Submitted:
12 November 2024
Posted:
13 November 2024
You are already at the latest version
Abstract
Monoclonal antibody (mAb) and cell-based immunotherapies represent cutting-edge strategies for cancer treatment. However, safety concerns persist due to the potential targeting of normal cells that express reactive antigens. Therefore, it is crucial to develop cancer-specific mAbs (CasMabs) that can bind to cancer-specific antigens and exhibit antitumor activity in vivo, thereby reducing the risk of adverse effects. We previously screened mAbs targeting human epidermal growth factor receptor 2 (HER2) and successfully developed a cancer-specific anti-HER2 mAb, H2Mab-250/H2CasMab-2 (mouse IgG1, kappa). In this study, we assessed both the in vitro and in vivo antitumor efficacy of the humanized H2Mab-250 (humH2Mab-250). Although humH2Mab-250 showed lower reactivity to HER2-overexpressed Chinese hamster ovary-K1 (CHO/HER2) and breast cancer cell lines (BT-474 and SK-BR-3) than trastuzumab in flow cytometry, both humH2Mab-250 and trastuzumab showed similar antibody-dependent cellular cytotoxicity (ADCC) against CHO/HER2 and the breast cancer cell lines in the presence of effector splenocytes. In addition, humH2Mab-250 exhibited significant complement-dependent cellular cytotoxicity (CDC) in CHO/HER2 cells compared to trastuzumab. Furthermore, humH2Mab-250 possesses compatible in vivo antitumor effects against breast cancer xenografts with trastuzumab. These findings highlight the distinct roles of ADCC and CDC in the antitumor effects of humH2Mab-250 and trastuzumab and suggest a potential direction for the clinical development of humH2Mab-250 for HER2-positive tumors.
Keywords:
cancer-specific monoclonal antibody
; HER2
; ADCC
; CDC
; xenograft
; breast cancer
1. Introduction
Monoclonal antibody (mAb)-based therapeutics are essential for treating various diseases. The U.S. Food and Drug Administration (FDA) approved the first therapeutic mAb, Orthoclone OKT3 (mouse anti-CD3 mAb), for kidney transplantation rejection in 1986 [1]. However, the first-generation mouse mAbs tested in clinical trials had limited effectiveness due to their immunogenicity and poor effector functions [2]. Patients developed human anti-mouse antibody responses, which caused the rapid clearance of the therapeutic mAbs from the body and restricted the number of possible treatment doses [2]. The creation of engineered chimeric, humanized, and fully human mAbs has uncovered several valuable applications for antibody-based therapies [3,4].
In mAb therapy for solid tumors, the FDA approved trastuzumab for human epidermal growth factor receptor 2 (HER2)-positive breast cancer in 1998 [5]. The HER2-positive breast cancer is defined by circumferential membrane staining that is complete, intense, and in >10% of tumor cells in immunohistochemistry (IHC 3+) and/or in situ hybridization (ISH)-positive [6]. Trastuzumab is a humanized mAb by inserting the complementary determining regions (CDRs) of mouse anti-HER2 mAb (clone 4D5) into the framework of a consensus human IgG1 [7]. Trastuzumab exhibited antitumor efficacy against HER2-positive breast cancer xenograft in monotherapy or combination therapy with chemotherapy [8,9]. The clinical efficacy of trastuzumab is mediated by the immunologic engagement [10]. Trastuzumab exerts antibody-dependent cellular cytotoxicity (ADCC) upon the binding of Fcγ receptors on natural killer cells or macrophages [10]. The combination therapy of trastuzumab with chemotherapy improves the progression-free survival and overall survival in HER2-positive breast cancer patients with metastasis [11]. Currently, HER2 overexpression and activating mutations have been observed in gastric and gastroesophageal cancers [12,13], endometrial cancer [14,15], non-small-cell lung cancer [16,17], and ovarian cancers [18].
Trastuzumab-deruxtecan (T-DXd), a trastuzumab-based antibody–drug conjugate (ADC), has been developed and received the FDA approval [19]. T-DXd has demonstrated superior efficacy not only in HER2-positive breast cancers [20,21] but also in HER2-low (IHC 1+ or IHC 2+/ISH-non-positive) advanced breast cancers [22] and HER2-mutant non-small-cell lung cancer [23]. Given that approximately half of all breast cancers are classified as HER2-low, a substantial number of patients are expected to benefit from T-DXd therapy [24]. Although T-DXd is generally well-tolerated and rarely causes severe toxicity, studies have consistently linked it to the development of cardiac toxicity. While this issue is not clinically significant in most cases, baseline cardiac evaluation, regular monitoring, and early detection of cardiac adverse events are still crucial for T-DXd. Since HER2 plays a critical role in normal heart development and homeostasis [25,26], on-target, off-tumor toxicity in the heart would cause adverse effects. Therefore, management of the specificity of mAb to tumors will be required for further optimization.
Activation of the complement-dependent cytotoxicity (CDC) pathway has been suggested as a mechanism to enhance the therapeutic effectiveness of antitumor mAbs [27]. It is one of the reported mechanisms for B-cell targeting anti-CD20 mAbs, such as ofatumumab and rituximab [28,29,30,31]. Trastuzumab mediates antitumor effects through various mechanisms but is unable to induce CDC in HER2-positive cells in the presence of human serum [32,33]. The activation of the classical complement pathway is regulated by various factors, including the size and density of the antigen, which influence the geometry of the antigen-antibody complex needed for effective C1q binding [27]. For optimal CDC activity, the Fc domains of antibodies within antigen-antibody clusters must be organized in a hexameric structure, which creates a geometry that enhances C1q binding and complement activation [27]. Approaches to improve CDC, such as antibody hexamerization [34,35] and Fc mutations [36], have shown promise in boosting antitumor activity in preclinical studies. For example, hexamerization was used to develop an anti-CD37 biparatopic antibody with enhanced in vitro CDC activity [37].
We previously generated cancer-specific anti-HER2 mAbs, H2Mab-214/H2CasMab-1 [38] and H2Mab-250/H2CasMab-2 [39], selected from 278 anti-HER2 clones, using HER2 expressed by glioblastoma LN229 cells as the target antigen. Interestingly, both H2Mab-214 and H2Mab-250 showed no reactivity toward spontaneously immortalized normal epithelial cells, such as HaCaT and MCF 10A [38,39]. Moreover, H2Mab-250 exhibited no binding to normal epithelial cells derived from various tissues, including the mammary gland, kidney proximal tubule, gingiva, colon, thymus, cornea, and lung bronchus [39]. In contrast, most anti-HER2 mAbs, including trastuzumab, reacted with both cancer and normal epithelial cells [39]. Furthermore, H2Mab-250 exhibited no reactivity with the normal heart in IHC [39]. Epitope mapping identified Trp614 in HER2 extracellular domain 4 (ECD4) as a critical determinant for H2Mab-250 recognition [39]. H2Mab-214 was also found to target a similar epitope as H2Mab-250, with structural analysis suggesting that H2Mab-214 binds to a misfolded region of the β-sheet in HER2-ECD4 [32]. This suggests that localized misfolding within the cysteine-rich portion of ECD4 contributes to the cancer specificity of H2Mab-214. Additionally, we engineered mouse IgG2a and mouse-human chimeric versions of H2Mab-250. Both antibodies demonstrated antitumor activity against breast cancer xenografts in vivo, performing comparably to trastuzumab despite lower binding affinity and effector function activation in vitro [40,41].
In this study, we evaluated both the in vitro and in vivo antitumor efficacy of the humanized version of H2Mab-250 (humH2Mab-250) compared with trastuzumab.
2. Materials and Methods
2.1. Cell Lines
CHO-K1, BT-474, and SK-BR-3 cell lines were sourced from the American Type Culture Collection (Manassas, VA, USA). CHO-K1 and CHO/HER2 (HER2-overexpressed CHO-K1) [39] were maintained in RPMI-1640 medium [Nacalai Tesque, Inc. (Nacalai), Kyoto, Japan], while BT-474 and SK-BR-3 were cultured in DMEM (Nacalai). All media were supplemented with 10% heat-inactivated fetal bovine serum [FBS; Thermo Fisher Scientific Inc. (Thermo), Waltham, MA, USA] and antibiotic-antimycotic mixed solution (Nacalai).
2.2. Recombinant mAb Production
The CDRs of H2Mab-250 VH, frame sequences of VH in human Ig, and CH of human IgG1 were cloned into the pCAG-Neo vector to generate a humanized anti-human HER2 mAb (humH2Mab-250). The CDRs of H2Mab-250 VL, frame sequences of VL in human Ig, and CL of human kappa chain were cloned into the pCAG-Ble vector. We transfected the antibody expression vectors of humH2Mab-250 into BINDS-09 (fucosyltransferase 8-knockout ExpiCHO-S) cells using the ExpiCHO-S Expression System (Thermo). Trastuzumab was produced as described previously [41]. As a control human IgG1 mAb, humCvMab-62 was produced from CvMab-62 [42] using the abovementioned method. To confirm the purity of mAbs, they were treated with sodium dodecyl sulfate sample buffer containing 2-mercaptoethanol, separated on 5%–20% polyacrylamide gel, and stained by Bio-Safe CBB G-250 (Bio-Rad Laboratories, Inc., Berkeley, CA).
2.3. Animal Experiments
To assess the antitumor effects of humH2Mab-250, animal experiments were authorized by the Institutional Committee for Experiments at the Institute of Microbial Chemistry (approval no. 2024-059).
2.4. Flow Cytometry
CHO-K1, CHO/HER2, BT-474, and SK-BR-3 cells were harvested using 0.25% trypsin and 1 mM ethylenediaminetetraacetic acid (EDTA; Nacalai). The cells (1 × 105 cells per sample) were incubated with blocking buffer (control) (0.1% BSA in PBS), trastuzumab, or humH2Mab-250 for 30 minutes at 4 °C. Following this, the cells were treated with fluorescein isothiocyanate (FITC)-conjugated anti-human IgG (1:2000; Sigma-Aldrich Corp., St. Louis, MO, USA) for 30 minutes at 4 °C. Fluorescence data were collected using the SA3800 Cell Analyzer (Sony Corp., Tokyo, Japan) and analyzed with FlowJo software [BD Biosciences (BD), Franklin Lakes, NJ, USA].
2.5. ADCC
The effector splenocytes were obtained from female BALB/c nude mice (Jackson Laboratory; Kanagawa, Japan). Target cells (CHO/HER2, BT-474, and SK-BR-3) were labeled with 10 µg/mL of Calcein AM (Thermo). The target cells were plated in 96-well plates at a density of 1 × 104 cells/well and combined with effector splenocytes (effector-to-target ratio, 50:1) and 100 μg/mL of either control human IgG1, trastuzumab, or humH2Mab-250. After incubating for 4.5 hours, the calcein released into the supernatant was measured as described previously [40].
2.6. CDC
The target cells labeled with Calcein AM (CHO/HER2, BT-474, and SK-BR-3) were seeded and combined with rabbit complement (final concentration 15%, Low-Tox-M Rabbit Complement; Cedarlane Laboratories, Hornby, ON, Canada) along with 100 μg/mL of either control human IgG1, trastuzumab, or humH2Mab-250. After a 4.5-hour incubation at 37 °C, the amount of calcein released into the medium was measured.
2.7. Antitumor Activity of humH2Mab-250 in Xenografts of CHO-K1, CHO/HER2, BT-474, and SK-BR-3
BALB/c nude mice were subcutaneously inoculated with 5 × 106 cells suspended in BD Matrigel Matrix Growth Factor Reduced (BD). On day 7 post-injection, the mice were treated with 100 μg of either control human IgG1 (n = 8), trastuzumab (n = 8), or humH2Mab-250 (n = 8) via intraperitoneal injection. The treatment was repeated on days 14 and 21. Tumor size was monitored on days 7, 10, 14, 21, 23, and 27, and tumor volume was calculated as described previously [40].
2.8. Statistical Analyses
Data are presented as the mean ± standard error of the mean (SEM). Statistical analyses for ADCC, CDC, and tumor weight were performed using one-way ANOVA followed by Tukey's multiple comparisons test. Two-way ANOVA with Tukey's multiple comparisons test was applied to measure tumor volume and mouse weight. A p-value of less than 0.05 was considered statistically significant.
3. Results
3.1. Humanized anti-HER2 mAb, humH2Mab-250
We previously established an anti-HER2 mAb (H2Mab-250; mouse IgG1, kappa) by immunization with the HER2 ectodomain produced by glioblastoma LN229 cells [39]. H2Mab-250 was shown to be useful for flow cytometry [39]. In this study, we engineered a humanized H2Mab-250 (humH2Mab-250) by fusing the VH and VL CDRs of H2Mab-250 with the CH and CL chains of human IgG1, respectively (Figure 1A). We also produced trastuzumab and humCvMab-62 from CvMab-62 (mouse IgG1) [42]. In reduced conditions, we confirmed the purity of mAbs by SDS-PAGE (Figure 1B).
As shown in Figure 2, humH2Mab-250 and trastuzumab were detected in CHO/HER2 cells in a dose-dependent manner (Figure 2A) but not in parental CHO-K1 cells (Figure 2B). Furthermore, humH2Mab-250 and trastuzumab reacted with HER2-positive breast cancer BT-474 (Figure 2C) and SK-BR-3 (Figure 2D). The reactivity of humH2Mab-250 to HER2-positive cells was similar compared to that of parental mAb, H2Mab-250 [39]. We also confirm that humCvMab-62 did not react with CHO-K1, CHO/HER2, BT-474, and SK-BR-3 at 10 µg/ml (Supplementary Figure S1). We used humCvMab-62 as a control human IgG1.
3.2. ADCC and CDC by humH2Mab-250 Against HER2-Positive Cells
We next examined whether humH2Mab-250 exerted ADCC activity against CHO/HER2 cells. As shown in Figure 3A, humH2Mab-250 and trastuzumab induced ADCC in the presence of effector splenocytes against CHO/HER2 (19.9 and 23.0% cytotoxicity, respectively) more effectively than the control human IgG1 (5.9% cytotoxicity; p < 0.05). Furthermore, both humH2Mab-250 and trastuzumab induced ADCC against BT-474 (8.8 and 9.9% cytotoxicity, respectively) more effectively than the control human IgG1 (1.7% cytotoxicity; p < 0.05, Figure 3B). Both humH2Mab-250 and trastuzumab also induced ADCC against SK-BR-3 (5.5 and 5.8% cytotoxicity, respectively) more effectively than the control human IgG1 (1.4% cytotoxicity; p < 0.05, Figure 3C).
We investigated CDC by humH2Mab-250 and trastuzumab against CHO/HER2. As shown in Figure 4A, humH2Mab-250 showed a significant CDC in the presence of complements against CHO/HER2 (14.4% cytotoxicity) more effectively than the control human IgG1 (3.5% cytotoxicity; p < 0.05). In contrast, trastuzumab did not (Figure 4A). Furthermore, both humH2Mab-250 and trastuzumab induced CDC against BT-474 (9.7 and 7.7% cytotoxicity, respectively) more effectively than the control human IgG1 (1.8% cytotoxicity; p < 0.05 [trastuzumab], p < 0.01 [humH2Mab-250], Figure 4B). In contrast, both humH2Mab-250 and trastuzumab did not induce CDC against SK-BR-3 significantly (Figure 4C).
3.3. Antitumor Effects of humH2Mab-250 Against BT-474 and SK-BR-3 Xenografts
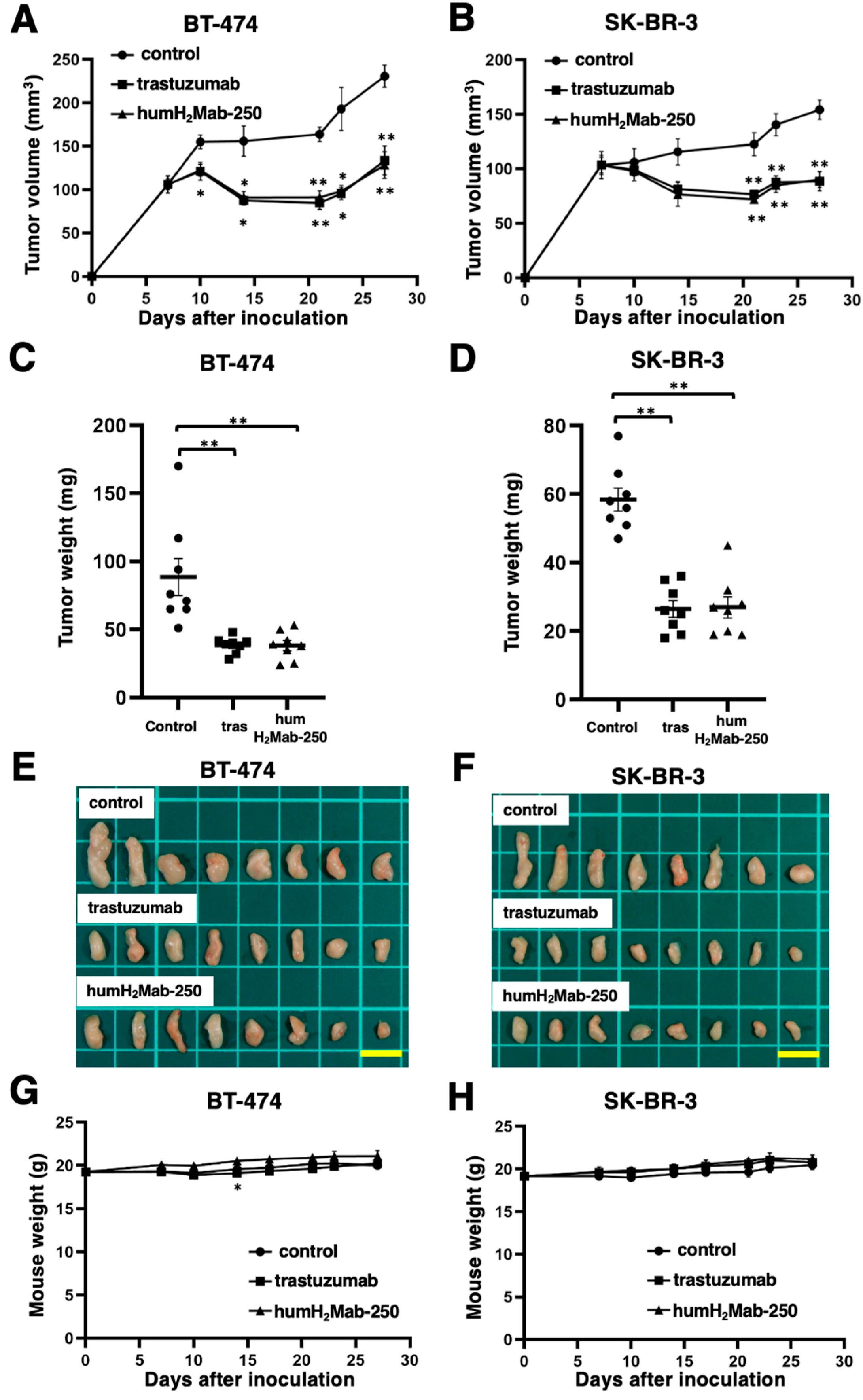
In the BT-474 and SK-BR-3 xenograft tumor-bearing mice, humH2Mab-250, trastuzumab, or control human IgG1 was intraperitoneally administered on days 7, 14, and 21. The humH2Mab-250 treatment significantly reduced the volume of BT-474 xenografts on days 10 (p < 0.05), 14 (p < 0.01), 21 (p < 0.01), 23 (p < 0.05), and 27 (p < 0.01) compared with that induced by the control human IgG1 (Figure 5A). The humH2Mab-250 treatment also caused a significant reduction in SK-BR-3 xenograft on days 21 (p < 0.01), 23 (p < 0.01), and 27 (p < 0.01) compared with that induced by the control human IgG1 (Figure 5B). Trastuzumab exhibited almost the same antitumor efficacy against BT-474 and SK-BR-3 xenografts with humH2Mab-250 (Figure 5A and 5B, respectively). The humH2Mab-250 and trastuzumab treatments resulted in similar decreases (57%) in BT-474 xenograft weight compared with that induced by the control human IgG1 on day 27 (Figure 5C). The humH2Mab-250 and trastuzumab treatments also resulted in 54% and 55% decrease in SK-BR-3 xenograft weight compared with that induced by the control human IgG1 on day 27 (Figure 5D).
Figures 5E and 5F demonstrate the BT-474 and SK-BR-3 xenografts resected on day 27. Body weight loss was rarely observed in BT-474 and SK-BR-3 xenograft-bearing mice treated with humH2Mab-250, trastuzumab, or control human IgG1 (Figure 5G and 5H). Supplementary Figure S2 presents the body appearance of BT-474 and SK-BR-3 xenograft-inoculated mice treated with humH2Mab-250, trastuzumab, or control human IgG1 on day 27.
4. Discussion
In the development of mAbs for cancer treatment, identifying and validating suitable antigenic targets is crucial [4]. To achieve a favorable therapeutic index and minimize on-target toxicity, the ideal target antigens should be highly expressed in tumors with minimal or no presence in normal tissues. However, finding such optimal targets remains a significant challenge. Technologies like bispecific antibodies, defucosylated antibodies, and ADCs have improved antibody efficacy and advanced cancer therapy. However, the issue of on-target toxicity—caused by antigen recognition in normal cells—still persists. For instance, 97 ADCs have been evaluated in clinical trials since 2000. However, 81 trials were terminated. The reason for the termination was a lack of efficacy (32 agents) and safety issues (32 agents) [43]. On-target, off-tumor toxicity is thought to be a cause of adverse effects when the target antigen is expressed in normal cells. Therefore, selecting mAbs that specifically recognize cancer-related epitopes is critical to reducing unwanted side effects.
In this study, humH2Mab-250 exhibited antitumor efficacy in mouse xenograft models (Figure 5). The humH2Mab-250 demonstrated enhanced CDC activity in the presence of complement (Figure 4). Therefore, the formation of the MAC (membrane attack complex) is thought to form efficiently on the cell surface. Various factors, such as antigen size and density, influence the activation of the classical complement pathway [44]. Moreover, the geometry of the antigen–mAb complex facilitates efficient binding of C1q, which initiates the classical complement activation pathway [27]. Since IgG antibodies can form ordered hexamers upon binding to their target antigen on cell surfaces [34,45], the structure of the humH2Mab-250-HER2 complex may allow sufficient access for complement proteins to trigger CDC. Further research is needed to understand better the mechanisms by which humH2Mab-250 induces CDC.
CasMabs targeting HER2 (clones H2Mab-214 [38] and H2Mab-250 [39]) were identified through screening for reactive with cancer and non-reactive with normal cells in flow cytometry. Both CasMabs demonstrated antitumor effects in mouse xenograft models with their recombinant mouse IgG2a or mouse-human chimeric IgG1 mAbs [38,40,41]. The recognition mechanism of H2Mab-214 was elucidated by X-ray crystallography, revealing that it binds to a locally misfolded structure in the extracellular domain IV of HER2, which typically forms a β-sheet [38]. Structural analysis of the H2Mab-250 and tumor-derived HER2 complex will be critical for further understanding the mechanism of cancer-specific recognition.
H2Mab-250 was also converted to a single chain variable fragment (scFv), developed to chimeric antigen receptor (CAR)-T cell therapy. A phase I clinical trial for patients with HER2-positive advanced solid tumors is underway in the US (NCT06241456). In CD19-positive relapsed/refractory B-cell leukemia patients who have previously been treated with CD19 CAR-T possessing mouse-derived scFv (mCD19 CAR-T), the reinfusion of mCD19 CAR-T cells may not be practical due to the development of antibodies against the anti-mouse scFv [46,47]. To address the immunogenicity, humanized CD19 CAR-T cell therapy was developed and showed a clinical benefit for the patients who had received mCD19 CAR-T therapy [48]. The scFv from humH2Mab-250 could be another option for CAR-T therapy targeting cancer-specific HER2.
Both trastuzumab and H2Mab-250 recognize the domain IV of HER2. Trastuzumab recognizes a wider epitope of HER2 (residues 579-625) [49]. In contrast, H2Mab-250 recognizes a narrow and membrane-proximal epitope of HER2 (residues 613-617) [39]. Significantly, the reactivity wholly disappeared in a HER2 (W614A) mutant [39]. Furthermore, H2Mab-250 showed a lower binding affinity (~10−9 M) than trastuzumab (~10−10 M) to HER2 ectodomain [41]. Several studies have shown that lower affinity CARs against CD19, glypican-3, and disialoganglioside (GD2) avoid excessive stimulation and exhaustion in the presence of low antigen burden, which leads to durable antitumor responses [50,51,52] Furthermore, a novel anti-CD19 mAb, h1218 that possesses a membrane-proximal epitope and exhibits faster on/off rates compared to clinically approved FMC63, was developed [53]. The h1218-CAR-T showed increased killing of B-cell malignancies compared to FMC63-CAR-T. Mechanistically, the h1218-CAR-T has reduced activation-induced cell death compared to FMC63-CAR-T owing to faster on/off rates [53]. These results support that the low affinity and membrane-proximal epitope possessing H2Mab-250 CAR-T exhibited effectiveness in a preclinical study [54]. Furthermore, the formation of the MAC at the membrane-proximal region is considered essential to attack the plasma membrane of tumor cells. Among mAbs targeting CD20, ofatumumab has been shown to possess potent CDC activity compared to rituximab [28,55], which might be due to the membrane-proximal epitope and kinetics of binding to CD20 for C1q binding[29,55]. Therefore, further studies are essential to reveal the relationship between the epitope and CDC in HER2-targeting mAbs.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org, Figure S1: Flow cytometry using humCvMab-62 against CHO/HER2, CHO-K1, BT474, and SK-BR-3; Figure S2: Body appearance in PC-10 and LN319 xenografts-implanted mice.
Author Contributions
Conceptualization, Y.K.; methodology, T.O.; formal analysis, T.T.; investigation, H.S., T.O., T.N., M.Y. and T.T.; data curation, H.S. and Y.K.; writing—original draft preparation, M.K.K., and H.S.; writing—review and editing, Y.K; project administration, Y.K.; funding acquisition, M.K.K., H.S., T.T., and Y.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported in part by the Japan Agency for Medical Research and Development (AMED) under Grant Numbers 24am0521010 (to Y.K.), JP23ama121008 (to Y.K.), JP23am0401013 (to Y.K.), JP23bm1123027 (to Y.K.), and JP23ck0106730 (to Y.K.), and by the Japan Society for the Promotion of Science (JSPS) Grants-in-Aid for Scientific Research (KAKENHI) grant nos. 22K06995 (to H.S.), 21K20789 (to T.T.), 21K07168 (to M.K.K.), and 22K07224 (to Y.K.).
Institutional Review Board Statement
The Institutional Committee for Experiments of the Institute of Microbial Chemistry approved animal experiments (approval nos. 2024-059).
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in the article and supplementary material.
Conflicts of Interest
The authors have no conflicts of interest to declare.
References
- Mullard, A. FDA approves 100th monoclonal antibody product. Nat Rev Drug Discov 2021, 20, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Pedrioli, A.; Oxenius, A. Single B cell technologies for monoclonal antibody discovery. Trends Immunol 2021, 42, 1143–1158. [Google Scholar] [CrossRef] [PubMed]
- Raja, A.; Kasana, A.; Verma, V. Next-Generation Therapeutic Antibodies for Cancer Treatment: Advancements, Applications, and Challenges. Mol Biotechnol 2024. [CrossRef]
- Paul, S.; Konig, M.F.; Pardoll, D.M.; et al. Cancer therapy with antibodies. Nat Rev Cancer 2024, 24, 399–426. [Google Scholar] [CrossRef]
- Oh, D.Y.; Bang, Y.J. HER2-targeted therapies - a role beyond breast cancer. Nat Rev Clin Oncol 2020, 17, 33–48. [Google Scholar] [CrossRef]
- Cardoso, F.; Paluch-Shimon, S.; Senkus, E.; et al. 5th ESO-ESMO international consensus guidelines for advanced breast cancer (ABC 5). Ann Oncol 2020, 31, 1623–1649. [Google Scholar] [CrossRef]
- Carter, P.; Presta, L.; Gorman, C.M.; et al. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc Natl Acad Sci U S A 1992, 89, 4285–4289. [Google Scholar] [CrossRef]
- Pietras, R.J.; Pegram, M.D.; Finn, R.S.; Maneval, D.A.; Slamon, D.J. Remission of human breast cancer xenografts on therapy with humanized monoclonal antibody to HER-2 receptor and DNA-reactive drugs. Oncogene 1998, 17, 2235–2249. [Google Scholar] [CrossRef]
- Baselga, J.; Norton, L.; Albanell, J.; Kim, Y.M.; Mendelsohn, J. Recombinant humanized anti-HER2 antibody (Herceptin) enhances the antitumor activity of paclitaxel and doxorubicin against HER2/neu overexpressing human breast cancer xenografts. Cancer Res 1998, 58, 2825–2831. [Google Scholar]
- Tsao, L.C.; Force, J.; Hartman, Z.C. Mechanisms of Therapeutic Antitumor Monoclonal Antibodies. Cancer Res 2021, 81, 4641–4651. [Google Scholar] [CrossRef]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001, 344, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Abrahao-Machado, L.F.; Scapulatempo-Neto, C. HER2 testing in gastric cancer: An update. World J Gastroenterol 2016, 22, 4619–4625. [Google Scholar] [CrossRef] [PubMed]
- Pous, A.; Notario, L.; Hierro, C.; Layos, L.; Bugés, C. HER2-Positive Gastric Cancer: The Role of Immunotherapy and Novel Therapeutic Strategies. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Balestra, A.; Larsimont, D.; Noël, J.C. HER2 Amplification in p53-Mutated Endometrial Carcinomas. Cancers (Basel) 2023, 15. [Google Scholar] [CrossRef]
- Diver, E.J.; Foster, R.; Rueda, B.R.; Growdon, W.B. The Therapeutic Challenge of Targeting HER2 in Endometrial Cancer. Oncologist 2015, 20, 1058–1068. [Google Scholar] [CrossRef]
- Garrido-Castro, A.C.; Felip, E. HER2 driven non-small cell lung cancer (NSCLC): potential therapeutic approaches. Transl Lung Cancer Res 2013, 2, 122–127. [Google Scholar] [CrossRef]
- Riudavets, M.; Sullivan, I.; Abdayem, P.; Planchard, D. Targeting HER2 in non-small-cell lung cancer (NSCLC): a glimpse of hope? An updated review on therapeutic strategies in NSCLC harbouring HER2 alterations. ESMO Open 2021, 6, 100260. [Google Scholar] [CrossRef]
- Nasioudis, D.; Gysler, S.; Latif, N.; et al. Molecular landscape of ERBB2/HER2 gene amplification among patients with gynecologic malignancies; clinical implications and future directions. Gynecol Oncol. [CrossRef]
- Mark, C.; Lee, J.S.; Cui, X.; Yuan, Y. Antibody-Drug Conjugates in Breast Cancer: Current Status and Future Directions. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Modi, S.; Saura, C.; Yamashita, T.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N Engl J Med 2020, 382, 610–621. [Google Scholar] [CrossRef]
- Shitara, K.; Bang, Y.J.; Iwasa, S.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Gastric Cancer. N Engl J Med 2020, 382, 2419–2430. [Google Scholar] [CrossRef]
- Modi, S.; Jacot, W.; Yamashita, T.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Low Advanced Breast Cancer. N Engl J Med 2022, 387, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Li, B.T.; Smit, E.F.; Goto, Y.; et al. Trastuzumab Deruxtecan in HER2-Mutant Non-Small-Cell Lung Cancer. N Engl J Med 2022, 386, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Mercogliano, M.F.; Bruni, S.; Mauro, F.L.; Schillaci, R. Emerging Targeted Therapies for HER2-Positive Breast Cancer. Cancers (Basel) 2023, 15. [Google Scholar] [CrossRef]
- Lee, K.F.; Simon, H.; Chen, H.; et al. Requirement for neuregulin receptor erbB2 in neural and cardiac development. Nature 1995, 378, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Crone, S.A.; Zhao, Y.Y.; Fan, L.; et al. ErbB2 is essential in the prevention of dilated cardiomyopathy. Nat Med 2002, 8, 459–465. [Google Scholar] [CrossRef]
- Reis, E.S.; Mastellos, D.C.; Ricklin, D.; Mantovani, A.; Lambris, J.D. Complement in cancer: untangling an intricate relationship. Nat Rev Immunol 2018, 18, 5–18. [Google Scholar] [CrossRef]
- Beurskens, F.J.; Lindorfer, M.A.; Farooqui, M.; et al. Exhaustion of cytotoxic effector systems may limit monoclonal antibody-based immunotherapy in cancer patients. J Immunol 2012, 188, 3532–3541. [Google Scholar] [CrossRef]
- Lin, T.S. Ofatumumab: a novel monoclonal anti-CD20 antibody. Pharmgenomics Pers Med. [CrossRef]
- Manches, O.; Lui, G.; Chaperot, L.; et al. In vitro mechanisms of action of rituximab on primary non-Hodgkin lymphomas. Blood 2003, 101, 949–954. [Google Scholar] [CrossRef]
- Di Gaetano, N.; Cittera, E.; Nota, R.; et al. Complement activation determines the therapeutic activity of rituximab in vivo. J Immunol 2003, 171, 1581–1587. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Y.J.; Wang, Z.; et al. CD55 and CD59 expression protects HER2-overexpressing breast cancer cells from trastuzumab-induced complement-dependent cytotoxicity. Oncol Lett 2017, 14, 2961–2969. [Google Scholar] [CrossRef]
- Mamidi, S.; Cinci, M.; Hasmann, M.; Fehring, V.; Kirschfink, M. Lipoplex mediated silencing of membrane regulators (CD46, CD55 and CD59) enhances complement-dependent anti-tumor activity of trastuzumab and pertuzumab. Mol Oncol 2013, 7, 580–594. [Google Scholar] [CrossRef] [PubMed]
- de Jong, R.N.; Beurskens, F.J.; Verploegen, S.; et al. A Novel Platform for the Potentiation of Therapeutic Antibodies Based on Antigen-Dependent Formation of IgG Hexamers at the Cell Surface. PLoS Biol 2016, 14, e1002344. [Google Scholar] [CrossRef] [PubMed]
- Diebolder, C.A.; Beurskens, F.J.; de Jong, R.N.; et al. Complement is activated by IgG hexamers assembled at the cell surface. Science 2014, 343, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Moore, G.L.; Chen, H.; Karki, S.; Lazar, G.A. Engineered Fc variant antibodies with enhanced ability to recruit complement and mediate effector functions. MAbs 2010, 2, 181–189. [Google Scholar] [CrossRef]
- Oostindie, S.C.; van der Horst, H.J.; Kil, L.P.; et al. DuoHexaBody-CD37(®), a novel biparatopic CD37 antibody with enhanced Fc-mediated hexamerization as a potential therapy for B-cell malignancies. Blood Cancer J 2020, 10, 30. [Google Scholar] [CrossRef]
- Arimori, T.; Mihara, E.; Suzuki, H.; et al. Locally misfolded HER2 expressed on cancer cells is a promising target for development of cancer-specific antibodies. Structure 2024, 32, 536–549.e535. [Google Scholar] [CrossRef]
- Kaneko, M.K.; Suzuki, H.; Kato, Y. Establishment of a Novel Cancer-Specific Anti-HER2 Monoclonal Antibody H(2)Mab-250/H(2)CasMab-2 for Breast Cancers. Monoclon Antib Immunodiagn Immunother 2024, 43, 35–43. [Google Scholar] [CrossRef]
- Suzuki, H.; Ohishi, T.; Tanaka, T.; Kaneko, M.K.; Kato, Y. Anti-HER2 Cancer-Specific mAb, H(2)Mab-250-hG(1), Possesses Higher Complement-Dependent Cytotoxicity than Trastuzumab. Int J Mol Sci 2024, 25. [Google Scholar] [CrossRef]
- Kaneko, M.K.; Suzuki, H.; Ohishi, T.; et al. A Cancer-Specific Monoclonal Antibody against HER2 Exerts Antitumor Activities in Human Breast Cancer Xenograft Models. Int J Mol Sci 2024, 25. [Google Scholar] [CrossRef]
- Inoue, T.; Yamamoto, Y.; Sato, K.; et al. Overcoming antibody-resistant SARS-CoV-2 variants with bispecific antibodies constructed using non-neutralizing antibodies. iScience 2024, 27, 109363. [Google Scholar] [CrossRef]
- Dumontet, C.; Reichert, J.M.; Senter, P.D.; Lambert, J.M.; Beck, A. Antibody-drug conjugates come of age in oncology. Nat Rev Drug Discov 2023, 22, 641–661. [Google Scholar] [CrossRef]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part I - Molecular Mechanisms of Activation and Regulation. Front Immunol 2015, 6, 262. [Google Scholar] [CrossRef] [PubMed]
- Hiemstra, I.H.; Santegoets, K.C.M.; Janmaat, M.L.; et al. Preclinical anti-tumour activity of HexaBody-CD38, a next-generation CD38 antibody with superior complement-dependent cytotoxic activity. EBioMedicine 2023, 93, 104663. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.L.; Fritsche, E.; Pulsipher, M.A.; et al. Immunogenicity of CAR T cells in cancer therapy. Nat Rev Clin Oncol 2021, 18, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Lu, W.; Chen, D.; et al. Mechanisms underlying CD19-positive ALL relapse after anti-CD19 CAR T cell therapy and associated strategies. Biomark Res 2020, 8, 18. [Google Scholar] [CrossRef]
- An, L.; Lin, Y.; Deng, B.; et al. Humanized CD19 CAR-T cells in relapsed/refractory B-ALL patients who relapsed after or failed murine CD19 CAR-T therapy. BMC Cancer 2022, 22, 393. [Google Scholar] [CrossRef]
- Diwanji, D.; Trenker, R.; Thaker, T.M.; et al. Structures of the HER2-HER3-NRG1beta complex reveal a dynamic dimer interface. Nature 2021, 600, 339–343. [Google Scholar] [CrossRef]
- Michelozzi, I.M.; Gomez-Castaneda, E.; Pohle, R.V.C.; et al. Activation priming and cytokine polyfunctionality modulate the enhanced functionality of low-affinity CD19 CAR T cells. Blood Adv 2023, 7, 1725–1738. [Google Scholar] [CrossRef]
- Caraballo Galva, L.D.; Jiang, X.; Hussein, M.S.; et al. Novel low-avidity glypican-3 specific CARTs resist exhaustion and mediate durable antitumor effects against HCC. Hepatology 2022, 76, 330–344. [Google Scholar] [CrossRef]
- Hoseini, S.S.; Dobrenkov, K.; Pankov, D.; Xu, X.L.; Cheung, N.K. Bispecific antibody does not induce T-cell death mediated by chimeric antigen receptor against disialoganglioside GD2. Oncoimmunology 2017, 6, e1320625. [Google Scholar] [CrossRef]
- Zhang, Y.; Patel, R.P.; Kim, K.H.; et al. Safety and efficacy of a novel anti-CD19 chimeric antigen receptor T cell product targeting a membrane-proximal domain of CD19 with fast on- and off-rates against non-Hodgkin lymphoma: a first-in-human study. Mol Cancer 2023, 22, 200. [Google Scholar] [CrossRef] [PubMed]
- Hosking, M.; Shirinbak, S.; Omilusik, K.; et al. 268 Development of FT825/ONO-8250: an off-the-shelf CAR-T cell with preferential HER2 targeting and engineered to enable multi-antigen targeting, improve trafficking, and overcome immunosuppression. Journal for ImmunoTherapy of Cancer 2023, 11 (Suppl 1), A307–A307. [Google Scholar] [CrossRef]
- Pawluczkowycz, A.W.; Beurskens, F.J.; Beum, P.V.; et al. Binding of submaximal C1q promotes complement-dependent cytotoxicity (CDC) of B cells opsonized with anti-CD20 mAbs ofatumumab (OFA) or rituximab (RTX): considerably higher levels of CDC are induced by OFA than by RTX. J Immunol 2009, 183, 749–758. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Generation of a humanized IgG1 mAb, humH2Mab-250. (A) The CDRs of H2Mab-250 VH and VL were cloned into human IgG1 and human kappa chain, respectively. The humH2Mab-250 was produced by BINDS-09 (fucosyltransferase 8-knockout ExpiCHO-S) cells, as described in materials and methods. (B) Confirmation of the purified mAbs. MAbs (2 µg) were treated with sodium dodecyl sulfate sample buffer containing 2-mercaptoethanol. Proteins were separated on 5%–20% polyacrylamide gel and stained by Bio-Safe CBB G-250.
Figure 1.
Generation of a humanized IgG1 mAb, humH2Mab-250. (A) The CDRs of H2Mab-250 VH and VL were cloned into human IgG1 and human kappa chain, respectively. The humH2Mab-250 was produced by BINDS-09 (fucosyltransferase 8-knockout ExpiCHO-S) cells, as described in materials and methods. (B) Confirmation of the purified mAbs. MAbs (2 µg) were treated with sodium dodecyl sulfate sample buffer containing 2-mercaptoethanol. Proteins were separated on 5%–20% polyacrylamide gel and stained by Bio-Safe CBB G-250.
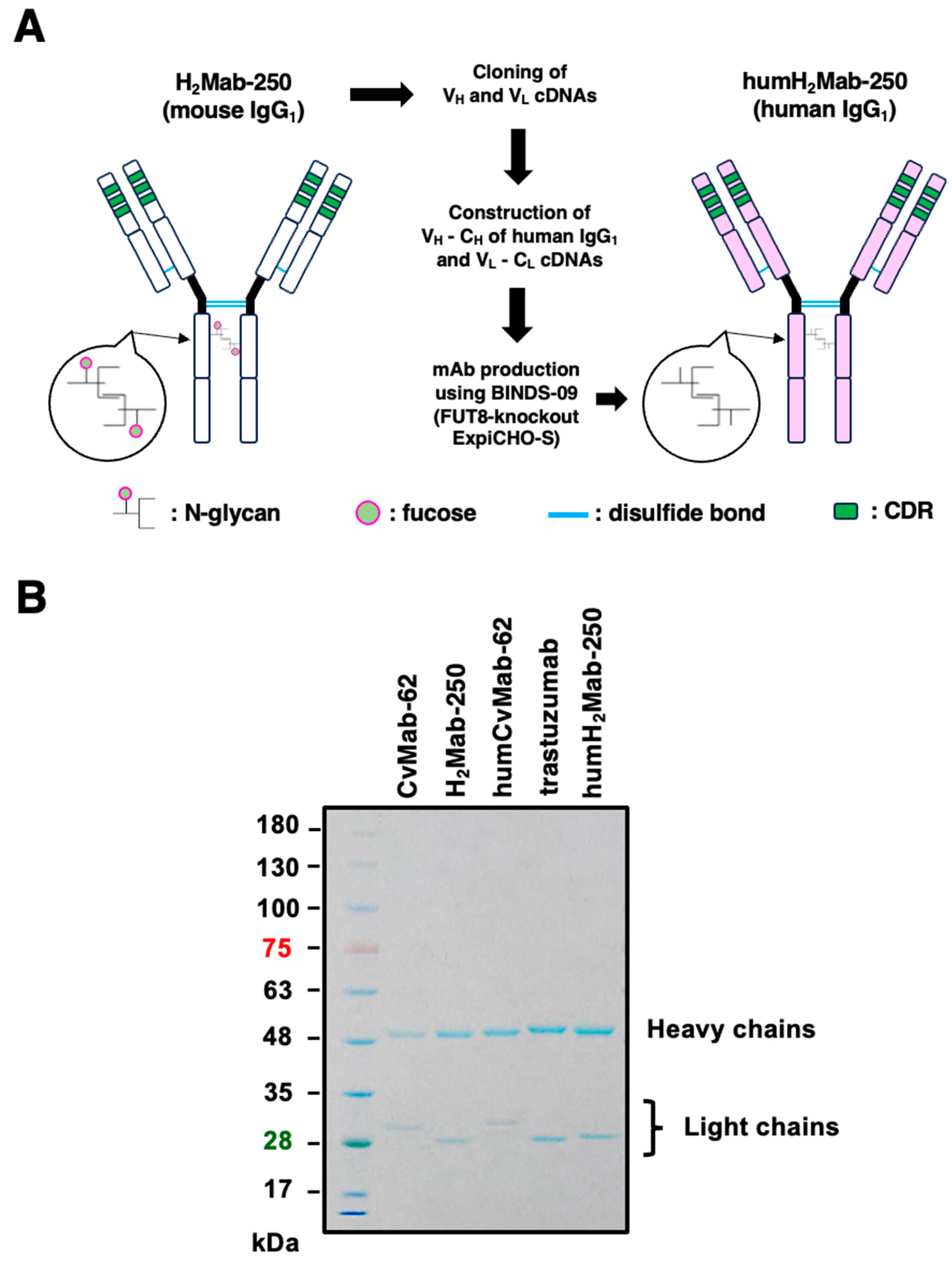
Figure 2.
Flow cytometry using humH2Mab-250 and trastuzumab. CHO/HER2(A), CHO-K1 (B), BT-474 (C), and SK-BR-3 (D) cells were treated with humH2Mab-250 (10 to 1.25 µg/mL), trastuzumab (10 to 1.25 µg/mL), or buffer control, followed by anti-human IgG conjugated with FITC. The SA3800 Cell Analyzer was used to analyze fluorescence data.
Figure 2.
Flow cytometry using humH2Mab-250 and trastuzumab. CHO/HER2(A), CHO-K1 (B), BT-474 (C), and SK-BR-3 (D) cells were treated with humH2Mab-250 (10 to 1.25 µg/mL), trastuzumab (10 to 1.25 µg/mL), or buffer control, followed by anti-human IgG conjugated with FITC. The SA3800 Cell Analyzer was used to analyze fluorescence data.
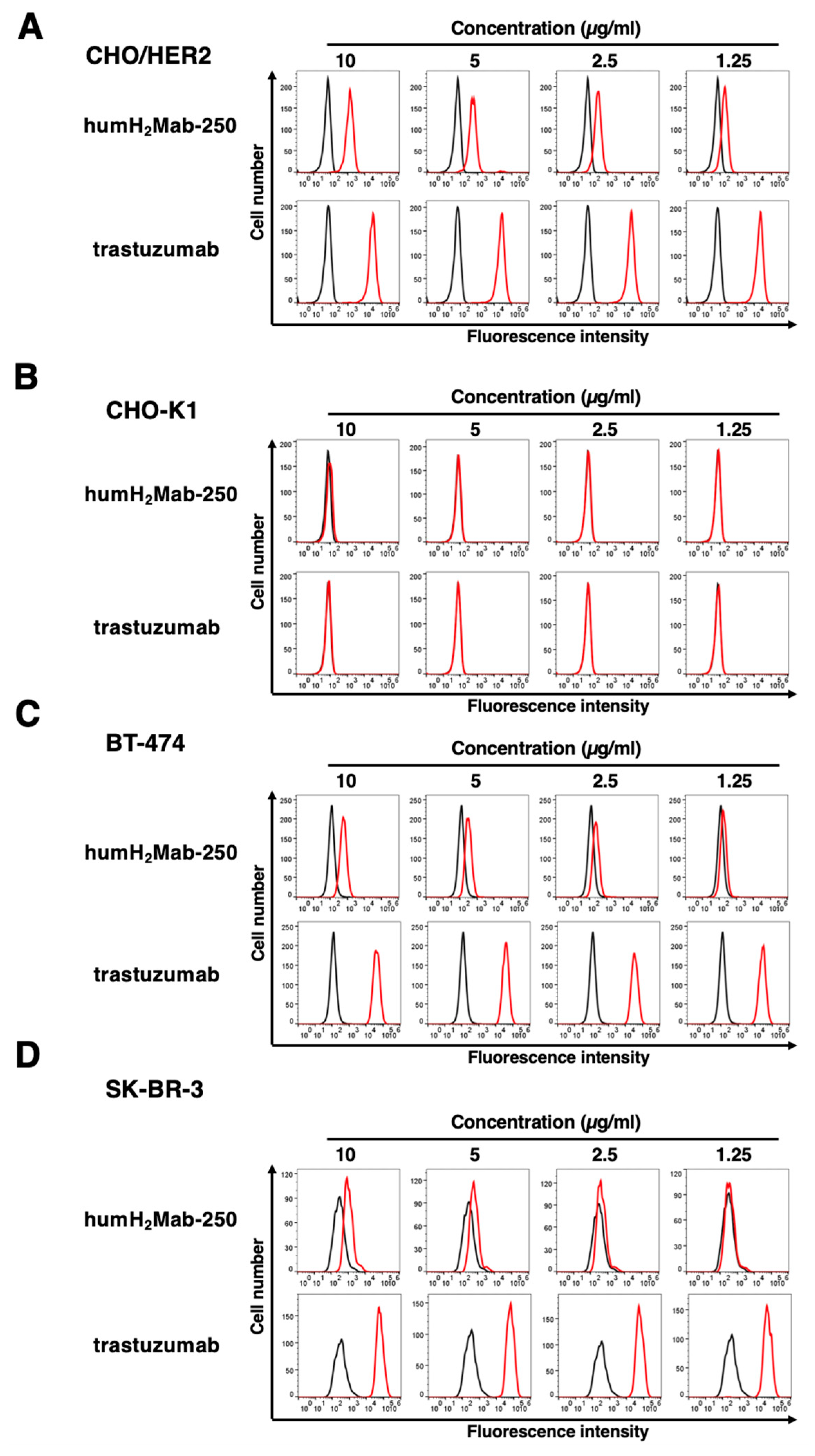
Figure 3.
The ADCC is mediated by humH2Mab-250 and trastuzumab. Calcein-labeled CHO/HER2 (A), BT-474 (B), and SK-BR-3 (C) were treated with trastuzumab (tras), humH2Mab-250 or control human IgG1 in the presence of effector splenocytes. The cytotoxicity was determined by the release of calcein into the medium. Values are shown as the mean ± SEM. Asterisks indicate statistical significance (* p < 0.05; one-way ANOVA Tukey’s multiple comparisons test).
Figure 3.
The ADCC is mediated by humH2Mab-250 and trastuzumab. Calcein-labeled CHO/HER2 (A), BT-474 (B), and SK-BR-3 (C) were treated with trastuzumab (tras), humH2Mab-250 or control human IgG1 in the presence of effector splenocytes. The cytotoxicity was determined by the release of calcein into the medium. Values are shown as the mean ± SEM. Asterisks indicate statistical significance (* p < 0.05; one-way ANOVA Tukey’s multiple comparisons test).
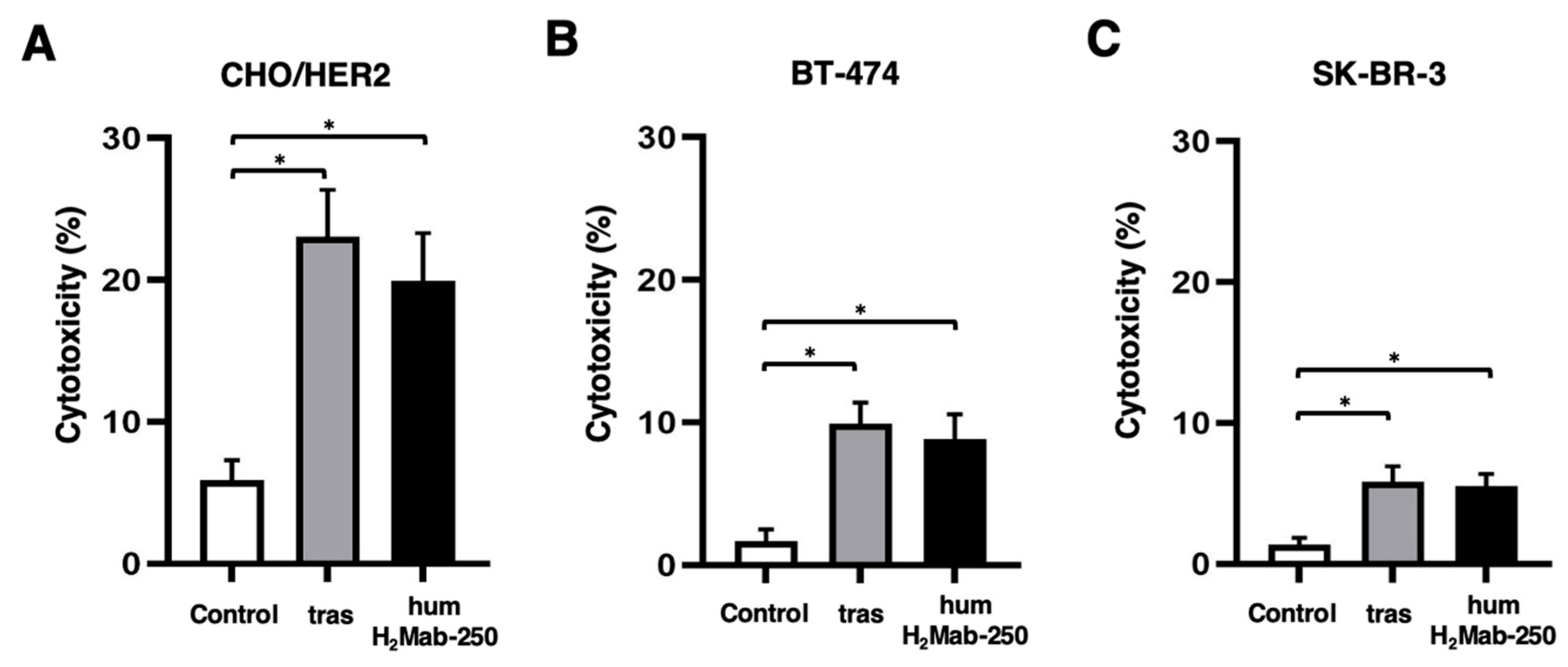
Figure 4.
The CDC is mediated by humH2Mab-250 and trastuzumab. Calcein-labeled CHO/HER2 (A), BT-474 (B), and SK-BR-3 (C) were treated with trastuzumab (tras), humH2Mab-250 or control human IgG1 in the presence of complements. The cytotoxicity was determined by the release of calcein into the medium. Values are shown as the mean ± SEM. Asterisks indicate statistical significance (* p < 0.05 and ** p < 0.01; one-way ANOVA Tukey’s multiple comparisons test).
Figure 4.
The CDC is mediated by humH2Mab-250 and trastuzumab. Calcein-labeled CHO/HER2 (A), BT-474 (B), and SK-BR-3 (C) were treated with trastuzumab (tras), humH2Mab-250 or control human IgG1 in the presence of complements. The cytotoxicity was determined by the release of calcein into the medium. Values are shown as the mean ± SEM. Asterisks indicate statistical significance (* p < 0.05 and ** p < 0.01; one-way ANOVA Tukey’s multiple comparisons test).
Figure 5.
Antitumor activity of humH2Mab-250 against BT-474 and SK-BR-3 xenografts. (A,B) BT-474 (A) and SK-BR-3 (B) cells were subcutaneously injected into BALB/c nude mice (day 0). On day 7, 100 μg of humH2Mab-250, trastuzumab, or control human IgG1 was administered. Additional antibodies were administered on days 14 and 21. The tumor volume was measured on days 7, 10, 14, 21, 23, and 27. Values are presented as the mean ± SEM. * p < 0.05 and ** p < 0.01; Two-way ANOVA Tukey’s multiple comparisons test. (C,D) BT-474 (C) and SK-BR-3 (D) xenograft tumor weight on day 27. Values are represented as the mean ± SEM. ** p < 0.01; one-way ANOVA Tukey’s multiple comparisons test). (E,F) The BT-474 (E) and SK-BR-3 (F) xenograft tumors on day 27 (scale bar, 1 cm). (G,H) Body weight of BT-474 (G) and SK-BR-3 (H) xenograft-bearing mice treated with humH2Mab-250, trastuzumab, or control human IgG1. Values are presented as the mean ± SEM. * p < 0.05 (Two-way ANOVA with Tukey’s multiple comparisons test).
Figure 5.
Antitumor activity of humH2Mab-250 against BT-474 and SK-BR-3 xenografts. (A,B) BT-474 (A) and SK-BR-3 (B) cells were subcutaneously injected into BALB/c nude mice (day 0). On day 7, 100 μg of humH2Mab-250, trastuzumab, or control human IgG1 was administered. Additional antibodies were administered on days 14 and 21. The tumor volume was measured on days 7, 10, 14, 21, 23, and 27. Values are presented as the mean ± SEM. * p < 0.05 and ** p < 0.01; Two-way ANOVA Tukey’s multiple comparisons test. (C,D) BT-474 (C) and SK-BR-3 (D) xenograft tumor weight on day 27. Values are represented as the mean ± SEM. ** p < 0.01; one-way ANOVA Tukey’s multiple comparisons test). (E,F) The BT-474 (E) and SK-BR-3 (F) xenograft tumors on day 27 (scale bar, 1 cm). (G,H) Body weight of BT-474 (G) and SK-BR-3 (H) xenograft-bearing mice treated with humH2Mab-250, trastuzumab, or control human IgG1. Values are presented as the mean ± SEM. * p < 0.05 (Two-way ANOVA with Tukey’s multiple comparisons test).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



