Introduction
Rheumatoid arthritis (RA) is a systemic autoimmune disease associated with a chronic inflammatory process that primarily affects synovial joints, causing irreversible bone erosions. The disease is characterised clinically by symmetric polyarticular inflammation, which can lead to progressive joint damage. As a result, it can be associated with substantial functional disability, morbidity, and accelerated mortality, which pose a substantial societal burden [
1,
2,
3]. RA affects approximately 0.4-1% of the global population, with higher rates in Northern Europe and North America than in Southern Europe.
Beyond its impact on joints, RA is a systemic disease that can affect the entire body and often leads to other health conditions. It can lead to complications involving the respiratory system (lung inflammation), the cardiovascular system (heart disease), and the nervous system. These complications contribute significantly to a reduced life expectancy for individuals with RA. [
4]. Compared to the healthy population, patients with RA have a 1.5-2 times increased risk of developing cardiovascular disease caused by a high proportion of systemic inflammation. The generally accepted theory is that chronic inflammation supports atherosclerosis, causing endothelial dysfunction, leading to earlier and faster progression of atherosclerotic plaques [
7] and leading to the progression of coronary heart disease, heart rhythm dysfunctions, and heart failure [
6].
The introduction of biological treatment of RA has opened new perspectives in understanding the involvement of inflammatory markers, cytokines, and the immune system in the pathology of this disease. In addition to their significant effects on remission and slowing disease progression, most biologics have demonstrated high efficacy in reducing cardiovascular risk [
10]. Although systemic autoimmunity is a defining characteristic of RA, the primary inflammatory site is the synovium. [
3]. The cause of RA is unclear, but some research suggests a possible genetic link. Susceptibility to the disease and external influences trigger an immune reaction with subsequent production of inflammatory cytokines in the initial stages of the disease. External factors include smoking, obesity or low levels of fertile hormones in women [
11].
Pathogenesis of RA has been extensively studied for decades and has revealed several risk factors and mechanisms involved in the early development of the disease. It is now well established that circulating autoantibodies, for example, rheumatoid factor (RF) and anti-citrullinated protein antibodies (ACPA), can be detected in the serum before RA symptom onset, often by many years. Before symptoms appear, individuals with RA may experience a pre-clinical phase characterised by autoimmunity. This phase usually involves markers of inflammation, such as elevated levels of C-reactive protein (CRP) in the blood and increased production of pro-inflammatory molecules like TNF-α, IL-6, IL-12, IFN-γ, and chemokines. Changes in these serum biomarkers can be predictors of the onset of the disease. For many patients, the appearance of synovitis detectable through a physical examination can signal the transition from the pre-clinical stage of the disease to its active phase. Advanced imaging modalities, such as MRI and musculoskeletal ultrasound (MSUS), provide critical insights regarding the transition to clinical RA (
Table 1).
The progression includes synovitis, bone marrow oedema, and tenosynovitis, particularly noticeable in the wrist, metacarpophalangeal (MCP) joints and metatarsophalangeal (MTP) joints. Clinically suspected arthralgia indicates future inflammatory arthritis but is independent of other clinical risk factors (such as age, CRP, ACPA), resulting in differences between seronegative and seropositive patients. Seronegative individuals have less involvement of the lower limbs, a shorter time from symptom onset to manifestation with arthralgias, more sensitive joints and a longer time to develop arthritis than seropositive individuals [
3].
Clinical manifestations of early rheumatoid disease include mild joint and mild systemic symptoms. Extraarticular signs are more commonly observed in later disease, although they may occasionally precede joint manifestations. Systemic changes manifest as weight loss or slightly elevated temperature. Joint pain often begins with stiffness and swelling of small joints of the hands and feet in a symmetrical distribution, including the MCP and MTP joints or the proximal interphalangeal (PIP) joint [
3,
12].
Although rheumatoid arthritis (RA) involves widespread inflammation throughout the body, it primarily targets the joints. Accurately diagnosing RA requires careful consideration of typical joint-related symptoms and the results of laboratory and imaging tests [
4]. Several other conditions can mimic RA, including osteoarthritis, lupus, Sjögren's syndrome, and viral infections like hepatitis B and C and HIV. These can also cause joint pain and inflammation and may even lead to a positive rheumatoid factor (RF) test, which is often seen in RA. However, the presence of anti-citrullinated protein antibodies (ACPAs) is a much more specific indicator of RA and significantly aids in diagnosis [
3].
Several clinical assessment tools help to monitor the activity of rheumatoid arthritis. Early diagnosis and treatment of new RA classification criteria were proposed as part of a joint ACR/European League Against Rheumatism (EULAR) effort in 2010. An updated recommendation from the American College of Rheumatology (ACR) in 2019 recommended the use of disease activity score (DAS28), clinical disease activity index (CDAI) and simplified disease activity index (SDAI). For their determination, in addition to the number of swollen and tender joints, erythrocyte sedimentation rate, CRP values and global assessment of the disease by the patient or doctor on a visual analogue scale are considered. Many studies have confirmed the relevance of the objectification of disease activity. Rheumatologists recommend monitoring the DAS28 index during routine examinations. The number 28 refers to the number of joints assessed. Doctors count painful and swollen joints; the resulting number defines the disease activity index or its change over time. [
4,
13,
14]. The scale of reference values of clinically monitored RA disease indicators is in
Table 2.
A deeper understanding of the pathogenesis of rheumatoid arthritis is critical to solving all the questions that this disease raises in its heterogeneity. Despite progress in RA research, we still need an overview of the mechanism of action of all signalling pathways involved in the pathogenesis of the disease. Significant findings in the broader spectrum of immune-mediated inflammatory conditions and various strategic procedures have proven themselves in clinical practice in recent decades. A critical step was the discovery of the complex regulatory roles of nuclear factor κB (NK-κB), which represents a family of inducible transcription factors that regulate many genes involved in immune processes. Most pharmacological interventions focus primarily on suppressing inflammation associated with the NK-κB and TNF-α pathways.
NF-κB Signalling in Inflammation
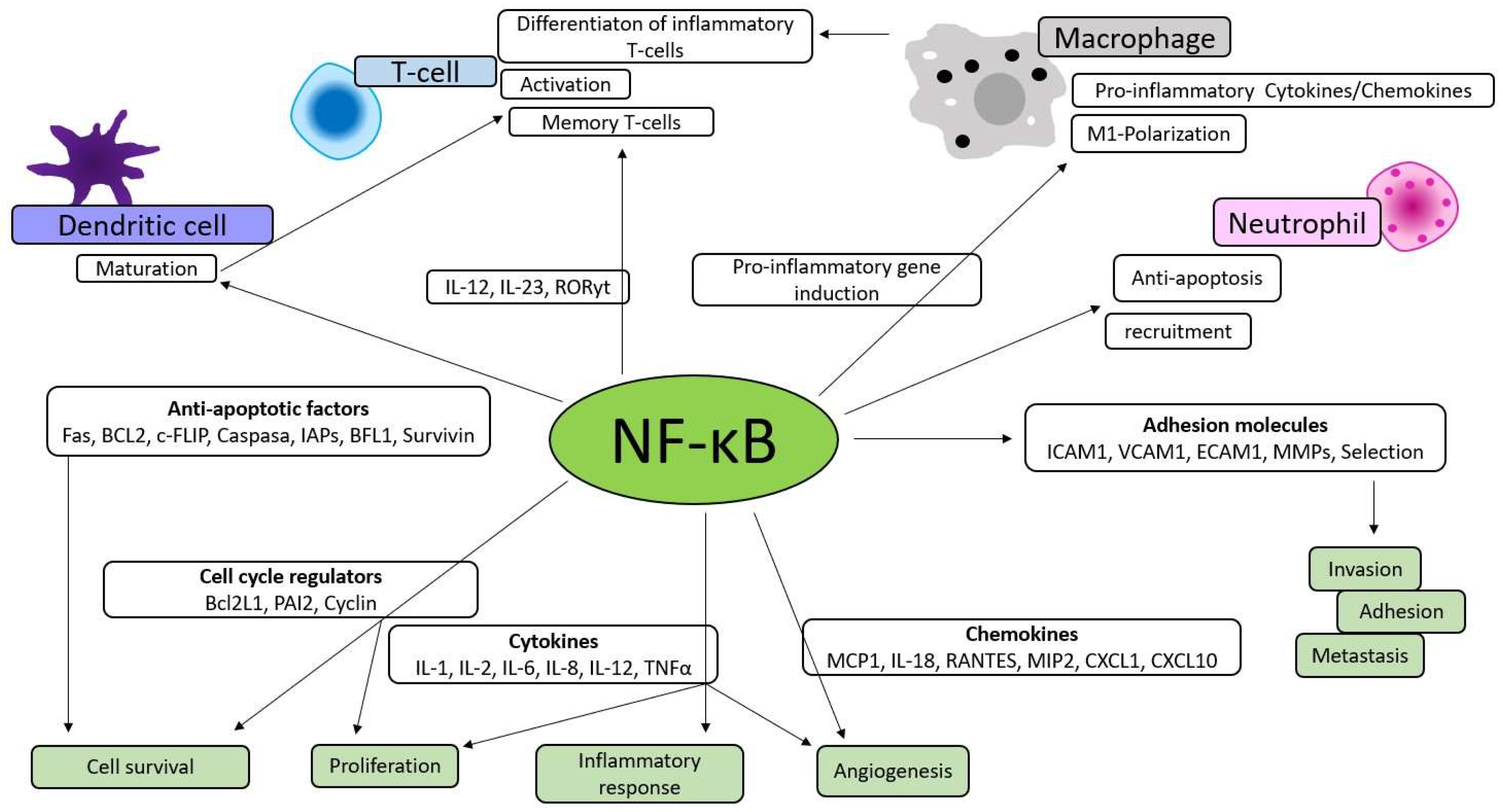
NK-κB is involved in the survival, activation and differentiation of innate immune cells and inflammatory T-cells, which implies that the reduced activity and deregulation of the factor directly contribute to the pathogenic processes of various inflammatory diseases. New and significant T-cell subgroups have been discovered, including peripheral helper T (Tph) cells, found in both synovial B-cell clusters and circulating in the bloodstream. These Tph cells stimulate B-cells to produce interleukin-21, which supports immunoglobulin affinity maturation, among other functions. Tph cells also play a role in B-cell proliferation and differentiation into antibody-producing plasma cells [
4].
In addition to directly affecting inflammation, it also increases the production of inflammatory cytokines, chemokines, and adhesion molecules, thereby participating in cell proliferation, apoptosis, morphogenesis, and differentiation, as shown in
Figure 1 [
22,
23].
NF-κB signalling responses in inflammation occur through two signalling pathways, the canonical and non-canonical. Membrane receptors (TNF, Il-1, Toll-like receptors), receptors for T-cells and B-cells and pro-inflammatory stimuli can activate the canonical signalling pathway. TNF superfamily, the receptor for lymphotoxin β, CD40, the activating factor of B-cells, or the receptor activator of NK-κB can activate the non-canonical signalling pathway [
22]. The canonical and non-canonical NF-κB signalling pathways mediate receptor activator of NF-κB ligand (RANKL) - induced differentiation of monocytes/macrophages into bone-resorbing osteoclasts, whose deregulation contributes to inflammatory loss of bone mass associated with rheumatoid arthritis [
24].
The most studied is an interaction between the pathological state of the bone mass and the immune system [
26]. The latest knowledge brings different therapeutic strategies to the fore, which consist not only of suppressing synovial inflammation and inhibiting bone erosion but also of the complex osteoimmunological understanding of rheumatoid arthritis [
24]. The interaction between RANKL and its activator RANK receptor promotes osteoclasts' differentiation, maturation and survival, leading to increased bone resorption and, thus, bone loss [
25].
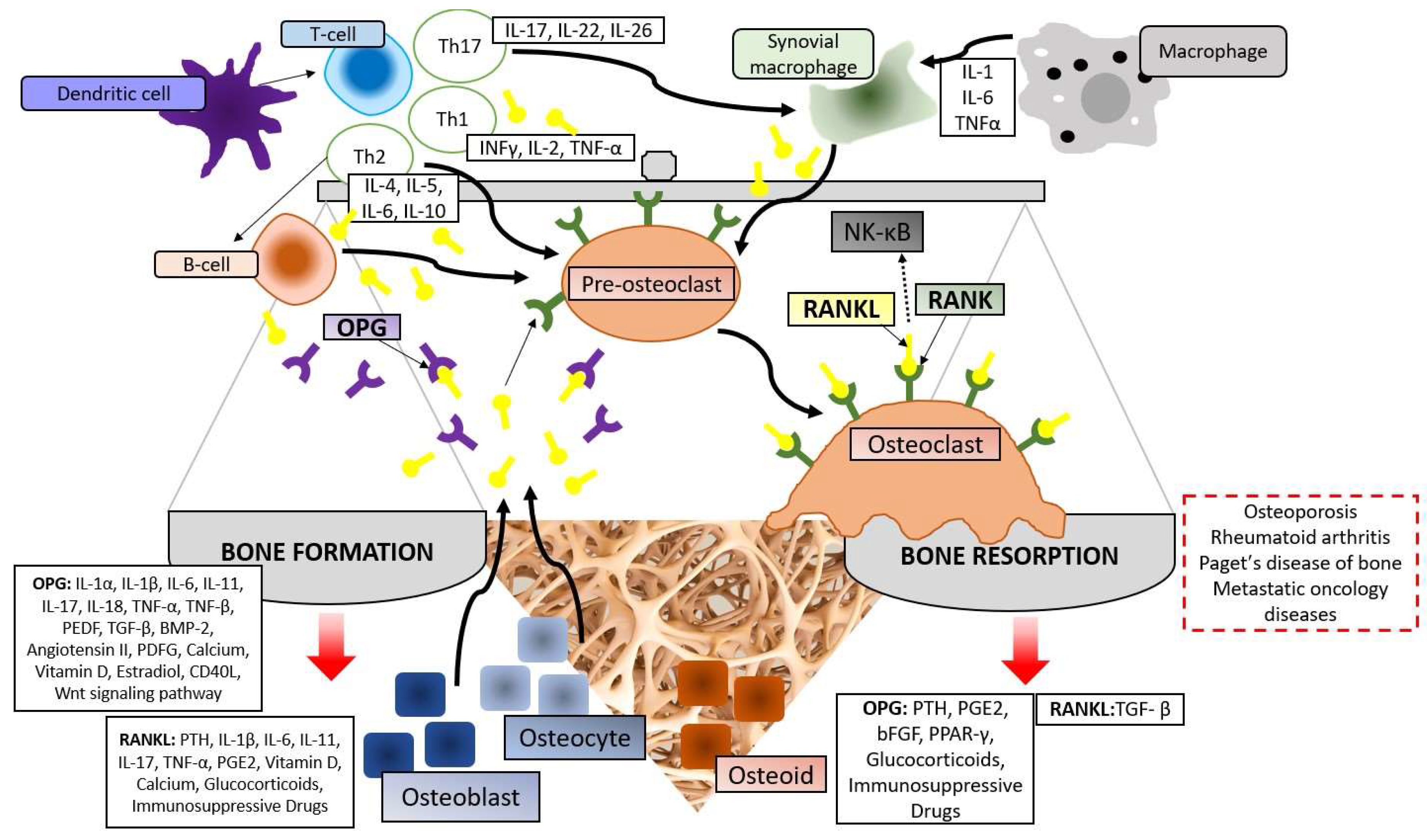
An important part of the RANKL/RANK signalling pathway is osteoprotegerin (OPG), which is known as an inhibitory factor of osteoclast genesis. The osteoblast lineage of bone, vascular endothelial cells, B-cells, and dendritic cells of the immune system express OPG. By inhibiting the interaction of RANKL and RANK, it prevents the formation of osteoclasts and, thereby, osteoclastic bone resorption [
27]. The discovery that cellular regulators of the immune and bone systems respond to the same cytokine systems and originate from common progenitors is the focus of the osteoimmunological mechanisms of diseases such as RA. The ratio of RANKL and OPG determines the physiological balance of bone formation and turnover in osteoimmunology (
Figure 2).
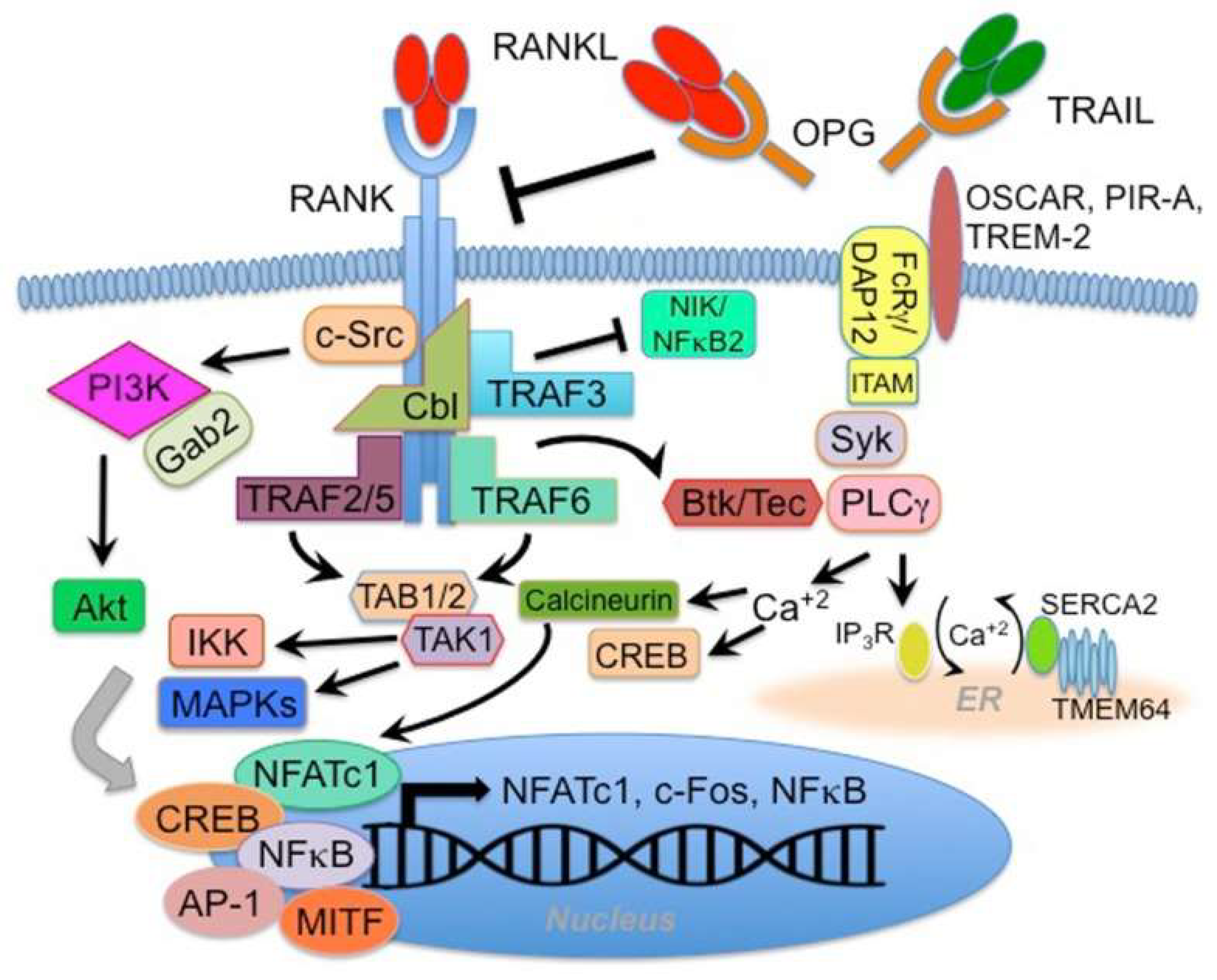
The RANK receptor has no intrinsic activity; therefore, its activation depends on docking and adapter proteins, such as TRAF 2, 3, 5, 6, Gab2 and Cbl. RANK-activated gene transcription varies in specific cells, but the typical result is the expression of NFATc1, c-fos, and NF-κB-related genes. The pathway via TRAF 2, 5 and 6 activates the TAB1/TAB2/TAK1 complex, which, in cooperation with other upstream kinases, leads to the activation of IKKβ and MAP kinases. In the nucleus, this results in the translocation and activation of transcription factors, including NFATc, CREB, NF-κB, AP-1 and MITF. RANK-associated TRAF 3 negatively regulates a non-canonical pathway by regulating the upstream kinase NIK. The TRAF 3 RING finger domain inhibits NIK and is overcome by RANK activation by RANKL. This, in turn, triggers linked TRAF 3. Gab2 and Cbl are bound to RANK-mediated activation of c-Src, PI3 kinase and Akt. A specific pathway for osteoclast cells is signalling between RANK and DAP12/FcRγ binding to the surface receptor OSCAR, PIR-A or TREM-2 to activate the Syk-PLCγ pathway and flux. Activation increases both NFATc1 and CREB activity. RANK also regulates calcium influx into osteoclast cells by interacting with the transmembrane protein TMEM64 with the sarcoplasmic reticulum ATPase (SERCA2), which promotes CREB and NFATc1 activity (
Figure 3) [
26].
Osteoprotegerin (OPG) captures RANKL molecules and prevents them from binding to RANK receptors. This interaction is critical to understanding complex diseases where the immune system plays a significant role. B cells increase RANKL production by activating Toll-like receptor (TLR) ligands such as LPS. Dendritic cells also activate T cells that display MHC/antigen complexes recognised by T cell receptors (TCRs). These activated T cells and other sources, such as osteoblasts and bone stromal cells, contribute to elevated RANKL levels. Detailed research into how RA develops has revealed that many dendritic cells gather in the synovium, the tissue lining the joints, and spread into the surrounding bone.
Chemotactically aggregated dendritic cells interact with T-cells via the RANK-RANKL pathway to activate an inflammatory response. Interactions of dendritic cells with helper T-cells influence their differentiation into Th1, Th2 and Th17 subsets. Th1 cells primarily secrete interferon-gamma, IL-2 and TNF-α to eradicate intracellular pathogens. Through TNF-α, they support the differentiation of osteoclast precursor cells, leading to bone tissue absorption and destruction. TNF-α suppresses osteoblast differentiation by inhibiting Wnt signalling and inhibits the osteogenic process by up-regulating the expression of the ubiquitin regulatory factor SMAD (a protein that mediates the canonical signalling cascade) in osteoblasts. The effect of Th1 cells is enhanced under conditions with low oestrogen levels or in the presence of inflammation. Th2 cells are involved in B-cell activation and elimination of extracellular pathogens by secretion of IL-4, IL-5, IL-6 and IL-10. IL-17, IL-22 and IL-26 are produced by Th17 cells, thereby inducing RANKL through synovial fibroblasts in the inflammatory process. Since synovial macrophages naturally increase the expression of RANKL through the secretion of IL-1, IL-6 and TNF-α, just like T-cells, the complexity of the osteoimmune system in RA is ensured [
26,
27,
28,
29].
Therefore, RANKL has become a subject of research interest in managing autoimmune diseases as a potential biomarker and, in the future, as a standard therapeutic tool. Denosumab is a fully human monoclonal antibody that binds RANKL with high affinity and selectivity. In this way, it suppresses the signalling pathway, blocks the association of RANKL with its RANK receptor in osteoclast precursor cells, and leads to the inhibition of osteoclast genesis. Denosumab is administered subcutaneously every six months at a dose of 60 mg to patients with osteoporosis. It is well known for its ability to increase BMD and decrease markers of bone turnover. Several clinical trials studied the role of denosumab in RA bone damage, summarised in a 2023 retrospective analysis of published studies. Denosumab has no anti-inflammatory properties but can suppress osteoporosis and joint destruction in patients taking csDMARDs (conventional synthetic disease-modifying antirheumatic drugs). Combination therapy may represent a future treatment strategy to inhibit the progression of bone erosion associated with RA [
30].
RANKL or OPG laboratory determination is expensive, requires expert scientific procedures, and is not routine. One proven method for determining these biomarkers is the ELISA method, an analytical method used for quantitatively determining various antigens. It is determined spectrophotometrically or based on fluorescence (fluorometric determination). The concentration of the product is proportional to the concentration of antigen or antibody in the sample. Another common feature of the ELISA method is anchoring (adsorption, covalent binding) of antigen or antibody to an insoluble carrier (often the surface of a reaction vessel or microtiter plate), which facilitates the separation of immunochemically bound molecules. The determination of the biomarkers RANKL and OPG is also based on the principle of sandwich immunoanalytical, while plasma obtained from the patient's whole blood is required to perform the test.
Rheumatoid Arthritis Therapy
Elucidation of the signalling pathways of NK-κB activation led to using it as a therapeutic target for chronic inflammatory diseases such as rheumatoid arthritis. Recent decades have seen significant progress in treating rheumatoid arthritis. Primarily pharmacological, secondary non-pharmacological methods like physiotherapy may prevent contractures and improve joint mobility. The European League Against Rheumatism, with the abbreviation EULAR (European League Against Rheumatism), governs Rheumatoid arthritis pharmacotherapy currently in Slovakia according to the recommendations, which issued five overarching principles and 11 recommendations regarding the use of antirheumatic drugs [
15].
According to the ACR and EULAR recommendations, the pharmacotherapy treatment of RA from two perspectives: symptomatic treatment based on the administration of nonsteroidal anti-inflammatory drugs (NSAIDs) and disease-modifying treatment based on the administration of antirheumatic medicines to promote remission by suppressing autoimmune activity and delaying or preventing joint destruction degeneration. The group of these pharmacologically effective drugs is generally called disease-modifying antirheumatic drugs with the designation DMARD, including conventional synthetic DMARDs (csDMARDs), targeted synthetic DMARDs (tsDMARDs) and biological DMARDs (bDMARDs) with the representation of specific drugs listed in
Table 3 [
1,
3,
15]. The prognosis of the disease depends on the early use of effective DMARDs, which can sufficiently suppress RA activity. Most patients newly diagnosed with RA can expect to experience remission or low disease activity if they are treated early by a skilled rheumatologist. A breakthrough in the field of RA treatment was the introduction of biological agents, which significantly improved patients' prognosis and quality of life [
3].
In the initial inflammatory phase of RA, as a symptomatic treatment to suppress pain and stiffness, NSAIDs are used. Their mechanism of action is the inhibition of cyclooxygenase 1 (COX-1) and cyclooxygenase 2 (COX-2); in the case of coxibs, it is a selective inhibition of COX-2 by blocking the synthesis of prostaglandins from arachidonic acid found in cell membranes. NSAIDs also inhibit the synthesis of pro-inflammatory cytokines and leukotrienes, prevent the release of lysosomal enzymes, and suppress the activity of polymorphonuclear cells and superoxide radicals [
16]. However, the disadvantage of this group of drugs still needs to be more suitable for long-term use due to the gastrointestinal problems and cardiovascular risks they pose to the patient. In the case of their involvement in the treatment of RA, they do not affect the overall disease's activity or its progression [
17].
Glucocorticoids (GC) (prednisone, hydrocortisone, prednisolone, and dexamethasone) have greater potential efficacy than NSAIDs due to their complex anti-inflammatory and immunosuppressive effects. Although NSAIDs have a better safety profile, GCs are given only for a short time as a bridge to DMARDs [
1].
The standard of care for the initial treatment of early rheumatoid arthritis is methotrexate. Black and his team 1964 published the first study on the use of methotrexate in patients with RA. Methotrexate is effective in patients with a severe disease course [
18]. Since its recognition as a potential treatment for RA, methotrexate has had the most significant impact on the management of RA to date and remains the mainstay of therapy. It has proven effective in monotherapy and as a basis for combination strategies with targeted conventional and biological disease-modifying antirheumatic drugs. These drugs are characterised by great effectiveness due to their targeted intervention in the etiopathogenetic processes in the organism [
13]. Biological drugs for autoimmune diseases have been developed based on knowledge of immune system abnormalities. That is why they are designed for precise targeting. They are administered either intravenously or subcutaneously directly.
The development of new biological drugs can specifically act on proteins and components of the immune system that are behind the development of autoimmune diseases [
19]. Biologic drugs used in the treatment of RA affect the biological response to various cytokines, especially TNF-α. It is the primary cytokine responsible for the systemic inflammation typical of RA, and its level is pathologically elevated. In general, bDMARDs can be subcategorised as anti-TNF biologics or non-anti-TNF-α biologics. Anti-TNF-α drugs act antagonistically by binding to receptor TNF-α. Non-anti-TNF-α drugs, which interfere with inflammatory factors and surface proteins of lymphocytes, have a similar effect.
Table 3 mentions the individual biological medicines used for the treatment of RA according to the influence of TNF-α. In clinical practice, they are used when csDMARDs do not work, requiring professional monitoring to control adverse events [
20].
The newest class of drugs in RA pharmacotherapy are Janus kinase (JAK) inhibitors, which have gained tremendous popularity quickly due to their oral form and targeting. The human JAK family was first described over twenty years ago and includes JAK1, JAK2, JAK3 and non-receptor tyrosine-protein kinase (TYK2). They are intracellular effectors of type I and II cytokine receptors, which mediate various cytokine signalling pathways through STAT transcription factors. The cytokine that binds to a receptor in the cell membrane initiates the signalling cascade. The receptors mutually form homodimers or heterodimers and undergo a subsequent structural conformational change, which enables the attachment of JAK during simultaneous phosphorylation of the tyrosine residues of the receptor. Subsequently, a docking site is created for the STAT protein, which is also phosphorylated. In this way, JAK activity accelerates the transport/translocation of STAT transcription factor proteins into the cell nucleus to trigger the expression of specific genes [
21].