
Submitted:
29 December 2024
Posted:
30 December 2024
You are already at the latest version
Abstract
Advancements in omics technologies have promoted the development of precision oncology. Lineage plasticity, a hallmark of cancer, incorporates molecular and histological aspects. Histological differentiation of adenocarcinoma, neuroendocrine, and squamous characteristics occurs in different anatomic locations. Lung cancer, which is highly heterogeneous, encompasses these differentiations, and therefore serves as a model for exploration. Data-driven understanding is critical in cancer differentiation research, with the two major differentiation pathways, squamous and neuroendocrine, supported by omics data. Here, genetic and non-genetic profiles are reviewed based on patient datasets, and shareable molecular features are described. This paper mainly discusses machine learning approaches to feature selection, where network modeling is effective for designing programmable differentiation. All methods are presented within the context of cancer lineage plasticity along with examples and hypotheses. It emphasizes that selected patient datasets combined with methods will ultimately lead to actionable cancer lineage. Chances for clinical translation are in the spotlight, including biomarkers, molecular subtypes, and targeted therapies.
Keywords:
neuroendocrine
; squamous
; lung
; machine learning
; overview
Introduction & Background
Conventional cancer therapies target organs or tissues. However, the advent of precision oncology has revolutionized treatment strategies through molecular targeting.[1] In theory, cancer cells deceive normal cells through similar molecules, or manipulate normal cells via specific molecules, i.e., any gene can potentially drive cancer. [2] Trans-differentiation pathways, including neuroendocrine and squamous, are supported by omics data and extend beyond the cellular level. [3] The concept of lineage plasticity, which merges molecular and histologic aspects, can guide drug discovery. [4] Nonetheless, targeting oncogenic lineage-restricted transcription factors (TFs) indiscriminately may lead to severe toxicity and failure to achieve desired tissue specificity. [5]
The accumulation of big data in cancer research has enabled the direct identification of lineage-restricted molecules. Omics studies aim to identify molecular subtypes with biological significance, revealing, for instance, distinct clusters of squamous carcinomas across different tissues. The TCGA consortium's integration of 33 cancer types has facilitated cross-tissue analyses, exploring anatomical systems and original lineages such as the pan-squamous phenotype. [6] Previous studies have strongly demonstrated the molecular and cellular aspects of the pan-squamous phenotype. [7]
Unlike the well-documented pan-squamous phenotype, the neuroendocrine phenotype remains relatively rare. Mechanisms underlying neuroendocrine phenotype lack consensus evidence and are often described as systemic disorders. Current diagnosis of neuroendocrine neoplasms (NENs) relies heavily on neuroendocrine markers, predominantly hormones and bioactive peptides (e.g., CHGA and SYP). Due to the occurrence of neuroendocrine phenotype in diverse anatomical locations, there remains an unmet need for reliable biomarkers. Progress in this field hinges on developing biomarkers through integrative approaches combining knowledge and omics analyses. [8,9]
The patient-centric model presents tailored opportunities to address rare diseases, a particularly pressing challenge given the large population of China. Data from cell lines or animal models may not always translate directly to human contexts, highlighting the importance of focusing on patient-derived data and utilizing machine learning with illustrative examples to generate initial hypotheses. The structure of this paper is outlined in Figure 1. It is anticipated that data providing access to key features will aid in the development of targeted therapies, and this mini-review aims to discuss the methodologies and their application to relevant diseases.
Review
Omics insights for neuroendocrine and squamous phenotypes
Currently, the characterization of the cancer genome, particularly with regard to coding gene drivers, is well-established. [10,11] Two key frameworks—evolutionary shaping and tissue specificity—have been highlighted in genomic studies. [5,10] This review focuses on shared characteristics between neuroendocrine and squamous phenotypes. First, somatic mutations and copy number variations were analyzed in parallel. Chromosome 3q amplification (such as TP63 and SOX2) was a significant feature of the squamous lineage, and cell cycle dysregulation including TP53 and CDKN2A mutations was also prevalent. [7] Compared to the squamous lineage, NENs are less well understood. [8,9] Recently, NENs have been divided into neuroendocrine tumors (NETs) and neuroendocrine carcinomas (NECs) based on the proliferative index, with point mutations showing tissue-specificity in NETs. [5] MEN1 is the most significantly mutated gene in NETs, while TP53 and RB1 are predominant drivers in NECs. [12,13,14,15] These results indicate that genetic events can drive phenotypic changes and influence tumor evolution. [10] Meanwhile, the experimental literature supported these mentioned driver mutations, which will not be expanded in detail here. Beyond that, germline mutations are noteworthy in NETs. And rare variants are expected to be captured in large datasets such as the AACR-GENIE, MSK series, Chinese-OrigiMed, and UK 100,000 genomes projects. Analyzing genomic data retrospectively in combination with NCG annotation can fully unlock the potential of gene therapeutics. [11]
The transcriptome and proteome are commonly applied for quantitative studies from a non-genetic perspective. To reduce off-target effects, widely expressed targets in tumors are preferred in proteome studies, focusing on essential genes rather than lineage specificity. [16,17,18] Cross-ethnic studies help obtain conservative results, with substantial data accumulated on lung adenocarcinoma. [19] While proteome studies offer the advantage of targetability, detecting low-abundance proteins (e.g., cell surface proteins) remains challenging. These proteins serve as markers for cellular differentiation and lineage. In contrast, the transcriptome does reveal convergent pathways. Neuroendocrine lineage based solely on transcriptome was not favored due to genetic discrepancies (e.g., RB1 mutations). However, recent data collection has allowed the molecular depiction of the pan-neuroendocrine landscape. [20,21] Meanwhile, convergent biologies between neuroendocrine and squamous phenotypes are also recognized by epigenetic inheritance. [22,23] Overall, non-genetic components reveal more shareable characteristics compared to genomics.
Despite the existence of convergent pathways against neuroendocrine and squamous phenotypes, there is room for improvement—one direction in neuroendocrine studies, such as small-cell cytology. An interesting finding is that small-cell carcinoma of the bladder is more similar to bladder carcinoma than to small-cell lung cancer, challenging the neuroendocrine phenotype concept. [24] Beyond neuroendocrine markers, the original lineage is the driver of neuroendocrine phenotype. Opinions on transcriptome suggest that neuroendocrine cells originate from the central and peripheral nervous systems, with shared transcription circuits (e.g., ASCL1 and NEUROD1 crosstalk) supporting this view. [8,25] In this regard, high-throughput screenings assist in the search for molecules, driven by data.
A bold hypothesis is that neuroendocrine and squamous molecules may regulate each other, possibly due to their mutually exclusive expression profiles. For example, smokers often exhibit neuroendocrine or squamous features in lung cancer. Recent studies also suggest that in advanced metastatic cancers of the bladder and colon, the proportion of neuroendocrine-like subtypes may be increased. [26,27] Another important aspect is that algorithms analyzing the data suggest that localized lineage molecules could represent the broader molecular profile. Given these complexities, discussing neuroendocrine and squamous lineages in isolation, based solely on transcriptomic data, may not be advisable.
Cancer Lineage Plasticity Guided by Machine Learning
Semi-supervised and unsupervised clustering methods
Most statistical methods assume linear fitting, and comparative studies screen to identify conservative features. [28,29] In this field, semi-supervised approaches have been extensively utilized in prognostic markers. The data format can be continuous or binary variables, and the patient's prognosis including follow-up time with survival events is also required. Generally used analyses include Cox regression and the shrinkage method, the latter of which minimize unimportant variables from high-dimensional data. Interestingly, lineage-restricted molecules are often accompanied by prognostic significance (e.g., NKX2-1). Apart from this, correlation analyses compare known lineage-restricted molecules with unknown ones, common in feature selection. [5]
Compared with semi-supervised methods, unsupervised clustering is less affected by feature selection. [30] Unsupervised clustering aims to uncover biological signals through dimensionality reduction, without requiring prior knowledge. Importantly, clustering is a population-based design that divides whole data into several groups, which can be complemented with studies of global correlation. How to combine omics and machine learning methods has been described in detail elsewhere, and will not be discussed here. Multi-omics may retain strong signals better than individual omics, as opposed to individual omics, which is suitable for external validation. Given the higher weight of underlying biological pathways, transcriptome data was often preferred in multi-omics analysis due to its high variability. [31] However, using TCGA consortium data alone underestimates protein levels, addressed by the CPTAC consortium. [17]
Subtype classifiers: training and external validation
Subclass classifiers are a method for solving classification issues that usually require given sample labels. The principle of "subtype classifiers" is training molecules concerning sample labels, and then using them for external validation. This methodology reduces the number of variables, and the assignment probability of individual samples is determined by a small number of feature comparisons. [32] Importantly, differences in technology must be considered (e.g., RNA sequencing vs. Microarrays). In cancer, molecular classifiers are mainly tissue-based. [33] Moreover, a new study suggests that original lineage is also practical by trained classifiers. [34] In classification tasks, DNA methylation arrays are more sensitive than RNA sequencing but struggle to detect detailed cluster information. [35] The available evidence suggests that both DNA methylation and transcriptome are feasible for classification, with transcriptome performing well in clustering scenarios.
Computational biology: focus on quantitative & qualitative
Compared to the other two approaches, computational biology can use high-throughput data combined with cellular experimental data. Key challenges in cancer include that lineage-restricted molecules might be undruggable and play essential roles in normal tissues. [36] Thus, constructing regulatory networks or targeting operative molecules is warranted. Network modeling to design gene circuits is gaining attention, with cell line data supporting signal transduction studies. Previous studies proved that programmable gene editing led to direct differentiation. [37] In lung cancer, genetic circuits can be engineered to block triple differentiation (adenocarcinoma-neuroendocrine-squamous). Candidates perhaps are the KRAB zinc finger protein family, which may persistently inhibit the differentiation program due to the fact that this family often exhibits transcriptional repression. [38] Furthermore, there may be some families or molecules that regulate differentiation in future studies.
Genetic circuits are designed as pre-simulated molecular networks that are later validated by specific molecules. The most classical approach is to combine network simulations and high-throughput screening to identify factors of direct differentiation. In large-scale level studies, it is preferable to study the entire gene family rather than a single member, emphasizing family characteristics. If this factor is untargetable, then the alternative strategy suggests to change to its collaborators. This review outlines the blocking of multiple differentiation through transcriptional repression and the establishment of regulatory networks for each lineage. As with all, computational biology is at the root of genetic circuits.
Computational biology embraced network modeling, which required a combination of quantitative and qualitative approaches, usually in the context of cell lines. Quantitative research found global transcription rates can be inferred using the network modeling method named GENIE3. [39] It can rapidly calculate linkages between genes, and tools like it include ARACNe, CellNet and Mogrify. Compared to quantitative approaches, Boolean modeling is a classic tool in qualitative studies. [40,41] Boolean models require existing phenotypes, such as taking ΔNp63 (Truncated isoform of TP63) as an input and squamous markers as an output. The relationship between genes is characterized as binary variables (activation or inhibition) based on experimental data, with middle regulations being molecules of interest. These can be kinases and chromatin regulatory factors, and there are already clinical trials underway. [3,4,16]
Clinical Applications and Future Directions
Lung cancer - a molecular subtype model
Lineage-restricted TFs were widely recognized diagnostic markers used in immunohistochemistry to distinguish histology. In contrast, molecular subtypes are emerging as an important focus. While molecular subtypes can be validated using clinical samples, translating these findings into routine clinical practice remains challenging. Each subtype may have a biological basis, but conserved signals across subtypes hold greater value. [2,15] The ideal subtype derived from a single tissue type should be mappable to anatomical systems or original lineages, meaning it can be applied in both horizontal and longitudinal comparative studies. For instance, overexpressed molecules associated with the three histological types of lung cancer can be replicated in lung adenocarcinoma (~10% Jaccard index, using GSE94601 as reference data; Supplementary Table S1). Similar findings were observed in Lund's advanced bladder cohort. [42] These results suggest that convergent pathways can be identified across different organs, potentially serving as candidate metagenes (Supplementary Table S2). It could be argued that the molecular drivers behind subtypes are more important than the subtypes themselves.
Modified subtypes in lung adenocarcinoma and lung squamous carcinoma can accomplish the above promises. This hypothesis is that lung adenocarcinoma is a subset of lung cancer, and lung squamous carcinoma is the pan-squamous miniature.
Assuming that genes with a high mRNA-protein correlation were taken as an assessment criterion for being activated, RNA processing and extracellular matrix pathways differed between subtypes in lung squamous carcinoma. [43] This observation aligns with findings in pan-squamous Chinese proteomic analysis. [44] Given the large differences in numbers between subtypes (LSQ1: 1090 vs LSQ2: 299, based on mRNA-protein correlation; |Δr | > 0.3), this may be caused by post-transcriptional regulation. Regarding post-transcriptional regulation, the coefficient of variation of five molecules associated with RNA processing, RBM10, SF1, CPSF6, SLTM and DDX5, were lower than those of KRT5. It is suggested that they may be less prone to off-target effects. [16] If this assumption is plausible, then extracellular matrix pathways should play an activated role in the cancer epithelium. Further, the understanding of epithelial mechanotransduction can be aided by high-dimensional analysis at spatial resolution. [45]
Cell-of-origin pairing for cancers of unknown origin and rare diseases
This section named "Cell-of-origin pairing for cancers of unknown origin and rare diseases" has some conceptual overlap with the "subtype classifiers" described above, as both involve scenario simulations of classification issues. Clinical translation of cell-of-origin pairing is appropriate in cancers of unknown origin and rare diseases. Consider platforms for sequencing, DNA methylation and transcriptome analyses performed well for categorization in both primary and unknown origin cancers. [28,46] A lack of approved therapies is a common problem in this field, including cancers of unknown origin and rare diseases. For this, tissue-agnostic therapies, such as genomics-guided and lineage-based therapies, are suggested in emerging viewpoints. [1] Lineage-based therapies allow for simultaneous consideration of diagnosis and treatment, offering broad applications. For some diseases, ethnicity differences are determinative; for instance, esophageal squamous carcinoma is highly prevalent in China, while esophageal adenocarcinoma is more prevalent in Caucasians. [9] This is why exploring the differences between adenocarcinoma and squamous carcinoma makes sense.
Lineage-based concepts also guide drug discovery in rare diseases. One hopeful chance is that olfactory neuroblastoma mimics small-cell lung cancer. Clustering analysis also appreciates the rationality of this measure because of convergent signaling. For rare diseases, the greatest problem is the lack of appropriate control samples, and outlier analysis may bring in targets although the efficacy needs to be improved. Theoretically, population-based N-of-1 trials can yield fast-track molecular insights. To support these, the Treehouse group proposes outlier analysis in pediatric cancer based on N-of-1 design. Firstly, the background data should be established, and individual patients were then compared against it to identify dysregulated molecules. [47] As an example, if a new patient has squamous cytology and has completed mass spectrometry, the aberrant targets can be recognized using the Chinese pan-squamous proteome. [44]
Targeted therapy design
Lineage-based targeted therapies have currently reached the preclinical stage, with early interception and refractory metastasis being prioritized. [3,4] Carcinoma in situ was a pre-invasive state that could further progress to invasive cancer, and early intervention may prevent cancer progression. Pre-invasive genetic drivers are few, but once formed, such conditions are often "irreversible." The key challenge is that directly targeting TFs may lead to de novo trans-differentiation, such as NKX2-1 in lung adenocarcinoma and ΔNp63 in lung squamous carcinoma. For trans-differentiation studies mainly including Pre- and post-transformation paired and direct collection of post-transformation samples, the former is temporarily limited by the lack of sufficient sample sizes. Refractory tumors collected directly, such as lung adenosquamous cell carcinoma, require pathological evidence, which has been well-studied by Ji's laboratory. [48] Early intervention is particularly emphasized, and lineage-restricted molecules serve as diagnostic markers. Furthermore, the therapeutic window needs comprehensive assessment, considering factors like the interaction of environment and genes. [49] Because disease regression took a long time, the results may not be sufficient for replication. Importantly,
squamous differentiation should intervention could be inferred from GSE108082 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE108082) and remained a lineage feature of squamous cell carcinoma. [50] It is desirable to obtain molecules that arrest the squamous differentiation and influence disease regression. In addition, identifying highly selective targets by gene circuits, such as Boolean modeling, is suggested. The choice of targeting design is crucial, and in addition, it should prioritize highly heterogeneous or transcription-driven cancers, such as some pediatric cancers.
Lung cancer is highly heterogeneous, consisting mainly of adenocarcinoma, squamous and neuroendocrine lineage, and small cell lung cancer being the most aggressive and representative type of neuroendocrine carcinoma. Lineage-based targeted therapies are particularly indicated for highly heterogeneous carcinoma in the precancerous stage. Precancerous patients benefit from early detection and intervention, which can boost survival or lower morbidity. In this regard, three therapeutic options have been proposed: 1) prevention of high-risk precancerous populations by block differentiation, available for instance in the KRAB zinc finger family; 2) direct targeting of TFs in the pre-invasive or refractory state by library screening, such as chemical probes and loss or gain of function; 3) programmable circuit design for highly selective targeting from downstream, upstream or co-factors. This is a universal design of lineage-restricted TFs, straightforward targeting. Exceptions include nuclear signaling receptors like AR, ER, MYC, etc. [37,51,52] For these, three possible options for targeting were summarised in Figure 2.
In all, the above analyses require identifying molecules with prognostic significance, incorporating convergent subtypes, etc., all of which require machine learning as a foundation. Data-driven initial understanding of molecular properties, such as whether they are associated with the cancer lineage, and from genes to diseases is the call of precision medicine.
Precision medicine vision
As interest and efforts in precision oncology escalate, recognizing the importance of biomarkers and their use in developing targeted therapies in clinical research is indispensable. Significant methodological advancements in genomics-guided clinical trial designs, such as basket and umbrella trials within the master protocol framework, have been made. However, umbrella trials like NCT02154490 and NCT03292250 have shown unsatisfactory results. [53] In contrast to genomics-guided clinical trial design, lineage-based concepts hold promise for enhancing outcomes for shared targets, with DLL3 as a successful example. Data-driven analysis not only enhances drug discovery for targeted therapies but also holds equal importance for immunotherapy and traditional treatment modalities. Lung cancer, being the most prevalent and fatal malignancy, already possesses a considerable amount of data that could be leveraged for clinical translation. [54]
To enhance the operational effectiveness of cancer lineage plasticity, patient-centric datasets, and illustrative examples have been curated. The utilization of freely available public resources, where academic advancements surpass financial incentives, remains crucial. Transcriptome has become routine in disease studies, with TFs likely playing a key role in driving cellular fate. In this context, the development of dedicated TFs platform (e.g., TFome™) may prove beneficial. Certain TFs exhibit cancer-specific expression patterns, which can be analyzed through regulated networks. [55] Up to now, clinical translation has successfully incorporated RNA-related products (e.g., Oncotype DX® and CancerTYPE ID®) primarily for prognostic stratification and origin classification. Looking ahead, liquid biopsies show promise in determining tumor origins compared to traditional tissue specimens. Additionally, integrating TF data with DNA methylation profiles represents a promising frontier. High-resolution data continues to enrich our understanding of life sciences, yet it's important to acknowledge limitations such as oversimplified assumptions, binary classifications, and the exclusive focus on TFs and transcriptomics.
Conclusions
Population-based data analysis enables the identification of cancer lineage factors, emphasizing the importance of sharing over differentiation. In genetics, the notable feature of the pan-squamous phenotype is chromosome 3q amplification. While mutations MEN1 and TP53- RB1 are predominantly enriched in neuroendocrine tumors and neuroendocrine carcinomas, respectively. The non-genetic part of the collection mainly consists of the transcriptome, proteome, and epigenome all associated with cancer lineage. For precision therapy, one possible approach is to design gene circuits to reduce tissue toxicity and induce direct differentiation. Furthermore, original lineage therapy should focus on early interception and provide insights into pre-invasive, refractory, rare, and unknown primary cancers. By the way, data collection involves assumptions beneath it; for example, follow-up data in cancer are often predicated on the use of radiochemotherapies, and aiming to develop novel therapies demands prospective validation.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.Org.
Author Contributions
Longjin Zeng wrote, conceptualized, and interpreted the manuscript.
Data Availability Statement
Data sharing is not applicable as no new data was generated.
Acknowledgments
Only articles with usable data or illustrations are cited; I sincerely apologize for not being able to discuss all authors and their respective studies. In addition, thanks to neuronal perspectives from Julien Sage, and Vân Anh Huynh-Thu, co-author of gene regulatory network tools. I am also deeply grateful for the support of familiar colleagues, and the inspiration drawn from Lund University. Most importantly, thanks to the data contributors.
Conflicts of Interest
No conflict of interest was declared.
References
- Subbiah V, Gouda MA, Ryll B, Burris HA, 3rd, et al. The evolving landscape of tissue-agnostic therapies in precision oncology. CA: a cancer journal for clinicians, 2024. [CrossRef]
- de Magalhães, JP. Every gene can (and possibly will) be associated with cancer. Trends in genetics 2022, 38, 216–217. [Google Scholar] [CrossRef]
- Davies, A.; Zoubeidi, A.; Beltran, H.; Selth, L.A. The Transcriptional and Epigenetic Landscape of Cancer Cell Lineage Plasticity. Cancer Discov. 2023, 13, 1771–1788. [Google Scholar] [CrossRef]
- Fujii, M.; Sekine, S.; Sato, T. Decoding the basis of histological variation in human cancer. Nat. Rev. Cancer 2023, 24, 141–158. [Google Scholar] [CrossRef] [PubMed]
- Haigis, K.M.; Cichowski, K.; Elledge, S.J. Tissue-specificity in cancer: The rule, not the exception. Science 2019, 363, 1150–1151. [Google Scholar] [CrossRef]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304.e296. [Google Scholar] [CrossRef]
- Guan, Y.; Wang, G.; Fails, D.; Nagarajan, P.; Ge, Y. Unraveling cancer lineage drivers in squamous cell carcinomas. Pharmacol. Ther. 2020, 206, 107448–107448. [Google Scholar] [CrossRef] [PubMed]
- Rindi, G.; Inzani, F. Neuroendocrine neoplasm update: toward universal nomenclature. Endocrine-Related Cancer 2020, 27, R211–R218. [Google Scholar] [CrossRef]
- Xue, J.; Lyu, Q. Challenges and opportunities in rare cancer research in China. Sci. China Life Sci. 2024, 67, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Martincorena I, Raine KM, Gerstung M, Dawson KJ, et al. Universal Patterns of Selection in Cancer and Somatic Tissues. Cell 2017, 171, 1029–1041.e21. [Google Scholar] [CrossRef] [PubMed]
- Dressler, L.; Bortolomeazzi, M.; Keddar, M.R.; Misetic, H.; Sartini, G.; Acha-Sagredo, A.; Montorsi, L.; Wijewardhane, N.; Repana, D.; Nulsen, J.; et al. Comparative assessment of genes driving cancer and somatic evolution in non-cancer tissues: an update of the Network of Cancer Genes (NCG) resource. Genome Biol. 2022, 23, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Zhou, W.; Li, L.; Wang, J.; Gao, Z.; Jiang, Y.; Jiang, X.; Shan, A.; Bailey, M.H.; Huang, K.-L.; et al. Pan-cancer analysis of somatic mutations across 21 neuroendocrine tumor types. Cell Res. 2018, 28, 601–604. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Yu, Z.; Liu, Y.; Guo, L.; Teng, L.; Guo, L.; Liang, L.; Wang, J.; Gao, J.; Li, R.; et al. Genomic characterization reveals distinct mutation landscapes and therapeutic implications in neuroendocrine carcinomas of the gastrointestinal tract. Cancer Commun. 2022, 42, 1367–1386. [Google Scholar] [CrossRef]
- van Riet, J.; van de Werken, H.J.G.; Cuppen, E.; Eskens, F.A.L.M.; Tesselaar, M.; van Veenendaal, L.M.; Klümpen, H.-J.; Dercksen, M.W.; Valk, G.D.; Lolkema, M.P.; et al. The genomic landscape of 85 advanced neuroendocrine neoplasms reveals subtype-heterogeneity and potential therapeutic targets. Nat. Commun. 2021, 12, 4612. [Google Scholar] [CrossRef] [PubMed]
- Yachida, S.; Totoki, Y.; Noë, M.; Nakatani, Y.; Horie, M.; Kawasaki, K.; Nakamura, H.; Saito-Adachi, M.; Suzuki, M.; Takai, E.; et al. Comprehensive Genomic Profiling of Neuroendocrine Carcinomas of the Gastrointestinal System. Cancer Discov. 2022, 12, 692–711. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Ruiz, P.; Ito, T.; Sellers, W.R. Targeting pan-essential genes in cancer: Challenges and opportunities. Cancer Cell 2021, 39, 466–479. [Google Scholar] [CrossRef]
- Savage SR, Yi X, Lei JT, Wen B, et al. Pan-cancer proteogenomics expands the landscape of therapeutic targets. Cell 2024, 187, 4389–4407.e15. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lih, T.M.; Pan, J.; Höti, N.; Dong, M.; Cao, L.; Hu, Y.; Cho, K.-C.; Chen, S.-Y.; Eguez, R.V.; et al. Proteomic signatures of 16 major types of human cancer reveal universal and cancer-type-specific proteins for the identification of potential therapeutic targets. J. Hematol. Oncol. 2020, 13, 1–15. [Google Scholar] [CrossRef]
- Vavilis, T.; Petre, M.L.; Vatsellas, G.; Ainatzoglou, A.; Stamoula, E.; Sachinidis, A.; Lamprinou, M.; Dardalas, I.; Vamvakaris, I.N.; Gkiozos, I.; et al. Lung Cancer Proteogenomics: Shaping the Future of Clinical Investigation. Cancers 2024, 16, 1236. [Google Scholar] [CrossRef] [PubMed]
- Chen F, Zhang Y, Gibbons DL, Deneen B, et al. Pan-Cancer Molecular Classes Transcending Tumor Lineage Across 32 Cancer Types, Multiple Data Platforms, and over 10,000 Cases. Clin Cancer Res 2018, 24, 2182–2193. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, C.; Zheng, S.; Yao, Y.; Wang, S.; Wang, X.; Yin, E.; Zeng, Q.; Zhang, C.; Zhang, G.; et al. Molecular subtypes of neuroendocrine carcinomas: A cross-tissue classification framework based on five transcriptional regulators. Cancer Cell 2024, 42, 1106–1125.e8. [Google Scholar] [CrossRef]
- Cejas, P.; Xie, Y.; Font-Tello, A.; Lim, K.; Syamala, S.; Qiu, X.; Tewari, A.K.; Shah, N.; Nguyen, H.M.; Patel, R.A.; et al. Subtype heterogeneity and epigenetic convergence in neuroendocrine prostate cancer. Nat. Commun. 2021, 12, 5775. [Google Scholar] [CrossRef]
- Terekhanova, N.V.; Karpova, A.; Liang, W.-W.; Strzalkowski, A.; Chen, S.; Li, Y.; Southard-Smith, A.N.; Iglesia, M.D.; Wendl, M.C.; Jayasinghe, R.G.; et al. Epigenetic regulation during cancer transitions across 11 tumour types. Nature 2023, 623, 432–441. [Google Scholar] [CrossRef]
- Chang MT, Penson A, Desai NB, Socci ND, et al. Small-Cell Carcinomas of the Bladder and Lung Are Characterized by a Convergent but Distinct Pathogenesis. Clin Cancer Res 2018, 24, 1965–1973. [Google Scholar] [CrossRef] [PubMed]
- Tsunemoto, R.; Lee, S.; Szűcs, A.; Chubukov, P.; Sokolova, I.; Blanchard, J.W.; Eade, K.T.; Bruggemann, J.; Wu, C.; Torkamani, A.; et al. Diverse reprogramming codes for neuronal identity. Nature 2018, 557, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Loriot, Y.; Kamal, M.; Syx, L.; Nicolle, R.; Dupain, C.; Menssouri, N.; Duquesne, I.; Lavaud, P.; Nicotra, C.; Ngocamus, M.; et al. The genomic and transcriptomic landscape of metastastic urothelial cancer. Nat. Commun. 2024, 15, 8603. [Google Scholar] [CrossRef] [PubMed]
- Moorman, A.R.; Benitez, E.K.; Cambuli, F.; Jiang, Q.; Mahmoud, A.; Lumish, M.; Hartner, S.; Balkaran, S.; Bermeo, J.; Asawa, S.; et al. Progressive plasticity during colorectal cancer metastasis. Nature 2024, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Heinze, G.; Wallisch, C.; Dunkler, D. Variable selection – A review and recommendations for the practicing statistician. Biom. J. 2018, 60, 431–449. [Google Scholar] [CrossRef]
- Zhang, J.Z.; Wang, C. A comparative study of clustering methods on gene expression data for lung cancer prognosis. BMC Res. Notes 2023, 16, 319. [Google Scholar] [CrossRef] [PubMed]
- Chalise P, Kwon D, Fridley BL, Mo Q. Statistical Methods for Integrative Clustering of Multi‐omics Data. Methods in molecular biology (Clifton, NJ) 2023, 2629, 73–93.
- Mamatjan Y, Agnihotri S, Goldenberg A, Tonge P, et al. Molecular Signatures for Tumor Classification: An Analysis of The Cancer Genome Atlas Data. The Journal of molecular diagnostics : JMD 2017, 19, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Huang, H.-C.; Qin, L.-X. Making External Validation Valid for Molecular Classifier Development. JCO Precis. Oncol. 2021, 5, 1250–1258. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Chen, J.; Ni, S.; Tan, C.; Xu, M.; Dong, L.; Yuan, L.; Wang, Q.; Du, X. Pan-cancer transcriptome analysis reveals a gene expression signature for the identification of tumor tissue origin. Mod. Pathol. 2016, 29, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Rydzewski, N.R.; Shi, Y.; Li, C.; Chrostek, M.R.; Bakhtiar, H.; Helzer, K.T.; Bootsma, M.L.; Berg, T.J.; Harari, P.M.; Floberg, J.M.; et al. A platform-independent AI tumor lineage and site (ATLAS) classifier. Commun. Biol. 2024, 7, 314. [Google Scholar] [CrossRef] [PubMed]
- Xia D, Leon AJ, Cabanero M, Pugh TJ, et al. Minimalist approaches to cancer tissue-of-origin classification by DNA methylation. Modern pathology 2020, 33, 1874–1888. [Google Scholar] [CrossRef]
- Cruz FD, Matushansky I. Solid tumor differentiation therapy - is it possible? Oncotarget 2012, 3, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.; Cross, R.S.; Jenkins, M.R. Synthetic biology, genetic circuits and machine learning: a new age of cancer therapy. Mol. Oncol. 2023, 17, 946–949. [Google Scholar] [CrossRef] [PubMed]
- Cappelluti, M.A.; Poeta, V.M.; Valsoni, S.; Quarato, P.; Merlin, S.; Merelli, I.; Lombardo, A. Durable and efficient gene silencing in vivo by hit-and-run epigenome editing. Nature 2024, 627, 416–423. [Google Scholar] [CrossRef]
- Zhang, J.; Han, X.; Ma, L.; Xu, S.; Lin, Y. Deciphering a global source of non-genetic heterogeneity in cancer cells. Nucleic Acids Res. 2023, 51, 9019–9038. [Google Scholar] [CrossRef]
- Kim N, Hwang CY, Kim T, Kim H, et al. A Cell-Fate Reprogramming Strategy Reverses Epithelial-to-Mesenchymal Transition of Lung Cancer Cells While Avoiding Hybrid States. Cancer research 2023, 83, 956–970. [Google Scholar] [CrossRef] [PubMed]
- Montagud, A.; Béal, J.; Tobalina, L.; Traynard, P.; Subramanian, V.; Szalai, B.; Alföldi, R.; Puskás, L.; Valencia, A.; Barillot, E.; et al. Patient-specific Boolean models of signalling networks guide personalised treatments. eLife 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Sjödahl, G.; Eriksson, P.; Liedberg, F.; Höglund, M. Molecular classification of urothelial carcinoma: global mRNA classification versus tumour-cell phenotype classification. J. Pathol. 2017, 242, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Arad, G.; Geiger, T. Functional Impact of Protein–RNA Variation in Clinical Cancer Analyses. Mol. Cell. Proteom. 2023, 22, 100587. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Yang, Y.; Jiang, D.; Qin, Z.; Xu, C.; Wang, H.; Huang, J.; Chen, L.; Luo, R.; Zhang, X.; et al. Proteomic analysis reveals key differences between squamous cell carcinomas and adenocarcinomas across multiple tissues. Nat. Commun. 2022, 13, 4167. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ma, J.; Zhang, Q.; Gong, H.; Gao, D.; Wang, Y.; Li, B.; Li, X.; Zheng, H.; Wu, Z.; et al. Spatially resolved proteomic map shows that extracellular matrix regulates epidermal growth. Nat. Commun. 2022, 13, 4012. [Google Scholar] [CrossRef]
- Möhrmann, L.; Werner, M.; Oleś, M.; Mock, A.; Uhrig, S.; Jahn, A.; Kreutzfeldt, S.; Fröhlich, M.; Hutter, B.; Paramasivam, N.; et al. Comprehensive genomic and epigenomic analysis in cancer of unknown primary guides molecularly-informed therapies despite heterogeneity. Nat. Commun. 2022, 13, 4485. [Google Scholar] [CrossRef] [PubMed]
- Vivian, J.; Eizenga, J.M.; Beale, H.C.; Vaske, O.M.; Paten, B. Bayesian Framework for Detecting Gene Expression Outliers in Individual Samples. JCO Clin. Cancer Informatics 2020, 4, 160–170. [Google Scholar] [CrossRef]
- Tang, S.; Xue, Y.; Qin, Z.; Fang, Z.; Sun, Y.; Yuan, C.; Pan, Y.; Zhao, Y.; Tong, X.; Zhang, J.; et al. Counteracting lineage-specific transcription factor network finely tunes lung adeno-to-squamous transdifferentiation through remodeling tumor immune microenvironment. Natl. Sci. Rev. 2023, 10, nwad028. [Google Scholar] [CrossRef]
- E Stanton, S.; E Castle, P.; Finn, O.J.; Sei, S.; A Emens, L. Advances and challenges in cancer immunoprevention and immune interception. J. Immunother. Cancer 2024, 12, e007815. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, V.H.; Pipinikas, C.P.; Pennycuick, A.; Lee-Six, H.; Chandrasekharan, D.; Beane, J.; Morris, T.J.; Karpathakis, A.; Feber, A.; E Breeze, C.; et al. Deciphering the genomic, epigenomic, and transcriptomic landscapes of pre-invasive lung cancer lesions. . 2019. [Google Scholar] [CrossRef]
- Chen, A.; Koehler, A.N. Transcription Factor Inhibition: Lessons Learned and Emerging Targets. Trends Mol. Med. 2020, 26, 508–518. [Google Scholar] [CrossRef]
- Shin, H.Y. Targeting Super-Enhancers for Disease Treatment and Diagnosis. Molecules and cells 2018, 41, 506–514. [Google Scholar] [CrossRef]
- Hayes DN, Oluoha O, Schwartz DL. For Squamous Cancers, the Streetlamps Shine on Occasional Keys, Most Baskets Are Empty, and the Umbrellas Cannot Keep Us Dry: A Call for New Models in Precision Oncology. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2024, 42, 487–490. [Google Scholar] [CrossRef]
- Cai, L.; Xiao, G.; Gerber, D.; Minna, J.D.; Xie, Y. Lung Cancer Computational Biology and Resources. Cold Spring Harb. Perspect. Med. 2021, 12, a038273. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Song, C.; Zhang, G.; Li, Y.; Zhao, Y.; Zhang, Q.; Zhang, Y.; Fan, S.; Zhao, J.; Xie, L.; et al. scGRN: a comprehensive single-cell gene regulatory network platform of human and mouse. Nucleic Acids Res. 2023, 52, D293–D303. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Overview of the "actionable cancer lineage," encompassing sections on omics insights, machine learning, and clinical applications. Cancer data can be used to identify lineage-related molecules as central hubs through feature selection, providing a theoretical foundation for therapeutic strategies and disease understanding. Abbreviation: CUP (cancer of unknown primary).
Figure 1.
Overview of the "actionable cancer lineage," encompassing sections on omics insights, machine learning, and clinical applications. Cancer data can be used to identify lineage-related molecules as central hubs through feature selection, providing a theoretical foundation for therapeutic strategies and disease understanding. Abbreviation: CUP (cancer of unknown primary).
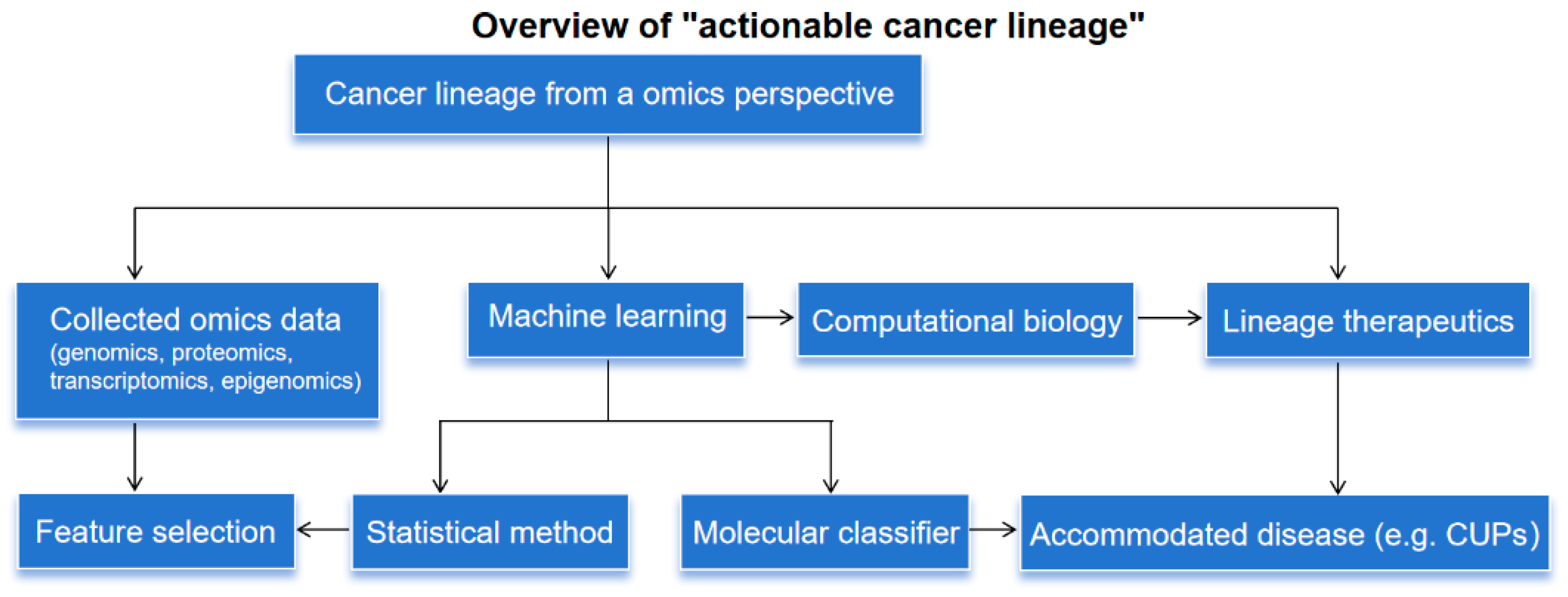
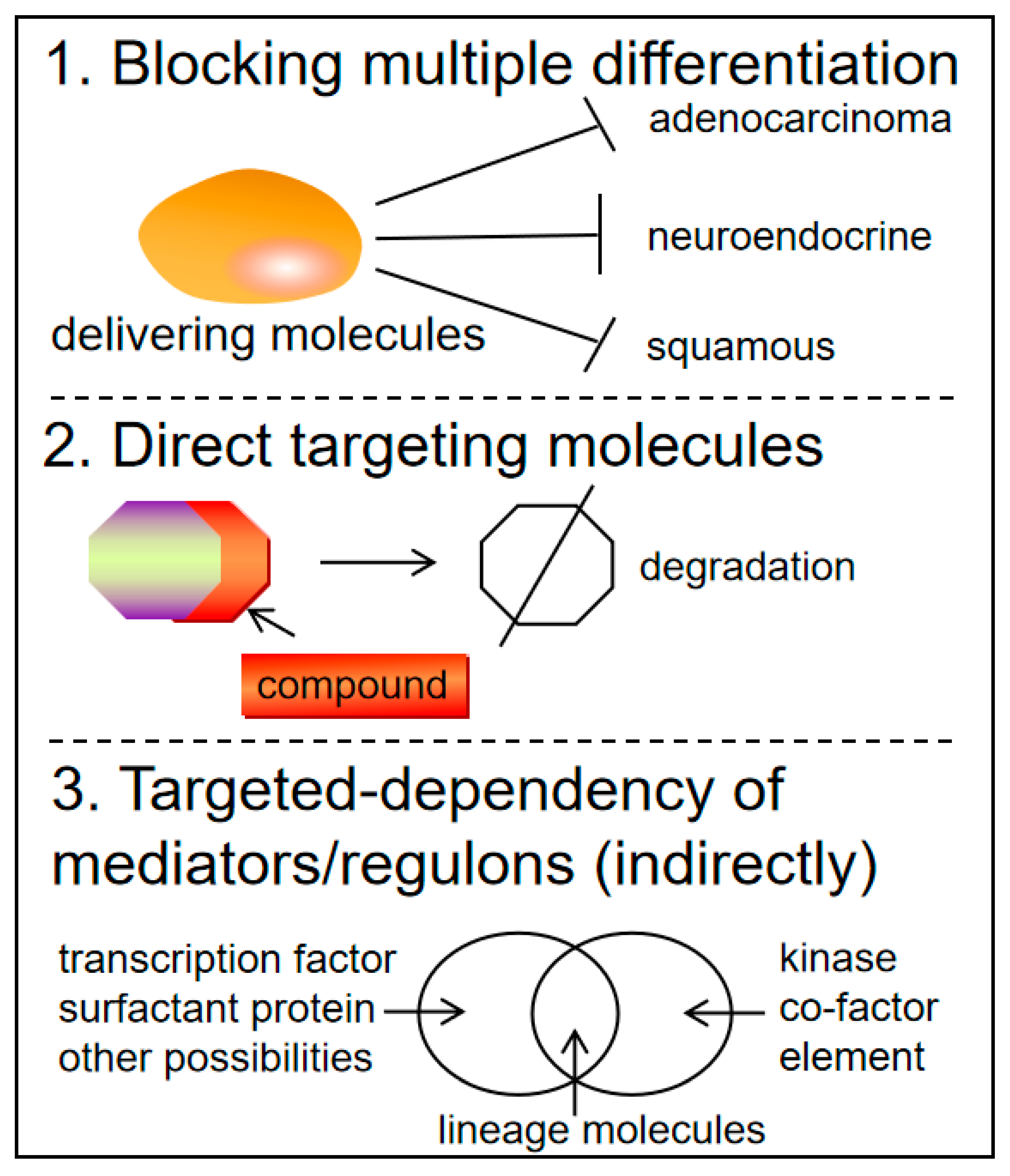
Figure 2.
Exploration of lineage-based therapeutics targeting both multiple and single differentiation pathways. One approach is based on the hypothesis that histologic phenotypes serve as a background. The other focuses on directly targeting compounds within monodifferentiation pathways or on mediators that regulate downstream molecules. For downstream markers, the keratin family may be involved. The key drivers with the greatest potential are transcription factors and cell surface proteins.
Figure 2.
Exploration of lineage-based therapeutics targeting both multiple and single differentiation pathways. One approach is based on the hypothesis that histologic phenotypes serve as a background. The other focuses on directly targeting compounds within monodifferentiation pathways or on mediators that regulate downstream molecules. For downstream markers, the keratin family may be involved. The key drivers with the greatest potential are transcription factors and cell surface proteins.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



