Elife 2024, 13. [Google Scholar] [CrossRef]
- Delogu, I.; Pastorino, B.; Baronti, C.; Nougairède, A.; Bonnet, E.; de Lamballerie, X. In Vitro Antiviral Activity of Arbidol against Chikungunya Virus and Characteristics of a Selected Resistant Mutant. Antiviral Res 2011, 90, 99–107. [Google Scholar] [CrossRef]
- Barrow, E.; Nicola, A. V.; Liu, J. Multiscale Perspectives of Virus Entry via Endocytosis. Virol J 2013, 10, 1–11. [Google Scholar] [CrossRef]
- Kim, A.S.; Diamond, M.S. A Molecular Understanding of Alphavirus Entry and Antibody Protection. Nature Reviews Microbiology 2022 21:6 2022, 21, 396–407. [Google Scholar] [CrossRef]
- Melton, J. V.; Ewart, G.D.; Weir, R.C.; Board, P.G.; Lee, E.; Gage, P.W. Alphavirus 6K Proteins Form Ion Channels. Journal of Biological Chemistry 2002, 277, 46923–46931. [Google Scholar] [CrossRef]
- Button, J.M.; Mukhopadhyay, S. Capsid-E2 Interactions Rescue Core Assembly in Viruses That Cannot Form Cytoplasmic Nucleocapsid Cores. J Virol 2021, 95. [Google Scholar] [CrossRef]
- Liu, D.X.; Liang, J.Q.; Fung, T.S. Human Coronavirus-229E, -OC43, -NL63, and -HKU1 (Coronaviridae). Encyclopedia of Virology: Volume 1-5, Fourth Edition 2021, 1–5, 428–440. [Google Scholar] [CrossRef]
- Schlicksup, C.J.; Zlotnick, A. Viral Structural Proteins as Targets for Antivirals. Curr Opin Virol 2020, 45, 43–50. [Google Scholar] [CrossRef]
- Chen, N.; Zhang, B.; Deng, L.; Liang, B.; Ping, J. Virus-Host Interaction Networks as New Antiviral Drug Targets for IAV and SARS-CoV-2. Emerg Microbes Infect 2022, 11, 1371–1389. [Google Scholar] [CrossRef] [PubMed]
- Burrell, C.J.; Howard, C.R.; Murphy, F.A. Virion Structure and Composition. Fenner and White’s Medical Virology 2017, 27–37. [Google Scholar] [CrossRef]
- Kaur, R.; Neetu; Mudgal, R.; Jose, J.; Kumar, P.; Tomar, S. Glycan-Dependent Chikungunya Viral Infection Divulged by Antiviral Activity of NAG Specific Chi-like Lectin. Virology 2019, 526, 91–98. [CrossRef]
- Kaur, R.; Neetu; Mudgal, R.; Jose, J.; Kumar, P.; Tomar, S. Glycan-Dependent Chikungunya Viral Infection Divulged by Antiviral Activity of NAG Specific Chi-like Lectin. Virology 2019, 526, 91–98. [CrossRef]
- Liu, D.X.; Liang, J.Q.; Fung, T.S. Human Coronavirus-229E, -OC43, -NL63, and -HKU1 (Coronaviridae). Encyclopedia of Virology: Volume 1-5, Fourth Edition 2021, 1–5, 428–440. [Google Scholar] [CrossRef]
- Kosik, I.; Yewdell, J.W. Influenza Hemagglutinin and Neuraminidase: Yin–Yang Proteins Coevolving to Thwart Immunity. Viruses 2019, Vol. 11, Page 346 2019, 11, 346. [Google Scholar] [CrossRef]
- Checkley, M.A.; Luttge, B.G.; Freed, E.O. HIV-1 Envelope Glycoprotein Biosynthesis, Trafficking, and Incorporation. J Mol Biol 2011, 410, 582–608. [Google Scholar] [CrossRef]
- Wrobel, A.G. Mechanism and Evolution of Human ACE2 Binding by SARS-CoV-2 Spike. Curr Opin Struct Biol 2023, 81, 102619. [Google Scholar] [CrossRef]
- Epand, R.M. Fusion Peptides and the Mechanism of Viral Fusion. Biochimica et Biophysica Acta (BBA) - Biomembranes 2003, 1614, 116–121. [Google Scholar] [CrossRef]
- Zhai, X.; Yuan, Y.; He, W.T.; Wu, Y.; Shi, Y.; Su, S.; Du, Q.; Mao, Y. Evolving Roles of Glycosylation in the Tug-of-War between Virus and Host. Natl Sci Rev 2024, 11. [Google Scholar] [CrossRef]
- Matsuyama, S.; Taguchi, F. Two-Step Conformational Changes in a Coronavirus Envelope Glycoprotein Mediated by Receptor Binding and Proteolysis. J Virol 2009, 83, 11133. [Google Scholar] [CrossRef]
- Marzinek, J.K.; Raghuvamsi Palur, V.; Salem, G.; Chen, F.-C.; Wu, S.-R.; Bond, P.J.; Chao, D.-Y. Uncovering the Conformational Dynamics of Dengue Virus and Its Virus-like Particles as Novel Vaccine Candidates. Biophys J 2023, 122, 508a–509a. [Google Scholar] [CrossRef]
- Katze, M.G.; He, Y.; Gale, M. Viruses and Interferon: A Fight for Supremacy. Nature Reviews Immunology 2002 2:9 2002, 2, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Xue, X.; Qiao, S.; Jia, L.; Wen, X.; Wang, Y.; Wang, C.; Li, H.; Cui, J. Umifenovir Epigenetically Targets the IL-10 Pathway in Therapy against Coxsackievirus B4 Infection. Microbiol Spectr 2023, 11. [Google Scholar] [CrossRef] [PubMed]
- Sargsyan, K.; Mazmanian, K.; Lim, C. A Strategy for Evaluating Potential Antiviral Resistance to Small Molecule Drugs and Application to SARS-CoV-2. Scientific Reports 2023 13:1 2023, 13, 1–12. [Google Scholar] [CrossRef]
- McCallum, M.; Czudnochowski, N.; Rosen, L.E.; Zepeda, S.K.; Bowen, J.E.; Walls, A.C.; Hauser, K.; Joshi, A.; Stewart, C.; Dillen, J.R.; et al. Structural Basis of SARS-CoV-2 Omicron Immune Evasion and Receptor Engagement. Science (1979) 2022, 375, 894–898. [Google Scholar] [CrossRef]
- Tang, H.; Ke, Y.; Liao, Y.; Bian, Y.; Yuan, Y.; Wang, Z.; Yang, L.; Ma, H.; Sun, T.; Zhang, B.; et al. Mutational Escape Prevention by Combination of Four Neutralizing Antibodies That Target RBD Conserved Regions and Stem Helix. Virol Sin 2022, 37, 860–873. [Google Scholar] [CrossRef]
- Shih, H.I.; Wang, Y.C.; Wang, Y.P.; Chi, C.Y.; Chien, Y.W. Risk of Severe Dengue during Secondary Infection: A Population-Based Cohort Study in Taiwan. Journal of Microbiology, Immunology and Infection 2024, 57, 730–738. [Google Scholar] [CrossRef]
- Wells, T.J.; Esposito, T.; Henderson, I.R.; Labzin, L.I. Mechanisms of Antibody-Dependent Enhancement of Infectious Disease. Nature Reviews Immunology 2024 25:1 2024, 25, 6–21. [Google Scholar] [CrossRef]
- Sarker, A.; Dhama, N.; Gupta, R.D. Dengue Virus Neutralizing Antibody: A Review of Targets, Cross-Reactivity, and Antibody-Dependent Enhancement. Front Immunol 2023, 14, 1200195. [Google Scholar] [CrossRef]
- Malik, S.; Ahsan, O.; Mumtaz, H.; Tahir Khan, M.; Sah, R.; Waheed, Y. Tracing down the Updates on Dengue Virus—Molecular Biology, Antivirals, and Vaccine Strategies. Vaccines 2023, Vol. 11, Page 1328 2023, 11, 1328. [Google Scholar] [CrossRef]
- Dengvaxia | European Medicines Agency (EMA) Available online:. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/dengvaxia (accessed on 10 January 2025).
- Ragonnet-Cronin, M.; Nutalai, R.; Huo, J.; Dijokaite-Guraliuc, A.; Das, R.; Tuekprakhon, A.; Supasa, P.; Liu, C.; Selvaraj, M.; Groves, N.; et al. Generation of SARS-CoV-2 Escape Mutations by Monoclonal Antibody Therapy. Nature Communications 2023 14:1 2023, 14, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.; Malik, Y.S.; Tomar, S. Identification of SARS-CoV-2 Cell Entry Inhibitors by Drug Repurposing Using in Silico Structure-Based Virtual Screening Approach. Front Immunol 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Hayden, F.G.; Osterhaus, A.D.M.E.; Treanor, J.J.; Fleming, D.M.; Aoki, F.Y.; Nicholson, K.G.; Bohnen, A.M.; Hirst, H.M.; Keene, O.; Wightman, K. Efficacy and Safety of the Neuraminidase Inhibitor Zanamivir in the Treatment of Influenzavirus Infections. New England Journal of Medicine 1997, 337, 874–880. [Google Scholar] [CrossRef]
- Collins, P.J.; Haire, L.F.; Lin, Y.P.; Liu, J.; Russell, R.J.; Walker, P.A.; Skehel, J.J.; Martin, S.R.; Hay, A.J.; Gamblin, S.J. Crystal Structures of Oseltamivir-Resistant Influenza Virus Neuraminidase Mutants. Nature 2008 453:7199 2008, 453, 1258–1261. [Google Scholar] [CrossRef] [PubMed]
- Vavricka, C.J.; Li, Q.; Wu, Y.; Qi, J.; Wang, M.; Liu, Y.; Gao, F.; Liu, J.; Feng, E.; He, J.; et al. Structural and Functional Analysis of Laninamivir and Its Octanoate Prodrug Reveals Group Specific Mechanisms for Influenza NA Inhibition. PLoS Pathog 2011, 7, e1002249. [Google Scholar] [CrossRef]
- Pattnaik, G.P.; Chakraborty, H. Entry Inhibitors: Efficient Means to Block Viral Infection. Journal of Membrane Biology 2020, 253, 425–444. [Google Scholar] [CrossRef]
- Beugeling, M.; De Zee, J.; Woerdenbag, H.J.; Frijlink, H.W.; Wilschut, J.C.; Hinrichs, W.L.J. Respiratory Syncytial Virus Subunit Vaccines Based on the Viral Envelope Glycoproteins Intended for Pregnant Women and the Elderly. Expert Rev Vaccines 2019, 18, 935–950. [Google Scholar] [CrossRef]
- Singh, V.A.; Nehul, S.; Kumar, C.S.; Banerjee, M.; Kumar, P.; Sharma, G.; Tomar, S. Chimeric Chikungunya Virus-like Particles with Surface Exposed SARS-CoV-2 RBD Elicits Potent Immunogenic Responses in Mice. bioRxiv 2023, 2023.01.29.526074. [CrossRef]
- Singh, V.A.; Kumar, C.S.; Khare, B.; Kuhn, R.J.; Banerjee, M.; Tomar, S. Surface Decorated Reporter-Tagged Chikungunya Virus-like Particles for Clinical Diagnostics and Identification of Virus Entry Inhibitors. Virology 2023, 578, 92–102. [Google Scholar] [CrossRef]
- Nieva, J.L.; Madan, V.; Carrasco, L. Viroporins: Structure and Biological Functions. Nature Reviews Microbiology 2012 10:8 2012, 10, 563–574. [Google Scholar] [CrossRef]
- Xia, X.; Cheng, A.; Wang, M.; Ou, X.; Sun, D.; Mao, S.; Huang, J.; Yang, Q.; Wu, Y.; Chen, S.; et al. Functions of Viroporins in the Viral Life Cycle and Their Regulation of Host Cell Responses. Front Immunol 2022, 13, 890549. [Google Scholar] [CrossRef]
- Devantier, K.; Kjær, V.M.S.; Griffin, S.; Kragelund, B.B.; Rosenkilde, M.M. Advancing the Field of Viroporins—Structure, Function and Pharmacology: IUPHAR Review 39. Br J Pharmacol 2024, 181, 4450–4490. [Google Scholar] [CrossRef] [PubMed]
- Pinto, L.H.; Holsinger, L.J.; Lamb, R.A. Influenza Virus M2 Protein Has Ion Channel Activity. Cell 1992, 69, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Nieva, J.L.; Madan, V.; Carrasco, L. Viroporins: Structure and Biological Functions. Nature Reviews Microbiology 2012 10:8 2012, 10, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Nieva, J.L.; Madan, V.; Carrasco, L. Viroporins: Structure and Biological Functions. Nature Reviews Microbiology 2012 10:8 2012, 10, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Surya, W.; Samsó, M.; Torres, J.; Surya, W.; Samsó, M.; Torres, J. Structural and Functional Aspects of Viroporins in Human Respiratory Viruses: Respiratory Syncytial Virus and Coronaviruses. Respiratory Disease and Infection - A New Insight 2013. [CrossRef]
- Das, K. Antivirals Targeting Influenza a Virus. J Med Chem 2012, 55, 6263–6277. [Google Scholar] [CrossRef]
- Thomaston, J.L.; Polizzi, N.F.; Konstantinidi, A.; Wang, J.; Kolocouris, A.; Degrado, W.F. Inhibitors of the M2 Proton Channel Engage and Disrupt Transmembrane Networks of Hydrogen-Bonded Waters. J Am Chem Soc 2018, 140, 15219–15226. [Google Scholar] [CrossRef]
- Nieto-Torres, J.L.; Verdiá-Báguena, C.; Castaño-Rodriguez, C.; Aguilella, V.M.; Enjuanes, L. Relevance of Viroporin Ion Channel Activity on Viral Replication and Pathogenesis. Viruses 2015, Vol. 7, Pages 3552-3573 2015, 7, 3552–3573. [Google Scholar] [CrossRef]
- Dey, D.; Siddiqui, S.I.; Mamidi, P.; Ghosh, S.; Kumar, C.S.; Chattopadhyay, S.; Ghosh, S.; Banerjee, M. The Effect of Amantadine on an Ion Channel Protein from Chikungunya Virus. PLoS Negl Trop Dis 2019, 13, e0007548. [Google Scholar] [CrossRef]
- Lamb, R.A. Influenza. Encyclopedia of Virology: Volume 1-5 2008, 1–5, 95–104. [Google Scholar] [CrossRef]
- Scott, C.; Griffin, S. Viroporins: Structure, Function and Potential as Antiviral Targets. Journal of General Virology 2015, 96, 2000–2027. [Google Scholar] [CrossRef]
- Nieva, J.L.; Madan, V.; Carrasco, L. Viroporins: Structure and Biological Functions. Nature Reviews Microbiology 2012 10:8 2012, 10, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Fatma, B.; Kumar, R.; Singh, V.A.; Nehul, S.; Sharma, R.; Kesari, P.; Kuhn, R.J.; Tomar, S. Alphavirus Capsid Protease Inhibitors as Potential Antiviral Agents for Chikungunya Infection. Antiviral Res 2020, 179, 104808. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, A.; Rao, A.L. The Interplay between Capsid Dynamics and Pathogenesis in Tripartite Bromoviruses. Curr Opin Virol 2021, 47, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Ghaemi, Z.; Gruebele, M.; Tajkhorshid, E. Molecular Mechanism of Capsid Disassembly in Hepatitis B Virus. Proc Natl Acad Sci U S A 2021, 118, e2102530118. [Google Scholar] [CrossRef]
- Mohajerani, F.; Tyukodi, B.; Schlicksup, C.J.; Hadden-Perilla, J.A.; Zlotnick, A.; Hagan, M.F. Multiscale Modeling of Hepatitis B Virus Capsid Assembly and Its Dimorphism. ACS Nano 2022, 16, 13845–13859. [Google Scholar] [CrossRef]
- Koehl, P.; Akopyan, A.; Edelsbrunner, H. Computing the Volume, Surface Area, Mean, and Gaussian Curvatures of Molecules and Their Derivatives. J Chem Inf Model 2023, 63, 973–985. [Google Scholar] [CrossRef]
- Mohajerani, F.; Tyukodi, B.; Schlicksup, C.J.; Hadden-Perilla, J.A.; Zlotnick, A.; Hagan, M.F. Multiscale Modeling of Hepatitis B Virus Capsid Assembly and Its Dimorphism. ACS Nano 2022, 16, 13845–13859. [Google Scholar] [CrossRef]
- Aggarwal, M.; Kaur, R.; Saha, A.; Mudgal, R.; Yadav, R.; Dash, P.K.; Parida, M.; Kumar, P.; Tomar, S. Evaluation of Antiviral Activity of Piperazine against Chikungunya Virus Targeting Hydrophobic Pocket of Alphavirus Capsid Protein. Antiviral Res 2017, 146, 102–111. [Google Scholar] [CrossRef]
- Fatma, B.; Kumar, R.; Singh, V.A.; Nehul, S.; Sharma, R.; Kesari, P.; Kuhn, R.J.; Tomar, S. Alphavirus Capsid Protease Inhibitors as Potential Antiviral Agents for Chikungunya Infection. Antiviral Res 2020, 179. [Google Scholar] [CrossRef]
- HIV-1 Capsid Inhibitors as Antiretroviral Agents.
- Segal-Maurer, S.; DeJesus, E.; Stellbrink, H.-J.; Castagna, A.; Richmond, G.J.; Sinclair, G.I.; Siripassorn, K.; Ruane, P.J.; Berhe, M.; Wang, H.; et al. Capsid Inhibition with Lenacapavir in Multidrug-Resistant HIV-1 Infection. New England Journal of Medicine 2022, 386, 1793–1803. [Google Scholar] [CrossRef]
- Klumpp, K.; Crépin, T. Capsid Proteins of Enveloped Viruses as Antiviral Drug Targets. Curr Opin Virol 2014, 5, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, Y.; Jia, R.; Wang, M.; Yin, Z.; Cheng, A. Structure and Function of Capsid Protein in Flavivirus Infection and Its Applications in the Development of Vaccines and Therapeutics. Veterinary Research 2021 52:1 2021, 52, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Jia, R.; Zhou, J.; Wang, M.; Yin, Z.; Cheng, A. Capsid-Targeted Viral Inactivation: A Novel Tactic for Inhibiting Replication in Viral Infections. Viruses 2016, Vol. 8, Page 258 2016, 8, 258. [Google Scholar] [CrossRef] [PubMed]
- Dhaka, P.; Mahto, J.K.; Singh, A.; Kumar, P.; Tomar, S. Structural Insights into the RNA Binding Inhibitors of the C-Terminal Domain of the SARS-CoV-2 Nucleocapsid 2024.
- Dhaka, P.; Singh, A.; Choudhary, S.; Peddinti, R.K.; Kumar, P.; Sharma, G.K.; Tomar, S. Mechanistic and Thermodynamic Characterization of Antiviral Inhibitors Targeting Nucleocapsid N-Terminal Domain of SARS-CoV-2. Arch Biochem Biophys 2023, 750, 109820. [Google Scholar] [CrossRef]
- The Isolation and Properties of Crystalline Tobacco Mosaic Virus.
- Namba, K.; Pattanayek, R.; Stubbs, G. Visualization of Protein-Nucleic Acid Interactions in a Virus: Refined Structure of Intact Tobacco Mosaic Virus at 2.9 Å Resolution by X-Ray Fiber Diffraction. J Mol Biol 1989, 208, 307–325. [Google Scholar] [CrossRef]
- Aggarwal, M.; Tapas, S.; Preeti; Siwach, A.; Kumar, P.; Kuhn, R.J.; Tomar, S. Crystal Structure of Aura Virus Capsid Protease and Its Complex with Dioxane: New Insights into Capsid-Glycoprotein Molecular Contacts. PLoS One 2012, 7. [CrossRef]
- Aggarwal, M.; Dhindwal, S.; Kumar, P.; Kuhn, R.J.; Tomar, S. Trans -Protease Activity and Structural Insights into the Active Form of the Alphavirus Capsid Protease. J Virol 2014, 88, 12242–12253. [Google Scholar] [CrossRef]
- Segal-Maurer, S.; DeJesus, E.; Stellbrink, H.-J.; Castagna, A.; Richmond, G.J.; Sinclair, G.I.; Siripassorn, K.; Ruane, P.J.; Berhe, M.; Wang, H.; et al. Capsid Inhibition with Lenacapavir in Multidrug-Resistant HIV-1 Infection. New England Journal of Medicine 2022, 386, 1793–1803. [Google Scholar] [CrossRef]
- Kanodia, S.; Da Silva, D.M.; Kast, W.M. Recent Advances in Strategies for Immunotherapy of Human Papillomavirus-Induced Lesions. Int J Cancer 2008, 122, 247–259. [Google Scholar] [CrossRef]
- Demmler-Harrison, G.J. ANTIVIRAL AGENTS. Feigin and Cherry’s Textbook of Pediatric Infectious Diseases, Sixth Edition 2009, 3245–3271. [CrossRef]
- Wlodawer, A.; Vondrasek, J. Inhibitors of HIV-1 Protease: A Major Success of Structure-Assisted Drug Design. Annu Rev Biophys Biomol Struct 1998, 27, 249–284. [Google Scholar] [CrossRef]
- FDA-Approved HIV Medicines | NIH Available online:. Available online: https://hivinfo.nih.gov/understanding-hiv/fact-sheets/fda-approved-hiv-medicines (accessed on 27 December 2024).
- Weber, I.T.; Miller, M.; Jaskólski, M.; Leis, J.; Skalka, A.M.; Wlodawer, A. Molecular Modeling of the HIV-1 Protease and Its Substrate Binding Site. Science 1989, 243, 928–931. [Google Scholar] [CrossRef]
- Lapatto, R.; Blundell, T.; Hemmings, A.; Overington, J.; Wilderspin, A.; Wood, S.; Merson, J.R.; Whittle, P.J.; Danley, D.E.; Geoghegan, K.F.; et al. X-Ray Analysis of HIV-1 Proteinase at 2.7 Å Resolution Confirms Structural Homology among Retroviral Enzymes. Nature 1989 342:6247 1989, 342, 299–302. [Google Scholar] [CrossRef] [PubMed]
- Roberts, N.A.; Martin, J.A.; Kinchington, D.; Broadhurst, A. V.; Craig, J.C.; Duncan, I.B.; Galpin, S.A.; Handa, B.K.; Kay, J.; Kröhn, A.; et al. Rational Design of Peptide-Based HIV Proteinase Inhibitors. Science (1979) 1990, 248, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Craig, J.C.; Duncan, I.B.; Hockley, D.; Grief, C.; Roberts, N.A.; Mills, J.S. Antiviral Properties of Ro 31-8959, an Inhibitor of Human Immunodeficiency Virus (HIV) Proteinase. Antiviral Res 1991, 16, 295–305. [Google Scholar] [CrossRef]
- Kempf, D.J.; Marsh, K.C.; Denissen, J.F.; McDonald, E.; Vasavanonda, S.; Flentge, C.A.; Green, B.E.; Fino, L.; Park, C.H.; Kong, X.P.; et al. ABT-538 Is a Potent Inhibitor of Human Immunodeficiency Virus Protease and Has High Oral Bioavailability in Humans. Proc Natl Acad Sci U S A 1995, 92, 2484–2488. [Google Scholar] [CrossRef]
- Erickson, J.W. Design and Structure of Symmetry-Based Inhibitors of HIV-1 Protease. Perspectives in Drug Discovery and Design 1993, 1, 109–128. [Google Scholar] [CrossRef]
- Dorsey, B.D.; Levin, R.B.; McDaniel, S.L.; Vacca, J.P.; Guare, J.P.; Anderson, P.S.; Huff, J.R.; Darke, P.L.; Zugay, J.A.; Emini, E.A.; et al. L-735,524: The Design of a Potent and Orally Bioavailable HIV Protease Inhibitor. J Med Chem 1994, 37, 3443–3451. [Google Scholar] [CrossRef]
- Vacca, J.P.; Dorsey, B.D.; Schleif, W.A.; Levin, R.B.; Mcdaniel, S.L.; Darke, P.L.; Zugay, J.; Quintero, J.C.; Blahy, O.M.; Roth, E.; et al. L-735,524: An Orally Bioavailable Human Immunodeficiency Virus Type 1 Protease Inhibitor. Proc Natl Acad Sci U S A 1994, 91, 4096–4100. [Google Scholar] [CrossRef]
- Wlodawer, A. Rational Approach to AIDS Drug Design through Structural Biology. Annu Rev Med 2002, 53, 595–614. [Google Scholar] [CrossRef]
- Varney, M.D.; Appelt, K.; Kalish, V.; Reddy, M.R.; Tatlock, J.; Palmer, C.L.; Romines, W.H.; Wu, B.W.; Musick, L. Crystal-Structure-Based Design and Synthesis of Novel C-Terminal Inhibitors of HIV Protease. J Med Chem 1994, 37, 2274–2284. [Google Scholar] [CrossRef]
- Kaldor, S.W.; Kalish, V.J.; Davies, J.F.; Shetty, B. V.; Fritz, J.E.; Appelt, K.; Burgess, J.A.; Campanale, K.M.; Chirgadze, N.Y.; Clawson, D.K.; et al. Viracept (Nelfinavir Mesylate, AG1343): A Potent, Orally Bioavailable Inhibitor of HIV-1 Protease. J Med Chem 1997, 40, 3979–3985. [Google Scholar] [CrossRef]
- Weber, I.T.; Waltman, M.J.; Mustyakimov, M.; Blakeley, M.P.; Keen, D.A.; Ghosh, A.K.; Langan, P.; Kovalevsky, A.Y. Joint X-Ray/Neutron Crystallographic Study of HIV-1 Protease with Clinical Inhibitor Amprenavir: Insights for Drug Design. J Med Chem 2013, 56, 5631–5635. [Google Scholar] [CrossRef] [PubMed]
- Chemburkar, S.R.; Bauer, J.; Deming, K.; Spiwek, H.; Patel, K.; Morris, J.; Henry, R.; Spanton, S.; Dziki, W.; Porter, W.; et al. Dealing with the Impact of Ritonavir Polymorphs on the Late Stages of Bulk Drug Process Development. Org Process Res Dev 2000, 4, 413–417. [Google Scholar] [CrossRef]
- McCauley, J.A.; Rudd, M.T. Hepatitis C Virus NS3/4a Protease Inhibitors. Curr Opin Pharmacol 2016, 30, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Love, R.A.; Parge, H.E.; Wickersham, J.A.; Hostomsky, Z.; Habuka, N.; Moomaw, E.W.; Adachi, T.; Hostomska, Z. The Crystal Structure of Hepatitis C Virus NS3 Proteinase Reveals a Trypsin-like Fold and a Structural Zinc Binding Site. Cell 1996, 87, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.L.; Morgenstern, K.A.; Lin, C.; Fox, T.; Dwyer, M.D.; Landro, J.A.; Chambers, S.P.; Markland, W.; Lepre, C.A.; O’Malley, E.T.; et al. Crystal Structure of the Hepatitis C Virus NS3 Protease Domain Complexed with a Synthetic NS4A Cofactor Peptide. Cell 1996, 87, 343–355. [Google Scholar] [CrossRef]
- Barbato, G.; Cicero, D.O.; Nardi, M.C.; Steinkühler, C.; Cortese, R.; De Francesco, R.; Bazzo, R. The Solution Structure of the N-Terminal Proteinase Domain of the Hepatitis C Virus (HCV) NS3 Protein Provides New Insights into Its Activation and Catalytic Mechanism. J Mol Biol 1999, 289, 371–384. [Google Scholar] [CrossRef]
- Llinàs-Brunet, M.; Bailey, M.; Fazal, G.; Goulet, S.; Halmos, T.; Laplante, S.; Maurice, R.; Poirier, M.; Poupart, M.A.; Thibeault, D.; et al. Peptide-Based Inhibitors of the Hepatitis C Virus Serine Protease. Bioorg Med Chem Lett 1998, 8, 1713–1718. [Google Scholar] [CrossRef]
- saalau-bethell, susanne; Woodhead, andrew J.; chessari, gianni; carr, maria; coyle, J.; graham, brent; hiscock, steven; murray, christopher W.; pathuri, puja; rich, sharna J.; et al. Discovery of an Allosteric Mechanism for the Regulation of HcV Ns3 Protein Function. 2012. [CrossRef]
- Choudhary, S.; Nehul, S.; Singh, A.; Panda, P.K.; Kumar, P.; Sharma, G.K.; Tomar, S. Unraveling Antiviral Efficacy of Multifunctional Immunomodulatory Triterpenoids against SARS-COV-2 Targeting Main Protease and Papain-like Protease. IUBMB Life 2024, 76, 228–241. [Google Scholar] [CrossRef]
- Choudhary, S.; Nehul, S.; Nagaraj, S.K.; Narayan, R.; Verma, S.; Sharma, S.; Kumari, A.; Rani, R.; Saha, A.; Sircar, D.; et al. Activity Profiling of Deubiquitinating Inhibitors-Bound to SARS-CoV-2 Papain like Protease with Antiviral Efficacy in Murine Infection Model 2022.
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An Oral SARS-CoV-2 Mpro Inhibitor Clinical Candidate for the Treatment of COVID-19. Science (1979) 2021, 374, 1586–1593. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal Structure of SARS-CoV-2 Main Protease Provides a Basis for Design of Improved a-Ketoamide Inhibitors. Science (1979) 2020, 368, 409–412. [Google Scholar] [CrossRef]
- FDA Approves First Oral Antiviral for Treatment of COVID-19 in Adults | FDA Available online:. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-oral-antiviral-treatment-covid-19-adults (accessed on 30 December 2024).
- Pagliano, P.; Spera, A.; Sellitto, C.; Scarpati, G.; Folliero, V.; Piazza, O.; Franci, G.; Conti, V.; Ascione, T. Preclinical Discovery and Development of Nirmatrelvir/Ritonavir Combinational Therapy for the Treatment of COVID-19 and the Lessons Learned from SARS-COV-2 Variants. Expert Opin Drug Discov 2023, 18, 1301–1311. [Google Scholar] [CrossRef] [PubMed]
- Unoh, Y.; Uehara, S.; Nakahara, K.; Nobori, H.; Yamatsu, Y.; Yamamoto, S.; Maruyama, Y.; Taoda, Y.; Kasamatsu, K.; Suto, T.; et al. Discovery of S-217622, a Noncovalent Oral SARS-CoV-2 3CL Protease Inhibitor Clinical Candidate for Treating COVID-19. J Med Chem 2022, 65, 6499–6512. [Google Scholar] [CrossRef] [PubMed]
- Narwal, M.; Armache, J.P.; Edwards, T.J.; Murakami, K.S. SARS-CoV-2 Polyprotein Substrate Regulates the Stepwise Mpro Cleavage Reaction. Journal of Biological Chemistry 2023, 299. [Google Scholar] [CrossRef]
- Pareek, A.; Kumar, R.; Mudgal, R.; Neetu, N.; Sharma, M.; Kumar, P.; Tomar, S. Alphavirus Antivirals Targeting RNA-Dependent RNA Polymerase Domain of NsP4 Divulged Using Surface Plasmon Resonance. FEBS J 2022, 289, 4901–4924. [Google Scholar] [CrossRef]
- Rani, R.; Long, S.; Pareek, A.; Dhaka, P.; Singh, A.; Kumar, P.; McInerney, G.; Tomar, S. Multi-Target Direct-Acting SARS-CoV-2 Antivirals against the Nucleotide-Binding Pockets of Virus-Specific Proteins. Virology 2022, 577, 1–15. [Google Scholar] [CrossRef]
- Rani, R.; Nehul, S.; Choudhary, S.; Upadhyay, A.; Kumar Sharma, G.; Kumar, P.; Tomar, S.; scientist, S. Revealing and Evaluation of Antivirals Targeting Multiple Druggable Sites of RdRp Complex in SARS-CoV-2. bioRxiv 2023, 2023.07.24.550324. [CrossRef]
- Choi, K.H. Viral Polymerases. Adv Exp Med Biol 2012, 726, 267–304. [Google Scholar] [CrossRef]
- Liu, S.; Knafels, J.D.; Chang, J.S.; Waszak, G.A.; Baldwin, E.T.; Deibel, M.R.; Thomsen, D.R.; Homa, F.L.; Wells, P.A.; Tory, M.C.; et al. Crystal Structure of the Herpes Simplex Virus 1 DNA Polymerase. Journal of Biological Chemistry 2006, 281, 18193–18200. [Google Scholar] [CrossRef]
- Zarrouk, K.; Piret, J.; Boivin, G. Herpesvirus DNA Polymerases: Structures, Functions and Inhibitors. Virus Res 2017, 234, 177–192. [Google Scholar] [CrossRef]
- Gustavsson, E.; Grünewald, K.; Elias, P.; Hällberg, B.M. Dynamics of the Herpes Simplex Virus DNA Polymerase Holoenzyme during DNA Synthesis and Proof-Reading Revealed by Cryo-EM. Nucleic Acids Res 2024, 52, 7292–7304. [Google Scholar] [CrossRef]
- Shankar, S.; Pan, J.; Yang, P.; Bian, Y.; Oroszlán, G.; Yu, Z.; Mukherjee, P.; Filman, D.J.; Hogle, J.M.; Shekhar, M.; et al. Viral DNA Polymerase Structures Reveal Mechanisms of Antiviral Drug Resistance. Cell 2024. [Google Scholar] [CrossRef]
- D’Cruz, O.J.; Uckun, F.M. Dawn of Non-Nucleoside Inhibitor-Based Anti-HIV Microbicides. Journal of Antimicrobial Chemotherapy 2006, 57, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Phenotypic Susceptibility to Nonnucleoside Inhibitors of... : JAIDS Journal of Acquired Immune Deficiency Syndromes.
- Smerdon, S.J.; Jäger, J.; Wang, J.; Kohlstaedt, L.A.; Chirino, A.J.; Friedman, J.M.; Rice, P.A.; Steitz, T.A. Structure of the Binding Site for Nonnucleoside Inhibitors of the Reverse Transcriptase of Human Immunodeficiency Virus Type 1. Proc Natl Acad Sci U S A 1994, 91, 3911–3915. [Google Scholar] [CrossRef]
- Merluzzi, V.J.; Hargrave, K.D.; Labadia, M.; Grozinger, K.; Skoog, M.; Wu, J.C.; Shih, C.-K.; Eckner, K.; Hattox, S.; Adams, J.; et al. Inhibition of HIV-1 Replication by a Nonnucleoside Reverse Transcriptase Inhibitor. Science 1990, 250, 1411–1413. [Google Scholar] [CrossRef] [PubMed]
- Omoto, S.; Speranzini, V.; Hashimoto, T.; Noshi, T.; Yamaguchi, H.; Kawai, M.; Kawaguchi, K.; Uehara, T.; Shishido, T.; Naito, A.; et al. Characterization of Influenza Virus Variants Induced by Treatment with the Endonuclease Inhibitor Baloxavir Marboxil. Scientific Reports 2018 8:1 2018, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Omoto, S.; Speranzini, V.; Hashimoto, T.; Noshi, T.; Yamaguchi, H.; Kawai, M.; Kawaguchi, K.; Uehara, T.; Shishido, T.; Naito, A.; et al. Characterization of Influenza Virus Variants Induced by Treatment with the Endonuclease Inhibitor Baloxavir Marboxil. Scientific Reports 2018 8:1 2018, 8, 1–15. [Google Scholar] [CrossRef]
- Chang, S.; Sun, D.; Liang, H.; Wang, J.; Li, J.; Guo, L.; Wang, X.; Guan, C.; Boruah, B.M.; Yuan, L.; et al. Cryo-EM Structure of Influenza Virus RNA Polymerase Complex at 4.3Å Resolution. Mol Cell 2015, 57, 925–935. [Google Scholar] [CrossRef]
- Kirchdoerfer, R.N.; Ward, A.B. Structure of the SARS-CoV Nsp12 Polymerase Bound to Nsp7 and Nsp8 Co-Factors. Nat Commun 2019, 10. [Google Scholar] [CrossRef]
- FDA Approves First Treatment for COVID-19.
- Yin, W.; Mao, C.; Luan, X.; Shen, D.D.; Shen, Q.; Su, H.; Wang, X.; Zhou, F.; Zhao, W.; Gao, M.; et al. Structural Basis for Inhibition of the RNA-Dependent RNA Polymerase from SARS-CoV-2 by Remdesivir. Science (1979) 2020, 368, 1499–1504. [Google Scholar] [CrossRef]
- Elfiky, A.A. Ribavirin, Remdesivir, Sofosbuvir, Galidesivir, and Tenofovir against SARS-CoV-2 RNA Dependent RNA Polymerase (RdRp): A Molecular Docking Study. Life Sci 2020, 253. [Google Scholar] [CrossRef]
- Sangawa, H.; Komeno, T.; Nishikawa, H.; Yoshida, A.; Takahashi, K.; Nomura, N.; Furuta, Y. Mechanism of Action of T-705 Ribosyl Triphosphate against Influenza Virus RNA Polymerase. Antimicrob Agents Chemother 2013, 57, 5202–5208. [Google Scholar] [CrossRef]
- Painter, G.R.; Natchus, M.G.; Cohen, O.; Holman, W.; Painter, W.P. Developing a Direct Acting, Orally Available Antiviral Agent in a Pandemic: The Evolution of Molnupiravir as a Potential Treatment for COVID-19. Curr Opin Virol 2021, 50, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.J.; Toots, M.; Lee, S.; Lee, M.E.; Ludeke, B.; Luczo, J.M.; Ganti, K.; Cox, R.M.; Sticher, Z.M.; Edpuganti, V.; et al. Orally Efficacious Broad-Spectrum Ribonucleoside Analog Inhibitor of Influenza and Respiratory Syncytial Viruses. Antimicrob Agents Chemother 2018, 62. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Peng, R.; Yuan, B.; Wang, M.; Zhao, J.; Fu, L.; Qi, J.; Shi, Y. Structural Basis of SARS-CoV-2 Polymerase Inhibition by Favipiravir. Innovation 2021, 2. [Google Scholar] [CrossRef] [PubMed]
- Renzi, G.; Carta, F.; Supuran, C.T. The Integrase: An Overview of a Key Player Enzyme in the Antiviral Scenario. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Pommier, Y.; Johnson, A.A.; Marchand, C. Integrase Inhibitors to Treat HIV/AIDS. Nat Rev Drug Discov 2005, 4, 236–248. [Google Scholar] [CrossRef]
- Cai, M.; Zheng, R.; Caffrey, M.; Craigie, R.; Marius Clore, G.; Gronenborn, A.M. Solution Structure of the N-Terminal Zinc Binding Domain of HIV-1 Integrase. Nat Struct Biol 1997, 4, 567–577. [Google Scholar] [CrossRef]
- Renzi, G.; Carta, F.; Supuran, C.T. The Integrase: An Overview of a Key Player Enzyme in the Antiviral Scenario. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Renzi, G.; Carta, F.; Supuran, C.T. The Integrase: An Overview of a Key Player Enzyme in the Antiviral Scenario. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Maertens, G.N.; Engelman, A.N.; Cherepanov, P. Structure and Function of Retroviral Integrase. Nat Rev Microbiol 2022, 20, 20–34. [Google Scholar] [CrossRef]
- Blanco, J.L.; Whitlock, G.; Milinkovic, A.; Moyle, G. HIV Integrase Inhibitors: A New Era in the Treatment of HIV. Expert Opin Pharmacother 2015, 16, 1313–1324. [Google Scholar] [CrossRef]
- Goldgur, Y.; Craigie, R.; Cohen, G.H.; Fujiwara, T.; Yoshinaga, T.; Fujishita, T.; Sugimoto, H.; Endo, T.; Murai, H.; Davies, D.R. Structure of the HIV-1 Integrase Catalytic Domain Complexed with an Inhibitor: A Platform for Antiviral Drug Design. Proc Natl Acad Sci U S A 1999, 96, 13040–13043. [Google Scholar] [CrossRef] [PubMed]
- Renzi, G.; Carta, F.; Supuran, C.T. The Integrase: An Overview of a Key Player Enzyme in the Antiviral Scenario. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Hare, S.; Smith, S.J.; Métifiot, M.; Jaxa-Chamiec, A.; Pommier, Y.; Hughes, S.H.; Cherepanov, P. Structural and Functional Analyses of the Second-Generation Integrase Strand Transfer Inhibitor Dolutegravir (S/GSK1349572). Mol Pharmacol 2011, 80, 565–572. [Google Scholar] [CrossRef]
- Ballantyne, A.D.; Perry, C.M. Dolutegravir: First Global Approval. Drugs 2013, 73, 1627–1637. [Google Scholar] [CrossRef]
- Renzi, G.; Carta, F.; Supuran, C.T. The Integrase: An Overview of a Key Player Enzyme in the Antiviral Scenario. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- New Report Documents Increase in HIV Drug Resistance to Dolutegravir.
- Li, M.; Passos, D.O.; Shan, Z.; Smith, S.J.; Sun, Q.; Biswas, A.; Choudhuri, I.; Strutzenberg, T.S.; Haldane, A.; Deng, N.; et al. Mechanisms of HIV-1 Integrase Resistance to Dolutegravir and Potent Inhibition of Drug-Resistant Variants. Sci Adv 2023, 9. [Google Scholar] [CrossRef]
- Passos, D.O.; Li, M.; Jóźwik, I.K.; Zhao, X.Z.; Santos-Martins, D.; Yang, R.; Smith, S.J.; Jeon, Y.; Forli, S.; Hughes, S.H.; et al. Structural Basis for Strand-Transfer Inhibitor Binding to HIV Intasomes. Science (1979) 2020, 367, 810–814. [Google Scholar] [CrossRef]
- Renzi, G.; Carta, F.; Supuran, C.T. The Integrase: An Overview of a Key Player Enzyme in the Antiviral Scenario. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Bonnard, D.; Le Rouzic, E.; Eiler, S.; Amadori, C.; Orlov, I.; Bruneau, J.M.; Brias, J.; Barbion, J.; Chevreuil, F.; Spehner, D.; et al. Structure-Function Analyses Unravel Distinct Effects of Allosteric Inhibitors of HIV-1 Integrase on Viral Maturation and Integration. Journal of Biological Chemistry 2018, 293, 6172–6186. [Google Scholar] [CrossRef]
- Maertens, G.N.; Engelman, A.N.; Cherepanov, P. Structure and Function of Retroviral Integrase. Nat Rev Microbiol 2022, 20, 20–34. [Google Scholar] [CrossRef]
- Christ, F.; Voet, A.; Marchand, A.; Nicolet, S.; Desimmie, B.A.; Marchand, D.; Bardiot, D.; Van Der Veken, N.J.; Van Remoortel, B.; Strelkov, S. V.; et al. Rational Design of Small-Molecule Inhibitors of the LEDGF/P75-Integrase Interaction and HIV Replication. Nat Chem Biol 2010, 6, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Wu, L.; Wang, M.; Cheng, A.; Yang, Q.; Wu, Y.; Jia, R.; Zhu, D.; Zhao, X.; Chen, S.; et al. Alpha-Herpesvirus Thymidine Kinase Genes Mediate Viral Virulence and Are Potential Therapeutic Targets. Front Microbiol 2019, 10, 941. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.G.; Visse, R.; Sandhu, G.; Davies, A.; Rizkallah, P.J.; Melitz, C.; Summers, W.C.; Sanderson, M.R. Crystal Structures of the Thymidine Kinase from Herpes Simplex Virus Type-I in Complex with Deoxythymidine and Ganciclovir. Nat Struct Biol 1995, 2, 876–881. [Google Scholar] [CrossRef] [PubMed]
- Wild, K.; Bohner, T.; Aubry, A.; Folkers, G.; Schulz, G.E. The Three-Dimensional Structure of Thymidine Kinase from Herpes Simplex Virus Type 1. FEBS Lett 1995, 368, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Frobert, E.; Ooka, T.; Cortay, J.C.; Lina, B.; Thouvenot, D.; Morfin, F. Herpes Simplex Virus Thymidine Kinase Mutations Associated with Resistance to Acyclovir: A Site-Directed Mutagenesis Study. Antimicrob Agents Chemother 2005, 49, 1055–1059. [Google Scholar] [CrossRef]
- Ramdhan, P.; Li, C. Targeting Viral Methyltransferases: An Approach to Antiviral Treatment for SsRNA Viruses. Viruses 2022, 14. [Google Scholar] [CrossRef]
- Jones, R.; Bragagnolo, G.; Arranz, R.; Reguera, J. Capping Pores of Alphavirus NsP1 Gate Membranous Viral Replication Factories. Nature 2021, 589, 615–619. [Google Scholar] [CrossRef]
- Lampio, A.; Kilpeläinen, I.; Pesonen, S.; Karhi, K.; Auvinen, P.; Somerharju, P.; Kääriäinen, L. Membrane Binding Mechanism of an RNA Virus-Capping Enzyme. Journal of Biological Chemistry 2000, 275, 37853–37859. [Google Scholar] [CrossRef]
- Jones, R.; Hons, M.; Rabah, N.; Zamarreño, N.; Arranz, R.; Reguera, J. Structural Basis and Dynamics of Chikungunya Alphavirus RNA Capping by NsP1 Capping Pores. Proc Natl Acad Sci U S A 2023, 120. [Google Scholar] [CrossRef]
- Bhutkar, M.; Saha, A.; Tomar, S. Viral Methyltransferase Inhibitors: Berbamine, Venetoclax, and Ponatinib as Efficacious Antivirals against Chikungunya Virus. Arch Biochem Biophys 2024, 759. [Google Scholar] [CrossRef]
- Mudgal, R.; Bharadwaj, C.; Dubey, A.; Choudhary, S.; Nagarajan, P.; Aggarwal, M.; Ratra, Y.; Basak, S.; Tomar, S. Selective Estrogen Receptor Modulators Limit Alphavirus Infection by Targeting the Viral Capping Enzyme NsP1. Antimicrob Agents Chemother 2022, 66. [Google Scholar] [CrossRef] [PubMed]
- Mudgal, R.; Mahajan, S.; Tomar, S. Inhibition of Chikungunya Virus by an Adenosine Analog Targeting the SAM-Dependent NsP1 Methyltransferase. FEBS Lett 2020, 594, 678–694. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Ray, D.; Zhao, Y.; Dong, H.; Ren, S.; Li, Z.; Guo, Y.; Bernard, K.A.; Shi, P.-Y.; Li, H. Structure and Function of Flavivirus NS5 Methyltransferase. J Virol 2007, 81, 3891–3903. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.P.; Sonntag, L.S.; Noble, C.; Nilar, S.H.; Ng, R.H.; Zou, G.; Monaghan, P.; Chung, K.Y.; Dong, H.; Liu, B.; et al. Small Molecule Inhibitors That Selectively Block Dengue Virus Methyltransferase. Journal of Biological Chemistry 2011, 286, 6233–6240. [Google Scholar] [CrossRef]
- Upadhyay, A.K.; Cyr, M.; Longenecker, K.; Tripathi, R.; Sun, C.; Kempf, D.J. Crystal Structure of Full-Length Zika Virus NS5 Protein Reveals a Conformation Similar to Japanese Encephalitis Virus NS5. Acta Crystallographica Section:F Structural Biology Communications 2017, 73, 116–122. [Google Scholar] [CrossRef]
- Jia, H.; Zhong, Y.; Peng, C.; Gong, P. Crystal Structures of Flavivirus NS5 Guanylyltransferase Reveal a GMP-Arginine Adduct. J Virol 2022, 96. [Google Scholar] [CrossRef]
- Chen, H.; Lin, S.; Yang, F.; Chen, Z.; Guo, L.; Yang, J.; Lin, X.; Wang, L.; Duan, Y.; Wen, A.; et al. Structural and Functional Basis of Low-Affinity SAM/SAH-Binding in the Conserved MTase of the Multi-Segmented Alongshan Virus Distantly Related to Canonical Unsegmented Flaviviruses. PLoS Pathog 2023, 19. [Google Scholar] [CrossRef]
- Bhutkar, M.; Kumar, A.; Rani, R.; Singh, V.; Pathak, A.; Kothiala, A.; Mahajan, S.; Waghmode, B.; Kumar, R.; Mudgal, R.; et al. SAM-Dependent Viral MTase Inhibitors: Herbacetin and Caffeic Acid Phenethyl Ester, Structural Insights into Dengue MTase. bioRxiv 2024, 2022.05.31.494145. [CrossRef]
- García, L.L.; Padilla, L.; Castaño, J.C. Inhibitors Compounds of the Flavivirus Replication Process. Virol J 2017, 14. [Google Scholar] [CrossRef]
- Benarroch, D.; Egloff, M.P.; Mulard, L.; Guerreiro, C.; Romette, J.L.; Canard, B. A Structural Basis for the Inhibition of the NS5 Dengue Virus MRNA 2′-O-Methyltransferase Domain by Ribavirin 5′-Triphosphate. Journal of Biological Chemistry 2004, 279, 35638–35643. [Google Scholar] [CrossRef]
- Milani, M.; Mastrangelo, E.; Bollati, M.; Selisko, B.; Decroly, E.; Bouvet, M.; Canard, B.; Bolognesi, M. Flaviviral Methyltransferase/RNA Interaction: Structural Basis for Enzyme Inhibition. Antiviral Res 2009, 83, 28–34. [Google Scholar] [CrossRef]
- Nencka, R.; Silhan, J.; Klima, M.; Otava, T.; Kocek, H.; Krafcikova, P.; Boura, E. Coronaviral RNA-Methyltransferases: Function, Structure and Inhibition. Nucleic Acids Res 2022, 50, 635–650. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Song, Y. Perspective for Drug Discovery Targeting SARS Coronavirus Methyltransferases: Function, Structure and Inhibition. J Med Chem 2024. [CrossRef] [PubMed]
- Pyle, A.M. Translocation and Unwinding Mechanisms of RNA and DNA Helicases. Annu Rev Biophys 2008, 37, 317–336. [Google Scholar] [CrossRef]
- Kolykhalov, A.A.; Mihalik, K.; Feinstone, S.M.; Rice, C.M. Hepatitis C Virus-Encoded Enzymatic Activities and Conserved RNA Elements in the 3′ Nontranslated Region Are Essential for Virus Replication In Vivo. J Virol 2000, 74, 2046–2051. [Google Scholar] [CrossRef]
- Jankowsky, E.; Gross, C.H.; Shuman, S.; Pyle, A.M. The DExH Protein NPH-II Is a Processive and Directional Motor for Unwinding RNA. Nature 2000, 403, 447–451. [Google Scholar] [CrossRef]
- Newman, J.A.; Douangamath, A.; Yadzani, S.; Yosaatmadja, Y.; Aimon, A.; Brandão-Neto, J.; Dunnett, L.; Gorrie-stone, T.; Skyner, R.; Fearon, D.; et al. Structure, Mechanism and Crystallographic Fragment Screening of the SARS-CoV-2 NSP13 Helicase. Nat Commun 2021, 12. [Google Scholar] [CrossRef]
- Smelkova, N. V; Borowiec, J.A. Dimerization of Simian Virus 40 T-Antigen Hexamers Activates T-Antigen DNA Helicase Activity. J Virol 1997, 71, 8766–8773. [Google Scholar] [CrossRef]
- Hughes, F.J.; Romanos, M.A. E1 Protein of Human Papillomavirus Is a DNA Helicase/ATPase. Nucleic Acids Res 1993, 21, 5817–5823. [Google Scholar] [CrossRef]
- Xu, T.; Sampath, A.; Chao, A.; Wen, D.; Nanao, M.; Chene, P.; Vasudevan, S.G.; Lescar, J. Structure of the Dengue Virus Helicase/Nucleoside Triphosphatase Catalytic Domain at a Resolution of 2.4 Å. J Virol 2005, 79, 10278–10288. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, Y.; Yang, M.; Yang, J.; Shao, Z.; Gao, Y.; Jiang, X.; Cui, R.; Zhang, Y.; Zhao, X.; et al. Structural and Functional Insights into the Helicase Protein E5 of Mpox Virus. Cell Discov 2024, 10. [Google Scholar] [CrossRef]
- Law, Y.-S.; Wang, S.; Tan, Y.B.; Shih, O.; Utt, A.; Goh, W.Y.; Lian, B.-J.; Chen, M.W.; Jeng, U.-S.; Merits, A.; et al. Interdomain Flexibility of Chikungunya Virus NsP2 Helicase-Protease Differentially Influences Viral RNA Replication and Infectivity. J Virol 2021, 95. [Google Scholar] [CrossRef] [PubMed]
- Lou, Z.; Sun, Y.; Rao, Z. Current Progress in Antiviral Strategies. Trends Pharmacol Sci 2014, 35, 86–102. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Bera, A.K.; Kuhn, R.J.; Smith, J.L. Structure of the Flavivirus Helicase: Implications for Catalytic Activity, Protein Interactions, and Proteolytic Processing. J Virol 2005, 79, 10268–10277. [Google Scholar] [CrossRef] [PubMed]
- Tortorici, M.A.; Duquerroy, S.; Kwok, J.; Vonrhein, C.; Perez, J.; Lamp, B.; Bricogne, G.; Rümenapf, T.; Vachette, P.; Rey, F.A. X-Ray Structure of the Pestivirus NS3 Helicase and Its Conformation in Solution. J Virol 2015, 89, 4356–4371. [Google Scholar] [CrossRef]
- Fang, X.; Lu, G.; Deng, Y.; Yang, S.; Hou, C.; Gong, P. Unusual Substructure Conformations Observed in Crystal Structures of a Dicistrovirus RNA-Dependent RNA Polymerase Suggest Contribution of the N-Terminal Extension in Proper Folding. Virol Sin 2023, 38, 531–540. [Google Scholar] [CrossRef]
- Anindita, P.D.; Halbeisen, M.; Řeha, D.; Tuma, R.; Franta, Z. Mechanistic Insight into the RNA-Stimulated ATPase Activity of Tick-Borne Encephalitis Virus Helicase. Journal of Biological Chemistry 2022, 298. [Google Scholar] [CrossRef]
- Shao, Z.; Su, S.; Yang, J.; Zhang, W.; Gao, Y.; Zhao, X.; Zhang, Y.; Shao, Q.; Cao, C.; Li, H.; et al. Structures and Implications of the C962R Protein of African Swine Fever Virus. Nucleic Acids Res 2023, 51, 9475–9490. [Google Scholar] [CrossRef]
- Xu, T.; Sampath, A.; Chao, A.; Wen, D.; Nanao, M.; Chene, P.; Vasudevan, S.G.; Lescar, J. Structure of the Dengue Virus Helicase/Nucleoside Triphosphatase Catalytic Domain at a Resolution of 2.4 Å. J Virol 2005, 79, 10278–10288. [Google Scholar] [CrossRef]
- Gu, M.; Rice, C.M. Three Conformational Snapshots of the Hepatitis C Virus NS3 Helicase Reveal a Ratchet Translocation Mechanism. Proc Natl Acad Sci U S A 2010, 107, 521–528. [Google Scholar] [CrossRef]
- Hutin, S.; Ling, W.L.; Tarbouriech, N.; Schoehn, G.; Grimm, C.; Fischer, U.; Burmeister, W.P. The Vaccinia Virus DNA Helicase Structure from Combined Single-Particle Cryo-Electron Microscopy and AlphaFold2 Prediction. Viruses 2022, 14. [Google Scholar] [CrossRef]
- Liu, D.; Wang, Y. Sen; Gesell, J.J.; Wyss, D.F. Solution Structure and Backbone Dynamics of an Engineered Arginine-Rich Subdomain 2 of the Hepatitis C Virus NS3 RNA Helicase. J Mol Biol 2001, 314, 543–561. [Google Scholar] [CrossRef] [PubMed]
- Hutin, S.; Ling, W.L.; Tarbouriech, N.; Schoehn, G.; Grimm, C.; Fischer, U.; Burmeister, W.P. The Vaccinia Virus DNA Helicase Structure from Combined Single-Particle Cryo-Electron Microscopy and AlphaFold2 Prediction. Viruses 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Frick, D.; Lam, A. Understanding Helicases as a Means of Virus Control. Curr Pharm Des 2006, 12, 1315–1338. [Google Scholar] [CrossRef]
- Tan, Q.; Zhu, Y.; Li, J.; Chen, Z.; Han, G.W.; Kufareva, I.; Li, T.; Ma, L.; Fenalti, G.; Li, J.; et al. Structure of the CCR5 Chemokine Receptor-HIV Entry Inhibitor Maraviroc Complex. Science (1979) 2013, 341, 1387–1390. [Google Scholar] [CrossRef]
- Wu, B.; Chien, E.Y.T.; Mol, C.D.; Fenalti, G.; Liu, W.; Katritch, V.; Abagyan, R.; Brooun, A.; Wells, P.; Bi, F.C.; et al. Structures of the CXCR4 Chemokine GPCR with Small-Molecule and Cyclic Peptide Antagonists. Science (1979) 2010, 330, 1066–1071. [Google Scholar] [CrossRef]
- Haqqani, A.A.; Tilton, J.C. Entry Inhibitors and Their Use in the Treatment of HIV-1 Infection. Antiviral Res 2013, 98, 158–170. [Google Scholar] [CrossRef]
- Tan, Q.; Zhu, Y.; Li, J.; Chen, Z.; Han, G.W.; Kufareva, I.; Li, T.; Ma, L.; Fenalti, G.; Li, J.; et al. Structure of the CCR5 Chemokine Receptor-HIV Entry Inhibitor Maraviroc Complex. Science (1979) 2013, 341, 1387–1390. [Google Scholar] [CrossRef]
- Blair, H.A. Ibalizumab: A Review in Multidrug-Resistant HIV-1 Infection. Drugs 2020, 80, 189–196. [Google Scholar] [CrossRef]
- Freeman, M.M.; Seaman, M.S.; Rits-Volloch, S.; Hong, X.; Kao, C.Y.; Ho, D.D.; Chen, B. Crystal Structure of HIV-1 Primary Receptor CD4 in Complex with a Potent Antiviral Antibody. Structure 2010, 18, 1632–1641. [Google Scholar] [CrossRef]
- Bhutkar, M.; Singh, V.; Dhaka, P.; Tomar, S. Virus-Host Protein-Protein Interactions as Molecular Drug Targets for Arboviral Infections. Frontiers in Virology 2022, 2. [Google Scholar] [CrossRef]
- Mahajan, S.; Choudhary, S.; Kumar, P.; Tomar, S. Antiviral Strategies Targeting Host Factors and Mechanisms Obliging +ssRNA Viral Pathogens. Bioorg Med Chem 2021, 46, 116356. [Google Scholar] [CrossRef] [PubMed]
- Dhaka, P.; Singh, A.; Nehul, S.; Choudhary, S.; Panda, P.K.; Sharma, G.K.; Kumar, P.; Tomar, S. Disruption of Molecular Interactions between the G3BP1 Stress Granule Host Protein and the Nucleocapsid (NTD-N) Protein Impedes SARS-CoV-2 Virus Replication. Biochemistry 2024. [Google Scholar] [CrossRef]
- Mahajan, S.; Kumar, R.; Singh, A.; Pareek, A.; Kumar, P.; Tomar, S. Targeting the Host Protein G3BP1 for the Discovery of Novel Antiviral Inhibitors against Chikungunya Virus. bioRxiv 2024, 2022.11.11.516135. [CrossRef]
- De Chassey, B.; Meyniel-Schicklin, L.; Aublin-Gex, A.; André, P.; Lotteau, V. New Horizons for Antiviral Drug Discovery from Virus–Host Protein Interaction Networks. Curr Opin Virol 2012, 2, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Idrees, S.; Chen, H.; Panth, N.; Paudel, K.R.; Hansbro, P.M. Exploring Viral–Host Protein Interactions as Antiviral Therapies: A Computational Perspective. Microorganisms 2024, 12. [Google Scholar] [CrossRef] [PubMed]
- Idrees, S.; Paudel, K.R.; Sadaf, T.; Hansbro, P.M. How Different Viruses Perturb Host Cellular Machinery via Short Linear Motifs. EXCLI J 2023, 22, 1113–1128. [Google Scholar] [CrossRef]
- de Chassey, B.; Meyniel-Schicklin, L.; Vonderscher, J.; André, P.; Lotteau, V. Virus-Host Interactomics: New Insights and Opportunities for Antiviral Drug Discovery. Genome Med 2014, 6. [Google Scholar] [CrossRef]
- Brito, A.F.; Pinney, J.W. Protein-Protein Interactions in Virus-Host Systems. Front Microbiol 2017, 8. [Google Scholar] [CrossRef]
- Kumar, N.; Sharma, S.; Kumar, R.; Tripathi, B.N.; Barua, S.; Ly, H.; Rouse, B.T. Host-Directed Antiviral Therapy. Clin Microbiol Rev 2020, 33. [Google Scholar] [CrossRef]
- Mahajan, S.; Choudhary, S.; Kumar, P.; Tomar, S. Antiviral Strategies Targeting Host Factors and Mechanisms Obliging +ssRNA Viral Pathogens. Bioorg Med Chem 2021, 46. [Google Scholar] [CrossRef]
- Eslami, N.; Aghbash, P.S.; Shamekh, A.; Entezari-Maleki, T.; Nahand, J.S.; Sales, A.J.; Baghi, H.B. SARS-CoV-2: Receptor and Co-Receptor Tropism Probability. Curr Microbiol 2022, 79, 1–13. [Google Scholar] [CrossRef]
- Baggen, J.; Vanstreels, E.; Jansen, S.; Daelemans, D. Cellular Host Factors for SARS-CoV-2 Infection. Nature Microbiology 2021 6:10 2021, 6, 1219–1232. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.S.; Diamond, M.S. A Molecular Understanding of Alphavirus Entry and Antibody Protection. Nature Reviews Microbiology 2022 21:6 2022, 21, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.F.; Wu, Y.S.; Poh, C.L. Molecular Mechanisms of Antiviral Agents against Dengue Virus. Viruses 2023, 15. [Google Scholar] [CrossRef] [PubMed]
- Anwar, M.N.; Akhtar, R.; Abid, M.; Khan, S.A.; Rehman, Z.U.; Tayyub, M.; Malik, M.I.; Shahzad, M.K.; Mubeen, H.; Qadir, M.S.; et al. The Interactions of Flaviviruses with Cellular Receptors: Implications for Virus Entry. Virology 2022, 568, 77–85. [Google Scholar] [CrossRef]
- Li, H.; Huang, Q.Z.; Zhang, H.; Liu, Z.X.; Chen, X.H.; Ye, L.L.; Luo, Y. The Land-Scape of Immune Response to Monkeypox Virus. EBioMedicine 2023, 87, 104424. [Google Scholar] [CrossRef]
- Chakraborty, C.; Sharma, A.R.; Bhattacharya, M.; Lee, S.S. A Detailed Overview of Immune Escape, Antibody Escape, Partial Vaccine Escape of SARS-CoV-2 and Their Emerging Variants With Escape Mutations. Front Immunol 2022, 13, 801522. [Google Scholar] [CrossRef]
- Lemaitre, J.C.; Grantz, K.H.; Kaminsky, J.; Meredith, H.R.; Truelove, S.A.; Lauer, S.A.; Keegan, L.T.; Shah, S.; Wills, J.; Kaminsky, K.; et al. A Scenario Modeling Pipeline for COVID-19 Emergency Planning. Scientific Reports 2021 11:1 2021, 11, 1–13. [Google Scholar] [CrossRef]
- Crucitti, D.; Pérez Míguez, C.; Ángel, J.; Arias, D.; Beltrá, D.; Prada, F.; Orgueira, A.M. De Novo Drug Design through Artificial Intelligence: An Introduction. Frontiers in Hematology 2024, 3, 1305741. [Google Scholar] [CrossRef]
- Floresta, G.; Zagni, C.; Patamia, V.; Rescifina, A. How Can Artificial Intelligence Be Utilized for de Novo Drug Design against COVID-19 (SARS-CoV-2)? Expert Opin Drug Discov 2023, 18, 1061–1064. [Google Scholar] [CrossRef]
- Patel, C.N.; Mall, R.; Bensmail, H. AI-Driven Drug Repurposing and Binding Pose Meta Dynamics Identifies Novel Targets for Monkeypox Virus. J Infect Public Health 2023, 16, 799–807. [Google Scholar] [CrossRef]
- Slater, A.; Nair, N.; Suétt, R.; Donnchadha, R. Mac; Bamford, C.; Jasim, S.; Livingstone, D.; Hutchinson, E. Visualising Viruses. Journal of General Virology 2022, 103, 001730. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Yang, Q.; Ran, T.; Zhang, G.; Li, W.; Zhou, P.; Tang, J.; Dai, M.; Zhong, J.; Chen, H.; et al. Discovery of Orally Bioavailable SARS-CoV-2 Papain-like Protease Inhibitor as a Potential Treatment for COVID-19. Nature Communications 2024 15:1 2024, 15, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Turzynski, V.; Monsees, I.; Moraru, C.; Probst, A.J. Imaging Techniques for Detecting Prokaryotic Viruses in Environmental Samples. Viruses 2021, Vol. 13, Page 2126 2021, 13, 2126. [Google Scholar] [CrossRef] [PubMed]
- Turzynski, V.; Monsees, I.; Moraru, C.; Probst, A.J. Imaging Techniques for Detecting Prokaryotic Viruses in Environmental Samples. Viruses 2021, Vol. 13, Page 2126 2021, 13, 2126. [Google Scholar] [CrossRef]
- Greasley, S.E.; Noell, S.; Plotnikova, O.; Ferre, R.A.; Liu, W.; Bolanos, B.; Fennell, K.; Nicki, J.; Craig, T.; Zhu, Y.; et al. Structural Basis for the in Vitro Efficacy of Nirmatrelvir against SARS-CoV-2 Variants. Journal of Biological Chemistry 2022, 298, 101972. [Google Scholar] [CrossRef]
- Rut, W.; Lv, Z.; Zmudzinski, M.; Patchett, S.; Nayak, D.; Snipas, S.J.; Oualid, F. El; Huang, T.T.; Bekes, M.; Drag, M.; et al. Activity Profiling and Crystal Structures of Inhibitor-Bound SARS-CoV-2 Papain-like Protease: A Framework for Anti–COVID-19 Drug Design. Sci Adv 2020, 6, 4596–4612. [Google Scholar] [CrossRef]
- Feng, Q.; Cheng, K.; Zhang, L.; Wang, D.; Gao, X.; Liang, J.; Liu, G.; Ma, N.; Xu, C.; Tang, M.; et al. Rationally Designed Multimeric Nanovaccines Using Icosahedral DNA Origami for Display of SARS-CoV-2 Receptor Binding Domain. Nature Communications 2024 15:1 2024, 15, 1–17. [Google Scholar] [CrossRef]
- Dokhale, S.; Garse, S.; Devarajan, S.; Thakur, V.; Kolhapure, S. Rational Design of Antiviral Therapeutics. Computational Methods for Rational Drug Design 2025, 423–443. [Google Scholar] [CrossRef]
- Al-Amran, F.G.; Hezam, A.M.; Rawaf, S.; Yousif, M.G. Genomic Analysis and Artificial Intelligence: Predicting Viral Mutations and Future Pandemics.
- Parvatikar, P.P.; Patil, S.; Khaparkhuntikar, K.; Patil, S.; Singh, P.K.; Sahana, R.; Kulkarni, R. V.; Raghu, A. V. Artificial Intelligence: Machine Learning Approach for Screening Large Database and Drug Discovery. Antiviral Res 2023, 220, 105740. [Google Scholar] [CrossRef]
- KP Jayatunga, M.; Ayers, M.; Bruens, L.; Jayanth, D.; Meier, C. How Successful Are AI-Discovered Drugs in Clinical Trials? A First Analysis and Emerging Lessons. Drug Discov Today 2024, 29, 104009. [Google Scholar] [CrossRef]
- Mouchlis, V.D.; Afantitis, A.; Serra, A.; Fratello, M.; Papadiamantis, A.G.; Aidinis, V.; Lynch, I.; Greco, D.; Melagraki, G. Advances in De Novo Drug Design: From Conventional to Machine Learning Methods. International Journal of Molecular Sciences 2021, Vol. 22, Page 1676 2021, 22, 1676. [Google Scholar] [CrossRef] [PubMed]
- Atz, K.; Cotos, L.; Isert, C.; Håkansson, M.; Focht, D.; Hilleke, M.; Nippa, D.F.; Iff, M.; Ledergerber, J.; Schiebroek, C.C.G.; et al. Prospective de Novo Drug Design with Deep Interactome Learning. Nature Communications 2024 15:1 2024, 15, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Bhutkar, M.; Choudhary, S.; Nehul, S.; Kumar, R.; Singla, J.; Kumar, P.; Tomar, S. Structure-Guided Mutations in CDRs for Enhancing the Affinity of Neutralizing SARS-CoV-2 Nanobody. Biochem Biophys Res Commun 2024, 734, 150746. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Choudhary, S.; Bhutkar, M.; Nehul, S.; Ali, S.; Singla, J.; Kumar, P.; Tomar, S. Designing and Bioengineering of CDRs with Higher Affinity against Receptor-Binding Domain (RBD) of SARS-CoV-2 Omicron Variant 2024.
- Sinha, S.; Vegesna, R.; Mukherjee, S.; Kammula, A. V.; Dhruba, S.R.; Wu, W.; Kerr, D.L.; Nair, N.U.; Jones, M.G.; Yosef, N.; et al. PERCEPTION Predicts Patient Response and Resistance to Treatment Using Single-Cell Transcriptomics of Their Tumors. Nature Cancer 2024 5:6 2024, 5, 938–952. [Google Scholar] [CrossRef]
- Mak, K.-K.; Wong, Y.-H.; Pichika, M.R. Artificial Intelligence in Drug Discovery and Development. Drug Discovery and Evaluation: Safety and Pharmacokinetic Assays 2024, 1461–1498. [CrossRef]
- He, H.; He, B.; Guan, L.; Zhao, Y.; Jiang, F.; Chen, G.; Zhu, Q.; Chen, C.Y.C.; Li, T.; Yao, J. De Novo Generation of SARS-CoV-2 Antibody CDRH3 with a Pre-Trained Generative Large Language Model. Nature Communications 2024 15:1 2024, 15, 1–19. [Google Scholar] [CrossRef]
- Dunbar, J.; Krawczyk, K.; Leem, J.; Marks, C.; Nowak, J.; Regep, C.; Georges, G.; Kelm, S.; Popovic, B.; Deane, C.M. SAbPred: A Structure-Based Antibody Prediction Server. Nucleic Acids Res 2016, 44, W474–W478. [Google Scholar] [CrossRef]
- Caradonna, T.M.; Schmidt, A.G. Protein Engineering Strategies for Rational Immunogen Design. npj Vaccines 2021 6:1 2021, 6, 1–11. [Google Scholar] [CrossRef]
- Shanehsazzadeh, A.; McPartlon, M.; Kasun, G.; Steiger, A.K.; Sutton, J.M.; Yassine, E.; McCloskey, C.; Haile, R.; Shuai, R.; Alverio, J.; et al. Unlocking de Novo Antibody Design with Generative Artificial Intelligence. bioRxiv 2024, 2023.01.08.523187. [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021 596:7873 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate Structure Prediction of Biomolecular Interactions with AlphaFold 3. Nature 2024 630:8016 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Watson, J.L.; Juergens, D.; Bennett, N.R.; Trippe, B.L.; Yim, J.; Eisenach, H.E.; Ahern, W.; Borst, A.J.; Ragotte, R.J.; Milles, L.F.; et al. De Novo Design of Protein Structure and Function with RFdiffusion. Nature 2023 620:7976 2023, 620, 1089–1100. [Google Scholar] [CrossRef]
- Greasley, S.E.; Noell, S.; Plotnikova, O.; Ferre, R.A.; Liu, W.; Bolanos, B.; Fennell, K.; Nicki, J.; Craig, T.; Zhu, Y.; et al. Structural Basis for the in Vitro Efficacy of Nirmatrelvir against SARS-CoV-2 Variants. Journal of Biological Chemistry 2022, 298, 101972. [Google Scholar] [CrossRef] [PubMed]
- Mohan, S.; Kerry, P.S.; Bance, N.; Niikura, M.; Pinto, B.M. Serendipitous Discovery of a Potent Influenza Virus A Neuraminidase Inhibitor. Angewandte Chemie International Edition 2014, 53, 1076–1080. [Google Scholar] [CrossRef] [PubMed]
- Rut, W.; Lv, Z.; Zmudzinski, M.; Patchett, S.; Nayak, D.; Snipas, S.J.; Oualid, F. El; Huang, T.T.; Bekes, M.; Drag, M.; et al. Activity Profiling and Crystal Structures of Inhibitor-Bound SARS-CoV-2 Papain-like Protease: A Framework for Anti–COVID-19 Drug Design. Sci Adv 2020, 6, 4596–4612. [Google Scholar] [CrossRef]
- Yuan, M.; Zhu, X.; He, W.T.; Zhou, P.; Kaku, C.I.; Capozzola, T.; Zhu, C.Y.; Yu, X.; Liu, H.; Yu, W.; et al. A Broad and Potent Neutralization Epitope in SARS-Related Coronaviruses. Proc Natl Acad Sci U S A 2022, 119, e2205784119. [Google Scholar] [CrossRef]
- Walls, A.C.; Fiala, B.; Schäfer, A.; Wrenn, S.; Pham, M.N.; Murphy, M.; Tse, L. V.; Shehata, L.; O’Connor, M.A.; Chen, C.; et al. Elicitation of Potent Neutralizing Antibody Responses by Designed Protein Nanoparticle Vaccines for SARS-CoV-2. Cell 2020, 183, 1367–1382.e17. [Google Scholar] [CrossRef]
- Feng, Q.; Cheng, K.; Zhang, L.; Wang, D.; Gao, X.; Liang, J.; Liu, G.; Ma, N.; Xu, C.; Tang, M.; et al. Rationally Designed Multimeric Nanovaccines Using Icosahedral DNA Origami for Display of SARS-CoV-2 Receptor Binding Domain. Nature Communications 2024 15:1 2024, 15, 1–17. [Google Scholar] [CrossRef]
- Brouwer, P.J.M.; Antanasijevic, A.; Ronk, A.J.; Müller-Kräuter, H.; Watanabe, Y.; Claireaux, M.; Perrett, H.R.; Bijl, T.P.L.; Grobben, M.; Umotoy, J.C.; et al. Lassa Virus Glycoprotein Nanoparticles Elicit Neutralizing Antibody Responses and Protection. Cell Host Microbe 2022, 30, 1759–1772.e12. [Google Scholar] [CrossRef]
- Sesterhenn, F.; Yang, C.; Bonet, J.; Cramer, J.T.; Wen, X.; Wang, Y.; Chiang, C.I.; Abriata, L.A.; Kucharska, I.; Castoro, G.; et al. De Novo Protein Design Enables the Precise Induction of RSV-Neutralizing Antibodies. Science (1979) 2020, 368. [Google Scholar] [CrossRef]
- Marcandalli, J.; Fiala, B.; Ols, S.; Perotti, M.; de van der Schueren, W.; Snijder, J.; Hodge, E.; Benhaim, M.; Ravichandran, R.; Carter, L.; et al. Induction of Potent Neutralizing Antibody Responses by a Designed Protein Nanoparticle Vaccine for Respiratory Syncytial Virus. Cell 2019, 176, 1420–1431.e17. [Google Scholar] [CrossRef]
- Mousa, J.J.; Sauer, M.F.; Sevy, A.M.; Finn, J.A.; Bates, J.T.; Alvarado, G.; King, H.G.; Loerinc, L.B.; Fong, R.H.; Doranz, B.J.; et al. Structural Basis for Nonneutralizing Antibody Competition at Antigenic Site II of the Respiratory Syncytial Virus Fusion Protein. Proc Natl Acad Sci U S A 2016, 113, E6849–E6858. [Google Scholar] [CrossRef]
- Baum, A.; Fulton, B.O.; Wloga, E.; Copin, R.; Pascal, K.E.; Russo, V.; Giordano, S.; Lanza, K.; Negron, N.; Ni, M.; et al. Antibody Cocktail to SARS-CoV-2 Spike Protein Prevents Rapid Mutational Escape Seen with Individual Antibodies. Science (1979) 2020, 369, 1014–1018. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Behre, G.; Hebert, C.; Kumar, P.; Farmer MacPherson, L.; Graham-Clarke, P.L.; De La Torre, I.; Nichols, R.M.; Hufford, M.M.; Patel, D.R.; et al. Bamlanivimab and Etesevimab Improve Symptoms and Associated Outcomes in Ambulatory Patients at Increased Risk for Severe Coronavirus Disease 2019: Results From the Placebo-Controlled Double-Blind Phase 3 BLAZE-1 Trial. Open Forum Infect Dis 2022, 9. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Zost, S.J.; Greaney, A.J.; Starr, T.N.; Dingens, A.S.; Chen, E.C.; Chen, R.E.; Case, J.B.; Sutton, R.E.; Gilchuk, P.; et al. Genetic and Structural Basis for SARS-CoV-2 Variant Neutralization by a Two-Antibody Cocktail. Nature Microbiology 2021 6:10 2021, 6, 1233–1244. [Google Scholar] [CrossRef] [PubMed]
- Focosi, D.; Casadevall, A.; Franchini, M.; Maggi, F. Sotrovimab: A Review of Its Efficacy against SARS-CoV-2 Variants. Viruses 2024, Vol. 16, Page 217 2024, 16, 217. [Google Scholar] [CrossRef] [PubMed]
- Vishweshwaraiah, Y.L.; Hnath, B.; Rackley, B.; Wang, J.; Gontu, A.; Chandler, M.; Afonin, K.A.; Kuchipudi, S. V; Christensen, N.; Yennawar, N.H.; et al. Adaptation-Proof SARS-CoV-2 Vaccine Design. Adv Funct Mater 2022, 32, 2206055. [Google Scholar] [CrossRef]
- Liu, Q.; Lu, Y.; Cai, C.; Huang, Y.; Zhou, L.; Guan, Y.; Fu, S.; Lin, Y.; Yan, H.; Zhang, Z.; et al. A Broad Neutralizing Nanobody against SARS-CoV-2 Engineered from an Approved Drug. Cell Death & Disease 2024 15:6 2024, 15, 1–9. [Google Scholar] [CrossRef]
- Pathogens Prioritization: A Scientific Framework for Epidemic and Pandemic Research Preparedness.
- Bardhan, M.; Ray, I.; Roy, S.; Roy, P.; Thanneeru, P.; Twayana, A.R.; Prasad, S.; Bardhan, M.; Anand, A. Disease X and COVID-19: Turning Lessons from India and the World into Policy Recommendations. Ann Med Surg (Lond) 2024, 86, 5914–5921. [Google Scholar] [CrossRef]
Figure 1.
Graphical representation of various viruses exhibiting diverse characteristics based on their structural composition and genetic organization. The figure above highlights examples of various virus types based on their diverse features.
Figure 1.
Graphical representation of various viruses exhibiting diverse characteristics based on their structural composition and genetic organization. The figure above highlights examples of various virus types based on their diverse features.
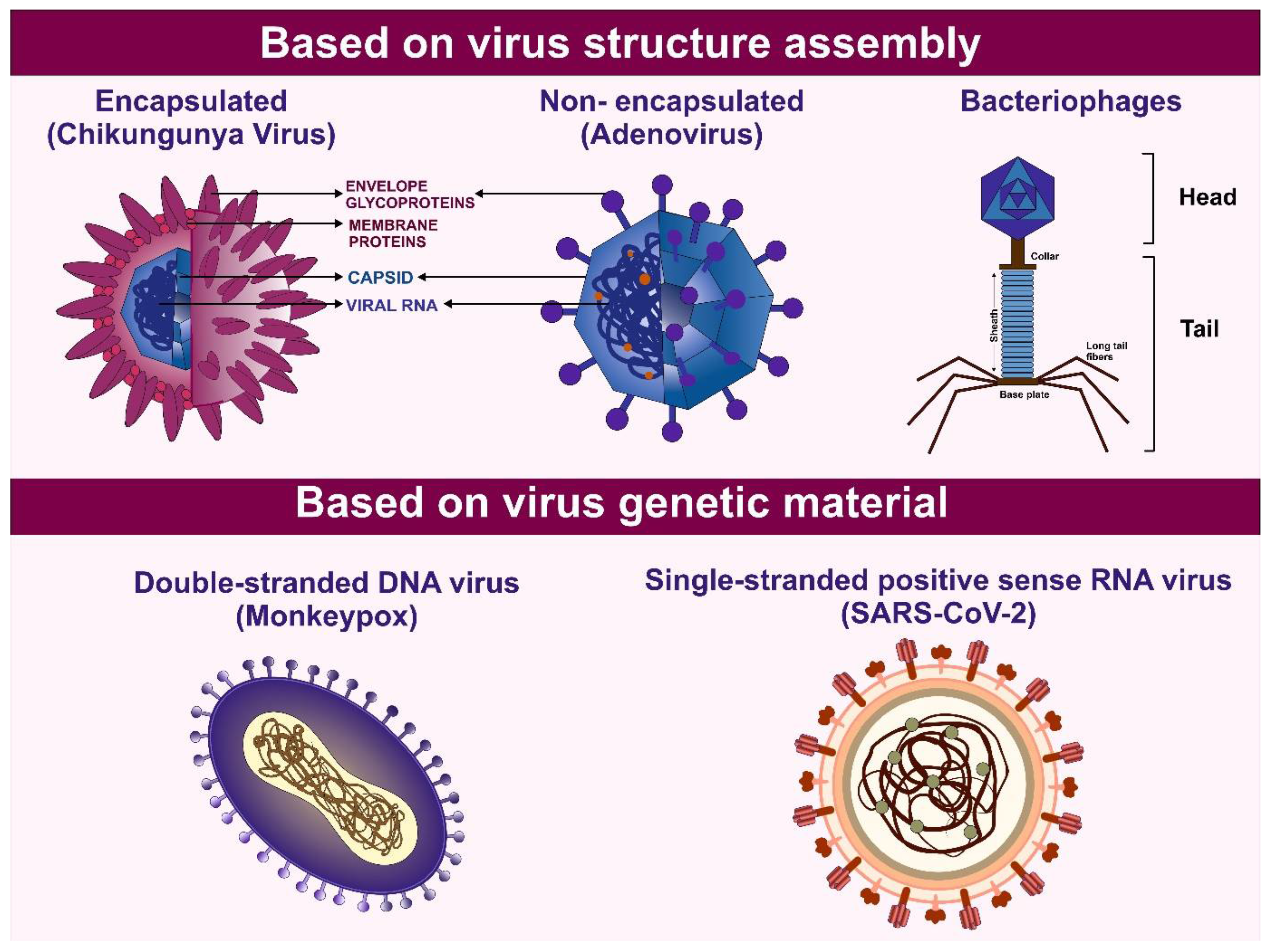

Table 1.
List of FDA approved therapeutics against viral structural proteins.
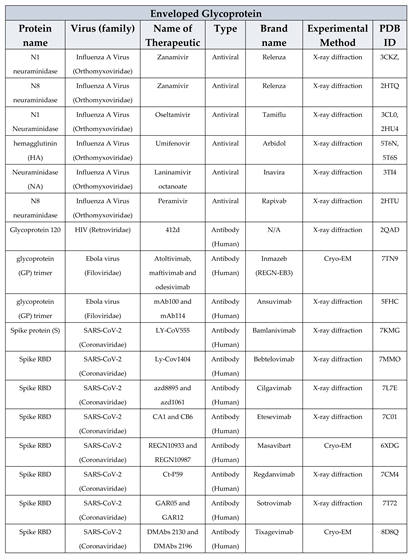
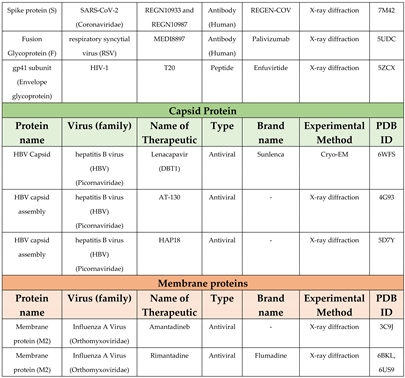
a Approved for use in Japan, not FDA approved; b Discontinued as a drug.
Table 2.
List of FDA approved drugs targeting viral replication enzymes.
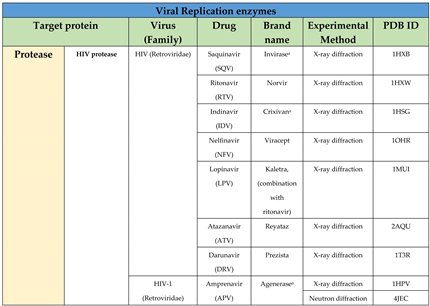
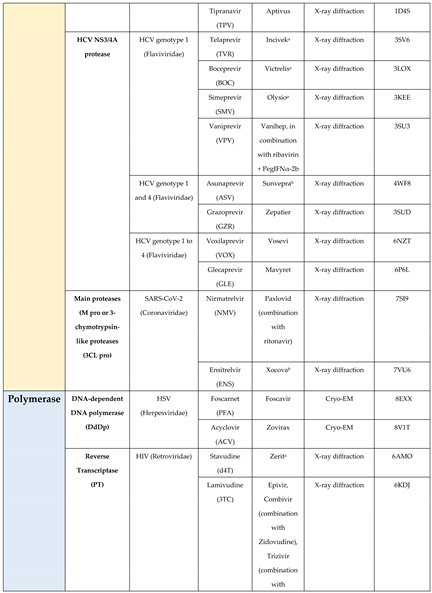
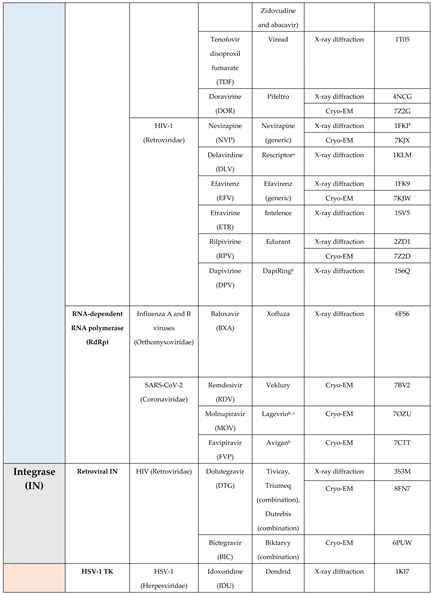
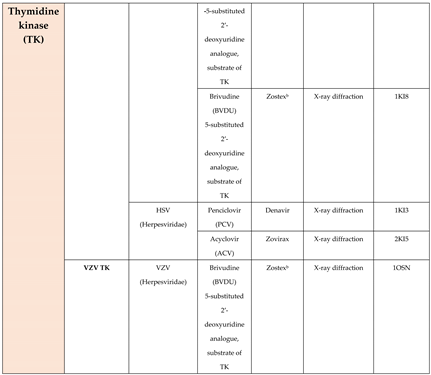
a Discontinued; b Approved in some countries, not FDA approved; c Emergency use authorization (EUA) by FDA.
Table 3.
List of therapeutics and the rational approaches used in their designing.
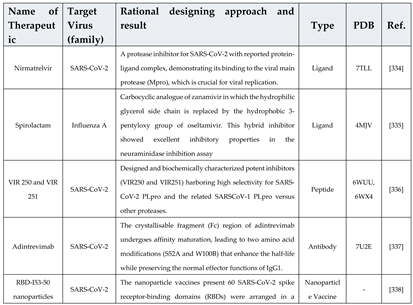
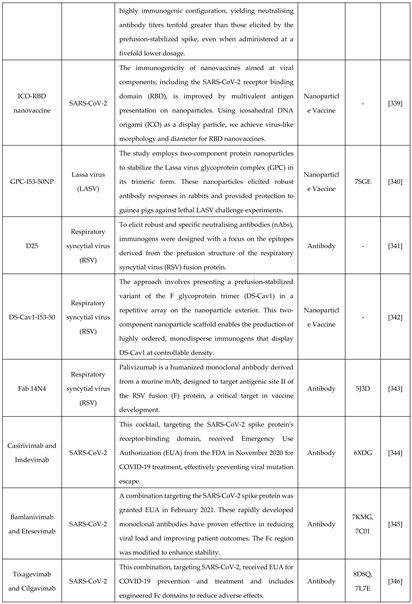
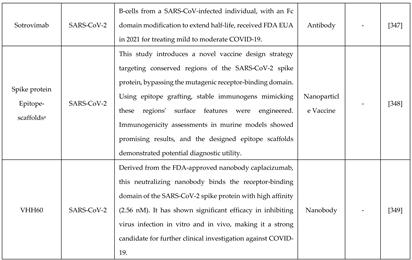
Not clinically approved.
Table 4.
Virus families and pathogens identified as high-priority Public Health Emergencies of International Concern (PHEICs) for 2024. The corresponding PDB IDs of structural and non-structural proteins for these "high-risk" viruses are provided, highlighting potential antiviral therapeutic targets. (Source: WHO Pathogen Prioritization Report, 2024).
Table 4.
Virus families and pathogens identified as high-priority Public Health Emergencies of International Concern (PHEICs) for 2024. The corresponding PDB IDs of structural and non-structural proteins for these "high-risk" viruses are provided, highlighting potential antiviral therapeutic targets. (Source: WHO Pathogen Prioritization Report, 2024).
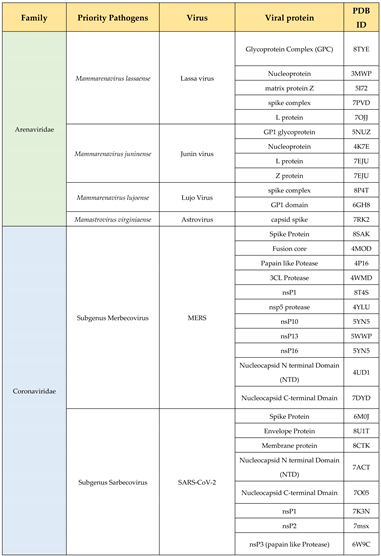
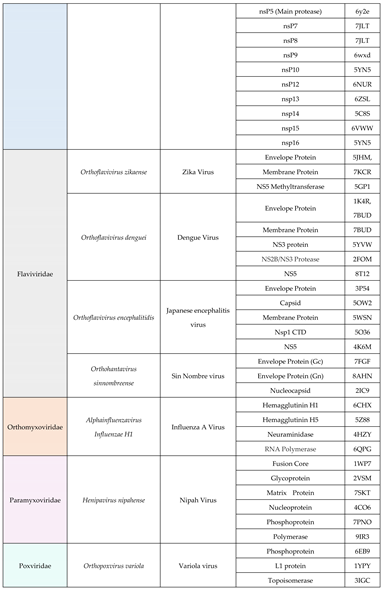
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



