Submitted:
13 November 2024
Posted:
15 November 2024
Read the latest preprint version here
Abstract
Peroxisome proliferator-activated receptors (PPARs) are a family of transcription factors that belong to the superfamily of nuclear hormone receptors expressed in various tissues, with each isoform having a distinct localization and function. In general, PPARs have an important role in fatty acid catabolism, the reduction of reactive oxygen species, and endothelial function. Specifically, PPAR functions through mechanisms such as transrepression of nuclear factor kappa B (NF-B), thereby modulating inflammatory responses and maintaining hepatic homeostasis. Together, PPAR and PPAR influence endothelial function and vascular health, which are critically affected in the context of MASLD. The complex interplay between PPARs, endothelial dysfunction, and MASLD highlights the potential of PPARs as a pharmacological target for therapeutic interventions. Recent advances in understanding the role of PPAR in modulating inflammation and endothelial function have led to the exploration of PPAR agonists in clinical trials and experimental studies. Agents such as lanifibranor, elafibranor, daidzein, and Iicarin have shown promise in improving metabolic, hepatic, and cardiovascular health in patients with MASLD. This review aimed to provide a comprehensive overview of the role of PPARs in endothelial dysfunction and MASLD, exploring their mechanisms in disease progression and potential pharmacological targeting.
Keywords:
1. The Function of the Endothelium
1.1. Endothelial Glycocalyx
1.2. Endothelial Functions
2. Metabolic Dysfunction-Associated Steatotic Liver Disease and Endothelial Dysfunction
2.1. MASLD Epidemiology and Pathogenesis
2.2. The Participation of Liver Sinusoidal Endothelial Cells in MASLD
2.3. The Relationship Between Endothelial Dysfunction and MASLD
3. Nuclear receptors: peroxisome proliferator-activated receptors
3.1. PPARs and Inflammation
3.2. PPARs and Endothelium
4. PPARs as Pharmacological Targets
5. Discussion
6. Conclusions
Author Contributions
Conflicts of Interest
References
- Clyne, A.M. Endothelial response to glucose: dysfunction, metabolism, and transport. Biochem. Soc. Trans. 2021, 49, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Wolinsky, H. A proposal linking clearance of circulating lipoproteins to tissue metabolic activity as a basis for understanding atherogenesis. Circ. Res. 1980, 47, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Gouverneur, M.; Spaan, J.A.E.; Pannekoek, H.; Fontijn, R.D.; Vink, H. Fluid shear stress stimulates incorporation of hyaluronan into endothelial cell glycocalyx. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H458–H452. [Google Scholar] [CrossRef] [PubMed]
- Lipowsky, H.H. Microvascular Rheology and Hemodynamics. Microcirculation 2005, 12, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Ugusman, A.; Kumar, J.; Aminuddin, A. Endothelial function and dysfunction: Impact of sodium-glucose cotransporter 2 inhibitors. Pharmacol. Ther. 2021, 224, 107832. [Google Scholar] [CrossRef] [PubMed]
- Saunders, S.; Jalkanen, M.; O'Farrell, S.; Bernfield, M. Molecular cloning of syndecan, an integral membrane proteoglycan. J. Cell Biol. 1989, 108, 1547–1556. [Google Scholar] [CrossRef]
- Leonova, E.I.; Galzitskaya, O.V. Structure and functions of syndecans in vertebrates. Biochemistry 2013, 78, 1071–1085. [Google Scholar] [CrossRef]
- Pahakis MY, Kosky JR, Dull RO, Tarbell JM. The role of endothelial glycocalyx components in mechanotransduction of fluid shear stress. Biochem Biophys Res Commun. 30 de marzo de 2007;355(1):228-33.
- Rosenberg RD, Shworak NW, Liu J, Schwartz JJ, Zhang L. Heparan sulfate proteoglycans of the cardiovascular system. Specific structures emerge but how is synthesis regulated? J Clin Invest. 1 de mayo de 1997;99(9):2062-70.
- Filmus J, Selleck SB. Glypicans: proteoglycans with a surprise. J Clin Invest. agosto de 2001;108(4):497-501.
- Reitsma, S.; Slaaf, D.W.; Vink, H.; van Zandvoort, M.A.M.J.; Oude Egbrink, M.G. The endothelial glycocalyx: composition, functions, and visualization. Pflug. Arch.-Eur J Physiol. 2007, 454, 345–359. [Google Scholar] [CrossRef]
- Henry, C.B.S.; Duling, B.R. Permeation of the luminal capillary glycocalyx is determined by hyaluronan. American Journal of Physiology-Heart and Circulatory Physiology 1999, 277, H508–H514. [Google Scholar] [CrossRef]
- Gaudette, S.; Hughes, D.; Boller, M. The endothelial glycocalyx: Structure and function in health and critical illness. J. Veter- Emerg. Crit. Care 2020, 30, 117–134. [Google Scholar] [CrossRef]
- Bevilacqua, M.P.; Nelson, R.M.; Mannori, G.; Cecconi, M.O. Endothelial–leukocyte adhesion molecules in human disease. Annu. Rev. Med. 1994, 45, 361–378. [Google Scholar] [CrossRef] [PubMed]
- Bergmeier, W.; Hynes, R.O. Extracellular Matrix Proteins in Hemostasis and Thrombosis. Cold Spring Harb. Perspect. Biol. 2011, 4, a005132–a005132. [Google Scholar] [CrossRef] [PubMed]
- Muller, W.A. Getting Leukocytes to the Site of Inflammation. Veter- Pathol. 2013, 50, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef] [PubMed]
- Barthel SR, Gavino JD, Descheny L, Dimitroff CJ. Targeting selectins and selectin ligands in inflammation and cancer. Expert Opinion on Therapeutic Targets. noviembre de 2007;11(11):1473-91.
- Kayal, S.; Jaïs, J.-P.; Aguini, N.; Chaudière, J.; Labrousse, J. Elevated Circulating E-Selectin, Intercellular Adhesion Molecule 1, and von Willebrand Factor in Patients with Severe Infection. Am. J. Respir. Crit. Care Med. 1998, 157, 776–784. [Google Scholar] [CrossRef]
- Radomski, M.W.; Palmer, R.M.J.; Moncada, S. Endogenous nitric oxide inhibits human platelet adhesion to vascular endothelium. Lancet 1987, 330, 1057–1058. [Google Scholar] [CrossRef]
- Ghosh, D.K.; Salerno, J.C. Nitric oxide synthases domain structure and alignment in enzyme function and control. Front. Biosci. 2003, 8, d193–209. [Google Scholar] [CrossRef]
- Neubauer, K.; Zieger, B. Endothelial cells and coagulation. Cell Tissue Res. 2021, 387, 391–398. [Google Scholar] [CrossRef]
- Badimón L, Martínez-González J. Disfunción endotelial. Revista Española de Cardiología Suplementos. enero de 2006;6(1):21A-30A.
- Lin, P.J.; Chang, C.H. Endothelium dysfunction in cardiovascular diseases. Changgeng Yi Xue Za Zhi 1994, 17. [Google Scholar]
- Widlansky, M.E.; Gokce, N.; Keaney, J.F., Jr.; Vita, J.A. The clinical implications of endothelial dysfunction. J. Am. Coll. Cardiol. 2003, 42, 1149–1160. [Google Scholar] [CrossRef]
- Wong, V.W.-S.; Ekstedt, M.; Wong, G.L.-H.; Hagström, H. Changing epidemiology, global trends and implications for outcomes of NAFLD. J. Hepatol. 2023, 79, 842–852. [Google Scholar] [CrossRef] [PubMed]
- Quek, J.; Chan, K.E.; Wong, Z.Y.; Tan, C.; Tan, B.; Lim, W.H.; Tan, D.J.H.; Tang, A.S.P.; Tay, P.; Xiao, J.; et al. Global prevalence of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in the overweight and obese population: a systematic review and meta-analysis. The Lancet Gastroenterology Hepatology 2022, 8, 20–30. [CrossRef]
- Karlsen, T.H.; Sheron, N.; Zelber-Sagi, S.; Carrieri, P.; Dusheiko, G.; Bugianesi, E.; Pryke, R.; Hutchinson, S.J.; Sangro, B.; Martin, N.K.; et al. The EASL–Lancet Liver Commission: protecting the next generation of Europeans against liver disease complications and premature mortality. Lancet 2021, 399, 61–116. [Google Scholar] [CrossRef] [PubMed]
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wong, V.W.-S.; Dufour, J.-F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. Journal of Hepatology 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Byrne, C.D.; Tilg, H. MASLD: a systemic metabolic disorder with cardiovascular and malignant complications. Gut 2024, 73, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Maldonado-Rojas, A.d.C.; Zuarth-Vázquez, J.M.; Uribe, M.; Barbero-Becerra, V.J. Insulin resistance and Metabolic dysfunction-associated steatotic liver disease (MASLD): Pathways of action of hypoglycemic agents. Ann. Hepatol. 2023, 29, 101182. [Google Scholar] [CrossRef]
- Koek, G.; Liedorp, P.; Bast, A. The role of oxidative stress in non-alcoholic steatohepatitis. Clin. Chim. Acta 2011, 412, 1297–1305. [Google Scholar] [CrossRef]
- Rinella, M.E.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; Romero, D.; Abdelmalek, M.F.; Anstee, Q.M.; Arab, J.P.; et al. A multi-society Delphi consensus statement on new fatty liver disease nomenclature. Hepatology 2023, 78, 1966–1986. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Samuel, V.T.; Shulman, G.I. Nonalcoholic Fatty Liver Disease as a Nexus of Metabolic and Hepatic Diseases. Cell Metab. 2018, 27, 22–41. [Google Scholar] [CrossRef]
- Wehmeyer, M.H.; Zyriax, B.-C.; Jagemann, B.; Roth, E.; Windler, E.; Wiesch, J.S.Z.; Lohse, A.W.; Kluwe, J. Nonalcoholic fatty liver disease is associated with excessive calorie intake rather than a distinctive dietary pattern. Medicine 2016, 95, e3887. [Google Scholar] [CrossRef] [PubMed]
- Vos, M.B.; Lavine, J.E. Dietary fructose in nonalcoholic fatty liver disease. Hepatology 2013, 57, 2525–2531. [Google Scholar] [CrossRef]
- Herman, M.A.; Samuel, V.T. The Sweet Path to Metabolic Demise: Fructose and Lipid Synthesis. Trends in Endocrinology & Metabolism 2016, 27, 719–730. [Google Scholar] [CrossRef]
- Schwärzler, J.; Grabherr, F.; Grander, C.; Adolph, T.E.; Tilg, H. The pathophysiology of MASLD: an immunometabolic perspective. Expert Rev. Clin. Immunol. 2023, 20, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Panciera, T.; Azzolin, L.; Cordenonsi, M.; Piccolo, S. Mechanobiology of YAP and TAZ in physiology and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-K.; Peng, Z.-G. Targeting Liver Sinusoidal Endothelial Cells: An Attractive Therapeutic Strategy to Control Inflammation in Nonalcoholic Fatty Liver Disease. Front. Pharmacol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Nasiri-Ansari, N.; Androutsakos, T.; Flessa, C.-M.; Kyrou, I.; Siasos, G.; Randeva, H.S.; Kassi, E.; Papavassiliou, A.G. Endothelial Cell Dysfunction and Nonalcoholic Fatty Liver Disease (NAFLD): A Concise Review. Cells 2022, 11, 2511. [Google Scholar] [CrossRef] [PubMed]
- Poisson, J.; Lemoinne, S.; Boulanger, C.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P.-E. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. Journal of Hepatology 2016, 66, 212–227. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.-S.; Cao, Z.; Lis, R.; Nolan, D.J.; Guo, P.; Simons, M.; Penfold, M.E.; Shido, K.; Rabbany, S.Y.; Rafii, S. Divergent angiocrine signals from vascular niche balance liver regeneration and fibrosis. Nature 2013, 505, 97–102. [Google Scholar] [CrossRef]
- DeLeve, L.D. Liver sinusoidal endothelial cells in hepatic fibrosis. Hepatology 2014, 61, 1740–1746. [Google Scholar] [CrossRef]
- Graupera, M.; March, S.; Engel, P.; Rodés, J.; Bosch, J.; García-Pagán, J.-C. Sinusoidal endothelial COX-1-derived prostanoids modulate the hepatic vascular tone of cirrhotic rat livers. Am. J. Physiol. Liver Physiol. 2005, 288, G763–G770. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Yadav, D.; Gupta, M.; Mishra, H.; Sharma, P. A Study of Carotid Atherosclerosis in Patients with Non-alcoholic Fatty Liver Disease. Indian J. Clin. Biochem. 2012, 28, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Schreiber R, Taschler U, Preiss-Landl K, Wongsiriroj N, Zimmermann R, Lass A. Retinyl ester hydrolases and their roles in vitamin A homeostasis. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. enero de 2012;1821(1):113-23.
- Tardelli, M.; Claudel, T.; Bruschi, F.V.; Moreno-Viedma, V.; Trauner, M. Adiponectin regulates AQP3 via PPARα in human hepatic stellate cells. Biochem. Biophys. Res. Commun. 2017, 490, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wu, Z.; Xia, M.; Salas, S.S.; Ospina, J.A.; Buist-Homan, M.; Harmsen, M.C.; Moshage, H. Extracellular vesicles derived from liver sinusoidal endothelial cells inhibit the activation of hepatic stellate cells and Kupffer cells in vitro. Biochim. et Biophys. Acta (BBA) - Mol. Basis Dis. 2024, 1870, 167020. [Google Scholar] [CrossRef]
- Gibert-Ramos, A.; Sanfeliu-Redondo, D.; Aristu-Zabalza, P.; Martínez-Alcocer, A.; Gracia-Sancho, J.; Guixé-Muntet, S.; Fernández-Iglesias, A. The Hepatic Sinusoid in Chronic Liver Disease: The Optimal Milieu for Cancer. Cancers 2021, 13, 5719. [Google Scholar] [CrossRef]
- Lin YC, Lo HM, Chen JD. Sonographic fatty liver, overweight and ischemic heart disease. World J Gastroenterol. 21 de agosto de 2005;11(31):4838-42.
- Anderson, T.J. Assessment and treatment of endothelial dysfunction in humans. J Am Coll Cardiol. 1999, 34, 631–638. [Google Scholar] [CrossRef]
- Kim J a, Montagnani M, Koh KK, Quon MJ. Reciprocal Relationships Between Insulin Resistance and Endothelial Dysfunction: Molecular and Pathophysiological Mechanisms. Circulation. 18 de abril de 2006;113(15):1888-904.
- Hadi, H.A.R.; Carr, C.S.; Al Suwaidi, J. Endothelial Dysfunction: Cardiovascular Risk Factors, Therapy, and Outcome. Vasc. Health Risk Manag. 2005, 1, 183–198. [Google Scholar]
- Targher, G.; Byrne, C.D.; Lonardo, A.; Zoppini, G.; Barbui, C. Non-alcoholic fatty liver disease and risk of incident cardiovascular disease: A meta-analysis. Journal of Hepatology 2016, 65, 589–600. [Google Scholar] [CrossRef]
- Matsuzawa Y, Kwon T, Lennon RJ, Lerman LO, Lerman A. Prognostic Value of Flow-Mediated Vasodilation in Brachial Artery and Fingertip Artery for Cardiovascular Events: A Systematic Review and Meta-Analysis. JAHA. 29 de octubre de 2015;4(11):e002270.
- Federico, A.; Dallio, M.; Masarone, M.; Persico, M.; Loguercio, C. The epidemiology of non-alcoholic fatty liver disease and its connection with cardiovascular disease: role of endothelial dysfunction. Eur Rev Med Pharmacol Sci. 2016, 20, 4731–4741. [Google Scholar]
- Qiu, Y.-Y.; Zhang, J.; Zeng, F.-Y.; Zhu, Y.Z. Roles of the peroxisome proliferator-activated receptors (PPARs) in the pathogenesis of nonalcoholic fatty liver disease (NAFLD). Pharmacol. Res. 2023, 192, 106786. [Google Scholar] [CrossRef]
- Evans RM, Mangelsdorf DJ. Nuclear Receptors, RXR, and the Big Bang. Cell. marzo de 2014;157(1):255-66.
- Tontonoz, P.; Spiegelman, B.M. Fat and Beyond: The Diverse Biology of PPARγ. Annu. Rev. Biochem. 2008, 77, 289–312. [Google Scholar] [CrossRef] [PubMed]
- Francque, S.; Szabo, G.; Abdelmalek, M.F.; Byrne, C.D.; Cusi, K.; Dufour, J.-F.; Roden, M.; Sacks, F.; Tacke, F. Nonalcoholic steatohepatitis: the role of peroxisome proliferator-activated receptors. Nat. Rev. Gastroenterol. Hepatol. 2020, 18, 24–39. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, L.; Chen, J.; Li, L.; Zheng, Z.; Ren, J.; Qiu, Y. Oleoylethanolamide, an endogenous PPAR-α ligand, attenuates liver fibrosis targeting hepatic stellate cells. Oncotarget 2015, 6, 42530–42540. [Google Scholar] [CrossRef] [PubMed]
- Wang Z, Tao Y, Qiu T, Yao X, Jiang L, Wang N, et al. Taurine protected As2O3-induced the activation of hepatic stellate cells through inhibiting PPARα-autophagy pathway. Chem Biol Interact. 25 de febrero de 2019;300:123-30.
- Chakravarthy MV, Lodhi IJ, Yin L, Malapaka RRV, Xu HE, Turk J, et al. Identification of a Physiologically Relevant Endogenous Ligand for PPARα in Liver. Cell. agosto de 2009;138(3):476-88.
- Todisco, S.; Santarsiero, A.; Convertini, P.; De Stefano, G.; Gilio, M.; Iacobazzi, V.; Infantino, V. PPAR Alpha as a Metabolic Modulator of the Liver: Role in the Pathogenesis of Nonalcoholic Steatohepatitis (NASH). Biology 2022, 11, 792. [Google Scholar] [CrossRef]
- Manickam, R.; Duszka, K.; Wahli, W. PPARs and Microbiota in Skeletal Muscle Health and Wasting. IJMS 2020, 21, 8056. [Google Scholar] [CrossRef] [PubMed]
- Naruhn, S.; Meissner, W.; Adhikary, T.; Kaddatz, K.; Klein, T.; Watzer, B.; Müller-Brüsselbach, S.; Müller, R. 15-Hydroxyeicosatetraenoic Acid Is a Preferential Peroxisome Proliferator-Activated Receptor β/δ Agonist. Mol. Pharmacol. 2009, 77, 171–184. [Google Scholar] [CrossRef]
- La Paglia, L.; Listì, A.; Caruso, S.; Amodeo, V.; Passiglia, F.; Bazan, V.; Fanale, D. Potential Role of ANGPTL4 in the Cross Talk between Metabolism and Cancer through PPAR Signaling Pathway. PPAR Res. 2017, 2017, 1–15. [Google Scholar] [CrossRef]
- Leone P, Solimando AG, Prete M, Malerba E, Susca N, Derakhshani A, et al. Unraveling the Role of Peroxisome Proliferator-Activated Receptor β/Δ (PPAR β/Δ) in Angiogenesis Associated with Multiple Myeloma. Cells. 25 de marzo de 2023;12(7):1011.
- Takada, I.; Makishima, M. Peroxisome proliferator-activated receptor agonists and antagonists: A patent review (2014-present). Expert Opin. Ther. Pat. 2020, 30, 1–13. [Google Scholar] [CrossRef]
- Berger, J.; Leibowitz, M.D.; Doebber, T.W.; Elbrecht, A.; Zhang, B.; Zhou, G.; Biswas, C.; Cullinan, C.A.; Hayes, N.S.; Li, Y.; et al. Novel Peroxisome Proliferator-activated Receptor (PPAR) γ and PPARδ Ligands Produce Distinct Biological Effects. J. Biol. Chem. 1999, 274, 6718–6725. [Google Scholar] [CrossRef]
- Chen, H.; Tan, H.; Wan, J.; Zeng, Y.; Wang, J.; Wang, H.; Lu, X. PPAR-γ signaling in nonalcoholic fatty liver disease: Pathogenesis and therapeutic targets. Pharmacol. Ther. 2023, 245, 108391. [Google Scholar] [CrossRef]
- Magliano, D.C.; Bargut, T.C.L.; de Carvalho, S.N.; Aguila, M.B.; Mandarim-De-Lacerda, C.A.; Souza-Mello, V. Peroxisome Proliferator-Activated Receptors-Alpha and Gamma Are Targets to Treat Offspring from Maternal Diet-Induced Obesity in Mice. PLOS ONE 2013, 8, e64258. [Google Scholar] [CrossRef] [PubMed]
- Patsouris D, Mandard S, Voshol PJ, Escher P, Tan NS, Havekes LM, et al. PPARα governs glycerol metabolism. J Clin Invest. 1 de julio de 2004;114(1):94-103.
- Dreyer, C.; Krey, G.; Keller, H.; Givel, F.; Helftenbein, G.; Wahli, W. Control of the peroxisomal β-oxidation pathway by a novel family of nuclear hormone receptors. Cell 1992, 68, 879–887. [CrossRef]
- Cheng, H.S.; Tan, W.R.; Low, Z.S.; Marvalim, C.; Lee, J.Y.H.; Tan, N.S. Exploration and Development of PPAR Modulators in Health and Disease: An Update of Clinical Evidence. IJMS 2019, 20, 5055. [Google Scholar] [CrossRef] [PubMed]
- Desai A, Yang Loureiro Z, DeSouza T, Yang Q, Solivan-Rivera J, Corvera S. PPARγ activation by lipolysis-generated ligands is required for cAMP dependent UCP1 induction in human thermogenic adipocytes. bioRxiv. 11 de agosto de 2024;2024.08.10.607465.
- Wang, W.; Zhou, X.; Kwong, J.S.W.; Li, L.; Li, Y.; Sun, X. Efficacy and safety of thiazolidinediones in diabetes patients with renal impairment: a systematic review and meta-analysis. Sci. Rep. 2017, 7, 1717. [Google Scholar] [CrossRef] [PubMed]
- Bougarne, N.; Weyers, B.; Desmet, S.J.; Deckers, J.; Ray, D.W.; Staels, B.; De Bosscher, K. Molecular actions of PPARα in lipid metabolism and inflammation. Endocr. Rev. 2018, 39, 760–802. [Google Scholar] [CrossRef]
- Stienstra, R.; Mandard, S.; Tan, N.S.; Wahli, W.; Trautwein, C.; Richardson, T.A.; Lichtenauer-Kaligis, E.; Kersten, S.; Müller, M. The Interleukin-1 receptor antagonist is a direct target gene of PPARα in liver. J. Hepatol. 2006, 46, 869–877. [Google Scholar] [CrossRef]
- Pawlak, M.; Baugé, E.; Bourguet, W.; De Bosscher, K.; Lalloyer, F.; Tailleux, A.; Lebherz, C.; Lefebvre, P.; Staels, B. The transrepressive activity of peroxisome proliferator-activated receptor alpha is necessary and sufficient to prevent liver fibrosis in mice. Hepatology 2014, 60, 1593–1606. [Google Scholar] [CrossRef]
- Devchand PR, Keller H, Peters JM, Vazquez M, Gonzalez FJ, Wahli W. The PPARα–leukotriene B4 pathway to inflammation control. Nature. noviembre de 1996;384(6604):39-43.
- Chen, J.; Montagner, A.; Tan, N.S.; Wahli, W. Insights into the Role of PPARβ/δ in NAFLD. IJMS 2018, 19, 1893. [Google Scholar] [CrossRef]
- Liu, S.; Brown, J.D.; Stanya, K.J.; Homan, E.; Leidl, M.; Inouye, K.; Bhargava, P.; Gangl, M.R.; Dai, L.; Hatano, B.; et al. A diurnal serum lipid integrates hepatic lipogenesis and peripheral fatty acid use. Nature 2013, 502, 550–554. [Google Scholar] [CrossRef]
- Qian B, Wang C, Li X, Ma P, Dong L, Shen B, et al. PPARβ/δ activation protects against hepatic ischaemia-reperfusion injury. Liver Int. diciembre de 2023;43(12):2808-23.
- Morán-Salvador E, López-Parra M, García-Alonso V, Titos E, Martínez-Clemente M, González-Périz A, et al. Role for PPARγ in obesity-induced hepatic steatosis as determined by hepatocyte- and macrophage-specific conditional knockouts. FASEB j. agosto de 2011;25(8):2538-50.
- Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, et al. PPARγ signaling and metabolism: the good, the bad and the future. Nat Med. mayo de 2013;19(5):557-66.
- Huang S, Jin Y, Zhang L, Zhou Y, Chen N, Wang W. PPAR gamma and PGC-1alpha activators protect against diabetic nephropathy by suppressing the inflammation and NF-kappaB activation. Nephrology (Carlton). 4 de septiembre de 2024.
- Abd-Elhamid TH, Althumairy D, Bani Ismail M, Abu Zahra H, Seleem HS, Hassanein EHM, et al. Neuroprotective effect of diosmin against chlorpyrifos-induced brain intoxication was mediated by regulating PPAR-γ and NF-κB/AP-1 signals. Food Chem Toxicol. 27 de agosto de 2024;193:114967.
- Geng, S.; Lu, R.; Zhang, Y.; Wu, Y.; Xie, L.; Caldwell, B.A.; Pradhan, K.; Yi, Z.; Hou, J.; Xu, F.; et al. Monocytes Reprogrammed by 4-PBA Potently Contribute to the Resolution of Inflammation and Atherosclerosis. Circ. Res. 2024, 135, 856–872. [Google Scholar] [CrossRef]
- Okayasu T, Tomizawa A, Suzuki K, Manaka K ichi, Hattori Y. PPARα activators upregulate eNOS activity and inhibit cytokine-induced NF-κB activation through AMP-activated protein kinase activation. Life Sciences. abril de 2008;82(15-16):884-91.
- Quintela AM, Jiménez R, Piqueras L, Gómez-Guzmán M, Haro J, Zarzuelo MJ, et al. PPAR β activation restores the high glucose-induced impairment of insulin signalling in endothelial cells. British J Pharmacology. junio de 2014;171(12):3089-102.
- Wakino, S.; Hayashi, K.; Kanda, T.; Tatematsu, S.; Homma, K.; Yoshioka, K.; Takamatsu, I.; Saruta, T. Peroxisome Proliferator-Activated Receptor γ Ligands Inhibit Rho/Rho Kinase Pathway by Inducing Protein Tyrosine Phosphatase SHP-2. Circ. Res. 2004, 95, e45–55. [Google Scholar] [CrossRef]
- Guixé-Muntet, S.; Biquard, L.; Szabo, G.; Dufour, J.; Tacke, F.; Francque, S.; Rautou, P.; Gracia-Sancho, J. Review article: vascular effects of PPARs in the context of NASH. Aliment. Pharmacol. Ther. 2022, 56, 209–223. [Google Scholar] [CrossRef]
- Xie, G.; Wang, X.; Wang, L.; Wang, L.; Atkinson, R.D.; Kanel, G.C.; Gaarde, W.A.; DeLeve, L.D. Role of Differentiation of Liver Sinusoidal Endothelial Cells in Progression and Regression of Hepatic Fibrosis in Rats. Gastroenterology 2012, 142, 918–927.e6. [Google Scholar] [CrossRef] [PubMed]
- De Silva TM, Li Y, Kinzenbaw DA, Sigmund CD, Faraci FM. Endothelial PPARγ (Peroxisome Proliferator–Activated Receptor-γ) Is Essential for Preventing Endothelial Dysfunction With Aging. Hypertension. julio de 2018;72(1):227-34.
- Gui, F.; You, Z.; Fu, S.; Wu, H.; Zhang, Y. Endothelial Dysfunction in Diabetic Retinopathy. Front. Endocrinol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Verna, L.; Chen, N.-G.; Chen, J.; Li, H.; Forman, B.M.; Stemerman, M.B. Constitutive Activation of Peroxisome Proliferator-activated Receptor-γ Suppresses Pro-inflammatory Adhesion Molecules in Human Vascular Endothelial Cells. 277, 3417. [Google Scholar] [CrossRef]
- Delerive, P.; Martin-Nizard, F.; Chinetti, G.; Trottein, F.; Fruchart, J.-C.; Najib, J.; Duriez, P.; Staels, B. Peroxisome Proliferator-Activated Receptor Activators Inhibit Thrombin-Induced Endothelin-1 Production in Human Vascular Endothelial Cells by Inhibiting the Activator Protein-1 Signaling Pathway. Circ. Res. 1999, 85, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Martin-Nizard F, Furman C, Delerive P, Kandoussi A, Fruchart JC, Staels B, et al. Peroxisome Proliferator–activated Receptor Activators Inhibit Oxidized Low-density Lipoprotein–induced Endothelin-1 Secretion in Endothelial Cells: Journal of Cardiovascular Pharmacology. diciembre de 2002;40(6):822-31.
- Berger, J.; Moller, D.E. The Mechanisms of Action of PPARs. Annu. Rev. Med. 2002, 53, 409–435. [Google Scholar] [CrossRef]
- Fougerat, A.; Montagner, A.; Loiseau, N.; Guillou, H.; Wahli, W. Peroxisome Proliferator-Activated Receptors and Their Novel Ligands as Candidates for the Treatment of Non-Alcoholic Fatty Liver Disease. Cells 2020, 9, 1638. [Google Scholar] [CrossRef] [PubMed]
- Cooreman, M.P.; Vonghia, L.; Francque, S.M. MASLD/MASH and type 2 diabetes: Two sides of the same coin? From single PPAR to pan-PPAR agonists. Diabetes Res. Clin. Pr. 2024, 212, 111688. [Google Scholar] [CrossRef]
- Francque, S.M.; Bedossa, P.; Ratziu, V.; Anstee, Q.M.; Bugianesi, E.; Sanyal, A.J.; Loomba, R.; Harrison, S.A.; Balabanska, R.; Mateva, L.; et al. A Randomized, Controlled Trial of the Pan-PPAR Agonist Lanifibranor in NASH. New Engl. J. Med. 2021, 385, 1547–1558. [Google Scholar] [CrossRef]
- Cooreman, M.P.; Butler, J.; Giugliano, R.P.; Zannad, F.; Dzen, L.; Huot-Marchand, P.; Baudin, M.; Beard, D.R.; Junien, J.-L.; Broqua, P.; et al. The pan-PPAR agonist lanifibranor improves cardiometabolic health in patients with metabolic dysfunction-associated steatohepatitis. Nat. Commun. 2024, 15, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Yao W, Wang K, Wang X, Li X, Dong J, Zhang Y, et al. Icariin ameliorates endothelial dysfunction in type 1 diabetic rats by suppressing ER stress via the PPARα/Sirt1/AMPKα pathway. Journal Cellular Physiology. marzo de 2021;236(3):1889-902.
- Das, D.; Sarkar, S.; Bordoloi, J.; Wann, S.B.; Kalita, J.; Manna, P. Daidzein, its effects on impaired glucose and lipid metabolism and vascular inflammation associated with type 2 diabetes. BioFactors 2018, 44, 407–417. [Google Scholar] [CrossRef]
- Yang, X.; Jiang, X.; Liu, C.; Yang, C.; Yao, S.; Qiu, H.; Yang, J.; Wu, K.; Liao, H.; Jiang, Q. Daidzein protects endothelial cells against high glucose-induced injury through the dual-activation of PPARα and PPARγ. Gen. Physiol. Biophys. 2024, 43, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Zhang M, Barroso E, Ruart M, Peña L, Peyman M, Aguilar-Recarte D, et al. Elafibranor upregulates the EMT-inducer S100A4 via PPARβ/δ. Biomedicine & Pharmacotherapy. noviembre de 2023;167:115623.

| Medication | Active Compound | PPAR targeted | Population | Study Design/Method | Results/observations | References |
|---|---|---|---|---|---|---|
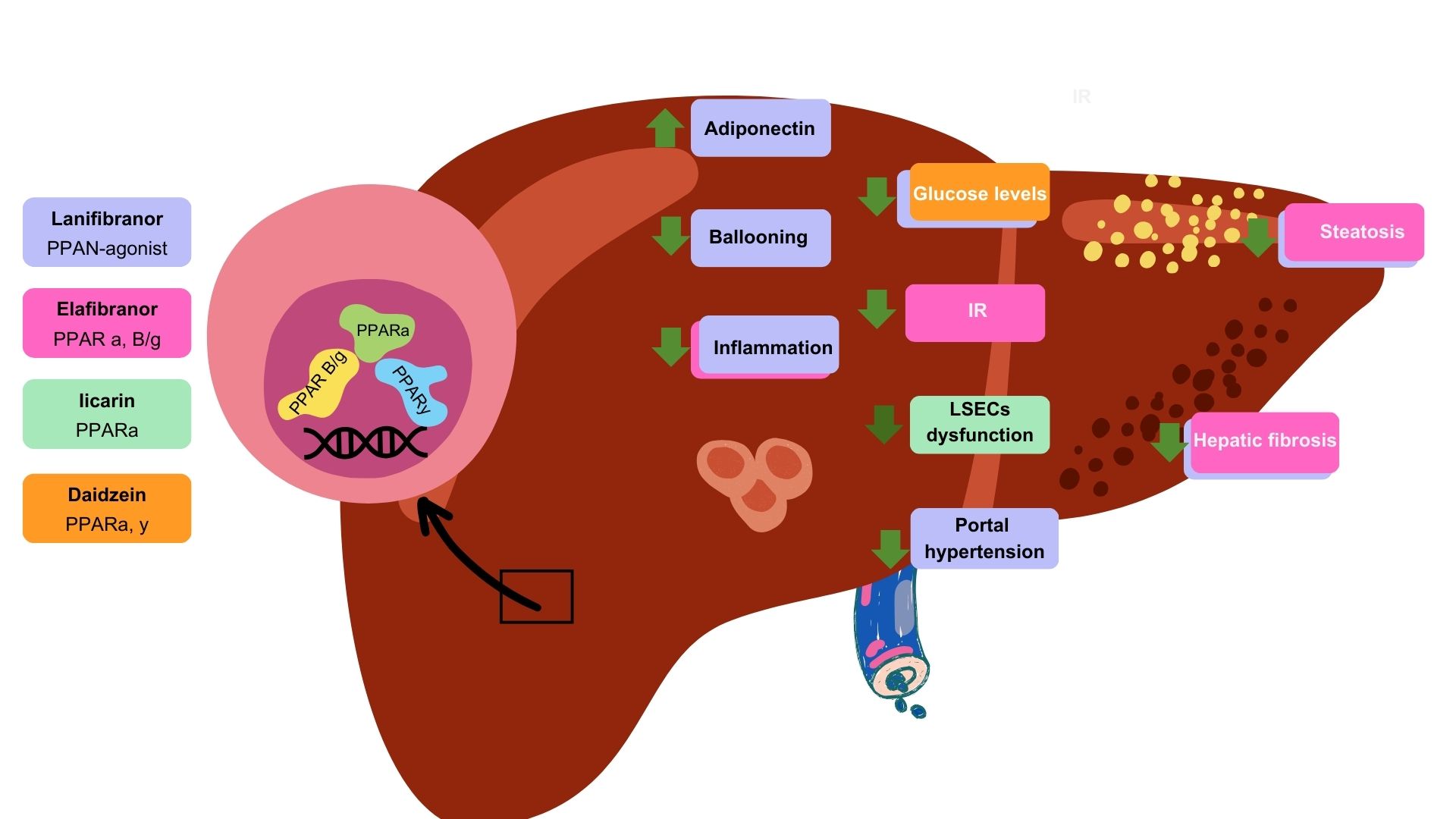
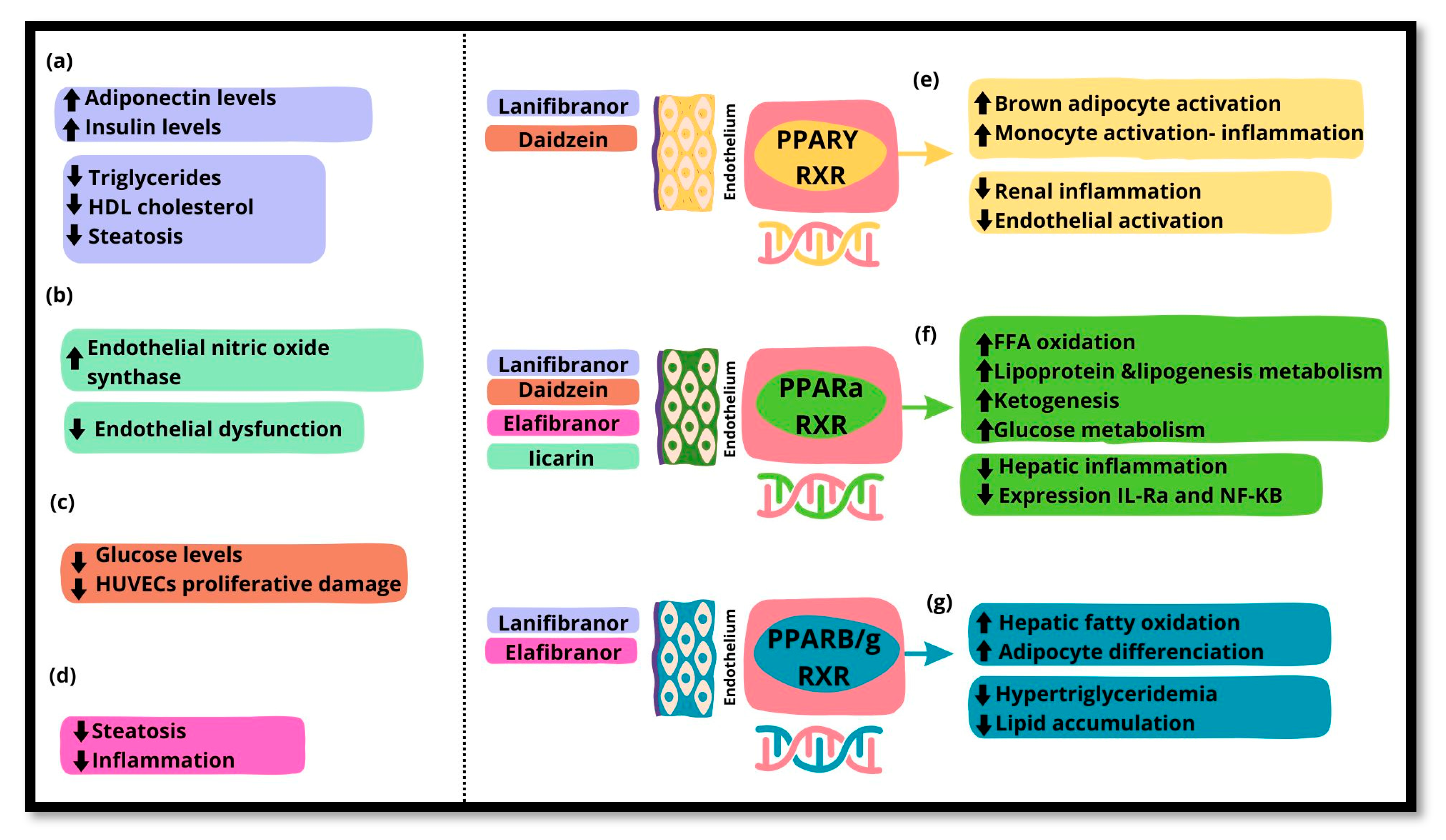
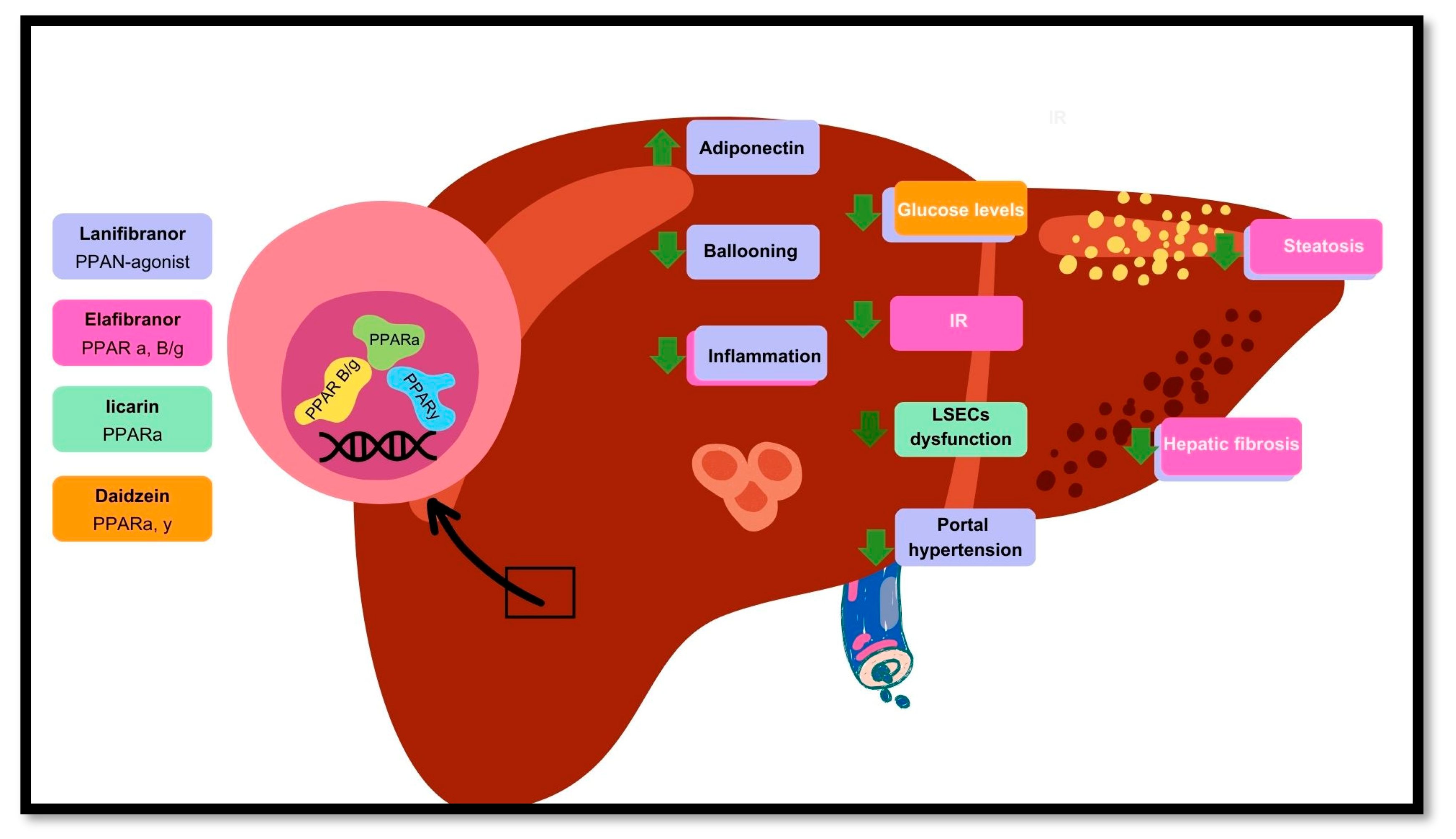
| Lanifibranor | PPAR agonist | PPARα and PPARδ, and partial activation of PPARɣ | 247 non-cirrhotic, highly active MASH patients | Doble blind randomized controlled trial | Significant improvement in triglycerides, HDL cholesterol, insulin levels and steatosis | Francque et al. 2021 |
| Lanifibranor | PPAR agonist | PPARα and PPARδ, and partial activation of PPARɣ | 247 MASH patients with a poor cardiometabolic health | Clinical trial | Increase adiponectin levels Improvement in hepatic and cardiovascular health Gain weight of 2.5 kg |
Cooreman et al. 2024 |
| Iicarin | Flavonoid glycoside | PPARα | Murine model (rat) - type 1 diabetes | Experimental study | Normalization endothelial dysfunction through inhibition of endoplasmic reticulum stress and activation of endothelial nitric oxide synthase | Yao et al. 2021 |
| Daidzein | Isoflavone | PPARα and PPARγ | HUVECS | In-vitro experimental study | Reversed high glucose levels Amelioration of HUVECs proliferative damage |
Yang et al. 2024 |
| Elafibranor | PPAR agonist | PPARα and PPARβ/δ | Murine MASH model (mice) | In vivo and in vitro experimental study | Amelioration of steatosis, inflammation Increased (EMT)-promoting proteins |
Zhang et al. 2023 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



