
Submitted:
30 December 2024
Posted:
31 December 2024
You are already at the latest version
Abstract
PCSK9 inhibitors (PCSK9i) represent a newer form of atherosclerosis treatment. Inflammation and haemostasis are key processes in the development of atherosclerosis. In this study, we investigated the influence of therapy with PCSK9i in patients with coronary artery disease (CAD) on regulators for lipoprotein homeostasis, inflammation and coagulation. Using quantitative polymerase chain reaction (qPCR) we measured the expression of the genes involved in lipoprotein homeostasis, namely for sterol regulatory element-binding protein 1 (SREBP1), SREBP2, low density lipoprotein receptor (LDLR), hepatic lipase type C (LIPC), LDLR-related protein (LRP8), and the genes associated with inflammation and coagulation, such as cluster of differentiation (CD) 36 (CD36), CD63 and CD14 in patients with CAD and healthy subjects. The results showed significant differences in the expression of the investigated genes between patients and healthy controls. Treatment with PCSK9i resulted in significant changes in the expression of all studied genes. Moreover, we found an association between changes in total cholesterol and SREBP1 expression, as well as a correlation between changes in HDL cholesterol and CD63 expression. To conclude, we established that PCSK9i may have a significant effect on the gene expression of lipid regulators, inflammatory markers and coagulation parameters, independent of their lipolytic effect.
Keywords:
PCSK9 inhibitors
; lipid regulators
; inflammation
; coagulation
; coronary artery disease
1. Introduction
PCSK9 inhibitors (PCSK9i) represent a new milestone in the treatment of the atherosclerotic process in particularly for the prevention of acute cardiovascular events [1]. The basic mechanism of their action is to reduce the concentration of low-density lipoprotein (LDL) cholesterol (LDL-C) by inhibiting the action of PCSK9 on the LDL-C receptor (LDLR) [2]. However, there is increasing research supporting their influence on platelet aggregation and inflammation, which present important factors in the development and complications of the atherosclerotic process [3]. Rare patients with familial hypercholesterolemia (FH) that possess mutations in both, the PCSK9 and LDLR gene, have a significantly higher risk of future coronary heart disease (CHD) compared to FH patients bearing mutation in only one of the two genes [4].
Clearly, the influence of PCSK9i on other factors involved in the regulation of LDL-C concentration needs to be considered. Members of the sterol regulatory element-binding protein (SREBP) group play a central role in the regulation of genes important for lipid biosynthesis and uptake [5]. In general, SREBP1 activates fatty acid and triglyceride synthesis, while SREBP2 initiates cholesterol synthesis [5]. The membrane-bound transcription factor SREBP2 is responsible for cholesterol homeostasis in cells and simultaneous gene expression of PCSK9 [6].
Lipase C, hepatic type (LIPC) is involved in the metabolism of triglycerides and high-density lipoprotein cholesterol (HDL-C). At the same time, its increased activity leads to the formation of smaller and denser LDL-C particles, which have a greater ability to penetrate through the endothelial barrier and are significantly more atherogenic than larger and less dense LDL-C particles [7].
Apolipoprotein E receptor-2 (apoER2), the protein encoded by LRP8 (LDL receptor-related protein-8) gene, is present in the cells involved in atherosclerosis pathogenesis such as platelets, endothelial cells [8] and monocytes/macrophages [9].
The binding of oxidized LDL (oxLDL) cholesterol to CD36 on the surface of platelets triggers a number of signalling pathways resulting in their activation, higher possibility of arterial thrombosis at the site of the atherosclerotic plaque rupture and ultimately acute cardiovascular event [10].
CD63 is known as a platelet activation molecule, which plays a role in the processes of haemostasis and atherosclerosis. In platelets, CD63 is mainly found in α granules, which fuse upon activation with the plasmalemma and expose CD63 to the platelet surface [11].
Previous studies have demonstrated the importance of the interactions between CD14 and the proatherogenic factors in the development of atherosclerosis. Macrophages within atherosclerotic lesions were shown to express above-average levels of CD14. Triggering of the CD14-dependent signalling pathways increases the expression of the scavenger receptor-AI (SR-AI) and accelerates the formation of foam cells. CD14 signalling also affects the increased adhesion and migration of the inflammatory cells to the atherosclerotic lesions [12], and promotes the cytokine expression and inflammation in macrophages. Increased levels of CD14 are associated with higher plasma cholesterol and a higher risk of development of CHD or other cardiovascular diseases [13].
The purpose of our study was to determine the effect of PCSK9i on the gene expression of the factors that participate, first, in the metabolism of atherogenic lipoproteins in addition to PCSK9, second, in the inflammation contributing to instability of the atherosclerotic plaques, and third, in the coagulation triggering the occurrence of thrombosis in the case of the atherosclerotic plaque rupture. For this purpose, we included high risk patients after an acute coronary syndrome (ACS) who had, despite the maximal tolerated dose of statins, insufficiently regulated LDL-C levels. In addition, they had greatly increased levels of lipoprotein (a) (Lp(a)), which is an independent lipid risk factor regardless of the LDL-C levels. The following genes SREBP1, SREBP2, LDLR, LIPC, LRP8, CD36, CD63 and CD14 were measured in patients before and after PCSK9i or placebo treatment along with matched heathy controls.
2. Materials and Methods
2.1. Patients and Controls
We included 96 patients and additional 25 healthy control subjects matched for sex and age. Inclusion and exclusion criteria are shown in Figure 1. All of the patients had been prescribed beta blockers and antiplatelet drugs and were receiving angiotensin-converting enzyme inhibitors/angiotensin II receptor blockers and statins at the highest tolerated doses, along with ezetimibe where needed. Their therapies had not been changed for at least 8 weeks before entering the study. Control subjects had no history of cardiovascular disease (CVD), no hypercholesterolemia, and Lp(a) levels less than 300 mg/L.
2.2. Clinical Examination
Systolic and diastolic blood pressures were measured in the sitting position after a minimum of 10 minutes rest, with the mean of three measurements recorded. Anthropometric parameters were recorded and body mass index was calculated.
2.3. Laboratory Analyses
The blood for laboratory analyses was taken in the morning after 12 h of fasting. Samples were collected from the antecubital vein into vacuum-sealed 5 mL tubes containing clot activator (Cacutube, LT Burnik, Slovenia). Serum was obtained by 15-min centrifugation at 2,000 × g. Total cholesterol, triglycerides, high-density lipoprotein cholesterol, apolipoproteins A1 and B were determined in the fresh serum by standard colorimetric or immunologic assays on an automated biochemistry analyser (Fusion 5.1; Ortho-Clinical Diagnostics, USA). The Friedewald formula [14] was used to calculate LDL-C. The same biochemistry analyser was used to determine Lp(a) with the Denka reagent (Randox, UK), which contains apo(a) isoform-insensitive antibodies, and therefore showed minimal apo(a) size-related bias.
The blood for laboratory analyses was taken in the morning after 12 h of fasting. Samples were collected from the antecubital vein into vacuum-sealed 5 mL tubes containing clot activator (Cacutube, LT Burnik, Slovenia). Serum was obtained by 15-min centrifugation at 2,000 × g. Total cholesterol, triglycerides, high-density lipoprotein cholesterol, apolipoproteins A1 and B were determined in the fresh serum by standard colorimetric or immunologic assays on an automated biochemistry analyser (Fusion 5.1; Ortho-Clinical Diagnostics, USA). The Friedewald formula [14] was used to calculate LDL-C. The same biochemistry analyser was used to determine Lp(a) with the Denka reagent (Randox, UK), which contains apo(a) isoform-insensitive antibodies, and therefore showed minimal apo(a) size-related bias.
2.4. Gene Expression Measurement
Total RNA was isolated from the samples collected in Tempus™ Blood RNA Tubes using Tempus™ Spin RNA Isolation Kit (Thermo Fisher Scientific) and evaluated as described previously [15]. Complementary DNA (cDNA) was synthesized using High-Capacity cDNA Archive kit (Thermo Fisher Scientific) according to the manufacturer instructions. Gene expression was measured according to MIQE guidelines [16]. Quantitative polymerase chain reaction (qPCR) was performed using 5× HOT FIREPol EvaGreen qPCR Supermix (Solis BioDyne) and LightCycler 480 II instrument (Roche). The sequences of the primers for the following genes CD14, LRP8, CD36, CD63 and SREBP2 were obtained from online sources [17,18,19,20,21] and validated on our samples. The primers for genes SREBP1, LDLR and LIPC (Supplemental Table S1) were self-designed and validated. The sequences for reference genes, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and ribosomal protein L13a (RPL13A) were taken from our previous studies. All of the data were normalized to geometric mean of GAPDH and RPL13A.
2.5. Statistical Analysis
Statistical analysis was performed using IBM SPSS Statistics, version 27.0 (IBM Corporation, New York, USA). First, the normality of the distribution of variables was tested using the Kolmogorov-Smirnov test. The values of variables that were normally distributed were presented with arithmetic mean and standard deviation. The median and the range between the lower and upper quartiles were used to display variables that were not normally distributed. For independent samples with normally distributed variables, the Student's t test was used to test differences between the groups. The Mann-Whitney U test was used to test the differences between the groups with non-normally distributed variables. The differences between the three groups were calculated using one-way ANOVA or Kruskal-Wallis test for the non-normally distributed variables. Differences between parameters at baseline and after 6 months of treatment were calculated using the Wilcoxon signed-rank test. Pearson and Spearman correlation analyses were used to determine the correlations between normally and non-normally distributed variables, respectively. The significance level was set at p<0.05.
3. Results
3.1. Subjects’ Characteristics
Clinical and laboratory parameters of the patients and controls are shown in Table 1. Amongst clinical parameters only body mass index (BMI) showed statistical significance between both groups, namely healthy subjects had significantly lower BMI (p=0.045). On the other hand, healthy subjects possessed significantly higher total cholesterol (p=0.035), LDL-C (p=0.023) and HDL-C (p=0.034) than patients. We also detected significantly lower Lp(a) concentrations in healthy subjects compared to patients (p<0.001).
Since there was no difference in any parameters in patients treated with PCSK9i or placebo, we decided to combine the groups for higher statistical power when comparing patients before treatment with healthy subjects.
3.2. The Results of Expression of the Tested Genes
As shown in Table 2, the expression of SREBP2, LDLR, LIPC, LRP8, CD36, CD63 and CD14 statistically significantly differed between patients and healthy subjects (**p<0.001), except for SREBP1 (p=0.976). LDLR, LIPC and LRP8 expressions were significantly higher, while expressions of SREBP2, CD36, CD63 and CD14 were significantly lower in the patient group.
All data are normalized to geometric mean of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and ribosomal protein L13a (RPL13A). Differences between the patients and controls were observed as indicated (**p<0.001).
Next, we investigated the expression of the tested genes in patients before and after 6 months of treatment with PCSK9i alirocumab and evolocumab. We demonstrated that changes in the expression of all genes except SREBP1 and LDLR were statistically significant after 6 months of treatment. Gene expression of LIPC and LRP8 was significantly lower, whereas the expression of SREBP2, CD36, CD63 and CD14 was significantly higher (Figure 2 and Figure 3).
Furthermore, we analysed the expression of the investigated genes in patient group receiving placebo. We found out that that the expression of SREBP2, LRP8 and CD36 significantly changed before and after placebo, while the others did not show any significant changes. The expressions of SREBP2 and CD36 have significantly increased (*p=0.031 and *p=0.001 respectively), while the expression of LRP8 has significantly decreased (*p=0.019).
Additionally, we compared the gene response after therapy with PCSK9i as opposed to placebo. Significantly different changes in the expression of CD36 (*p=0.002) and CD63 (*p=0.002) were observed, namely the expression of both genes was significantly lower in the group of patients receiving placebo (Figure 4 and Figure 5).
Additionally, we analysed changes in lipid profile after PCSK9i treatment, since alirocumab and evolocumab are medications for treatment of dyslipidaemias. In placebo group a minimal increase in total cholesterol concentrations (3%) was detected, while in PCSK9i treated patients a significant decrease (35%, **p<0.001) was observed. In placebo and PCSK9i group the changes were as followed: 4% and -64% (**p<0.001) for LDL-C, 7% and 8% (p=0.725) for HDL cholesterol, 11% and -11% (*p=0.005) for triglycerides, and 2% and -21% (**p<0.001) for Lp(a).
Having identified the changes in lipid profile in patients treated with PCSK9i, we further aimed to investigate whether changes in lipid profile correlate with changes in the expression of the tested genes (Table 3). We found a statistically significant positive correlation between change in total cholesterol and change in SREBP1 (*p=0.045; ρ=0.252) and a statistically significant negative correlation between change in HDL-C and change in CD63 (*p=0.019; ρ=-0.294). The correlations between changes in HDL-C and CD14 (p=0.066), and changes in LDL-C and CD63 (p=0.069) were borderline significant.
4. Discussion
To the best of our knowledge this is the first study to investigate the effect of PCSK9i on factors participating in the metabolism of atherogenic lipoproteins, inflammation and coagulation in high-risk CHD patients. The gene expressions of all investigated regulators of the lipid metabolism except for SREBP1 were significantly different in healthy controls compared to patients. The same is true for the gene expression of the regulators of inflammation and coagulation. At the same time our results showed that treatment with PCSK9i changed the expression of all genes except SREBP1 and LDLR.
At first glance it is surprising that healthy controls possessed increased values of total and LDL-C compared to patients, however we have to bear in mind that all our patients were treated with the highest tolerated dose of statin and ezetimibe if needed. We examined the expression of several genes encoding the proteins involved in the LDL-C metabolism. SREBP-1 is responsible for both fatty acid and cholesterol synthesis, whereas SREBP-2 exclusively regulates cholesterol synthesis [22]. Drug-induced reduction in intracellular cholesterol levels triggers the activation of SREBPs, including SREBP-1 and SREBP-2, to restore the cholesterol balance [23]. Unfortunately, data for humans are not available. However, it was shown that high doses of statins in mice without hypercholesterolemia increase not only the production of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, but also the gene expression for SREBP-2 [24]. This is in contrast to our results, as we found no differences in SREBP1 expression between healthy controls and patients, all of whom were treated with the highest tolerated doses of statins. However, SREBP2 gene expression was even higher in healthy controls than in patients. Yet, we have to be aware of the two facts; first, the patients had significantly lower cholesterol levels compared to the healthy controls, which could have reversed the relationship in the case of SREBP1 and SREBP2, and second, all patients in our study had significantly increased Lp(a) levels before starting treatment with PCSK9i. It is well-recognised that Lp(a) includes large amounts of apo(B), which might also have influenced these results.
In healthy individuals, a three-week low carbohydrate/high fat (LCHF) diet significantly increased both LDL-C and apo(B), as well as SREBP1 gene expression. However, it is unclear whether the increase in SREBP1 gene expression and apo(B)-containing lipoproteins is causally related [25]. We have no data on the association between Lp(a) concentration and the gene expression for SREBP1 and 2.
In our study, treatment with PCSK9i did not alter SREBP1 expression, while there was a significant increase in SREBP2 expression. There are no clinical studies investigating the effect of PCSK9i on these proteins. However, the IB20 antibody against PCSK9 in mice reduced the gene expression of SREBP in hepatocytes consistent with a reduction in LDL-C levels. Consistent with this observation in mice, in statin-responsive human primary hepatocytes, IB20 lowers the transcription of several SREBP regulated genes involved in cholesterol and fatty-acid synthesis [26]. These results are expected, as we assume that therapy with PCSK9i decreases intracellular cholesterol levels to the extent of increasing SREBP2 transcription. Hence our results are comparable to previous studies in mice.
Statins decrease plasma LDL-C by inhibiting HMG-CoA reductase, the rate-limiting enzyme in cholesterol synthesis. This inhibition induces intracellular cholesterol depletion, which results in upregulation of the hepatic LDLR [27]. Therefore, it is not unexpected that patients in our study had significantly higher LDLR expression compared to the control group. Interestingly, treatment with PCSK9i had no effect on LDLR expression, a result that is somewhat surprising, given that treatment with PCSK9i was presumed to lead to a significant reduction in LDL-C levels.
The LIPC gene encodes the enzyme hepatic lipase, which functions to hydrolyse triglycerides and phospholipids present in plasma lipoproteins. In addition, it acts as a ligand to cellular receptors and proteoglycans on the surface of hepatocytes to facilitate the uptake of lipoproteins into the liver. The results of previous studies in humans and animals demonstrate both proatherogenic and antiatherogenic roles of hepatic lipase. Its proatherogenic effects are the result of an inverse relationship between increased hepatic lipase activity and plasma concentrations of antiatherogenic HDL-C, and a positive correlation with plasma concentrations of proatherogenic LDL-C. The lipolytic function of hepatic lipase, which leads to lower levels of proatherogenic apolipoprotein B-containing lipoproteins in plasma, reduces the risk of developing atherosclerosis [28]. The influence of hepatic lipase activity on the risk of CHD is not clear and mainly depends on the concentration and type of lipoprotein(s) that predominate in a particular lipid metabolic disorder [29]. In general, serum hepatic lipase concentrations are very low, as most hepatic lipase is bound to proteoglycans on the surface of hepatocytes, and high serum concentrations are therefore considered a risk factor for the development of CHD [30]. Several studies have demonstrated significantly higher serum hepatic lipase levels in patients with CHD compared to healthy subjects, suggesting that serum hepatic lipase levels could be used as a potential marker of coronary artery disease progression [30,31]. Given the above, it is not surprising that the patients in our study had significantly higher LIPC expression compared to the control group. Our results also showed that after six months of treatment with PCSK9i, lipid levels decrease to such an extent that LIPC expression also decreases. However, the results of numerous studies regarding the role of hepatic lipase in lipid homeostasis are contradictory, suggesting its proatherogenic and antiatherogenic role. Based on the results of our study we merely suggest that a positive correlation between lipid concentration and LIPC gene expression exists.
Similar to our study, Shen et al. found that LRP8 expression is significantly increased in patients with premature coronary artery disease compared to healthy individuals [32]. Unfortunately, this study lacks the information whether the patients were previously treated with statins and, if so, at what dose. To the best of our knowledge there are no other studies evaluating the impact of statin treatment on LRP8 expression. We were also not able to find any data on the association between LRP8 and PCSK9 in the literature. Considering that Shen et al. reported increased LRP8 levels in patients with premature coronary artery disease [32], we could assume that the down-regulation of LRP8 expression observed after treatment with PCSK9i in our study may have a beneficial effect on inhibiting the atherosclerotic process.
CD36 expression at the protein level has been shown to be significantly increased in patients with carotid atherosclerosis, particularly in the advanced stages of the disease [33]. All of our patients had an advanced form of atherosclerosis, as they had all suffered from ACS. However, our results showed lower CD36 expression in patients compared to healthy subjects. This discrepancy suggests that the gene and protein levels may differ. Increased expression of surface marker CD36 is thought to activate macrophages, leading to the formation of unstable atherosclerotic plaques that are more prone to rupture, thereby resulting in subsequent thrombosis [34]. Additionally, increased CD36 levels contribute to accelerated oxidation of LDL-C, which is considered to be even more atherogenic than LDL-C itself [35]. Treatment with statins has been shown to decrease the CD36 expression on platelet surfaces and thus inhibiting the progression of atherosclerosis. This reduction occurs independently of LDL-C lowering, suggesting the so-called pleiotropic effects of statins [36]. In our study, treatment with both PCSK9i and placebo significantly increased CD36 expression. Given the established association between increased CD36 expression on platelets and the progression of the atherosclerotic process [37], one might speculate that treatment with PCSK9i does not appear to beneficially affect the process of atherosclerosis. However, it is important to note that in our study, CD36 expression was measured at the gene level, which might not accurately reflect the levels of the CD36 protein on the platelet surfaces. On the other hand, CD36 expression in our healthy subjects was higher than in patients receiving either PCSK9i or placebo. Of note, CD36 expression was comparable between healthy controls and patients after PCSK9i treatment, but this was not the case for the patients receiving placebo. One potential mechanism by which PCSK9i may influence CD36 production could involve inhibition of its degradation in hepatocytes and adipocytes. While PCSK9 has been shown to promote CD36 degradation in mouse hepatocytes [38], data on its effects in humans are lacking.
Moreover, the results of our study showed significantly higher CD63 expression in healthy controls compared to patients with CHD. On the contrary, Murakami et al. using flow cytometry found no differences in CD63 expression on platelets between healthy controls and patients with CHD [39]. At the same time, they found no correlation between CD63 expression and the extent of coronary disease. However, it is important to note that the subjects in their study were not treated with statins prior to the study. Cha et al. demonstrated significantly higher CD63 expression on platelets in patients with hyperlipidaemia after ischemic stroke compared to healthy controls. Treatment with simvastatin in these patients resulted in significant reduction in CD63 expression on platelets [40]. Their findings are similar to ours, as all the patients in our study had been previously treated with statins and had significantly lower CD63 expression than healthy controls. There is currently no data on the association between CD63 and PCSK9 levels, and even less on the potential effects of treatment with PCSK9i on either the protein levels or the expression of the CD63 gene. In line with the expression of the CD36 gene, our study showed that treatment with PCSK9i led to a significant increase in the expression of the CD63 gene. Similar to CD36, CD63 gene expression in the control group was comparable to that of patients treated with PCSK9i, but not to those who received the placebo.
In patients with CHD who had not previously been treated with statins, CD14+ monocyte expression was reduced compared to healthy controls [41]. Although all of our CHD patients had received statin treatment prior to study, they also had lower CD14 expression. In our study, treatment with PCSK9i led to an increase in CD14 expression, whereas no statistically significant changes were observed in the placebo group. Given that CD14 is expressed on monocytes, which typically have an opposing effect, it is difficult to suspect a potential antiatherosclerotic effect of PCSK9i. It is important to consider that the role of the monocytes in the atherosclerosis is strongly influenced by both surface markers, CD14 and CD16, as well as their ratio [42]. However, since CD14 levels are generally reduced in patients with advanced atherosclerosis [41], the observed increase in CD14 expression might have a beneficial effect on the atherosclerotic process. In patients with stable coronary artery disease, a statistically significant correlation was found between monocyte concentration and Lp(a)-PCSK9 complex concentration, mainly due to monocytes expressing the highest CD14 levels on their surface and exhibiting the most pronounced proinflammatory effects [43]. This association might have been even more significant in our patients, who had highly elevated Lp(a) levels prior to treatment. Despite a significant decrease in Lp(a) following treatment with PCSK9i, their Lp(a) levels remained considerably higher than the desired target levels according to current treatment guidelines [44,45]. Furthermore, oxidized phospholipids, which are a component of Lp(a) and have a strong pro-inflammatory effect, are significantly associated with the concentration of pro-inflammatory monocytes expressing high levels of CD14 on their surface [46].
In our study, we observed that only the change in CD63 gene expression was associated with a change in lipid parameters, namely HDL-C. All other changes could be attributed to pleotropic effects, i.e. effects independent on the effect on lipid parameters. In addition to lowering LDL-C, PCSK9i also lower Lp(a) by 20-35%, but the mechanism of the latter is not fully elucidated [47]. The peculiarity of our study is, as previously mentioned, that the patients experienced an expected decrease in Lp(a) concentration by approximately 20%, but at the end of the study the Lp(a) concentrations were still strongly in the atherogenic range. On the other hand, the LDL-C levels were very low, as all patients had concentrations below 1.4 mmol/L. Given that drugs that specifically reduce Lp(a) concentration by up to 90%, are currently in the clinical trial phase (NCT04023552), it would be interesting to see whether the addition of these drugs to our patients would have an additional effect on the expression of the studied genes.
There are two shortcomings of this study. The first one is the relatively small number of subjects, which is a consequence of the very strict inclusion criteria, which can, on the other hand, be an advantage due to the very homogeneous population. The second limitation is that all the measurements were performed at the level of gene expression, which does not necessarily correlate with the levels of proteins that are a result of the expression of these genes. The process of translation, i.e. gene expression until the formation of a functional protein, is influenced by a large number of factors that are mostly unknown and therefore could not be taken into account in our analyses.
5. Conclusions
Inflammation and haemostasis as crucial processes in the development of atherosclerosis are targets of the novel treatment with PCSK9i. Our study observed a significant difference in the gene expression of the factors playing role in lipoprotein homeostasis, inflammation and coagulation between healthy subjects and patients. Moreover, we demonstrated that treatment with PCSK9i influences the gene expression of most of these factors. However, gene expression of SREBP2, LRP8 and CD36 also significantly changed after placebo. When comparing response after therapy with PCSK9i as opposed to response after placebo, changes in expression of CD36 and CD63 were both significantly lower after placebo. Given that the primary effect of PCSK9i is to reduce LDL-C and, to a lesser extent, Lp(a), we can argue that the effects on the expression of the studied genes are independent of the lipolytic effect of PCSK9i. Of course, to confirm that these results are also clinically significant, a study with a much larger number of subjects would be needed, where in addition to gene expression, the result of their expression at the protein level would also be measured in relation to the effects on cardiovascular events.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1: Primer pair sequences used for the gene expression references.
Author Contributions
Conceptualization, P.L., H.M., M.Š., J.Z.; Methodology, All authors; Formal Analysis, All authors; Investigation, All authors; Data Curation, All authors; Writing—Original Draft Preparation, P.L., H.M., M.Š., J.Z.; Writing—Review and Editing, All authors; Visualization, P.L., H.M., M.Š.; Supervision, J.Z. and M.Š.; Project Administration, M.Š.; Funding Acquisition, M.Š. All authors have read and agreed to the published version of the manuscript.
Funding
This study was funded by University Medical Centre Ljubljana (funding number 20240051) and Research Program P3-0308 of the Slovenian Research Agency. The funding source did not have any role in the study design, the collection, analysis and interpretation of data, the writing of the manuscript, or the decision to submit the manuscript for publication.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Slovenian Ethics Committee for Research in Medicine (0120-357/2018/8; date of approval: 23 April 2019).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The original contributions presented in this study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Acknowledgments
The authors thank nurses of the Clinical Department of Angiology, University Clinical Centre of Ljubljana for their valuable technical assistance. We are also grateful to the staff of the Laboratory for Haemostasis and Atherothrombosis at the Clinical Department of Angiology, University Clinical Centre Ljubljana for laboratory measurements.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. New England Journal of Medicine 2017, 376, 1713–1722. [CrossRef]
- Roth, E.M.; Davidson, M.H. PCSK9 Inhibitors: Mechanism of Action, Efficacy, and Safety. Rev Cardiovasc Med 2018, 19, 31–46. [CrossRef]
- Paciullo, F.; Momi, S.; Gresele, P. PCSK9 in Haemostasis and Thrombosis: Possible Pleiotropic Effects of PCSK9 Inhibitors in Cardiovascular Prevention. Thromb Haemost 2019, 119, 359–367. [CrossRef]
- Doi, T.; Hori, M.; Harada-Shiba, M.; Kataoka, Y.; Onozuka, D.; Nishimura, K.; Nishikawa, R.; Tsuda, K.; Ogura, M.; Son, C.; et al. Patients With LDLR and PCSK9 Gene Variants Experienced Higher Incidence of Cardiovascular Outcomes in Heterozygous Familial Hypercholesterolemia. J Am Heart Assoc 2021, 10. [CrossRef]
- Peng, C.; Lei, P.; Li, X.; Xie, H.; Yang, X.; Zhang, T.; Cao, Z.; Zhang, J. Down-Regulated of SREBP-1 in Circulating Leukocyte Is a Risk Factor for Atherosclerosis: A Case Control Study. Lipids Health Dis 2019, 18, 177. [CrossRef]
- Sobati, S.; Shakouri, A.; Edalati, M.; Mohammadnejad, D.; Parvan, R.; Masoumi, J.; Abdolalizadeh, J. PCSK9: A Key Target for the Treatment of Cardiovascular Disease (CVD). Adv Pharm Bull 2020, 10, 502–511. [CrossRef]
- Dijk, W.; Di Filippo, M.; Kooijman, S.; van Eenige, R.; Rimbert, A.; Caillaud, A.; Thedrez, A.; Arnaud, L.; Pronk, A.; Garçon, D.; et al. Identification of a Gain-of-Function LIPC Variant as a Novel Cause of Familial Combined Hypocholesterolemia. Circulation 2022, 146, 724–739. [CrossRef]
- Ramesh, S.; Morrell, C.N.; Tarango, C.; Thomas, G.D.; Yuhanna, I.S.; Girardi, G.; Herz, J.; Urbanus, R.T.; de Groot, P.G.; Thorpe, P.E.; et al. Antiphospholipid Antibodies Promote Leukocyte–Endothelial Cell Adhesion and Thrombosis in Mice by Antagonizing ENOS via Β2GPI and ApoER2. Journal of Clinical Investigation 2011, 121, 120–131. [CrossRef]
- Yang, X. V.; Banerjee, Y.; Fernández, J.A.; Deguchi, H.; Xu, X.; Mosnier, L.O.; Urbanus, Rolf.T.; de Groot, P.G.; White-Adams, T.C.; McCarty, O.J.T.; et al. Activated Protein C Ligation of ApoER2 (LRP8) Causes Dab1-Dependent Signaling in U937 Cells. Proceedings of the National Academy of Sciences 2009, 106, 274–279. [CrossRef]
- Yang, M.; Li, W.; Harberg, C.; Chen, W.; Yue, H.; Ferreira, R.B.; Wynia-Smith, S.L.; Carroll, K.S.; Zielonka, J.; Flaumenhaft, R.; et al. Cysteine Sulfenylation by CD36 Signaling Promotes Arterial Thrombosis in Dyslipidemia. Blood Adv 2020, 4, 4494–4507. [CrossRef]
- Hamamoto, K.; Ohga, S.; Nomura, S.; Yasunaga, K. Cellular Distribution of CD63 Antigen in Platelets and in Three Megakaryocytic Cell Lines. Histochem J 1994, 26, 367–375. [CrossRef]
- Wu, Z.; Zhang, Z.; Lei, Z.; Lei, P. CD14: Biology and Role in the Pathogenesis of Disease. Cytokine Growth Factor Rev 2019, 48, 24–31. [CrossRef]
- Sharygin, D.; Koniaris, L.G.; Wells, C.; Zimmers, T.A.; Hamidi, T. Role of <scp>CD14</Scp> in Human Disease. Immunology 2023, 169, 260–270. [CrossRef]
- Friedewald, W.T.; Levy, R.I.; Fredrickson, D.S. Estimation of the Concentration of Low-Density Lipoprotein Cholesterol in Plasma, Without Use of the Preparative Ultracentrifuge. Clin Chem 1972, 18, 499–502. [CrossRef]
- Hrovat, K.; Rehberger Likozar, A.; Zupan, J.; Šebeštjen, M. Gene Expression Profiling of Markers of Inflammation, Angiogenesis, Coagulation and Fibrinolysis in Patients with Coronary Artery Disease with Very High Lipoprotein(a) Levels Treated with PCSK9 Inhibitors. J Cardiovasc Dev Dis 2022, 9, 211. [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin Chem 2009, 55, 611–622. [CrossRef]
- SREBP2: Https://Www.Origene.Com/Catalog/Gene-Expression/Qpcr-Primer-Pairs/Hp207891-Srebp2-Srebf2-Human-Qpcr-Primer-Pair-Nm-004599.
- ApoER2 (LRP8): Https://Www.Origene.Com/Catalog/Gene-Expression/Qpcr-Primer-Pairs/Hp231155-Apoer2-Lrp8-Human-Qpcr-Primer-Pair-Nm-004631.
- CD36: Https://Www.Origene.Com/Catalog/Gene-Expression/Qpcr-Primer-Pairs/Hp200058-Cd36-Human-Qpcr-Primer-Pair-Nm-000072.
- CD63: Https://Www.Origene.Com/Catalog/Gene-Expression/Qpcr-Primer-Pairs/Hp227481-Cd63-Human-Qpcr-Primer-Pair-Nm-001780.
- CD14: Https://Www.Origene.Com/Catalog/Gene-Expression/Qpcr-Primer-Pairs/Hp200558-Cd14-Human-Qpcr-Primer-Pair-Nm-000591.
- Varghese, J.F.; Patel, R.; Yadav, U.C.S. Sterol Regulatory Element Binding Protein (SREBP) -1 Mediates Oxidized Low-Density Lipoprotein (OxLDL) Induced Macrophage Foam Cell Formation through NLRP3 Inflammasome Activation. Cell Signal 2019, 53, 316–326. [CrossRef]
- Horton, J.D.; Shimomura, I. Sterol Regulatory Element-Binding Proteins. Curr Opin Lipidol 1999, 10, 143–150. [CrossRef]
- Roglans, N.; Verd, J.C.; Peris, C.; Alegret, M.; Vázquez, M.; Adzet, T.; Diaz, C.; Hernández, G.; Laguna, J.C.; Sánchez, R.M. High Doses of Atorvastatin and Simvastatin Induce Key Enzymes Involved in VLDL Production. Lipids 2002, 37, 445–454. [CrossRef]
- Retterstøl, K.; Svendsen, M.; Narverud, I.; Holven, K.B. Effect of Low Carbohydrate High Fat Diet on LDL Cholesterol and Gene Expression in Normal-Weight, Young Adults: A Randomized Controlled Study. Atherosclerosis 2018, 279, 52–61. [CrossRef]
- Zhang, L.; McCabe, T.; Condra, J.H.; Ni, Y.G.; Peterson, L.B.; Wang, W.; Strack, A.M.; Wang, F.; Pandit, S.; Hammond, H.; et al. An Anti-PCSK9 Antibody Reduces LDL-Cholesterol On Top Of A Statin And Suppresses Hepatocyte SREBP-Regulated Genes. Int J Biol Sci 2012, 8, 310–327. [CrossRef]
- Young, S.G.; Fong, L.G. Lowering Plasma Cholesterol by Raising LDL Receptors — Revisited. New England Journal of Medicine 2012, 366, 1154–1155. [CrossRef]
- Santamarina-Fojo, S.; González-Navarro, H.; Freeman, L.; Wagner, E.; Nong, Z. Hepatic Lipase, Lipoprotein Metabolism, and Atherogenesis. Arterioscler Thromb Vasc Biol 2004, 24, 1750–1754. [CrossRef]
- Brunzell, J.D.; Zambon, A.; Deeb, S.S. The Effect of Hepatic Lipase on Coronary Artery Disease in Humans Is Influenced by the Underlying Lipoprotein Phenotype. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids 2012, 1821, 365–372. [CrossRef]
- Han, H.; Dai, D.; Wang, W.; Zhu, J.; Zhu, Z.; Lu, L.; Zhang, R. Impact of Serum Levels of Lipoprotein Lipase, Hepatic Lipase, and Endothelial Lipase on the Progression of Coronary Artery Disease. Journal of Interventional Medicine 2019, 2, 16–20. [CrossRef]
- Yu, X.; Lu, J.; Li, J.; Guan, W.; Deng, S.; Deng, Q.; Ye, H.; Han, W.; Yu, Y.; Zhang, R. Serum Triglyceride Lipase Concentrations Are Independent Risk Factors for Coronary Artery Disease and In-Stent Restenosis. J Atheroscler Thromb 2019, 26, 762–774. [CrossRef]
- Shen, G.-Q.; Li, L.; Girelli, D.; Seidelmann, S.B.; Rao, S.; Fan, C.; Park, J.E.; Xi, Q.; Li, J.; Hu, Y.; et al. An LRP8 Variant Is Associated with Familial and Premature Coronary Artery Disease and Myocardial Infarction. The American Journal of Human Genetics 2007, 81, 780–791. [CrossRef]
- Ackers, I.; Szymanski, C.; Duckett, K.J.; Consitt, L.A.; Silver, M.J.; Malgor, R. Blocking Wnt5a Signaling Decreases CD36 Expression and Foam Cell Formation in Atherosclerosis. Cardiovascular Pathology 2018, 34, 1–8. [CrossRef]
- Shu, H.; Peng, Y.; Hang, W.; Nie, J.; Zhou, N.; Wang, D.W. The Role of CD36 in Cardiovascular Disease. Cardiovasc Res 2022, 118, 115–129. [CrossRef]
- Li, N. Platelets as an Inter-player between Hyperlipidaemia and Atherosclerosis. J Intern Med 2024, 296, 39–52. [CrossRef]
- Schrör, K.; Verheugt, F.W.A.; Trenk, D. Drug–Drug Interaction between Antiplatelet Therapy and Lipid-Lowering Agents (Statins and PCSK9 Inhibitors). Thromb Haemost 2023, 123, 166–176. [CrossRef]
- Ackers, I.; Szymanski, C.; Duckett, K.J.; Consitt, L.A.; Silver, M.J.; Malgor, R. Blocking Wnt5a Signaling Decreases CD36 Expression and Foam Cell Formation in Atherosclerosis. Cardiovasc Pathol 2018, 34, 1–8. [CrossRef]
- Demers, A.; Samami, S.; Lauzier, B.; Des Rosiers, C.; Ngo Sock, E.T.; Ong, H.; Mayer, G. PCSK9 Induces CD36 Degradation and Affects Long-Chain Fatty Acid Uptake and Triglyceride Metabolism in Adipocytes and in Mouse Liver. Arterioscler Thromb Vasc Biol 2015, 35, 2517–2525. [CrossRef]
- MURAKAMI, T.; KOMIYAMA, Y.; MASUDA, M.; KIDO, H.; NOMURA, S.; FUKUHARA, S.; KARAKAWA, M.; IWASAKA, T.; TAKAHASHI, H. Flow Cytometric Analysis of Platelet Activation Markers CD62P and CD63 in Patients with Coronary Artery Disease. Eur J Clin Invest 1996, 26, 996–1003. [CrossRef]
- Cha, J.-K.; Jeong, M.-H.; Jang, J.-Y.; Bae, H.-R.; Lim, Y.-J.; Kim, J.S.; Kim, S.-H.; Kim, J.W. Serial Measurement of Surface Expressions of CD63, P-Selectin and CD40 Ligand on Platelets in Atherosclerotic Ischemic Stroke. Cerebrovascular Diseases 2003, 16, 376–382. [CrossRef]
- Du, P.; Guo, R.; Gao, K.; Yang, S.; Yao, B.; Cui, H.; Zhao, M.; Jia, S. Identification of Differentially Expressed Genes and the Role of PDK4 in CD14+ Monocytes of Coronary Artery Disease. Biosci Rep 2021, 41. [CrossRef]
- Krychtiuk, K.A.; Lenz, M.; Hohensinner, P.; Distelmaier, K.; Schrutka, L.; Kastl, S.P.; Huber, K.; Dostal, E.; Oravec, S.; Hengstenberg, C.; et al. Circulating Levels of Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) Are Associated with Monocyte Subsets in Patients with Stable Coronary Artery Disease. J Clin Lipidol 2021, 15, 512–521. [CrossRef]
- Filatova, A.Y.; Afanasieva, O.I.; Arefieva, T.I.; Potekhina, A. V; Tyurina, A. V; Klesareva, E.A.; Razova, O.A.; Ezhov, M. V; Pokrovsky, S.N. The Concentration of PCSK9-Lp(a) Complexes and the Level of Blood Monocytes in Males with Coronary Atherosclerosis. J Pers Med 2023, 13. [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the Management of Dyslipidaemias: Lipid Modification to Reduce Cardiovascular Risk. Eur Heart J 2020, 41, 111–188. [CrossRef]
- Corrigendum to: 2019 ESC/EAS Guidelines for the Management of Dyslipidaemias: Lipid Modification to Reduce Cardiovascular Risk. Eur Heart J 2020, 41, 4255–4255. [CrossRef]
- Krychtiuk, K.A.; Kastl, S.P.; Hofbauer, S.L.; Wonnerth, A.; Goliasch, G.; Ozsvar-Kozma, M.; Katsaros, K.M.; Maurer, G.; Huber, K.; Dostal, E.; et al. Monocyte Subset Distribution in Patients with Stable Atherosclerosis and Elevated Levels of Lipoprotein(a). J Clin Lipidol 2015, 9, 533–541. [CrossRef]
- O’Donoghue, M.L.; Fazio, S.; Giugliano, R.P.; Stroes, E.S.G.; Kanevsky, E.; Gouni-Berthold, I.; Im, K.; Lira Pineda, A.; Wasserman, S.M.; Češka, R.; et al. Lipoprotein(a), PCSK9 Inhibition, and Cardiovascular Risk. Circulation 2019, 139, 1483–1492. [CrossRef]
Figure 1.
Inclusion and exclusion criteria for the patient cohort. ACS – acute coronary syndrome. LDL-C – low density lipoprotein cholesterol. Lp(a) – lipoprotein(a).
Figure 1.
Inclusion and exclusion criteria for the patient cohort. ACS – acute coronary syndrome. LDL-C – low density lipoprotein cholesterol. Lp(a) – lipoprotein(a).
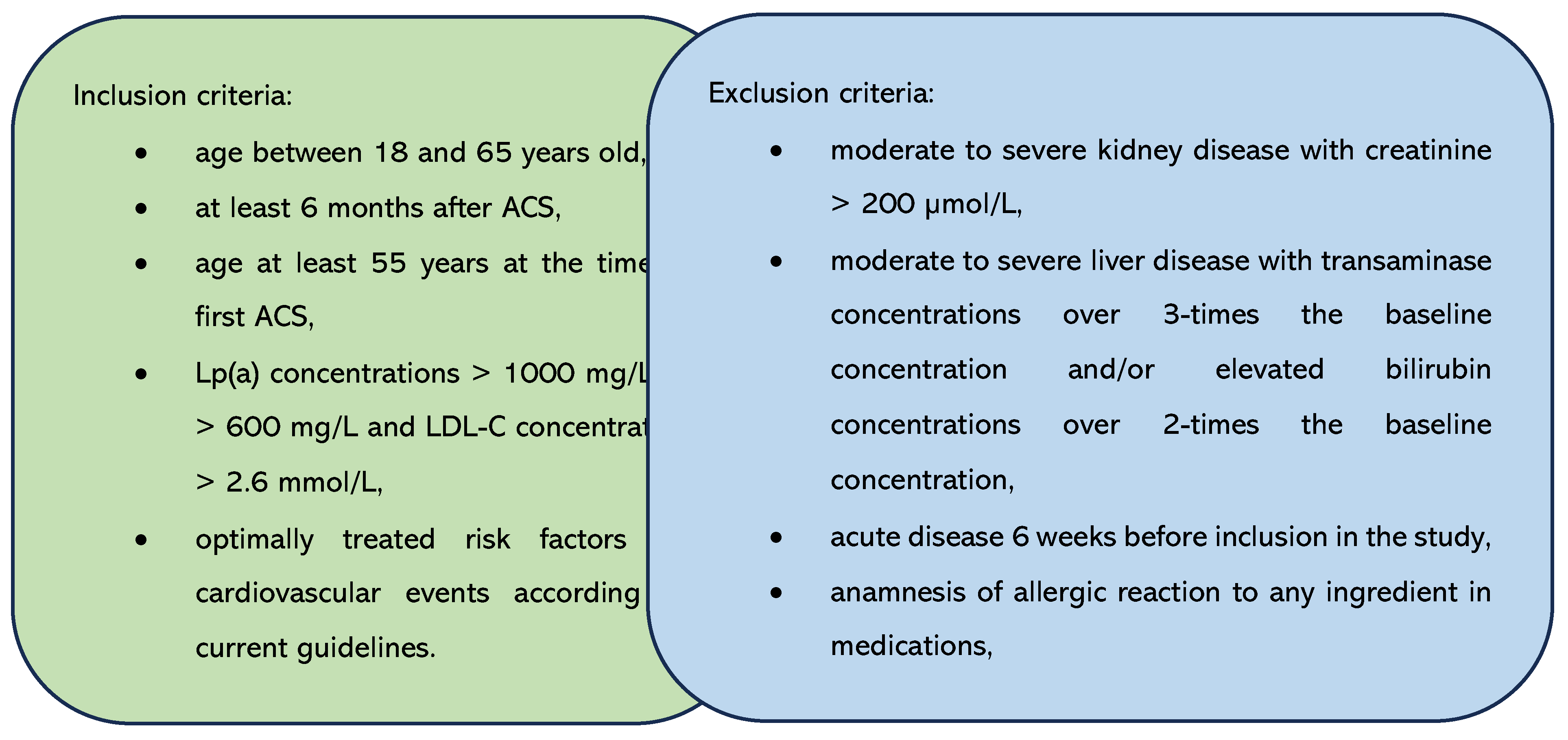
Figure 2.
The expression of genes encoding lipoprotein homeostasis regulators’ in patients before and after therapy with PCSK9i. SREBP – gene encoding sterol regulatory element-binding protein. LDLR – gene encoding LDL receptor. LIPC – gene encoding hepatic lipase type C. LRP8 – gene encoding LDLR-related protein 8. Differences before and after PCSK9i therapy were observed as indicated (**p<0.001, *p<0.05).
Figure 2.
The expression of genes encoding lipoprotein homeostasis regulators’ in patients before and after therapy with PCSK9i. SREBP – gene encoding sterol regulatory element-binding protein. LDLR – gene encoding LDL receptor. LIPC – gene encoding hepatic lipase type C. LRP8 – gene encoding LDLR-related protein 8. Differences before and after PCSK9i therapy were observed as indicated (**p<0.001, *p<0.05).
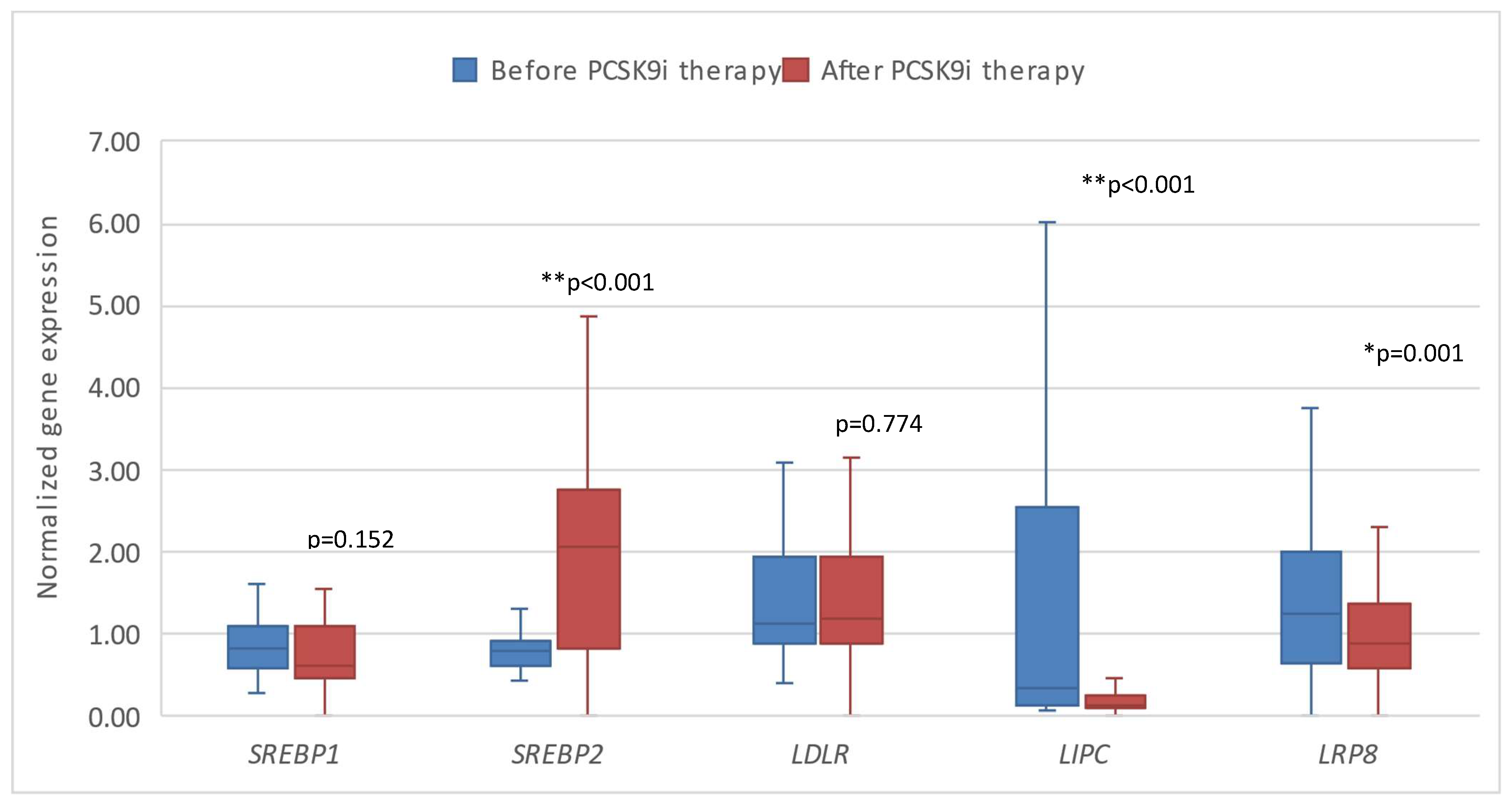
Figure 3.
The expression of genes encoding inflammatory factors in patients before and after therapy with PCSK9i. CD36 – gene encoding cluster of differentiation 36. CD63 – gene encoding cluster of differentiation 63. CD14 – gene encoding cluster of differentiation 14. Differences before and after PCSK9i therapy were observed as indicated (*p<0.05).
Figure 3.
The expression of genes encoding inflammatory factors in patients before and after therapy with PCSK9i. CD36 – gene encoding cluster of differentiation 36. CD63 – gene encoding cluster of differentiation 63. CD14 – gene encoding cluster of differentiation 14. Differences before and after PCSK9i therapy were observed as indicated (*p<0.05).
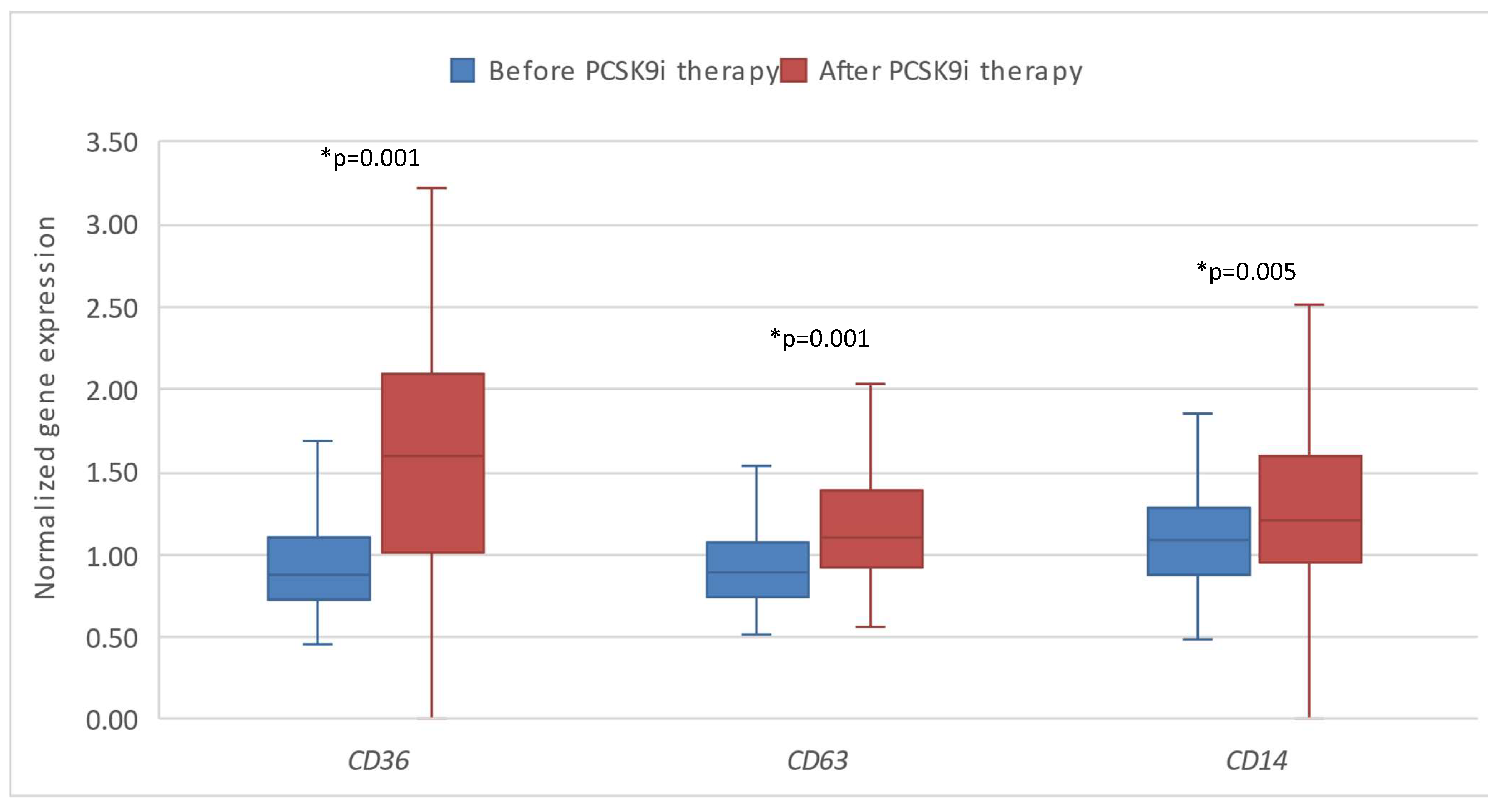
Figure 4.
The expression of genes encoding lipoprotein homeostasis regulators’ in patients after therapy with PCSK9i and after placebo. SREBP – gene encoding sterol regulatory element-binding protein. LDLR – gene encoding LDL receptor. LIPC – gene encoding hepatic lipase type C. LRP8 – gene encoding LDLR-related protein 8.
Figure 4.
The expression of genes encoding lipoprotein homeostasis regulators’ in patients after therapy with PCSK9i and after placebo. SREBP – gene encoding sterol regulatory element-binding protein. LDLR – gene encoding LDL receptor. LIPC – gene encoding hepatic lipase type C. LRP8 – gene encoding LDLR-related protein 8.
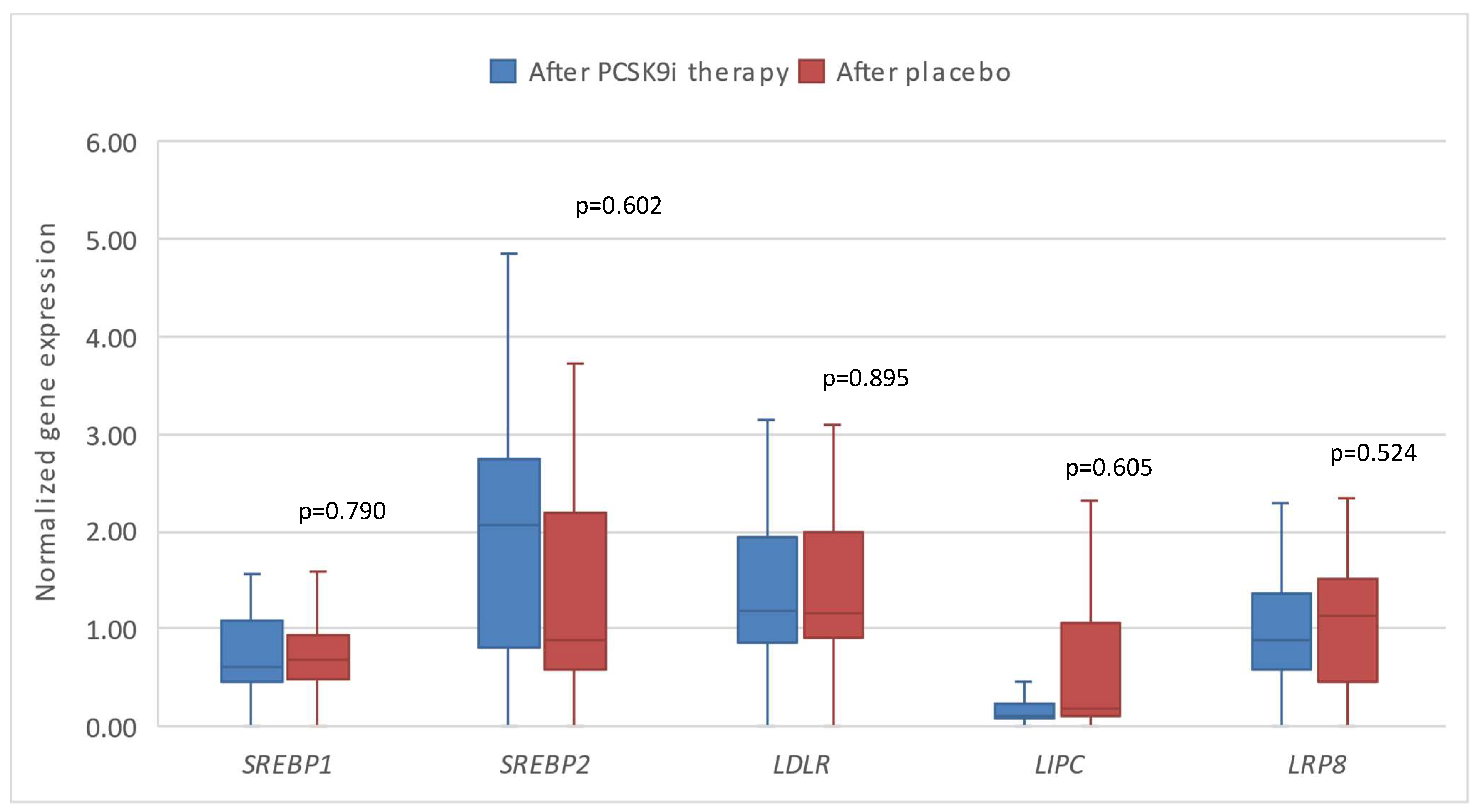
Figure 5.
The expression of genes encoding inflammatory factors in patients after therapy with PCSK9i and after placebo. CD36 – gene encoding cluster of differentiation 36. CD63 – gene encoding cluster of differentiation 63. CD14 – gene encoding cluster of differentiation 14. Differences before and after PCSK9i therapy were observed as indicated (*p<0.05).
Figure 5.
The expression of genes encoding inflammatory factors in patients after therapy with PCSK9i and after placebo. CD36 – gene encoding cluster of differentiation 36. CD63 – gene encoding cluster of differentiation 63. CD14 – gene encoding cluster of differentiation 14. Differences before and after PCSK9i therapy were observed as indicated (*p<0.05).
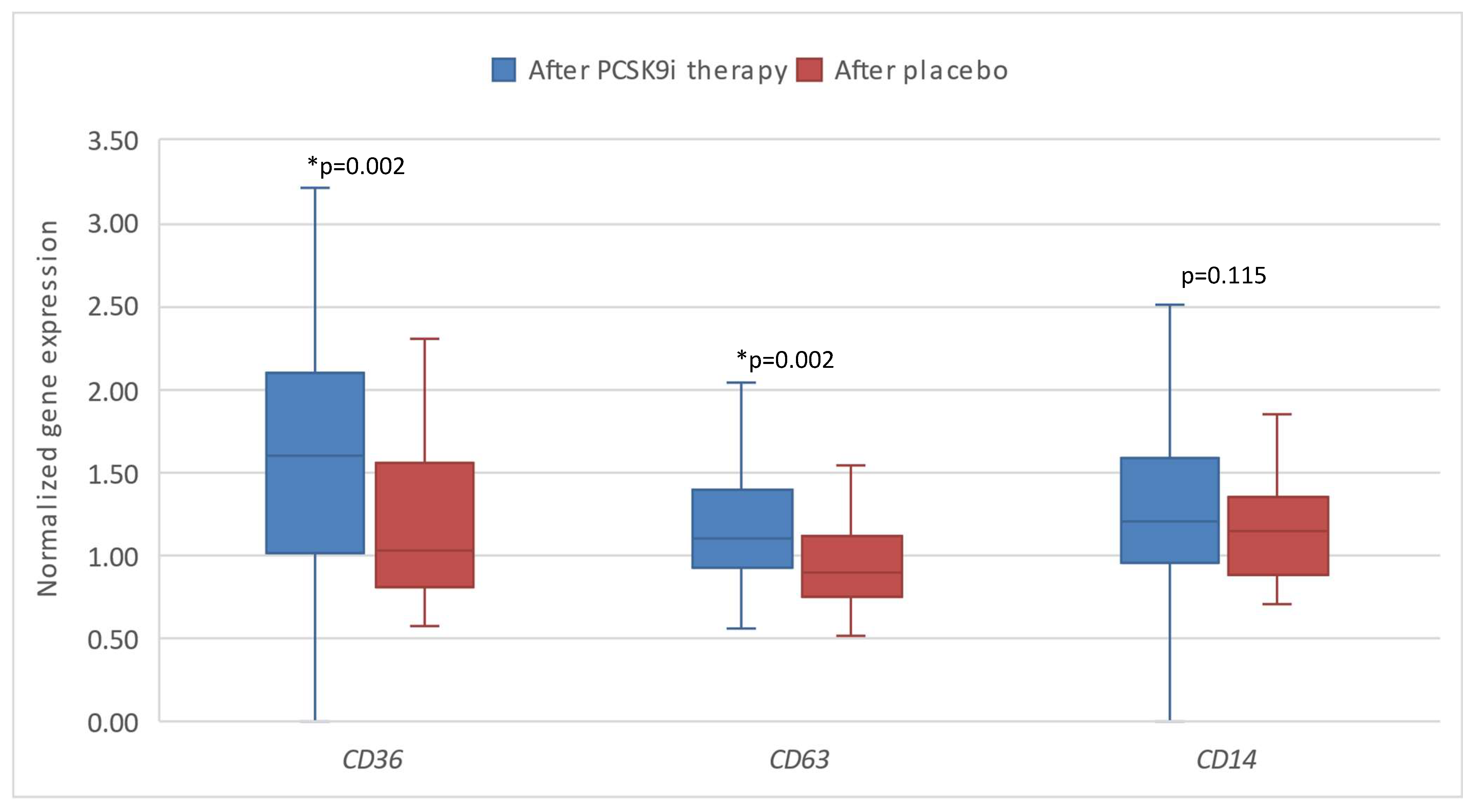

Table 1.
Clinical and laboratory parameters in patients and healthy controls.
| Patients (n=96) |
Controls (n=25) |
p Value | |
|---|---|---|---|
| Gender (M/F) | 91/5 | 23/2 | 0.641 |
| Age (years) | 50.46 ± 8.74 | 48.92 ± 7.14 | 0.076 |
| BMI (kg/m2) | 28.57 ± 3.79 | 25.40 ± 3.32 | *0.045 |
| Total cholesterol [mmol/L] | 4.25 ± 0.88 | 5.80 ± 0.67 | *0.034 |
| LDL-C [mmol/L] | 2.33 ± 0.77 | 3.56 ± 0.63 | *0.023 |
| HDL-C [mmol/L] | 1.17 ± 0.27 | 1.52 ± 0.42 | *0.034 |
| TG [mmol/L] | 1.47 (1.04-2.10) | 1.31 (0.99 – 2.01) | 0.096 |
| Lp(a) [mg/L] | 1431.00 (1203.00-1658.00) | 11.00 (4.00 – 18.50) | **<0.001 |
BMI – body mass index. M/F – male/female. LDL-C – low density lipoprotein cholesterol. HDL-C – high density lipoprotein cholesterol. TG – triglycerides. Lp(a) – lipoprotein(a). Differences between the patients and controls were observed as indicated (**p<0.001, *p<0.05).
Table 2.
The results of gene expression of in patients and controls.
| Patients (n=96) |
Controls (n=25) |
p Value |
|
|---|---|---|---|
| SREBP1 | 0.82 (0.60-1.11) | 0.80 (0.49-1.72) | 0.976 |
| SREBP2 | 0.77 (0.63-0.89) | 2.33 (1.90-2.87) | **<0.001 |
| LDLR | 1.18 (0.89-2.24) | 1.32 (1.04-2.33) | **<0.001 |
| LIPC | 0.35 (0.14-2.60) | 0.08 (0.06-0.11) | **<0.001 |
| LRP8 | 1.56 (0.94-2.73) | 0.75 (0.52 – 1.20) | **<0.001 |
| CD36 | 0.86 (0.72-1.02) | 1.69 (1.34 – 2.14) | **<0.001 |
| CD63 | 0.87 (0.76-1.02) | 1.23 (1.03 – 1.48) | **<0.001 |
| CD14 | 1.07 (0.85-1.25) | 1.31 (1.13 – 1.74) | **<0.001 |
SREBP – gene encoding sterol regulatory element-binding protein. LDLR – gene encoding LDL receptor. LIPC – gene encoding hepatic lipase type C. LRP8 – gene encoding LDLR-related protein 8. CD36 – gene encoding cluster of differentiation 36. CD63 – gene encoding cluster of differentiation 63. CD14 – gene encoding cluster of differentiation 14. .
Table 3.
The correlations between changes in lipid profile and the expression of the investigated genes.
Table 3.
The correlations between changes in lipid profile and the expression of the investigated genes.
| ΔSREBP1 | ΔSREBP2 | ΔLDLR | ΔLIPC | ΔLRP8 | ΔCD36 | ΔCD63 | ΔCD14 | |
|---|---|---|---|---|---|---|---|---|
| ΔTC | ρ=0.252 p=0.045* |
ρ=-0.077 p=0.544 |
ρ=-0.023 p=0.859 |
ρ=0.163 p=0.198 |
ρ=-0.062 p=0.641 |
ρ=0.072 p=0.576 |
ρ=0.218 p=0.086 |
ρ=-0.065 p=0.610 |
| ΔLDL-C | ρ=0.082 p=0.521 |
ρ=-0.031 p=0.806 |
ρ=-0.136 p=0.282 |
ρ=0.091 p=0.477 |
ρ=-0.015 p=0.910 |
ρ=0.144 p=0.263 |
ρ=0.230 p=0.069 |
ρ=0.050 p=0.694 |
| ΔHDL-C | ρ=-0.094 p=0.461 |
ρ=-0.075 p=0.554 |
ρ=-0.043 p=0.734 |
ρ=-0.172 p=0.173 |
ρ=-0.123 p=0.354 |
ρ=-0.055 p=0.672 |
ρ=-0.294 p=0.019* |
ρ=-0.231 p=0.066 |
| ΔTG | ρ=0.108 p=0.397 |
ρ=-0.043 p=0.735 |
ρ=0.089 p=0.483 |
ρ=-0.044 p=0.728 |
ρ=-0.156 p=0.237 |
ρ=-0.081 p=0.531 |
ρ=0.179 p=0.160 |
ρ=-0.021 p=0.871 |
| ΔLp(a) | ρ=-0.013 p=0.921 |
ρ=-0.096 p=0.457 |
ρ=-0.002 p=0.985 |
ρ=0.098 p=0.450 |
ρ=0.050 p=0.715 |
ρ=0.105 p=0.427 |
ρ=0.187 p=0.149 |
ρ=0.053 p=0.684 |
Values with symbol Δ represent the differences between values at the beginning of the study and after 6 months of PCSK9i treatment. The Δ values were calculated using the following equation: ((value after 6 months-value before treatment)/value before treatment) *100%. Values are expressed as correlation coefficient (ρ) and p value (*p<0.05). TC – total cholesterol. LDL-C – low density lipoprotein cholesterol. HDL-C – high density lipoprotein cholesterol. TG – triglycerides. SREBP1/2 –sterol regulatory element-binding protein 1/2 gene. LDLR – LDL receptor gene. LIPC – hepatic lipase type C gene. LRP8 – LDLR-related protein 8 gene. CD36 – cluster of differentiation 36 gene. CD63 – cluster of differentiation 63 gene. CD14 – cluster of differentiation 14 gene.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



