
Submitted:
08 January 2025
Posted:
09 January 2025
You are already at the latest version
Abstract
Solid organ transplantation remains a life-saving treatment for patients worldwide. Unfortunately, the supply of donor organs cannot meet the current need, making the search for alternative sources even more essential. Xenotransplantation using sophisticated genetic engineering techniques to delete and overexpress specific genes in the donor animal has been investigated as a possible option. However, the use of exogenous tissue presents another host of obstacles, particularly regarding organ rejection. Given these limitations, interspecies blastocyst complementation in combination with precise gene knockouts presents a unique, promising pathway for the transplant organ shortage. In recent years, great advancements have been made in the field, with encouraging results in producing a donor-derived organ in a chimeric host. That said, one of the major barriers to successful interspecies chimerism is the mismatch in the developmental stages of the donor and the host cells in the chimeric embryo. Another major barrier to successful chimerism is the mismatch in developmental speeds between the donor and host cells in the chimeric embryos. This review outlines 22 studies in which blastocyst complementation was used to generate solid organs. In particular, the genesis of liver, lung, kidney, pancreas, heart, thyroid, thymus and parathyroids was investigated. Of the 22 studies, 7 included an interspecies model. Of the 7, one was done using human donor cells in a pig host, and all others were rat-mouse chimeras. While very promising results have been demonstrated, with great advancements in the field, several challenges continue to persist. In particular, successful chimerism, organ generation and donor contribution, synchronized donor-host development, as well as ethical concerns regarding human-animal chimeras remain important aspects that will need to be addressed in future research.
Keywords:
blastocyst complementation
; chimerism
; genetic engineering
; intra-/interspecies
; pluripotent stem cells
; porcine
; transplantation
; xenotransplantation
1. Introduction
In the United States alone, the total number of solid organ transplants has increased from 83 transplants per million to almost 140 transplants per million since 2000 [1]. Unfortunately, despite increasing deceased organ donors, it is estimated that 17 people still die each day while on the organ transplant waiting list [1,2]. The supply of living and deceased organ donors cannot cover the current requirements, making the search for alternatives even more important.
One such alternative involves the production of exogenic organs. Historically, this has been attempted with xenotransplantation of solid organs. A more recent example of this was in 2022, whereby a gene-edited porcine heart was transplanted in a patient with severe heart failure and allowed for the longest recorded survival of 7 weeks following xenotransplantation [3]. Further advancements have opened the pathway to blastocyst complementation, an important technique that would allow for interspecies tissue and organ production.
The first requirement for successful blastocyst complementation involves genetic modification of a host blastocyst. The host must undergo knockout or gene modification that leads to a developmental defect of the organ of choice and thereby opens a niche for wild-type stem cells to occupy and develop into the desired organ. Techniques used to achieve this often include CRISPR/Cas9 or TALEN, as they have shown most success in targeted genomic modification. Donor embryonic stem cells (ESC) or induced pluripotent stem cells (iPSC) are then introduced and injected into the host blastocyst. During subsequent embryonic in vivo organ development, the organ of choice is primarily made up of donor cells, which can then be transplanted back into the donor (See Figure 1) [4]. First attempts at this were done in 1993, whereby embryonic stem cells were injected into RAG2 deficient mice to generate chimeric B and T cells [5]. Subsequent studies have shown that interspecies chimerism could be possible. For example, Xiang et al used two distant rodent species to demonstrate that injection of donor embryonic stem cells into a host resulted in most organs containing donor-derived cells, with some tissues showing up to 40% contribution [6]. This innovative technique offers a promising pathway to help with the continuing transplant organ shortage. As such, this review aims to discuss the advances made in blastocyst complementation for solid organs. A summary of results from all 22 studies can be found under Table 1.
Xenotransplantation. The host (pig) is modified to reduce risk of rejection. This includes knockout of growth hormone (to reduce intrinsic host growth) and xenoantigens, in addition to inactivation of porcine endogenous retroviruses (PERVs). Human transgenes are also expressed to improve compatibility. The new organ is mostly made up of host cells, then transplanted into the human.
Blastocyst Complementation. The host, pig embryo for example, is first genetically modified, often using TALEN or CRISPR for organ agenesis to produce an organ niche. Donor human pluripotent stem cells are then introduced in the host blastocyst, allowing for development of a chimeric host with an organ of choice. The organ, which is mostly made up of donor/human cells, is then re-transplanted into the donor.
2. Liver
Per the CDC, the number of deaths due to chronic liver disease surpassed 50,000 in 2022 [7]. For patients with chronic liver disease or liver failure, orthotopic liver transplantation remains a critical, life-saving form of treatment. In 2022, over 10,000 patients required liver transplantation in the United States, making it the second most frequent solid organ transplant [1]. The need for viable transplants therefore remains crucial for the survival of many patients. The potential of iPSCs in post-natal hosts has been evaluated. Several studies have used human iPSCs, which were induced to differentiate into functional hepatocyte-like cells. These cells were then successfully transplanted into injured mice livers, or livers in hepatic failure (usually induced by CCl4). Results from these studies showed success in improving survival or function (measured via increased albumin, or decreased bilirubin and LDH) [8,9,10]. Successes in this area, and subsequent novel uses of human embryonic stem cells or iPSCs have shown that human organ development in a non-human host could be achieved via blastocyst complementation.
2.1. Hepatogenesis and Elimination of Hepatic Development
The liver is a complex organ. While certain resident cells, such as Kupffer, Ito or stromal cells are of mesodermal origin, the liver’s primary cells, i.e., hepatocytes and cholangiocytes, stem from the endoderm. From the endoderm, the foregut, midgut and hindgut are established. The ventral foregut is what eventually develops into the liver bud. Development from the liver bud into hepatoblasts requires inductive signaling from the neighboring cardiac mesoderm [11]. Final differentiation is in part determined by the localization of the hepatoblasts. Differentiation into cholangiocytes occurs in cells next to portal veins (the caudal section of the liver bud), while cells located in the parenchyma give rise to hepatocytes (the cranial section of the liver bud) [12]. Naturally, there are several transcription regulators that have been found to be crucial for the successful development of the liver, including FoxA1-3, GATA4/6, HNF1 alpha/beta, HNF4alpha, HNF6, OC-2, C/EBPalpha/beta, Hex, Prox1 [13,14]. Of those, Hhex, in particular, has shown great promise as a target for preventing hepatogenesis. Hhex is a homebox gene and is critical for liver differentiation and hepatobiliary development, as its absence prevents successful migration to the pseudostratified epithelium, and subsequent hepatoblast differentiation [15].
Hhex mutations have been shown to affect development of several systems, including the hematopoietic system, vascular system, forebrain and thyroid [16,17,18]. As such, different targets have been investigated, including FAH. Deficiency in fumarylacetoacetate hydrolase (FAH) has shown to lead to apoptosis and mutagenesis in hepatocytes due to accumulation in the toxic metabolic fumaryl-acetoacetate, particularly in hepatocytes [19]. FAH deficient mice have also been shown to successfully be repopulated with hepatocytes when injected with human hepatocytes [20].
2.2. Application of Blastocyst Complementation
While iPSCs have shown promising potential for several applications, including regenerative medicine, disease modeling and gene therapy for inherited liver diseases, blastocyst complementation for organ development has also shown great promise [14]. Matsunari et al demonstrated two key elements. Firstly, they showed that bi-allelic H-hex-knockout using TALEN in pigs successfully led to severe liver dysplasia and developmental retardation. Initial blastocyst complementation produced 37 blastocysts; of those, only 4 were chimeric fetuses. While 3 of the chimeric fetuses continued to demonstrate high developmental retardation, one of the chimeric fetuses showed organogenesis similar to that of wild-type fetuses. Further work with 95 chimeric, blastocyst complemented fetuses resulted in 3 alive fetuses with normal liver development at the time of cesarean section [21].
Ruiz-Estevez et al similarly produced H-hex knockouts in pigs and mice using CRISPR/Cas9, with absence in hepatogenesis in both species [22]. Knockout of H-HEX was embryonically lethal around E10.5 for both species. Intra-species blastocyst complementation was then performed, with eGFP-labeled donor-derived cells. Their study showed a very high contribution of donor-derived cells (evidenced by positive eGFP signal) in several tissues, including the liver. In mice, 22/32 embryos demonstrated eGFP positive signal, with 50% surviving past E10.5. In pigs only 2 embryos were recovered of the 46 and 48 complemented embryos. However, both these embryos demonstrated strong eGFP signal, with visible liver tissue, and entirely H-HEX wildtype sequences in the liver.
Most recently, Simpson et al demonstrated conditional H-hex knockout in pigs [23]. As discussed previously, knockout of H-hex has widespread consequences outside of just liver development in the body, such as arrested forebrain development and changes in endothelial cell differentiation and lymphatic vessel formation. As such, a more targeted knockout would be of interest. In this case, by using a conditional H-hex knockout under FOXA3 promoter, the fetuses showed lack of hepatogenesis, as well as absent mesonephros and developmental retardation. During the 2 rounds of blastocyst complementation, 2 healthy fetuses were rescued on day 28 from 120 chimeric blastocysts. Immunohistochemistry confirmed all hepatocytes were donor-derived, with enhanced GFP-labelled cells.
The above-mentioned examples are very recent examples of successful blastocyst complementation for liver development. While rates of chimerism and viable chimeras continue to pose a significant obstacle, advancements in this field, particularly using larger animals such as pigs, hold great promise for future interspecies blastocyst complementation.
3. Lung
Lung disease remains a significant cause of mortality and morbidity. The number of lung transplantations has progressively increased over past decades, with over 3,000 transplants being done in the US alone in 2023 [1]. Main causes for transplantation continue to be COPD, cystic fibrosis and idiopathic pulmonary fibrosis, with the last one showing an increase for transplantation in North America [24]. Given the increasing demand for transplants, finding sources for lung tissue remains crucial.
3.1. Lung Development and Its Elimination
Lung development is a highly complex process with several steps resulting in significant cellular diversity. Initial development of the respiratory system starts from the primitive foregut endoderm. Initial elongation and bifurcation results in two primary bronchial buds that will eventually form the right and left lung [25]. As the trachea develops and separates from the esophagus, mesenchymal cells surround and differentiate into cartilage precursors. The visceral and parietal pleura forms from the splanchnic and somatic mesodermal layer respectively. Functional lung tissue requires specific mesenchymal and epithelial cells, and complex interaction between the two as the epithelial components of endodermal origin descend and undergo repetitive branching and growth into neighboring mesenchyme [25]. Wnt/beta-catenin has been shown to be crucial for this step, as inactivation has led to deviation in epithelial branching and differentiation between distal and proximal lung, as well as decreased endothelial differentiation and mesenchymal growth [27,28]. Similarly, Nkx2-1 has been shown to be crucial for branching of the bronchial tree [26]. Further branching, vascularization and differentiation (including eventual alveolarization) occurs in the following weeks, allowing progressive formation of future airways [25].
Several key players have been identified in lung development. Fgf10, and its receptor major receptor Fgfr2b are one example. Specifically, Fgf10 has been shown to be essential for lung as well as fore-and hindlimb formation. Interestingly, in Fgf deficient mice, while lung development was disrupted, the trachea remained intact [29]. Furthermore, it has been shown to be significant for branching morphogenesis [31]. While NKx2-1 is essential for thyroid, forebrain and pituitary development, it has also been shown to be crucial in several steps of lung development [26]. Firstly, it plays an important role in distinguishing trachea and lung from the neighboring esophagus [31]. Furthermore, it is highly involved in lung morphogenesis, particularly distal branching, as well as differentiation of specific epithelial cells in the lung [26,32,33]. Additional important factors include Sox17, BMP and Foxa2. BMP has been shown to regulate proximal-distal differentiation [34], while Sox 17 influences pulmonary vascular morphogenesis and differentiation of respiratory epithelial cells [35,36]. Finally, Foxa2 has been shown to be pivotal for alveolarization and respiratory goblet cell expansion [37].
3.2. Application of Blastocyst Complementation
While the use of blastocyst complementation to generate lungs has remained a challenge for several years due to a lack of a definite gene target to prevent lung development, the past couple of years have shown great advancements in the area.
Mori et al used conditional blastocyst complementation in two different mouse models [39]. The first targeted fgfr2 gene deletion, resulting in the absence of lungs. Importantly, fgfr2 is activated by several ligands, including fgf10, and mice deficient in both have been shown to have similar lung phenotype and limb defects [29,38]. The second mouse model investigated Wnt/beta catenin (Ctnnb) knockout. Knockout of this pathway has been shown to lead to lung and trachea agenesis, compared to just lung agenesis with fgfr2 [28,29]. They used donor pluripotent stem cells engineered to express a fluorescent protein (GFP) marking. In both models, the chimeric mice (4 fgfr2-null mice, 3 Ctnnb1-null mice) showed normal lung development with pulmonary function tests demonstrating comparable results to their wild-type counterparts. As expected, the GFP reporter demonstrated strong signals in trachea and lung for the Ctnnb1-null model, while the fgfr2-null model only showed strong signals in the lung. Interestingly, in Fgfr2-null mice, while alveolar type 1 and 2, secretory, multiciliate cells showed significantly high GFP signals; and those in endothelial or mesenchymal cells were much more variable, with over half being of host origin.
Further work has also been done by Kitahara et al using Fgf-10 deficient mice, more specifically Fgf10 Ex1mut/Ex3mut heterozygous mutant mice [40]. As previously outlined, fgf-10 is crucial for lung development. Once again, the embryonic stem cells that were injected for blastocyst complementation were marked with GFP. Initial work on neonate mice was promising, and further work on mice that reached adulthood was done. A total of 153 neonates were obtained from 638 microinjected embryos, with only 16 live chimeras surviving to weaning. All 5 fgf10 Ex1mut/Ex3mut chimeric mice survived and were eventually sacrificed at 4 months. No significant histologically or morphological changes were observed in the adult fgf-10 Ex1mut/Ex3mut chimeras. Similarly to Mori et al, Kitahara et al showed strong GFP signals across the lung, especially in parenchymal cells. However, they also demonstrated strong reporter expression in interstitial regions, as well as vascular endothelial and smooth muscle cells. Overall, while a substantial number of cells were GFP positive, and therefore from donor cells, most cell types in the chimeric lungs showed a mixture of donor and host cells.
Miura et al further highlighted the potential of blastocyst complementation using the Foxa2-driven Fgfr2 pathway as a knockout target [41]. Their aim was to find a single lineage that would encompass lung epithelium and mesenchyme. They showed that using this pathway, instead of systemic Fgfr2 depletion, avoided agenesis in several other systems such as kidneys, limbs and more. Their initial work demonstrated that Foxa2 seemed to be involved in the majority of lung epithelium, and half of lung mesenchymal development. The lungs generated in the Foxa2-driven Fgfr2 knockout mice showed rescue of the lung phenotype, with almost the entirety of the lung epithelium and mesenchyme being composed of the GFP labelled iPSCs. Interestingly, knock-out of Fgfr2 in the Foxa2 lineage led to decreased proliferation of host cells compared to donor cells in mesenchyme, leading to increasing lung complementation during development. Follow up was done 4 weeks post birth, and the proportion of host-derived cells was significantly low in both epithelium, endothelium and mesenchyme. Pulmonary function showed no significant difference in tidal volume, airway resistance, frequency and expiratory flow at 50% expired tidal volume.
A different approach to blastocyst complementation has been examined, particularly via the Nkx2-1 pathway. Wen et al first demonstrated promising results in 2020 in rescuing lung and thyroid tissue in Nkx2-1 homozygous knockout mice [42]. GFP-labelled mouse embryonic stem cells were injected into blastocysts. Histological analysis showed that rescued chimeric lung lobes contained bronchioles, blood vessels and alveolar saccules. Electron microscopy also showed the presence of AT2 cells with surfactant secretion, and AT1 cells expressing T1alpha, both of which indicate successful differentiation during development. Furthermore, all respiratory epithelial cell subtypes showed similar gene expression, whether they were derived from embryonic stem cells or from endogenous cells. Importantly, most epithelial cells expressed GFP, while other cell types (endothelial, hematopoietic, fibroblasts and pericytes) showed more variable GFP signals. This demonstrated that high embryonic stem cell contribution remains a challenge in non-epithelial respiratory cells. Unfortunately, while lung and thyroid tissue could be rescued, tracheo-esophageal fusion was still present and complementation was inefficient in the forebrain. Furthermore, all Nkx2-1 homozygous knockout mice died at birth.
In 2024, Wen et al showed further progress by using interspecies blastocyst complementation [43]. As expected, they showed that Nkx2-1 homozygous knockout rats lacked lung and thyroid, with tracheo-esophageal fusion. Subsequent interspecies blastocyst complementation was performed using mice wild type embryonic stem cells, and analysis was done on chimeras aged E20.5. Similar to their previous study, the Nkx2-1 knockout mouse-rat chimeras showed that the mouse embryonic stem cell-derived cells expressed AT1 and AT2 markers (T1alpha and Pro-SPC, SPB, respectively). Furthermore, and in contrast to their previous work, in 30% of the Nkx2-1 chimeras, the mouse embryonic stem cells contributed highly to mesenchymal and vascular cells (including endothelial cells, smooth muscle cells, immune cells, pericytes and fibroblasts), with 98.5% of all cells in the new lung being mouse-derived. As before, however, the blastocyst complementation did not rescue the tracheo-esophageal phenotype. High mouse contribution was found in both the forebrain and the thyroid. Finally, it was noted that the mouse-rat chimeras had both smaller body and lung sizes.
4. Kidney
Since 2000, the number of kidney transplants has progressively increased, with over 26,000 being done in the U.S. alone. Despite the increased number of procedures, the number of new people on the kidney transplant waiting list has increased to over 44,000 in the U.S. Of those on the waiting list, approximately 12% have been waiting 5 years or longer. The most common primary causes for kidney failure of adults on the waiting list have continued to be diabetes and hypertension [44]. While dialysis continues to be an important alternative for many patients, the increased quality of life and increased freedom in patients’ lives cannot be ignored. As such, blastocyst complementation offers up a unique opportunity to meet the continued demand for transplants.
4.1. Nephrogenesis Elimination of Kidney Development
Kidney development undergoes several stages, with the formation of the pro-nephros starting as early as the third week, with subsequent development of the mesonephros, then metanephros. From there, the ureteric bud and metanephric mesenchyme develop, with the former eventually undergoing branching to form basic renal structure. Both essential structures are of mesodermal origin. Interaction between the ureteric bud and the metanephric mesenchyme is crucial for nephron formation. The Wnt pathway is particularly important for this interaction and allows for mesenchymal-epithelial transformation and differentiation of nephron epithelia [45,46].
Other major signaling pathways for nephrogenesis and ureteric bud branching include Sonic hedgehog, bone morphogenic proteins and fibroblast growth factors [46]. Further complex interactions between factors are required for nephrogenesis, including Lim1, Pax2, Eya1, Six1,2,4, Sall1 and WT-1. Pax2 is essential for intermediate mesoderm differentiation, and Pax2 knockout, for example, leads to agenesis of kidneys, ureters and genital tracts [47]. Both Eya1 knockout and Sal1 knockout lead to defects in the ureteric bud growth, and therefore renal agenesis [48,49]. Wt1 knockout causes metanephric mesenchyme apoptosis, and hence no further kidney development [50]. Six1 knockout leads to metanephric induction failure and has been shown to interact with both Pax2 and Eya1, with the former likely acting downstream and the latter acting upstream to Six1. Six1 has also been shown to be required for both Six2 and Sall1 expression [51].
4.2. Application of Blastocyst Complementation
Usui et al first used Sall1-/- knockout mice in 2012, with mouse embryonic or induced pluripotent stem cells injected into the blastocyst cavity [52]. Interestingly, while the newly formed kidneys were entirely derived from the injected stem cells, contribution was minimal in the bladder and ureter. Of the 9 neonate pups obtained, 3 were Sall1-/-. Of note, in the ESC complemented mice, nephron epithelia and renal stroma was almost fully made up of ESC-derived cells, while the collecting tubule showed a mix of cells. In the iPSC complemented mice, of the 37 neonate pups retrieved, 5 were Sall1-/-. iPSC-derived cells largely contributed to all kidney epithelial cells, except for collecting ducts. Furthermore, kidney stromal elements including vessels and nerves showed a mix of cell origin. Contribution in other organ systems varied for both ESC or iPSC complemented mice, however most non-kidney tissue did show some level of chimerism. In terms of development, kidneys were histologically and morphologically normal in ESC or iPSC complemented mice. Unfortunately, even complemented Sall1 knockout mice were not able to survive to adulthood, with many demonstrating similar issues with nursing that non-complemented, Sall1 knockout mice. Moreover, they did not have success generating kidneys when injecting rat iPSCs into Sall1-/- mice.
While the above-mentioned study did not find success with interspecies kidney generation, using the opposite configuration, i.e. injecting GFP-labelled mouse iPSC and ESCs in rats seemed to show promising results. Goto et al demonstrated this, similarly, using Sall1 as a knockout target [53]. They firstly concluded that the failure in successful rat complementation in Sall1-/- mice was due to insufficient and decreased efficiency in rat iPSC contribution to the metanephric mesenchyme. In the Sall1-/- chimeric mice kidneys showed uniform GFP expression. Notably, kidney size in mouse-complemented Sall1-/- rats was smaller than wildtype or heterozygous rats, although they were similar in size to wild type mice. They also demonstrated similar issues with mouse-derived contributions. While the metanephric mesenchymal cells (e.g. glomerular basal membrane, proximal tubule, loop of Henle) were entirely of mouse origin, and in the correct localization, the collecting tubules and blood vessels contained a mix of cells. Importantly, successful connection between ureter and bladder was shown when injecting intra-urethral dye. Furthermore, they encountered similar difficulty with postnatal survival, and all complemented Sall1-/0 rats had the previously mentioned nursing defect. This may be due to defects in olfactory development, potentially leading to anosmic phenotype.
Finally, both Matsunari et al and Wang et al recently demonstrated promising results using human cells in pig embryos. Given their size, pigs represent an attractive option for growing human organs. Matsunari et al used a Sall1 knockout model. While their first attempt using cloned embryos did not succeed, using IVF-derived embryos led to one chimeric fetus of 12 with morphologically and histologically normal kidney, with positive donor-cell expression [21].
Given the difficulties in synchronizing development, Wang et al opted to target both Six1 and Sall-1 as knockouts to ensure a large window in developmental progression is achieved [54]. Following injection of DsRed-labelled human iPSCs, gestation was terminated at either E25 or E28, particularly due to concerns about potential contribution to brain tissue. While no fully developed kidneys were obtained, when compared to wildtype embryos, the mesonephros obtained in the chimeric embryos showed similar mesonephric density and were histologically similar. Furthermore, over 50% of the mesonephric cells showed DsRed expression, with higher rates of DsRed expression in mesonephric tubules, and lower rates in mesenchymal cells. Importantly, DsRed labelled mesonephric tubules expressed Sall1, Six1, Pax2 and WT1, all important kidney developmental markers, suggesting the potential for further successful development.
5. Pancreas
In 2021, there were almost 4,000 people on the pancreas transplant waiting list in the U.S., with just under 1,000 pancreases being transplanted that year. Most patients awaiting a pancreas transplant were patients with type 1 diabetes, with type 2 diabetes being the second most common cause. While oral medications and insulin delivery systems continue to be a mainstay for both these patient populations, the discrepancy between people on the waitlist compared to number of transplants performed highlights the importance of pancreas transplants in patients with severe complications or unstable glycemic control [55].
5.1. Pancreas Development and Its Elimination
The pancreas develops from a ventral and a dorsal bud [56]. The dorsal bud is of endodermal foregut origin and will form the tail, body and neck of the pancreas, as well as the duct of Santorini. The ventral bud arises from the hepatic diverticulum, and forms the head, the uncinate process and the duct of Wirsung. Importantly, there are initially two ventral buds, however the left one must regress to prevent the formation of annular pancreas [57,58]. Fusion of the remaining ventral bud with the dorsal bud occurs around week 6-7 during gut rotation to form the whole pancreas. Endocrine cells are initially identified shortly after this process, and islet formation slowly progresses with insulin-expressing cells developing first. The proportion of insulin, glucagon and somatostatin-producing cells resembling that of adult pancreases by week 21[58].
Signaling for pancreas development involves several pathways. Studies have detected PDX11 and SHH around week 4. SOX9, GATA4 and 6 have also been identified around week 5. Expression of certain factors becomes much more important in differentiation different cell types, with NEUROG3 being a hallmark of endocrine cells, GATA4 of acinar exocrine cells, and SOX9, PDX1 and FOXA2 being found in pancreatic duct cells. While NEUROG3 seems to be required to determine an endocrine cell fate, it is only transiently increased and no longer detected by 35 weeks [56,59]. Inactivation of PDX1 causes pancreatic agenesis and exocrine deficiency [60]. Similarly, changes in downstream regions of PTF1A have also led to pancreatic agenesis [61]. Further potential targets that may cause defects in pancreatic development include GATA4 and 6, HNF1B, SOx9 and UBR1 [59].
5.2. Application of Blastocyst Complementation
Current diabetes treatment, particularly for type 1 diabetics, requires lifelong glycemic control and insulin injection. As such, pancreas and pancreatic islet transplantation have become an important aspect of treatment for patients with difficult glycemic control, as well as patients with severe complications. For type 2 diabetics, while islet transplantation is not currently available due to their increased need for insulin, pancreas transplantation remains an option for a subset of these patients, with reported successes [62]. As such, there is a continued need for pancreatic organs or tissue, which cannot be met with current supply [55].
Some success has been shown using xenotransplantation of islets. This was first done in 1994, where 10 type 1 diabetic patients with kidney transplant (and therefore immune suppression) received porcine pancreatic tissue. Porcine C-peptide was detected for 200-400 days in the urine of four patients [63]. Interestingly, another case study demonstrated that in a patient with type 1 diabetes who underwent porcine islet transplantation, live cells were found and retrieved 9.5 years following transplant, with insulin production being confirmed in in vitro glucose stimulation [64]. Therefore, while xenotransplantation shows potential, the use of blastocyst complementation provides a unique opportunity, as it allows for a decreased need for immune suppression and increased overall functionality may be achieved.
Kobayashi et al were some of the first to demonstrate success with blastocyst complementation back in 2010 [64]. They first demonstrated that blastocyst complementation could be used for organ generation. Using Pdx1-/- blastocyst, they developed a niche for pancreas development. Mouse iPSC or ESCs were then injected, which led to morphologically and histologically normal pancreas generation, with pancreatic islets, duct epithelia and exocrine tissue being entirely derived from the mouse donor cells. However, as with other organs, stromal elements such as vessels, fibrocytes or nerves showed a mix of host and donor cells. Functionally, glucose tolerance testing showed successful insulin secretion in response to glucose with maintenance of normal glucose levels. They further highlighted the potential of blastocyst complementation by transplanting the iPSC derived islets into mice with induced diabetes. No immune suppression was required, and the transplanted mice showed normal blood glucose levels and normal response to glucose tolerance testing. Most importantly, then they went on to produce interspecies pancreases. Rat iPSCs were injected into mouse blastocysts. Of the 10 Pdx1-/- mice, pancreatic epithelia were entirely made up of the rat-derived cells. While the number of chimeric mice reaching adulthood was low, in the 2 that did fully mature, their pancreas was histologically and morphologically normal with normal serum glucose levels and response to glucose loading. Their work was critical in highlighting that blastocyst complementation could be successful for the pancreas, even in the case of inter-species complementation.
The same group went on to demonstrate that the opposite was possible, that is, using EGFP-labelled mouse iPSC or ESCs to rescue pancreatic phenotype in PDX-1 -/- rats. The pancreatic endocrine, exocrine and duct epithelium showed EGFP expression. The PDX-1 knockout chimeric rats responded more slowly in the glucose tolerance test but otherwise showed no significant functional changes. They then transplanted the mouse-derived pancreases into diabetic mice. As has been previously discussed in other organs, they did find that a notable amount of tissue, such as endothelial cells, were rat derived. Therefore, while the transplanted mice were given immune suppression for the first five days, the transplanted mice showed normal glycemic levels for over 370 days even without continued immune suppression. Transplanted islets expressed insulin, glucagon, and somatostatin underscoring the potential for blastocyst complementation as a treatment modality [65].
Matsunari et al further demonstrated success in the area, however they used a larger pig model [66]. Firstly, they demonstrated that Hes1 overexpression under Pdx1 promotion led to inhibition of pancreatic development. They then showed that blastocyst complementation could rescue pancreas development in these pigs. Of the 14 full term fetuses obtained, 5 were chimeric, with histologically normal pancreas, and almost all pancreatic cells derived from donor cells. They went on to successfully generate chimeric adult pigs, with normal serum glucose levels. Oral glucose tolerance test results were also normal in one of the chimeric pigs. Necropsy of one chimeric pig showed macroscopic normal intestinal organs. While all tissue examined in the study showed donor-derived cells, progeny sired by the chimeric male pigs all demonstrated apancreatogenic phenotype, suggesting that sperm were derived from host, not donor cells.
Finally, more recently Matsunari et al further continued their work in another pig model [21]. However, whereas their work in 2013 was done using Pdx1-Hes1 overexpression, in this case, they used a PDX1 knockout model. Of the 10 fetuses obtained, 4 were chimeric, with half of the chimeras showing histologically and morphologically normal pancreatic phenotype. Compared to the chimeras with non-rescued pancreases, these pigs showed high levels of chimerism in the pancreas.
The above-mentioned studies have not only shown that blastocyst complementation could be successful for pancreas generation, be it in small or large animal models, but that interspecies transplantation of generated organs could also improve glycemic control in diabetic animals.
6. Heart
Heart transplantation is a life-saving operation for many people. There continues to be an unmet need for transplant donors, with over 8,000 people in the USA on the waiting list in 2022 alone. The leading cause for requiring a heart transplant remains cardiomyopathy, with coronary heart disease coming in second. Approximately 400 transplants have been done in the pediatric population, and congenital defects continue to be the leading cause of heart transplants in this patient population. [67] The need for donor hearts therefore remains critical for many of these patients, and as such, finding possible alternatives continues to be of great importance.
6.1. Cardiogenesis and Elimination of Cardiac Development
Cardiac development starts from mesodermal cells, with growth factor secretion (such as BMP) from neighboring endodermal and ectodermal cells allowing for differentiation to occur [68]. Concomitant to cardiomyocyte development, the endocardium develops from a specific subset of mesodermal progenitors. This process seems to be largely dependent on Tie and Tie2 expression, with knockout of these genes leading to normal vascular structures except for the endocardium [69,70]. Further development leads to folding, with the heart tube organizing into a 2-3 cardiomyocyte layer and one inner endocardial layer. Subsequent looping allows for the formation and spatial relation of a four chambered heart, with cardiomyocytes differentiation into either atrial, ventricular or conduction-specific cells [71].
Several transcription factors have been identified to be critical for cardiac development from the mesoderm, including Nkx2-5, GATA4, MesP1. In regard to Nkx2-5, while its deletion did not lead to agenesis, looping was disrupted, as well as trabeculation and endocardial cushion formation [72]. Further differentiation also seems to be greatly affected by GATA transcription factors, with GATA4 inhibition causing defects in cardiomyocyte differentiation and further development [73,74]. Finally, MesP1 and P2 have been shown to be important for mesodermal migration from the primitive streak, with MesP1 deficiency leading to inhibition of cardiac mesoderm [75,76].
6.2. Application of Blastocyst Complementation
The first hurdle in applying blastocyst complementation for cardiogenesis is that no single knockout target has been identified that fully inhibits cardiogenesis. While Coppiello et al showed success in the area, this required depletion of both the heart and vascular system using Cre dependent DTA (diphtheria toxin subunit A) to mediate agenesis [77]. In order to induce cardiomyocyte agenesis, they used a Nkx2.5-Cre strain, and to mediate endothelial agenesis, they used a Tie2-Cre strain. Firstly, they demonstrated that cardiac complementation could be achieved when injecting mouse iPSCs in the Nkx2.5-Cre mice. The hearts in all 14 live embryos obtained on embryonic day 14 showed normal beating and morphology, with 100% of the cardiomyocytes being donor derived. Per vascular complementation in the Tie2-Cre strain, of the 42 chimeras produced, 5 were successfully Tie-Cre complemented with normal morphology, and practically all endothelial cells being of donor origin (99.3%). Combining both Tie2-Cre and Nkx2.5-Cre strain complementation led to the successful development of chimeric mice with a donor-derived heart and vascular system. Heart function (via echocardiogram), as well as vascular and cardiomyocyte density in 8 and 3 chimeras respectively showed no difference when compared to control animals. Interspecies complementation of rat ESC in mouse blastocyst proved to be difficult. While they achieved rat heart complementation in 19/47 Nkx2.5-Cre strain mice at E10.5, with practically all cardiomyocytes being donor-derived, they were unable to show any success with heart or vascular system complementation in Tie2-Cre; and Nkx2.5-Cre mice at E11.5 or E14.5. Difficulty in this area seems to be in part due to improper vascularization and vascular development to allow for appropriate oxygenation.
7. Thyroid
Hypothyroidism continues to be one of the most common endocrine diseases. More recent data analysis suggests just over 10% of the U.S. population suffers from this disorder [79]. The mainstay of current treatment for hypothyroidism continues to be thyroid hormone replacement therapy. While inexpensive, and relatively accessible, this is not without its own challenges, as treatment is lifelong, and requires continued thyroid testing and monitoring. Particularly in older patients, over or under treatment with thyroid replacement therapy can have significant effects. In one study, only 42.8% of patients under thyroid hormone treatment had a TSH in the euthyroid range, with 41% having low TSH and 16% having high TSH [80]. This can have clinical significance, particularly in older patients, where it has been shown that patients under replacement therapy with TSH outside of the euthyroid range can lead to adverse health [81,82]. Thyroid transplantation, or thyroid tissue transplantation has become an area of interest, especially for patients where thyroid hormone replacement therapy can prove challenging [83]. As such, the use of blastocyst complementation also offers up an interesting treatment alternative for this patient population.
7.1. Thyroid Embryogenesis and Elimination of Thyroid Development
The thyroid gland is of endodermal origin from the ventral pharynx wall near the base of the tongue. The primitive thyroid diverticulum eventually migrates down to take its spot right above the trachea, as well as bifurcating to form 2 lobes [84,85]. Thyrocytes, which are of endodermal origin, start forming follicles as the left and right lobe are established. The calcitonin-producing C cells, which were initially thought to be of neural crest origin, may in fact also be endoderm-derived [86]. Transcription factors that have been found to be crucial for thyroid development include Nkx2-1, Pax8, Foxe1 and Hhex [87]. Nkx2-1 and Pax8 have been shown to be enough to determine differentiation of thyroid follicular cells, although deficiency in either one of the four above-mentioned transcription factors will lead to severely impaired morphogenesis or agenesis [88,89].
7.2. Application of Blastocyst Complementation
Ran et al already demonstrated that blastocyst complementation to generate lungs was possible using fgf10 Ex1mut./Ex3mut mice [40]. They once again used this model, which led to the development of severely hypoplastic thyroids, with only remnant tissue being present. Of note, targeting the fgf10 pathway did not seem to influence calcitonin expression. GFP-labelled mouse iPSCs were then injected into the embryos. Initial analysis of neonatal chimeras showed morphological and histologically normal thyroids with high GFP expression and comparable Tg, T3 and Nkx2-1 expression when compared to wildtype mice. Five complemented chimeric mice that survived to adulthood were then further analyzed. Expression levels of Nkx2-1, Foxe1, and Pax8 in the newly generated thyroid follicular cells were similar to levels found in wildtype mice. Histology was also normal. The proportion of donor-derived cells amongst thyroid follicular cells was greater than 85%. Mesenchymal and C-cells did not show similar donor-derived predominance. Finally, functional testing demonstrated comparable levels of T3 and T4 between wild type and chimeric mice [90].
Wen et al more recently also demonstrated that blastocyst complementation could rescue the thyroid phenotype in Nkx2-1 knockout mice [42]. They initially investigated this pathway to determine whether blastocyst complementation could rescue mice with lung agenesis, however, since this pathway also leads to thyroid agenesis, both organ systems were evaluated together. Nkx2-1 -/- mice were injected with GFP-labelled mouse ESCs. At embryonic day E17.5, the chimeras showed thyroid tissue, with the majority of thyrocytes expressing donor-derived GFP. Unfortunately, no functional testing could be performed in adult mice, as tracheo-esophageal separation could not be achieved in the chimeric mice, and all died at birth [42].
8. Other: Parathyroid, Thymus
Finally, there are a variety of further solid organs that have also been investigated. Application of blastocyst complementation for these has, however, been more limited compared to liver, lung, kidney, heart and pancreas. The organs discussed below include the parathyroid and the thymus. The successful use of blastocyst complementation in these instances continue to demonstrate the potential that this technique can have in a multitude of systems.
8.1. Development of Thymus and Parathyroid
The thymus and parathyroid both originate from the endodermal gut tube, although the ectoderm also contributes to thymic epithelium. The thymus and one pair of parathyroid glands will eventually arise from the third pharyngeal pouch, with specification of cell fate leading to parathyroid cells assuming GCM2-positivity, and thymus cells assuming FOXN1-positivity. The second pair of parathyroids originating from the fourth pharyngeal pouch [91]. The two thymic lobes eventually gravitate toward the midline in order to fuse together and then reaches its final position in the anterior superior mediastinum. Hematopoietic progenitor cells eventually migrate to colonize the thymus, with stem cells from the bone marrow and fetal liver, at which point the thymus assumes its critical role in T cell maturation and development [92,93].
The parathyroids from the third pharyngeal pouch similarly undergo caudal migration to eventually become the inferior parathyroids, while the parathyroids from the fourth pharyngeal pouch only minimally move and will form the superior parathyroids. The chief cells of the parathyroid will ultimately start producing parathyroid hormone (PTH) [91,93].
GCM2 is crucial for parathyroid cell differentiation as well as preventing apoptosis. GCM2 activation is also largely dependent on SHH signaling from the endoderm or neighboring mesenchyme, which likely acts via GATA3 and TBX1 downstream activation. Other transcription factors that seem to play a critical role in parathyroid organogenesis include HOX3, which may work together with PBX1, PAX, EYA and SIX transcriptional regulators to enable parathyroid development. Of note, MafB in particular seems to be required for eventual production and expression of PTH [91].
The thymus requires many of the same transcription factors mentioned above for successful development. The earliest marker for specific thymic differentiation is FOXN1, and it has been shown to be important for branching and thymic colonization, although it does not seem that it is required for initial thymus formation or migration. Early thymus development seems to involve Hoxa3, Eya1, Pax1/9, Six1/4 and Tbx1 networks, with clear involvement of Wnt and BMP signaling, although there is likely a still unclear driving force for very initial thymus fate development [92,93].
8.2. Application of Blastocyst Complementation
Given the importance of the FOXN1 pathway in thymic epithelial cell differentiation, Yamazaki et al used a FOXN1 knockout model to generate athymic mice [94]. The knockout mice were complemented with mouse embryonic stem cells. The extent of chimerism did not seem to influence thymic rescue, although lower chimerism seemed to result in smaller thymus. Of note, almost all thymic epithelial cells were donor-derived, with a higher percentage of peripheral T cells being donor-derived if chimerism levels were higher. When compared to normal mice, there was no significant difference in the number of peripheral T cells or in the gene-expression profile of complemented thymic epithelial cells. Of note, neither donor nor host-derived thymic cells showed significant differences in the proliferation of CD4+ and CD8+ T cells upon stimulation with antibodies, and chimeric mice showed similar production of IFN gamma, IL-2 and Granzyme B by splenic T cells. Finally, Anti-PDL1 antibody treatment in the chimeric mice increased T cell activation and IFN gamma production, as well as suppressing tumor growth. The results suggested that blastocyst complementation led to the rescue of a functional thymus.
Miura et al, while investigating Foxa2 driven Fgfr2 depletion for lung agenesis, also found that blastocyst complementation successfully rescued the thymus agenesis phenotype [41]. In 5 of the Foxa2 driven fgf2r knockout mice, an average of 92.4% of the thymic epithelium and 52.9% of the thymic mesenchyme was chimeric (although the latter showed much greater variability in the extent of chimerism).
Kano et al very recently demonstrated functional parathyroid gland generation using blastocyst complementation with a GCM2 knockout model and complementation with mouse ESCs [95]. Histologically normal parathyroid glands were achieved, with donor-derived chief cells. Other cell lineages, such as endothelial or mesenchymal cells showed a mix of donor and host-derived cells. Functionally, compared to control mice, complemented mice showed similar plasma calcium levels, basal PTH values and PTH stimulation response. Gene expression profiles showed either higher or comparable expression levels. Transplantation of the donor-derived parathyroid glands into GCM2 post-natal knockout mice led to survival in the transplanted group. Finally, they achieved interspecies blastocyst complementation using rat hosts and mouse ESCs. The GMC2 knockout rats were injected with mouse ESC, leading to the development of parathyroid glands which exhibited transcription factors required for further development and PTH expression. Unfortunately, due to the development of umbilical hernias, survival after birth was not possible.
9. Conclusions and Challenges of Interspecies Chimerism
Blastocyst complementation is a promising option for organ generation, especially for transplant use. This review underlines the many encouraging results for several solid organs. However, there are certainly challenges that must be overcome in this field.
9.1. Inefficient Chimerism and Survival to Adulthood
In all studies discussed, there were difficulties in generating living chimeras that survived to adulthood. Such survival can be very challenging when knockouts of major pathways leads to phenotypes that cannot be rescued with blastocyst complementation and simply do not allow for postnatal survival (e.g. tracheoesophageal fistula in Nkx2-1 knockouts, nursing issues in Sall-1 knockouts, or umbilical hernias in GMC2 knockouts) [42,43,52,53,95]. Success with interspecies chimerism, seems to be inversely proportional to the evolutionary distance between the donor and host species [96]. The most successful interspecies chimerism to date has been observed between evolutionarily closely related species such as mouse and rat, which have been outlined in this review [43,53,64,65,77,95].
This was particularly apparent with respect to donor contribution in endothelial and vascular systems. For most of the solid organs, host tissue still largely made up these anatomical areas. This has great significance for interspecies chimerism, particularly when considering the risk of rejection on transplantation of the chimeric organ into the donor recipient. Being able to combine vasculature depletion with other organ-of choice depletion would allow for overall higher levels of chimerism in the newly generated organ, particularly in supporting tissue. Matsunari et al, for example, demonstrated that blastocyst complementation of dual knockout KDR (to inhibit vasculogenesis) and PDX1 (to inhibit pancreatic development) led to fully developed pancreas with donor-derived endothelial and hematopoietic cells in four full term pigs [21].
Successful interspecies chimerism becomes even more evident with more evolutionarily distant donor-host species’ pairs such as human-mouse, and human-pig. To date, these combinations have exhibited low levels of donor contributions to host, and low success rates of chimerism. As example, with human donor cells in a MYOD/MYF6-KO pig model, <5% of skeletal muscle was derived from human cells [97]. Certain measures have been shown to improve human contribution in interspecies models, such as inhibition of apoptosis. BMI1 and BCL2 over-expression to inhibit apoptosis allowed for increased efficiency of human contribution in mouse, rabbit and pig models [98,99].
9.2. Barriers to Interspecies Chimerism During Development
Some of the major barriers for successful interspecies chimerism include (i) cell competition between donor and host cells, leading to limited contribution and cell death of donor cells in the chimeric embryo; (ii) issues with donor-host ligand and receptor compatibility; (iii) asynchrony in developmental speeds between the donor and host cells in the developing chimera; and (iv) mismatch in developmental stages of donor and host cells at the time of introduction of donor cells into the host embryo [100,101,102]. Multiple strategies have been employed to overcome these barriers, with infrequent success.
Limited contribution and cell death of donor cells in chimeric embryos has been discussed above, including the use of overexpression of anti-apoptotic genes allowing for increased human contribution in chimeras. Other strategies like matching developmental stages of donor and host species, during introduction of donor cells into the host embryo, have been explored. Stage-matching chimerism studies have demonstrated that successful chimerism was observed when (i) mouse embryonic stem cells (mESCs), a pluripotent cell type, was injected into the mouse early blastocyst (E3.5); and (ii) mouse neural crest cells (mNCCs), a multipotent cell type, was injected into the late mouse E8.5 gastrulating embryos [103,104]. However, when the donor and host stages were interchanged and the early mESCs were injected into the late gastrulating embryo, or the multipotent mNCCs were injected into the early E3.5 embryo, successful chimerism was not observed. This mouse-mouse stage-matching study demonstrated the importance of matching developmental stages between donor cells and host embryos. The naïve mouse ESCs were more compatible with the early-stage mouse blastocyst, and more differentiated donor cells contributed better to later stage gastrulating embryos where the germ layers were defined.
Another interspecies chimerism demonstrated that human pluripotent stem cells (PSCs) injected into the E6.5-7 gastrulating mouse embryo successfully contributed to the in vitro cultured chimeric embryo and had cell-type specific gene expression [105]. Since human PSCs are more ‘primed’ than the ‘naïve’ mESCs, the h-PSCs are at a later stage of development and are more like the primed cells from the mouse post-implantation embryos [106,107]. The authors observed successful proliferation of human cells in the mouse embryos in 70% of the chimeras [105]. However, these embryos were cultured in vitro for only 2 days and hence any further inferences could not be made.
While these experimental strategies have been useful in demonstrating interspecies barriers and overcoming them to some extent, these trial-and-error ex vivo and in vivo procedures involving embryos are expensive, time consuming, and low-throughput. Very few studies have interrogated molecular factors that are crucial for enhancing chimerism, identifying targets to ‘synchronize’ donor-host cells, and overcoming barriers in the less successful chimerism pairs. There is a need to understand the cellular and molecular signaling mechanisms of the donor and host cells, as they develop together within the chimeric embryos. Further, there is a need to be able to do so in an affordable, high-throughput manner.
9.2.1. Single Cell Molecular Approaches to Understand Donor and Host Cell Mechanisms in Chimeric Embryos
The development of single cell (sc) sequencing technologies, such as sc-transcriptome and sc-genome sequencing, have revolutionized the way in which we are able to gain insight into the genome features. Sc-RNA sequencing has allowed us to analyze gene expression within each cell and compare it with other cells within the same cell type or across different cells. With sc-genome sequencing techniques, we are now able to gain insight into genome coverage, copy number variation at the level of individual genes, and single nucleotide variations. Hence, being able to utilize these single cell sequencing techniques in the context of interspecies chimerism, would provide us greater insight into donor cells and host cells in the developing chimeric embryos, and understand differences in replication patterns and gene expression between donor and host cells.
Single Cell RNA Sequencing for Enhancing Interspecies Chimerism Efficiency
One of the major barriers to successful interspecies chimerism is the differences in developmental stages between the donor and host cells [102]. For example, the donor and host cells might not match with regards to level of differentiation, embryonic stage, or donor stem cell status (naïve versus primed). Using sc-RNA sequencing, we are able to analyze gene expression across early embryonic stages and stem cells, and computationally stage-match donor and host species, based on gene expression [108]. Sc-RNA sequencing has been performed on multiple cell types, including early developing embryos of humans, mice, pigs, marmoset, and other species [109,110,111,112]. Access to these datasets allows us to perform in silico stage-matching of multiple donor-host species [108]. In Shetty et al. 2023, the authors used an in silico analysis approach, based on similarities in gene expression, to stage match stem cells and early embryos of commonly used donor and host species, including human, marmoset, mouse, and pig [108]. They identified that the stages that best matched with each other were the human blastocyst (E6/E7), the gastrulating mouse embryo (E6–E6.75), the marmoset late inner cell mass, and the pig late blastocyst. They also found that the human SCs best matched with the mouse late gastrulating embryo, in line with the results of the ex vivo and in vivo stage-matching experiments previously performed [105,113,114]. This is a high-throughput, fast, and cost-efficient analysis that narrows down multiple donor cell-host embryo combinations that have the most similar gene expression and hence are most likely to produce successful chimeras when tested using ex vivo and in vivo techniques.
Replication Timing to Determine Developmental Speeds of Donor and Host Species
In addition to sc-transcriptomics, sc-genomic DNA (sc-gDNA) sequencing allows us to analyze patterns of genome replication, through a technique called ‘replication timing’ [115,116]. Replication timing (RT) provides us with information on how the cell’s genome replicates during the S-phase of the cell cycle. This allows us to gain insight into different genes, and if they replicate early or late during the S-phase. Replication timing (RT) patterns are conserved within cell types, mitotically inherited, and correlated with chromosomal organization and gene expression [117,118]. Using RT, one can also gain insight into not only how the cells replicate, but also how it coordinates with cell-type specific speed of replication.
One of the major barriers to successful chimerism is the differential developmental speeds of the donor and host cells, in the chimeric embryo [101,108] (Figure 2). Currently, there is no established way to define ‘developmental speed’, other than the metric of hours and days. Using RT, it would be possible to define cell-type specific developmental speed at the molecular level and further analyze differences in donor-host developmental speeds at the genomic level. This will enable us to understand the current developmental speed barriers in the interspecies chimerism field and allow us to identify molecular targets to overcome these barriers.
9.3. Ethical Considerations
Finally, a particularly important note about using human interspecies models must take into account human cell contribution in a developing embryo, and in particular, brain tissue. As such, timing must consider the development of off-target tissues and identify the best genes to target for minimal extra-organ contributions. In models where major pathways such as Nkx or HHEx are targets for knock-out, levels of chimerism in other organs, particularly in neural or germ-line tissue is particularly important to consider. Kobayashi et al for example induced Mixl1 expression to attempt to restrict injected stem cell differentiation to endodermal tissue. They found that at a sufficient level, this did result in a significant reduction in donor pluripotent stem cell contribution to organs that were not of endodermal origin [119]. Hashimoto et al further demonstrated that knockout of Prdm14 and Otx2 inhibited contribution of donor cells to germ line spermatozoa, oocytes, testes, ovaries, as well as brain [120]. These studies underscore that certain measures can be taken to address ethical concerns for human interspecies chimeras.
In conclusion, while many challenges and questions remain in the field of blastocyst complementation significant progress has been made. A number of solid organs have been successfully generated using this technique, with significant efforts dedicated to improving chimera generation and addressing ethical concerns. As a result, blastocyst complementation continues to present a promising avenue to meet the ongoing demand for organ tissue in the transplant and medical field.
Author Contributions
Conceptualization, methodology, formal analysis, E.B., W.C.L., and C.J.S.; writing—original draft preparation, E.B.; writing—review and editing, E.B., A.V.S, W.C.L., and C.J.S.; visualization, E.B., A.V.S., W.C.L., and C.J.S.; funding acquisition, W.C.L. and C.J.S. All authors have read and agreed to the final submitted version of the manuscript.
Funding
This work was funded, in part, by the National Institutes of Health grant R01 AI173804-01 (CJS and WCL).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
None.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| iPSC | induced pluripotent stem cells |
| ESC | embryonic stem cells |
| KO | knock-out |
References
- Global Observatory on Donation and Transplantation. Data Charts and Tables. Updated 2024. Accessed November 5 2024, November 30, 2024. https://www.transplant-observatory.org/data-charts-and-tables/chart/.
- Health Resources & Services Administration. Organ Donation Statistics. OrganDonor.gov. Updated July 2023. Accessed November 5, 2024. https://www.organdonor.gov/learn/organ-donation-statistics.
- Griffith BP, Goerlich CE, Singh AK, et al. Genetically modified porcine-to-human cardiac xenotransplantation. N Engl J Med. 2022;387(1):35-44. [CrossRef]
- Founta KM, Papanayotou C. In Vivo generation of organs by blastocyst complementation: Advances and challenges. Int J Stem Cells. 2022;15(2):113-121. [CrossRef]
- Chen J, Lansford R, Stewart V, Young F, Alt FW. RAG-2-deficient blastocyst complementation: an assay of gene function in lymphocyte development. Proc Natl Acad Sci U S A. 1993;90(10):4528-4532. [CrossRef]
- Xiang AP, Mao FF, Li WQ, et al. Extensive contribution of embryonic stem cells to the development of an evolutionarily divergent host. Hum Mol Genet. 2008;17(1):27-37. [CrossRef]
- Centers for Disease Control and Prevention. Liver Disease. CDC. Updated June 7, 2023. Accessed November 14, 2024. https://www.cdc.gov/nchs/fastats/liver-disease.htm.
- Asgari S, Moslem M, Bagheri-Lankarani K, Pournasr B, Miryounesi M, Baharvand H. Differentiation and transplantation of human induced pluripotent stem cell-derived hepatocyte-like cells. Stem Cell Rev Rep. 2013;9(4):493-504. [CrossRef]
- Chen YF, Tseng CY, Wang HW, Kuo HC, Yang VW, Lee OK. Rapid generation of mature hepatocyte-like cells from human induced pluripotent stem cells by an efficient three-step protocol. Hepatology. 2012;55(4):1193-1203. [CrossRef]
- Nagamoto Y, Takayama K, Ohashi K, et al. Transplantation of a human iPSC-derived hepatocyte sheet increases survival in mice with acute liver failure. J Hepatol. 2016;64(5):1068-1075. [CrossRef]
- Zaret KS. From Endoderm to Liver Bud: Paradigms of Cell Type Specification and Tissue Morphogenesis. Curr Top Dev Biol. 2016;117:647-669. [CrossRef]
- Zorn AM. Liver development. 2008 Oct 31. In: StemBook [Internet]. Cambridge (MA): Harvard Stem Cell Institute; 2008-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK27068/. [CrossRef]
- Tachmatzidi EC, Galanopoulou O, Talianidis I. Transcription Control of Liver Development. Cells. 2021;10(8):2026. [CrossRef]
- Zhou W, Nelson ED, Abu Rmilah AA, Amiot BP, Nyberg SL. Stem cell-related studies and stem cell-based therapies in liver diseases. Cell Transplant. 2019;28(9-10):1116-1122. [CrossRef]
- Bort R, Signore M, Tremblay K, Martinez Barbera JP, Zaret KS. Hex homeobox gene controls the transition of the endoderm to a pseudostratified, cell emergent epithelium for liver bud development. Dev Biol. 2006;290(1):44-56. [CrossRef]
- Gauvrit S, Villasenor A, Strilic B, et al. HHEX is a transcriptional regulator of the VEGFC/FLT4/PROX1 signaling axis during vascular development. Nat Commun. 2018;9(1):2704. [CrossRef]
- Martinez Barbera JP, Clements M, Thomas P, et al. The homeobox gene Hex is required in definitive endodermal tissues for normal forebrain, liver and thyroid formation. Development. 2000;127(11):2433-2445. [CrossRef]
- Goodings C, Smith E, Mathias E, et al. Hhex is required at multiple stages of adult hematopoietic stem and progenitor cell differentiation. Stem Cells. 2015;33(8):2628-2641. [CrossRef]
- Kubo S, Sun M, Miyahara M, et al. Hepatocyte injury in tyrosinemia type 1 is induced by fumarylacetoacetate and is inhibited by caspase inhibitors. Proc Natl Acad Sci U S A. 1998;95(16):9552-9557. [CrossRef]
- Azuma H, Paulk N, Ranade A, et al. Robust expansion of human hepatocytes in Fah-/-/Rag2-/-/Il2rg-/- mice. Nat Biotechnol. 2007;25(8):903-910. [CrossRef]
- Matsunari H, Watanabe M, Hasegawa K, et al. Compensation of disabled organogeneses in genetically modified pig fetuses by blastocyst complementation. Stem Cell Reports. 2020;14(1):21-33. [CrossRef]
- Ruiz-Estevez M, Crane AT, Rodriguez-Villamil P, et al. Liver development is restored by blastocyst complementation of HHEX knockout in mice and pigs. Stem Cell Res Ther. 2021;12(1):292. [CrossRef]
- Simpson SG, Park K-E, Yeddula SGR, et al. Blastocyst complementation generates exogenous donor-derived liver in ahepatic pigs. FASEB J. 2024;38:e70161. [CrossRef]
- Chambers DC, Perch M, Zuckermann A, et al. The International Thoracic Organ Transplant Registry of the International Society for Heart and Lung Transplantation: Thirty-eighth adult lung transplantation report - 2021; Focus on recipient characteristics. J Heart Lung Transplant. 2021;40(10):1060-1072. [CrossRef]
- Schittny JC. Development of the lung. Cell Tissue Res. 2017;367(3):427-444. [CrossRef]
- Kimura S, Hara Y, Pineau T, et al. The T/ebp null mouse: thyroid-specific enhancer-binding protein is essential for the organogenesis of the thyroid, lung, ventral forebrain, and pituitary. Genes Dev. 1996;10(1):60-69. [CrossRef]
- Mucenski ML, Wert SE, Nation JM, et al. β-Catenin is required for specification of proximal/distal cell fate during lung morphogenesis. J Biol Chem. 2003;278(41):40231-40238. [CrossRef]
- Goss AM, Tian Y, Tsukiyama T, et al. Wnt2/2b and beta-catenin signaling are necessary and sufficient to specify lung progenitors in the foregut. Dev Cell. 2009;17(2):290-298. [CrossRef]
- Sekine K, Ohuchi H, Fujiwara M, et al. Fgf10 is essential for limb and lung formation [published correction appears in Nat Genet. 2019;51(5):921. doi: 10.1038/s41588-019-0396-9]. Nat Genet. 1999;21(1):138-141. [CrossRef]
- Bellusci S, Grindley J, Emoto H, Itoh N, Hogan BL. Fibroblast growth factor 10 (FGF10) and branching morphogenesis in the embryonic mouse lung. Development. 1997;124(23):4867-4878. [CrossRef]
- Minoo P, Su G, Drum H, Bringas P, Kimura S. Defects in tracheoesophageal and lung morphogenesis in Nkx2.1(-/-) mouse embryos. Dev Biol. 1999;209(1):60-71. [CrossRef]
- Little DR, Lynch AM, Yan Y, Akiyama H, Kimura S, Chen J. Differential chromatin binding of the lung lineage transcription factor NKX2-1 resolves opposing murine alveolar cell fates in vivo. Nat Commun. 2021;12(1):2509. [CrossRef]
- Herriges M, Morrisey EE. Lung development: orchestrating the generation and regeneration of a complex organ. Development. 2014;141(3):502-513. [CrossRef]
- Weaver M, Yingling JM, Dunn NR, Bellusci S, Hogan BL. Bmp signaling regulates proximal-distal differentiation of endoderm in mouse lung development. Development. 1999;126(18):4005-4015. [CrossRef]
- Park KS, Wells JM, Zorn AM, Wert SE, Whitsett JA. Sox17 influences the differentiation of respiratory epithelial cells. Dev Biol. 2006;294(1):192-202. [CrossRef]
- Lange AW, Haitchi HM, LeCras TD, et al. Sox17 is required for normal pulmonary vascular morphogenesis. Dev Biol. 2014;387(1):109-120. [CrossRef]
- Wan H, Kaestner KH, Ang SL, et al. Foxa2 regulates alveolarization and goblet cell hyperplasia. Development. 2004;131(4):953-964. [CrossRef]
- De Moerlooze L, Spencer-Dene B, Revest JM, et al. An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal-epithelial signalling during mouse organogenesis. Development. 2000;127(3):483-492. [CrossRef]
- Mori M, Furuhashi K, Danielsson JA, et al. Generation of functional lungs via conditional blastocyst complementation using pluripotent stem cells. Nat Med. 2019;25(11):1691-1698. [CrossRef]
- Kitahara A, Ran Q, Oda K, et al. Generation of lungs by blastocyst complementation in apneumic Fgf10-deficient mice. Cell Rep. 2020;31(6):107626. [CrossRef]
- Miura A, Sarmah H, Tanaka J, et al. Conditional blastocyst complementation of a defective Foxa2 lineage efficiently promotes the generation of the whole lung. Elife. 2023;12:e86105. [CrossRef]
- Wen B, Li E, Ustiyan V, et al. In vivo generation of lung and thyroid tissues from embryonic stem cells using blastocyst complementation. Am J Respir Crit Care Med. 2021;203(4):471-483. [CrossRef]
- Wen B, Li E, Wang G, et al. CRISPR-Cas9 genome editing allows generation of the mouse lung in a rat. Am J Respir Crit Care Med. 2024;210(2):167-177. [CrossRef]
- Organ Procurement and Transplantation Network (OPTN) and Scientific Registry of Transplant Recipients (SRTR). OPTN/SRTR 2022 Annual Data Report. U.S. Department of Health and Human Services, Health Resources and Services Administration; 2024. Accessed 12/4/2024.
- Stark K, Vainio S, Vassileva G, McMahon AP. Epithelial transformation of metanephric mesenchyme in the developing kidney regulated by Wnt-4. Nature. 1994;372(6507):679-683. [CrossRef]
- Reidy KJ, Rosenblum ND. Cell and molecular biology of kidney development. Semin Nephrol. 2009;29(4):321-337. [CrossRef]
- Torres M, Gómez-Pardo E, Dressler GR, Gruss P. Pax-2 controls multiple steps of urogenital development. Development. 1995;121(12):4057-4065. [CrossRef]
- Xu, PX., Adams, J., Peters, H. et al. Eya1-deficient mice lack ears and kidneys and show abnormal apoptosis of organ primordia. Nat Genet. 1999;23,113-117. [CrossRef]
- Nishinakamura R, Matsumoto Y, Nakao K, et al. Murine homolog of SALL1 is essential for ureteric bud invasion in kidney development. Development. 2001;128(16):3105-3115. [CrossRef]
- Kreidberg JA, Sariola H, Loring JM, et al. WT-1 is required for early kidney development. Cell. 1993;74:679-691. [CrossRef]
- Xu PX, Zheng W, Huang L, et al. Six1 is required for the early organogenesis of mammalian kidney. Development. 2003;130(14):3085-3094. [CrossRef]
- Usui J, Kobayashi T, Yamaguchi T, et al. Generation of kidney from pluripotent stem cells via blastocyst complementation. Am J Pathol. 2012;180(6):2417-2426. [CrossRef]
- Goto T, Hara H, Sanbo M, et al. Generation of pluripotent stem cell-derived mouse kidneys in Sall1-targeted anephric rats. Nat Commun. 2019;10(1):451. [CrossRef]
- Wang J, Xie W, Li N, et al. Generation of a humanized mesonephros in pigs from induced pluripotent stem cells via embryo complementation. Cell Stem Cell. 2023;30(9):1235-1245.e6. [CrossRef]
- OPTN/SRTR 2021 Annual Data Report: Pancreas,; Kandaswamy, Raja et al., Amer J Transplant. Volume 23, Issue 2, S121-S177.
- Henry BM, Skinningsrud B, Saganiak K, et al. Development of the human pancreas and its vasculature - An integrated review covering anatomical, embryological, histological, and molecular aspects. Ann Anat. 2019;221:115-124. [CrossRef]
- Adda G, Hannoun L, Loygue J. Development of the human pancreas: variations and pathology. A tentative classification. Anat Clin. 1984;5:275-283. [CrossRef]
- Pan FC, Brissova M. Pancreas development in humans. Curr Opin Endocrinol Diabetes Obes. 2014;21(2):77-82. [CrossRef]
- Jennings RE, Berry AA, Strutt JP, Gerrard DT, Hanley NA. Human pancreas development. Development. 2015;142(18):3126-3137. [CrossRef]
- Stoffers DA, Zinkin NT, Stanojevic V, Clarke WL, Habener JF. Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat Genet. 1997;15(1):106-110. [CrossRef]
- Weedon MN, Cebola I, Patch A-M, et al. Recessive mutations in a distal PTF1A enhancer cause isolated pancreatic agenesis. Nat Genet. 2014;46(1):61-64. [CrossRef]
- Amara D, Hansen KS, Kupiec-Weglinski SA, et al. Pancreas transplantation for type 2 diabetes: A systematic review, critical gaps in the literature, and a path forward. Transplantation. 2022;106(10):1916-1934. [CrossRef]
- Groth CG, Korsgren O, Tibell A, et al. Transplantation of porcine fetal pancreas to diabetic patients. Lancet. 1994;344(8934):1402-1404. [CrossRef]
- Kobayashi T, Yamaguchi T, Hamanaka S, et al. Generation of rat pancreas in mouse by interspecific blastocyst injection of pluripotent stem cells. Cell. 2010;142(5):787-799. [CrossRef]
- Yamaguchi T, Sato H, Kato-Itoh M, et al. Interspecies organogenesis generates autologous functional islets. Nature. 2017;542(7640):191-196. [CrossRef]
- Matsunari H, Nagashima H, Watanabe M, et al. Blastocyst complementation generates exogenic pancreas in vivo in apancreatic cloned pigs. Proc Natl Acad Sci U S A. 2013;110(12):4557-4562. [CrossRef]
- OPTN/SRTR 2022 Annual Data Report: Heart. Colvin, Monica M. et al. Amer J Transplant. Volume 24, Issue 2, S305-S393.
- Buijtendijk MFJ, Barnett P, van den Hoff MJB. Development of the human heart. Am J Med Genet C Semin Med Genet. 2020;184(1):7-22. [CrossRef]
- Harris IS, Black BL. Development of the endocardium. Pediatr Cardiol. 2010;31(3):391-399. [CrossRef]
- Puri MC, Partanen J, Rossant J, Bernstein A. Interaction of the TEK and TIE receptor tyrosine kinases during cardiovascular development. Development. 1999;126(20):4569-4580. [CrossRef]
- Wu SM, Fujiwara Y, Cibulsky SM, et al. Developmental origin of a bipotential myocardial and smooth muscle cell precursor in the mammalian heart. Cell. 2006;127(6):1137-1150. [CrossRef]
- Lyons I, Parsons LM, Hartley L, et al. Myogenic and morphogenetic defects in the heart tubes of murine embryos lacking the homeo box gene Nkx2-5. Genes Dev. 1995;9(13):1654-1666.
- Grépin C, Robitaille L, Antakly T, Nemer M. Inhibition of transcription factor GATA-4 expression blocks in vitro cardiac muscle differentiation. Mol Cell Biol. 1995;15(8):4095-4102. [CrossRef]
- Charron F, Paradis P, Bronchain O, Nemer G, Nemer M. Cooperative interaction between GATA-4 and GATA-6 regulates myocardial gene expression. Mol Cell Biol. 1999;19(6):4355-4365. [CrossRef]
- Saga Y, Miyagawa-Tomita S, Takagi A, Kitajima S, Miyazaki Ji, Inoue T. MesP1 is expressed in the heart precursor cells and required for the formation of a single heart tube. Development. 1999;126(15):3437-3447. [CrossRef]
- Kitajima S, Takagi A, Inoue T, Saga Y. MesP1 and MesP2 are essential for the development of cardiac mesoderm. Development. 2000;127(15):3215-3226. [CrossRef]
- Coppiello G, Barlabé P, Moya-Jódar M, et al. Generation of heart and vascular system in rodents by blastocyst complementation. Dev Cell. 2023;58(24):2881-2895.e7. [CrossRef]
- Das S, Koyano-Nakagawa N, Gafni O, et al. Generation of human endothelium in pig embryos deficient in ETV2. Nat Biotechnol. 2020;38(3):297-302. [CrossRef]
- Wyne KL, Nair L, Schneiderman CP, et al. Hypothyroidism prevalence in the United States: A retrospective study combining national health and nutrition examination survey and claims data, 2009-2019. J Endocr Soc. 2022;7(1):bvac172. [CrossRef]
- Somwaru LL, Arnold AM, Joshi N, Fried LP, Cappola AR. High frequency of and factors associated with thyroid hormone over-replacement and under-replacement in men and women aged 65 and over. J Clin Endocrinol Metab. 2009;94(4):1342-1345. [CrossRef]
- Flynn RW, Bonellie SR, Jung RT, et al. Serum thyroid-stimulating hormone concentration and morbidity from cardiovascular disease and fractures in patients on long-term thyroxine therapy. J Clin Endocrinol Metab. 2010;95(1):186-193. [CrossRef]
- Thayakaran R, Adderley NJ, Sainsbury C, et al. Thyroid replacement therapy, thyroid stimulating hormone concentrations, and long term health outcomes in patients with hypothyroidism: longitudinal study. BMJ. 2019;366:l4892. [CrossRef]
- Romitti M, Costagliola S. Progress toward and challenges remaining for thyroid tissue regeneration. Endocrinology. 2023;164(10):bqad136. [CrossRef]
- Rosen RD, Sapra A. Embryology, Thyroid. In: StatPearls. Treasure Island (FL): StatPearls Publishing; May 1, 2023.
- Policeni BA, Smoker WR, Reede DL. Anatomy and embryology of the thyroid and parathyroid glands. Semin Ultrasound CT MR. 2012;33(2):104-114. [CrossRef]
- Nilsson M, Williams D. On the origin of cells and derivation of thyroid cancer: C Cell Story Revisited. Eur Thyroid J. 2016;5(2):79-93. [CrossRef]
- Nilsson M, Fagman H. Mechanisms of thyroid development and dysgenesis: an analysis based on developmental stages and concurrent embryonic anatomy. Curr Top Dev Biol. 2013;106:123-170. [CrossRef]
- Antonica, F., Kasprzyk, D., Opitz, R. et al. Generation of functional thyroid from embryonic stem cells. Nature. 2012;491(7422):66-71. [CrossRef]
- Parlato R, Rosica A, Rodriguez-Mallon A, et al. An integrated regulatory network controlling survival and migration in thyroid organogenesis. Dev Biol. 2004;276(2):464-475. [CrossRef]
- Ran Q, Zhou Q, Oda K, et al. Generation of thyroid tissues From embryonic stem cells via blastocyst complementation in vivo. Front Endocrinol (Lausanne). 2020;11:609697. [CrossRef]
- Peissig K, Condie BG, Manley NR. Embryology of the parathyroid glands. Endocrinol Metab Clin North Am. 2018;47(4):733-742. [CrossRef]
- Figueiredo M, Zilhão R, Neves H. Thymus inception: Molecular network in the early stages of thymus organogenesis. Int J Mol Sci. 2020;21(16):5765. [CrossRef]
- Gordon J, Manley NR. Mechanisms of thymus organogenesis and morphogenesis. Development. 2011;138(18):3865-3878. [CrossRef]
- Yamazaki, K., Kubara, K., Ishii, S. et al. In vitro and in vivo functions of T cells produced in complemented thymi of chimeric mice generated by blastocyst complementation. Sci Rep. 2022;12(1):3242. [CrossRef]
- Kano M, Mizuno N, Sato H, et al. Functional calcium-responsive parathyroid glands generated using single-step blastocyst complementation. Proc Natl Acad Sci U S A. 2023;120(28):e2216564120. [CrossRef]
- De Los Angeles A, Pho N, Redmond DE Jr. Generating human organs via interspecies chimera formation: Advances and barriers. Yale J Biol Med. 2018;91(3):333-342.
- Maeng G, Das S, Greising SM, et al. Humanized skeletal muscle in MYF5/MYOD/MYF6-null pig embryos. Nat Biomed Eng. 2021;5(8):805-814. [CrossRef]
- Huang K, Zhu Y, Ma Y, et al. BMI1 enables interspecies chimerism with human pluripotent stem cells. Nat Commun. 2018;9(1):4649. [CrossRef]
- Wang X, Li T, Cui T, et al. Human embryonic stem cells contribute to embryonic and extraembryonic lineages in mouse embryos upon inhibition of apoptosis. Cell Res. 2018;28(1):126-129. [CrossRef]
- Zheng C, Hu Y, Sakurai M, et al. Cell competition constitutes a barrier for interspecies chimerism. Nature. 2021;592(7853):272-276. [CrossRef]
- Strell P, Shetty A, Steer CJ, Low WC. Interspecies chimeric barriers for generating exogenic organs and cells for transplantation. Cell Transplant. 2022;31. [CrossRef]
- Cohen MA, Markoulaki S, Jaenisch R. Matched developmental timing of donor cells with the host Is crucial for chimera formation. Stem Cell Reports. 2018;10(5):1445-1452. [CrossRef]
- Cohen MA, Wert JK, Goldman J, et al. Human neural crest cells contribute to coat pigmentation in interspecies chimeras after in utero injection into mouse embryos. Proc Natl Acad Sci U S A. 2016;113(6):1570-1575. [CrossRef]
- Mascetti, VL, Pedersen RA. Contributions of mammalian chimeras to pluripotent stem cell research. Cell Stem Cell. 2016;19(2):163-175. [CrossRef]
- Mascetti VL, Pedersen RA. Human-mouse chimerism validates human stem cell pluripotency. Cell Stem Cell. 2016;18(1):67-72. [CrossRef]
- Weinberger L, Ayyash M, Novershtern N, Hanna JH. Dynamic stem cell states: naive to primed pluripotency in rodents and humans. Nat Rev Mol Cell Biol. 2016;17(3):155-169. [CrossRef]
- Nichols J, Smith A. Naive and primed pluripotent states. Cell Stem Cell. 2009;4(6):487-492. [CrossRef]
- Shetty A, Lim S, Strell P, et al. In silico stage-matching of human, marmoset, mouse, and pig embryos to enhance organ development through interspecies chimerism. Cell Transplant. 2023;32. [CrossRef]
- Stirparo GG, Boroviak T, Guo G, et al. Erratum: Correction: Integrated analysis of single-cell embryo data yields a unified transcriptome signature for the human pre-implantation epiblast (doi:10.1242/dev.158501) Development. 2018;145(15):dev169672). [CrossRef]
- Xue Z, Huang K, Cai C, et al. Genetic programs in human and mouse early embryos revealed by single-cell RNA sequencing. Nature. 2013;500(7464):593-597. [CrossRef]
- Deng Q, Ramsköld D, Reinius B, Sandberg R. Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science. 2014;343(6167):193-196. [CrossRef]
- Boroviak T, Stirparo GG, Dietmann S, et al. Single cell transcriptome analysis of human, marmoset and mouse embryos reveals common and divergent features of preimplantation development. Development. 2018;145(21):dev167833. [CrossRef]
- Ramos-Ibeas, P, Sang F, Zhu Q, et al. Pluripotency and X chromosome dynamics revealed in pig pre-gastrulating embryos by single cell analysis. Nat Commun. 2019;10(1):500. [CrossRef]
- Fu R, Yu D, Ren J, et al. Domesticated cynomolgus monkey embryonic stem cells allow the generation of neonatal interspecies chimeric pigs. Protein Cell. 2020;11(2):97-107. [CrossRef]
- Miura H, Takahashi S, Shibata T, et al. Mapping replication timing domains genome wide in single mammalian cells with single-cell DNA replication sequencing. Nat Protoc. 2020;15(12):4058-4100. [CrossRef]
- Pope BD, Ruba T, Dileep V, et al. Topologically associating domains are stable units of replication-timing regulation. Nature. 2014;515 (7527):402-405. [CrossRef]
- Rivera-Mulia JC, Buckley Q, Sasaki T, et al. Dynamic changes in replication timing and gene expression during lineage specification of human pluripotent stem cells. Genome Res. 2015;25(8):1091-1103. [CrossRef]
- Ma J, Duan Z. Replication timing becomes intertwined with 3D genome organization. Cell. 2019;176(4):681-684. [CrossRef]
- Kobayashi T, Kato-Itoh M, Nakauchi H. Targeted organ generation using Mixl1-inducible mouse pluripotent stem cells in blastocyst complementation. Stem Cells Dev. 2015;24(2):182-189. [CrossRef]
- Hashimoto H, Eto T, Yamamoto M, et al. Development of blastocyst complementation technology without contributions to gametes and the brain. Exp Anim. 2019;68(3):361-370. [CrossRef]
Figure 1.
Two Distinct Genetic Approaches to Chimeric Organ Transplantation.
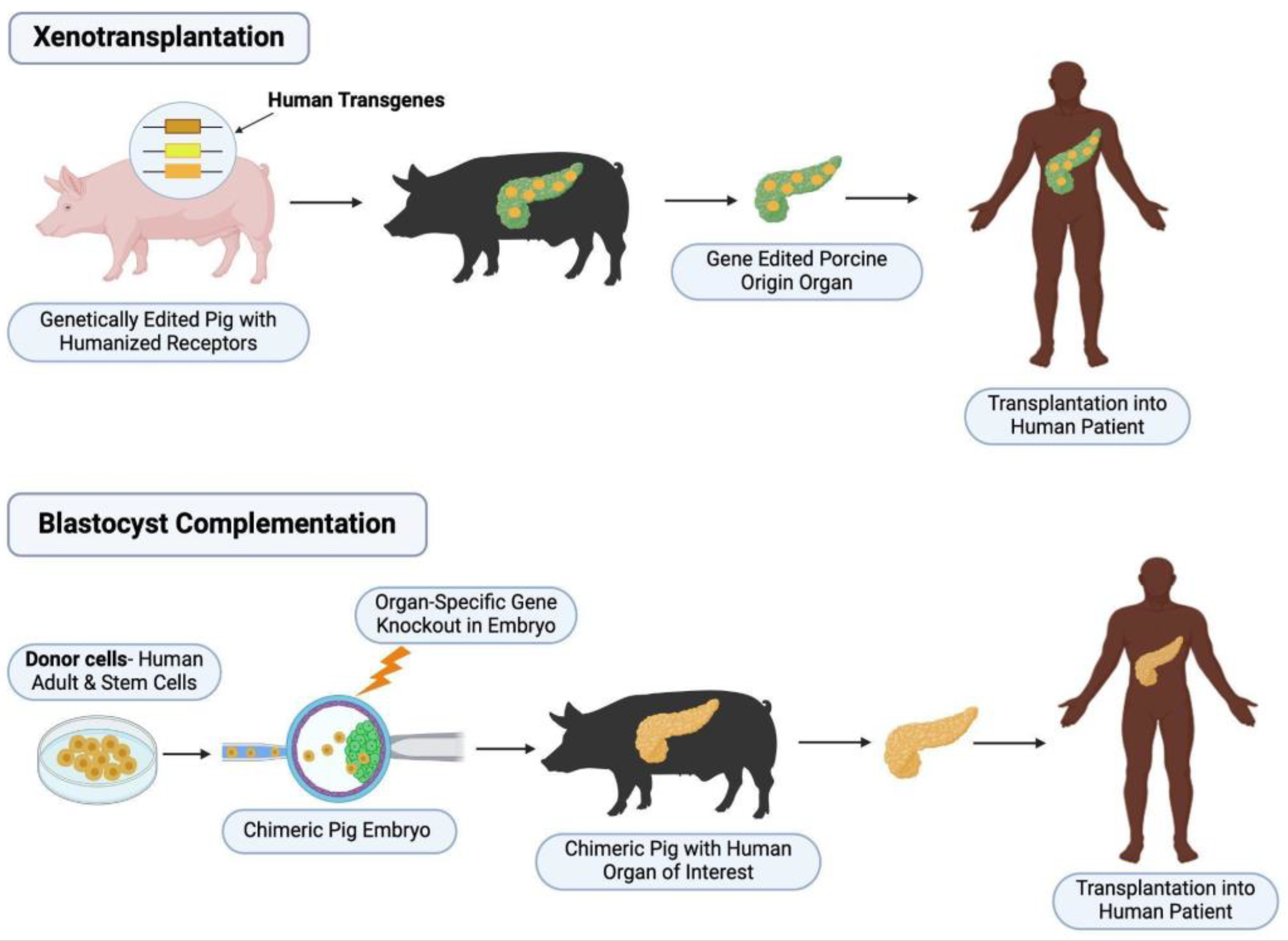
Figure 2.
Barrier to Interspecies Chimerism – Differences in Developmental Speeds Across Donor and Host Species. Early preimplantation embryos of human, marmoset mouse, and pig embryos are depicted from the zygote to the late blastocyst stage.
Figure 2.
Barrier to Interspecies Chimerism – Differences in Developmental Speeds Across Donor and Host Species. Early preimplantation embryos of human, marmoset mouse, and pig embryos are depicted from the zygote to the late blastocyst stage.
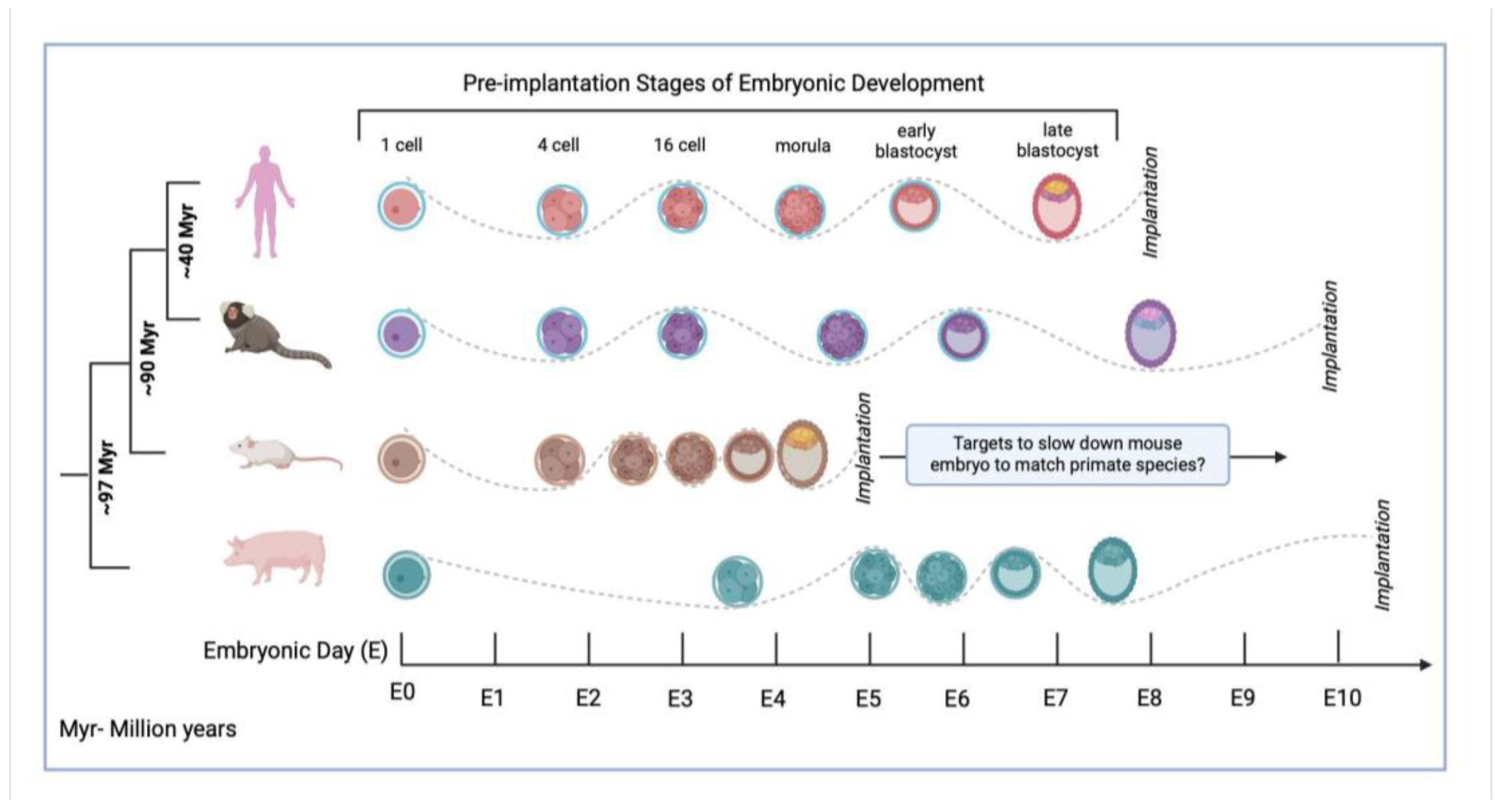

Table 1.
Summary of results of all 22 studies, including organ generated, species of host and donor cells used for complementation, target gene modification to achieve organ agenesis-including knock-out (KO) or gene introduction, functionality of de-novo organ, as well as survival or follow up timeline done on chimeric animals.
Table 1.
Summary of results of all 22 studies, including organ generated, species of host and donor cells used for complementation, target gene modification to achieve organ agenesis-including knock-out (KO) or gene introduction, functionality of de-novo organ, as well as survival or follow up timeline done on chimeric animals.
| Organ | Species | Gene target | Function | Survival | Reference |
|---|---|---|---|---|---|
| Liver | Pig host, pig donor | H-hex KO | Of the 4 chimeric fetuses achieved in the first round of blastocyst complementation, 1 showed normal liver. In the second round, 3 further chimeric fetuses from 95 complemented blastocysts alive at cesarean | Full-term development, alive at time of cesarean | Matsunari et al, 2020 [21] |
| Liver | Pig host, pig donor, Mouse host, mouse donor |
H-hex KO | In mice: increased survival past the embryonically lethal stage in H-hex knockout, with retarded to normal growth when compared to age-matched wild-type embryos. High degrees of chimerism present in complemented embryos. In pigs: restoration of H-hex and AFP liver-protein expression in liver cells, with high donor eGFP+ signaling in liver tissue. |
Mice: E12.5 Pig: E25 |
Ruiz-Estevez et al, 2021 [22] |
| Liver | Pig host, pig donor | Conditional H-hex KO with FOXA3 promoter | Two rounds with each 2/120 healthy fetuses with all hepatocytes of donor origin. | Fetuses collected after 21 days | Simpson et al, 2024 [23] |
| Lung | Mouse host, mouse donor | Ctnnb, Fgfr2 KO | Pulmonary function tests (Resistance, compliance, elastance, methacholine challenge) showed non-significant differences between wild-type and Ctnnb-null and Fgfr2-null. GFP+ signals were very strong in epithelial tissue, but variable in mesenchymal and endothelial cells |
For Fgfr2-null: Day 80 For Ctnnb-null: Day 50 |
Mori et al, 2019 [39] |
| Lung | Mouse host, mouse donor | Fgf10 Ex1mut./ Ex3mut | Histologically and morphologically normal lungs compared to wildtype, cells were a mix of GFP+ve embryonic stem cells and host cells | 4 months | Kitahara et al, 2020 [40] |
| Lung | Mouse host, mouse donor | Foxa2 driven fgfr2 KO | No significant difference in pulmonary function test (airway resistance, frequency, tidal volume, expiratory flow at 50% expired tidal volume). | 4 weeks | Miura et al, 2023 [41] |
| Lung | Mouse host, mouse donor | NKx2-1 KO | Rescued lung and thyroid tissue, with embryonic stem cell-derived cells expressing similar gene expression and differentiation characteristics (such as surfactant production, T1alpha expression). | Death at birth for all Nkx2-1 homogenous knockout mice | Wen et al, 2020 [42] |
| Lung | Mouse host, rat donor | NKx2-1 KO | Rescued lung tissue, with 30% of all Nkx2-1 homozygous knockout mouse-rat chimeras demonstrating 98.5% cell contribution from mouse embryonic stem cells. RNAseq showed normal gene expression profiles and cell signaling pathways in the chimeras. | Embryos harvested at E20.5 | Wen et al, 2024 [43] |
| Kidney | Mouse host, Mouse donor | Sall1 KO | Histologically and morphologically normal kidneys. Both ESC and iPSC-complemented mice showed high contribution in kidney epithelia (except collection tubules). Stromal elements (vessels, nerves) were a mix of host and donor cells. | No survival of Sall1-/- mice to adulthood | Usui et al, 2012 [52] |
| Kidney | Rat host, mouse donor | Sall1 KO | Morphologically rescued kidneys. High mouse contribution in metanephric mesenchymal cells but collecting tubules and blood vessels showed a mix of donor mouse and host rat cells. Successful connection between ureter and bladder. Decreased kidney size compared to control rats, but similar to wildtype mice. Size of glomeruli like control rats, number of glomeruli similar to control mice. | No survival of Sall1-/- mice to adulthood | Goto et al, 2019 [53] |
| Kidney | Pig host, pig donor | Sall1 KO | First attempt did not result in successful kidney development. Second attempt led to 1 chimera from 97 complemented blastocysts with histologically and morphologically normal kidney. | Fetus, Day 43 | Matsunari et al, 2020 [21] |
| Kidney | Pig host, human donor | Six1, Sall1 KO | Histologically similar mesonephros to wildtype embryos, and similar mesonephric tubule density. DsRed-labelled human-derived contribution was around 50-65% for all mesonephric cells, with over 60% in mesonephric tubules, but under 40% in mesenchyme. | Gestation was terminated at E25 or E28 | Wang et al, 2023 [54] |
| Pancreas | Mouse host, mouse donor Mouse host, rat donor |
PDX1 KO | Mouse-Mouse chimeras: Functional, histologically and morphologically normal pancreas. Pancreatic islets, ducts and exocrine tissue entirely derived from mouse donor cells in PDX1 -/- mice. When transplanted in diabetic mice, normal serum glucose and normal response to glucose tolerance test Mouse-rat chimeras: Pancreatic epithelia was fully composed of rat-derived cells. Of the 2 chimeras that reached full maturity, histological and morphological analysis was normal, with normal serum glucose levels and glucose tolerance testing. |
PDx1-/-Mouse-Mouse: 60 days post transplantation of iPSC-derived pancreas into diabetic mice PDX1 -/- Mouse-Rat: 8 weeks (only 2/10 survived to adulthood) |
Kobayashi et al, 2010 [64] |
| Pancreas | Pig host, pig donor | Introduction of Pdx1-Hes1 transgene | Histologically normal pancreas, almost all pancreatic cells derived from donor cells. Normal serum glucose levels, with 1 chimeric pig showing normal oral glucose tolerance test. | Minimum of 12 months | Matsunari et al, 2013 [66] |
| Pancreas | Rat host, mouse donor | Pdx1 KO | Morphologically normal pancreas, homogeneously expressing mouse-derived cells. Supporting tissue did demonstrate host rat-derived cells. Mouse-derived pancreatic islets were then re-transplanted in mice, with normal glycemic levels during follow up. Islet cells showed successful hormone secretion with insulin, glucagon, somatostatin expression. | Normal glycemic levels for up to 370 days following transplantation of pancreas derived from blastocyst complementation | Yamaguchi et al, 2017 [65] |
| Pancreas | Pig host, pig donor | Pdx1 KO | Histologically, morphologically normal pancreas in 2/4 chimeric animals. In pigs with successful pancreatic rescue, high levels of chimerism could be shown. | Full-term fetuses | Matsunari et al, 2020 [21] |
| Heart | Mouse host, mouse donor Mouse host, rat donor |
Nkx2.5-Cre and Tie2-Cre dependent DTA (diptheria toxin A) | Mouse-mouse chimera: Donor-derived endothelial cells and cardiomyocytes, with normal functioning hearts in 8 chimeras. No signs of fibrosis. In 3 chimeras, cardiomyocyte area and vascular density comparable to control. Rat-mouse chimera: Heart complementation with almost complete donor derived cardiomyocytes in Nkx2.5-Cre mice at E10.5. Unsuccessful heart or vascular system complementation in Nkx2.5-Cre;Tie-2Cre mice in later developmental stages (E11.5, E14.5). |
Mouse-mouse chimera: up to adulthood Rat-mouse chimera: E10.5 (Nkx2.5-Cre), E10.5, E11.5, E14.5 (Nkx2.5-Cre;Tie2-Cre) |
Cappiello et al, 2023 [77] |
| Thyroid | Mouse host, mouse donor | Fgf10 Ex1mut./ Ex3mut | Morphologically and histologically normal thyroid in neonatal and adult mice. GFP expression was 86.4% +/- 7.9% in thyroid follicular cells. GFP expression did not dominate in C-cells, vasculature and connective tissue. T3 and T4 levels comparable to wildtype. | adulthood | Ran et al, 2020 [90] |
| Thyroid | Mouse host, mouse donor | NKx2-1 KO | Rescued thyroid tissue in chimeric mice, with efficient donor contribution to thyrocyte progenitor cells. | E17.5 | Wen et al, 2021 [42] |
| Thymus | Mouse host, mouse donor | FOXN1 KO | Rescued thymus in 11 mice. In 2 mice examined, 98 and 96.9% of thymic epithelial cells were donor derived. Compared to normal mice: no significant difference in number of peripheral T cells, or gene-expression profile. In splenic T cells, no significant difference in CD4+ or CD8+ T cell proliferation or production of IFN gamma, IL-2, Granzyme B with anti-CD3 stimulation. Under anti-PDL1 treatment: suppression of MC38 tumor growth and increased IFNgamma production and T cell activation (via decrease in PD1 expression). | Up to 42 weeks | Yamazaki et al, 2022 [94] |
| Thymus | Mouse host, mouse donor | Foxa2 -driven Fgfr2 KO | Rescued thymic phenotype. Chimerism in thymus: average 92.4% (SD 5.1) in thymic epithelium, average 52.9% (SD 20) in thymic mesenchyme. | Up to 4 weeks | Miura et al, 2023 [41] |
| Parathyroid | Mouse host, mouse donor Rat host, mouse donor |
GCM2 KO |
Mouse-mouse: histologically normal parathyroids. GFP donor-derived signal was 94.6% in chief cells, 65,2% in endothelial cells and 45.6% in mesenchymal cells. Function: compared to control mice-similar plasma Calcium levels, basal PTH levels and PTH stimulation response. Gene expression level: Compared to control mice-increased GATA3, GCM2, similar levels of Mafb, Casr, PTH. Rat-mouse: rescued parathyroid phenotype, successful expression of transcription factors necessary for further development and PTH. |
Mouse-mouse: adulthood Rat-mouse: death soon after birth |
Kano et al, 2023 [95] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



