Submitted:
01 January 2025
Posted:
02 January 2025
You are already at the latest version
Abstract
Metabolic stress on the heart can cause dilation of coronary arterioles for blood flow recruitment. Although potassium ions (K+) released from the myocardium are a major mediator for this response, the underlying signaling pathways for vasodilation are incompletely understood. Herein, roles of smooth muscle inward-rectifier K+ channel subtype 2.1 (Kir2.1) and Na+/K+ ATPase were examined. Porcine coronary arterioles were isolated, cannulated, and pressurized for vasomotor study. Vessels developed basal tone and dilated concentration-dependently to extraluminal K+ from 7 to 20 mM. Higher K+ concentrations (25-40 mM) caused graded vasoconstriction. Vasodilation to K+ (10 mM) was not altered by endothelial removal, and blockade of ATP-sensitive K+ channels, voltage-sensitive K+ channels, or calcium-activated K+ channels did not affect K+-induced vasodilation. However, sustained but not abrupt transient vasodilation to K+ was reduced by nonspecific Kir channel inhibitor Ba2+ or specific Kir2.1 channel blocker chloroethylclonidine. The Na+/K+ ATPase inhibitor ouabain attenuated K+-elicited vasodilation, and ouabain with Ba2+ abolished the response. Transfection of arterioles with Kir2.1 antisense oligonucleotides abolished sustained but not transient dilation. It is concluded that extraluminal K+ elevation within the physiological range induces initial transient dilation of porcine coronary arterioles by activating Na+/K+ ATPase and sustained dilation via smooth muscle Kir2.1 channels.
Keywords:
Na+/K+ ATPase
; inward-rectifier potassium channels
; resistance arterioles
; potassium
; vasodilation
1. Introduction
Coronary blood flow is closely regulated to meet the metabolic demands of the heart by changes in the diameter of coronary arterioles. The coronary arteriolar diameter is influenced by myriad factors including the locally released metabolites from active cardiomyocytes. Potassium ions (K+) are one of the major metabolites released into the interstitial fluid and have been implicated to participate in the regulation of vascular resistance in the coronary circulation [1,2,3]. During periods of acute myocardial ischemia [4,5,6,7,8] or increase in myocardial activity [2] interstitial K+ can increase from basal levels of 3-5 mM up to about 10-15 mM. The direct impact of K+ on vascular tone was supported by previous studies in isolated vessel preparations demonstrating that small modest increases in extraluminal K+ from 3 to 7 mM [9] or 14 mM [10] elicit dilation of pharmacologically preconstricted coronary arteries (260-700 µm). Although these in vitro studies demonstrated the capability of coronary conduit vessels in response to elevated K+, the sensitivity and reactivity of coronary resistance arterioles <100 µm in resting diameter, the major site for coronary flow regulation, to small elevations of extracellular K+ within the physiological range (6 to 15 mM) has not been systematically investigated.
The underlying mechanisms implicated for the K+-induced vasodilation include activation of the Na+/K+ ATPase pump or the inward-rectifier K+ (Kir) channels. Early in-situ studies [11] and in isolated perfused heart [12] preparations indicate that intra-arterial infusion of isotonic KCl (at levels estimated to raise interstitial K+ concentration about 1-2 mM) causes coronary vasodilation, which is partially reduced by pharmacological blockade of Na+/K+ ATPase. By contrast, the activation of barium (Ba2+)-sensitive Kir channels appears to be the predominant mechanism contributing to K+-induced vasodilation of isolated rat small coronary arteries (about 100-150 µm in resting diameter) [13]. However, it remains unclear whether activation of Na+/K+ ATPase and/or Kir channels is attributable to the dilation of small coronary resistance arterioles to elevated K+. In addition, the relative cellular roles, i.e., the endothelium versus smooth muscle, of Na+/K+ ATPase and specific Kir channel subtypes in coronary arterioles remain unknown. Since vascular dilation in response to K+ was absent in mice lacking the gene encoding for Kir2.1 channels [14] and electrogenic Na+/K+ pump activity can alter cell membrane potential and influence vascular activity [15], herein we test the hypothesis that elevation of K+ within the physiological range triggers the smooth muscle Kir2.1 channel subtype, in addition to the activation of Na+/K+ ATPase, to elicit coronary arteriolar dilation. An isolated and pressurized microvessel preparation [16], coupled with videomicroscopic techniques [17,18], was utilized to eliminate the confounding influences from changes in hemodynamics and neurohumoral factors. The sensitivity and reactivity of porcine coronary arterioles to small increases in extracellular K+ and the cellular and molecular mechanisms underlying the vasodilation to K+ were characterized.
2. Results
2.1. Vasomotor Responses to Elevated Extraluminal K+
Within 60 min of equilibration, all isolated porcine coronary arterioles (n = 69) developed a similar level of basal tone (constricted to 60 ± 1% of maximum passive diameter 103 ± 4 μm; range 42-192 μm). Figure 1A shows the representative tracing of the arteriolar response to increases in the extraluminal concentrations of K+ from the baseline level of 5 mM up to 40 mM.
Administration of KCl at various increments to the vessel bath (from 5 to 6, 7, 10, and 15 mM) caused a rapid cumulative increase in the vessel diameter (Figure 1A). No further increase in diameter was observed at 20 mM K+. Elevation of K+ to 25, 30, and 35 mM caused constriction of coronary arterioles back towards their resting diameter. At 40 mM K+, coronary arterioles constricted significantly below their resting diameter (Figure 1A). The changes in diameters were sustained at each concentration of K+ for 2 to 5 min in the observation period. The vasomotor reaction of coronary arterioles to K+ (range from 5 to 40 mM) is summarized in Figure 1B. In another series of experiments, to examine whether the vasodilation to K+ is vessel-size dependent, the vessels were exposed to 10 mM KCl only. The dilation of small arterioles (resting diameter 17-43 µm) and intermediate arterioles (resting diameter 45-77 µm) to 10 mM KCl were comparable, but both responses were greater than those of large arterioles (resting diameter 81-110 µm) (Figure 1C). Further studies were conducted below with 10 mM KCl to characterize the mechanism underlying the vasodilation.
2.2. Role of Endothelium
To explore the possible role of the endothelium in mediating the vasodilator response to K+, the vasomotor activity was assessed before and after removal of the endothelium. The efficacy of endothelial disruption by 3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate (CHAPS) was verified by the absence of vasodilator response to 1 nM endothelium-dependent vasodilator bradykinin (Control: 59 ± 5% dilation vs. CHAPS: 5 ± 2% dilation, P < 0.05, n = 5). Endothelial removal did not affect resting vascular diameter and the vasodilations to elevated K+ (Table 1, Group 1).
2.3. Role of K+ Channels
The relative role of smooth muscle K+ channels in the coronary arteriolar dilation to K+ was assessed in the presence of their respective inhibitors. As shown in Figure 2A, administration of 10 mM K+ caused dilation of arterioles with the increase in diameter reaching a plateau within 3 min that was sustained for at least 2 min. The arteriole regained tone after washing with PSS. Following treatment with Ba2+, administration of 10 mM K+ evoked an abrupt vasodilation within 1 min with the diameter returning towards the resting diameter and maintaining a small steady-state dilation for at least 3 min. These dynamic changes in diameters in response to 10 mM K+ are summarized in Figure 2B. Ba2+ did not alter the initial phase of K+-induced dilation within 1 min but significantly reduced the steady-state dilation at 5 min (Figure 2). By contrast, blockade of ATP-sensitive K+ (KATP) channels with glibenclamide (Table 1, Group 2), large-conductance calcium-activated K+ (KCa) channels with iberiotoxin (Table 1, Group 3), or voltage-dependent K+ (KV) channels with 4-aminopyridine (4-AP) (Table 1, Group 4) did not influence the initial or steady-state components of the K+-induced response.
2.4. Role of Kir2.1 Channels
Similar to the influence of Ba2+, treatment with Kir2.1 channel inhibitor chloroethylclonidine (CEC) did not affect the extent of abrupt dilation at 1 min but inhibited the steady-state dilation at 5 min in response to 10 mM K+ (Figure 3A). The CEC blockade appeared to be specific because the inhibitor did not alter vasodilation to 1 µM pinacidil (Control: 53 ± 5% dilation vs. CEC: 58 ± 10% dilation; n = 3) or 1 µM sodium nitroprusside (Control: 52 ± 4% vs. CEC: 55 ± 4% dilation; n = 3). To support the functional studies above, the expression of Kir2.1 channels in porcine coronary arterioles was examined using reverse transcription-polymerase chain reaction (RT-PCR) and western blot analyses. As shown in Figure 3B, Kir 2.1 mRNA was detected in isolated coronary arterioles. Both Kir2.1 channel protein and cardiac alpha-tropomyosin were detected in cardiomyocytes. In contrast, Kir2.1 channel, but not cardiac alpha-tropomyosin, was detected in coronary arterioles (Figure 3C).
We determined the involvement of Kir2.1 channels in K+-induced vasodilation by corroborating the pharmacological studies with molecular knockdown of Kir2.1. Following a 24-hr treatment of coronary arterioles with Kir2.1 sense oligonucleotides, the abrupt (1 min) and steady-state (5 min) dilations to 10 mM K+ were comparable (Figure 4A). By contrast, only an abrupt dilation was observed in the vessels treated with Kir2.1 antisense oligonucleotides followed by sustained vasoconstriction (Figure 4A). The vasodilator response to 1 µM sodium nitroprusside (Sense: 54 ± 7% dilation, n = 3; Antisense: 51 ± 15% dilation, n = 3) remained intact in the antisense-treated vessels. We also confirmed the fidelity of this molecular approach by specifically measuring Kir2.1 mRNA. As shown in Figure 4B, Kir2.1 and Kir2.3 mRNAs were detected in a single isolated coronary arteriole. Moreover, pretreatment with Kir2.1 antisense (As), but not Kir2.1 sense (Se), oligonucleotides reduced Kir2.1 transcripts without altering the expression of Kir2.3 and GAPDH (Kir2.1/GAPDH: Sense, 0.39±0.03 vs. Antisense: 0.19±0.08).
2.5. Role of Na+/K+ ATPase
The effect of Na+/K+ ATPase inhibitor ouabain on K+-induced vasodilation was examined in a separate group of vessels. As shown in Figure 5A, pretreatment of the arterioles with ouabain significantly decreased the resting diameter (Control: 50 ± 2 µm vs. Ouabain: 42 ± 3 µm, P = 0.03, n = 6) and attenuated the abrupt dilation at 1 min and the sustained vasodilator response at 5 min in response to 10 mM KCl (Figure 5A). It does not appear that ouabain elicited a nonspecific effect attributed to an increase in resting tone, because incubation of arterioles with 4-AP also significantly decreased resting diameter but did not alter K+-induced vasodilation (Table 1, Group 4). Pretreatment with both ouabain and Ba2+ abolished the arteriolar dilation to 10 mM K+ (Figure 5B).
3. Discussion
In the present study, five salient findings were noted regarding the vasomotor influence of K+ on isolated porcine coronary arterioles: (1) physiological concentrations of extracellular K+ in the range of 7 to 15 mM elicited a sustained, endothelium-independent coronary arteriolar dilation, with small and intermediate arterioles dilating to a greater extent than large arterioles; (2) both Ba2+ and CEC treatments inhibit the sustained vasodilation to K+, indicating a partial role for Kir channels in the sustained vasodilator response to extraluminal K+; (3) molecular evidence of mRNA confirmed the presence of Kir2.1 in porcine coronary arterioles; (4) antisense knockdown of Kir2.1 channel mRNA abolishes the sustained vasodilator response to K+, thus corroborating the pharmacological data; and (5) ouabain treatment blocked both the abrupt onset and sustained vasodilation to K+, suggesting that activation of the Na+/K+ ATPase pump plays a significant role in initiating the robust vasodilator response to K+.
In 1938, Katz and Linder reported that intra-arterial administration of K+ into the coronary artery of dogs produces coronary vasodilation, suggesting that coronary blood flow and metabolic demand may be closely related via K+ signaling in the heart [1]. Subsequent studies have shown that the byproducts of tissue metabolism, including extracellular K+ ions, dilate blood vessels to match perfusion to metabolism in several tissues, including the heart [19,20]. Experimental studies in animals have demonstrated that coronary arterial occlusion triggers the dilation of small coronary arterioles, and this dilation causes blood flow recruitment immediately after the release of the occlusion. Thus, coronary blood flow is abruptly increased to compensate for the oxygen debt during occlusion. This phenomenon of reactive hyperemia is thought to be mediated via several metabolites. Interestingly, the extracellular concentration of K+ in the heart rises immediately after the onset of acute coronary artery occlusion and can reach 10-15 mM in the ischemic region within 6 to 15 min of myocardial ischemia [4,5,6,7,8]. The present results demonstrated that extracellular K+ concentration in the 10-15 mM range causes abrupt coronary arteriolar dilation and could possibly contribute to this local reactive hyperemic response. Likewise, increases in extracellular K+ may trigger functional hyperemia during increased heart rate. An earlier study has shown that myocardial pacing in the dog caused a nearly 1-2-mM increase in the coronary interstitial level of K+ that preceded the increase in coronary blood flow [2]. The authors suggested that this small elevation in the K+ concentration might initiate rather than maintain the functional hyperemic response. However, they were also uncertain whether a 1-2 mM increase in the local level of K+ is sufficient to evoke dilation of coronary arterioles. Our present findings support this concept and provide the first direct evidence that an increase in the extraluminal concentration of K+ from the normal level of 5 to 7 mM can elicit a nearly 20% increase in the resting diameter of porcine coronary arterioles. Because the flow is proportional to 4th power of vessel radius changes, a 20% increase in caliber can elicit a 2-fold increase in blood flow. Taken together with the previous in vivo studies, it appears that small elevations in the extracellular concentration of K+ in the physiological range of 7 to 15 mM can have a significant impact on local coronary vascular resistance, and thus blood flow in vivo.
The endothelium is well known to mediate and/or modulate smooth muscle activity, especially in the microvessels because the ratio of endothelial to smooth muscle cells is markedly increased in the microvascular network and the vascular cells are intimately exposed to the tissue metabolites. Endothelial denudation significantly reduced relaxation of isolated small mesenteric arteries (100-200 µm) in response to 7.8 to 13.8 mM K+ [21], suggesting a role for the endothelium in this K+-induced response. However, it is unclear whether these observations can be applied to the regulation of vasomotor activity by K+ in the coronary arterioles. In the isolated coronary arteriolar preparation, we found that the endothelium is unlikely to be the target of the K+ for vasodilation, because the K+-elicited response was not altered by endothelial removal, a result consistent with other studies in the small coronary and cerebral arteries [13,22]. Our results in coronary arterioles provide further evidence that the K+-induced dilation, at least in the coronary microcirculation, originates from the smooth muscle cell.
In the present study, the dilation of coronary arterioles to K+ was sensitive to a low concentration of nonspecific Kir channel inhibitor Ba2+ and the Kir2.1 channel-specific blocker CEC (Figs. 2 and 3). Indeed, several other studies in the cerebral [13,22], skeletal muscle [23], cremasteric [24], and small coronary [13] arteries have shown that Ba2+ inhibits the K+-induced vasodilator response. In the present study, as well as other previous studies, Ba2+-sensitive K+-induced dilation was resistant to inhibitors of KATP, KCa and KV channels, suggesting that K+ was acting through the Kir channel [13,25]. In support of an unequivocal role for the Kir channels in the regulation of coronary microvascular blood flow, the density of Kir current has been shown to be inversely related to blood vessel size [26,27,28]. Our findings show that small and intermediate arterioles about 30-60 µm in resting diameter exhibit greater dilation in response to 10 mM K+ than their upstream parent vessels (>100 µm in resting diameter) (Figure 1C), supporting the concept of the dominant role of small resistance arterioles in metabolic regulation of coronary blood flow [29]. Although the pharmacologic data pointed to the important role of Kir in mediating K+-induced coronary arteriolar dilation, the limitation of selectivity and specificity of pharmacologic blockers must be considered. Importantly, our functional data were also corroborated with molecular evidence at the level of mRNA expression, ascertaining that Kir2.1 channel subtypes are not only present in the coronary arterioles but also mediate the vasodilation to K+. This was specifically demonstrated by the knockdown of Kir2.1 channel mRNA using antisense oligonucleotides, which correspondingly matches the elimination of K+-induced vasodilation. Although it was not studied in the coronary microvasculature, the importance of Kir channels was shown with Kir2.1 gene disruption in mice, resulting in the loss of inwardly rectifying K+ currents and K+-induced vasorelaxation in murine cerebral arteries [14]. Without Kir2.1, the coronary arterioles constricted to a low concentration of KCl (10 mM) (Figure 4A). It is worth noting that Kir2.1 deficiency may contribute, in part, to the development of hypertension [30] and possibly the impairment of coronary [31] and cerebral [32] flow regulation under increased metabolic activity of the tissue. It appears that Kir2.1 channels contribute significantly to the vasomotor influence by K+ in the organs that predominantly rely on metabolic flow regulation.
In our study, inhibition of Kir2.1 revealed two components of vasodilation elicited by K+, i.e., an abrupt/transient phase of initial dilation (e.g., ~1 min after addition of KCl) and a sustained/prolonged phase of dilation (e.g., 2-5 min after addition of KCl). For example, antisense Kir2.1 channel knockdown, as well as pretreatment of the coronary arterioles with Ba2+ or CEC, allowed for an abrupt transient dilation but reduced the sustained component (Figs. 2, 3, and 4). Therefore, Kir channel blockade did not abolish the initial dilation response, pointing to another putative mechanism that contributed to the activation of Kir channels in K+-induced vasodilation. We found that ouabain alone partially inhibited the K+-induced vasodilation in both abrupt and sustained phases but abolished the vasodilator response when it was combined with Ba2+ (Figure 5). These data, along with our molecular findings, support the idea that a small elevation of K+ activates Na+/K+ ATPase for abrupt and transient vasodilation and then triggers the opening of Kir2.1 channels for the sustained dilation. It should be noted that ouabain slightly but significantly caused constriction of coronary arterioles under resting conditions, indicating a tonic inhibition of Na+/K+ ATPase in the development of basal vascular tone in these vessels. However, the increased vascular tone did not appear to affect vasomotor activity, because administrating Kv channel inhibitor 4-AP significantly increased vascular tone to a level similar to that of ouabain, but the K+-induced vasodilation (10 mM) was no different than untreated controls. In small cerebral arteries, K+ concentrations from 0 to 5 mM caused activation of Na+/K+ ATPase pump, hyperpolarization, and dilation [22]. The transient nature of the response was attributed to K+ saturation of the Na+/K+ ATPase pump, Na+ extrusion and hyperpolarization, followed by reaching a new steady state [13,22,33,34]. Since there are at least four isoforms of Na+/K+ ATPase with varying sensitivity to external K+ [35], it remains unclear about the specific molecular role of each Na+/K+ ATPase isoform in mediating coronary arteriolar dilation to K+. For instance, a previous study has shown expression of Na+/K+ ATPase α1, α2, and α3 isoforms in rat mesenteric artery myocytes, where only α2 and α3 subtypes are responsible for hyperpolarization due to increments in external K+ concentrations [35]. In our study, the identity of Na+/K+ ATPase isoform(s) that may account for the initiation of vasodilation to K+ remains to be probed.
In summary, the present study shows that elevated extraluminal K+ concentrations, within the physiologic levels, are associated with marked dilation of coronary arterioles inversely related to vessel size. The vasodilator response to K+ is independent of other K+ channels and the endothelium. The induction of initial transient dilation of porcine coronary arterioles by activating Na+/K+ ATPase appears to trigger sustained vasodilation via smooth muscle Kir2.1 channels, although the precise molecular signaling between Na+/K+ ATPase activity and activation of Kir remains unclear. These vasodilator mechanisms are expected to play important roles in response to the elevated K+ during metabolic activation or stress, including ischemia and hypoxia.
4. Materials and Methods
4.1. Animals and Chemicals
All animal procedures adhered to the approved guidelines set by the Texas A&M University and Baylor Scott & White Health Institutional Animal Care and Use Committees. Pigs (8-12 wk old of either sex; 10-20 kg; n = 54) purchased from Barfield Farms (Rogers, TX) or Oak Hill Genetics (Ewing, IL) were sedated with Telazol (4.4 mg/kg) and Xylazine (2.2 mg/kg) and then anesthetized and anticoagulated with an intravenous administration of pentobarbital (30 mg/kg) and heparin (1000 U/kg), respectively. Following a thoracotomy, the heart was excised and immediately placed in cold (5°C) saline. Drugs were obtained from Sigma (St. Louis, MO), except as otherwise stated. Rauwolscine and CEC were dissolved in water. Glibenclamide was dissolved in dimethyl sulfoxide, whereas pinacidil, ouabain, and 4-AP were dissolved in absolute ethanol. Subsequent concentrations of these drugs and all other drugs were dissolved in PSS. The final concentrations of dimethyl sulfoxide and ethanol in the tissue bath were 0.03% and 0.1%, respectively. Vehicle control studies indicated that these final concentrations of solvent had no effect on arteriolar function.
4.2. Isolation and Cannulation of Coronary Microvessels
Because coronary arterioles are sensitive to changes in local hemodynamics [16,17,36] and neurohumoral factors [37], the individual arterioles were studied in vitro in the present study to eliminate these confounding influences that are inevitable in in-vivo preparations. Subepicardial arteriolar branches (about 1 mm in length; 30 to 100 µm in internal diameter in situ) were dissected from the surrounding cardiac tissue and were cannulated with glass micropipettes as previously described [29,38]. The vessels were pressurized to 60 cmH2O intraluminal pressure, consistent with the in vivo level of coronary arteriolar pressure [39,40] and bathed in physiological salt solution (PSS) (mM: NaCl 145.0, KCl 5.0, CaCl2 2.0, MgCl2 1.17, NaH2PO4 1.2, glucose 5.0, pyruvate 2.0, EDTA 0.02, and MOPS 3.0) containing 1% albumin (USB Corporation, Cleveland, OH) as described previously [36]. The inner diameter of coronary arterioles was measured using video microscopic techniques and recorded with a PowerLab data acquisition system (ADInstruments, Colorado Springs, CO) [16,36].
4.3. Pharmacological Assessment of K+-induced Dilation
Cannulated coronary arterioles were bathed in PSS-albumin at 36-37°C to allow development of basal tone. The vessels constricted to about 50-70% of maximum diameter within a 60-min equilibration period and maintained a stable resting diameter. After stable basal tone was developed, the concentration-dependent responses to incremental increases in the extraluminal K+ concentration (5 to 40 mM) were assessed. The extraluminal K+ concentration (5 mM in PSS) was increased by adding KCl to the vessel chamber with final concentrations of 6, 7, 10, 15, 20, 25, 30, 35, and 40 mM in the vessel bath. The osmolality of the solution was maintained constant by equimolar decreases in the NaCl concentration (5 mM K+: 300 ± 1.4 mOsmol vs. 20 mM K+: 300 ± 1.0 mOsmol) [41]. Further studies on the evaluation of signaling pathways were performed at 10 mM because this concentration of K+ is consistent with that found in the interstitium of the heart under metabolic stress [4,5,6]. In some vessels, the K+-induced dilation was examined after endothelial removal by perfusion of a nonionic detergent, CHAPS (0.4%), as previously described [38]. Only vessels that exhibited normal basal tone, did not dilate to the endothelium-dependent agonist bradykinin (1 nM), and showed unaltered vasodilation to endothelium-independent agonist sodium nitroprusside (1 nM to 100 µM) after endothelial removal were accepted for data analysis.
In other vessels, the contributions of K+ channels and Na+-K+ ATPase to the K+-induced dilation were evaluated in a series of experiments by administration of their cognate inhibitors to the extraluminal bath for at least 30 min. The roles of KV channels, KATP channels, and KCa channels were assessed in the presence of their respective inhibitors 4-AP (1 mM) [42,43], glibenclamide (5 µM) [18], and iberiotoxin (0.1 µM) [38]. The role of Kir channels in K+-induced dilation was assessed in the presence of 30 µM BaCl2 (a specific inhibitory concentration for Kir channels) [13,26,41]. Furthermore, the contribution of Kir2.1 channels was assessed in the presence of its inhibitor CEC (30 µM) [25,44]. Since CEC can also activate α2-adrenoceptors, which can cause dilation of coronary arterioles [45], vessels were incubated with both CEC and an α2-adrenoceptor antagonist rauwolscine (5 µM) [46]. Our preliminary studies showed that rauwolscine alone prevented vasodilation to CEC but did not alter vasodilation to 10 mM KCl (Control: 45 ± 3% change in resting diameter vs. Rauwolscine: 49 ± 10% change in resting diameter, n = 2). The role of the Na+/K+ ATPase in the vasodilator response to K+ was determined in the presence of its specific inhibitor ouabain (1.5 µM) [43]. To examine the specificity of pharmacological inhibitors in the K+-induced effect, vasodilations to sodium nitroprusside and KATP channel opener pinacidil in the presence of these inhibitors were studied in some vessels.
4.4. Antisense Knockdown of Kir2.1 Channels
To directly identify a functional role for Kir2.1 channels, the coronary arteriolar dilation to K+ was examined in the presence of antisense or sense oligonucleotides according to the Superfect Transfection Reagent protocol (Qiagen) [47,48]. Coronary arterioles (60-100 µm in diameter in situ) were carefully dissected and incubated in Dulbecco’s Modified Eagles Medium containing 10% fetal bovine serum, 1% antibiotic-antimycotic (Sigma), and Superfect Transfection Reagent (20 µl/ml) for 3 hr at 37°C in the absence and presence of antisense or sense oligonucleotides (2.5 µg/ml) for Kir2.1 channels (22 base pairs corresponding to bases 381-402 of gene accession no. AF153820; synthesized by Sigma-Genosys) [49]. After this incubation period with the transfection-oligonucleotide complexes, the media was removed, and the vessels were then incubated with media alone for 24 hr. The vessels were subsequently cannulated and pressurized for functional assessment in response to K+. RT-PCR analysis was performed as described below to verify the efficacy of Kir2.1 blockade by antisense.
4.5. RNA Isolation and RT-PCR
Total RNA was isolated from individual porcine coronary arterioles (60-100 μm in diameter in situ, 2-3 mm in length) as previously described [50,51]. Briefly, the vessel was homogenized in 1 mL Trizol Reagent (Life Technologies, Rockville, MO), and total RNA was isolated according to the manufacturer’s instructions with slight modification. The total RNA sample (400-500 µl) isolated from Trizol/chloroform extraction was then coprecipitated with 5 µl of glycogen (25 µg/ml; Invitrogen, #AM9510) to optimize total RNA recovery during the overnight precipitation step with isopropyl alcohol plus 50 µl sodium acetate (5 M) [52]. Sets of primers specific for Kir2.1 (gene accession no. AF153820, sense: 5'-TGA CAA CGC AGA CTT TGA AAT CGT-3', antisense: 5'-TCT GGA ACT CCA TTT TCA CTG TCG-3'), Kir2.3 (gene accession no. NM0044981 sense: 5'-ACG AGA ACG AGC TGG CCC TTA TGA-3', antisense: 5'-ACT CCC TGC GGT AGG AGA TGT TGT-3'), and GAPDH [50,51] were used. Specifically, 0.3 and 0.1 µg of total RNA for Kir and GAPDH samples, respectively, were annealed to the 3’- specific primers and the RT reaction was performed using Thermoscript reverse transcriptase (Life Technologies). Five microliters of RT cDNA were subsequently used to perform PCR for 30 (GAPDH) or 35 (Kir channels) cycles with Expand High Fidelity PCR enzyme (Roche, Indianapolis, IN). Electrophoresis of PCR amplified products on a 1.8% agarose gel followed by treatment with ethidium bromide allowed for band visualization. Images of stained products were acquired with the Gel Doc 2000 system (Bio-Rad Laboratories, Hercules, CA). The level of Kir channel transcripts was normalized to that of GAPDH transcripts.
4.6. Western Blot Analysis
Isolated coronary arterioles (5 vessels pooled, 40-100 μm in diameter in situ, 1-3 mm in length) and myocardial tissue were sonicated in lysis buffer containing 50 mM Tris-HCl, pH 7.5, 0.1 mM EDTA, 0.1 mM EGTA, 1 μg/mL leupeptin, 1 μg/mL aprotinin and 0.1 mM phenylmethylsulfonyl fluoride as previously described [50]. The protein content of each tissue lysate was determined using BCA protein assay kit (Pierce, Rockford, IL). Briefly, 4 μg of protein per lane were separated by 10% SDS-PAGE under reducing conditions. After electrophoresis, proteins were transferred onto a nitrocellulose membrane (Bio-Rad Laboratories), followed by blocking for overnight at 4°C with 5% dry milk in PBS. Immunodetection was then achieved by allowing the membranes to react at room temperature with primary antibodies against Kir2.1 (rabbit anti Kir2.1, 1:500, Alomone Labs, Jerusalem, Israel) for 3 hr or cardiac alpha-tropomyosin (mouse anti-cardiac alpha-tropomyosin, 1:10,000, Developmental Studies Hybridoma Bank at the University of Iowa) for 2 hr. Cardiac alpha-tropomyosin was used to test for contamination of microvessel samples with cardiomyocytes. After washing with TBS for 30 min, the secondary antibody labeled with horseradish peroxidase was administered for 1 hr at room temperature. The blots were revealed with horseradish peroxidase-labeled goat anti-rabbit (1:2000) or anti-mouse (1:25,000) IgG secondary antibodies (Jackson Laboratories) by an enhanced chemiluminescence assay (Amersham Pharmacia, Piscataway, NJ).
4.7. Data Analysis
At the end of each functional experiment, the vessel was dilated with sodium nitroprusside (100 µM) in an ethylenediaminetetraacetic acid (1 mM)-calcium-free PSS after the washout of pharmacological inhibitors to obtain its maximum diameter at 60 cmH2O intraluminal pressure [53]. For the analysis of vasomotor responses, internal diameter changes were normalized to the resting basal diameter and expressed as a percent change in resting diameter. Data are reported as mean ± SEM and n value represents the number of vessels studied (one vessel per pig for each treatment). Statistical comparisons of vasomotor responses and resting vascular tone before and after various treatments were performed using Student’s t-test or analysis of variance with the Bonferonni multiple-range test as deemed appropriate. A value of P < 0.05 was considered significant.
Author Contributions
Conceptualization, T.W.H and L.K.; Conducting Experiments, T.W.H., H.M.R., X.X., and S.S.; Methodology, T.W.H., L.K., and M.M.; Data Analysis, T.W.H. ,M.M. and L.K. Writing-Original Draft Preparation, H.M.R..; Writing – Review & Editing, T.W.H. and L.K.; Funding Acquisition, L.K. and T.W.H.; All authors read and approved the final version of the manuscript.
Funding
This work was supported by the American Heart Association (0235258N to T.W.H) and the National Institutes of Health (EY018420 to T.W.H. and HL71761 to L.K.).
Institutional Review Board Statement
The experimental procedures and protocols were carried out under the guidance of the Animal Care and Use Committee at the Texas A&M University Health Science Center and Baylor Scott & White Health.
Informed Consent Statement
Not applicable.
Data Availability Statement
The datasets generated and/or analyzed for the current study are available from the corresponding authors upon reasonable request.
Acknowledgments
Authors gratefully acknowledge Wenjuan Xu for her technical support and assistance in the collection of heart samples.
Conflicts of Interest
All authors declare that they have no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Katz, L.N.; Lindner, E. The action of excess Na, Ca and K on the coronary vessels. Am J Physiol 1938, 124, 155–160. [Google Scholar] [CrossRef]
- Murray, P.A.; Belloni, F.L.; Sparks, H.V. The role of potassium in the metabolic control of coronary vascular resistance of the dog. Circ Res 1979, 44, 767–780. [Google Scholar] [CrossRef] [PubMed]
- Haddy, F.J. Potassium and blood vessels. Life Sci 1975, 16, 1489–1497. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.L.; Gettes, L.S. Effect of acute coronary artery occlusion on local myocardial extracellular K+ activity in swine. Circulation 1980, 61, 768–778. [Google Scholar] [CrossRef]
- Hirche, H.; Franz, C.; Bos, L.; Bissig, R.; Lang, R.; Schramm, M. Myocardial extracellular K+ and H+ increase and noradrenaline release as possible cause of early arrhythmias following acute coronary artery occlusion in pigs. J Mol Cell Cardiol 1980, 12, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, V.; Guggi, M.; Meesmann, W.; Kessler, M.; Greitschus, F. Extracellular potassium activity changes in the canine myocardium after acute coronary occlusion and the influence of beta-blockade. Cardiovasc Res 1979, 13, 297–302. [Google Scholar] [CrossRef]
- Weiss, J.N.; Lamp, S.T.; Shine, K.I. Cellular K+ loss and anion efflux during myocardial ischemia and metabolic inhibition. Am J Physiol 1989, 256, H1165–H1175. [Google Scholar] [CrossRef]
- Kleber, A.G. Resting membrane potential, extracellular potassium activity, and intracellular sodium activity during acute global ischemia in isolated perfused guinea pig hearts. Circ Res 1983, 52, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Gellai, M.; Detar, R. Evidence in support of hypoxia but against high potassium and hyperosmolarity as possible mediators of sustained vasodilation in rabbit cardiac and skeletal muscle. Circ Res 1974, 35, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Konold, P.; Gebert, G.; Brecht, K. The mechanical response of isolated arteries to potassium. Experientia 1968, 24, 247–248. [Google Scholar] [CrossRef]
- Murray, P.A.; Sparks, H.V. The mechanism of K+-induced vasodilation of the coronary vascular bed of the dog. Circ Res 1978, 42, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Bunger, R.; Haddy, R.J.; Querengasser, A.; Gerlach, E. Studies on potassium induced coronary dilation in the isolated guinea pig heart. Pflugers Arch 1976, 363, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Knot, H.J.; Zimmermann, P.A.; Nelson, M.T. Extracellular K+-induced hyperpolarizations and dilatations of rat coronary and cerebral arteries involve inward rectifier K+ channels. J Physiol 1996, 492, 419–430. [Google Scholar] [CrossRef]
- Zaritsky, J.J.; Eckman, D.M.; Wellman, G.C.; Nelson, M.T.; Schwarz, T.L. Targeted disruption of Kir2. 1 and Kir2.2 genes reveals the essential role of the inwardly rectifying K+ current in K+-mediated vasodilation. Circ Res 2000, 87, 160–166. [Google Scholar]
- Haddy, F.J. Potassium effects on contraction in arterial smooth muscle mediated by Na+, K+-ATPase. Fed Proc 1983, 42, 239–245. [Google Scholar] [PubMed]
- Kuo, L.; Davis, M.J.; Chilian, W.M. Myogenic activity in isolated subepicardial and subendocardial coronary arterioles. Am J Physiol Heart Circ Physiol 1988, 255, H1558–H1562. [Google Scholar] [CrossRef]
- Kuo, L.; Davis, M.J.; Chilian, W.M. Endothelium-dependent, flow-induced dilation of isolated coronary arterioles. Am J Physiol 1990, 259, H1063–H1070. [Google Scholar] [CrossRef]
- Hein, T.W.; Kuo, L. cAMP-independent dilation of coronary arterioles to adenosine: role of nitric oxide, G proteins, and KATP channels. Circ Res 1999, 85, 634–642. [Google Scholar] [CrossRef]
- Haddy, F.J.; Scott, J.B. Metabolically linked vasoactive chemicals in local regulation of blood flow. Physiol Rev 1968, 48, 688–707. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.B.; Daugherty, R.M., Jr.; Overbeck, H.W.; Haddy, F.J. Vascular effects of ions. Fed Proc 1968, 27, 1403–1407. [Google Scholar]
- Dora, K.A.; Garland, C.J. Properties of smooth muscle hyperpolarization and relaxation to K+ in the rat isolated mesenteric artery. Am J Physiol Heart Circ Physiol 2001, 280, H2424–H2429. [Google Scholar] [CrossRef] [PubMed]
- McCarron, J.G.; Halpern, W. Potassium dilates rat cerebral arteries by two independent mechanisms. Am J Physiol 1990, 259, H902–H908. [Google Scholar] [CrossRef]
- De Clerck, I.; Boussery, K.; Pannier, J.L.; Van De Voorde, J. Potassium potently relaxes small rat skeletal muscle arteries. Med Sci Sports Exerc 2003, 35, 2005–2012. [Google Scholar] [CrossRef] [PubMed]
- Burns, W.R.; Cohen, K.D.; Jackson, W.F. K+-induced dilation of hamster cremasteric arterioles involves both the Na+/K+-ATPase and inward-rectifier K+ channels. Microcirculation 2004, 11, 279–293. [Google Scholar] [CrossRef]
- Chilton, L.; Loutzenhiser, R. Functional evidence for an inward rectifier potassium current in rat renal afferent arterioles. Circ Res 2001, 88, 152–158. [Google Scholar] [CrossRef]
- Quayle, J.M.; Dart, C.; Standen, N.B. The properties and distribution of inward rectifier potassium currents in pig coronary arterial smooth muscle. J Physiol 1996, 494, 715–726. [Google Scholar] [CrossRef]
- Quayle, J.M.; McCarron, J.G.; Brayden, J.E.; Nelson, M.T. Inward rectifier K+ currents in smooth muscle cells from rat resistance-sized cerebral arteries. Am J Physiol 1993, 265, C1363–C1370. [Google Scholar] [CrossRef]
- Hirst, G.D.; Edwards, F.R. Sympathetic neuroeffector transmission in arteries and arterioles. Physiol Rev 1989, 69, 546–604. [Google Scholar] [CrossRef]
- Kuo, L.; Davis, M.J.; Chilian, W.M. Longitudinal gradients for endothelium-dependent and -independent vascular responses in the coronary microcirculation. Circulation 1995, 92, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Gradel, A.K.J.; Salomonsson, M.; Sorensen, C.M.; Holstein-Rathlou, N.H.; Jensen, L.J. Long-term diet-induced hypertension in rats is associated with reduced expression and function of small artery SKCa, IKCa, and Kir2. 1 channels. Clin Sci (Lond) 2018, 132, 461–474. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.E.; Yin, M.Z.; Kim, H.J.; Vorn, R.; Yoo, H.Y.; Kim, S.J. Decreased inward rectifier and voltage-gated K+ currents of the right septal coronary artery smooth muscle cells in pulmonary arterial hypertensive rats. Korean J Physiol Pharmacol 2020, 24, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Sancho, M.; Gao, Y.; Hald, B.O.; Yin, H.; Boulton, M.; Steven, D.A.; MacDougall, K.W.; Parrent, A.G.; Pickering, J.G.; Welsh, D.G. An assessment of KIR channel function in human cerebral arteries. Am J Physiol Heart Circ Physiol 2019, 316, H794–H800. [Google Scholar] [CrossRef]
- Nakao, M.; Gadsby, D.C. [Na] and [K] dependence of the Na/K pump current-voltage relationship in guinea pig ventricular myocytes. J Gen Physiol 1989, 94, 539–565. [Google Scholar] [CrossRef]
- Hexum, T.D. Characterization of NaK-ATPase from vascular smooth muscle. Gen Pharmacol 1981, 12, 393–396. [Google Scholar] [CrossRef]
- Weston, A.H.; Richards, G.R.; Burnham, M.P.; Feletou, M.; Vanhoutte, P.M.; Edwards, G. K+-induced hyperpolarization in rat mesenteric artery: identification, localization and role of Na+/K+-ATPases. Br J Pharmacol 2002, 136, 918–926. [Google Scholar] [CrossRef]
- Kuo, L.; Chilian, W.M.; Davis, M.J. Interaction of pressure- and flow-induced responses in porcine coronary resistance vessels. Am J Physiol Heart Circ Physiol 1991, 261, H1706–H1715. [Google Scholar] [CrossRef]
- Davis, M.J.; Hill, M.; Kuo, L. Local Regulation of Blood Flow. In Handbook of Physiology, 2nd ed.; Tuma, R.F., Duran, W.N., Ley, K., Eds.; The American Physiological Society and Elsevier: Bethesda, MD, 2008; pp. 159–284. [Google Scholar]
- Hein, T.W.; Liao, J.C.; Kuo, L. oxLDL specifically impairs endothelium-dependent, NO-mediated dilation of coronary arterioles. Am J Physiol Heart Circ Physiol 2000, 278, H175–H183. [Google Scholar] [CrossRef]
- Chilian, W.M.; Eastham, C.L.; Marcus, M.L. Microvascular distribution of coronary vascular resistance in beating left ventricle. Am J Physiol 1986, 251, H779–H788. [Google Scholar] [CrossRef]
- Jones, C.J.H.; Kuo, L.; Davis, M.J.; Chilian, W.M. Distribution and control of coronary microvascular resistance. Adv Exp Med Biol 1993, 346, 181–188. [Google Scholar] [PubMed]
- Rivers, R.J.; Hein, T.W.; Zhang, C.; Kuo, L. Activation of barium-sensitive inward rectifier potassium channels mediates remote dilation of coronary arterioles. Circulation 2001, 104, 1749–1753. [Google Scholar] [CrossRef] [PubMed]
- Heaps, C.L.; Bowles, D.K. Gender-specific K+-channel contribution to adenosine-induced relaxation in coronary arterioles. J Appl Physiol 2002, 92, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Qamirani, E.; Razavi, H.M.; Wu, X.; Davis, M.J.; Kuo, L.; Hein, T.W. Sodium azide dilates coronary arterioles via activation of inward rectifier K+ channels and Na+-K+-ATPase. Am J Physiol Heart Circ Physiol 2006, 290, H1617–H1623. [Google Scholar] [CrossRef]
- Barrett-Jolley, R.; Dart, C.; Standen, N.B. Direct block of native and cloned (Kir2. 1) inward rectifier K+ channels by chloroethylclonidine. Br J Pharmacol 1999, 128, 760–766. [Google Scholar]
- Wang, S.Y.; Friedman, M.; Johnson, R.G.; Weintraub, R.M.; Sellke, F.W. Adrenergic regulation of coronary microcirculation after extracorporeal circulation and crystalloid cardioplegia. Am J Physiol Heart Circ Physiol 1994, 267, H2462–H2470. [Google Scholar] [CrossRef] [PubMed]
- Rosa Jr, R.H.; Hein, T.W.; Yuan, Z.; Xu, W.; Pechal, M.I.; Geraets, R.L.; Newman, J.M.; Kuo, L. Brimonidine evokes heterogeneous vasomotor response of retinal arterioles: diminished nitric oxide-mediated vasodilation when size goes small. Am J Physiol Heart Circ Physiol 2006, H231–H238. [Google Scholar] [CrossRef] [PubMed]
- Bolz, S.S.; Fisslthaler, B.; Pieperhoff, S.; De Wit, C.; Fleming, I.; Busse, R.; Pohl, U. Antisense oligonucleotides against cytochrome P450 2C8 attenuate EDHF-mediated Ca2+ changes and dilation in isolated resistance arteries. FASEB J 2000, 14, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Fisslthaler, B.; Popp, R.; Kiss, L.; Potente, M.; Harder, D.R.; Fleming, I.; Busse, R. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature 1999, 401, 493–497. [Google Scholar] [CrossRef]
- Nakamura, T.Y.; Artman, M.; Rudy, B.; Coetzee, W.A. Inhibition of rat ventricular IK1 with antisense oligonucleotides targeted to Kir2.1. mRNA. Am J Physiol 1998, 274, H892–H900. [Google Scholar] [PubMed]
- Hein, T.W.; Wang, W.; Zoghi, B.; Muthuchamy, M.; Kuo, L. Functional and molecular characterization of receptor subtypes mediating coronary microvascular dilation to adenosine. J Mol Cell Cardiol 2001, 33, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Hein, T.W.; Wang, W.; Chang, C.I.; Kuo, L. Constitutive expression of arginase in microvascular endothelial cells counteracts nitric oxide-mediated vasodilatory function. FASEB J 2001, 15, 1264–1266. [Google Scholar] [CrossRef]
- Monteiro, M.B.; Santos-Bezerra, D.P.; Thieme, K.; Passarelli, M.; Machado, U.F.; Lin, C.J.; Correa-Giannella, M.L. Optimization of total RNA isolation from human urinary sediment. Clin Chim Acta 2016, 462, 158–161. [Google Scholar] [CrossRef]
- Hein, T.W.; Kuo, L. LDLs impair vasomotor function of the coronary microcirculation: role of superoxide anions. Circ Res 1998, 83, 404–414. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Vasomotor response of isolated coronary arterioles to elevations in extraluminal KCl. (A) Representative tracing shows concentration-dependent response of an arteriole exposed to various concentrations of KCl in the extraluminal bath from 5 to 40 mM. (B) Summary data show that extraluminal K+ concentrations of 6 (n = 13), 7 (n = 13), 10 (n = 19), 15 (n = 17), 20 (n = 15), 25 (n = 9), and 30 (n = 8) mM caused an increase in vessel resting diameter (vasodilation), whereas 35 mM K+ (n = 8) did not significantly alter resting diameter. Vasoconstriction occurred at 40 mM K+ (n = 9). *P < 0.05 versus the resting values. (C) Summary data of percent change in resting diameter of small (34 ± 5 µm resting diameter, n = 9), intermediate (57 ± 2 resting diameter, n = 24), and large (98 ± 4 µm resting diameter, n = 8) coronary arterioles to 10 mM KCl. *P < 0.05 versus the Small and Intermediate Arterioles; n, number of vessels.
Figure 1.
Vasomotor response of isolated coronary arterioles to elevations in extraluminal KCl. (A) Representative tracing shows concentration-dependent response of an arteriole exposed to various concentrations of KCl in the extraluminal bath from 5 to 40 mM. (B) Summary data show that extraluminal K+ concentrations of 6 (n = 13), 7 (n = 13), 10 (n = 19), 15 (n = 17), 20 (n = 15), 25 (n = 9), and 30 (n = 8) mM caused an increase in vessel resting diameter (vasodilation), whereas 35 mM K+ (n = 8) did not significantly alter resting diameter. Vasoconstriction occurred at 40 mM K+ (n = 9). *P < 0.05 versus the resting values. (C) Summary data of percent change in resting diameter of small (34 ± 5 µm resting diameter, n = 9), intermediate (57 ± 2 resting diameter, n = 24), and large (98 ± 4 µm resting diameter, n = 8) coronary arterioles to 10 mM KCl. *P < 0.05 versus the Small and Intermediate Arterioles; n, number of vessels.
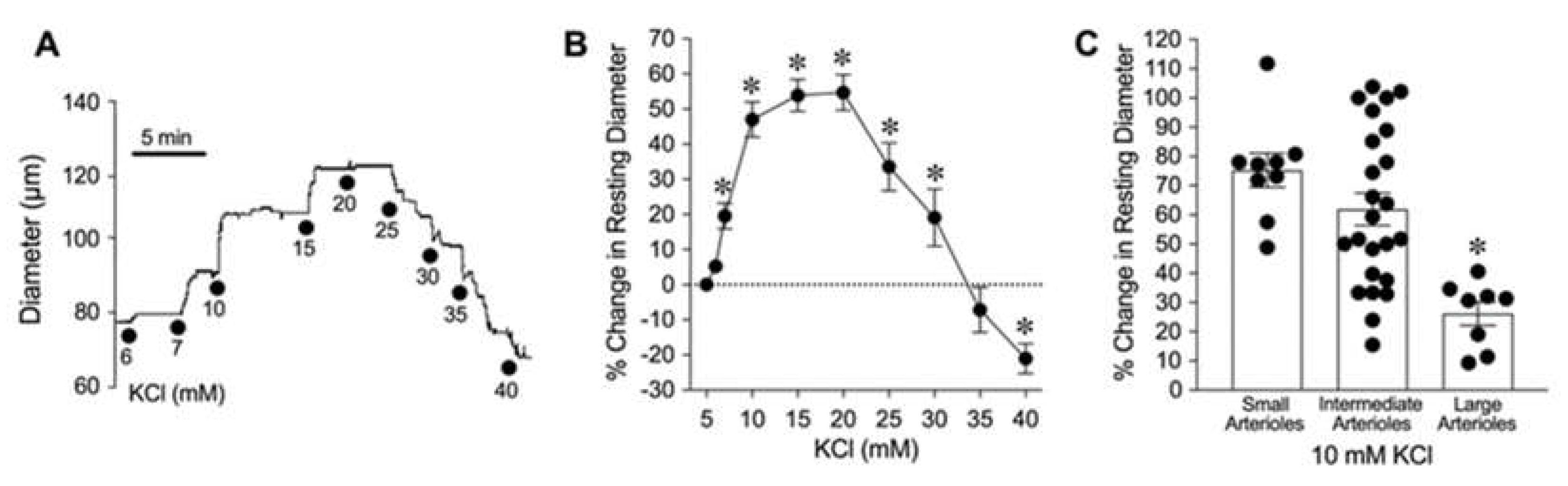
Figure 2.
Role of Kir channels in K+-induced dilation of isolated coronary arterioles. (A) The changes in diameter in response to 10 mM KCl were evaluated before (Control) and after pharmacological blockade with Kir channel inhibitor Ba2+ (30 µM, n = 9). (B) Summary data show the percent change in resting diameter at 1 min and 5 min after administration of 10 mM KCl in the absence (Control) and the presence of Ba2+ (n = 9). *P < 0.05 vs. Control response at 5 min.
Figure 2.
Role of Kir channels in K+-induced dilation of isolated coronary arterioles. (A) The changes in diameter in response to 10 mM KCl were evaluated before (Control) and after pharmacological blockade with Kir channel inhibitor Ba2+ (30 µM, n = 9). (B) Summary data show the percent change in resting diameter at 1 min and 5 min after administration of 10 mM KCl in the absence (Control) and the presence of Ba2+ (n = 9). *P < 0.05 vs. Control response at 5 min.
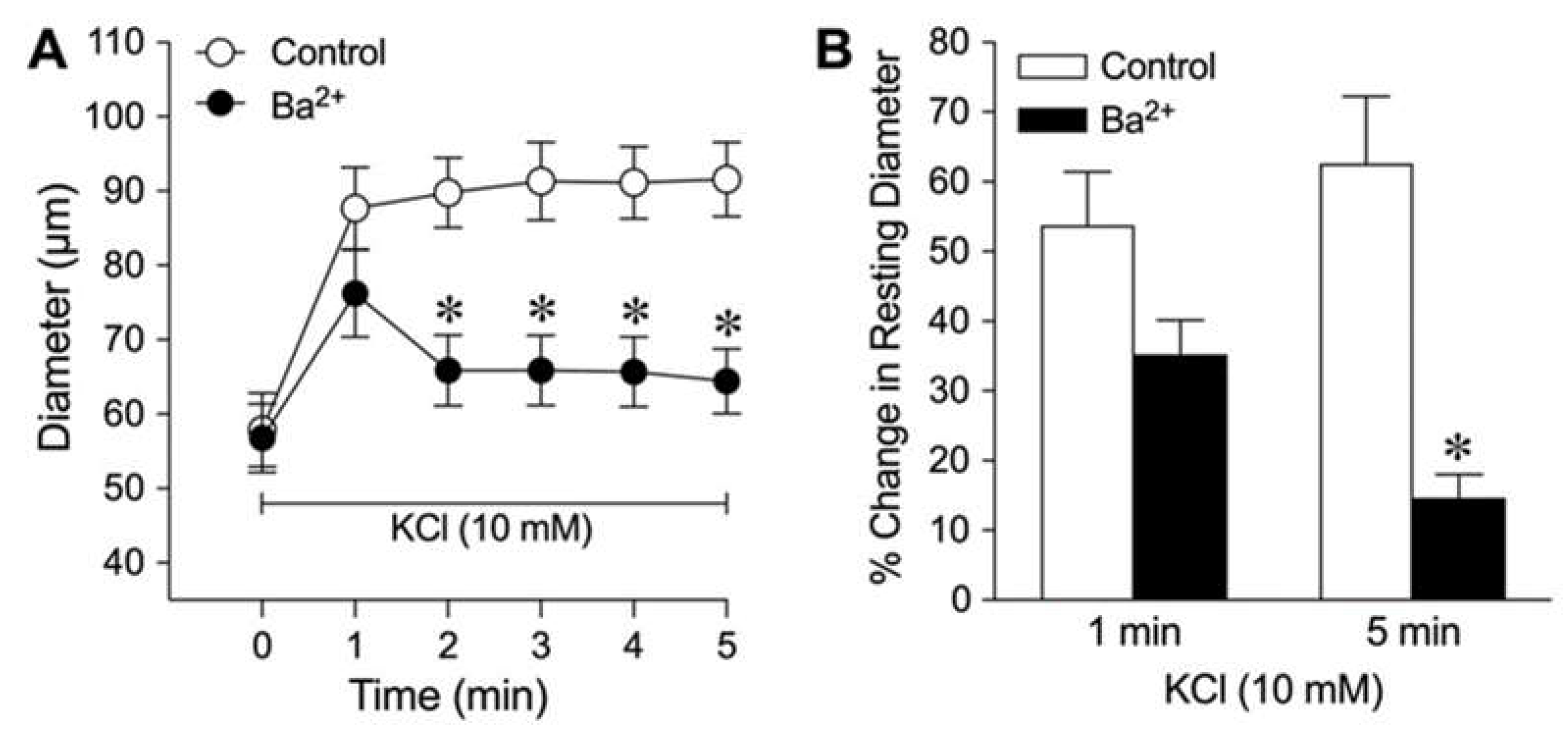
Figure 3.
Role of Kir2.1 channels in K+-induced dilation of isolated coronary arterioles. (A) Summary data show the percent change in resting diameter at 1 min and 5 min after administration of 10 mM KCl in the absence (Control) and presence of pharmacological Kir2.1 inhibitor CEC (n = 6). *P < 0.05 vs. Control response at 5 min. (B) Total RNA in a single isolated coronary arteriole (CA, 60-80 µm in diameter in situ) from three different pigs (CA1, CA2, and CA3) was reversed transcribed using gene-specific primers for Kir2.1 (326 bp) and GAPDH (311 bp) mRNAs. After the PCR reaction, gene products were electrophoresed on a 1.8% agarose gel and visualized with ethidium bromide staining. φX174 RF DNA/Hae III fragments were used as a size marker. (C) Western immunoblots were performed with protein from cardiomyocytes (CM) and coronary arterioles (CA) using anti-Kir2.1 and anti-alpha-tropomyosin (alpha-TM) antibodies. .
Figure 3.
Role of Kir2.1 channels in K+-induced dilation of isolated coronary arterioles. (A) Summary data show the percent change in resting diameter at 1 min and 5 min after administration of 10 mM KCl in the absence (Control) and presence of pharmacological Kir2.1 inhibitor CEC (n = 6). *P < 0.05 vs. Control response at 5 min. (B) Total RNA in a single isolated coronary arteriole (CA, 60-80 µm in diameter in situ) from three different pigs (CA1, CA2, and CA3) was reversed transcribed using gene-specific primers for Kir2.1 (326 bp) and GAPDH (311 bp) mRNAs. After the PCR reaction, gene products were electrophoresed on a 1.8% agarose gel and visualized with ethidium bromide staining. φX174 RF DNA/Hae III fragments were used as a size marker. (C) Western immunoblots were performed with protein from cardiomyocytes (CM) and coronary arterioles (CA) using anti-Kir2.1 and anti-alpha-tropomyosin (alpha-TM) antibodies. .
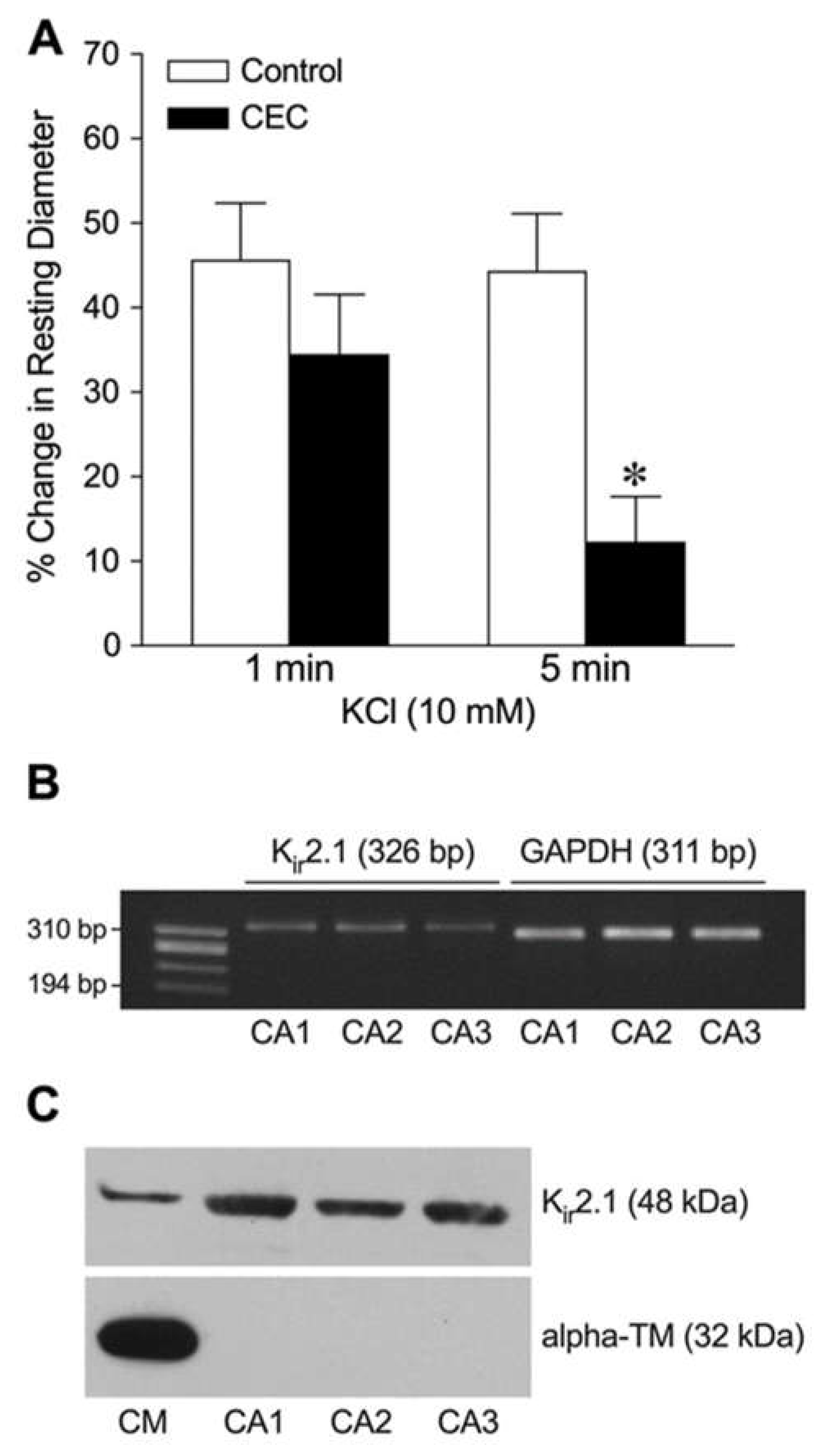
Figure 4.
Assessment of K+-induced dilation of isolated coronary arterioles after Kir2.1 channel knockdown. Summary data show the percent change in resting diameter at 1 min and 5 min after administration of 10 mM KCl in vessels transfected with sense (n = 5) or antisense (n = 5) oligonucleotides. *P < 0.05 vs. Sense response. (B) Total RNA from a single coronary arteriole (100 µm in diameter in situ) transfected with Kir2.1 sense (Se) or Kir2.1 antisense (AS) was reversed transcribed using gene-specific primers for Kir2.1 (326 bp), Kir2.3 (210 bp), and GAPDH (311 bp) mRNAs. After the PCR reaction, gene products were electrophoresed on a 1.8% agarose gel and visualized with ethidium bromide staining. φX174 RF DNA/Hae III fragments were used as a size marker.
Figure 4.
Assessment of K+-induced dilation of isolated coronary arterioles after Kir2.1 channel knockdown. Summary data show the percent change in resting diameter at 1 min and 5 min after administration of 10 mM KCl in vessels transfected with sense (n = 5) or antisense (n = 5) oligonucleotides. *P < 0.05 vs. Sense response. (B) Total RNA from a single coronary arteriole (100 µm in diameter in situ) transfected with Kir2.1 sense (Se) or Kir2.1 antisense (AS) was reversed transcribed using gene-specific primers for Kir2.1 (326 bp), Kir2.3 (210 bp), and GAPDH (311 bp) mRNAs. After the PCR reaction, gene products were electrophoresed on a 1.8% agarose gel and visualized with ethidium bromide staining. φX174 RF DNA/Hae III fragments were used as a size marker.
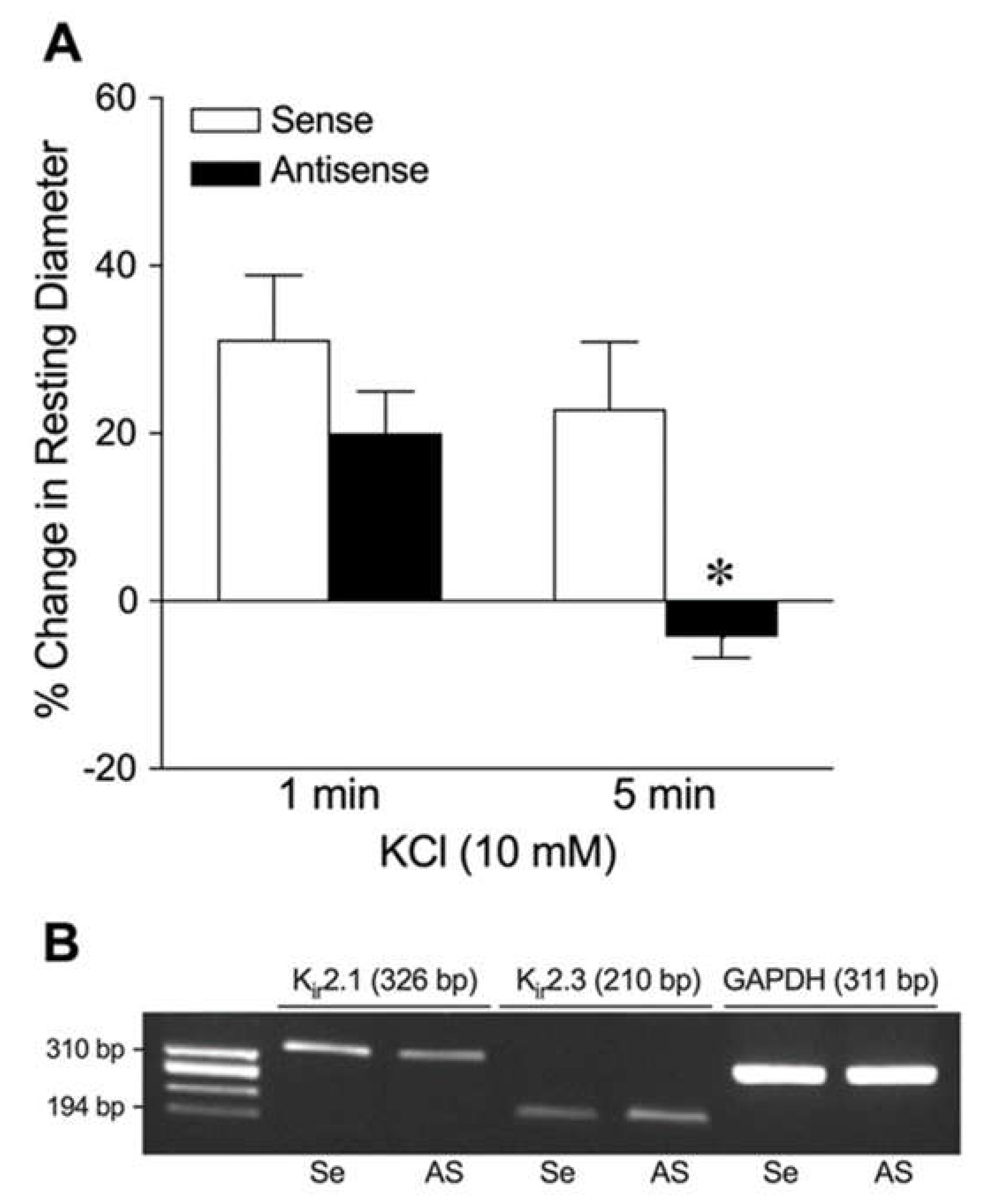
Figure 5.
Roles of Na+/K+ ATPase and Kir channels in K+-induced dilation of isolated coronary arterioles. The changes in diameter in response to 10 mM KCl were evaluated before (Control) and after pharmacological blockade with (A) Na+/K+ ATPase inhibitor ouabain (n = 6) or (B) combination of ouabain and Kir channel inhibitor Ba2+ (n = 7). *P < 0.05 vs. Control response.
Figure 5.
Roles of Na+/K+ ATPase and Kir channels in K+-induced dilation of isolated coronary arterioles. The changes in diameter in response to 10 mM KCl were evaluated before (Control) and after pharmacological blockade with (A) Na+/K+ ATPase inhibitor ouabain (n = 6) or (B) combination of ouabain and Kir channel inhibitor Ba2+ (n = 7). *P < 0.05 vs. Control response.
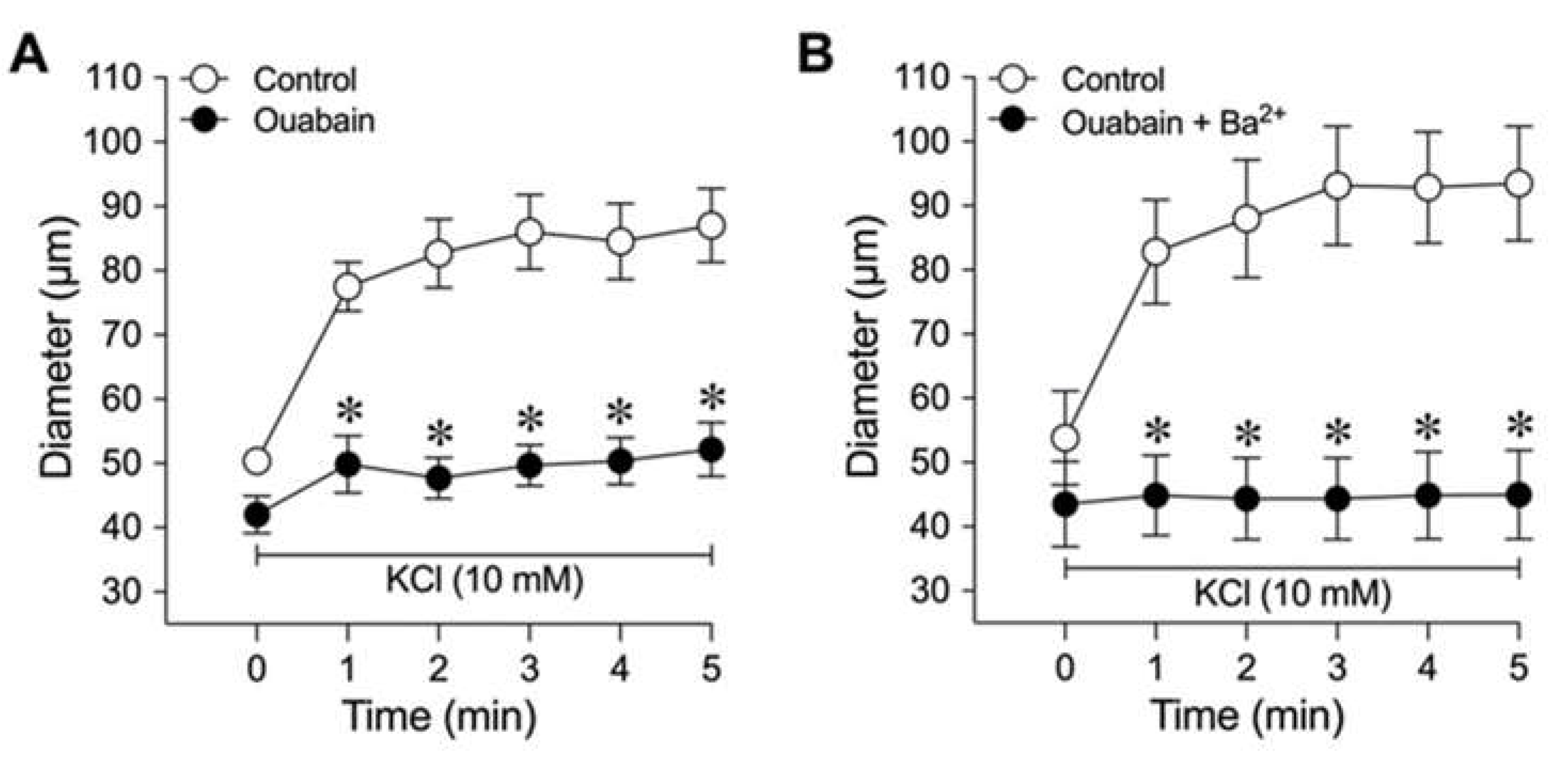

Table 1.
Effect of endothelial denudation and K+ channel blockers on dilation of porcine coronary arterioles to an increase in extraluminal KCl.
Table 1.
Effect of endothelial denudation and K+ channel blockers on dilation of porcine coronary arterioles to an increase in extraluminal KCl.
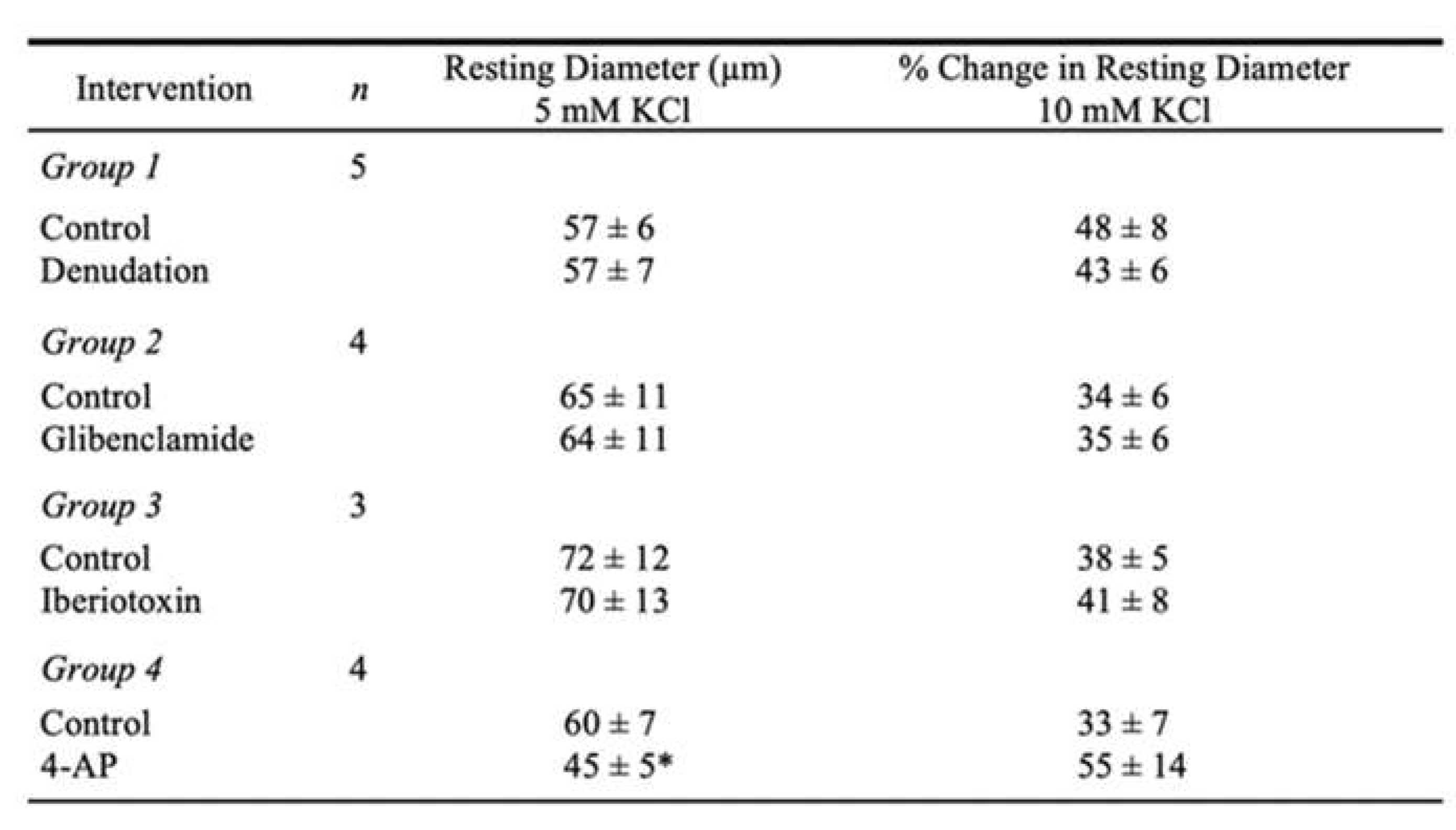 |
Values are mean ± SEM; n = number of vessels. Diameter changes were normalized to resting diameter at 5 mM KCl and expressed as % change in resting diameter at 10 mM KCl. *P < 0.05 vs. Control.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



