
Submitted:
29 November 2024
Posted:
02 December 2024
You are already at the latest version
Abstract
Background. Antibodies against Ku have been described in patients with various connective tissue diseases. The objective of this study was to describe the clinical, functional and imaging characteristics of interstitial lung disease in patients with anti-Ku antibodies.
Methods. This single-center, retrospective observational study was conducted at a tertiary referral institution. Patients with positive anti-Ku antibodies and interstitial lung disease, identified between 2007 and 2022 were included. Clinical, immunological, functional, and imaging data were systematically reviewed.
Results. Nineteen patients (10 females) with a mean age of 59 ± 12.6 years were included. The most frequent associated diagnosis was systemic sclerosis (42%), followed by rheumatoid arthritis (26%), Sjögren syndrome, undifferentiated connective tissue disease, and overlap between systemic sclerosis and idiopathic inflammatory myopathy (scleromyositis). Imaging revealed frequent septal and intralobular reticulations, and ground-glass opacities, with nonspecific interstitial pneumonia as the predominant pattern (53%). The mean forced vital capacity was 82% ± 26 of the predicted value, and the mean diffusing capacity for carbon monoxide was 55% ± 21. Over the first year of follow-up, the mean annual forced vital capacity decline was 140 mL/year (range: 0–1610 mL/year). The overall survival rate was 82% at 5 years and 67% at 10 years.
Conclusion. Most patients with interstitial lung disease and anti-Ku antibodies presented with autoimmune diseases, particularly systemic sclerosis, and exhibited a CT pattern consistent with nonspecific interstitial pneumonia. These findings underscore the importance of recognizing anti-Ku antibodies as a marker for interstitial lung disease in the context of systemic autoimmune diseases.
Keywords:
autoimmunity
; pulmonary fibrosis
; connective tissue disease
1. Introduction
Interstitial lung diseases (ILDs) encompass a wide spectrum of conditions with diverse etiologies, clinical presentations, imaging features and outcomes [1]. ILDs can be classified into disease categories based on the mode of onset, presumed pathophysiology, or management purposes. The ILDs can be further divided into known and unknown causes or etiological contexts. Those occurring on the basis of a known underlying disease include connective tissue disease (CTD)-associated ILD, also known as systemic autoimmune rheumatic diseases, hypersensitivity pneumonitis, drug-induced ILD, and other ILDs associated with occupational and environmental exposures.
ILD is one of the more severe forms of organ involvement in patients with CTD. It is present in approximately 11% of patients with rheumatoid arthritis, 47% of those with systemic sclerosis (SSc), 41% of those with idiopathic inflammatory myopathy (IIM), 17% of those with primary Sjögren syndrome, 6% with mixed CTD, and 6% with systemic lupus erythematosus (SLE) [2]. In all CTDs, ILD is associated with an excess of morbidity and mortality [3]. The imaging and histopathologic patterns of ILD vary according to the underlying CTD. Non-specific interstitial pneumonia (NSIP) is the most common pattern in CTD-ILD, particularly in SSc, IIM, and mixed CTD [2].
The diagnostic approach of ILD is complex and best conducted by a multidisciplinary team in specialized centers [4] to integrate clinical, radiological, physiological, biological, and sometimes histological findings. The nature and severity of lung disease may vary according to the CTD category and autoantibody [3,5], as specific serological autoantibodies are often associated with the occurrence and clinical course of ILD in people with CTD [6,7]. The identification of autoantibodies also facilitates the diagnosis and classification of CTDs, especially when ILD precedes extra-respiratory manifestations, as often in the case of IIM [8,9,10]. Autoantibodies found in patients with SSc include antitopoisomerase-1 (anti-Scl-70) antibodies, anti-centromere antibodies, anti-RNA polymerase-3 antibodies, and more rare ones, including anti-Ku antibodies [11]. Numerous autoantibodies may be associated with IIM, e.g. anti-synthetase antibodies, myositis-specific antibodies, and myositis-associated autoantibodies, including anti-Ro/SSa, anti-U1RNP 70 kDa, anti-PM/Scl 75 and 100 kDa, and anti-Ku antibodies [12]. Anti-PM/Scl and anti-Ku autoantibodies are frequently associated with IIM overlapping with SSc (scleromyositis) [9,10,13,14].
Anti-Ku antibodies are autoantibodies targeting the Ku protein, which is an 80- and 70-kDa DNA-binding protein involved in the DNA repair pathway for double-strand breaks and preventing telomere shortening [15]. Telomere degradation results in cellular replication arrest and premature cell death. This mechanism has been described in familial pulmonary fibrosis linked to TERC and TERT gene mutations, among others [16]. Anti-Ku antibodies were first identified in overlap syndromes involving IIM/SSc in 1981 by Mimori et al. [17], but they are also found in other autoimmune diseases such as SSc, SLE, IIM, and mixed CTD [18]. The prevalence of anti-Ku antibodies in each of these conditions varies greatly across studies, ranging from 1% to 16% in SSc, up to 20% in Sjögren syndrome, and as high as 26% in IIM [19], likely due to heterogeneity of study population and immunologic detection methods across studies [20,21].
Although previous studies have explored the clinical and systemic phenotype associated with anti-Ku antibodies [18,22,23,24,25,26,27,28,29,30,31,32,33], limited data are available on the specific features and progression of lung involvement in patients with these antibodies. The present study aims to characterize in patients with anti-Ku antibodies, with a focus on clinical, imaging, and functional aspects of lung disease.
2. Patients and Methods
2.1. Study Design and Eligibility Criteria
This single-center retrospective observational study was conducted at a referral institution (Hospices Civils de Lyon, HCL). The inclusion criteria were the identification of anti-Ku antibodies between 2007 and August 2022 confirmed in the immunology department of the institution, and the presence of ILD on chest CT scans confirmed by a chest radiologist (DG). Cases with a positive anti-Ku antibody test between 2007 and 2022 were identified using the HCL centralized immunology laboratory's computerized records. Patients under 18 years old at data collection (January 2021–August 2022) were excluded.
2.2. Antibody Testing
Autoantibody testing was performed by two of the authors (NF, FC) in the immunology laboratory of the HCL. Anti-Ku antibody identification was performed using the Dot immunoassay (Euroimmun, Bussy-St Martin, France), with a positive threshold set at 15. Nuclear antibodies fluorescence patterns were assessed using indirect immunofluorescence on Hep-2 cells (Kallestadt, Biorad, Marnes la Coquette, France) as anti-Ku antibodies typically exhibit a speckled fluorescence pattern, with fluorescence around the chromatin and negative mitoses in metaphase and telophase. Patients with false-positive anti-Ku results, i.e. weak intensity and/or inconsistent immunofluorescence findings were excluded. In all cases, the clinical diagnosis of the autoimmune disease was established by a consultant rheumatologist or an internal medicine specialist.
2.3. Clinical and Radiological Data Collection
Clinical, biological, functional, and radiological data were extracted from electronic medical records. Pulmonary function tests were consistently performed using the same spirometer in each individual. Reference standards used were those of the Global Lung Initiative.
Chest CT scans were analyzed by a chest radiologist with 20 years of experience (DG) and a pulmonologist specializing in ILD (KA, 10 years of experience, from the National coordinating Reference Center for Rare Pulmonary Diseases). Discrepancies were adjudicated by another thoracic radiologist (SSM). Collected imaging data included confirmation of ILD presence, ILD features, distribution of the lesions, and the ILD pattern. CT scans were performed with thin sections without contrast injection.
2.4. Ethics
In accordance with French regulations, each patient was informed through a notice letter about his/her right to oppose the collection of his/her personal data. This study was conducted according to the guidelines of the Declaration of Helsinki and was approved by the Institutional Review Board of HCL (22-1789, on February 3, 2023).
2.5. Statistical Analysis
Qualitative variables are reported as frequencies and percentages, and quantitative variables are expressed as median (interquartile range [IQR]) or means ± standard deviation counts (proportion (%), whichever was appropriate, along with range. The statistical analyses were performed using Excel software (version 2410). No imputation was performed for missing data.
3. Results
3.1. Study Population
Among 144 serum samples with anti-Ku antibodies detected using the dot assay, 31 were associated with patients presenting diffuse abnormalities on chest CT scan (Figure 1). Upon further review of the immunology data, 10 cases were considered false positives based on inconsistent indirect immunofluorescence pattern on HEp-2 cells, (Figure 2) or low-intensity close to the positivity threshold. However, one patient with an atypical homogeneous fluorescence pattern was maintained in the cohort due to the presence of an anti-chromatin antibody, which may have masked the speckled nuclear typically associated with anti-Ku antibodies. Additionally, two samples exhibited both nuclear and cytoplasmic fluorescence without a clearly identified target.
Two cases were further excluded upon review of the clinical data and chest CT scans because of the absence of definite ILD. In one case, the final diagnosis was asthma with bronchial dilation and micronodules related to an intercurrent infection. The other patient had interstitial abnormalities secondary to SARS-CoV-2 infection, which resolved on follow-up scan.
In total, 19 patients with confirmed anti-Ku antibodies, typical fluorescence pattern, and confirmed ILD on chest CT were included in the final cohort.
3.2. Patient Characteristics
The sex ratio was balanced, with 53% of the participants being female (n = 10) (Table 1). The mean age at diagnosis of autoimmune disease was 59 years ± 12.6 (range: 26–76 years), while the mean age at diagnosis of ILD was 61 years ± 11.7 (range: 40–82 years). The average time interval between autoimmune disease diagnosis and ILD onset was 2 years (range: 0–15 years). ILD was diagnosed before the onset of autoimmune disease in none of the patients. The mean duration of ILD follow-up was 7 years ± 4.9 (range: 0–15.3 years).
Eight patients were current or former smokers (42%). Moderate occupational or environmental exposure was noted in two patients (11%), but it was not considered causative of ILD. At the time of autoimmune disease diagnosis, most patients presented with dyspnea (79%) with a median modified Medical Research Council score of 2 and crackles on auscultation (53%). Four patients reported coughing (21%), and 6 patients (32%) reported general decline in health status. One patient exhibited finger clubbing.
3.3. Characteristics of Autoimmune Disease
The diagnosis of autoimmune disease is summarized in Table 2. The most frequent diagnosis was SSc (42%), followed by rheumatoid arthritis (26%). Idiopathic inflammatory myopathy was diagnosed in 10% of the cases, and an overlap of SSc and IIM (scleromyositis) was observed in 14% of the cases.
Arthralgia was reported in 12 patients (63%), with only one patient showing radiographic evidence of joint destruction (Table 3). Muscle weakness was present in six patients (32%), including three with muscle atrophy (16%) and two experiencing loss of ambulation (11%). Creatine phosphokinase levels were measured in six patients, with a mean of 681 IU/L ± 420 (range: 233–1177 IU/L). Four patients (21%) exhibited a myogenic pattern on electromyography, and muscle biopsy revealed myositis in two patients (11%).
Scleroderma-related symptoms included Raynaud’s phenomenon in 11 patients (58%), sclerodactyly in 8 (42%), limited mouth opening in 5 (26%), telangiectasia in 1 (5%), and subcutaneous calcinosis in 1 (5%). Capillaroscopy, when performed, was positive in 7 patients (37%) with findings of giant capillaries, hemorrhage, or capillary rarefaction. Skin ulcers or necrosis were present in three patients (16%) although no patient exhibited vasculitic purpura. Ocular or oral dryness was noted in four patients (26%), with positive salivary gland biopsy findings in four (21%). Schirmer tests were negative in the entire cohort.
Seven patients (37%) had gastroesophageal reflux, and three reported epigastric pain (16%). Esophageal dilation and dysphagia were documented in five patients (26%), and dysphagia in three (16%). Peripheral neuropathy of various etiologies was noted in five patients (26%). The central nervous system was not involved. Pericarditis associated with scleroderma was identified in two patients (11%).
3.4. Immunology
The mean intensity of anti-Ku antibody was 156 UFR ± 49 (range: 84-223 UFR). Antinuclear antibodies were found in all cases, with titers of 640 in two patients, 1,280 in sixteen patients, and 160 in one patient). The typical speckled pattern of antinuclear antibodies on HEp-2 cells was detected in 18 cases (95%), while one patient exhibited a homogeneous immunofluorescence pattern. Indeed, the presence of anti-chromatin antibodies, may have masked the typical pattern associated with anti-Ku antibodies. Thirteen patients (70%) had additional antibody identified (Table 2). Patients with isolated anti-Ku antibodies or additional antibodies exhibited similar characteristics (data not shown).
3.5. Pulmonary Function Tests
The pulmonary function tests at baseline are presented in Table 4. At diagnosis, the mean forced vital capacity (FVC) was 82% of the predicted value, the mean total lung capacity was 77% of the predicted value, and the mean diffusing capacity for carbon monoxide (DLCO) was 55% ± 21. Almost the entire cohort (n = 17; 89%) exhibited diffusion impairment, with decreased DLCO and/or KCO below 70%, and half of the patients (n = 10; 53%) showed a restrictive ventilatory pattern. Only one patient had an obstructive ventilatory defect associated with COPD. Two patients were receiving supplemental oxygen therapy.
Follow-up assessments were conducted annually. The average annual decline in FVC was 140 mL/year (range: 0–1610 mL/year), and the mean annual decrease in DLCO was 0.97%/year (range: 0–4.9%/year).
3.6. Imaging Findings
The most common imaging findings were septal reticulations, intralobular reticulations, and ground-glass opacities in most patients (Table 5). Traction bronchiectasis and bronchiolectasis were present in approximately half of the patients (53%). Honeycombing was observed in only three patients (16%). The typical subpleural sparing of the NSIP pattern was observed in only two patients (11%).
The most frequent radiological pattern was nonspecific interstitial pneumonia (NSIP), identified in 10 cases (53%); two additional cases had a pattern of NSIP / organizing pneumonia overlap (Figure 3). No significant difference was observed in the distribution of patterns according to the underlying diagnosis of autoimmune disease (data not shown).
The extent of ILD comprised 0-25% of the total lung volume in 10 patients (53%), 25-50% of the lung volume in 7 (37%), and >50% of the lung volume in 2 patients (11%). The distribution was predominantly peripheral in 14 cases (74%), and most commonly affected the lower lobes in 9 cases (47%).
To assess the progression of interstitial lung disease (ILD), the initial diagnostic CT scan was compared with the most recent CT scan, with an average follow-up duration of 7 years. A significant majority of patients (n=13; 68%) showed worsening CT lesions, whereas two patients (11%) had stable disease, and one patient experienced improvement, which was attributed to the initiation of oral glucocorticoid therapy. Three patients did not have follow-up CT scan available for comparison.
3.7. Bronchoalveolar Lavage
Six patients (32%) underwent bronchoscopy with bronchoalveolar lavage at ILD diagnosis. The bronchoalveolar lavage profile was predominantly neutrophilic in five patients (83%) and mixed neutrophilic and lymphocytic in one patient. None of the patients underwent biopsies, whether endoscopic or surgical.
3.8. Treatment and Outcome
During the follow-up, treatments administered to the patients included: oral glucocorticoids in 17 (89%), mycophenolate mofetil in seven patients (37%), methotrexate in six patients (32%), azathioprine in six patients (32%), hydroxychloroquine in five patients (26%), intravenous pulse cyclophosphamide in four patients (21%), intravenous immunoglobulins in two patients (11%), and rituximab, abatacept, adalimumab, and tocilizumab in one patient each. In total, nearly all patients (89%) received glucocorticoids for more than one month, and 15 patients (79%) were treated with at least one immunosuppressant during follow-up.
Control of both systemic disease and ILD was achieved with glucocorticoids alone in three patients (16%), another immunosuppressive drug alone in four patients (21%), and a combination of glucocorticoids and other immunosuppressants in three patients (16%). Three patients remained clinically stable without specific treatment (16%). Four patients (21%) showed uncontrolled disease progression despite immunosuppressant and/or glucocorticoid treatment, requiring intensified immunosuppressive therapy, and two of these patients subsequently died. Of note, due to the large time span of this retrospective study, none of the patients received antifibrotic therapy. Two patients were recently diagnosed and have not yet undergone treatment reassessment.
Four patients developed group 3 pulmonary hypertension, including three with SSc and one with IIM. Two patients experienced severe respiratory involvement with hypoxemia, necessitating long-term supplemental oxygen therapy. No patient in this cohort underwent lung transplantation.
Over the follow-up period, seven patients died (37%), with an average time from diagnosis to death of 6.7 years ± 5.5 (range 0–15.3 years). The causes of death were acute respiratory distress in three patients (16%), septic shock in two patients (11%), and macrophage activation syndrome related to lymphoma in one patient. The cause of death was unreported in one patient. The overall survival rate was 82% at 5 years and 67% at 10 years (Figure 4). No patient was lost to follow-up.
4. Discussion
In this case series, we described the characteristics of ILD associated with the presence of anti-Ku antibodies. In all cases, ILD onset was chronic, occurring at the onset of the autoimmune disease or shortly thereafter during follow-up. The mean delay between the diagnosis of autoimmune disease and ILD was 2 years. This disease course is consistent with previous series, with SSc and rheumatoid arthritis being the most frequent diagnoses [11,34], with onset of lung involvement within the first few years of the disease course of systemic autoimmune disease. ILD preceded systemic disease in none of the patients.
Clinical respiratory manifestations were nonspecific. Lung function tests were consistent with mild to moderate severity in the majority of the cases. The imaging pattern of ILD was that of NSIP in half of the cases, which is the most frequent pattern observed in both SSc and IIM [2]; in two cases, a CT pattern of NSIP / organizing pneumonia overlap was seen, as frequently seen in patients with IIM and especially the antisynthetase syndrome [10,35,36]. In five cases (26%), ILD was fibrotic on chest CT, but the pattern was indeterminate for usual interstitial pneumonia (UIP); only two cases had a pattern of probable UIP and no definite UIP pattern was seen, consistent with SSc being the most frequent diagnostic category in this series [2,11,37].
The most common extra-respiratory symptoms observed in our study were arthralgia (62%) and Raynaud’s phenomenon (58%), similar to a previous case series of patients with anti-Ku antibodies [26,28]. Patients with ILD and anti-Ku antibodies were diagnosed with a spectrum of autoimmune diseases based on rheumatologic evaluation. SSc was the most frequently diagnosis in our study, but other conditions such as rheumatoid arthritis, Sjögren syndrome, undifferentiated CTD, and IIM were also diagnosed. In a study by Rigolet et al. [28] conducted in a referral center for internal medicine, IIM (37% of cases) was more frequent than SSc (23%), and SLE was observed in 23% of cases. ILD was present in only 11/34 patients (37%) [28]. In a study by Spielmann et al. [26] from a tertiary rheumatology department, the predominant diagnosis associated with anti-Ku antibodies was Sjögren syndrome (36%), followed by IIM (24%), overlapping CTD (26%), and SLE (19%), whereas only 5% of their population consisted of SSc. These findings illustrate that anti-Ku antibodies may be associated with a variety of autoimmune diseases, with the frequency distribution likely reflecting patient recruitment patterns across centers. Cases of anti-Ku antibody with a diagnosis of SSc or IIM seem to be the most frequently associated with ILD. Interestingly, Spielmann et al. identified two subsets of patients with anti-Ku antibodies [26]; in our series, none corresponded to the subset associated with antibodies against double-stranded DNA and a risk of glomerulonephritis, and most if not all our cases corresponded to the other subset described by Spielmann et al., with elevated serum level of creatine phosphokinase and associated with a risk of ILD. Notably, all 6 patients tested in our cohort had elevated serum creatine phosphokinase levels.
Seventy percent of the patients in the current series had another antibody identified in addition to the anti-Ku antibody. This finding is consistent with previous studies [26,28]. Anti-Ku antibodies were associated with other antibodies in 77% of cases in the study by Rigolet et al [28], primarily rheumatoid factor and anti-SSA/SSB. Anti-Ku antibodies were similarly associated with rheumatoid factor and anti-SSA antibodies in 31% of cases in the study by Spielmann et al [26]. This high rate of multiple autoantibody positivity underscores the complex autoimmune background often seen in patients positive for anti-Ku antibodies.
Interestingly, of the 29 patients initially identified as anti-Ku-positive, 10 were excluded because of inconsistent indirect immunofluorescence patterns for typical anti-Ku antinuclear antibodies or because of low-intensity of dot immunoassay close to the positivity threshold. Autoantibody testing using dot immunoassay is commonly performed when systemic autoimmune disease is suspected, and anti-Ku antibodies may be included in the myositis panel. However, it is essential to confirm that patients who are positive for anti-Ku antibodies truly have anti-Ku antibodies. Indeed, it has been already demonstrated that for dot immunoassay, false positive results are frequent for anti-Ku antibodies which show generally a low intensity [38]. The confirmation on HEp2 cells is also essential. When a specific antibody test is inconsistent with the antinuclear pattern, a false positive test result may be considered [39,40]. Finding a specific HEp-2 immunofluorescence pattern confers specificity. This added value of immunofluorescence pattern compared to dot immunoassay has been already described for other autoantibodies such as anti-SRP antibodies [41]. Furthermore, the reactivity for Ku can be due to the presence of antibodies to dsDNA or DNA-binding proteins that are complexed with dsDNA/dsDNA-binding proteins in the serum of patients that then secondarily bind to Ku or dsDNA/dsDNA-binding proteins in an immunoassay[42].
Although the sample size limited an in-depth analysis of disease evolution, our standpoint, our findings on lung function are consistent with progressive ILD patterns in scleroderma observed in larger cohorts, such as the EUSTAR registry [43]. Over the first year of follow-up, we documented various patterns of disease evolution, with lung function improvement (defined as a FVC absolute increase of 5% or more) in five patients (36%), stable disease (FVC variation less than 5%) in six patients (43%), moderate decline (FVC decrease of 5–10%) in two patients (14%), and significant deterioration (FVC decrease >10%) in one patient (7%). Overall, the mean decline in FVC (140 mL/year) was slightly greater than that observed in the placebo arm of the Senscis trial (93.3 mL/year) that enrolled patients with SSc and ILD involving at least 10% of the lung volume [44].
The management of ILD in our patients was at the discretion of the treating physicians, who followed the current practices. Given the wide treatment period covered by the study and the absence of guidelines until very recently for CTD-ILD [45,46], the treatments received by the patients were extremely heterogeneous. Nevertheless, glucocorticoids and immunosuppressants, including mycophenolate mofetil and cyclophosphamide, are frequently used as first-line therapy for patients with SSc. Alternative immunosuppressants, such as rituximab and tocilizumab, are now being considered in subsets of patients with SSc [47,48]. Methotrexate use was relatively frequent in our cohort, likely due to the high prevalence of arthralgia in our population and not solely in patients with rheumatoid arthritis. None of the patients received antifibrotic therapy in this cohort because of the study period.
The limitations of this study include its retrospective design and the inherent risk of missing data. Nevertheless, most patients were consistently monitored in a pneumology department specialized in ILD and had nearly complete data for pulmonary function tests and chest CT scans. Importantly, no patient was lost to follow-up. The single-center nature of our study may be seen as a limitation; however, it facilitated the thorough identification of anti-Ku-positive patients through centralized testing within our institution (HCL), which encompasses four large public hospitals serving a large catchment area. Although the sample size was relatively small, anti-Ku antibody-associated ILD is rare and even nationally, the affected population remains limited. Patients were followed up at a national reference center for ILD and other rare lung diseases, ensuring accurate ILD diagnosis and phenotyping. Another limitation was the lack of histopathologic confirmation of ILD, as lung biopsy indications for ILD diagnosis have become increasingly restricted because of the minimal impact on management in the setting of autoimmune disease and increasing attention to morbidity and mortality risks. In this relatively small cohort, the pattern of ILD progression could not be assessed according to current standards [46,47] ; further studies are warranted to assess the proportion of patients with ILD and anti-Ku antibodies who develop progressive pulmonary fibrosis and may benefit from anti-fibrotic therapy.
5. Conclusions
In conclusion, in this series of 19 well-characterized patients with ILD and anti-Ku antibodies, we found that the majority of the patients had an autoimmune disease corresponding to SSc and a CT pattern of NSIP, yet with a range of other CTDs and CT patterns. ILD associated with anti-Ku antibodies is a heterogeneous condition, and its management needs to be individualized and discussed among multidisciplinary teams [49].
Author Contributions
Laure Petitgrand: Original Draft Preparation, Formal Analysis, Investigation, Resources, Data Curation, Review & Editing, Approval. Kais Ahmad: Conception and Design, Data acquisition, Formal Analysis, Investigation, Review & Editing, Approval. Delphine Gamondès: Data acquisition, Formal Analysis, Investigation, Review & Editing, Approval. Rémi Diesler: Data acquisition, Investigation, Review & Editing, Approval. Frédéric Coutant: Data acquisition, Investigation, Review & Editing, Approval. Laure Gallay: Data acquisition, Investigation, Review & Editing, Approval. Romain Fort: Data acquisition, Investigation, Review & Editing, Approval. Julie Traclet: Data acquisition, Investigation, Review & Editing, Approval. Francois Lestelle: Data acquisition, Investigation, Review & Editing, Approval. Arnaud Hot: Data acquisition, Investigation, Review & Editing, Approval. Roland Chapurlat: Data acquisition, Investigation, Review & Editing, Approval. Cyrille Confavreux: Data acquisition, Investigation, Review & Editing, Approval. Stéphane Durupt: Data acquisition, Investigation, Review & Editing, Approval. Ségolène Turquier: Data acquisition, Investigation, Review & Editing, Approval. Salim Si-Mohamed: Data acquisition, Investigation, Review & Editing, Approval. Nicole Fabien: Data acquisition, Investigation, Review & Editing, Approval. Vincent Cottin: Conceptualization, Methodology, Validation, Formal Analysis, Investigation, Resources, Data Curation, Writing, Review & Editing, Visualization, Supervision.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki and was approved by the Institutional Review Board of Hospices Civils de Lyon (22-1789, on February 3, 2023).
Informed Consent Statement
Each patient was informed through a notice letter about their right to object to the collection of their personal data, in line with French regulations.
Data Availability Statement
The data supporting the reported results can be obtained from the first author upon reasonable request.
Acknowledgments
We thank Drs Magali Aubineau, Annick Charhon, Fabienne Coury-Lucas, Elodie Feurer, Deborah Gensburger, Jean-Christophe Lega, Raphaele Nove-Josserand, Laurent Perrard, Solene Poutrel, Gaelle Richard-Colmant, Emilie Virot, for patient care.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wijsenbeek M, Cottin V. Spectrum of Fibrotic Lung Diseases. N Engl J Med 2020; 383: 958-968. [CrossRef]
- Joy GM, Arbiv OA, Wong CK, Lok SD, Adderley NA, Dobosz KM, Johannson KA, Ryerson CJ. Prevalence, imaging patterns and risk factors of interstitial lung disease in connective tissue disease: a systematic review and meta-analysis. Eur Respir Rev 2023; 32. [CrossRef]
- Wijsenbeek M, Suzuki A, Maher TM. Interstitial lung diseases. Lancet 2022; 400: 769-786.
- Lynch DA, Sverzellati N, Travis WD, Brown KK, Colby TV, Galvin JR, Goldin JG, Hansell DM, Inoue Y, Johkoh T, Nicholson AG, Knight SL, Raoof S, Richeldi L, Ryerson CJ, Ryu JH, Wells AU. Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner Society White Paper. Lancet Respir Med 2018; 6: 138-153.
- Fischer A, Antoniou KM, Brown KK, Cadranel J, Corte TJ, du Bois RM, Lee JS, Leslie KO, Lynch DA, Matteson EL, Mosca M, Noth I, Richeldi L, Strek ME, Swigris JJ, Wells AU, West SG, Collard HR, Cottin V. An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J 2015; 46: 976-987. [CrossRef]
- Mariampillai K, Granger B, Amelin D, Guiguet M, Hachulla E, Maurier F, Meyer A, Tohme A, Charuel JL, Musset L, Allenbach Y, Benveniste O. Development of a New Classification System for Idiopathic Inflammatory Myopathies Based on Clinical Manifestations and Myositis-Specific Autoantibodies. JAMA Neurol 2018; 75: 1528-1537. [CrossRef]
- Patterson KA, Roberts-Thomson PJ, Lester S, Tan JA, Hakendorf P, Rischmueller M, Zochling J, Sahhar J, Nash P, Roddy J, Hill C, Nikpour M, Stevens W, Proudman SM, Walker JG. Interpretation of an Extended Autoantibody Profile in a Well-Characterized Australian Systemic Sclerosis (Scleroderma) Cohort Using Principal Components Analysis. Arthritis Rheumatol 2015; 67: 3234-3244. [CrossRef]
- Cottin V, Thivolet-Bejui F, Reynaud-Gaubert M, Cadranel J, Delaval P, Ternamian PJ, Cordier JF. Interstitial lung disease in amyopathic dermatomyositis, dermatomyositis and polymyositis. Eur Respir J 2003; 22: 245-250. [CrossRef]
- Hervier B, Devilliers H, Stanciu R, Meyer A, Uzunhan Y, Masseau A, Dubucquoi S, Hatron PY, Musset L, Wallaert B, Nunes H, Maisonobe T, Olsson NO, Adoue D, Arlet P, Sibilia J, Guiguet M, Lauque D, Amoura Z, Hachulla E, Hamidou M, Benveniste O. Hierarchical cluster and survival analyses of antisynthetase syndrome: phenotype and outcome are correlated with anti-tRNA synthetase antibody specificity. Autoimmun Rev 2012; 12: 210-217.
- Lega JC, Cottin V, Fabien N, Thivolet-Bejui F, Cordier JF. Interstitial Lung Disease Associated with Anti-PM/Scl or Anti-Aminoacyl-tRNA Synthetase Autoantibodies: A Similar Condition? The Journal of Rheumatology 2010; 37: 1000-1009.
- Mismetti V, Si-Mohamed S, Cottin V. Interstitial Lung Disease Associated with Systemic Sclerosis. Semin Respir Crit Care Med 2024. [CrossRef]
- Barba T, Mainbourg S, Nasser M, Lega JC, Cottin V. Lung Diseases in Inflammatory Myopathies. Semin Respir Crit Care Med 2019; 40: 255-270. [CrossRef]
- Marie I, Lahaxe L, Benveniste O, Delavigne K, Adoue D, Mouthon L, Hachulla E, Constans J, Tiev K, Diot E, Levesque H, Boyer O, Jouen F. Long-term outcome of patients with polymyositis/ dermatomyositis and anti-PM-Scl antibody. Br J Dermatol 2010; 162: 337-344. [CrossRef]
- Guillen-Del Castillo A, Pilar Simeon-Aznar C, Fonollosa-Pla V, Alonso-Vila S, Reverte-Vinaixa MM, Munoz X, Pallisa E, Selva-O'allaghan A, Fernandez-Codina A, Vilardell-Tarres M. Good outcome of interstitial lung disease in patients with scleroderma associated to anti-PM/Scl antibody. Semin Arthritis Rheum 2014; 44: 331-337. [CrossRef]
- Fell VL, Schild-Poulter C. The Ku heterodimer: function in DNA repair and beyond. Mutation research Reviews in mutation research 2015; 763: 15-29. [CrossRef]
- Borie R, Kannengiesser C, Antoniou K, Bonella F, Crestani B, Fabre A, Froidure A, Galvin L, Griese M, Grutters JC, Molina-Molina M, Poletti V, Prasse A, Renzoni E, van der Smagt J, van Moorsel CHM. European Respiratory Society statement on familial pulmonary fibrosis. Eur Respir J 2023; 61. [CrossRef]
- Mimori T, Akizuki M, Yamagata H, Inada S, Yoshida S, Homma M. Characterization of a high molecular weight acidic nuclear protein recognized by autoantibodies in sera from patients with polymyositis-scleroderma overlap. J Clin Invest 1981; 68: 611-620. [CrossRef]
- Franceschini F, Cavazzana I, Generali D, Quinzanini M, Viardi L, Ghirardello A, Doria A, Cattaneo R. Anti-Ku antibodies in connective tissue diseases: clinical and serological evaluation of 14 patients. J Rheumatol 2002; 29: 1393-1397.
- Hoa S, Hudson M, Troyanov Y, Proudman S, Walker J, Stevens W, Nikpour M, Assassi S, Mayes MD, Wang M, Baron M, Fritzler MJ. Single-specificity anti-Ku antibodies in an international cohort of 2140 systemic sclerosis subjects: clinical associations. Medicine (Baltimore) 2016; 95: e4713.
- Rozman B, Cucnik S, Sodin-Semrl S, Czirják L, Varjú C, Distler O, Huscher D, Aringer M, Steiner G, Matucci-Cerinic M, Guiducci S, Stamenkovic B, Stankovic A, Kveder T. Prevalence and clinical associations of anti-Ku antibodies in patients with systemic sclerosis: a European EUSTAR-initiated multi-centre case-control study. Ann Rheum Dis 2008; 67: 1282-1286. [CrossRef]
- Cruellas MG, Viana Vdos S, Levy-Neto M, Souza FH, Shinjo SK. Myositis-specific and myositis-associated autoantibody profiles and their clinical associations in a large series of patients with polymyositis and dermatomyositis. Clinics (Sao Paulo, Brazil) 2013; 68: 909-914. [CrossRef]
- Cavazzana I, Ceribelli A, Quinzanini M, Scarsi M, Airò P, Cattaneo R, Franceschini F. Prevalence and clinical associations of anti-Ku antibodies in systemic autoimmune diseases. Lupus 2008; 17: 727-732. [CrossRef]
- Tyndall AJ, Bannert B, Vonk M, Airo P, Cozzi F, Carreira PE, Bancel DF, Allanore Y, Muller-Ladner U, Distler O, Iannone F, Pellerito R, Pileckyte M, Miniati I, Ananieva L, Gurman AB, Damjanov N, Mueller A, Valentini G, Riemekasten G, Tikly M, Hummers L, Henriques MJ, Caramaschi P, Scheja A, Rozman B, Ton E, Kumanovics G, Coleiro B, Feierl E, Szucs G, Von Muhlen CA, Riccieri V, Novak S, Chizzolini C, Kotulska A, Denton C, Coelho PC, Kotter I, Simsek I, de la Pena Lefebvre PG, Hachulla E, Seibold JR, Rednic S, Stork J, Morovic-Vergles J, Walker UA. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis 2010; 69: 1809-1815. [CrossRef]
- Belizna C, Henrion D, Beucher A, Lavigne C, Ghaali A, Lévesque H. Anti-Ku antibodies: Clinical, genetic and diagnostic insights. Autoimmun Rev 2010; 9: 691-694. [CrossRef]
- Cooley HM, Melny BJ, Gleeson R, Greco T, Kay TW. Clinical and serological associations of anti-Ku antibody. J Rheumatol 1999; 26: 563-567.
- Spielmann L, Nespola B, Séverac F, Andres E, Kessler R, Guffroy A, Poindron V, Martin T, Geny B, Sibilia J, Meyer A. Anti-Ku syndrome with elevated CK and anti-Ku syndrome with anti-dsDNA are two distinct entities with different outcomes. Ann Rheum Dis 2019; 78: 1101-1106.
- Yang H, Li W, Tian X, Wang G, Shu X, Peng Q, Lu X. Immune-mediated necrotizing myopathies and interstitial lung disease are predominant characteristics in anti-Ku positive patients with idiopathic inflammatory myopathies. Ann Rheum Dis 2022; 81: e48. [CrossRef]
- Rigolet A, Musset L, Dubourg O, Maisonobe T, Grenier P, Charuel J-L, Behin A, Herson S, Amoura Z, Benveniste O. Inflammatory Myopathies With Anti-Ku Antibodies. Medicine 2012; 91: 95-102. [CrossRef]
- Casal-Dominguez M, Pinal-Fernandez I, Derfoul A, Graf R, Michelle H, Albayda J, Tiniakou E, Adler B, Danoff SK, Lloyd TE, Christoper-Stine L, Paik JJ, Mammen AL. The phenotype of myositis patients with anti-Ku autoantibodies. Semin Arthritis Rheum 2021; 51: 728-734. [CrossRef]
- Holzer MT, Uruha A, Roos A, Hentschel A, Schänzer A, Weis J, Claeys KG, Schoser B, Montagnese F, Goebel HH, Huber M, Léonard-Louis S, Kötter I, Streichenberger N, Gallay L, Benveniste O, Schneider U, Preusse C, Krusche M, Stenzel W. Anti-Ku + myositis: an acquired inflammatory protein-aggregate myopathy. Acta neuropathologica 2024; 148: 6. [CrossRef]
- Oyama M, Holzer MT, Ohnuki Y, Saito Y, Nishimori Y, Suzuki S, Shiina T, Leonard-Louis S, Benveniste O, Schneider U, Stenzel W, Nishino I, Suzuki S, Uruha A. Pathologic Features of Anti-Ku Myositis. Neurology 2024; 102: e209268. [CrossRef]
- Bhalodia A, Bermea K, Schmidt J, Gilotra N, Barth AS, Adamo L, Paik JJ. Increased risk of myocarditis and arrythmias in anti-Ku-positive scleroderma-myositis overlap patients: a case series. Rheumatology (Oxford) 2024; 63: e268-e269. [CrossRef]
- Sousa M, Martins P, Santos B, Costa E, Santos FC, Freitas R, Faria M, Martins F, Rodrigues T, Santiago T, Silva JAP, Inês LS. Anti-Ku antibody syndrome: is it a distinct clinical entity? A cross-sectional study of 75 patients. Rheumatology (Oxford) 2023; 62: e213-e215. [CrossRef]
- Pugashetti JV, Lee JS. Overview of Rheumatoid Arthritis-Associated Interstitial Lung Disease and Its Treatment. Semin Respir Crit Care Med 2024; 45: 329-341. [CrossRef]
- Kambouchner M, Levy P, Nicholson AG, Schubel K, Magois E, Feuillet S, Valeyre D, Bernaudin JF, Nunes H. Prognostic relevance of histological variants in nonspecific interstitial pneumonia. Histopathology 2014; 65: 549-560. [CrossRef]
- Enomoto N, Sumikawa H, Sugiura H, Kitani M, Tanaka T, Hozumi H, Fujisawa T, Suda T. Clinical, radiological, and pathological evaluation of "NSIP with OP overlap" pattern compared with NSIP in patients with idiopathic interstitial pneumonias. Respir Med 2020; 174: 106201. [CrossRef]
- Desai SR, Veeraraghavan S, Hansell DM, Nikolakopolou A, Goh NS, Nicholson AG, Colby TV, Denton CP, Black CM, du Bois RM, Wells AU. CT features of lung disease in patients with systemic sclerosis: comparison with idiopathic pulmonary fibrosis and nonspecific interstitial pneumonia. Radiology 2004; 232: 560-567.
- Tansley SL, Li D, Betteridge ZE, McHugh NJ. The reliability of immunoassays to detect autoantibodies in patients with myositis is dependent on autoantibody specificity. Rheumatology (Oxford) 2020; 59: 2109-2114. [CrossRef]
- Infantino M, Tampoia M, Fabris M, Alessio MG, Previtali G, Pesce G, Deleonardi G, Porcelli B, Musso M, Grossi V, Benucci M, Manfredi M, Bizzaro N. Combining immunofluorescence with immunoblot assay improves the specificity of autoantibody testing for myositis. Rheumatology (Oxford) 2019; 58: 1239-1244. [CrossRef]
- Damoiseaux J, Vulsteke JB, Tseng CW, Platteel ACM, Piette Y, Shovman O, Bonroy C, Hamann D, De Langhe E, Musset L, Chen YH, Shoenfeld Y, Allenbach Y, Bossuyt X. Autoantibodies in idiopathic inflammatory myopathies: Clinical associations and laboratory evaluation by mono- and multispecific immunoassays. Autoimmun Rev 2019; 18: 293-305. [CrossRef]
- Picard C, Vincent T, Lega JC, Hue S, Fortenfant F, Lakomy D, Humbel RL, Goetz J, Molinari N, Bardin N, Bertin D, Johanet C, Chretien P, Dubucquoi S, Streichenberger N, Desplat-Jégo S, Bossuyt X, Sibilia J, Abreu I, Chevailler A, Fabien N. Heterogeneous clinical spectrum of anti-SRP myositis and importance of the methods of detection of anti-SRP autoantibodies: a multicentric study. Immunologic research 2016; 64: 677-686. [CrossRef]
- Mahler M, Satoh M, Fritzler MJ. Anti-Ku antibodies: important points to consider. Ann Rheum Dis 2021; 80: e182. [CrossRef]
- Hoffmann-Vold AM, Allanore Y, Alves M, Brunborg C, Airó P, Ananieva LP, Czirják L, Guiducci S, Hachulla E, Li M, Mihai C, Riemekasten G, Sfikakis PP, Kowal-Bielecka O, Riccardi A, Distler O. Progressive interstitial lung disease in patients with systemic sclerosis-associated interstitial lung disease in the EUSTAR database. Ann Rheum Dis 2021; 80: 219-227. [CrossRef]
- Distler O, Highland KB, Gahlemann M, Azuma A, Fischer A, Mayes MD, Raghu G, Sauter W, Girard M, Alves M, Clerisme-Beaty E, Stowasser S, Tetzlaff K, Kuwana M, Maher TM, Investigators ST. Nintedanib for Systemic Sclerosis-Associated Interstitial Lung Disease. N Engl J Med 2019; 380: 2518-2528. [CrossRef]
- Raghu G, Montesi SB, Silver RM, Hossain T, Macrea M, Herman D, Barnes H, Adegunsoye A, Azuma A, Chung L, Gardner GC, Highland KB, Hudson M, Kaner RJ, Kolb M, Scholand MB, Steen V, Thomson CC, Volkmann ER, Wigley FM, Burlile D, Kemper KA, Knight SL, Ghazipura M. Treatment of Systemic Sclerosis-associated Interstitial Lung Disease: Evidence-based Recommendations. An Official American Thoracic Society Clinical Practice Guideline. Am J Respir Crit Care Med 2024; 209: 137-152.
- Johnson SR, Bernstein EJ, Bolster MB, Chung JH, Danoff SK, George MD, Khanna D, Guyatt G, Mirza RD, Aggarwal R, Allen A, Jr., Assassi S, Buckley L, Chami HA, Corwin DS, Dellaripa PF, Domsic RT, Doyle TJ, Falardeau CM, Frech TM, Gibbons FK, Hinchcliff M, Johnson C, Kanne JP, Kim JS, Lim SY, Matson S, McMahan ZH, Merck SJ, Nesbitt K, Scholand MB, Shapiro L, Sharkey CD, Summer R, Varga J, Warrier A, Agarwal SK, Antin-Ozerkis D, Bemiss B, Chowdhary V, Dematte D'Amico JE, Hallowell R, Hinze AM, Injean PA, Jiwrajka N, Joerns EK, Lee JS, Makol A, McDermott GC, Natalini JG, Oldham JM, Saygin D, Lakin KS, Singh N, Solomon JJ, Sparks JA, Turgunbaev M, Vaseer S, Turner A, Uhl S, Ivlev I. 2023 American College of Rheumatology (ACR)/American College of Chest Physicians (CHEST) Guideline for the Treatment of Interstitial Lung Disease in People with Systemic Autoimmune Rheumatic Diseases. Arthritis Rheumatol 2024; 76: 1182-1200.
- Khanna D, Lin CJF, Furst DE, Goldin J, Kim G, Kuwana M, Allanore Y, Matucci-Cerinic M, Distler O, Shima Y, van Laar JM, Spotswood H, Wagner B, Siegel J, Jahreis A, Denton CP. Tocilizumab in systemic sclerosis: a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Respir Med 2020; 8: 963-974. [CrossRef]
- Ebata E, Yoshizaki A, Oba K, Kashiwabara K, Ueda K, Uemura Y, Watadani T, Fukasawa T, Miura S, Yoshizaki-Ogawa A, Asano Y, Okiyama N, Kodera M, Hasagawa M, Sato S. Safety and efficacy of rituximab in systemic sclerosis (DESIRES): a double-blind, investigator-initiated, randomised, placebo-controlled trial. Lancet Rheumatology 2021; 3: e489-e-497. [CrossRef]
- Cottin V, Martinez FJ, Smith V, F. WSL. Multidisciplinary teams in the clinical care of fibrotic interstitial lung disease: current perspectives. Eur Respir Rev 2022; 31: 220003. [CrossRef]
Figure 1.
Flowchart of the study.
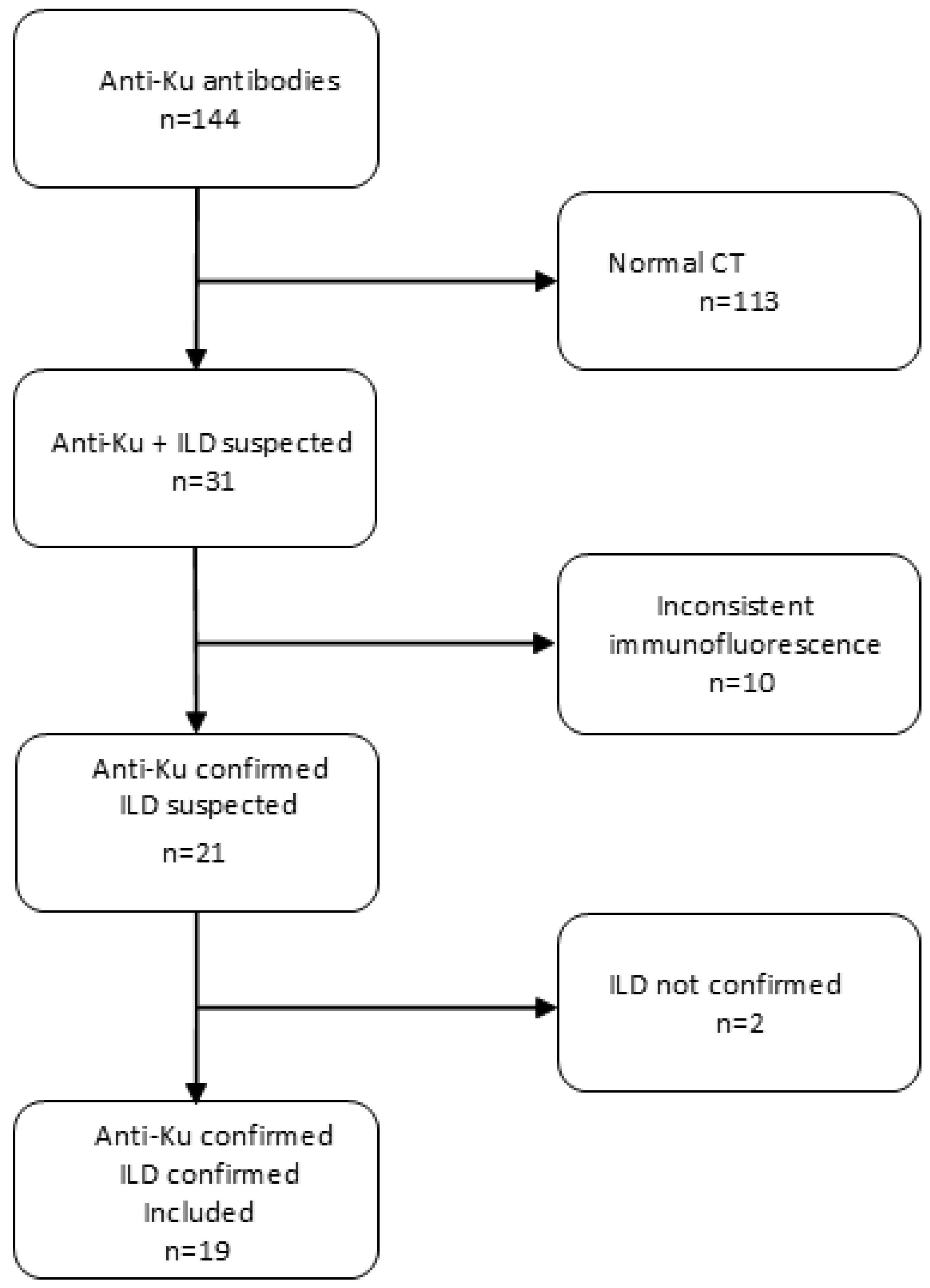
Figure 2.
Characteristic speckled nuclear fluorescence pattern of anti-Ku antibodies observed on HEp-2 cells by indirect immunofluorescence. The white arrow highlights the nucleus of a cell in metaphase with fluorescence around the chromatin and negative mitoses.
Figure 2.
Characteristic speckled nuclear fluorescence pattern of anti-Ku antibodies observed on HEp-2 cells by indirect immunofluorescence. The white arrow highlights the nucleus of a cell in metaphase with fluorescence around the chromatin and negative mitoses.
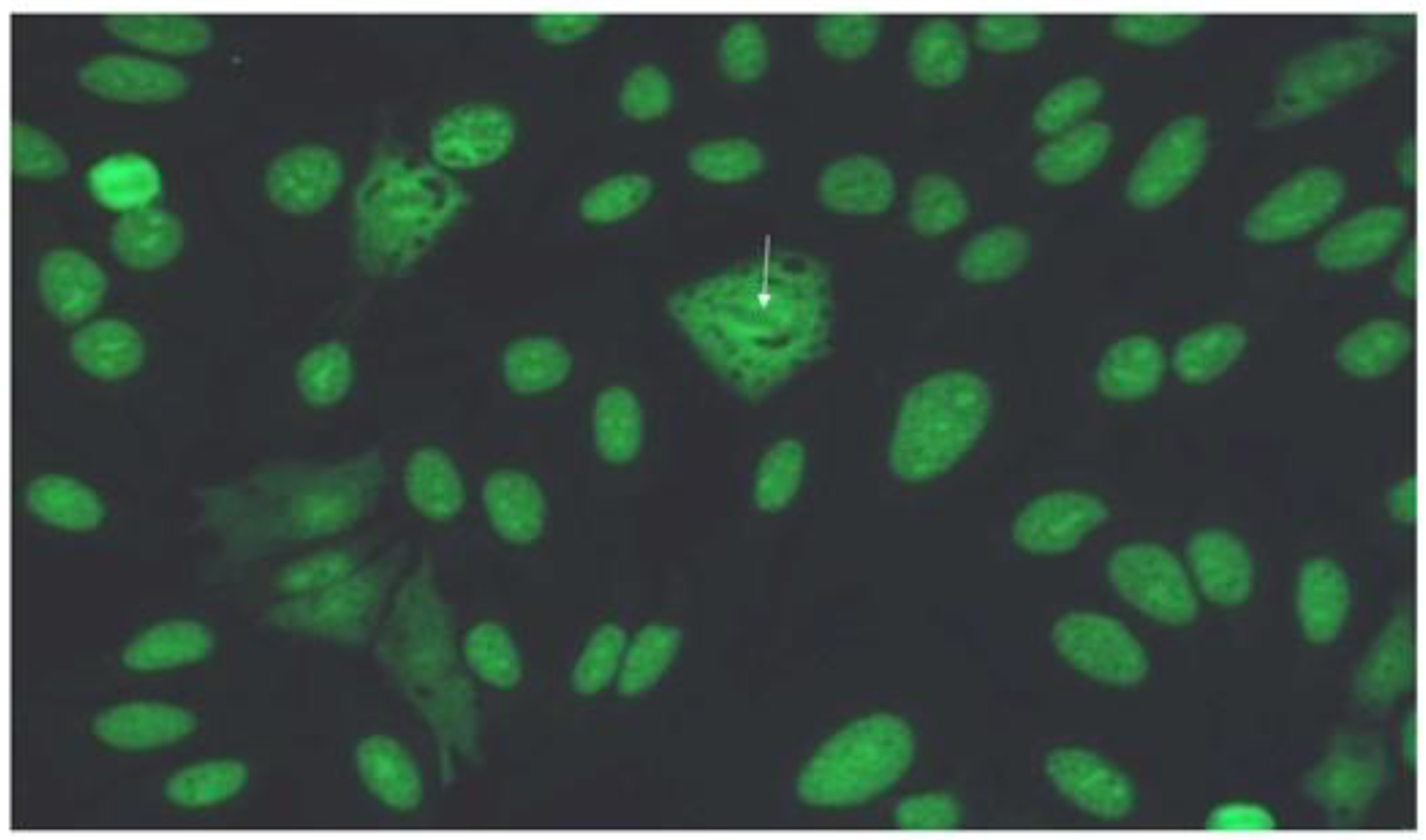
Figure 3.
Computed tomography of a male patient with anti-Ku antibodies demonstrating a pattern of fibrotic nonspecific interstitial pneumonia. A: axial view of lower part of the lungs, demonstrating diffuse distribution of ground glass opacities, reticulation, bronchiectasis and bronchiolectasis. B: sagittal view showing apicobasal distribution; C: coronal view, minimal intensity projection 13 mm. D: sagittal view, minimal intensity projection 13 mm.
Figure 3.
Computed tomography of a male patient with anti-Ku antibodies demonstrating a pattern of fibrotic nonspecific interstitial pneumonia. A: axial view of lower part of the lungs, demonstrating diffuse distribution of ground glass opacities, reticulation, bronchiectasis and bronchiolectasis. B: sagittal view showing apicobasal distribution; C: coronal view, minimal intensity projection 13 mm. D: sagittal view, minimal intensity projection 13 mm.
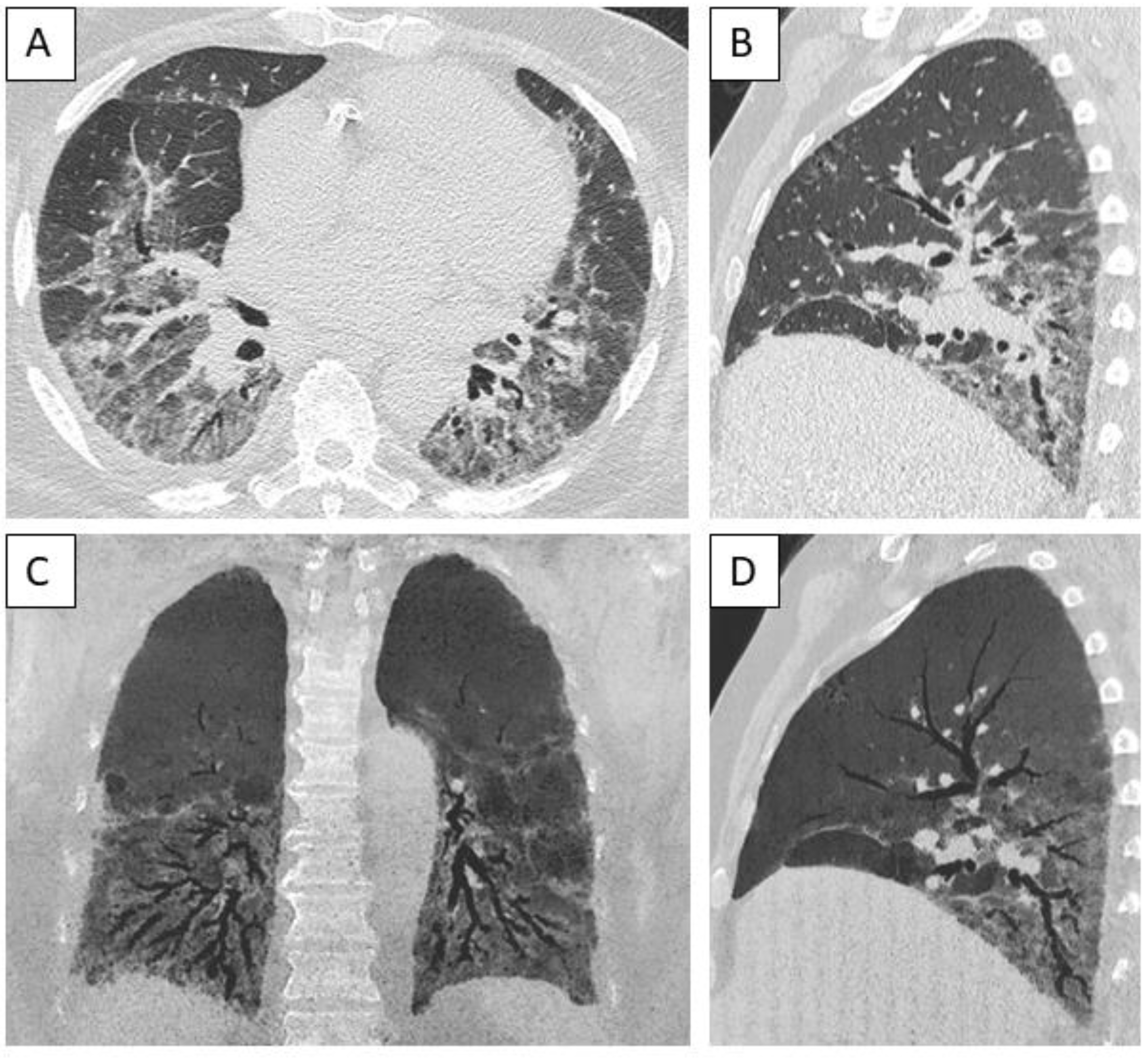
Figure 4.
Computed tomography of a male patient with anti-Ku antibodies demonstrating a pattern of fibrotic nonspecific interstitial pneumonia, demonstrating apicobasal distribution of ground glass opacities and reticulation with subpleural sparing, bronchiectasis and bronchiolectasis, associated with paraseptal emphysema. A: Upper part of the lungs, axial view. B: Middle part of the lungs, axial view. C: Lower part of the lungs, axial view. D: Sagittal view, minimal intensity projection 5 mm.
Figure 4.
Computed tomography of a male patient with anti-Ku antibodies demonstrating a pattern of fibrotic nonspecific interstitial pneumonia, demonstrating apicobasal distribution of ground glass opacities and reticulation with subpleural sparing, bronchiectasis and bronchiolectasis, associated with paraseptal emphysema. A: Upper part of the lungs, axial view. B: Middle part of the lungs, axial view. C: Lower part of the lungs, axial view. D: Sagittal view, minimal intensity projection 5 mm.
Figure 5.
Kaplan-Meier estimates of overall survival.
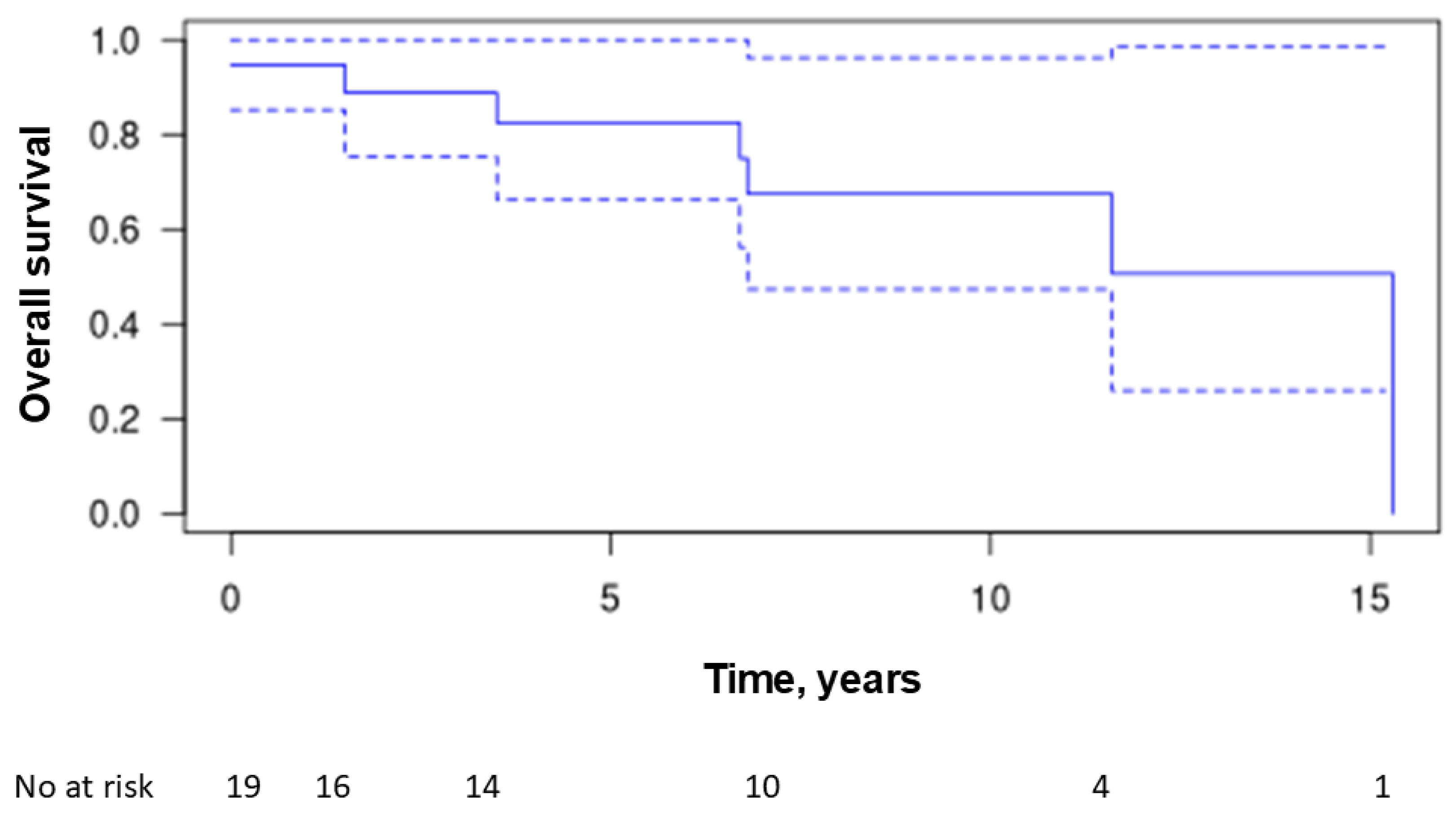

Table 1.
Main individual characteristics of 19 patients with ILD and anti-Ku antibodies.
| SEX | AGE | TOBACCO SMOKING | CTD | ANA TITER | ANA PATTERN |
FVC, % | DLCO, % | CT PATTERN | FOLLOW-UP, YRS | DEATH | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 62 | 1 | IIM | 1280 | Speckled | 96 | 52 | Indeterm. UIP | 1,1 | N |
| 2 | M | 68 | 1 | SSc | 160 | Speckled | 48 | 28 | NSIP | 0,0 | N |
| 3 | F | 61 | 0 | SM, RA, SjS | 1280 | Speckled | 50 | N/A | Indeterm. UIP | 6,8 | Y |
| 4 | F | 61 | 1 | SSc, RA, SjS | 1280 | Speckled | 106 | 51 | Indeterm. UIP | 11,1 | N |
| 5 | F | 65 | 0 | SSc | 1280 | Speckled | 81 | 23 | NSIP | 11,6 | Y |
| 6 | F | 40 | 1 | SSc | 1280 | Speckled | 96 | 75 | Indeterm. UIP | 9,6 | N |
| 7 | M | 76 | 0 | UCTD, PMR | 1280 | Speckled | 61 | N/A | NSIP/OP | 6,7 | Y |
| 8 | F | 69 | 0 | UCTD | 1600 | Speckled | 81 | 54 | NSIP | 3,5 | Y |
| 9 | M | 59 | 1 | IIM | 1280 | Speckled | 76 | 70 | Indeterm. UIP | 1,7 | N |
| 10 | M | 73 | 0 | UCTD | 1280 | Speckled | 86 | 75 | Probable UIP | 9,9 | N |
| 11 | F | 26 | 0 | RA | 1280 | Speckled | 75 | 77 | Probable UIP | 15,2 | N |
| 12 | F | 55 | 0 | UCTD, SjS | 1280 | Speckled | 55 | 70 | NSIP/OP | 11,8 | N |
| 13 | M | 61 | 0 | SSc | 1280 | Speckled | 33 | 24 | NSIP | 0,0 | Y |
| 14 | F | 66 | 0 | SM | 1600 | Speckled | 87 | 35 | NSIP | 15,3 | Y |
| 15 | M | 45 | 1 | SSc | 1280 | Speckled | 99 | 58 | NSIP | 5,4 | N |
| 16 | M | 75 | 1 | SM | 1280 | Speckled | 90 | 26 | NSIP | 1,5 | Y |
| 17 | M | 50 | 0 | SSc, RA | 1280 | Speckled | 119 | 81 | NSIP | 7,6 | N |
| 18 | F | 66 | 0 | RA, SjS | 1280 | Homogeneous | 133 | 82 | NSIP | 9,2 | N |
| 19 | F | 53 | 1 | SSc | 1280 | Speckled | 79 | 47 | NSIP | 4,1 | N |
F : female ; Indeterm.UIP : indterminate for usual interstitial pneuomonia ; IIM : idiopathic inflammatory myopathy ; M : male ; NSIP : nonspecific interstitial pneumonia ; OP : organizig pneumonia ; PMR : polymyalgia rheumatica ; RA : rheumatoid arthritis ; SjS : Sjögren syndrome ; SM : scleromyositis ; SSc : systemic sclerosis ; UCTD : undifferenciated connective tissue disease.
Table 2.
Diagnosis of autoimmune disease and autoantibodies in 19 patients with ILD and anti-KU antibodies. Eight patients (42%) were classified into more than one category due to various overlap syndromes.
Table 2.
Diagnosis of autoimmune disease and autoantibodies in 19 patients with ILD and anti-KU antibodies. Eight patients (42%) were classified into more than one category due to various overlap syndromes.
| Clinical autoimmune disease diagnosis | N (%) |
|---|---|
| Systemic sclerosis | 8 (42) |
| Rheumatoid arthritis | 5 (26) |
| Sjögren syndrome | 4 (21) |
| Undifferentiated connective tissue disease | 4 (21) |
| Scleromyositis | 3 (14) |
| Idiopathic inflammatory myopathy | 2 (10) |
| Polymyalgia rheumatica | 1 (5) |
| Auto-antibodies | N (%) |
| Anti-Ku | 19 (100) |
| Anti-nuclear antibodies | 19 (100) |
| Typical speckled pattern | 18 (95) |
| Homogeneous pattern | 1 (5) |
| Other auto-antibodies | |
| Rheumatoid factor | 4 (21) |
| Anti-SSA | 2 (11) |
| Anti-TRIM21 | 2 (11) |
| Anti-PMScl | 1 (5) |
| Anti-RNA-Polymerase-3 | 1 (5) |
| Anti-JO1 | 1 (5) |
| Anti-EJ | 1 (5) |
| Anti-PL7 | 1 (5) |
| Anti-fibrillarin | 1 (5) |
| Anti-chromatin | 1 (5) |
| Anti-NOR90 | 1 (5) |
| Anti-cyclic citrullinated peptide | 1 (5) |
Table 3.
Clinical characteristics at ILD diagnosis in the 19 patients.
| Mean ± SD or N (%) | |
|---|---|
| Age at ILD diagnosis | 61 ± 11.7 |
| Sex, F | 10 (53) |
| Medical history | |
| Hypertension | 5 (26) |
| Atrial fibrillation | 5 (26) |
| Venous thromboembolism | 4 (21) |
| Cancer | 4 (21) |
| Hypothyroidism | 3 (16) |
| Type-2 diabetes | 2 (11) |
| Ischemic cardiomyopathy | 3 (16) |
| Chronic renal failure | 1 (5) |
| Obstructive sleep apnea syndrome | 1 (5) |
| Respiratory manifestations | |
| Shortness of breath | 6 (32) |
| Cough | 4 (21) |
| Crackles | 10 (53) |
| Finger clubbing | 1 (5) |
| Other organ manifestations | |
| Arthralgia | 12 (63) |
| Myalgia | 6 (32) |
| Muscle weakness | 6 (32) |
| Skin ulcers or necrosis | 3 (16) |
| Calcinosis | 1 (5) |
| Raynaud’ phenomenon | 11 (58) |
| Sclerodactyly | 8 (42) |
| Mouth-opening limitation | 5 (26) |
| Telangiectasiae | 1 (5) |
| Gastroesophageal reflux | 7 (37) |
| Epigastric pain | 3 (16) |
| Dysphagia | 3 (16) |
| Pericarditis | 2 (11) |
| Sicca syndrome | 4 (21) |
| Peripheral neuropathy | 5 (26) |
Table 4.
Pulmonary function tests of 19 patients with ILD and anti-Ku antibodies.
| Parameter | mean ± SD |
|---|---|
| Forced vital capacity (L) | 3.04 ± 0.82 |
| Forced vital capacity, % of predicted | 82 ± 26 |
| Forced expiratory volume in 1 s | 2.22 L ± 0.89 |
| Forced expiratory volume in 1 s, % of predicted | 80 ± 26 |
| Forced expiratory volume in 1 s/ forced vital capacity, absolute | 0.80 ± 0.096 |
| Total lung capacity (L) | 4.27 L ± 1.59 |
| Total lung capacity, % of predicted | 77 ± 23 |
| Diffusion capacity of carbon monoxide, % of predicted | 55 ± 21 |
| Diffusion coefficient, % of predicted | 80 ± 22 |
Table 5.
CT findings of 19 patients with ILD and anti-Ku antibodies.
| CT features | n (%) |
|---|---|
| Reticulation | 16 (84) |
| Intralobular reticulation | 14 (74) |
| Ground-glass opacities | 9 (68) |
| Traction bronchiectasis | 10 (53) |
| Traction bronchiolectasis | 10 (53) |
| Honeycombing | 3 (16) |
| Cysts | 3 (16) |
| Emphysema | 3 (16) |
| Micronodules | 1 (5) |
| Consolidations | 1 (5) |
| Mediastinal lymphadenopathy | 7 (37) |
| Subpleural sparing | 2 (11) |
| CT patterns | |
| Nonspecific interstitial pneumonia | 10 (53) |
| Nonspecific interstitial pneumonia/organizing pneumonia | 2 (11) |
| Indeterminate for usual interstitial pneumonia | 5 (26) |
| Probable usual interstitial pneumonia | 2 (11) |
| Definite usual interstitial pneumonia | 0 (0) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



