
Submitted:
13 January 2025
Posted:
14 January 2025
You are already at the latest version
Abstract
Cardiovascular drugs are widely used for the prevention and treatment of various cardiac and vascular disorders. However, some of these drugs can also cause adverse effects on the kidney, leading to acute or chronic renal dysfunction, electrolyte imbalances, and increased mortality. The mechanisms of drug-induced renal toxicity vary depending on the type and class of the drug, the dose and duration of exposure, and the patient’s characteristics and comorbidities. In this review, we summarize the current knowledge on the renal effects of some common cardiovascular drugs, such as diuretics, angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, calcium channel blockers, beta-blockers, antiplatelet agents, anticoagulants, and statins and proton-pump inhibitors. We also discuss the clinical implications and management strategies for preventing or minimizing drug-induced nephrotoxicity, as well as the potential role of oxidative stress in its pathogenesis.
Keywords:
drugs for the heart
; nephrotoxicity
; glomerulopathies
1. Introduction
The kidney plays a vital role in maintaining fluid and electrolyte homeostasis, regulating blood pressure, excreting metabolic waste products, and producing hormones. All the waste products in particular can reach a high concentration in the kidneys for 3 main reasons: 1)very high perfusion, 2)the presence of a variety of xenobiotic transporters and metabolizing enzymes, and 3) concentration of solutes during urine production [1]. Regarding the first point, the perfusion is as high as from 800 to 1200 ml/min that means almost 20% of the cardiac output. Since the total plasm is almost 3 liters, in 24 hours all the plasma is filtered 20 times. Such high perfusion is driven by a very unique type of resistance that are arranged in parallel and not in series, so the total resistance is decreased. This accounts for the higher blood flow and high hydrostatic pressure in the glomerular capillary network. Then the glomerular blood is connected by means of the efferent arteriole with a second capillary network: the peritubular vasa recta capillary network. Such low pressure capillary network is the vascular highway for the immunocompetent cells moving to e from the interstitium and nephron tubular section [2]. Regarding the second point, the tubular cells are furnished with all the of xenobiotic transporter and metabolizing enzymes and are intermixed with resident dendritic cells (macrophages)[3]. Therefore any xenobiotic has a profound biochemical and also immunological contact with the nephrons and renal interstitium. Regarding the last point, since proximal tubules reabsorbs almost all the glomeral filtration in terms of Na+ and water, high concentration of toxic substances/drugs are reached in the tubules and after all in the interstitium [4]. For all these reasons the kidney is a major target organ for many drugs, as it is involved in their glomerular filtration, tubular secretion/reabsorption and metabolic transformation and elimination in the urine. Therefore, the kidney is susceptible to injury by various drugs that can alter its structure and function [5].
Drug-induced nephrotoxicity is a common problem in clinical medicine and the incidence of drug-related acute kidney injury (AKI) may be as high as 60 percent [6]. The condition can be costly and may require multiple interventions, including hospitalization. Drug-induced nephrotoxicity can also lead to chronic kidney disease (CKD), end-stage renal disease (ESRD), and increased cardiovascular morbidity and mortality [7].
Cardiovascular drugs are among the most frequently prescribed medications worldwide, as they are used for the prevention and treatment of various cardiac and vascular disorders, such as hypertension, heart failure, ischemic heart disease, arrhythmias, stroke, and peripheral arterial disease. However, some of these drugs can also cause adverse effects on the kidney, either directly or indirectly. The mechanisms of drug-induced renal toxicity vary depending on the type and class of the drug, the dose and duration of exposure, and the patient’s characteristics and comorbidities [8].
In this review, we summarize the current knowledge on the renal effects of some common cardiovascular drugs, such as diuretics, angiotensin-converting enzyme inhibitors (ACEIs), angiotensin receptor blockers (ARBs), calcium channel blockers (CCBs), beta-blockers (BBs), antiplatelet agents (APAs), anticoagulants (ACs), statins and proton-pump inhibitors (PPIs). We also discuss the clinical implications and management strategies for preventing or minimizing drug-induced renal toxicity. In Figure 1 the structures of each class of cardiovascular drugs are reported.
2. Mechanisms of Drug-Induced Nephrotoxicity
The mechanisms of drug-induced nephrotoxicity may differ between various drugs or drug classes, and they are generally categorized based on the histological component of the kidney that is affected. Some common ways that damage the kidneys are changes in glomerular hemodynamics, tubular cell toxicity, inflammation, crystal nephropathy, rhabdomyolysis, and thrombotic microangiopathy [9].
2.1. Changes in Glomerular Hemodynamics
The glomerular filtration rate (GFR) of healthy young individuals is 120 ml per minute [10]. The kidneys can regulate the blood flow in the afferent and efferent arterioles to keep a constant filtration rate and to ensure the proper urine output. Glomerular hemodynamics refers to the blood flow and pressure within the glomerulus, the network of capillaries that filters blood and forms urine. Changes in glomerular hemodynamics can affect the filtration rate and the permeability of the glomerular membrane, leading to proteinuria, hematuria, or reduced renal function [11]. Some drugs can alter glomerular hemodynamics by affecting the balance between vasoconstrictors and vasodilators, such as angiotensin II, prostaglandins, nitric oxide, and endothelin. For example, nonsteroidal anti-inflammatory drugs (NSAIDs) can inhibit prostaglandin synthesis and cause afferent arteriole constriction, reducing renal blood flow and glomerular filtration rate [12]. Similarly, angiotensin-converting enzyme inhibitors (ACEIs) and angiotensin receptor blockers (ARBs) can block angiotensin II-mediated efferent arteriole constriction and cause glomerular capillary pressure to drop, resulting in decreased filtration fraction [13]. Other drugs that can cause changes in glomerular hemodynamics include calcineurin inhibitors (cyclosporine and tacrolimus), contrast media, amphotericin B, cisplatin, and gentamicin [8].
2.2. Tubular Cell Toxicity
Tubular cells are responsible for reabsorbing water and solutes from the filtrate and secreting waste products into the urine. They also express various transporters, enzymes, and receptors that are involved in drug uptake, metabolism, and excretion. The renal tubules, especially the proximal tubule cells, are exposed to drugs that undergo concentration and reabsorption in the tubules, and therefore are highly susceptible to drug toxicity [14]. Some drugs can directly or indirectly impair the function or integrity of tubular cells, leading to acute tubular necrosis, tubulointerstitial inflammation, or obstructive nephropathy. The drug-induced cytotoxicity results from the damage to the mitochondria in the tubules, the disruption of the tubular transport system, and the increase in oxidative stress due to free radical production [15]. One of the most well known drugs that exert -related tubular toxicity are aminoglycosides, amphotericin and some antivirals. They disrupt cell membrane of the tubular cells but also mitochondria organelles. In particular aminoglycosides can bind to phospholipids in the cell membrane and inhibit protein synthesis, resulting in cell swelling, vacuolization, and apoptosis [16]. Amphotericin B can disrupt the cell membrane and increase the permeability to cations, causing cell dysfunction and necrosis [17,18]. Antivirals such as adefovir and foscarnet can inhibit mitochondrial DNA polymerase and cause mitochondrial toxicity [19,20]. Other drugs that can cause tubular cell toxicity include cisplatin, methotrexate, acyclovir, vancomycin, and contrast media [6,21].
2.3. Inflammation
Many drugs that are nephrotoxic can cause inflammation in the glomerulus, the proximal tubules, and the surrounding extracellular matrix, and then lead to fibrosis of the kidney tissue. Inflammation that impairs normal kidney functions and causes toxicity includes glomerulonephritis, acute and chronic interstitial nephritis, or vasculitis [22]. Some drugs can induce inflammation by triggering a hypersensitivity reaction that involves T cells, antibodies, or complement activation. For example, penicillins, cephalosporins, sulfonamides, and rifampin can cause acute interstitial nephritis (AIN), which is characterized by fever, rash, eosinophilia, and hematuria [23]. NSAIDs can also cause interstitial nephritis by inhibiting prostaglandin synthesis and increasing leukotriene production [24]. Chronic interstitial nephritis occurs frequently with long-term use of calcineurin inhibitors, lithium, some anticancer drugs or analgesics [25,26,27,28]. In case of chronic interstitial nephritis, early detection is especially important because it is difficult to diagnose until most of the kidney function is lost.
2.4. Crystal Nephropathy
Crystal nephropathy refers to the formation and deposition of insoluble crystals within the renal tubules, causing obstruction, inflammation, and injury. Crystal nephropathy can occur as a result of endogenous or exogenous factors, such as hyperuricemia, hyperoxaluria, or drug administration [29]. The risk factors for developing crystal nephropathy include dehydration, high dose or prolonged use of the drug, renal impairment, and metabolic acidosis [30]. Some drugs can form crystals in the urine due to their low solubility, high dose, or acidic pH. For example, acyclovir can precipitate as needle-shaped crystals in the distal tubules, especially when given as rapid intravenous bolus or in dehydrated patients [31,32]. Sulfonamides can form crystals that are associated with casts and interstitial nephritis [33,34]. Methotrexate can cause intratubular precipitation of 7-hydroxymethotrexate, which is a metabolite with low solubility [35]. Other drugs that can cause crystal nephropathy include indinavir, triamterene, atazanavir, and ciprofloxacin [36].
2.5. Rhabdomyolysis
Rhabdomyolysis is a syndrome of skeletal muscle breakdown that releases intracellular contents, such as myoglobin, creatine kinase, and electrolytes, into the circulation. Rhabdomyolysis can cause AKI by several mechanisms, such as direct tubular toxicity of myoglobin, tubular obstruction by myoglobin casts, vasoconstriction and ischemia, oxidative stress, and inflammation [37]. Rhabdomyolysis can be triggered by various factors, such as trauma, ischemia, infection, exercise, or drugs. Some drugs can cause rhabdomyolysis directly by inducing muscle damage or indirectly by enhancing the effects of other factors. For example, statins can cause rhabdomyolysis by inhibiting the synthesis of coenzyme Q10 and impairing mitochondrial function [38,39]. Cocaine can cause rhabdomyolysis by inducing vasoconstriction, hyperthermia, and seizures [40]. Colchicine can cause rhabdomyolysis by disrupting microtubule function and inhibiting cellular transport [41]. Other drugs that can cause rhabdomyolysis include antipsychotics, antidepressants, antihistamines, antimalarials, and opioids [42,43,44,45].
2.6. Thrombotic Microangiopathy
Thrombotic microangiopathy (TMA) is a disorder characterized by microvascular thrombosis, hemolytic anemia, thrombocytopenia, and organ damage, especially in the kidneys. TMA can be caused by various conditions, such as genetic defects, infections, autoimmune diseases, or drugs [46]. Drug-induced TMA (DITMA) can be classified into immune-mediated and dose-related/toxic mechanisms, depending on the type of drug and the timing of onset. Immune-mediated DITMA involves drug-dependent antibodies that activate platelets or endothelial cells, leading to thrombosis and inflammation. Dose-related/toxic DITMA involves drugs that directly damage the endothelium or interfere with its function, leading to exposure of subendothelial matrix and activation of coagulation [47]. For example, clopidogrel and ticlopidine can cause TMA by inducing antibodies against platelet glycoproteins or endothelial cells [48]. Cyclosporine and mitomycin-C can cause TMA by damaging the endothelium and activating the coagulation cascade [49]. Quinine can cause TMA by binding to platelets and inducing complement activation [50]. Other drugs that can cause TMA include gemcitabine, bevacizumab and sunitinib [47,51,52,53].
3. Renal Effects of Cardiovascular Drugs
3.1. Diuretics
Diuretics are drugs that increase urine output by inhibiting sodium reabsorption in different segments of the nephron. They are widely used for the treatment of hypertension, heart failure, edema, and other conditions associated with fluid overload. Diuretics can be classified into different groups according to their site of action: loop diuretics (e.g., furosemide), thiazide diuretics (e.g., hydrochlorothiazide), potassium-sparing diuretics (e.g., spironolactone), osmotic diuretics (e.g., mannitol), and carbonic anhydrase inhibitors (e.g., acetazolamide) [54].
Loop diuretics, in particular, are drugs that increase the elimination of water and electrolytes by blocking the sodium/potassium/chloride cotransporter in the thick ascending limb of Henle’s loop. They are often used in patients with or at risk of acute kidney injury (AKI) for various indications, such as volume overload, hyperkalemia, hypercalcemia, and hyperazotemia. They may have important benefits in critically ill patients who receive large volumes of fluids and can prevent or treat fluid overload and pulmonary edema, which can improve oxygenation and hemodynamics [55]. However, they can also have negative effects on renal function and outcomes in AKI patients.
Loop diuretics can cause a reduction in the effective circulating volume by inducing venodilation or diuresis, which can lead to a reduction in renal blood flow and glomerular filtration rate through the stimulation of the renin-angiotensin-aldosterone system [56]. Although some studies suggest that loop diuretics can prevent tubular obstruction by increasing urine flow and flushing out tubular debris, other studies suggest that loop diuretics can worsen tubular obstruction by acidifying the urine and enhancing the aggregation of Tamm-Horsfall protein in the tubules [57]. Loop diuretics can also cause electrolyte disturbances and metabolic alkalosis [58]. High doses of loop diuretics can be ototoxic and cause hearing impairment or ringing in the ears [59]. Loop diuretics may also affect mucociliary clearance in the respiratory tract and have some immune-suppressive effects [60].
Diuretics are usually used in CHF to relieve symptoms of pulmonary edema. Many clinical trials have suggested that in those patients with heart failure who developed worsening renal failure had a higher central venous pressure (CVP) or intra-abdominal pressure (often caused by ascites or edema of internal organs), so that treatment of reducing CVP and intra-abdominal pressure (mostly using diuretics), were given to CHF patients to prevent worsening renal failure, so as to reduce mortality [61,62,63]. However, growing evidence shows that heavy dependence on the strategy of diuretics to achieve this goal may adversely affect renal function and outcome [64]. Moreover, loop diuretics block sodium chloride uptake in the macula densa, independent of any effect on sodium and water balance, thereby stimulating the RAAS, and leading to AKI. Recently, some trials have suggested that excessive diuresis is harmful since it worsens renal function in HF patients, so as to increase the mortality [65,66].
Sometimes, AKI were caused by the combination of diuretics and other agents, such as antibiotics, ACEI/ARB and NSAIDs. Diuretics are often used in combination with ACE inhibitors to control blood pressure, but ACEIs, by causing efferent arteriolar dilatation, can further reduce intraglomerular pressure, eventually leading to AKI [67]. We recommend cautious use of diuretics and ACE inhibitors in patients especially those with underlying kidney disease. NSAIDs could inhibit PGI2 synthesis so as to affect intraglomerular hemodynamics, and it is riskier in causing AKI when combined with diuretics [24].
Despite their widespread use in AKI, there is no clear evidence that loop diuretics improve outcomes in AKI. Loop diuretics do not decrease mortality, dialysis requirement, or ICU length of stay in AKI patients. Therefore, the use of loop diuretics should not be based solely on urine output, but on careful assessment of volume status, renal function, and electrolyte balance [57].
Finally, it is useful to mention a rare but serious complication of treatment with triamterene. Triamterene is a potassium-sparing diuretic used in combination with thiazide diuretics, which is frequently associated with crystalluria, even if it can rarely cause a serious nosological entity defined as crystalline nephropathy [68]. Crystalline nephropathy is a type of kidney disease characterized by the histologic finding of crystal deposition within the kidney parenchyma, which can lead to renal tubular injury, inflammation, fibrosis, and dysfunction [29]. The best way to prevent crystalline nephropathy is to check serum creatinine and urine sediment regularly, avoid volume depletion, nonsteroidal anti-inflammatory drug use, and acid urine, in patients who are at high risk. Urine alkalinization may help to prevent urinary crystal and cast formation; however, the treatment also involves stopping triamterene and alkalinizing urine in patients who do not have oliguria [29].
3.2. Angiotensin-Converting Enzyme Inhibitors and Angiotensin Receptor Blockers
ACEIs and ARBs are drugs that inhibit the renin-angiotensin-aldosterone system (RAAS), a hormonal system that regulates blood pressure, fluid and electrolyte balance, and vascular tone. ACEIs block the conversion of angiotensin I to angiotensin II, a potent vasoconstrictor and pro-inflammatory peptide, while ARBs block the binding of angiotensin II to its receptors. ACEIs and ARBs are widely used for the treatment of hypertension, heart failure, diabetic nephropathy, and other cardiovascular and renal diseases. They have been shown to reduce cardiovascular morbidity and mortality, as well as to slow the progression of CKD and ESRD [69,70].
ACE inhibitor therapy can often improve renal blood flow (RBF) and sodium excretion rates in congestive heart failure (CHF) and slow down the progression of chronic renal disease, but it can also cause a syndrome of “functional renal insufficiency” and/or hyperkalemia. This type of AKI usually occurs soon after starting ACE inhibitor therapy but can also happen after months or years of therapy, even without any previous adverse effects [71,72,73].
Various mechanisms are implicated in the development of AKI in patients undergoing ACE inhibitor therapy. One primary cause is a reduction in renal perfusion due to a fall in mean arterial pressure (MAP) that is insufficient to maintain adequate renal perfusion or triggers significant reflex activation of renal sympathetic nerves [74]. Additionally, ACE inhibitor therapy can lead to hypotension through other potential mechanisms, such as an increase in vasodilatory prostaglandins or a decrease in total peripheral resistance, particularly in cases where cardiac output remains unchanged due to heart failure [75,76,77]. Another mechanism is in patients who are volume depleted due to diuretic therapy. In this case, ACE inhibitors can cause ARF in patients who have CHF and are undergoing diuretic therapy. Studies have shown that patients undergoing diuretic therapy who are given ACE inhibitors have a higher risk of developing AKI compared to those who are not taking diuretics [78,79]. The reason for this is that diuretics cause a decrease in blood volume, leading to reduced blood flow to the kidneys. ACE inhibitors further reduce blood flow to the kidneys by causing vasodilation of the efferent arterioles, which can result in a decrease in GFR and an increase in serum creatinine levels [80]. Another condition includes patients with high-grade bilateral renal artery stenosis or patients with atherosclerotic disease in smaller preglomerular vessels or with afferent arteriolar narrowing due to hypertension [81,82,83,84]. Moreover, ACE inhibitors may precipitate AKI in patients taking vasoconstrictor agents, such as nonsteroidal anti-inflammatory agents. This is because these agents can cause vasoconstriction of the afferent arteriole, which reduces the renal perfusion and exacerbates the previously well-described effects of diuretics and ACE inhibitors. This combination has been known as the " Triple whammy" and can result in a significant reduction of the GFR and glomerular perfusion [85,86,87].
The management of AKI from ACE inhibitors involves prompt recognition, discontinuation of the offending drug, and a close follow-up for patients with a higher risk such as those with chronic kidney disease, heart failure or volume depletion. Therefore, these patients should be monitored closely for changes in serum creatinine and potassium levels when starting or adjusting ACE inhibitor therapy. If AKI occurs, the ACE inhibitor should be stopped immediately and the patient should be assessed for volume status, blood pressure, electrolytes, and urine output. Fluid resuscitation may be needed to restore renal perfusion pressure and improve GFR. However, excessive fluid administration should be avoided in patients with heart failure or pulmonary edema [88]. In some cases, vasopressors may be required to maintain adequate blood pressure [89]. Hyperkalemia may occur as a result of reduced potassium excretion and should be treated with appropriate measures, such as sodium bicarbonate, insulin and glucose, calcium gluconate, or potassium binders [90]. Dialysis may be necessary in severe cases of AKI or hyperkalemia that are refractory to medical therapy [91].
3.3. Calcium Channel Blockers
CCBs are drugs that inhibit the influx of calcium ions into vascular smooth muscle cells and cardiac myocytes, resulting in vasodilation and negative inotropic and chronotropic effects.
Numerous studies have demonstrated that CCBs are less effective than other medications in reducing proteinuria and preventing kidney damage in patients with proteinuric kidney diseases, regardless of whether they have diabetes or not. This is due to the fact that CCBs can compromise the kidney's ability to regulate its own blood pressure, resulting in increased pressure in the glomeruli [92]. CCBs may cause, in fact, glomerular autoregulation impairment by dilating the afferent glomerular artery, thereby eliminating the potent mechanism that shields the glomerular capillaries from systemic pressure transmission. This effect can lead to increased proteinuria and potentially progressive damage to the glomerular capillary network, especially when a hypertensive condition persists [93]. This clarifies why CCBs have been found to be less effective at safeguarding the kidneys in clinical trials than RAS blockers, not because RAS blockers have additional advantages [94].
Moreover, some CCBs may also interact with other drugs metabolized by the cytochrome P450 3A4 enzyme, such as clarithromycin, and increase the risk of acute kidney injury due to hypotension and hypoperfusion [95]. The risk is higher for nifedipine, felodipine and amlodipine, and lower for verapamil and diltiazem [96]. Therefore, possible approaches could involve discontinuing the use of the calcium-channel blocker for the duration of clarithromycin treatment or selecting an antibiotic that does not inhibit CYP3A4 when it is clinically appropriate.
3.4. Beta-Blockers
BBs are drugs that inhibit the binding of catecholamines to beta-adrenergic receptors in the heart, blood vessels, and other tissues, resulting in negative inotropic and chronotropic effects, vasodilation, and reduced renin secretion. BBs are widely used for the treatment of hypertension, angina pectoris, arrhythmias, heart failure, and other cardiovascular disorders. However, BBs may also have an impact on renal function, as alpha-, beta 1- and beta 2-adrenergic receptors in the kidney mediate vasoconstriction, renin secretion and vasodilatation, respectively [97].
The effects of beta blockers on renal function may depend on several factors, such as the degree of cardioselectivity (i.e., the selectivity for beta-1 receptors over beta-2 receptors), the intrinsic sympathomimetic activity (i.e., the partial agonist activity at beta receptors), the lipid solubility (i.e., the ability to cross the blood-brain barrier), and the route of administration (i.e., oral or intravenous) of the drug [97]. The administration of beta blockers, regardless of whether they are cardio-selective or have intrinsic sympathomimetic activity, typically leads to a decrease in GFR [98,99,100]. This could be explained by several mechanisms such as by lowering cardiac output, blocking beta 2-receptors, or stimulating alpha-receptors [101]. However, it is widely known that sympathetic over-activity is a component of CKD and plays a fundamental role in sustaining hypertension and the resulting cardiac complications [102]. Even though the administration of BBs can result in statistically significant changes in renal function, these changes are typically not deemed clinically significant, even in patients who have pre-existing renal disease [97,103]. For these reasons, it is unfortunate that β-blockers are not being utilized to their maximum potential due to apprehensions regarding their possible adverse effects on renal function and glycemic control, although it is important to remember that some BBs, such as atenolol, nadolol and sotalol, are not metabolized and are excreted by the kidney, so their dose must be adjusted according to the level of renal function [104].
3.5. Antiplatelet Agents
APAs are drugs that interfere with platelet activation, clumping, and clot formation. Aspirin is the most commonly used antiplatelet drug, and it permanently inhibits cyclooxygenase (COX), which, at low doses, is used to treat or prevent cardiovascular problems and reduce the risk of cardiovascular disease. P2Y12 receptor blockers, including clopidogrel, prasugrel, and ticagrelor, are mainly used in combination with aspirin to treat acute coronary syndrome and prevent clotting in stents after percutaneous coronary intervention. However, these drugs can also cause kidney damage, especially in patients with chronic kidney disease [105].
Low-dose aspirin, with a dosage of 100 mg, is classified as a nonsteroidal anti-inflammatory drug (NSAID). Historically, NSAIDs have been considered unsafe for use in patients with chronic kidney disease (CKD) due to several mechanisms [106]. Firstly, aspirin and NSAIDs are drugs that block the production of prostaglandins, which are lipid mediators that regulate various physiological processes, including inflammation, pain, and blood flow [107]. Prostaglandins are synthesized from arachidonic acid by the enzyme COX, which has two isoforms: COX-1 and COX-2. COX-1 is constitutively expressed in most tissues and is responsible for the production of prostaglandins that protect the gastric mucosa, regulate platelet aggregation, and maintain renal perfusion. COX-2 is inducible by inflammatory stimuli and is responsible for the production of prostaglandins that mediate inflammation, pain, and fever [108]. Aspirin and NSAIDs inhibit both COX-1 and COX-2, but with different degrees of selectivity. Aspirin irreversibly acetylates COX-1 and COX-2, while most NSAIDs reversibly bind to the active site of both isoforms [109]. By inhibiting COX-1 and COX-2, aspirin and NSAIDs reduce inflammation and pain, but they also cause side effects in the gastrointestinal tract, kidney, and platelets [110]. In the kidneys, prostaglandins act as vasodilators of the afferent arteriole, increasing renal blood flow and GFR, but they can also modulate the activity of the renin-angiotensin-aldosterone system and the sympathetic nervous system to ensure sufficient blood flow to the organs [111,112]. Prostaglandins also regulate sodium and water excretion by influencing tubular reabsorption and have an antagonistic effect on the receptors for antidiuretic hormone (ADH), thereby promoting diuresis [113,114]. By blocking the production of prostaglandins, aspirin and NSAIDs impair these renal functions, resulting in a reduction of total renal perfusion and GFR and sodium and water retention, leading to renal vasoconstriction and medullary ischemia and finally culminating in AKI or, in the long term, worsen CKD [24,115,116]. The risk of aspirin- and NSAID-induced kidney damage is higher in patients who have dehydration, heart failure, liver cirrhosis, sepsis, or use of other nephrotoxic drugs, such as diuretics and ACEIs [117,118].
So, the main form of AKI caused by aspirin and NSAIDs is hemodynamically mediated, due to reduced renal perfusion and ischemia. However, there exists a second form of AKI caused by aspirin and NSAIDs which is AIN. AIN is based on a delayed hypersensitivity reaction to these drugs that causes inflammation and edema of the renal interstitium, which leads to nephrotic proteinuria or acute tubular necrosis a few days after the initiation of treatment, typically restored after discontinuation of the drug [119,120,121,122]. Instead, long-term use of aspirin and NSAIDs can lead to CKD by causing chronic interstitial nephritis or papillary necrosis [24,123].
The prevention of aspirin and NSAIDs induced kidney damage involves careful selection and monitoring of these drugs in patients who are at high risk of renal injury, especially those with advanced age and preexisting renal disease. The lowest effective dose and shortest duration of treatment should be used, and alternative analgesic or anti-inflammatory agents should be considered if possible.
Although there is limited evidence that P2Y12 inhibitors can cause renal damage, some studies have suggested that these drugs may be potentially hazardous, particularly when combined with other nephrotoxic medications such as statins [93]. For instance, the combination of ticagrelor and rosuvastatin is commonly used in patients with acute coronary syndrome and has been associated with AKI in some studies [124,125]. The underlying mechanism of this association is unknown and is not likely to be related to hepatic metabolism interactions, given that ticagrelor is mainly metabolized by CYP3A4 while rosuvastatin is primarily metabolized by CYP2C9 [124]. It is possible that ticagrelor may cause a transient worsening of renal function through an unknown mechanism, thereby enhancing the ability of rosuvastatin to induce rhabdomyolysis and subsequent AKI. Therefore, caution should be exercised when combining P2Y12 inhibitors with other nephrotoxic drugs, and close monitoring of renal function is recommended in patients receiving such combination therapy.
Finally, some studies have shown that clopidogrel may be a cause of thrombotic microangiopathy, a condition characterized by renal failure, hemolytic anemia, and thrombocytopenia [126,127]. Due to the widespread use of clopidogrel, clinicians should exercise caution and remain vigilant regarding this infrequent but potentially serious complication.
3.6. Anticoagulants
ACs are drugs that inhibit the coagulation cascade, which is responsible for blood clotting and hemostasis. They are used to prevent or treat thromboembolic disorders, such as deep vein thrombosis, pulmonary embolism, atrial fibrillation, or stroke. There are different types of anticoagulants, such as vitamin K antagonists (e.g., warfarin), direct thrombin inhibitors (e.g., dabigatran), and direct factor Xa inhibitors (e.g., rivaroxaban, apixaban). Studies have demonstrated the involvement of these drugs in a new nosological entity called Anticoagulant-related nephropathy (ARN).
ARN, previously known as warfarin nephropathy, is a relatively recently described entity despite the fact that warfarin has been used for several decades. ARN is a form of AKI that can occur in patients with supratherapeutic international normalized ratio (INR), as well as those taking newer anticoagulants [128,129]. When there is no apparent cause of AKI, and the patient has recently had a supratherapeutic INR and hematuria, ARN may be considered as a possible diagnosis. Renal biopsy can help to confirm the presence of ARN in such cases: the absence of active inflammatory lesions and the presence of RBCs and RBC occlusive casts tubules and Bowman space are indicative of ARN [130]. Recent research has tried to shed light on the complex and multifactorial mechanism underlying ARN. One of the key factors is a reduction in the number of functional nephrons, which can lead to over-proliferation of the surviving glomeruli and glomerular hypertension, rendering them vulnerable to glomerular hemorrhage [131]. It has been proposed that the combination of mild glomerular disease and ARN may lead to glomerular hematuria and a significant accumulation of RBCs within nephrons. If urinary flow is diminished due to interstitial inflammation or changes in blood pressure and most of all dehydration, intratubular RBCs may form occlusive casts, leading to the development of AKI [132]. This could explain why patients with pre-existing chronic kidney disease are at higher risk of developing this condition. In addition, studies have described intricate interactions involving these molecules, which can trigger a cascade of oxidative stress and inflammation in the renal tubular epithelium and surrounding interstitium [133]. Finally, recent studies have proposed alternative pathways involving a decrease in protein C levels and abnormal signaling of endothelial protein C receptors [134].
Therefore, when a patient presents with unexplained AKI that does not resolve, ARN should be considered, and a renal biopsy should be proposed to confirm the diagnosis. If ARN is diagnosed, a switch from warfarin to DOACs should be considered, or a reduction in DOAC dosage if the patient is already on DOACs [130]. However, further studies are needed to establish guidelines for the management of ARN, as current guidelines are lacking. Additionally, close monitoring of kidney function and anticoagulant therapy is essential for the management of these patients.
3.7. Statins
Statins, also known as 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors, are a commonly prescribed class of drugs used for the management and treatment of hypercholesteremia. They work by reducing the levels of low-density lipoprotein (LDL), which is a major contributor to atherosclerosis and coronary heart disease total cholesterol, and triglycerides, while increasing high-density lipoprotein (HDL) concentrations. As such, they are widely used for both primary and secondary prevention of coronary heart disease. Statins, however, can cause numerous unwanted side effects that can cause discontinuation of the drug. Guidelines on cholesterol and statins are often created by experts who have conflicts of interest so we think that the officialy reported data on statins toxicity are downplayed [135]. The main reported collateral effects are the statin-associated muscle symptoms (SAMSs) such as myalgia, myopathy, myositis and even worse rhabdomyolysis; also new onset type 2 diabetes mellitus, neuropsychiatric effects such as depression, sleep problems etc., hepatotoxicity, microbiome-mediated effects and finally renal toxicity just to name only some [136].
Statin-induced kidney injury is a rare but serious complication of statin therapy. The statins more directing impacting the kidneys are the hydrophilic ones like pravastatin and rosuvastain especially at high dosage [135,137]. But even the less hydrophillic ones like simvastatins and atorvastatin, more extensively metabolized by the liver with minimal clearing by the kidneys, can exert significant nephrotoxicity [93,138,139,140]. Statin induced nephrotoxicity can involve several mechanisms. One possible mechanism involves the potential of statins, especially at high dosage, to cause rhabdomyolysis, that is a condition characterized by the breakdown of skeletal muscle, which can lead to the release of sarcoplasmic proteins and electrolytes, that can lead to AKI and other potentially life-threatening complications, such as hyperkaliemia and cardiac arrhythmias [141,142]. Another less known but more clinically relevant mechanism is an acute or subacute tubulo-interstitial nephritis, provoked by statins at high dosage, which can lead to a direct tubular damage related to cumulative metabolic effects of the statin on tubular cells [138]. Last but not least, there is a growing body of evidence suggesting that statin toxicity may be closely linked to oxidative stress as it can be deduced by the numerous functional reactive groups in the molecule (Figure 1). It has been found that statins can generate reactive oxygen species (ROS) during metabolism, leading to oxidative stress and varying levels of toxicity, including damage to skeletal muscle, the liver and kidneys [143]. Moreover, statin-induced suppression of coenzyme Q10 (CoQ-10) production, which has antioxidant properties, has been proposed as another potential cause of AKI [144]. CoQ-10 is an enzyme that plays a role in generating adenosine triphosphate (ATP) in the mitochondria, and its suppression can impair the mitochondrial respiratory chain, leading to mitochondrial dysfunction [145]. This effect could also be supported by the ability of statins to directly inhibit complexes I and III and to trigger mitochondria-induced calcium signaling alteration, enhancing ROS and promoting inflammation and apoptosis [146,147,148]. Overall statin-induced tubulo-interstitial nephritis is probably underreported because evolves insidiously in patients who are prone to develop AKI for other reasons (e.g., comorbid conditions such as diabetes and arterial hypertension, concomitant nephrotoxic drug treatment, etc.) [93,149].
Identifying statin-induced kidney injury is crucial, and physicians should exercise caution in dosing statins. Patients using high doses of statins should be educated about potential side effects. Although clinical rhabdomyolysis with statins is rare, it should be considered in patients who present with weakness and renal failure after a cardiac intervention [150]. A thorough drug history analysis can aid in early detection and treatment of statins adverse effects. Treatment typically involves stopping the medication and potentially switching to an alternative regimen. Apart from rhabdomyolysis the renal toxic effect by statin in particular the tubular interstitial damage should be carefully monitored. The best way to pursue this aim is by evaluating for progressive augmentation of protein in the urine essentially by searching abnormal casts only detectable on direct urine microscopy by an experienced observer. In case such protein elevation is documented, the statin suspension is warranted; you don’t have to wait for the decline of kidney filtration function (reduction of creatine clearance) that indicates an already irreversible and conspicuous statin driven damage [93,138].
3.8. Proton-Pump Inhibitors
PPIs are a class of drugs that are commonly used to treat acid-related disorders such as peptic ulcers, gastroesophageal reflux disease, and Helicobacter pylori infection by reducing the production of gastric acid through inhibition of the enzyme H+/K+ATPase in the stomach. Although not primarily cardiovascular drugs, PPIs are also widely used in cardiology as gastroprotective agents for patients taking antiplatelet and anticoagulant medications, as these medications increase the risk of gastrointestinal bleeding. However, PPIs have also been associated with several adverse effects, including AIN, AKI and CKD [151]. An immune-mediated reaction to the deposition of PPIs or their metabolites in the tubulo-interstitium is responsible for the formation of an interstitial inflammatory infiltrate and edema, which leads to the development of AIN and AKI. This results in acute inflammation and tubulointerstitial damage, which can ultimately progress to interstitial fibrosis and chronic interstitial nephritis, that, if left untreated, can lead to CKD and, in severe cases, even renal failure [152]. Another potential side effect associated with the use of PPIs is hypomagnesemia, which may affect kidney function by dysregulating vascular and endothelial function, contributing to the progression of CKD [153,154]. Moreover, numerous studies have shown that the use of PPIs is linked to a higher risk of enteric infections, such as C. difficile infection, which, in turn, can lead to dehydration-associated AKI [155,156]. Finally, studies suggest that mitochondrial injury may play a key role in inducing necrotic cell death in proximal tubular cells, and the promotion of such necrotic cell death by PPIs was found to be facilitated by ROS [157]. The necrosis induced by PPIs may lead to the release of cell debris, which could trigger an immune response and contribute to the development of acute tubulointerstitial nephritis observed in PPIs-treated patients [158]. Additionally, the risk of PPIs-induced nephrotoxicity may be increased by additional environmental factors, including comorbidities and concomitant medications such as oral anticoagulants that can lead to iron overload in proximal tubular cells. Iron overload was found to facilitate PPIs-induced nephrotoxicity in cultured cells. The combined effect of these factors may explain why GFR is lost at a faster rate in patients on chronic PPIs therapy [157].
Considering the medical relevance of PPIs, it is crucial to ensure that these medications are used properly in accordance with therapeutic guidelines. Close monitoring of the benefits derived from PPIs use is necessary, and discontinuation (with gradual tapering) of the drug therapy should occur promptly once it is no longer required. As part of routine clinical practice, monitoring of GFR on monthly basis may be helpful in detecting potential adverse effects of PPIs use.
3.9. Contrast Media
Although contrast media (CM) is not strictly a cardiovascular drug, it has an increasing importance in the context of cardiac procedures, such as coronary angiography, percutaneous coronary intervention and the escalating use of coronary computed tomography [159], where it is used to enhance the visualization of the vascular structures.
Contrast-induced nephropathy (CIN) has been defined as the occurrence of acute renal impairment within 2-7 days after iodinated contrast media (CM) administration [160]. It is a serious adverse effect that can lead to acute AKI and increased morbidity and mortality. Several mechanisms interact in a complex manner to contribute to the pathophysiology of CIN [161]. After an initial phase of vasodilatation, contrast media (CM) triggers intense vasoconstriction due to the inhibition of nitric oxide-mediated vasodilatation, changes in intracellular calcium concentration, and the release of adenosine and endothelin [162,163,164]. Then the major damaging renal mechanism comes into play: the massive oxidative interaction of CM with renal tubular cells and endothelial cells with consequent release of ROS. However, according to Li et al, the increase in ROS synthesis appears to be more a consequence of direct CM toxicity on tubular cells than the cause of cellular damage. This process is favored by the sustained reduction in renal blood flow with the consequent ischemia even of the outer regions of the medulla, resulting finally in acute tubular necrosis [165]. The route of administration and the chemical properties of CM play a crucial role in the development of CIN, with intra-arterial administration being more nephrotoxic than intravenous injection [161,166]. Other factors that contribute to the development of AKI include hypotension, microembolization of atheromatous debris, or bleeding complications, leading to ischemic acute tubular necrosis [167]. The risk factors for CIN can be exacerbated by hemodynamic alterations and medications, such as metformin, which can lead to lactic acidosis in the presence of kidney dysfunction and AKI [168,169].
The prevention of CIN is crucial as there is currently no targeted treatment available. Some common general measures that can be taken to prevent CIN include limiting the volume of CM and discontinuing the use of nephrotoxic drugs at least 48 hours before exposure to CM [161]. Moreover, hydration with intravenous fluids is recommended before and after the procedure, with a sliding scale protocol based on left ventricular end-diastolic pressure [168]. Bicarbonate infusion can also help to prevent renal tubular fluid injury by alkalinizing the environment and scavenging ROS [168].
4. Role of Oxidative Stress in Drug-Induced Nephrotoxicity
Oxidative stress is a condition of imbalance between the production and elimination of ROS, and/or decreased antioxidant defense activity, which can cause damage to cellular components such as lipids, proteins, and DNA [170]. Oxidative stress is involved in the pathogenesis of various diseases, including drug-induced nephrotoxicity. Several drugs can induce oxidative stress in the kidney by different mechanisms, such as increasing ROS generation, decreasing antioxidant defense, or impairing mitochondrial function [171]. Oxidative stress can contribute to renal injury by activating inflammatory responses, inducing apoptosis, modulating redox signaling, and altering gene expression [172].
It is widely recognized that CKD is characterized by increased levels of oxidative stress [173]. The kidneys are highly susceptible to damage caused by oxidative stress due to the intense oxidative activity of their mitochondria. CKD is characterized by increased levels of oxidative stress, which result from both a depletion of antioxidants and the over-production of ROS. It has been demonstrated that his excessive ROS generation leads to the oxidation of biomolecules like lipids, proteins, and DNA, which can further aggravate renal injury [174,175]. Oxidative stress in CKD is also linked to impaired mitochondrial function and the heightened release of mitochondrial ROS, which contributes to the progression of renal injury and the development of atherosclerotic diseases [176]. Furthermore, patients with CKD may experience complications such as hypertension, atherosclerosis, inflammation, and anemia, which are associated with increased oxidative stress [177].
Drug-induced AKI typically manifests as two distinct patterns of renal injury: acute tubular necrosis (ATN) and acute interstitial nephritis (AIN). While AIN results from medications that trigger an allergic reaction, ATN arises from direct toxicity to tubular epithelial cells [178]. The development and progression of ATN are influenced by various cellular mechanisms, with oxidative stress being a critical factor that can trigger an inflammatory response through the release of proinflammatory cytokines and the accumulation of inflammatory cells in tissues. Specifically, oxidative stress can activate signaling pathways that contribute to the development of ATN by inducing mitochondrial dysfunction, lysosomal hydrolase inhibition, phospholipid damage, and increased intracellular calcium concentration. This leads to the overproduction of ROS and the depletion or inactivation of cellular antioxidants such as glutathione, which further exacerbate renal tubular cell death [179]. The resulting inflammation can contribute to the pathogenesis of ATN and its progression to AKI. These processes have been adequately described for drugs such as cisplatin and aminoglycosides, which can induce renal injury through mechanisms that are not related to their systemic pharmacological effects [180,181,182]. Instead, the accumulation of these drugs in proximal tubular cells promotes oxidative stress and mitochondrial damage, leading to apoptosis and necrosis. This process can trigger the development of fibrosis and inflammation, which further promotes the production of ROS and proinflammatory cytokines. As a result, a vicious cycle is created that is extremely detrimental to the kidney [179].
At this point, it is plausible to consider that other drugs, including cardiovascular medications, could induce renal injury through similar mechanisms, although there is limited literature on the topic. As extensively described in the preceding paragraphs, studies have suggested a potential involvement of ROS in renal injury mediated by statins and PPIs. It has also been hypothesized that CIN may involve excessive ROS production as one of its mechanisms of renal injury [166,183,184,185]. Conversely, drugs that do not cause renal damage, such as beta-blockers, have been widely recognized for their antioxidant properties, as it could be deduced by the presence of the OH-groups in the molecule (Figure 1). This may help explain the beta blockers protective role for both the kidneys and the cardiovascular system [186,187,188,189].
Preclinical studies have demonstrated the significant and promising nephroprotective activity of multiple antioxidants, especially those derived from natural food sources [190], indicating their potential as effective sources of nephroprotective agents [191,192]. Therefore, targeting oxidative stress may offer a promising strategy to prevent or treat drug-induced nephrotoxicity and its associated morbidity and mortality. However, the current knowledge of the mechanisms and biomarkers of oxidative stress in drug-induced nephrotoxicity is still limited and requires further investigation. Moreover, the clinical efficacy and safety of antioxidant interventions in kidney disease are not well established and need to be evaluated in large-scale randomized controlled trials. Some of the future perspectives of oxidative stress and drug-induced nephrotoxicity include:
- developing novel and specific biomarkers of oxidative stress and drug-induced nephrotoxicity that can reflect the degree and location of renal injury, predict the risk and outcome of drug-induced nephrotoxicity, monitor the response to treatment, and guide personalized therapy [9];
- identifying novel and effective antioxidants that can target specific sources or pathways of oxidative stress and drug-induced nephrotoxicity, modulate redox signaling, protect renal cells and tissues from oxidative damage, and preserve or restore renal function [192];
- designing personalized and precision antioxidant therapy based on individual characteristics and needs, such as genetic background, epigenetic modifications, co-morbidities, co-administered drugs, environmental factors, and oxidative stress status.
- exploring the potential synergistic or additive effects of combining antioxidant therapy with other therapeutic modalities, such beta-blockers [193].
- assessing the long-term benefits and risks of antioxidant therapy for drug-induced nephrotoxicity on various clinical outcomes, such as renal function preservation, cardiovascular protection, quality of life improvement, and survival extension.
5. Conclusions
Drug-induced nephrotoxicity is a serious complication that can affect the prognosis and quality of life of patients treated with various medications and diagnostic agents. The mechanisms of drug-induced nephrotoxicity vary depending on the type and class of the drug, the dose and duration of exposure, and the patient’s characteristics and comorbidities. Analyzing the structural characteristics of the various drugs, it is evident that they are all small molecules; therefore, it is entirely plausible to hypothesize that, especially in the presence of high dosages, these drugs can bind in a non-specific manner to other molecules different from their target, which can be the reason for the secondary effects of cytotoxicity. The prevention and management of drug-induced nephrotoxicity require a comprehensive approach that involves knowledge of the mechanisms of drug-induced nephrotoxicity, understanding of the patient and drug-related risk factors, and therapeutic intervention by correcting risk factors, assessing baseline renal function before initiation of therapy, adjusting the drug dosage and avoiding use of nephrotoxic drug combinations. Targeting oxidative stress could be a potential future solution for drug-induced nephrotoxicity, although further studies are needed.
Author Contributions
Conceptualization, C.C.; methodology, C.C.; writing—original draft preparation, C.C.; writing—review and editing, C.C.; A.S.; R.A.; M.E.L.; supervision, M.E.L.; project administration, M.E.L.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
No acknowledgments have to be added.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Van Vleet, T.R.; Schnellmann, R.G. Toxic nephropathy: environmental chemicals. Seminars in nephrology 2003, 23, 500–508. [Google Scholar] [CrossRef]
- Dalal, R.; Bruss, Z.S.; Sehdev, J.S. Physiology, Renal Blood Flow and Filtration. Updated 2023 Jul 24. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. Available online: https://www.ncbi.nlm.nih.gov/books/NBK482248/.
- Sanchez-Alamo, B.; Cases-Corona, C.; Fernandez-Juarez, G. Facing the Challenge of Drug-Induced Acute Interstitial Nephritis. Nephron 2023, 147, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Rennke, H.G.; Bradley, M.D. Renal Pathophysiology. The essentials. fourth ed. Baltimore, MD: Wolters Kluwer- Lippincot Williams & Wilkins. 2014. [Google Scholar]
- Petejova, N.; Martinek, A.; Zadrazil, J.; Teplan, V. Acute toxic kidney injury. Renal failure 2019, 41, 576–594. [Google Scholar] [CrossRef]
- Ghane Shahrbaf, F.; Assadi, F. Drug-induced renal disorders. Journal of renal injury prevention 2015, 4, 57–60. [Google Scholar] [CrossRef]
- Pazhayattil, G.S.; Shirali, A.C. Drug-induced impairment of renal function. International journal of nephrology and renovascular disease 2014, 7, 457–468. [Google Scholar] [CrossRef]
- Dobrek, L. A Synopsis of Current Theories on Drug-Induced Nephrotoxicity. Life (Basel, Switzerland) 2023, 13. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Moon, A. Drug-induced nephrotoxicity and its biomarkers. Biomolecules & therapeutics 2012, 20, 268–272. [Google Scholar] [CrossRef]
- Kaufman, D.P.; Basit, H.; Knohl, S.J. Physiology, Glomerular Filtration Rate. StatPearls. Treasure Island (FL) ineligible companies. Disclosure: Hajira Basit declares no relevant financial relationships with ineligible companies. Disclosure: Stephen Knohl declares no relevant financial relationships with ineligible companies.: StatPearls Publishing. Copyright © 2023, StatPearls Publishing LLC.; 2023.
- Hostetter, T.H.; Rosenberg, M.E. Hemodynamic effects of glomerular permselectivity. American journal of nephrology 1990, 10 Suppl 1, 24–27. [Google Scholar] [CrossRef]
- Horl, W.H. Nonsteroidal Anti-Inflammatory Drugs and the Kidney. Pharmaceuticals (Basel) 2010, 3, 2291–2321. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.J.; Vaughan, D.E. Angiotensin-Converting Enzyme Inhibitors. 1998, 97, 1411–1420. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.M.; Trepiccione, F.; Unwin, R.J. Drug toxicity in the proximal tubule: new models, methods and mechanisms. Pediatric nephrology (Berlin, Germany) 2022, 37, 973–982. [Google Scholar] [CrossRef]
- Gai, Z.; Gui, T.; Kullak-Ublick, G.A.; Li, Y.; Visentin, M. The Role of Mitochondria in Drug-Induced Kidney Injury. Frontiers in physiology 2020, 11, 1079. [Google Scholar] [CrossRef]
- Krause, K.M.; Serio, A.W.; Kane, T.R.; Connolly, L.E. Aminoglycosides: An Overview. Cold Spring Harbor perspectives in medicine 2016, 6. [Google Scholar] [CrossRef]
- Yano, T.; Itoh, Y.; Kawamura, E.; Maeda, A.; Egashira, N.; Nishida, M.; Kurose, H.; Oishi, R. Amphotericin B-induced renal tubular cell injury is mediated by Na+ Influx through ion-permeable pores and subsequent activation of mitogen-activated protein kinases and elevation of intracellular Ca2+ concentration. Antimicrobial agents and chemotherapy 2009, 53, 1420–1426. [Google Scholar] [CrossRef] [PubMed]
- Varlam, D.E.; Siddiq, M.M.; Parton, L.A.; Rüssmann, H. Apoptosis contributes to amphotericin B-induced nephrotoxicity. Antimicrobial agents and chemotherapy 2001, 45, 679–685. [Google Scholar] [CrossRef]
- Tanji, N.; Tanji, K.; Kambham, N.; Markowitz, G.S.; Bell, A.; D'Agati V, D. Adefovir nephrotoxicity: possible role of mitochondrial DNA depletion. Human pathology 2001, 32, 734–740. [Google Scholar] [CrossRef]
- Deray, G.; Martinez, F.; Katlama, C.; Levaltier, B.; Beaufils, H.; Danis, M.; Rozenheim, M.; Baumelou, A.; Dohin, E.; Gentilini, M.; et al. Foscarnet nephrotoxicity: mechanism, incidence and prevention. American journal of nephrology 1989, 9, 316–321. [Google Scholar] [CrossRef]
- Caiazza, A.; Russo, L.; Sabbatini, M.; Russo, D. Hemodynamic and tubular changes induced by contrast media. BioMed research international 2014, 2014, 578974. [Google Scholar] [CrossRef] [PubMed]
- Imig, J.D.; Ryan, M.J. Immune and inflammatory role in renal disease. Comprehensive Physiology 2013, 3, 957–976. [Google Scholar] [CrossRef] [PubMed]
- Barreto, E.F.; Rule, A.D. Management of Drug-Associated Acute Interstitial Nephritis. Kidney360 2020, 1, 62–64. [Google Scholar] [CrossRef] [PubMed]
- Lucas, G.N.C.; Leitão, A.C.C.; Alencar, R.L.; Xavier, R.M.F.; Daher, E.F.; Silva Junior, G.B.D. Pathophysiological aspects of nephropathy caused by non-steroidal anti-inflammatory drugs. Jornal brasileiro de nefrologia 2019, 41, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Farouk, S.S.; Rein, J.L. The Many Faces of Calcineurin Inhibitor Toxicity-What the FK? Advances in chronic kidney disease 2020, 27, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Gong, R.; Wang, P.; Dworkin, L. What we need to know about the effect of lithium on the kidney. American journal of physiology. Renal physiology 2016, 311, F1168–f1171. [Google Scholar] [CrossRef]
- Santos, M.L.C.; de Brito, B.B.; da Silva, F.A.F.; Botelho, A.; de Melo, F.F. Nephrotoxicity in cancer treatment: An overview. World journal of clinical oncology 2020, 11, 190–204. [Google Scholar] [CrossRef]
- Keen, M.U.; Aeddula, N.R. Analgesic Nephropathy. StatPearls. Treasure Island (FL) ineligible companies. Disclosure: Narothama Aeddula declares no relevant financial relationships with ineligible companies.: StatPearls Publishing. Copyright © 2023, StatPearls Publishing LLC.; 2023.
- Perazella, M.A.; Herlitz, L.C. The Crystalline Nephropathies. Kidney international reports 2021, 6, 2942–2957. [Google Scholar] [CrossRef]
- Mulay, S.R.; Shi, C.; Ma, X.; Anders, H.J. Novel Insights into Crystal-Induced Kidney Injury. Kidney diseases (Basel, Switzerland) 2018, 4, 49–57. [Google Scholar] [CrossRef]
- Genc, G.; Ozkaya, O.; Acikgöz, Y.; Yapici, O.; Bek, K.; Gülnar Sensoy, S.; Ozyürek, E. Acute renal failure with acyclovir treatment in a child with leukemia. Drug and chemical toxicology 2010, 33, 217–219. [Google Scholar] [CrossRef]
- Izzedine, H.; Launay-Vacher, V.; Deray, G. Antiviral drug-induced nephrotoxicity. American journal of kidney diseases: the official journal of the National Kidney Foundation 2005, 45, 804–817. [Google Scholar] [CrossRef]
- Derebail, V.K.; McGregor, J.G.; Colindres, R.E.; Singh, H.K.; Kshirsagar, A.V. The Case: Acute kidney injury in a patient with P. carinii pneumonia. Kidney international 2009, 75, 865–866. [Google Scholar] [CrossRef]
- Perazella, M.A. Crystal-induced acute renal failure. The American journal of medicine 1999, 106, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Hamed, K.M.; Dighriri, I.M.; Baomar, A.F.; Alharthy, B.T.; Alenazi, F.E.; Alali, G.H.; Alenazy, R.H.; Alhumaidi, N.T.; Alhulayfi, D.H.; Alotaibi, Y.B.; et al. Overview of Methotrexate Toxicity: A Comprehensive Literature Review. Cureus 2022, 14, e29518. [Google Scholar] [CrossRef] [PubMed]
- Yarlagadda, S.G.; Perazella, M.A. Drug-induced crystal nephropathy: an update. Expert opinion on drug safety 2008, 7, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Hebert, J.F.; Burfeind, K.G.; Malinoski, D.; Hutchens, M.P. Molecular Mechanisms of Rhabdomyolysis-Induced Kidney Injury: From Bench to Bedside. Kidney international reports 2023, 8, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Marcoff, L.; Thompson, P.D. The Role of Coenzyme Q10 in Statin-Associated Myopathy: A Systematic Review. Journal of the American College of Cardiology 2007, 49, 2231–2237. [Google Scholar] [CrossRef] [PubMed]
- Mollazadeh, H.; Tavana, E.; Fanni, G.; Bo, S.; Banach, M.; Pirro, M.; von Haehling, S.; Jamialahmadi, T.; Sahebkar, A. Effects of statins on mitochondrial pathways. Journal of cachexia, sarcopenia and muscle 2021, 12, 237–251. [Google Scholar] [CrossRef]
- Daras, M.; Kakkouras, L.; Tuchman, A.J.; Koppel, B.S. Rhabdomyolysis and hyperthermia after cocaine abuse: a variant of the neuroleptic malignant syndrome? Acta neurologica Scandinavica 1995, 92, 161–165. [Google Scholar] [CrossRef]
- Fernández-Cuadros, M.E.; Goizueta-San-Martin, G.; Varas-de-Dios, B.; Casique-Bocanegra, L.O.; Manrique-de-Lara-Cadiñanos, P.; Albaladejo-Florin, M.J.; Algarra-López, R.; Pérez-Moro, O.S. Colchicine-Induced Rhabdomyolysis: Clinical, Biochemical, and Neurophysiological Features and Review of the Literature. Clinical medicine insights. Arthritis and musculoskeletal disorders 2019, 12, 1179544119849883. [Google Scholar] [CrossRef]
- Packard, K.; Price, P.; Hanson, A. Antipsychotic use and the risk of rhabdomyolysis. Journal of pharmacy practice 2014, 27, 501–512. [Google Scholar] [CrossRef]
- Khosla, U.; Ruel, K.S.; Hunt, D.P. Antihistamine-induced rhabdomyolysis. Southern medical journal 2003, 96, 1023–1026. [Google Scholar] [CrossRef] [PubMed]
- Comelli, I.; Lippi, G.; Magnacavallo, A.; Cervellin, G. Mefloquine-associated rhabdomyolysis. The American journal of emergency medicine 2016, 34, 2250.e2255–2250.e2256. [Google Scholar] [CrossRef] [PubMed]
- Blain, P.G.; Lane, R.J.; Bateman, D.N.; Rawlins, M.D. Opiate-induced rhabdomyolysis. Human toxicology 1985, 4, 71–74. [Google Scholar] [CrossRef]
- Arnold, D.M.; Patriquin, C.J.; Nazy, I. Thrombotic microangiopathies: a general approach to diagnosis and management. CMAJ: Canadian Medical Association journal = journal de l'Association medicale canadienne 2017, 189, E153–e159. [Google Scholar] [CrossRef] [PubMed]
- Mazzierli, T.; Allegretta, F.; Maffini, E.; Allinovi, M. Drug-induced thrombotic microangiopathy: An updated review of causative drugs, pathophysiology, and management. Frontiers in pharmacology 2022, 13, 1088031. [Google Scholar] [CrossRef] [PubMed]
- Zakarija, A.; Kwaan, H.C.; Moake, J.L.; Bandarenko, N.; Pandey, D.K.; McKoy, J.M.; Yarnold, P.R.; Raisch, D.W.; Winters, J.L.; Raife, T.J.; et al. Ticlopidine- and clopidogrel-associated thrombotic thrombocytopenic purpura (TTP): review of clinical, laboratory, epidemiological, and pharmacovigilance findings (1989-2008). Kidney international. Supplement, 2009; S20–S24. [Google Scholar] [CrossRef]
- Zakarija, A.; Bennett, C. Drug-induced thrombotic microangiopathy. Seminars in thrombosis and hemostasis 2005, 31, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Page, E.E.; Little, D.J.; Vesely, S.K.; George, J.N. Quinine-Induced Thrombotic Microangiopathy: A Report of 19 Patients. American journal of kidney diseases: the official journal of the National Kidney Foundation 2017, 70, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Daviet, F.; Rouby, F.; Poullin, P.; Moussi-Francès, J.; Sallée, M.; Burtey, S.; Mancini, J.; Duffaud, F.; Sabatier, R.; Pourroy, B.; et al. Thrombotic microangiopathy associated with gemcitabine use: Presentation and outcome in a national French retrospective cohort. British journal of clinical pharmacology 2019, 85, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Hilburg, R.; Geara, A.S.; Qiu, M.K.; Palmer, M.B.; Chiang, E.Y.; Burger, R.A.; Hogan, J.J. Bevacizumab-associated thrombotic microangiopathy treated with eculizumab: A case series and systematic review of the literature. Clinical nephrology 2021, 96, 51–59. [Google Scholar] [CrossRef]
- Noronha, V.; Punatar, S.; Joshi, A.; Desphande, R.V.; Prabhash, K. Sunitinib-induced thrombotic microangiopathy. Journal of cancer research and therapeutics 2016, 12, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Arumugham, V.B.; Shahin, M.H. Therapeutic Uses of Diuretic Agents. StatPearls. Treasure Island (FL) ineligible companies. Disclosure: Mohamed Shahin declares no relevant financial relationships with ineligible companies.: StatPearls Publishing. Copyright © 2023, StatPearls Publishing LLC.; 2023.
- Oh, S.W.; Han, S.Y. Loop Diuretics in Clinical Practice. Electrolyte & blood pressure: E & BP 2015, 13, 17–21. [Google Scholar] [CrossRef]
- Ho, K.M.; Power, B.M. Benefits and risks of furosemide in acute kidney injury. Anaesthesia 2010, 65, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Hegde, A. Diuretics in Acute Kidney Injury. Indian journal of critical care medicine: peer-reviewed, official publication of Indian Society of Critical Care Medicine 2020, 24, S98–s99. [Google Scholar] [CrossRef] [PubMed]
- Miltiadous, G.; Mikhailidis, D.P.; Elisaf, M. Acid-base and electrolyte abnormalities observed in patients receiving cardiovascular drugs. Journal of cardiovascular pharmacology and therapeutics 2003, 8, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.; Liu, H.; Qi, W.; Jiang, H.; Li, Y.; Wu, X.; Sun, H.; Gross, K.; Salvi, R. Ototoxic effects and mechanisms of loop diuretics. Journal of otology 2016, 11, 145–156. [Google Scholar] [CrossRef]
- Kondo, C.S.; Macchionne, M.; Nakagawa, N.K.; de Carvalho, C.R.; King, M.; Saldiva, P.H.; Lorenzi-Filho, G. Effects of intravenous furosemide on mucociliary transport and rheological properties of patients under mechanical ventilation. Critical care (London, England) 2002, 6, 81–87. [Google Scholar] [CrossRef]
- Mullens, W.; Abrahams, Z.; Francis, G.S.; Sokos, G.; Taylor, D.O.; Starling, R.C.; Young, J.B.; Tang, W.H.W. Importance of venous congestion for worsening of renal function in advanced decompensated heart failure. J Am Coll Cardiol 2009, 53, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Damman, K.; van Deursen, V.M.; Navis, G.; Voors, A.A.; van Veldhuisen, D.J.; Hillege, H.L. Increased central venous pressure is associated with impaired renal function and mortality in a broad spectrum of patients with cardiovascular disease. J Am Coll Cardiol 2009, 53, 582–588. [Google Scholar] [CrossRef]
- Mullens, W.; Abrahams, Z.; Skouri, H.N.; Francis, G.S.; Taylor, D.O.; Starling, R.C.; Paganini, E.; Tang, W.H. Elevated intra-abdominal pressure in acute decompensated heart failure: a potential contributor to worsening renal function? J Am Coll Cardiol 2008, 51, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Jessup, M.; Costanzo, M.R. The cardiorenal syndrome: do we need a change of strategy or a change of tactics? J Am Coll Cardiol 2009, 53, 597–599. [Google Scholar] [CrossRef] [PubMed]
- Palazzuoli, A.; Ruocco, G.; Ronco, C.; McCullough, P.A. Loop diuretics in acute heart failure: beyond the decongestive relief for the kidney. Critical care (London, England) 2015, 19, 296. [Google Scholar] [CrossRef]
- Hasselblad, V.; Gattis Stough, W.; Shah, M.R.; Lokhnygina, Y.; O'Connor, C.M.; Califf, R.M.; Adams, K.F., Jr. Relation between dose of loop diuretics and outcomes in a heart failure population: results of the ESCAPE trial. European journal of heart failure 2007, 9, 1064–1069. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, W.; Ren, H.; Chen, X.; Xie, J.; Chen, N. Diuretics associated acute kidney injury: clinical and pathological analysis. Renal failure 2014, 36, 1051–1055. [Google Scholar] [CrossRef] [PubMed]
- Nasr, S.H.; Milliner, D.S.; Wooldridge, T.D.; Sethi, S. Triamterene crystalline nephropathy. American journal of kidney diseases: the official journal of the National Kidney Foundation 2014, 63, 148–152. [Google Scholar] [CrossRef]
- Zhang, Y.; He, D.; Zhang, W.; Xing, Y.; Guo, Y.; Wang, F.; Jia, J.; Yan, T.; Liu, Y.; Lin, S. ACE Inhibitor Benefit to Kidney and Cardiovascular Outcomes for Patients with Non-Dialysis Chronic Kidney Disease Stages 3-5: A Network Meta-Analysis of Randomised Clinical Trials. Drugs 2020, 80, 797–811. [Google Scholar] [CrossRef]
- Baltatzi, M.; Savopoulos, C.; Hatzitolios, A. Role of angiotensin converting enzyme inhibitors and angiotensin receptor blockers in hypertension of chronic kidney disease and renoprotection. Study results. Hippokratia 2011, 15, 27–32. [Google Scholar] [PubMed]
- Schoolwerth, A.C.; Sica, D.A.; Ballermann, B.J.; Wilcox, C.S. Renal Considerations in Angiotensin Converting Enzyme Inhibitor Therapy. 104 2001, 104, 1985–1991. [Google Scholar] [CrossRef]
- Knight, E.L.; Glynn, R.J.; McIntyre, K.M.; Mogun, H.; Avorn, J. Predictors of decreased renal function in patients with heart failure during angiotensin-converting enzyme inhibitor therapy: Results from the Studies of Left Ventricular Dysfunction (SOLVD). American Heart Journal 1999, 138, 849–855. [Google Scholar] [CrossRef]
- Weinfeld, M.S.; Chertow, G.M.; Stevenson, L.W. Aggravated renal dysfunction during intensive therapy for advanced chronic heart failure. American Heart Journal 1999, 138, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Muslih, A.I. Reduction of mean arterial pressure and proteinuria by the effect of ACEIs (Lisinopril) in Kurdish hypertensive patients in Hawler City. Global journal of health science 2012, 4, 14–19. [Google Scholar] [CrossRef]
- Swartz, S.L. The role of prostaglandins in mediating the effects of angiotensin converting enzyme inhibitors and other antihypertensive drugs. Cardiovascular drugs and therapy 1987, 1, 39–43. [Google Scholar] [CrossRef]
- Banas, J.S., Jr. Effects of inhibitors of angiotensin-converting enzyme on regional hemodynamics. The American journal of cardiology 1992, 69, 40c–45c. [Google Scholar] [CrossRef]
- Lant, A.F. Evolution of diuretics and ACE inhibitors, their renal and antihypertensive actions--parallels and contrasts. British journal of clinical pharmacology 1987, 23 Suppl 1, 27s–41s. [Google Scholar] [CrossRef]
- Mandal, A.K.; Markert, R.J.; Saklayen, M.G.; Mankus, R.A.; Yokokawa, K. Diuretics potentiate angiotensin converting enzyme inhibitor-induced acute renal failure. Clinical nephrology 1994, 42, 170–174. [Google Scholar]
- Packer, M.; Lee, W.H.; Medina, N.; Yushak, M.; Kessler, P.D. Functional Renal Insufficiency During Long-Term Therapy with Captopril and Enalapril in Severe Chronic Heart Failure. Annals of Internal Medicine 1987, 106, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Wynckel, A.; Ebikili, B.; Melin, J.-P.; Randoux, C.; Lavaud, S.; Chanard, J. Long-term follow-up of acute renal failure caused by angiotensin converting enzyme inhibitors. American Journal of Hypertension 1998, 11, 1080–1086. [Google Scholar] [CrossRef] [PubMed]
- Main, J. Atherosclerotic renal artery stenosis, ACE inhibitors, and avoiding cardiovascular death. Heart (British Cardiac Society) 2005, 91, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Schoolwerth, A.C.; Sica, D.A.; Ballermann, B.J.; Wilcox, C.S. Renal considerations in angiotensin converting enzyme inhibitor therapy: a statement for healthcare professionals from the Council on the Kidney in Cardiovascular Disease and the Council for High Blood Pressure Research of the American Heart Association. Circulation 2001, 104, 1985–1991. [Google Scholar] [CrossRef]
- Sica, D.A. Angiotensin-Converting Enzyme Inhibitors' Side Effects—Physiologic and Non-Physiologic Considerations. J Clin Hypertens (Greenwich) 2007, 7 (Suppl 8), 17–23. [Google Scholar] [CrossRef]
- Ponticelli, C.; Cucchiari, D. Renin-angiotensin system inhibitors in kidney transplantation: a benefit-risk assessment. Journal of Nephrology 2017, 30, 155–157. [Google Scholar] [CrossRef]
- Calvo Barbado, D.M.; Saiz Fernández, L.C.; Leache Alegría, L.; Celaya Lecea, M.C.; Gutiérrez-Valencia, M. Acute Kidney Injury associated with "Triple whammy" combination: a protocol for a systematic review. F1000Res 2022, 11, 496. [Google Scholar] [CrossRef]
- Leete, J.; Wang, C.; López-Hernández, F.J.; Layton, A.T. Determining risk factors for triple whammy acute kidney injury. Mathematical Biosciences 2022, 347, 108809. [Google Scholar] [CrossRef] [PubMed]
- Loboz, K.K.; Shenfield, G.M. Drug combinations and impaired renal function -- the 'triple whammy'. British journal of clinical pharmacology 2005, 59, 239–243. [Google Scholar] [CrossRef]
- Patil, V.P.; Salunke, B.G. Fluid Overload and Acute Kidney Injury. Indian journal of critical care medicine: peer-reviewed, official publication of Indian Society of Critical Care Medicine 2020, 24, S94–s97. [Google Scholar] [CrossRef] [PubMed]
- Busse, L.W.; Ostermann, M. Vasopressor Therapy and Blood Pressure Management in the Setting of Acute Kidney Injury. Seminars in nephrology 2019, 39, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Mushiyakh, Y.; Dangaria, H.; Qavi, S.; Ali, N.; Pannone, J.; Tompkins, D. Treatment and pathogenesis of acute hyperkalemia. Journal of community hospital internal medicine perspectives 2011, 1. [Google Scholar] [CrossRef]
- Bianchi, S.; Aucella, F.; De Nicola, L.; Genovesi, S.; Paoletti, E.; Regolisti, G. Management of hyperkalemia in patients with kidney disease: a position paper endorsed by the Italian Society of Nephrology. J Nephrol 2019, 32, 499–516. [Google Scholar] [CrossRef] [PubMed]
- Griffin, K.A.; Bidani, A.K. Potential risks of calcium channel blockers in chronic kidney disease. Curr Cardiol Rep 2008, 10, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Caiati, C.; Argentiero, A.; Favale, S.; Lepera, M.E. Cardiorenal Syndrome Triggered by Slowly Progressive Drugs Toxicity-Induced Renal Failure along with Minimal Mitral Disease: A Case Report. Endocrine, metabolic & immune disorders drug targets 2022, 22, 970–977. [Google Scholar] [CrossRef]
- Griffin, K.A.; Abu-Amarah, I.; Picken, M.; Bidani, A.K. Renoprotection by ACE Inhibition or Aldosterone Blockade Is Blood Pressure–Dependent. Hypertension 2003, 41, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Mishima, E.; Maruyama, K.; Nakazawa, T.; Abe, T.; Ito, S. Acute Kidney Injury from Excessive Potentiation of Calcium-channel Blocker via Synergistic CYP3A4 Inhibition by Clarithromycin Plus Voriconazole. Internal medicine (Tokyo, Japan) 2017, 56, 1687–1690. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, S.; Fleet, J.L.; Bailey, D.G.; McArthur, E.; Wald, R.; Rehman, F.; Garg, A.X. Calcium-channel blocker-clarithromycin drug interactions and acute kidney injury. Jama 2013, 310, 2544–2553. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, R. Beta-blockers and renal function. Drugs 1982, 23, 195–206. [Google Scholar] [CrossRef]
- Zech, P.; Pozet, N.; Labeeuw, M.; Laville, M.; Hadj-Aissa, A.; Arkouche, W.; Poncet, J.F. Acute renal effects of beta-blockers. American journal of nephrology 1986, 6 Suppl 2, 15–19. [Google Scholar] [CrossRef]
- Sullivan, J.M.; Adams, D.F.; Hollenberg, N.K. beta-adrenergic blockade in essential hypertension: reduced renin release despite renal vasoconstriction. Circulation Research 1976, 39, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Epstein, M.; Oster, J.R. Beta blockers and renal function: a reappraisal. Journal of clinical hypertension 1985, 1, 85–99. [Google Scholar] [PubMed]
- Bakris, G.L.; Hart, P.; Ritz, E. Beta blockers in the management of chronic kidney disease. Kidney international 2006, 70, 1905–1913. [Google Scholar] [CrossRef]
- Kalaitzidis, R.; Bakris, G. Should nephrologists use beta-blockers? A perspective. Nephrology Dialysis Transplantation 2009, 24, 701–702. [Google Scholar] [CrossRef]
- Hall, M.E.; Rocco, M.V.; Morgan, T.M.; Hamilton, C.A.; Jordan, J.H.; Edwards, M.S.; Hall, J.E.; Hundley, W.G. Beta-Blocker Use Is Associated with Higher Renal Tissue Oxygenation in Hypertensive Patients Suspected of Renal Artery Stenosis. Cardiorenal medicine 2016, 6, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Borchard, U. Pharmacokinetics of beta-adrenoceptor blocking agents: clinical significance of hepatic and/or renal clearance. Clinical physiology and biochemistry 1990, 8 Suppl 2, 28–34. [Google Scholar]
- Capodanno, D.; Angiolillo, D.J. Antithrombotic Therapy in Patients With Chronic Kidney Disease. Circulation 2012, 125, 2649–2661. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.H.; Liou, H.H.; Huang, Y.C.; Lee, T.S.; Chen, M.; Fang, Y.W. Hazardous Effect of Low-Dose Aspirin in Patients with Predialysis Advanced Chronic Kidney Disease Assessed by Machine Learning Method Feature Selection. Healthcare (Basel, Switzerland) 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arteriosclerosis, thrombosis, and vascular biology 2011, 31, 986–1000. [Google Scholar] [CrossRef]
- Crofford, L.J. COX-1 and COX-2 tissue expression: implications and predictions. The Journal of rheumatology. Supplement 1997, 49, 15–19. [Google Scholar]
- Shibata, K.; Akagi, Y.; Nozawa, N.; Shimomura, H.; Aoyama, T. Influence of nonsteroidal anti-inflammatory drugs on aspirin's antiplatelet effects and suggestion of the most suitable time for administration of both agents without resulting in interaction. Journal of pharmaceutical health care and sciences 2017, 3, 9. [Google Scholar] [CrossRef]
- Bjorkman, D.J. The effect of aspirin and nonsteroidal anti-inflammatory drugs on prostaglandins. The American journal of medicine 1998, 105, 8S–12S. [Google Scholar] [CrossRef]
- Packer, M. Interaction of prostaglandins and angiotensin II in the modulation of renal function in congestive heart failure. Circulation 1988, 77, I64–73. [Google Scholar] [PubMed]
- Kramer, H.J.; Stinnesbeck, B.; Klautke, G.; Kipnowski, J.; Klingmueller, D.; Glaenzer, K.; Duesing, R. Interaction of renal prostaglandins with the renin-angiotensin and renal adrenergic nervous systems in healthy subjects during dietary changes in sodium intake. Clinical science (London, England: 1979) 1985, 68, 387–393. [Google Scholar] [CrossRef]
- Kim, G.H. Renal effects of prostaglandins and cyclooxygenase-2 inhibitors. Electrolyte & blood pressure: E & BP 2008, 6, 35–41. [Google Scholar] [CrossRef]
- Culpepper, R.M.; Andreoli, T.E. Interactions among prostaglandin E2, antidiuretic hormone, and cyclic adenosine monophosphate in modulating Cl- absorption in single mouse medullary thick ascending limbs of Henle. The Journal of clinical investigation 1983, 71, 1588–1601. [Google Scholar] [CrossRef] [PubMed]
- Ejaz, P.; Bhojani, K.; Joshi, V.R. NSAIDs and kidney. The Journal of the Association of Physicians of India 2004, 52, 632–640. [Google Scholar] [PubMed]
- Burukoglu, D.; Baycu, C.; Taplamacioglu, F.; Sahin, E.; Bektur, E. Effects of nonsteroidal anti-inflammatory meloxicam on stomach, kidney, and liver of rats. Toxicology and industrial health 2016, 32, 980–986. [Google Scholar] [CrossRef] [PubMed]
- Drożdżal, S.; Lechowicz, K.; Szostak, B.; Rosik, J.; Kotfis, K.; Machoy-Mokrzyńska, A.; Białecka, M.; Ciechanowski, K.; Gawrońska-Szklarz, B. Kidney damage from nonsteroidal anti-inflammatory drugs-Myth or truth? Review of selected literature. Pharmacology research & perspectives 2021, 9, e00817. [Google Scholar] [CrossRef]
- Dreischulte, T.; Morales, D.R.; Bell, S.; Guthrie, B. Combined use of nonsteroidal anti-inflammatory drugs with diuretics and/or renin-angiotensin system inhibitors in the community increases the risk of acute kidney injury. Kidney international 2015, 88, 396–403. [Google Scholar] [CrossRef] [PubMed]
- McLeish, K.R.; Senitzer, D.; Gohara, A.F. Acute interstitial nephritis in a patient with aspirin hypersensitivity. Clinical immunology and immunopathology 1979, 14, 64–69. [Google Scholar] [CrossRef]
- Dixit, M.P.; Nguyen, C.; Carson, T.; Guedes, B.; Dixit, N.M.; Bell, J.M.; Wang, Y. Non-steroidal anti-inflammatory drugs-associated acute interstitial nephritis with granular tubular basement membrane deposits. Pediatric nephrology (Berlin, Germany) 2008, 23, 145–148. [Google Scholar] [CrossRef]
- Ravnskov, U. Glomerular, tubular and interstitial nephritis associated with non-steroidal antiinflammatory drugs. Evidence of a common mechanism. British journal of clinical pharmacology 1999, 47, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Harirforoosh, S.; Asghar, W.; Jamali, F. Adverse effects of nonsteroidal antiinflammatory drugs: an update of gastrointestinal, cardiovascular and renal complications. Journal of pharmacy & pharmaceutical sciences: a publication of the Canadian Society for Pharmaceutical Sciences, Societe canadienne des sciences pharmaceutiques 2013, 16, 821–847. [Google Scholar] [CrossRef]
- Leung, S.J.; Cisu, T.; Grob, B.M. Bilateral Ureteral Obstruction Secondary to Papillary Necrosis From Non-Steroidal Anti-Inflammatory Drug Use in an Adult Patient. Cureus 2021, 13, e16926. [Google Scholar] [CrossRef] [PubMed]
- Samuel, G.; Atanda, A.C.; Onyemeh, A.; Awan, A.; Ajiboye, O. A Unique Case of Drug Interaction between Ticagrelor and Statin Leading to Acute Renal Failure. Cureus 2017, 9, e1633. [Google Scholar] [CrossRef] [PubMed]
- Park, I.S.; Lee, S.B.; Song, S.H.; Seong, E.Y.; Kim, I.Y.; Rhee, H.; Kim, M.J.; Lee, D.W. Ticagrelor-induced acute kidney injury can increase serum concentration of statin and lead to concurrence of rhabdomyolysis. Anatolian journal of cardiology 2018, 19, 225–226. [Google Scholar] [CrossRef]
- Tada, K.; Ito, K.; Hamauchi, A.; Takahashi, K.; Watanabe, R.; Uchida, A.; Abe, Y.; Yasuno, T.; Miyake, K.; Sasatomi, Y.; et al. Clopidogrel-induced Thrombotic Microangiopathy in a Patient with Hypocomplementemia. Internal medicine (Tokyo, Japan) 2016, 55, 969–973. [Google Scholar] [CrossRef] [PubMed]
- Etta, P.K.; Gowrishankar, S. Clopidogrel Induced Thrombotic Microangiopathy Successfully Treated with Conservative Approach. Indian journal of nephrology 2020, 30, 209–210. [Google Scholar] [CrossRef]
- Afiari, A.; Drekolias, D.; Jacob, J. Anticoagulant-Related Nephropathy: A Common, Under-Diagnosed Clinical Entity. Cureus 2022, 14, e22038. [Google Scholar] [CrossRef] [PubMed]
- Zakrocka, I.; Załuska, W. Anticoagulant-related nephropathy: Focus on novel agents. A review. Advances in clinical and experimental medicine: official organ Wroclaw Medical University 2022, 31, 165–173. [Google Scholar] [CrossRef]
- Brodsky, S.; Eikelboom, J.; Hebert, L.A. Anticoagulant-Related Nephropathy. Journal of the American Society of Nephrology: JASN 2018, 29, 2787–2793. [Google Scholar] [CrossRef]
- Oliver, T.; Salman, L.A.; Ciaudelli, B.; Cohen, D.A. Anticoagulation-Related Nephropathy: The Most Common Diagnosis You've Never Heard Of. The American journal of medicine 2019, 132, e631–e633. [Google Scholar] [CrossRef]
- Brodsky, S.V.; Satoskar, A.; Chen, J.; Nadasdy, G.; Eagen, J.W.; Hamirani, M.; Hebert, L.; Calomeni, E.; Nadasdy, T. Acute kidney injury during warfarin therapy associated with obstructive tubular red blood cell casts: a report of 9 cases. American journal of kidney diseases: the official journal of the National Kidney Foundation 2009, 54, 1121–1126. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, S.V.; Mhaskar, N.S.; Thiruveedi, S.; Dhingra, R.; Reuben, S.C.; Calomeni, E.; Ivanov, I.; Satoskar, A.; Hemminger, J.; Nadasdy, G.; et al. Acute kidney injury aggravated by treatment initiation with apixaban: Another twist of anticoagulant-related nephropathy. Kidney research and clinical practice 2017, 36, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Mezue, K.; Ram, P.; Egbuche, O.; Menezes, R.G.; Lerma, E.; Rangaswami, J. Anticoagulation-related nephropathy for the internist: a concise review. American journal of cardiovascular disease 2020, 10, 301–305. [Google Scholar]
- Mach, F.; Ray, K.K.; Wiklund, O.; Corsini, A.; Catapano, A.L.; Bruckert, E.; De Backer, G.; Hegele, R.A.; Hovingh, G.K.; Jacobson, T.A.; et al. Adverse effects of statin therapy: perception vs. the evidence - focus on glucose homeostasis, cognitive, renal and hepatic function, haemorrhagic stroke and cataract. Eur Heart J 2018, 39, 2526–2539. [Google Scholar] [CrossRef]
- Ward, N.C.; Watts, G.F.; Eckel, R.H. Statin Toxicity. Circ Res 2019, 124, 328–350. [Google Scholar] [CrossRef]
- Wolfe, S.M. Dangers of rosuvastatin identified before and after FDA approval. The Lancet 2004, 363, 2189–2190. [Google Scholar] [CrossRef] [PubMed]
- van Zyl-Smit, R.; Firth, J.C.; Duffield, M.; Marais, A.D. Renal tubular toxicity of HMG-CoA reductase inhibitors. Nephrol Dial Transplant 2004, 19, 3176–3179. [Google Scholar] [CrossRef]
- Londrino, F.; Zattera, T.; Falqui, V.; Corbani, V.; Cavallini, M.; Stefanini, T.; Chiappini, N.; Ardini, M.; Martina, V.; Rombolà, G. Rosuvastatin-induced acute interstitial nephritis. Case Rep Nephrol Urol 2013, 3, 87–90. [Google Scholar] [CrossRef]
- Annigeri, R.A.; Mani, R.M. Acute interstitial nephritis due to statin and its class effect. Indian journal of nephrology 2015, 25, 54–56. [Google Scholar] [CrossRef] [PubMed]
- Ezad, S.; Cheema, H.; Collins, N. Statin-induced rhabdomyolysis: a complication of a commonly overlooked drug interaction. Oxford medical case reports 2018, 2018, omx104. [Google Scholar] [CrossRef]
- Botshekan, S.; Yalameha, B. Are statins toxic or safe for kidney diseases? An updated mini-review study. Journal of Nephropathology 2020, 9, e38–e38. [Google Scholar] [CrossRef]
- Liu, A.; Wu, Q.; Guo, J.; Ares, I.; Rodríguez, J.-L.; Martínez-Larrañaga, M.-R.; Yuan, Z.; Anadón, A.; Wang, X.; Martínez, M.-A. Statins: Adverse reactions, oxidative stress and metabolic interactions. Pharmacology & Therapeutics 2019, 195, 54–84. [Google Scholar] [CrossRef]
- McMurray, J.J.; Dunselman, P.; Wedel, H.; Cleland, J.G.; Lindberg, M.; Hjalmarson, A.; Kjekshus, J.; Waagstein, F.; Apetrei, E.; Barrios, V.; et al. Coenzyme Q10, rosuvastatin, and clinical outcomes in heart failure: a pre-specified substudy of CORONA (controlled rosuvastatin multinational study in heart failure). J Am Coll Cardiol 2010, 56, 1196–1204. [Google Scholar] [CrossRef] [PubMed]
- Deichmann, R.; Lavie, C.; Andrews, S. Coenzyme q10 and statin-induced mitochondrial dysfunction. The Ochsner journal 2010, 10, 16–21. [Google Scholar]
- Liu, A.; Wu, Q.; Guo, J.; Ares, I.; Rodríguez, J.L.; Martínez-Larrañaga, M.R.; Yuan, Z.; Anadón, A.; Wang, X.; Martínez, M.A. Statins: Adverse reactions, oxidative stress and metabolic interactions. Pharmacol Ther 2019, 195, 54–84. [Google Scholar] [CrossRef]
- Sirvent, P.; Mercier, J.; Vassort, G.; Lacampagne, A. Simvastatin triggers mitochondria-induced Ca2+ signaling alteration in skeletal muscle. Biochemical and biophysical research communications 2005, 329, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, Y.; Ghorbanihaghjo, A.; Naghi-Zadeh, M.; Yagin, N.L. Oxidative stress as a possible mechanism of statin-induced myopathy. Inflammopharmacology 2018, 26, 667–674. [Google Scholar] [CrossRef]
- Verdoodt, A.; Honore, P.M.; Jacobs, R.; De Waele, E.; Van Gorp, V.; De Regt, J.; Spapen, H.D. Do Statins Induce or Protect from Acute Kidney Injury and Chronic Kidney Disease: An Update Review in 2018. J Transl Int Med 2018, 6, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Chitralli, D.; Raheja, R.; Br, K. Clinical Rhabdomyolysis With Acute Kidney Injury Secondary to High-Intensity Rosuvastatin Use: A Case Report. Cureus 2020, 12, e10932. [Google Scholar] [CrossRef] [PubMed]
- Morschel, C.F.; Mafra, D.; Eduardo, J.C.C. The relationship between proton pump inhibitors and renal disease. Jornal brasileiro de nefrologia 2018, 40, 301–306. [Google Scholar] [CrossRef]
- Moledina, D.G.; Perazella, M.A. PPIs and kidney disease: from AIN to CKD. J Nephrol 2016, 29, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Van Laecke, S.; Van Biesen, W.; Vanholder, R. Hypomagnesaemia, the kidney and the vessels. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association 2012, 27, 4003–4010. [Google Scholar] [CrossRef] [PubMed]
- Malavade, P.; Hiremath, S. Proton Pump Inhibitors: More Indigestion than Relief? Indian journal of nephrology 2017, 27, 249–257. [Google Scholar] [CrossRef]
- Leonard, J.; Marshall, J.K.; Moayyedi, P. Systematic review of the risk of enteric infection in patients taking acid suppression. The American journal of gastroenterology 2007, 102, 2047–2056. [Google Scholar] [CrossRef] [PubMed]
- Arrich, J.; Sodeck, G.H.; Sengolge, G.; Konnaris, C.; Mullner, M.; Laggner, A.N.; Domanovits, H. Clostridium difficile causing acute renal failure: case presentation and review. World journal of gastroenterology 2005, 11, 1245–1247. [Google Scholar] [CrossRef]
- Fontecha-Barriuso, M.; Martín-Sanchez, D.; Martinez-Moreno, J.M.; Cardenas-Villacres, D.; Carrasco, S.; Sanchez-Niño, M.D.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. Molecular pathways driving omeprazole nephrotoxicity. Redox biology 2020, 32, 101464. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Gong, X. Research Progress on the Potential Mechanisms of Acute Kidney Injury and Chronic Kidney Disease Induced by Proton Pump Inhibitors. Integrative Medicine in Nephrology and Andrology 2023, 10. [Google Scholar] [CrossRef]
- Caiati, C.; Scardapane, A.; Iacovelli, F.; Pollice, P.; Achille, T.I.; Favale, S.; Lepera, M.E. Coronary Flow and Reserve by Enhanced Transthoracic Doppler Trumps Coronary Anatomy by Computed Tomography in Assessing Coronary Artery Stenosis. Diagnostics (Basel) 2021, 11, 245. [Google Scholar] [CrossRef] [PubMed]
- McCullough, P.A.; Soman, S.S. Contrast-induced nephropathy. Crit Care Clin 2005, 21, 261–280. [Google Scholar] [CrossRef] [PubMed]
- Azzalini, L.; Spagnoli, V.; Ly, H.Q. Contrast-Induced Nephropathy: From Pathophysiology to Preventive Strategies. The Canadian journal of cardiology 2016, 32, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Morcos, S.K.; Dawson, P.; Pearson, J.D.; Jeremy, J.Y.; Davenport, A.P.; Yates, M.S.; Tirone, P.; Cipolla, P.; de Haën, C.; Muschick, P.; et al. The haemodynamic effects of iodinated water soluble radiographic contrast media: a review. European Journal of Radiology 1998, 29, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Tumlin, J.; Stacul, F.; Adam, A.; Becker, C.R.; Davidson, C.; Lameire, N.; McCullough, P.A. Pathophysiology of contrast-induced nephropathy. The American journal of cardiology 2006, 98, 14k–20k. [Google Scholar] [CrossRef]
- Persson, P.B.; Hansell, P.; Liss, P. Pathophysiology of contrast medium-induced nephropathy. Kidney international 2005, 68, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ren, K. The Mechanism of Contrast-Induced Acute Kidney Injury and Its Association with Diabetes Mellitus. Contrast media & molecular imaging 2020, 2020, 3295176. [Google Scholar] [CrossRef]
- Seeliger, E.; Sendeski, M.; Rihal, C.S.; Persson, P.B. Contrast-induced kidney injury: mechanisms, risk factors, and prevention. European heart journal 2012, 33, 2007–2015. [Google Scholar] [CrossRef] [PubMed]
- McCullough, P.A. Contrast-induced acute kidney injury. J Am Coll Cardiol 2008, 51, 1419–1428. [Google Scholar] [CrossRef] [PubMed]
- Modi, K.; Padala, S.A.; Gupta, M. Contrast-Induced Nephropathy. StatPearls. Treasure Island (FL) ineligible companies. Disclosure: Sandeep Padala declares no relevant financial relationships with ineligible companies. Disclosure: Mohit Gupta declares no relevant financial relationships with ineligible companies.: StatPearls Publishing. Copyright © 2023, StatPearls Publishing LLC.; 2023.
- Iftikhar, H.; Saleem, M.; Kaji, A. Metformin-associated Severe Lactic Acidosis in the Setting of Acute Kidney Injury. Cureus 2019, 11, e3897. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxidative medicine and cellular longevity 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Deavall, D.G.; Martin, E.A.; Horner, J.M.; Roberts, R. Drug-induced oxidative stress and toxicity. Journal of toxicology 2012, 2012, 645460. [Google Scholar] [CrossRef] [PubMed]
- Tomsa, A.M.; Alexa, A.L.; Junie, M.L.; Rachisan, A.L.; Ciumarnean, L. Oxidative stress as a potential target in acute kidney injury. PeerJ 2019, 7, e8046. [Google Scholar] [CrossRef]
- Tirichen, H.; Yaigoub, H.; Xu, W.; Wu, C.; Li, R.; Li, Y. Mitochondrial Reactive Oxygen Species and Their Contribution in Chronic Kidney Disease Progression Through Oxidative Stress. Frontiers in physiology 2021, 12, 627837. [Google Scholar] [CrossRef] [PubMed]
- Che, R.; Yuan, Y.; Huang, S.; Zhang, A. Mitochondrial dysfunction in the pathophysiology of renal diseases. American journal of physiology. Renal physiology 2014, 306, F367–378. [Google Scholar] [CrossRef] [PubMed]
- Daenen, K.; Andries, A.; Mekahli, D.; Van Schepdael, A.; Jouret, F.; Bammens, B. Oxidative stress in chronic kidney disease. Pediatric nephrology (Berlin, Germany) 2019, 34, 975–991. [Google Scholar] [CrossRef]
- Ho, H.J.; Shirakawa, H. Oxidative Stress and Mitochondrial Dysfunction in Chronic Kidney Disease. Cells 2022, 12. [Google Scholar] [CrossRef]
- Podkowińska, A.; Formanowicz, D. Chronic Kidney Disease as Oxidative Stress- and Inflammatory-Mediated Cardiovascular Disease. Antioxidants (Basel, Switzerland) 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Moledina, D.G.; Parikh, C.R. Differentiating Acute Interstitial Nephritis from Acute Tubular Injury: A Challenge for Clinicians. Nephron 2019, 143, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Hosohata, K. Role of Oxidative Stress in Drug-Induced Kidney Injury. International journal of molecular sciences 2016, 17. [Google Scholar] [CrossRef]
- Arany, I.; Safirstein, R.L. Cisplatin nephrotoxicity. Seminars in nephrology 2003, 23, 460–464. [Google Scholar] [CrossRef]
- Santos, N.A.; Catão, C.S.; Martins, N.M.; Curti, C.; Bianchi, M.L.; Santos, A.C. Cisplatin-induced nephrotoxicity is associated with oxidative stress, redox state unbalance, impairment of energetic metabolism and apoptosis in rat kidney mitochondria. Archives of toxicology 2007, 81, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Novoa, J.M.; Quiros, Y.; Vicente, L.; Morales, A.I.; Lopez-Hernandez, F.J. New insights into the mechanism of aminoglycoside nephrotoxicity: an integrative point of view. Kidney international 2011, 79, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Kusirisin, P.; Chattipakorn, S.C.; Chattipakorn, N. Contrast-induced nephropathy and oxidative stress: mechanistic insights for better interventional approaches. Journal of translational medicine 2020, 18, 400. [Google Scholar] [CrossRef] [PubMed]
- Manda, P.; Srinivasa Rao, P.; Bitla, A.R.; Vinapamula, K.S.; Jeyaseelan, L.; Rajasekhar, D.; Vishnubhotla, S. Study of contrast-induced oxidative stress in nondiabetic patients undergoing coronary angiography. Saudi journal of kidney diseases and transplantation: an official publication of the Saudi Center for Organ Transplantation, Saudi Arabia 2019, 30, 45–52. [Google Scholar] [PubMed]
- Hizoh, I.; Haller, C. Radiocontrast-induced renal tubular cell apoptosis: hypertonic versus oxidative stress. Investigative radiology 2002, 37, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, M.M.; Bernardo, D.R.D.; de Bragança, A.C.; Massola Shimizu, M.H.; Seguro, A.C.; Volpini, R.A.; Canale, D. Treatment with β-blocker nebivolol ameliorates oxidative stress and endothelial dysfunction in tenofovir-induced nephrotoxicity in rats. Frontiers in medicine 2022, 9, 953749. [Google Scholar] [CrossRef] [PubMed]
- Gokturk, H.; Ulusu, N.N.; Gok, M.; Tuncay, E.; Can, B.; Turan, B. Long-term treatment with a beta-blocker timolol attenuates renal-damage in diabetic rats via enhancing kidney antioxidant-defense system. Molecular and cellular biochemistry 2014, 395, 177–186. [Google Scholar] [CrossRef]
- Coats, A.; Jain, S. Protective effects of nebivolol from oxidative stress to prevent hypertension-related target organ damage. Journal of Human Hypertension 2017, 31, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Murakami, M.; Miura, D.; Yunoki, K.; Enko, K.; Tanaka, M.; Saito, Y.; Nishii, N.; Miyoshi, T.; Yoshida, M.; et al. Beta-Blockers and Oxidative Stress in Patients with Heart Failure. Pharmaceuticals (Basel, Switzerland) 2011, 4, 1088–1100. [Google Scholar] [CrossRef] [PubMed]
- Caiati, C.; Stanca, A.; Lepera, M.E. Free Radicals and Obesity-Related Chronic Inflammation Contrasted by Antioxidants: A New Perspective in Coronary Artery Disease. 2023, 13, 712. [Google Scholar]
- Ayza, M.A.; Zewdie, K.A.; Yigzaw, E.F.; Ayele, S.G.; Tesfaye, B.A.; Tafere, G.G.; Abrha, M.G. Potential Protective Effects of Antioxidants against Cyclophosphamide-Induced Nephrotoxicity. International journal of nephrology 2022, 2022, 5096825. [Google Scholar] [CrossRef]
- Ranasinghe, R.; Mathai, M.; Zulli, A. Cytoprotective remedies for ameliorating nephrotoxicity induced by renal oxidative stress. Life Sciences 2023, 318, 121466. [Google Scholar] [CrossRef] [PubMed]
- Tylicki, L.; Rutkowski, B.; Hörl, W.H. Antioxidants: a possible role in kidney protection. Kidney & blood pressure research 2003, 26, 303–314. [Google Scholar] [CrossRef]
Figure 1.
Schematic representation of the chemical structure of each different class of cardiovascular drugs. Chemical structures of Bumetanide (diuretics), Clopidogrel (antiplatelet agents - APAs), Olmesartan (angiotensin receptor blockers - ARBs), Atenolol (beta blockers), Amlodipine (calcium channel blockers - CCBs), Warfarin (anticoagulants - ACs), Lovastatin (statins), Captopril (angiotensin-converting enzyme inhibitors -ACEIs), and Esomeprazole (proton-pump inhibitors - PPIs) are depicted. Thered dashed boxes highlight the parts of the molecule that interact with its target.
Figure 1.
Schematic representation of the chemical structure of each different class of cardiovascular drugs. Chemical structures of Bumetanide (diuretics), Clopidogrel (antiplatelet agents - APAs), Olmesartan (angiotensin receptor blockers - ARBs), Atenolol (beta blockers), Amlodipine (calcium channel blockers - CCBs), Warfarin (anticoagulants - ACs), Lovastatin (statins), Captopril (angiotensin-converting enzyme inhibitors -ACEIs), and Esomeprazole (proton-pump inhibitors - PPIs) are depicted. Thered dashed boxes highlight the parts of the molecule that interact with its target.
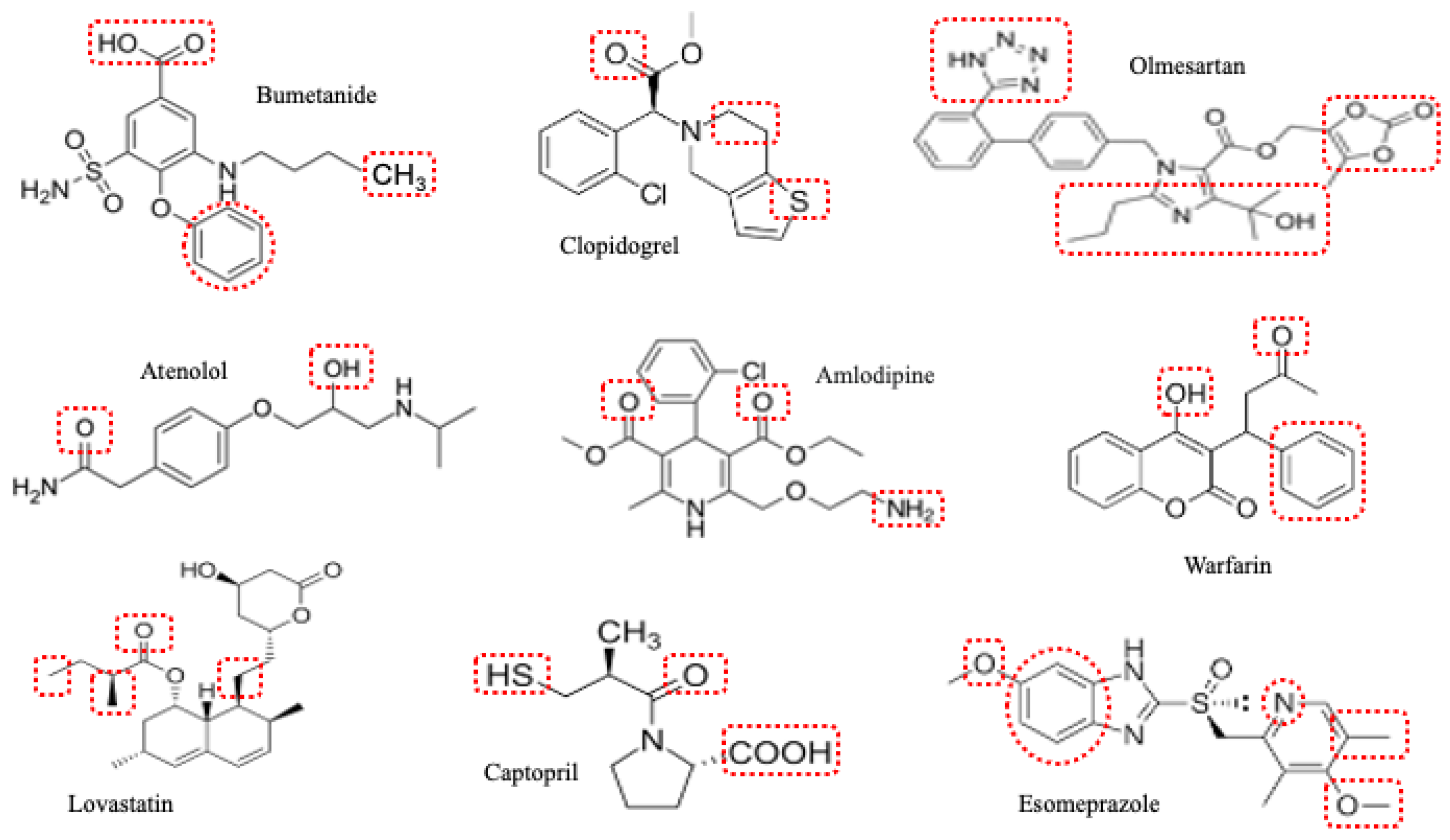
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



