Submitted:
02 January 2025
Posted:
04 January 2025
You are already at the latest version
Abstract
Iodine(III) reagents became a highly relevant tool in organic synthesis due to the great versatility as strong but green oxidants. Several transformations involving the cyclizations as well as functionalization of different organic cores have been broadly described and reviewed. Herein a new facet which involves the participation of these reagents in photochemical transformations exclusively by direct irradition or in photoredox cycles using some transition metals, will be briefly described as well as some plausible further transformations that potentially can be developed.
Keywords:
photochemistry
; iodine(III) based reagents
; photoredox
; photocatalysis
& These authors contributed equally to this work.
1. Introduction
Iodine(III) reagents have been an excellent tool in organic synthesis. They have found exceptional applications as non-transition-metal based-oxidants which have been broadly reviewed [1-6]. Their low-toxicity and green oxidative features makes them good candidates for reactions such as halogenation [7-13], nitration [14], arylation [15-16], amination among some of the most representative.
The previously mentioned transformations concern polar or pericyclic reactions, which in general require either heat or a metal for their activation. Herein, we will summarize in a brief and concise manner, the most representative reactions involving the use of iodine(III) reagents in photochemistry from 1984 up to now. This short overview covers the activation by direct photoexcitation as well as photocatalysis using the most reported metals. Other photochemical reactions using iodine(V) reagents are out of this review scope [17].
The aim of this document is contextualizing the great advances of this synergic combination of iodine(III) reagents and photochemistry and give a plausible perspective for the future on this synthetic strategy.
2. General Considerations
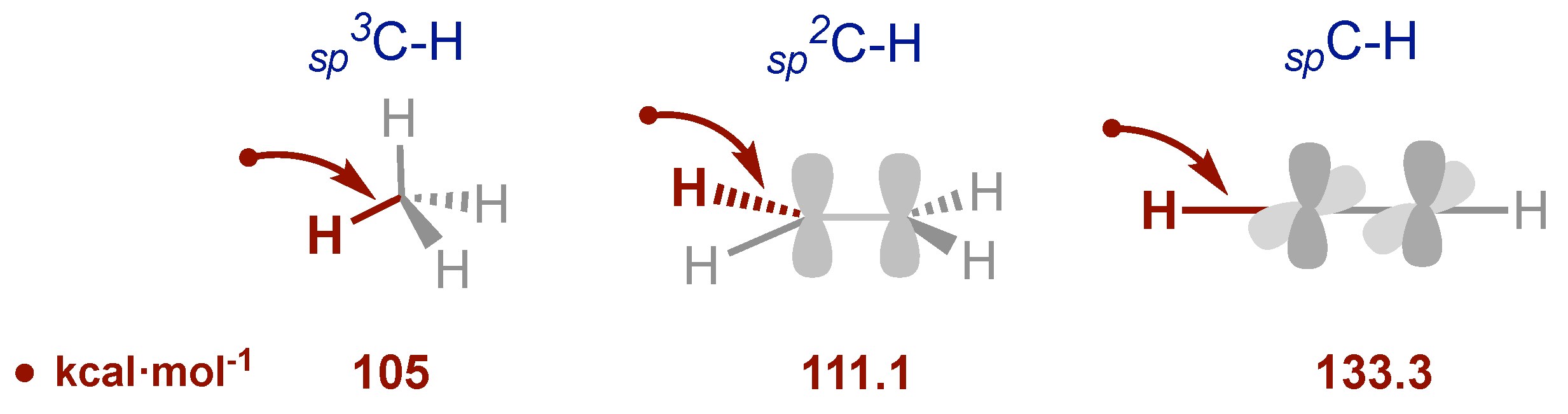
Strategies in organic synthesis for accessing the different structural motifs and relevant frameworks for keeping all the living beings, are broad and diversified [18-35]. Among some of the most important, it is possible to highlight the activation and sequential functionalization of inert and stable C-H bonds [36-37], which is a straightforward strategy for introducing different groups in a molecule. However, the process typically requires strong reaction conditions such as high temperatures or high catalytic charges for completing the cycle in metal-catalyzed transformations. Depending on the groups present, this issue could represent an inconvenience to consider. The challenge regarding the direct C-H activation, could be envisioned considering the following bond energies [38] (Figure 1).
Few synthetic tools can provide these high energy levels to carry out the previous and direct functionalization. One of the most relevant, involves the use of the innate energy from light. In this context, the energy in kcal·mol-1 supplied by a specific wavelength can be calculated according to the fundamental equation (Eq. 1):
E = 2.86 x 104 / λ
Where λ is the wavelength expressed in nanometers. Thus, it is easy to calculate that, light of λ= 210 nm gives 136.2 kcal/mol, which is enough energy to break even the strongest spC-H bond [39]. In consequence, the use of light in chemistry has been broadly used in combination with other synthetic tools. Herein we will review in a brief manner, the use of photochemistry in combination with iodine(III) reagents organized for a best understand in direct photoexcitation and photocatalysis.
3. Synthetic Methods Using Direct Photoexitation and Iodine(III) Reagents
According to our proposed organization, following it will be reviewed the most representative synthetic methods using iodine(III) regents involved in direct photoexitation.
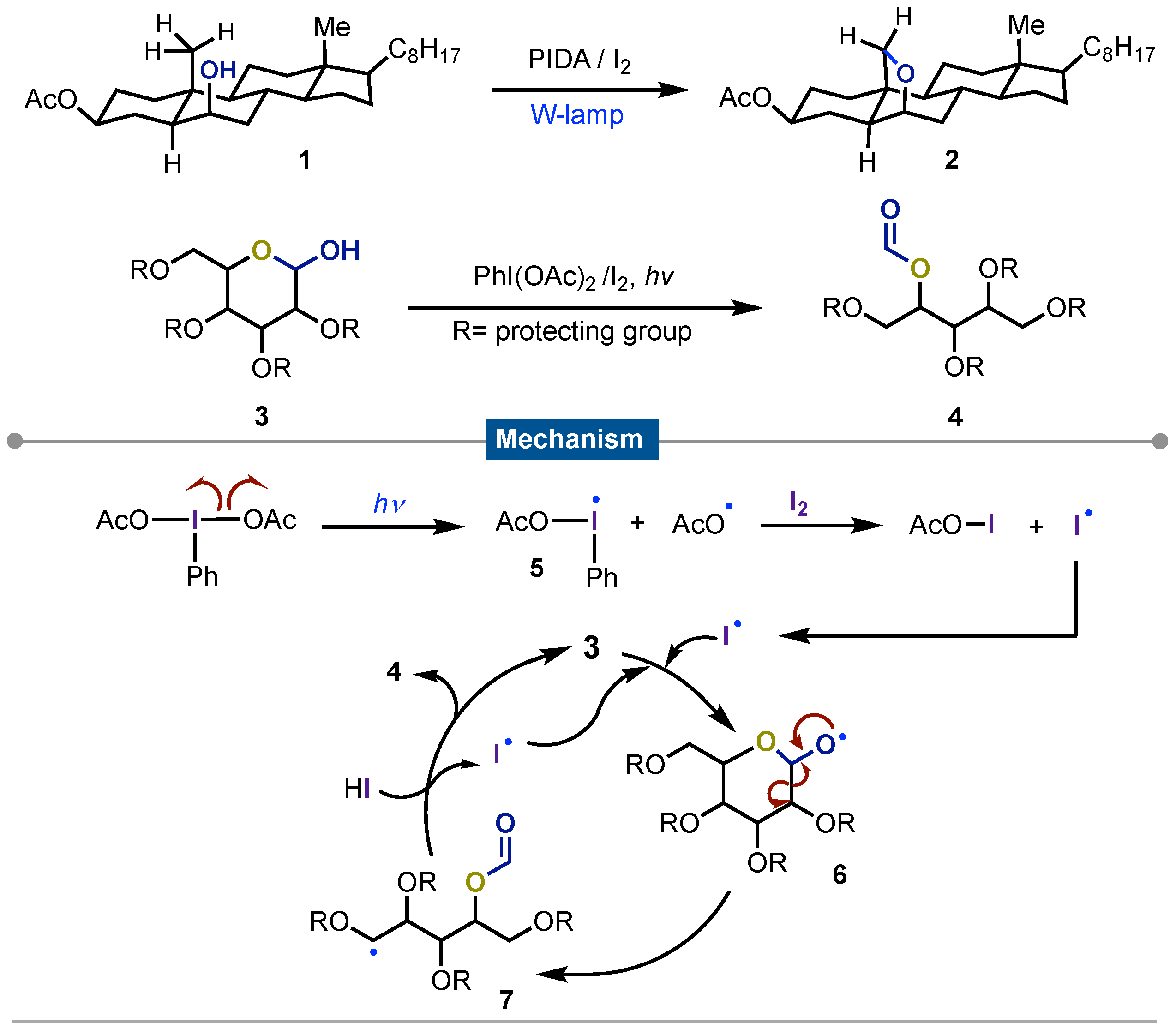
The most representative start for the combination of iodine(III) and photochemistry was described in 1984 by Suarez [40]. He reported the earliest procedure by irradiating diacetoxyiodobenzene (PIDA) in presence of molecular iodine under white light with a W-lamp for the homolysis of alcohols 1 to form new C-O bonds via [1,5]-H shift which was applied originally to get cholesterol derivatives 2. The reaction is nowadays known as Suarez-cleavage. This procedure also allowed the formation of carbonyl derivatives from cyclic carbohydrates 3 to get the ring opening products 4 [41]. On the other hand, due to the great method´s versatility, the procedure has been broadly exploited and applied to the synthesis of cyclic ethers [42] as well as for chiral spiroacetals from pyranose or furanose via alkoxy radical intermediate [43]. Several other uses have been described with this radical fragmentation. Concerning the mechanism to make C-O bonds in steroids, this started with homolysis of PIDA under light irradiation, forming iodanyl 5 and the acetoxyl radical. The former reacted with molecular iodine to form acetyl hypoiodite along with an iodine radical that reacted with sugar 3 generating oxygen-centered radical 6. This formed the radical formate 7 through hexose ring opening. Final reaction with hydriodic acid yielded the observed product 4 with the regeneration of the iodine radical that continues in nother cycle (Scheme 1).
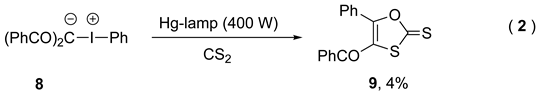
The following procedure was described by Varvoglis [44] who described the UV-photoexcitation of zwitterionic iodonium salts 7 which gave 1,3-oxathiole-2-thione 8 in reaction with carbon disulfide. Even though the yields were low, the irradiation decreased dramatically the reaction time from weeks to hours, indicating a different activation of the iodonium yilde (Eq. 2).
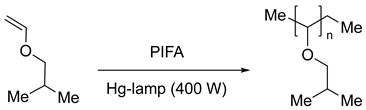
Varvoglis [45] also described the photoexcitation of PIDA and [bis(trifluoroacetoxy)iodo]benzene PIFA under the 400 W Hg-lamp irradiation that were used as initiators in the ibutyl vinyl ether polymerization (Eq. 3).
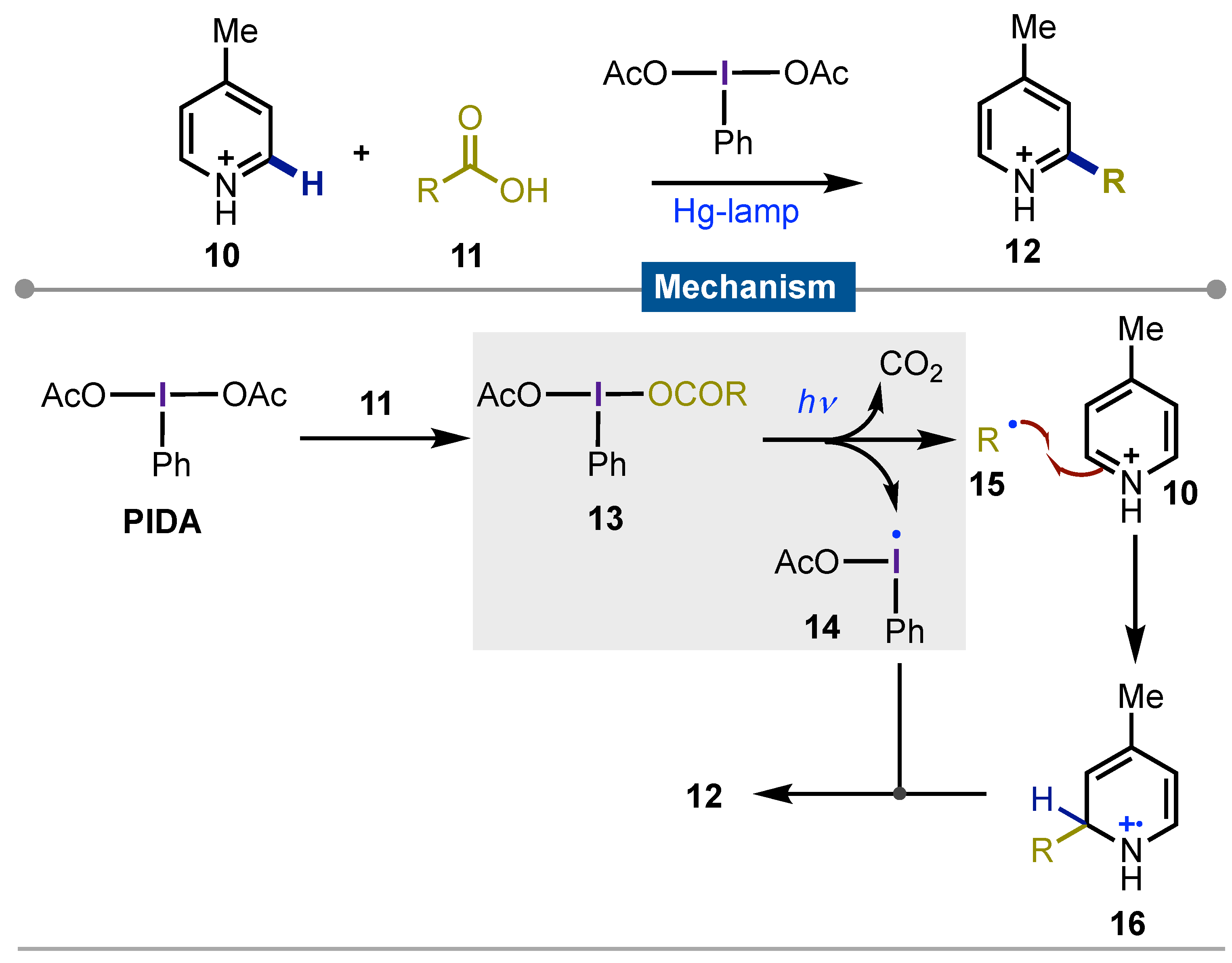
Later, Minisci [46] described the alkyl radical generation via decarboxylation of carboxylic acids 11 that reacted with pyridinium salts 10, using a low-pressure Hg-lamp to get C2 functionalized products 12. Herein the direct irradiation on PIDA derivatives 13 gave rise to iodine-centered radical 14 that after release of carbon dioxide, easily homolyzed to give alkyl radicals 15. These reacted with pyridine derivatives 10 to give C2-functionalized carbon-centered radical 16 which is oxidized by 14. During this process, aromaticity is recovered yielding the observed final pyridines in their salt form (Scheme 2).
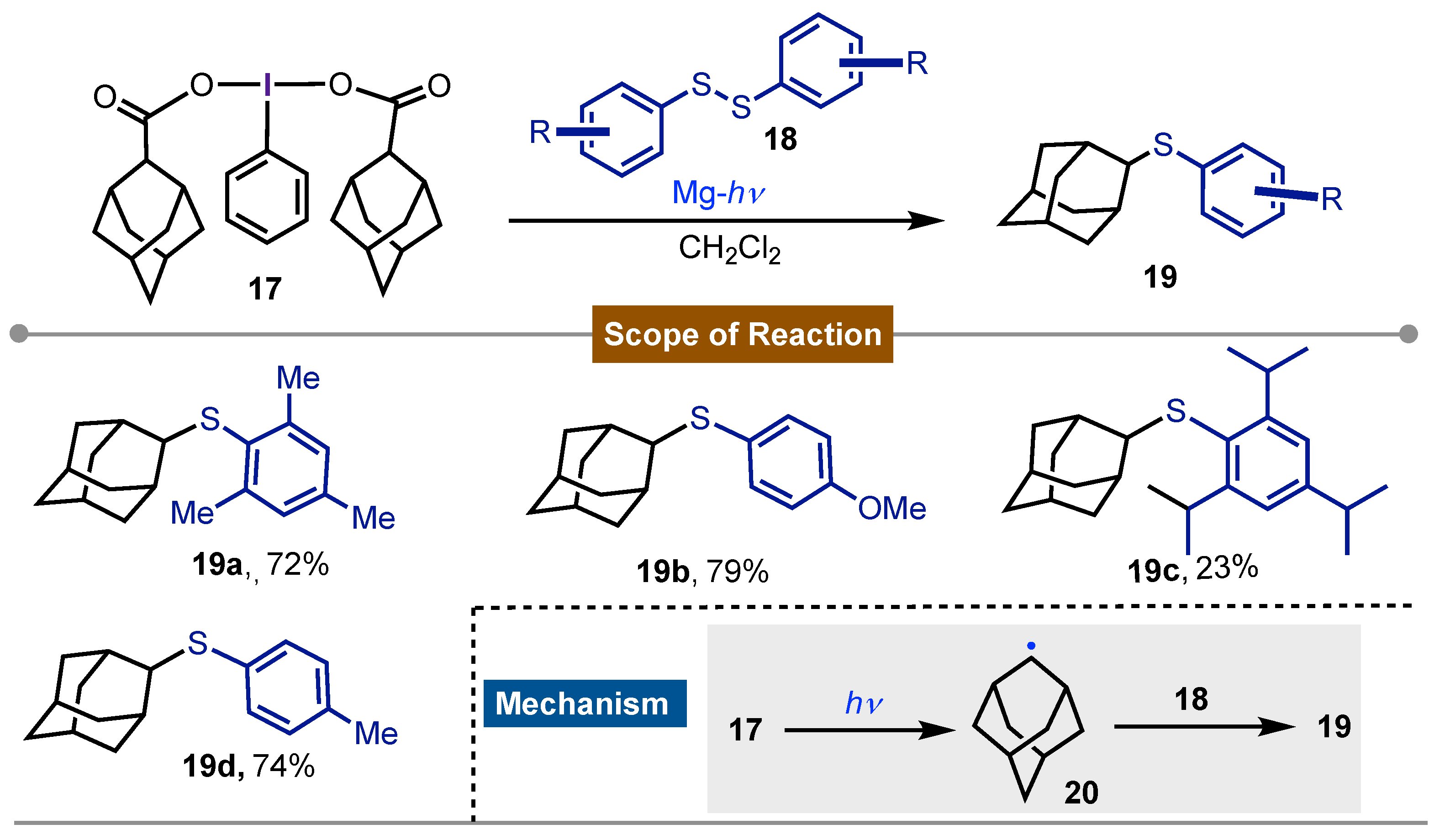
Then, in 1995 Togo and Yokoyama [47] described the synthesis of adamantyl sulfides 19 by direct irradiation of [bis(1-adamantanecarboxy)iodo]arenes 17 with a high-pressure Hg-lamp in presence of disulfides 18. The developed procedure took place via radical decarboxylation of the adamantyl-containing iodine(III) reagent 17 to give a carbon-centered radical 20 which reacted with the disulfide 18 to finally give the formation of sulfides 19a-f. The protocol tolerates a good functional group at the aryl moiety and proceeded in mild reaction conditions (Scheme 3).
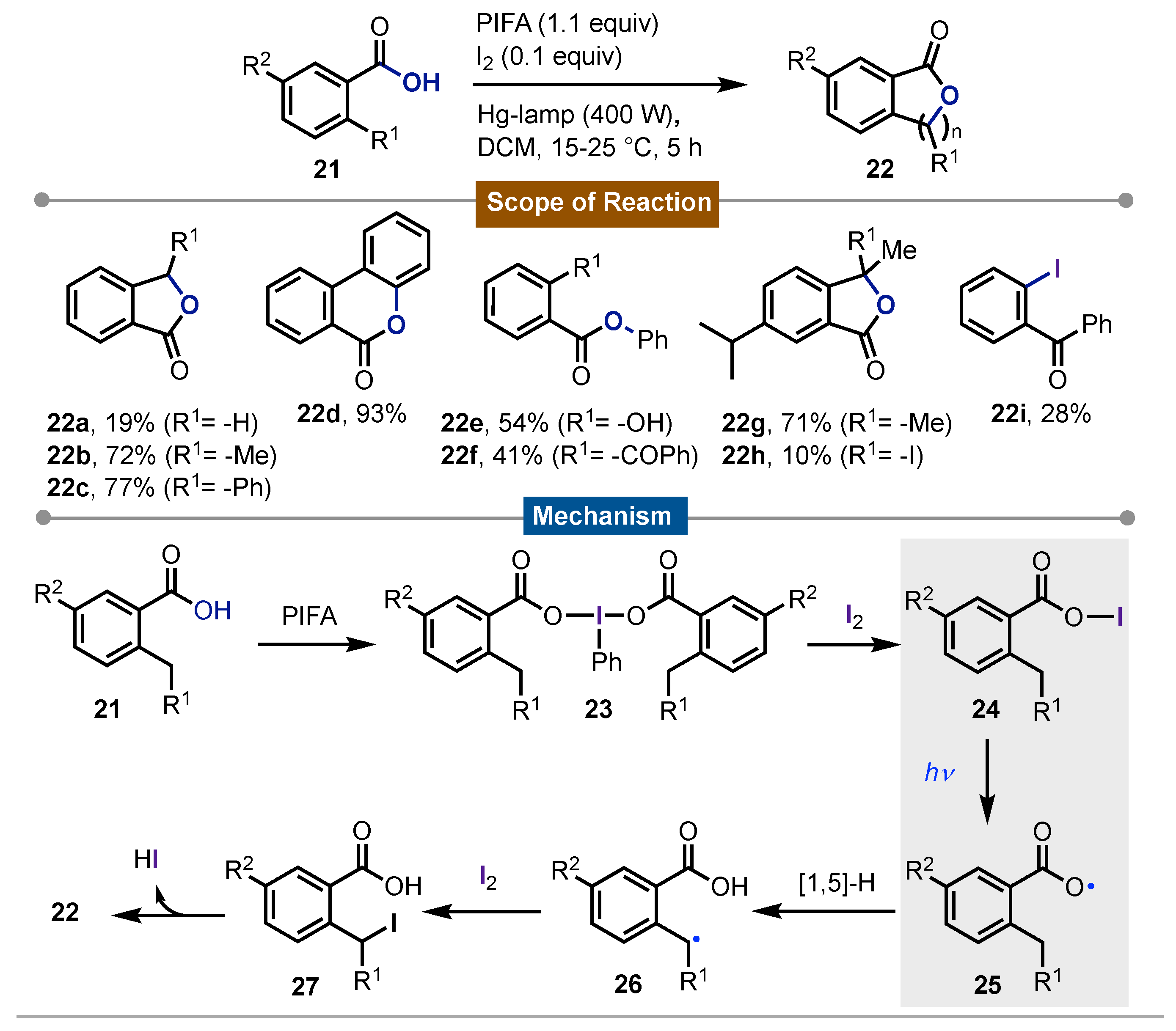
Two years later, the same authors reported [48] another photochemical homolytic cleavage on iodine(III) reagents. In this development some o-alkyl and o-aryl-benzenecarboxylic acids 21 and alcohols were subjected to Suarez´s conditions to yield heterocycles such as 22. The direct irradiation of the hypoiodite 24 coming from the formed in situ iodine(III) derivatives 23, led to the formation of several oxygen-centered radicals 25 that intramolecularly reacted via [1,5]-HAT in a direct sp3C-H bond activation of the o-alkyl group to form the corresponding benzylic carbon-centered radical 26. Then reaction with molecular iodine gave 27. The final intramolecular cyclization gave rise to the formation of different cumarine, phtalide and benzocumarine derivatives 22a-i with the concomitant hydroiodic acid release. An excellent scope was presented in this work (Scheme 4).
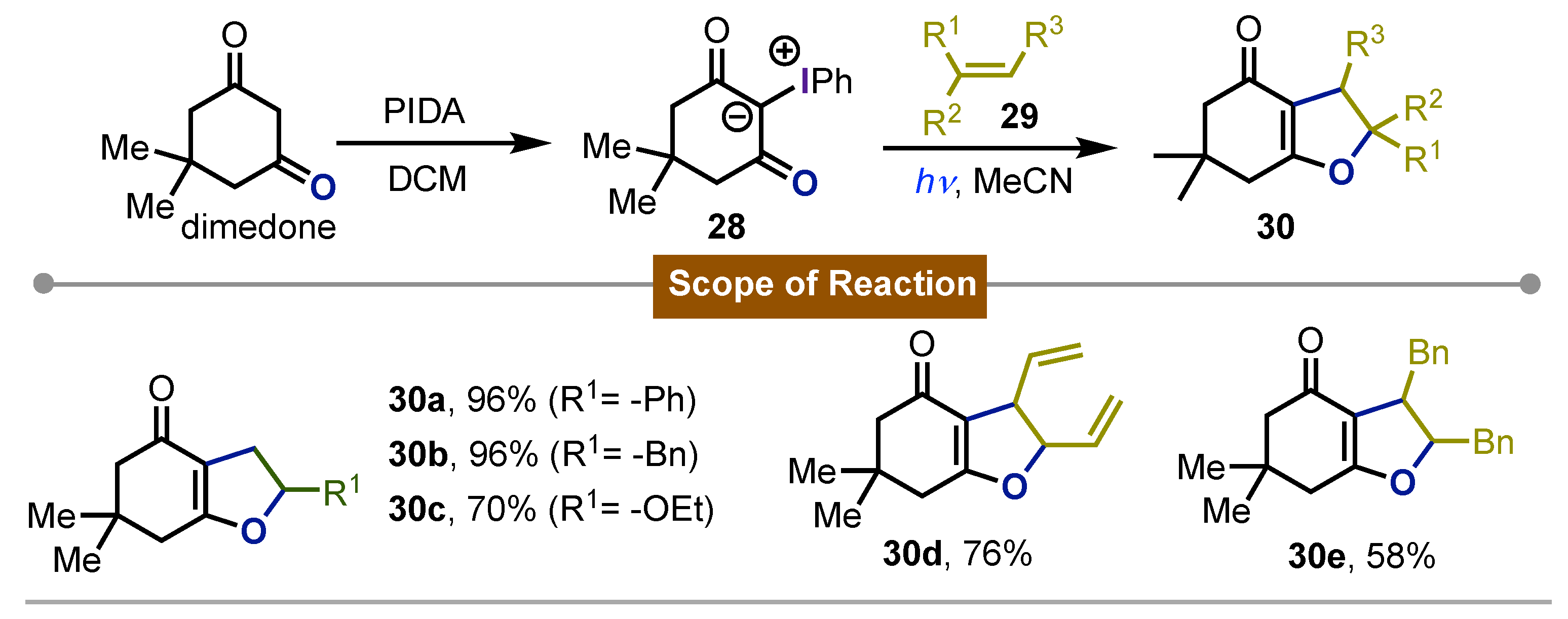
In 2000, Hadjiarapoglou [49] carried out the direct photoexcitation of phenyl iodonium ylide 28 obtained from dimedone using a 400 W medium pressure Hg-lamp; in the presence of different alkenes 29. The reaction led to the formation of dihydrofuranes 30 in good to high yields via cycloaddition. This process was regio- and stereoselective, highlighting the isolation of a single isomer where the oxygen of the ylide bonded exclusively at the more substituted side of the alkene in the final products 30a-e. Interestingly the olefin stereochemistry was not preserved, which could probably imply a stepwise reaction mechanism. Several other transformations involving iodonium ylides have also been reviewed (Scheme 5).
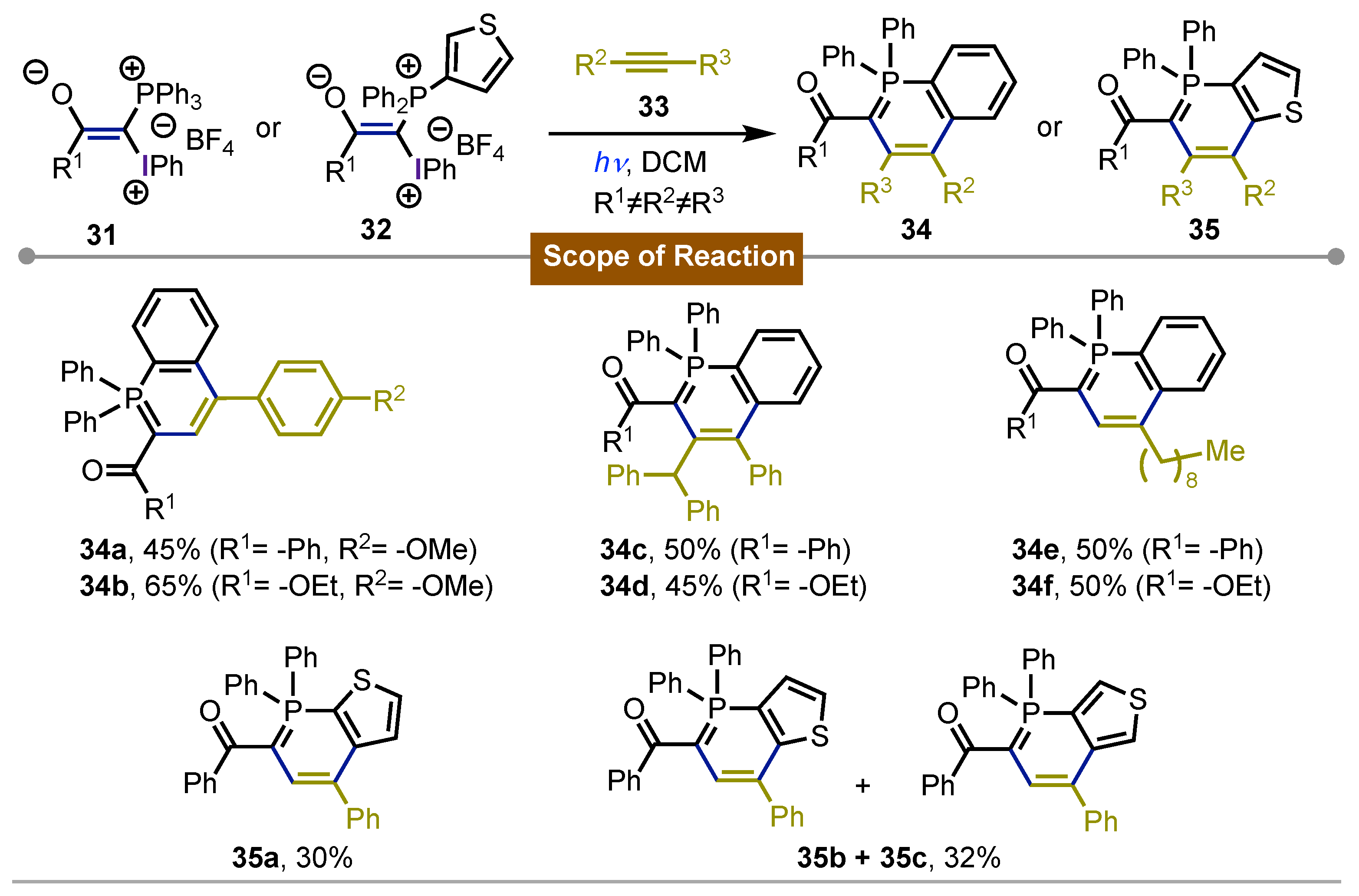
In 2009, Matveeva [50, 51] carried out the synthesis of several λ5-phposhinolines 34 and 35 by the photochemical reaction between acetylenes 33 and mixed phosphonium-iodonium ylides 31 or 32. This rare class of heterocycles were obtained under the direct irradiation with a Hg-lamp at 366 nm. The few protocols to obtain such aromatic phosphorus-contained derivatives 34a-f in very mild reaction conditions were the key features in this protocol. The incorporation of a thiophene ring in the mixed ylide 31 is a variant of the method that enabled the formation of the corresponding thiophene-fused λ5-phoshinolines 35a-c. The full mechanistic investigation regarding this photochemical reactivity was reported in the following year [52, 53] (Scheme 6).
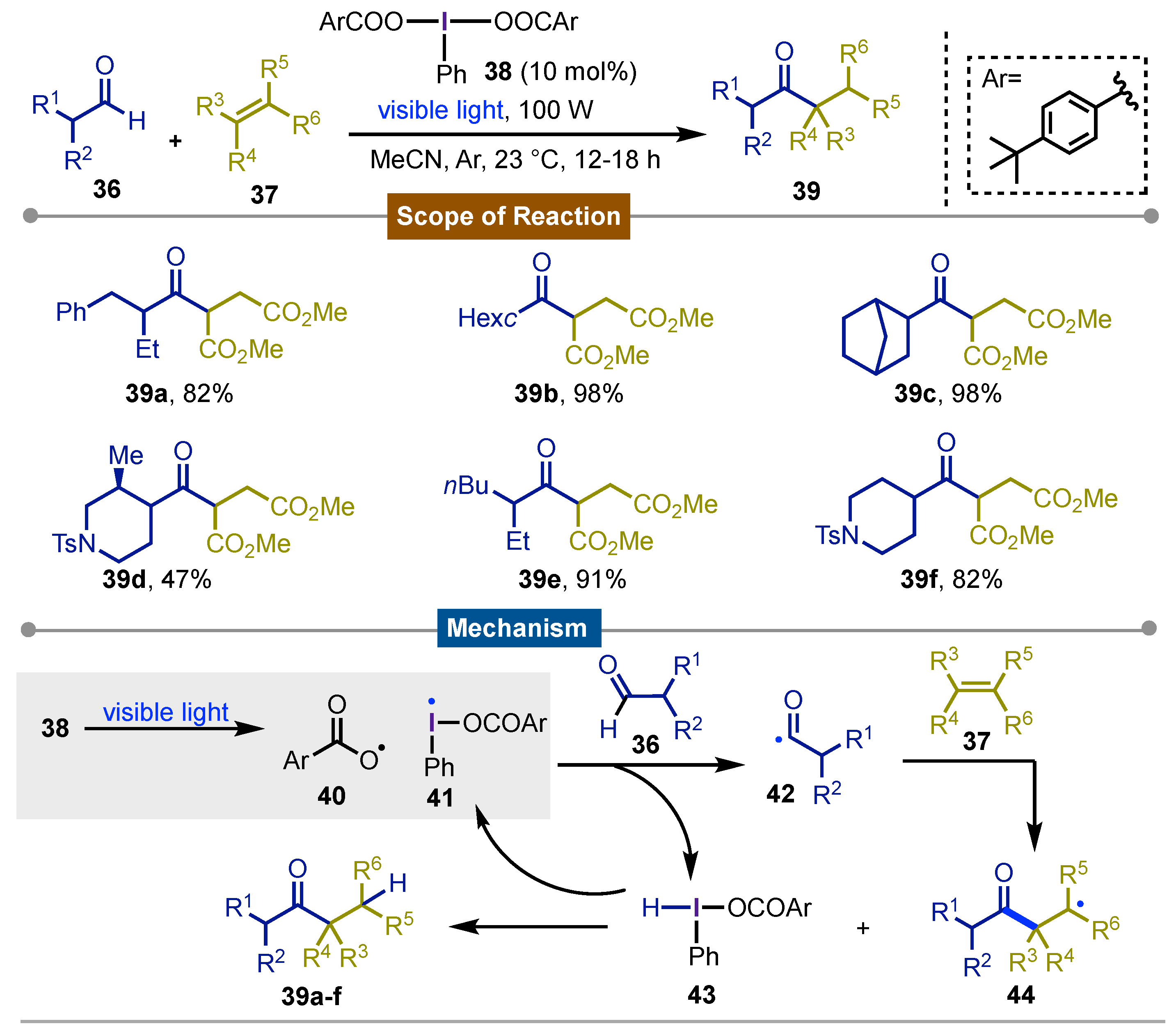
The direct hydroacylation using branched aldehydes as starting materials via acyl radical formation, is a tremendous challenge due to the easy decarbonylation of the formyl substrates. Accordingly, a practical solution to this problem was described by Maruoka in 2014 [54]. In this work, different acyl radicals 42 were softly generated from the reaction of branched aldehydes 36 with iodanyl 41 and/or acyloxy radicals 40 which were obtained from the photochemical homolysis of iodine(III) derivatives 38 under visible light irradiation (400 nm). Then, a protonated iodine(III) derivative 43 was formed along a transient acyl radical 42 which was trapped by dimethyl maleate derivatives 37. The former reaction led to the formation of the corresponding adduct with the radical transfer to the maleate moiety to get a carbon-centered radical 44. Finally, in a radical pathway, 43 carried out a HAT regenerating iodanyl radical 41 which continued in another cycle with the concomitant formation of observed products 39a-f.
Scheme 7.
Iodine(III) catalyzed, photochemical hydroacylation of aldehydes.
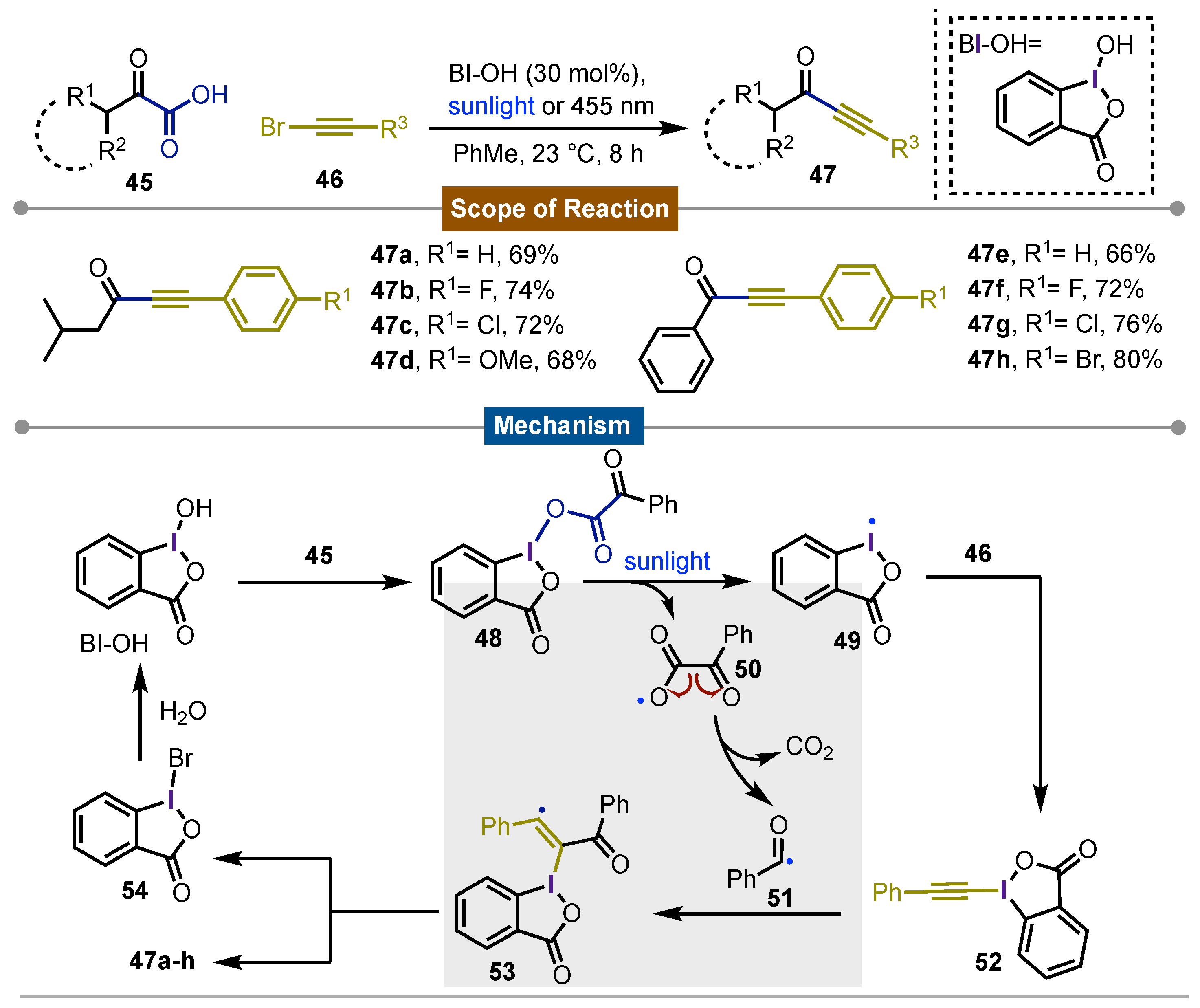
The following work presented by Li and Wang [55], described the synthesis of ynones 47 using 2-oxo-2-phenylacetic acid derivatives 45 and terminal bromoacetylenes 46 in presence of catalytic amounts (30 mol%) of BI-OH. This iodine(III) reagent was photoexcited using sunlight or a blue led (455 nm) at room temperature. The reaction mechanism started with the interaction between BI-OH and 45 to bond the oxalic functionality to iodine(III) reagent obtaining 48. This intermediate homolyzed giving iodanyl radical 49 and oxygen-centered radical 50. In one side 49 reacted with bromoacetylenes 46 forming new iodine(III) reagent 52. On the other hand, homolytically fragmentation of 50 released carbon dioxide and acyl radical 51 which reacted with 52 leading to the formation of iodine(III) adduct 53. Next, a radical cleavage released the observed products 47a-h with the concomitant formation of 54 after the reaction with radical bromine. Final hydrolysis regenerated BI-OH. In general, this protocol represented a novel approach in organic synthesis in terms of the energetic economy considering the endless sunlight. The broad scope of this procedure and the good chemical yields were the most representative features (Scheme 8).
In 2016, Wang [56] described the decarboxylative acylarylation of acrylamides 55 using α-oxocarboxylic acids 56 in presence of catalytic amounts of BI-OH as the iodine(III) reagent. This procedure was applied to the synthesis of several 2-oxoindiolines 57 with different α-acyl aromatic derivatives. The optimal conditions implied the irradiation of BI-OH with a blue led obtaining good to excellent yields. Limitations include the presence of amino and some ester functionalities in the aromatic ring. In general, the reaction mechanism involved the formation of acyl radical 59 via blue led irradiation-promoted decarboxylation of iodine(III) oxalic derivatives 58 formed by reaction of BI-OAc with 56. On one side, acyl radical 59 was formed which reacted with 55 in a Markovnikov fashion, leading to the acyl group bonded to the double bond with the concomitant indoline formation. Ring closing ensued with the formation of non-aromatic aryl radical 60. On the other side, after decarboxylation, iodanyl radical 49 was also formed which aromatized the previously formed radical to continue with another cycle along the formation of the observed products 57a-e (Scheme 9).
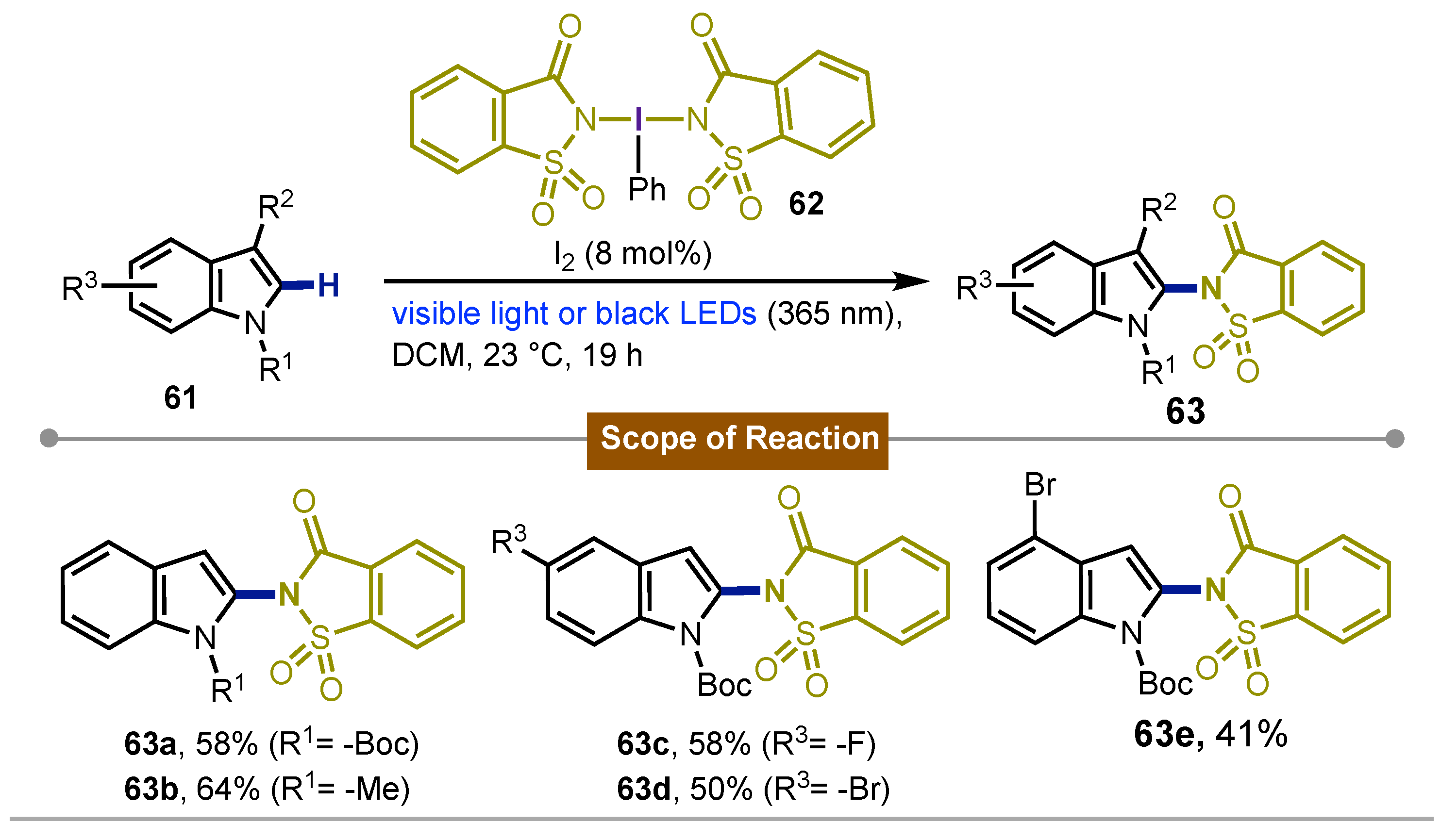
In 2018 Muñiz [57] described an interesting C-2 selective amination of indoles 61 as well as the amination of several heterocycles. Iodobenzene bis(saccharinato) 62 and 8 mol% of molecular iodine were used under black light irradiation (365 nm). The process yielded several C2 aminated indoles 63a-e with a big scope obtaining chemical yields ranging from poor to excellent (26-98%). A radical process was proposed in this development (Scheme 10).
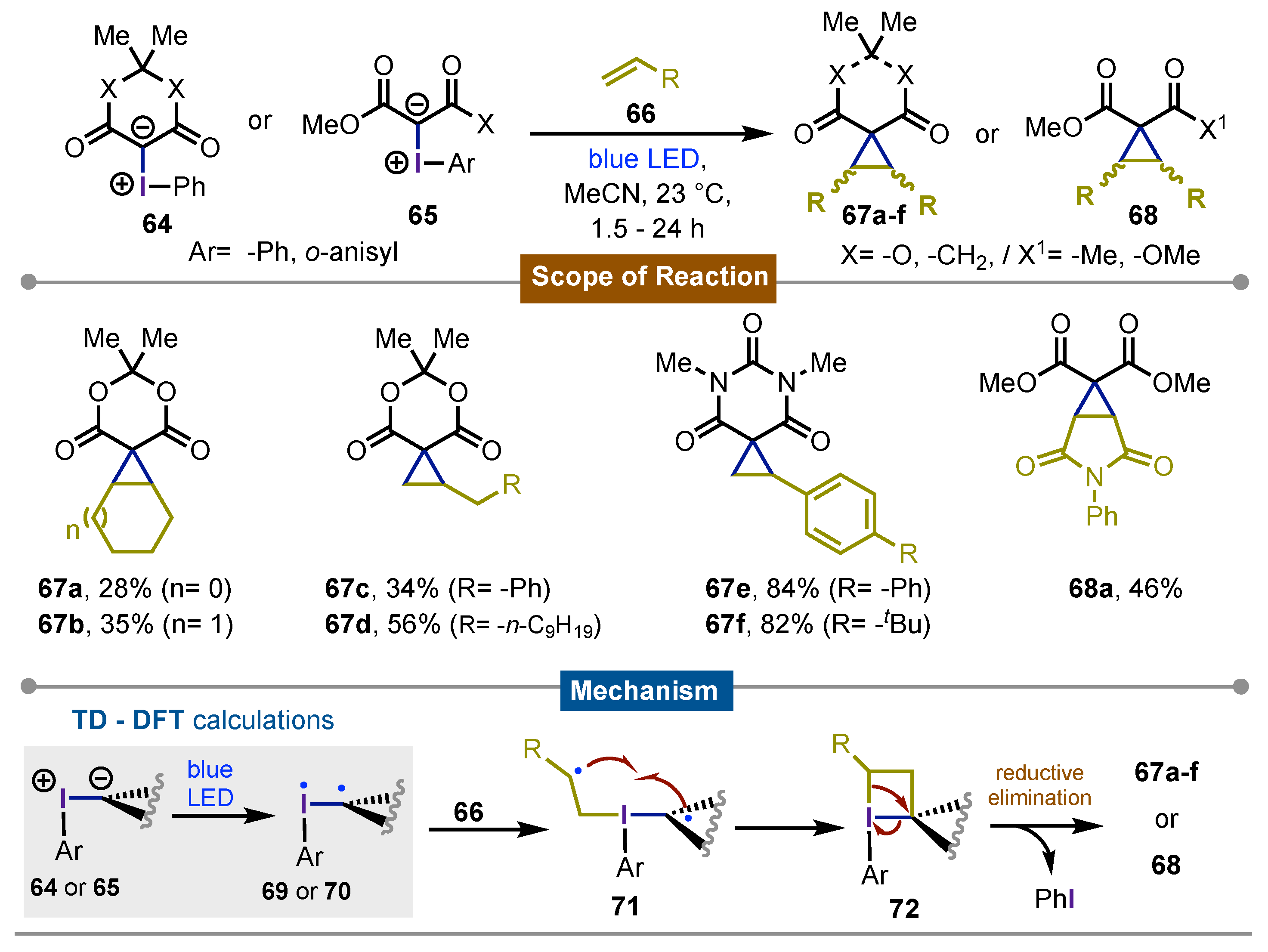
In 2019, Quideau, Hopkins and Murphy [58], described representative an interesting cyclopropanation reaction using iodine(III)-based dimedone-derived ylides 64 or ylides 65 upon direct irradiation with blue LEDs. The development was systematically conducted starting with (time-dependent) TD-DFT calculations of the HOMO and LUMO to determine the electronic population of the molecule during the time irradiation. Therein it was identified the biradical nature at the iodine(III) and the carbon to which it was bonded. These results indicated that the carbene formation by an heterolytic cleavage was not operating.
Then, several reactions of iodonium ylides and different styrenes 66, gave rise to the formation of dimedone-derived 67 as well as some open 1,3-dicarbonyl-based cyclopropane derivatives 68 in excellent yields. The proposed reaction mechanism started with ylide irradiation to give the biradical intermediate 69 or 70, this upon reaction with styrenes 66 generated intermediate 71, that in a step-way fashion intramolecularly closed via radical pairing to get the four-membered iodane 72. Final reductive elimination of iodobenzene yielded the observed cyclopropanes 67a-f and 68 (Scheme 11).
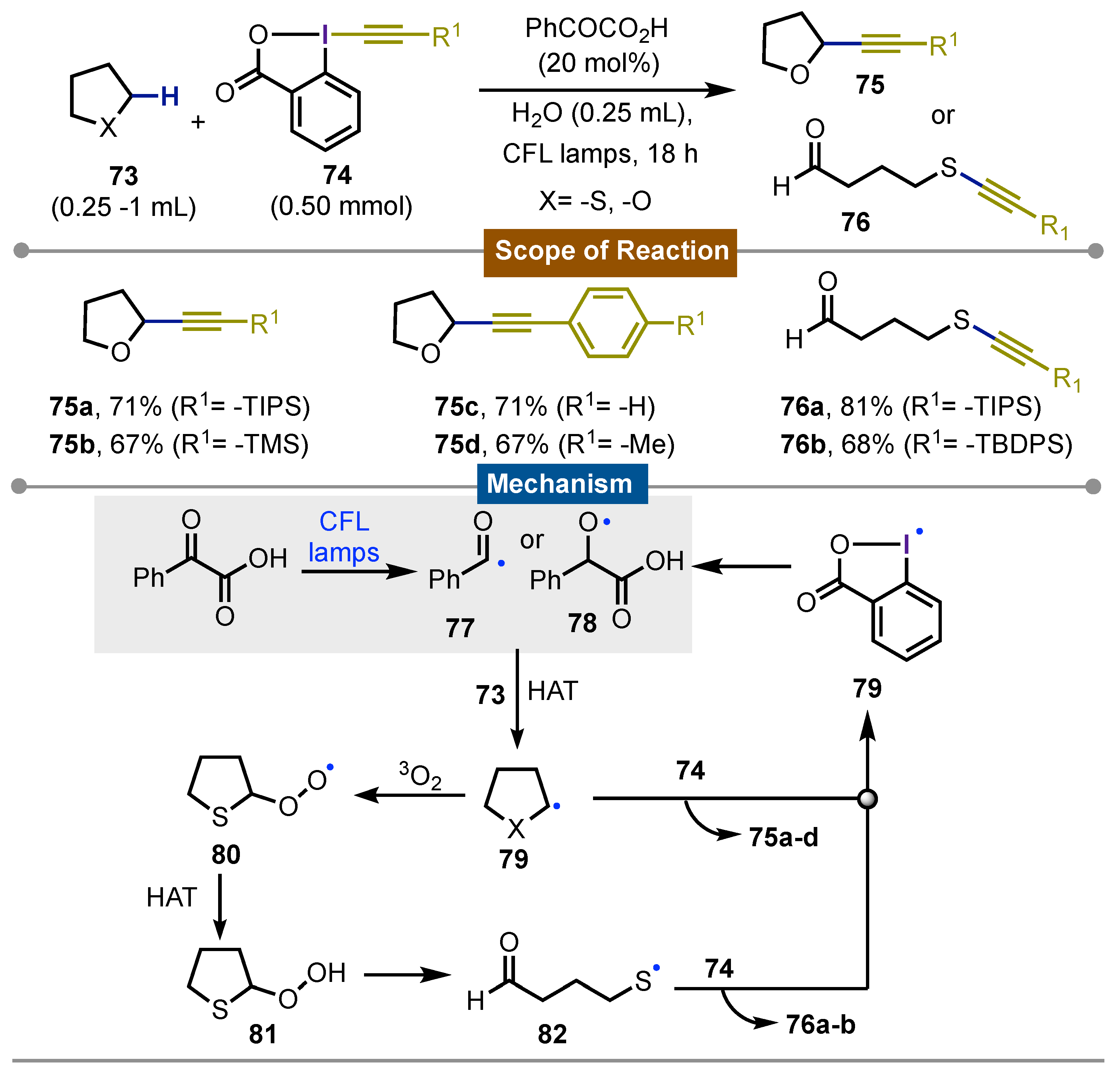
In early 2020, Waser and Kokotos [59], described a versatile and useful method for the direct sp3C-H alkynylation of aliphatic cyclic ethers and thioethers 73. The process took place with irradiation of a CFL household bulb using 20 mol% of phenylglyoxalic acid as catalyst and alkynylbenziodoxolones (EBX) reagents 74 as the alkynyl transferring counterpart. The protocol gave rise to the formation of C2-alkynylated cyclic ethers 75. Overall the procedure was efficient and with an interesting result when it was applied to cyclic thioethers. In this case, alkynylation at the sulfur atom occurred with the concomitant ring opening, giving to the linear fragment with the formation of an alkynyl thioether in one side and with an aldehyde in another side of molecule 76. Remarkably no oxidation of the sulfur atom was observed as described by other procedures. Concerning the reaction mechanism, a complex proposal was described. However, the most relevant steps included acyl radical 77 formation from phenylglyoxalic acid or its triplet biradical 78 state formation under the CFL light. This carries out a HAT at the sp3C2-H of cyclic ethers to form the corresponding alkyl radical 79. This reacts with the EBX derivatives 74 releasing the observed alkynylated product 75a-d along to an iodanyl radical 79 which continues the radical propagation with phenyl phenylglyoxalic acid. On the other side, the same path is proposed for the cyclic thioethers, up to the radical 79 formation. Therein, an initial reaction with 3O2 led to the formation of peroxide radical 80 that protonates to get 81 which enabled ring opening to give an aldehyde moiety and sulfur centred radical 82 that continued its reaction with EBX 74 to yield the observed products 76a-b (Scheme 12).
In 2023, Chen and Yang [60] developed a visible-light-induced radical strategy for accessing 2-oxoindoles containing the fluoromethyelensufonyl moiety. Photochemical homolysis of PhI(OCOCH2F)2 83 gave rise to the ·CH2F radical 85 and DABSO as the SO2 source. The obtained C3-functionalized oxindoles 84, were synthesized starting from N-arylacrylamides 82 in overall good yields. Some relevant features such as catalyst-free, photochemical activation, mild reaction conditions, broad functional group compatibility, and good to excellent yields, highlighted this work. The proposed mechanism started with the generation of fluoromethylene radical 85 by homolysis of a single ligand in the iodine(III) reagent 83 giving also the iodanyl radical 86. Formed radical 85 reacted with DABSO to get a new fluoromethylene sulfonyl radical 87 which interacted with the N-arylacrylamide 82 to close the indoline ring giving rise to non-aromatic radical 89 via intermediate 88. Final aromatization with iodanyl radical 86 yielded the observed final products 84a-e (Scheme 13).
In 2023, Ma and Liu [61], described the fluorosulfonylation of terminal alkanes using the corresponding carboxylic acids 90 and their carboxyiodobenzene derivatives formed in situ by mixing with PIDA. Photochemical irradiation of these formed reagents with blue LEDs using 4CzIPN (2,4,5,6-tetra-(9H)-carbazol-9-yl) as photocatalyst in combination with DABSO and KHF2 as sulfonyl and fluorine sources respectively gave rise to the corresponding fluorosulfonylated alkyl derivatives 91. The observed chemical yields for 91a-d were modest to good while the reaction mechanism is similar to proposed by Chen and Yang (Scheme 14).
A recent report in this section was described by Zhu [62] in 2024. Herein the direct azolation of inert sp3C-H bonds from cyclic ethers was developed. The rationalization of this process was carried out through a systematic radical polarity analysis framework for the projection of the radical reactivity patterns. Accordingly, blue LED irradiation of PIDA in the presence of benzo[b]pyrazoles 92 and different ring-size cyclic ethers 93 lead to the formation of the observed direct azolation products 94 in good yields. The protocol displayed in general good regiochemistry at N1 (Scheme 15).
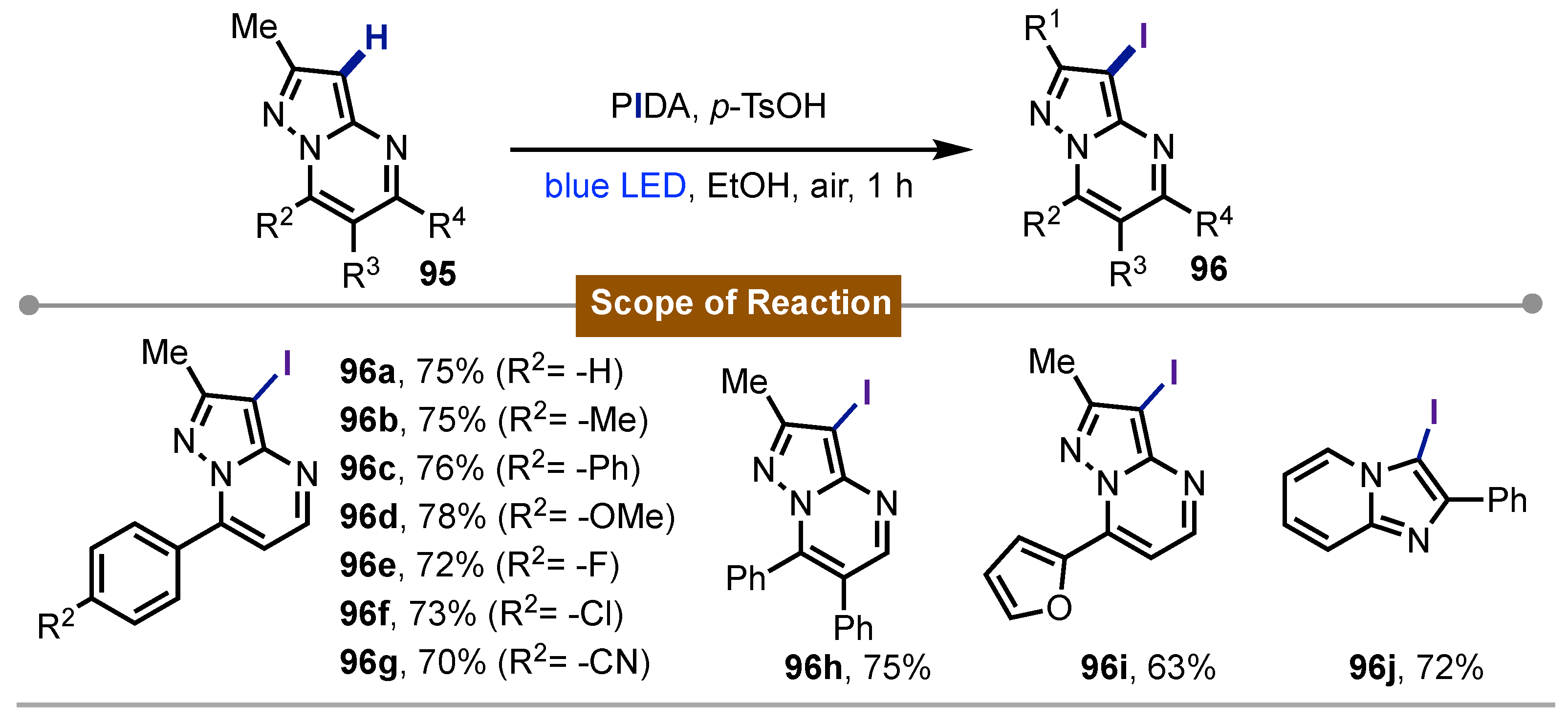
The most recent work at this time was published by Bagdi [63] in 2024. In this study, is described the iodination of imidazopyridines 95 derivatives, using PIDA as iodine(III) reagent, which acted as a source of the iodine atom in this methodology. The system formed by PIDA and p-TsOH, under irradiation with blue LED light in the presence of oxygen led to the formation of iodinated products 96. This novel methodology was photocatalyst-free, and its scope includeed the iodination of electron-donating and electron-withdrawing groups, with iodinated products obtained in moderate to good yields (Scheme 16).
4. Photocatalytic Synthetic Methods using Iodine(III) Reagents
Following our work organization, it will be reviewed the most relevant synthetic protocols with iodine(III) regents involved in direct photoredox cycles using transition metals.
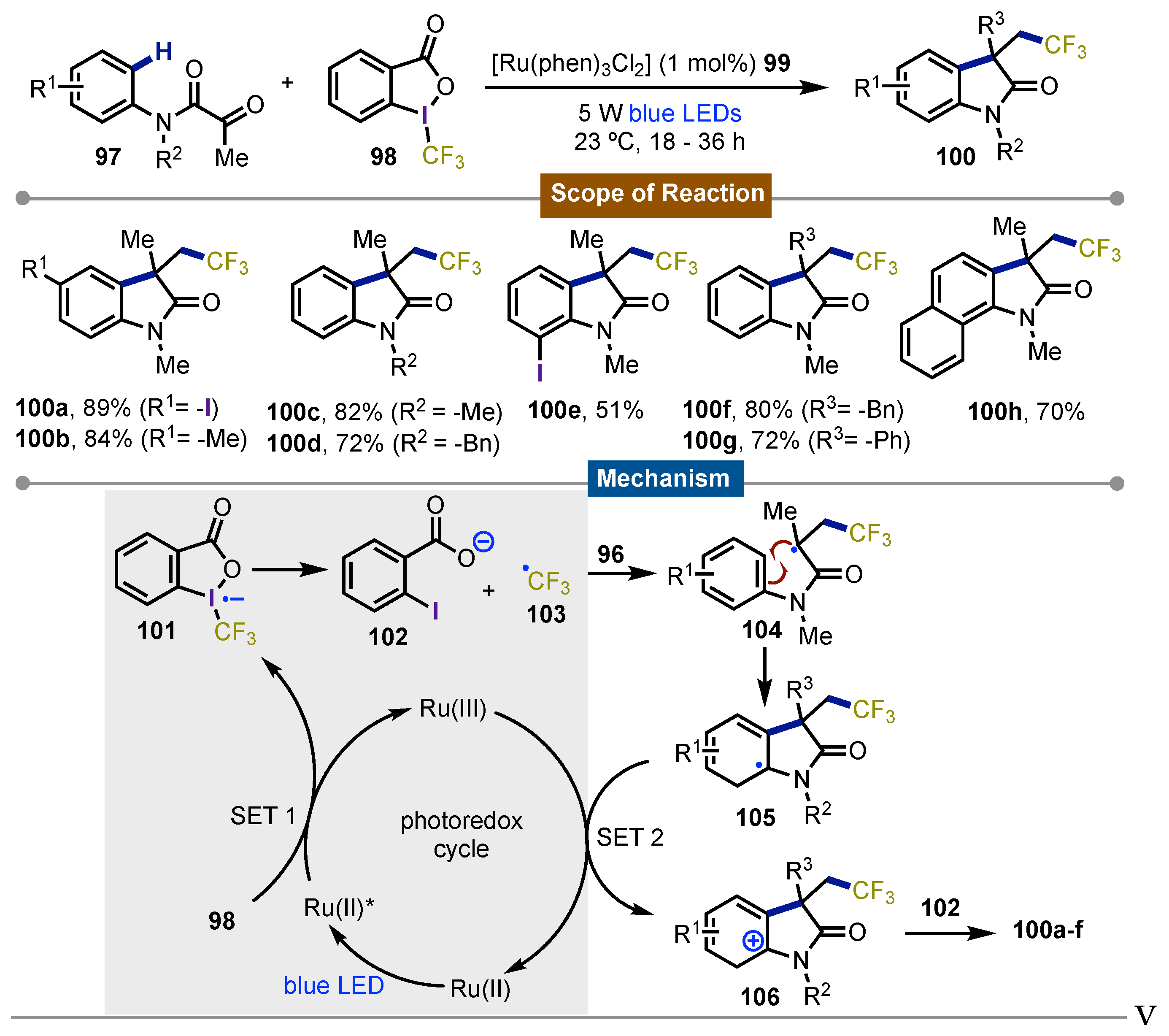
The pursuit of the trifluoromethylation of molecules lead Zhu [64] in 2013 to develop a protocol for the visible-light-promoted carbotrifluoromethylation of electron-withdrawing alkenes for the preparation of oxindoles bearing a quaternary center. Using Togni’s reagent 98 as the CF3 source, N-aryl acrylamide derivatives 97 were transformed into their corresponding oxindoles 100 using [Ru(phen)3Cl2] (phen=phenanthroline) 99 as the photocatalyst. The N-acrylamide derivatives were protected on the nitrogen atom in order the reaction to proceed. The protocol tolerated substituents on the aromatic ring, and even some heterocycles. Also, different α-substituted olefins gave the desired product in moderate to good yields (51-91%). Addition of TEMPO as radical scavenger hinhibeted the reaction and just small amount of product was obtained. This supported the hypothesis that a radical pathway was operative in the process. In the presence of electron-donating groups on the meta position of the aromatic ring, the reaction was regioselective for the most hindered carbon, suggesting that the rate-limiting step was the formation of a cationic intermediate stabilized by the substituents present in the aromatic ring. The proposed mechanism involved the formation of the radical anion of Togni’s reagent 101 via reductive SET enabled by Ru(byp)32+. This intermediate collapsed giving rise to formation of a ·CF3 radical 103 and 2-iodobenzoate 102. Then the reaction of ·CF3 with N-aryl acrylamide derivatives 97, forms the carbon-centered radical intermediate 104. Next, a radical C-H functionalization cascade ensued forming a radical oxindole 105 which was oxidized through a second SET process catalyzed by Ru(III) to give the key cationic intermediate 106. Final deprotonation of 106 assisted by 102 gave rise to the observed final products 100a-h (Scheme 17).
v
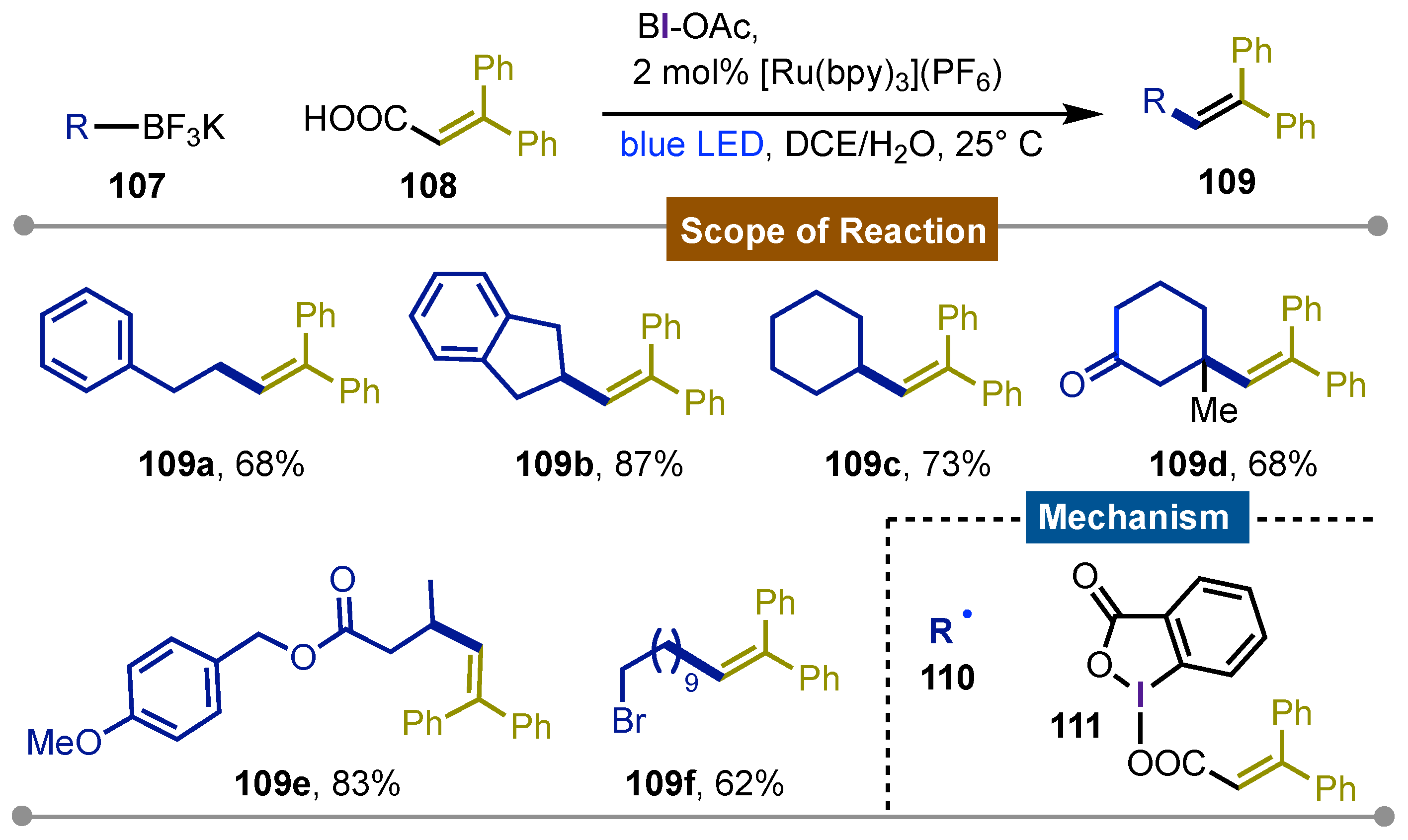
Chen and co-workers reported in 2014 [65] a photoredox system for the chemoselective sp3C-sp2C coupling of alkyltrifluoroborates 107 and vinyl carboxylic acids 108 by decarboxylative alkenylation using visible light to get the corresponding products 109. The best results were achieved using the [Ru(bpy)3](PF6)2/acetoxybenziodoxole (BI-OAc) system in DCE/H2O. The reaction conditions tolerated alkylboronates with different degrees of substitution and bearing different functional groups such as ketones, esters and alkyl bromides giving the desired alkenes 109a-f in good yields (62-83%). The vinyl carboxylic acids could bear a wide variety of substituents on the aryl groups of the β-position producing the final alkene with moderate to good yields (58 to 82%). Reactions with TEMPO confirmed the alkyl radical formation instead of an alkenyl radical. Monitoring the reaction by NMR showed the formation of complex BI-OOCCH=CHR’ 111, an unprecedented stable species. This was proposed as the first step of the mechanism followed by oxidation of the photoexcited Ru-catalyst which was responsible for the deboronation of the trifluoroalkyl substrate to form the alkyl radical 110. The latter was added to the α-carbon of complex 111 which underwent decarboxylation to generate the benziodoxole radical that gives the final alkenes 109a-f. Comparing this protocol to the common Heck-type reaction, this reaction not only had better chemoselectivity but the milder reaction conditions opened the possibility to further applications especially for biomolecules (Scheme 18).
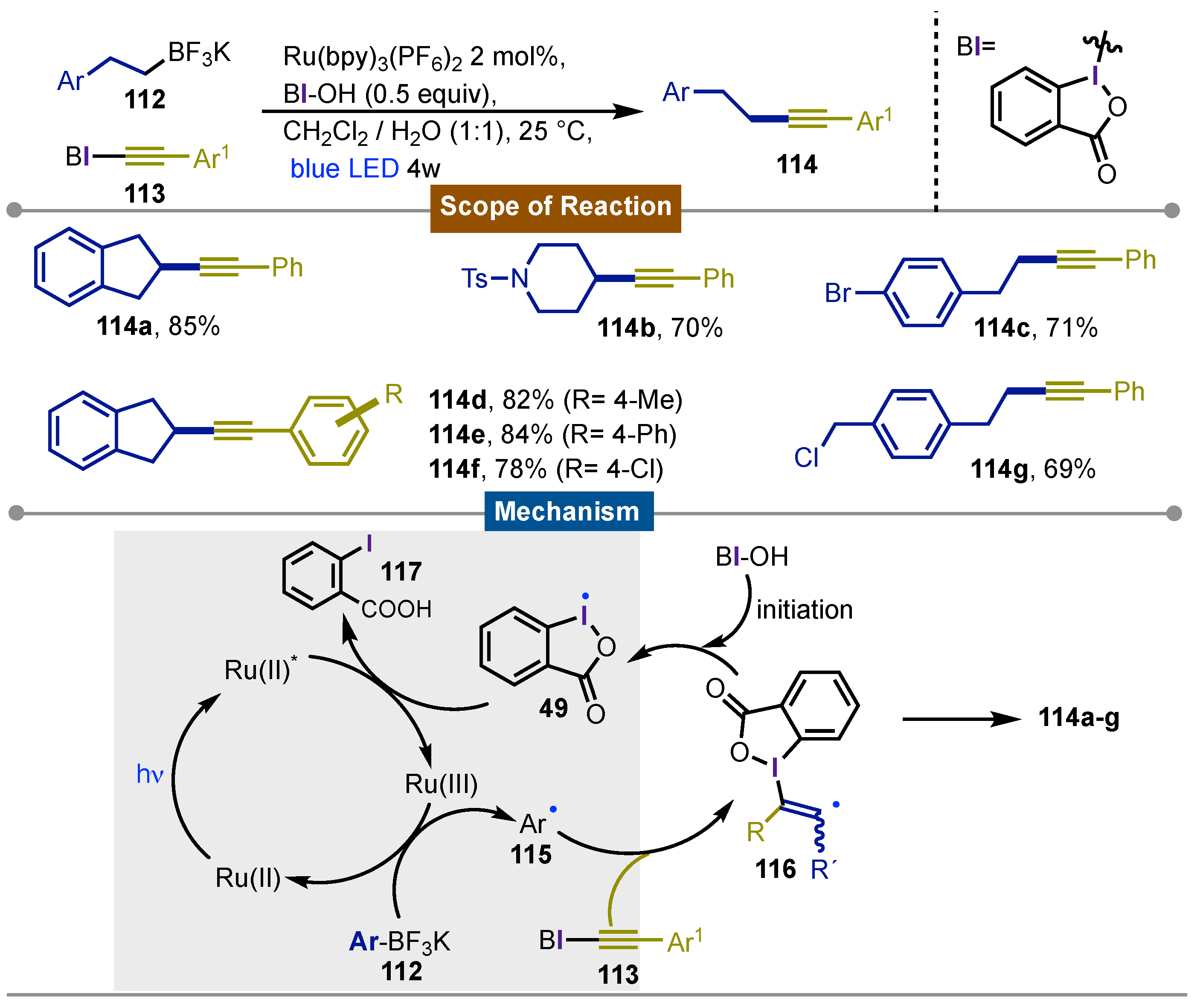
Indeed one year later in 2014 [66], Chen’s group expanded their findings to the deborative alkynylation of alkyl boronates. As its predecessor, the reaction proceeded with a wide variety of alkyl boronates 112, alkynyl benziodoxole 113 and hydroxybenziodoxole (BI−OH) in the presence of Na2CO3 under blue LED (λmax = 468 ± 25 nm) irradiation using [Ru-(bpy)3](PF6)2 as the photocatalyst. The reaction was compatible for alkynes bearing electron-donating and electron-withdrawing groups on the aryl. This was especially relevant for functional groups that were sensitive to other metal-catalyzed reactions, such as aryl halides and azides among others. The desired alkynes 114a-g were obtained with moderate to good yields (59-82%). Tests with TEMPO were carried out as well as isotopic labeling and on−off light experiments to elucidate the reaction mechanism. The mechanism started by the formation of the radical 115 enabled by the reduction of Ru(byp)33+ to Ru(byp)32+. The former radical get an α-addition into 113 giving rise to the -radical 116 which reacted with BI-OH to produce the benziodoxole radical 49 along to the formation of the obdserved products 114. Radical 49 oxidized to [Ru(II)]* to Ru(III) allowing the get into a new cycle while 2-iodobenzoic acid 117 is also released. For biomolecule compatibility, it was demonstrated that the reaction could be carried out in pH 7.4 phosphate saline buffer with comparable yields to the DCM/H2O system. The presence of aminoacids, nucleosides, oligosaccharides, nucleic acids, proteins and even bacterial cell lysates didn’t affect the outcome of the reaction, broadening the application of this protocol to biomolecule research (Scheme 19).
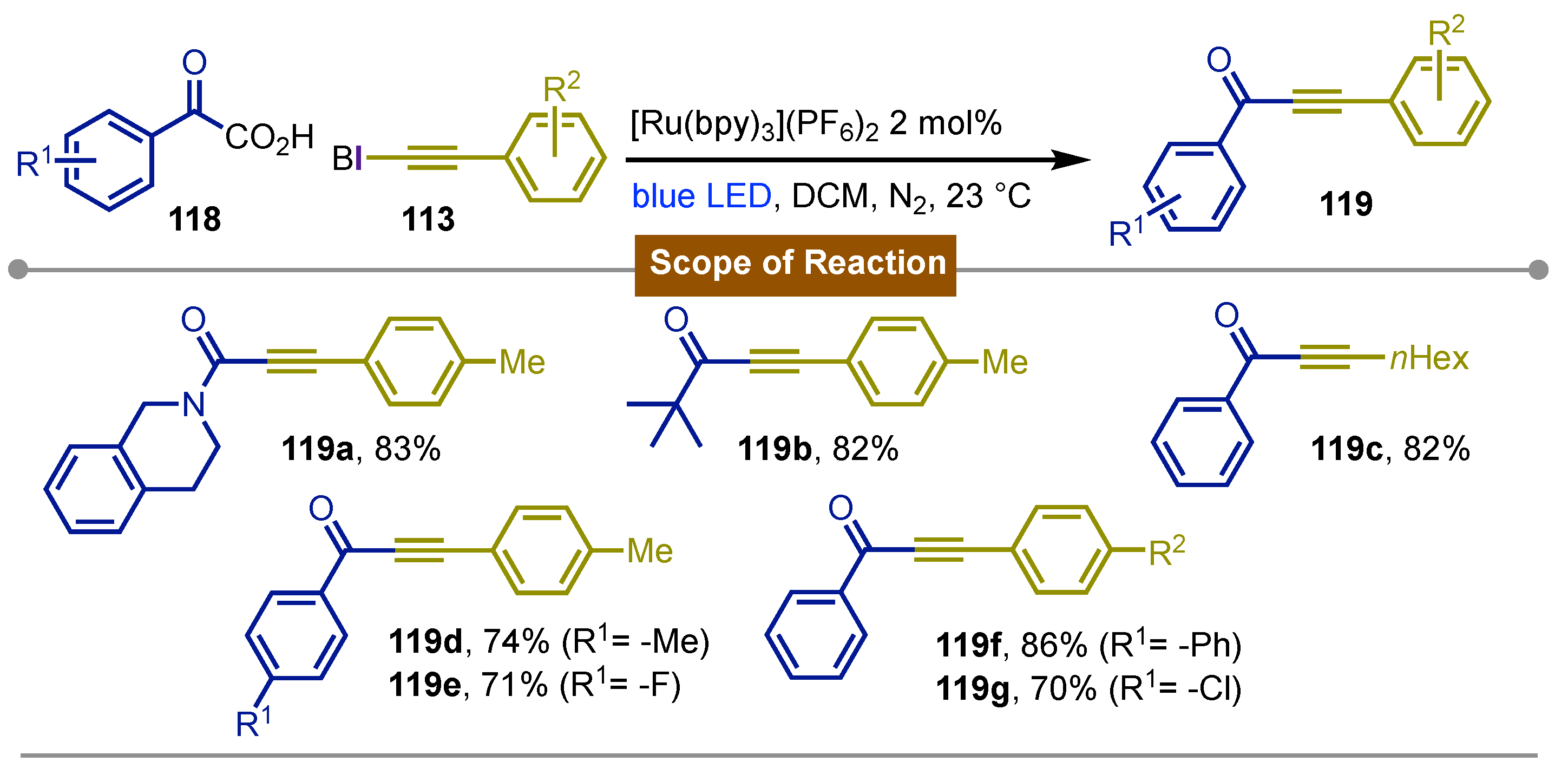
Their next report came in 2015 [67] on the decarboxylative ynonylation of alkynes with a combination of hypervalent iodine(III) reagents for the synthesis of ynones, ynamides and ynoates. The reaction take place using α-ketoacids 118 and alkynyl benziodoxole 113 under the use of the [Ru(bpy)3](PF6)2/acetoxybenziodoxole photoredox system in dichloromethane at room temperature for 5 hours to give variety of yinones 119a-g. The yields ranged from moderate to excellent tolerating a wide variety of groups. Herein not only the α-keto acid but also on the alkyne worked, which was especially interesting for groups such as azides which are usually reactive towards transition metal-catalyzed reactions. Experiments carried out shown that BI-OAc was not only an oxidant for the photoexcited [Ru(byp)32+]* species but it also had a role in the activation of the α-keto acid forming a benziodoxole/ketoacid complex (BI–O2CCOR’). This protocol could also be modified to run under neutral aqueous reaction conditions, allowing the gram-scale synthesis (Scheme 20).
Another important development in Chen’s [68] group research was the generation of alkoxy radicals by cyclic iodine(III) reagent catalysis reported in 2016. Due to their properties similar to transition-metal reactivity, cyclic iodine(III) reagents were tested as a milder alternative to the harsher conditions necessary when these metal catalysts were used. Specifically for the formation of alkoxy radicals, it was interesting to promote the sp3C−sp3C cleavage via β-fragmentation. This process, that was usually limited to strained cycloalkanols, could be expanded to even linear alcohols yielding stable ketones and alkyl radicals that could undergo alkenylation/alkynylation. Thus alcohols 120 or 121 were transformed into adducts 123 or 124 respectively in moderate to good yields (44 to 86%) using acetoylbenziodoxole (BI-OAc) under blue LED (λmax = 468 ± 25 nm) irradiation and [Ru(bpy)3](PF6)2 as the photocatalyst. Mechanistic studies inferred that the process initiated by oxidation of the photoexcited [Ru(bpy)32+]* to Ru(bpy)33+ with BI-OAc or its decomposing benziodoxole radical 49. The Ru(bpy)33+ then oxidizes the benziodoxole/alcohol complex 125 formed in situ and releases BI-OAc for a new catalytic cycle. On the other hand the alkoxyl radical 126 generated undergoes β-fragmentation to yield the C−C bond-cleavage forming the alkyl radical 127 which reacted with the alkynyl or vinyl carboxylate-bound benziodoxole 128 or 129. Finally, the alkynylation or alkenylation products 123 or 124 were obtained, and the benziodoxole radical 49 is released for further [Ru(bpy)32+]* oxidation (Scheme 21).
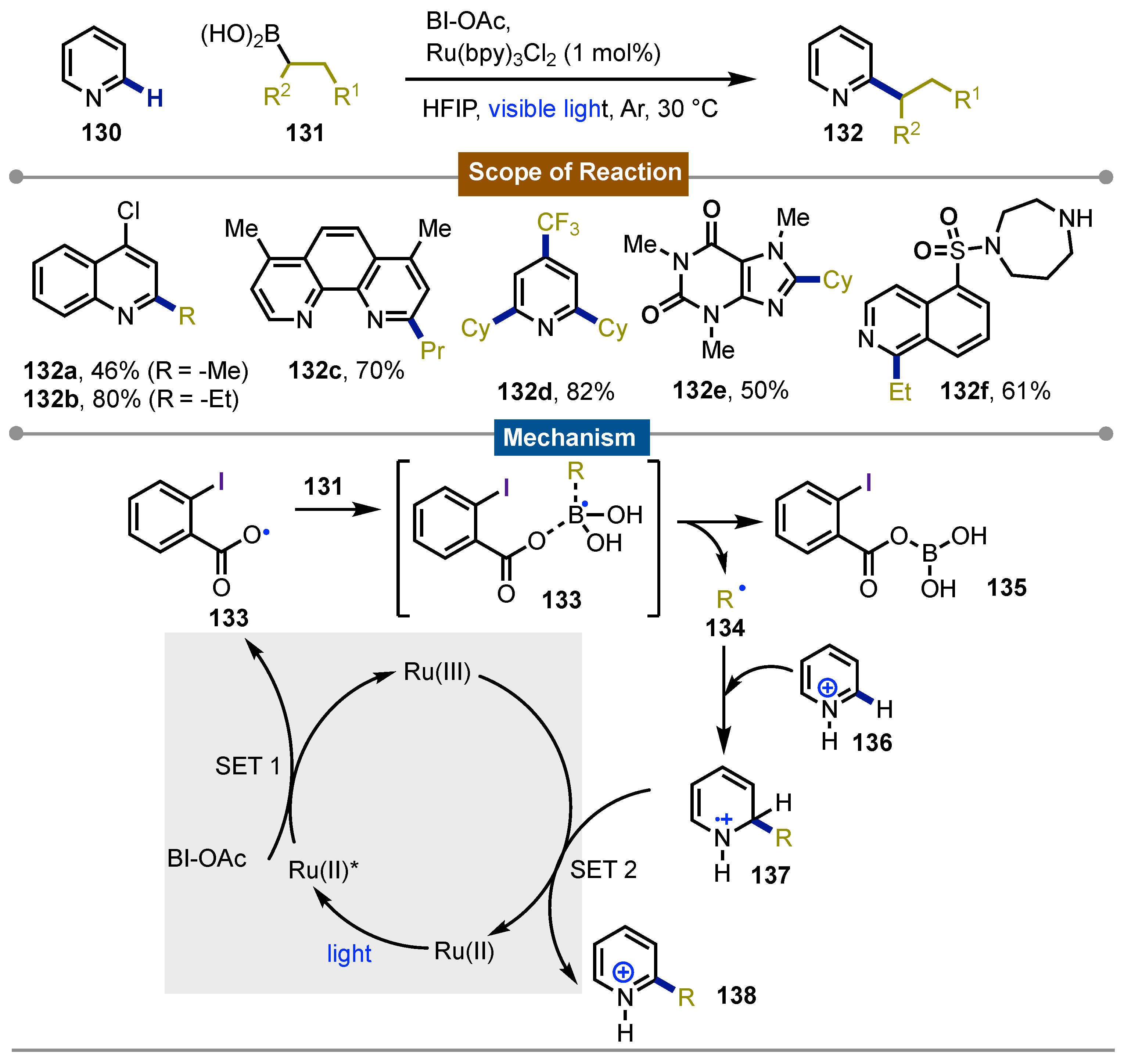
That same year Li [69] and co-workers published the photoredox-mediated Minisci C-H activation of heteroarenes 130 with alkyl boronic acids 131 using BI-OAc as the oxidant. The reaction proceeded in presence of 1 mol% of Ru(bpy)3Cl2. Visible light was used for the photoredox system and the reaction was kept under argon. The reaction showed a broad substrate scope 132a-f, with even primary alkyl boronic acids forming the more challenging primary alkyl radicals 134. The conditions allowed a great functional group tolerance including substrates such as alkyl and aryl halides, esters and carbamates among others, which opened the possibility for late stage functionalization for complex molecules. The reaction occurred at the C2 or C4 of the heteroarenes with electron-deficient N-heteroarenes being more reactive towards alkylation. DFT calculations proposed that after reduction of BI-OAc by photoexcited [Ru(bpy)3Cl2]* an O-centered radical 133 was formed after I–O bond cleavage. This species reacted with the alkyl boronic acid 131 to form the alkyl radical 134 via 133 which underwent the nucleophilic addition reaction with the protonated N-heteroarenes 136 to form a σ-complex 137. SET oxidation provided the final products 138 which were neutralized and closed the photoredox cycle (Scheme 22).
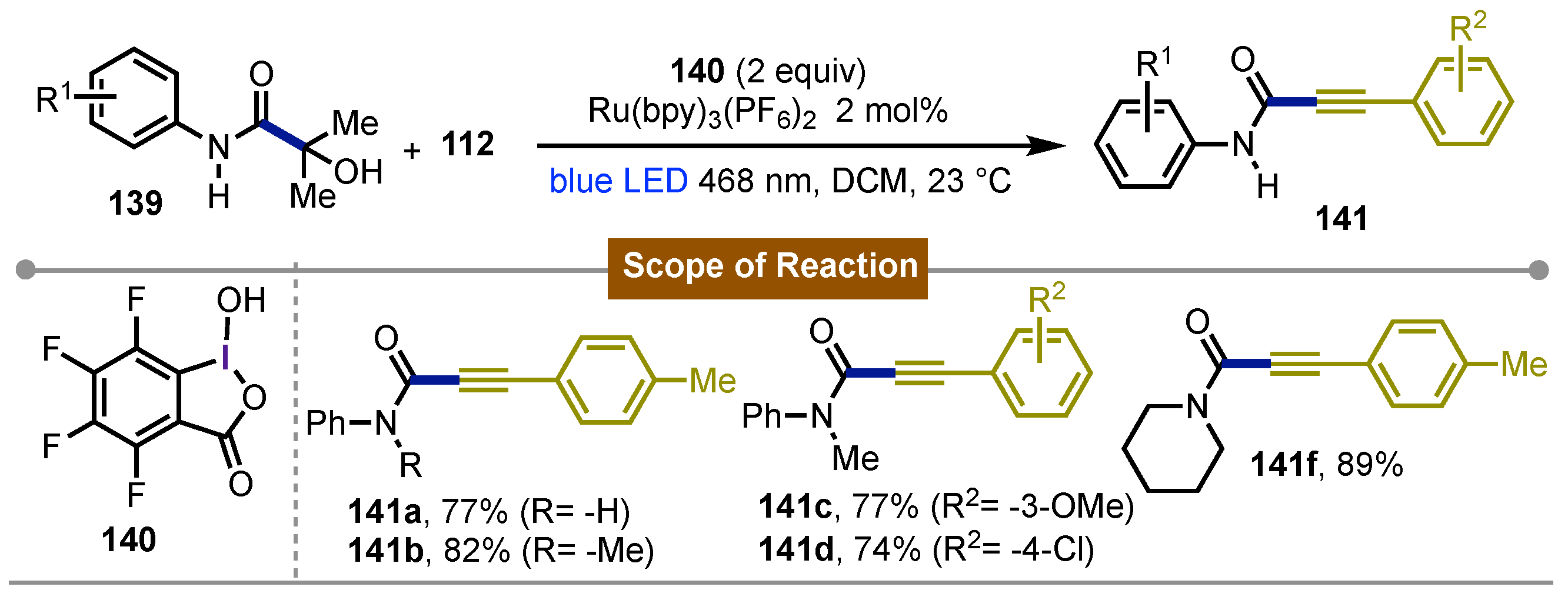
Also in 2017 [70], Chen and collaborators described the synthesis of ynamides, ynoates, and ynones from β-amide alcohol 139 and alkynyl benziodoxole 128. This alkynylation is promoted by the tetrafluoro-BI-OH 140 system and Ru(bpy)₃(PF₆)₂ as photocatalysis, which enabled regio- and chemoselective carbonyl-sp³C bond cleavage. Irradiation with blue LED light (468 nm) gets the [Ru(II)]* to Ru(III) oxidation. This photoredox procedure allowed to the photocatalyst the generation of radicals in the medium, leading to the key carbonyl-sp³C bond cleavage bond cleavage. The scope of the system enabled the formation of the corresponding products 141a-f, where it is observed that both EDG (electron-donating groups) and EWG (electron-withdrawing groups) were well tolerated (Scheme 23).
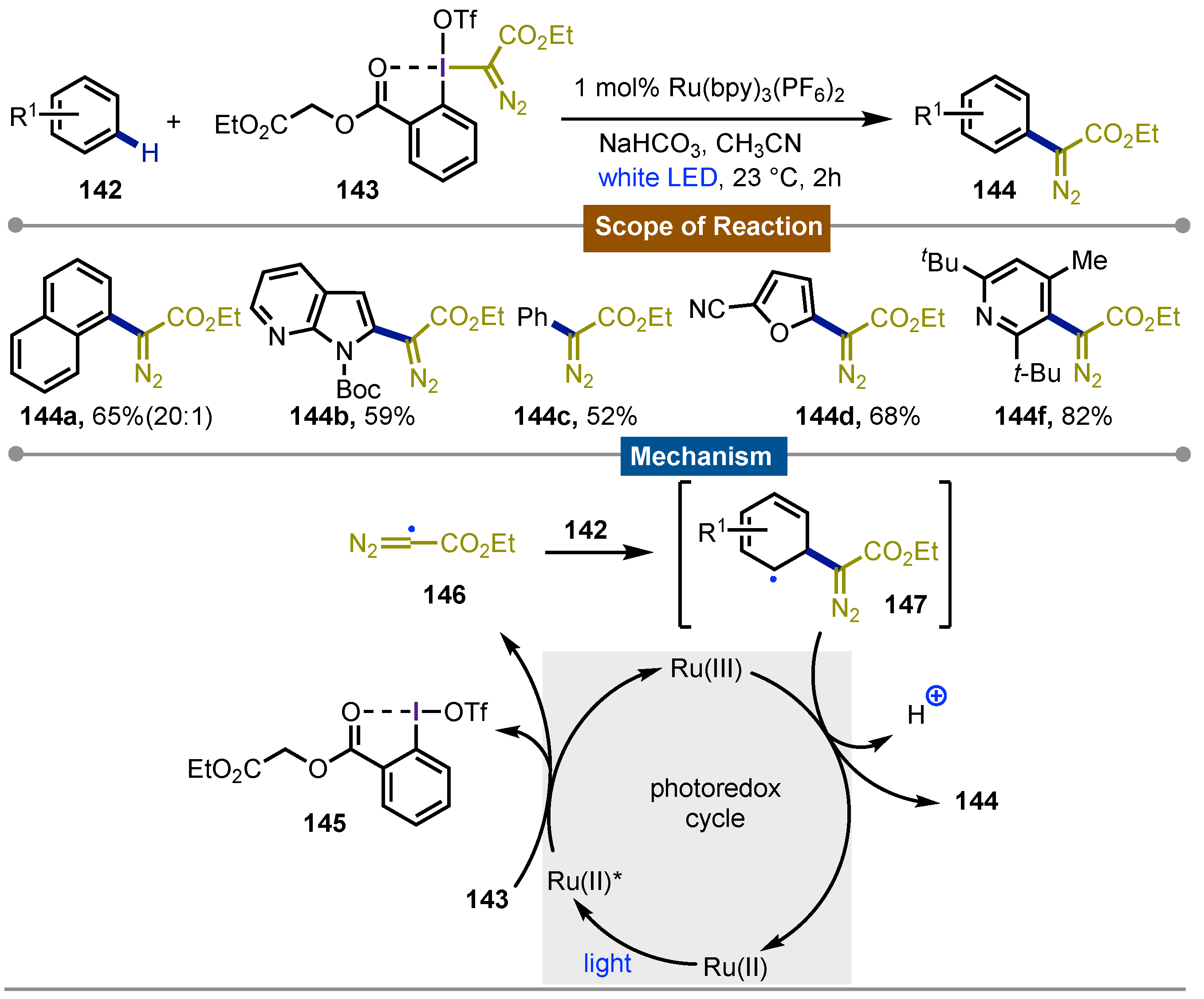
Interested in carbyne chemistry, Suero’s [71] group described in 2018 the generation of carbyne equivalents via photoredox catalysis with hypervalent iodine reagents. Benziodoxolone and its pseudocyclic analogue 143 were tested as diazomethyl radical precursors. By mixing this iodine(III) precursor with the arene 142, and 1 mol% of Ru(bpy)3(PF6)2 in CH3CN under blue LED irradiation gave the desired C–H diazo methylation. The reaction showed a broad scope, yielding a wide variety diazomethyl arenes 144a-f. The products were obtained in moderate to good yields highlighting a successful late stage functionalization on a wide variety of drugs. Even when the yields were poor, recovery of the original drug was possible while de novo synthesis for those products would be more challenging. Their proposed mechanism involved the formation of the diazo methyl radical 146 from the hypervalent iodine reagent 143 via SET enabled by the oxidation of [Ru(II)]* to Ru(III) which also releases the iodane 145. Next, this carbyne radical is added to the arene 142 leading to the formation of the non-aromatic carbon-centered radical 147 which has the -diazo ester functionality incorporated. Final oxidative deprotonation of 147 from Ru(III) catalyst give rise to the formation of the observed products 144a-f with the concomitant regeneration of Ru(II) which enter in another catalytic cycle (Scheme 24).
Furthermore, Bolm’s [72] group worked on the preparation of new hypervalent iodine(III) reagents to test their reactivity. In 2020, they reported that mixing p-tolyldifluoro iodobenzene 149 with sulfoximines 148 and styrenes 151 yielded products 149 under blue LED irradiation and 1 mol% of Ru(bpy)3(PF6)2 as photocatalyst. Taking into consideration that both sulfoximine and fluoro groups are of synthetic interest, they tested the reaction of the in situ formed hypervalent-iodine(III) reagent 150. A step-wise approach gave the best results when 149 and NH-sulfoximine 148 were stirred in DCM for 20 min under argon at room temperature. Styrene 151 were then added to the reaction mixture and finally irradiated with blue LED (24 W) for 12 hours to give the desired product. The reaction showed a broad scope not only for the sulfoximine where different groups were tolerated on the aryl, but also on the styrene. The reaction showed high regioselectivity for 1,2-disubstituted styrenes due to the benzylic stabilization of the radical 153. Further experiments to elucidate the mechanism were carried out. It was stablished that the N-I bond cleavage in 150 by single-electron transfer, is the first step in the photoredox cycle which generates N-centred radical 153, iodoaryl 154 and a fluride anion. Next, the addition of 151 to the double bond gives rise to the C-centered radical 155 that was followed by SET oxidation to form the benzylic cation 156. Final reaction with the fluoride anion, yielded the observed prodcuts 152 (Scheme 25).
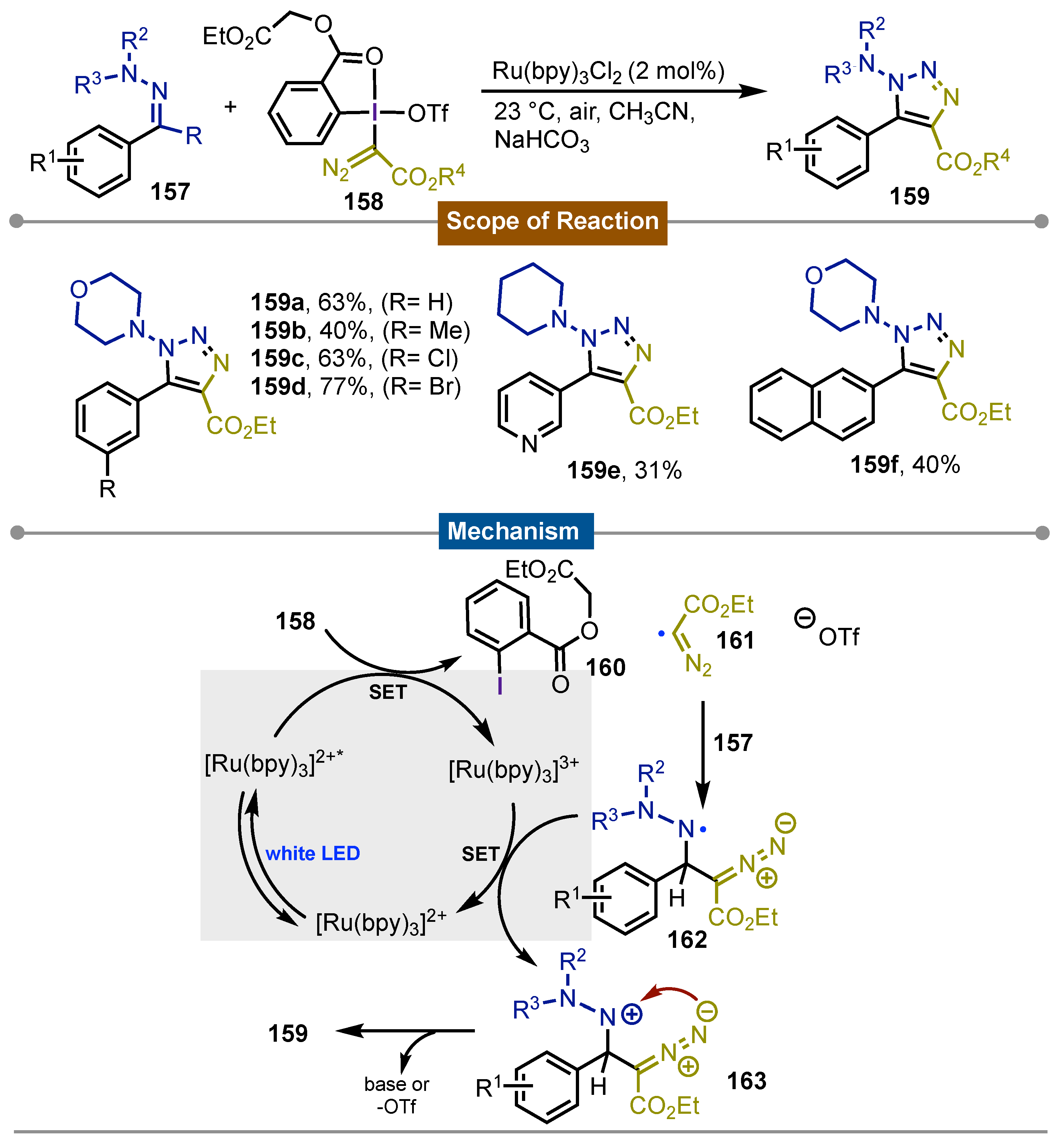
Inspired by Suero’s work on the generation of diazomethyl radicals, Li’s [73] group described in 2021 a photoredox [3+2] cyclization reaction for the synthesis of 1-amino-1,2,3-triazoles 159 using hypervalent iodine(III) diazo reagents 158. N,N-dialkylhydrazones 157 were transformed into the desired 1,2,3-triazole derivatives under the optimized conditions. Hydrazones could bear a wide variety of substituents in the aromatic ring and on the sp3N atom giving the products in moderate to good yields. The synthetic utility was demonstrated by the late-stage functionalization of natural product derivatives. Experimental studies supported a reaction pathway that intiates by the photochemical escition of diazo-containign iodine(III) reagent 158 enabled by photoexcited [Ru(bpy)3]2+* which generates iodoarene 160 and carbon-centered radical 161 containing the diazo moiety. The former radical reacted with hydrazone derivatives 157 giving the homologated nitrogen centered radical 162 which is oxidized by [Ru(bpy)3]3+ leading to the formation of the nitrenium 163. Final attack form the diazo fragment closes the ring yielding the observed final triazoles 159 (Scheme 26).
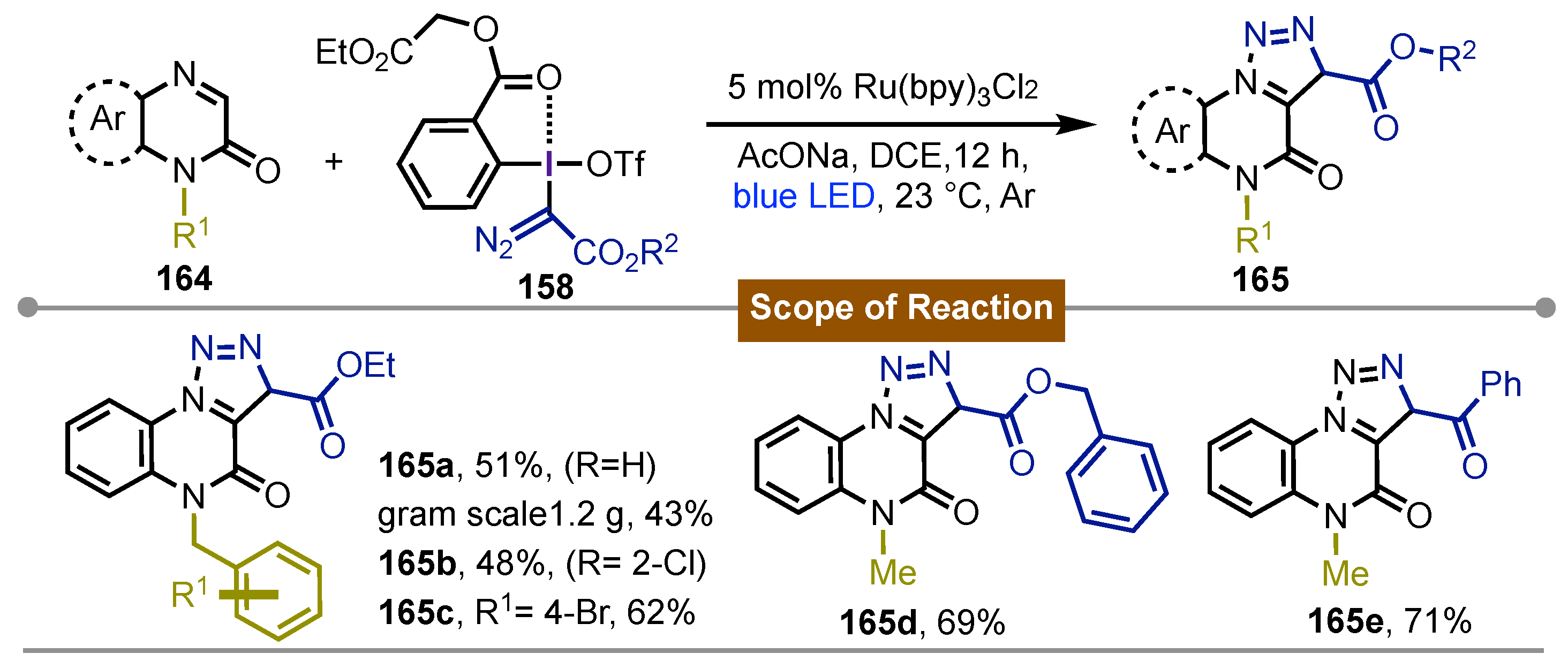
In their follow-up work, Li [74] and co-workers estended their photoredox-catalyzed [3 + 2] cyclization using hypervalent iodine(III) reagents to afford [1,2,3]-triazolo-[1,5-a]quinoxalin-4(5H)-ones 165. Their approach was especially attractive since these quinoxalinones were found in many bioactive products but few protocols were available for the synthesis of the tricyclic scaffold with the 1,2,3-triazole moiety. Based on their previous report, hypervalent iodine diazo reagent 158 was used as a diazomethyl radical precursor. Quinoxalinone 164 and 1.5 equivalents of the iodine(III) reagent were mixed with 2 equivalents of sodium acetate and 2.5 mol% of Ru(bpy)3Cl2 in DCE at room temperature under the irradiation of a blue LED (24 W) for 12 hours. The conditions tolerated a wide variety of N-subtitution on the quinoxalinone core giving the tricyclic compounds 165 with moderate to good yields. Limitations of the process imply the free nitrogen didn’t afford the desired product. The reaction proceeded with different substituents of the aromatic ring except when a NO2 group was present at the C6-position. Different ester and benzoyl groups were introduced in the hypervalent iodine(III) diazoderivative 158 reagents showing good performance. Further experiments to elucidate the mechanism supported the formation of the diazoacetate radical which could attack the C=N bond of the quinoxalinone forming another radical that upon oxidation with [Ru(bpy)3]3+ would form the cation that would provide the tricyclic product in the presence of base. In general quite similar to mechanism of previous work (Scheme 27).
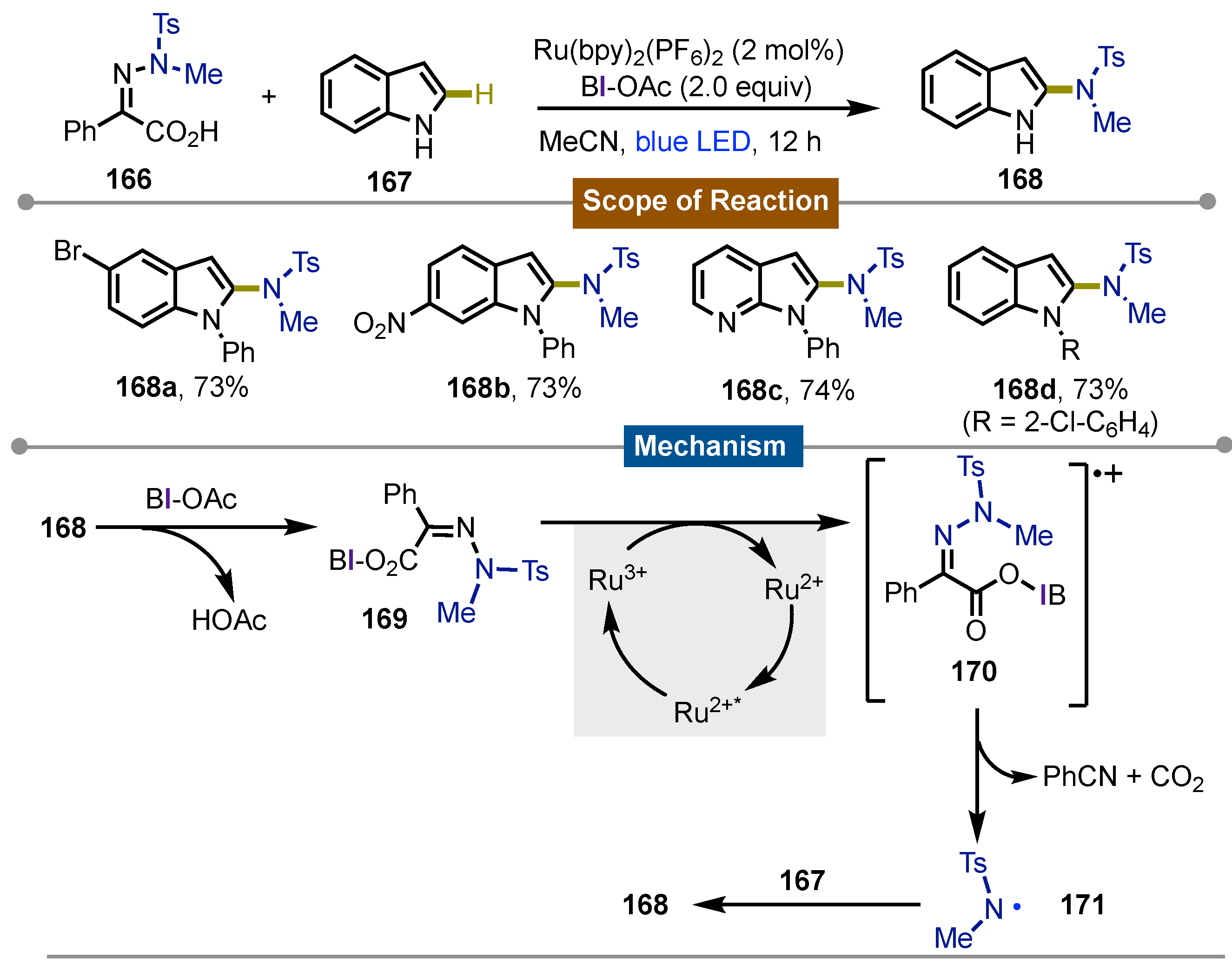
In 2022, Chen’s [75] group proposed that BI-OAc could generate amidyl radicals via the N−N bond cleavage of amidyl-iminophenyl-acetic acids (IPA-NR2) that in a Minisci-type reaction, promote the sp2C−H amination of arenes and heteroarenes. IPA-NR2 166 were easily accessible through condensation of hidrazines with methyl benzoylformates. The reaction between them and heteroarenes 167 proceeded in the presence of 2 equivalents of BI-OAc, 2 mol% of Ru(bpy)3(PF6)2 in acetonitrile under blue LED irradiation for 12 hours to give 2-aminoindoles 168. In such way, the products were obtained with moderate to good yields. Electron-deficient and electron-rich arenes and heteroarenes performed in a similar way under the reaction conditions and N-substitution even favored it. On the other hand, simple benzenes, pyridines, or isoquinolines weren’t suitable and gave the hydrogenation adduct TsNHMe as the product. Different sulfonamide substituents on the IPA were well tolerated. The importance of BI-OAc coordination to the IPA-NR2 was demonstrated by carrying out the reaction with the corresponding methyl ester which didn’t react. The mechanistic studies were consistent with previous reports about cyclic iodine activation of carboxylates.Thus, initial interaction between 168 and BI-OAc gets iodine(III) intermediate BI-IPA-NTsMe 169 which is oxidized to the corresponding iminophenylcarboxyl radical cation 170 by Ru(III) photocatlyst which underwent concerted carbon dioxide and phenylnitrile elimination to form the sulfonamidyl radical 171. Then, radical addition to the arene 167, followed by oxidation with BI radical afforded the final products 168a-d (Scheme 28).
Most recently, in 2024, Suero’s [76] group reported the first photoredox-catalyzed alkoxy diazomethylation of alkenes yielding compounds from the elusive β-alkoxydiazo family. Following a similar protocol previously described by his group, the use of alkenes 172 as substrates could allow the introduction of the ether moiety by trapping the final cation with an alcohol in multicomponent disconnection approach. Indeed the reaction proceeded using diazo-containing hypervlent iodine(III) reagent 173 and Ru(dtbbpy)3(PF6)2 which had a better reductive capability than Ru(bpy)3(PF6)2 in its excited state hence increasing the efficiency of the radical generation to get adducts 174. The reaction tolerated a wide variety of styrenes with different functionalities in every position of the aromatic ring with moderate to excellent yields, even α-alkyl substituted styrenes worked well in the conditions of the reaction. The reaction with α,β-di-substituted styrenes gave E/Z mixtures with high yields and excellent diastereoconvergence. Alcohols with different degrees of substitution were introduced with high yields and even an intramolecular version was achieved. Finally the evaluation of the scope on the iodine reagent showed that derivatives with benzyl esters and even phosphonate and sulfonate were suitable in these conditions. The rection mechanism is similar to previously described (Scheme 29).
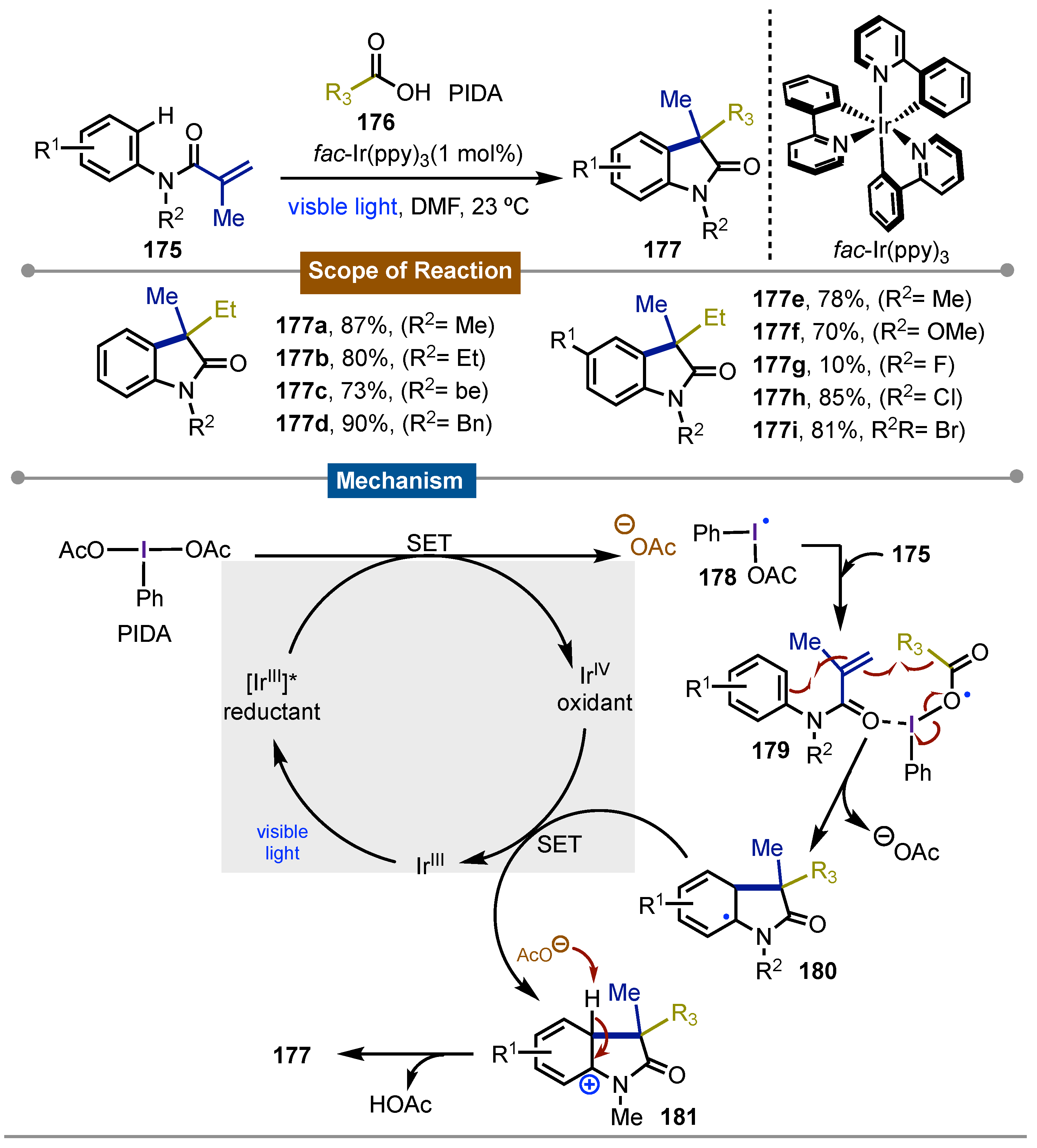
The most recent example using iodine(III) reagents in a photocatalytic cycle using iridium, came from Zhu [77] in 2013. Herein, it was used N-methyl-N-phenylmethacrylamide 175 and different alkyl carboxylic acids 176 for a secuential decarboxylative/C-H functionalization rection that produced C3-disubstituded 2-oxoindoles 177. The scope covered the funtionalization of acrylamides with different alkyls. The proposal of the reaction mechanism started with the [IrIII]*-promoted photochemical reduction of the iodine(III)-containing alkyl group to get a iodanyl radical 178. The following reaction of this iodine-centered radical with the acrylamide gives rise to the adduct 179 which homolytically broken and transfer the alkyl fragment to the double bond of amide, leading to the carbon dioxide and iodobenzene release, along to the formation of the carbon-centered non-aromatic 2-oxoindol radical 180. This intermediate is oxidized by [IrIV] catalyst to its corresponding carbocation 181. Final acetate assisted aromatization yielded the observed products 177a-k (Scheme 30).
5. Summary and Perspectives
In summary, several synthetic protocols have been developed mainly the last twenty years using this poweful combination of iodine(III) reagents and photochemistry. Several functional groups have been indroduced in a radical way such as trifluoromethyl, alkyl, alkenyl, alkynyl, fluoromethylen or fluorosulfonyl among others, in different cores including aromatics, alkenyls or alkyls. This strategy resulted unique in terms of innovation and easiness to directly broke and make high enegy bonds, which would result unviable or require several steps using other protocols.
Due to the efectiviness of this synergic combination (iodine(III)-photochemistry), an interesting perspective could involve the aryl-aryl and aryl-heteroaryl corss-bond formation. This plausible development would represent the full emulation of common transition-metal chemistry.
Author Contributions
All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by The Office of Support to Research and Postgraduate of Guanajuato University (UG-DAIP).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the author.
Acknowledgments
We acknowledge the facilities of the DCNyE, the Chemistry Department and the National Laboratory UG-CONACyT (LACAPFEM) at the University of Guanajuato. We also thank CONACyT for fellowship support to J.G.I.G., E.D.H.V., A.K.G.D., J.A.M.C., K.M.H.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Segura-Quezada, L. A.; Torres-Carbajal, K. R.; Satkar, Y.; Juárez Ornelas, K. A.; Mali, N.; Patil, D. B.; Gámez-Montaño, R.; Zapata-Morales, J. R.; Lagunas-Rivera, S.; Ortíz-Alvarado, R.; Solorio-Alvarado, C. R. Oxidative Halogenation of Arenes, Olefins and Alkynes Mediated by Iodine(III) Reagents. Mini-Rev. Org. Chem. 2021, 18, 159–172. [Google Scholar] [CrossRef]
- Segura-Quezada, L. A.; Torres-Carbajal, K. R.; Juárez-Ornelas, K. A.; Alonso-Castro, A. J.; Ortiz-Alvarado, R.; Dohi, T.; Solorio-Alvarado, C. R. Iodine (III) reagents for oxidative aromatic halogenation. Org. Biomol. Chem. 2022, 20, 5009–5034. [Google Scholar] [CrossRef] [PubMed]
- Kikushima, K.; Elboray, E. E.; Jiménez-Halla, J. O. C.; Solorio-Alvarado, C. R.; Dohi, T. Diaryliodonium (III) salts in one-pot double functionalization of C–IIII and ortho C–H bonds. Org. Biomol. Chem. 2022, 20, 3231–3248. [Google Scholar] [CrossRef] [PubMed]
- Segura-Quezada, L. A.; Torres-Carbajal, K. R.; Juárez-Ornelas, K. A.; Navarro-Santos, P.; Granados-López, A. J.; González-García, G.; Ortiz-Alvarado, R.; de León-Solis, C.; Solorio-Alvarado, C. R. Iodine(III)-Mediate Oxidative Cyanation, Azidation, Nitration, Sulfenylation and Slenization in Olefins and Aromatic Systems. Curr. Org. Chem. 2022, 26, 1954–1968. [Google Scholar] [CrossRef]
- Chávez-Rivera, R.; Navarro-Santos, P.; Chacón-García, L.; Ortiz-Alvarado, R.; Solorio Alvarado, C. R. Iodine(III)-Aluminum or -Ammonium Salts for the Oxidative Aromatic Inorganic Functionalization. ChemisrtySelect. 2023, 8. [Google Scholar] [CrossRef]
- Barrera-Nava, M. P.; Segura-Quezada, L. A.; Ibarra-Gutiérrez, J. G.; Chávez-Rivera, R.; Ortiz-Alvarado, R.; Solorio-Alvarado, C. R. Iodine(III) reagents for the aromatic functionalization with inorganic groups. Tetrahedron Lett. 2024, 166, 134203. [Google Scholar] [CrossRef]
- Nahide, P. D.; Ramadoss, V.; Juárez-Ornelas, K. A.; Satkar, Y.; Ortiz-Alvarado, R.; Cervera-Villanueva, J. M. J.; Alonso-Castro, Á. J.; Zapata-Morales, J. R.; Ramírez-Morales, M. A.; Ruiz-Padilla, A. J.; Deveze-Álvarez, M. A.; Solorio-Alvarado, C. R. In Situ Formed IIII-Based Reagent for the Electrophilic Ortho-Chlorination of Phenols and Phenol Ethers: The Use of PIFA-AlCl3 System. Eur. J. Org. Chem. 2018, 485–493. [Google Scholar] [CrossRef]
- Satkar, Y.; Ramadoss, V.; Nahide, P. D.; García-Medina, E.; Juárez-Ornelas, K. A.; Alonso-Castro, A. J.; Chávez-Rivera, R.; Jiménez-Halla, J. O. C.; Solorio-Alvarado, C. R. Practical, Mild and Efficient Electrophilic Bromination of Phenols by a New I(III)-based reagent: The PIDA–AlBr3 System. RSC Adv. 2018, 8, 17806–17812. [Google Scholar] [CrossRef]
- Satkar, Y.; Yera-Ledesma, L. F.; Mali, N.; Patil, D.; Navarro-Santos, P.; Segura-Quezada, L. A.; Ramírez-Morales, P. I.; Solorio-Alvarado, C. R. Iodine(III)-Mediated, Controlled Di- or Monoiodination of Phenols. J. Org. Chem. 2019, 84, 4149–4164. [Google Scholar] [CrossRef] [PubMed]
- Segura-Quezada, A.; Satkar, Y.; Patil, D.; Mali, N.; Wrobel, K.; González, G.; Zárraga, R.; Ortiz-Alvarado, R.; Solorio-Alvarado, C. R. Iodine(III)/AlX3-mediated electrophilic chlorination and bromination of arenes. Dual Role of AlX3 (X = Cl, Br) for (PhIO)n depolymerization and as the halogen source. Tetrahedron Lett. 2019, 60, 1551–1555. [Google Scholar] [CrossRef]
- Patil, D. B.; Gámez-Montaño, R.; Ordoñez, M.; Solis-Santos, M.; Jiménez-Halla, J. O. C.; Solorio-Alvarado, C. R. Iodine(III)-Mediated Electrophilic Chlorination and Catalytic Nitration of N-Tosyl Anilines. Eur. J. Org. Chem. 2022, e202201295. [Google Scholar] [CrossRef]
- Mali, N.; Ibarra-Gutiérrez, J. G.; Lugo Fuentes, L. I.; Ortíz-Alvarado, R.; Chacón-García, L.; Navarro-Santos, P.; Jiménez-Halla, J. O. C.; Solorio-Alvarado, C. R. Iodine(III)-Mediated Free-Aniline Iodination through Acetyl Hypoiodite Formation: Study of the Reaction Pathway. Eur. J. Org. Chem. 2022, e202201067. [Google Scholar] [CrossRef]
- Juárez-Ornelas, K. A.; Solís-Hernández, M.; Navarro-Santos, P.; Jiménez-Halla, J. O. C.; Solorio-Alvarado, C. R. Divergent role of PIDA and PIFA in the AlX3 (X = Cl, Br) halogenation of 2-naphthol: a mechanistic study. Beilstein J. Org. Chem. 2024, 20, 1580–1589. [Google Scholar] [CrossRef]
- Juárez-Ornelas, K. A.; Jiménez-Halla, J. O. C.; Kato, T.; Solorio-Alvarado, C. R.; Maruoka, K. Iodine(III)-Catalyzed Electrophilic Nitration of Phenols via Non-Brønsted Acidic NO2+ Generation. Org. Lett. 2019, 21(5), 1315–1319. [Google Scholar] [CrossRef]
- Nahide, P. D.; Solorio-Alvarado, C. R. Mild, Rapid and Efficient Metal-Free Synthesis of 2-Aryl-4-Aryloxyquinolines via Direct Csp2O Bond Formation by Using Diaryliodonium Salts. Tetrahedron Lett. 2017, 58, 279–284. [Google Scholar] [CrossRef]
- Satkar, Y.; Wrobel, K.; Trujillo-González, D. E.; Ortiz-Alvarado, R.; Jiménez-Halla, J. O. C.; Solorio-Alvarado, C. R. The Diaryliodonium(III) Salts Reaction With Free-Radicals Enables One-Pot Double Arylation of Naphthols. Front. Chem. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Naakajima, M.; Nagaswaa, S.; Matsumoto, K.; Matsuda, Y.; Nemoto, T. Synthesis of Visible-Light–Activated Hypervalent Iodine and Photooxidation under Visible Light Irradiation via a Direct S0→Tn Transition. Chem. Pharm. Bull. 2022, 70, 235–239. [Google Scholar] [CrossRef]
- Gutierrez-Cano, J. R.; Nahide, P. D.; Ramadoss, V.; Satkar, Y.; Ortiz-Alvarado, R.; Alba-Betancourt, C.; Mendoza-Macías, C. L.; Solorio-Alvarado, C. R. Synthesis and Biological Evaluation of New 3,4-Diarylmaleimides as Enhancers (Modulators) of Doxorubicin Cytotoxic Activity on Cultured Tumor Cells from a Real Case of Breast Cancer. J. Mex.Chem. Soc. 2017, 61. [Google Scholar] [CrossRef]
- Ramadoss, V.; Alonso-Castro, A. J.; Campos-Xolalpa, N.; Solorio-Alvarado, C. R. Protecting-Group-Free Total Synthesis and Biological Evaluation of 3-Methylkealiiquinone and Structural Analogues. J. Org. Chem. 2018, 83, 10627–10635. [Google Scholar] [CrossRef]
- Ramadoss, V.; Alonso-Castro, A. J.; Campos-Xolalpa, N.; Ortiz-Alvarado, R.; Yahuaca-Juárez, B.; Solorio-Alvarado, C. R. Total synthesis of kealiiquinone: the regio-controlled strategy for accessing its 1-methyl-4-arylbenzimidazolone core. RSC Adv. 2018, 8, 30761–30776. [Google Scholar] [CrossRef]
- Solorio-Alvarado, C. R.; Ramadoss, V.; Gámez-Montaño, R.; Zapata-Morales, J. R.; Alonso-Castro, A. J. Total synthesis of the linear and angular 3-methylated regioisomers of the marine natural product kealiiquinone and biological evaluation of related Leucetta sp. alkaloids on human breast cancer. Med. Chem. Res. 2019, 28, 473–484. [Google Scholar] [CrossRef]
- Nahide, P. D.; Alba-Betancourt, C.; Chávez-Rivera, R.; Romo-Rodríguez, P.; Solís-Hernández, M.; Segura-Quezada, L. A.; Torres-Carbajal, K. R.; Gámez-Montaño, R.; Deveze-Álvarez, M. A.; Ramírez-Morales, M. A.; Alonso-Castro, A. J.; Zapata-Morales, J. R.; Ruiz-Padilla, A. J.; Mendoza-Macías, C. L.; Meza-Carmen, V.; Cortés-García, C. J.; Corrales-Escobosa, A. R.; Núñez-Anita, R. E.; Ortíz-Alvarado, R.; Chacón-García, L.; Solorio-Alvarado, C. R. Novel 2-aryl-4-aryloxyquinoline-based fungistatics for Mucor circinelloides. Biological evaluation of activity, QSAR and docking study. Bioorg. Med. Chem. Lett. 2022, 63, 128649. [Google Scholar] [CrossRef]
- Torres-Carbajal, K. R.; Segura-Quezada, L. A.; Ortíz-Alvarado, R.; Chávez-Rivera, R.; Tapia-Juárez, M.; González-Domínguez, M. I.; Ruiz-Padilla, A. J.; Zapata-Morales, J. R.; de León-Solís, C.; Solorio Alvarado, C. R. Indomethacin Synthesis, Historical Overview of Their Structural Modifications. ChemistrySelect. 2022, 7. [Google Scholar] [CrossRef]
- Hernández-Velázquez, E. D.; Alba-Betancourt, C.; Alonso-Castro, Á. J.; Ortiz-Alvarado, R.; López, J. A.; Meza-Carmen, V.; Solorio-Alvarado, C. R. Metformin, a biological and synthetic overview. Bioorg. Med. Chem. Lett. 2023, 86, 129241. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Velázquez, E. D.; Herrera, M. D.; Alba-Betancourt, C.; Navarro-Santos, P.; Ortíz-Alvarado, R.; Solorio-Alvarado, C. R. Synthesis and in vivo Evaluation of Fluorobenzyl Metformin Derivatives as Potential Drugs in The Diabetes Treatment. Asian J. Org. Chem. 2023, 12. [Google Scholar] [CrossRef]
- Segura-Quezada, L. A.; Hernández-Velázquez, E. D.; Corrales-Escobosa, A. R.; de León-Solis, C.; Solorio-Alvarado, C. R. Ningalins, Pyrrole-Bearing Metabolites Isolated from Didemnum spp. Synthesis and MDR-Reversion Activity in Cancer Therapy. Chem. Biodiversity. 2024, 21. [Google Scholar] [CrossRef]
- Segura-Quezada, L. A.; Alba-Betancourt, C.; Chacón-García, L.; Chávez-Rivera, R.; Navarro-Santos, P.; Ortiz-Alvarado, R.; Tapia-Juárez, M.; Negrete-Díaz, J. V.; Martínez-Morales, J. F.; Deveze-Álvarez, M. A.; Zapata-Morales, J. R.; Solorio Alvarado, C. R. Synthesis and Anti-inflammatory Effect of Simple 2,3-Diarylindoles. On Route to New NSAID Scaffolds. ChemistrySelect. 2024, 9. [Google Scholar] [CrossRef]
- Fraire-Soto, I.; Araujo-Huitrado, J. G.; Granados-López, A. J.; Segura-Quezada, L. A.; Ortiz-Alvarado, R.; Herrera, M. D.; Gutiérrez-Hernández, R.; Reyes-Hernández, C. A.; López-Hernández, Y.; Tapia-Juárez, M.; Negrete-Díaz, J. V.; Chacón-García, L.; Solorio-Alvarado, C. R.; López, J. A. Differential Effect of 4H-Benzo[d] [1, 3]oxazines on the Proliferation of Breast Cancer Cell Lines. Curr. Med. Chem. 2024, 31, 6306–6318. [Google Scholar] [CrossRef]
- Ramadoss, V.; Nahide, P. D.; Juárez-Ornelas, K. A.; Rentería-Gómez, M.; Ortiz-Alvarado, R.; Solorio-Alvarado, C. R. A four-step scalable formal synthesis of ningalin C. ARKIVOC. 2016, 2016, 385–394. [Google Scholar] [CrossRef]
- Nahide, P. D.; Jiménez-Halla, J. O. C.; Wrobel, K.; Solorio-Alvarado, C. R.; Ortiz Alvarado, R.; Yahuaca-Juárez, B. Gold(I)-catalysed high-yielding synthesis of indenes by direct Csp3–H bond activation. Org. Biomol. Chem. 2018, 16, 7330–7335. [Google Scholar] [CrossRef] [PubMed]
- Segura-Quezada, L. A.; Torres-Carbajal, K. R.; Mali, N.; Patil, D. B.; Luna-Chagolla, M.; Ortiz-Alvarado, R.; Tapia-Juárez, M.; Fraire-Soto, I.; Araujo-Huitrado, J. G.; Granados-López, A. J.; Gutiérrez-Hernández, R.; Reyes-Estrada, C. A.; López-Hernández, Y.; López, J. A.; Chacón-García, L.; Solorio-Alvarado, C. R. Gold(I)-Catalyzed Synthesis of 4H-Benzo[d][1,3]oxazines and Biological Evaluation of Activity in Breast Cancer Cells. ACS Omega. 2022, 7, 6944–6955. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Núñez, E.; Raducan, M.; Lauterbach, T.; Molawi, K.; Solorio, C. R.; Echavarren, A. M. Evolution of Propargyl Ethers into Allylgold Cations in the Cyclization of Enynes. Angew. Chem, Int. Ed. 2009, 48, 6152–6155. [Google Scholar] [CrossRef] [PubMed]
- Solorio-Alvarado, C. R.; Echavarren, A. M. Gold-Catalyzed Annulation/Fragmentation: Formation of Free Gold Carbenes by Retro-Cyclopropanation. J. Am. Chem. Soc. 2010, 132, 11881–11883. [Google Scholar] [CrossRef]
- Solorio-Alvarado, C. R.; Wang, Y.; Echavarren, A. M. Cyclopropanation with Gold(I) Carbenes by Retro-Buchner Reaction from Cycloheptatrienes. J. Am. Chem. Soc. 2011, 133, 11952–11955. [Google Scholar] [CrossRef]
- McGonigal, P. R.; de León, C.; Wang, Y.; Homs, A.; Solorio-Alvarado, C. R.; Echavarren, A. M. Gold for the Generation and Control of Fluxional Barbaralyl Cations. Angew. Chem. Int. Ed. 2012, 51, 13093–13096. [Google Scholar] [CrossRef] [PubMed]
- Moteki, S. A.; Usui, A.; Zhang, T.; Solorio Alvarado, C. R.; Maruoka, K. Site-Selective Oxidation of Unactivated Csp3-H Bonds with Hypervalent Iodine(III) Reagents. Angew. Chem. Int. Ed. 2013, 52, 13093–13096. [Google Scholar] [CrossRef]
- Yahuaca-Juárez, B.; González, G.; Ramírez-Morales, M. A.; Alba-Betancourt, C.; Deveze-Álvarez, M. A.; Mendoza-Macías, C. L.; Ortiz-Alvarado, R.; Juárez-Ornelas, K. A.; Solorio-Alvarado, C. R.; Maruoka, K. Iodine(III)-catalyzed benzylic oxidation by using the (PhIO)n/Al(NO3)3 system. Synth. Commun. 2020, 50, 539–548. [Google Scholar] [CrossRef]
- Xue, X.-S.; Ji, P.; Zhou, B.; Cheng, J. P. The Essential Role of Bond Energetics in C–H Activation/Functionalization. Chem. Rev. 2017; 117, 8622–8648. [Google Scholar]
- Francis, A. C.; Richard, J. S. Advanced Organic Chemistry, 5th ed.; Springer US, 2007; p. 1074. [Google Scholar] [CrossRef]
- Concepión, J. I.; Francisco, C. G.; Hern´ andez, R.; Salazar, J. A. and Suárez, E., Tetrahedron Lett., 1984, 25, 1953-1956.
- Hernández, R.; León, E. I.; Moreno, P.; Riesco-Fagundo, C. and Suárez, E., J. Org. Chem., 2004, 69, 8437-8444.
- Boto, A.; Hernández, D.; Hernández, R. and Suárez, E., J. Org. Chem., 2003, 68, 5310-5319.
- Francisco, C. G.; González, C. C.; Paz, N. R. and Suárez, E., Org. Lett., 2003, 5, 4171-4173.
- Papadopoulou, M.; Spyroudis, S.; Varvoglis, A. 1,3-Oxathiole-2-Thiones from the Reaction of Carbon Disulfide with Zwitterionic Iodonium Compounds. J. Org. Chem, 1985, 50, 1509–1511. [Google Scholar] [CrossRef]
- Georgiev, G.; Spyroudis, S.; Varvoglis, A. Diacetoxyiodobenzene and Bis(Trifluoroacetoxy)Iodobenzene as Photoinitiators for Cationic Polymerisations. Polym. Bull. [CrossRef]
- Minisci, F.; Vismara, E.; Fontana, F.; Claudia Nogueira Barbosa, M. A New General Method of Homolytic Alkylation of Protonated Heteroaromatic Bases by Carboxylic Acids and Iodosobenzene Diacetate. Tetrahedron Lett, 1989, 30, 4569–4572. [Google Scholar] [CrossRef]
- Togo, H.; Muraki, T.; Yokoyama, M. Preparation of Adamantyl Sulfides with [Bis(1-Adamantanecarboxy)Iodo]Arenes and Disulfides. Synthesis 1995, 1995, 155–157. [Google Scholar] [CrossRef]
- Togo, H.; Muraki, T.; Hoshina, Y.; Yamaguchi, K.; Yokoyama, M. Formation and Synthetic Use of Oxygen-Centred Radicals with (Diacetoxyiodo)Arenes. J. Chem. Soc., Perkin Trans. 1, 1997, 787–794. [CrossRef]
- Gogonas, E. P.; Hadjiarapoglou, L. P. [3+2]-Cycloaddition Reactions of 2-Phenyliodonio-5,5-Dimethyl-1,3-Dioxacyclohexanemethylide. Tetrahedron Lett, 2000, 41, 9299–9303. [Google Scholar] [CrossRef]
- Matveeva, E. D.; Podrugina, T. A.; Pavlova, A. S.; Mironov, A. V.; Borisenko, A. A.; Gleiter, R.; Zefirov, N. S. Heterocycles from Phosphonium−Iodonium Ylides. Photochemical Synthesis of λ5-Phosphinolines. J. Org. Chem 2009, 74, 9428–9432. [Google Scholar] [CrossRef] [PubMed]
- Matveeva, E. D.; Podrugina, T. A.; Taranova, M. A.; Borisenko, A. A.; Mironov, A. V.; Gleiter, R.; Zefirov, N. S. Photochemical Synthesis of Phosphinolines from Phosphonium−Iodonium Ylides. J. Org. Chem 2010, 76, 566–572. [Google Scholar] [CrossRef] [PubMed]
- Nekipelova, T. D.; Kuzmin, V. A.; Matveeva, E. D.; Gleiter, R.; Zefirov, N. S. On the Mechanism of Photoinduced Addition of Acetonitrile to Phosphonium–Iodonium Ylides. J. Phys. Org. Chem 2012, 26, 137–143. [Google Scholar] [CrossRef]
- Nekipelova, T. D.; Taranova, M. A.; Matveeva, E. D.; Podrugina, T. A.; Kuzmin, V. A.; Zefirov, N. S. Mechanism and Remarkable Features of Photoinduced Cycloaddition of Phenylacetylene to Mixed Phosphonium-Iodonium Ylide. Dokl. Akad. Nauk 2012, 447, 262–265. [Google Scholar] [CrossRef]
- Moteki, S. A.; Usui, A.; Selvakumar, S.; Zhang, T.; Maruoka, K. Metal-Free CH Bond Activation of Branched Aldehydes with a Hypervalent Iodine(III) Catalyst under Visible-Light Photolysis: Successful Trapping with Electron-Deficient Olefins. Angew. Chem. Int. Ed 2014, 53, 11060–11064. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Li, H.; Ji, W.; Wang, L. Sunlight-Driven Decarboxylative Alkynylation of α-Keto Acids with Bromoacetylenes by Hypervalent Iodine Reagent Catalysis: A Facile Approach to Ynones. Angew. Chem. Int. Ed 2015, 54, 8374–8377. [Google Scholar] [CrossRef]
- Ji, W.; Tan, H.; Wang, M.; Li, P.; Wang, L. Photocatalyst-Free Hypervalent Iodine Reagent Catalyzed Decarboxylative Acylarylation of Acrylamides with α-Oxocarboxylic Acids Driven by Visible-Light Irradiation. Chem. Commun 2016, 52, 1462–1465. [Google Scholar] [CrossRef] [PubMed]
- Lucchetti, N.; Tkacheva, A.; Fantasia, S.; Muñiz, K. Radical C−H-Amination of Heteroarenes Using Dual Initiation by Visible Light and Iodine. Adv. Synth. Catal 2018, 360, 3889–3893. [Google Scholar] [CrossRef]
- Chidley, T.; Jameel, I.; Rizwan, S.; Peixoto, P. A.; Pouységu, L.; Quideau, S.; Hopkins, W. S.; Murphy, G. K. Blue LED Irradiation of Iodonium Ylides Gives Diradical Intermediates for Efficient Metal-free Cyclopropanation with Alkenes. Angew. Chem. Int. Ed 2019, 58, 16959–16965. [Google Scholar] [CrossRef] [PubMed]
- Voutyritsa, E.; Garreau, M.; Kokotou, M. G.; Triandafillidi, I.; Waser, J.; Kokotos, C. G. Photochemical Functionalization of Heterocycles with EBX Reagents: C−H Alkynylation versus Deconstructive Ring Cleavage. Chem. Eur. J 2020, 26, 14453–14460. [Google Scholar] [CrossRef] [PubMed]
- Induced, Catalyst-Free Monofluoromethyl Sulfonylation of Alkenes with Iodine(III) Reagent and DABSO. Org. Lett 2023, 25, 7062–7066. [CrossRef]
- Ou, C.; Cai, Y.; Ma, Y.; Zhang, H.; Ma, X.; Liu, C. Aliphatic Sulfonyl Fluoride Synthesis via Decarboxylative Fluorosulfonylation of Hypervalent Iodine(III) Carboxylates. Org. Lett. 2023, 25, 6751–6756. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Yue, W.; Wang, Z.; Xu, H.; Yang, M.; Zhu, J. Iodine(III)-Mediated Photochemical C–H Azolation. Org. Lett. 2024, 26, 9305–9310. [Google Scholar] [CrossRef]
- Paul, S.; Das, S.; Choudhuri, T.; Sikdar, P.; Bagdi, A. K. PIDA as an Iodinating Reagent: Visible-Light-Induced Iodination of Pyrazolo[1,5-a]Pyrimidines and Other Heteroarenes. Chem. Asian J. 2024, e202401101. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Xie, J.; Xue, Q.; Pan, C.; Cheng, Y.; Zhu, C. Visible-Light-Induced Trifluoromethylation of N-Aryl Acrylamides: A Convenient and Effective Method To Synthesize CF3-Containing Oxindoles Bearing a Quaternary Carbon Center. Chem. Eur. J 2013, 19, 14039–14042. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Jia, K.; Chen, Y. Hypervalent Iodine Reagents Enable Chemoselective Deboronative/Decarboxylative Alkenylation by Photoredox Catalysis. Angew. Chem 2014, 127, 1901–1904. [Google Scholar] [CrossRef]
- Huang, H.; Zhang, G.; Gong, L.; Zhang, S.; Chen, Y. Visible-Light-Induced Chemoselective Deboronative Alkynylation under Biomolecule-Compatible Conditions. J. Am. Chem. Soc., 2014, 136, 2280–2283. [CrossRef]
- Huang, H.; Zhang, G.; Chen, Y. Dual Hypervalent Iodine(III) Reagents and Photoredox Catalysis Enable Decarboxylative Ynonylation under Mild Conditions. Angew. Chem. Int. Ed 2015, 54, 7872–7876. [Google Scholar] [CrossRef]
- Jia, K.; Zhang, F.; Huang, H.; Chen, Y. Visible-Light-Induced Alkoxyl Radical Generation Enables Selective C(sp3)–C(sp3) Bond Cleavage and Functionalizations. J. Am. Chem. Soc 2016, 138, 1514–1517. [Google Scholar] [CrossRef]
- Li, G.-X.; Morales-Rivera, C. A.; Wang, Y.; Gao, F.; He, G.; Liu, P.; Chen, G. Photoredox-Mediated Minisci C–H Alkylation of N-Heteroarenes Using Boronic Acids and Hypervalent Iodine. Chem. Sci 2016, 7, 6407–6412. [Google Scholar] [CrossRef] [PubMed]
- Jia, K.; Pan, Y.; Chen, Y. Selective Carbonyl−C(Sp3) Bond Cleavage To Construct Ynamides, Ynoates, and Ynones by Photoredox Catalysis. Angew. Chem 2017, 129, 2518–2521. [Google Scholar] [CrossRef]
- Wang, Z.; Herraiz, A. G.; del Hoyo, A. M.; Suero, M. G. Generating carbyne equivalents with photoredox catalysis. Nature 2018, 554, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Tu, Y.; Ma, D.; Bolm, C. Photocatalytic Fluoro Sulfoximidations of Styrenes. Angew. Chem. Int. Ed., 2020, 59, 14134–14137. [CrossRef]
- Dong, J.-Y.; Wang, H.; Mao, S.; Wang, X.; Zhou, M.-D.; Li, L. Visible Light-Induced [3+2] Cyclization Reactions of Hydrazones with Hypervalent Iodine Diazo Reagents for the Synthesis of 1-Amino-1,2,3-Triazoles. Advanced Synthesis & Catalysis 2021, 363, 2133–2139. [Google Scholar] [CrossRef]
- Wen, J.; Zhao, W.; Gao, X.; Ren, X.; Dong, C.; Wang, C.; Liu, L.; Li, J. Synthesis of [1,2,3]Triazolo-[1,5-a]quinoxalin-4(5H)-ones through Photoredox-Catalyzed [3 + 2] Cyclization Reactions with Hypervalent Iodine(III) Reagents. The Journal of Organic Chemistry 2022, 87(6), 4415–4423. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Liu, Z.; Zou, P.; Chen, Y.; Chen, Y. Hypervalent Iodine Reagents Enable C(sp2)–H Amidation of (Hetero)Arenes with Iminophenylacetic Acids. Org. Lett. 2022, 24, 6681–6685. [Google Scholar] [CrossRef]
- He, Q.; Zhang, Q.; Rolka, A. B.; Suero, M. G. Alkoxy Diazomethylation of Alkenes by Photoredox-Catalyzed Oxidative Radical-Polar Crossover. J. Am. Chem. Soc 2024, 146, 12294–12299. [Google Scholar] [CrossRef]
- Xie, J.; Xu, P.; Li, H.; Xue, Q.; Jin, H.; Cheng, Y.; Zhu, C. A Room Temperature Decarboxylation/C–H Functionalization Cascade by Visible-Light Photoredox Catalysis. Chem. Commun 2013, 49, 5672. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Calculated energies for the stronger C-H bonds.
Scheme 1.
Suarez´s cleavage.
Scheme 2.
Piridine´s core 2-alkyl functionalization by photo-decarboxylative radical generation.
Scheme 3.
Iodine(III) mediated synthesis of aryl-adamantyl thioethers.
Scheme 4.
Synthesis of cumarine, phtalide and benzocumarine using Surez´s strategy.
Scheme 5.
Cycloaddition on photoexcited iodonium ylides for the synthesis of hydrobenzofuranes.
Scheme 6.
Synthesis of λ5-phoshinolines using iodine(III) ylides and acetilenes under photochemical conditions.
Scheme 6.
Synthesis of λ5-phoshinolines using iodine(III) ylides and acetilenes under photochemical conditions.
Scheme 8.
Iodine(III)-catlyzed photochemical synthesis of ynones by decrboxylative functionalization of alkynes.
Scheme 8.
Iodine(III)-catlyzed photochemical synthesis of ynones by decrboxylative functionalization of alkynes.
Scheme 9.
Photochemical synthesis of C3-disubstituted 2-oxoindoles catlyzed by cyclic iodine(III) reagents.
Scheme 9.
Photochemical synthesis of C3-disubstituted 2-oxoindoles catlyzed by cyclic iodine(III) reagents.
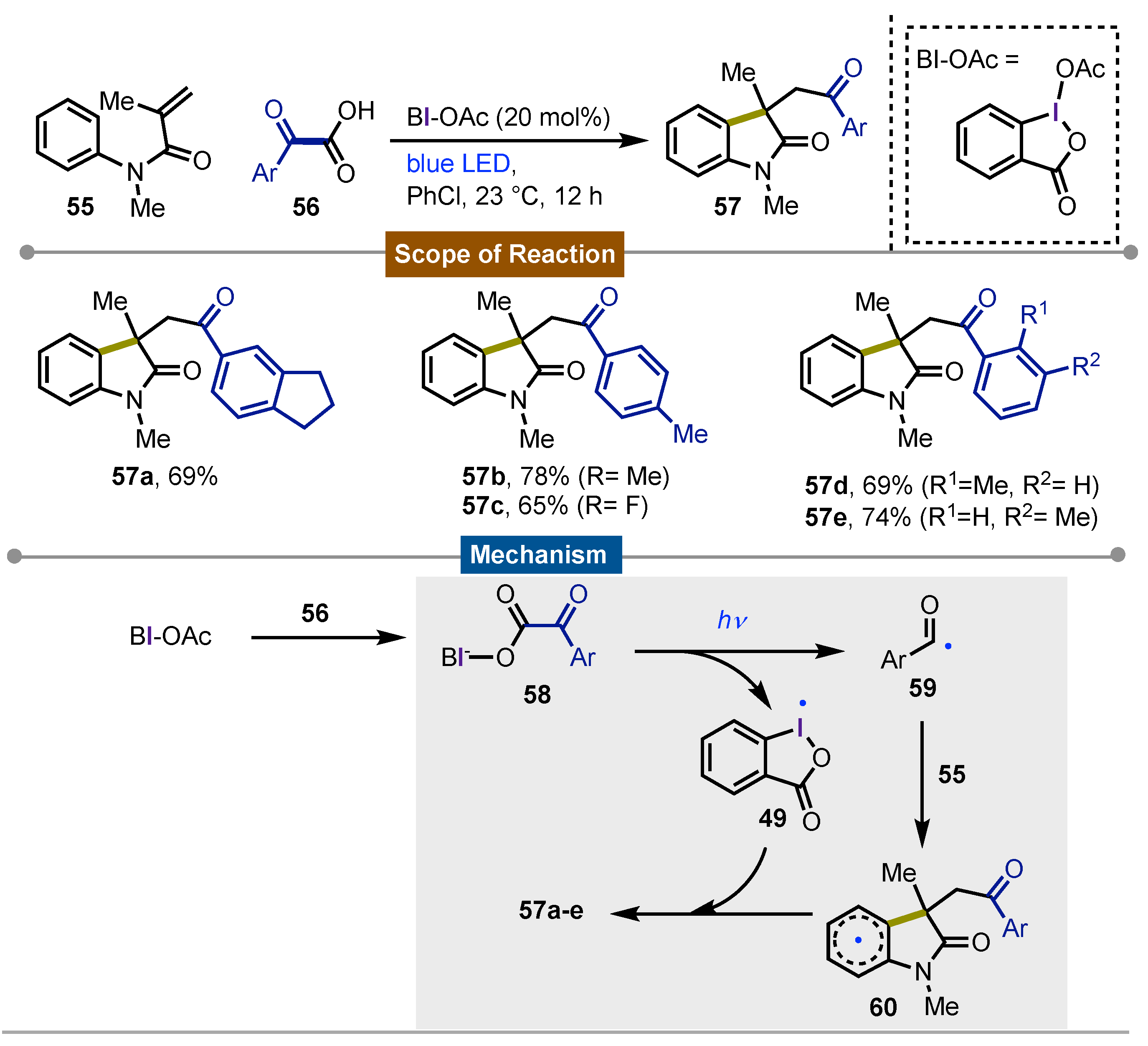
Scheme 10.
Iodine(III)-mediated photochemical C2-amination of indoles.
Scheme 11.
Photochemical cycloporoanation of styrenes with iodine(III)-dimedone ylides.
Scheme 12.
Photochemical alkynyltion of cyclic ethers and thioethers using EBX reagents.
Scheme 13.
Photochemical synthesis of C3-fluoromethylensulfonyl 2-oxoindoles.
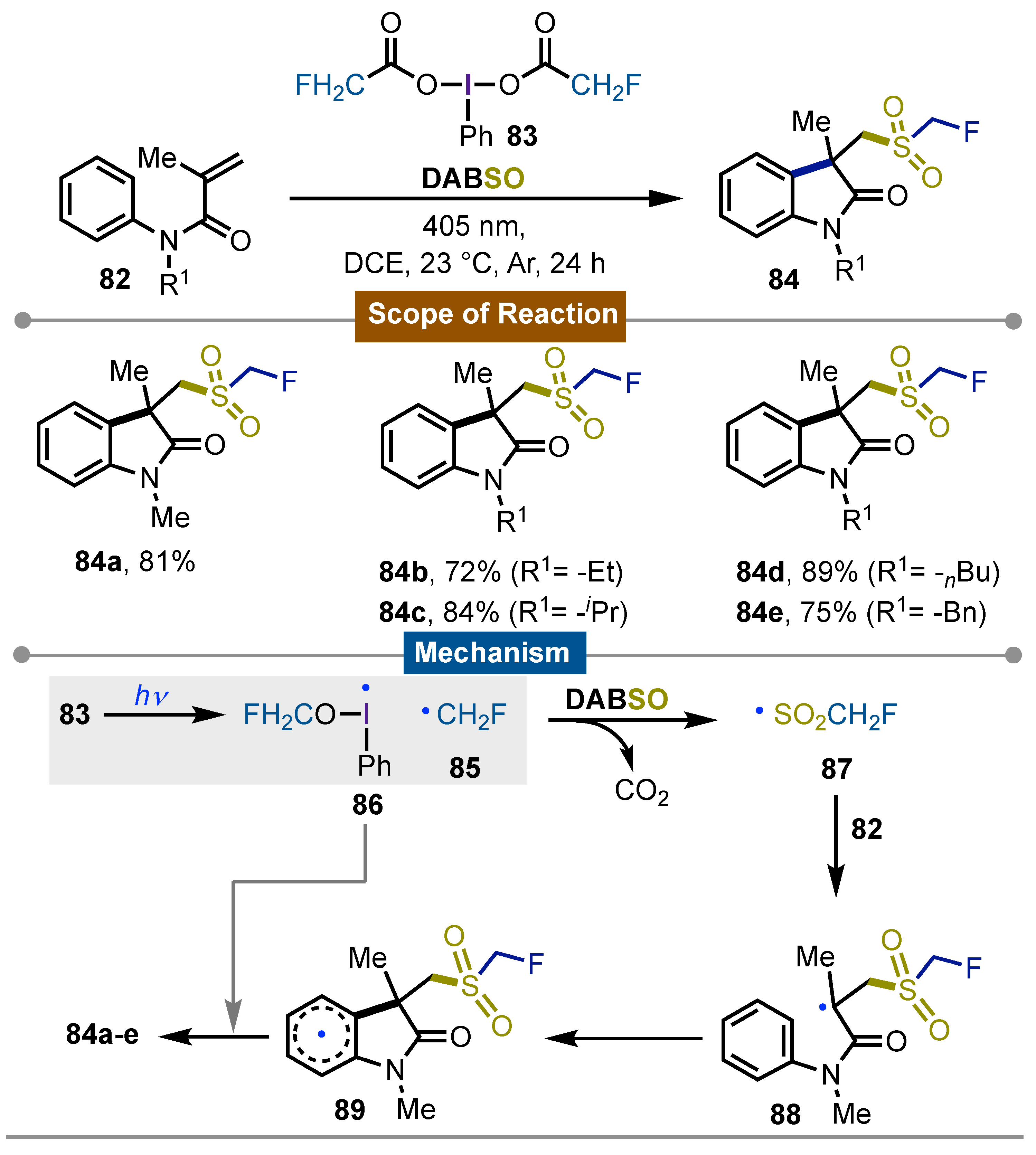
Scheme 14.
Photochemical fluorosulfonylation of homobenzylic derivatives.
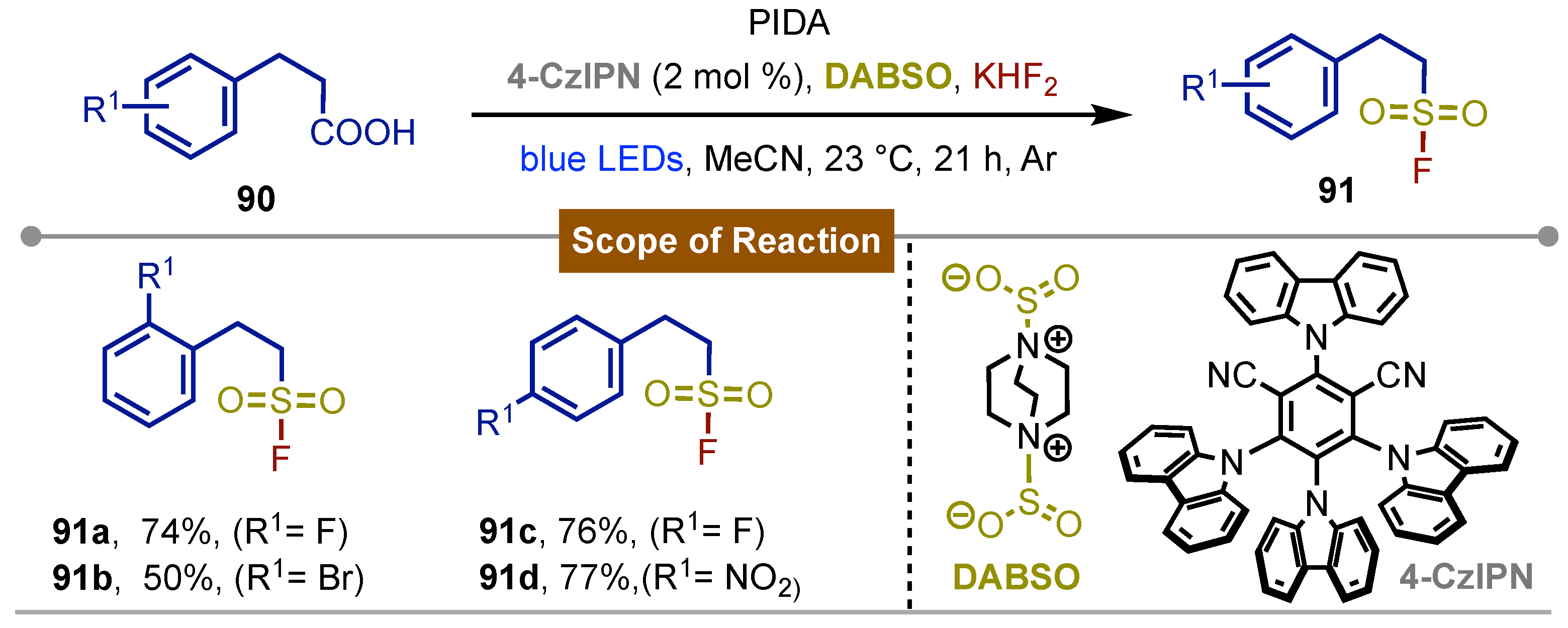
Scheme 15.
Photochemicl and direct sp3C-H azolation of cyclic ethers medited by PIDA.
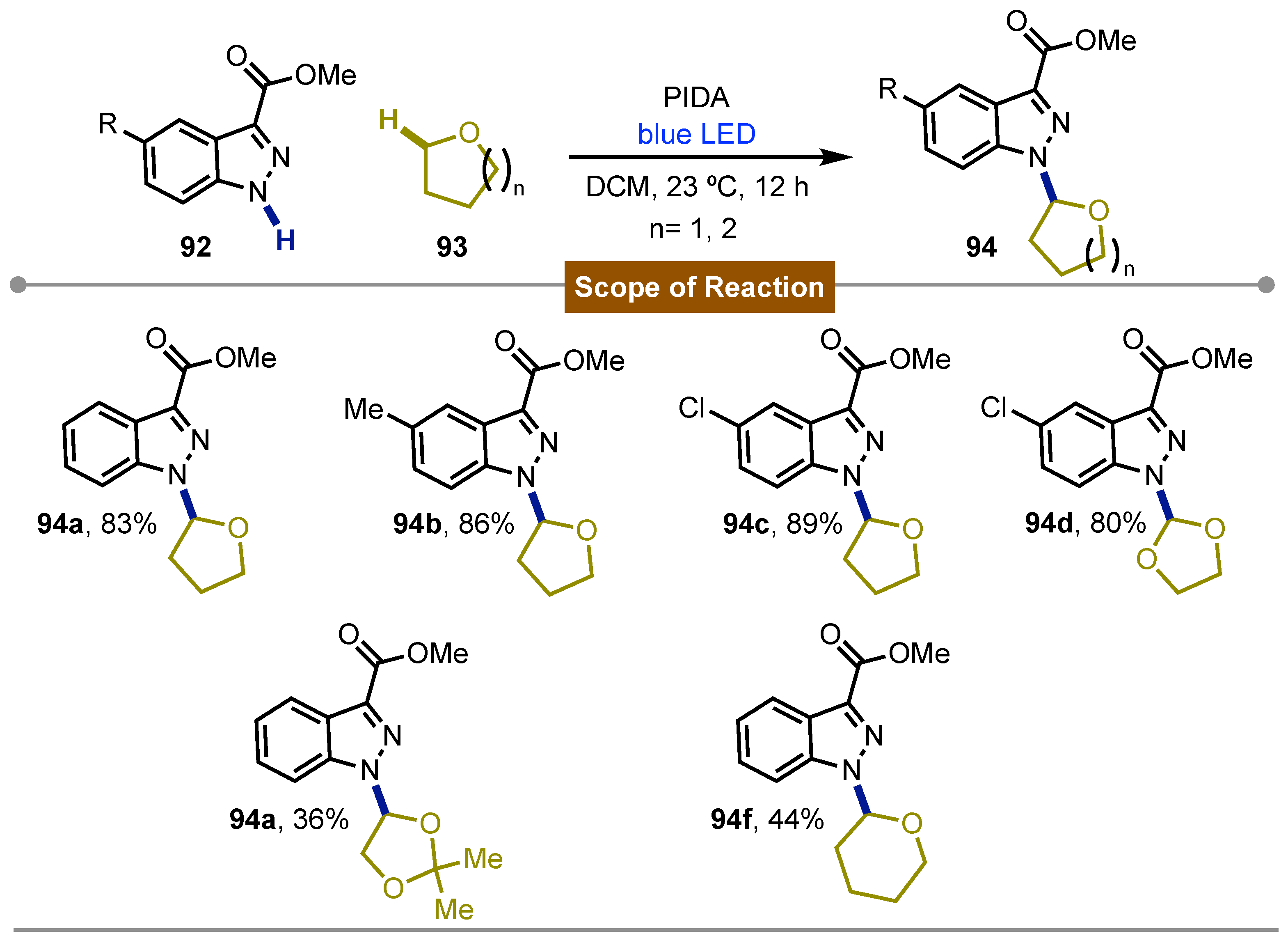
Scheme 16.
Photochemical iodination of imidazopiridines mediated by PIDA.
Scheme 17.
Synthesis 2-oxindoles by visible-light-promoted carbotrifluoromethylation of electron-withdrawing alkenes developed.
Scheme 17.
Synthesis 2-oxindoles by visible-light-promoted carbotrifluoromethylation of electron-withdrawing alkenes developed.
Scheme 18.
Chemoselective sp3C-sp2C photoredox coupling of alkyltrifluoroborates with alkenyl hypervalent iodine reagents formed in situ for the sintesis of substituted olefines.
Scheme 18.
Chemoselective sp3C-sp2C photoredox coupling of alkyltrifluoroborates with alkenyl hypervalent iodine reagents formed in situ for the sintesis of substituted olefines.
Scheme 19.
Photocatalytic deboronative alkynylation of alkyl boronates using EBX reagents.
Scheme 20.
Photocatalytic decarboxylative ynonylation of alkynes using EBX reagents.
Scheme 21.
β-fragmentation of tertiary alcohols for the photocatalytic synthesis of b-alkynyl ketones using acrylic acids and EBX reagents.
Scheme 21.
β-fragmentation of tertiary alcohols for the photocatalytic synthesis of b-alkynyl ketones using acrylic acids and EBX reagents.
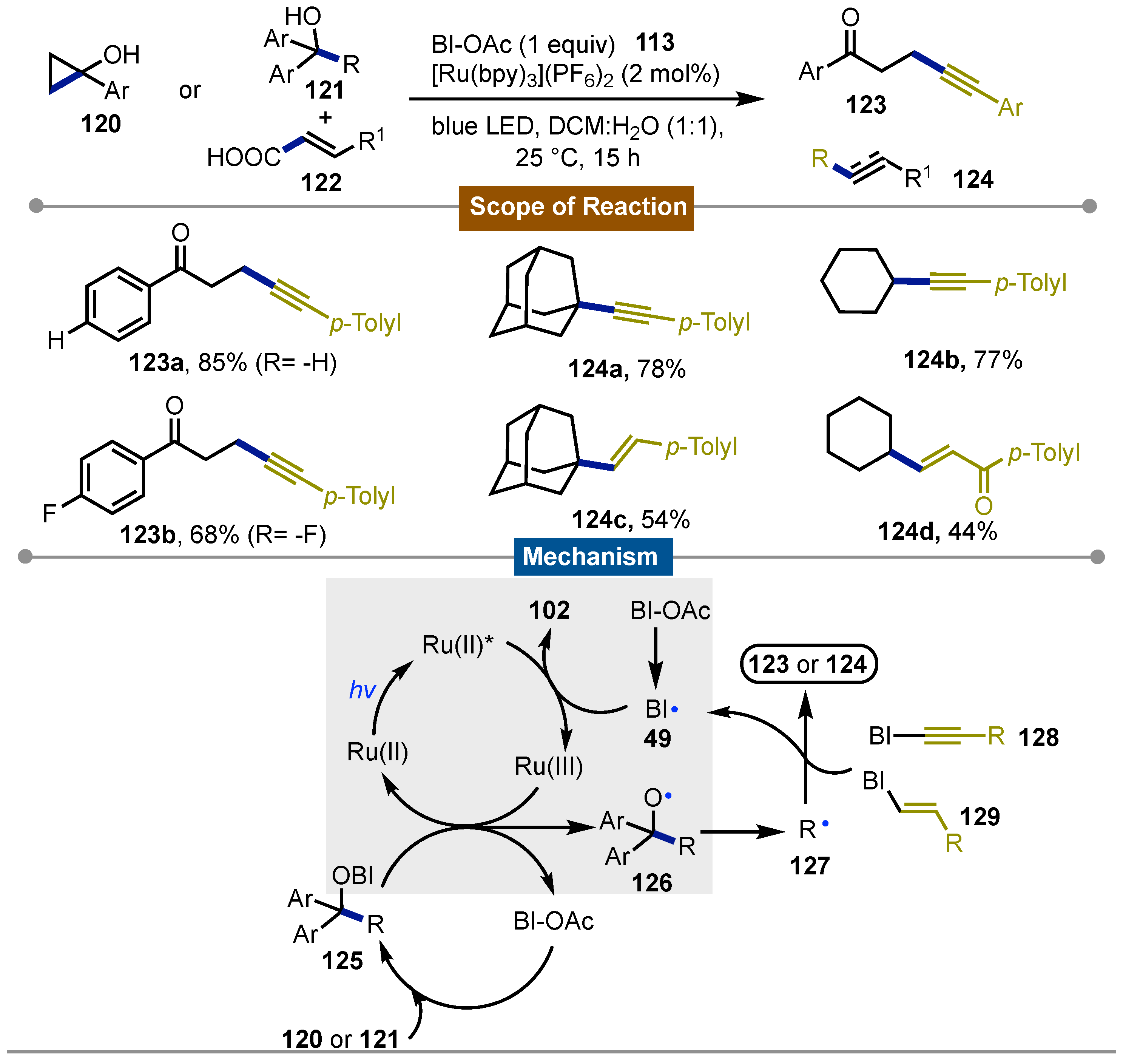
Scheme 22.
Photocatalytic Minisci-type C-H activation of heteroarenes with alkyl boronic acids.
Scheme 23.
Synthesis of ynamides, using a hypervalent iodine(III)-[Ru] photoredox system.
Scheme 24.
Diazomethylation of arenes with carbyne-equivalents obtained by photoredox catalysis.
Scheme 25.
Photocatalytic fluorosulfoximidations of styrenes mediated by l3-difluoroiodanes.
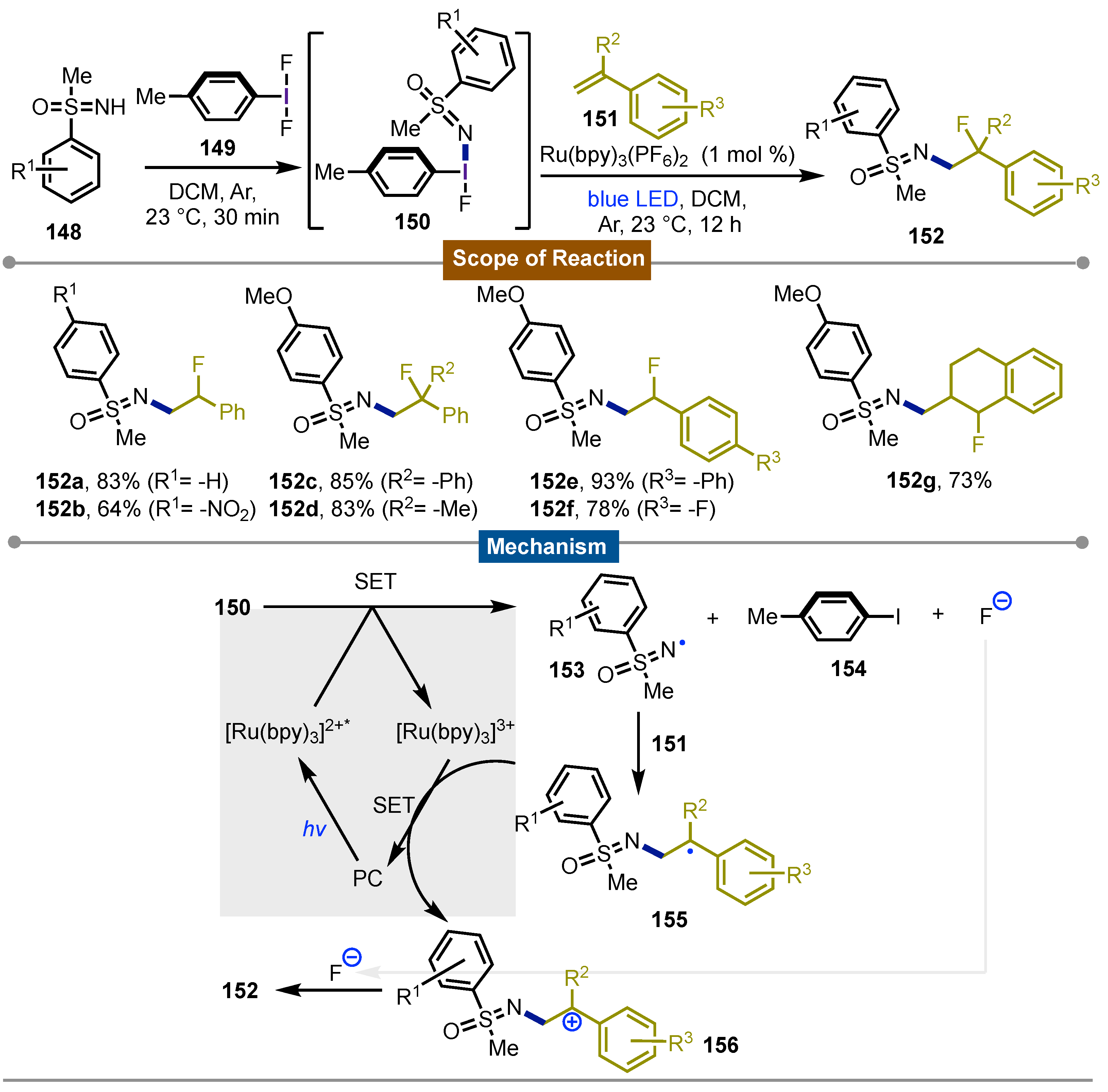
Scheme 26.
Mechanism and scope of the photoredox [3+2] cyclization reaction for the synthesis of 1-amino-1,2,3-triazoles.
Scheme 26.
Mechanism and scope of the photoredox [3+2] cyclization reaction for the synthesis of 1-amino-1,2,3-triazoles.
Scheme 27.
Photoredox-catalyzed [3 + 2] cyclization with hypervalent iodine(III)reagents for the synthesis of [1,2,3]triazolo-[1,5-]quinoxalin-4(5H)-ones.
Scheme 28.
Photoredox sp2C−H amidation of arenes and heteroarenes with amidyl-iminophenylacetic acids.
Scheme 28.
Photoredox sp2C−H amidation of arenes and heteroarenes with amidyl-iminophenylacetic acids.
Scheme 29.
Photoredox alkoxy diazomethylation of alkenes using iodine(III)-containing diazo derivtives.
Scheme 29.
Photoredox alkoxy diazomethylation of alkenes using iodine(III)-containing diazo derivtives.
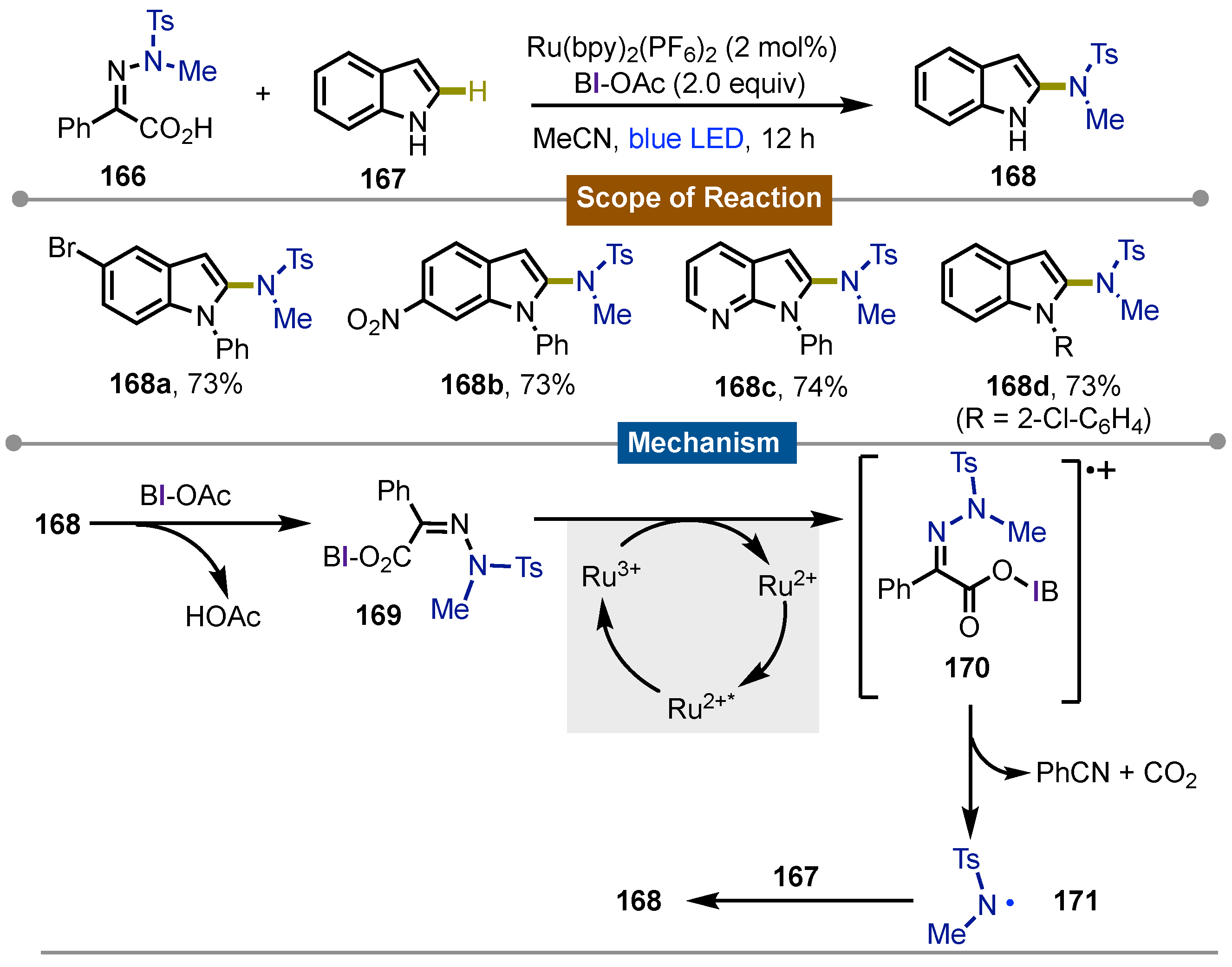
Scheme 30.
Photocatalytic C3-alkylation/cyclization of acrylamides mediated by iodine(III) reagents for the synthesis of 2-oxoindoles.
Scheme 30.
Photocatalytic C3-alkylation/cyclization of acrylamides mediated by iodine(III) reagents for the synthesis of 2-oxoindoles.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



