
Submitted:
26 November 2024
Posted:
27 November 2024
You are already at the latest version
Abstract
The synthesis of prostaglandylinositol cyclic phosphate (cyclic PIP) is stimulated by insulin and by adrenaline via its a1- and a2- adrenoceptors. Cyclic PIP inhibits protein kinase A and activates protein ser/thr phosphatase holoenzymes. Consequently, it inhibits glycogen phosphorylase and glucose-release. On extra-corporal rat liver perfusion, 0.1 μM cyclic PIP triggers a rapid glucose-release, comparable to the one generated by the calcium ionophore A23187, confirming reports that phenylephrine stimulates glucose-release via a1-adrenoceptors. 10–9 M glucagon stimulates glucose-release nearly 2-fold in male rat livers. Simultaneous stimulation with glucagon and 2 x 10–9 M insulin causes a 65% reduced glucose-release. 10–6 M adrenaline stimulates a rapid first phase and a second, cyclic AMP-triggered phase of glucose-release. Insulin triggers no rapid glucose- release, though both hormones stimulate the synthesis of cyclic PIP. The synthesis of cyclic PIP peaks 1 min after stimulation with adrenaline, but 3–4 min after stimulation with insulin, and cyclic PIP synthesis increases approximately 6-fold slower on insulin than on adrenaline stimulation. This can explain the too low Ca2+ increase on insulin-stimulation in order to “flash- activate” glycogen phosphorylase. In summary, cyclic PIP first “supports” and thereafter antagonizes the cyclic AMP-triggered glucose-release from liver.
Keywords:
alpha-adrenoceptor-action
; adrenaline
; calcium ion
; cyclic AMP
; cyclic PIP
; insulin action
; prostaglandylinositol cyclic phosphate
; prostaglandin E action-mechanism
1. Introduction
Presently, the dominant opinion is that adrenaline via α1A- and α1B-adrenoceptors activates intracellular phospholipase Cβ (PLC), which splits off inositol 1,4,5-trisphosphate (IP3) from membrane-bound phosphoinositides. IP3 triggers then Ca2+ release from intracellular stores as the endoplasmic reticulum [1,2,3,4]. However, adrenaline stimulates also Ca2+ influx into cells [5], and inhibits adenylate cyclase type 5 in HL-1 cardiomyocytes via its α1A-adrenoceptors [6]. These results indicate that α1A-adrenoceptor action is more complex than presently assumed. Furthermore, there are more published results, which do not match with the present view of α1- and α2-adrenoceptor action. First, α1- and α2-adrenoceptors activate phospholipase A2 (PLA2), which releases unsaturated C20-fatty acids as dihomo-γ-linolenic acid and arachidonic acid from membrane-bound lipids for the synthesis of prostaglandins [7,8]. Second, phospholipase C is activated also by α2-adrenoceptors [9], though α2-adrenoceptors should only signal inhibition of adenylate cyclase via the Gi protein [10]. Third, in the years 1985 to 1987 several research-groups reported that stimulation of α1- and α2-adrenoceptors triggers the same physiological effects as vasoconstriction, for instance, of the coronary bed and the pulmonary arteries, which is similarly inhibited by Ca2+-channel blockade [11,37,38,39,40,41,42]. However, other scientists started to suggest that α1- and α2-adrenoceptor actions are different in their biochemistry and mechanism of action [10].
In recent years the existence and action of the natural cyclic AMP antagonist prostaglandyl-inositol cyclic phosphate (cyclic PIP) has been reported [11,12,13]. Its synthesis is stimulated by adrenaline via α1- and α2-adrenoceptors. More specifically, phenylephrine increases via α1-adrenoceptors cyclic PIP synthesis, which is inhibited by the α1-adrenoceptor antagonist prazosin. Likewise, clonidine increases cyclic PIP synthesis via α2-adrenoceptors, which is inhibited by the α2-adrenoceptor antagonist yohimbine. This has been shown in brain, heart and liver of rats. In the heart cyclic PIP synthesis is stimulated by α1-adrenoceptors, in liver more by α1- than by α2-adrenoceptors, and in brain predominantly by α2-adrenoceptors [11]. Consistent with α-adrenoceptor action, cyclic PIP triggers the following effects: It inhibits insulin release from pancreatic β-cells [12], an α2-adrenoceptor effect [14]; it triggers a 2.7-fold positive inotropic effect on the papillary muscle of the kitten heart, an α1-adrenoceptor effect [15]; it increases glucose uptake into adipocytes 10-fold [16], an α1-adrenoceptor effect [17].
The synthesis of cyclic PIP is stimulated, so far discovered, by insulin and adrenaline and also by vasopressin and angiotensin II [13]. The chemical structure of cyclic PIP was elaborated by mass spectrometry of the dephosphorylated cyclic PIP. It is composed of prostaglandin E (PGE) and myo-inositol (1:2-cyclic)-phosphate, which is bound by its C4-hydroxyl group to the C15-hydroxyl group of the PGE [12]. The substrates for cyclic PIP biosynthesis are PGE and activated inositol phosphate [12,13]. Cyclic PIP synthase is a membrane-bound enzyme, which is activated by tyrosine phosphorylation in case of insulin stimulation and most likely by a G protein in case of adrenaline stimulation of its synthesis (Figure 1). The primary regulatory actions of cyclic PIP are the dose-dependent, 7-fold activation of protein ser/thr phosphatase holoenzyme and the 100% inhibition of protein kinase A (PKA) [12].
The prevailing opinion that α1-adrenoceptors stimulate IP3 synthesis and α2-adrenoceptors inhibit adenylate cyclase via the Gi protein [18], found its way into textbooks. However, the above enumerated, published results indicate that this description of the mechanism of α1- and α2-adrenoceptor action is more complex than this present interpretation suggests. One has to take note that all these, above mentioned results, concerning α1- and α2-adrenoceptor action, are consistent with the biosynthesis and action of cyclic PIP. The stimulation of both, α1- and α2-adrenoceptors increases (1) the activity of PLA2, leading to the biosynthesis of prostaglandins as PGE and (2) the activity of PLC, leading to the biosynthesis of activated inositol phosphate (n-Ins-P) [13]. Both compounds are the substrates of cyclic PIP synthesis (Figure 1). Importantly, PLA2 and PLC are activated by tyr-phosphorylation and also by G proteins, related to stimulation of cyclic PIP synthase by insulin or adrenaline, respectively [19].
The substrates of cyclic PIP synthase: (1) PGE. Years ago, after a biochemical action mechanism for prostaglandins was not found, and reports on the existence and action of cyclic PIP were not taken into account [11,16], prostaglandins were suggested to be “tissue hormones” because all cells of an organism can synthesize this class of compounds. This interpretation led to the current notion that prostaglandins are synthesized intracellularly, then excreted, and prostaglandin transporters (PTG) bring the prostaglandins for their degradation back into cells [30,31]. Extracellularly prostaglandins bind to different, prostanoid-receptors, which are predominantly coupled to Gq- and Gi/o-proteins, which bring the signal to various effectors [32,33,34]. Years ago, the argument has been made that this reaction path looks more like a detour, but not like a straight forward action-mechanism of prostaglandins, whereas the need of PGE for the synthesis of cyclic PIP (Figure 1) is a straight reaction path [16]. In support of this argument is that the Km-values of the substrates of cyclic PIP synthase, PGE and activated inositol phosphate, are 1.8 and 3.0 x 10–6 M, respectively [19]. These low Km-values warrant that already small amounts of these substrates are fast converted to cyclic PIP. Further research will have to show, whether both modes of action coexist or, if just one of these two ways of action will be found to meet the biological reality (see chapter 3). (2) Activated inositol phosphate, the second substrate for cyclic PIP synthesis. PLC, activated by adrenaline, liberates apart from IP3, various inositol phosphates from phosphoinositides. Inositol (1:2-cyclic,4)-bisphosphate (pr-Ins-P) was isolated in a 65% yield from aortic myocytes and then converted in the presence of GTP to activated inositol phosphate, which is suggested to be guanosine diphospho-4-inositol (1:2-cyclic)-phosphate (n-Ins-P) (Figure 1) [13].
The mechanism of α2-adrenoceptor-triggered inhibition of adenylate cyclase by the Gi protein is, presently, not finally solved, because: (1) the Gi protein inhibits adenylate cyclase only by 30 to 40%; (2) cyclic PIP inhibits adenylate cyclase by 100%. This inhibition most likely results from protein phosphorylation, but unsolved is, if ser/thr- or tyr-residues of adenylate cyclase or the Gi protein were phosphorylated [35], or if β-arrestins could be involved [36]; (3) Pradipta Gosh and his group reported that the tyr-phosphorylated Gαi protein completely inhibits adenylate cyclase. Insulin, the epidermal growth factor, and the platelet-derived growth factor, stimulate tyrosine kinases, which phosphorylate Gαi in intact cells but not very well in homogenates [37]. Very likely these three different kinds of inhibition of adenylate cyclase are parts of one complex way of inhibition [35].
Why is the synthesis of cyclic PIP rather complex and interference-prone? It is tempting to compare the synthesis of cyclic PIP with the one of cyclic AMP, which switches on catabolism and which is fast synthesized from always available ATP. On the one hand it is logic that the substrates for the synthesis of cyclic PIP are only synthesized when they are needed. On the other hand, why are the precursors for the synthesis of the substrates PGE and n-Ins-P stored in and liberated from membrane-bound lipids, whose stored amounts can be limiting, especially regarding the supply with unsaturated C20 fatty acids.
On α1- and α2-adrenoceptor stimulation contrary effects are triggered. This finding was most likely the reason why the intracellular increase of Ca2+-levels, triggered by IP3 was ascribed to α1- and the inhibition of adenylate cyclase to α2-adrenoceptor action [10]. However, existence and regulatory action of cyclic PIP do not match with this interpretation. To start solving this contradiction, the effect of cyclic PIP on the glucose release from liver was determined. On the one hand it was to expect that cyclic PIP, which inhibits PKA, inhibits accordingly the cyclic AMP-stimulated glucose release from liver. But on the other hand, intracorporal liver-perfusion of rodents with phenylephrine increases the glucose release, which needs increased Ca2+ levels, and which is ascribed to α1-adrenoceptor action [38]. Thus, it had to be determined which role cyclic PIP plays in the release of glucose from liver.
2. Results
Experiments on glucose release from liver have been performed applying intracorporal, recirculating liver perfusion in rodents [38]. With this method the glucose content of the perfusion buffer could be determined, from which the actual amount of glucose release from liver at any time of an experiment had to be calculated. In the following experiments rat livers were extra-corporally and nonrecirculating perfused, enabling direct measurement of the released glucose at any time. This experimental design should warrant that no interferences occur with the animal body, because the liver is disconnected, for instance, from nerves and skeletal muscles, and from the pancreas and the adrenal gland. Basal glucose release from unstimulated, male rat livers was about 300 μg glucose per min per g liver wet weight during the initial 15 min. In all experiments this level was set to 100%. Within 60 min the glucose release decreased by approximately 30%.
Addition of a half-maximally effective amount of cyclic PIP (0.1 μM) to the perfusion buffer triggered instantly a spike-shaped peak of glucose release (Figure 2). This result was not expected, since so far discovered, cyclic PIP antagonizes cyclic AMP’s actions. It inhibits dose-dependently the PKA and activates the counter-regulatory enzyme of the PKA, the protein ser/thr phosphatase holoenzyme [12]. Thus, cyclic PIP should prevent the phosphorylation and thus activation of phosphorylase kinase and subsequently of glycogen phosphorylase. On incubation of purified phosphorylase kinase, which still contained PKA [39], p 109, the phosphorylation and thus activation of glycogen phosphorylase was increased approximately 2-fold in the presence of cyclic AMP. The addition of cyclic PIP reduced this phosphorylation in both the absence and the presence of cyclic AMP by 66% and 39%, respectively, as calculated with the values obtained at 5 min (Figure 3). This inhibition is due to the finding that approximately four times more cyclic PIP is needed to inhibit PKA to an equal amount in the presence than in the absence of cyclic AMP [11].
In order to find an explanation for this obviously contradictory result, the glucose release was determined on stimulation with glucagon, insulin and adrenaline, though these experiments have been performed already years ago by different research groups. The only difference is that the livers were now extra-corporally and nonrecirculating perfused. On glucagon-stimulation (10–9 M) the glucose release increased by about 70% within 13 min and started to decrease after 30 – 40 min (Figure 4). Dibutyryl-cyclic AMP (10-4 M), added to the perfusion buffer, triggered a similar increase in glucose release. On co-stimulation of the liver with glucagon (10–9 M) and insulin (2 x 10–9 M) the glucose release was decreased by 65%. The initial increase was only half that seen with glucagon alone and the decline occurred earlier, only 15 min after the hormone addition (Figure 4). In short, glucagon increases the level of cyclic AMP, which activates PKA. This leads to activation of phosphorylase kinase and then glycogen phosphorylase b, which degrades glycogen, and leads to glucose release from the liver. In the additional presence of insulin, the synthesis of cyclic PIP is increased. On the one hand this leads to less activated PKA and thus less activated glycogen phosphorylase, and on the other hand cyclic PIP activates protein ser/thr phosphatase. This increases the dephosphorylation and inhibition of phosphorylase, causing together a lower release of glucose [12].
Compared to glucagon, adrenaline stimulated the glucose release in a rather different time-profile in male and female rats (Figure 5). The glucose release was determined in male and female rat livers, since in male livers the α-adrenoceptors prevail, whereas in female livers, α- and β-adrenoceptors are present in approximately equal amounts [40]. Glucose was released in two phases. The first one peaked in 1 to 2 min, followed by a more prolonged second phase primarily in the female livers (Figure 5, closed square), which is comparable to the profile of glucose release on stimulation with glucagon (Figure 4), but maximum glucose release was reached within 7 min and it began to decline after 15 min, comparable to the stimulation of glucose release with glucagon plus insulin. In male rat livers (Figure 5, open square) almost no second phase of glucose release was found. The first phase of glucose release looks comparable to the glucose release triggered by cyclic PIP (Figure 2), indicating that the first phase of glucose release on adrenergic stimulation is most likely stimulated by cyclic PIP and thus by α-adrenoceptors. In support of this conclusion is that liver perfusion with 10-6 M phentolamine, an α-adrenoceptor antagonist, 5 min prior to stimulation with 10-6 M adrenaline prevents the spike of glucose release (Figure S2 in Supplementary Material). And the second, more prolonged phase is ascribed to cyclic AMP and thus β-adrenoceptor action.
The low level of β-adrenoceptors in male rats and the action of cyclic PIP may explain why the second phase of glucose release is reduced in male rats [35]. The early decline of glucose release after adrenergic stimulation is striking. The slope of decline is minus 6.3 %/min 20 to 30 min after adrenergic stimulation (Figure 5). On glucagon stimulation the slope of decline of glucose release is minus 0.4 %/min 20 to 30 min after hormonal stimulation, and in case of stimulation with glucagon plus insulin (Figure 4) the slope of decline is minus 5.7 %/min, which is obviously the result of cyclic PIP action (Figure 3).
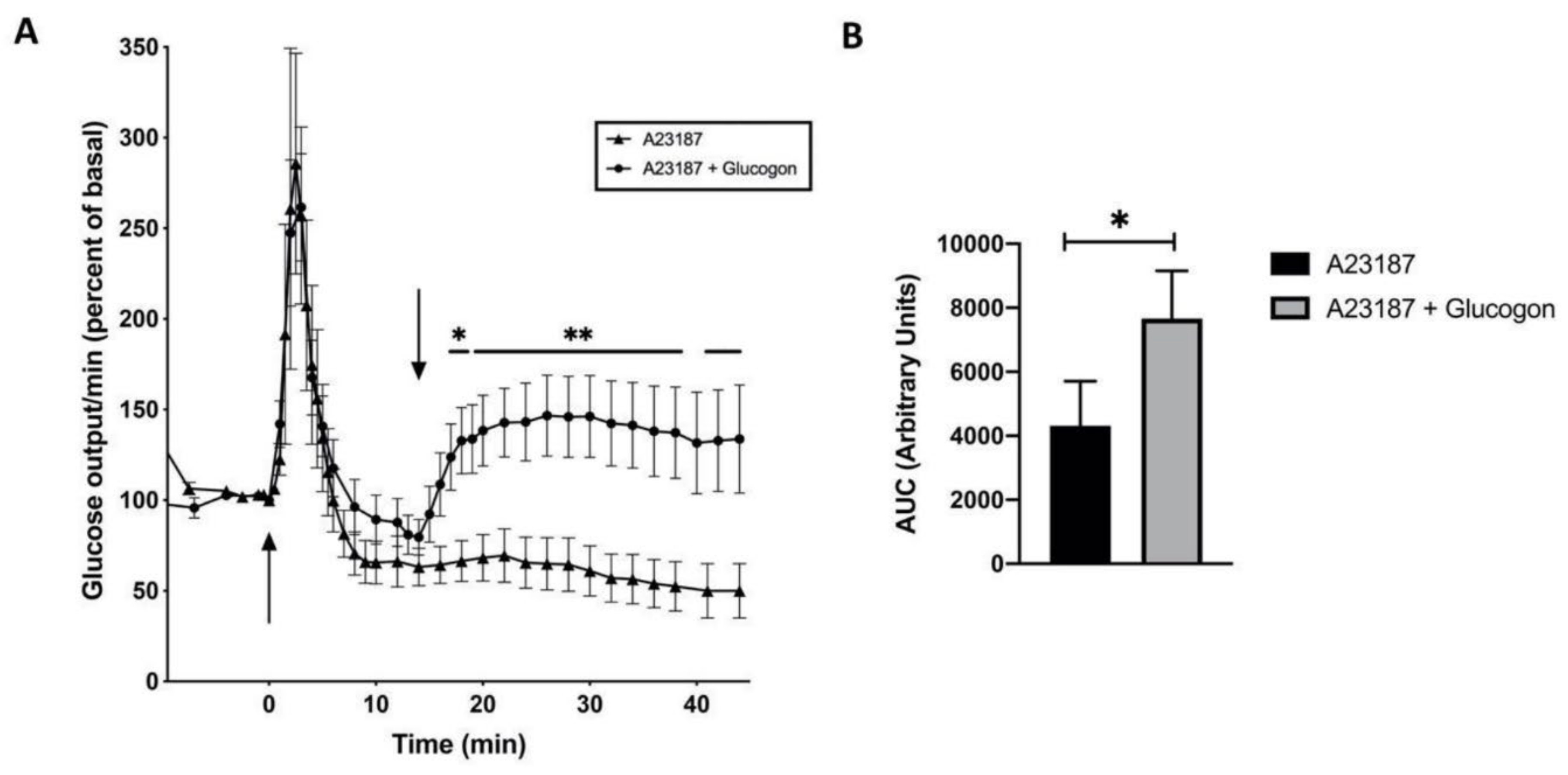
Ca2+ is involved in the α1-adrenoceptor triggered glucose release [38]. Therefore, perfusion of male rat livers with the Ca2+ ionophore A23187 (10–5 M) was performed. A spike-shaped glucose release was found (Figure 6, closed triangle), which increased 2.5 to 3.5-fold within 2 min and then decreased rapidly, confirming that the increase of cytosolic Ca2+ is essential for the α1-adrenoceptor stimulated glucose release. A successive stimulation with glucagon at 15 min triggered a glucose release (Figure 6, closed circle), which is comparable to that seen on glucagon stimulation (Figure 4), indicating that Ca2+ is essential for the first phase of glucose release on stimulation with adrenaline, however, it is not involved in the decrease of the glucose release, which is found on stimulation with glucagon plus insulin (Figure 4), or adrenaline 20 to 30 min after hormonal stimulation (Figure 5).
The glycogen content of all livers was determined after liver perfusion experiments in order to rule out the possibility that limiting glycogen stores accounted for lowered glucose release. The remaining glycogen content of the livers was 5 to 11 mg glycogen per g liver, indicating that any reduced glucose release is not a result of an exhaustion of glycogen stores.
Insulin and adrenaline stimulate the synthesis of cyclic PIP [12], but only adrenaline and cyclic PIP, as intracellular executor of these hormones, trigger a spike-shaped peak of glucose release (Figure 2 and Figure 5). However, cyclic AMP-mediated glucose release is inhibited by insulin and adrenaline stimulation (Figure 4 and Figure 5), which can be best explained by the action of cyclic PIP (Figure 3). Furthermore, adrenaline and insulin are suggested to increase intracellular Ca2+ levels [38,41]. A possible explanation for the difference between adrenaline and insulin stimulated glucose release may be obtained by comparing the synthesis rate of cyclic PIP after stimulation with insulin or adrenaline. A maximal concentration of insulin (10–7 M) or adrenaline (10–4 M), stimulate the synthesis of 43 pmol or 74 pmol cyclic PIP per g rat liver, respectively. In the presence of the β-adrenoceptor antagonist propranolol, which reduces the synthesis-stimulation of cyclic AMP, cyclic PIP synthesis reached a maximum within 1 min in male rat hepatocytes, but on insulin stimulation maximum synthesis was reached after 3 – 4 min [12]. Thus, in the case of adrenaline the mean value of increase of cyclic PIP synthesis is approximately 74 pmol/min and in the case of insulin 12 pmol/min. The slower rate of insulin-stimulated cyclic PIP synthesis could well explain why the increase of the Ca2+ levels triggered by insulin is too low to activate glycogen phosphorylase. This consideration recalls the result of Bruton et al. [42] that the Ca2+ increase caused by insulin is confined to the membrane and the cellular Ca2+ level does not reach the necessary concentration for the “flash-activation” of glycogen phosphorylase [43,44].
3. Discussion
Glucose release from liver is triggered by cyclic AMP and by cyclic PIP. Exton et al. found that glucose release is increased better by stimulation of α1- than β-adrenoceptors [38]. Studer and Borle reported that glucose release in male rat livers is predominantly triggered via α-adrenoceptors, but in female rat livers by α- and β-adrenoceptors [40]. Complementary to these reports, the present report shows that adrenaline stimulates glucose release (1) in a faster, cyclic PIP-triggered way, and (2) a slower β-adrenergic, cyclic AMP-triggered way. In support of this conclusion is that in the presence of the α-adrenoceptor-antagonist phentolamine no fast, spike-shaped glucose release was found, but the cyclic AMP triggered glucose release persists (Supplemental Material, Figure S2). The finding of two separate modes of α-adrenoceptor action, namely the spike-shaped, fast glucose release followed by an inhibition of the cyclic AMP-mediated glucose release, which are both triggered by cyclic PIP, is explicitly shown in this report. Assimacopoulos-Jeannet et al. had observed that “glucagon action is somewhat inhibited by α-adrenoceptor action”, but they had no explanation for this observation [45]. Concerning the applied kind of liver-perfusion, the intracorporal liver perfusion is certainly closer to the physiological situation and the extra-corporal liver perfusion gives more accurate results on the glucose release from liver at a specific time.
Not finally resolved is, if the slower synthesis rate of cyclic PIP on insulin stimulation could result from a slower binding of insulin to its receptor when compared with adrenalins binding to its receptor. The 1000-fold lower concentration at which insulin acts could be a possible reason. Interesting is that the stimulation of glucose release by glucagon and adrenalin via its β-receptors is comparable, since glucose release reaches a maximum after 13 min after stimulation with glucagon but only 7 min after stimulation with adrenalin concerning its second phase of glucose release (Figure 4 and Figure 5). It cannot ruled out that a difference in the speed of the signal transduction could be a reason too. In case of adrenaline’s binding to its receptor the involved G protein separates its α-subunit, which then activates cyclic PIP synthase. In case of insulin’s binding to its receptor, the tyrosine kinase of the β-subunits of this receptor, after its auto-phosphorylation, phosphorylates and activates the insulin receptor substrates (IRS) and the cyclic PIP synthase [12].
It is well documented that prostaglandin E (PGE) is one of the two substrates of cyclic PIP synthesis [11,12,13,16]. As mentioned in the introduction, adrenaline sends via it’s α-adrenoceptors a complex signal into cells, which leads to the synthesis of cyclic PIP (Figure 1). The different, further path of prostaglandin action, involving prostanoid receptors, is not yet finally characterized, though of most of the prostanoid-receptors the binding affinity and also their amino acid sequence is determined [46,47,48,49]. Nevertheless, confusing is that PGE is found to be excreted unmetabolized (see below) from the cells of its synthesis and binds then on surrounding cells to prostanoid receptors. These receptors are suggested to send signals into cells in part via the Gq-, Gi-, and also Gs-proteins, which are used by adrenaline for its actions. This means that adrenaline triggers via G-proteins (Figure 1) the synthesis of PGE [7,8], which then also acts via these G proteins. In this context the question remains unsolved as to whether the same signal-transduction-path could lead to different effects. In case there is no proof for an intracellular mechanism that controls, which intracellular effects are modulated, then one could assume that prostaglandins via its prostanoid receptors trigger again intracellular prostaglandin synthesis. And, however you take it, biochemical experiments are missing, as for instance, (1) which different regulations could occur when the same G proteins are stimulated either by adrenoceptors or prostanoid receptors, or (2) which regulatory mechanisms have not yet been found, which determine if PGE activates or inhibits adenylate cyclase [46]. Years ago, it was found that at physiological concentrations PGE inhibits adenylate cyclase in intact cells but not of purified plasma membranes [39] pp 301 –304, and it activates cyclic AMP synthesis of hepatocytes approximately two-fold at concentrations higher than 10–7 M [16], which was then seen as a pharmacological effect. Furthermore, one has to be aware that on the isolation of cyclic PIP from rat liver a major amount of its PGE-part is already degraded to dinor-PGE (the C18-homolog of PGE), because of its fast degradation in liver [12], and the same fast degradation rate of PGE is to be expected also in case of its excretion.
Intermediate synthesis products of a synthesis path, which leads, for instance, to a regulatory active compound, are expected to be inactive. However, in the case of cyclic PIP synthesis this appears to be different. One has to be aware that prostaglandins can enter cells as hepatocytes. This increases cyclic PIP synthesis [11], and additionally – as an essential molecule part of cyclic PIP – PGE (4 x 10–6 M), for instance, activates PKA by 15 to 20% (Wasner, unpublished). Certainly, this is a marginal effect when one compares it with the more than 10-fold activation of PKA by cyclic AMP. In summary, PGE is one of the two substrates of cyclic PIP synthesis. Additionally, PGE, as all prostaglandins, is seen as “tissue hormone” and the unsolved question is, if both these kinds of actions can physiologically be relevant. Further on, one may ask if these prostanoid-receptors could have still other, not identified, different functions. For instance, the regulatory subunit of PKA binds cyclic AMP very well, but this subunit is nevertheless not a receptor.
On α-adrenoceptor stimulation, intracellular Ca2+-levels are increased by IP3 and cyclic PIP. Most likely they represent two distinct ways of regulation of intracellular Ca2+ levels. However, peculiar is that both reaction paths are triggered, for instance, by adrenaline via it’s α-adrenoceptors. IP3 triggers release of Ca2+ from intracellular stores as the endoplasmic reticulum [50,51,52], and cyclic PIP stimulates Ca2+ influx into cells [12].
Ca2+ is involved in different reaction sequences with respect to triggering glucose release from liver. (1) Ca2+ is necessary for cyclic AMP action, owing to the requirement of Ca2+ for phosphorylase kinase activity [53], and (2) Ca2+ is needed for the “flash activation” of glycogen phosphorylase in skeletal muscle [43,44], and it appears that glycogen phosphorylase of liver is comparably activated. The role of Ca2+ in the mechanism of insulin action was a matter of controversy, which was settled by Bruton et al. [42], showing that insulin triggers a slower increase of Ca2+ than adrenaline. Assuming a similar Ca2+ increase also in hepatocytes, the slower increase of cyclic PIP synthesis after insulin stimulation could be an explanation why no burst of glucose release is observed on insulin stimulation.
Earl Sutherland declared that calcium is a cofactor of different enzyme-reactions, but it is not a second messenger [39], pp 18,143,223. Nevertheless, other scientists like to see Ca2+ as a second messenger [54]. In case of the intracellular need of Ca2+, cells open specific gates on hormonal stimulation to allow Ca2+ influx [55,56]. The increase of Ca2+ in the cells is triggered by the second messenger IP3, and also by the intracellular regulator cyclic PIP. This makes it rather unrealistic that Ca2+ can be a second messenger too. The question which needs to be reconsidered is: What are optimal credentials, which allow a compound or intracellular regulator to be called a second messenger? Sutherland had 4 credentials coined, because of which he called cyclic AMP a second messenger [39, pp 22–29]. Cofactors are essential for various enzyme-catalyzed reactions to occur, but a second messenger only increases or decreases the activity of regulatory enzymes. Cyclic AMP, for instance, switches on catabolism by moving the equilibrium between the phospho- and the de-phospho-form of interconvertible enzymes to the phospho-form [12]. Cyclic AMP was for many years the only accepted second messenger. Then many compounds obtained this label, since the credentials were decreased because of which a compound can be called a second messenger. Presently, in the beating heart Ca2+ is seen as the essential regulator [57,58], whereas existence and action of cyclic PIP is scarcely recognized, though it is the intracellular regulator which stimulates anabolic regulations. However, unsolved is presently, if the effects of increased Ca2+ levels support the anabolic regulations triggered by cyclic PIP.
4. Materials and Methods
4.1. Materials
(–)-adrenaline bitartrate, glucagon, insulin, cyclic AMP, dibutyryl cyclic AMP, bovine serum albumin (BSA fraction V), A23187 and EGTA (ethylene glycol-bis(β-aminoethyl ether) N,N,N’,N’-tetra-acetic acid) were obtained from Millipore-Sigma (Darmstadt, Germany); ATP, glycogen from rabbit liver, collagenase from Clostridium histolyticum and amylo-α-1,4-α-1,6-glucosidase from Aspergillus niger and gluco-quant were from Roche Diagnostics (Mannheim, Germany); [γ-32P]ATP from Amersham Pharmacia Biotech (now GE-Healthcare, Solingen, Germany); and sodium pentobarbital from Wirtschaftsgenossenschaft Deutscher Tierärzte (Darmstadt, Germany). All other chemicals of reagent grade were from E. Merck (Darmstadt, Germany).
4.2. Animals
Sprague-Dawley rats (200–350 g) had free access to water and food (standard diet, Ssniff). They were anesthetized with an overdose of sodium pentobarbital (30 – 40 mg/kg body weight) prior to use.
4.3. Liver Perfusion
Isolated liver was perfused in situ as described previously [59] in a non-recirculating manner in the physiological antegrade direction from the portal vein to the hepatic vein with Krebs-Ringer-bicarbonate buffer (118.46 mM NaCl; 4.74 mM KCl; 2.57 mM CaCl2; 1.12 mM KH2PO4; 1.185 mM MgSO4; 24.4 mM NaHCO3; 2% fatty acid free BSA). The influent perfusate was gassed with O2/CO2 (95/5% by vol). The temperature was 37 0C and the constant flow rate was 6 ml/min. The extracorporeal, non-recirculating perfusion of the liver had the advantage that the perfusate only contains the released glucose at a given time. (No cyclic AMP phosphodiesterase inhibitors to the perfusion buffer were added, in order to prevent an imbalance between stimulatory and inhibitory hormone signals, because no agents are presently known, which block the degradation of cyclic PIP comparable to the methylxanthines, which inhibit the cyclic AMP phosphodiesterase.) In case of hormonal stimulation (indicated by arrows in the Figures>), the hormones were injected into the Krebs-Ringer bicarbonate perfusion buffer. The effluent perfusate was collected in 0.5 min or 1 min samples and assayed for its glucose content.
4.4. Statistical Analysis
All glucose values of the experiments are expressed as mean + SEM (standard error of the mean). The differences between two groups were analyzed using an unpaired t-test (Graph Pad Prism Software Inc., San Diego, CA, USA). Significance was accepted at p < 0.05.
4.5. Glucose Determination
Glucose was determined according to Schmidt [60]. The glucose concentration of the perfusate is converted into mg glucose per g liver and min. For determination of the glucose content of liver, an aliquot of the liver was homogenized and its glucose content determined after denaturation of proteins.
4.6. Liver Glycogen
Glycogen content of rat liver was determined according to Passonneau and Lauderdale [61]. A weighed amount of rat liver, generally after a perfusion experiment, was homogenized in 0.25 M KOH and boiled for 10 min. 30 µl of the supernatant was incubated with 120 µl 0.1 M acetate buffer of pH 4.7 and 30 µl amylo-α-1,4-α-1,6-glucosidase (10 mg/10 ml of 20 mM Tris/HCl buffer of pH 7.5) for 3 h at 25 0C and glucose was determined.
4.7. Phosphorylation of Glycogen Phosphorylase
The assay was performed analog to the protein kinase A (PKA) assay [11], but the histone type IIa of the kinase assay was replaced by glycogen phosphorylase b and the PKA was replaced by phosphorylase kinase (0.25 mg/ml), which still contained PKA. Incubation temp. was 30 0C, the buffer pH was 6.8. Glycogen phosphorylase b was purified according to [62].
5. Conclusions
In summary, glucose release from male and female rat liver is triggered (1) by cyclic AMP, which is increased by glucagon or adrenaline via its β-adrenoceptors, and (2) by cyclic PIP, whose synthesis is stimulated by adrenaline via its α1- and α2-adrenoceptors [12]. Glycogen phosphorylase is activated by 2 independent ways [63], (1) by cyclic PIP action, involving high Ca2+ levels and (2) by protein ser/thr-phosphorylation, induced by cyclic AMP. This view of two independent ways of activation of glycogen phosphorylase is supported by the result that after a maximal stimulation of glucose release by 10–9 M glucagon [39], pp. 237–238, a successive stimulation with 10–7 M adrenaline further increases the glucose release, showing additivity of both effects (Supplementary Material, Figure S1). Furthermore, cyclic PIP triggers two independent effects, first the fast, spike-shaped glucose release (Figure 2), which precedes the β-adrenergic glucose release, and then the inhibition of the cyclic AMP triggered signal, leading to inhibition of glycogen phosphorylase (Figure 3). Stimulation of rat livers with insulin increases cyclic PIP synthesis, which only inhibits the cyclic AMP-triggered signal, most likely because the insulin stimulated Ca2+ increase is too slow. The peculiarity of the adrenergic stimulated glucose release is that α- and β-adrenoceptors show at first a concerted action, which is triggered by cyclic PIP and cyclic AMP. Once the rapid calcium effect has ended, the inhibition of the cyclic AMP triggered glucose release by cyclic PIP becomes apparent. Effects of cyclic PIP, resulting from increased Ca2+ influx, has to be determined also in other cell types of a body. Most likely then the logic will be found why α-adrenoceptors trigger in hepatocytes two opposing effects time-dependently.
The increase of intracellular Ca2+-levels by cyclic PIP reveals a mechanism of cyclic PIP action which appears to be distinct from its antagonism to cyclic AMP. A more detailed characterization of the cyclic PIP-triggered Ca2+ influx into cells needs to be performed. This will most likely happen when the chemical synthesis of cyclic PIP is reached. One has to be aware that cyclic PIP is difficult to handle under the generally mild conditions of biochemical experiments [12,13], and cyclic PIP is more difficult to handle under the harsher conditions of chemical synthesis. Further on, it has to be determined, if all or which one of the six α-adrenoceptor subtypes, the α1A-, α1B-, α1D-, and α2A-, α2B-, α2C-adrenoceptor, trigger the synthesis of cyclic PIP.
A follow-up question of his report is to ask, how is the influx of glucose into the liver regulated and which role cyclic PIP could play. In recent reports is shown that insulin stimulates the postprandial glucose uptake in the liver [64]. Cyclic PIP stimulates the glucose uptake in adipocytes [16], and it can be assumed that cyclic PIP is involved also in the postprandial glucose uptake into cells of a body. Insulin and adrenaline stimulate the synthesis of cyclic PIP [12], thus the effect of both hormones on glucose uptake by the liver should be determined.
Supplementary Materials
The following are available online at www.preprints.org, Figure S1. Time course of glucose release from male rat liver on successive stimulation with glucagon and adrenaline. (A) Stimulation with glucagon (10–9 M) at 0 min and with adrenaline (10–7 M) at 27 min; (B) Stimulation with adrenaline (10–6 M) at 0 min and with glucagon (10–9 M) at 15 min. (The arrows indicate the times of hormone addition.) C) The glucagonstimulated glucose release after adrenaline stimulation (Figure S1B) is significantly smaller (Figure S1 C, black bar) than the glucose release on a first stimulation with glucagon (Figure S1 C, grey bar). The baseline used was the glucose release on adrenaline stimulation (Figure 5, male rat). ** p < 0.005 with versus without preceding adrenaline stimulation. Figure S2. Adrenaline (10–6 M) stimulated glucose release in the absence (gray curve) and presence (black curve) of 10–6 M phentolamine (alpha-adrenoceptor antagonist) from female rat livers. Phentolamine prevented the a-adrenoceptor triggered, spike-shaped glucose release, but not the ß-adrenoceptor triggered component. However, because of a small affinity of phentolamine to the ß-adrenoceptor, the cyclic AMP triggered glucose release is lower in the presence than in the absence of phentolamine.
Author Contributions
S.W. performed the liver perfusions and prepared the figures; L.C. performed the statistical calculations; H.K.W., the project leader, prepared cyclic PIP and wrote the manuscript.
Institutional Review Board
All experiments were conducted in accordance with the Principles of Laboratory Animal Care (GV-SOLAS, Society for Laboratory Animal Care) and were approved by the ethics committee on animal welfare of the State of North Rhine Westfalia (file number 26.4203.3; 1998).
Informed Consent Statement
Not applicable.
Data Availability Statement
We were engaged to report all experimental details in the Section 4. In case of further questions, please contact the corresponding author.
Acknowledgments
H.K.W. is thankful that he could pursue the research on cyclic PIP at the German Diabetes Center (DDZ) up to 2004. S.W. performed his thesis in the lab of H.K.W., who thanks Knut Feldmann for the support with glycogen phosphorylase, Jochen Schulz and especially Kathy Rundell for valuable suggestions and improvements of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bartok, A.; Weaver, D.; Golenar, T.; Nichtova, Z.; Katona, M.; Bansaghi, S.; Alzayady, K.J.; Thomas, V.K.; Ando, H.; Mikoshiba, K.; et al. IP3 receptor isoforms differently regulate ER-mitochondrial contacts and local calcium transfer. Nat. Commun. 2019, 10, 3726. [Google Scholar] [CrossRef] [PubMed]
- Young, M.P.; Schug, Z.T.; Booth, D.M.; Yule, D.I.; Mikoshiba, K.; Hajnoczky, G.; Joseph, S.K. Metabolic adaptation to the chronic loss of Ca2+ signaling induced by KO of IP3 receptors or the mitochondrial Ca2+ uniporter. J. Biol. Chem. 2022, 298, 101436. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.; Deb, B.K.; Arige, V.; Musthafa, T.; Malik, S.; Yule, D.I.; Taylor, C.W.; Hasan, G. Regulation of store-operated Ca2+ entry by IP3 receptors independent of their ability to release Ca2+. eLife 2023, 12, e80447. [Google Scholar] [CrossRef] [PubMed]
- Gomez, L.C.; Kawaguchi, S.; Collin, T.; Jalil, A.; del Pilar Gomez, M.; Nasi, E.; Marty, A.; Llano, J. Influence of spatially segregated IP3-producing pathways on spike generation and transmitter release in Purkinje cell axons. Proc. Natl. Acad. Sci. USA 2020, 117, 11097–11108. [Google Scholar] [CrossRef]
- Bylund, D.B. Subtypes of α1- and α2-adrenergic receptors. FASEB J. 1992, 6, 832–839. [Google Scholar] [CrossRef]
- Garro, M.A.; Urizar, E.; Lazkano, A.; Zubillaga, E.; Querejeta, R. The α1A adrenergic receptor inhibits type 5 adenylyl cyclase in HL-1 cardiomyocytes. Clin. J. Pharmacol. Pharmacother. 2018, 1, 1003. [Google Scholar]
- Burch, R.M.; Luini, A.; Axelrod, J. Phospholipase A2 and phospholipase C are activated by distinct GTP-binding proteins in response to a1-adrenergic stimulation of FRTL5 thyroid cells. Proc. Natl. Acad. Sci USA 1986, 83, 7201–7205. [Google Scholar] [CrossRef]
- Felder, C.S.; Williams, H.L.; Axelrod, J. A transduction pathway associated with receptors coupled to the inhibitory guanine nucleotide binding protein Gi that amplifies ATP-mediated arachidonic acid release. Proc. Natl. Acad. Sci. USA 1991, 88, 6477–6480. [Google Scholar] [CrossRef]
- Gesek, F.A. Alpha2-adrenergic receptors activate phospholipase C in renal epithelial cells. Mol. Pharmacol. 1996, 50, 407–414. [Google Scholar]
- Fain, J.N.; Garcia-Sainz, J.A. Role of phosphatidylinositol turnover in alpha1 and of adenylate cyclase inhibition in alpha2 effects of catecholamines. Life Sci. 1980, 26, 1183–1194. [Google Scholar] [CrossRef]
- Wasner, H.K.; Salge, U.; Gebel, M. The endogenous cyclic AMP antagonist, cyclic PIP: its ubiquity, hormone stimulated synthesis and identification as prostaglandylinositol cyclic phosphate. Acta. Diabetol. 1993, 30, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Wasner, H.K. Prostaglandylinositol cyclic phosphate, the natural antagonist to cyclic AMP. IUBMB Life 2020, 72, 2282–2289. [Google Scholar] [CrossRef] [PubMed]
- Gypakis, A.; Adelt, S.; Lemoine, H.; Vogel, G.; Wasner, H.K. Activated inositol phosphate, substrate for synthesis of prostaglandylinositol cyclic phosphate (cyclic PIP)–the key for the effectiveness of inositol feeding. Int. J. Mol. Sci. 2024, 25(3), 1362. [Google Scholar] [CrossRef] [PubMed]
- Morgan, N.G.; Montague, W. Studies on the mechanism of inhibition of glucose-stimulated insulin secretion by noradrenaline in rat islets of Langerhans. Biochem. J. 1985, 226, 571–576. [Google Scholar] [CrossRef]
- Wasner, H.K.; Lemoine, H.; Junger, E.; Leßmann, M.; Kaufmann, R. Prostaglandylinositol cyclic phosphate, a new second messenger. In Prostaglandins, Leukotrienes, Lipoxins and PAF. Bailey, J.M., Ed., Plenum Press: New York, USA, 1991, pp. 153–168.
- Wasner, H.K.; Weber, S.; Partke, H.J.; Amini-Hadi-Kiashar, H. Indomethancin treatment causes loss of insulin action in rats: Involvement of prostaglandins in the mechanism of insulin action. Acta Diabetol. 1994, 31, 175–182. [Google Scholar] [CrossRef]
- Flechtner-Mors, M.; Jenkinson, C.P.; Alt, A.; Biesalski, H.K.; Adler, G.; Ditschuneit, H.H. Sympathetic regulation of glucose uptake by the α1-adrenoceptor in human obesity. Obes. Res. 2004, 12, 612–620. [Google Scholar] [CrossRef]
- Taylor, B.N.; Cassagnol, M. Alpha-adrenergic receptors. In: StatPearls, Treasure Island, Fl; StatPearls Publishing; 2024. Available online: www.ncbi.nlm.nih.gov/books/NBK539830/.
- Wasner, H.K.; Gebel, M.; Hucken, S.; Schaefer, M.; Kincses, M. Two separate mechanisms for activation of cyclic PIP synthase: By a G protein or by protein tyrosine phosphorylation. Biol. Chem. 2000, 381, 145–153. [Google Scholar] [CrossRef]
- Kanemaru, K.; Nakamura, Y. Activation mechanisms and diverse functions of mammalian phospholipase C. Biomolecules 2023, 13, 915. [Google Scholar] [CrossRef]
- Boi, R.; Ebefors, K.; Henricsson, M.; Boren, J.; Nystroem, J. Modified lipid metabolism and cytosolic phospholipase A2 activation in mesangial cells under pro-inflammatory conditions. Sci. Rep. 2022, 12, 7233. [Google Scholar] [CrossRef]
- Khan, S.A.; Ilies, M.A. The phospholipase A2 superfamily: structure, isozymes, catalysis, physiologic and pathologic roles. Int. J. Mol. Sci. 2023, 24, 1353. [Google Scholar] [CrossRef]
- Dabral, D.; van den Bogaart, G. The roles of phospholipase A2 in phagocytes. Front. Cell Dev. Biol. 2021, 9, 673502. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Zhou, Q.; Labroska, V.; Qin, S.; Darbalaei, S.; Wu, Y.; Yuliantie, E.; Xie, L.; Tao, H.; Cheng, J.; et al. G protein-coupled receptors: structure- and function-based drug dis-covery. Sig. Transduct. Target. Ther. 2021, 6, 7. [Google Scholar] [CrossRef]
- Myagmar, B.-E.; Ismaili, T.; Swigart, P.M.; Raghunathan, A.; Baker, A.J.; Sahdeo, S.; Blevitt, J.M. Milla, M.E.; Simpson, P.C. Coupling of Gq signaling is required for cardioprotection by an alpha-1A-adrenergic receptor agonist. Circulation Research 2019, 125, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Kostenis, E.; Pfeil, E.M.; Annala, S. Heterotrimeric Gq proteins as therapeutic targets? J. Biol. Chem. 2020, 295, 5206–5215. [Google Scholar] [CrossRef] [PubMed]
- Durkee, C.A.; Cavelo, A.; Lines, J.; Kofuji, P.; Aguilar, J.; Araque, A. Gi/o protein-coupled receptors inhibit neurons but activate astrocytes and stimulate gliotransmission. Glia 2019, 67, 1076–1093. [Google Scholar] [CrossRef]
- Dickerson, M.T.; Dadi, P.K.; Zaborska, K.E.; Nakhe, A.Y.; Schaub, C.M.; Dobson, J.R.; Wright, N.M.; Lynch, J.C.; Scott, C.F.; Robison, L.D.; et al. Gi/o protein-coupled receptor inhibition of beta-cell electrical excitability and insulin secretion on Na+/K+ ATPase activation. Nat. Commun. 2022, 13, 6461. [Google Scholar] [CrossRef]
- Hunt, T.W.; Carroll, R.C.; Peralta, E.G. Heterotrimeric G proteins containing Gαi3 regulate multiple effector enzymes in the same cell. J. Biol. Chem. 1994, 269, 29565–29570. [Google Scholar] [CrossRef]
- Pai, V.J.; Lu, R.; Wu, L.; Garcia Macia, M.; Koba, W.R.; Chi, Y.; Singh, R.; Schwartz, G.J.; Schuster, V.L. Inhibiting the prostaglandin transporter PGT induces non-canonical thermogenesis at thermoneutrality. bioRxiv 2019. [Google Scholar] [CrossRef]
- Schuster, V.L. Molecular mechanisms of prostaglandin transport. Annu. Rev. Physiol. 1998, 60, 221–242. [Google Scholar] [CrossRef]
- Fischer, D.P.; Griffiths, A.L. Lui, S.; Sabar, U.J.; Farrar, D.; O’Donovan, P.J.; Woodward, D.F.; Marshall, K.M. Distribution and function of prostaglandin E2 receptors in mouse uterus translational value for human reproduction. J. Pharmacol. Exp. Ther. 2020, 373, 381–390. [Google Scholar] [CrossRef]
- Ye, Y.; Lin, P.; Zhu, J.; Jeschke, U.; von Schoenfeldt, V. Multiple roles of prostaglandin E2 receptors in female reproduction. Endocrines 2020, 1, 22–34. [Google Scholar] [CrossRef]
- Lv, X.; Gao, K.; Nie, J.; Zhang, X.; Zhang, S.; Ren, Y.; Sun, X.; Li, Q.; Huang, J.; Liu, L.; et al. Structures of human prostaglandin F2α receptors reveal the mechanism of ligand and G protein selectivity. Nat. Commun. 2023, 14, 8136. [Google Scholar] [CrossRef] [PubMed]
- Wasner, H.K. Insulin resistance develops due to an imbalance in the synthesis of cyclic AMP and the natural cyclic AMP antagonist prostaglandylinositol cyclic phosphate (cyclic PIP). Stresses 2023, 3, 762–772. [Google Scholar] [CrossRef]
- Kee, T.R.; Khan, S.A.; Neidhart, M.B.; Masters, B.M.; Zhao, V.K.; Kim, Y.K.; McGill Percy, K.C.; Woo, J.-A.A. The multifaceted functions of β-arrestins and their therapeutic potential in neurodegenerative diseases. Exp. Mol. Med. 2024, 56, 129–141. [Google Scholar] [CrossRef]
- Kalogriopoulos, N.A.; Lopez-Sanchez, I.; Lin, C.; Ngo, T.; Midde, K.K.; Roy, S.; Aznar, N.; Murray, F.; Garcia-Marcos, M.; Kufareva, I.; et al. Receptor tyrosine kinases activate heterotrimeric G proteins via phosphorylation within the interdomain cleft of Gαi. Proc. Natl. Acad. Sci. USA 2020, 117, 28763–28774. [Google Scholar] [CrossRef]
- Charest, R.; Blackmore, P.F.; Berthon, B.; Exton, J.H. Changes in free cytosolic Ca2+ in hepatocytes following α1-adrenergic stimulation. J. Biol. Chem. 1983, 258, 8769–8773. [Google Scholar] [CrossRef]
- Robison, G.A.; Butcher, R.W.; Sutherland, E.W. Cyclic AMP.; Academic Press: New York, NY, USA, 1971. [Google Scholar]
- Studer, R.K.; Borle, A.B. Differences between male and female rats in the regulation of the hepatic glycogenolysis. J. Biol. Chem. 1982, 257, 7987–7993. [Google Scholar] [CrossRef]
- Pershadsingh, H.A.; Shade, D.L.; Delfert, D.M.; McDonald, J.M. Chelation of intracellular calcium blocks insulin action in the adipocyte. Proc. Natl. Acad. Sci. US 1987, 84, 1025–1029. [Google Scholar] [CrossRef]
- Bruton, J.D.; Katz, A.; Westerblad, H. Insulin increases near-membrane but not global Ca2+ in isolated skeletal muscle. Proc. Natl. Acad. Sci. USA 1999, 96, 3281–3286. [Google Scholar] [CrossRef]
- Heilmeyer, L.M.G.; Meyer, F.; Haschke, R.H.; Fischer, E.H. Control of phosphorylase activity in a muscle glycogen particle. II. Activation by calcium. J. Biol. Chem. 1970, 245, 6649–6656. [Google Scholar] [CrossRef]
- Brautigan, D.L. Phosphorylase phosphatase and flash activation of skeletal muscle glycogen phosphorylase–a tribute to Edmond, H. Fischer. IUBMB Life 2023, 75, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Assimacopoulos-Jeannet, F.D.; Blackmore, P.F.; Exton, J.H. Studies of the interaction between glucagon and α-adrenergic agonists in the control of hepatic glucose output. J. Biol. Chem. 1982, 257, 3759–3765. [Google Scholar] [CrossRef] [PubMed]
- Biringer, R.G. A review of prostanoid receptors: expression, characterization, regulation and mechanism of actions. J. Cell Commun. Signal. 2021, 15, 155–184. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Xu, Y.; He, Q.; Li, D.; Duan, J.; Li, C.; You, C.; Chen, H.; Fan, W.; Jiang, Y.; et al. Ligand-induced activation and G protein coupling of prostaglandin-F2α receptor. Nat. Commun. 2023, 14, 2668. [Google Scholar] [CrossRef]
- Kurz, M.; Krett, A.-L.; Buenemann, M. Voltage dependence of prostanoid receptors. Mol. Pharmacol. 2020, 97, 267–277. [Google Scholar] [CrossRef]
- Herrera, M.; Yang, T.; Sparks, M.A.; Manning, M.W.; Koller, B.H.; Coffman, T.M. Complex role for E-prostanoid 4 receptors in hypertension. J. Am. Heart Association 2019, 8, e010745. [Google Scholar] [CrossRef]
- Schmitz, E.A.; Takahashi, H.; Karakas, E. Structural basis for activation and gating of IP3 receptors. Nat. Commun. 2022, 13, 1408. [Google Scholar] [CrossRef]
- Lebeau, P.F.; Platko, K.; Byun, J.H.; Austin, R.C. Calcium as a reliable marker for the quantitative assessment of endoplasmic reticulum stress in live cells. J. Biol. Chem. 2021, 296, 100779. [Google Scholar] [CrossRef]
- Woll, K.A.; Van Petegem, F. Calcium-release channels: structure and function of IP3 receptors and ryanodine receptors. Physiol. Rev. 2022, 102, 209–268. [Google Scholar] [CrossRef]
- Yang, X.; Zhu, M.; Lu, X.; Wang, Y.; Xiao, J. Architecture and activation of human muscle phosphorylase kinase. Nat. Commun. 2024, 15, 2719. [Google Scholar] [CrossRef]
- Baudet, S.; Zagar, Y.; Roche, F.; Gomez-Bravo, C.; Couvet, S.; Becret, J.; Belle, M.; Vougny, J.; Uthayasuthan, S.; Ros, O.; et al. Subcellular second messenger networks drive distinct repellent-induced axon behaviors. Nat. Commun. 2023, 14, 3809. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.S. Store-operated calcium channels: From function to structure and back again. Cold Spring Harb. Perspect. Biol. 2020, 12, a035055. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A.; Lenaeus, M.J.; Jamal El-Din, T.M. Structure and pharmacology of voltage-gated sodium and calcium channels. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 133–154. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Lu, K.; Kamla, C.; Kameritsch, P.; Seidel, T.; Dendorfer, A. Synchronous force and Ca2+ measurements for repeated characterization of excitation-contraction coupling in human myocardium. Commun. Biol. 2024, 7, 220. [Google Scholar] [CrossRef]
- Zhang, J.; Simpson, P.C.; Jensen, B.C. Cardiac α1A-adrenergic receptors: emerging protective roles in cardiovascular diseases. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H725–H733. [Google Scholar] [CrossRef]
- Sies, H. The use of perfusion of liver and other organs for the study of microsomal electron transport and cytochrome P-450 systems. Meth. Enzymol. 1978, 52, 48–59. [Google Scholar]
- Schmidt, F.H. Die enzymatische Bestimmung von Glucose und Fructose nebeneinander. Klin. Wochenschr. 1961, 39, 1244–1247. [Google Scholar] [CrossRef]
- Passonneau, J.V.; Lauderdale, V.R. A comparison of three methods of glycogen measurement in tissue. Analyt. Biochem. 1974, 60, 405–412. [Google Scholar] [CrossRef]
- Feldmann, K. and Hull, W.F. 31P nuclear magnetic resonance studies of glycogen phosphorylase from rabbit skeletal muscle: Ionization status of pyridoxal 5‘-phosphate. Proc. Natl. Acad. Sci. USA 1977, 74, 856–860. [Google Scholar] [CrossRef]
- Soderling, T.R.; Park, C.R. Recent advances in glycogen metabolism. Adv. Cyclic Nucleotide Res. 1974, 4, 283–333. [Google Scholar]
- Kraft, G.; Coate, K.C.; Smith, M.; Farmer, B.; Scott, M.; Cherrington, A.D.; Edgerton, D.S. The importance of the mechanisms by which insulin regulates meal-associated liver glucose uptake in the dog. Diabetes 2021, 70, 1292–1302. [Google Scholar] [CrossRef]
Figure 1.
Scheme of the biosynthesis of cyclic PIP and its substrates prostaglandin E (PGE) and activated inositol phosphate (n-Ins-P) on α1↑- or α2-adrenoceptor stimulation. Phospholipase Cβ (PLC) [20], phospholipase A2 (PLA2) [21,22,23], and cyclic PIP synthase are activated by G proteins (GPLA, GPLC, Gcyclic PIP), transmitting the signal from the α-adrenoceptors to the effector enzymes. It appears that it is, presently, not finally determined which G protein activates which one of these three enzymes: α1A-adrenoceptor action is connected with Gq protein and α2-adrenoceptor action with Gi/o proteins [24,25,26,27,28]; Axelrod reported that PLA2 and PLC are activated by different G proteins [7], and Hunt et al. suggested that these enzymes are activated by one type of G protein [29].
Figure 1.
Scheme of the biosynthesis of cyclic PIP and its substrates prostaglandin E (PGE) and activated inositol phosphate (n-Ins-P) on α1↑- or α2-adrenoceptor stimulation. Phospholipase Cβ (PLC) [20], phospholipase A2 (PLA2) [21,22,23], and cyclic PIP synthase are activated by G proteins (GPLA, GPLC, Gcyclic PIP), transmitting the signal from the α-adrenoceptors to the effector enzymes. It appears that it is, presently, not finally determined which G protein activates which one of these three enzymes: α1A-adrenoceptor action is connected with Gq protein and α2-adrenoceptor action with Gi/o proteins [24,25,26,27,28]; Axelrod reported that PLA2 and PLC are activated by different G proteins [7], and Hunt et al. suggested that these enzymes are activated by one type of G protein [29].
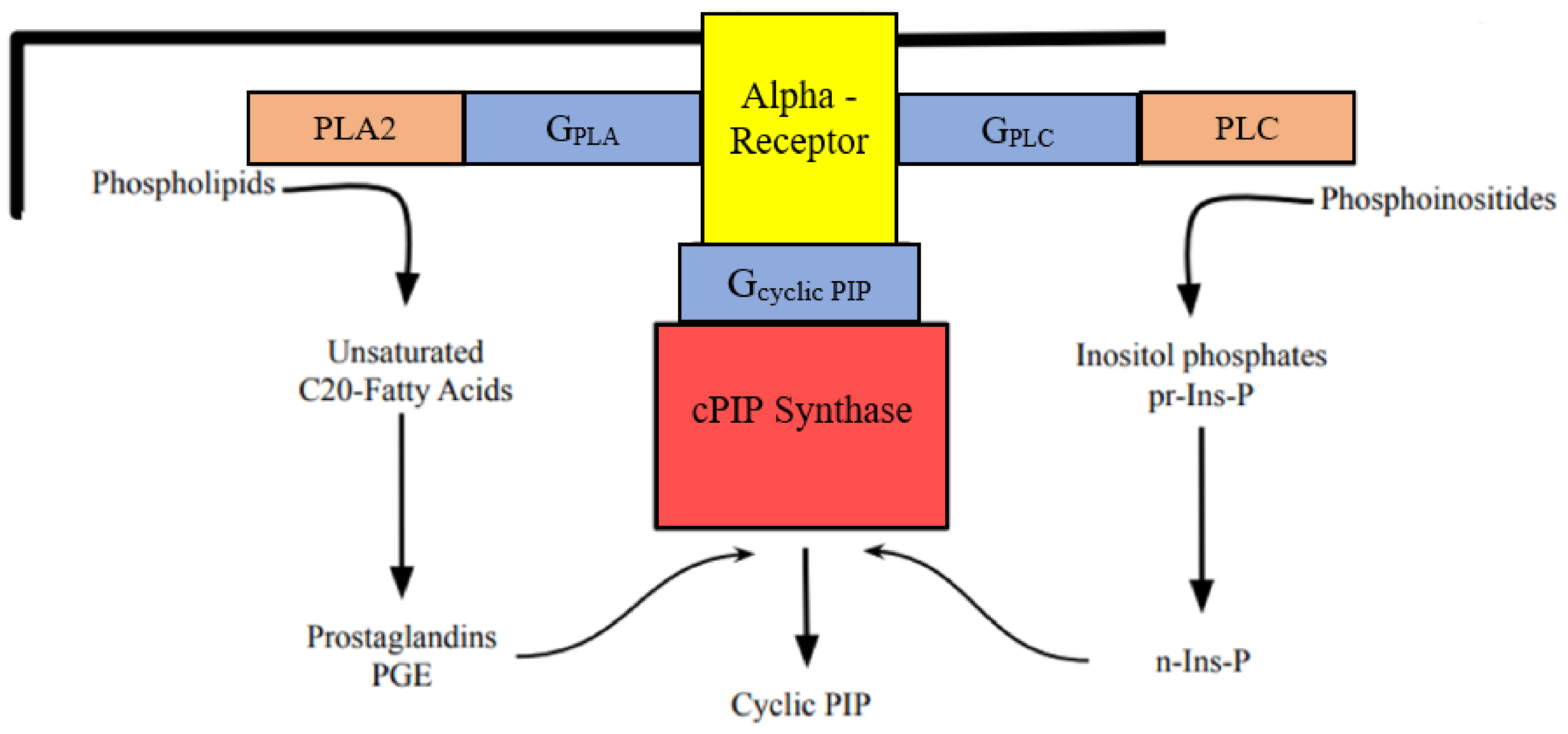
Figure 2.
Time course of glucose release (% of basal; means ± SEM) from male rat liver on perfusion with Krebs-Ringer-buffer containing cyclic PIP (0.1 μΜ) (curve with open triangle; n = 4) and the baseline (curve with closed triangle; n = 5). The arrow indicates the addition of cyclic PIP.
Figure 2.
Time course of glucose release (% of basal; means ± SEM) from male rat liver on perfusion with Krebs-Ringer-buffer containing cyclic PIP (0.1 μΜ) (curve with open triangle; n = 4) and the baseline (curve with closed triangle; n = 5). The arrow indicates the addition of cyclic PIP.
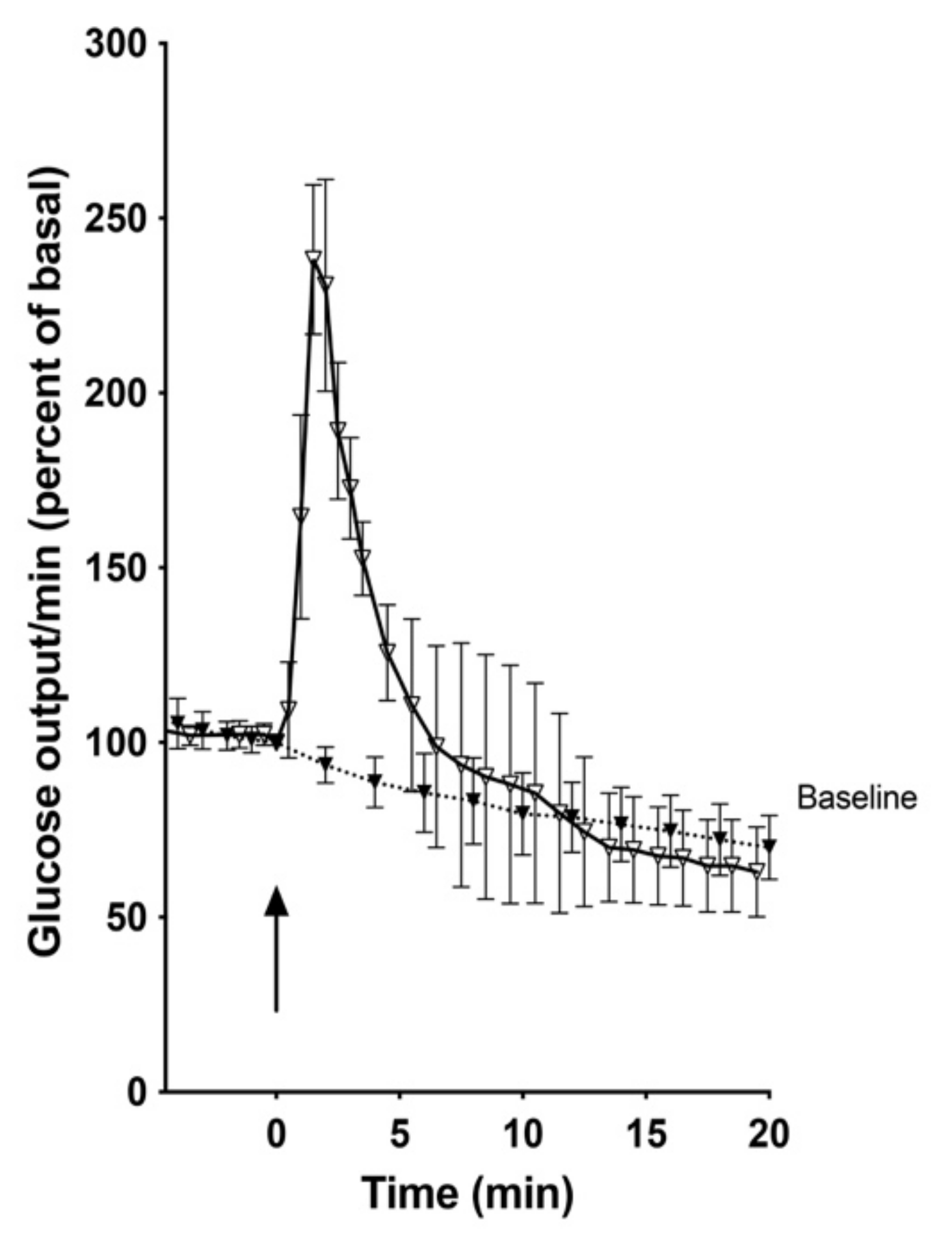
Figure 3.
Phosphorylation of glycogen phosphorylase b by phosphorylase kinase in the presence of protein kinase A (open circle) and in the additional presence of cyclic AMP (10–6M) (open square) or the additional presence of cyclic PIP (0.08μM) (closed triangle) and in the presence of cyclic AMP plus cyclic PIP (closed square). Two independent and two technical replicates were performed. The error range of the enzyme assay is below 7%.
Figure 3.
Phosphorylation of glycogen phosphorylase b by phosphorylase kinase in the presence of protein kinase A (open circle) and in the additional presence of cyclic AMP (10–6M) (open square) or the additional presence of cyclic PIP (0.08μM) (closed triangle) and in the presence of cyclic AMP plus cyclic PIP (closed square). Two independent and two technical replicates were performed. The error range of the enzyme assay is below 7%.
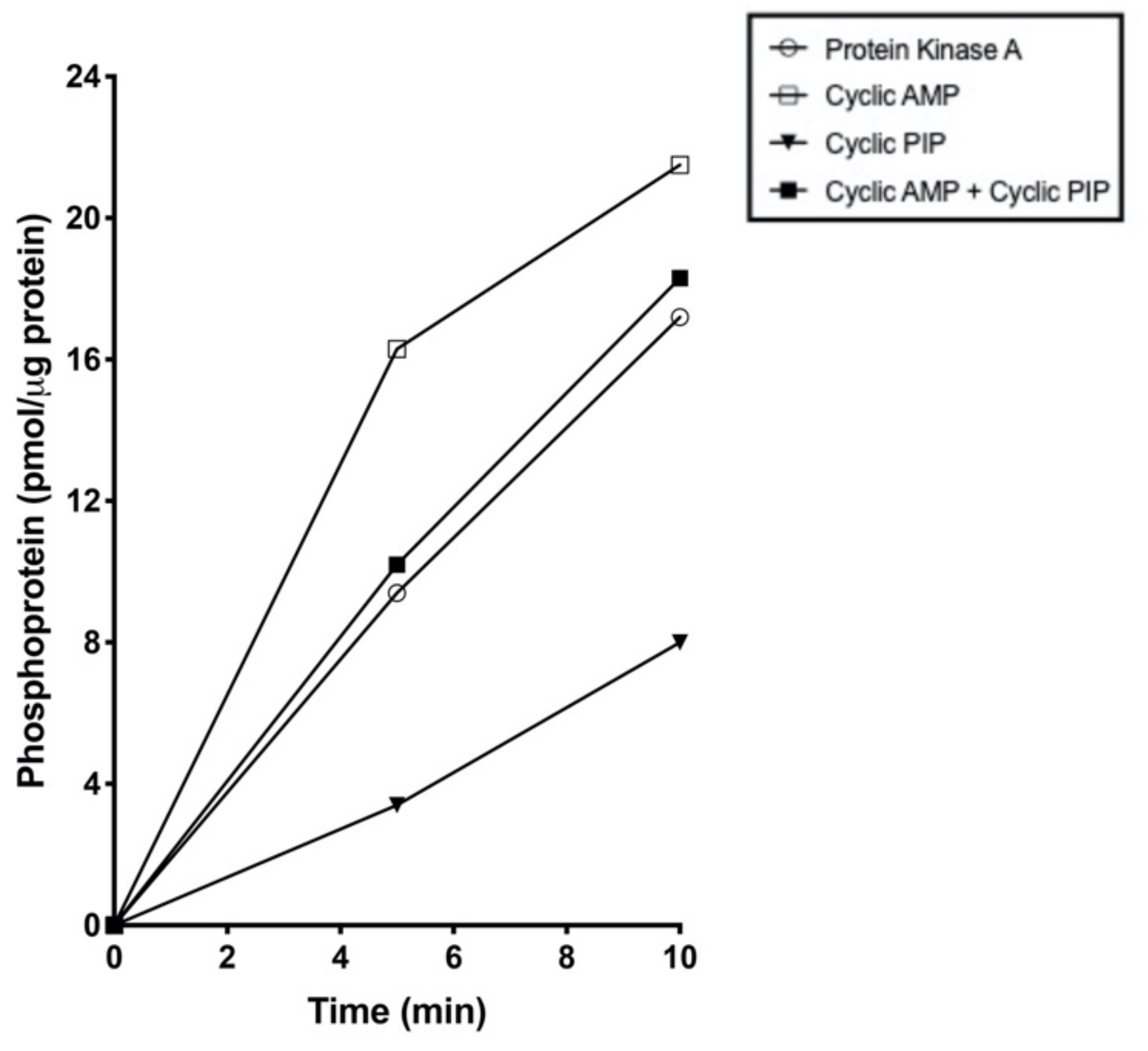
Figure 4.
(A) Time course of glucose release (% of basal; means ± SEM) from male rat liver stimulated with glucagon (10–9 M) (closed circle; n = 5), or with glucagon plus insulin (2 x 10–9 M) (open circle; n = 3), and the baseline (closed triangle; n = 5). (B) Area under the curve (AUC) between 0 and 40 min of the experimental groups after baseline was subtracted. ** p < 0.005 of glucagon vs glucagon plus insulin.
Figure 4.
(A) Time course of glucose release (% of basal; means ± SEM) from male rat liver stimulated with glucagon (10–9 M) (closed circle; n = 5), or with glucagon plus insulin (2 x 10–9 M) (open circle; n = 3), and the baseline (closed triangle; n = 5). (B) Area under the curve (AUC) between 0 and 40 min of the experimental groups after baseline was subtracted. ** p < 0.005 of glucagon vs glucagon plus insulin.
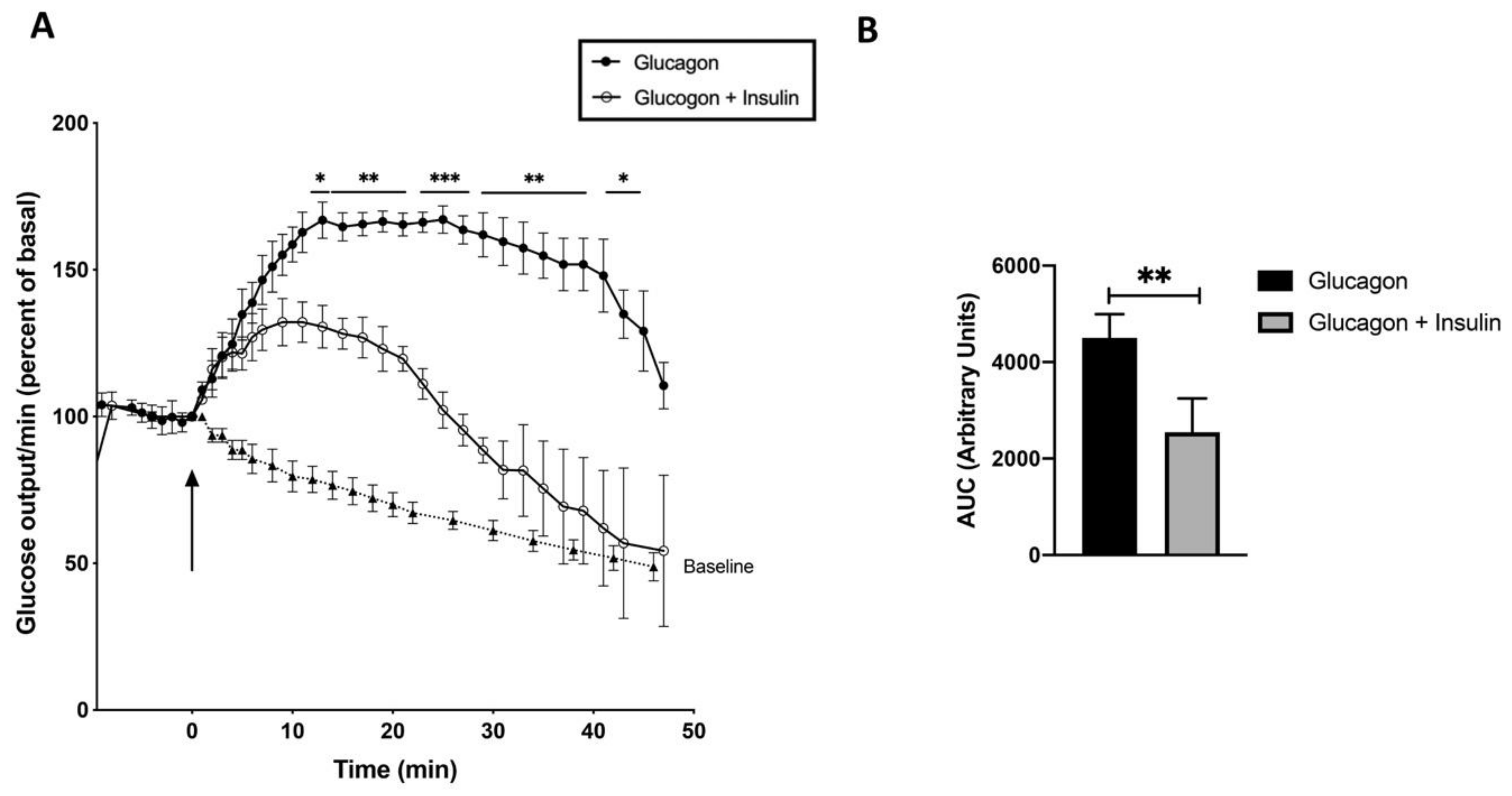
Figure 5.
Time course of glucose release (% of basal; means ± SEM) from male and female rat liver stimulated with adrenaline (10–6 M). (A) Livers of male rats (open square; n = 6), of female rats (closed square; n = 5), and the baseline (closed triangle; n = 4). (B) Area under the curve (AUC) between 0 and 30 min of the experimental groups after baseline was subtracted. **p < 0.005 of male vs female rats.
Figure 5.
Time course of glucose release (% of basal; means ± SEM) from male and female rat liver stimulated with adrenaline (10–6 M). (A) Livers of male rats (open square; n = 6), of female rats (closed square; n = 5), and the baseline (closed triangle; n = 4). (B) Area under the curve (AUC) between 0 and 30 min of the experimental groups after baseline was subtracted. **p < 0.005 of male vs female rats.
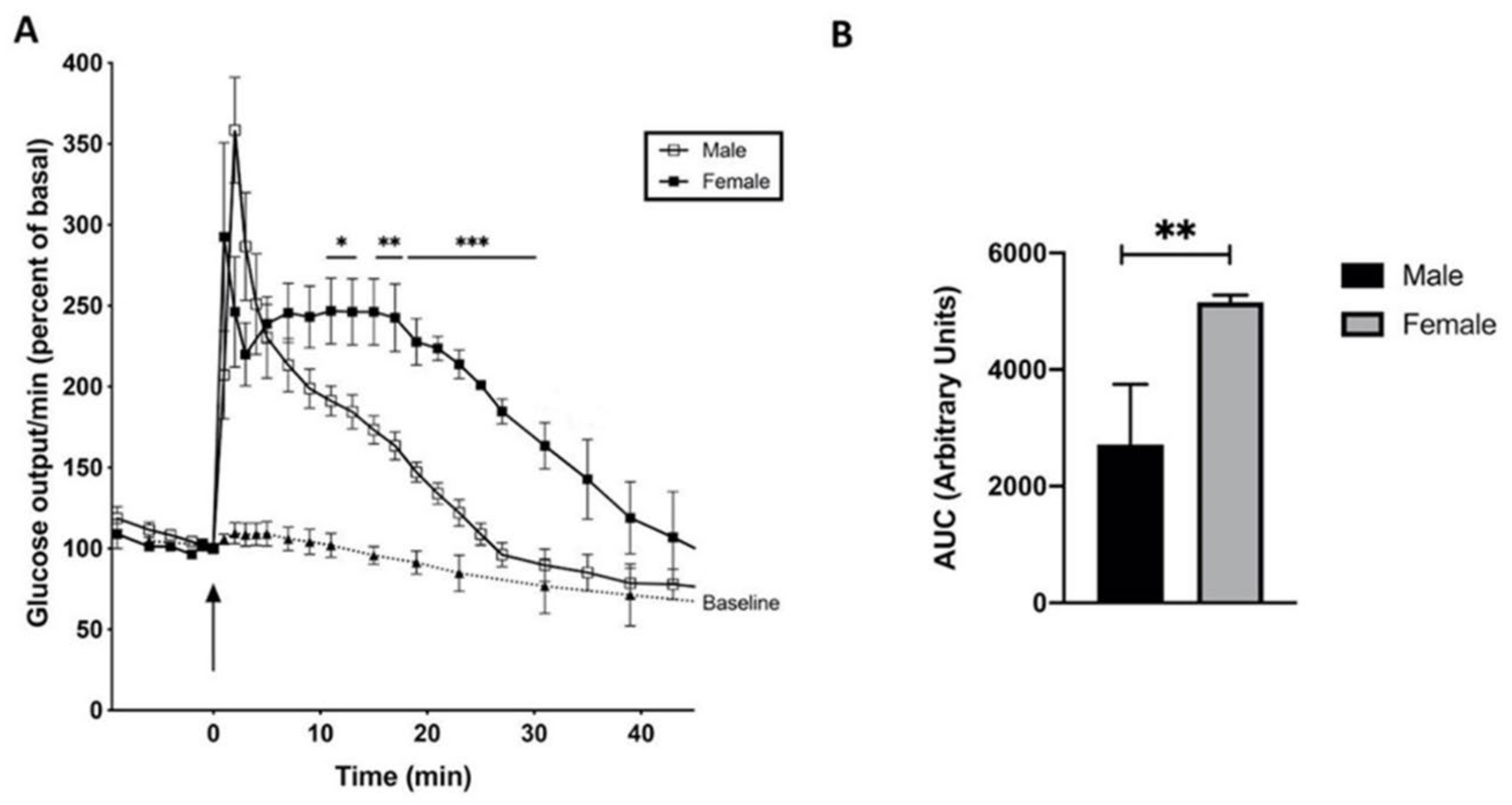
Figure 6.
Time course of glucose release (% of basal; means ± SEM) from male rat livers. (A) The arrow at 0 min indicates the addition of the Ca2+ ionophore A23187 (10–5 M) to the Krebs-Ringer buffer (closed triangle; n = 3), and the arrow at 15 min indicates the successive stimulation with glucagon (closed circle; n = 3). (B) Area under the curve (AUC) of the experimental groups between 15 and 40 min. * p < 0.05 without vs with glucagon stimulation.
Figure 6.
Time course of glucose release (% of basal; means ± SEM) from male rat livers. (A) The arrow at 0 min indicates the addition of the Ca2+ ionophore A23187 (10–5 M) to the Krebs-Ringer buffer (closed triangle; n = 3), and the arrow at 15 min indicates the successive stimulation with glucagon (closed circle; n = 3). (B) Area under the curve (AUC) of the experimental groups between 15 and 40 min. * p < 0.05 without vs with glucagon stimulation.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



