
Submitted:
31 December 2024
Posted:
03 January 2025
You are already at the latest version
Abstract
Hereditary spherocytosis (HS) is an erythrocytic membranopathy that belongs to a group of rare genetic disorders. Mutations in five genes, including ANK1, cause clinical manifestations of the disease. Identified variations in individual families provide a better understanding of the molecular basis of the disease. In this study, we used two sequencing methods, WES and Sanger sequencing, analyzing gDNA and cDNA as templates, to detect and verify the variants putatively responsible for the clinical symptoms observed in a Polish family diagnosed with hereditary spherocytosis. We detected two variants that occur in cis in the ANK1 gene, a known missense mutation (NP_000028.3:p.V463I) and a novel frameshift mutation (NP_000028.3: p.V1626fs*64) that ap-pears to be crucial for the probands. As shown by transcriptome studies, the mutant allele is not present at a detectable level. We conclude that the molecular basis of this case is related to an un-stable transcript of the mutant allele and that the direct cause of the spherocytosis is a deficiency of erythrocyte ankyrin leading to a disruption of the AE1-erythrocyte ankyrin-spectrin complex in the erythrocyte membrane.
Keywords:
hereditary spherocytosis
; ankyrin-1
; erythrocyte membrane protein
; whole exome sequencing
1. Introduction
The most common erythrocyte membrane disorder in the world is hereditary spherocytosis, a hemolytic anemia that affects one in 2,000–5,000 people in northern European countries [1,2]. Disruption of the connections between the membrane skeleton and the lipid bilayer containing integral proteins is the molecular basis of hereditary spherocytosis [3,4]. In HS cases described in the literature, dysregulation or loss of erythrocyte membrane proteins can reduce the stability and deformability of erythrocytes [1]. The result is increased osmotic fragility of erythrocytes, leading to erythrocyte rupture, premature hemolysis, and anemia [5]. The lifespan of red blood cells is reduced from 120 to a few days.
Compensation for hemolysis by increased erythropoiesis usually depends on the type of defect, general health, and the individual patients' abilities. However, in most cases it is insufficient, ultimately leading to anemia. Other diagnostic criteria include jaundice, reticulocytosis, splenomegaly, gallstones, spherocytes in the peripheral blood smear, and decreased osmotic resistance of erythrocytes [6,7]. Within the same family, a similar clinical picture can be observed, but significant differences occur. These are due to individual differences, including age and general health, but also to additional genetic variants present in selected family members [8]. The uniqueness of the variants underlying HS (often identified only within a single family), the presence of additional genetic variants, similar clinical manifestations of other RBC defects, as well as the general health of the patients, make the diagnosis of hereditary spherocytosis a challenge for hematologists [9].
Interesting research by Huisjes et al. shows that in patients from one family in which the defect correlates with moderate/severe spherocytosis, a greater diversity of clinical symptoms is observed [5]. In these patients, the lower density (correlated with lower MCHC) and heterogeneity of the erythrocytes caused relatively little membrane loss. Nevertheless, these cells were rapidly removed by the spleen due to a significant loss of membrane deformation capacity. In HS patients after splenectomy and with a mild form of HS, erythrocytes that remained in circulation for a longer time were characterized by a higher density associated with significant membrane loss. The greatest diversity in severity and clinical manifestations of the HS was seen in patients with ANK1 gene mutations.
It is currently recognized that mutations in five proteins responsible for these interactions correlate with the RBC defect discussed here. These are proteins encoded by the following genes: ANK1 (ankyrin-1), SPTA1 (α-spectrin), SPTB (β-spectrin), SLC4A1 (solute carrier family 4 member 1, Diego blood group, AE1), and EPB42 (protein 4.2) [10,11]. Autosomal dominant inheritance is common in HS and specific variants have been identified in the genes ANK1 (spherocytosis, type 1; MIM#182900), SPTB (spherocytosis, type 2; MIM#616649), and SLC4A1 (spherocytosis, type 4; MIM#612653) in about two-thirds of cases [2,12]. However, biallelic pathogenic variants (a recessive form of spherocytosis) have been observed mainly in the SPTA1 gene (spherocytosis, type 3; MIM#270970), and less frequently in the EPB42 (spherocytosis, type 5; MIM#612690) and ANK1 genes. α-Spectrin is a protein with high expression in human and mouse erythrocytes which results in normal protein levels in heterozygotes. It is regulated by regulatory elements (GATA-1- and NF-E2) that determine high erythroid-specific expression [13]. Heterozygotes of the EPB42 gene are also clinically normal, whereas homozygotes have reduced levels of the AE1 protein, which is essential for erythrocyte membrane stability [14,15].
Among de novo mutations, changes in the ANK1 gene predominate in patients with HS [16,17,18]. The gene encoding ankyrin-1 appears to be particularly prone to slipped strand mispairing during DNA replication [19]. This is due to the high GC content in the nucleotide sequence encoding the exons, especially in the ANK1 gene promoter (77% between positions -1 to -306). The GC-rich ankyrin-1 gene promotor, like housekeeping promoters, does not contain consensus TATA, InR, or CCAAT sequences [20,21].
The ANK1 gene encodes ankyrin-1, which links the spectrin-actin cytoskeleton to integral membrane proteins of the red blood cell (RBC) and is essential for cell membrane integrity. The protein was first detected in erythrocytes, hence the name erythrocyte ankyrin is often used [22]. Further discoveries have shown their presence also in the muscle and brain [23,24]. Mutations in the ANK1 gene can affect the expression of ankyrin-1 and disrupt the function of key membrane proteins. The absence or decrease of normal ankyrin-1 in the cell membrane also leads to a loss of β-spectrin [25]. Ankyrin mRNA deficiency resulting from mutations in the ANK1 gene leads to reduced ankyrin synthesis, which in turn leads to reduced spectrin assembly at the membrane. The deficiency of these two crucial proteins affects the structure and flexibility of the red blood cell membrane, causing changes in the shape of the cell.
Here we report a novel p.V1626fs*64 frameshift mutation and a known missense p.V463I mutation in the ANK1 gene, detected by WES and confirmed by standard Sanger sequencing, related to the phenotype in a Polish family with autosomal dominant HS. Transcript analysis of this gene revealed only an unmutated sequence. The most probable cause of the molecular basis is the unstable transcript of the mutant allele and decreased ankyrin-1 in the erythrocytes of two HS patients from the studied AM family.
2. Materials and Methods
2.1. Patients
Present study included two patients from a Polish family referred to Department and Clinic of Hematology, Cellular Therapies and Internal Medicine at the Medical University of Wroclaw, clinically diagnosed with HS. Moderate disease symptoms were seen in the unsplenectomized subjects, father (AM175, age 82) and daughter (AM174, age 52). Other family members, except for AM173 (daughter of patient AM174, 16 years old, who was in the study and in a clinically healthy state), were not available. Typical features such as anemia, spherocytes on peripheral blood smear, splenomegaly, elevated bilirubin and reticulocytosis were used as diagnostic criteria for HS. Clinical manifestations were similar between patients. The research had the approval of the Wroclaw Medical University Ethics Committee (protocol KB-199/2017). Prior to inclusion in the protocol, informed consent was obtained from all family members.
2.2. DNA and RNA Isolation
Whole blood was obtained by venipuncture from two HS patients (AM175 and AM174), and one asymptomatic family member (AM173) and collected in tubes containing EDTA. Genomic DNA extraction was carried out using the standard method (QIAamp DNA Blood Mini Kit, Qiagen, Hilden, Germany). The miRNeasy Mini Kit (Qiagen, Hilden, Germany) was used to purify total RNA from fresh whole blood. The manufacturer's recommendations have been followed for the above procedures. The isolated gDNA was stored at -20 °C and the RNA at -80 °C until analysis. Absorbance in a Cary 60 UV spectrophotometer at 260 nm was used to determine the concentration and quality of purified DNA and RNA.
2.3. Whole Exome Sequencing
The whole exome sequencing of the genomic DNA samples from three family members was performed at the Heflin Center for Genomic Science Core Laboratories at the University of Alabama at Birmingham (AL, USA). Briefly, the sequencing library was prepared using the SureSelect Target Enrichment System (Agilent Technologies, Inc., CA, USA) and captured using the Agilent SureSelect Human Clinical Research Exome (CRE) capture kit (Agilent Technologies, Inc., CA, USA). The read length of each sample was 100 bp on an Illumina NextSeq500 (Illumina, Inc., CA, USA), and the average coverage depth was at least 100×.
2.4. Whole-Exome Sequencing Data Analysis
The WES data were trimmed for low quality sequences and mapped against the UCSC Human Reference Genome (human genome 19/GRCh37.13) using Burrows-Wheeler Alignment (BWA). The Heflin Centre for Genomic Science Core Laboratories at the University of Alabama at Birmingham (UAB), AL, USA performed bioinformatics analysis to detect single nucleotide variants (SNVs) and insertions/deletions. All variants were then filtered, annotated and evaluated using Ingenuity Variant Analysis (IVA; QIAGEN, CA, USA).
A strategy was developed to select the rare and potentially deleterious variants present in both HS patients and lacking in a healthy family member. We mainly considered heterozygous and homozygous in-exon variations with predicted consequences (splicing, non-synonymous, frameshift, stop). The clinical significance of the selected changes was assessed using 1000 Genome Browser database (https://www.internationalgenome.org), ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/), HGMD (http://www.hgmd.cf.ac.uk/), Online Mendelian Inheritance in Man (OMIM) (http://www.omim.org/), Single Nucleotide Polymorphism (dbSNP) (https://www.ncbi.nlm.nih.gov/snp/), GeneCards (https://www.genecards.org/), The Universal Protein Resource (UniProt) (https://www.uniprot.org/) databases (each accessed on 27 November 2024), and literature data.
2.5. Variants Validation
Targeted Sanger sequencing of genomic DNA and/or cDNA was applied to validate potentially pathogenic relevant variants selected by WES. Total RNA was reversely transcribed into cDNA for sequencing using the Maxima H Minus Double-Stranded cDNA Synthesis Kit (Thermo Fisher Scientific, Waltham, MA, USA). Genomed S.A. (Warsaw, Poland) performed the Sanger sequencing and synthesized all primers. Primers used were designed with Gene Runner version 6.5.52x64 Beta. The sequences of all primers are listed in Tables S1.1 and S1.2 (see Supplementary material for details). The PCR amplification parameters are available upon request. All the genetic variants reported in the manuscript have been lifted to GRCh38.
3. Results
3.1. Hematological Parameters
Clinical manifestations, family history, laboratory test results, and morphological review of the peripheral blood smear were made to diagnose hereditary spherocytosis in the investigated family. The two studied probands (AM174 and AM175), who underwent splenectomy, presented similar clinical presentation resembling those of HS patients, namely, jaundice, moderate chronic anemia, reticulocytosis, bilirubinemia, and splenomegaly. The hematological parameters for probands are listed in Table 1.
3.2. Validation of WES Results
For each available AM family member, whole exome sequencing was performed, including two with symptoms of HS, the father of patient AM174 (AM175), the daughter of patient AM175 (AM174), and one asymptomatic family member - the daughter of patient AM174 (AM173). All the detected variants, that were filtered and annotated by Ingenuity Variant Analysis software, were classified according to their clinical significance using the ClinVar database and/or the ACMG classification [26]. Briefly, 93,162 nucleotide sequence changes were detected in the genetic material of AM family members using the WES method. We began the search of the obtained data by reviewing the pool of known and well-characterized genetic variants. The 1,916 variants classified by the Human Gene Mutation Database (HGMD) were identified, including 1,052 that were present simultaneously in both studied HS patients (AM174 and AM175). The only variant classified as pathogenic and detected as heterozygous in both HS patients was frameshift mutation in the ENAM gene (p.V1626fs*64; HGMD CI033730 (DM)), which correlates with responsible for autosomal recessive amelogenesis imperfecta and localized enamel defects. This change cannot be the basis of the analysed HS case due to the inheritance pattern. None of these known variants (classified by HGMD) appear to be crucial for the HS phenotype observed in the AM family.
Secondly, to investigate the correlation between the phenotype and genetic variations, we analyzed genes associated with the phenotype of known hereditary hemolytic anemias [27,28,29]. Data obtained by the WES method were compared with data collected from biomedical databases and the world literature regarding erythrocyte defects: membranopathies, enzymopathies, and hemoglobinopathies. In the panel of genes recommended for RBC pathologies, more than three hundred and sixty variants (including 11 unknown) have been identified (see Additional file 2 for details) [27]. Among these variants, we selected 12 potentially important changes in 8 genes (correlated with membranopaties and enzymopaties as well as other potentially pathogenic variants) and we verified their presence in the gDNA (Table S1.3). Selection criteria were inheritance pattern, frequency, clinical manifestation and significance as classified by ClinVar, CADD score, and SIFT function prediction, as well as other data deposited in biomedical databases including HGMD and literature data. We applied IVA-recommended filtering algorithms (Common Variants, Predicted Deleterious, Phenotype Driven Ranking, and Biological Context). Among the detected variants, only one was crucial and classified as likely pathogenic according to ACMG recommendations. A novel ANK1 gene c. 4959del (p.V1626fs*64) frameshift mutation was detected only in HS patients from the AM family (Figure 1).
Five genes associated with clinical phenotypes of hereditary spherocytosis were of particular interest for our research (ANK1, SPTA1, SPTB, SLC4A1, and EPB42). In four of these genes, 92 variants were found (see Additional file 3 for details). From these, we selected 5 potentially crucial changes (including the new variant (p.V1626fs*64), described above) in two genes (ANK1 and SPTB), which we verified the presence in the gDNA (Additional file 1: Table S1.). Two variants of the SPTB gene were only confirmed in a single AM family member: AM175 (heterozygous, NP_001342365.1:p.His1691Tyr, rs2082307149; frequency: A=0.000007 (1/140244, GnomAD)) and AM173 (homozygous, asymptomatic) /AM 174 (heterozygous, HS patient) (NP_001342365.1:p.Arg1403Gln, rs17180350, CM187439 (DM?)).
Two variants were confirmed only in HS patients first AM174 and AM175 in the ANK1 gene: a known missense mutation (NM_000037.4:c.1387G>A, NP_000028.3:p.V463I, rs140085544) which was heterozygous in both HS patients. Second, was a new single nucleotide deletion (NM_000037.4:c. 4959del, NP_000028.3: p.V1626fs*64), also heterozygous, in both HS patients, resulting in a frameshift and a premature stop codon, 64 codons downstream this change (see Figure 1). The mentioned above missense mutation was first described by Eber et al. [30] and deposited in the HGMD database (CM960064), and is now classified in ClinVar (variant-condition record (RCV)) as: “Conflicting pathogenicity classifications” (uncertain significance(2); benign(1); probably benign(1); variant ID: 719959)[26]. The new variant (p.V1626fs*64) detected by WES was automatically classified as likely pathogenic using the results of the Ingenuity Variant Analysis plugin results based on the guidelines of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology [25]. Criteria for classification of the p.V1626fs*64 variant: PVS1 null variant (frameshift) in a gene where LOF is a known mechanism of disease (Very strong); PM2 Absent from controls in Exome Sequencing Project, 1000 Genomes Project, or Exome Aggregation Consortium (Moderate); PM4 Protein length changes as a result of in-frame deletions in a nonrepeat region (Moderate); PP1 Cosegregation with disease (hereditary spherocytosis) in multiple affected family members in a gene definitively known to cause the disease (Supporting); PP4 Patient’s phenotype or family history is highly specific for a disease with a single genetic etiology (Supporting)”. Conservation phyloP p-value IVA plugin (QIAGEN, CA, USA) was predicted for the variant conservation phyloP p-value: 5.998E-10 (see Supplementary file S3 for details).
The detected deletion (NM_000037.4:c.4959del, NP_000028.3: p.V1626fs*64) is in the sequence encoding the regulatory domain of erythrocyte ankyrin (1383-1881 amino acid residues). A transcript of this gene was examined to verify allelic expression. Analysis of the double-stranded cDNA based on Sanger sequencing revealed the expression of only one unmutated allele of the ANK1 gene in both HS patients. Therefore, both mutations detected in the ANK1 gene were verified: a frameshift mutation (p.V1626fs*64) and a missense mutation (p.V463I) (see Figure 2 and Figure S1).
4. Discussion
Our previous research into the molecular basis of hereditary hemolytic anemias has allowed us to detect new variants and describe the molecular basis of these rare diseases in the Polish population [31,32,33,34,35,36]. Here we report a Polish family with two variants in the ANK1 gene, one a known missense mutation and the other a newly discovered frameshift mutation.
We decided to use WES, which is currently the most effective method for identifying the molecular mechanism of inherited disorders that may underline the clinical symptoms seen in probands from the AM family studied [28,29,37]. We were able to filter the variants identified by WES and narrow the search to changes associated with red cell pathology using the Ingenuity Variant Analysis plugin. The analysis allowed the selection of 8 variants correlated with the phenotype of hereditary hemolytic anemias and 4 other potentially pathogenic changes that may have affected the health status of AM family members (see Table S2). These variations were verified in the gDNA by Sanger sequencing. Five changes in membranopathy-related genes were confirmed, including two each in the ANK1 and SPTB genes. These four changes are in genes encoding proteins whose defects underlie hereditary spherocytosis, which is consistent with the initial clinical diagnosis. After analysis of all the data collected on these membranopathy-associated variants, two located in the ANK1 gene were selected as primary candidates in the HS patients from the AM family. The four other potentially pathogenic variations listed in Table S2 appeared irrelevant to the hematological abnormalities observed in the probands.
In the studied family members (AM174 and AM175) initially diagnosed with HS, the analysis of clinical symptoms and hematological data are in accordance with the whole exome sequencing results. In addition, the typical autosomal dominant type of inheritance of ankyrin-1 defects was observed in this family (OMIM #182900). Mutations in the ANK1 gene also cause a recessive form of the disease [38], usually with a mild to severe clinical course [1,39]. Ankyrin-1 binds spectrin to AE1 and plays a key role in stabilizing the erythrocyte membrane, therefore the ankyrin deficiency observed in HS patients leads to a reduction in spectrin assembly at the membrane [36]. Usually, a combined decrease of spectrin and ankyrin is detected (15–50% [1]), which is much easier to prove in HS patients after splenectomy. The ankyrin-1 deficiency in the erythrocyte membranes (that is usually detected by the SDS-PAGE method) can be masked in non-splenectomized patients by excessive hemolysis of the defective erythrocytes and subsequent reticulocytosis [6,40]. As shown by Miraglia et al., normal or even higher-than-normal levels of ankyrin-1 have been observed in patients with HS, which, according to the authors, indicates the inactivation of one ankyrin allele [40]. Two variants in the ANK1 gene were confirmed to be heterozygous only in HS patients and absent in an asymptomatic AM family member. The detected variants in the ANK1 gene, a new single nucleotide deletion resulting in a frameshift and a premature stop codon 64 codons downstream (p.V1626fs*64) and a known missense mutation (p.V463I, rs140085544) are most likely to occur in cis in both probands according to the inheritance pattern in the investigated AM family (Figure 3).
Variant p.V463I was found by Eber et al. in a patient with recessive HS, where the second change was detected in the promoter of the ANK1 gene [30]. The single nucleotide substitution (NM_000037.4:c.-108T>C, rs77173848) is silent in heterozygotes and always co-occurs with another variant of the ANK1 gene in other HS patients. Interestingly, the missense mutation p.V463I detected by Eber et al. was associated with a decrease in AE1 protein level [30]This was linked to the variant's location in the domain of ankyrin, which is responsible for binding the AE1 protein. This most likely involves disrupting the AE1-erythrocyte ankyrin-spectrin complex in the erythrocyte membrane.
In the AM family, the changes in the transcript are due to the coexistence of the two detected variants (frameshift mutation p.V1626fs*64 and missense mutation p.V463I) that occur in the cis. Loss of the double-mutated allele in the cDNA was observed only in the studied patients with spherocytosis as was shown for both detected variants (see Figure 2 and Figure S1). This indicates that the mutant transcript was not present at a level that could be detected. Therefore, the physiological effects are a consequence of reduced ankyrin-1 expression, leading to membrane dysfunction and premature removal of erythrocytes in the spleen. The clinical manifestation is the hemolytic anemia observed in probands from AM family. Previously, we have described a similar physiological effect for another frameshift mutation in the SPTB gene in HS patients in the Polish population [41]. In other known cases of HS patients with mutations in the SPTB, ANK1, and SLC4A1 genes with dominant inheritance, comparable observations have been made [42,43,44,45,46].
5. Conclusions
The identification of genetic variants that cause rare genetic diseases using high-throughput genomic analysis is now a highly accurate diagnostic tool. It allowed us to detect a novel frameshift mutation (p.V1626fs*64) and known missense mutation (p.V463I) in the ANK1 gene in individuals diagnosed with HS. However, using the classical Sanger sequencing method, we were able to confirm this change in the gDNA and cDNA of probands. This allowed us to suggest that the mutant transcript is unstable and that the cause of HS symptoms is the decrease in ankyrin-1 protein. This diagnostic approach allows us to verify the reliability of genome analysis methods using NGS and to deepen our knowledge of the molecular basis of a rare genetic disorder.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Additional File S1: Supplementary tables and figure. This file contains supplementary material: Table S1 Sequences of the PCR primers designed to verify the variants detected in the ANK1 gene (NC_000007.14). Table S2: The PCR primer sequences used to amplify ANK1 gene transcript variant 3 (NM_000037.4, MANE). Table S1.3 Variants identified by Sanger sequencing of all genes studied in AM family members. *Accessed on 27 November 2024 Figure S1.1 Fragment of sequencing traces showing localization of the missense mutation (p.V463I, rs140085544). The mutation is not present in a healthy individual (AM173). A heterozygous single nucleotide substitution G/A is identified in HS patient AM174 (genomic DNA was used as a template). Loss of the mutant allele is shown in the cDNA relative to the genomic DNA (cDNA from patient AM174 was used as a template). Additional File S2: Supplementary tables. Detailed data on the analyzed variants detected using WES (in the panel of genes recommended for RBC pathologies and analyzed using the Ingenuity Variant Analysis plugin (IVA; QIAGEN, CA, USA). Additional File S3: Detailed data on the analyzed variants detected by WES in the genes whose mutations result in HS and analyzed using the Ingenuity Variant Analysis plugin (IVA; QIAGEN, CA, USA).
Author Contributions
D.M.B. designed the experiments, performed the PCR experiments (cDNA and gDNA), evaluated the WES analyses, analyzed and interpreted the experimental data, prepared the figures, and wrote the manuscript. J.R. and K.K. were responsible for the recruitment, clinical care of the patients, and ethical aspects of the study. P.K. performed the PCR experiments (gDNA) and evaluated the WES analyses. A.F.S. supervised the study, did the final editing of the manuscript, and acquired financing. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported in part by a grant from the National Science Centre, Poland, Project No. 2015/19/B/NZ5/03469
Institutional Review Board Statement
The study was conducted under the Declaration of Helsinki and approved by the Ethics Committee of Wroclaw Medical University (protocol code KB-199/2017). The patients provided informed consent to participate in the study.
Informed Consent Statement
The patients provided informed consent for the publication of anonymized data on their genetic material and blood parameters.
Data Availability Statement
All data generated or analyzed during this study are included in this article and its additional files. WES datasets used and/or analyzed during this study are available from the corresponding author upon reasonable request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Perrotta, S.; Gallagher, P.G.; Mohandas, N. Hereditary Spherocytosis. Lancet 2008, 372, 1411–1426. [Google Scholar] [CrossRef] [PubMed]
- Eber, S.; Lux, S.E. Hereditary Spherocytosis—Defects in Proteins That Connect the Membrane Skeleton to the Lipid Bilayer. Semin Hematol 2004, 41, 118–141. [Google Scholar] [CrossRef] [PubMed]
- Ciepiela, O. Old and New Insights into the Diagnosis of Hereditary Spherocytosis. 2018, i, 1–10. [CrossRef]
- Iolascon, A.; Andolfo, I.; Russo, R. Advances in Understanding the Pathogenesis of Red Cell Membrane Disorders. Br J Haematol 2019, 187, 13–24. [Google Scholar] [CrossRef]
- Huisjes, R.; Makhro, A.; Llaudet-Planas, E.; Hertz, L.; Petkova-Kirova, P.; Verhagen, L.P.; Pignatelli, S.; Rab, M.A.E.; Schiffelers, R.M.; Seiler, E.; et al. Density, Heterogeneity and Deformability of Red Cells as Markers of Clinical Severity in Hereditary Spherocytosis. Haematologica 2020, 105, 338–347. [Google Scholar] [CrossRef]
- Bolton-Maggs, P.H.B.; Langer, J.C.; Iolascon, A.; Tittensor, P.; King, M.J. Guidelines for the Diagnosis and Management of Hereditary Spherocytosis - 2011 Update. Br J Haematol 2012, 156, 37–49. [Google Scholar] [CrossRef]
- Wu, Y.; Liao, L.; Lin, F. The Diagnostic Protocol for Hereditary Spherocytosis-2021 Update. J Clin Lab Anal 2021, 35. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Huang, H.; Luo, K.; Yi, Y.; Shi, X. Two Different Pathogenic Gene Mutations Coexisted in the Same Hereditary Spherocytosis Family Manifested with Heterogeneous Phenotypes. BMC Med Genet 2019, 20. [Google Scholar] [CrossRef]
- Liang, G.; Lin, Z.; Zhang, Y.; Zhang, Q.; Zhu, D.; Liang, X.; Xie, H.; Wei, X.; Shang, X. Precise Diagnosis of a Hereditary Spherocytosis Patient with Complicated Hematological Phenotype. Molecular Genetics and Genomics 2024, 299, 57. [Google Scholar] [CrossRef] [PubMed]
- Häuser, F.; Rossmann, H.; Adenaeuer, A.; Shrestha, A.; Marandiuc, D.; Paret, C.; Faber, J.; Lackner, K.J.; Lämmle, B.; Beck, O. Hereditary Spherocytosis: Can Next-Generation Sequencing of the Five Most Frequently Affected Genes Replace Time-Consuming Functional Investigations? Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Shu, H.; Zhou, M.; Gong, Y. Literature Review on Genotype–Phenotype Correlation in Patients with Hereditary Spherocytosis. Clin Genet 2022, 102, 474–482. [Google Scholar] [CrossRef]
- Risinger, M.; Kalfa, T.A. Red Cell Membrane Disorders: Structure Meets Function. Blood 2020, 136, 1250–1261. [Google Scholar] [CrossRef]
- Boulanger, L.; Sabatino, D.E.; Wong, E.Y.; Cline, A.P.; Garrett, L.J.; Garbarz, M.; Dhermy, D.; Bodine, D.M.; Gallagher, P.G. Erythroid Expression of the Human α-Spectrin Gene Promoter Is Mediated by GATA-1- and NF-E2-Binding Proteins. Journal of Biological Chemistry 2002, 277, 41563–41570. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, S.; Kajii, E.; Omi, T.; Kamesaki, T.; Akifuji, Y.; Ikemoto, S. Point Mutation in the Band 4.2 Gene Associated with Autosomal Recessively Inherited Erythrocyte Band 4.2 Deficiency. Eur J Haematol 1993, 50, 286–291. [Google Scholar] [CrossRef]
- Peters, L.L.; Jindel, H.K.; Gwynn, B.; Korsgren, C.; John, K.M.; Lux, S.E.; Mohandas, N.; Cohen, C.M.; Cho, M.R.; Golan, D.E.; et al. Mild Spherocytosis and Altered Red Cell Ion Transport in Protein 4.2–Null Mice. Journal of Clinical Investigation 1999, 103, 1527–1537. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.L.; Sang, B.H.; Lei, Q.L.; Song, C.Y.; Lin, Y.B.; Lv, Y.; Yang, C.H.; Li, N.; Yang, Y.H.; Zhang, X.W.; et al. A de Novo ANK1 Mutation Associated to Hereditary Spherocytosis: A Case Report. BMC Pediatr 2019, 19. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Xie, Y.; Wu, P.; Li, S.; Hua, Y.; Lu, X.; Zhao, W. Targeted Next-Generation Sequencing Identified a Novel ANK1 Mutation Associated with Hereditary Spherocytosis in a Chinese Family. Hematology (United Kingdom) 2019, 24, 583–587. [Google Scholar] [CrossRef]
- Xie, L.; Xing, Z.; Li, C.; Liu, S. xi; Wen, F. qiu Identification of a De Novoc.1000delA ANK1 Mutation Associated to Hereditary Spherocytosis in a Neonate with Coombs-Negative Hemolytic Jaundice-Case Reports and Review of the Literature. BMC Med Genomics 2021, 14. [Google Scholar] [CrossRef]
- Iolascon, a; Miraglia del Giudice, E. ; Perrotta, S.; Alloisio, N.; Morlé, L.; Delaunay, J. Hereditary Spherocytosis: From Clinical to Molecular Defects. Haematologica 1998, 83, 240–257. [Google Scholar]
- Gallagher, P.G.; Romana, M.; Tse, W.T.; Lux, S.E.; Forget, B.G. The Human Ankyrin-1 Gene Is Selectively Transcribed in Erythroid Cell Lines despite the Presence of a Housekeeping-like Promoter. Blood 2000, 96, 1136–1143. [Google Scholar] [CrossRef] [PubMed]
- Sabatino, D.E.; Wong, C.; Cline, A.P.; Pyle, L.; Garrett, L.J.; Gallagher, P.G.; Bodine, D.M. A Minimal Ankyrin Promoter Linked to a Human γ-Globin Gene Demonstrates Erythroid Specific Copy Number Dependent Expression with Minimal Position or Enhancer Dependence in Transgenic Mice. Journal of Biological Chemistry 2000, 275, 28549–28554. [Google Scholar] [CrossRef] [PubMed]
- Bennett, V.; Stenbuck, P.J. The Membrane Attachment Protein for Spectrin Is Associated with Band 3 in Human Erythrocyte Membranes. Nature 1979, 280. [Google Scholar] [CrossRef]
- Sharma, N.; Bham, K.; Senapati, S. Human Ankyrins and Their Contribution to Disease Biology: An Update. J Biosci 2020, 45, 146. [Google Scholar] [CrossRef]
- Stevens, S.R.; Rasband, M.N. Pleiotropic Ankyrins: Scaffolds for Ion Channels and Transporters. Channels 2022, 16, 216–229. [Google Scholar] [CrossRef] [PubMed]
- Hanspal, M.; Yoon, S.; Yu, H.; Hanspal, J.; Lambert, S.; Palek, J.; Prchal, J. Molecular Basis of Spectrin and Ankyrin Deficiencies in Severe Hereditary Spherocytosis: Evidence Implicating a Primary Defect of Ankyrin. Blood 1991, 77, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Russo, R.; Andolfo, I.; Manna, F.; Gambale, A.; Marra, R.; Rosato, B.E.; Caforio, P.; Pinto, V.; Pignataro, P.; Radhakrishnan, K.; et al. Multi-Gene Panel Testing Improves Diagnosis and Management of Patients with Hereditary Anemias. Am J Hematol 2018, 93, 672–682. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Kim, H.; Ahn, W.K.; Lee, S.T.; Han, J.W.; Choi, J.R.; Lyu, C.J.; Hahn, S.; Shin, S. Diagnostic Yield of Targeted Next-Generation Sequencing for Pediatric Hereditary Hemolytic Anemia. BMC Med Genomics 2023, 16. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Park, J.; Kim, M. Diagnostic Approaches for Inherited Hemolytic Anemia in the Genetic Era. Blood Res 2017, 52, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Eber, S.W.; Gonzalez, J.M.; Lux, M.L.; Scarpa, A.L.; Tse, W.T.; Dornwell, M.; Herbers, J.; Kugler, W.; Ozcan, R.; Pekrun, A.; et al. Ankyrin–1 Mutations Are a Major Cause of Dominant and Recessive Hereditary Spherocytosis. Nat Genet 1996, 13, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Bogusławska, D.M.; Skulski, M.; Machnicka, B.; Potoczek, S.; Kraszewski, S.; Kuliczkowski, K.; Sikorski, A.F. Identification of a Novel Mutation of β-Spectrin in Hereditary Spherocytosis Using Whole Exome Sequencing. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Bogusławska, D.M.; Heger, E.; Machnicka, B.; Skulski, M.; Kuliczkowski, K.; Sikorski, A.F. A New Frameshift Mutation of the β-Spectrin Gene Associated with Hereditary Spherocytosis. Ann Hematol 2017, 96, 163–165. [Google Scholar] [CrossRef] [PubMed]
- Boguslawska, D.M.; Heger, E.; Listowski, M.; Wasiński, D.; Kuliczkowski, K.; Machnicka, B.; Sikorski, A.F. A Novel L1340P Mutation in the ANK1 Gene Is Associated with Hereditary Spherocytosis? Br J Haematol 2014, 167, 269–271. [Google Scholar] [CrossRef]
- Bogusławska, D.M.; Skulski, M.; Bartoszewski, R.; Machnicka, B.; Heger, E.; Kuliczkowski, K.; Sikorski, A.F. A Rare Mutation (p.F149del) of the NT5C3A Gene Is Associated with Pyrimidine 5′-Nucleotidase Deficiency. Cell Mol Biol Lett 2022, 27, 104. [Google Scholar] [CrossRef] [PubMed]
- Bogusławska, D.M.; Kraszewski, S.; Skulski, M.; Potoczek, S.; Kuliczkowski, K.; Sikorski, A.F. Novel Variant of the SLC4A1 Gene Associated with Hereditary Spherocytosis. Biomedicines 2023, 11. [Google Scholar] [CrossRef] [PubMed]
- Machnicka, B.; Czogalla, A.; Bogusławska, D.M.; Stasiak, P.; Sikorski, A.F. Spherocytosis-Related L1340P Mutation in Ankyrin Affects Its Interactions with Spectrin. Life 2023, 13. [Google Scholar] [CrossRef] [PubMed]
- Meienberg, J.; Bruggmann, R.; Oexle, K. ; Matyas, · Gabor Clinical Sequencing: Is WGS the Better WES? Hum Genet 2016, 135, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Edelman, E.J.; Maksimova, Y.; Duru, F.; Altay, C.; Gallagher, P.G. A Complex Splicing Defect Associated with Homozygous Ankyrin-Deficient Hereditary Spherocytosis. Blood 2007, 109, 5491–5493. [Google Scholar] [CrossRef]
- Delaunay, J. The Molecular Basis of Hereditary Red Cell Membrane Disorders. Blood Rev 2007, 21, 1–20. [Google Scholar] [CrossRef]
- Miraglia Del Giudice, E.; Francese, M.; Polito, R.; Nobili, B.; Iolascon, A.; Perrotta, S. Apparently Normal Ankyrin Content in Unsplenectomized Hereditary Spherocytosis Patients with the Inactivation of One Ankyrin (ANK1) Allele. Haematologica 1997, 82. [Google Scholar]
- Bogusławska, D.M.; Heger, E.; Machnicka, B.; Skulski, M.; Kuliczkowski, K.; Sikorski, A.F. A New Frameshift Mutation of the β-Spectrin Gene Associated with Hereditary Spherocytosis. Ann Hematol 2017, 96, 163–165. [Google Scholar] [CrossRef] [PubMed]
- Bassères, D.S.; Tavares, A.C.; Costa, F.F.; Saad, S.T.O. SS-Spectrin São PauloII, a Novel Frameshift Mutation of the ß-Spectrin Gene Associated with Hereditary Spherocytosis and Instability of the Mutant MRNA. Brazilian Journal of Medical and Biological Research 2002, 35, 921–925. [Google Scholar] [CrossRef] [PubMed]
- Miraglia Del Giudice, E.; Lombardi, C.; Francesie, M.; Nobili, B.; Conte, M.L.; Amendola, G.; Cutillo, S.; Iolascon, A.; Perrotta, S. Frequent de Novo Monoallelic Expression of β-Spectrin Gene (SPTB) in Children with Hereditary Spherocytosis and Isolated Spectrin Deficiency. Br J Haematol 1998, 101. [Google Scholar] [CrossRef] [PubMed]
- Jarolim, P.; Rubin, H.L.; Brabec, V.; Palek, J. Comparison of the Ankyrin (AC)n Microsatellites in Genomic DNA and MRNA Reveals Absence of One Ankyrin MRNA Allele in 20% of Patients with Hereditary Spherocytosis. Blood 1995, 85, 3278–3282. [Google Scholar] [CrossRef] [PubMed]
- Jarolim, P.; Murray, J.L.; Rubin, H.L.; Taylor, W.M.; Prchal, J.T.; Ballas, S.K.; Snyder, L.M.; Chrobak, L.; Melrose, W.D.; Brabec, V.; et al. Characterization of 13 Novel Band 3 Gene Defects in Hereditary Spherocytosis with Band 3 Deficiency. Blood 1996, 88. [Google Scholar] [CrossRef]
- Dhermy, D.; Bournier, O.; Bourgeois, M.; Grandchamp, B. The Red Blood Cell Band 3 Variant (Band 3(Bicetrel):R490C) Associated with Dominant Hereditary Spherocytosis Causes Defective Membrane Targeting of the Molecule and a Dominant Negative Effect. Mol Membr Biol 1999, 16. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The novel frameshift mutation of the ANK1 gene is related to hereditary spherocytosis. Sequence fragments of the ANK1 gene in affected patients (AM174 and AM175) as well as one asymptomatic family member (AM173). Genomic DNA sequence analysis revealed a single nucleotide deletion only in HS patients (NM_000037.4:c.4959del causing the frameshift mutation NP_000028.3: p.V1626fs*64 resulting in a premature stop codon).
Figure 1.
The novel frameshift mutation of the ANK1 gene is related to hereditary spherocytosis. Sequence fragments of the ANK1 gene in affected patients (AM174 and AM175) as well as one asymptomatic family member (AM173). Genomic DNA sequence analysis revealed a single nucleotide deletion only in HS patients (NM_000037.4:c.4959del causing the frameshift mutation NP_000028.3: p.V1626fs*64 resulting in a premature stop codon).
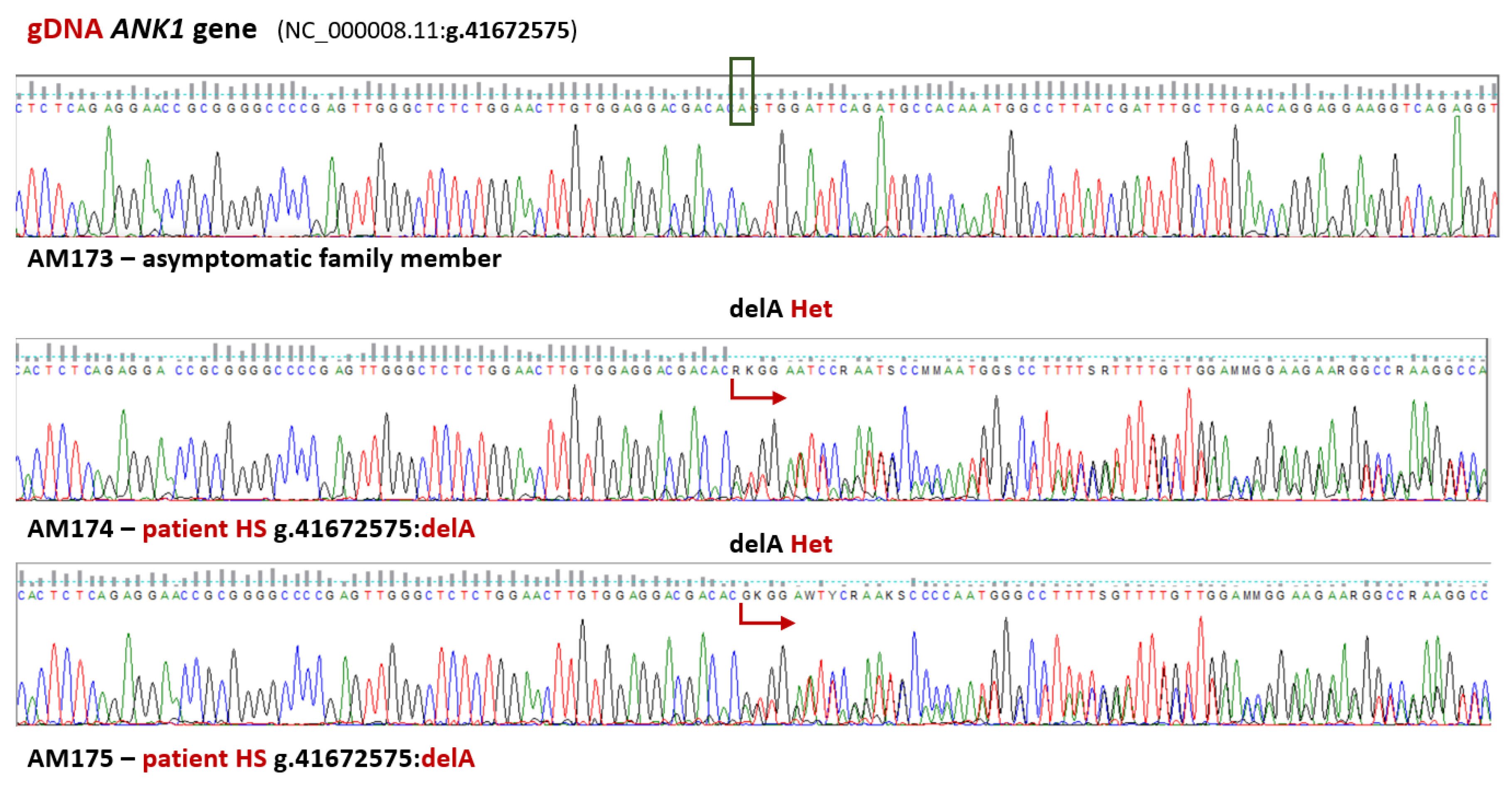
Figure 2.
Mutated transcripts are absent in patients’ transcriptomes. Fragment of sequencing traces detected in AM family members showing loss of the mutant allele (NM_000037.4:c.4959del) in the cDNA compared to the genomic DNA.
Figure 2.
Mutated transcripts are absent in patients’ transcriptomes. Fragment of sequencing traces detected in AM family members showing loss of the mutant allele (NM_000037.4:c.4959del) in the cDNA compared to the genomic DNA.
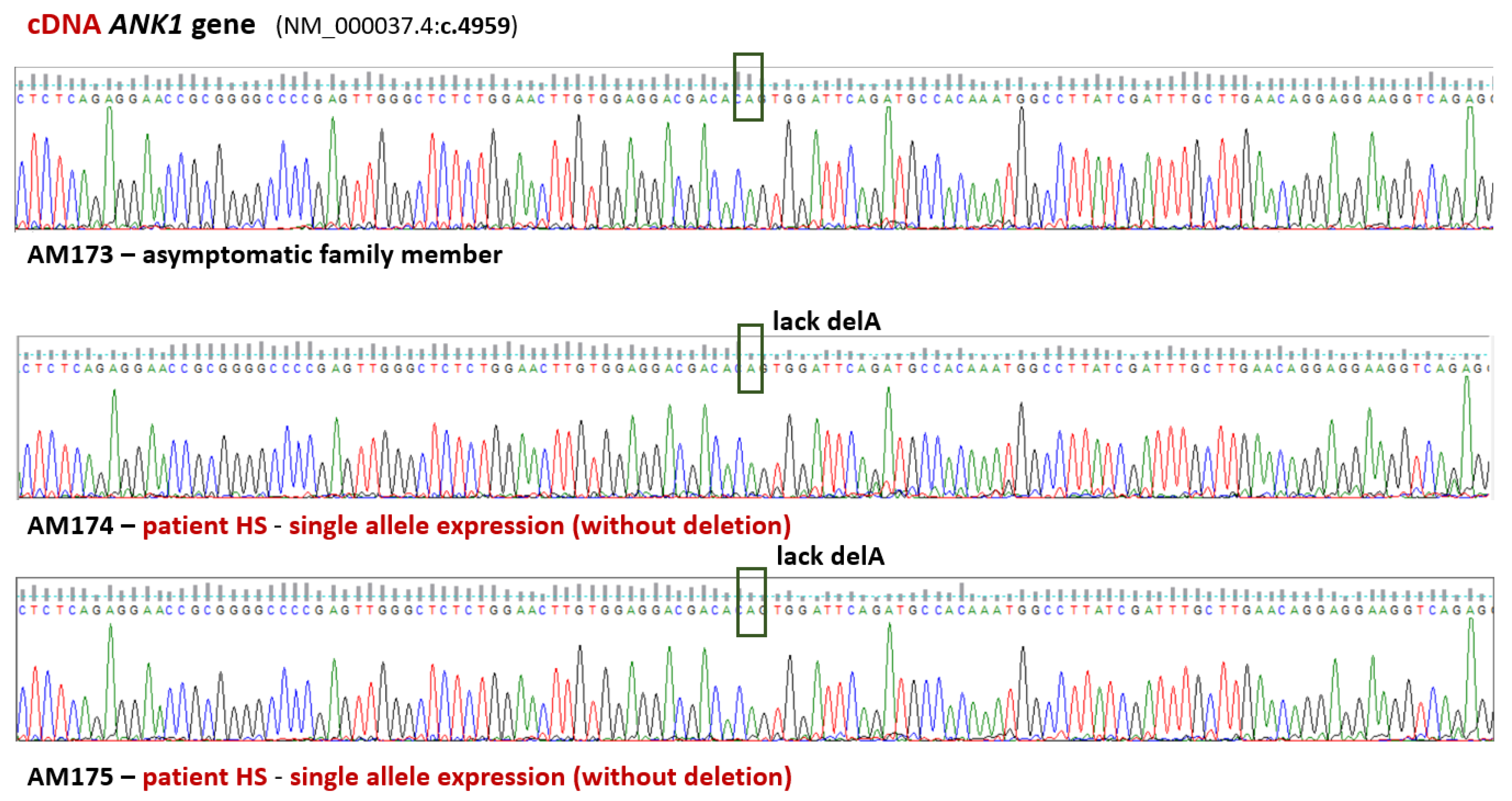
Figure 3.
Two variants detected in the ANK1 gene in the AM family appear to be crucial for the probands. A. The known missense mutation (NP_000028.3:p.V463I) was found in the sequence encoding the 89kDa domain, in the region of the ankyrin repeats. The new variant (p.V1626fs*64) is in the erythrocyte ankyrin regulatory domain sequence. B. A new single nucleotide deletion (NM_000037.4:c.4959del), resulting in a frameshift and a premature stop codon, 64 codons downstream of this change (NP_000028.3:p.V1626fs*64).
Figure 3.
Two variants detected in the ANK1 gene in the AM family appear to be crucial for the probands. A. The known missense mutation (NP_000028.3:p.V463I) was found in the sequence encoding the 89kDa domain, in the region of the ankyrin repeats. The new variant (p.V1626fs*64) is in the erythrocyte ankyrin regulatory domain sequence. B. A new single nucleotide deletion (NM_000037.4:c.4959del), resulting in a frameshift and a premature stop codon, 64 codons downstream of this change (NP_000028.3:p.V1626fs*64).
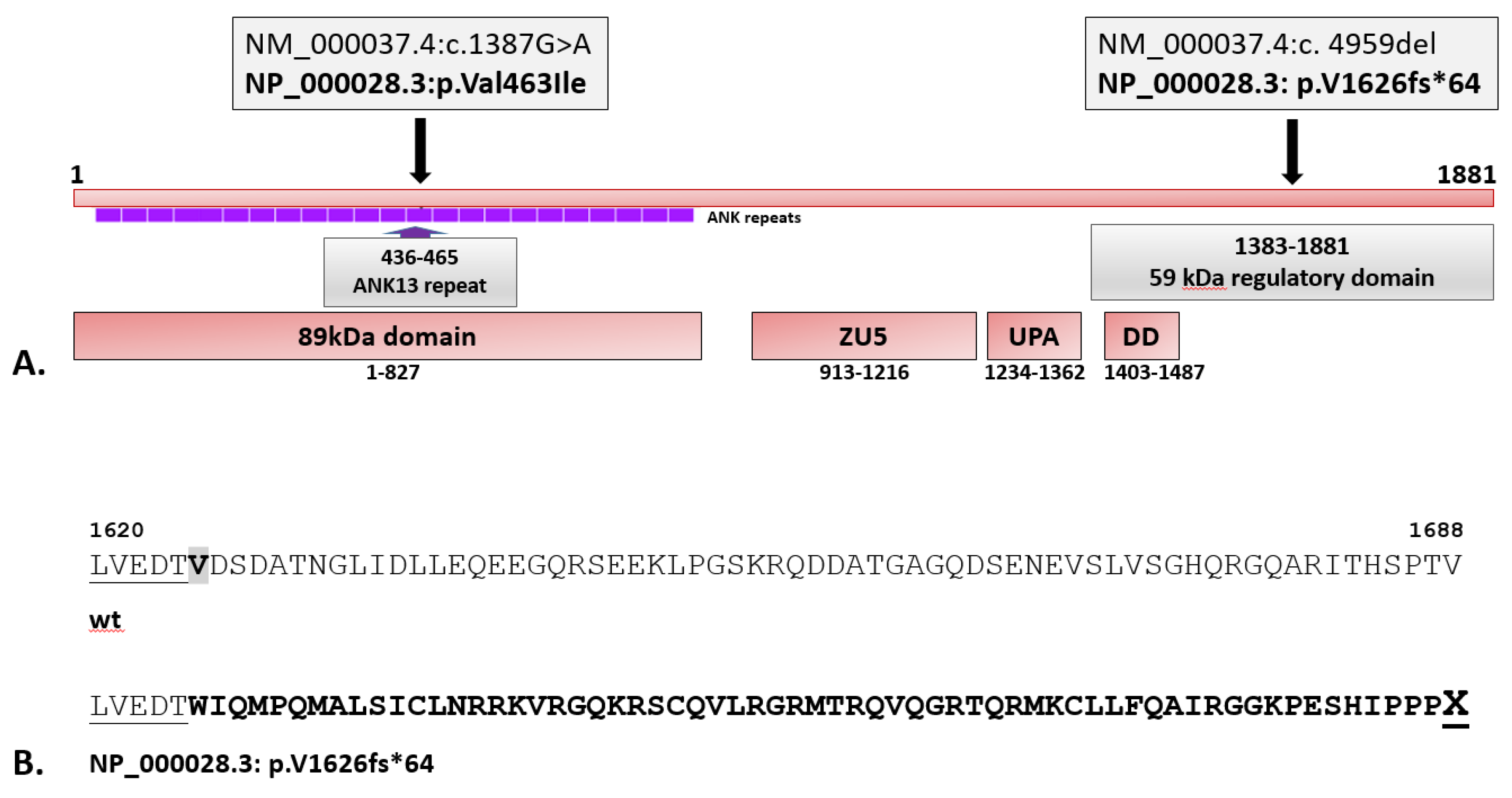

Table 1.
Hematological parameters of probands from the AM family: white blood cells (WBCs); red blood cells (RBC); hemoglobin (Hb); hematocrit (HCT); platelets (PLT); mean corpuscular volume (MCV); mean corpuscular hemoglobin (MCH); mean corpuscular hemoglobin concentration (MCHC).
Table 1.
Hematological parameters of probands from the AM family: white blood cells (WBCs); red blood cells (RBC); hemoglobin (Hb); hematocrit (HCT); platelets (PLT); mean corpuscular volume (MCV); mean corpuscular hemoglobin (MCH); mean corpuscular hemoglobin concentration (MCHC).
| Laboratory tests | Units | HS patients |
Reference range (female) |
Reference range (male) |
|
| AM174 (female) | AM175 (male) | ||||
| WBC | (G/L) | 11.37±3.02 | 11.80±2.05 | 4-10 | 4-10 |
| RBC | (T/L) | 3.58±0.35 | 3.58±0.35 | 4.0-5.0 | 4.5-5.9 |
| Hb | (mmol/L) | 7.21±0.50 | 7.65±0.52 | 7.54-9.93 | 8.69 -11.17 |
| HCT | (L/L) | 0.32±0.02 | 0.34±0.02 | 0.37-0.47 | 0.37-0.53 |
| PLT | (G/L) | 321±20 | 222±19 | 140-440 | 140-440 |
| MCV | (fL) | 88±2 | 95±3 | 81-98 | 81-98 |
| MCH | (fmol) | 2.02±0.06 | 2.17±0.13 | 1.61-2.11 | 1.61-2.11 |
| MCHC | (mmol/L) | 22.88±0.11 | 21.16±2.14 | 19.24-22.96 | 19.24-22.96 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



