
Submitted:
13 January 2025
Posted:
14 January 2025
You are already at the latest version
Abstract
Palatogenesis is a complex developmental process requiring temporospatially coordinated cellular and molecular events. The following review focuses on genetic, epigenetic, and environmental aspects directing palatal formation and their implication in orofacial clefting genesis. Essential for palatal shelf development and elevation (TGF-β, BMP, FGF, and WNT), the subsequent processes of fusion (SHH) and proliferation, migration, differentiation, and apoptosis of neural crest-derived cells are controlled through signaling pathways. Interruptions to these processes may result in the birth defect cleft lip and/or palate (CL/P), which happens in approximately 1 in every 700 live births worldwide. Recent progress has emphasized epigenetic regulations via the class of non-coding RNAs with microRNAs based on critically important biological processes, such as proliferation, apoptosis, and epithelial-mesenchymal transition. These environmental risks (maternal smoking, alcohol, retinoic acid, and folate deficiency) interact with genetic and epigenetic factors during palatogenesis, while teratogens like dexamethasone and TCDD inhibit palatal fusion. Orofacial cleft: genetic, epigenetic, and environmental impact on the complex epidemiology This is an extensive review, offering current perspectives on gene-environment interactions, as well as non-coding RNAs, in palatogenesis and emphasizing open questions regarding these interactions in palatal development.
Keywords:
palate development
; congenital disorder
; cleft palate/lip
; genetic network
; epigenetics
; environmental factors
1. Introduction
Craniofacial development, especially palatogenesis, is among the intricate processes in vertebrate embryogenesis and requires the precise coordination of numerous cellular and molecular events. Secondary palate formation is critical because its disruption results in cleft lip and/or palate (CL/P), a prevalent congenital anomaly in approximately 1/700 live births across populations. Development of the palate is a complicated process that requires neural crest-derived cells to coordinate cellular processes such as growth, movement, differentiation, and cell death. Palatogenesis is controlled by conserved signaling pathways such as TGF-β, BMP, FGF, WNT, and SHH. These pathways help the palatal shelf grow, rise, and fuse, similar to those of other organ systems. These processes function as part of a network that controls genes and epigenetics. Epigenetic processes, including DNA methylation, histone changes, and non-coding RNAs, have been linked to essential aspects of palatal development owing to progress in molecular biology. When a woman is early in her pregnancy and is exposed to factors such as smoking, drinking, retinoic acid, and some teratogens, they can change the genetic and epigenetic processes that affect palatal development. It is essential to know their complex interactions to design more efficient preventive and therapeutic strategies for orofacial clefts. This review provides a thorough overview of the biology of secondary palate development, emphasizing the synergy and interaction between genetic, epigenetic, and environmental factors. We highlight recent progress in key signaling pathways relevant to palatogenesis, their regulation by epigenetic mechanisms, and the newly appreciated role of non-coding RNAs in palatogenesis.
2. Craniofacial Development: Molecular and Genetic Basis
2.1. Anatomical Development and Classification of Cleft Lip and/or Palate (CL/P)
2.1.1. Anatomical Overview of Palate Formation
The hard palate (bony part of the front) and soft palate (muscular part of the back) separate the oral and nasal cavities during embryogenesis. During palate development, shelves elevate, contact, and fuse at the midline to form the hard (anterior) and soft palates (posterior). This process is essential for both speech and swallowing. Palatal fusion begins at the back and progresses toward the front. Facial development begins early in pregnancy [1]. During development, the formation of the face and palate requires the spatiotemporal coordination of diverse cellular processes, including, but not limited to, growth, migration, differentiation, and apoptosis [2]. Palate development is a highly complex process that is regulated by transcription factors, growth factors, signaling molecules, and epigenetic regulators. An imbalance in fine-tuning can result in craniofacial defects such as cleft lip and/or palate. This process can be severely affected by environmental exposures, such as maternal smoking, addiction to medication, or environmental toxins, leading to the development of these congenital alterations [1].
The upper lip, philtrum, and primary palate develop from the fusion of the medial nasal and maxillary processes (Figure 1A) [3]; however, disruption of these processes can result in cleft lip and/or cleft palate. The secondary palate fuses with the primary palate in its front and nasal septum in its anterodorsal region, both developing simultaneously (Figure 1B-D). This fusion forms a complete palate in the oral cavity and separates it from the nasal cavity (Figure 1E). Palatal shelf elevation, contact, or fusion failure results in secondary cleft palate. In humans, palatal development commences at approximately week 6 of gestation and is completed by week 12 [1]. In mice, this process starts at approximately E11.5 and is essentially completed by E17 (Figure. 1) [2] [3]. The complex process of palate formation depends on the precise spatiotemporal regulation of multiple factors, such as transcription factors, growth factors, signaling molecules, and epigenetic factors, for normal development. Disruption of this process owing to maternal smoking, medication abuse, or exposure to environmental factors can result in craniofacial deformities with cleft lip and/or palate. All of these are taken into an intricate balance to form and fuse the palatal shelves correctly. Slight disturbance of this fine balance leads to developmental anomalies, often cleft palate, and other craniofacial abnormalities. Understanding the molecular mechanisms involved in palate formation is essential for developing effective preventive strategies and treatments for orofacial cleft disorders [3 138].
2.1.2. Classification of Human Cleft Lip and/or Palate
A cleft lip and/or palate is a common birth defect that is distinguished by the location and extent of the cleft palate. A cleft lip occurs when the tissues of the upper lip do not fuse properly during fetal development, resulting in a gap or opening on one (unilateral) or both sides (bilateral) (Figure 2) [1].
There are three subtypes of unilateral cleft lip.
- -
- Incomplete cleft lip (smaller gap)
- -
- Complete cleft lip (full width of upper lip)
- -
- Median cleft lip (rarely middle upper lip)
Bilateral cleft lip is less common and more difficult to treat due to the severity of the deformity.
A cleft palate is a deformity of the roof of the mouth caused by inappropriate fusion of the maxillary palatal processes during the early stages of pregnancy, resulting in either a complete (hard or soft) or incomplete palate (Figure 2).
- -
- Complete cleft palate involves both hard and soft palates.
- -
- An incomplete cleft palate encompasses both hard and soft palates.
- -
- Submucous cleft palate: involves a small opening in the soft palate, with the mucous membrane remaining intact [1].
A submucosal cleft involves a soft palate defect; however, the overlying mucosal layer is intact. Diagnosis may be delayed until speech or hearing issues arise. The cleft lip and palate frequently occur together (Figure 2) [1].
Asymmetric clefts affecting one or both sides of the lip or palate may affect classification and management [4]. Although evidence from mouse models of orofacial cleft asymmetry is limited, craniofacial structure formation can be regulated by genetic, epigenetic, and environmental factors similar to those in humans. Although many genetic causes and mutations associated with orofacial clefts have been identified, gaps in knowledge regarding the cellular and molecular mechanisms involved mean that clinical care and even prevention strategies have not changed significantly [5]. Further studies using mouse models are essential to develop effective treatments.
3. The Pathogenesis of Orofacial Clefts in Humans Involves Genetic and Environmental Factors
3.1. Overview of Syndromic/Non-Syndromic Associated with Cleft Lip and/or Palate
CL/P (orofacial cleft) is the most common congenital craniofacial malformation [6]. The prevalence of cleft lip is approximately 1 in every 700 live births (Table 1) [6] [1]. In the entire population, there are significantly more males than females born with cleft lip [7]. The variation in occurrence shows a wide disparity among various racial and ethnic groups, while the African-American population has a lower incidence (approximately 0.5/1000) [7]. A cleft lip contains one or more clefts that extend from the upper lip to one or both nostrils. A cleft palate is a type of fissure that forms on the roof of the mouth, upper lip, or both when the bones do not properly fuse during embryogenesis. This may affect one side, both sides or the central region (Figure 2) [1]. Although CL/P is not fatal, CL/P-affected patients suffer from dental, occlusal, functional, and aesthetic problems, along with secondary complications such as auditory, respiratory, and nutritional problems. The etiology of cleft palate is multifactorial and involves both genetic and environmental components [1]. Mutations in various genes, including monogenic disorders and chromosomal rearrangements, have been observed in patients with CL/P and are present in numerous genetic syndromes (Table 1,2) [8].
With or without cleft palate, cleft lips are normally divided into isolated (non-syndromic) and Mendelian syndromic forms. Non-syndromic CL/P (93–97% of cases) has a complex etiology that is attributed to the interaction of genes with environmental factors (Table 1). The recurrence risk of non-syndromic CL/P is estimated to be 4–10. Syndromic forms of CL/P account for approximately 5–7% of cases, encompass more than 200 different conditions and have been recognized by varying patterns and prevalence of congenital malformations [9].
3.2. The Genetic and Epigenetic Basis of Craniofacial Abnormalities: Non-Syndromic and Syndromic Forms
3.2.1. Non-Syndromic Craniofacial Anomalies
Non-syndromic oral clefts (NSOFC) account for 70% of all clefts and are among humans' most common birth defects. Its global prevalence is approximately 1 in 700 live births (Table 1) [8]. Non-syndromic clefts, defined as isolated deformities, comprise approximately 45% of cleft palate alone and 85% of non-syndromic cleft lip and/or palate (NSCLP) (Table 1) [9]. NSCLP is associated with distinct genetic factors such as single nucleotide polymorphisms (SNPs), single gene mutations, environmental factors, and microRNA patterns (Table 1) [9]. The WNT pathway is critical for craniofacial development, and WNT pathway genes, including AXIN1 and WNT9B, have been associated with NSOFC [10] [9]. Genes in this pathway, specifically FGFR1 and FGF2, have been associated with NSOFC [9]. Other genes associated with NSOFCs via linkage studies include COL11A1, IRF6, EGF, MSX1, PTCH, TGFB1, ROR2, FOXE1, TGFB3, RARA, APOC2, BCL3, and PVRL2 [9]. Linkage analysis revealed that more than 20 chromosomal regions were linked to the NSOFC. Notable examples include chr 1p, 1q21, 1q32-42.3, 6p, 2p, 4q, and 17q [9].
Several epigenetic-wide association studies (EWAS) have been conducted on orofacial clefts (Table 1). A study in the UK conducted EWAS using blood and lip tissues to test the association between methylation at each site and cleft subtype (cleft lip only (CLO) n = 50; cleft palate only (CPO) n = 50; cleft lip and palate (CLP) n = 50) [11]. Recently, [12] demonstrated that eight genes (ABCB1, ALKBH8, CENPF, CSAD, EXPH5, PDZD8, SLC16A9, and TTC28) were consistently expressed in relevant mouse and human craniofacial tissues during facial formation and three genes (ABCB1, TTC28, and PDZD8) showed statistically significant mutation constraints. These findings highlight the role of rare variants in identifying candidate genes for NSOFCs.
3.2.2. Syndromic Craniofacial Anomalies
While syndromic OFCs are more often attributable to one congenital cause or disrupted gene than NSOFCs, other challenges arise in determining the underlying mechanisms [9,13]. The underlying etiology associated with the OFC phenotype may be more difficult to determine for many syndromic conditions in which clefts are mild or uncommon. Often, multiple disease-causing genes and factors can exist, and the role of specific genes in cleft-associated cases may not be described thoroughly, particularly if clefts are a minor feature and not the primary focus of studies under particular conditions [13]. In addition, many syndromes are caused by deletions that disrupt several genes, further complicating the association between specific loci and lip and/or palate fusion (Table 2) [13].
DiGeorge Syndrome
DiGeorge syndrome (DGS) is a congenital disorder with a broad phenotypic presentation that results predominantly from the microdeletion of chromosome 22 at a location known as 22q11.2 [14]. More than half of patients with 22q11.2DS (DiGeorge syndrome/velo-cardio-facial syndrome) exhibit craniofacial malformations, of which CP is the most frequently observed defect. Many of the developmental anomalies observed with this syndrome could be attributed to a decrease in copy number of genes located within the deleted region at 22q11.2, including the possible aberrant expression of the T-box TF (TBX1), whose role in palatogenesis has been well documented [15]. The loss and gain of Tbx1 function suggests that Tbx1 dosage is a critical determinant of normal palatogenesis. Tbx1 regulates craniofacial development through miR-96–5p, which represses Tbx1 expression by binding to the 3’-UTR of its mRNA [15,16].
Van der Woude Syndrome
Van der Woude syndrome (VWS) is the most common form of syndromic clefting, accounting for approximately 2% of all CL/P cases, with a prevalence of 1/34,000 live births. Van der Woude syndrome (VWS) is an autosomal dominant disorder in which affected individuals have one or more of the following manifestations: cleft lip, cleft palate, hypodontia, or paramedian lower lip pits [13]. In this review, analysis of the van der Woude syndrome (VWS) family identified an SNP, rs539075, located within intron 2 of the cadherin gene (CDH2) that may be associated with CL/P[17]. This group also identified an intronic variant of NOL4 in patients with VWS, which co-segregated with CL/P [15].
Stickler Syndrome
Stickler (STL) syndrome is a heterogeneous disease characterized by collagen abnormalities (particularly collagen types II, IX, and XI). The prevalence of Stickler syndrome is estimated to be 1:7500–9000 [18]. STL is a disorder that includes congenital myopia, possibly coexisting with cataracts, retinal damage, cleft uvula, submucosal cleft palate, changes in craniofacial structure, changes in joints and bones, joint hypermobility, and progressive hearing loss [18].
STL is molecularly diagnosed based on the presence of pathogenic variants in six collagen-type genes (COL2A1, COL11A1, COL11A2, COL9A1, COL9A2, and COL9A3) and two non-collagen genes (LRP2 and LOXL3) [19,20], following a predominantly autosomal dominant inheritance pattern. Mutations in COL2A1 located on chromosome 12 (12q13.11) can cause SLT1 mutations [18]. STL2 occurs due to mutations in the gene encoding the α1 chain of collagen XI in COL11A1, which is located on the short arm of chromosome 1 (1p21.1). The main cause of Stickler syndrome type III (STL3) and non-ocular Stickler syndrome is deletions of the COL11A2-located on chromosome 6 (6p21.3), which encodes the α2 chain of collagen type XI [19].
Pierre-Robin Syndrome (PRS)
The Pierre-Robin syndrome (PRS) refers to a set of characteristic craniofacial phenotypes commonly observed: glossoptosis, cleft palate, micrognathia, and upper airway obstruction [13]. The Pierre-Robin syndrome occurs in 1/8500–1/14,000 births [21]. Cleft palate is associated with deletions in 2q and 4p, and duplications in 3p, 3q, 7q, 8q, 10 p, 14q, 16p, and 22q [21]. The dominant model suggests that mandibular hypoplasia causes a highly retropositioned tongue, blocking palatal shelf elevation and the airway. Alternatively, intrauterine mandibular compression or delayed neuromuscular development may restrict mandibular and tongue growth, leading to palate and airway obstruction [21]. Isolated PRS has been associated with mutations in or near the SRY-related HMG box 9 (SOX9) gene [22]. Mutations in BMPR1B have also been reported to cause PRS in two unrelated families [23]. Several genes encoding ECM components and ECM-interacting proteins have also been associated with syndromes, including clefts [13].
Kabuki Syndrome
Kabuki syndrome is a genetic disorder primarily characterized by distinct facial features, including midfacial hypoplasia, broad nasal tip, elongated palpebral fissures, and large abnormal earlobes. Other features include cleft or a high-arched palate, growth retardation, cognitive disabilities, and congenital heart defects [24] [25]. Kabuki syndrome is caused by mutations in the KMT2D gene, which encodes an H3K4 histone methylase that promotes active gene transcription, or in the KDM6A gene, an X-linked histone H3K27 demethylase. Approximately 60–70% of cases are attributed to KMT2D mutations [24]. This mutated gene was the first pathogenic gene recognized in Kabuki syndrome and is also known as MLL2 [26]. Over 50 mutations have been identified at different sites of KMT2D, including nonsense, missense, frameshift, small deletions, and splice-site variants (Barry et al., 2022).
Wolf-Hirschhorn Syndrome
Wolf-Hirschhorn Syndrome (WHS) is a developmental disorder characterized by intellectual disability, growth delays, heart and skeletal defects, seizures, and sometimes midline issues, such as cleft palate and facial asymmetry. [13]. Wolf–Hirschhorn syndrome (WHS) is a rare contiguous gene deletion syndrome (prevalence of 1:20,000–50,000 births, with a female-to-male ratio of 2:1) induced by the absence of the distal portion of the short arm of chromosome 4 [27]. WHS typically arises from deletions in chromosome 4p16.3, which vary in size and position. Genetic defects are usually partial deletions of the distal short arm of chromosome 4, but the WHS phenotype can also be generated by complex chromosomal rearrangements, such as translocations or ring chromosomes [27]. Unbalanced translocations can be de novo or inherited from a parent with balanced rearrangement. The most frequently observed translocations are (1) those involving a rearrangement t(4p;8p), t(4p;7p), t(4p;11p), t(4p;20q), t(4p;21q), and t(4p;12p); (2) inverted duplications associated with terminal deletions in the same 4p arm; or (3) unbalanced pericentric inversions [27,28].
CHARGE Syndrome
CHARGE(Coloboma, Heart, Atresia of the choanae, Retarded growth and development, Genital abnormalities, Ear abnormalities, and hearing loss) syndrome is a complex genetic disorder that affects multiple systems of the body. CHARGE syndrome is a multiple congenital malformation syndrome with an estimated birth prevalence of 1 in 15,000–17,000 newborns [13]. Mutations in chromatin remodeling and the gene expression regulator CHD7 account for most cases. CHD7, located on chromosome 8, is responsible for CHARGE syndrome and was first discovered in a study that uncovered mutations in CHD7 in individuals with this disorder [29]. This leads to a protein that plays a role in chromatin remodeling, both of which are important for gene expression during developmental processes. Disruption of embryonic development due to mutations in CHD7 results in a complex phenotype involving various organs [29].
Apert Syndrome
Apert syndrome (AS) is one of the most common craniosynostosis syndromes worldwide. The prevalence of AS is 1/ 65,000 in the general population [30]. Patients with AS may develop oral problems such as severe maxillary hypoplasia, cleft lip, and cleft palate [31]. The cleft palate occurs in approximately 30% of patients with Apert syndrome, with soft palate clefts being more common than hard palate clefts [32]. AS follows dominant genetic patterns and most patients are de novo cases caused by mutations in FGFR2 [33]. FGFR2 is the receptor for fibroblast growth factor (FGF) and is encoded by a gene at locus 10q26. FGFR2 is activated by binding to FGF and plays a role in cell proliferation, angiogenesis, and bone differentiation [34]. Mutation of exon IIIa of FGFR2 can cause AS because it leads to an increased bone differentiation rate of Mesenchymal Stem Cells (MSCs) and the development of craniosynostosis. Common types of gene mutations are FGFR2 p.Ser252Trp (S252W) of 755C > G and p.Pro253Arg (P253R) of 758C > G. The S252W mutation of FGFR2 is usually accompanied by severe skeletal malformations of the craniofacial region and a higher incidence of cleft palate, but the P253R mutation of FGFR2 is often accompanied by more prominent syndactyly of the hands and feet [35].
Tatton-Brown-Rahman Syndrome
Mutations in DNMT3B cause immunodeficiency-centromeric instability-facial anomaly (ICF) syndrome, which is characterized by facial abnormalities, neurological dysfunction, and immunodeficiency, with mouse models showing similar craniofacial defects. Mutations in DNMT3A are linked to the Tatton–Brown–Rahman syndrome (TBRS) or DNMT3A overgrowth syndrome (DOS), which is characterized by overgrowth, macrocephaly, facial dysmorphism, intellectual disability, and autism [36]. Whole-genome bisulfite sequencing of the patients revealed focal hypomethylation [37]. Several genes have been implicated in WHS pathology, including WHSC1, which encodes a histone methyltransferase [38]; WHSC2, which encodes a protein involved in RNA polymerase II regulation [39]; LETM1, a mitochondrial ion transporter [40]; and TACC3, which regulates microtubule growth [41].
Arboleda-Tham Syndrome
Arboleda-Tham Syndrome (ARTHS, OMIM #616268) is characterized by intellectual disability, developmental and speech delays, hypotonia, and congenital heart defects. Fewer common features include seizures, microcephaly, and autism spectrum disorder [42]. ARTHS is a rare genetic disorder caused by de novo heterozygous mutations in KAT6A, also known as MYST3 or MOZ [42]. Most identified pathogenic mutations are protein-truncating and occur throughout the gene, constituting more than half of the gene with a particular concentration in the last exon. KAT6A is a member of the MYST family of highly conserved histone acetyltransferases that activate gene expression by catalyzing the addition of acetyl groups to the histone tail [43]. This enhances chromatin accessibility, efficiently assembling transcription factors and transcriptional machinery [44]. Other reports have demonstrated that KAT6A specifically acetylates lysine residues in histone H3, including lysine 9 [45] and lysine 23 [46].
3.3. Key Genes Involved in Craniofacial Development
3.3.1. Morphological and Molecular Control of Palatal Shelf Growth and Patterning
In humans, lip closure and palatal fusion occur at 6 and 12 weeks gestation [47]. Because of these precise timings, animal models, particularly mice, are essential for studying craniofacial development [2] [3]. Mice are key model organisms for investigating cleft lip and palate because they are genetically similar to humans and share similar facial developmental processes [3].
Palatal shelves are mostly composed of neural crest-derived mesenchyme [48]. A thin layer of oral epithelium borders them with a distinct anterior-posterior (A-P) axis. In mice, the embryonic development of the palate begins at approximately E11.5 (Figure 1A). Although neural crest cells begin to migrate to established positions, the palate, frontonasal projections, and palatal shelves have not yet been developed. E12.5 shows no obvious formation of frontonasal projections or palatal shelves (Figure 3A-B). By E13.5, the palatal shelves and vertical tissue plates of the palate began to increase and move toward each other (Figure 1B, 3C-D). They eventually fuse along the midline between E13.5 and E15.5 (Figure 1C-E, 4) [2]. The growth and fusion of these shelves are regulated by interactions between epithelial and mesenchymal tissues along the anterior-posterior axis [49] [50] [51].
SHH signaling is crucial for palatal shelf outgrowth, as it regulates cell proliferation and promotes the development of the palate [50]. SHH interacts with other signaling pathways, such as FGF and BMP, to ensure proper palatogenesis. The inactivation of Shh in the epithelium or mesodermal-specific inactivation of smoothened (Smo) can impair palatal cell proliferation and growth (Figure 3E) [52]. In addition, mice with co-mutations in Hhat and Ptch1 show Shh gradient disruption during frontonasal protrusion development, resulting in hypoplasia of the central and lateral protrusions, ultimately resulting in cleft lip and residual midline epithelial junctions [53]. Primary cilia are small hair-like projections extending from various tissues' surfaces [54]. Essential for transmitting Shh signaling. Reduced expression of forkhead box F1 (Foxf1) in the palatal mesenchyme suggests that primary cilia are downstream effectors of Shh signaling (Figure 3E) [55].
Essential for palatal shelf outgrowth, with its absence leading to a cleft palate owing to impaired proliferation [56]. Although Fgf10 is expressed in the mesenchyme, its receptor, Fgfr2b, is crucial in the epithelium, and epithelial-specific deletion of Fgfr2 causes cleft palate (Figure 3E) [57]. Shh signaling, which is dependent on Fgf10, is reduced in Fgf10−/− and Fgfr2b−/− embryos, highlighting a positive feedback loop between Shh and FGF signaling that regulates palatal proliferation [56] [58]. These two signaling pathways and transcription factors work together to activate mesenchymal signaling to ensure proper palatogenesis and the establishment of the oral and nasal cavities. Fgf10 maintains Shh expression in the palatal epithelium, whereas Fgf7 suppresses Shh expression that is regulated by Dlx5 (Figure 3E) [59]. Recent studies have demonstrated a complex regulatory network involving Shh, Foxf1/2, and Fgf18 in developing the palatal shelves (Figure 3E). The ablation of Foxf1 and Foxf2 in mouse embryos interferes with palatal outgrowth, which affects the expression of Fgf18 and Shh [51]. This coordination between transcription factors and FGF ligands, which is controlled by Shh signaling, regulates the growth and patterning of palatal shelves (Figure 3E).
Fgf9, a critical FGF ligand in craniofacial development, is expressed in the palatal epithelium and mesenchyme during palatogenesis in mice (Figure 3E) [60]. Additionally, Sox11 null mice, with decreased Fgf9 expression, exhibit an undersized mandible and cleft palate, resembling cleft palate caused by micrognathia and tongue malposition in the Pierre Robin Sequence (PRS) [61]. In a recent study, elevated Fgf9 levels also induced TMJ dysplasia, impairing the spatial coordination between tongue descent and palatal shelf elevation, thereby exacerbating cleft palate formation. TMJ dysplasia restricts the posterior dimension of the mandible and adds stress to the posterior palate, thereby increasing the likelihood of cleft formation. These findings suggest that TMJ dysplasia, which also co-occurs with cleft palate in human syndromes such as achondroplasia and Muenke syndrome, may contribute to cleft palate, even without reducing mandibular length [62].
Expression of the LIM homeobox genes Lhx6 and Lhx8 negatively regulates proliferation of the maxillary arch and palatal mesenchyme by repressing FOX family transcription factors and the cell cycle inhibitor Cdkn1c (p57Kip2) (Figure 3E) [63]. The maintenance of mitochondrial homeostasis by Lhx6 is mediated through PINK1/ Parkin-mediated mitophagy and the MAPK signaling pathway. The transcriptional downregulation of Lhx6 by retinoic acid (RA) impairs the maintenance of mitochondrial homeostasis at the transcriptional level; hence, it causes defects in the proliferation and migration of HEPM cells and cleft palate formation [64]. Although the Shh-Foxf1/2-Fgf18-Shh molecular circuit is known to be involved in early palatal development (Figure 3E), it is unclear whether Lhx6/8 also influences the Shh and FGF signaling network during palatal shelf formation. Additionally, transforming growth factor-β (Tgf-β) signaling affects Shh signaling in the palatal mesenchyme by regulating lipid metabolism [65].
The Bmp (Bone Morphogenetic Protein) signaling pathway regulates cell proliferation, cell differentiation and apoptosis, which are critical steps in the morphogenesis of the face [66] [67]. The BMP pathway may interact with other cellular pathways, such as the Shh signaling pathway, which plays a crucial role in the development of the craniofacial [68] and interacts with the palatal mesenchyme, where loss of Smo results in increased Bmp4 expression and decreased levels of Bmp2 (Figure 3E) [58]. Shh signaling promotes the activity of Bmp2 to stimulate cell proliferation in the palatal mesenchyme [69]. The role of Bmp2 during the development of the facial process in craniofacial morphogenesis and the essential role of Bmp4 in tissue differentiation, along with the establishment of facial prominence [70]. Although complete inactivation of Bmp4 is lethal during early embryonic stages, its targeted deletion in the maxillary mesenchyme and oral epithelium results in cleft lip without affecting the secondary palate [66]. Overexpression of the BMP antagonist Noggin in the palatal mesenchyme causes delayed palatal growth and cleft palate [71]. This highlights the essential role of Bmp signaling in normal palatogenesis, and dysregulation of this process results in cleft lip or cleft palate [72]. Studies have shown that Bmpr1a, a type I Bmp receptor, is essential for palate formation (Figure 3F) [66]. Deletion of Bmpr1a in the maxillary mesenchyme and oral epithelium of mice causes cleft lip and palate, whereas its loss in the oral epithelium alone does not [73]. This suggests that Bmpr1a signaling in the mesenchyme is crucial for palatogenesis. Conditional deletion of Bmpr1a in neural crest cells leads to severe craniofacial defects [74], whereas its inactivation in the palatal mesenchyme results in anteriorly restricted cleft palate and reduced cell proliferation. Loss of Bmpr1a also disrupts Shh expression, indicating that BMP-SHH interactions regulate palate growth [75]. Additionally, loss of the BMP antagonist Noggin causes cleft palate with increased apoptosis and decreased cell proliferation [76], highlighting the need for tightly regulated Bmp signaling during palate development.
WNT signaling is crucial for Pax9-mediated secondary palate development [77] [78] [79] and regulates cell proliferation, migration, and differentiation [80]. In Pax9−/− mice, decreased Axin2 and β-catenin levels and increased Dkk2 expression (Figure 3F-G) disrupted WNT signaling, but pharmacological inhibition of DKK partially rescued palate morphology. Inactivation of Sostdc1 restores WNT signaling and rescues cleft palate (Figure 3G) [77]. Pathogenic variants in WNT pathway genes, such as Wnt3a, are linked to non-syndromic cleft lip and/or palate [81]. WNT signaling disruptions can lead to cleft lip and/or palate and are also associated with other conditions [8], including cancer [82] and skeletal disorders [83].
In Pax9−/− mice, EDA/EDAR signaling downstream of WNT signaling is reduced but is not essential for palatogenesis [79]. In utero stimulation with EDAR agonists restored cleft palate in these mice, and the creases appeared disorganized and did not influence the expression of Bmp4, Msx1, Fgf10, or Osr2. These studies indicate that Pax9 acts through the WNT signaling pathway by regulating antagonists of WNT in the palatal mesenchyme (Figure 3E). However, further studies are needed to understand how Pax9 regulates WNT target genes.
3.3.2. Molecular Regulation and Regional Patterning Along the Anterior-Posterior Axis of Palatal Development
The developing palatal shelves are molecularly and morphologically regionalized along the A-P axis, where regions of the anterior express transcription factors different from those of the posterior parts [84]. Msx1 and Shox2 are required for anterior palatal mesenchymal cell proliferation (Figure 3F), whereas the posterior region expresses Meox2 and Tbx22 (Figure 3G). Msx1 acts via Bmp4 in the mesenchyme to regulate Shh expression in the anterior palatal epithelium (Figure 3F) [69], whereas Mn1 and Barx1 are expressed more posteriorly (Figure 3G) [85]. Msx1 maintains Shh expression in the anterior palatal epithelium by regulating Bmp4 in the mesenchyme (Figure 3F), whereas Mn1 and Barx1 are primarily expressed in the posterior region (Figure 3G). Mice with disrupted Msx1 or Mn1 genes exhibited complete cleft palate, but the defects were region-specific. Msx1−/− mice have proliferation defects only in the anterior palate, whereas Mn1−/− mice have growth defects in the middle and posterior palates [69] [85]. Shox2−/− mice have a cleft restricted to the anterior palate, whereas the posterior palate develops normally, demonstrating the role of Shox2 in anterior palatal expansion [86]. In contrast, Tbx22−/− mice experience varying cleft severity, from full cleft palate to submucous cleft palate, with Tbx22 acting downstream of Mn1 in posterior palatal outgrowth [85]. Msx1 and Shox2 expression in the anterior palate is regulated by BMP signaling, as evidenced by the decreased expression in Wnt1-Cre; Bmpr1af/− mice [74]. In palatal explant cultures, Msx1 expression was specifically induced in the anterior palatal mesenchyme by Bmp4 [84], while exogenous Bmp4 could not stimulate Shox2 expression. However, the anterior palatal epithelium was able to induce Shox2 expression in the posterior mesenchyme, revealing distinct differences between the epithelium and mesenchyme along the anterior-posterior (A-P) axis (Figure 3F) [86]. Furthermore, canonical Wnt signaling is restricted to the anterior palatal mesenchyme and depends on Gpr177 for Wnt secretion. In particular, Wnt5a expression is high in the anterior mesenchyme. It regulates mesenchymal migration and elongation of the palatal shelf and its transcription is controlled by Msx1 (Figure 3F) [87]. In particular, LIM domain transcription factors, along with the cofactor Ldb1, have been identified in palatal growth and patterning, and their chemical and genetic inactivation leads to posterior mesenchymal ectopic expression of Wnt5a (Figure 3F) [88]. These findings highlight the distinct molecular mechanisms involved in A-P patterning of the palate.
3.3.3. Regulatory Networks and Patterning Along the Mediolateral Axis
SHH signaling plays a key role in palatal development because its disruption reduces its expression in the palatal mesenchyme [58]. The expression of Osr2 is dependent on Pax9, and embryos lacking both Osr2 and Pax9 exhibit cleft palate, along with decreased Fgf10 expression in the palatal mesenchyme (Figure 3E). This suggests the importance of Osr2 and Pax9 in palatal development and regulating Fgf10 levels [89]. Patterning along the mediolateral axis of the palate is critical for establishing the gene expression domains that provide proper growth and fusion. Around E12, the lateral side of the palatal shelves begins to form palatal rugae, and Shh expression is restricted to this region [55]. The zinc-finger transcription factors Osr1 and Osr2 exhibit graded expression along the mediolateral axis of the developing palatal mesenchyme (Figure 3E). By E13.5, Osr1 expression was confined to the lateral side, whereas Osr2 was strongly expressed in the lateral mesenchyme, tapering medially. Deletion of Osr2 leads to cleft palate because of reduced cell proliferation on the medial side and disrupted patterning. Osr2 partly compensates for the role of Osr1, as evidenced by the repair of cleft palate in Osr2-deficient mice with Osr1 cDNA [90].
Increased expression of osteogenesis-related genes, such as Mef2c, Sox6, Sp7, and several BMP ligands (Bmp3, Bmp5, and Bmp7), as well as ectopic expression of class-3 Semaphorins (Sema3a, Sema3d, and Sema3e), was observed in Osr2−/− mice. This highlights the role of Osr2 in the repression of mesenchymal cell proliferation and the prevention of premature osteogenesis. The function of Semaphorins in palatogenesis is yet to be determined [91]. A Dlx5-dependent transcriptional pathway regulates mediolateral patterning and palatal expansion (Figure 3E). Dlx5 is co-expressed in the medial mesenchyme of the palatal shelf with Fgf7; the expression of the latter gene is dramatically downregulated in the palates of Dlx5 mutant embryos. This reduction in Fgf7 expression may cause the expansion of Shh expression into the medial palatal epithelium, as exogenous Fgf7 can inhibit Shh expression in palatal explant cultures. Although palatal shelves in Dlx5-deficient mice are elevated and fused, the oral palate is significantly enlarged and the soft palate deformed [2].
Interestingly, although Msx1-deficient mice show reduced expression of Shh in the anterior palate, compound mutants that lack both Dlx5 and Msx1 express Shh within the medial epithelium, compensating for cell proliferation defects caused by Msx1 [59]. This study identified a new pathway involving Dlx5 and Fgf7 in the mediolateral patterning and palate growth. However, because Fgf7-deficient mice do not display overt palatal defects, another signaling molecule could act downstream of Dlx5 to modulate Shh expression [1].
3.3.4. Genetic Network Controlling Palatal Shelf Adhesion and Fusion
Concomitantly, the growth of the palatal shelves supports the development of the maxillary and mandibular processes; however, this only occurs because of downward and forward movement of the tongue. This is required to elevate the palatal shelves, which then contact and fuse the midline (Figure 1, 4B-C) [2]. An elaborate interaction of signaling pathways is engaged in the process that promotes the adhesion and fusion of shelves. Mesenchymal integrity in the fused palate is compromised by the removal of the mesenchyme between the shelves (Figure 4F). Disruption of midline marginal epithelial differentiation, adhesion capacity, and loss of mesenchymal-epithelial transition can lead to a cleft palate. Mutations or dysfunction in genes such as Jag2, Fgf10, Irf6, and Grhl3 result in inadequate adhesion or fusion of palatal shelves, leading to cleft palate [47] [92]. The absence of Jag2, a Notch ligand, causes cleft palate in Jag2zΔDSL/ΔDSL mice mainly because of abnormal adhesion of the palatal shelves to the tongue. Jag2 is expressed in the oral epithelium and maintains periderm cells, which are essential for regulating fusion competence (Figure 4D) [47]. Fgf10−/− embryos also showed reduced Jag2 expression and palatal-tongue fusion defects, suggesting that Fgf10 regulates palatal development upstream of Jag2-Notch signaling (Figure 4D). Mice with functional interferon regulatory factor 6 (Irf6) mutations due to homologous splicing null or R84C point mutations exhibit undifferentiated hyperproliferative epidermis, resulting in various developmental abnormalities including cleft palate and inappropriate oral adhesions [92]. Irf6 cooperates with Jag2 to regulate epidermal differentiation as demonstrated by severe defects in Irf6R84C/+; Jag2ΔDSL/+ mice [93]. This phenotype was similar to that observed in mice with homozygous Irf6 or Jag2 alleles, highlighting the importance of these genes in palatal development (Figure 4D). Expression of either gene is unaffected in individual mutants, indicating that Irf6 does not directly regulate Jag2 expression (Figure 4D) [92]. Mice lacking the p63 transcription factor show cleft palate and undifferentiated epidermis [1], with reduced Irf6 expression in the palatal epithelium [94]. Compound mutant mice, p63+/–; Irf6R84C/+, also exhibited failed palatal shelf fusion due to improper periderm cell maintenance. p63 may positively regulate Jag2 and Fgfr2 expression, although its relationship with the Jag2-Notch and Fgf10-Fgfr2b pathways in palatal epithelial differentiation is not fully understood [95]. The absence of Ikk-α or Tbx1 in mouse embryos leads to abnormal oral adhesions between the tongue and palatal shelves, indicating that palatal epithelial differentiation is controlled by a genetic network that includes Irf6, Jag2, p63, Ikk-α, Tbx1, and Fgf10-Fgfr2b signaling pathways (Figure 4D) [96].
Periderm removal and disappearance of the medial edge of the palatal shelf are two important events in determining whether palatal fusion can occur, and abnormalities such as abnormal oral adhesions do not occur. However, the mechanisms controlling periderm removal and Midline epithelial seam (MES) disappearance need to be determined. There are three dominating hypotheses on the way in which the MES disappears [97] [98]. According to one hypothesis, this may involve epithelial-mesenchymal transition (EMT). EMT may enable the MES epithelium to integrate into the mesenchyme of the non-cleft palate. Several in vivo lineage analyses have been performed using epithelial-restricted Cre-expressing transgenic lines and ROSA26R reporter lines to trace MES cell fate. For example, a study examining lacZ expression in ShhGFPCre or K14-Cre mice crossed with R26R reporter mice did not report the presence of lacZ-expressing mesenchymal cells. It thus concluded that EMT was not a major mechanism for MES regression [97]. In contrast, another study reported mesenchymal β-galactosidase activity in K14-Cre; R26R embryos before and during MES regression [99], which may be related to different Cre levels or expression patterns between the various K14-Cre transgenic mouse lines.
ESRP1 and its paralog ESRP2 are epithelial splicing regulatory proteins that co-localize with Irf6 and function in the embryonic epithelium to regulate craniofacial development and epithelial-mesenchymal transition during embryogenesis (Figure 4D) [100]. Functional studies have shown that Esrp1/2 mutations result in defective splicing of pre-mRNA, which in turn causes aberrant isoforms of CTNND1, leading to weakened epithelial integrity and orofacial cleft anomalies. In addition, studies have identified ESRP1/2-controlled isoforms of CTNND1 that regulate epithelial adhesion and WNT signaling, implicating disrupted splicing in craniofacial anomalies (Figure 4D) [101].
Apoptosis plays a crucial role in the dissolution of the medial edge of the palatal shelf during palatal fusion, allowing the mesenchyme to become connected. The usual signs of apoptosis, including TUNEL positivity and active caspase 3, are commonly detected in MES cells during this process, with very few proliferating cells observed in this region [97] [102]. However, recent research examining the role of the Apaf1 gene, which is involved in caspase 3-mediated apoptosis, found that Apaf1 deficiency does not affect palatal fusion or MES dissolution [103], contradicting earlier studies that reported fusion issues in Apaf1-deficient embryos [102]. This may be due to the fact that, in most of the earlier studies, the palate evaluation was incomplete. While apoptosis is one of the major mechanisms of MES breakdown, further studies are necessary to clarify the molecular mechanism of palate fusion, including Tgf-β signaling. Among these, Tgf-β3, exclusively expressed in the medial-edge epithelium (MEE), plays an important role in the removal of MES (Figure 4E). The absence of Tgf-β3 in embryonic mice leads to improper midline contact between the palatal shelves and the persistence of MES [1].
Tgf-β signaling is essential for palatal fusion and is activated through type I and type II receptor dimers, leading to phosphorylation of R-Smads and transcriptional regulation. Smad2 is crucial for MES breakdown and Smad2 overexpression can partially restore fusion in Tgf-β3 deficient mice. However, the deletion of Smad4 does not affect fusion, suggesting that other pathways, such as the p38 MAPK pathway, are involved [104]. Tgf-β signaling activates Tak1, which works independently of the Smad pathway, promoting palatal fusion through both Smad and p38 MAPK-dependent mechanisms [2]. Irf6 regulates periderm differentiation and is activated in periderm and basal MEE cells before fusion (Figure 4E). Its absence in mutant embryos resulted in failed fusion; however, Irf6 overexpression restored this process. Irf6 downregulates p63 and increases p21 expression, facilitating cell cycle exit and MEE degeneration (Figure 4E) [105] [106]. Tgf-β3 downregulates Jag2 in MEE, and blocking Notch signaling can partially restore palatal fusion in Tgf-β3-deficient cultures [107]. Oral periderm integrity is maintained by Jag2-Notch signaling [47]. A reduction in Jag2 expression within the medial edge epithelium (MEE) is a key mechanism through which Tgf-β3 disrupts periderm function and promotes palatal shelf adhesion. Beta-catenin (Ctnnb1) also plays a role in palatal fusion by regulating Tgf-β3 expression in MEE. Destruction of β-catenin epithelial cells (Ctnnb1) results in decreased apoptotic MES cells in the MEE, loss of Tgf-β3 expression, and failure of palatal shelf fusion and cleft palate. However, β-catenin can function in adherent junctions or in the canonical Wnt signaling pathway [108], and the exact mechanism of its involvement in MES dissolution requires further research.
Several transcription factors are crucial for palatal fusion. The Snail family, including Snai1 and Snai2, plays a key role, as fusion fails in Snai1+/–; Snai2+/– compound mutants along with reduced MES apoptosis (Figure 4E). Interestingly, although Tgfβ-3 expression remains unaffected in these mutants [44], exogenous Tgfβ-3 can induce Snai1 expression through a Smad-independent pathway, suggesting that Snail factors may act downstream or in parallel with Tgfβ-3 signaling. Runx1 is a transcription factor involved in palate development and is expressed throughout the MEE during palatal fusion (Figure 4E) [1]. Disruption of Runx1 results in anterior-specific failure of palatal shelf fusion and a cleft between the primary and secondary palates. This failure is linked to a distinct region in the anterior MEE, with less TUNEL staining and unique behavior [52]. In contrast, the Meox2 transcription factor is crucial for maintaining the integrity of the posterior palate after fusion. Meox2−/− embryos show a post-fusion split in the posterior palate [103].
Irf6 is essential for Snai2 expression in MEE cells, and Snai2 knockdown slows palatal fusion in explant culture [109]. Reverse signaling of ephrin enhances Snai1 expression in MEE cells and can partially rescue fusion in the presence of Tgf-β3-blocking antibodies, suggesting cooperation between ephrin and Tgf-β3 signaling in regulating palatal fusion [110]. Snai1 and Snai2, acting downstream of Tgf-β3, downregulate E-cadherin, which may loosen MEE and periderm cell adhesion, leading to periderm desquamation (Figure 4E). Tgf-β3 and Irf6 also induce MMP13, which is involved in basement membrane degradation in the MEE. CEACAM1, expressed in the periderm before fusion, is also involved in the process of palatal fusion because Ceacam1−/− embryos present a delay in fusion, whereas its relationship with Tgf-β3 signaling is unknown [111]. Further studies are needed to determine how desquamation, apoptosis, and Tgf-β3-mediated periderm cell death are interrelated. The TGF-β signaling pathway is important in several biological and cellular processes, including cell growth regulation, immune responses, and embryonic development [112]. Shh signaling modulates lipid metabolism in the palatal mesenchyme. In facial morphogenesis, Tgf-β signaling is essential for palatal fusion through its interaction with other signaling pathways, such as WNT, FGF, and BMP. Tgf-β is involved in epithelial-mesenchymal transition, which is a vital step for successful palatal shelf migration and fusion [113]. The Tgf-β signaling pathway involves several genes, and pathogenic variants in some of these genes have been shown to be associated with the development of orofacial clefts, such as the variants in the Interferon Regulatory Factor 6 (IRF6) gene associated with Van der Woude syndrome (VWS) [114], the SMAD gene family, which also cross-interacts with the BMP signaling pathway, and variants in these genes are associated with an increased risk of cleft lip development [113]. The genetic process of palatal development involves both genetic and epigenetic factors, including microRNAs (miRNAs) that regulate gene expression during palatal fusion.
3.4. Epigenetic Mechanisms Landscape in Palatogenesis: Molecular Dynamics and Developmental Regulation
3.4.1. Overall Epigenetic Modifications in Development
Epigenetics refers to mechanisms that alter gene expression by modifying the chromatin structure rather than changing the DNA sequence itself [115]. DNA is tightly wrapped around histone proteins to form nucleosomes, which are basic units of chromatin [116]. The arrangement of chromatin determines whether DNA is transcriptionally active; loosely packed chromatin is typically active (euchromatin), while densely packed chromatin is generally inactive (heterochromatin) Epigenetic modifiers, which include "writers," "erasers," and "readers," regulate chromatin structure through various mechanisms such as DNA and histone modifications, large protein complexes, and non-coding RNAs DNA methylation generally occurs in CpG islands near promoter regions, while histone modifications involve the addition of chemical marks to histone tails, influencing the recruitment of transcriptional machinery. Protein complexes, such as polycomb repressive complexes and chromatin remodeling complexes, alter chromatin architecture and DNA accessibility. Non-coding RNAs, including microRNAs and long non-coding RNAs, are involved in gene silencing and chromatin regulation (Figure 5) [117]. When these epigenetic regulators are disrupted by mutations, gene expression can become aberrantly activated or repressed, leading to diseases such as cancer and neural crest-related disorders. Epigenetic modifications provide additional genetic information that can be inherited across generations. DNA methylation and histone modification in mammals regulate gene expression and affect cell fate during development [118]. These dynamic modifications can vary during developmental processes such as craniofacial and neural tube development, tissue regeneration, and senescence [119]. DNA methylation and histone modifications also regulate genomic imprinting, which is the parent of the origin of certain genes. Epigenetic modifications are influenced by environmental factors, such as folates and retinoids, which contribute to altered developmental outcomes, including non-syndromic orofacial clefts (OFCs), cleft lip and palate (CLP), and cleft palate only (CPO) [120]. Epigenetic modifications may explain variations in OFC prevalence across populations, which cannot be attributed solely to genetic differences [121].
3.4.2. DNA Methylation Dynamics in Palatogenesis and Craniofacial Development
DNA methylation involves adding a methyl group to cytosine nucleotides in CpG sequences [122], often in CpG islands within promoter regions. This process recruits transcriptional repressors that inhibit gene expression by blocking transcription factors (Figure 5) [123]. In mammals and other vertebrates, methylation of cytosine (C) at the C5 position, leading to the formation of 5-methylcytosine (5mC), is widely accepted as the only epigenetic form of DNA methylation [8]. The methylation of adenine (A) in vertebrates remains controversial, whereas the methylation of guanine (G) and thymine (T) has not been reported. DNA methylation is catalyzed by DNA methyltransferases (DNMTs), which use S-adenosylmethionine (SAM) as an exclusive donor for methyl groups. Thus, the activity of DNMTs (histone-modifying enzymes) depends on an adequate supply of SAM, which is produced through folate and methionine cycles [121].
Methylation is thought to promote gene silencing by preventing the binding of transcription machinery or activators to DNA through spatial interference. In addition, methyl-CpG binding proteins recruited to 5-methylcytosine (5mC) can activate transcriptional repressors such as histone deacetylases [124]. Methylation generally occurs at cis-regulatory elements, particularly promoters and enhancers, to control variability in gene expression [125]. Promoters adjacent to the 5'-untranslated region (5'-UTR) are where the transcription machinery binds and initiates transcription. Enhancers located near the promoter or in distant regions, including the 3'-untranslated region (3'-UTR), interact with transcription factors to promote gene expression. They can physically form loops interacting with promoters and promoting transcriptional activation [126]. Enhancer activity is highly tissue-specific and plays a key role in spatial and temporal gene expression dynamics during embryonic development [127]. Although many studies have focused on promoter methylation [125], recent evidence suggests that enhancer and gene body methylation may play equal or even greater roles in regulating gene expression during development [128]. Methylation occurs at specific base sequences or motifs, and regulates gene expression across generations. Epigenetic methylation is commonly found in regions enriched with 5′-CpG-3′ motifs known as CpG islands [125]. Methylation at CpG islands is symmetrically maintained on both strands, making it a heritable epigenetic marker that does not require de novo methylation for reestablishment. Promoters are strongly linked to CpG islands, whereas enhancers may or may not be associated with them. Because of the correlation between promoters and CpG islands, early studies primarily focused on promoter methylation as the major epigenetic control of gene expression regulation. Another significant epigenetic marker is the pentanucleotide motif, 5′-CCWGG-3′ (where W can be A or T), which undergoes methylation inside C. Methylation of 5′-CWG-3′ has been conserved across generations in mammalian cells, likely because of its recognition as 5′-CCWGG-3′ by methyltransferases. The stable methylation of this motif challenges the previous notion that only CpG islands contain C bases suitable for transgenerational marking. The 5′-CCWGG-3′ motif is crucial for enhancer methylation dynamics in orofacial development [8] and is gaining increasing attention.
Adenine methylation, specifically the conversion of adenine nucleobases into N6-methyldeoxyadenine (N6mA), is a known modification of RNA that plays the role of N6-methyladenosine (m6A) in mammalian RNA processing [129]. However, their roles in mammalian DNA methylation remain controversial. Although N6mA is a well-established modification in prokaryotes and some eukaryotes, such as Caenorhabditis elegans, where it influences mitochondrial stress adaptation [130], its presence and function in mammalian DNA is disputed. Recent studies have questioned earlier claims regarding N6mA as a functional epigenetic marker of mammalian DNA. This suggests that the supposed evidence of N6mA in mammalian DNA may be due to RNA contamination or technical issues [131]. N6mA may be misincorporated into DNA by polymerase activity during the processing of ribo-N6mA, rather than functioning as a true epigenetic marker [132]. In RNA, N6mA modifications have been well established, especially in the development context. The potential role of N6mA in RNA during orofacial development was also discussed. RNA methylation, often mediated by Nsun family genes [121], is highly expressed in mouse embryonic tissues involved in craniofacial development, suggesting that RNA modifications may play a role in the etiology of orofacial clefts (OFCs).
DNA methylation is catalyzed by a family of DNA methyltransferases (DNMTs) [133], including DNMT1, DNMT3A, and DNMT3B, which play important roles in cell fate determination and tissue specification by modulating gene expression. DNMT1 is a methyltransferase that maintains methylation patterns through DNA replication and repair. The de novo methylation functions, on the other hand are provided by DNMT3A and DNMT3B, which stablish new methylation marks mainly at the promoter region. These enzymes play significant roles in regulating gene expression, especially early in development and in tissue-specific GRNs. In mice, Dnmt3a and Dnmt3b are highly expressed in undifferentiated embryonic stem cells and their expression decreases as cells differentiate [115]. DNMT3A represses neural genes such as Sox2 and Sox3 to promote neural crest specification during chicken development [134]. Zebrafish Dnmt3b and methyltransferase G9a regulate neurogenesis and craniofacial skeletal element formation [135]. Studies in human embryonic stem cells have shown that knockdown of DNMT3B accelerates neural and neural crest differentiation by upregulating neural crest specification genes such as PAX3, PAX7, FOXD3, SOX10, and SNAIL2 [136].
Studies have shown that DNMT3B is essential for neural crest and craniofacial development; however, conditional loss of Dnmt3b in mouse neural crest cells results in only mild neural crest migration defects and no significant craniofacial phenotypes [137]. This suggests that DNMT3B may function earlier in development than previously thought or may affect other tissues that are secondary to neural crest development. Recent studies have shown that DNMT3B can function without a catalytic domain and may act as a secondary cofactor to support the enzymatic activity of other DNMTs, such as DNMT3A [138]. Future studies should explore the dual role of DNMT3B better to understand its role in craniofacial and neural crest development.
Several studies using mouse models have explored the role of differential gene methylation in orofacial development, particularly in palatogenesis. CpG methylation in the palate is significantly higher at embryonic day E14.5 than at E13.5 and E18.5, a critical time when the palatal shelves are elevated above the tongue, just before medial epithelial seam formation [8]. A microarray approach was used to examine DNA methylation in mouse palates from E12 to E14. They found that 73% of the detected genes were methylated, mostly within gene bodies rather than promoters, with 30% of methylation occurring in CpG islands [139]. These findings align with those of previous studies on retinoic acid (RA) exposure, where differentially methylated regions (DMRs) were located at intronic enhancers of genes linked to palatogenesis. Sox4, a key gene in palatal development, showed decreased expression at E13 and E14 because of methylation in the CpG-poor promoter region. Sox4 plays a role in integrating several signaling pathways, including Tgfβ, Wnt/β-catenin, BMP, FGF, and Hedgehog, which regulate palatal fusion and extension [8].
Mutant mouse strains have also shed light on the role of methylation in orofacial development. The A/WySn strain, which has a 15-20% risk of CL/P, involves an epistatic interaction between Clf1 (an IAP retrotransposon at the 3′ end of Wnt9b), Clf2, and a maternal effect. Clf2 suppresses Clf1 IAP via DNA methylation. Clf1 was later identified as a metastable epiallele with stochastic methylation during embryogenesis, indicating that some individuals lack Clf1 methylation, leaving them vulnerable to CL/P [140].
3.4.3. Epigenome-Wide Association Studies (EWAS) in Orofacial Clefts (OFCs)
Epigenome-wide association studies (EWAS) have identified significant DNA methylation differences linked to orofacial clefts (OFCs), particularly NSCLP. For example, a study in the UK found differentially methylated regions in the blood and lip tissue across cleft subtypes, including well-known cleft-related genes TBX1, COL11A2, HOXA2, and PDGFRA, and identified 250 new loci [11]. A later study conducted in Brazil revealed 578 methylation variable positions associated with NSCLP that were highly enriched for regulatory regions involved in craniofacial development [141]. Long interspersed nucleotide element-1 (LINE-1), a marker of global DNA methylation, was found to be differentially methylated in non-syndromic orofacial clefts compared to controls [142]. Mutations in the 5,10-methylenetetrahydrofolate reductase (MTHFR) gene, such as c.C677T and cA1298C, have been reported to reduce DNA methylation levels [9], whereas increased methylation levels of LINE-1 have been observed in the center of cleft lips in the c.C677T mutation [130]. These DMRs may explain the absence of hereditary patterns in cleft lips. With environmental pressures, population-specific epigenetic modifications can be anticipated, and worldwide investigation of cleft populations is needed to understand their epigenetic contribution to cleft palate etiology.
In the last decade, methylation profiling has identified epigenetic modifications as key players in the etiology of orofacial clefts (OFC). These modifications are particularly appealing as mechanisms for the environmental causes of OFCs. For example, maternal smoking has been shown to differentially methylate genes previously associated with OFCs in children, including MSX1, PDGFRA, GRHL3, ZIC2, and HOXA2 [143]. Other methylation profiling studies have identified genes with variable methylation levels that may influence the incidence of OFCs. These include transcription factors (LHX8, PRDM16, PBX1, GSC, VAX1, and MYC), growth factors and modulators (WNT9B, BMP4, EPHB2, BICC1, and DHRS2), extracellular matrix genes (CRISPLD2, NTN1, and CDH1), and miRNAs (MIR140 and MIR300) [141]. Some of these genes, including PRDM16, BHMT2, and WHSC1, encode proteins involved in methyltransferase activity [144]. Further studies have explored the variable methylation positions across different OFC subtypes. Identified hundreds of methylation variable positions distinguishing between cleft lip and palate (CLP), cleft lip only (CLO), and cleft palate only (CPO) [11]. These findings suggest that DNA methylation profiling is a promising approach that could offer a more detailed understanding of the etiology of OFC.
3.4.4. Impacts of Histone Modifications in Craniofacial Development
Histone modifications, the chemical modifications of histone proteins, are complex in nature and important in regulating chromatin structure and gene expression, especially development (Figure 5). Specifically, PTMs, such as methylation, acetylation, deacetylation, phosphorylation, ubiquitination, and sumoylation dynamically modulate gene expression by compaction and relaxation of chromatin [117], and therefore chromatin accessibility. These changes are tightly linked to cell fate decisions during neural crest cell (NCC) development, and regulation of these alterations is especially critical [145]. Another essential PTM that affects chromatin accessibility and gene expression is histone acetylation, which is regulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs) (Figure 5).
HAT KAT6A is related to Arboleda-Tham syndrome, which is characterized by developmental delays and craniofacial abnormalities [146]. NCC migration and differentiation also rely on HDAC function, and mutations in HDAC1, HDAC2, and HDAC4 have been linked to craniofacial defects [147]. In addition, NCCs depend on specific histone modifications, such as H3K4me1 and H3K27ac, in enhancer regions to regulate chromatin structure and gene expression during development [148]. DNA methylation and histone modifications often work together, and environmental factors such as 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) exposure can disrupt histone acetylation, thereby affecting developmental processes such as cleft palate formation in mice [149]. Mutations in histone-modifying enzyme genes are commonly associated with developmental disorders, such as orofacial cleft (OFC) [13]. Taken together, the functions of enzymes such as histone methyltransferases, demethylases, HATs, and HDACs are crucial for the proper development of the neural crest. The disruption of these processes can affect neural crest cell proliferation, migration, and differentiation, resulting in congenital craniofacial birth defects.
Histone H3K27me3 Demethylase KDM6A, KDM6B
Kabuki syndrome, caused by mutations in KDM6A or KMT2D, leads to developmental abnormalities. KDM6A, an X-linked H3K27 demethylase, is associated with stunted growth and cleft palate, as observed in one patient with haploinsufficiency [150]. Zebrafish studies have confirmed that reduced kdm6a expression leads to craniofacial defects, supporting its role in cleft palate formation [151]. Conditional knockout of Kdm6a in neural crest cells (NCCs) using Wnt1-Cre in mice revealed sex-specific effects, with females showing more severe phenotypes, including cleft palate. Males may compensate for Kdm6a loss through a Y-linked homolog without demethylase activity. Despite the importance of Kdm6a in neural crest development, no changes in H3K27 or H3K4 trimethylation were observed, suggesting that Kdm6a regulates development through mechanisms independent of its demethylase activity [152].
Kdm6b is a critical player in cranial neural crest development, and loss of Kdm6b disrupts P53 pathway-mediated activity, resulting in a complete cleft palate, along with cell proliferation and differentiation defects in mice. Kdm6b and Ezh2 antagonistically control H3K27me3 activity in the Trp53 promoter in cranial neural crest cells. More importantly, in the absence of Kdm6b, the transcription factor Tfdp1, which normally binds to the Trp53 promoter, failed to activate the expression of Trp53 in palatal mesenchymal cells. Furthermore, the expression of Trp53 in such cells cannot be compensated for by the highly homologous histone demethylase Kdm6a [153]
Histone H3 Lysine 4 Methyltransferase KMT2D
Mutations in KMT2D, similar to those in KDM6A, are associated with Kabuki syndrome. A Xenopus model study showed that the knockdown of kmt2d affected neural crest cell (NCC) dispersion but not other migratory behaviors, confirming the role of KMT2D in H3K4 methylation [154]. This study identified sema3f, a gene necessary for cranial NCC migration, as a target, and its overexpression partially rescued this phenotype. In contrast, a mouse conditional knockout (cKO) of Kmt2d showed a fully penetrant cleft palate but did not affect NCC migration. This observation is consistent with that observed in patients with Kabuki syndrome [155]. This cleft palate is associated with abnormal expression of extracellular matrix members. Kmt2d mutations may affect other cis genes, with downstream effects involving RAP1A dysfunction and impaired RAS/MAPK activation [156]. MAPK signaling inhibitors, such as desmethyl-dabrafenib, can prevent structural defects during embryogenesis in a zebrafish model of Kabuki syndrome, with any toxic effects [157], and may represent a future therapeutic strategy.
Histone-Lysine Demethylase PHF8
Mutations in histone demethylases such as PHF8, which demethylates H4K20 and H3K9, can lead to severe developmental disorders. PHF8 mutations are linked to cleft lip and/or palate (CL/P) and X-linked intellectual disabilities [158]. PHF8's demethylase activity is essential for ribosomal RNA (rRNA) transcription [159] and neural differentiation [160]. Its catalytic domain, 2OG oxygenase, suggests a link between hypoxia and orofacial clefts, particularly in children of mothers exposed to tobacco smoke during conception [161]. In zebrafish, phf8 regulates msx1 expression, which is associated with neural-crest induction and craniofacial development [160]. Phf8 overexpression in mice also promotes bone regeneration, indicating potential therapeutic applications for craniofacial defects through regulation of SATB2 [162].
Histone-Lysine N-Methyltransferase MECOM (PRDM3)
N-methyltransferase MECOM (PRDM3) regulates the methylation of H3K4 and H3K9 residues. During zebrafish development, prdm3 is highly expressed in pharyngeal arches [163]. Morpholino knockdown results in neural and stellate defects and reduced expression of the NCC markers dlx2a and barx1. In another study, similar effects of prdm3 knockdown were observed in developing zebrafish, with decreased methylation of H3K4 and H3K9 [164]. Conditional knockout of Prdm3 by Sox2-Cre in developing mice results in mid-gestational lethality.
Histone-Lysine N-Methyltransferase PRDM16
Prdm16 has overlapping functions with prdm3 in zebrafish, as its knockdown reduces the expression of neural crest markers dlx2a and barx1 and lowers H3K4 and H3K9 methylation [164]. Prdm16 is crucial for palatogenesis in mice, as shown by loss-of-function studies using mutagenesis, RNA interference, and gene trapping [165]. During palate development, Prdm16 regulates several target genes involved in myogenesis, chondrogenesis, and osteogenesis [166], and its loss perturbs the expression of Tgf-β and Bmp signaling pathways. Conditional knockouts revealed its role in H3K9 methylation but not H3K4 methylation [164], and it may also regulate orofacial development through Smad transcription factors [8].
Arginine Methyltransferase PRMT1
PRMT1 encodes an arginine methyltransferase responsible for H4R3me2a modification and regulates over 85% of arginine methylation activity, some of which targets non-histone proteins [167]. Conditional knockout of Prmt1 in neural crest cells (NCCs) using Wnt1-Cre leads to craniofacial malformations, including cleft palate, similar to defects observed in Msx1-null mice [168]. Prmt1 cKO reduces Msx1 expression in critical craniofacial regions at embryonic day 12.5. A follow-up study found disrupted BMP signaling in knockouts linked to PRMT1 methylation of Smad6, a BMP inhibitor. Reduced H4R3me2a suggests PRMT1’s role in development also involves histone modification [169].
Histone Methyltransferase WHSC1
WHSC1 is a methyltransferase gene linked to Wolf-Hirschhorn syndrome and is expressed in both the epithelium and mesenchyme during mouse palate development. [170] found that its expression was reduced when pregnant mice were treated with all-trans retinoic acid (RA), a treatment that causes cleft palate. Researchers have suggested that whsc1 plays a role in promoting cell proliferation. In a separate study, the knockdown of whsc1 in Xenopus led to reduced facial width and smaller midfacial areas. It decreased the migratory distance and total area of the cranial neural crest cells (NCCs) [171].
Histone Deacetylases HDAC3 and HDAC4
HDAC3 is essential for mouse development, and conditional knockout in neural crest cells (NCCs) leads to craniofacial defects, including cleft palate [172]. HDAC3 regulates the transcription factors Msx1, Msx2, and Bmp4. In conditional knockouts, these genes showed increased expression, whereas cell proliferation decreased and apoptosis increased at E12.5. Histone acetylation is likely important for balancing gene expression in NCCs.
HDAC4, a class II histone deacetylase, plays a role in osteogenesis by interacting with MEF2 and regulating endochondral ossification [8]. During zebrafish development, hdac4 is expressed in pre-migratory and migrating cranial NCCs. hdac4 knockdown results in reduced or absent cranial NCCs, leading to palatal defects such as shortened, clefted, or missing ethmoid plates [173].
Histone Acetyltransferase KAT6A
Microdeletion or mutation of TBX1 in humans causes DiGeorge Syndrome, which includes symptoms such as submucous cleft palate (SMCP), heart defects, and thymic dysfunction [174]. In mice, the deletion of the acetyltransferase KAT6A, which regulates Tbx1 expression, partially mimicked DiGeorge Syndrome, including SMCP. The study found that an extra copy of Tbx1 did not rescue the palatal defects caused by Kat6a deficiency, suggesting that either a higher dosage of Tbx1 is needed or that Kat6a influences additional genes beyond Tbx1.
3.4.5. Non-Coding RNAs in Craniofacial Development and Orofacial Clefts
MicroRNAs (miRNAs) are small (~22-nucleotide) non-coding RNA molecules that regulate various developmental processes. miRNAs typically bind to the 3′ untranslated regions (3′UTRs) of target mRNAs through complementarity between the miRNA seed sequence and miRNA response element (MRE) [15]. Perfect binding results in mRNA degradation, whereas partial binding leads to suppression of transcription. miRNAs can regulate multiple mRNAs, and a single mRNA can be targeted by several miRNAs. In addition to gene silencing, miRNAs can activate transcription, upregulate protein expression, and target mitochondrial transcripts [15]. miRNAs play important roles in embryonic orofacial tissues by targeting genes involved in cell proliferation, apoptosis, differentiation, cell adhesion, and epithelial-mesenchymal transition (EMT) (Table 3) [175].
The formation of craniofacial structures begins with migration and patterning of neural crest cells (NCCs). Dicer, an enzyme essential for miRNA processing, is critical for the survival of post-migratory NCCs, although it is not required for their initial migration into facial primordia [176]. NCC-specific Dicer knockout mice exhibit various craniofacial defects, with some showing microcephaly, facial hypoplasia, and cleft palate (CP) [176]. However, palatal development is halted in Dicer knockout (KO) models because NCC-derived skeletal structures remain immature or absent [176]. Most of these defects result from extensive apoptosis induced by NCC derivatives and are associated with defective MAPK/ERK signaling [176]. Functional analysis has revealed that miR-21 and miR-181a repress Sprouty2, a negative regulator of the MAPK/ERK signaling pathway essential for cell proliferation, differentiation, and apoptosis [177]. Due to abnormal NCC patterning, Murine Dicer disruption leads to severe defects in the maxillary, mandibular, and frontonasal processes. miR-452 is key in regulating EMT and NCC patterning by targeting Wnt5a [178]. Loss of miR-452 increases Wnt5a expression and reduces Shh and Fgf8 signaling, thereby decreasing Dlx2 expression, a key regulator of NCC patterning in the first pharyngeal arch [139]. Recently, miR-149 has been implicated in the etiology of non-syndromic cleft lip with or without palate through its role in migrating human neural crest cells (hNCCs) derived from human induced pluripotent stem cells (Figure 6). Using 3′ RNA-Seq, 604 differentially expressed genes were identified in hNCCs overexpressing miR-149 compared with untreated cells, highlighting their involvement in this process[179].
During palatogenesis and orofacial cleft development, specific miRNAs play crucial roles in regulating cell proliferation (Figure 6). Overexpression of miR-133b, miR-374a-5p, and miR-4680-3p inhibits cell proliferation in human embryonic palate mesenchymal (HEPM) cells, probably by downregulating GCH1, PAX7, FGFR2, and ERBB2 [15] [180]. Similarly, miR-497-5p and miR-655-3p reduce the proliferation of human lip fibroblasts by targeting multiple genes implicated in orofacial clefts in CL/P studies. miR-124-3p reduces myeloma cell line (MELM) cell proliferation by downregulating Bmpr1a, Cdc42, and Tgfbr1 [181]. In particular, strong expression was found in the maxillary process of miR-124-3p, particularly at GD13.5. In addition, the conserved human and mouse regulatory subnetwork with five transcription factors, including GLI2, PAX3, PAX7, PAX9, and SATB2 [182]; three non-transcription factor genes, FGFR1, RARA, and SUMO1; and five miRNAs, including miR-27b, miR-133b, miR-205, miR-376b, and miR-376c, control cell proliferation of lip mesenchymal cells via the following specific gene targets: miR-27b targets PAX9 and RARA; miR-133b targets FGFR1, PAX7, and SUMO1; and miR-205 targets PAX9 and RARA [183].
In addition, [184] identified miRNAs (hsa-miR-133b, hsa-miR-140-5p, hsa-miR-374a-5p, hsa-miR-381a-3p, and hsa-miR-4680-3p) associated with CP development in humans through systematic reviews, bioinformatics analyses, and cell proliferation assays in human embryonic palatal mesenchymal(HEPM) cells[180,184]. Using data from patients with CP and HEPM cells, [185] demonstrated that hsa-let-7c-5p and hsa-miR-193a-3p are involved in the development of CP using the data of CP patients and HEPM cells. Among the seven miRNAs, we found that PB specifically induced let-7c-5p expression and that the let-7c-5p specific inhibitor alleviated the PB-induced suppression of HEPM cell proliferation, indicating that let-7c-5p plays a crucial role in PB-induced toxicity. Let-7c-5p is highly expressed in craniofacial tissues of embryonic mice [185].
Studies of the mir-17-92 cluster provided the first genetic evidence that specific miRNAs are functionally associated with mammalian CL/P[186]. The mir-17-92 cluster, which consists of 6 highly conserved miRNAs (miR-17, miR-18a, miR-19a, miR-19b-1, miR-20a, and miR-92a-1) belonging to four families (miR-17, miR-18, miR-19, and miR-92), is located on mouse chromosome 14 (chromosome 13 in humans) [8]. The expression of mir-17-92 and its two paralogs follows a similar pattern in mouse embryos, decreasing from E12 to E14 and concentrated in the distal tips of the PS during palatogenesis [175] [186]. Direct targets of miR-17-92 include T-box factors such as Tbx1 and Tbx3, which harbor functional MREs in their transcripts. These genes are upregulated in miR-17-92 mutant craniofacial structures, and studies have shown that miR-17-92 directly represses Fgf10 expression, which is crucial for proper maturation of the palate epithelium [8] [186]. ChIP and ChIP-Seq data demonstrated the binding of AP-2α and Smad1/2/5 to miR-17-92 chromatin, suggesting that AP-2α and BMP signaling regulate mir-17-92 expression. It is evident from these studies that the downregulation of specific progenitor genes, such as T-box factors, by miR-17-92, is critical for normal midfacial development [186].
The miR-17-92 cluster also targets the TGF-β signaling pathway in the palatal mesenchyme [8]. A decrease in the expression of miR-17-92 from GD12-14, with a concomitant increase in the expression of key components of the pathway, such as TGFBR2, SMAD2, and SMAD4, was evident in the palatal shelves. Luciferase assays in palate mesenchymal cells (PMCs) revealed that TGFBR2 is directly targeted by miR-17 and miR-20a, whereas SMAD2 and SMAD4 are targets of miR-18a [8]. It is hypothesized that the mir-17-92 cluster regulates palatal shelf elongation and elevation by regulating the TGF-β induced inhibition of proliferation and collagen synthesis. E2F1 directly binds to the miR-17-92 promoter, facilitating its transcription. The findings in palatal mesenchymal cells (PMCs) include the expression of E2F1 and E2F3 in palatal tissue on GD12-14, significant inhibition of cell proliferation upon E2F1 knockdown, and upregulation of the miR-17-92 cluster with E2F1 overexpression [187]. Excessive cell proliferation is curtailed when miR-17 and miR-20a bind to the 3-UTR of E2F1, thereby forming a negative feedback loop that helps regulate the cell cycle. Abnormal regulation of this negative feedback loop may lead to palatal cleft formation [187].
Palatal fusion proceeds through the resolution of a medial edge seam (MES), which is consistent with a mechanism of "convergence and extrusion" during the final stages of palatal fusion [188]. It involves the creation of temporary epithelial bridges over the opposing palatal shelves, followed by a thick epithelial layer that converges into a unified monolayer through cellular intercalation and oronasal translocation of MES epithelium [188]. [189] has been identified miR-22–3p, an actomyosin contractility regulator during palatal fusion. Inhibition of miR-22 activity in palate organ cultures using anti-miR-22 resulted in the failure of MEE dissolution and MES removal, supporting a key role for miR-22 in palatogenesis. Several potential mRNA targets of miR-22 are transcripts encoding two myosin-heavy chains (Myh9 and Myh10) and essential actomyosin contractility. Although functional interactions between miR-22 and these targets have not been demonstrated in palatal tissues, mRNAs Myh9 and Myh10 represent functionally validated targets of miR-22 in other biological settings [189].
The epithelial-to-mesenchymal (EMT) transcription factor (TFs) balance is important for efficient palatal closure. Balanced regulation of epithelial and mesenchymal TFs, such as Grhl2 and Zeb1, is necessary for palatal closure [190]. Zeb1 is considered responsible for mesenchymal identity, whereas Grhl2 and its targets (e.g., Ovol1, Ovol2, and the miR-200 family of miRNAs) promote epithelial identity [190]. The results of this study indicated that Grhl2/miR-200 and Zeb1/Zeb2 antagonize each other and that Grhl2 transactivates the miR-200 family of miRNAs to repress Zeb1/Zeb2 [190]. In this respect, miR-200b was shown to target Smad2, Snail, Zeb1, and Zeb2, all of which encode transcription factors that function as mediators of the Tgf-β signaling pathway. In response to TGF-β, SMAD2/3 is activated. It forms a complex with SMAD4, which then interacts with either ZEB1, ZEB2, or SNAIL to repress epithelial markers, stimulate mesenchymal markers and induce migration and apoptosis [191] [15]. miR-200b expression in MES gradually diminishes as palatal fusion proceeds, as the inhibition of cell proliferation in MES is necessary before palates can fuse. These results indicated that miR-200b overexpression induces abnormal palatogenesis in MES by inhibiting Tgf-β-mediated Smad2 and Snail expression [191] [15].
Analysis of the MEE of Tgf-β3-/- mouse fetuses at GD13.5, indicated that the expression of miR-206 in the developing palate was significantly diminished compared to that in their wild-type counterparts, suggesting an important role of miR-206 in palatal ontogeny [15] [192]. Expression profiling of mouse embryonic maxillary mesenchymal (MEMM) cells treated with anti-miR-206 revealed significant changes in the expression of ~ 230 genes, including several members of the TGF-β and Wnt/β-catenin superfamilies [192]. Aberrant regulation of this pathway can cause various disorders, such as orofacial clefts, Kallman syndrome, Crouzon syndrome, and Apert syndrome [13].
Additionally, maternal miRNA expression influences cleft risk. For example, overexpression of miR-152 has been associated with craniofacial dysmorphism, and maternal circulating miR-let7-3p may be a diagnostic biomarker for non-syndromic CL/P (Table 3). SNPs in miRNA biogenesis enzymes or within miRNA-binding regions of cleft-related genes (e.g., FGF2, FGF9, and MSX1) have been associated with cleft risk [183]. A genetic association study showed that an SNP (rs7205289:C>A) located in the precursor of miR-140 (pre-mir-140) contributes to non-syndromic cleft palate susceptibility by influencing the processing of miR-140 [193]. The minor A allele of rs7205289, with a higher frequency in patients, is associated with decreased miR-140-5p expression and increased miR-140-3p expression [184]. In addition, this allele is conserved in primates and functionally important. Therefore, dysregulation of miR-140 may be implicated in the etiology of cleft palate, which is supported by genetic evidence. miRNAs are essential for properly regulating craniofacial development, and this biomarker is supported by numerous craniofacial developmental defects, such as cleft palate and other orofacial defects resulting from miRNA dysregulation[15].
Long non-coding RNAs (lncRNAs) act as miRNA sponges by binding to specific miRNAs via MREs to reduce miRNA levels [194]. The function of long non-coding RNAs (lncRNAs) is closely related to their subcellular localization. In the cytoplasm, lncRNAs play regulatory roles by functioning as miRNA sponges and engaging in a competing endogenous RNA (ceRNA) mechanism. Furthermore, cytoplasmic lncRNAs can interact with RNA-binding proteins (RBPs), thereby exerting biological effects [195]. In contrast, the nuclear localization of lncRNAs enables them to modulate gene transcription or pre-transcriptional processes through interactions with DNA promoter regions or transcription factors, referred to as cis- and trans-regulatory mechanisms (Table 3) [196] [197].
The SNP rs2262251 (G > C) located in lncRNA RP11-462G12.2, was found to be specifically associated with CL/P but not CP. Overexpression of the G allele inhibits apoptosis and promotes the proliferation of HEK-293 and HEPM cells [15,198]. Transfection studies using luciferase reporters indicated that the C allele, but not the G allele, specifically binds to miR-744–5p. Transfection of miR-744–5p into HEPM cells decreased lncRNA expression of the C-allele, and this decrease was rescued using miR-744–5p inhibitors. Conversely, overexpression of lncRNA decreased miR-744–5p levels, thus confirming the sponging effect of lncRNA with the C-allele [198]. Both miR-744–5p and IQSEC2 exhibited a significant reverse correlation and were expressed in lip tissues of patients with CL/P. Thus, the C-allele of the lncRNA regulates IQSEC2 expression by sponging miR-744–5p, enhancing apoptosis, and suppressing proliferation[198]. LncRNA-NONMMUT100923.1 was found to regulate Cdsn expression by competitively binding to miR-200a-3p in a ceRNA network during palatogenesis, potentially inhibiting medial edge epithelial cell adhesion by preventing desmosome junction disintegration [196]. For instance, one study highlighted the potential regulatory role of NONMMUT004850.2/NONMMUT024276.2-miR-741-3p/miR-465b-5p-Prkar1α in palatal fusion during cleft palate development[199]. A complex regulatory association involving miR-483-3p, miR-4690-3p, miR-654-3p, miR-6515-5p, lncRNA RP11-731F5.2, lncRNA XIST, lncRNA RP11-591C20.9, RARA, and SMPD1 was also revealed in the CL/P and CPO groups [200,201].
Some studies utilizing the same lncRNA dataset GSE183527 as our study identified that ceRNA networks (MALAT1-hsa-miR-1224-3p-SP1, MALAT1-hsa-miR-6734-5p/hsa-miR-1224-3p-WNT10A, NEAT1-hsa-miR-140-3p.1-CXCR4, NEAT1-hsa-miR-3129-5p/hsa-miR-199a-3p/hsa-miR-199b-3p-ZEB1[202], and NEAT1-hsa-miR-130b-3p/hsa-miR-212–3p/hsa-miR-200b-3p-SMAD2) may contribute to the etiology of non-syndromic orofacial clefts [196]. lncRNA TPT1-AS1 has been reported to regulate various biological processes, including cell proliferation, apoptosis, autophagy, invasion, migration, and epithelial-mesenchymal transition (EMT), all of which are implicated in the progression of NSCL/P[196]. The potential roles of the lncRNAs FENDRR and TPT1-AS1 and the mRNAs EIF3H, RBBP6, and SRSF1 in NSCL/P development were investigated. Further investigation into the regulatory mechanisms of these lncRNAs and their interactions with other factors, such as environmental exposure, could provide additional insights into the development of cleft palate [195] [196].
3.4.6. Epigenetic Regulation in Chromatin Organization and Craniofacial Development
The Polycomb repressive complexes PRC1 and PRC2 are essential transcriptional repressors that suppress gene expression [203]. EZH2, a catalytic subunit of PRC2, undergoes H3K27 methylation. Conditional knockout of Ezh2 in murine neural crest cells (NCCs) causes Hox gene derepression, maintains NCCs in a pre-differentiation state, and impairs osteochondroprogenitor programs, which affect cartilage and bone formation [204]. EZH2 mutations are also linked to Weaver Overgrowth syndrome, a genetic disorder that causes bone overgrowth [205]. Ring1b/Rnf2, an E3 ubiquitin ligase in PRC1, regulates craniofacial chondrocyte differentiation. Zebrafish ring1b mutants exhibit impaired cranial cartilage and bone development [206]. PHC1 and PHC2 are key regulators of HOX gene expression and craniofacial patterning, and genetic mutations affecting these proteins have been shown to underlie CATCH-22 syndrome associated with DiGeorge syndrome-like phenotypes and non-syndromic cleft lip and palate [207]. ASXL proteins (ASXL1/2/3), scaffolding components of the PRC complex, are critical for early neural crest development. Mutations in ASXL cause neurodevelopmental disorders such as Bohring-Opitz, Shashi-Pena, and Bainbridge-Ropers syndromes, all of which feature craniofacial defects and developmental delays [208].
ATP-dependent chromatin remodeling complexes alter chromatin structure in an ATP-dependent manner to control gene expression, providing access to transcription factors and machinery associated with DNA. The SWI/SNF complex, which contains subunits including ARID1A, ARID1B, BRG1 (SMARCA4), BRM, BAF155, BAF170, SMARCA2 (SNF5), SMARCB1 (INI1), and SMARCE1, is an important regulator of gene expression. Mutations in these factors are associated with syndromic developmental defects, such as Coffin-Siris syndrome, which presents as a developmental disability, characteristic facial features, and malformations of the fifth finger or toe (Figure 5). Coffin-Siris syndrome patients frequently have mutations affecting the SWI/SNF complex subunits ARID1A or ARID1B. In mice, loss of Arid1a in neural crest cells (NCCs) causes severe craniofacial defects. In human studies, ARID1B mutations disrupt the switch between ARID1A and ARID1B during neural crest development, impairing differentiation [209]. Brg1 mutations are also linked to Coffin-Siris syndrome, with loss of Brg1 impairing NCC survival and differentiation. Brg1 is essential for conserving a pool of multipotent NCCs, and its loss leads to craniofacial defects in zebrafish and mice [210]. Moreover, the loss of some core SWI/SNF subunits, such as BAF155 and BAF170, leads to abnormalities in the survival, migration, and differentiation of neural crest cells, thereby leading to craniofacial defects [211]. Other SWI/SNF complex subunits, including SMARCA2, SMARCB1, and SMARCE1, also play similar roles in neural crest specification and craniofacial development [212]. Another chromatin remodeler, CHD7, also plays a crucial role in neural crest development, and its mutations cause the CHARGE syndrome. CHD7 interacts with the PBAF complex to regulate the expression and migration of the neural crest. Animal models, including zebrafish and mice, show that mutations in Chd7 recapitulate craniofacial and cardiovascular defects found in CHARGE syndrome [29].
Other epigenetic regulators, such as special AT-rich sequence binding protein 2(SATB2), are also important in craniofacial development besides ATP-dependent chromatin remodeling complexes. SATB2 is a chromatin reader that binds to the nuclear matrix attachment regions to activate transcription. In humans, translocations involving the 2q32–q33 chromosomal region, which encompasses SATB2, are associated with cleft palate [115]. Mutations in SATB2 are linked to SATB2-associated syndromes characterized by intellectual disability, developmental delays, and craniofacial and dental abnormalities [213]. In mice, SATB2 mutations result in craniofacial abnormalities resembling human phenotypes and osteoblast differentiation defects [115]. SATB2 regulates skeletal patterning by controlling Hox genes and later drives osteogenesis by regulating genes such as Runx2 and ATF4, as well as craniofacial patterning genes including Pax9, Msx1, Alx4, and Lhx7. These findings show that SATB2 plays a dual role in neural crest development: it promotes neural crest cell (NCC) survival early on and later drives osteogenic differentiation during craniofacial structure formation.
3.4.7. Environmental Influences on Epigenetics and Craniofacial Development
Exposure to environmental factors, especially those that take place in utero, is associated with a higher risk. These include smoking tobacco, drinking alcohol, and/or using several drugs such as benzodiazepines, corticosteroids, antibiotics, and antiepileptic drugs [214]. Additionally, organic solvents and pesticides have been implicated in occupational exposure [215]. It has been postulated that the interaction between genetic predispositions and environmental factors leads to cleft palate development (Figure 6) [214,216]. A recent study demonstrated an increased risk of orofacial clefts (OFCs) due to the high concentrations of toxic elements in diverse biological matrices. However, contiguous relationships were found for elevating the concentrations of essential trace elements (ETEs) to reduce the risk. Dietary intake of maternal foods containing ETEs, such as zinc (Zn), selenium (Se), copper (Cu), cobalt (Co), and molybdenum (Mo), is associated with a marked decrease in the risk of OFCs [215]. These results imply that exposure to toxic elements, both environmental contaminants and food, contributes to the risk of OFCs. In contrast, ETEs are inversely associated with OFCs, indicating a protective role against OFCs [215].
RA, a metabolic derivative produced through the retinol (vitamin A) metabolic pathway, is an important signaling molecule required for both morphogenesis and differentiation during embryonic development, including craniofacial morphogenesis. Deficiencies in vitamin A and its metabolites have long been known to cause congenital craniofacial defects, and excess retinoids induce defects in rodents, including CL/P [217] [13]. In rodents, studies on the role of RA signaling have utilized various approaches, such as targeted knockout of retinoic acid receptors (RARs) and retinoid X receptors (RXRs), knockout or inhibition of enzymes involved in RA metabolism, and addition of exogenous RA [214]. Circulating retinol is bound by a retinol-binding protein (RBP) and taken up by target cells via the RBP receptor, STRA6 [218]. Cellular retinol-binding protein (CRBP) facilitates the conversion of retinol to retinaldehyde by alcohol dehydrogenase (ADH) or retinol dehydrogenase (RDH) enzymes, which are then oxidized to retinoic acid (RA) by retinaldehyde dehydrogenases (RALDH1-3) [219]. RA activates RARs and RXRs, which regulate gene transcription[214]. Mutations in RARs (α, γ) result in cleft palate and midfacial clefts, whereas Aldh1a3-deficient mice exhibit reduced RA activity leading to lethal choanal atresia [220]. Reciprocal regulation between CYP26 enzymes and Tbx1 is crucial for palatogenesis, as Tbx1 controls oral epithelial differentiation, and its deficiency leads to cleft palate [221]. All three Cyp26 genes (Cyp26a1, Cyp26b1, and Cyp26c1) that metabolize RA are dysregulated in Tbx1 mutants [222], leading to RA accumulation and reduced expression of the key regulators Bmp2 and Fgf10, resulting in cleft palate[223]. RA negatively modulates Tgf-β signal transduction by enhancing Smad7 expression and downregulating Smad2 activity, whereas Tgf-β3 represses RA signaling. Two pathways mutually regulate the common co-repressors of TGIF1, RA, and TGF-β signaling [224] [214]. All-trans retinoic acid (ATRA), a form of RA, also mediates its effects through Notch signaling in palatal cells to drive apoptosis and cell proliferation. In previous studies, ATRA exposure increased Notch1 expression in epithelial cells and Notch2 in mesenchymal cells while reducing CyclinD1, inhibiting proliferation [225]. It also decreases the expression of key genes (Lhx8, Msx1, and Msx2) in chick embryos, affecting orofacial development [226]. Mutual repression of RA and Wnt signaling is crucial for craniofacial development [227]. Wnt-deficient mice show increased RA activity, whereas RA suppresses Wnt/β-catenin signaling and disrupts cell cycle regulation [214]. RA also maintains Shh and Fgf8 expression, but excess RA inhibits Shh signaling and increases apoptosis in mouse maxillary tissue. Shh regulates RA levels through Cyp26 gene activation, highlighting the complex interactions between the RA, Shh, and Wnt pathways in craniofacial development [217,227]. Notch signaling in both the epithelium and mesenchyme may be altered after ATRA exposure, thus altering p21 signaling. For example, ATRA exposure at E10 increased Notch2 and decreased CyclinD1 in mesenchymal cells between E12.5-14.5, inhibiting proliferation [225]. Another study found that ATRA exposure at E12 increased Notch1 expression in MEE cells at E15, concomitantly inhibiting normal epithelial apoptosis [228]. ATRA also reduced the expression of Lhx8, Msx1, and Msx2 in the upper jaw of chick embryos and genes regulated by Fgf and RA signaling [226]. ATRA may influence Lef1-mediated periderm EMT in mice by decreasing Ambra1 expression, whereas Cyp26b1 is essential for proper palatogenesis and tongue depression [229]. The cleft palate (CP) in atRA-treated mice has been linked to the upregulation of miR-124–3p and miR-4680–3p (Figure 6). In atRA-treated mouse embryonic palatal mesenchymal (MEPM) cells and embryos, miR-124–3p levels are increased, leading to the downregulation of target genes such as Axin1, Fst, Vcan, and Zeb1 [184]. Likewise, overexpression of miR-4680–3p inhibits the proliferation of atRA-exposed HEPM cells by suppressing the expression of CP-associated genes, such as ERBB2 and JADE1 [180]. MiR-106a-5p was significantly (∼8.9-fold) upregulated in atRA-induced CP tissues and negatively correlated with Tgfbr2 protein expression levels. The overexpression of miR-106a-5p downregulated the expression of Tgfbr2 and altered the levels of pSMAD2 and pSMAD3 [230]. In addition, miR-106a-5p was strongly negatively correlated with cholesterol metabolites, suggesting that miR-106a-5p may play a putative role in cholesterol metabolism through TGF-β signaling in the defective palatogenesis of atRA-induced CP [230]. Thus, the ability of miR-106a-5p to modulate apoptosis may occur via the TGFβ/SMAD pathway [230] [15]. Recently, a ceRNA regulatory network was elucidated, where LncRNA-NONMMUT100923.1 regulates Cdsn expression by competitively binding to endogenous miR-200a-3p during palatogenesis in an all-trans retinoic acid (ATRA)-induced murine model [231]. Recently, RNA sequencing of mouse embryonic palatal shelf (MEPS) tissue revealed that all-trans retinoic acid (ATRA) treatment significantly upregulated miRNA-470-5p expression while downregulating Fgfr1 expression compared to controls. The results consistently demonstrated that miRNA-470-5p inhibits epithelial-mesenchymal transition (EMT) in MEPS epithelial cells, primarily by suppressing Fgfr1 expression [232].
Alcohol drinking has been reported in studies from China, the Democratic Republic of the Congo, and Mexico to be associated with an increased risk of OFC [233] [214]. Notably, a pooled study indicated an increased risk of cleft lip only (CLO) associated with binge drinking during pregnancy [234] [214]. The exact mechanisms by which alcohol increases the risk of OFCs are not fully understood; however, several teratological hypotheses have been proposed. These include antagonism of retinoic acid (RA) signaling, altered epigenetics, and oxidative stress, with ethanol (EtOH) oxidized to the toxic intermediate acetaldehyde (AcAL) [235]. These mechanisms involve the oxidation of EtOH into a toxic intermediate, acetaldehyde (AcAL), which is ultimately metabolized to acetyl-CoA by aldehyde dehydrogenase (ALDH) [235].
In the RA-EtOH competition model, AcAL competes with retinaldehyde for the enzyme synthesizing RA. Research using Xenopus has shown that EtOH exposure inhibits RA synthesis by preventing the activity of Aldh1a2, which converts retinaldehyde into RA [236]. Kinetic analysis of human ALDH1A2 revealed a preference for AcAL over retinaldehyde as a substrate, providing a biochemical basis for the RA-EtOH competition model. Another zebrafish study found that craniofacial malformations in pdgfra mutants were exacerbated by EtOH and that pdgfra protected against EtOH through the PI3K-mTOR pathway [237]. EtOH can alter the epigenetic state of neural stem cells, affecting the methylation of cell cycle genes and exacerbating malformations in genetic mutants [238]. Additionally, EtOH metabolism to AcAL by CYP2E1 can lead to oxidative stress, apoptosis, and activation of signaling pathways that could disrupt orofacial development [239]. In murine embryonic stem cells, EtOH and AcAL promote differentiation through transcriptional activities of the RA receptor, RAR-γ, which induces the expression of target genes such as Hoxa1 and Cyp26a1 [240]. Thus, EtOH exposure may alter cell stemness and/or fate during embryogenesis [241]. Although these effects are seemingly consistent with the disruption of orofacial development, there is little experimental evidence linking them. Much work remains to be done regarding EtOH exposure and the mechanisms of OFC, but there is sufficient knowledge to begin well-planned investigations [214].
Dioxins and related compounds are a group of chemically similar substances consisting of two coplanar benzene rings, known to induce various toxicity phenotypes with differing potencies [15]. Each compound was assigned a toxic equivalency factor (TEF) based on its toxicity relative to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), which is the most toxic compound in the group. TCDD serves as a prototypical compound in studies of toxicity mechanisms, with teratogenicity being a sensitive indicator of its effects in experimental animals. TCDD exposure disrupts gene expression in both epithelial and mesenchymal tissues, affecting multiple stages of palatogenesis, including growth and fusion [214]. Experimental studies have shown that TCDD administration to pregnant hamsters, mice, and rats induces cleft palate in their fetuses [214]. The aryl hydrocarbon receptor (AhR), also known as dioxin receptor, is a crucial ligand-activated transcription factor for the teratogenic effects of TCDD [242]. AhR is highly active in the epithelial cells of the palatal shelves and is found in the bone and muscle tissues of the palate. In TCDD-exposed fetuses, the medial edge epithelium (MEE) is covered by a thinner epithelial monolayer with fewer filopodia, impairing proper fusion of the palatal shelves, unlike control fetuses, which have thicker epithelial layers [243]. Decreases in filopodia and fusion failure have also been observed in ex vivo cultures of palatal shelves exposed to TCDD [244]. Notably, TGF-β3 supplementation rescued the fusion of these cultures, supporting the implication of its insufficiency in TCDD-induced cleft palate. However, the expression of TGF-β3 is increased in the palates of TCDD-exposed mice, highlighting the need for different experimental systems [245]. Additionally, TCDD exposure reduces the expression of several crucial factors involved in craniofacial development, including FGFR1, Runx2, osteopontin (OPN), MyoD, and desmin. It alters the expression of E-cadherin and Sox9, which may further contribute to cleft palate formation [15,214]. Moreover, TCDD disrupts growth factor signaling during palatogenesis via aryl hydrocarbon receptor (AhR) signaling, particularly by reducing the expression of transforming growth factor alpha (TGF-α) in human palatal epithelial cells [246]. This suggests that TCDD targets the epidermal growth factor (Egf) pathway, a conclusion supported by knockout studies, despite the seemingly counterintuitive decrease in ligand expression [214]. In human fetal palatal epithelial cells (hFPECs), TCDD exposure stimulates EGF receptor phosphorylation, leading to ERK/p38 phosphorylation and increased cell proliferation [247]. TCDD also promotes cell cycle progression to S and G2/M phases via the PI3K/AKT pathway [247]. Notably, exposure to TCDD disrupts the epithelial-to-mesenchymal transition by decreasing epithelial markers, such as E-cadherin and keratin-14, and increasing the expression of mesenchymal markers, such as vimentin and fibronectin. This is mediated by the induction of Slug, an inducer of EMT and a repressor of E-cadherin, which contains DREs in its promoter, modulated by AHR. This suggests that inappropriate EMT through Slug is a key mechanism in AHR-mediated cleft palate [247] [214]. This aligns with previous findings that TGF-β3 protects against cleft palate following TCDD exposure[248]. [249] revealed that TCDD function is mainly related to the metabolic processes of intracellular compounds, including the metabolic processes of cellular aromatic compounds and the metabolism of exogenous drugs by cytochrome P450. Furthermore, circRNA_1781/miR-30c-1-3p/PKIB and XR_380026.2/miR-1249-3p/DNAH10 ceRNA networks were hypothesized to be involved in palatal development, suggesting that circRNA_1781/miR-30c-1-3p/PKIB and XR_380026.2/miR-1249-3p/DNAH10 ceRNA networks may be critical for palatogenesis, providing a foundation for the investigation of cleft palate [249].
Dexamethasone (DEX) is a synthetic glucocorticoid used clinically in many applications owing to its anti-inflammatory and immunosuppressive properties. Such effects are mediated through interactions with various signaling pathways and molecules, including toll-like receptors and mitogen-activated protein kinases (Figure 6) [250]. The mechanism of action of GCs involves the diffusion of extracellular GCs into the cytoplasm, where they bind to the cytosolic GC receptor (GR). Without GCs, GR forms a complex with heat shock proteins (HSP70 and HSP90), FKBP52, and p23. The binding of GCs results in the dissociation of this complex, and the resultant GC-GR complex forms a dimer. The activated dimer then translocates to the nucleus and binds to glucocorticoid response elements (GREs) in the promoter region of target genes, thus activating transcription (transactivation) [250]. Alternatively, the active GC-GR complex can directly bind as a monomer to NF-κB (p50/p65), without dimerization. This monomeric complex, bound to NF-κB, subsequently binds to the NF-κB response elements to repress transcription. Despite their therapeutic benefits, GCs exhibit teratogenic and toxic effects. For example, the risk of CL/P, preterm birth, and low birth weight is increased two- to nine-fold after exposure to oral or systemic corticosteroids during pregnancy [183]. DEX can cross the blood-placental barrier and bind to cytoplasmic GR, inducing cleft palate in mice by inhibiting cell proliferation in the palatal mesenchyme [250]. Recently, Analysis of miRNA expression in developing mouse palatal shelves revealed distinct changes between embryonic days E13.5 and E14.5. The miR-449 family (including miR-449a-3p, miR-449a-5p, miR-449b, miR-449c-3p, and miR-449c-5p) showed increased expression on E4.5, whereas miR-19a-3p, miR-130a-3p, miR-301a-3p, and miR-486b-5p showed decreased expression. The functional role of these miRNAs in cell proliferation was further investigated, demonstrating that overexpression of the miR-449 family and miR-486b-5p represses cell proliferation in primary mouse embryonic palatal mesenchymal cells and the O9-1 cranial neural crest cell line. In contrast, when miR-130a-3p and miR-301a-3p were inhibited, the opposite effect was observed; these miRNAs' lower expression also reduced cell proliferation in the same cell types [250]. These findings suggest that miR-130a-3p plays an important role in dexamethasone-induced CP in mice [250] [183].
Folate metabolism is an important step during embryogenesis. Folate metabolism involves complex mechanisms of regulation and interaction with the methionine cycle. Food folic acid is subjected to metabolic processing in the small intestine and liver by first reducing it to dihydrofolate and then to tetrahydrofolate (THF) by dihydrofolate reductase (DHFR). Under the action of serine hydroxymethyltransferase (SHMT), vitamin B6 is required to produce 5,10-methylene-THF, followed by 5-methyl-THF by methylenetetrahydrofolate reductase (MTHFR). 5-Methyl-THF enters target cells via folate carriers and is converted to THF by methionine synthetase, using vitamin B12 as a cofactor. In this reaction, Hcy is converted into methionine. Methionine is then converted to S-adenosylmethionine, the major methyl donor in biomolecule methylation, which includes DNA and histone modifications. After donating a methyl group, SAM becomes SAH, which is recycled to homocysteine and completes the methionine cycle [214]. A systematic review showed an inverse association between folic acid supplementation and OFCs [214]. A cross-sectional study among the California population found a decreased prevalence of both CL/P (PR = 0.91, 95% CI: 0.82, 1.00) and CPO (PR = 0.81, 95% CI: 0.70, 0.93) following mandatory grain fortification with folic acid [251]. A recent meta-analysis found that folic acid supplementation was only protective when administered periconceptionally (OR = 0.64, 95% CI: 0.56, 0.74) versus during pregnancy (OR = 0.90, 95% CI: 0.71, 1.14) [252]. Recent studies have highlighted the gene-environment (G × E) interactions affecting the Chilean population's orofacial clefts (OFCs) and folate metabolism. A polymorphism in SHTM1, less frequent in cleft lip and/or palate (CL/P) cases, may protect against CL/P by reducing enzymatic activity and increasing cellular folate levels [253]. Folate is critical for DNA methylation because the folate cycle provides methyl groups to SAM, a molecule known to methylate DNA and histones via DNMTs and histone methyltransferases, respectively [121]. Similarly, three intronic MTR alleles involved in SAM metabolism might be protective [253]. The usual MTHFR c.677C>T polymorphism decreases the enzymatic activity of MTHFR and increases CL/P risk, particularly when combined with low maternal folic acid intake [254]. This result was corroborated by a meta-analysis that pooled 15 studies; maternal 677C>T mutations confer susceptibility to CL/P, confirming that maternal folate metabolism plays an important role in the development of OFCs [254]. Some theories have focused on folate facilitating correct DNA and histone methylation during development [255], whereas others believe it aids cell proliferation [256].
Polymorphisms in other folate-related genes have also been linked to OFCs. Folate deficiency impairs DNA repair owing to uracil misincorporation and contributes to genomic instability[214]. Folate also affects processes dependent on S-adenosylmethionine (SAM), including the synthesis of polyamines for cell proliferation, differentiation, apoptosis, and DNA methylation with respect to epigenetic regulation [214]. Folate deficiency can result in toxic homocysteine accumulation, with hyperhomocysteinemia associated with OFCs. Elevated homocysteine levels increase asymmetric dimethylarginine, reactive oxygen species (ROS), and oxidative stress [257]. Thus, folate exerts its protective effects by its involvement in cell growth and differentiation and in maintaining physiological balance.
Phenytoin is also known to be a teratogen. Between 1993 and 2007, approximately 1% of the population received antiepileptic drug prescriptions, and approximately 20% of these prescriptions were for phenytoin (brand name: Dilantin or Phenytek). This percentage increases to approximately 5% in women of childbearing age, emphasizing this well-recognized teratogen's importance. Women with epilepsy who take phenytoin either alone or in combination with other medications have a two- to threefold increased risk of having children with congenital malformations The fetal hydantoin syndrome, associated with phenytoin exposure, includes growth restriction, typical facial features that include midfacial hypoplasia, increased risk for cleft lip, limb abnormalities that most commonly consist of hypoplasia of the distal phalanges with small nails, and an increased incidence of heart defects. First-trimester exposure to phenytoin increases the risk of maxillary hypoplasia. In one study, the prevalence was 16.7%. The relationship between maxillary hypoplasia and cleft lip is considered an interrelated abnormality arising from the underdevelopment of the maxillary process. Experimental examination of fetal hydantoin syndrome in rats revealed that the litter contained not only fetuses with cleft lip but also those with maxillary hypoplasia. Severe underdevelopment of the maxillary process is expressed as cleft lip, while less severe expressions are present as maxillary hypoplasia. Growth retardation of the maxillary process is common in all cleft lip cases. Maxillary hypoplasia and cleft lip are manifestations of impaired development of the maxillary process. Investigations in rat models of fetal hydantoin syndrome have consistently observed both cleft lip and maxillary hypoplasia within the same litter, suggesting a continuum in severity [258]. In a mouse study, increased expression of miR-196a-5p subsequently inhibited cell proliferation in cultured mouse embryonic lip mesenchymal (MELM) cells by suppressing Pbx1, Pbx3, and Rpgrip1l expression (Figure 6) [183]. Notably, a substantial decrease in miR-196a-5p expression occurs in the maxillary and nasal processes (MxPs and NPs) between embryonic days E10.5 and E12.5 [183]. These collective findings suggest a pleiotropic role for miR-196a-5p in diverse developmental processes, including those critical for palate formation.
Maternal smoking was recognized as a risk factor in a meta-analysis of 24 case-control and cohort studies published in the World Health Organization Bulletin in 2004, reporting associations between maternal smoking and either CL/P [RR = 1.34; 95% CI: 1.25, 1.44] or CPO (RR = 1.22; 95% CI: 1.10, 1.35) [259]. Ten years later, the 50th Anniversary United States Surgeon General's report declared sufficient evidence for a causal link between maternal active smoking and OFC [260]. Humans possess two NAT genes, NAT1 and NAT2, each with over two dozen known polymorphisms that may affect arylamine detoxification and OFC risk. A study on a California population linked two fetal polymorphisms of NAT1 that increased the risk of CL/P fourfold when mothers smoked during early pregnancy (compared to reference genotype infants with nonsmoking mothers [261]. Smoke is known to contain endogenous tobacco plant compounds such as nicotine, pyrolysis products, and added chemicals. Correspondingly, there are several hypothesized mechanisms by which tobacco smoke increases the risk of OFCs. Genetic susceptibility significantly influences the risk of tobacco smoke-induced orofacial clefts (OFCs). Polymorphisms in genes encoding developmental signaling ligands, such as transforming growth factor alpha (TGFA), transforming growth factor beta 3 (TGFB3), and bone morphogenetic protein 4 (BMP4), have been linked to OFCs in relation to maternal smoke exposure [214]. Variants affecting the interaction between TGFA and smoking (gene-environment interactions, GxE) have been observed in several populations [262] [214]. TGF-B3 encodes a ligand crucial for TGF-β signaling, essential for lip and palate development. Polymorphisms in TGFB3 associated with smoking have been linked to cleft lip and/or palate (CL/P), cleft palate only (CPO), and submucosal cleft palate (SMCP) [263]. BMP4 is a ligand for BMP signaling, has polymorphisms associated with CL/P, and plays a role in the fusion of medial and lateral nasal processes. Variants of BMP4 in conjunction with smoking have also been associated with CL/P [264]. The gene expression in cell cycle regulation, DNA repair, and oxidative stress response is affected by tobacco smoke in fetal mouse tissues [265]. Additionally, tobacco smoke can induce proteasome-mediated degradation of proteins that mediate DNA methylation in 1st branchial arch (BA1) cells [266]. Nicotine is a vasoconstrictor that can impair uterine vascular function and affect the blood flow and oxygen delivery to the fetus [267]. Several teratogens, such as polycyclic aromatic hydrocarbons (PAHs), dioxins, carbon monoxide, pesticides, and heavy metals such as cadmium, may be present in tobacco smoke [268]. Exposure to heavy metals can cause OFCs in rodent models and is believed to act teratogenically through the induction of oxidative stress and perturbation of redox-sensitive signaling pathways [269].
4. Conclusions
This comprehensive review illustrates the complex interaction of genetic, epigenetic, and environmental risk factors in regulating palatogenesis and etiology of cleft lip and/or palate (CL/P). These studies have also elucidated the complex molecular networks associated with critical signaling pathways, including TGF-β, BMP, FGF, Wnt, and SHH, as well as epigenetic mechanisms (e.g., non-coding RNAs, microRNAs, and long non-coding RNAs) that contribute to craniofacial development. Recent evidence has elucidated the necessity for the precise regulation of various developmental stages, including proliferation, migration, differentiation, and apoptosis.
An additional layer of complexity arises from environmental factors, including maternal tobacco use, alcohol exposure, folate deficiency, and teratogens, such as retinoic acid and TCDD. This disruption leads to an imbalance between genetic and environmental factors that initiate normal development, resulting in various congenital malformations, including CL/P. Recent advances in gene-environment interactions and epigenetic regulation will illuminate future prevention and treatment methodologies. The remaining gaps need to be resolved in future studies. In particular, the role of non-coding RNAs and chromatin remodeling in craniofacial development awaits further investigation. Additionally, it is important to identify precise molecular targets to translate these findings into clinical applications. This review integrates the genetic, epigenetic, and environmental perspectives to establish a framework for future multidisciplinary strategies to reduce the burden of orofacial clefts.
Author Contributions
H. I., Y. S., Y. -H. J: Investigation, Writing-original draft. J.-K. K., D.-K. P., D.-S. K.: Writing review and editing. H. K.: Conceptualization, Project administration, Writing-original draft, Writing-review, and editing. J.-O. S.: Conceptualization, Funding acquisition, Project administration, Writing-original draft, writing review, and editing.
Funding
This study was supported by Soonchunhyang University Fund (No. 20220446). This work was supported by the National Research Foundation of Korea(NRF) grant funded by the Korean government(MSIT) (RS-2024-00457696).
Declaration of Competing Interest
The authors declare that they have no conflicts of interest.
References
- Won, H.-J.; Kim, J.-W.; Won, H.-S.; Shin, J.-O., Gene regulatory networks and signaling pathways in palatogenesis and cleft palate: a comprehensive review. Cells 2023, 12, (15), 1954. [CrossRef]
- Bush, J. O.; Jiang, R., Palatogenesis: morphogenetic and molecular mechanisms of secondary palate development. Development 2012, 139, (2), 231-243. [CrossRef]
- Jiang, R.; Bush, J. O.; Lidral, A. C., Development of the upper lip: morphogenetic and molecular mechanisms. Developmental dynamics: an official publication of the American Association of Anatomists 2006, 235, (5), 1152-1166.
- Yuzuriha, S.; Oh, A. K.; Mulliken, J. B., Asymmetrical bilateral cleft lip: Complete or incomplete and contralateral lesser defect (minor-form, microform, or mini-microform). Plastic and reconstructive surgery 2008, 122, (5), 1494-1504. [CrossRef] [PubMed]
- Nasreddine, G.; El Hajj, J.; Ghassibe-Sabbagh, M., Orofacial clefts embryology, classification, epidemiology, and genetics. Mutation Research/Reviews in Mutation Research 2021, 787, 108373.
- Rahimov, F.; Jugessur, A.; Murray, J. C., Genetics of nonsyndromic orofacial clefts. The Cleft palate-craniofacial journal 2012, 49, (1), 73-91.
- Yılmaz, H. N.; Özbilen, E. Ö.; Üstün, T., The prevalence of cleft lip and palate patients: a single-center experience for 17 years. Turkish journal of orthodontics 2019, 32, (3), 139. [CrossRef]
- Garland, M. A.; Sun, B.; Zhang, S.; Reynolds, K.; Ji, Y.; Zhou, C. J., Role of epigenetics and miRNAs in orofacial clefts. Birth Defects Res 2020, 112, (19), 1635-1659. [CrossRef]
- Alade, A.; Awotoye, W.; Butali, A., Genetic and epigenetic studies in non-syndromic oral clefts. Oral Dis 2022, 28, (5), 1339-1350. [CrossRef] [PubMed]
- Mani, P.; Jarrell, A.; Myers, J.; Atit, R., Visualizing canonical Wnt signaling during mouse craniofacial development. Developmental dynamics: an official publication of the American Association of Anatomists 2010, 239, (1), 354-363.
- Sharp, G.; Ho, K.; Davies, A.; Stergiakouli, E.; Humphries, K.; McArdle, W.; Relton, C., Distinct DNA methylation profiles in subtypes of orofacial cleft. Clinical Epigenetics, 9, 63. In 2017.
- Alade, A.; Mossey, P.; Awotoye, W.; Busch, T.; Oladayo, A. M.; Aladenika, E.; Olujitan, M.; Wentworth, E.; Anand, D.; Naicker, T.; Gowans, L. J. J.; Eshete, M. A.; Adeyemo, W. L.; Zeng, E.; Van Otterloo, E.; O’Rorke, M.; Adeyemo, A.; Murray, J. C.; Cotney, J.; Lachke, S. A.; Romitti, P.; Butali, A., Rare variants analyses suggest novel cleft genes in the African population. Scientific Reports 2024, 14, (1), 14279. [CrossRef]
- Reynolds, K.; Zhang, S.; Sun, B.; Garland, M. A.; Ji, Y.; Zhou, C. J., Genetics and signaling mechanisms of orofacial clefts. Birth Defects Research 2020, 112, (19), 1588-1634. [CrossRef]
- McDonald-McGinn, D. M.; Sullivan, K. E.; Marino, B.; Philip, N.; Swillen, A.; Vorstman, J. A.; Zackai, E. H.; Emanuel, B. S.; Vermeesch, J. R.; Morrow, B. E.; Scambler, P. J.; Bassett, A. S., 22q11.2 deletion syndrome. Nat Rev Dis Primers 2015, 1, 15071. [CrossRef]
- Seelan, R. S.; Pisano, M. M.; Greene, R. M., MicroRNAs as epigenetic regulators of orofacial development. Differentiation 2022, 124, 1-16. [CrossRef] [PubMed]
- Gao, S.; Moreno, M.; Eliason, S.; Cao, H.; Li, X.; Yu, W.; Bidlack, F. B.; Margolis, H. C.; Baldini, A.; Amendt, B. A., TBX1 protein interactions and microRNA-96-5p regulation controls cell proliferation during craniofacial and dental development: implications for 22q11. 2 deletion syndrome. Human molecular genetics 2015, 24, (8), 2330-2348. [CrossRef] [PubMed]
- Kumari, P.; Singh, S. K.; Raman, R., A novel non-coding RNA within an intron of CDH2 and association of its SNP with non-syndromic cleft lip and palate. Gene 2018, 658, 123-128. [CrossRef] [PubMed]
- Snead, M.; McNinch, A.; Poulson, A.; Bearcroft, P.; Silverman, B.; Gomersall, P.; Parfect, V.; Richards, A., Stickler syndrome, ocular-only variants and a key diagnostic role for the ophthalmologist. Eye 2011, 25, (11), 1389-1400. [CrossRef] [PubMed]
- Alzahrani, F.; Al Hazzaa, S. A.; Tayeb, H.; Alkuraya, F. S., LOXL3, encoding lysyl oxidase-like 3, is mutated in a family with autosomal recessive Stickler syndrome. Human genetics 2015, 134, 451-453. [CrossRef]
- Schrauwen, I.; Sommen, M.; Claes, C.; Pinner, J.; Flaherty, M.; Collins, F.; Van Camp, G., Broadening the phenotype of LRP2 mutations: a new mutation in LRP2 causes a predominantly ocular phenotype suggestive of Stickler syndrome. Clinical Genetics 2014, 86, (3), 282-286. [CrossRef]
- Giudice, A.; Barone, S.; Belhous, K.; Morice, A.; Soupre, V.; Bennardo, F.; Boddaert, N.; Vazquez, M.-P.; Abadie, V.; Picard, A., Pierre Robin sequence: A comprehensive narrative review of the literature over time. Journal of stomatology, oral and maxillofacial surgery 2018, 119, (5), 419-428.
- Gordon, C. T.; Attanasio, C.; Bhatia, S.; Benko, S.; Ansari, M.; Tan, T. Y.; Munnich, A.; Pennacchio, L. A.; Abadie, V.; Temple, I. K., Identification of novel craniofacial regulatory domains located far upstream of sox 9 and disrupted in pierre robin sequence. Human mutation 2014, 35, (8), 1011-1020. [CrossRef]
- Yang, Y.; Yuan, J.; Yao, X.; Zhang, R.; Yang, H.; Zhao, R.; Guo, J.; Jin, K.; Mei, H.; Luo, Y., BMPR1B mutation causes Pierre Robin sequence. Oncotarget 2017, 8, (16), 25864. [CrossRef]
- Murakami, H.; Tsurusaki, Y.; Enomoto, K.; Kuroda, Y.; Yokoi, T.; Furuya, N.; Yoshihashi, H.; Minatogawa, M.; Abe-Hatano, C.; Ohashi, I., Update of the genotype and phenotype of KMT2D and KDM6A by genetic screening of 100 patients with clinically suspected Kabuki syndrome. American journal of medical genetics Part A 2020, 182, (10), 2333-2344. [CrossRef] [PubMed]
- Barry, K. K.; Tsaparlis, M.; Hoffman, D.; Hartman, D.; Adam, M. P.; Hung, C.; Bodamer, O. A., From Genotype to Phenotype-A Review of Kabuki Syndrome. Genes (Basel) 2022, 13, (10).
- Lederer, D.; Grisart, B.; Digilio, M. C.; Benoit, V.; Crespin, M.; Ghariani, S. C.; Maystadt, I.; Dallapiccola, B.; Verellen-Dumoulin, C., Deletion of KDM6A, a histone demethylase interacting with MLL2, in three patients with Kabuki syndrome. The American Journal of Human Genetics 2012, 90, (1), 119-124. [CrossRef] [PubMed]
- Gavril, E. C.; Luca, A. C.; Curpan, A. S.; Popescu, R.; Resmerita, I.; Panzaru, M. C.; Butnariu, L. I.; Gorduza, E. V.; Gramescu, M.; Rusu, C., Wolf-Hirschhorn Syndrome: Clinical and Genetic Study of 7 New Cases, and Mini Review. Children (Basel) 2021, 8, (9).
- Nevado, J.; Ho, K. S.; Zollino, M.; Blanco, R.; Cobaleda, C.; Golzio, C.; Beaudry-Bellefeuille, I.; Berrocoso, S.; Limeres, J.; Barrúz, P., International meeting on Wolf-Hirschhorn syndrome: Update on the nosology and new insights on the pathogenic mechanisms for seizures and growth delay. American Journal of Medical Genetics Part A 2020, 182, (1), 257-267. [CrossRef] [PubMed]
- Sperry, E. D.; Hurd, E. A.; Durham, M. A.; Reamer, E. N.; Stein, A. B.; Martin, D. M., The chromatin remodeling protein CHD7, mutated in CHARGE syndrome, is necessary for proper craniofacial and tracheal development. Developmental Dynamics 2014, 243, (9), 1055-1066. [CrossRef] [PubMed]
- Giancotti, A.; D’Ambrosio, V.; De Filippis, A.; Aliberti, C.; Pasquali, G.; Bernardo, S.; Manganaro, L.; Group, P. S., Comparison of ultrasound and magnetic resonance imaging in the prenatal diagnosis of Apert syndrome: report of a case. Child's Nervous System 2014, 30, 1445-1448.
- Kakutani, H.; Sato, Y.; Tsukamoto-Takakusagi, Y.; Saito, F.; Oyama, A.; Iida, J., Evaluation of the maxillofacial morphological characteristics of Apert syndrome infants. Congenital Anomalies 2017, 57, (1), 15-23. [CrossRef] [PubMed]
- Letra, A.; de Almeida, A. L.; Kaizer, R.; Esper, L. A.; Sgarbosa, S.; Granjeiro, J. M., Intraoral features of Apert's syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2007, 103, (5), e38-41. [CrossRef] [PubMed]
- Liu, C.; Cui, Y.; Luan, J.; Zhou, X.; Han, J., The molecular and cellular basis of Apert syndrome. Intractable & Rare Diseases Research 2013, 2, (4), 115-122.
- Yeh, E.; Atique, R.; Fanganiello, R. D.; Sunaga, D. Y.; Ishiy, F. A. A.; Passos-Bueno, M. R., Cell type-dependent nonspecific fibroblast growth factor signaling in Apert syndrome. Stem Cells and Development 2016, 25, (16), 1249-1260. [CrossRef] [PubMed]
- Li, Y.; Ma, D.; Sun, Y.; Meng, L.; Wang, Y.; Jiang, T., Apert Syndrome With FGFR2 758 C > G Mutation: A Chinese Case Report. Front Genet 2018, 9, 181. [CrossRef] [PubMed]
- Yokoi, T.; Enomoto, Y.; Naruto, T.; Kurosawa, K.; Higurashi, N., Tatton-Brown-Rahman syndrome with a novel DNMT3A mutation presented severe intellectual disability and autism spectrum disorder. Human genome variation 2020, 7, (1), 15. [CrossRef]
- Smith, A. M.; LaValle, T. A.; Shinawi, M.; Ramakrishnan, S. M.; Abel, H. J.; Hill, C. A.; Kirkland, N. M.; Rettig, M. P.; Helton, N. M.; Heath, S. E., Functional and epigenetic phenotypes of humans and mice with DNMT3A Overgrowth Syndrome. Nature communications 2021, 12, (1), 4549. [CrossRef] [PubMed]
- Nimura, K.; Ura, K.; Shiratori, H.; Ikawa, M.; Okabe, M.; Schwartz, R. J.; Kaneda, Y., A histone H3 lysine 36 trimethyltransferase links Nkx2-5 to Wolf–Hirschhorn syndrome. Nature 2009, 460, (7252), 287-291. [CrossRef] [PubMed]
- Mariotti, M.; Manganini, M.; Maier, J., Modulation of WHSC2 expression in human endothelial cells. FEBS letters 2000, 487, (2), 166-170. [CrossRef]
- Schneider, C. A.; Rasband, W. S.; Eliceiri, K. W., NIH Image to ImageJ: 25 years of image analysis. Nature methods 2012, 9, (7), 671-675. [CrossRef] [PubMed]
- Gergely, F.; Karlsson, C.; Still, I.; Cowell, J.; Kilmartin, J.; Raff, J. W., The TACC domain identifies a family of centrosomal proteins that can interact with microtubules. Proceedings of the National Academy of Sciences 2000, 97, (26), 14352-14357. [CrossRef] [PubMed]
- Kennedy, J.; Goudie, D.; Blair, E.; Chandler, K.; Joss, S.; McKay, V.; Green, A.; Armstrong, R.; Lees, M.; Kamien, B.; Hopper, B.; Tan, T. Y.; Yap, P.; Stark, Z.; Okamoto, N.; Miyake, N.; Matsumoto, N.; Macnamara, E.; Murphy, J. L.; McCormick, E.; Hakonarson, H.; Falk, M. J.; Li, D.; Blackburn, P.; Klee, E.; Babovic-Vuksanovic, D.; Schelley, S.; Hudgins, L.; Kant, S.; Isidor, B.; Cogne, B.; Bradbury, K.; Williams, M.; Patel, C.; Heussler, H.; Duff-Farrier, C.; Lakeman, P.; Scurr, I.; Kini, U.; Elting, M.; Reijnders, M.; Schuurs-Hoeijmakers, J.; Wafik, M.; Blomhoff, A.; Ruivenkamp, C. A. L.; Nibbeling, E.; Dingemans, A. J. M.; Douine, E. D.; Nelson, S. F.; Hempel, M.; Bierhals, T.; Lessel, D.; Johannsen, J.; Arboleda, V. A.; Newbury-Ecob, R., KAT6A Syndrome: genotype–phenotype correlation in 76 patients with pathogenic KAT6A variants. Genetics in Medicine 2019, 21, (4), 850-860. [CrossRef]
- Singh, M.; Spendlove, S. J.; Wei, A.; Bondhus, L. M.; Nava, A. A.; de, L. V. F. N.; Amano, S.; Lee, J.; Echeverria, G.; Gomez, D.; Garcia, B. A.; Arboleda, V. A., KAT6A mutations in Arboleda-Tham syndrome drive epigenetic regulation of posterior HOXC cluster. Hum Genet 2023, 142, (12), 1705-1720. [CrossRef] [PubMed]
- Murray, S. A.; Oram, K. F.; Gridley, T., Multiple functions of Snail family genes during palate development in mice. 2007.
- Thomas, T.; Voss, A. K., The diverse biological roles of MYST histone acetyltransferase family proteins. Cell Cycle 2007, 6, (6), 696-704. [CrossRef] [PubMed]
- Yan, K.; Rousseau, J.; Machol, K.; Cross, L. A.; Agre, K. E.; Gibson, C. F.; Goverde, A.; Engleman, K. L.; Verdin, H.; De Baere, E.; Potocki, L.; Zhou, D.; Cadieux-Dion, M.; Bellus, G. A.; Wagner, M. D.; Hale, R. J.; Esber, N.; Riley, A. F.; Solomon, B. D.; Cho, M. T.; McWalter, K.; Eyal, R.; Hainlen, M. K.; Mendelsohn, B. A.; Porter, H. M.; Lanpher, B. C.; Lewis, A. M.; Savatt, J.; Thiffault, I.; Callewaert, B.; Campeau, P. M.; Yang, X. J., Deficient histone H3 propionylation by BRPF1-KAT6 complexes in neurodevelopmental disorders and cancer. Sci Adv 2020, 6, (4), eaax0021. [CrossRef] [PubMed]
- Casey, L. M.; Lan, Y.; Cho, E. S.; Maltby, K. M.; Gridley, T.; Jiang, R., Jag2-Notch1 signaling regulates oral epithelial differentiation and palate development. Developmental dynamics: an official publication of the American Association of Anatomists 2006, 235, (7), 1830-1844.
- Ito, Y.; Yeo, J. Y.; Chytil, A.; Han, J.; Bringas Jr, P.; Nakajima, A.; Shuler, C. F.; Moses, H. L.; Chai, Y., Conditional inactivation of Tgfbr2 in cranial neural crest causes cleft palate and calvaria defects. 2003.
- Lan, Y.; Jiang, R., Mouse models in palate development and orofacial cleft research: Understanding the crucial role and regulation of epithelial integrity in facial and palate morphogenesis. In Current topics in developmental biology, Elsevier: 2022; Vol. 148, pp 13-50.
- Hammond, N. L.; Dixon, M. J., Revisiting the embryogenesis of lip and palate development. Oral Diseases 2022, 28, (5), 1306-1326. [CrossRef] [PubMed]
- Xu, J.; Liu, H.; Lan, Y.; Aronow, B. J.; Kalinichenko, V. V.; Jiang, R., A Shh-Foxf-Fgf18-Shh molecular circuit regulating palate development. PLoS genetics 2016, 12, (1), e1005769. [CrossRef]
- Charoenchaikorn, K.; Yokomizo, T.; Rice, D. P.; Honjo, T.; Matsuzaki, K.; Shintaku, Y.; Imai, Y.; Wakamatsu, A.; Takahashi, S.; Ito, Y., Runx1 is involved in the fusion of the primary and the secondary palatal shelves. Developmental biology 2009, 326, (2), 392-402. [CrossRef] [PubMed]
- Kurosaka, H.; Iulianella, A.; Williams, T.; Trainor, P. A., Disrupting hedgehog and WNT signaling interactions promotes cleft lip pathogenesis. The Journal of clinical investigation 2014, 124, (4), 1660-1671. [CrossRef]
- Goetz, S. C.; Anderson, K. V., The primary cilium: a signalling centre during vertebrate development. Nature Reviews Genetics 2010, 11, (5), 331-344. [CrossRef]
- Shin, J.-O.; Song, J.; Choi, H. S.; Lee, J.; Lee, K.; Ko, H. W.; Bok, J., Activation of sonic hedgehog signaling by a Smoothened agonist restores congenital defects in mouse models of endocrine-cerebro-osteodysplasia syndrome. EBioMedicine 2019, 49, 305-317. [CrossRef] [PubMed]
- Rice, R.; Spencer-Dene, B.; Connor, E. C.; Gritli-Linde, A.; McMahon, A. P.; Dickson, C.; Thesleff, I.; Rice, D. P., Disruption of Fgf10/Fgfr2b-coordinated epithelial-mesenchymal interactions causes cleft palate. The Journal of clinical investigation 2004, 113, (12), 1692-1700. [CrossRef] [PubMed]
- Hosokawa, R.; Deng, X.; Takamori, K.; Xu, X.; Urata, M.; Bringas Jr, P.; Chai, Y., Epithelial-specific requirement of FGFR2 signaling during tooth and palate development. Journal of Experimental Zoology Part B: Molecular and Developmental Evolution 2009, 312, (4), 343-350.
- Lan, Y.; Jiang, R., Sonic hedgehog signaling regulates reciprocal epithelial-mesenchymal interactions controlling palatal outgrowth. 2009.
- Han, J.; Mayo, J.; Xu, X.; Li, J.; Bringas Jr, P.; Maas, R. L.; Rubenstein, J. L.; Chai, Y., Indirect modulation of Shh signaling by Dlx5 affects the oral-nasal patterning of palate and rescues cleft palate in Msx1-null mice. Development 2009, 136, (24), 4225-4233. [CrossRef] [PubMed]
- Iwata, J.-i.; Tung, L.; Urata, M.; Hacia, J. G.; Pelikan, R.; Suzuki, A.; Ramenzoni, L.; Chaudhry, O.; Parada, C.; Sanchez-Lara, P. A., Fibroblast growth factor 9 (FGF9)-pituitary homeobox 2 (PITX2) pathway mediates transforming growth factor β (TGFβ) signaling to regulate cell proliferation in palatal mesenchyme during mouse palatogenesis. Journal of Biological Chemistry 2012, 287, (4), 2353-2363. [CrossRef] [PubMed]
- Li, R.; Sun, Y.; Chen, Z.; Zheng, M.; Shan, Y.; Ying, X.; Weng, M.; Chen, Z., The fibroblast growth factor 9 (Fgf9) participates in palatogenesis by promoting palatal growth and elevation. Frontiers in Physiology 2021, 12, 653040. [CrossRef] [PubMed]
- Lin, C.; Liu, S.; Ruan, N.; Chen, J.; Chen, Y.; Zhang, Y.; Zhang, J., Cleft Palate Induced by Augmented Fibroblast Growth Factor-9 Signaling in Cranial Neural Crest Cells in Mice. Stem Cells and Development 2024, 33, (19-20), 562-573.
- Cesario, J. M.; Landin Malt, A.; Deacon, L. J.; Sandberg, M.; Vogt, D.; Tang, Z.; Zhao, Y.; Brown, S.; Rubenstein, J. L.; Jeong, J., Lhx6 and Lhx8 promote palate development through negative regulation of a cell cycle inhibitor gene, p57Kip2. Human molecular genetics 2015, 24, (17), 5024-5039. [CrossRef]
- Luo, H.; Ieong, H. C.; Li, R.; Huang, D.; Chen, D.; Chen, X.; Guo, Y.; Qing, Y.; Guo, B.; Li, R.; Teng, Y.; Li, W.; Cao, Y.; Zhou, C.; Wang, W., Lhx6 deficiency causes human embryonic palatal mesenchymal cell mitophagy dysfunction in cleft palate. Mol Med 2024, 30, (1), 183. [CrossRef]
- Iwata, J.-i.; Suzuki, A.; Yokota, T.; Ho, T.-V.; Pelikan, R.; Urata, M.; Sanchez-Lara, P. A.; Chai, Y., TGFβ regulates epithelial-mesenchymal interactions through WNT signaling activity to control muscle development in the soft palate. Development 2014, 141, (4), 909-917. [CrossRef] [PubMed]
- Liu, W.; Sun, X.; Braut, A.; Mishina, Y.; Behringer, R. R.; Mina, M.; Martin, J. F., Distinct functions for Bmp signaling in lip and palate fusion in mice. 2005.
- Ueharu, H.; Mishina, Y., BMP signaling during craniofacial development: new insights into pathological mechanisms leading to craniofacial anomalies. Frontiers in Physiology 2023, 14, 1170511. [CrossRef] [PubMed]
- Hammond, N. L.; Brookes, K. J.; Dixon, M. J., Ectopic hedgehog signaling causes cleft palate and defective osteogenesis. Journal of dental research 2018, 97, (13), 1485-1493. [CrossRef] [PubMed]
- Zhang, Z.; Song, Y.; Zhao, X.; Zhang, X.; Fermin, C.; Chen, Y., Rescue of cleft palate in Msx1-deficient mice by transgenic Bmp4 reveals a network of BMP and Shh signaling in the regulation of mammalian palatogenesis. 2002.
- Saket, M.; Saliminejad, K.; Kamali, K.; Moghadam, F. A.; Anvar, N. E.; Khorshid, H. R. K., BMP2 and BMP4 variations and risk of non-syndromic cleft lip and palate. Archives of Oral Biology 2016, 72, 134-137. [CrossRef]
- Xiong, W.; He, F.; Morikawa, Y.; Yu, X.; Zhang, Z.; Lan, Y.; Jiang, R.; Cserjesi, P.; Chen, Y., Hand2 is required in the epithelium for palatogenesis in mice. Developmental biology 2009, 330, (1), 131-141. [CrossRef]
- Parada, C.; Chai, Y., Roles of BMP signaling pathway in lip and palate development. Cleft Lip and Palate 2012, 16, 60-70.
- Andl, T.; Ahn, K.; Kairo, A.; Chu, E. Y.; Wine-Lee, L.; Reddy, S. T.; Croft, N. J.; Cebra-Thomas, J. A.; Metzger, D.; Chambon, P., Epithelial Bmpr1a regulates differentiation and proliferation in postnatal hair follicles and is essential for tooth development. 2004.
- Li, L.; Lin, M.; Wang, Y.; Cserjesi, P.; Chen, Z.; Chen, Y., BmprIa is required in mesenchymal tissue and has limited redundant function with BmprIb in tooth and palate development. Developmental biology 2011, 349, (2), 451-461. [CrossRef]
- Baek, J.-A.; Lan, Y.; Liu, H.; Maltby, K. M.; Mishina, Y.; Jiang, R., Bmpr1a signaling plays critical roles in palatal shelf growth and palatal bone formation. Developmental biology 2011, 350, (2), 520-531. [CrossRef] [PubMed]
- He, F.; Xiong, W.; Wang, Y.; Matsui, M.; Yu, X.; Chai, Y.; Klingensmith, J.; Chen, Y., Modulation of BMP signaling by Noggin is required for the maintenance of palatal epithelial integrity during palatogenesis. Developmental biology 2010, 347, (1), 109-121. [CrossRef] [PubMed]
- Li, C.; Lan, Y.; Krumlauf, R.; Jiang, R., Modulating Wnt signaling rescues palate morphogenesis in Pax9 mutant mice. Journal of dental research 2017, 96, (11), 1273-1281. [CrossRef] [PubMed]
- Jia, S.; Zhou, J.; Fanelli, C.; Wee, Y.; Bonds, J.; Schneider, P.; Mues, G.; D'Souza, R. N., Small-molecule Wnt agonists correct cleft palates in Pax9 mutant mice in utero. Development 2017, 144, (20), 3819-3828. [CrossRef]
- Jia, S.; Zhou, J.; Wee, Y.; Mikkola, M.; Schneider, P.; D’souza, R., Anti-EDAR agonist antibody therapy resolves palate defects in Pax9-/-mice. Journal of dental research 2017, 96, (11), 1282-1289. [CrossRef]
- Reynolds, K.; Kumari, P.; Sepulveda Rincon, L.; Gu, R.; Ji, Y.; Kumar, S.; Zhou, C. J., Wnt signaling in orofacial clefts: crosstalk, pathogenesis and models. Disease models & mechanisms 2019, 12, (2), dmm037051.
- Yao, T.; Yang, L.; Li, P.-q.; Wu, H.; Xie, H.-b.; Shen, X.; Xie, X.-d., Association of Wnt3A gene variants with non-syndromic cleft lip with or without cleft palate in Chinese population. Archives of Oral Biology 2011, 56, (1), 73-78. [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M., Wnt signaling in cancer. Oncogene 2017, 36, (11), 1461-1473. [CrossRef] [PubMed]
- Huybrechts, Y.; Mortier, G.; Boudin, E.; Van Hul, W., WNT signaling and bone: lessons from skeletal dysplasias and disorders. Frontiers in Endocrinology 2020, 11, 165. [CrossRef] [PubMed]
- Hilliard, S. A.; Yu, L.; Gu, S.; Zhang, Z.; Chen, Y. P., Regional regulation of palatal growth and patterning along the anterior–posterior axis in mice. Journal of anatomy 2005, 207, (5), 655-667. [CrossRef] [PubMed]
- Liu, W.; Lan, Y.; Pauws, E.; Meester-Smoor, M. A.; Stanier, P.; Zwarthoff, E. C.; Jiang, R., The Mn1 transcription factor acts upstream of Tbx22 and preferentially regulates posterior palate growth in mice. 2008.
- Yu, L.; Gu, S.; Alappat, S.; Song, Y.; Yan, M.; Zhang, X.; Zhang, G.; Jiang, Y.; Zhang, Z.; Zhang, Y., Shox2-deficient mice exhibit a rare type of incomplete clefting of the secondary palate. 2005.
- Nishihara, H.; Kobayashi, N.; Kimura-Yoshida, C.; Yan, K.; Bormuth, O.; Ding, Q.; Nakanishi, A.; Sasaki, T.; Hirakawa, M.; Sumiyama, K., Coordinately co-opted multiple transposable elements constitute an enhancer for wnt5a expression in the mammalian secondary palate. PLoS genetics 2016, 12, (10), e1006380. [CrossRef] [PubMed]
- Almaidhan, A.; Cesario, J.; Landin Malt, A.; Zhao, Y.; Sharma, N.; Choi, V.; Jeong, J., Neural crest-specific deletion of Ldb1 leads to cleft secondary palate with impaired palatal shelf elevation. BMC Developmental Biology 2014, 14, 1-10. [CrossRef] [PubMed]
- Zhou, J.; Gao, Y.; Lan, Y.; Jia, S.; Jiang, R., Pax9 regulates a molecular network involving Bmp4, Fgf10, Shh signaling and the Osr2 transcription factor to control palate morphogenesis. Development 2013, 140, (23), 4709-4718. [CrossRef] [PubMed]
- Gao, Y.; Lan, Y.; Ovitt, C. E.; Jiang, R., Functional equivalence of the zinc finger transcription factors Osr1 and Osr2 in mouse development. Developmental biology 2009, 328, (2), 200-209. [CrossRef]
- Fu, X.; Xu, J.; Chaturvedi, P.; Liu, H.; Jiang, R.; Lan, Y., Identification of Osr2 transcriptional target genes in palate development. Journal of dental research 2017, 96, (12), 1451-1458. [CrossRef] [PubMed]
- Richardson, R. J.; Dixon, J.; Malhotra, S.; Hardman, M. J.; Knowles, L.; Boot-Handford, R. P.; Shore, P.; Whitmarsh, A.; Dixon, M. J., Irf6 is a key determinant of the keratinocyte proliferation-differentiation switch. Nature genetics 2006, 38, (11), 1329-1334. [CrossRef]
- Richardson, R. J.; Dixon, J.; Jiang, R.; Dixon, M. J., Integration of IRF6 and Jagged2 signalling is essential for controlling palatal adhesion and fusion competence. Human molecular genetics 2009, 18, (14), 2632-2642. [CrossRef] [PubMed]
- Thomason, H. A.; Zhou, H.; Kouwenhoven, E. N.; Dotto, G.-P.; Restivo, G.; Nguyen, B.-C.; Little, H.; Dixon, M. J.; Van Bokhoven, H.; Dixon, J., Cooperation between the transcription factors p63 and IRF6 is essential to prevent cleft palate in mice. The Journal of clinical investigation 2010, 120, (5), 1561-1569. [CrossRef] [PubMed]
- Candi, E.; Rufini, A.; Terrinoni, A.; Giamboi-Miraglia, A.; Lena, A. M.; Mantovani, R.; Knight, R.; Melino, G., ΔNp63 regulates thymic development through enhanced expression of FgfR2 and Jag2. Proceedings of the National Academy of Sciences 2007, 104, (29), 11999-12004. [CrossRef] [PubMed]
- Richardson, R. J.; Hammond, N. L.; Coulombe, P. A.; Saloranta, C.; Nousiainen, H. O.; Salonen, R.; Berry, A.; Hanley, N.; Headon, D.; Karikoski, R., Periderm prevents pathological epithelial adhesions during embryogenesis. The Journal of clinical investigation 2014, 124, (9), 3891-3900. [CrossRef] [PubMed]
- Sani, F. V.; Hallberg, K.; Harfe, B. D.; McMahon, A. P.; Linde, A.; Gritli-Linde, A., Fate-mapping of the epithelial seam during palatal fusion rules out epithelial–mesenchymal transformation. Developmental biology 2005, 285, (2), 490-495. [CrossRef] [PubMed]
- Xu, X.; Han, J.; Ito, Y.; Bringas Jr, P.; Urata, M. M.; Chai, Y., Cell autonomous requirement for Tgfbr2 in the disappearance of medial edge epithelium during palatal fusion. Developmental biology 2006, 297, (1), 238-248. [CrossRef]
- Jin, J.-Z.; Ding, J., Analysis of cell migration, transdifferentiation and apoptosis during mouse secondary palate fusion. 2006.
- Lee, S.; Sears, M. J.; Zhang, Z.; Li, H.; Salhab, I.; Krebs, P.; Xing, Y.; Nah, H. D.; Williams, T.; Carstens, R. P., Cleft lip and cleft palate in Esrp1 knockout mice is associated with alterations in epithelial-mesenchymal crosstalk. Development 2020, 147, (21).
- Caetano da Silva, C.; Macias Trevino, C.; Mitchell, J.; Murali, H.; Tsimbal, C.; Dalessandro, E.; Carroll, S. H.; Kochhar, S.; Curtis, S. W.; Cheng, C. H. E.; Wang, F.; Kutschera, E.; Carstens, R. P.; Xing, Y.; Wang, K.; Leslie, E. J.; Liao, E. C., Functional analysis of ESRP1/2 gene variants and CTNND1 isoforms in orofacial cleft pathogenesis. Communications Biology 2024, 7, (1), 1040. [CrossRef] [PubMed]
- Cecconi, F.; Alvarez-Bolado, G.; Meyer, B. I.; Roth, K. A.; Gruss, P., Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell 1998, 94, (6), 727-737. [CrossRef]
- Jin, J. Z.; Ding, J., Analysis of Meox-2 mutant mice reveals a novel postfusion-based cleft palate. Developmental dynamics: an official publication of the American Association of Anatomists 2006, 235, (2), 539-546.
- Shiomi, N.; Cui, X. M.; Yamamoto, T.; Saito, T.; Shuler, C. F., Inhibition of SMAD2 expression prevents murine palatal fusion. Developmental Dynamics: An Official Publication of the American Association of Anatomists 2006, 235, (7), 1785-1793.
- Iwata, J.-i.; Suzuki, A.; Pelikan, R. C.; Ho, T.-V.; Sanchez-Lara, P. A.; Urata, M.; Dixon, M. J.; Chai, Y., Smad4-Irf6 genetic interaction and TGFβ-mediated IRF6 signaling cascade are crucial for palatal fusion in mice. Development 2013, 140, (6), 1220-1230. [CrossRef]
- Shin, J.-O.; Lee, J.-M.; Bok, J.; Jung, H.-S., Inhibition of the Zeb family prevents murine palatogenesis through regulation of apoptosis and the cell cycle. Biochemical and biophysical research communications 2018, 506, (1), 223-230. [CrossRef] [PubMed]
- Jin, J. Z.; Warner, D. R.; Lu, Q.; Pisano, M. M.; Greene, R. M.; Ding, J., Deciphering TGF-β3 function in medial edge epithelium specification and fusion during mouse secondary palate development. Developmental Dynamics 2014, 243, (12), 1536-1543. [CrossRef] [PubMed]
- He, F.; Xiong, W.; Wang, Y.; Li, L.; Liu, C.; Yamagami, T.; Taketo, M. M.; Zhou, C.; Chen, Y., Epithelial Wnt/β-catenin signaling regulates palatal shelf fusion through regulation of Tgfβ3 expression. Developmental biology 2011, 350, (2), 511-519. [CrossRef]
- Ke, C.-Y.; Xiao, W.-L.; Chen, C.-M.; Lo, L.-J.; Wong, F.-H., IRF6 is the mediator of TGFβ3 during regulation of the epithelial mesenchymal transition and palatal fusion. Scientific reports 2015, 5, (1), 12791. [CrossRef] [PubMed]
- Serrano, M. J.; Liu, J.; Svoboda, K. K.; Nawshad, A.; Benson, M. D., Ephrin reverse signaling mediates palatal fusion and epithelial-to-mesenchymal transition independently of Tgfss3. Journal of cellular physiology 2015, 230, (12), 2961-2972. [CrossRef]
- Mima, J.; Koshino, A.; Oka, K.; Uchida, H.; Hieda, Y.; Nohara, K.; Kogo, M.; Chai, Y.; Sakai, T., Regulation of the epithelial adhesion molecule CEACAM1 is important for palate formation. PloS one 2013, 8, (4), e61653. [CrossRef]
- Nakajima, A.; F. Shuler, C.; Gulka, A. O.; Hanai, J.-i., TGF-β signaling and the epithelial-mesenchymal transition during palatal fusion. International journal of molecular sciences 2018, 19, (11), 3638. [CrossRef]
- Iwata, J.-i.; Parada, C.; Chai, Y., The mechanism of TGF-β signaling during palate development. Oral diseases 2011, 17, (8), 733-744. [CrossRef] [PubMed]
- Seaberg, A.; Awotoye, W.; Qian, F.; Machado-Paula, L. A.; Dunlay, L.; Butali, A.; Murray, J.; Moreno-Uribe, L.; Petrin, A. L., DNA Methylation Effects on Van der Woude Syndrome Phenotypic Variability. Cleft Palate Craniofac J 2024, 10556656241269495. [CrossRef] [PubMed]
- Shull, L. C.; Artinger, K. B., Epigenetic regulation of craniofacial development and disease. Birth Defects Research 2024, 116, (1), e2271. [CrossRef] [PubMed]
- Allis, C. D.; Jenuwein, T., The molecular hallmarks of epigenetic control. Nature Reviews Genetics 2016, 17, (8), 487-500. [CrossRef]
- Bannister, A. J.; Kouzarides, T., Regulation of chromatin by histone modifications. Cell research 2011, 21, (3), 381-395. [CrossRef]
- Godini, R.; Lafta, H. Y.; Fallahi, H., Epigenetic modifications in the embryonic and induced pluripotent stem cells. Gene Expression Patterns 2018, 29, 1-9. [CrossRef] [PubMed]
- Horvath, S., DNA methylation age of human tissues and cell types. Genome biology 2013, 14, 1-20. [CrossRef] [PubMed]
- Jambhekar, A.; Dhall, A.; Shi, Y., Roles and regulation of histone methylation in animal development. Nature reviews Molecular cell biology 2019, 20, (10), 625-641. [CrossRef]
- Seelan, R. S.; Pisano, M.; Greene, R. M., Nucleic acid methylation and orofacial morphogenesis. Birth Defects Research 2019, 111, (20), 1593-1610. [CrossRef]
- Cheng, X.; Blumenthal, R. M., Mediating and maintaining methylation while minimizing mutation: Recent advances on mammalian DNA methyltransferases. Current opinion in structural biology 2022, 75, 102433. [CrossRef]
- Moore, L. D.; Le, T.; Fan, G., DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, (1), 23-38. [CrossRef] [PubMed]
- Du, Q.; Luu, P.-L.; Stirzaker, C.; Clark, S. J., Methyl-CpG-binding domain proteins: readers of the epigenome. Epigenomics 2015, 7, (6), 1051-1073. [CrossRef] [PubMed]
- Jones, P. A., Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nature reviews genetics 2012, 13, (7), 484-492. [CrossRef] [PubMed]
- Whalen, S.; Truty, R. M.; Pollard, K. S., Enhancer–promoter interactions are encoded by complex genomic signatures on looping chromatin. Nature genetics 2016, 48, (5), 488-496. [CrossRef] [PubMed]
- Ong, C. T.; Corces, V. G., Enhancers: emerging roles in cell fate specification. EMBO reports 2012, 13, (5), 423-430. [CrossRef]
- Blattler, A.; Yao, L.; Witt, H.; Guo, Y.; Nicolet, C. M.; Berman, B. P.; Farnham, P. J., Global loss of DNA methylation uncovers intronic enhancers in genes showing expression changes. Genome Biology 2014, 15, (9), 469. [CrossRef] [PubMed]
- Geula, S.; Moshitch-Moshkovitz, S.; Dominissini, D.; Mansour, A. A.; Kol, N.; Salmon-Divon, M.; Hershkovitz, V.; Peer, E.; Mor, N.; Manor, Y. S., m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science 2015, 347, (6225), 1002-1006. [CrossRef]
- Khan, M. F. J.; Little, J.; Aleotti, V.; Mossey, P. A.; Steegers-Theunissen, R. P.; Autelitano, L.; Meazzini, M. C.; Ravaei, A.; Rubini, M., LINE-1 methylation in cleft lip tissues: Influence of infant MTHFR c. 677C> T genotype. Oral Diseases 2019, 25, (6), 1668-1671.
- Douvlataniotis, K.; Bensberg, M.; Lentini, A.; Gylemo, B.; Nestor, C. E., No evidence for DNA N 6-methyladenine in mammals. Science advances 2020, 6, (12), eaay3335. [CrossRef] [PubMed]
- Musheev, M. U.; Baumgärtner, A.; Krebs, L.; Niehrs, C., The origin of genomic N 6-methyl-deoxyadenosine in mammalian cells. Nature chemical biology 2020, 16, (6), 630-634. [CrossRef]
- Altun, G.; Loring, J. F.; Laurent, L. C., DNA methylation in embryonic stem cells. Journal of cellular biochemistry 2010, 109, (1), 1-6. [CrossRef] [PubMed]
- Hu, N.; Strobl-Mazzulla, P.; Sauka-Spengler, T.; Bronner, M. E., DNA methyltransferase3A as a molecular switch mediating the neural tube-to-neural crest fate transition. Genes & development 2012, 26, (21), 2380-2385.
- Rai, K.; Jafri, I. F.; Chidester, S.; James, S. R.; Karpf, A. R.; Cairns, B. R.; Jones, D. A., Dnmt3 and G9a Cooperate for Tissue-specific Development in Zebrafish 2. Journal of Biological Chemistry 2010, 285, (6), 4110-4121. [CrossRef]
- Martins-Taylor, K.; Schroeder, D. I.; LaSalle, J. M.; Lalande, M.; Xu, R.-H., Role of DNMT3B in the regulation of early neural and neural crest specifiers. Epigenetics 2012, 7, (1), 71-82. [CrossRef] [PubMed]
- Jacques-Fricke, B. T.; Roffers-Agarwal, J.; Gammill, L. S., DNA methyltransferase 3b is dispensable for mouse neural crest development. 2012.
- Nowialis, P.; Lopusna, K.; Opavska, J.; Haney, S. L.; Abraham, A.; Sheng, P.; Riva, A.; Natarajan, A.; Guryanova, O.; Simpson, M., Catalytically inactive Dnmt3b rescues mouse embryonic development by accessory and repressive functions. Nature communications 2019, 10, (1), 4374. [CrossRef] [PubMed]
- Seelan, R. S.; Appana, S. N.; Mukhopadhyay, P.; Warner, D. R.; Brock, G. N.; Pisano, M. M.; Greene, R. M., Developmental profiles of the murine palatal methylome. Birth Defects Research Part A: Clinical and Molecular Teratology 2013, 97, (4), 171-186.
- Juriloff, D. M.; Harris, M. J.; Mager, D. L.; Gagnier, L., Epigenetic mechanism causes Wnt9b deficiency and nonsyndromic cleft lip and palate in the A/WySn mouse strain. Birth Defects Research Part A: Clinical and Molecular Teratology 2014, 100, (10), 772-788.
- Alvizi, L.; Ke, X.; Brito, L. A.; Seselgyte, R.; Moore, G. E.; Stanier, P.; Passos-Bueno, M. R., Differential methylation is associated with non-syndromic cleft lip and palate and contributes to penetrance effects. Scientific reports 2017, 7, (1), 2441. [CrossRef]
- Cáceres-Rojas, G.; Salamanca, C.; Krause, B. J.; Recabarren, A. S.; Recabarren, P. A.; Pantoja, R.; Leiva, N.; Pardo, R.; Santos, J. L.; Suazo, J., Nonsyndromic orofacial clefts in Chile: LINE-1 methylation and MTHFR variants. Epigenomics 2020, 12, (20), 1783-1791. [CrossRef]
- Joubert, B. R.; Felix, J. F.; Yousefi, P.; Bakulski, K. M.; Just, A. C.; Breton, C.; Reese, S. E.; Markunas, C. A.; Richmond, R. C.; Xu, C.-J., DNA methylation in newborns and maternal smoking in pregnancy: genome-wide consortium meta-analysis. The American Journal of Human Genetics 2016, 98, (4), 680-696. [CrossRef] [PubMed]
- Pinheiro, I.; Margueron, R.; Shukeir, N.; Eisold, M.; Fritzsch, C.; Richter, F. M.; Mittler, G.; Genoud, C.; Goyama, S.; Kurokawa, M., Prdm3 and Prdm16 are H3K9me1 methyltransferases required for mammalian heterochromatin integrity. Cell 2012, 150, (5), 948-960. [CrossRef] [PubMed]
- Hu, N.; Strobl-Mazzulla, P. H.; Bronner, M. E., Epigenetic regulation in neural crest development. Developmental biology 2014, 396, (2), 159-168. [CrossRef]
- Arboleda, V. A.; Lee, H.; Dorrani, N.; Zadeh, N.; Willis, M.; Macmurdo, C. F.; Manning, M. A.; Kwan, A.; Hudgins, L.; Barthelemy, F., De novo nonsense mutations in KAT6A, a lysine acetyl-transferase gene, cause a syndrome including microcephaly and global developmental delay. The American Journal of Human Genetics 2015, 96, (3), 498-506. [CrossRef] [PubMed]
- Milstone, Z. J.; Lawson, G.; Trivedi, C. M., Histone deacetylase 1 and 2 are essential for murine neural crest proliferation, pharyngeal arch development, and craniofacial morphogenesis. Developmental Dynamics 2017, 246, (12), 1015-1026. [CrossRef] [PubMed]
- Rada-Iglesias, A.; Bajpai, R.; Prescott, S.; Brugmann, S. A.; Swigut, T.; Wysocka, J., Epigenomic annotation of enhancers predicts transcriptional regulators of human neural crest. Cell stem cell 2012, 11, (5), 633-648. [CrossRef] [PubMed]
- Cuiping, L.; Xingang, Y.; Yuexian, F.; Lin, Q.; Xiaofei, T.; Yan, L.; Guanghui, W., The role of histone H3 acetylation on cleft palate in mice induced by 2, 3, 7, 8-tetrachlorodibenzopdioxin. Zhonghua Zheng Xing wai ke za zhi= Zhonghua Zhengxing Waike Zazhi= Chinese Journal of Plastic Surgery 2014, 30, (5), 369-372.
- Lindgren, A. M.; Hoyos, T.; Talkowski, M. E.; Hanscom, C.; Blumenthal, I.; Chiang, C.; Ernst, C.; Pereira, S.; Ordulu, Z.; Clericuzio, C., Haploinsufficiency of KDM6A is associated with severe psychomotor retardation, global growth restriction, seizures and cleft palate. Human genetics 2013, 132, 537-552. [CrossRef]
- Van Laarhoven, P. M.; Neitzel, L. R.; Quintana, A. M.; Geiger, E. A.; Zackai, E. H.; Clouthier, D. E.; Artinger, K. B.; Ming, J. E.; Shaikh, T. H., Kabuki syndrome genes KMT2D and KDM6A: functional analyses demonstrate critical roles in craniofacial, heart and brain development. Human molecular genetics 2015, 24, (15), 4443-4453. [CrossRef]
- Shpargel, K. B.; Starmer, J.; Wang, C.; Ge, K.; Magnuson, T., UTX-guided neural crest function underlies craniofacial features of Kabuki syndrome. Proceedings of the National Academy of Sciences 2017, 114, (43), E9046-E9055. [CrossRef] [PubMed]
- Guo, T.; Han, X.; He, J.; Feng, J.; Jing, J.; Janečková, E.; Lei, J.; Ho, T. V.; Xu, J.; Chai, Y., KDM6B interacts with TFDP1 to activate P53 signaling in regulating mouse palatogenesis. Elife 2022, 11. [CrossRef]
- Schwenty-Lara, J.; Nehl, D.; Borchers, A., The histone methyltransferase KMT2D, mutated in Kabuki syndrome patients, is required for neural crest cell formation and migration. Human Molecular Genetics 2020, 29, (2), 305-319. [CrossRef]
- Lee, J. M.; Jung, H.; de Paula Machado Pasqua, B.; Park, Y.; Jeon, S.; Lee, S. K.; Lee, J. W.; Kwon, H. E., Mll4 regulates postnatal palate growth and midpalatal suture development. bioRxiv 2024. [CrossRef] [PubMed]
- Bögershausen, N.; Tsai, I.-C.; Pohl, E.; Kiper, P. Ö. S.; Beleggia, F.; Percin, E. F.; Keupp, K.; Matchan, A.; Milz, E.; Alanay, Y., RAP1-mediated MEK/ERK pathway defects in Kabuki syndrome. The Journal of clinical investigation 2015, 125, (9), 3585-3599. [CrossRef]
- Tsai, I.-C.; McKnight, K.; McKinstry, S. U.; Maynard, A. T.; Tan, P. L.; Golzio, C.; White, C. T.; Price, D. J.; Davis, E. E.; Amrine-Madsen, H., Small molecule inhibition of RAS/MAPK signaling ameliorates developmental pathologies of Kabuki Syndrome. Scientific reports 2018, 8, (1), 10779. [CrossRef]
- Fortschegger, K.; de Graaf, P.; Outchkourov, N. S.; van Schaik, F. M.; Timmers, H. M.; Shiekhattar, R., PHF8 targets histone methylation and RNA polymerase II to activate transcription. Molecular and cellular biology 2010. [CrossRef]
- Feng, W.; Yonezawa, M.; Ye, J.; Jenuwein, T.; Grummt, I., PHF8 activates transcription of rRNA genes through H3K4me3 binding and H3K9me1/2 demethylation. Nature structural & molecular biology 2010, 17, (4), 445-450.
- Qi, H. H.; Sarkissian, M.; Hu, G.-Q.; Wang, Z.; Bhattacharjee, A.; Gordon, D. B.; Gonzales, M.; Lan, F.; Ongusaha, P. P.; Huarte, M., Histone H4K20/H3K9 demethylase PHF8 regulates zebrafish brain and craniofacial development. Nature 2010, 466, (7305), 503-507. [CrossRef] [PubMed]
- Loenarz, C.; Ge, W.; Coleman, M. L.; Rose, N. R.; Cooper, C. D.; Klose, R. J.; Ratcliffe, P. J.; Schofield, C. J., PHF8, a gene associated with cleft lip/palate and mental retardation, encodes for an N ε-dimethyl lysine demethylase. Human molecular genetics 2010, 19, (2), 217-222. [CrossRef] [PubMed]
- Han, Q.; Yang, P.; Wu, Y.; Meng, S.; Sui, L.; Zhang, L.; Yu, L.; Tang, Y.; Jiang, H.; Xuan, D., Epigenetically modified bone marrow stromal cells in silk scaffolds promote craniofacial bone repair and wound healing. Tissue engineering Part A 2015, 21, (15-16), 2156-2165.
- Ding, H. L.; Clouthier, D. E.; Artinger, K. B., Redundant roles of PRDM family members in zebrafish craniofacial development. Developmental Dynamics 2013, 242, (1), 67-79. [CrossRef]
- Shull, L. C.; Sen, R.; Menzel, J.; Goyama, S.; Kurokawa, M.; Artinger, K. B., The conserved and divergent roles of Prdm3 and Prdm16 in zebrafish and mouse craniofacial development. Developmental biology 2020, 461, (2), 132-144. [CrossRef]
- Strassman, A.; Schnütgen, F.; Dai, Q.; Jones, J. C.; Gomez, A. C.; Pitstick, L.; Holton, N. E.; Moskal, R.; Leslie, E. R.; von Melchner, H., Generation of a multipurpose Prdm16 mouse allele by targeted gene trapping. Disease models & mechanisms 2017, 10, (7), 909-922.
- Warner, D. R.; Mukhopadhyay, P.; Webb, C. L.; Greene, R. M.; Pisano, M. M., Chromatin immunoprecipitation-promoter microarray identification of genes regulated by PRDM16 in murine embryonic palate mesenchymal cells. Experimental Biology and Medicine 2012, 237, (4), 387-394. [CrossRef]
- Xu, J.; Wang, A. H.; Oses-Prieto, J.; Makhijani, K.; Katsuno, Y.; Pei, M.; Yan, L.; Zheng, Y. G.; Burlingame, A.; Brückner, K., Arginine methylation initiates BMP-induced Smad signaling. Molecular cell 2013, 51, (1), 5-19. [CrossRef] [PubMed]
- Gou, Y.; Li, J.; Wu, J.; Gupta, R.; Cho, I.; Ho, T. V.; Chai, Y.; Merrill, A.; Wang, J.; Xu, J., Prmt1 regulates craniofacial bone formation upstream of Msx1. Mech Dev 2018, 152, 13-20. [CrossRef] [PubMed]
- Gou, Y.; Li, J.; Jackson-Weaver, O.; Wu, J.; Zhang, T.; Gupta, R.; Cho, I.; Ho, T. V.; Chen, Y.; Li, M.; Richard, S.; Wang, J.; Chai, Y.; Xu, J., Protein Arginine Methyltransferase PRMT1 Is Essential for Palatogenesis. J Dent Res 2018, 97, (13), 1510-1518. [CrossRef]
- Liu, S.; Higashihori, N.; Yahiro, K.; Moriyama, K., Retinoic acid inhibits histone methyltransferase Whsc1 during palatogenesis. Biochem Biophys Res Commun 2015, 458, (3), 525-530. [CrossRef]
- Mills, A.; Bearce, E.; Cella, R.; Kim, S. W.; Selig, M.; Lee, S.; Lowery, L. A., Wolf-Hirschhorn Syndrome-Associated Genes Are Enriched in Motile Neural Crest Cells and Affect Craniofacial Development in Xenopus laevis. Front Physiol 2019, 10, 431. [CrossRef] [PubMed]
- Singh, N.; Gupta, M.; Trivedi, C. M.; Singh, M. K.; Li, L.; Epstein, J. A., Murine craniofacial development requires Hdac3-mediated repression of Msx gene expression. Dev Biol 2013, 377, (2), 333-44. [CrossRef] [PubMed]
- DeLaurier, A.; Nakamura, Y.; Braasch, I.; Khanna, V.; Kato, H.; Wakitani, S.; Postlethwait, J. H.; Kimmel, C. B., Histone deacetylase-4 is required during early cranial neural crest development for generation of the zebrafish palatal skeleton. BMC Dev Biol 2012, 12, 16. [CrossRef]
- Voss, A. K.; Vanyai, H. K.; Collin, C.; Dixon, M. P.; McLennan, T. J.; Sheikh, B. N.; Scambler, P.; Thomas, T., MOZ regulates the Tbx1 locus, and Moz mutation partially phenocopies DiGeorge syndrome. Dev Cell 2012, 23, (3), 652-63. [CrossRef]
- Mukhopadhyay, P.; Brock, G.; Pihur, V.; Webb, C.; Pisano, M. M.; Greene, R. M., Developmental microRNA expression profiling of murine embryonic orofacial tissue. Birth Defects Research Part A: Clinical and Molecular Teratology 2010, 88, (7), 511-534.
- Nie, X.; Wang, Q.; Jiao, K., Dicer activity in neural crest cells is essential for craniofacial organogenesis and pharyngeal arch artery morphogenesis. Mechanisms of development 2011, 128, (3-4), 200-207.
- Huang, Z.-P.; Chen, J.-F.; Regan, J. N.; Maguire, C. T.; Tang, R.-H.; Dong, X. R.; Majesky, M. W.; Wang, D.-Z., Loss of microRNAs in neural crest leads to cardiovascular syndromes resembling human congenital heart defects. Arteriosclerosis, thrombosis, and vascular biology 2010, 30, (12), 2575-2586.
- Sheehy, N. T.; Cordes, K. R.; White, M. P.; Ivey, K. N.; Srivastava, D., The neural crest-enriched microRNA miR-452 regulates epithelial-mesenchymal signaling in the first pharyngeal arch. Development 2010, 137, (24), 4307-4316. [CrossRef] [PubMed]
- Stüssel, L. G.; Hollstein, R.; Laugsch, M.; Hochfeld, L. M.; Welzenbach, J.; Schröder, J.; Thieme, F.; Ishorst, N.; Romero, R. O.; Weinhold, L.; Hess, T.; Gehlen, J.; Mostowska, A.; Heilmann-Heimbach, S.; Mangold, E.; Rada-Iglesias, A.; Knapp, M.; Schaaf, C. P.; Ludwig, K. U., MiRNA-149 as a Candidate for Facial Clefting and Neural Crest Cell Migration. Journal of Dental Research 2022, 101, (3), 323-330. [CrossRef]
- Suzuki, A.; Li, A.; Gajera, M.; Abdallah, N.; Zhang, M.; Zhao, Z.; Iwata, J., MicroRNA-374a,-4680, and-133b suppress cell proliferation through the regulation of genes associated with human cleft palate in cultured human palate cells. BMC medical genomics 2019, 12, 1-13. [CrossRef] [PubMed]
- Suzuki, A.; Yoshioka, H.; Summakia, D.; Desai, N. G.; Jun, G.; Jia, P.; Loose, D. S.; Ogata, K.; Gajera, M. V.; Zhao, Z., MicroRNA-124-3p suppresses mouse lip mesenchymal cell proliferation through the regulation of genes associated with cleft lip in the mouse. Bmc Genomics 2019, 20, 1-17. [CrossRef]
- Gajera, M.; Desai, N.; Suzuki, A.; Li, A.; Zhang, M.; Jun, G.; Jia, P.; Zhao, Z.; Iwata, J., MicroRNA-655-3p and microRNA-497-5p inhibit cell proliferation in cultured human lip cells through the regulation of genes related to human cleft lip. BMC medical genomics 2019, 12, 1-18. [CrossRef] [PubMed]
- Yoshioka, H.; Li, A.; Suzuki, A.; Ramakrishnan, S. S.; Zhao, Z.; Iwata, J., Identification of microRNAs and gene regulatory networks in cleft lip common in humans and mice. Human Molecular Genetics 2021, 30, (19), 1881-1893. [CrossRef]
- Li, A.; Jia, P.; Mallik, S.; Fei, R.; Yoshioka, H.; Suzuki, A.; Iwata, J.; Zhao, Z., Critical microRNAs and regulatory motifs in cleft palate identified by a conserved miRNA–TF–gene network approach in humans and mice. Briefings in bioinformatics 2020, 21, (4), 1465-1478. [CrossRef] [PubMed]
- Fu, C.; Lou, S.; Zhu, G.; Fan, L.; Yu, X.; Zhu, W.; Ma, L.; Wang, L.; Pan, Y., Identification of new miRNA-mRNA networks in the development of non-syndromic cleft lip with or without cleft palate. Frontiers in cell and developmental biology 2021, 9, 631057. [CrossRef]
- Wang, J.; Bai, Y.; Li, H.; Greene, S. B.; Klysik, E.; Yu, W.; Schwartz, R. J.; Williams, T. J.; Martin, J. F., MicroRNA-17-92, a Direct Ap-2α Transcriptional Target, Modulates T-Box Factor Activity in Orofacial Clefting. PLOS Genetics 2013, 9, (9), e1003785.
- Li, Y.-H.; Yang, J.; Zhang, J.-L.; Liu, J.-Q.; Zheng, Z.; Hu, D.-H., BMP4 rs17563 polymorphism and nonsyndromic cleft lip with or without cleft palate: A meta-analysis. Medicine 2017, 96, (31), e7676. [CrossRef] [PubMed]
- Kim, S.; Lewis, A. E.; Singh, V.; Ma, X.; Adelstein, R.; Bush, J. O., Convergence and extrusion are required for normal fusion of the mammalian secondary palate. PLoS biology 2015, 13, (4), e1002122. [CrossRef] [PubMed]
- Mukhopadhyay, P.; Smolenkova, I.; Seelan, R. S.; Pisano, M. M.; Greene, R. M., Spatiotemporal Expression and Functional Analysis of miRNA-22 in the Developing Secondary Palate. The Cleft Palate Craniofacial Journal 2023, 60, (1), 27-38. [CrossRef] [PubMed]
- Carpinelli, M. R.; De Vries, M. E.; Auden, A.; Butt, T.; Deng, Z.; Partridge, D. D.; Miles, L. B.; Georgy, S. R.; Haigh, J. J.; Darido, C., Inactivation of Zeb1 in GRHL2-deficient mouse embryos rescues mid-gestation viability and secondary palate closure. Disease models & mechanisms 2020, 13, (3), dmm042218.
- Shin, J.-O.; Lee, J.-M.; Cho, K.-W.; Kwak, S.; Kwon, H.-J.; Lee, M.-J.; Cho, S.-W.; Kim, K.-S.; Jung, H.-S., MiR-200b is involved in Tgf-β signaling to regulate mammalian palate development. Histochemistry and Cell Biology 2012, 137, (1), 67-78. [CrossRef] [PubMed]
- Mukhopadhyay, P.; Smolenkova, I.; Warner, D.; Pisano, M. M.; Greene, R. M., Spatio-temporal expression and functional analysis of miR-206 in developing orofacial tissue. MicroRNA 2019, 8, (1), 43-60. [CrossRef] [PubMed]
- Li, L.; Meng, T.; Jia, Z.; Zhu, G.; Shi, B., Single nucleotide polymorphism associated with nonsyndromic cleft palate influences the processing of miR-140. Am J Med Genet A 2010, 152a, (4), 856-62. [CrossRef] [PubMed]
- Cesana, M.; Cacchiarelli, D.; Legnini, I.; Santini, T.; Sthandier, O.; Chinappi, M.; Tramontano, A.; Bozzoni, I., A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 2011, 147, (2), 358-369. [CrossRef] [PubMed]
- Ma, B.; Wang, S.; Wu, W.; Shan, P.; Chen, Y.; Meng, J.; Xing, L.; Yun, J.; Hao, L.; Wang, X.; Li, S.; Guo, Y., Mechanisms of circRNA/lncRNA-miRNA interactions and applications in disease and drug research. Biomedicine & Pharmacotherapy 2023, 162, 114672.
- Wu, C.; Liu, H.; Zhan, Z.; Zhang, X.; Zhang, M.; You, J.; Ma, J., Unveiling dysregulated lncRNAs and networks in non-syndromic cleft lip with or without cleft palate pathogenesis. Scientific Reports 2024, 14, (1), 1047. [CrossRef] [PubMed]
- Geisler, S.; Coller, J., RNA in unexpected places: long non-coding RNA functions in diverse cellular contexts. Nature reviews Molecular cell biology 2013, 14, (11), 699-712. [CrossRef]
- Yun, L.; Ma, L.; Wang, M.; Yang, F.; Kan, S.; Zhang, C.; Xu, M.; Li, D.; Du, Y.; Zhang, W., Rs2262251 in lncRNA RP11-462G12. 2 is associated with nonsyndromic cleft lip with/without cleft palate. Human Mutation 2019, 40, (11), 2057-2067.
- Shu, X.; Dong, Z.; Zhang, M.; Shu, S., Integrated analysis identifying long non-coding RNAs (lncRNAs) for competing endogenous RNAs (ceRNAs) network-regulated palatal shelf fusion in the development of mouse cleft palate. Annals of translational medicine 2019, 7, (23).
- Ito, Y.; Yeo, J. Y.; Chytil, A.; Han, J.; Bringas, P., Jr; Nakajima, A.; Shuler, C. F.; Moses, H. L.; Chai, Y., Conditional inactivation of Tgfbr2 in cranial neural crest causes cleft palate and calvaria defects. Development 2003, 130, (21), 5269-5280. [CrossRef]
- Gao, Y.; Zang, Q.; Song, H.; Fu, S.; Sun, W.; Zhang, W.; Wang, X.; Li, Y.; Jiao, X., Comprehensive analysis of differentially expressed profiles of non-coding RNAs in peripheral blood and ceRNA regulatory networks in non-syndromic orofacial clefts. Molecular medicine reports 2019, 20, (1), 513-528. [CrossRef]
- Wang, E.; Guo, Y.; Gao, S.; Zhou, Y.; Liu, B.; Dissanayaka, W. L.; Zheng, Y.; Zhou, Q.; Zhai, J.; Gao, Z., Long non-coding RNAs MALAT1 and NEAT1 in non-syndromic orofacial clefts. Oral Diseases 2023, 29, (4), 1668-1679. [CrossRef]
- Piunti, A.; Shilatifard, A., Author Correction: The roles of Polycomb repressive complexes in mammalian development and cancer. Nat Rev Mol Cell Biol 2022, 23, (6), 444. [CrossRef] [PubMed]
- Kim, H.; Langohr, I. M.; Faisal, M.; McNulty, M.; Thorn, C.; Kim, J., Ablation of Ezh2 in neural crest cells leads to aberrant enteric nervous system development in mice. PLoS One 2018, 13, (8), e0203391. [CrossRef]
- Lui, J. C.; Barnes, K. M.; Dong, L.; Yue, S.; Graber, E.; Rapaport, R.; Dauber, A.; Nilsson, O.; Baron, J., Ezh2 Mutations Found in the Weaver Overgrowth Syndrome Cause a Partial Loss of H3K27 Histone Methyltransferase Activity. J Clin Endocrinol Metab 2018, 103, (4), 1470-1478. [CrossRef] [PubMed]
- van der Velden, Y. U.; Wang, L.; Querol Cano, L.; Haramis, A.-P. G., The polycomb group protein ring1b/rnf2 is specifically required for craniofacial development. PloS one 2013, 8, (9), e73997. [CrossRef] [PubMed]
- Marc, S.; Mizeranschi, A. E.; Paul, C.; Otavă, G.; Savici, J.; Sicoe, B.; Torda, I.; Huțu, I.; Mircu, C.; Ilie, D. E., Simultaneous occurrence of hypospadias and bilateral cleft lip and jaw in a crossbred calf: Clinical, computer tomographic, and genomic characterization. Animals 2023, 13, (10), 1709. [CrossRef] [PubMed]
- Lichtig, H.; Artamonov, A.; Polevoy, H.; Reid, C. D.; Bielas, S. L.; Frank, D., Modeling Bainbridge-Ropers syndrome in Xenopus laevis embryos. Frontiers in physiology 2020, 11, 75. [CrossRef] [PubMed]
- Pagliaroli, L.; Porazzi, P.; Curtis, A. T.; Scopa, C.; Mikkers, H. M.; Freund, C.; Daxinger, L.; Deliard, S.; Welsh, S. A.; Offley, S., Inability to switch from ARID1A-BAF to ARID1B-BAF impairs exit from pluripotency and commitment towards neural crest formation in ARID1B-related neurodevelopmental disorders. Nature communications 2021, 12, (1), 6469. [CrossRef] [PubMed]
- Miyake, N.; Tsurusaki, Y.; Matsumoto, N. In Numerous BAF complex genes are mutated in Coffin–Siris syndrome, American Journal of Medical Genetics Part C: Seminars in Medical Genetics, 2014; Wiley Online Library: 2014; pp 257-261.
- Bi-Lin, K. W.; Seshachalam, P. V.; Tuoc, T.; Stoykova, A.; Ghosh, S.; Singh, M. K., Critical role of the BAF chromatin remodeling complex during murine neural crest development. PLoS genetics 2021, 17, (3), e1009446. [CrossRef] [PubMed]
- Diets, I. J.; Prescott, T.; Champaigne, N. L.; Mancini, G. M.; Krossnes, B.; Frič, R.; Kocsis, K.; Jongmans, M. C.; Kleefstra, T., A recurrent de novo missense pathogenic variant in SMARCB1 causes severe intellectual disability and choroid plexus hyperplasia with resultant hydrocephalus. Genetics in Medicine 2019, 21, (3), 572-579. [CrossRef] [PubMed]
- Huang, X.; Chen, Q.; Luo, W.; Pakvasa, M.; Zhang, Y.; Zheng, L.; Li, S.; Yang, Z.; Zeng, H.; Liang, F., SATB2: A versatile transcriptional regulator of craniofacial and skeleton development, neurogenesis and tumorigenesis, and its applications in regenerative medicine. Genes & Diseases 2022, 9, (1), 95-107.
- Garland, M. A.; Reynolds, K.; Zhou, C. J., Environmental mechanisms of orofacial clefts. Birth Defects Research 2020, 112, (19), 1660-1698. [CrossRef] [PubMed]
- Shiani, A.; Sharafi, K.; Omer, A. K.; Kiani, A.; Matin, B. K.; Heydari, M. B.; Massahi, T., A Systematic Literature Review on the Association Between Toxic and Essential Trace Elements and the Risk of Orofacial Clefts in Infants. Biol Trace Elem Res 2024, 202, (8), 3504-3516. [CrossRef]
- Heydari, M.-H.; Sadeghian, A.; Khadivi, G.; Mustafa, H. J.; Javinani, A.; Nadjmi, N.; Khojasteh, A., Prevalence, trend, and associated risk factors for cleft lip with/without cleft palate: a national study on live births from 2016 to 2021. BMC Oral Health 2024, 24, (1), 36. [CrossRef]
- Cunningham, T. J.; Duester, G., Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nat Rev Mol Cell Biol 2015, 16, (2), 110-23. [CrossRef]
- Kawaguchi, R.; Yu, J.; Honda, J.; Hu, J.; Whitelegge, J.; Ping, P.; Wiita, P.; Bok, D.; Sun, H., A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science 2007, 315, (5813), 820-825. [CrossRef]
- Niederreither, K.; Dollé, P., Retinoic acid in development: towards an integrated view. Nature Reviews Genetics 2008, 9, (7), 541-553. [CrossRef] [PubMed]
- Dupé, V.; Matt, N.; Garnier, J.-M.; Chambon, P.; Mark, M.; Ghyselinck, N. B., A newborn lethal defect due to inactivation of retinaldehyde dehydrogenase type 3 is prevented by maternal retinoic acid treatment. Proceedings of the National Academy of Sciences 2003, 100, (24), 14036-14041. [CrossRef] [PubMed]
- Funato, N.; Nakamura, M.; Richardson, J. A.; Srivastava, D.; Yanagisawa, H., Tbx1 regulates oral epithelial adhesion and palatal development. Hum Mol Genet 2012, 21, (11), 2524-37. [CrossRef]
- Roberts, C.; Ivins, S.; Cook, A. C.; Baldini, A.; Scambler, P. J., Cyp26 genes a1, b1 and c1 are down-regulated in Tbx1 null mice and inhibition of Cyp26 enzyme function produces a phenocopy of DiGeorge Syndrome in the chick. Human molecular genetics 2006, 15, (23), 3394-3410. [CrossRef] [PubMed]
- Okano, J.; Kimura, W.; Papaionnou, V. E.; Miura, N.; Yamada, G.; Shiota, K.; Sakai, Y., The regulation of endogenous retinoic acid level through CYP26B1 is required for elevation of palatal shelves. Developmental Dynamics 2012, 241, (11), 1744-1756. [CrossRef] [PubMed]
- Liu, X.; Zhang, H.; Gao, L.; Yin, Y.; Pan, X.; Li, Z.; Li, N.; Li, H.; Yu, Z., Negative Interplay of Retinoic Acid and TGF-β Signaling Mediated by TG-Interacting Factor to Modulate Mouse Embryonic Palate Mesenchymal–Cell Proliferation. Birth Defects Research Part B: Developmental and Reproductive Toxicology 2014, 101, (6), 403-409.
- Zhang, Y.; Dong, S.; Wang, J.; Wang, M.; Chen, M.; Huang, H., Involvement of Notch2 in all-trans retinoic acid-induced inhibition of mouse embryonic palate mesenchymal cell proliferation. Molecular Medicine Reports 2017, 16, (3), 2538-2546. [CrossRef] [PubMed]
- Shimomura, T.; Kawakami, M.; Okuda, H.; Tatsumi, K.; Morita, S.; Nochioka, K.; Kirita, T.; Wanaka, A., Retinoic acid regulates Lhx8 expression via FGF-8b to the upper jaw development of chick embryo. Journal of bioscience and bioengineering 2015, 119, (3), 260-266. [CrossRef]
- Liu, J.; Xiao, Q.; Xiao, J.; Niu, C.; Li, Y.; Zhang, X.; Zhou, Z.; Shu, G.; Yin, G., Wnt/β-catenin signalling: function, biological mechanisms, and therapeutic opportunities. Signal Transduction and Targeted Therapy 2022, 7, (1), 3. [CrossRef] [PubMed]
- Zhang, Y.; Dong, S.; Wang, W.; Wang, J.; Wang, M.; Chen, M.; Hou, J.; Huang, H., Activation of Notch1 inhibits medial edge epithelium apoptosis in all-trans retinoic acid-induced cleft palate in mice. Biochemical and biophysical research communications 2016, 477, (3), 322-328. [CrossRef]
- Shu, X.; Dong, Z.; Shu, S., AMBRA1-mediated autophagy and apoptosis associated with an epithelial-mesenchymal transition in the development of cleft palate induced by all-trans retinoic acid. Annals of translational medicine 2019, 7, (7).
- Zhang, W.; Shen, Z.; Xing, Y.; Zhao, H.; Liang, Y.; Chen, J.; Zhong, X.; Shi, L.; Wan, X.; Zhou, J., MiR-106a-5p modulates apoptosis and metabonomics changes by TGF-β/Smad signaling pathway in cleft palate. Experimental cell research 2020, 386, (2), 111734. [CrossRef]
- Zhang, M.; Zhou, J.; Ji, Y.; Shu, S.; Zhang, M.; Liang, Y., LncRNA-NONMMUT100923.1 regulates mouse embryonic palatal shelf adhesion by sponging miR-200a-3p to modulate medial epithelial cell desmosome junction during palatogenesis. Heliyon 2023, 9, (5), e16329.
- Zhou, J.; Zhang, M.; Zhang, M.; Tan, M.; Ji, Y.; Shu, S.; Liang, Y., MiRNA-470-5p suppresses epithelial-mesenchymal transition of embryonic palatal shelf epithelial cells by targeting Fgfr1 during palatogenesis. Experimental Biology and Medicine 2023, 248, (13), 1124-1133. [CrossRef] [PubMed]
- Cho, E. J.; Chung, G. E.; Yoo, J. J.; Cho, Y.; Shin, D. W.; Kim, Y. J.; Yoon, J. H.; Han, K.; Yu, S. J., The association between alcohol consumption and the risk of hepatocellular carcinoma according to glycemic status in Korea: A nationwide population-based study. PLoS Med 2023, 20, (6), e1004244. [CrossRef] [PubMed]
- Maranhão, S. C.; Sá, J.; Cangussú, M. C. T.; Coletta, R. D.; Reis, S. R.; Medrado, A. R., Nonsyndromic oral clefts and associated risk factors in the state of Bahia, Brazil. European Archives of Paediatric Dentistry 2021, 22, 121-127. [CrossRef]
- Deitrich, R.; Zimatkin, S.; Pronko, S., Oxidation of ethanol in the brain and its consequences. Alcohol Res Health 2006, 29, (4), 266-73.
- Shabtai, Y.; Bendelac, L.; Jubran, H.; Hirschberg, J.; Fainsod, A., Acetaldehyde inhibits retinoic acid biosynthesis to mediate alcohol teratogenicity. Sci Rep 2018, 8, (1), 347. [CrossRef] [PubMed]
- McCarthy, N.; Wetherill, L.; Lovely, C. B.; Swartz, M. E.; Foroud, T. M.; Eberhart, J. K., Pdgfra protects against ethanol-induced craniofacial defects in a zebrafish model of FASD. Development 2013, 140, (15), 3254-3265. [CrossRef]
- Hicks, S. D.; Middleton, F. A.; Miller, M. W., Ethanol-induced methylation of cell cycle genes in neural stem cells. Journal of neurochemistry 2010, 114, (6), 1767-1780. [CrossRef]
- Jin, M.; Ande, A.; Kumar, A.; Kumar, S., Regulation of cytochrome P450 2e1 expression by ethanol: role of oxidative stress-mediated pkc/jnk/sp1 pathway. Cell death & disease 2013, 4, (3), e554-e554.
- Serio, R. N.; Laursen, K. B.; Urvalek, A. M.; Gross, S. S.; Gudas, L. J., Ethanol promotes differentiation of embryonic stem cells through retinoic acid receptor-γ. J Biol Chem 2019, 294, (14), 5536-5548. [CrossRef]
- Serio, R. N.; Gudas, L. J., Modification of stem cell states by alcohol and acetaldehyde. Chem Biol Interact 2020, 316, 108919. [CrossRef] [PubMed]
- Hahn, M. E.; Karchner, S. I.; Merson, R. R., Diversity as opportunity: insights from 600 million years of AHR evolution. Current opinion in toxicology 2017, 2, 58-71. [CrossRef] [PubMed]
- Fujiwara, K.; Yamada, T.; Mishima, K.; Imura, H.; Sugahara, T., Morphological and immunohistochemical studies on cleft palates induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin in mice. Congenit Anom (Kyoto) 2008, 48, (2), 68-73. [CrossRef] [PubMed]
- Takagi, T. N.; Matsui, K. A.; Yamashita, K.; Ohmori, H.; Yasuda, M., Pathogenesis of cleft palate in mouse embryos exposed to 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin (TCDD). Teratogenesis, carcinogenesis, and mutagenesis 2000, 20, (2), 73-86.
- Yuan, X.; Qiu, L.; Pu, Y.; Liu, C.; Zhang, X.; Wang, C.; Pu, W.; Fu, Y., Histone acetylation is involved in TCDD-induced cleft palate formation in fetal mice. Molecular medicine reports 2016, 14, (2), 1139-1145. [CrossRef] [PubMed]
- Liu, X.; Li, X.; Tao, Y.; Li, N.; Ji, M.; Zhang, X.; Chen, Y.; He, Z.; Yu, K.; Yu, Z., TCDD inhibited the osteogenic differentiation of human fetal palatal mesenchymal cells through AhR and BMP-2/TGF-β/Smad signaling. Toxicology 2020, 431, 152353. [CrossRef] [PubMed]
- Gao, Z.; Bu, Y.; Zhang, G.; Liu, X.; Wang, X.; Ding, S.; Wang, E.; Shi, R.; Li, Q.; Fu, J., Effect of TCDD on the fate of epithelial cells isolated from human fetal palatal shelves (hFPECs). Toxicology and Applied Pharmacology 2016, 305, 186-193. [CrossRef]
- Thomae, T. L.; Stevens, E. A.; Bradfield, C. A., Transforming growth factor-β3 restores fusion in palatal shelves exposed to 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin. Journal of Biological Chemistry 2005, 280, (13), 12742-12746. [CrossRef] [PubMed]
- Yu, Z.; Zhang, Y.; Wang, G.; Song, S.; Su, H.; Duan, W.; Wu, Y.; Zhang, Y.; Liu, X., Identification of competing endogenous RNA networks associated with circRNA and lncRNA in TCDD-induced cleft palate development. Toxicol Lett 2024, 401, 71-81. [CrossRef]
- Yoshioka, H.; Jun, G.; Suzuki, A.; Iwata, J., Dexamethasone Suppresses Palatal Cell Proliferation through miR-130a-3p. Int J Mol Sci 2021, 22, (22).
- Yang, W.; Carmichael, S. L.; Shaw, G. M., Folic acid fortification and prevalences of neural tube defects, orofacial clefts, and gastroschisis in California, 1989 to 2010. Birth Defects Research Part A: Clinical and Molecular Teratology 2016, 106, (12), 1032-1041.
- Jahanbin, A.; Shadkam, E.; Miri, H. H.; Shirazi, A. S.; Abtahi, M., Maternal folic acid supplementation and the risk of oral clefts in offspring. Journal of Craniofacial Surgery 2018, 29, (6), e534-e541. [CrossRef]
- Salamanca, C.; González-Hormazábal, P.; Recabarren, A. S.; Recabarren, P. A.; Pantoja, R.; Leiva, N.; Pardo, R.; Suazo, J., A SHMT1 variant decreases the risk of nonsyndromic cleft lip with or without cleft palate in Chile. Oral Diseases 2020, 26, (1), 159-165. [CrossRef]
- Ramírez-Chau, C.; Blanco, R.; Colombo, A.; Pardo, R.; Suazo, J., MTHFR c. 677C> T is a risk factor for non-syndromic cleft lip with or without cleft palate in Chile. Oral Diseases 2016, 22, (7), 703-708. [CrossRef] [PubMed]
- Padmanabhan, N.; Jia, D.; Geary-Joo, C.; Wu, X.; Ferguson-Smith, A. C.; Fung, E.; Bieda, M. C.; Snyder, F. F.; Gravel, R. A.; Cross, J. C., Mutation in folate metabolism causes epigenetic instability and transgenerational effects on development. Cell 2013, 155, (1), 81-93. [CrossRef]
- Wahl, S. E.; Kennedy, A. E.; Wyatt, B. H.; Moore, A. D.; Pridgen, D. E.; Cherry, A. M.; Mavila, C. B.; Dickinson, A. J., The role of folate metabolism in orofacial development and clefting. Developmental biology 2015, 405, (1), 108-122. [CrossRef]
- Tyagi, N.; Sedoris, K. C.; Steed, M.; Ovechkin, A. V.; Moshal, K. S.; Tyagi, S. C., Mechanisms of homocysteine-induced oxidative stress. American Journal of Physiology-Heart and Circulatory Physiology 2005, 289, (6), H2649-H2656.
- Ritchie, H. E.; Oakes, D.; Farrell, E.; Ababneh, D.; Howe, A., Fetal hypoxia and hyperglycemia in the formation of phenytoin-induced cleft lip and maxillary hypoplasia. Epilepsia Open 2019, 4, (3), 443-451. [CrossRef] [PubMed]
- Little, J.; Cardy, A.; Munger, R. G., Tobacco smoking and oral clefts: a meta-analysis. Bulletin of the World Health Organization 2004, 82, (3), 213-218.
- Honein, M. A.; Devine, O.; Grosse, S. D.; Reefhuis, J., Prevention of orofacial clefts caused by smoking: implications of the Surgeon General's report. Birth Defects Research Part A: Clinical and Molecular Teratology 2014, 100, (11), 822-825.
- Lammer, E. J.; Shaw, G. M.; Iovannisci, D. M.; Van Waes, J.; Finnell, R. H., Maternal smoking and the risk of orofacial clefts: Susceptibility with NAT1 and NAT2 polymorphisms. Epidemiology 2004, 15, (2), 150-6. [CrossRef] [PubMed]
- Junaid, M.; Narayanan, M. A.; Jayanthi, D.; Kumar, S. R.; Selvamary, A. L., Association between maternal exposure to tobacco, presence of TGFA gene, and the occurrence of oral clefts. A case control study. Clinical Oral Investigations 2018, 22, 217-223. [CrossRef]
- Ebadifar, A.; Hamedi, R.; KhorramKhorshid, H. R.; Kamali, K.; Moghadam, F. A., Parental cigarette smoking, transforming growth factor-alpha gene variant and the risk of orofacial cleft in Iranian infants. Iran J Basic Med Sci 2016, 19, (4), 366-73.
- Lin, J.-y.; Luan, R.-s.; Guo, Z.-q.; Lin, X.-q.; Tang, H.-y.; Chen, Y.-p., [The correlation analysis between environmental factors, bone morphogenetic protein-4 and transforming growth factor beta-3 polymorphisms in nonsyndromic cleft lip with or without cleft palate]. Zhonghua Kou Qiang Yi Xue Za Zhi 2010, 45, (10), 596-600.
- Izzotti, A.; Balansky, R. M.; Cartiglia, C.; Camoirano, A.; Longobardi, M.; De Flora, S., Genomic and transcriptional alterations in mouse fetus liver after transplacental exposure to cigarette smoke. The FASEB journal 2003, 17, (9), 1127-1129. [CrossRef] [PubMed]
- Mukhopadhyay, P.; Greene, R. M.; Pisano, M. M., Cigarette smoke induces proteasomal-mediated degradation of DNA methyltransferases and methyl CpG-/CpG domain-binding proteins in embryonic orofacial cells. Reproductive Toxicology 2015, 58, 140-148. [CrossRef] [PubMed]
- Xiao, D.; Huang, X.; Yang, S.; Zhang, L., Direct effects of nicotine on contractility of the uterine artery in pregnancy. Journal of Pharmacology and Experimental Therapeutics 2007, 322, (1), 180-185. [CrossRef] [PubMed]
- Carroquino, M. J.; Posada, M.; Landrigan, P. J., Environmental Toxicology: Children at Risk. Environmental Toxicology. 2012 Dec 4:239-91. [CrossRef]
- Ni, W.; Yang, W.; Yu, J.; Li, Z.; Jin, L.; Liu, J.; Zhang, Y.; Wang, L.; Ren, A., Umbilical cord concentrations of selected heavy metals and risk for orofacial clefts. Environmental science & technology 2018, 52, (18), 10787-10795.
Figure 1.
Developmental progression of secondary palate formation in mouse embryos from E11.5 to E15.5. (A-E) Frontal views show the developmental sequence of palatal shelf elevation and fusion. At E11.5 (A), the medial nasal process (MNP) and maxillary processes (MxP) are visible with the initial formation of the primary palate (PP). From E13.5 to E15.5 (B-E), the palatal shelves (PS) undergo vertical-to-horizontal elevation, with concurrent development of the nasal septum (NS). Progressive fusion of the PS occurs, resulting in the formation of the secondary palate (SP) by E15.5. (F-T) Frontal sections through the developing palate's anterior, middle, and posterior regions at corresponding developmental stages. The sections demonstrate the progressive growth, elevation, and fusion of the PS around the tongue (T). The medial edge epithelium (MEE) is visible in anterior sections at early stages. The dynamic process of palatal shelf reorientation and fusion proceeds in an anterior-to-posterior sequence, with complete fusion achieved by E15.5. Color code: Pink - Medial nasal process (MNP); Turquoise - Maxillary process (MxP) (E11.5) & Maxillary tissue (E13.5-E15.5) & lip; Yellow - Primary palate (PP); Light blue - Nasal septum (NS); Light pink - Palatal shelves (PS); black arrowhead indicate the gap between the primary and secondary palates, which will close following fusion between these tissues.
Figure 1.
Developmental progression of secondary palate formation in mouse embryos from E11.5 to E15.5. (A-E) Frontal views show the developmental sequence of palatal shelf elevation and fusion. At E11.5 (A), the medial nasal process (MNP) and maxillary processes (MxP) are visible with the initial formation of the primary palate (PP). From E13.5 to E15.5 (B-E), the palatal shelves (PS) undergo vertical-to-horizontal elevation, with concurrent development of the nasal septum (NS). Progressive fusion of the PS occurs, resulting in the formation of the secondary palate (SP) by E15.5. (F-T) Frontal sections through the developing palate's anterior, middle, and posterior regions at corresponding developmental stages. The sections demonstrate the progressive growth, elevation, and fusion of the PS around the tongue (T). The medial edge epithelium (MEE) is visible in anterior sections at early stages. The dynamic process of palatal shelf reorientation and fusion proceeds in an anterior-to-posterior sequence, with complete fusion achieved by E15.5. Color code: Pink - Medial nasal process (MNP); Turquoise - Maxillary process (MxP) (E11.5) & Maxillary tissue (E13.5-E15.5) & lip; Yellow - Primary palate (PP); Light blue - Nasal septum (NS); Light pink - Palatal shelves (PS); black arrowhead indicate the gap between the primary and secondary palates, which will close following fusion between these tissues.
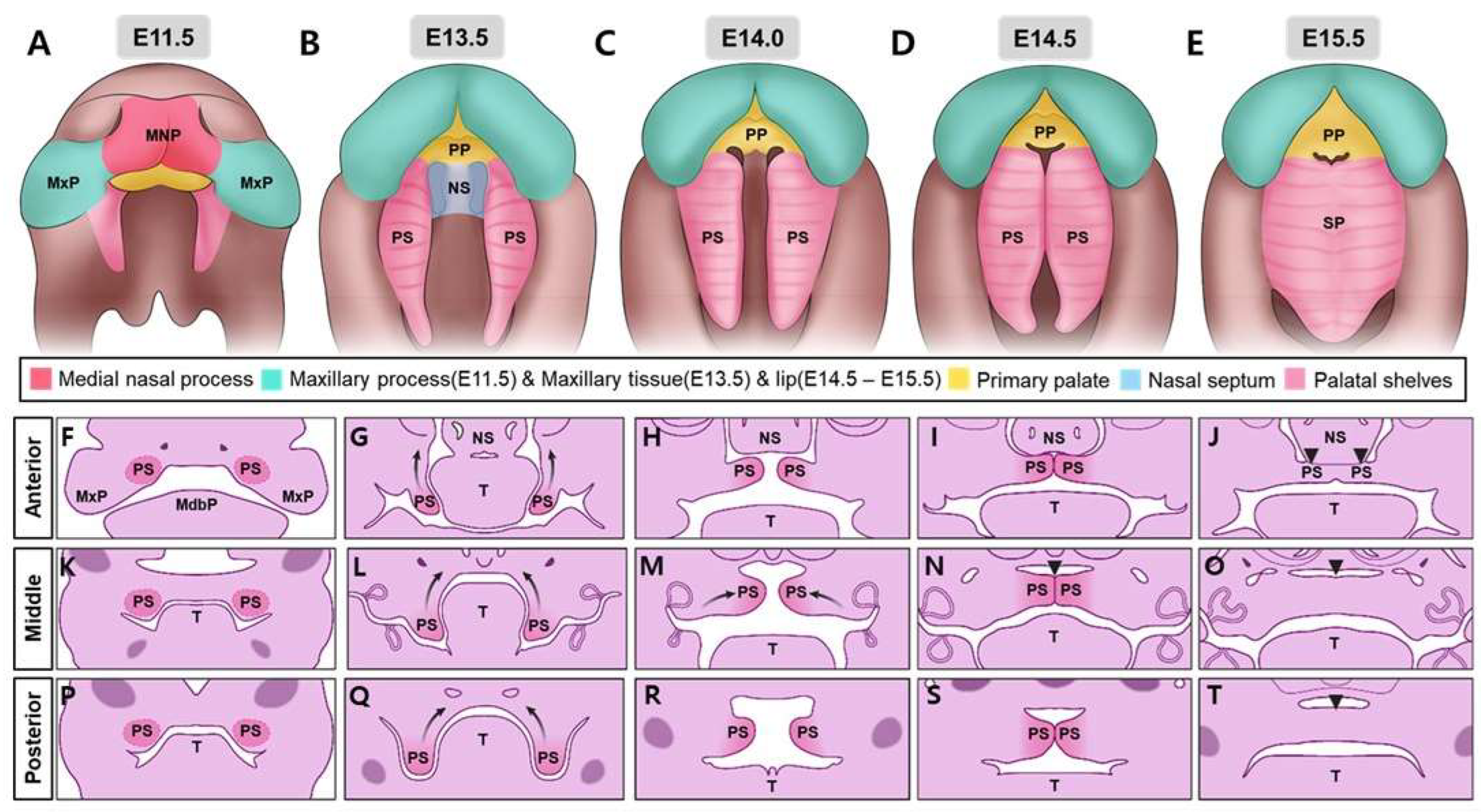
Figure 2.
The anatomical spectrum of normal and cleft lip and/or palate malformations in humans. (A-C) Frontal facial views show variations in lip formation. (A) Normal lip morphology with complete fusion. (B) The unilateral cleft lip is showing incomplete fusion on one side. (C) Bilateral cleft lip presenting incomplete fusion on both sides of the upper lip. (D-F) Oral views of the palatal region depicting normal and cleft phenotypes. (D) Normal palate showing complete fusion of palatal shelves. (E) Unilateral cleft lip and palate with the incomplete fusion of the palatal shelf on one side. (F) Bilateral cleft lip and palate showing incomplete fusion of palatal shelves on both sides. Color code: Turquoise - Maxillary tissue; Yellow - Primary palate; Pink - Palatal and soft tissues.
Figure 2.
The anatomical spectrum of normal and cleft lip and/or palate malformations in humans. (A-C) Frontal facial views show variations in lip formation. (A) Normal lip morphology with complete fusion. (B) The unilateral cleft lip is showing incomplete fusion on one side. (C) Bilateral cleft lip presenting incomplete fusion on both sides of the upper lip. (D-F) Oral views of the palatal region depicting normal and cleft phenotypes. (D) Normal palate showing complete fusion of palatal shelves. (E) Unilateral cleft lip and palate with the incomplete fusion of the palatal shelf on one side. (F) Bilateral cleft lip and palate showing incomplete fusion of palatal shelves on both sides. Color code: Turquoise - Maxillary tissue; Yellow - Primary palate; Pink - Palatal and soft tissues.
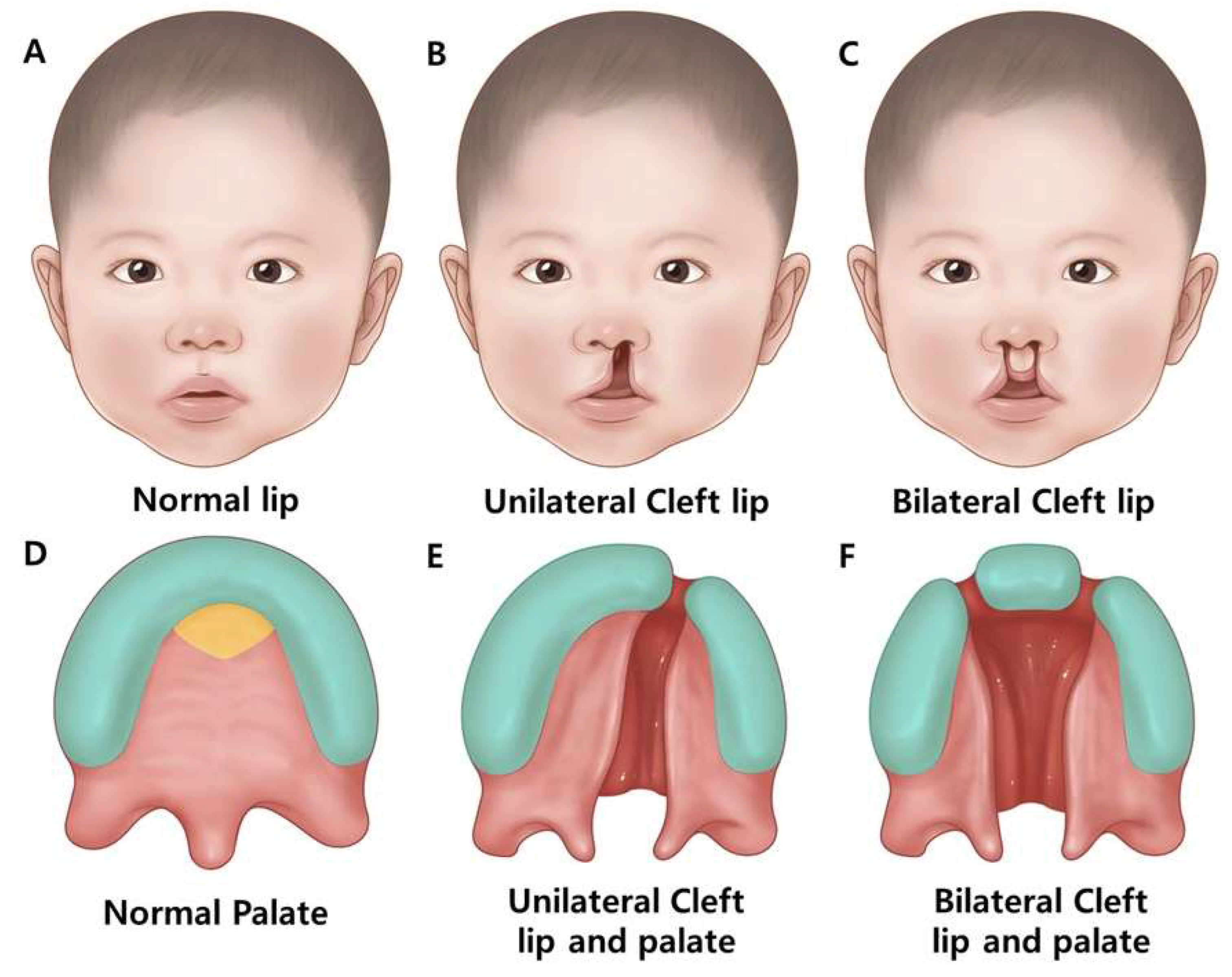
Figure 3.
Complex molecular networks that coordinate palatal shelf growth, patterning, and morphogenesis along both the anterior-posterior and medial-lateral axes during palatogenesis. (A-D) Lateral and oral views of developing mouse embryos at E12.5 and E13.5 with corresponding schematic diagram The anterior and posterior orientation is indicated by dashed lines. (E-G) Schematic representations of molecular networks controlling palate development: (E) Key factors involved in palate growth and patterning along the anterior-posterior axis, showing interactions between epithelial and mesenchymal factors and their downstream targets. (F) Molecular regulation of anterior palatal shelf development. (G) Posterior palatal shelf patterning network showing interactions. Color code: Pink – Palatal Epithelium; Apricot - Mesenchyme; Blue - Transcription factors; Green - Receptors; Yellow - Ligands; Orange - Other regulatory molecules.
Figure 3.
Complex molecular networks that coordinate palatal shelf growth, patterning, and morphogenesis along both the anterior-posterior and medial-lateral axes during palatogenesis. (A-D) Lateral and oral views of developing mouse embryos at E12.5 and E13.5 with corresponding schematic diagram The anterior and posterior orientation is indicated by dashed lines. (E-G) Schematic representations of molecular networks controlling palate development: (E) Key factors involved in palate growth and patterning along the anterior-posterior axis, showing interactions between epithelial and mesenchymal factors and their downstream targets. (F) Molecular regulation of anterior palatal shelf development. (G) Posterior palatal shelf patterning network showing interactions. Color code: Pink – Palatal Epithelium; Apricot - Mesenchyme; Blue - Transcription factors; Green - Receptors; Yellow - Ligands; Orange - Other regulatory molecules.
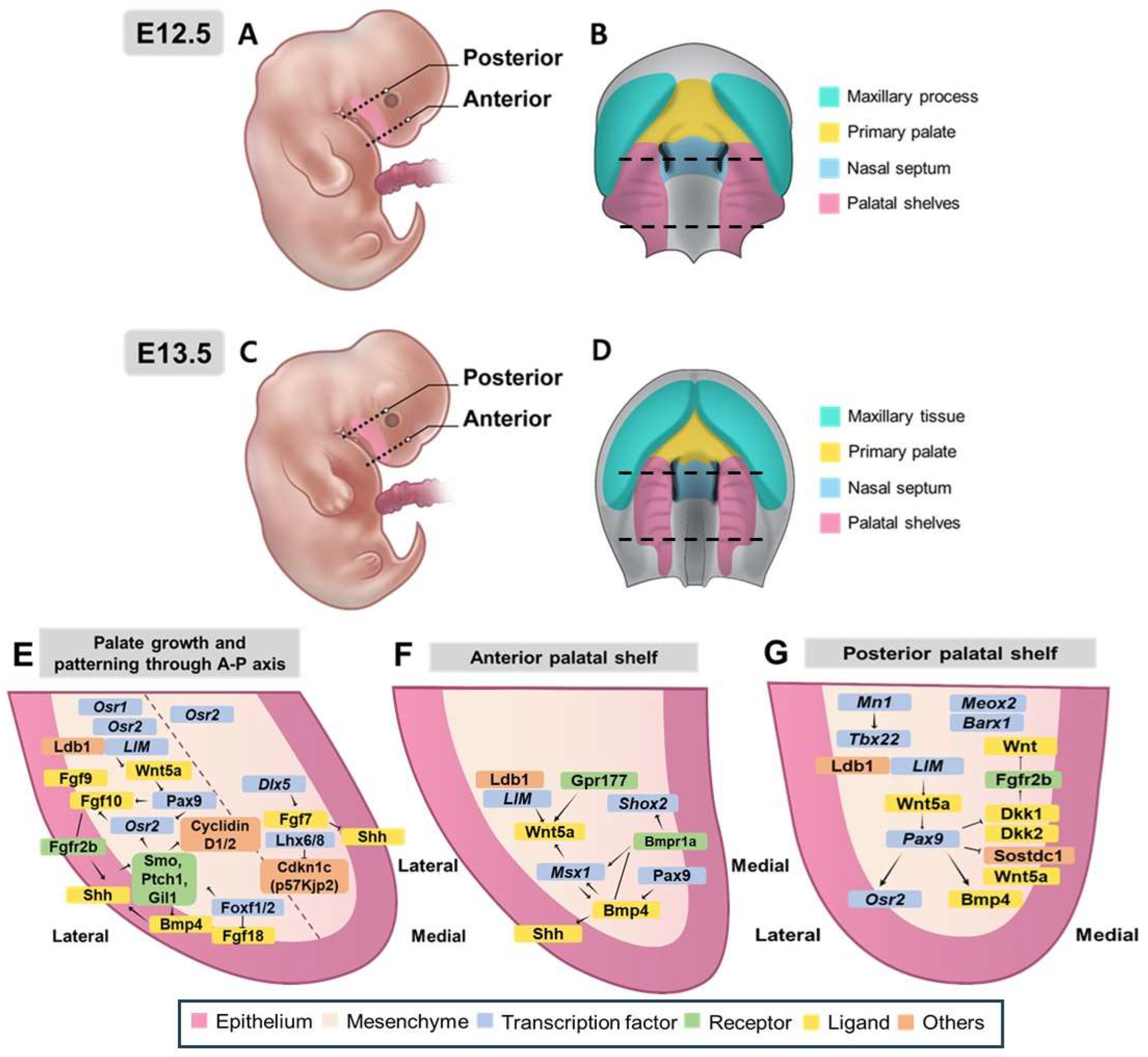
Figure 4.
Development and molecular regulation of palatal shelf adhesion and fusion. (A-C) Schematic representation of mouse embryo development from E14.5 to E15.5. (A) Lateral view of E12.5 mouse embryo showing the anterior-posterior axis of palatal development. (B) Oral view at E14.5 showing elevated palatal shelves before fusion. (C) Oral view at E15.5 showing palatal fusing. (D-F) Molecular pathways controlling three key stages of palatal fusion. (D) Epithelial differentiation and periderm maintenance pathway showing genetic interactions. (E) Palatal adhesion and medial edge epithelium (MEE) formation pathway involving β-catenin, Tgf-β3, and downstream effectors. (F) Midline epithelial seam (MES) degeneration process leading to palatal fusion. Color code: Pink - Epithelium; Apricot - Mesenchyme; Blue - Transcription factors; Green - Receptors; Yellow - Ligands; Orange - Other regulatory molecules.
Figure 4.
Development and molecular regulation of palatal shelf adhesion and fusion. (A-C) Schematic representation of mouse embryo development from E14.5 to E15.5. (A) Lateral view of E12.5 mouse embryo showing the anterior-posterior axis of palatal development. (B) Oral view at E14.5 showing elevated palatal shelves before fusion. (C) Oral view at E15.5 showing palatal fusing. (D-F) Molecular pathways controlling three key stages of palatal fusion. (D) Epithelial differentiation and periderm maintenance pathway showing genetic interactions. (E) Palatal adhesion and medial edge epithelium (MEE) formation pathway involving β-catenin, Tgf-β3, and downstream effectors. (F) Midline epithelial seam (MES) degeneration process leading to palatal fusion. Color code: Pink - Epithelium; Apricot - Mesenchyme; Blue - Transcription factors; Green - Receptors; Yellow - Ligands; Orange - Other regulatory molecules.
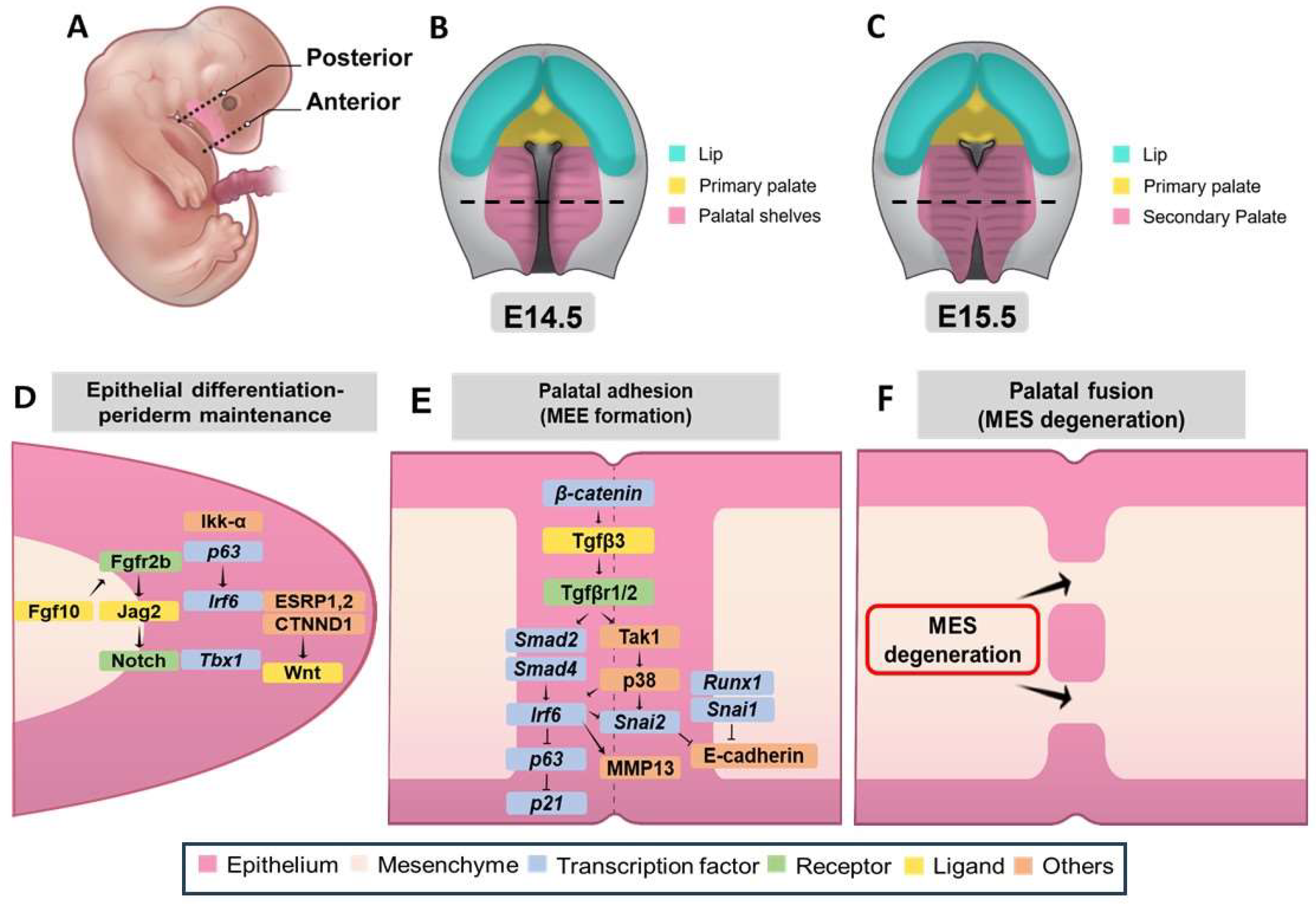
Figure 5.
Epigenetic regulation during craniofacial development. (Top, A-B) Schematic representation of early craniofacial development showing the morphological changes from initial facial prominences to their fusion. (A) The left panel shows the initial facial prominences, including midbrain, forebrain, lateral and medial nasal processes, nasal pit, maxillary and mandibular processes, and second arch. (B) The right panel shows the subsequent development of the frontonasal region, maxillary region, and the mandibular region. (Bottom, C) Four major epigenetic mechanisms regulating craniofacial development: DNA Methylation: Addition of methyl groups to promoter regions controlling target gene expression. Histone Modification: Post-translational modifications, including methylation (Me) and acetylation (Ac) of histone proteins. Non-coding RNAs: Involvement of microRNAs (miRNA) and long non-coding RNAs (lncRNA) in gene regulation. Chromatin Remodeling: ATP-dependent nucleosome ejection and sliding mediated by SWI/SNF complexes. Color code: Dark blue - Midbrain; Light blue - Forebrain; Green - Lateral nasal process; Red - Medial nasal process; Navy blue - Nasal pit; Turquoise - Maxillary process; Purple - Mandibular process and second arch; Orange - Frontonasal region; Grey – Other facial region behind maxillary/mandibular regions.
Figure 5.
Epigenetic regulation during craniofacial development. (Top, A-B) Schematic representation of early craniofacial development showing the morphological changes from initial facial prominences to their fusion. (A) The left panel shows the initial facial prominences, including midbrain, forebrain, lateral and medial nasal processes, nasal pit, maxillary and mandibular processes, and second arch. (B) The right panel shows the subsequent development of the frontonasal region, maxillary region, and the mandibular region. (Bottom, C) Four major epigenetic mechanisms regulating craniofacial development: DNA Methylation: Addition of methyl groups to promoter regions controlling target gene expression. Histone Modification: Post-translational modifications, including methylation (Me) and acetylation (Ac) of histone proteins. Non-coding RNAs: Involvement of microRNAs (miRNA) and long non-coding RNAs (lncRNA) in gene regulation. Chromatin Remodeling: ATP-dependent nucleosome ejection and sliding mediated by SWI/SNF complexes. Color code: Dark blue - Midbrain; Light blue - Forebrain; Green - Lateral nasal process; Red - Medial nasal process; Navy blue - Nasal pit; Turquoise - Maxillary process; Purple - Mandibular process and second arch; Orange - Frontonasal region; Grey – Other facial region behind maxillary/mandibular regions.
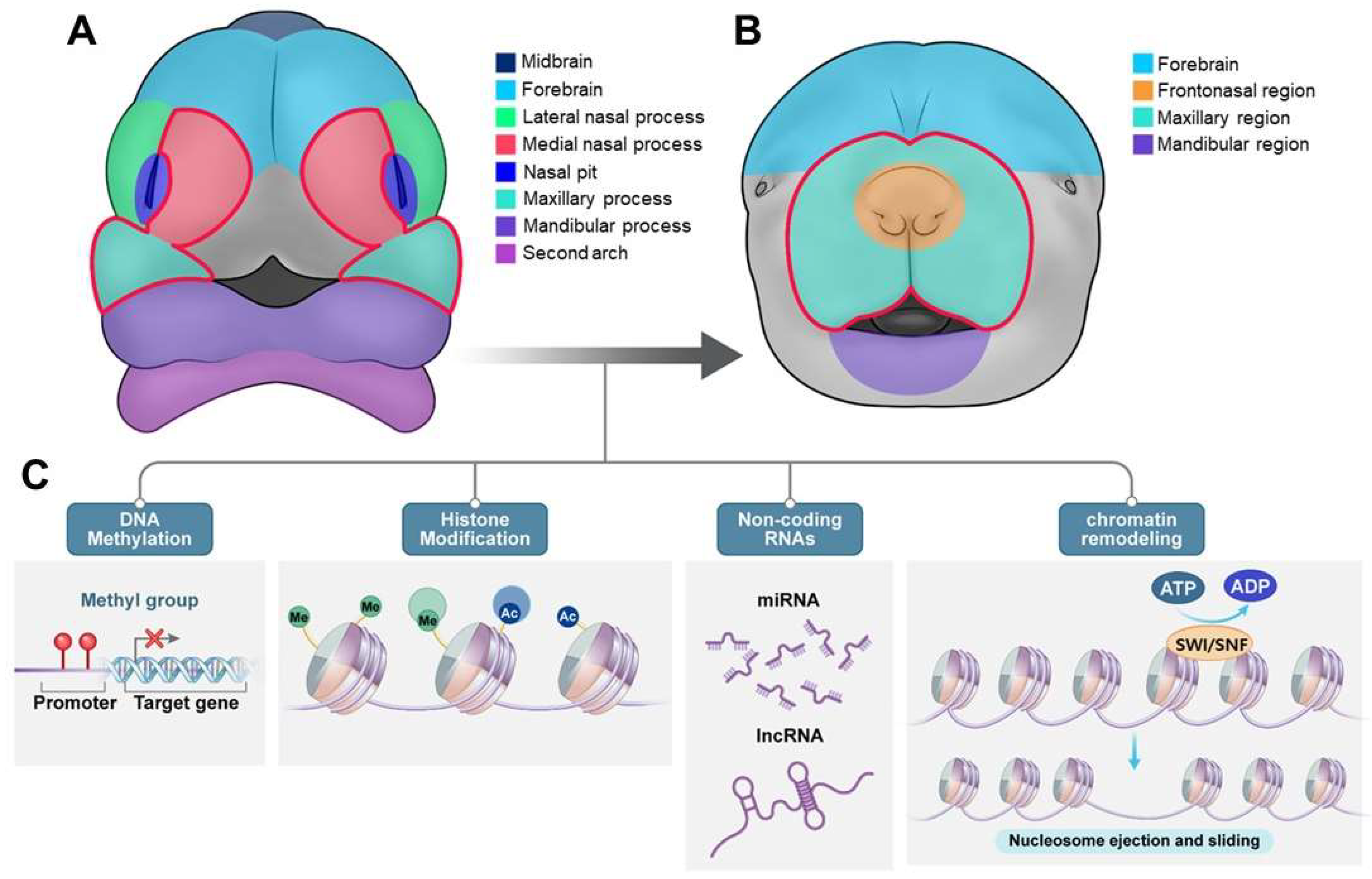
Figure 6.
Summary of the miRNAs and genes associated with cleft lip and/or palate(CL/P) in mice and human. (A-B) The complex miRNA-mediated regulatory networks involved in cleft lip and/or palate(CL/P). Environmental factors also affect cleft palate development by modulating miRNA activity. and how their dysregulation contributes to cleft formation through cell proliferation defects, differentiation defects, and cell death. CL, cleft lip; CP, cleft palate.
Figure 6.
Summary of the miRNAs and genes associated with cleft lip and/or palate(CL/P) in mice and human. (A-B) The complex miRNA-mediated regulatory networks involved in cleft lip and/or palate(CL/P). Environmental factors also affect cleft palate development by modulating miRNA activity. and how their dysregulation contributes to cleft formation through cell proliferation defects, differentiation defects, and cell death. CL, cleft lip; CP, cleft palate.
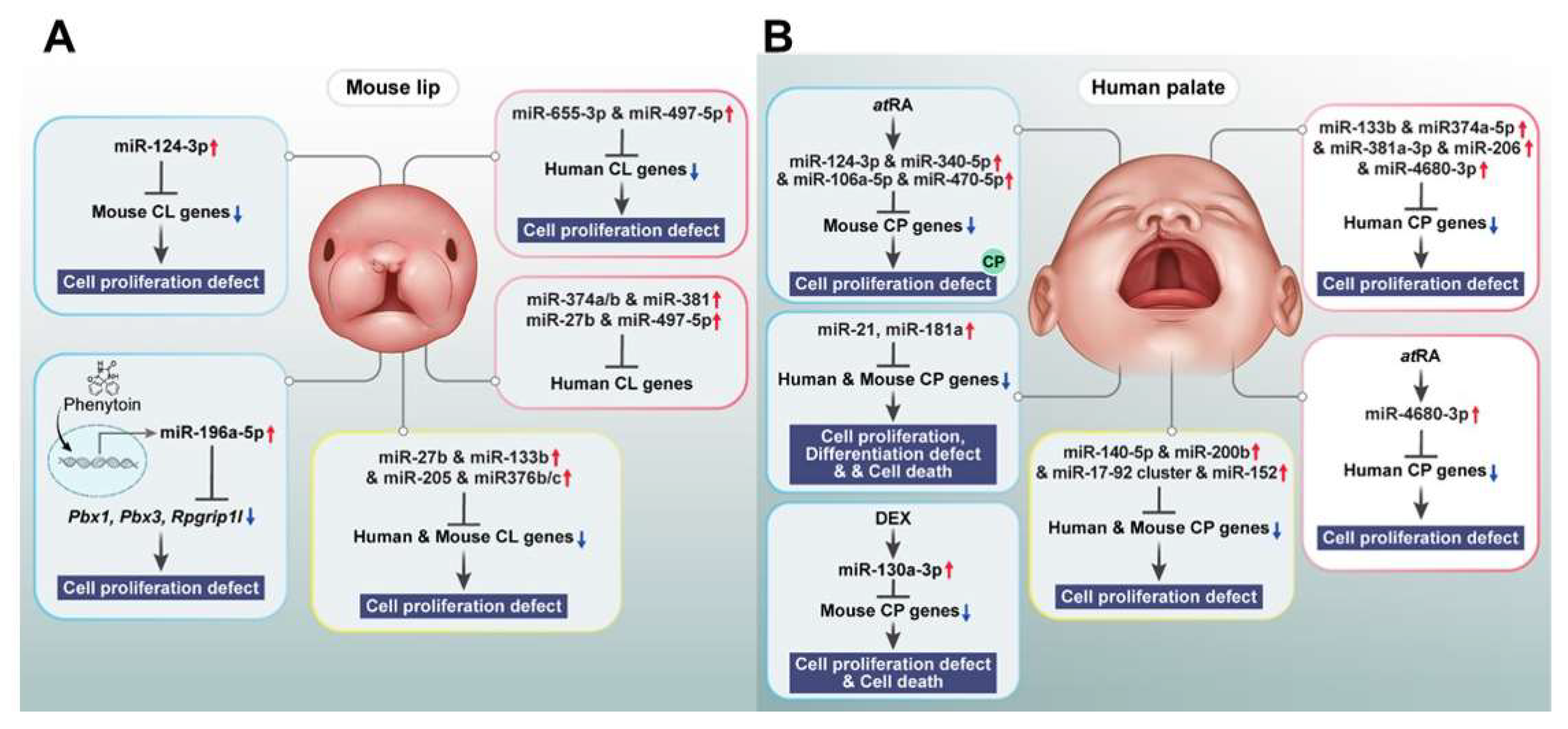

Table 1.
Comprehensive genetic information of non-syndromic orofacial clefts: Prevalence, Subtypes, and Associated Factors.
Table 1.
Comprehensive genetic information of non-syndromic orofacial clefts: Prevalence, Subtypes, and Associated Factors.
| Feature | Description | Associated Genes/Regions/Pathways | Reference |
| Prevalence | ~70% of all clefts; ~1 in 700 live births globally | N/A | [70] |
| Subtypes | 45% cleft palate alone; 85% nonsyndromic cleft lip with or without palate |
N/A | [105] |
| Genetic Factors | SNPs, single gene mutations, microRNA patterns | WNT pathway (AXIN1, WNT9B), Fgf10/Fgfr2/Shh pathway (FGFR1, FGF2), COL11A1, IRF6, EGF, MSX1, PTCH, TGFB1, ROR2, FOXE1, TGFB3, RARA, APOC2, BCL3, PVRL2 | [105], [182] |
| Chromosomal Regions | Linkage to >20 regions | chr 1p, 1q21, 1q32-42.3, 6p, 2p, 4q, 17q | [105] |
| Epigenetic Factors | DNA methylation | EWAS studies identify differentially methylated regions | [102] |
| Rare Variants | Mutations in specific genes | ABCB1, ALKBH8, CENPF, CSAD, EXPH5, PDZD8, SLC16A9, TTC28 (ABCB1, TTC28, and PDZD8 show significant mutation constraint) | [183] |
Table 2.
Genetic Syndromes Associated with Orofacial Clefts and Their Key Features.
| Syndrome |
Key Features (Including OFCs) |
Associated Genes /Chromosomal Regions |
Reference |
| DiGeorge Syndrome (DGS) | Cleft palate (most frequent), cardiac defects, immune deficiency, characteristic facial features | TBX1 (within the 22q11.2 deletion), miR-96-5p | [139], [184], [185] |
| Van der Woude Syndrome (VWS) | Cleft lip, cleft palate, hypodontia, paramedian lower lip pits | IRF6 (most common), GRHL3, CDH2 (SNP rs539075), NOL4 | [113], [139], [186] |
| Stickler Syndrome (STL) | Cleft palate/uvula, myopia, retinal detachment, joint problems, hearing loss | COL2A1 (STL1), COL11A1 (STL2), COL11A2 (STL3), COL9A1, COL9A2, COL9A3, LRP2, LOXL3 | [187], [188], [189] |
| Pierre-Robin Sequence (PRS) | Micrognathia, glossoptosis (posterior displacement of the tongue), cleft palate, airway obstruction | SOX9, BMPR1B, deletions on 2q and 4p, duplications on 3p, 3q, 7q, 8q, 10p, 14q, 16p, and 22q | [113], [190], [191], [192] |
| Kabuki Syndrome | Distinct facial features (midfacial hypoplasia, broad nasal tip, elongated palpebral fissures, large ears), cleft palate/high-arched palate, growth retardation, intellectual disability, congenital heart defects | KMT2D (most common), KDM6A |
[193], [194], [195] |
| Wolf-Hirschhorn Syndrome (WHS) | Intellectual disability, growth delays, heart and skeletal defects, seizures, cleft palate, facial asymmetry | WHSC1, WHSC2, LETM1, TACC3 | [113,196,197] |
| CHARGE Syndrome | Coloboma, Heart defects, Atresia of the choanae, Retarded growth/development, Genital abnormalities, Ear abnormalities/hearing loss, cleft palate | CHD7 | [113,178] |
| Apert Syndrome (AS) | Craniosynostosis, midface hypoplasia, cleft palate (more commonly soft palate), syndactyly of hands and feet | FGFR2 (p.Ser252Trp, p.Pro253Arg) |
[198,199,200,201,202,203] |
| Tatton-Brown-Rahman Syndrome (TBRS) | Overgrowth, macrocephaly, facial dysmorphism, intellectual disability, autism | DNMT3A | [204,205] |
| Arboleda-Tham Syndrome (ARTHS) | Intellectual disability, developmental/speech delays, hypotonia, congenital heart defects | KAT6A | [210,211,212,213] |
Table 3.
Regulatory Roles of microRNAs(miRNAs) and Long Non-Coding RNAs(lncRNAs) in Orofacial Development.
Table 3.
Regulatory Roles of microRNAs(miRNAs) and Long Non-Coding RNAs(lncRNAs) in Orofacial Development.
| Molecule Type | Specific Molecule | Target /Function | Effect on Orofacial Cleft Development | Reference |
| miRNA | miR-21, miR-181a | Sprouty2 (MAPK/ERK pathway) | cell proliferation, differentiation, and survival of neural crest cells | [177] |
| miR-452 | Wnt5a | EMT and neural crest cells patterning | [178], [139] | |
| miR-149 | hNCC migration | neural crest cells | [179] | |
| miR-133b, miR-374a-5p, miR-4680-3p | GCH1, PAX7, FGFR2, ERBB2 | cell proliferation | [15], [180] | |
| miR-497-5p | mTOR | cell proliferation | [181] | |
| miR-655-3p | TGF-β, Wnt | cell proliferation | [181] | |
| miR-124-3p | Bmpr1a, Cdc42, Tgfbr1 | proliferation in embryonic lip mesenchymal cells | [182], [183] | |
| miR-27b | PAX9, RARA | cell proliferation of lip mesenchymal cells | [185] | |
| miR-133b | FGFR1, PAX7, SUMO1 | cell proliferation of lip mesenchymal cells | [186], [8] | |
| miR-205 | PAX9, RARA | cell proliferation of lip mesenchymal cells | [189] | |
| hsa-let-7c-5p,hsa-miR-193a-3p | HEPM cells | cell proliferation | [190], [191], [15] | |
| miR-17, miR-18a, miR-19a, miR-19b-1, miR-20a, miR-92a-1 (mir-17-92 cluster) | Tbx1, Tbx3, Fgf10, TGFBR2, SMAD2, SMAD4 | midfacial development | [192], [15] | |
| miR-22-3p | Myh9, Myh10 | MES dissolution and palatal fusion | [193], [184] | |
| miR-200b | Smad2, Snail, Zeb1, Zeb2 (mediators of TGFβ signaling) | MES | [198],[15] | |
| miR-206 | TGFβ, Wnt/β-catenin | palatal fusion | [198], [15] | |
| miR-140 | SNP rs7205289, TGF-β | cell migration | [196] | |
| miR-744-5p | lncRNA RP11-462G12.2 (C-allele) | cell apoptosis, proliferation | [199] | |
| lncRNA | RP11-462G12.2(C-allele) | miR-744-5p, IQSEC2 | C-allele binds miR-744-5p | [202], [196] |
| NONMMUT100923.1 | miR-200a-3p | medial edge epithelial cell adhesion | [196], [195] | |
| NONMMUT004850.2/NONMMUT024276.2 | miR-741-3p,miR-465b-5p | palatal fusion | [199] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



