Submitted:
01 November 2024
Posted:
05 November 2024
You are already at the latest version
Abstract
Leishmaniasis is a serious infectious disease caused by Leishmania parasites, predominantly affecting tropical and subtropical regions. These parasites replicate within macrophages, manipulating the host immune response and facilitating infection progression. This study examined the expression profiles of long non-coding RNAs (lncRNAs), potential cis-target genes, and hub genes at different time points in human macrophages infected with Leishmania major. RNA-Seq analysis identified 39,828 lncRNAs, with 2,903 showing differential expression at one or more time points. As the infection progressed (4, 24, 48, and 72 hours), the number of up- and down-regulated lncRNAs gradually decreased. Six lncRNAs (lnc-UNC5D-8, lnc-TENM3-1, DIRC3-1, lnc-MTRNR2L12-10, lnc-FAM43A-6, and AKAP2-1) were consistently differentially expressed across all time points, potentially playing key roles in regulating the host immune response. Time-specific hub genes were also identified, regulating critical processes such as keratinization, epigenetic modifications, and immune responses. In particular, these genes were pivotal during the later stages of infection in maintaining tissue integrity and regulating immune responses. Early immune responses were dominated by immunoglobulin receptor activity and adaptive immune system activation. These findings highlight the critical roles of lncRNAs and hub genes in macrophage responses to Leishmania infection, offering potential targets for future therapeutic strategies.
Keywords:
long noncoding RNAs (lncRNAs)
; Leishmania major
; gene expression
; macrophages
; cis-target genes
1. Introduction
Leishmaniasis is an infectious disease caused by Leishmania parasites that predominantly affects tropical and subtropical regions and has significant effects on human health [1]. These parasites multiply in host macrophage cells and cause the disease to progress. Macrophages are an important component of the host immune system and regulate various immune responses when they encounter pathogens to fight infection [2]. However, Leishmania parasites are capable of manipulating the defense mechanisms of macrophages, facilitating their own replication and contributing to the advancement of the infection.
During Leishmania infection, macrophages undergo significant changes in various biological processes. Both host and parasite experience alterations in gene expression throughout the course of the infection [3]. Identifying the epigenetic changes induced by the parasite over time in the host can provide valuable insights into the progression of the disease. In particular, elucidating the mechanisms involved in regulating gene expression within macrophages can enhance our understanding of the pathogenesis of the infection and contribute to the development of novel therapeutic strategies [4].
In recent years, non-coding RNAs (ncRNAs) have emerged as promising tools in both the diagnosis and treatment of infectious and non-infectious diseases, making them a focal point of diagnostic research [5]. In eukaryotic cells, ncRNAs can be classified into three groups based on their nucleotide sequence length, function, and structure: small RNAs (<50 nucleotides), RNA polymerase III transcripts (e.g., 5S rRNA, tRNA), and long non-coding RNAs (lncRNAs; >200 nucleotides) [6]. LncRNAs are distinguished from other non-coding RNAs by their characteristics, including small open reading frames, low GC content, tissue-specific expression, and poorly conserved sequences across species [7]. Based on their genomic location, lncRNAs can be categorized into four groups: sense, antisense, intronic, and intergenic [8].
In mammals, thousands of lncRNAs have been shown to play regulatory roles in various biological functions, such as cell development [9], chromatin modification [10], and immune regulation [11]. LncRNAs can interact with RNA, DNA, proteins, or microRNAs (miRNAs) to regulate processes including transcription, splicing mechanisms, nucleic acid degradation, and translation. Additionally, lncRNAs are involved in regulating gene expression in developmental processes, cell proliferation, inflammation, and host-pathogen interactions [12].
Although several transcriptomic studies have examined the relationship between mRNAs and lncRNAs in macrophages infected by Leishmania species [13], these studies have not identified the target genes of lncRNAs involved during a specific time course of L. major infection. In this study, we aimed to identify the target genes of lncRNAs displaying differential expression patterns during continuous assessment of the control, 4hpi, 24hpi, 48hpi, and 72hpi stages of L. major infection, as well as the miRNAs interacting with these lncRNAs.
2. Materials and Methods
2.1. RNA-Seq Data Retrieval from Macrophages Infected with Leishmania Major
To investigate and compare the lncRNA expressions in L. major (Friedlin strain) infected human macrophages previously generated public RNA-Seq data were downloaded from the SRA-NCBI database under BioProject number PRJNA290995 [14]. The libraries were sequenced using the Illumina HiSeq 1500 platform. The datasets covered different time periods control groups and after infection of macrophages. In time-course experiments, a total of 32 data sets were collected from infected and uninfected human macrophages at four different time points at 4h, 24h, 48h, and 72h post-infection (Supplementary Table S1).
2.2. Bioinformatic Identification of lncRNAs and Gene Expression Analysis
RNA-Seq files for both control and treated groups were downloaded from the Sequence Read Archive (SRA) database at NCBI (https://www.ncbi.nlm.nih.gov/). The raw reads adapters and low-quality reads were filtered out by fastqc v 0.11.9 [15]. After, the transcripts were mapped to human genome assembly GRCh38.p13 provided by GENCODE using the HISAT2 software v2.2.1 [16]. Whether the determined lncRNAs consisted of annotated lncRNAs or not, the structures of lncRNAs and mRNAs were compared according to transcript length, number of exons and ORFs. The coding potential of the filtered transcripts were predicted using lncRNA prediction tool CPC. The crossover data were coupled with known lncRNAs for further research. The abundances of transcripts were calculated as transcripts per million (TPM) units [17]. Differentially expressed lncRNAs (DElncRNAs) across different time points were identified using edgeR. During the p value calculation, the model was created with the generalized linear model in the edgeR package [18]. While creating this model, first a matrix was created on the basis of transcripts and groups, this matrix was also normalized and the model that calculates its distribution in groups was calculated. Then the fold change between time periods was compared and p value calculation was made for each transcript [19]. DElncRNAs were considered significant with a p-value < 0.05 and log2 fold change (log2FC) ≤ -1.5 and ≥ 1.5.
2.3. The Cis-Target Gene Identification and Functional Enrichment Analysis
For each differentially expressed lncRNA, 300 kb upstream or downstream of the given lncRNA were scanned to find “cis-regulated mRNAs. Using the g:Profiler (https://biit.cs.ut.ee/gprofiler/gost, accessed on 7 August 2024), differentially expressed lncRNAs cis-target genes were submitted to Gene Ontology (GO) and Pathway analysis to assess their roles in biological processes, molecular function, and cellular component terms. The parameter was filtered at a adjusted p-value of <0.05.
2.4. PPI Network Construction and Hub Gene Identification
3. Results
We re-analyzed RNA-Seq data generated by Fernandes et al. to investigate the differentially expressed long non-coding RNAs (DElncRNAs) in human macrophages at 4-, 24-, 48-, and 72-hours post-infection with L. major. After processing control and time-point datasets, clean reads were aligned to the GRCh38.p13 genome assembly.
3.1. Differentially Expressed lncRNAs Between Time Point Comparisons in Human Macrophages
In total, we detected 39,828 lncRNAs and classified them based on genomic location, with 77.54% identified as intergenic (Supplementary Table S1). Among these, 2,903 DElncRNAs were expressed in at least one of seven comparisons (4hpi vs control; 24hpi vs control; 48hpi vs control; 72hpi vs control; 4hpi vs 24hpi; 24hpi vs 48hpi; 48hpi vs 72hpi). Specifically, 202, 342, 281, and 495 DElncRNAs were detected at 4-, 24-, 48-, and 72-hours post-infection, respectively, compared to the control (Figure 1, Supplementary Table S2). Additionally, 935, 251, and 397 DElncRNAs were identified in the time-point comparisons of 4hpi vs 24hpi, 24hpi vs 48hpi, and 48hpi vs 72hpi, respectively.
There was no consistent pattern in the number of up- or down-regulated DElncRNAs over time when comparing each time point with its control. However, within time-point comparisons, the number of DElncRNAs steadily decreased from 4 hours to 72 hours post-infection. Overlaps in DElncRNAs between comparisons are shown in Figure 2a and Figure 2b. Notably, six lncRNAs (lnc-UNC5D-8, lnc-TENM3-1, DIRC3-1, lnc-MTRNR2L12-10, lnc-FAM43A-6, and AKAP2-1) were differentially expressed at all time points, with all but AKAP2-1 being consistently down-regulated.
Additionally, lnc-CMPK2-2 and lnc-TMEM121-26 showed expression changes across all time points. Table 1 illustrates the fold changes for both lncRNAs at different time intervals.
3.2. Cis-Target Genes of DElncRNAs and Functional Enrichment Analysis
Cis-target genes of the identified DElncRNAs were located within 300 kb upstream or downstream of each lncRNA’s transcription start and stop sites. A total of 58,446 cis-acting target genes were identified for the 2,903 DElncRNAs (Supplementary Table S3).
Most lncRNAs’ functions have not been fully examined till now, and analysing cis-target genes of them using GO functional analysis and pathway analysis may help to identify lncRNA roles in the response upon L. major infection in human macrophages. Cis-target genes were examined for KEGG pathway and GO enrichment in molecular function, biological processes and cellular components. Our aim in this study was to provide clues regarding the epigenetic regulation of human macrophages following L. major infection across time points. As expected, we identified similarities as well as differences in the molecular response of the cell to the pathogen over time points. GO terms significantly enriched for cis-target genes of DElncRNAs at each time-point comparison are presented in Supplementary Table S4.
3.3. Temporal Dynamics of lncRNA Expression and Immune Response
In our study, we examined the expression profiles of long non-coding RNAs (lncRNAs) in human macrophages infected with L. major at different time points of infection (4, 24, 48 and 72 hours) and identified potential cis-target genes of these lncRNAs. Comparisons at different time points revealed significant changes in biological processes (BP), cellular components (CC) and molecular functions (MF). These changes helped us understand how lncRNAs play a dynamic role in infected macrophages over time.
Firstly, a rapid activation of the immune response was observed in the early stages of infection compared to the control group at 4 hours of infection. During this period, processes associated with the activation of the adaptive immune system, such as the biological process immunoglobulin-mediated immune response and B cell-mediated immunity, were found to be prominent. In terms of cellular components, the plasma membrane and immunoglobulin complexes were active. In terms of molecular functions, immune response triggering functions such as antigen binding and immunoglobulin receptor binding were found to be important. Enrichment analysis also highlighted pathways such as Estrogen signaling (KEGG:04915) and Immunoregulatory interactions (REAC:R-HSA-198933). This early response may be regulated by 74 transcription factors (TFs) and 3 microRNAs (hsa-miR-9500, hsa-miR-6813-5p, hsa-miR-6085) (Figure 3, Supplementary Table S4).
Between 4- and 24-hours post-infection, lncRNAs played increasingly complex roles in immune regulation. Molecular functions related to chemokine receptors and proinflammatory cytokine release were activated, while biological processes involving inflammatory responses became prominent. Several immune-related pathways, including those associated with Leishmaniasis (KEGG:05140, REAC:R-HSA-9664433, REAC:R-HSA-9662851, REAC:R-HSA-9664417, REAC:R-HSA-9658195) and immune signaling pathways, became statistically significant for the first time during this interval (Figure 4, Supplementary Table S4).
From 24 to 48 hours, significant changes in immune response were observed. Biological processes such as adaptive immune response and post-transcriptional gene regulation were activated, and cellular components including the immunoglobulin complex and keratin filaments were identified. Pathways related to B cell receptor signaling (KEGG:04662) and Leishmania-related phagocytosis were significantly enriched (Figure 5, Supplementary Table S4).
Finally, between 48 and 72 hours, a more complex immune response emerged, with biological processes related to tissue remodeling, cell differentiation, and inflammation resolution being prominent. Cellular components such as the extracellular matrix and cell surface receptors were active, and molecular functions including chemokine receptor activity and signal transduction were highlighted (Figure 6, Supplementary Table S4).
3.4. Hub Gene Identification
Using the STRING tool, cis-target gene-based PPI network was created. Hub genes were identified for each time point using CytoHubba. In all comparisons except 4hpi vs 24hpi, 10 hub genes were identified. In all comparisons except 4hpi vs 24hpi, 10 hub genes were identified. In the 0 to 4-hour interval, hub genes were associated with cytoskeleton organization and immune response. Between 4 and 24 hours, 8 of the identified hub genes were core components of the nucleosome, with KDM6B identified as a lysine-specific demethylase involved in transcriptional regulation. Interestingly, all hub genes identified between 24 and 48 hours were related to keratinization, while those between 48 and 72 hours were associated with histone modifications and immune response (Figure 7).
4. Discussion
In this study, the temporal expression profiles of differentially expressed lncRNAs (DElncRNAs) were examined in human macrophages infected with L. major, and the potential cis-target genes of these DElncRNAs were identified. The RNA-Seq analysis revealed the regulatory profiles of lncRNAs showing differential expression at 4-, 24-, 48-, and 72-hours post-infection, along with their associated target genes. Our findings demonstrate significant variations in lncRNA expression in macrophages during infection, suggesting that these lncRNAs influence various biological processes and pathways over the course of the infection, playing key roles in regulating the host immune response against the pathogen.
Differentially expressed lncRNAs were identified at 4-, 24-, 48-, and 72-hours post-infection, showing significant changes in the number of DElncRNAs compared to control groups. Additionally, comparisons between different time points revealed a gradual decrease in both upregulated and downregulated lncRNAs as the infection progressed. This finding suggests that the macrophage response to infection, mediated by lncRNAs, undergoes dynamic changes over time.
Six lncRNAs (lnc-UNC5D-8, lnc-TENM3-1, DIRC3-1, lnc-MTRNR2L12-10, lnc-FAM43A-6, and AKAP2-1) were consistently expressed across all time points, which is particularly noteworthy. All lncRNAs, except AKAP2-1, were downregulated. This suggests that the suppression of certain lncRNAs may play a crucial role in sustaining the macrophage response to L. major infection. Additionally, common DElncRNAs were identified across the different time points, indicating their potential involvement in regulating macrophage functions at various stages of infection. Understanding the role of these lncRNAs could be important for deciphering the pathophysiology of the infection.
In our study, the cis-target genes located within 300 kb upstream and downstream of the differentially expressed lncRNAs were also identified. This analysis suggests that lncRNAs may control the expression of neighboring protein-coding genes through cis-regulatory mechanisms. A functional enrichment analysis of the cis-target genes was performed to predict the biological processes and pathways in which these lncRNAs might be epigenetically involved at different time points during the infection.
The rapid activation of the immune response at 4 hours post-infection, compared to the control group, suggests an immediate defense mechanism triggered in response to the pathogen. In the early stages of infection, macrophages recognize pathogens using pattern recognition receptors (PRRs) and bind antigens, a process accelerated by immunoglobulin receptors. The swift engagement of immunoglobulin-mediated responses aims to limit the spread of the parasite and reduce the severity of infection [22,23,24]. Our data highlight that antigen binding and immunoglobulin-mediated mechanisms were quickly activated in the first hours of infection, strongly regulated by infected macrophages. The enrichment of processes such as immunoglobulin receptor binding and the prominent involvement of B cell-mediated immunity further underscores the pivotal role of the adaptive immune system during this early phase. These observations align with findings that early immune responses, particularly those involving antibody-mediated pathways, play a critical role in containing parasitic infections [25].
The enrichment analysis also revealed pathways like estrogen signaling and immunoregulatory interactions, suggesting that these pathways might contribute to modulating the host immune response. Interestingly, estrogens, a class of sex hormones, have been linked to immune regulation [26]. The early immune response may also be shaped by transcriptional regulation involving 74 transcription factors and 3 microRNAs (hsa-miR-9500, hsa-miR-6813-5p, hsa-miR-6085), highlighting the complex regulatory networks that control the host’s defense mechanisms at the molecular level.
Between 4- and 24-hours post-infection, the activation of processes related to chemokine receptors and proinflammatory cytokine release indicates an enhanced inflammatory response by infected macrophages aimed at restricting the parasite. This finding aligns with reports in the literature that chemokines and proinflammatory cytokines regulate the infection response, supporting phagocytosis and limiting pathogen spread [27,28]. These molecules not only play roles in recruiting immune cells to the site of infection but also contribute to the activation and maturation of macrophages, reinforcing their capacity to eliminate pathogens. Moreover, during this period, lymphocyte and leukocyte-mediated processes, crucial components of the cellular basis of adaptive immunity, were observed to be active. This suggests that the immune response was transitioning from an innate to a more adaptive phase, where lymphocytes play a critical role in pathogen clearance. The involvement of structures such as the immunoglobulin complex and plasma membrane, identified as key cellular components, further supports their known roles in pathogen elimination. The significance of immune-related pathways associated with Leishmaniasis during this period highlights the critical transition in immune signaling as the infection progresses. These pathways are likely involved in coordinating the host’s defense mechanisms, facilitating the recognition and targeting of Leishmania parasites, and guiding the immune response towards resolution of infection.
Between 24- and 48-hours post-infection, significant changes in immune responses were observed, particularly involving the activation of immunoglobulin-mediated immune responses and miRNA-mediated post-transcriptional gene silencing. This indicates an ongoing regulation of immune responses and modulation of transcriptional processes. Studies have shown that during later stages of Leishmania infection in macrophages, gene expression undergoes substantial changes, with proinflammatory responses playing dual roles in controlling the parasite and aiding its survival [29,30,31,32]. In the study conducted by Fernandes and colleagues, it was demonstrated that the transcriptome profile of human macrophages infected with Leishmania was reprogrammed post-infection, leading to significant changes in cellular responses [33]. Additionally, the identification of cellular components like the immunoglobulin complex and keratin filaments points to their crucial role in immune response regulation. Immunoglobulin complexes, essential for antigen recognition and immune signaling, are likely involved in enhancing phagocytic activity against the parasite. The presence of keratin filaments, typically linked to structural support, could suggest a role in maintaining cellular integrity during the immune response. The enrichment of pathways such as B cell receptor signaling (KEGG:04662) and Leishmania-related phagocytosis emphasizes the involvement of adaptive immune mechanisms in these later stages of infection. B cell receptor signaling plays a critical role in antibody-mediated immunity, facilitating the recognition and clearance of pathogens.
Between 48- and 72-hours post-infection, processes related to the adaptive immune response, immunoglobulin-mediated immune response, and humoral immune response became prominent. This suggests that macrophages may mount a more robust and targeted immune response against the parasite at this stage. While the early phases of infection were dominated by immune activation and inflammatory processes, later stages show the predominance of humoral responses and adaptive immune mechanisms. During this period, cellular components such as the IgG immunoglobulin complex and extracellular region were active, and molecular functions like immunoglobulin receptor binding and antigen binding were highlighted.
In the 0 to 4-hour interval, the hub genes were primarily associated with cytoskeleton organization and immune response. This suggests that early host responses are geared towards maintaining cellular integrity and potentially to limit the spread of the parasite.
Between 4- and 24-hours post-infection, 8 of the identified hub genes were core components of the nucleosome, indicating a significant shift towards chromatin remodeling and gene regulation. The identification of KDM6B, a lysine-specific demethylase involved in transcriptional regulation, is particularly noteworthy as it points to the modulation of epigenetic marks during infection. In earlier study, they demonstrated that the functional knockdown of KDM6B in an experimental model of visceral leishmaniasis led to a substantial reduction in the parasite burden within infected organs [34].
In the 24 to 48-hour interval, the hub genes were all related to keratinization. This process is closely linked to the function of keratinocytes, which play a pivotal role in the immune response and wound healing in cutaneous leishmaniasis [35]. Furthermore, keratinocyte apoptosis and necrosis have been implicated in the formation of ulcers, a hallmark of cutaneous leishmaniasis pathology. Beyond their involvement in ulceration, keratinocytes also may contribute to the healing of Leishmania-related cutaneous wounds by modulating the inflammatory environment at the infection site. Keratinocytes have been shown to either initiate or suppress proinflammatory responses, creating a microenvironment that is uniquely tailored to each Leishmania species [36]. The ability to modulate this inflammatory process may significantly influence the course of the disease. Thus, the activation of keratinization-related genes during the 24 to 48-hour interval suggests that keratinocytes play an important role not only in the structural defense of the infected area but also in regulating the immune response and potentially influencing disease progression through species-specific interactions.
Finally, between 48 and 72 hours, hub genes were predominantly associated with histone modifications and immune response. This further highlights the role of epigenetic regulation in shaping the immune response in the later stages of infection. Histone modifications may facilitate gene expression, allowing for a sustained immune response as the infection progresses. These dynamic changes in hub gene activity reflect the complex interplay between cellular processes and immune defenses throughout the course of infection, emphasizing the importance of both structural and regulatory mechanisms in the host’s response to Leishmania.
5. Conclusions
In conclusion, this study provides insights into the time-dependent expression profiles of lncRNAs during L. major infection and their relationships with potential cis-target genes, enhancing our understanding of how immune responses are regulated at different stages of infection in infected macrophages. The identification of hub genes related to cytoskeleton organization and immune response in the early stages suggests that macrophages mount a rapid and defensive response to the infection. In later stages, processes such as keratinization, histone modifications, and epigenetic regulators play a more significant role. These findings offer valuable insights for future therapeutic approaches and represent a critical step toward understanding the immune-regulatory roles of lncRNAs in infectious diseases.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1: Overview of RNA-seq Libraries and Associated Metadata for L. major-Infected Macrophages at Different Time Points; Table S2: Basic Characteristics of Genes and Transcripts, and Differentially Expressed lncRNAs Across Various Time Points During L. major Infection; Table S3: Cis-Target Genes of Differentially Expressed lncRNAs Located Within 300 kb Upstream or Downstream of Transcription Start and Stop Sites; Table S4: Enrichment Analysis of Biological Processes, Cellular Components, and Molecular Functions in L. major-Infected Macrophages at Different Time Points.
Author Contributions
Conceptualization, S.S. and T.G.T.; methodology, S.S. and T.G.T.; software, T.G.T.; validation, S.S. and T.G.T.; formal analysis, S.S.; investigation, T.G.T.; resources, S.S.; data curation, T.G.T.; writing—original draft preparation, T.G.T.; writing—review and editing, S.S.; visualization, T.G.T.; supervision, T.G.T.; project administration, S.S.; funding acquisition, S.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Çankırı Karatekin University Scientific Research Projects Coordination Unit, grant number EYO080120B25.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.
Acknowledgments
Thanks to the Çankırı Karatekin University Scientific Research Projects Coordination Unit for supporting this work. Thanks to the anonymous reviewers, academic editors and editors for their comments and suggestions.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Kaye, P.; Scott, P. Leishmaniasis: Complexity at the Host-Pathogen Interface. Nat. Rev. Microbiol. 2011, 9, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Sacks, D.; Sher, A. Evasion of Innate Immunity by Parasitic Protozoa. Nat. Immunol. 2002, 3, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- Ovalle-Bracho, C.; Franco-Muñoz, C.; Londoño-Barbosa, D.; Restrepo-Montoya, D.; Clavijo-Ramírez, C. Changes in Macrophage Gene Expression Associated with Leishmania (Viannia) braziliensis Infection. PLoS ONE 2015, 10, e0128934. [Google Scholar] [CrossRef]
- Liu, D.; Uzonna, J.E. The Early Interaction of Leishmania with Macrophages and Dendritic Cells and Its Influence on the Host Immune Response. Front. Cell. Infect. Microbiol. 2012, 2, 83. [Google Scholar] [CrossRef]
- Yang, J.X.; Rastetter, R.H.; Wilhelm, D. Non-Coding RNAs: An Introduction. Adv. Exp. Med. Biol. 2016, 886, 13–32. [Google Scholar] [PubMed]
- Deng, W.; Zhu, X.; Skogerbø, G.; Zhao, Y.; Fu, Z.; Wang, Y.; He, H.; Cai, L.; Sun, H.; Liu, C.; et al. Organization of the Caenorhabditis elegans Small Non-Coding Transcriptome: Genomic Features, Biogenesis, and Expression. Genome Res. 2006, 16, 20–29. [Google Scholar] [CrossRef]
- Petrella, V.; Aceto, S.; Musacchia, F.; Colonna, V.; Robinson, M.; Benes, V.; Cicotti, G.; Bongiorno, G.; Gradoni, L.; Volf, P.; et al. De Novo Assembly and Sex-Specific Transcriptome Profiling in the Sand Fly Phlebotomus perniciosus (Diptera, Phlebotominae), a Major Old World Vector of Leishmania infantum. BMC Genomics 2015, 16, 847. [Google Scholar] [CrossRef]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and Functions of Long Noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef]
- Peng, L.; Paulson, A.; Li, H.; Piekos, S.; He, X.; Li, L.; Zhong, X.B. Developmental Programming of Long Non-Coding RNAs during Postnatal Liver Maturation in Mice. PLoS One 2014, 9, e114917. [Google Scholar] [CrossRef]
- Angrand, P.O.; Vennin, C.; Le Bourhis, X.; Adriaenssens, E. The Role of Long Non-Coding RNAs in Genome Formatting and Expression. Front. Genet. 2015, 6, 165. [Google Scholar] [CrossRef]
- Imamura, K.; Akimitsu, N. Long Non-Coding RNAs Involved in Immune Responses. Front. Immunol. 2014, 5, 573. [Google Scholar] [CrossRef] [PubMed]
- Cao, J. The Functional Role of Long Non-Coding RNAs and Epigenetics. Biol. Proced. 2014, 16, 11. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, J.C.R.; Gonçalves, A.N.A.; Floeter-Winter, L.M.; Nakaya, H.I.; Muxel, S.M. Comparative Transcriptomic Analysis of Long Noncoding RNAs in Leishmania-Infected Human Macrophages. Front. Genet. 2023, 13, 1051568. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, M.C.; Dillon, L.A.; Belew, A.T.; Bravo, H.C.; Mosser, D.M.; El-Sayed, N.M. Dual Transcriptome Profiling of Leishmania-Infected Human Macrophages Reveals Distinct Reprogramming Signatures. mBio 2016, 7, e00027–16. [Google Scholar] [CrossRef]
- Andrews, S. A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 November 2023).
- Kim, J.S.; Lee, K.T.; Bahn, Y.S. Deciphering the Regulatory Mechanisms of the cAMP/Protein Kinase A Pathway and Their Roles in the Pathogenicity of Candida auris. Microbiol. Spectr. 2023, 11, e0215223. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-Optimal Probabilistic RNA-Seq Quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Lund, S.P.; Nettleton, D.; McCarthy, D.J.; Smyth, G.K. Detecting Differential Expression in RNA-Sequence Data Using Quasi-Likelihood with Shrunken Dispersion Estimates. Stat. Appl. Genet. Mol. Biol. 2012, 11, 1544–6115.1826. [Google Scholar]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-Protein Interaction Networks, Integrated over the Tree of Life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma Receptors as Regulators of Immune Responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Murray, H.W.; Berman, J.D.; Davies, C.R.; Saravia, N.G. Advances in Leishmaniasis. Lancet 2005, 366, 1561–1577. [Google Scholar] [CrossRef] [PubMed]
- Wen, T.H.; Tsai, K.W.; Wu, Y.J.; Liao, M.T.; Lu, K.C.; Hu, W.C. The Framework for Human Host Immune Responses to Four Types of Parasitic Infections and Relevant Key JAK/STAT Signaling. Int. J. Mol. Sci. 2021, 22, 13310. [Google Scholar] [CrossRef]
- Harding, A.T.; Heaton, N.S. The Impact of Estrogens and Their Receptors on Immunity and Inflammation during Infection. Cancers 2022, 14, 909. [Google Scholar] [CrossRef]
- Oghumu, S.; Lezama-Dávila, C.M.; Isaac-Márquez, A.P.; Satoskar, A.R. Role of Chemokines in Regulation of Immunity against Leishmaniasis. Exp. Parasitol. 2010, 126, 389–396. [Google Scholar] [CrossRef]
- de Araújo, F.F.; Costa-Silva, M.F.; Pereira, A.A.S.; Rêgo, F.D.; Pereira, V.H.S.; de Souza, J.P.; Fernandes, L.O.B.; Martins-Filho, O.A.; Gontijo, C.M.F.; Peruhype-Magalhães, V.; et al. Chemokines in Leishmaniasis: Map of Cell Movements Highlights the Landscape of Infection and Pathogenesis. Cytokine 2021, 147, 155339. [Google Scholar] [CrossRef]
- Rodriguez, N.E.; Chang, H.K.; Wilson, M.E. Novel Program of Macrophage Gene Expression Induced by Phagocytosis of Leishmania chagasi. Infect. Immun. 2004, 72, 2111–2122. [Google Scholar] [CrossRef]
- Pandey, S.P.; Doyen, N.; Mishra, G.C.; Saha, B.; Chandel, H.S. TLR9-Deficiency Reduces TLR1, TLR2 and TLR3 Expressions in Leishmania major-Infected Macrophages. Exp. Parasitol. 2015, 154, 82–86. [Google Scholar] [CrossRef]
- Buates, S.; Matlashewski, G. General Suppression of Macrophage Gene Expression during Leishmania donovani Infection. J. Immunol. 2001, 166, 3416–3422. [Google Scholar] [CrossRef]
- Dillon, L.A.; Okrah, K.; Hughitt, V.K.; Suresh, R.; Li, Y.; Fernandes, M.C.; Belew, A.T.; Corrada Bravo, H.; Mosser, D.M.; El-Sayed, N.M. Transcriptomic Profiling of Gene Expression and RNA Processing during Leishmania major Differentiation. Nucleic Acids Res. 2015, 43, 6799–6813. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, M.C.; Dillon, L.A.; Belew, A.T.; Bravo, H.C.; Mosser, D.M.; El-Sayed, N.M. Dual Transcriptome Profiling of Leishmania-Infected Human Macrophages Reveals Distinct Reprogramming Signatures. mBio 2016, 7, e00027–16. [Google Scholar] [CrossRef] [PubMed]
- Dutta, M.; Qamar, T.; Kushavah, U.; Siddiqi, M.I.; Kar, S. Exploring Host Epigenetic Enzymes as Targeted Therapies for Visceral Leishmaniasis: In Silico Design and In Vitro Efficacy of KDM6B and ASH1L Inhibitors. Mol. Divers. 2024, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Jafarzadeh, A.; Nair, A.; Jafarzadeh, S.; Nemati, M.; Sharifi, I.; Saha, B. Immunological Role of Keratinocytes in Leishmaniasis. Parasite Immunol. 2021, 43, e12870. [Google Scholar] [CrossRef]
- Scorza, B.M.; Wacker, M.A.; Messingham, K.; Kim, P.; Klingelhutz, A.; Fairley, J.; Wilson, M.E. Differential Activation of Human Keratinocytes by Leishmania Species Causing Localized or Disseminated Disease. J. Investig. Dermatol. 2017, 137, 2149–2156. [Google Scholar] [CrossRef]
Figure 1.
Temporal Dynamics of DElncRNAs in Leishmania-Infected Macrophages.
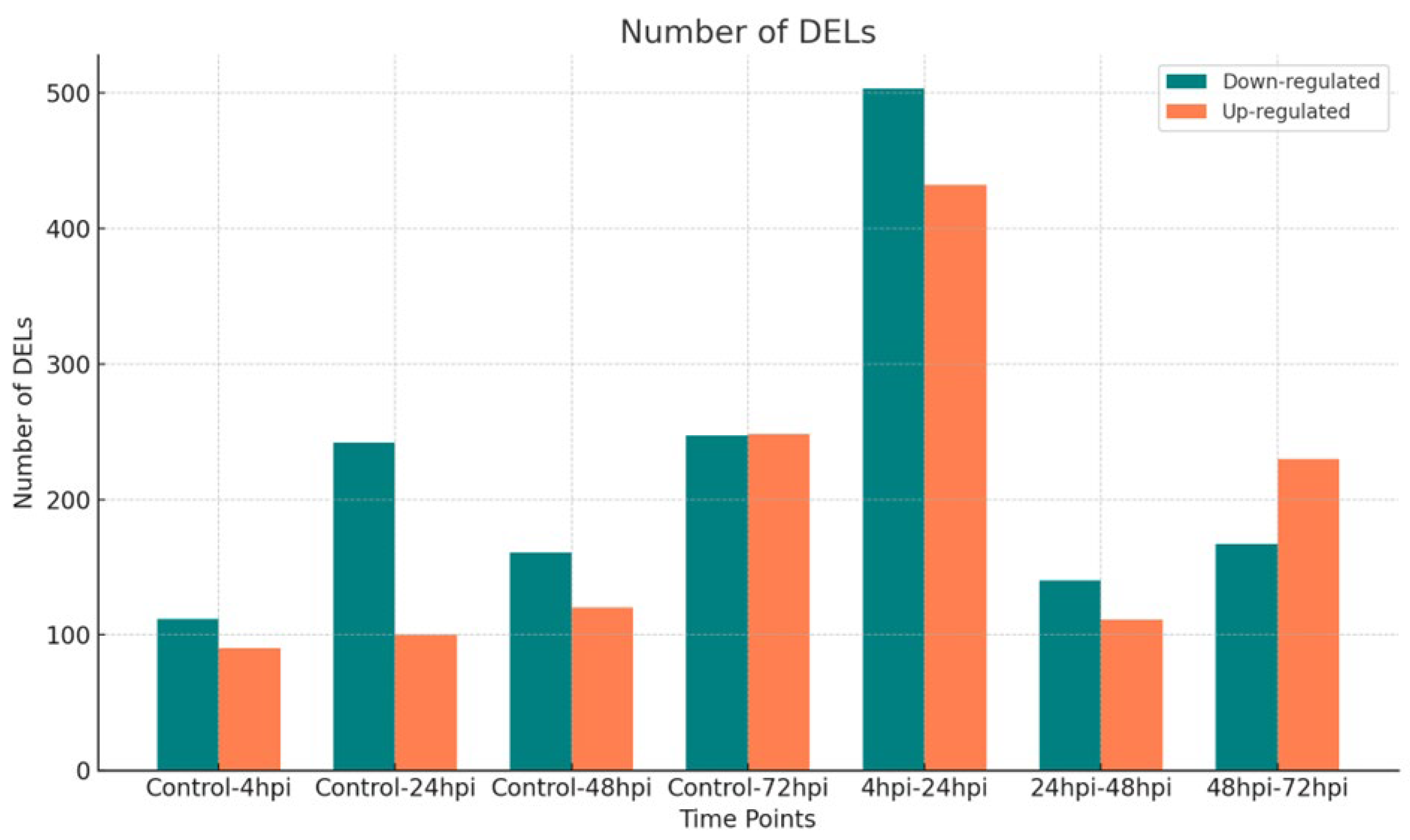
Figure 2.
Venn Diagrams of Differentially Expressed lncRNAs in Leishmania-Infected Macrophages (a) Venn diagram illustrating the overlap of differentially expressed lncRNAs at 4hpi, 24hpi, 48hpi, and 72hpi time points in Leishmania-infected macrophages; (b) Venn diagram comparing the differentially expressed lncRNAs between the time intervals of 4hpi-24hpi, 24hpi-48hpi, and 48hpi-72hpi in Leishmania-infected macrophages.
Figure 2.
Venn Diagrams of Differentially Expressed lncRNAs in Leishmania-Infected Macrophages (a) Venn diagram illustrating the overlap of differentially expressed lncRNAs at 4hpi, 24hpi, 48hpi, and 72hpi time points in Leishmania-infected macrophages; (b) Venn diagram comparing the differentially expressed lncRNAs between the time intervals of 4hpi-24hpi, 24hpi-48hpi, and 48hpi-72hpi in Leishmania-infected macrophages.
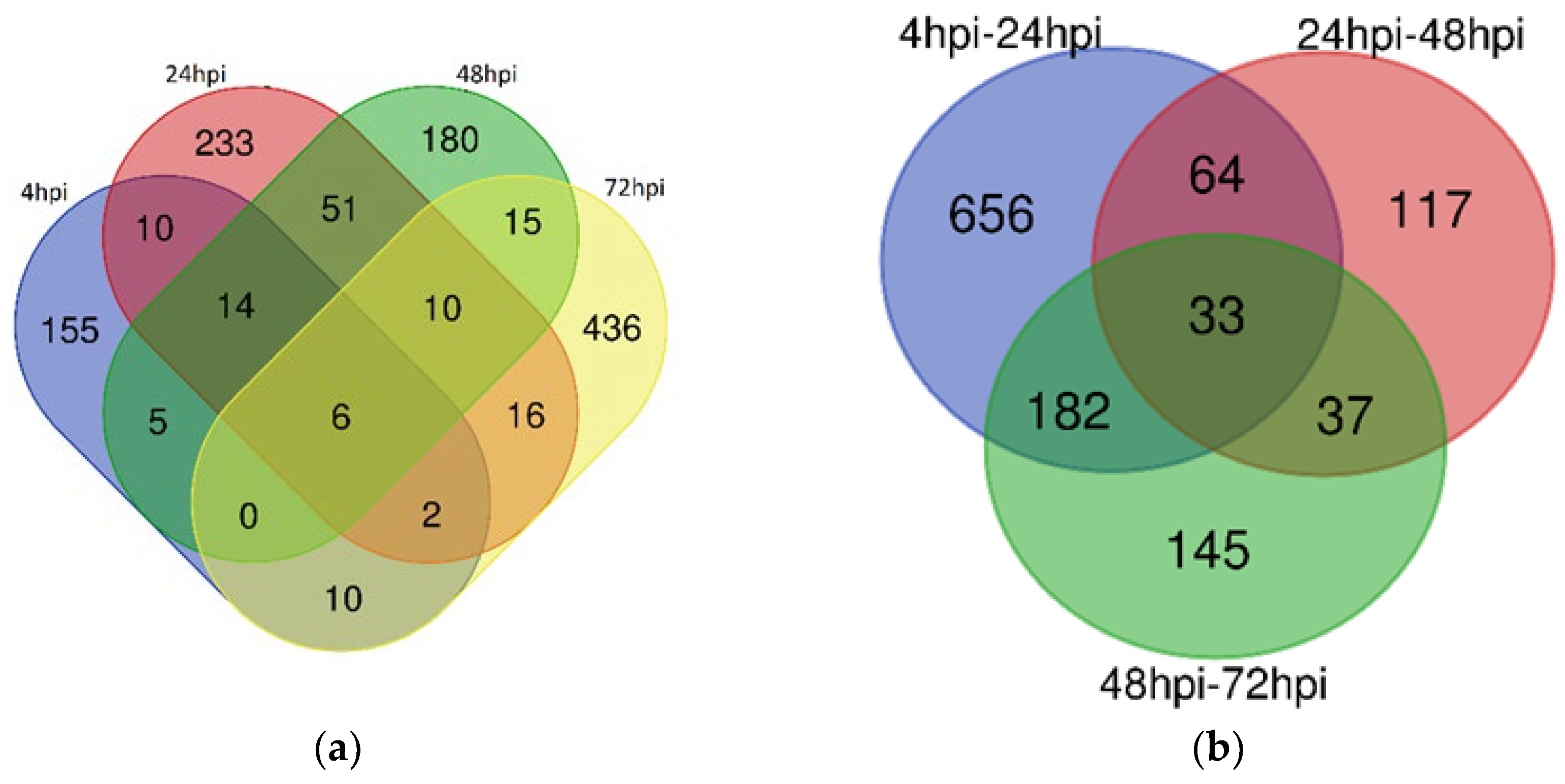
Figure 3.
Enrichment Analysis of Immune Response Pathways in Leishmania-Infected Macrophages at 0-4 Hours Post-Infection.
Figure 3.
Enrichment Analysis of Immune Response Pathways in Leishmania-Infected Macrophages at 0-4 Hours Post-Infection.
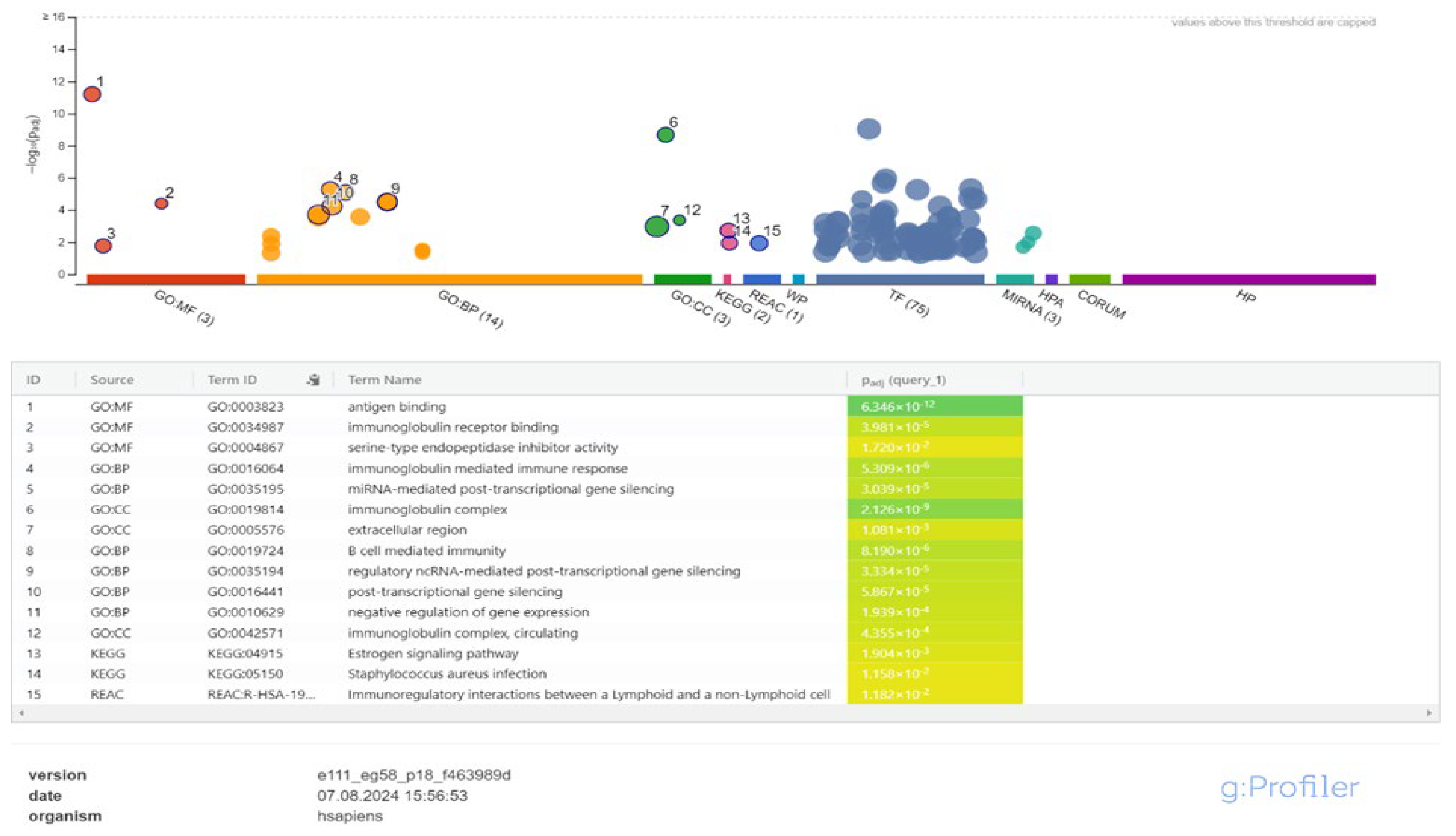
Figure 4.
Enrichment Analysis of Immune Pathways in Leishmania-Infected Macrophages Between 4- and 24-Hours Post-Infection.
Figure 4.
Enrichment Analysis of Immune Pathways in Leishmania-Infected Macrophages Between 4- and 24-Hours Post-Infection.
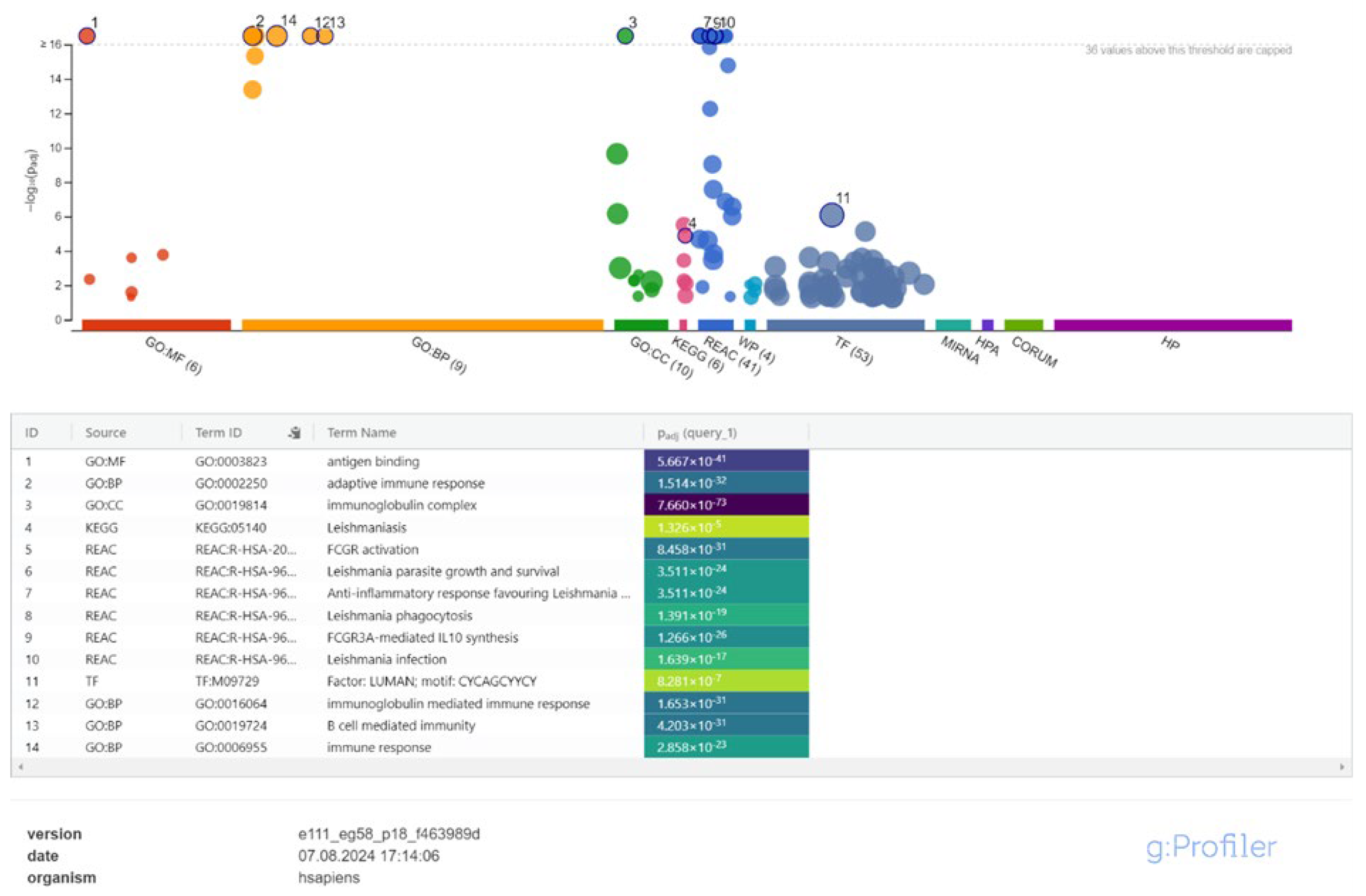
Figure 5.
Enrichment Analysis of Immune Pathways in Leishmania-Infected Macrophages Between 24- and 48-Hours Post-Infection.
Figure 5.
Enrichment Analysis of Immune Pathways in Leishmania-Infected Macrophages Between 24- and 48-Hours Post-Infection.
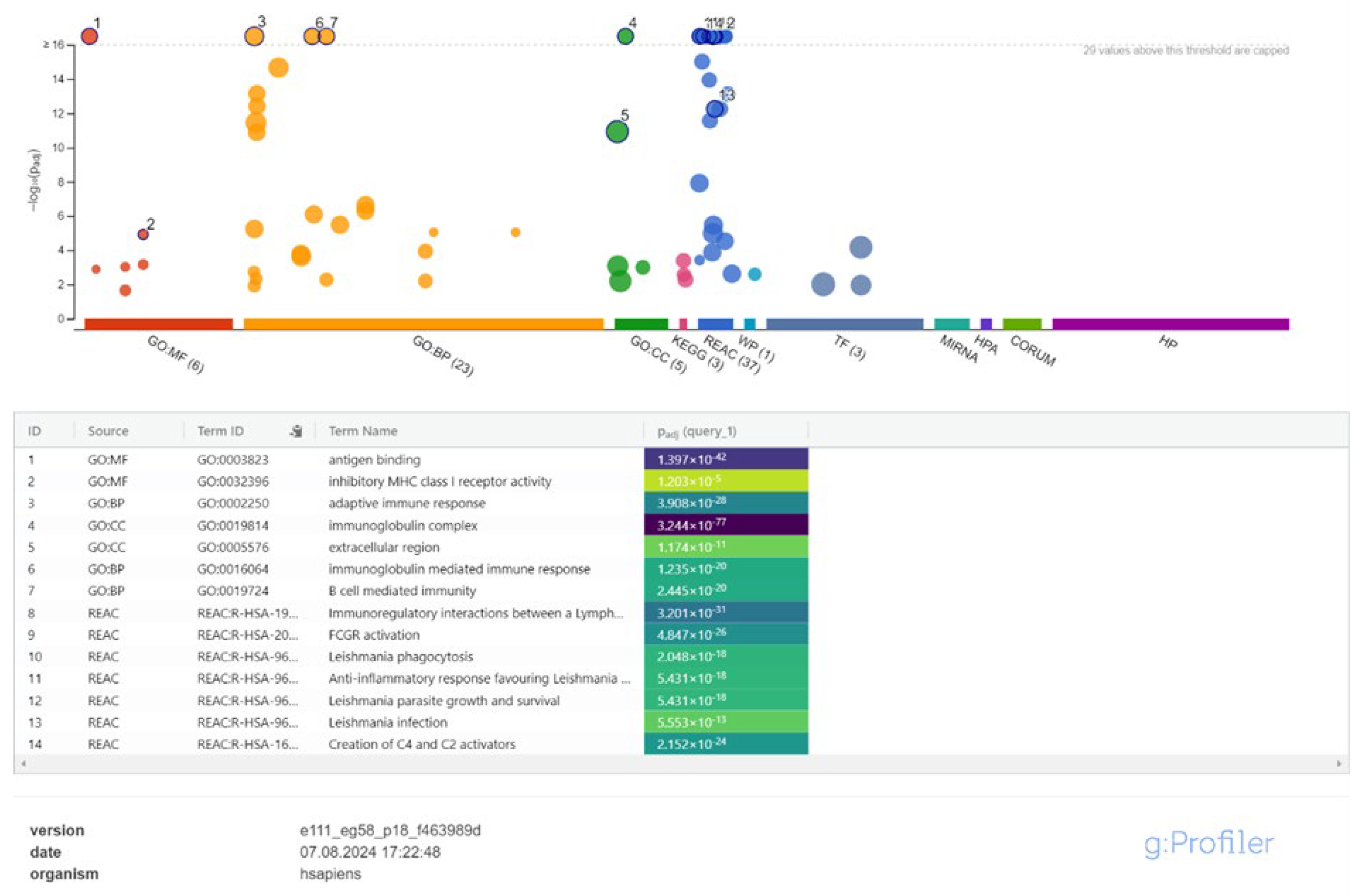
Figure 6.
Enrichment Analysis of Immune Pathways in Leishmania-Infected Macrophages Between 48- and 72-Hours Post-Infection.
Figure 6.
Enrichment Analysis of Immune Pathways in Leishmania-Infected Macrophages Between 48- and 72-Hours Post-Infection.
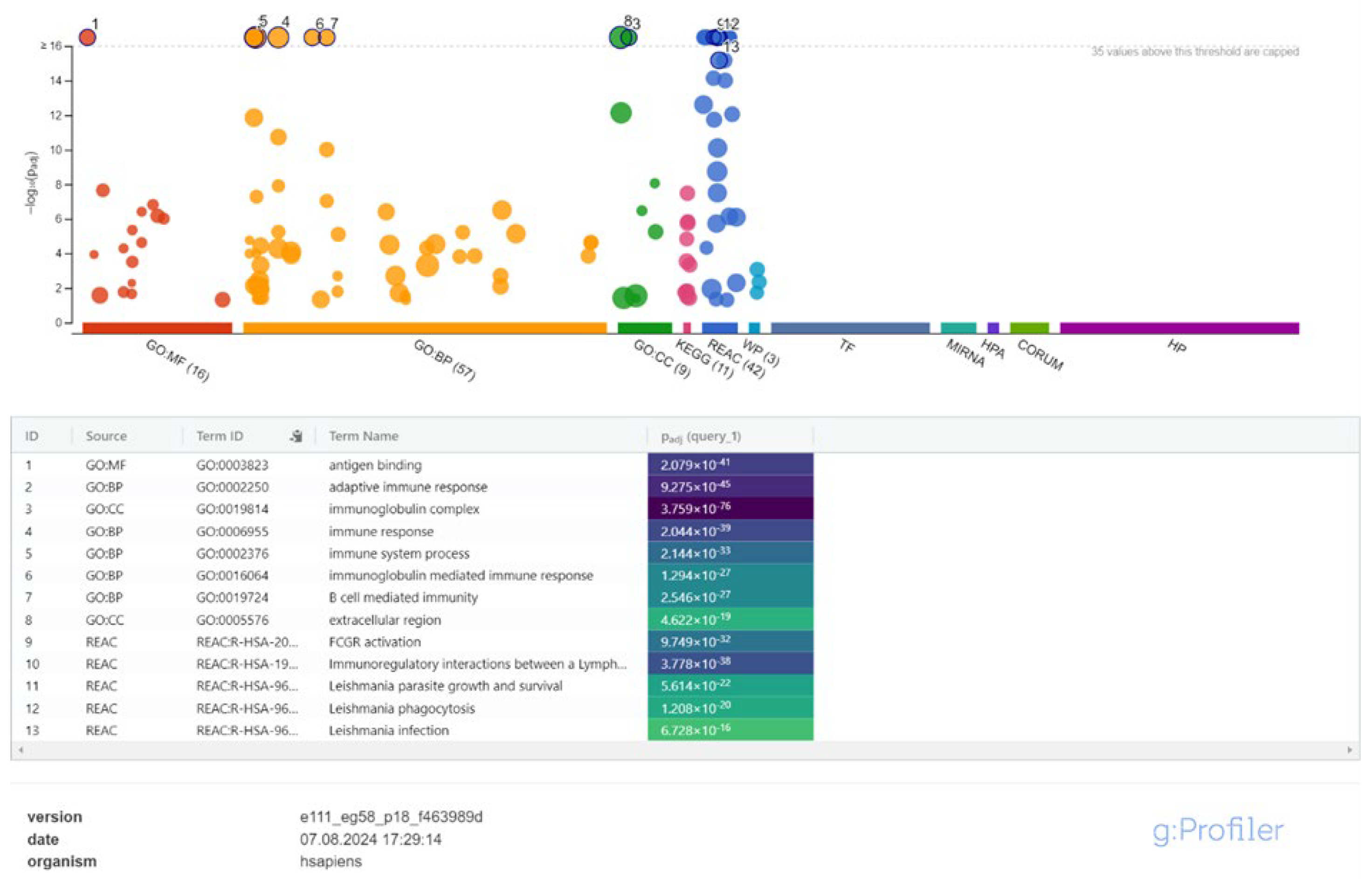
Figure 7.
Hub Gene Networks in Leishmania-Infected Macrophages at Different Time Points (a) Hub gene network for the 0 to 4-hour interval, showing genes related to cytoskeleton organization and immune response; (b) Hub gene network between 4 and 24 hours, where 8 hub genes are core nucleosome components, and KDM6B is a lysine-specific demethylase involved in transcriptional regulation; (c) Hub gene network between 24 and 48 hours, where all hub genes are associated with keratinization; (d) Hub gene network between 48 and 72 hours, showing genes related to histone modifications and immune response.
Figure 7.
Hub Gene Networks in Leishmania-Infected Macrophages at Different Time Points (a) Hub gene network for the 0 to 4-hour interval, showing genes related to cytoskeleton organization and immune response; (b) Hub gene network between 4 and 24 hours, where 8 hub genes are core nucleosome components, and KDM6B is a lysine-specific demethylase involved in transcriptional regulation; (c) Hub gene network between 24 and 48 hours, where all hub genes are associated with keratinization; (d) Hub gene network between 48 and 72 hours, showing genes related to histone modifications and immune response.
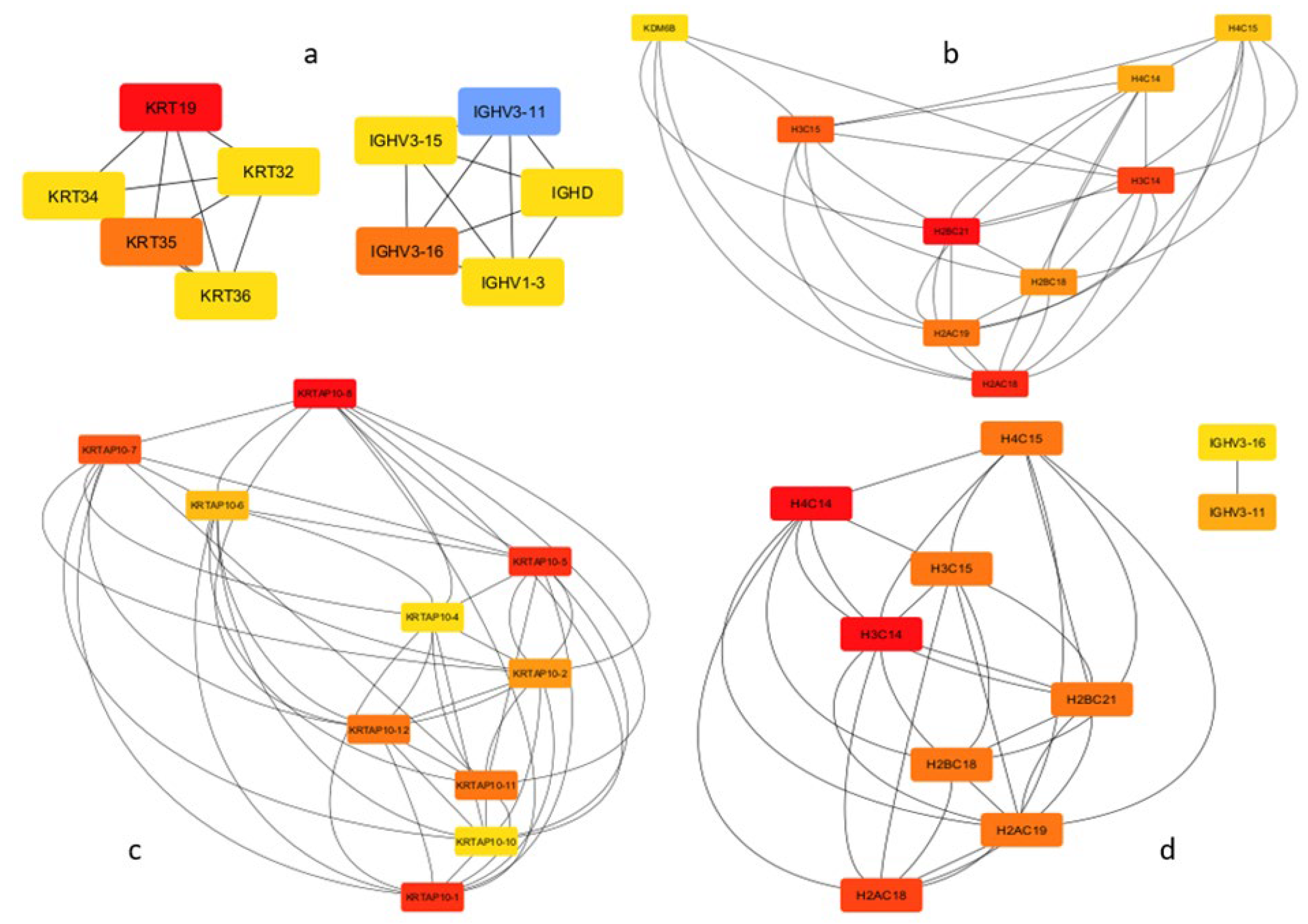

Table 1.
Fold Change in Expression of lnc-CMPK2-2 and lnc-TMEM121-26 Across Different Time Intervals During L. major Infection.
Table 1.
Fold Change in Expression of lnc-CMPK2-2 and lnc-TMEM121-26 Across Different Time Intervals During L. major Infection.
| Time Interval | lnc-CMPK2-2 | lnc-TMEM121-26 |
|---|---|---|
| 0hpi - 4hpi | 2.71 | -2.48 |
| 4hpi - 24hpi | 3.61 | 5.38 |
| 24hpi - 48hpi | 2.36 | -3.51 |
| 48hpi - 72hpi | -4.10 | -3.19 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



