
Submitted:
20 November 2024
Posted:
20 November 2024
You are already at the latest version
Abstract
In this review, we focus on the one-electron oxidation of DNA, which is a multipart event controlled by several competing factors. We will discuss the oxidation free energies of the four nucleobases and the electron detachment from DNA influenced by specific interactions, like hydrogen bonding and stacking interactions with neighboring sites in the double strand. The formation of radical cation (hole), which can migrate through DNA (hole transport), depending on the sequence-specific effects and the allocation of the final oxidative damage, is also addressed. Particular attention is given to the one-electron oxidation of ds-ODN containing G:C pairs, including the complex mechanism of the deprotonation vs. hydration steps of G:C•+ pair, as well as to the modes of formation of the two guanyl radical tautomers after deprotonation. Among the reactive oxygen species (ROS) generated in aerobic organisms by cellular metabolisms, several oxidants react with DNA. The mechanism of stable product formation and their use as biomarkers of guanine oxidation in DNA damage are also addressed.
Keywords:
DNA
; nucleobase
; voltammetry
; oxidation
; radical cation
; hole transport
; reaction mechanism
; oxidative stress
; biomarker
1. Oxidation Free Energy of Nucleobases
Oxidative damage of DNA is a complex phenomenon controlled by several competing factors of both kinetic and thermodynamic nature. The hole-trapping efficiency of a DNA sequence and the location of the final oxidative damage in the double helix depends on the oxidation free energies of the different nucleobases, which are the primary sites for electron detachment from DNA [1]. The gas-phase ionization energies of DNA nucleobases have been well-established for a long time [2,3]. In solution, the oxidation free energies of nucleobases, nucleosides, and nucleotides have been investigated by several techniques: cyclic voltammetry [4,5], photoelectron spectroscopy [6,7], and pulse radiolysis [8,9]. All the methods yield guanine (G) as the most easily oxidizable nucleobase, followed by adenine (A), while cytosine (C) and thymine (T) exhibit significantly higher oxidation free energies. Although all techniques almost respect the above trend, for the ionization levels of DNA constituents in solution, no consensus has been reached. That is mainly because all the employed techniques are unable to provide unambiguous data. Electrochemical measurements are affected by the low solubility of nucleobases, the scarce activity of working electrodes, and narrow electrochemical windows [4,10]; furthermore, nucleobase oxidations are irreversible processes in most solvents used in electrochemistry [5,11], so the electrode response can be affected by collateral unknown reactions. Pulse radiolysis cannot provide a direct measurement of oxidation free energies, the latter are inferred by measuring the rates of electron transfer processes between the sample and a reference system with a known reduction potential. The results will largely depend on the reliability and completeness of the adopted reaction scheme of the redox processes occurring in the solution. Photoelectron spectroscopy allows the measurement of only the vertical ionization potential (VIP) of a given species, oxidation free energies are then obtained by adding vibrational relaxation energies, which are usually estimated by resorting to theoretical computations in solution. The cumulative error has been estimated to amount to ca. 0.2-0.3 eV.
The inherent difficulties in the measure of one-electron oxidation potentials of nucleobases by electrochemical methods have sparked a large scientific debate on the reliability of those experimental values. Several computational studies based on either implicit solvation models or hybrid QM/MM solvation schemes have addressed the problem [6,12,13]. However, the approximate nature of such models does not ensure robust quantitative results. Very recently, by combining state-of-the-art ab initio molecular dynamics simulations with a grand-canonical formulation of solutes in aqueous solutions, the oxidation potentials of some nucleobases and related nucleosides have been calculated by considering explicitly the nucleobases immersed into the bulk of solvent molecules (water) treated at full quantum mechanical level [14]. Noteworthy, this state-of-the-art computational study points out that voltammetry estimates of oxidation free energies inferred by raw peak potentials provide very reliable relative hole-site energies for DNA nucleobases, notwithstanding the irreversible nature of nucleobase oxidation in water [14].
2. In situ Hole Energies in DNA
In DNA, oxidation-free energies can be significantly different from single molecule ones: specific interactions among nucleobases, i.e., hydrogen bonds with their complementary ones in the double helix and with solvent [15,16,17,18,19,20], as well as stacking interactions with neighboring sites [21,22,23,24,25,26,27], significantly affect the oxidation free energies of DNA constituents, making them dependent on the primary and higher order structure. Because of that, each DNA sequence is characterized by its series of oxidation-free energies, often called in situ hole energies [28].
The effects of the formation of H-bonds with the complementary base on the ionization energies of G have been first estimated by theoretical computations [15,16], and later on experimentally observed by Kaway et al. [17], who measured the quenching of triplet transient absorption of N,N’-Dibutylnaphthaldiimide (NDI) by electron transfer to G and G:C complex, viz.,
3NDI* + G → NDI•– + G•+
Measurements were carried out in dichloromethane, where the association constants for the base-pair formation were measured by NMR to be higher than 104 M–1, ensuring that a high percentage of guanine derivatives were engaged in the formation of the G:C complex. Nucleosides were properly derivatized with a tert-butyldimethylsilyl group on the ribose unit to increase their solubility in chlorinated organic solvents. The rate of electron transfer (ET) reaction (1) significantly increases for the G:C complex concerning G one, showing that the one-electron oxidation rate of G is controlled by base pairing with C [17,18]. Using Marcus equation with estimated values of reorganization energy and the preexponential factors, it was estimated that base pairing lowers the oxidation potential of G by ca. 100 mV [18].
The influence of base-pair formation on the oxidation energy of G has also been studied by differential pulse voltammetry using the same silyl derivatives of guanosine (Guo) and deoxycytidine (dCyd) employed by Kawai in chloroform [19]. The results are summarized in Figure 1a. The differential pulse voltammogram of a solution containing equimolar amounts of Guo and dCyd shows two well-resolved peaks, one occurring at the same potential observed for solutions containing only Guo, which has been assigned to the free fraction of Guo in solution, the other occurring at lower potential, 0.34 V, which has been assigned to the formation of the base pair. The large shift of the peak potential observed upon base-pair formation (0.34 eV) is clearly due to the scarce ability of chloroform to solvate the guanosine cation; in a more polar solvent, the shift is expected to be much lower. Similar results have been found for the H-bonded complex between silyl derivatives of adenosine (Ado’) and deoxythymidine (dThd’), see Figure 1b [20].
Voltammetric measurements are not able to provide information on the effect of base pair formation on the hole site energies of pyrimidine nucleobases. A rough estimate for deoxycytidine has been obtained using spectroelectrochemistry [29]. Spectroelectrochemistry is a powerful technique for investigating the fate of intermediate species produced during an electrochemical process, but, in the case irreversible redox processes occur, it must be used with great caution: reliable information can be obtained only on conditions that all the processes occurring at the electrode are properly identified and their spectra well characterized. Spectroelectrochemistry of Guo in CHCl3, integrated by a careful analysis of the products of the oxidation process, yielded interesting results. Although Guo oxidation is an irreversible process, so the guanosine radical cation should be rapidly consumed [19], the electronic absorption spectra recorded under oxidation at controlled potential (in an electrochemical cell equipped with an optically transparent thin-layer electrode (OTTLE) in different solvents) show the three characteristic absorption peaks of Guo radical cation (Guo•+) in water (320, 390, and 500 nm) [8,30,31]. In DMSO, the electronic absorption spectrum is very similar to that observed for an aqueous solution of Guo by pulse radiolysis at acidic and neutral pH, with the three absorption peaks exhibiting the same intensity ratios of the spectrum obtained by pulse radiolysis. In less polar solvents (CHCl3), peak frequencies are slightly blue-shifted, and the relative intensity of the peak at 310 nm is higher and increases as electrooxidation proceeds, which suggests that an oxidation product is also absorbing at that wavelength region. Indeed, upon switching the potential off, only a signal peaked at 316 nm persisted. The product was identified by HPLC–MS and NMR spectroscopy to be the 8-(8-guanosyl)guanosine derivative, which exhibits a strong absorption at 320 nm in water [32]. The fact that the radical cation Guo•+ is the prevalent species at the electrode during oxidation at controlled potential was also confirmed by the FTIR absorption spectrum of Guo recorded in CHCl3. The difference spectrum (reported in Figure 2b, dashed line) was the same obtained by time-dependent spectroscopy and unequivocally assigned to Guo•+ [33].
The electronic spectra of solutions containing Guo:dCyd mixtures were recorded in a OTTLE cell at a controlled potential of +0.57 V versus Fc+/Fc in CH2Cl2. At such potential, only the H-bonded Guo:dCyd can be oxidized. The electronic difference spectrum (evaluated for that recorded without any applied potential) exhibits a positive broadband not observed during the oxidation of solutions containing only Guo. The electronic transition was, therefore, assigned to a charge-transfer transition localizing the hole on the cytidine unit. Indeed, replacing dCyd with 5-mCyd, the band was red-shifted by ca. 1200 cm–1, as expected from the fact that the ionization energy of 5-mCyd is lower than that of cytidine by ca. 0.2 eV [3]. Time-dependent density functional theory (TDDFT) computations further supported the above assignment [29]. Those results provided a rough estimate of the vertical ionization energy of dCyd forming the Watson-Crick complex with G.
Spectroelectrochemical measurements also provided interesting information about the processes occurring during adenosine oxidation at the electrode. The UV-vis and the IR spectra of derivativized adenosine in dichloromethane were recorded during potentiostatic oxidation at an optically transparent thin-layer electrode. In the IR region, oxidized Ado shows a broad Zundel-like absorption [34], extending from 2800 up to 3600 cm−1, induced by self-association, possibly followed by PT from the exocyclic amine group of Ado+ [35]. To the best of our knowledge, no information is currently available for the hole site energy of thymidine H-bonded to its complementary base.
Another important factor that affects hole site energies is intra-strand π–π stacking interactions. Stacking interactions are of outstanding importance in the chemistry of DNA. Apart from providing the main driving force for the establishment of coiled conformations both in single strand and duplex, they play a major role in the photophysics of DNA [36] and finely tune the redox potential of DNA nucleobases in a way that depends on the primary and higher-order structures [28]. Indeed, DNA oxidative damage exhibits base sequence selectivity: guanines with adjacent purine nucleobases are more reactive than those with pyrimidine neighbors, the effect being larger for GG sequences than GA ones [21,22,23]. Even greater selectivity is observed for cleavage at GGG sequences [24,25,26,27].
That observation can be easily rationalized based on the simplest but physically well-sound two-state quantum model: upon ionization of two consecutive Gs, a G1G2 step, two states can be formed G1.+G2 and G1G2.+, which are approximately degenerate. If the two Gs are in a stacked configuration, the energies of the two states are shifted up and down compared to those of unstacked ones by a quantity related to the “strength” of the stacking interaction, see Figure 3 [37].
Computational studies yielded results largely in line with the predictions of the simple two-state model and showed that the ionization potential of 5’-GG-3’ is ca. 0.5 eV lower than monomeric 5’-G [38,39]. However, experimental studies have shown that the selectivity observed for oxidative damage at GG or GGG sites compared to single G ones is quite modest [22,23,24,26], suggesting that either the relative energies of these sites are more similar to each other or that more stable sites are less reactive. Those computational studies neglected the contribution of the solvent reorganization energy. Kurnikov et al. have reported that solvation energies level down oxidation-free energies for the calculated gas-phase ionization potentials of G, GG, and GGG sequences. The solvent stabilization energy is larger for a hole localized on a single G and decreases with increasing delocalization. The net effect is a modest stabilization energy, less than 0.1 eV, for GG versus G [40], more in line with the observed oxidative damage selectivity.
Lewis and coworkers approached the problem of the stabilization energy due to stacking interactions from a different point of view. They have investigated the dynamics of hole transport in several DNA hairpins possessing a stilbenedicarboxamide electron acceptor and G, GG, and GGG donors and determined the energetics of hole transfer from the observed rates [41,42]. Forward and backward rate constants for photo-induced hole transfer were fitted from time-dependent transient absorption spectroscopical measurements, and the results were employed in a kinetic model for the evaluation of equilibrium constants and free energies for hole transport [42]. GG and GGG donors have been found to form slightly deeper hole traps than a single G, the experimental values found for the relative hole-free energies of G, GG, and GGG are 0, -0.052, and -0.077 eV [41,42], in line with those estimated by electrochemical measurements, see infra.
The π−π stacking interaction energy between two nucleobases is expected to be in the range of 0.1−0.3 eV for a neutral pair [42,43,44], such an energy difference can be easily detected by differential pulse voltammetry, which turned out to be a powerful experimental tool for detecting the presence of stacking interactions [45], and for estimating their strengths in oxidized samples. Indeed, differential pulse voltammetry has provided precious information concerning the effect of stacking interactions on the hole site energies of DNA nucleobases [37,46,47]. The first voltammetric measurements were carried out on A-rich oligonucleotides [37]. That choice was dictated by the well-known ability of consecutive As to confer structural rigidity to DNA by forming strong stacking interactions [48,49]. Two sets of oligonucleotides were considered, the first one consisting of 5′-ACCCCA-3′ and 5′-AACCAA-3′, the second one including 5′-TTAATT-3′, 5′-TTAAAT-3′, and 5′-TAAAAT-3′ examers, differing for the outer or inner locations of consecutive As inside the oligonucleotide. Differential pulse voltammograms, recorded in water at room temperature, with 0.05 M phosphate buffer, showed a progressive lowering of the potential of the first anodic peak as the number of A bases increases for both sets of oligonucleotides. These results were interpreted according to the simple two-state model discussed above in terms of hole delocalization over two or more A nucleobases, in line with previous theoretical computations [50], see Figure 3. Noteworthy, the above results refer to single-strand oligonucleotides and point toward a well-organized structure of such oligomers in solution due to strong stacking interactions. The analysis of spin distributions indicates that the observed oxidation potential shifts can be assigned to orbital mixing effects among stacked nucleobases, which lead to the formation of delocalized polarons [51]. Aside from evidence provided by voltammetric measurements, the formation of delocalized charge domains has been later on observed also by time-dependent spectroscopy measurements carried out for oxidized DNA hairpins possessing two or more intervening A-T steps [52].
Similar results have also been obtained for G-rich oligo: a progressive lowering of the oxidation potential was observed in single and double-stranded oligonucleotide examers possessing an increasing number of consecutive guanines; hole site energies can be lowered up to 0.3 eV for sites composed of six consecutive guanines [53]. The effect of hole delocalization in G-rich oligonucleotides is significantly less relevant than in A-rich oligos, in line with the results obtained by transient absorption spectroscopy, indicating a lower extent of hole delocalization on GG steps. Theoretical studies have concluded that the positive charge is almost entirely localized on 5’-G in GG steps, possibly due to strong electrostatic interactions [54,55]. Significant hole delocalization has also been found in an alternating GC doubled-stranded oligodeoxynucleotide, i.e., the palindromic 5’-d(GCGCGC)-3’. The oligomer, upon oxidation by SO4 •– produced by ionizing radiation in aqueous solutions under anoxic conditions, exhibits novel time-dependent spectral shapes and kinetic behavior, which were assigned using theoretical computations to the formation of the species [G:C/C:G]•+, in which the electron-hole is delocalized over the Gs of the two base pairs (see Figure 4) [56]. Although in line with the direct conductance measurements of single DNA oligomers in aqueous solution [57], that result is somewhat surprising because intrastrand interactions are known to be extremely more effective than interstrand ones [58].
Once again, those results suggest that the formation of hole-delocalized domains in DNA is a complex phenomenon where several factors could come into play [40,50,59,60,61,62]. Indeed, in DNA hole solvation energy, which favors charge localization on a single nucleobase, is likely comparable with the quantum delocalization energy [40,60], so that small factors can tip the scales to one side or the other. That occurs particularly in the case of stacked Gs, for which both time-resolved spectroscopic and electrochemical measurements have shown that hole site energy is slightly affected by delocalization, ca. 0.1 eV [41,42,47]. Contrarily, hole delocalization appears to be favored in tracts of stacked As [37,52], highly favoring charge transport along DNA double strands, see infra [61,62,63].
3. Charge Transport
The other peculiarity that further complicates the understanding of DNA oxidative damage is the possibility that the radical cation migrates away from the site of its initial formation. Charge transport in duplex DNA was first observed in the late 1960s by Eley and Spivey, who measured the conductivity of a few samples of DNA and RNA in the dry state [64]. They suggested that efficient charge transport occurs along paths provided by the stacked aromatic base pairs of duplex DNA. This observation went almost unnoticed until time-dependent spectroscopic observations by Barton and co-workers revealed efficient photoinduced electron transfer over a distance greater than 40 Å, between metal intercalators that are tethered to the 5' termini of a 15-base pair DNA duplex [65].
That observation triggered a large body of experimental and theoretical studies focused not only on the biologically relevant topic of oxidative DNA damages but also on the possibility that DNA could serve as a one-dimensional molecular wire and its potential implications in nanoelectronics, where the self-organizing properties of DNA could be exploited [66,67,68,69,70].
Given the considerations on hole site energies discussed above, charge transport in DNA must strongly depend on the specific sequence of nucleosides and DNA conformation. The sequence dependence of charge (mainly hole) transport has been deeply investigated in DNA oligomers, and several excellent reviews are available in the literature [28,71,72,73,74,75]. It was soon realized that electron transfer (ET) in DNA is extremely different from other ET processes occurring in biosystems because of the tiny energy difference between acceptor and donor groups and because of strong intrastrand interactions between stacked nucleobases and, probably smaller but still significant, interstrand interactions [56,63,76,77]. In most of the oligos used for measuring hole transfer rates, the hole is injected into a G site because G is the most easily oxidizable nucleobase, and the acceptor site is usually constituted by GG or GGG steps. As discussed above, the driving force for hole transport in these systems amounts to a few tenths of mV. Furthermore, in studying the distance dependence of hole transfer rates, a bridge of consecutive A:T steps is usually interposed between the donor and acceptor groups. Consecutive A:T steps are the most prone to form delocalized domains, leading to a significant stabilization of the hole energy. Therefore, increasing hole tunneling distances by increasing the number of interposed A:T steps could also cause a decrease in the energy barrier for hole transport; the two effects could balance each other, leading to a very peculiar regime in which hole transfer rates weakly depend on the donor-acceptor distances. A large body of experimental works has clearly shown that ET in DNA is characterized by two regimes, one for shorter donor distance between the hole donor and the hole acceptor nucleobases in which the rate exponentially depends on distance, the other for longer donor-acceptor distances, characterized by a much weaker distance dependence [46,63,76,77,78,79,80,81,82,83,84].
That behavior is well exemplified by Giese’s experiment on ds- G(T)nGGG oligonucleotides, in which a hole is injected onto the single G site via photoexcitation of a suitably modified nucleotide and the yields of oxidation products formed at the initial site (PG) and at the trap site (PGGG) are measured [63]. For shorter sequences, up to n = 3, the product ratio PGGG/PG drops by a factor of ca. 8 for each A:T step. In longer sequences, for n = 4–7, the yield ratio exhibits a much weaker distance dependence, whereas, for n = 7–16, no substantial change in PGGG/PG was detected. Experimental results were interpreted by admitting a switch of HT mechanism from coherent super-exchange in the short-range regime to a thermally induced multistep or multirange hopping for the long-range regime [76,77,78,79,80,81,82,83,84].
In Giese’s experiment, the sequence of consecutive A:T steps acts as a shuttle for hole hopping, allowing for efficient electron tunneling up to at least four A:T steps. Curiously, the same structural motif behaves as an unsurmountable barrier for hole transport in other oligomers, realized and characterized by Schuster’s group, whose structures are reported in Figure 5 [77], see the caption of Figure 5 for more details. The discordant behavior has been recently rationalized by introducing a multistep electron transfer mechanism, in which a manifold of fast coherent elementary ET processes takes place in resonance conditions, which are triggered by environmental motions [77,85]. The mechanism is schematized in Figure 6; it essentially consists of four steps: an activation step, which brings a donor and an acceptor group into vibronic degeneracy, step 1, triggering elementary electron transfer between resonant donor and acceptor groups, step 2. The elementary ET step is then followed by a relaxation step mainly consisting of solvent relaxation triggered by the nonequilibrium charge distribution created by ET, step 3. The final step is the irreversible formation of oxidative damage products.
The ds-G(T)nGGG series of oligomers studied by Giese and coworkers represents a very peculiar case. Simple quantum dynamics simulations showed these are two-state systems since the time-dependent probability amplitude for hole motion simply oscillates between the donor initial (ground) vibronic state and a final state of the G triplets, which is quasi-degenerate with the initial state. Notwithstanding the simplicity of the adopted model, but noteworthy, not of the computational task which required the inclusion of a huge number of basis states [86], the results are in very good agreement with the observed PGGG/PG, testifying that the essential features of charge transport are caught by the model, on conditions that a very large region of the Hilbert space is properly included into computations [87].
By contrast, the set of oligomers considered by Schuster and coworkers contains several sites very close in energy to each other. In this latter case, interference among probability amplitudes could arise, resulting in a much more complex charge transport behavior. Modeling charge transport as an incoherent sequential hopping, in which transient pairwise resonances promote comparatively faster hole motion, satisfying results have been obtained for the oligos 1 and 2 of Figure 5, the computed yields of oxidative damage, reported in blue in Figure 5, are in reasonable agreement with the observed one (gray histogram). However, when the incoherent charge transport model is applied to oligo 3 and 4, significant discordances appear, especially for 4, for which a significant population is predicted on the 8oxo-G trap site of oligomer 4 in Figure 5, much higher than the observed one. That result would be in line with those obtained for Giese’s oligos, which have shown that a (T:A)4 step works as a shuttle for hole transport, but in 4, the experimental result is different, and we must accept the experimental observation. Thus, for those oligos, a fully coherent model in which vibronic states of several sites are involved at once has been adopted. In such a model, all the possible low-lying hole sites are brought together in resonant conditions so that the mobile hole can exploit different paths. Such paths interfere with each other, leading to a complex time dependence of probability amplitudes for each site. In this scenario, the kinetic constant to be used in the mechanism of Figure 6 will become time-dependent, and calculating the final yields a too-hard computational task. In ref. [76] authors have chosen to feed the kinetic model of Figure 6 with a time-independent rate constant obtained by averaging time-dependent rates over a half period of the coherent transition times, namely at complete depopulation of the initial state. That empirical choice has led to the computed oxidative damage yields shown in Figure 5, green panel. Now, the computed oxidative damage at the 8oxo-G site is about 1%, to be compared with the 25% yielded by the incoherent model.
The emerging mechanistic picture shares many common points with other mechanisms previously proposed in the literature, such as the flickering resonance charge transport mechanism or the unfurling mechanism [83,84], but adds another important feature: coherent effects can manifest themselves even in the presence of fast dephasing mechanisms.
The appearance of coherent effects on macroscopic yield ratios is somewhat surprising because irreversible damage at nucleobases occurs on a much longer time scale than coherent hole motion. Coherent oscillations are indeed rapidly destroyed by fast solvent deactivation, and that, in some cases, can lead to the formation of an equilibrium hole population. That is the cases of oligomers 1, 2, and, to a lesser extent, 3. For oligomer 4, the situation is very different; coherent effects are crucial for reproducing the observed formation of two different spatial regions: a kinetically allowed region, within which a quasi-equilibrium population is reached on comparatively longer time scales, and an almost “kinetically forbidden” region, in which hole populations are always too low for establishing an equilibrium regime. The incoherent model works very well for Giese’s oligomers as well as for 1 and 2, so it would be hard to attribute the poor agreement with experimental data obtained for 4 to its inherent deficiencies. 4 has peculiarities, i.e., the possibility that a given event occurs in different indistinguishable ways, which are not present at all in Giese’s oligomers or whose effects are smeared out in 1 and 2. The coherent model takes into account indistinguishable pathways, yielding more satisfying results than the incoherent one, suggesting that charge transport in DNA is characterized by different facets that overall cannot be accounted for by a simple sequential hopping mechanism.
4. Deprotonation vs. Hydrolysis of Guanyl Radical Cation (G•+): From Nucleoside to DNA
We showed in the previous section that one-electron oxidation of DNA brings to the guanyl radical cation moiety (G•+). The fate of this species follows two main pathways: deprotonation and hydration. The deprotonation process has been studied in detail by various spectroscopic techniques and, in particular, by time-resolved absorption experiments. The position of the proton released is depicted in Figure 7, in the form G(N1-H)• that lost H+ from the N1 position and the form G(N2-H)• that lost H+ from the N2 position. G(N1-H)• and G(N2-H)• are two tautomers, and for evidencing the two tautomeric structures, we draw resonance forms where the unpaired electron is placed in the C5 position [88].
First of all, the well-understood behavior of guanyl radicals in the building block of DNA, i.e., 2’-deoxyguanosine (dGuo) must be evidenced The G in dGuo can be oxidized to G•+ by a variety of oxidants like SO4•–, Br2•–, Cl2•–, CO3•– and many others, including various metal complexes [88,89,90]. The subsequent deprotonation affords exclusively the G(N1-H)• with a rate constant of 1.5 × 107 s–1 and an activation energy of 15.1 ± 1.5 kJ mol–1 at pH 7 [91]. The reaction of HO• with dGuo occurs mainly by H-atom abstraction from the exocyclic NH2 group affording G(N2-H)•. The initially formed G(N2-H)• radical undergoes a water-assisted tautomerization with a rate constant of 2.3 × 104 s–1 to give the most stable tautomer G(N1-H)• [92,93]. The G(N2-H)• has a characteristic band >600 nm that is missing in its tautomer G(N1-H)• [88,92,93]. When N1-H moiety is replaced by N1-Me, the deprotonation occurs from the exocyclic NH2 group [8]. Theoretical calculations suggested that deprotonation from N2-H moiety is competitive with deprotonation from N1-H and indicated that G(N1-H)• is favored over G(N2-H)• in environments with high dielectric constants, such as water [94]. Furthermore, the two conformational isomers of G(N2-H)• in which the remaining N2–H is either syn or anti to the guanine N3 atom were calculated, finding that the syn-conformer is lower in energy than the anti-conformer [94].
The fate of G•+ in ds-ODNs or DNA is more complicated [88]. In the early works, it was suggested that in DNA, the H+ is not directly lost from G•+ to the aqueous phase but remains within the hydrogen-bonded G:C•+ pair [89]. ESR studies showed that the H+ is transferred to the adjacent cytosine at 77 K, whereas a prototropic equilibrium should be established at ambient temperature (see in Figure 8 the structures highlighted in the colored box) [95]. However, the dynamics of G:C•+ pair deprotonation depend strongly on ODN secondary structures. The prototropic equilibria in one electron oxidized G:C and the subsequent deprotonation step are quite important since they control the hole transfer in DNA.
The one-electron oxidation of various ds-ODNs containing G:C pairs by SO4•– has been reported by time-resolved spectroscopies. In the pulse radiolysis study, eleven ds-ODNs with different sequences (11mer to 13mer ds-ODNs) were reported [96]. The monophasic decay of the transient species associated with the release of the proton into solution was observed, the rate constant varying in the range 0.3–2 x 107 s–1, depending on the ds-ODN sequence. For example, the transient from the ds-ODN of the 13mer A5G3A5 decays with k = 4.5 x 106 s–1 in water. The proposed mechanism consists of the formation of G(N1-H)•:C (see Figure 8). In the laser flash photolysis study, the one-electron oxidation of 30mer ODN: 5’-CGT ACT CTT TGG TGG GTC GGT TCT TTC TAT-3’ by SO4•– was studied in ss-ODNs and ds-ODN [97]. In both cases, the observed transient species and their formation were very similar and assigned to G(N1-H)•:C, whereas the kinetics of G(N1-H)•:C decay were quite different. In ds-ODN, the decay is biphasic, with one component decaying with a lifetime of ∼2.2 ms and the other one with a lifetime of ∼0.18 s, whereas in ss-ODN the decay is monophasic with a ∼0.28 s lifetime. The ms decay component in ds-ODN is correlated with the higher yield of 8-oxo-G with respect to ss-ODN (see also Section 5).
The deprotonation process of G•+ produced by low-energy/low-intensity photoionization of ODNs was also studied by time-resolved absorption experiments [98,99,100,101]. The deprotonation in duplex DNA leads to G(N1-H)• and is completed within 2 μs, whereas in guanine quadruplexes the G(N2-H)• is observed, and it spans from at least 30 ns to over 50 μs connected with the anisotropic structure of DNA and the mobility of its hydration shell.
The alternating guanine cytosine duplexes have attracted considerable interest, obviously due to the multiple G:C pairs that facilitate specific studies. It is reported that the transient species of one-electron oxidation of the 11-mer ds-ODN 5′-d(CGCGCGCGCGC)-3′ release of the proton into solution with a rate constant of 2 × 107 s−1, but no data are shown for this ds-ODN [96]. The ds-ODN d(G*CG*CG*CG*C), where G* is selectively deuterated at C8 on guanine moieties, was used for ESR experiments; it is established that at 77 K one-electron oxidized guanine in ds-ODN exists as the deprotonated neutral radical G(-H)• as a result of facile proton transfer to the hydrogen-bonded cytosine [95]. The transient species generated with the direct absorption of low-energy UV irradiation of ds-ODN palindromic 5′-d(GCGCGCGCGC)-3′ and register at 100 μs was assigned at G(N1-H)• [98]. The one-oxidation of the 8-mer ds-ODN d(TGCGCGCA) has been studied by ESR and UV-visible spectral analysis; at pH ≥7, the initial site of deprotonation is found to be at N1 forming G(N1–H)•:C at 155 K and upon annealing to 175 K, the site of deprotonation to the solvent shifts to an equilibrium mixture of G(N1–H)•:C and G(N2–H)•:C [102].
As referred above, evidence has suggested that HO• and one-electron oxidants may partly induce common degradation pathways [103]. We recently reported the reaction of SO4•– and HO• radicals with an alternating GC ds-ODN in aqueous solutions for comparison [104]. In particular, the transient absorption spectra and associated kinetic data in the range of ns to ms were obtained by pulse radiolysis using the palindromic 5′-d(GCGCGC)-3′. The addition of HO• to the G:C pair moiety affording the adduct 8-HO-G•:C (k = 2.3 × 109 M−1 s−1 calculated for a G:C pair), whereas the one-electron oxidation by SO4•– (k = 13.6 × 109 M−1 s−1 calculated for a G:C pair) is different from the previously reported spectra of one-electron oxidation of a variety of ds-ODN after deprotonation, i.e., G(N1-H)•:C (Figure 9). The transient spectrum showed a dominant absorption band with λmax = 330 nm, which decayed by a first-order rate constant of 1.5 × 105 s−1 by hydration affording the 8-HO-G•:C adduct. In summary, the neutral radical 8-HO-G•:C is a common transient of both oxidizing species (Figure 9). Computational studies employing density functional theory (DFT) for structural and time-dependent DFT for spectroscopic features were performed on 5′-d(GCGC)-3′, predicting that the electron hole is delocalized on the two stacked base pairs (Figure 4) [56]. This type of electron-hole stabilization in CG-rich sequences of DNA rather prefers the reaction with water instead of deprotonation.
A pulse radiolysis study reported the reaction of four 12mer ODNs with four or two Gs subjected, either as ss-ODNs or ds-ODNs, to the reaction with HO• radicals. The characteristic band >600 nm of guanyl radical G(N2-H)• was present in ss-ODNs and disappeared in μs time scale, but was absent in ds-ODNs [104].
Numerous articles also address the properties of G•+:C pair fragments of DNA by theoretical methods, including solvent effects [105,106,107]. The calculations provide reference absorption spectra for guanine radicals in duplexes and predict changes in transient absorption spectra for hole localization, hydration of the radical cation, and deprotonation steps [98,108]. The separation of charge from spin [G(N1-H)•:CH+] and the deprotonation from the exocyclic NH2 group of cytosine [G(N1-H)•:C] were addressed. Moreover, it was predicted that at ambient temperature both structures (G(N1–H)•:C and G(N2–H)•:C) should be present in equilibrium in near equal amounts as shown by ESR results [102,108]. The reactivity of HO• with the G:C pair was also addressed theoretically by mapping the energy profiles of all possible addition and hydrogen abstraction reactions [56,109,110]. The HO• addition to C8 of G is predicted to be the most favored process. The reactivity of HO• with the triple forming oligonucleotides C(H+)G:C favored an ambident reactivity: the C8 addition leading to 8-HO-G• and the H-abstraction from the NH2 group leading to G(N2–H)• [111].
5. The Mechanism of the End Products Formation Used as Biomarkers of Oxidative Stress
Guanine can be oxidized to G•+ by a variety of oxidants, including various metal complexes. For example, carbonate radical anion (CO3•−) is an important reactive species under physiological conditions. The pair CO2/HCO3– is an active buffer maintaining physiological pH, and HCO3– exists in mM levels in vivo. Figure 10 shows the reaction of CO2 with peroxynitrite (ONOO–) generating CO3•− through the unstable nitrosoperoxycarbonate [112,113]; the Fenton reaction in the presence of HCO3– also affords CO3•– rather than HO• radical [114,115]. The reduction potential of CO3•−/CO3 is 1.59 V. Therefore, CO3•− is a milder single-electron oxidant that abstracts electrons from guanine moieties [116,117].
The main guanine lesions observed in vitro and in vivo from oxidatively generated DNA damage are outlined in Figure 11 [90]. They are collected in two groups: (i) upper part, 7,8-dihydro-8-oxo-2′-deoxyguanosine (8-oxo-G), formamidopyrimidine-2′-deoxyguanosine (Fapy-G) characterized by the presence of an open imidazole ring, mainly detected as the free base modification due to stability reasons; spiroiminodihydantoin (Sp) and 5-guanidinohydantoin-2’-deoxyribose (Gh) which are further oxidation products of 8-oxo-dG, and (ii) lower part, in the presence of molecular oxygen, the lesions 5-carboxamido-5-formamido-2- iminohydantoin-2’-deoxyribonucleoside (2Ih) and 2-amino-5-[2-deoxyribose]-4H-imidazol-4-one (Iz) that rapidly convers to 2,2-diamino-4-[(2- deoxyribose)amino]-5(2H)-oxazolone (Z) by hydrolysis. The same end-products are also observed from the oxidation of the building block 2’-deoxyguanosine. However, the different observed distribution of products depends on the substrate, oxidant, and experimental conditions.
To discuss the main mechanistic feature of the guanine oxidation, Figure 12 shows the reaction 2’-deoxyguanosine (G) by CO3•– and HO• radicals [88,118,119]. These oxidants give (N1-H)• and 8-HO-G• radicals as common intermediates but generated by different routes. 8-HO-G• is the common precursor of 8-oxo-G and Fapy-G (Figure 12, upper-left side). The Fapy-G formation requires ring-opening followed by one-electron reduction or vice versa (i.e., one-electron reduction followed by ring-opening). The formation of Fapy-G is dependent on the oxygen concentration and the redox environment [120,121,122,123]. The reduction potential of 8-oxo-G is 0.55 V lower than that of G [124], and therefore, it is easily oxidized in the presence of one-electron oxidants, affording the by-products Sp and Gh [125,126].
The 8-oxo-G lesions are formed at higher levels in ds-ODNs compared to the other lesions and ss-ODNs. For example, in the one-electron oxidation of 30mer ODNs by SO4•– study described above, the quantification of the 8-oxo-G lesion as the final product was also measured in both ss-ODNs and ds-ODN [97]. The yield of 8-oxo-G in ds-ODN is ~7 times higher than in ss-ODNs, indicating that the secondary structure of ds-ODN plays an important role. In ds-ODN, the equilibrium [G•+:C ⇆ G(N1-H)•:CH+] facilitates the hydration with the formation of [8-HO-G•:C] that further oxidizes to give the 8-oxoG:C (see Figure 8).
From oxidation of G under aerobic conditions or in the presence of CO3•–, two other products are formed via C5 paths (Figure 6, lower-right side), viz., 2Ih and Iz; the latter is further hydrolyzed to Z [127,128]. The oxidation mechanism and kinetics of G by carbonate radical anion (CO3•−) have also been investigated theoretically [129]. Other oxyl-type radicals like O2•–, and NO2• react with G(N1-H)• of ss-ODNs or ds-ODNs by combining either C8 or C5 paths (left and right side of Figure 8, respectively) with the formation of expected end-products [130,131,132]. Computational investigation using density functional theory into the oxidation of guanine to form Iz through C5 or C8 paths has been reported [133]A few studies at a macromolecular level in vitro report the simultaneous formation of various lesions using LC-MS/MS and isotopomeric internal standards. For example, a comparative analysis of four oxidized G lesions (8-oxo-G, Sp, Gh, and Z) from the reaction of calf thymus DNA with ONOO–, singlet oxygen derived from photoactivated rose bengal, and HO• radical from γ-radiation was examined [134]. In all cases, the most abundant product was 8-oxo-G, and its accumulation was dependent on the nature of the oxidizing agent with the subsequent conversion to Sp and Gh. Another example is a comparative analysis of three oxidized G lesions (8-oxo-G, Z, and 8-NO2-G) from the reaction of 18-mer ds-ODNs with HO• from Fenton reaction or NO2•/CO3•– from the decomposition of ONOOCO2– (see Figure 4) [135]. Their findings suggest that the patterns of these lesions’ formation in the genome are distinct and are influenced by oxidant identity and the secondary structure of DNA. The oxidatively derived products 2Ih, Sp, and Gh in cellular studies, as well as their recognition by the DNA glycosylases and removal by the base excision repair (BER) pathway, are the focus of a review [136]. Fapy-G, due to its instability, is mainly detected as the free base modification (i.e., Fapy-Gua), and both GC-MS/MS and LC-MS/MS techniques with isotope dilution approach have been used for the measurements in vitro and in vivo [120,121,122]. It is also worth mentioning that increasing concentrations of oxygen lead to elevated levels of 8-oxo-G in the reaction of HO• with the 21-mer ODN 5’-GGGTTAGGGTTAGG G TTAGGG-3’ in ds-ODN [137,138]. Three pathways contribute to the formation of 8-oxo-G in the presence of O2: (i) one-electron oxidation (probably by a variety of reactions not very well understood) followed by hydration reaction (~45%), (ii) an intramolecular addition of a transiently generated pyrimidine peroxyl radical onto the C8 of a vicinal guanine base (~50%), and (iii) direct addition of HO• to the C8 position of G as the minor path (~5%) [139].
Author Contributions
writing—original draft preparation, C.C and A.P.; writing—review and editing, C.C and A.P.; authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The financial support of the University of Salerno is gratefully acknowledged.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Paleček, E.; Bartošík, M. Electrochemistry of Nucleic Acids. Chem. Rev. 2012, 112, 3427–3481. [Google Scholar] [CrossRef] [PubMed]
- Hush, N. S.; Cheung, A. S. Ionization Potentials and Donor Properties of Nucleic Acid Bases and Related Compounds. Chem. Phys. Lett. 1975, 34, 11–13. [Google Scholar] [CrossRef]
- Orlov, V.M.; Smirnov, A.N.; Varshavsky, Y.M. Ionization Potentials and Electron-Donor Ability of Nucleic Acid Bases and Their Analogues. Tetrahedron Lett. 1976, 48, 4377–4378. [Google Scholar] [CrossRef]
- Seidel, C.A.M.; Schulz, A.; Sauer, M.H.M. Nucleobase-Specific Quenching of Fluorescent Dyes. I. Nucleobase One-Electron Redox Potentials and Their Correlation with Static and Dynamic Quenching Efficiencies. J. Phys. Chem. 1996, 100, 5541–5553. [Google Scholar] [CrossRef]
- Oliveira-Brett, A.M.; Piedade, J.A.P.; Silva, L.A.; Diculescu, V.C. Voltammetric Determination of All DNA Nucleotides. Anal. Biochem. 2004, 332, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, C. A. ; Pluhařov. , E.; Seidel, R.; Schroeder, W. P.; Faubel, M.; Slav.ček, P.; Winter, B.; Jungwirth, P.; Bradforth, S. E. Oxidation Half-Reaction of Aqueous Nucleosides and Nucleotides via Photoelectron Spectroscopy Augmented by ab Initio Calculations. J. Am. Chem. Soc. 2015, 137, 201–209. [Google Scholar]
- Slavìček, P.; Winter, B.; Faubel, M.; Bradforth, S. E.; Jungwirth, P. Ionization Energies of Aqueous Nucleic Acids: Photoelectron Spectroscopy of Pyrimidine Nucleosides and ab Initio Calculations. J. Am. Chem. Soc. 2009, 131, 6460–6467. [Google Scholar] [CrossRef]
- Candeias, L. P.; Steenken, S. Structure and acid-base properties of one-electron-oxidized deoxyguanosine, guanosine, and 1-methylguanosine. J. Am. Chem. Soc. 1989, 111, 1094–1099. [Google Scholar] [CrossRef]
- Steenken, S.; Jovanovic, S.V. How Easily Oxidizable Is DNA? One-Electron Reduction Potentials of Adenosine and Guanosine Radicals in Aqueous Solution. J. Am. Chem. Soc. 1997, 119, 617–618. [Google Scholar] [CrossRef]
- Faraggi, M.; Broitman, F.; Trent, J. B.; Klapper, M. H. One-Electron Oxidation Reactions of Some Purine and Pyrimidine Bases in Aqueous Solutions. Electrochemical and Pulse Radiolysis Studies. J. Phys. Chem. 1996, 100, 14751–14761. [Google Scholar] [CrossRef]
- Oliveira-Brett, A. M.; Matysik, J. M. Voltammetric and Sonovoltammetric Studies on the Oxidation of Thymine and Cytosine at a Glassy Carbon Electrode. J. Electroanal. Chem. 1997, 429, 95–99. [Google Scholar] [CrossRef]
- D’Annibale, V.; Nardi, A. N.; Amadei, A.; D’Abramo, M. Theoretical Characterization of the Reduction Potentials of Nucleic Acids in Solution. J. Chem. Theory Comput. 2021, 17, 1301–1307. [Google Scholar] [CrossRef] [PubMed]
- Tòth, Z.; Kubečka, J.; Muchov, E.; Slavìček, P. Ionization Energies in Solution with the QM:QM Approach. Phys. Chem. Chem. Phys. 2020, 22, 10550–10560. [Google Scholar] [CrossRef] [PubMed]
- Ambrosio, F.; Landi, A; Peluso, A.; Capobianco, A. Quantum Chemical Insights into DNA Nucleobase Oxidation: Bridging Theory and Experiment J. Chem. Theory Comp. 2024, 20, 9708–9719.
- Colson, A.O.; Besler, B.; Sevilla, M.D. Ab Initio Molecular Orbital Calculation of DNA Base Pair Radical Ions: Effects of Base Pairing on Proton Transfer Energies, Electron Affinities and Ionization Potentials. J. Phys. Chem. 1992, 96, 9787–9794. [Google Scholar] [CrossRef]
- Hutter, M.; Clark, T. On the Enhanced Stability of the Guanine−Cytosine Base-Pair Radical Cation J. Am. Chem. Soc. 1996, 118, 7574–7577. [Google Scholar] [CrossRef]
- Kawai, K.; Wata, Y.; Ichinose, N.; Majima, T. Selective Enhancement of the One-Electron Oxidation of Guanine by Base Pairing with Cytosine. Angew. Chem. Int. Ed. 2000, 39, 4327–4329. [Google Scholar] [CrossRef]
- Kawai, K.; Wata, Y.; Hara, M.; Toyo, S.; Majima, T. Regulation of One-Electron Oxidation Rate of Guanine by Base Pairing with Cytosine Derivatives. J. Am. Chem. Soc. 2002, 124, 3586–3590. [Google Scholar] [CrossRef]
- Caruso, T.; Carotenuto, M.; Vasca, E.; Peluso, A. Direct Experimental Observation of the Effect of the Base Pairing on the Oxidation Potential of Guanine. J. Am. Chem. Soc. 2005, 127, 15040–15041. [Google Scholar] [CrossRef]
- Caruso, T.; Capobianco, A.; Peluso, A. The Oxidation Potential of Adenosine and Adenosine-Thymidine Base-Pair in Chloroform Solution. J. Am. Chem. Soc. 2007, 129, 15347–15353. [Google Scholar] [CrossRef]
- Kovalsky, O. I.; Panyutin, I. G.; Budowsky, E. I. Sequence-Specificity of Alkali-Sensitive Lesions Induced in DNA by High-Intensity Ultraviolet Laser Radiation. Photochem. Photobiol. 1990, 52, 509–517. [Google Scholar] [CrossRef]
- Saito, I.; Takayama, M.; Sugiyama, H.; Nakatani, K. Photoinduced DNA Cleavage via Electron Transfer: Demonstration That Guanine Residues Located 5′ Guanine Are the Most Electron-Donating Sites. J. Am. Chem.Soc. 1995, 117, 6406–6407. [Google Scholar] [CrossRef]
- Muller, J. G.; Hickerson, R. P.; Perez, R. J.; Burrows, C. J. Damage from Sulfite Autoxidation Catalyzed by a Nickel(II) Peptide J. Am. Chem. Soc. 1997, 119, 1501–1506. [Google Scholar] [CrossRef]
- Nakatani, K.; Fujisawa, K.; Dohno, C.; Nakamura, T.; Saito, I. p-Cyano Substituted 5-Benzoyldeoxyuridine as a Novel Electron-Accepting Nucleobase for One-Electron Oxidation of DNA. Tetrahedron Lett. 1998, 39, 5995–5998. [Google Scholar] [CrossRef]
- Yoshioka, Y.; Kitagawa, Y.; Takano, Y.; Yamaguchi, K.; Nakamura, T.; Saito, I. Experimental and Theoretical Studies on the Selectivity of GGG Triplets toward One-Electron Oxidation in B-Form DNA. J. Am. Chem. Soc. 1999, 121, 8712–8719. [Google Scholar] [CrossRef]
- Hickerson, R. P.; Prat, F.; Muller, J. G.; Foote, C. S.; Burrows, C. J. Sequence and Stacking Dependence of 8-Oxoguanine Oxidation: Comparison of One-Electron vs Singlet Oxygen Mechanisms J. Am. Chem. Soc. 1999, 121, 9423–9428. [Google Scholar] [CrossRef]
- Nakatani, K.; Dohno, C.; Saito, I. Modulation of DNA-Mediated Hole-Transport Efficiency by Changing Superexchange Electronic Interaction J. Am. Chem. Soc. 2000, 122, 5893–5894. [Google Scholar] [CrossRef]
- Kanvah, S.; Joseph, J.; Schuster, G. B.; Barnett, R. N.; Cleveland, C. L.; Landman, U. Oxidation of DNA: Damage to Nucleobases. Acc. Chem. Res. 2010, 43, 280–287. [Google Scholar] [CrossRef]
- Capobianco, A.; Carotenuto, M.; Caruso, T.; Peluso, A. The Charge-Transfer Band of an Oxidized Watson-Crick Guanosine-Cytidine Complex. Angew. Chem. Int. Ed. 2009, 48, 9526–9528. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; Caminal, C.; Guerra, M.; Mulazzani, Q. G. Tautomers of one-electron oxidized guanosine. Angew. Chem. Int. Ed. 2005, 44, 6030–6032. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; Caminal, C.; Altieri, A.; Vougioukalakis, G. C.; Mulazzani, Q. G.; Gimisis, T.; Guerra, M. Tautomerism in the guanyl radicals. J. Am. Chem. Soc. 2006, 128, 13796–13805. [Google Scholar] [CrossRef]
- Bose, S. N.; Davies, R. J. H.; Anderson, D.W.; van Niekerk, J. C.; Nassimbeni, L. R.; MacFarlane, R. D. Photochemical (8→8) coupling of purine nucleosides to guanosine. Nature 1978, 271, 783–784. [Google Scholar] [CrossRef] [PubMed]
- Kuimova, M. K.; Cowan, A. J.; Matousek, P.; Parker, A.W.; Sun, X. Z.; Towrie, M.; George, M.W. Monitoring the direct and indirect damage of DNA bases and polynucleotides by using time-resolved infrared spectroscopy. Proc. Natl. Acad. Sci. USA 2006, 103, 2150–2153. [Google Scholar] [CrossRef]
- Zundel, G. Hydrogen bonds with large proton polarizability and proton transfer processes in electrochemistry and biology. Adv. Chem. Phys. 2007, 1–217. [Google Scholar]
- Capobianco, A.; Caruso, T.; Celentano, M.; La Rocca, M.V.; Peluso, A. Proton transfer in oxidized adenosine self-aggregates. J. Chem. Phys. 2013, 139, 145101. [Google Scholar] [CrossRef] [PubMed]
- Schwalb, K.; Temps, F. Base Sequence and Higher-Order Structure Induce the Complex Excited-State Dynamics in DNA. Science 2008, 322, 243–245. [Google Scholar] [CrossRef]
- Capobianco, A.; Caruso, T.; Celentano, M.; D’Ursi, A.M.; Scrima, M.; Peluso, A. Stacking Interactions between Adenines in Oxidized Oligonucleotides. J. Phys. Chem. B 2013, 117, 8947–8953. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, H.; Saito, I. Theoretical Studies of GG-Specific Photocleavage of DNA via Electron Transfer: Significant Lowering of Ionization Potential and 5′-Localization of HOMO of Stacked GG Bases in B-Form DNA. J. Am. Chem. Soc. 1996, 118, 7063–7068. [Google Scholar] [CrossRef]
- Prat, F.; Houk, K. N.; Foote, C. S. Effect of Guanine Stacking on the Oxidation of 8-Oxoguanine in B-DNA. J. Am. Chem. Soc. 1998, 120, 845–846. [Google Scholar] [CrossRef]
- Kurnikov, I. V.; Tong, G. S. M.; Madrid, M.; Beratan, D. N. Hole Size and Energetics in Double Helical DNA: Competition between Quantum Delocalization and Solvation Localization. J. Phys. Chem. B 2002, 106, 7–10. [Google Scholar] [CrossRef]
- Lewis, F. D.; Liu, X.; Liu, J.; Hayes, R. T.; Wasielewski, M. R. Dynamics and Equilibria for Oxidation of G, GG, and GGG Sequences in DNA Hairpins. J. Am. Chem. Soc. 2000, 122, 12037–12038. [Google Scholar] [CrossRef]
- Lewis, F. D.; Liu, J.; Zuo, Z.; Hayes, R. T.; Wasielewski, M. R. Dynamics and Eergetics of Single-Step Hole Transport in DNA Hairpins. J. Am. Chem. Soc. 2003, 125, 4850–4861. [Google Scholar] [CrossRef] [PubMed]
- Ke, C.; Humeniuk, M.; S-Gracz, H.; Marszalek, P. E. Direct Measurements of Base Stacking Interactions in DNA by Single-Molecule Atomic-Force Spectroscopy. Phys. Rev. Lett. 2007, 99, 018302–4. [Google Scholar] [CrossRef] [PubMed]
- Cysewski, P.; Czyzṅ ikowska, Z.; Zalesń y, R.; Czeleń, P. The Post-SCF Quantum Chemistry Characteristics of the Guanine−Guanine Stacking in B-DNA. Phys. Chem. Chem. Phys. 2008, 10, 2665–2672. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Chang, J.; Abdelwahed, S. H.; Thakur, K.; Rathore, R.; Bard, A. J. Electrochemistry and Electrogenerated Chemiluminescence of π-Stacked Poly(fluorenemethylene) Oligomers. Multiple, Interacting Electron Transfers. J. Am. Chem. Soc. 2012, 134, 16265–16274. [Google Scholar] [CrossRef] [PubMed]
- Peluso, A.; Caruso, T.; Landi, A.; Capobianco, A. The dynamics of hole transfer in DNA. Molecules 2019, 24, 4044. [Google Scholar] [CrossRef]
- Capobianco, A.; Caruso, T.; D’Ursi, A. M.; Fusco, S.; Masi, A.; Scrima, M.; Chatgilialoglu, C.; Peluso, A. Delocalized Hole Domains in Guanine-Rich DNA Oligonucleotides. J. Phys. Chem. B 2015, 119, 5462–5466. [Google Scholar] [CrossRef]
- El Hassan, M.A.; Calladine, C.R. Conformational Characteristics of DNA: Empirical Classifications and a Hypothesis for the Conformational Behaviour of Dinucleotide Steps. Philos. Trans. R. Soc. A 1997, 355, 43–100. [Google Scholar] [CrossRef]
- Calladine, C.R.; Drew, H.; Luisi, B.; Travers, A. Understanding DNA: The Molecule and How it Works, 3rd ed.; Elsevier Academic Press: Oxford, UK, 2004; Chapter 3. [Google Scholar]
- Kumar, A.; Sevilla, M. D. Density Functional Theory Studies of the Extent of Hole Delocalization in One-Electron Oxidized Adenine and Guanine Base Stacks. J. Phys. Chem. B 2011, 115, 4990–5000. [Google Scholar] [CrossRef]
- Capobianco, A.; Caruso, T.; Peluso, A. Hole Delocalization over Adenine Tracts in Single Stranded DNA Oligonucleotides. Phys. Chem. Chem. Phys. 2015, 17, 4750–4756. [Google Scholar] [CrossRef]
- Harris, M.A.; Mishra, A.K.; Young, R.M.; Brown, K.E.; Wasielewski, M.R.; Lewis, F.D. Direct Observation of the Hole Carriers in DNA Photoinduced Charge Transport. J. Am. Chem. Soc. 2016, 138, 5491–5494. [Google Scholar] [CrossRef]
- Sugiyama, H.; Saito, I. Theoretical Studies of GG-Specific Photocleavage of DNA via Electron Transfer: Significant Lowering of Ionization Potential and 50-Localization of HOMO of Stacked GG Bases in B-form DNA. J. Am. Chem. Soc. 1996, 118, 7063–7068. [Google Scholar] [CrossRef]
- Senthilkumar, K.; Grozema, F.C.; Fonseca Guerra, C.; Bickelhaupt, F.M.; Siebbeles, L.D.A. Mapping the Sites for Selective Oxidation of Guanines in DNA. J. Am. Chem. Soc. 2003, 125, 13658–13659. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Sevilla, M.D. Photoexcitation of Dinucleoside Radical Cations: A Time-Dependent Density Functional Study. J. Phys. Chem. B 2006, 110, 24181–24188. [Google Scholar] [CrossRef] [PubMed]
- Masi, A.; Capobianco, A.; Bobrowski, K.; Peluso, A.; Chatgilialoglu, C. Hydroxyl Radical vs. One-Electron Oxidation Reactivities in an Alternating GC Double-Stranded Oligonucleotide: A New Type Electron Hole Stabilization. Biomolecules 2023, 13, 1493. [Google Scholar] [CrossRef]
- Xu, B.; Zhang, P.; Li, X.; Tao, N. Direct Conductance Measurement of Single DNA Molecules in Aqueous Solution. Nano Lett. 2004, 4, 1105–1108. [Google Scholar] [CrossRef]
- Mallajosyula, S.S.; Pati, S.K. Toward DNA Conductivity: A Theoretical Perspective. J. Phys. Chem. Lett. 2010, 1, 1881–1894. [Google Scholar] [CrossRef]
- Capobianco, A.; Peluso, A. The oxidization potential of AA steps in single strand DNA oligomers. RSC Adv. 2014, 4, 47887–47893. [Google Scholar] [CrossRef]
- Basko, D.M.; Conwell, E.M. Effect of Solvation on Hole Motion in DNA. Phys. Rev. Lett. 2002, 88, 098102. [Google Scholar] [CrossRef]
- Conwell, E.M.; Bloch, S.M.; McLaughlin, P.M.; Basko, D.M. Duplex Polarons in DNA. J. Am. Chem. Soc. 2007, 129, 9175–9181. [Google Scholar] [CrossRef]
- O’Neill, M.A.; Barton, J.K. DNA Charge Transport: Conformationally Gated Hopping through Stacked Domains. J. Am. Chem. Soc. 2004, 126, 11471–11483. [Google Scholar] [CrossRef]
- Giese, B.; Amaudrut, J.; Köhler, A.K.; Spormann, M.; Wessely, S. Direct Observation of Hole Transfer through DNA by Hopping between Adenine Bases and by Tunneling. Nature 2001, 412, 318–320. [Google Scholar] [CrossRef] [PubMed]
- Eley, D. D.; Spivey, D. I. Semiconductivity of Organic Substances Part 9. Nucleic Acid in Dry State. Trans. Faraday Soc. 1962, 58, 411–415. [Google Scholar] [CrossRef]
- Murphy, C. J.; Arkin, M. R.; Jenkins, Y.; Ghatlia, N. D.; Bossmann, S. H.; Turro, N. J.; Barton, J. K. Long-Range Photoinduced Electron-Transfer through a DNA Helix. Science 1993, 262, 1025–1029. [Google Scholar] [CrossRef] [PubMed]
- Endres, R.G.; Cox, D.L.; Singh, R.R.P. The Quest for High-Conductance DNA. Rev. Mod. Phys. 2004, 76, 195–214. [Google Scholar] [CrossRef]
- Singh, B.; Sariciftci, N.S.; Grote, J.G.; Hopkins, F.K. Bio-Organic-Semiconductor-Field-Effect-Transistor Based on Deoxyribonucleic Acid Gate Dielectric. J. Appl. Phys. 2006, 100, 024514. [Google Scholar] [CrossRef]
- Zalar, P.; Kamkar, D.; Naik, R.; Ouchen, F.; Grote, J.G.; Bazan, G.C.; Nguyen, T.Q. DNA Electron Injection Interlayers for Polymer Light-Emitting Diodes. J. Am. Chem. Soc. 2011, 133, 11010–11013. [Google Scholar] [CrossRef]
- Zhang, Y.; Zalar, P.; Kim, C.; Collins, S.; Bazan, G.C.; Nguyen, T.Q. DNA Interlayers Enhance Charge Injection in Organic Field-Effect Transistors. Adv. Mater. 2012, 24, 4255–4260. [Google Scholar] [CrossRef]
- Gomez, E.F.; Venkatraman, V.; Grote, J.G.; Steckl, A.J. Exploring the Potential of Nucleic Acid Bases in Organic Light Emitting Diodes. Adv. Mater. 2015, 27, 7552–7562. [Google Scholar] [CrossRef]
- Giese, B. Long Distance Electron Transfer through DNA. Annu. Rev. Biochem. 2002, 71, 51–70. [Google Scholar] [CrossRef]
- Lewis, F.D.; Letsinger, R.L.; Wasielewski, M.R. Dynamics of Photoinduced Charge Transfer and Hole Transfer in Synthetic DNA Hairpins. Acc. Chem Res. 2001, 34, 159–170. [Google Scholar] [CrossRef]
- Genereux, J.C.; Barton, J.K. Mechanisms for DNA Charge Transport. Chem Rev. 2010, 110, 1642–1662. [Google Scholar] [CrossRef] [PubMed]
- Kawai, K.; Majima, T. Hole Transfer Kinetics of DNA. Acc. Chem. Res. 2013, 46, 2616–2625. [Google Scholar] [CrossRef] [PubMed]
- Arnold, A.R.; Grodick, M.A.; Barton, J.K. DNA Charge Transport: Principles to the Cell. Cell Chem. Biol. 2016, 23, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Landi, A.; Borrelli, R.; Capobianco, A.; Peluso, A. Transient and Enduring Electronic Resonances Drive Coherent Long Distance Charge Transport in Molecular Wires. J. Phys. Chem. Lett. 2019, 10, 1845–1851. [Google Scholar] [CrossRef] [PubMed]
- Landi, A.; Capobianco, A.; Peluso, A. Coherent Effects in Charge Transport in Molecular Wires: Toward a Unifying Picture of Long-Range Hole Transfer in DNA. J. Phys. Chem. Lett. 2020, 11, 7769–7777. [Google Scholar] [CrossRef]
- Bixon, M.; Giese, B.; Wessely, S.; Langenbacher, T.; Michel-Beyerle, M.E.; Jortner, J. Long-Range Charge Hopping in DNA. Proc. Natl. Acad. Sci. USA 1999, 96, 11713–11716. [Google Scholar] [CrossRef]
- Renaud, N.; Berlin, Y.A.; Lewis, F.D.; Ratner, M.A. Between Superexchange and Hopping: An Intermediate Charge-Transfer Mechanism in polyA-polyT DNA Hairpins. J. Am. Chem. Soc. 2013, 135, 3953–3963. [Google Scholar] [CrossRef]
- Renger, T.; Marcus, R.A. Variable Range Hopping Electron Transfer Through Disordered Bridge States: Application to DNA. J. Phys. Chem. A 2003, 107, 8404–8419. [Google Scholar] [CrossRef]
- Bixon, M.; Jortner, J. Incoherent Charge Hopping and Conduction in DNA and Long Molecular Chains. Chem. Phys. 2005, 319, 273–282. [Google Scholar] [CrossRef]
- Grozema, F. C.; Tonzani, S.; Berlin, Y. A.; Schatz, G. C.; Siebbeles, L. D. A.; Ratner, M. A. Effect of Structural Dynamics on Charge Transfer in DNA Hairpins. J. Am. Chem. Soc. 2008, 130, 5157−5166.
- Zhang, Y.; Liu, C.; Balaeff, A.; Skourtis, S. S.; Beratan, D. N. Biological Charge Transfer via Flickering Resonance. Proc. Natl. Acad. Sci. USA 2014, 111, 10049–10054. [Google Scholar] [CrossRef]
- Levine, A. D.; Iv, M.; Peskin, U. Length-Independent Transport Rates in Biomolecules by Quantum Mechanical Unfurling. Chem. Sci. 2016, 7, 1535–1542. [Google Scholar] [CrossRef] [PubMed]
- Leo,A.; Peluso, A. Electron Transfer Rates in Polar and Non-Polar Environments: a Generalization of Marcus’ Theory to Include an Effective Treatment of Tunneling Effects. J. Phys. Chem. Lett. 2022, 13, 9148–9155.
- Borrelli, R.; Capobianco, A.; Landi, A.; Peluso, A. Vibronic couplings and coherent electron transfer in bridged systems. Phys. Chem. Chem. Phys. 2015, 17, 30937–30945. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, R.; Di Donato, M. Peluso, A. Role of intramolecular vibrations in long-range electron transfer between pheophytin and ubiquinone in bacterial photosynthetic reaction centers. Biophys. J. 2005, 89, 830–841. [Google Scholar] [CrossRef] [PubMed]
- Chatgilialoglu, C. The Two Faces of the Guanyl Radical: Molecular Context and Behavior. Molecules 2021, 26, 3511. [Google Scholar] [CrossRef] [PubMed]
- Steenken, S. Purine bases, nucleosides, and nucleotides: aqueous solution redox chemistry and transformation reactions of their radical cations and e– and OH adducts. Chem. Rev. 1989, 89, 503–520. [Google Scholar] [CrossRef]
- DNA Damage, DNA Repair and Disease; Dizdaroglu, M., Lloyd, R. S., Eds.; Royal Society of Chemistry: Croydon, UK, 2021.
- Zhang, X.; Jie, J.; Song, D.; Su, H. Deprotonation of Guanine Radical Cation G•+ Mediated by the Protonated Water Cluster. J. Phys. Chem. A 2020, 124, 6076–6083. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; D’Angelantonio, M.; Guerra, M.; Kaloudis, P.; Mulazzani, Q. G. A reevaluation of the ambident reactivity of guanine moiety towards hydroxyl radicals. Angew. Chem. Int. Ed. 2009, 48, 2214–2217. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; D’Angelantonio, M.; Kciuk, G.; Bobrowski, K. New insights into the reaction paths of hydroxyl radicals with 2’-deoxyguanosine. Chem. Res. Toxicol. 2011, 24, 2200–2206. [Google Scholar] [CrossRef]
- Adhikary, A.; Kumar, A.; Becker, D.; Sevilla, M. D. The Guanine Cation Radical: Investigation of Deprotonation States by ESR and DFT. J. Phys. Chem. B 2006, 110, 24171–24180. [Google Scholar] [CrossRef]
- Adhikary, A.; Khanduri, D.; Sevilla, M. D. Direct observation of the hole protonation state and hole localization site in DNA-oligomers. J. Am. Chem. Soc. 2009, 131, 8614–8619. [Google Scholar] [CrossRef]
- Kobayashi, K.; Yamagami, R.; Tagawa, S. Effect of base sequence and deprotonation of guanine cation radical in DNA. J. Phys. Chem. B 2008, 112, 10752–10757. [Google Scholar] [CrossRef] [PubMed]
- Rokhlenko, Y.; Cadet, J.; Geacintov, N.E.; Shafirovich, V. Mechanistic aspects of hydration of guanine radical cations in DNA. J. Am. Chem. Soc. 2014, 136, 5956–5962. [Google Scholar] [CrossRef] [PubMed]
- Banyasz, A.; Martínez-Fernández, L.; Improta, R.; Ketola, T.-M.; Balty, C.; Markovitsi, D. Radicals generated in alternating guanine cytosine duplexes by direct absorption of low-energy UV radiation. Phys. Chem. Chem. Phys. 2018, 20, 21381–21389. [Google Scholar] [CrossRef] [PubMed]
- Balanikas, E.; Banyasz, A.; Baldacchino, G.; Markovitsi, D. Populations and Dynamics of Guanine Radicals in DNA strands— Direct versus Indirect Generation. Molecules 2019, 24, 2347. [Google Scholar] [CrossRef]
- Balanikas, E.; Banyasz, A.; Douki, T.; Baldacchino, G.; Markovitsi, D. Guanine Radicals Induced in DNA by Low-Energy Pho- toionization. Acc. Chem. Res. 2020, 53, 1511–1519. [Google Scholar] [CrossRef]
- Balanikas, E.; Banyasz, A.; Baldacchino, G.; Markovitsi, D. Deprotonation Dynamics of Guanine Radical Cations. Photochem. Photobiol. 2022, 98, 523–531. [Google Scholar] [CrossRef]
- Adhikary, A.; Kumar, A.; Munafo, S.A.; Khanduri, D.; Sevilla, M.D. Prototropic equilibria in DNA containing one-electron oxidized GC:intra-duplex vs. duplex to solvent deprotonation. Phys. Chem. Chem. Phys. 2010, 12, 5353–5368. [Google Scholar] [CrossRef]
- Cadet, J.; Wagner, J.R. Oxidatively generated base damage to cellular DNA by hydroxyl radical and one-electron oxidants: Similarities and differences. Arch. Biochem. Biophys. 2014, 557, 47–54. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; Krokidis, M.G.; Masi, A.; Barata-Vallejo, S.; Ferreri, C.; Terzidis, M.A.; Szreder, T.; Bobrowski, K. New insights into the reaction paths of hydroxyl radicals with purine moieties in DNA and double-stranded oligonucleotides. Molecules 2019, 24, 3860. [Google Scholar] [CrossRef]
- Kumar, A.; Sevilla, M. D. Influence of Hydration on Proton Transfer in the Guanine—Cytosine Radical Cation (G•+—C) Base P air: A Density Functional Theory Study. J. Phys. Chem. B 2009, 113, 11359–11361. [Google Scholar] [CrossRef]
- Steenken, S.; Reynisson, J. DFT calculations on the deprotonation site of the one-electron oxidised guanine–cytosine base pair. Phys. Chem. Chem. Phys. 2010, 12, 9088–9093. [Google Scholar] [CrossRef] [PubMed]
- Cerón-Carrasco, J.P.; Requena, A.; Perpète, E.A.; Michaux, C.; Jacquemin, D. Theoretical Study of the Tautomerism in the One-Electron Oxidized Guanine-Cytosine Base Pair. J. Phys. Chem. B 2010, 114, 13439–13445. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Sevilla, M. D. Excited States of One-Electron Oxidized Guanine-Cytosine Base Pair Radicals: A Time Dependent Density Functional Theory Study. J. Phys. Chem. A 2019, 123, 3098–3108. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Eriksson, L.A. Effects of OH Radical Addition on Proton Transfer in the Guanine-Cytosine Base Pair. J Phys. Chem. B 2007, 111, 6571–6576. [Google Scholar] [CrossRef] [PubMed]
- Cerón-Carrasco, J.P.; Jacquemin, D. Interplay between hydroxyl radical attack and H-bond stability in guanine–cytosine. RCS Adv. 2012, 2, 11867–11875. [Google Scholar] [CrossRef]
- Wang, Y.; Wei, S. Influence of hydrogen bonds on the reaction of guanine and hydroxyl radical: DFT calculations in C(H+)GC motif. Phys. Chem. Chem. Phys. 2024, 26, 5683–5692. [Google Scholar] [CrossRef]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef]
- Möller, M.N.; Denicola, A. Diffusion of peroxynitrite, its precursors, and derived reactive species, and the effect of cell membranes. Redox Biochem. Chem. 2024, 9, 100033. [Google Scholar] [CrossRef]
- Illes, E.; Patra, S.G.; Marks, V.; Mizrahi, A.; Meyerstein, D. The FeII(citrate) Fenton reaction under physiological conditions. J. Inorg. Biochem. 2020, 206, 111018. [Google Scholar] [CrossRef]
- Bounds, P.L.; Koppenol, W.H. Peroxynitrite: A tale of two radicals, Redox Biochem. Chem. 2024, 10, 100038. [Google Scholar]
- Shafirovich, V.; Dourandin, A.; Huang, W.; Geacintov, N.E. The carbonate radical is a site-selective oxidizing agent of guanine in double-stranded oligonucleotides. J. Biol. Chem. 2001, 276, 24621–24626. [Google Scholar] [CrossRef] [PubMed]
- Crean, C.; E. Geacintov, N.E.; Shafirovich, V. Oxidation of Guanine and 8-oxo-7,8-Dihydroguanine by Carbonate Radical Anions: Insight from Oxygen-18 Labeling Experiments. Angew. Chem. Int. Ed. 2005, 44, 5057–5060. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.M.; Burrows, C.J. Iron Fenton oxidation of 2′-deoxyguanosine in physiological bicarbonate buffer yields products consistent with the reactive oxygen species carbonate radical anion not hydroxyl radical. Chem. Commun. 2020, 56, 9779–9782. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.M.; Burrows, C. Chemistry of ROS-mediated oxidation to the guanine base in DNA and its biological consequences. Int. J. Radiat. Biol. 2022, 98, 452–460. [Google Scholar] [CrossRef]
- Jaruga, P.; Kirkali, G.; Dizdaroglu, M. Measurement of formamidopyrimidines in DNA. Free Radic. Biol. Med. 2008, 45, 1601–1609. [Google Scholar] [CrossRef]
- Dizdaroglu, M.; Kirkali, G.; Jaruga, P. Formamidopyrimidines in DNA: mechanisms of formation, repair, and biological effects. Free Radic. Biol Med. 2008, 45, 1610–1621. [Google Scholar] [CrossRef]
- Dizdaroglu, M. Oxidatively induced DNA damage and its repair in cancer. Mutat. Res. Rev. Mutat. Res. 2015, 763, 212–245. [Google Scholar] [CrossRef]
- Greenberg, M. M. The formamidopyrimidines: purine lesions formed in competition with 8-oxopurines from oxidative stress. Acc. Chem. Res. 2012, 45, 588–597. [Google Scholar] [CrossRef]
- Steenken, S.; Jovanovic, S. V.; Bietti, M.; Bernhard, K. The trap depth (in DNA) of 8-oxo-7,8-dihydro-2’deoxyguanosine as derived from electron-transfer equilibria in aqueous solution. J. Am. Chem. Soc. 2000, 122, 2373–2374. [Google Scholar] [CrossRef]
- Fleming, A. M.; Muller, J. G.; Dlouhy, A. C.; Burrows, C. J. Structural context effects in the oxidation of 8-oxo-7,8-dihydro-2’-deoxyguanosine to hydantoin products: electrostatics, base stacking, and base pairing. J. Am. Chem. Soc. 2012, 134, 15091–15102. [Google Scholar] [CrossRef]
- Chabot, M.B.; Fleming, A.M.; Burrows, C. J. Insights into the 5-carboxamido-5-formamido-2-iminohydantoin structural isomerization equilibria. J. Org. Chem. 2022, 87, 11865–11870. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A. M.; Muller, J. G.; Ji, I.; Burrows, C. J. Characterization of 2’-deoxyguanosine oxidation products observed in the Fenton-like system Cu(II)/H2O2/reductant in nucleoside and oligodeoxynucleotide contexts. Org. Biomol. Chem. 2011, 9, 3338–3348. [Google Scholar] [CrossRef] [PubMed]
- Alshykhly, O. R.; Fleming, A. M.; Burrows, C. J. 5-Carboxamido-5-formamido-2-iminohydantoin, in Addition to 8-oxo-7,8-Di-hydroguanine, Is the Major Product of the Iron-Fenton or X-ray Radiation-Induced Oxidation of Guanine under Aerobic Reducing Conditions in Nucleoside and DNA Contexts. J. Org. Chem. 2015, 80, 6996–7007. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; An, P.; Li, S.; Zhou, L. The oxidation mechanism and kinetics of 2′-deoxyguanosine by carbonate radical anion. Chem. Phys. Lett. 2020, 739, 136982. [Google Scholar] [CrossRef]
- Misiaszek, R.; Crean, C.; Joffe, A.; Geacintov, N. E.; Shafirovich, V. Oxidative DNA damage associated with combination of guanine and superoxide radicals and repair mechanisms via radical trapping. J. Biol. Chem. 2004, 279, 32106–32115. [Google Scholar] [CrossRef]
- Misiaszek, R.; Crean, C.; Geacintov, N. E.; Shafirovich, V. Combination of Nitrogen Dioxide Radicals with 8-Oxo-7,8-dihydroguanine and Guanine Radicals in DNA: Oxidation and Nitration End-Products. J. Am. Chem. Soc. 2005, 127, 2191–2200. [Google Scholar] [CrossRef]
- Joffe, A.; Mock, S.; Yun, B. H.; Kolbanovskiy, A.; Geacintov, N. E.; Shafirovich, V. Oxidative Generation of Guanine Radicals by Carbonate Radicals and Their Reactions with Nitrogen Dioxide to Form Site Specific 5-Guanidino-4-nitroimidazole Lesions in Oligodeoxynucleotides. Chem. Res. Toxicol. 2003, 16, 966–973. [Google Scholar] [CrossRef]
- Hebert, S.P.; Bernhard Schlegel, H.B. Computational Investigation into the Oxidation of Guanine to Form Imidazolone (Iz) and Related Degradation Products. Chem. Res. Toxicol. 2020, 33, 1010–1027. [Google Scholar] [CrossRef]
- Cui, L.; Ye, W.; Prestwich, E.G.; Wishnok, J.S.; Taghizadeh, K.; Dedon, P.C.; Tannenbaum, S.R. Comparative analysis of four oxidized guanine lesions from reactions of DNA with peroxynitrite, single oxygen, and γ-radiation. Chem. Res. Toxicol. 2013, 26, 195–202. [Google Scholar] [CrossRef]
- Matter, B.; Seiler, C. L.; Murphy, K.; Ming, X.; Zhao, J.; Lindgren, B.; Jones, R.; Tretyakova, N. Mapping three guanine oxidation products along DNA following exposure to three types of reactive oxygen species. Free Radic. Biol. Med. 2018, 121, 180–189. [Google Scholar] [CrossRef]
- Fleming, A.M.; Burrows, C.J. Formation and processing of DNA damage substrates for the hNEIL enzymes. Free Radic. Biol. Med. 2017, 107, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Chatgilialoglu, C.; Eriksson, L.A.; Krokidis, M.G.; Masi, A.; Wang, S.-D.; Zhang, R. Oxygen dependent purine lesions in double-stranded oligodeoxynucleotides: Kinetic and computational studies highlight the mechanism for 5’,8-cyplopurine formation. J. Am. Chem. Soc. 2020, 142, 5825–5833. [Google Scholar] [CrossRef] [PubMed]
- Chatgilialoglu, C.; Ferreri, C.; Krokidis, M.G.; Masi, A.; Terzidis, M.A. On the relevance of hydroxyl radical to purine DNA damage. Free Radic. Res. 2021, 55, 384–404. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, F.; Auvre, F.; Radicella, J.P.; Ravanat, J.-L. HO• radicals induce an unexpected high proportion of tandem base lesions refractory to repair by DNA glycosylases. Proc. Natl. Acad. Sci. USA 2010, 107, 5528–5533. [Google Scholar] [CrossRef]
Figure 1.
Differential pulse voltammetry of: a) 2’,3’-O-isopropylidene 5’-O-(tert-butyldimethylsilyl)guanosine (Guo) (red line), 3’,5’-bis-O-(tertbutyldimethylsilyl)-2’-deoxycytidine (dCyd) (gray dashed line), and their base pair complex Guo:dCyd (blue line) in CHCl3; b) solution containing 2’,3’-O-isopropylidene 5’-O-(tert-butyldimethylsilyl)-adenosine (Ado’) and 3’,5’-bis-O-(tert-butyldimethylsilyl)thymidine (Thd’) in CHCl3, red line Ado’ 2.0 mM and Thd’ 2.0 mM, dashed blue line Ado’ 2.0 mM and Thd’ 20.0 mM. Adapted with permission from: Caruso, T.; Carotenuto, M.; Vasca, E.; Peluso, A. J. Am. Chem. Soc. 2005, 127, 15040–15041, and Caruso, T.; Capobianco, A.; Peluso, A. J. Am. Chem. Soc. 2007, 129, 15347–15353. Copyright (2005 and 2007) American Chemical Society.
Figure 1.
Differential pulse voltammetry of: a) 2’,3’-O-isopropylidene 5’-O-(tert-butyldimethylsilyl)guanosine (Guo) (red line), 3’,5’-bis-O-(tertbutyldimethylsilyl)-2’-deoxycytidine (dCyd) (gray dashed line), and their base pair complex Guo:dCyd (blue line) in CHCl3; b) solution containing 2’,3’-O-isopropylidene 5’-O-(tert-butyldimethylsilyl)-adenosine (Ado’) and 3’,5’-bis-O-(tert-butyldimethylsilyl)thymidine (Thd’) in CHCl3, red line Ado’ 2.0 mM and Thd’ 2.0 mM, dashed blue line Ado’ 2.0 mM and Thd’ 20.0 mM. Adapted with permission from: Caruso, T.; Carotenuto, M.; Vasca, E.; Peluso, A. J. Am. Chem. Soc. 2005, 127, 15040–15041, and Caruso, T.; Capobianco, A.; Peluso, A. J. Am. Chem. Soc. 2007, 129, 15347–15353. Copyright (2005 and 2007) American Chemical Society.
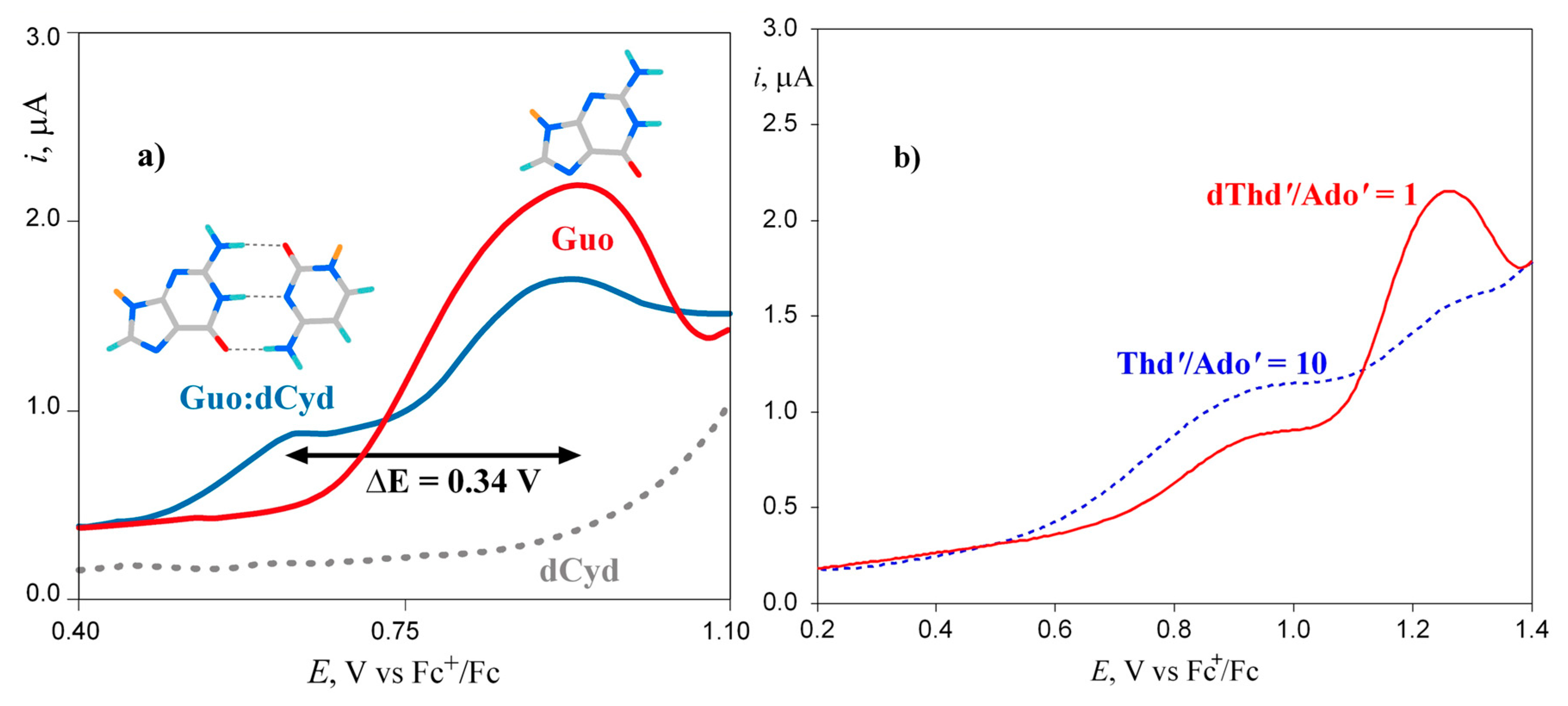
Figure 2.
Spectroelectrochemistry characterization of the Guo:dCyd base pair complex: (a) Cyclic voltammogram recorded in the OTTLE cell containing 20 mM Guo and 20 mM dCyd in CHCl3; working electrode, Pt, supporting electrolyte tetrabutylammonium perchlorate (TBAP) 0.1 M, scan rate 50 mV/s, ferrocenium/ferrocene (Fc+/Fc) as internal reference half-couple. (b) IR spectrum of Guo in CHCl3 (full line), and difference spectrum obtained by subtracting the spectrum of Guo from that recorded at 0.91 V vs. Fc+/Fc (dashed line). (c) UV/vis absorption spectral changes recorded during the electrooxidation at controlled potential of a Guo solution 10 mM in chloroform at different times; the intense red curve was recorded after the potential was switched off. (d) NIR absorption spectra of the Guo:dCyd base pair complex and Guo:5mCyd complexes in CH2Cl2, recorded at +0.57 V vs. Fc+/Fc. Guo, C: 10 mm; TBAP: 0.1m; 5mCyd= 2’,3’,5’-tri-O-(tert-butyldimethylsilyl)-5-methylcytidine. Adapted with permission from: Capobianco, A.; Carotenuto, M.; Caruso, T.; Peluso, A. The Charge-Transfer Band of an Oxidized Watson-Crick Guanosine-Cytidine Complex. Angew. Chem. Int. Ed. 2009, 48, 9526–9528, Copyright (2009) Wiley.
Figure 2.
Spectroelectrochemistry characterization of the Guo:dCyd base pair complex: (a) Cyclic voltammogram recorded in the OTTLE cell containing 20 mM Guo and 20 mM dCyd in CHCl3; working electrode, Pt, supporting electrolyte tetrabutylammonium perchlorate (TBAP) 0.1 M, scan rate 50 mV/s, ferrocenium/ferrocene (Fc+/Fc) as internal reference half-couple. (b) IR spectrum of Guo in CHCl3 (full line), and difference spectrum obtained by subtracting the spectrum of Guo from that recorded at 0.91 V vs. Fc+/Fc (dashed line). (c) UV/vis absorption spectral changes recorded during the electrooxidation at controlled potential of a Guo solution 10 mM in chloroform at different times; the intense red curve was recorded after the potential was switched off. (d) NIR absorption spectra of the Guo:dCyd base pair complex and Guo:5mCyd complexes in CH2Cl2, recorded at +0.57 V vs. Fc+/Fc. Guo, C: 10 mm; TBAP: 0.1m; 5mCyd= 2’,3’,5’-tri-O-(tert-butyldimethylsilyl)-5-methylcytidine. Adapted with permission from: Capobianco, A.; Carotenuto, M.; Caruso, T.; Peluso, A. The Charge-Transfer Band of an Oxidized Watson-Crick Guanosine-Cytidine Complex. Angew. Chem. Int. Ed. 2009, 48, 9526–9528, Copyright (2009) Wiley.
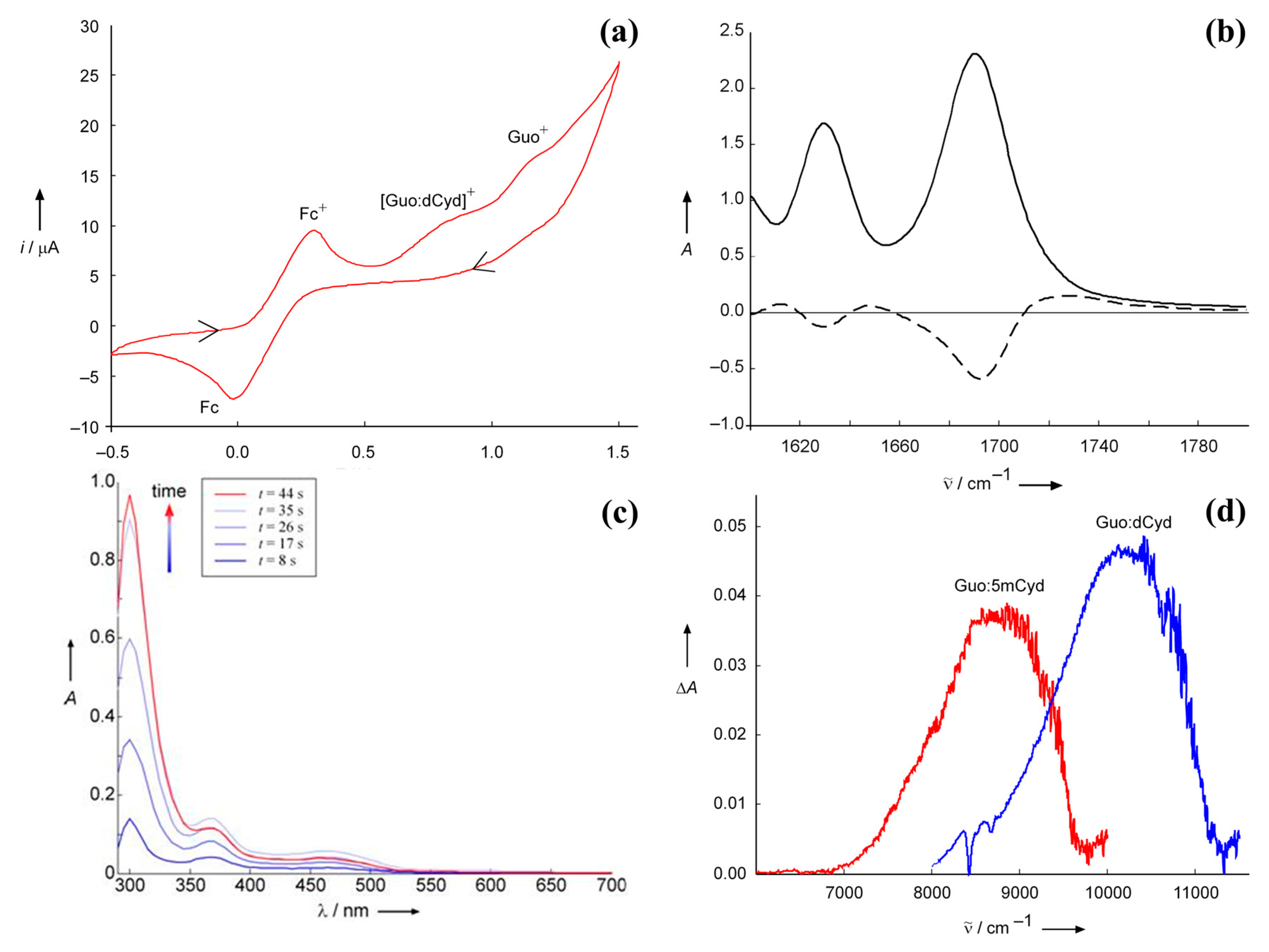
Figure 3.
The effects of π stacking interactions upon the electronic states of one-electron-oxidized DNA: a perturbation, as for instance excitation, removal or addition of an electron, applied to the A or B unstacked nucleobase gives rise to two states |A*B⟩ and |AB*⟩ with energies ɛA and ɛB; upon formation of a π stacking interaction, the two states are coupled with each other, and the energy levels of the electron-hole are shifted up and down for those of the unstacked pair by a factor depending on the strength of the π stacking interaction, β, and on the difference between nucleobase oxidation potentials, as shown on the right side of the panel. Adapted with permission from Capobianco, A.; Caruso, T.; Celentano, M.; D’Ursi, A.M.; Scrima, M.; Peluso, A. J. Phys. Chem. B 2013, 117, 8947–8953.Copyright 2013 American Chemical Society.
Figure 3.
The effects of π stacking interactions upon the electronic states of one-electron-oxidized DNA: a perturbation, as for instance excitation, removal or addition of an electron, applied to the A or B unstacked nucleobase gives rise to two states |A*B⟩ and |AB*⟩ with energies ɛA and ɛB; upon formation of a π stacking interaction, the two states are coupled with each other, and the energy levels of the electron-hole are shifted up and down for those of the unstacked pair by a factor depending on the strength of the π stacking interaction, β, and on the difference between nucleobase oxidation potentials, as shown on the right side of the panel. Adapted with permission from Capobianco, A.; Caruso, T.; Celentano, M.; D’Ursi, A.M.; Scrima, M.; Peluso, A. J. Phys. Chem. B 2013, 117, 8947–8953.Copyright 2013 American Chemical Society.
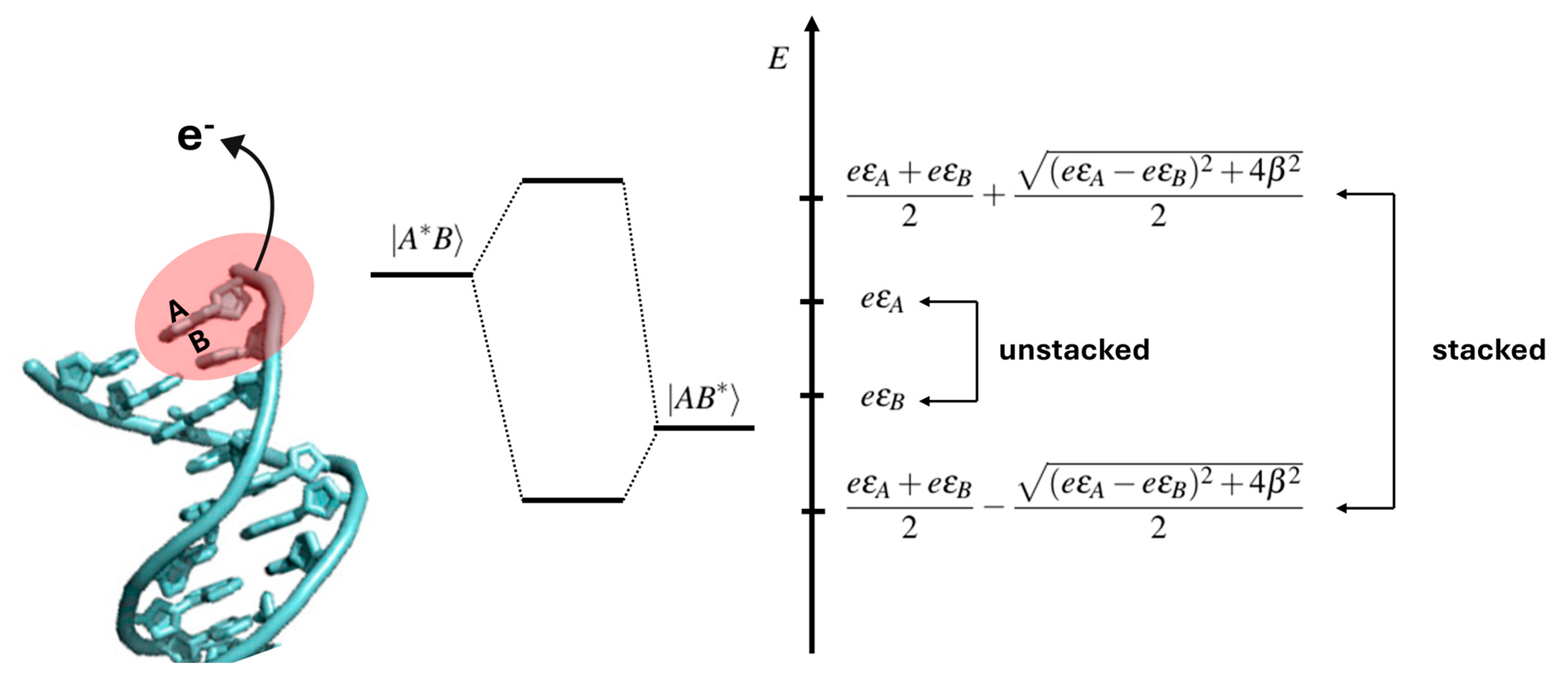
Figure 4.
Optimized geometry and spin density plot (PCM/B3LYP-D3BJ/TZVP) of [ds-5′-GCGC-3′]•+. Hydrogen atoms and sodium counterions have been omitted for clarity.
Figure 4.
Optimized geometry and spin density plot (PCM/B3LYP-D3BJ/TZVP) of [ds-5′-GCGC-3′]•+. Hydrogen atoms and sodium counterions have been omitted for clarity.
Figure 5.
Structures and predicted and observed oxidative damages of the four DNA oligomers studied in ref.s 28 and 77. A hole is injected on the leftmost G or GG sites, producing oxidative damages at G, GG, and 8-oxo-G. The predicted (blue incoherent and green coherent model) and experimental (gray) product ratios are shown in the panel below each oligonucleotide. Oligomer 1 consists only of single G sites, oxidative damage is observed to almost the same extent at all G sites. Oligomer 2 contains two trap sites at both ends, connected by several TGT steps acting as efficient hole shuttles; the injected hole easily migrates along the strand, splitting to a larger extent over the two trap sites (>80%) and a lesser extent onto single G sites. Oligomer 3 possesses a deeper 8-oxo-G trap on which most of the oxidative damage (>90%) is observed. In oligomer 4, a T quadruplet is interposed between the donor GG and the acceptor 8-oxo-G; the hole is not able to freely move along the strand and localizes to a larger extent on the G doublet (>90%) and to a lesser extent on single G’s without crossing the T quadruplet bridge. For oligomers 3 and 4 the coherent model yields more satisfying results than the incoherent one, testifying that charge transport cannot occur by a simple sequential hopping mechanism. Adapted with permission from Landi, A.; Capobianco, A.; Peluso, A. J. Phys. Chem. Lett. 2020, 11, 7769−7777.
Figure 5.
Structures and predicted and observed oxidative damages of the four DNA oligomers studied in ref.s 28 and 77. A hole is injected on the leftmost G or GG sites, producing oxidative damages at G, GG, and 8-oxo-G. The predicted (blue incoherent and green coherent model) and experimental (gray) product ratios are shown in the panel below each oligonucleotide. Oligomer 1 consists only of single G sites, oxidative damage is observed to almost the same extent at all G sites. Oligomer 2 contains two trap sites at both ends, connected by several TGT steps acting as efficient hole shuttles; the injected hole easily migrates along the strand, splitting to a larger extent over the two trap sites (>80%) and a lesser extent onto single G sites. Oligomer 3 possesses a deeper 8-oxo-G trap on which most of the oxidative damage (>90%) is observed. In oligomer 4, a T quadruplet is interposed between the donor GG and the acceptor 8-oxo-G; the hole is not able to freely move along the strand and localizes to a larger extent on the G doublet (>90%) and to a lesser extent on single G’s without crossing the T quadruplet bridge. For oligomers 3 and 4 the coherent model yields more satisfying results than the incoherent one, testifying that charge transport cannot occur by a simple sequential hopping mechanism. Adapted with permission from Landi, A.; Capobianco, A.; Peluso, A. J. Phys. Chem. Lett. 2020, 11, 7769−7777.
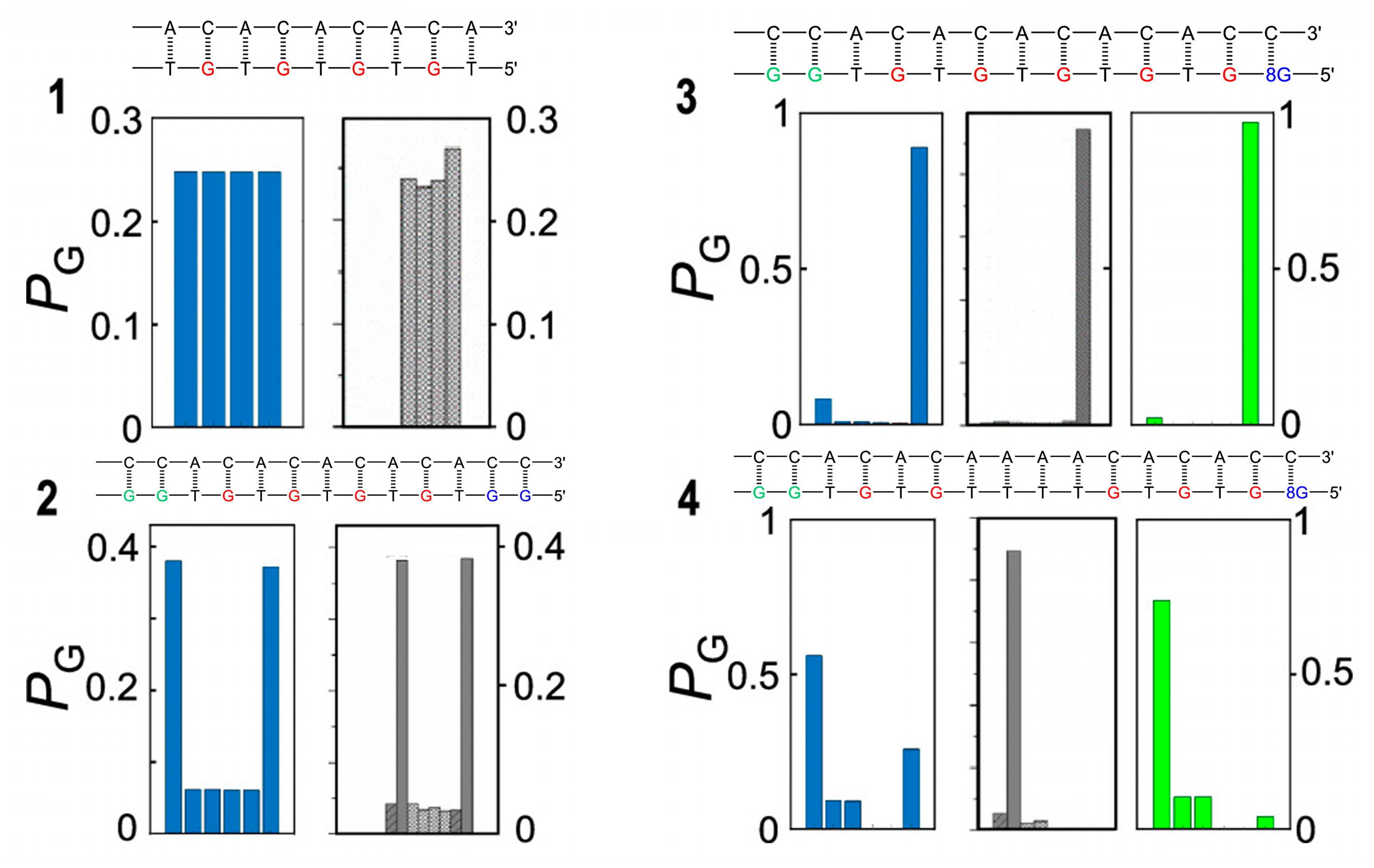
Figure 6.
The hole transport mechanism adopted in ref. 77 for predicting the yields of oxidative damages for Giese’s ds-G(T)nGGG series and Schuster’s oligomers of Figure 5.
Figure 6.
The hole transport mechanism adopted in ref. 77 for predicting the yields of oxidative damages for Giese’s ds-G(T)nGGG series and Schuster’s oligomers of Figure 5.
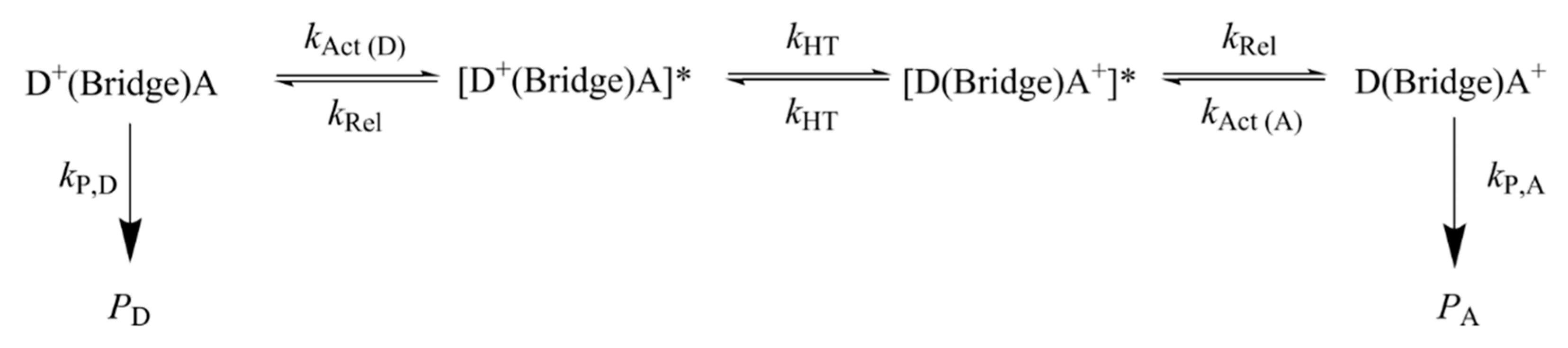
Figure 7.
Guanyl radical cation moiety (G•+) and the corresponding guanyl radicals G(N1-H)• or G(N2-H)• obtained by loss of protons. Both G(N1-H)• and G(N2-H)• have various resonance forms, including those where the unpaired electron is placed in the C5 position for evidencing the two tautomeric structures (highlighted in red); for more detail, see reference [88].
Figure 7.
Guanyl radical cation moiety (G•+) and the corresponding guanyl radicals G(N1-H)• or G(N2-H)• obtained by loss of protons. Both G(N1-H)• and G(N2-H)• have various resonance forms, including those where the unpaired electron is placed in the C5 position for evidencing the two tautomeric structures (highlighted in red); for more detail, see reference [88].
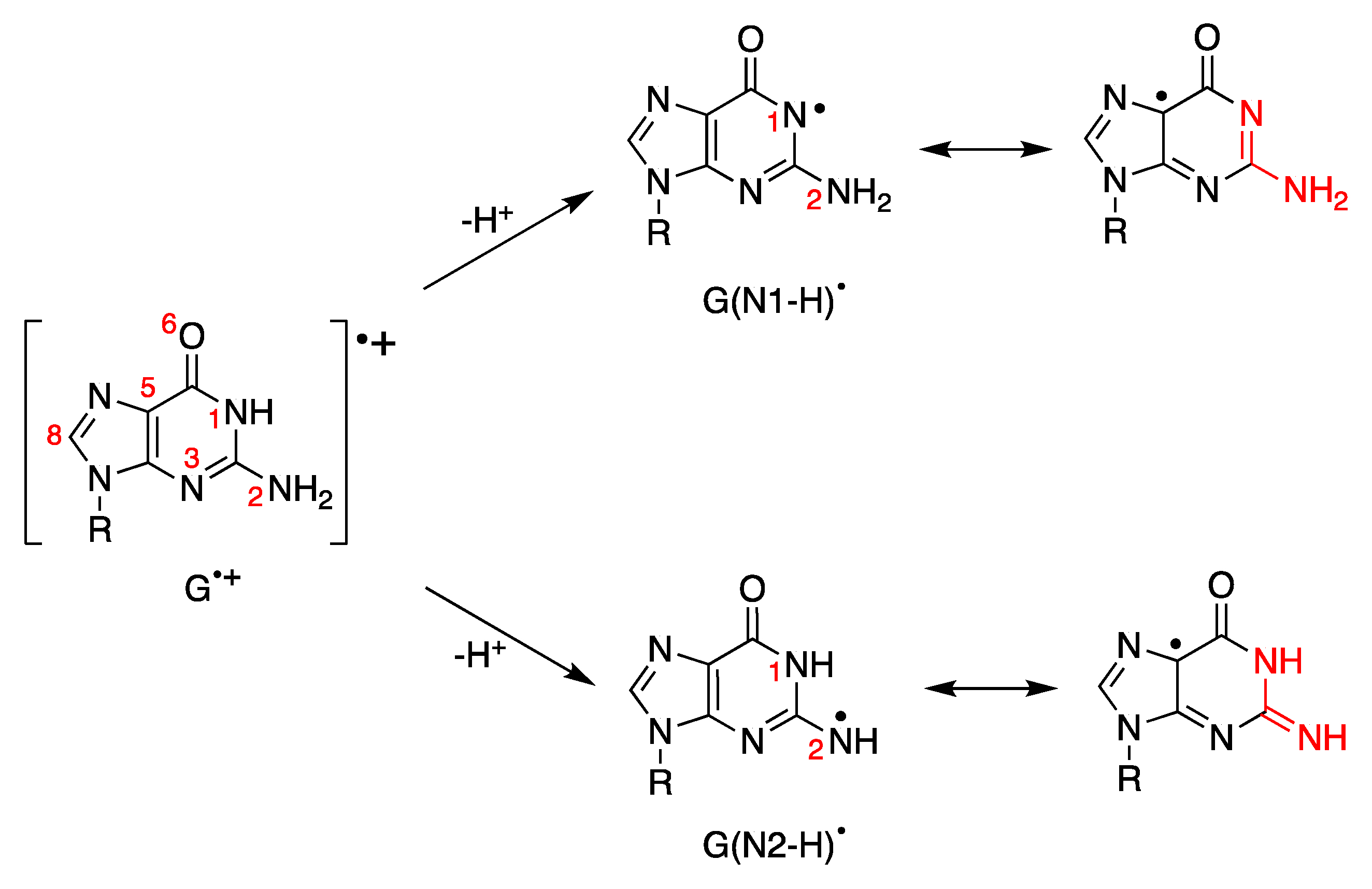
Figure 8.
Deprotonation vs. hydration of G•+ in ds-ODNs. The fragment G•+:C is in equilibrium with G(N1-H)•:CH+] and both of them may lose H+ affording the two tautomers of guanyl moiety, i.e., G(N2-H)• or G(N1-H)•, respectively. Hydrolysis of G•+ affords 8-HO-G• radical which follows a further oxidation step to give 8-oxoG:C fragment as a stable product.
Figure 8.
Deprotonation vs. hydration of G•+ in ds-ODNs. The fragment G•+:C is in equilibrium with G(N1-H)•:CH+] and both of them may lose H+ affording the two tautomers of guanyl moiety, i.e., G(N2-H)• or G(N1-H)•, respectively. Hydrolysis of G•+ affords 8-HO-G• radical which follows a further oxidation step to give 8-oxoG:C fragment as a stable product.
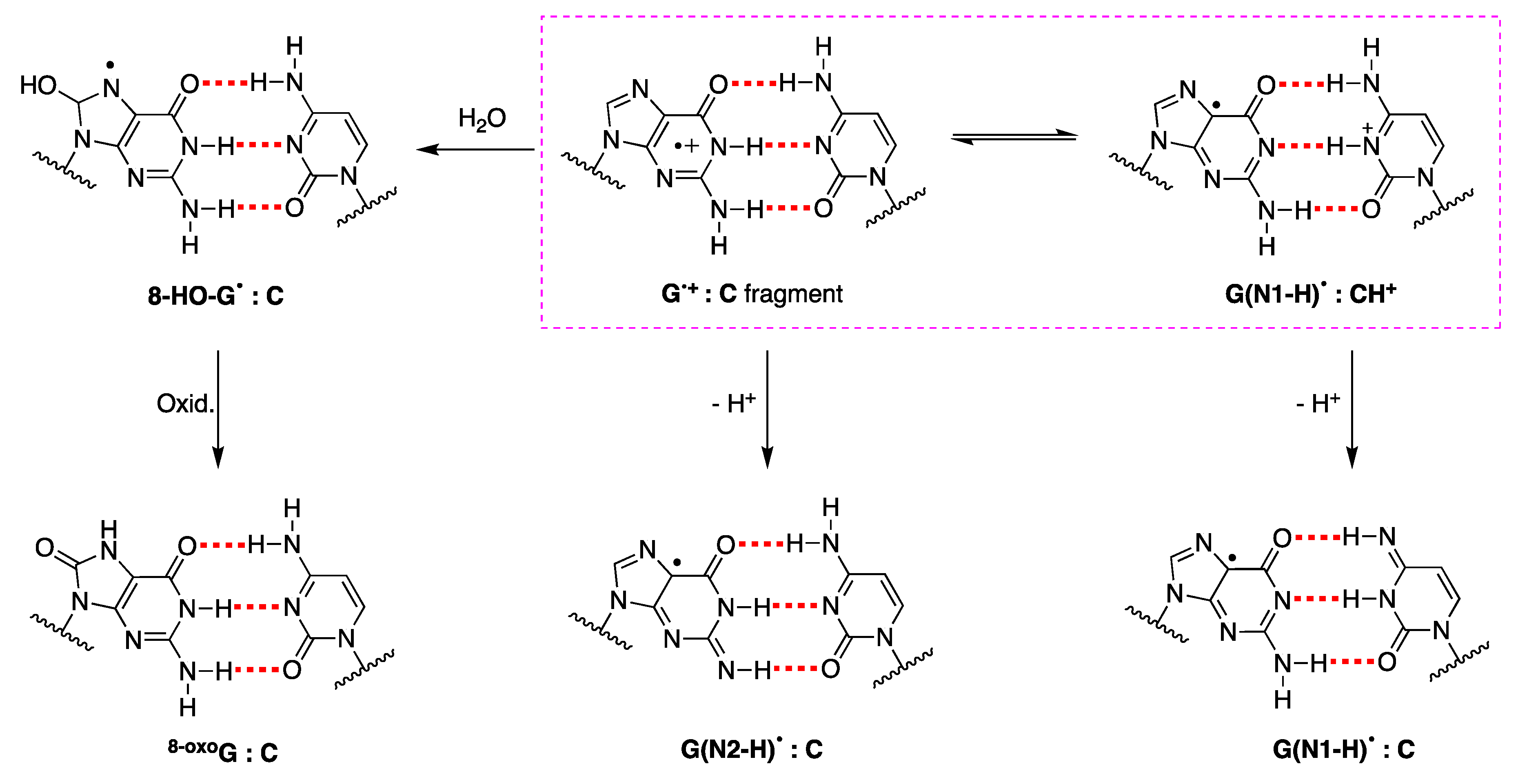
Figure 9.
The reported reaction mechanism for the reaction of HO• and SO4•− with the ds-ODN of the palindromic 5′-d(GCGCGC)-3′, in deoxygenated aqueous solutions.
Figure 9.
The reported reaction mechanism for the reaction of HO• and SO4•− with the ds-ODN of the palindromic 5′-d(GCGCGC)-3′, in deoxygenated aqueous solutions.
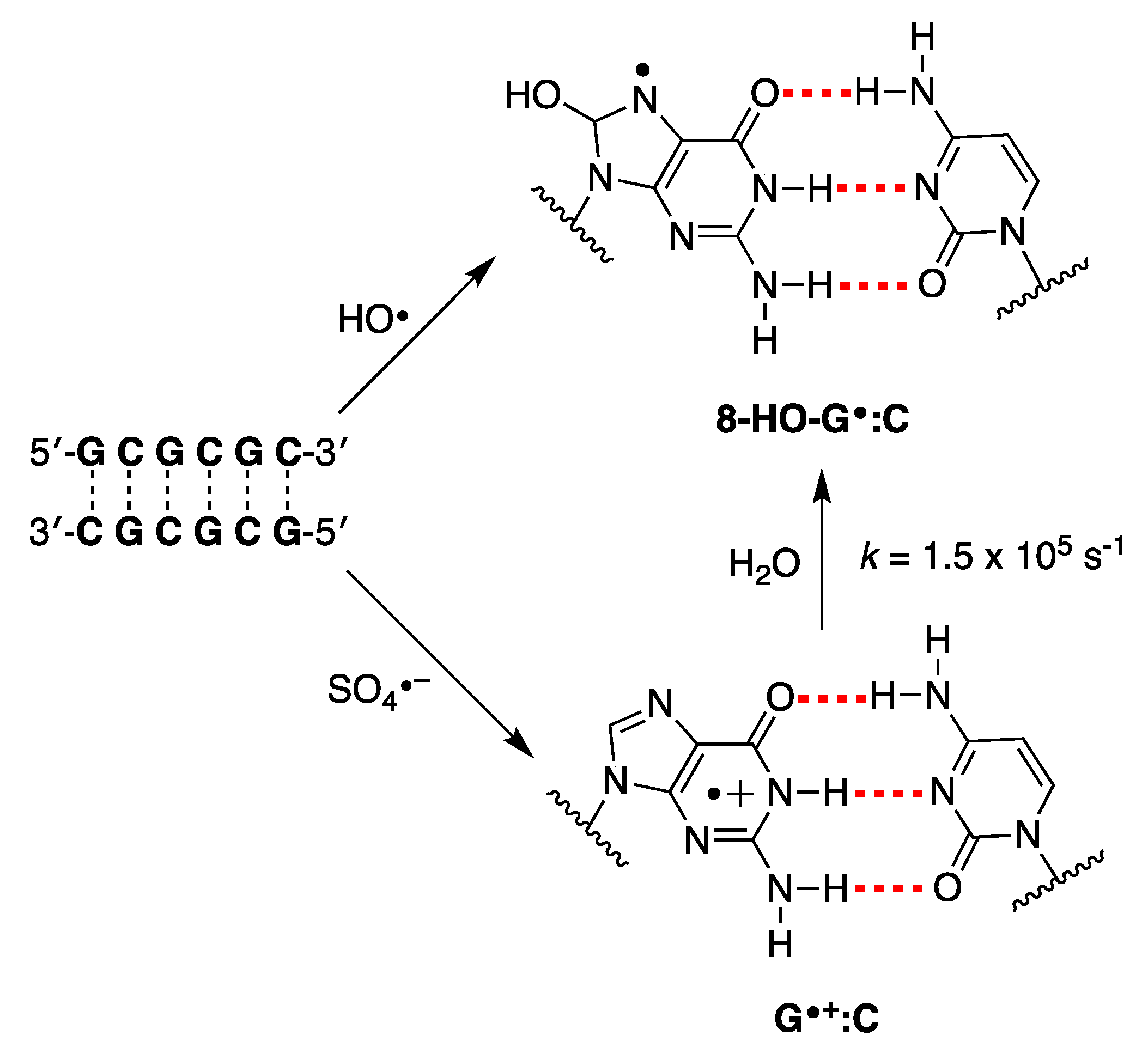
Figure 10.
Schematic flow shows the formation of carbonate radical anion (CO3•−) in vivo. The superoxide radical anion (O2•–) is the most important radical formed in aerobic organisms and the main entry to the ROS network. Under physiological conditions, superoxide dismutase (SOD) catalyzes O2•– to H2O2, and O2•– may react with nitric oxide (NO•) to give ONOO–. The Fenton reaction in the presence of HCO3– and the reaction of ONOO– with CO2 produce CO3•−.
Figure 10.
Schematic flow shows the formation of carbonate radical anion (CO3•−) in vivo. The superoxide radical anion (O2•–) is the most important radical formed in aerobic organisms and the main entry to the ROS network. Under physiological conditions, superoxide dismutase (SOD) catalyzes O2•– to H2O2, and O2•– may react with nitric oxide (NO•) to give ONOO–. The Fenton reaction in the presence of HCO3– and the reaction of ONOO– with CO2 produce CO3•−.
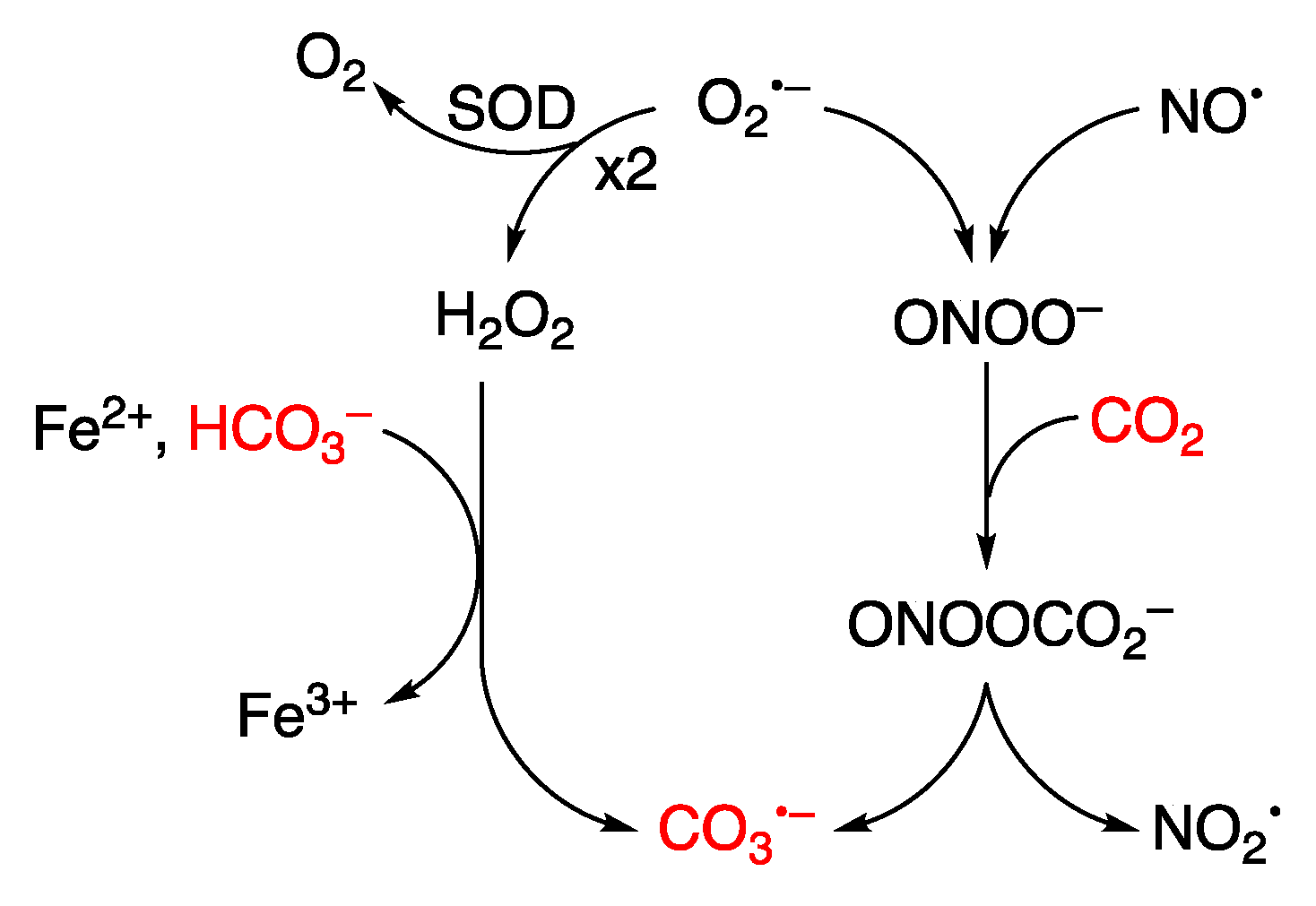
Figure 11.
Guanine lesions are detected after oxidatively induced DNA damage in vivo; these lesions are mainly repaired by the base excision repair (BER) pathway. The same lesions have also been found in vitro experiments after reaction with some reactive oxygen species (ROS) through one-electron oxidation and HO• with naked DNA or double-stranded model oligonucleotides (ds-ODNs).
Figure 11.
Guanine lesions are detected after oxidatively induced DNA damage in vivo; these lesions are mainly repaired by the base excision repair (BER) pathway. The same lesions have also been found in vitro experiments after reaction with some reactive oxygen species (ROS) through one-electron oxidation and HO• with naked DNA or double-stranded model oligonucleotides (ds-ODNs).
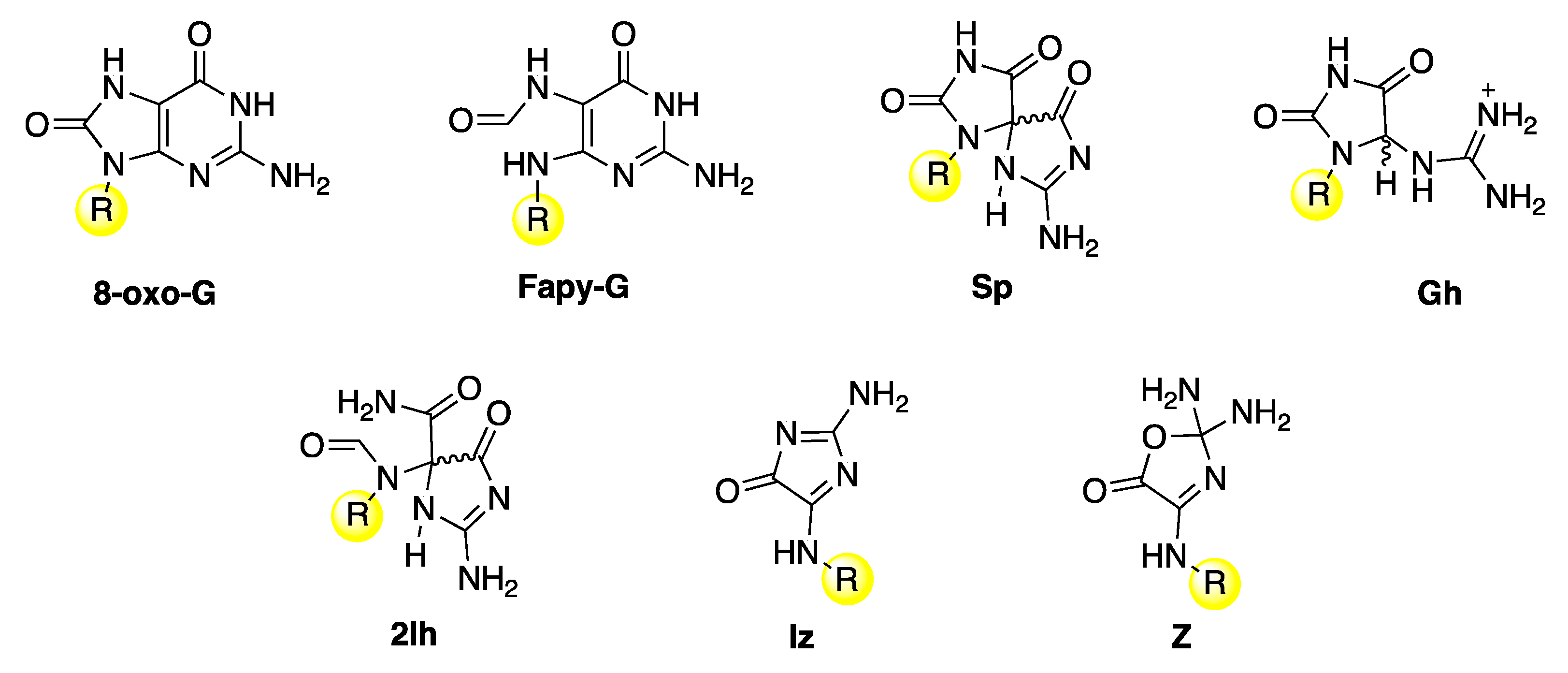
Figure 12.
The mechanism of guanine oxidation by carbonate radical anion (CO3•–) or hydroxyl radical (HO•). For the chemical structures of the end products: 8-oxo-G, Fapy-G, Sp, Gh, 2Ih, Iz and Z, see Figure 5.
Figure 12.
The mechanism of guanine oxidation by carbonate radical anion (CO3•–) or hydroxyl radical (HO•). For the chemical structures of the end products: 8-oxo-G, Fapy-G, Sp, Gh, 2Ih, Iz and Z, see Figure 5.
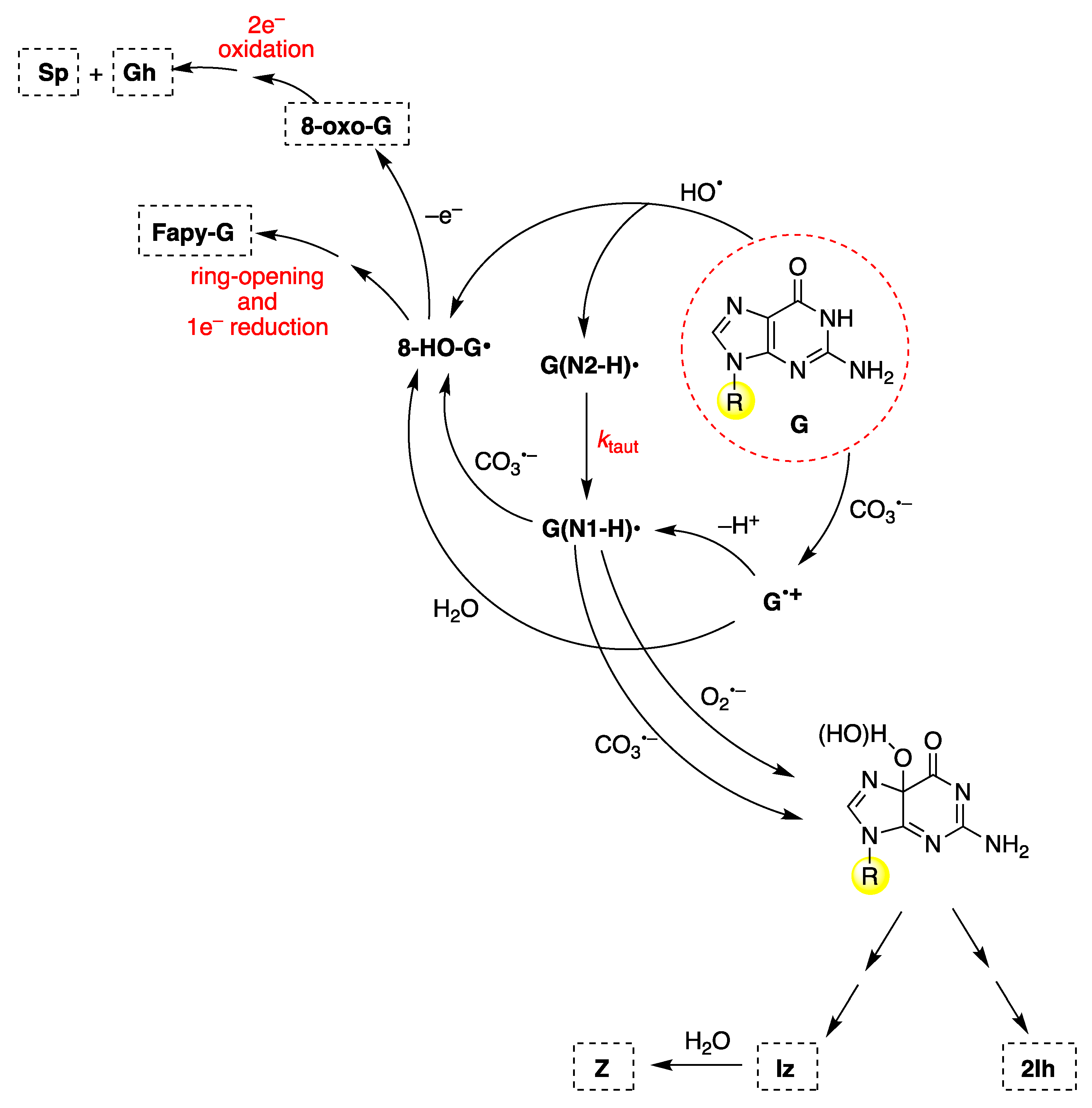
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



