
Submitted:
27 November 2024
Posted:
28 November 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Before the Coronavirus disease 2019 (COVID-19) era, the global prevalence of pulmonary arterial hypertension (PAH) was between 0.4 and 1.4 per 100,000 people. The long-term effects of protracted COVID associated with pulmonary vascular disease (PVD) risk factors such as obesity, cardiovascular disease, and some infectious diseases are expected to increase this prevalence. According to preliminary data, the exact prevalence of early estimates put the prevalence of PVD in patients with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection at 22%, although its predictive value is unknown. PVD caused by COVID-19 co-infections is understudied and underreported during COVID-19, and its impact on the future is unknown. However, due to COVID-19/co-infection pathophysiological effects on pulmonary vascularization, PVD mortality and morbidity may be a genuine worry now and in the future. Based on reported studies, this literature looks at the potential link between COVID-19, parasitic coinfection, and PVD. This review also highlights hypothetical pathophysiological mechanisms between COVID-19 and parasitic coinfection that could trigger PVD. This review hypothesized that multiple pathways associated with schistosomiasis, human immunodeficiency virus (HIV), pulmonary tuberculosis (PTB), pulmonary aspergillosis, Wuchereria bancrofti, Clonorchis sinensis, paracoccidioidomycosis, Human Herpesvirus 8, and Scrub typhus coupled with acute or long COVID-19 may increase the burden of PVD and worsen its mortality in the future. This is due to acute and/or chronic inflammation, which contributes to endothelial dysfunctions and coagulation abnormalities in pulmonary vascularization. This review advises that PVD with a history of COVID-19/co-infections be screened systematically to enhance the epidemiology, clinical profiles, and prognostics. Further experimental studies are also needed to determine pathophysiological pathways between PVD and with history of COVID-19/co-infections.
Keywords:
Co-infections
; COVID-19
; long COVID
; PVD
; Pulmonary Vascular Remodelling and hypothesis
Peter S Nyasulu 1,*,†, Jacques L. Tamuzi 2,†, Rudolf KF Oliveira 3, Suellen Dos Santos Oliveira 4, Nicola Petrosillo 5, Vinicio de Jesus Perez 6, Navneet Dhillon 7 and Ghazwan Butrous 8
1 Division of Epidemiology & Biostatistics, Department of Global Health, Faculty of Medicine & Health Sciences, Stellenbosch University, France Van Zijl Drive, Tygerberg, Cape Town, South Africa
2 Department of Global Health, Faculty of Medicine and Health Sciences, Stellenbosch University, South Africa; drjacques.tamuzi@gmail.com
3 Universidade Federal de São Paulo, Brazil; rudolf.oliveira@unifesp.br
4 University of Illino, USA; suelleno@uic.edu
5 Policlinico Universitario, Italy; n.petrosillo@policlinicocampus.it
6 14Division of Pulmonary and Critical Care Medicine and Department of Medicine Stanford University Stanford 94305 California, USA; vdejesus@stanford.edu
7 Division of Pulmonary and Critical Care Medicine, Department of Internal Medicine, University of Kansas Medical Center, Mail Stop 3007, 3901 Rainbow Blvd, Kansas City, KS, 66160, USA; ndhillon@kumc.edu
8 University of Kent, Canterbury, UK; gbutrous@gmail.com
1. Introduction
Pulmonary vascular diseases (PVDs) is a progressive, severe, and hemodynamic disorder that may cause high mortality if not well treated [1]. Increased pulmonary vascular resistance (PVR) is caused by diseases of the pulmonary vasculature in pulmonary embolism, chronic thromboembolic pulmonary hypertension (CTEPH), and pulmonary arterial hypertension (PAH) [2]. Since the World Health Organization (WHO) conducted the first World Symposium on Pulmonary Hypertension (WSPH) in Geneva in 1973, PAH has been defined as mean pulmonary arterial pressure (mPAP) ≥ 25 mmHg assessed by right heart catheterisation (RHC) in the supine position at rest [3]. At rest, mPAP is 14.0±3.3 mmHg; this value is independent of gender and ethnicity and may be only slightly altered by age and posture [4]. PAH prevalence ranged from 0.4 to 1.4 per 100,000 people worldwide [5]. PAH prevalence is projected to rise before the COVID-19 event due to the long-term impacts of protracted COVID related with PVDs risk factors such as obesity, cardiovascular disease, and various infectious diseases (schistosomiasis, HIV, pulmonary tuberculosis...). PAH may rise more than expected due to COVID-19 pulmonary hemodynamics via endothelial dysfunction, vascular leak, and thrombotic microangiopathy mechanisms similar to those contributing to pulmonary vascular disease [6,7]. Lung parenchymal damage and altered pulmonary haemodynamics may determine PAH and secondary right ventricular (RV) involvement in patients with COVID-19, even in non-advanced disease stages [8].
A major source of concern is the occurrence and persistence of pulmonary vasculature pathology in post-acute COVID-19 stage and long COVID associated with potential infectious diseases associated with PVDs, primarily schistosomiasis, HIV, tuberculosis, pulmonary aspergillosis, Wuchereria bancrofti, Clonorchis sinensis, paracoccidioidomycosis, Human Herpesvirus 8, and Scrub typhus. The likelihood of observing a high prevalence of PVDs due to co-infections in post-COVID era is pronounced. This could be explained by the long COVID persistently impaired physiological gas exchange caused by damaged lung architecture, including vasculature, hypoxic vasoconstriction, thrombotic events, direct viral damage, pro-inflammatory cytokines, and microthrombi [9].
PVDs are caused by a combination of genetic and environmental variables, as well as non-communicable and infectious diseases. Co-infection-related PVDs are yet poorly described. The association of COVID-19 with other possible viral, bacterial, protozoal, and helminthic agents that may cause PVDs should be cause for worry. According to one study, 19% of COVID-19 patients have co-infections, and 24% have superinfections [10]. The pooled prevalence of pathogen type stratified by co- or superinfection was 10% for viral co-infections and 4% for viral superinfections; 8% for bacterial co-infections and 20% for bacterial superinfections; 4% for fungus co-infections and 8% for fungal superinfections [10]. Studies have revealed that most of the potential co-infections inducing PVDs may be associated COVID-19. Early estimates suggested that the prevalence of PH is estimated at 22% in patients with SARS-COV-2 infection[11], and another study estimated that The incidence of PVDs in post-COVID-19 patients with suspected manifestations of PH is 70% [12]. The authors found that the prevalence of PVDs and right ventricular dysfunction in COVID-19 was 22% and 19%, respectively and these were associated with increased hospital morbidity and mortality [13]. This prevalence may be higher in protracted COVID-19 coupled with PVDs related co-infections. Even though PVDs from noncommunicable diseases are more documented and reported, infectious diseases such as schistosomiasis, human immunodeficiency virus (HIV), pulmonary tuberculosis (PTB), filariasis, pulmonary aspergillosis, and others are significant sources of PVDs in respective high burden regions. PVDs associated with COVID-19 co-infections is understudied and underreported during the COVID-19 and post-COVID era, and its impact on future PVDs is unknown. However, due to the pathophysiological effects of COVID-19/co-infections on pulmonary vascularization and their impacts in inducing PVDs, COVID-19 infections may be a genuine worry now and in the future.
Interestingly, of the co-infections that cause PVDs, it was reported that many HIV patients in Africa are also co-infected with schistosomiasis [14]. Early, this study stipulated that both HIV proteins and schistosoma eggs-inducing granulomas implicated in the release of inflammatory mediators known to cause adverse pulmonary arterial remodelling and endothelial cell injury [14]. Later, this hypothesis was confirmed by an experimental study that demonstrated that combined pulmonary persistence of HIV proteins and Schistosoma eggs, as it may occur in co-infected people, alters the cytokine landscape and targets the vascular endothelium for aggravated pulmonary vascular pathology [15]. This paper reviews the current literature on the potential link between COVID-19, parasitic coinfection, and PVDs. The authors acknowledge their findings are hypothetical and need more research to confirm this link. However, the authors believe this review is essential because it highlights a potential area of concern and encourages further research.
2. COVID-19 and Parasitic Co-Infection
2.1. Schistosomiasis and Other Helminthic Diseases
Over 230 million people (80% of which are in Africa) are affected by schistosomiasis, which may cause PVDs in half of them [16,17] and the clinical presentation of pulmonary hypertension in 7–15% of them [16,18,19]. Schistosomiasis is endemic in more than 78 countries and poses a serious disease burden to society, representing a major public health problem [20]. Approximately 10–20% of those with hepatosplenic disease, or 2–5 million people worldwide, develop PVDs, a progressive and fatal illness [21,22]. This high prevalence makes schistosomiasis one of the most common causes of PAH worldwide [19]. Initial symptoms include dyspnoea, dry cough, lower extremity oedema, fatigue and exercise intolerance [21]. As the disease progresses, patients can experience chest pain from right ventricular angina and syncope caused by depressed cardiac output, post-inflammatory cardiomyopathy, complex ventricular arrhythmias, and low systolic blood pressure [21,23]. Patients develop myocarditis, progressive right heart failure, and heart failure [23] . Physical examination may reveal a prominent pulmonic component, second heart sound (P2), right ventricular heave and digital clubbing. Patients with frank right heart failure manifest cyanosis and peripheral oedema or anasarca [21]. Radiographs may reveal cardiomegaly, particularly dilatation of the right ventricle and right atrium, and enlarged pulmonary trunk and arteries, with pruning of the distal vasculature. Electrocardiography typically shows right ventricular hypertrophy or strain and right atrial enlargement and may also reveal a right bundle branch block [21]. Echocardiography demonstrates right ventricular dilatation, potentially compressing the left ventricle with septal bowing, usually accompanied by right atrial dilation, tricuspid valve regurgitation and an increased pressure gradient across the tricuspid valve. Finally, right heart catheterization (when available) is used to confirm the diagnosis of PVDs in the absence of an elevated pulmonary artery occlusion pressure [21]. Other Helminthic diseases can induce PVDs such as Wuchereria bancrofti, a threadlike worm that causes filariasis (elephantiasis) and Clonorchis sinensis (Chinese liver fluke), which is a widespread parasite in southeast Asia, has been associated with cases of PVDs.
The literature review has revealed cases of helminthic diseases associated with COVID-19. Two cohort studies conducted in Ethiopia reported COVID-19/schistosomiasis co-infection of 4.5% [24] and 6.2% [25], respectively. COVID-19 infections and induced PVDs may potentialize multiple other co-infections. Among them, we found co-infections with Wuchereria bancrofti, a threadlike worm that causes filariasis (elephantiasis) [16,26,27], Clonorchis sinensis (Chinese liver fluke), which is a widespread parasite in southeast Asia, has been associated with cases of pulmonary hypertension [28] A case report of SARS-CoV-2/Wuchereria bancrofti co-infection was also reported [19].
The effect of COVID-19 on schistosomiasis-inducing PVD (COVID-Sch-PVD) is not well described. It is not clear if COVID-19 infection could trigger a host’s immune response to chronic indolent schistosomiasis infections or increase COVID-Sch-PVD morbidity and mortality of patients who are chronically exposed to the schistosoma parasite. Chronic portal hypertension can result in the opening of portocaval shunts, allowing schistosoma eggs to migrate from the portal system to the pulmonary parenchyma in which acute or long COVID may increase the likelihood of COVID-Sch-PVDs. Within the lungs, the eggs induce an immune response, which similarly results in severe granulomas which may be associated with severe inflammation related to COVID-19. This pathology of pulmonary arterial vascular remodeling could also be manifested, resulting in clinical COVID-Sch-PVD [29]. Severe COVID-19 has multisystem manifestations, including systemic thrombosis, cardiac injury, renal failure, and hepatic dysfunction. Following the acute phase, many patients with COVID-19 had symptoms due to persistent damage in several organs, including the lung vasculature and the sequelae of this damage such as chronic lung fibrosis and possibly PVDs [30]. The patho-immunological interactions of both COVID-19 and helminthic diseases are complex. However, acute COVID-19 phase, as well as long COVID, may interact in multiple pathways with chronic schistosomiasis and potentialize COVID-Sch-PVD (Figure 1).
Clinical study on SARS-CoV-2 reveals that the virus may cause alterations in pulmonary hemodynamics via processes comparable to those that cause PVDs , such as endothelial dysfunction, vascular leak, and thrombotic microangiopathy [6,7]. While most acute pulmonary embolisms and clots resolve with anticoagulation, clot persistence can lead to continued post-embolic symptoms of shortness of breath and the development of PVDs. About 30–50% of the patients have persistent defects up to 1 year after diagnosis [31,32]. The lumen can contain both fresh microthrombi and chronic thrombotic lesions with acute or chronic inflammatory cells and fibroblasts with variable degrees of organization. Increased PAH may be caused by a combination of pulmonary vasoconstriction, inward vascular wall remodelling, and in situ thrombosis, increasing vascular stiffness and narrowing of the vascular lumen [33,34]. The endothelium in PVDs is thought to be activated or affected by chronic hypoxia, inflammation, viral infection, mechanical stretch, shear stress, and/or unknown causes [33]. This results in altered production of endothelial mediators and the release of inflammatory mediators, which have been increasingly implicated in PVDs [33,35]. These inflammatory mediators, which include cell adhesion molecules, cytokines, chemokines, and growth factors, direct inflammatory cell recruitment and propagate a number of inflammatory pathways that lead to vascular cell proliferation, migration, and extracellular matrix deposition, all of which contribute to PH's structural remodelling [33,36]. Bone morphogenic protein (BMP) and transforming growth factor-beta (TGF-β) are known to synergistically activate regulatory T cells to reduce inflammation and prevent autoimmune disease. Therefore, a loss of bone morphogenetic protein receptor type 2 (BMPR2) function can result in dysregulated immune cell recruitment, increased cytokine expression, and vascular infiltration by inflammatory cells into the intima [9,37]. Loss-of-function mutations in the BMPR2 are responsible for heritable pulmonary arterial hypertension in most (>80%) patients[38]. Loss of pericytes during severe cases of COVID-19 is associated with vascular inflammation [39]. Inflammatory processes are prominent in various forms of PVD. They are increasingly recognized as major pathogenic components of pulmonary vascular remodelling in PVDs related to more classical forms of inflammatory syndromes, fibrosis and microvascular clotting such as schistosomiasis (Figure 1).
Helminths are typically associated with Th2-mediated immune responses through various regulatory mechanisms [40,41]. The downregulation of the inflammation associated with Th2 immune response in schistosomiasis infection in endemic countries may impair immunity to COVID-19, thus resulting in increased susceptibility and higher occurrence of COVID-19 in schistosomiasis-endemic areas (Figure 1). In Sch-PVD, the release of this cytokine is probably a consequence of the Th2 inflammation elicited by eggs deposited into lungs through a series of cellular and signaling events [42,43]. An interesting aspect is the fact that TGF-β activation seems to become autonomous and independent of schistosome antigen resulting in a persistent vascular disease despite parasite eradication [42]. Schistosoma infections are associated with a strong CD4 T-helper 2 (Th2) response [44]. The Th2 response follows in response to the egg and causes the production of a battery of cytokines such as IL-4, IL-6, IL-10 IL-13, IL-21, IL-31, AMCase, Ym1, and FIZZ1, as well as various chemokines [44,45]. The Th2 cells suppress the Th1 pro-inflammatory response and produce protective eosinophil-rich granulomatous lesions around newly deposited eggs, but they allow the development of fibrosis [44,46]. The pathogenesis of severe disease in COVID-19 has been linked to the phenomenon of immune hyperactivation, that resembles that of a chronic inflammatory condition [25]. In the acute phase, SARS-CoV-2 infection drastically increases the production of pro-inflammatory cytokines including IL2, IL7, IP10, MIP1α, MCP1, and TNFα, resulting in severe lung damage and fibrosis [47]. Furthermore, patients with long COVID-19 also showed dysregulated levels of Matrix metalloproteinases (MMP)-2/MMP-9 [48], showing the substantial role of SARS-CoV-2 infection on lung fibrosis. Since PAH developing secondarily in pulmonary fibrosis patients (PF-PH) is a frequent co-morbidity, COVID-19/schistosomiasis inducing PVD should be viewed as a serious threat. This could explain the rapid lung damage that could be observed in COVID-19/Schistosoma co-infection [49] as both conditions may activate TGF-β, inducing an additive effect and consequently rapid lung fibrosis that could induce COVID-Sch-PVDs. However, this association may be underdiagnosed, particularly in endemic areas.
However, PVDs may be found in case of SARS-CoV-2 associated with other helminthic co-infection in acute or long COVID periods due to the immunological interaction of both COVID-19 and helminthic PVDs complications associated with SARS-CoV-2/ Wuchereria bancrofti may be also found in case of Clonorchis sinensis/SARS-CoV-2 and hydatid cysts/SARS-CoV-2. Chronic helminthic infection suppresses both Th1 and Th2 responses by actively inducing the expansion of FOXP3+ regulatory T cells, IL-10 producing B cells, and alternatively activated macrophages (AAMs), which together promote the release of regulatory cytokines such as TGF-β and IL-10 [50]. On the one hand, helminthiasis escapes immune surveillance and survive in the human body for prolonged periods via antigen-specific T cell hypoactivity [51]. A muted T helper 1 response with the expansion of T helper 2 and an increase in interleukin (IL)-10 (the cytokines synthesis inhibitor) are characteristic. For filariasis phenotype to manifest (such as hydrocele, lymphedema, and elephantiasis), a CD4+ T cell response is typically triggered, and phenotype absence suggests T cell hypo responsiveness. On the other hand, COVID-19 includes dysregulation of various pro-inflammatory cytokines such as the type I interferon pathway, activation of the humoral immune pathway, and increased secretion of IL-6. This dysregulated immune response is thought to contribute to a severe form of COVID-19. Overall, COVID-19 associated chronic helminthic infections may be associated with increased risk of PVDs. This could be explained by the pathophysiology of COVID-Sch-PVD, which could potentialize each other. Due to these potential pathophysiological mechanisms, PVDs may raise more than expected in affected COVID-19 areas and endemic schistosomiasis and other helminthic diseases inducing PVDs. This could be explained by short- and long-term COVID-19 physiopathology in previous and future schistosomiasis cases.
2.2. HIV and Viral Infections like Other Human Herpesviruses
The prevalence of PAH due to HIV infection is estimated at 8 % (3.65%-12.34%) [52]. The most significant burden of HIV lies in sub-Saharan Africa where more than 20.9 million people were affected in 2022 [53]. Here, the prevalence of PAH due to HIV might be up to 0·5 per 1000 individuals, reaching 20–50 times higher than the prevalence of all PAH subtypes together in the developed world [54]. The incidence of PAH seems to be 1000 times higher in HIV-infected patients than in the general population[44]. Among 131 cases with HIV-PAH, a study found a progressive shortness of breath was the most common presenting symptom (85% of cases), followed by pedal oedema (30%), non-productive cough (19%), fatigue (13%), syncope or near syncope (12%), and chest pain (7%) [55] associated with chest pain, dyspnea, and palpitations [12] Right ventricular failure, myocarditis, progressive right heart failure, and heart failure may constitute the complications related to HIV-PAH. Experimental studies had shown that nicotinamide adenine dinucleotide phosphate (NADPH) oxidases as one of the leading players in the oxidative stress-mediated endothelial dysfunction on the dual hit of HIV-viral protein(s) induced PAH [56] and the presence of HIV-1 proteins likely impact pulmonary vascular resistance and exacerbate hypoxia-induced PH [57] (Figure 2).
COVID-19 has been found to be associated with PVDs and pulmonary embolism [58]. The pooled prevalence of HIV among COVID-19 patients was 26.9% and was significantly higher in studies conducted in Africa compared to those conducted elsewhere [59]. Knowing that the two conditions may be associated with PVDs, it is unknown if they could interact or aggravate PVDs. A study showed that average time interval between the diagnosis of HIV infection and the diagnosis of pulmonary hypertension was 33 months, while in 6% of cases, the diagnosis of HIV infection was established after the diagnosis of pulmonary hypertension [60]. Because of the probable effects of HIV and COVID-19 on the pathogenesis of PVDs, we estimated that this time would be shorter in the case of COVID-HIV-PVD. The pathobiology of COVID-19/HIV inducing COVID-HIV-PVD may be the subject of more attention more particularly in high burden HIV countries where COVID-19 cases were more recorded. Duration for how long after SARS-CoV-2 infection this impaired HIV viral control might persist is not clear, but in conjunction with innates immune dysregulation, it is possible that SARS-CoV-2 infection might allow for HIV provirus to be reactivated and replicate, increasing the risk for developing COVID-HIV-PVD initiated by HIV viral and COVID-19 protein-mediated pulmonary vascular remodelling [9]. Coincidentally, both HIV and COVID-19 are viruses whose membrane fusion protein gp120 and spike protein as well as other HIV proteins such as Tat and Nef, may trigger cell signaling that may promote pulmonary vascular remodelling, in addition to predisposing infected individuals to developing COVID-HIV-PVDs [61] (Figure 2). In severe PVDs, macrophages, lymphocytes and dendritic cells are important inflammatory cellular components in the perivasculature of HIV tissues [44]. Hence, HIV-induced chronic inflammation and immune hyperactivation may enrich the pro-inflammatory milieu implicated in HIV-PVD. Inflammatory cells (macrophages, T and B lymphocytes) have been detected in various parts of the remodelled small pulmonary arteries and in plexiform lesions in many forms of PVDs. Furthermore, elevated serum levels of pro-inflammatory cytokines IL-1 and IL-6, 10, chemokines, and various types of autoantibodies (anti-nuclear, anti-endothelial, anti-fibroblast, and anti–fibrillin-1 have been detected in severe PVDs associated with HIV [44,62] (Figure 2) . In the same line, COVID-19-induced changes in cells of the lung vascular wall, including endothelial cells, pericytes, smooth muscle cells (SMCs) and fibroblasts have been observed [30].The histopathological picture shows vascular intimal and medial hyperplasia, endarteritis obliterans, and severe inflammatory reactions [14,63]. The typical plexogenic arteriopathy is present in >80% of PLWH [14,63]. Based on the acute pathophysiology of SARS-COV-2 connected to PVDs, as well as the protracted COVID pathophysiology associated with HIV, PVDs may pose a major concern in PLWH.
Other viral infections, such as human herpesvirus-8, showed evidence of PVD [64]. Cool et al. found that 62% of cells within and around the plexiform lesions from lung tissue of patients with various causes of PVDs had evidence of infection with human herpesvirus 8 (HHV-8) [44,64]. Herpes virus infection may exacerbate other pulmonary fibrosis-associated pathologies, and this may be hypothesized as more likely in case of COVID-19/herpesvirus co-infections. PVDs are common in pulmonary fibrosis, affecting up to 40% of patients and associated with worse outcomes [65]. Gamma herpes viruses are vasculotropic and there is some evidence of their direct involvement in PVDs [66,67]. Herpes virus, particularly Epstein–Barr virus (EBV), in explant lungs of patients with pulmonary fibrosis undergoing transplantation was associated with vascular remodeling. There was increased TGFβ expression and arterial intimal thickening, compared with disease and healthy control lungs, along with higher mean pulmonary arterial pressure and worse clinical outcomes. Patients with post-COVID manifestations presented with reactivation of EBV in 42.6%, human herpesvirus-8 (HHV-6) in 25.0%, and EBV plus HHV6 in 32.4% [68]. A case of COVID-19/co-infection was described with left ventricular thrombus (LVT) [69]. Even though we did not find a case of COVID-19/ HHV-8 inducing PVDs, it is therefore imperative to assess the role of HHV-8 in PVDs in endemic areas to provide further information on the role of both COVID-19 and herpes viruses that can bring more clarifications on the pathogenesis and interactions on acute stage and long-term stages of both viruses on PVDs.
2.3. Tuberculosis and Other Bacterial Infections
PVDs were incidental to find approximately 15% of them having a preceding history of pulmonary tuberculosis (PTB) without any other forthcoming cause for PVDs [70]. The mean PASP was 29 mmHg and RAP was estimated in 99 subjects with values ≤5 mmHg in 57 (57%), 5–10 mmHg in 34 (34%); and >10 mmHg in 8 (8%). Probable PVD-post TB was observed in 9 subjects, yielding a prevalence of 9% (95% CI: 4.4%–16.7%)[71].This entity of “tuberculosis-related PVDs,” is not well-recognized and has not been given any space in all the classifications of PVDs [72]. Recently, Patel et al. have described PVDs in six out of 50 (12%) cases of PVDs develop from tuberculosis [11]. An association was found between PVDs and the number of previous PTB episodes, with each additional episode of PTB increasing the odds of PH-post PTB 2.13-fold [71]. A systematic review estimated the prevalence of COVID-19/PTB at 3% [2%-5%]. However, this prevalence may be lower due to high case fatality found in post-mortem 25% [3%-47%] compared to clinical PTB diagnostics [73]. In a study assessing PAH among 14 cases treated for PTB, estimated pulmonary artery systolic pressure (PASP) of 51 to 80 mm/Hg was found in 9 patients (64.3%). In contrast, PASP of 40 to 50 mm/Hg was found in 4 patients (28.6%) and one patient had PASP more than 80 mm/Hg after 9 years[74]. This finding suggests that PVD will more likely develop with more extensively destroyed lung tissues and progress to poorer outcomes regardless of the regional distribution of PTB [74]. PAH in patients with TB-destroyed lung (TDL) was associated with the severity of lung destruction and led to more frequent exacerbation than that of TDL without PAH. In the same, Ryu et al. reviewed the clinical outcomes of 169 patients with TDL and reported more extensive lung destruction was revealed as a risk factor for a poorer prognosis [75]. In fact, PTB can result in TDL, which is caused when parenchyma is damaged due to excessive M1 activity. Furthermore, macrophages unleash reactive oxygen (ROS), mount oxidative stress, and cause bystander damage to surrounding tissue. Other molecules, such as cathelicidins defensins, cathepsins, matrix metalloproteases and S100 proteins drive connective tissue damage, fibrosis, and angiogenesis. During the process of granuloma formation, activated alveolar macrophages (AMs) (realizing specific cytokines such as TGF-β and IL-10) invade subtending epithelium and attract mononuclear cells from neighboring blood vessels through chemotaxis and form the cellular matrix of the early granuloma [76]. As the result, PTB may trigger PVDs. Some bacterial infections, such as Bordetella Pertussis, may also trigger PVDs through its toxins. The mechanism of development of PVDs is thought to result from residual pulmonary structural damage and pulmonary function abnormalities leading to gas exchange abnormalities and chronic hypoxia [74,77]. In the literature review, case studies have shown the association between pulmonary embolism, TB and COVID-19 [78,79].
Results showed that 50% of COVID-19 patients were coinfected or carried bacterial pathogens [80]. A global meta-analysis estimated the prevalence of COVID-19/PTB at 7.1% (95%CI: 4.0% ~ 10.8%) [81]. However, the incidence of active PTB among COVID-19 patients was higher in high tuberculosis burden countries. Bordetella pertussis infection rate was significantly higher in COVID-19 positive patients [80]. PVDs associated with B. pertussis infection in infants is strongly correlated with disease severity, with 75% of infants that succumb to infection displaying features of pulmonary hypertension compared with just 6% of those that survive infection [82]. Other plausible COVID-19/ bacterial co-infections that could be associated with PVDs are numerous. In fact, COVID-19, and Pneumocystis jirovecii pulmonary co-infection was associated with extensive vascular thromboses in the pulmonary venous territory and a pulmonary edema focus below a subpleural haemorrhage area. Peri bronchial pulmonary artery thromboses [83]. Scrub typhus/COVID-19 co-infections were associated with associated with a transient increase in the risk of vascular events, including pulmonary embolism [84].
TDL may be associated with COVID-19 as reactivation of latent TB in the setting of COVID-19 infection is plausible, given that the two diseases augment each other with a transient decrease in cellular immunity [73,85]. Furthermore, previous or active PTB was a risk factor for COVID-19 both in terms of severity and mortality irrespective of HIV status [86]. In fact, the TGF-β activation pathways in both SARS-CoV-2 and PTB contribute to the production of fibrin, collagen, and secreted proteases (Matrix metalloproteinases) associated with human cavities involved in the formation of fibrosis and tissue remodelling [73,87]. In addition to transforming growth factor beta (TGF- β), and ACE2, other pathways can contribute to SARS-CoV-2 mediated lung fibrosis. In the same line, monocyte chemoattractant protein 1 (MCP-1) is a chemokine that causes lung fibrosis [73]. Studies reported that pulmonary thromboembolism (PTE) has been associated with TB [88]. As current or previous TB was associated with severe COVID-19, this can cause the development of COVID-19–associated coagulopathy, with features of both disseminated intravascular coagulation and thrombotic microangiopathy and PTE [89]. Alveolar and endothelial damage of smaller vessels may be followed by microvascular pulmonary thrombosis, which could then extend to larger vessels. As demonstrated in studies of COVID-19/TB co-infection producing pulmonary embolism, this may increase the risk of PTE [78,88]. In addition, there are permanent changes in lung architecture after TB as shown in TDL pathophysiology described above. These changes may co-exist with long COVID bother PTB and COVID-19 may also induce permanent fibrotic changes. Based on this previous and current PTB associated with COVID-19 may be in risk of PVDs. In the same line, Bordetella pertussis infection as a consequence of PVD and the associated Bordetella pertussis and COVID-19 is well established. How Bordetella pertussis may induce PVDs remains unknown. Reports have described the identification of hyperleukocyte thrombi in small pulmonary blood vessels and speculated that vascular occlusion caused by leukocytosis induced by pertussis toxin results in increased pulmonary arterial pressure and establishment of PVDs [90,91]. In addition, pertussis pneumonia may trigger hypoxia, acute pulmonary vasoconstriction, microcirculation disturbances, and clotting dysfunction [92]. Previous or present Bordetella pertussis infection in conjunction with severe COVID-19 may precipitate the development of PVDs since the pathophysiological mechanisms of both conditions may exacerbate each other.
We estimated that the impact of COVID-19 on TB inducing PVDs (COVID-TB-PVD) may be high with more severe clinical due to the interactions of these conditions in acute phase and long-term stages inducing vasculature, hypoxic vasoconstriction, thrombotic events, pro-inflammatory cytokines, microthrombi and pulmonary fibrosis. However, COVID-19 or long COVID may induced COVID-19-TB-PVD should be subject of investigations to improve its epidemiology and clinical profiles. Figure 3 depicts a hypothetical pathophysiology for acute SARS-CoV-2 and long COVID coupled with PTB and other bacterial infections that cause PVDs. Previous PTB history or GeneXpert associated with D-dimer echocardiogram are suggested in case of COVID-TB-PVD.
2.4. Pulmonary aspergillosis and Other Fungal Infections
Aspergillus is a fungus with a wide spectrum of pulmonary diseases, ranging from noninvasive to an invasive infection, and, because of its angio-trophism, may form microthrombus and pulmonary embolism with an imbalance in the ventilation/perfusion ratio, which worsens hypoxia. As with most chronic granulomatous disease, chronic pulmonary aspergillosis (CPA) may increase the risk of PVDs. However, the association between pulmonary aspergillosis and PVDs is not well established.
COVID-19-associated pulmonary aspergillosis (CAPA) is considered a potentially life-threatening secondary infection in a large number of critically ill COVID-19 patients. Presumed CAPA may be present in as much as 19% of ICU patients [93,94]. Studies have shown that CAPA has a high risk of pulmonary embolism [94,95]. CAPA cumulative incidence of venous thromboembolism reported was 49% in patients admitted to the ICU [8]. Further, cases of extensive pulmonary fibrosis and chronic respiratory failure were reported in CAPA cases [96,97]. Even though the literature did establish the association between CAPA and PVDs, we hypothesize that CAPA may trigger PVDs as the pathophysiological mechanisms have shown extended pulmonary fibrosis and thromboembolism as well as fungal angio-trophism. Thereby facilitating SARS-CoV-2 entry into the target cell via angiotensin-converting enzyme 2 (ACE2). Endothelial cells express the receptors required for SARS-CoV-2 entry into the cells such as ACE2 and transmembrane protease serine subtype 2 (TMPRSS2) is known to be associated with endothelial injury that may potentialize Aspergillus angio-tropism which may aggravate the SARS-CoV-2 infection. This could be one of the plausible explanations high CAPA incidence and severity in the ICU. Lastly, the early inflammatory hyperactivation pathway induced by the SARS-CoV-2 infection may be a major factor in establishing a highly permissive inflammatory environment that favors fungal pathogenesis.
Besides, fungal infections, like paracoccidioidomycosis can cause PVDs in patients. A case reported PTB, COVID-19, and Paragonimus westermani co-infection was also reported [98]. It is unknown how paracoccidioidomycosis associated to COVID-19 may trigger PVDs. Knowing that its pathophysiology may be more similar to CAPA-induced PVDs; as stated above SARS-CoV-2 infection may be a primary component in creating a highly permissive inflammatory way that promotes fungal pathogenesis.
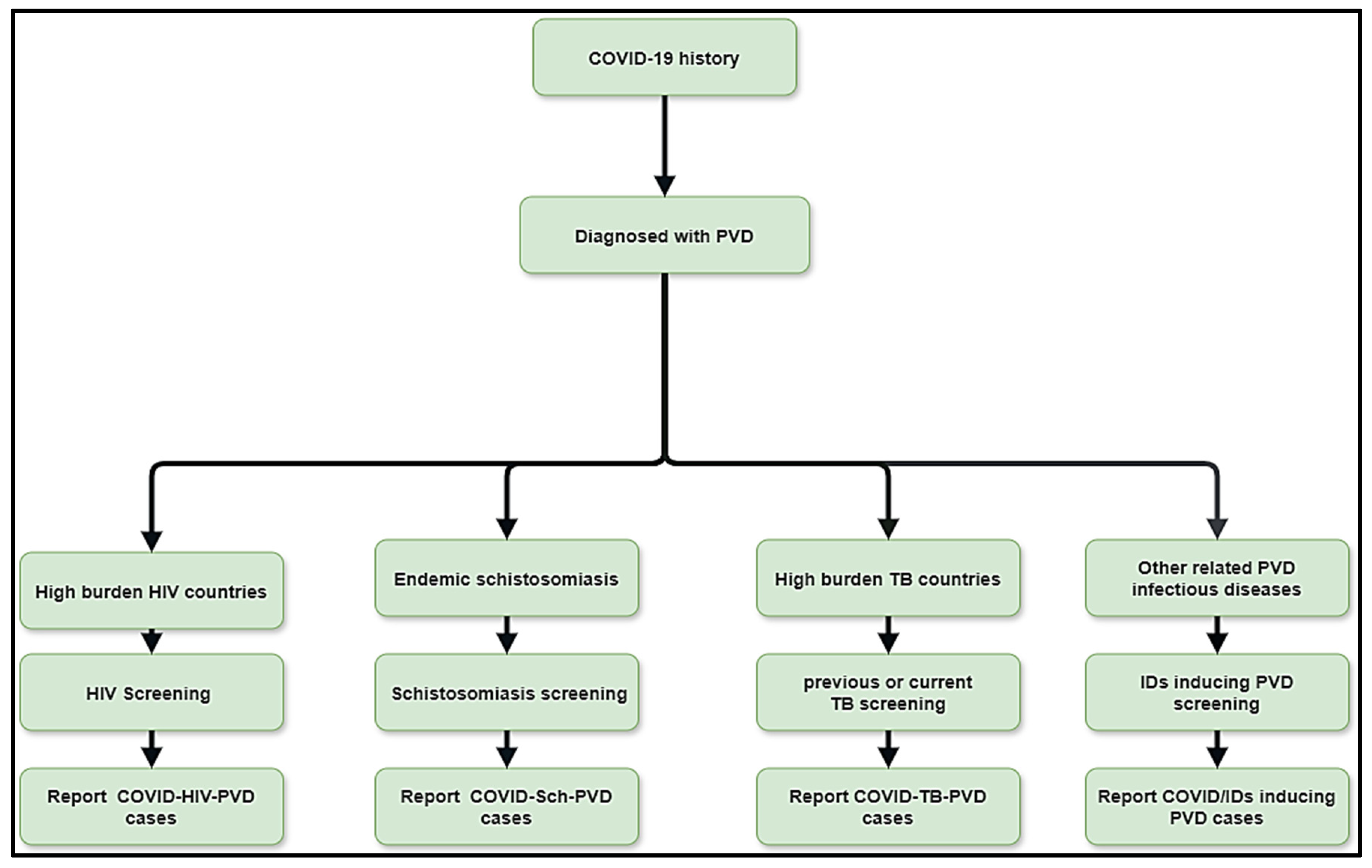
3. A Multidisciplinary Diagnostic and Management Approach of PVDs Related to COVID-19 Co-Infections Is Important
The interactions between possible COVID-19 co-infections that cause PVDs are a novel subject that needs additional research to have a better understanding of epidemiology, clinical profile, diagnostics, management, and prognostics. This multidisciplinary approach includes schistosomiasis, HIV, PTB, pulmonary aspergillosis, Wuchereria bancrofti, Clonorchis sinensis, paracoccidioidomycosis, and other infections that occur before or after acute COVID-19 or during long COVID phases. Human Herpesvirus 8, and Scrub typhus. This review suggests that the diagnostics of the leading co-infections inducing PVDs should be undertaken in the context of COVID-Sch-PVD, COVID-HIV-PVD and COVID-TB-PVD or other related IDs in high burden or endemic regions (Figure 4). In case of suspicion of COVID-Sch-PVD, COVID-HIV-PVD, COVID-TB-PVD, or other co-infections, the effects of COVID-19 and long COVID-19 on coagulation, inflammatory, and lung fibrosis biomarkers should be measured. Furthermore, cytological, histological, and medicinal imaging should be highlighted in cases of such co-infections. These assessments are critical since epidemiology, clinical profile, diagnostics, treatment, and prognostics are all unknown. As a result, patients with a history of COVID-19 and high levels of D-dimer should undergo CT angiography in settings with a high burden of schistosomiasis, HIV, PTB, pulmonary aspergillosis, Wuchereria bancrofti, Clonorchis sinensis, paracoccidioidomycosis, Human Herpesvirus 8, and Scrub typhus. Elevated D-dimer has been observed in patients with COVID-19 and long COVID. It is well known that elevated D-dimer levels are linked to acute pulmonary emboli and inflammatory disorders. D-dimer could be regarded as a simple, reliable, and low-cost diagnostic for tracking and following patients who have recovered from COVID-19. In the presence of the co-infections, COVID-19 or lengthy COVID may produce inflammation, which leads to endothelial function and coagulation abnormalities. In situations of suspicion of COVID-Sch-PVD, COVID-HIV-PVD, COVID-TB-PVD, or other co-infections, D-dimer with echocardiography should be used in screening for PVD (Figure 4). Other tests include HIV Elisa, TB GeneXpert, herpes simplex serology, immunochromatographic test (ICT) for f, and immunochromatographic test (ICT) for helminthiasis.
4. Conclusion
A wide range of infectious diseases can contribute to the development of PVDs during the acute COVID-19 and protracted COVID phases, increasing the global PVD burden. Based on previous research and pathophysiological pathways, this review identified COVID-19 and long COVID associated with parasites inducing PVDs as potent risks of increasing the burden of PVDs. Parasites inducing PVD such as schistosomiasis, HIV, PTB, pulmonary aspergillosis, Wuchereria bancrofti, Clonorchis sinensis, paracoccidioidomycosis, Human Herpesvirus 8, and Scrub typhus were found among the COVID-19 and long COVID. However, it is hypothetical that this association may increase the risk of PVDs. Known that these potential associations could increase the PVD, PVDs systematic screening are needed in post-COVID era. By the way, the epidemiology, clinical profiles, and prognostics of COVID-Sch-PVD, COVID-HIV-PVD, COVID-TB-PVD, and others remain unknown. Screening for COVID-Sch-PVD, COVID-HIV-PVD, COVID-TB-PVD, and other co-infections should be done in suspect patients to better the epidemiology, clinical profiles, and prognostics of these co-infections. This will boost research and allow more experimental studies to light this field.
Abbreviation
References
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019, 53. [Google Scholar] [CrossRef] [PubMed]
- Melot, C.; Naeije, R. Pulmonary vascular diseases. Compr Physiol. 2011, 1, 593–619. [Google Scholar] [PubMed]
- Hatano S, Strasser T, World Health Organization. Primary pulmonary hypertension: report on a WHO meeting, Geneva, 15-17 October 1973. World Health Organization; 1975.
- Kovacs, G.; Maier, R.; Aberer, E.; Brodmann, M.; Scheidl, S.; Tröster, N.; et al. Borderline pulmonary arterial pressure is associated with decreased exercise capacity in scleroderma. Am J Respir Crit Care Med. 2009, 180, 881–6. [Google Scholar] [CrossRef] [PubMed]
- Emmons-Bell, S.; Johnson, C.; Boon-Dooley, A.; Corris, P.A.; Leary, P.J.; Rich, S.; et al. Prevalence, incidence, and survival of pulmonary arterial hypertension: A systematic review for the global burden of disease 2020 study. Pulm Circ. 2022, 12, e12020. [Google Scholar] [CrossRef] [PubMed]
- Castiglione, L.; Droppa, M. Pulmonary Hypertension and COVID-19. Hamostaseologie. 2022, 42, 230–8. [Google Scholar] [CrossRef]
- Potus, F.; Mai, V.; Lebret, M.; Malenfant, S.; Breton-Gagnon, E.; Lajoie, A.C.; et al. Novel insights on the pulmonary vascular consequences of COVID-19. Am J Physiol Lung Cell Mol Physiol. 2020, 319, L277–88. [Google Scholar] [CrossRef]
- Pagnesi, M.; Baldetti, L.; Beneduce, A.; Calvo, F.; Gramegna, M.; Pazzanese, V.; et al. Pulmonary hypertension and right ventricular involvement in hospitalised patients with COVID-19. Heart. 2020, 106, 1324–31. [Google Scholar] [CrossRef]
- Kumar R, Aktay-Cetin Ö, Craddock V, Morales-Cano D, Kosanovic D, Cogolludo A. ; et al. Potential long-term effects of SARS-CoV-2 infection on the pulmonary vasculature: Multilayered cross-talks in the setting of coinfections and comorbidities. PLoS Pathog. 2023, 19, e1011063.
- Musuuza, J.S.; Watson, L.; Parmasad, V.; Putman-Buehler, N.; Christensen, L.; Safdar, N. Prevalence and outcomes of co-infection and superinfection with SARS-CoV-2 and other pathogens: a systematic review and meta-analysis. PloS One. 2021, 16, e0251170. [Google Scholar] [CrossRef]
- Egom, E.À.; Shiwani, H.A.; Nouthe, B. Egom EÀ, Shiwani HA, Nouthe B. From acute SARS-CoV-2 infection to pulmonary hypertension. Front Physiol. 2022, 13, 1023758. [Google Scholar]
- Taha, H.A.; Elshafey, B.I.; Abdullah, T.M.; Salem, H.A. Study of pulmonary hypertension in post-COVID-19 patients by transthoracic echocardiography. Egypt J Bronchol. 2023, 17, 1–8. [Google Scholar] [CrossRef]
- Oktaviono, Y.H.; Mulia, E.P.B.; Luke, K.; Nugraha, D.; Maghfirah, I.; Subagjo, A. Right ventricular dysfunction and pulmonary hypertension in COVID-19: a meta-analysis of prevalence and its association with clinical outcome. Arch Med Sci AMS. 2022, 18, 1169. [Google Scholar] [CrossRef] [PubMed]
- Butrous, G. Human immunodeficiency virus–associated pulmonary arterial hypertension: considerations for pulmonary vascular diseases in the developing world. Circulation. 2015, 131, 1361–70. [Google Scholar] [CrossRef] [PubMed]
- Medrano-Garcia, S.; Morales-Cano, D.; Barreira, B.; Vera-Zambrano, A.; Kumar, R.; Kosanovic, D.; et al. HIV and Schistosoma Co-Exposure Leads to Exacerbated Pulmonary Endothelial Remodeling and Dysfunction Associated with Altered Cytokine Landscape. Cells. 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Butrous, G.; Mathie, A. Infection in pulmonary vascular diseases: Would another consortium really be the way to go? Glob Cardiol Sci Pract. 2019, 2019, 1. [Google Scholar] [CrossRef]
- Butrous, G. The global challenge of pulmonary vascular diseases and its forgotten impact in the developing world. Adv Pulm Hypertens. 2012, 11, 117–8. [Google Scholar] [CrossRef]
- Butrous, G. Pulmonary Vascular Diseases Secondary to Schistosomiasis. Adv Pulm Hypertens. 2017, 15. [Google Scholar] [CrossRef]
- Kolosionek, E.; Crosby, A.; Harhay, M.O.; Morrell, N.; Butrous, G. Pulmonary vascular disease associated with schistosomiasis. Expert Rev Anti Infect Ther. 2010, 8, 1467–73. [Google Scholar] [CrossRef]
- Xue, Q.; Deng, Y.; Liu, Y.; Wang, Y.; Hu, W.; Huang, Y.; et al. A retrospective analysis of schistosomiasis related literature from 2011-2020: Focusing on the next decade. Acta Trop. 2023, 238, 106750. [Google Scholar] [CrossRef]
- Kolosionek, E.; Crosby, A.; Harhay, M.O.; Morrell, N.; Butrous, G. Pulmonary vascular disease associated with schistosomiasis. Expert Rev Anti Infect Ther. 2010, 8, 1467–73. [Google Scholar] [CrossRef]
- de Cleva R, Herman P, Pugliese V, Zilberstein B, Saad WA, Rodrigues J. ; et al. Prevalence of pulmonary hypertension in patients with hepatosplenic Mansonic schistosomiasis--prospective study. Hepatogastroenterology. 2003, 50, 2028–30.
- Rohun, J.; Dorniak, K.; Faran, A.; Kochańska, A.; Zacharek, D.; Daniłowicz-Szymanowicz, L. Long COVID-19 myocarditis and various heart failure presentations: a case series. J Cardiovasc Dev Dis. 2022, 9, 427. [Google Scholar] [CrossRef] [PubMed]
- Wolday, D.; Gebrecherkos, T.; Arefaine, Z.G.; Kiros, Y.K.; Gebreegzabher, A.; Tasew, G.; et al. Effect of co-infection with intestinal parasites on COVID-19 severity: a prospective observational cohort study. EClinicalMedicine. 2021, 39. [Google Scholar]
- Gebrecherkos, T.; Gessesse, Z.; Kebede, Y.; Gebreegzabher, A.; Tasew, G.; Abdulkader, M.; et al. Effect of co-infection with parasites on severity of COVID-19. medRxiv. 2021, 2021–02. [Google Scholar]
- Obeyesekere, I.; Peiris, D. Pulmonary hypertension and filariasis. Br Heart J. 1974, 36, 676–81. [Google Scholar] [CrossRef]
- Walloopillai, N. Primary pulmonary hypertension, an unexplained epidemic in Sri Lanka. Pathobiology. 1975, 43, 248–50. [Google Scholar] [CrossRef]
- Lai, K.; McFadzean, A.; Yeung, R. Microembolic pulmonary hypertension in pyogenic cholangitis. Br Med J. 1968, 1, 22. [Google Scholar] [CrossRef]
- Ross, A.G.; Sleigh, A.C.; Li, Y.; Davis, G.M.; Williams, G.M.; Jiang, Z.; et al. Schistosomiasis in the People’s Republic of China: prospects and challenges for the 21st century. Clin Microbiol Rev. 2001, 14, 270–95. [Google Scholar] [CrossRef] [PubMed]
- Halawa, S.; Pullamsetti, S.S.; Bangham, C.R.; Stenmark, K.R.; Dorfmüller, P.; Frid, M.G.; et al. Potential long-term effects of SARS-CoV-2 infection on the pulmonary vasculature: a global perspective. Nat Rev Cardiol. 2022, 19, 314–31. [Google Scholar] [CrossRef]
- Oliveira, R.K.; Nyasulu, P.S.; Iqbal, A.A.; Hamdan Gul, M.; Ferreira, E.V.; Leclair, J.W.; et al. Cardiopulmonary disease as sequelae of long-term COVID-19: Current perspectives and challenges. Front Med. 2022, 9, 1041236. [Google Scholar] [CrossRef]
- Nijkeuter, M.; Hovens, M.M.; Davidson, B.L.; Huisman, M.V. Resolution of thromboemboli in patients with acute pulmonary embolism: a systematic review. Chest. 2006, 129, 192–7. [Google Scholar] [CrossRef] [PubMed]
- Pullamsetti, S.; Savai, R.; Janssen, W.; Dahal, B.; Seeger, W.; Grimminger, F.; et al. Inflammation, immunological reaction and role of infection in pulmonary hypertension. Clin Microbiol Infect. 2011, 17, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Rubin, L.J. Pulmonary arterial hypertension. Proc Am Thorac Soc. 2006, 3, 111–5. [Google Scholar] [CrossRef] [PubMed]
- Budhiraja, R.; Tuder, R.M.; Hassoun, P.M. Endothelial dysfunction in pulmonary hypertension. Circulation. 2004, 109, 159–65. [Google Scholar] [CrossRef] [PubMed]
- Hassoun, P.M.; Mouthon, L.; Barberà, J.A.; Eddahibi, S.; Flores, S.C.; Grimminger, F.; et al. Inflammation, growth factors, and pulmonary vascular remodeling. J Am Coll Cardiol. 2009, 54, S10–S19. [Google Scholar] [CrossRef]
- Andruska, A.; Spiekerkoetter, E. Consequences of BMPR2 deficiency in the pulmonary vasculature and beyond: contributions to pulmonary arterial hypertension. Int J Mol Sci. 2018, 19, 2499. [Google Scholar] [CrossRef]
- Ogo, T.; Chowdhury, H.; Yang, J.; Long, L.; Li, X.; Torres Cleuren, Y.N.; et al. Inhibition of overactive transforming growth factor–β signaling by prostacyclin analogs in pulmonary arterial hypertension. Am J Respir Cell Mol Biol. 2013, 48, 733–41. [Google Scholar] [CrossRef]
- Burel-Vandenbos, F.; Cardot-Leccia, N.; Passeron, T. Apoptosis and pericyte loss in alveolar capillaries in COVID-19 infection: choice of markers matters. Author’s reply. Intensive Care Med. 2020, 46, 1967–8. [Google Scholar] [CrossRef]
- Oyeyemi, O.; Okunlola, O.; Adebayo, A. Assessment of schistosomiasis endemicity and preventive treatment on coronavirus disease 2019 outcomes in Africa. New Microbes New Infect. 2020, 38, 100821. [Google Scholar] [CrossRef]
- Maizels, R.M.; McSorley, H.J. Regulation of the host immune system by helminth parasites. J Allergy Clin Immunol. 2016, 138, 666–75. [Google Scholar] [CrossRef]
- Ferrari, T.C.A.; Albricker, A.C.L.; Gonçalves, I.M.; Freire, C.M.V. Schistosome-associated pulmonary arterial hypertension: a review emphasizing pathogenesis. Front Cardiovasc Med. 2021, 8, 724254. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Mickael, C.; Chabon, J.; Gebreab, L.; Rutebemberwa, A.; Garcia, A.R.; et al. The causal role of IL-4 and IL-13 in Schistosoma mansoni pulmonary hypertension. Am J Respir Crit Care Med. 2015, 192, 998–1008. [Google Scholar] [CrossRef]
- Butrous, G.; Ghofrani, H.A.; Grimminger, F. Pulmonary vascular disease in the developing world. Circulation. 2008, 118, 1758–66. [Google Scholar] [CrossRef] [PubMed]
- Chiu, B.C.; Freeman, C.M.; Stolberg, V.R.; Komuniecki, E.; Lincoln, P.M.; Kunkel, S.L.; et al. Cytokine–chemokine networks in experimental mycobacterial and schistosomal pulmonary granuloma formation. Am J Respir Cell Mol Biol. 2003, 29, 106–16. [Google Scholar] [CrossRef] [PubMed]
- Boros, D.L.; Whitfield, J.R. Enhanced Th1 and dampened Th2 responses synergize to inhibit acute granulomatous and fibrotic responses in murine schistosomiasis mansoni. Infect Immun. 1999, 67, 1187–93. [Google Scholar] [CrossRef]
- Noor, R. How do the severe acute respiratory coronavirus 2 (SARS-CoV-2) and its variants escape the host protective immunity and mediate pathogenesis? Bull Natl Res Cent. 2022, 46, 1–14. [Google Scholar] [CrossRef]
- DAvila-Mesquita, C.; Couto, A.E.S.; Campos, L.C.B.; Vasconcelos, T.F.; Michelon-Barbosa, J.; Corsi, C.A.C.; et al. MMP-2 and MMP-9 levels in plasma are altered and associated with mortality in COVID-19 patients. Biomed Pharmacother Biomedecine Pharmacother. 2021, 142, 112067. [Google Scholar]
- Laveaux, S.; Vandecasteele, S.; Van De Moortele, K. Chronic Schistosomiasis Presenting with Migrating Pulmonary Manifestation after Recent COVID-19 Infection: HRCT Findings. J Belg Soc Radiol. 2022, 106. [Google Scholar] [CrossRef]
- Turner, J.D.; Jackson, J.A.; Faulkner, H.; Behnke, J.; Else, K.J.; Kamgno, J.; et al. Intensity of intestinal infection with multiple worm species is related to regulatory cytokine output and immune hyporesponsiveness. J Infect Dis. 2008, 197, 1204–12. [Google Scholar] [CrossRef]
- Babu, S.; Nutman, T.B. Immunology of lymphatic filariasis. Parasite Immunol. 2014, 36, 338–46. [Google Scholar] [CrossRef]
- Mahajan, G.; Barjatya, H.; Bhakar, B.; Gothwal, S.K.; Jangir, T. To estimate prevalence of pulmonary arterial hypertension in HIV patients and its association with CD4 cell count. Clin Epidemiol Glob Health. 2024, 25, 101479. [Google Scholar] [CrossRef]
- UNAIDS Global HIV statistics [Internet]. 2023. Available from: https://www.unaids.org/sites/default/files/media_asset/UNAIDS_FactSheet_en.pdf.
- Hoeper, M.M.; Humbert, M.; Souza, R.; Idrees, M.; Kawut, S.M.; Sliwa-Hahnle, K.; et al. A global view of pulmonary hypertension. Lancet Respir Med. 2016, 4, 306–22. [Google Scholar] [CrossRef] [PubMed]
- Almodovar, S.; Cicalini, S.; Petrosillo, N.; Flores, S.C. Pulmonary hypertension associated with HIV infection: pulmonary vascular disease: the global perspective. Chest. 2010, 137, 6S–12S. [Google Scholar] [CrossRef] [PubMed]
- Agarwal S, Sharma H, Chen L, Dhillon NK. NADPH oxidase-mediated endothelial injury in HIV- and opioid-induced pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2020, 318, L1097–108.
- Porter, K.M.; Walp, E.R.; Elms, S.C.; Raynor, R.; Mitchell, P.O.; Guidot, D.M.; et al. Human immunodeficiency virus-1 transgene expression increases pulmonary vascular resistance and exacerbates hypoxia-induced pulmonary hypertension development. Pulm Circ. 2013, 3, 58–67. [Google Scholar] [CrossRef]
- Nuche, J.; Pérez-Olivares, C.; de la Cal, T.S.; López-Guarch, C.J.; Ynsaurriaga, F.A.; Subías, P.E. Clinical course of COVID-19 in pulmonary arterial hypertension patients. Rev Espanola Cardiol Engl Ed. 2020, 73, 775. [Google Scholar] [CrossRef]
- Danwang, C.; Noubiap, J.J.; Robert, A.; Yombi, J.C. Outcomes of patients with HIV and COVID-19 co-infection: a systematic review and meta-analysis. AIDS Res Ther. 2022, 19, 1–12. [Google Scholar] [CrossRef]
- Mehta NJ, Khan IA, Mehta RN, Sepkowitz DA. HIV-related pulmonary hypertension: analytic review of 131 cases. Chest. 2000, 118, 1133–41.
- Suresh SJ, Suzuki YJ. SARS-CoV-2 spike protein and lung vascular cells. J Respir. 2020, 1, 40–8.
- Dorfmüller, P.; Perros, F.; Balabanian, K.; Humbert, M. Inflammation in pulmonary arterial hypertension. Eur Respir J. 2003, 22, 358–63. [Google Scholar] [CrossRef]
- Butrous, G. Human immunodeficiency viruses and their effect on the pulmonary vascular bed. Am J Physiol-Lung Cell Mol Physiol. 2021, 321, L1062–6. [Google Scholar] [CrossRef] [PubMed]
- Cool, C.D.; Rai, P.R.; Yeager, M.E.; Hernandez-Saavedra, D.; Serls, A.E.; Bull, T.M.; et al. Expression of human herpesvirus 8 in primary pulmonary hypertension. N Engl J Med. 2003, 349, 1113–22. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.M.; Lederer, D.J.; Borczuk, A.C.; Kawut, S.M. Pulmonary hypertension in idiopathic pulmonary fibrosis. Chest. 2007, 132, 998–1006. [Google Scholar] [CrossRef]
- Duckworth, A.; Longhurst, H.J.; Paxton, J.K.; Scotton, C.J. The role of herpes viruses in pulmonary fibrosis. Front Med. 2021, 8, 704222. [Google Scholar] [CrossRef]
- Calabrese, F.; Kipar, A.; Lunardi, F.; Balestro, E.; Perissinotto, E.; Rossi, E.; et al. Herpes virus infection is associated with vascular remodeling and pulmonary hypertension in idiopathic pulmonary fibrosis. PLoS One. 2013, 8, e55715. [Google Scholar] [CrossRef] [PubMed]
- Zubchenko, S.; Kril, I.; Nadizhko, O.; Matsyura, O.; Chopyak, V. Herpesvirus infections and post-COVID-19 manifestations: a pilot observational study. Rheumatol Int. 2022, 42, 1523–30. [Google Scholar] [CrossRef]
- Mohamed, A.; Gidda, H.; Zavoshi, S.; Mahmood, R. A Case of Left Ventricular Thrombus and Herpetic Esophagitis in an Immunocompetent Patient With COVID-19. Cureus. 2023, 15. [Google Scholar] [CrossRef]
- Bhattacharyya, P.; Saha, D.; Bhattacherjee, P.D.; Das, S.K.; Bhattacharyya, P.P.; Dey, R. Tuberculosis associated pulmonary hypertension: The revelation of a clinical observation. Lung India Off Organ Indian Chest Soc. 2016, 33, 135. [Google Scholar] [CrossRef]
- Louw, E.; Baines, N.; Maarman, G.; Osman, M.; Sigwadhi, L.; Irusen, E.; et al. The prevalence of pulmonary hypertension after successful tuberculosis treatment in a community sample of adult patients. Pulm Circ. 2023, 13, e12184. [Google Scholar] [CrossRef]
- Galie, N.; Hoeper, M.M.; Humbert, M.; Torbicki, A.; Vachiery, J.L.; Barbera, J.A.; et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J. 2009, 30, 2493–537. [Google Scholar]
- Tamuzi, J.L.; Lulendo, G.; Mbuesse, P.; Nyasulu, P.S. The incidence and mortality of COVID-19 related TB disease in Sub-Saharan Africa: A systematic review and meta-analysis. medRxiv. 2022, 2022–01. [Google Scholar]
- Ahmed, A.E.H.; Ibrahim, A.S.; Elshafie, S.M. Pulmonary hypertension in patients with treated pulmonary tuberculosis: analysis of 14 consecutive cases. Clin Med Insights Circ Respir Pulm Med. 2011, 5, CCRPM–S6437. [Google Scholar] [CrossRef]
- Jo, Y.S.; Park, J.H.; Lee, J.K.; Heo, E.Y.; Chung, H.S.; Kim, D.K. Risk factors for pulmonary arterial hypertension in patients with tuberculosis-destroyed lungs and their clinical characteristics compared with patients with chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2017, 2433–43. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.G.; Cardona, P.J.; Kim, M.J.; Allain, S.; Altare, F. Foamy macrophages and the progression of the human tuberculosis granuloma. Nat Immunol. 2009, 10, 943–8. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, S. Pathogenesis of cor pulmonale in pulmonary tuberculosis. 1986.
- Zouaki I, Chahbi Z, Raiteb M, Zyani M. COVID-19 and Pulmonary Tuberculosis Coinfection in a Moroccan Patient with Pulmonary Embolism: A Case Report and Literature Review. Case Rep Infect Dis. 2022, 2022.
- Parolina, L.; Pshenichnaya, N.; Vasilyeva, I.; Lizinfed, I.; Urushadze, N.; Guseva, V.; et al. Clinical characteristics of COVID-19 in patients with tuberculosis and factors associated with the disease severity. Int J Infect Dis. 2022, 124, S82–S89. [Google Scholar] [CrossRef]
- He, F.; Xia, X.; Nie, D.; Yang, H.; Jiang, Y.; Huo, X.; et al. Respiratory bacterial pathogen spectrum among COVID-19 infected and non-COVID-19 virus infected pneumonia patients. Diagn Microbiol Infect Dis. 2020, 98, 115199. [Google Scholar] [CrossRef]
- Wang, Q.; Cao, Y.; Liu, X.; Fu, Y.; Zhang, J.; Zhang, Y.; et al. Systematic review and meta-analysis of Tuberculosis and COVID-19 Co-infection: Prevalence, fatality, and treatment considerations. PLoS Negl Trop Dis. 2024, 18, e0012136. [Google Scholar] [CrossRef]
- Berger, J.T.; Carcillo, J.A.; Shanley, T.P.; Wessel, D.L.; Clark, A.; Holubkov, R.; et al. Critical pertussis illness in children: a multicenter prospective cohort study. Pediatr Crit Care Med J Soc Crit Care Med World Fed Pediatr Intensive Crit Care Soc. 2013, 14, 356–65. [Google Scholar] [CrossRef]
- Jeican, I.I.; Inișca, P.; Gheban, D.; Tăbăran, F.; Aluaș, M.; Trombitas, V.; et al. COVID-19 and Pneumocystis jirovecii pulmonary coinfection—the first case confirmed through autopsy. Medicina (Mex). 2021, 57, 302. [Google Scholar] [CrossRef]
- Saibaba, J.; Selvaraj, J.; Viswanathan, S.; Pillai, V.; Saibaba Jr, J. Scrub Typhus and COVID-19 Coinfection Unmasking Antiphospholipid Antibody Syndrome. Cureus. 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Udwadia ZF, Vora A, Tripathi AR, Malu KN, Lange C, Raju RS. COVID-19-Tuberculosis interactions: When dark forces collide. Indian J Tuberc. 2020, 67, S155–62.
- Tamuzi, J.L.; Ayele, B.T.; Shumba, C.S.; Adetokunboh, O.O.; Uwimana-Nicol, J.; Haile, Z.T.; et al. Implications of COVID-19 in high burden countries for HIV/TB: A systematic review of evidence. BMC Infect Dis. 2020, 20, 744. [Google Scholar] [CrossRef] [PubMed]
- Tsenova, L.; Singhal, A. Effects of host-directed therapies on the pathology of tuberculosis. J Pathol. 2020, 250, 636–46. [Google Scholar] [CrossRef]
- Di Bari, V.; Gualano, G.; Musso, M.; Libertone, R.; Nisii, C.; Ianniello, S.; et al. Increased association of pulmonary thromboembolism and tuberculosis during COVID-19 pandemic: data from an Italian infectious disease referral hospital. Antibiotics. 2022, 11, 398. [Google Scholar] [CrossRef]
- Thachil, J.; Tang, N.; Gando, S.; Falanga, A.; Cattaneo, M.; Levi, M.; et al. ISTH interim guidance on recognition and management of coagulopathy in COVID-19. J Thromb Haemost. 2020, 18, 1023–6. [Google Scholar] [CrossRef] [PubMed]
- Paddock, C.D.; Sanden, G.N.; Cherry, J.D.; Gal, A.A.; Langston, C.; Tatti, K.M.; et al. Pathology and pathogenesis of fatal Bordetella pertussis infection in infants. Clin Infect Dis Off Publ Infect Dis Soc Am. 2008, 47, 328–38. [Google Scholar] [CrossRef]
- Yeung, K.H.T.; Duclos, P.; Nelson, E.A.S.; Hutubessy, R.C.W. An update of the global burden of pertussis in children younger than 5 years: a modelling study. Lancet Infect Dis. 2017, 17, 974–80. [Google Scholar] [CrossRef]
- Shi, T.; Wang, L.; Du, S.; Fan, H.; Yu, M.; Ding, T.; et al. Mortality risk factors among hospitalized children with severe pertussis. BMC Infect Dis. 2021, 21, 1057. [Google Scholar] [CrossRef]
- Flikweert, A.W.; Grootenboers, M.J.; Yick, D.C.; du Mee, A.W.; van der Meer, N.J.; Rettig, T.C.; et al. Late histopathologic characteristics of critically ill COVID-19 patients: Different phenotypes without evidence of invasive aspergillosis, a case series. J Crit Care. 2020, 59, 149–55. [Google Scholar] [CrossRef]
- van Arkel AL, Rijpstra TA, Belderbos HN, Van Wijngaarden P, Verweij PE, Bentvelsen RG. COVID-19–associated pulmonary aspergillosis. Am J Respir Crit Care Med. 2020, 202, 132–135.
- Chethan, M.; Devi, H.; Deshmukh, S. Pleural aspergillosis with pulmonary artery thrombosis as a complication of Covid-19. Lung India. 2022, S214–S214. [Google Scholar]
- Lamoth, F.; Glampedakis, E.; Boillat-Blanco, N.; Oddo, M.; Pagani, J.L. Incidence of invasive pulmonary aspergillosis among critically ill COVID-19 patients. Clin Microbiol Infect Off Publ Eur Soc Clin Microbiol Infect Dis. 2020, 26, 1706–8. [Google Scholar] [CrossRef] [PubMed]
- Chauvet, P.; Mallat, J.; Arumadura, C.; Vangrunderbeek, N.; Dupre, C.; Pauquet, P.; et al. Risk Factors for Invasive Pulmonary Aspergillosis in Critically Ill Patients With Coronavirus Disease 2019-Induced Acute Respiratory Distress Syndrome. Crit Care Explor. 2020, 2, e0244. [Google Scholar] [CrossRef] [PubMed]
- DYee, C.; Aung, H.K.K.; Mg Mg, B.; Htun, W.P.P.; Janurian, N.; Pyae Phyo, A.; et al. Case Report: A case report of multiple co-infections (melioidosis, paragonimiasis, Covid-19 and tuberculosis) in a patient with diabetes mellitus and thalassemia-trait in Myanmar. Wellcome Open Res. 2022, 7. [Google Scholar]
Figure 1.
Hypothetical pathophysiology explaining acute SARS-CoV-2 and long COVID associated with schistosomiasis and other helminthic diseases inducing PVDs.
Figure 1.
Hypothetical pathophysiology explaining acute SARS-CoV-2 and long COVID associated with schistosomiasis and other helminthic diseases inducing PVDs.
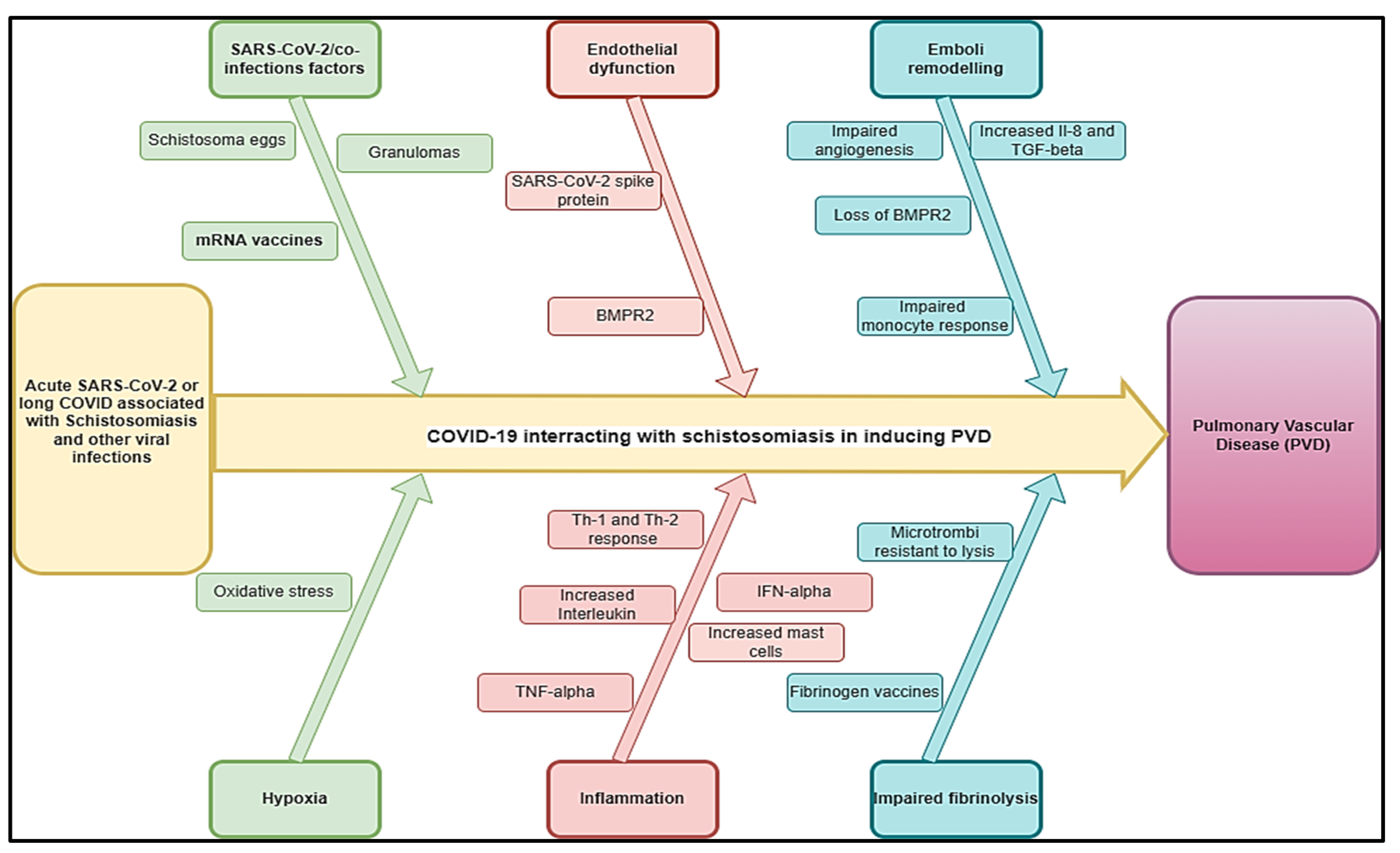
Figure 2.
Hypothetical pathophysiology explaining acute SARS-CoV-2 and long COVID associated with HIV and other viral diseases inducing PVDs.
Figure 2.
Hypothetical pathophysiology explaining acute SARS-CoV-2 and long COVID associated with HIV and other viral diseases inducing PVDs.
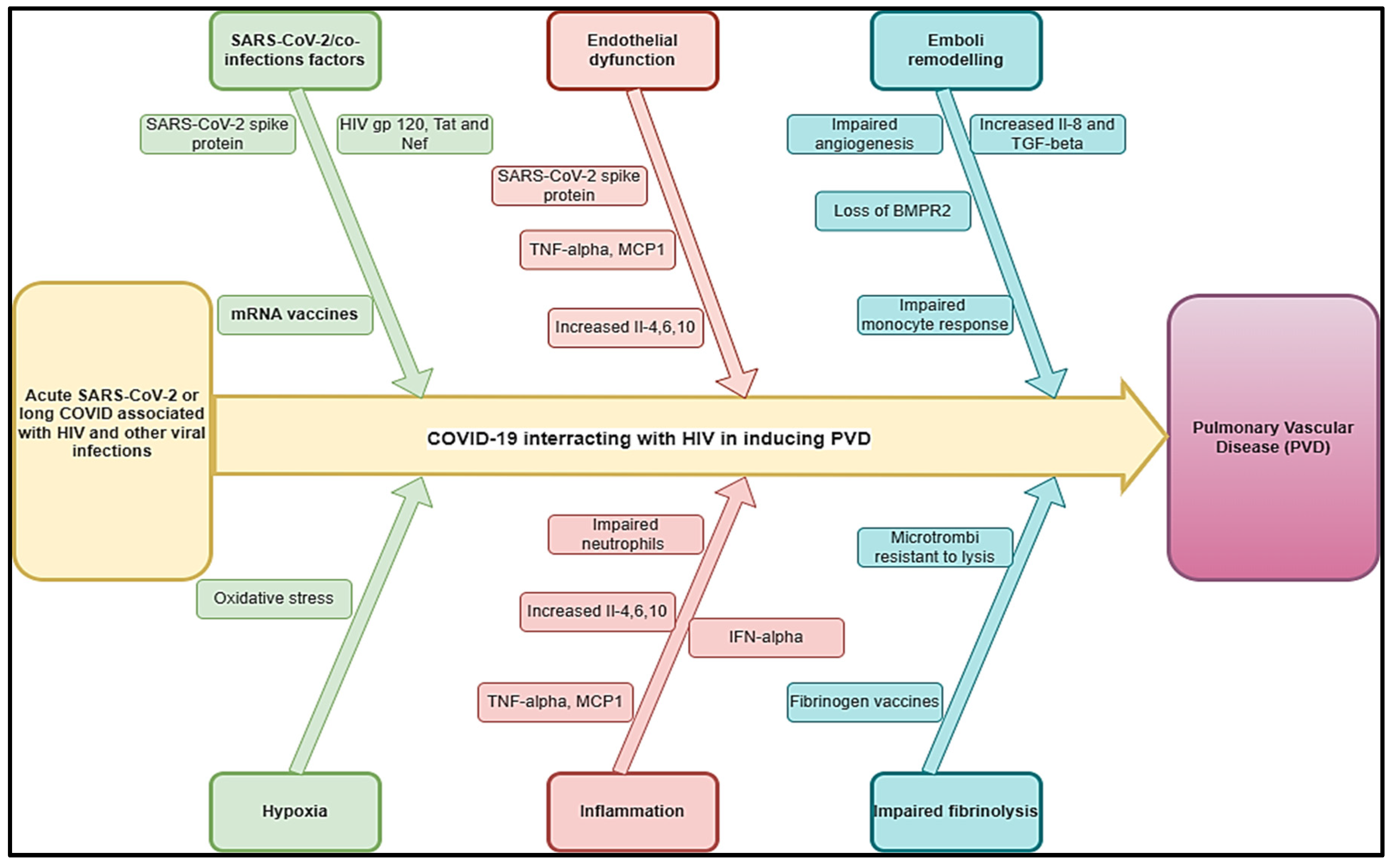
Figure 3.
Hypothetical pathophysiology explaining acute SARS-CoV-2 and long COVID associated with PTB and other bacterial diseases inducing PVDs.
Figure 3.
Hypothetical pathophysiology explaining acute SARS-CoV-2 and long COVID associated with PTB and other bacterial diseases inducing PVDs.
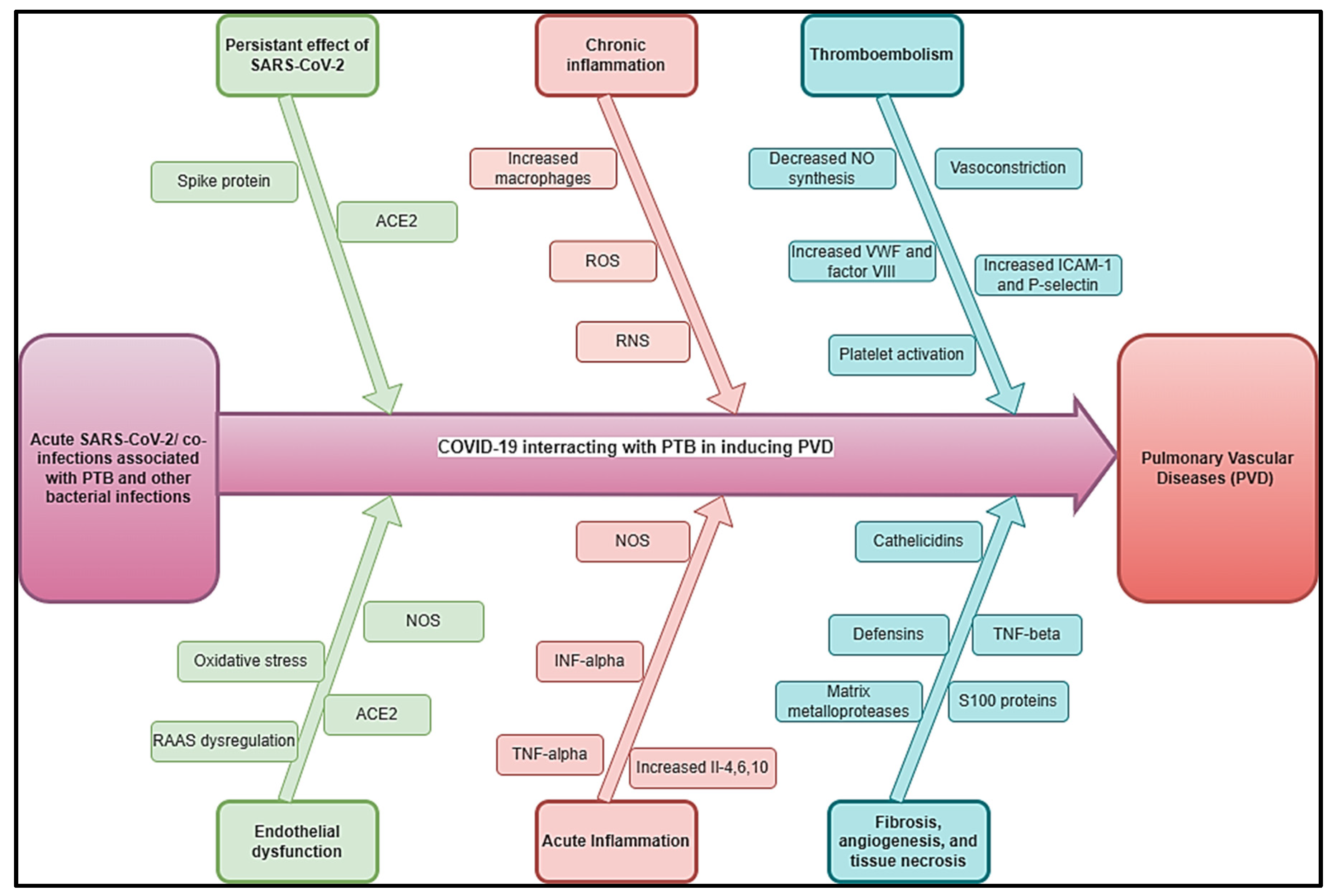
Figure 4.
Flow diagram showing the methods of improving post COVID/co-infections inducing PVDs.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



