Submitted:
24 October 2024
Posted:
25 October 2024
You are already at the latest version
Abstract
Candida glabrata (Nakaseomyces glabratus), the second most prevalent Candida pathogen globally, has emerged as a major clinical threat due to its ability to develop high-level azole resistance. In this study, two new 5,6-dihydrotetrazolo[1,5-c]quinazoline derivatives (c11 and c12) were synthesized and characterized using IR, LC-MS, 1Н and 13C NMR spectra. Along with 13 previously reported analogues, these compounds underwent in vitro antifungal testing against clinical C. glabrata isolates using a serial dilution method (0.125-64 mg/L). Remarkably, compounds c5 and c1 exhibited potent antifungal activity, with minimum inhibitory concentrations of 0.37 μM and 0.47 μM, respectively – about 20-fold improvement over standard drugs like amphotericin B, caspofungin, and micafungin. A detailed structure-activity relationship analysis revealed crucial molecular features enhancing antifungal potency. Extensive molecular docking studies across 18 protein targets explored potential binding pockets and affinities of the lead compounds. A robust 3D-QSAR model, incorporating molecular descriptors Mor26m and Mor29e, displayed good predictive ability for antifungal activity. In silico predictions indicated an absence of herbicidal effect, negligible environmental toxicity (to honeybees, avian species, and aquatic organisms), and mild human toxicity concerns for these compounds. This comprehensive approach aims to develop novel and effective antifungal compounds against the clinically relevant pathogen C. glabrata.
Keywords:
antifungal activity
; Candida glabrata
; 5
; 6-dihydrotetrazolo[1
; 5-c]quinazolines
; molecular docking
; toxicity
; QSAR
1. Introduction
Candida glabrata (Nakaseomyces glabratus) is a nonhyphae-producing haploid yeast described in 1917 by Harry Warren Anderson as part of the intestinal biota called Cryptococcus glabratus [1]. However, it was not until 1995 that Kevin C. Hazen recognized C. glabrata as an emerging pathogenic yeast commonly found in patients with diabetes mellitus, solid tumors, malnutrition, in neonates, and sometimes in patients with hematologic neoplasms [2]. Moreover, this haploid yeast species is known for its ability to cause invasive candidiasis [3,4,5,6,7,8,9,10]. Managing C. glabrata infections poses significant challenges, as evidenced by data from candidemia cases in Atlanta and Baltimore between 2008-2013, where it was the second most prevalent species, accounting for 27% of cases [11]. During this period, there was an increase in multidrug-resistant Candida cases from 1.8% to 2.6%. Similarly, a European study from 2018-2022 across 17 countries showed high proportions of C. glabrata (25-33%) in France, Czech Republic, and the UK [12]. However, the in vitro antifungal susceptibility among the three common Candida species (C. albicans, C. tropicalis, and C. glabrata) obtained before and during the era of COVID-19 did not change significantly [13]. While its colonization was uncommon in community-dwelling individuals, regardless of age, it was much more common in those hospitalized or residing in extended care facilities, additionally in those with dentures [14].
Recent studies have reported the development of various classes of compounds that have shown promising antifungal activity against Candida species: hesperetins (A), imidazopyrimidines (B), quinoxaline-triazoles (C), piperazine-tetrazoles (D, G), tetrazole derivatives (E, H), pyridine-tetrazoles (F), isoxazole-tetrazoles (J), and thiobenzoylimidazoles (K), etc. (Figure 1).
Molecular hybridization has emerged as a promising strategy for developing new antifungal compounds with enhanced activity and selectivity. And by combining two pharmacophore groups or two rings with known activity, synergistic effects can be achieved [15,16,17]. So, hesperetin derivatives A are expected to interact in close proximity with the critical active site of adhesin-like protein AWP1 structure of C. glabrata [18,19]. The 3-benzoyl imidazo[1,2-a]pyrimidine derivatives B were the most active against C. guilliermondii and C. glabrata, indicating an important role in biological activity for the benzene ring with electron-withdrawing substituents [15]. The 2-((5-(2,3-diethylquinoxalin-6-yl)-4-ethyl-4H-1,2,4-triazol-3-yl)thio)-1-(4-nitrophenyl)ethan-1-one (C) outperformed fluconazole as a control towards C. krusei strain, and was at the same level against C. glabrata [16].
1-(4-(4-Chlorobenzyl)piperazin-1-yl)-2-((1-methyl-1H-tetrazol-5-yl)thio)ethan-1-one D was found effective against C. krusei and C. parapsilosis [17]. 2-(2-(1H-Tetrazol-5-yl)ethyl)isoindoline-1,3-dione (E) was also among other tetrazole derivatives active against C. glabrata [20]. Compound VT-1598 (F) effectively controlled in vitro growth of mucosally derived C. abicans, C. glabrata, C. utilis and C. krusei clinical isolates, including fluconazole-resistant strains [10]. Tetrazole derivative G, having 3-trifluoromethyl substitution on the phenyl ring of piperazine was the most active in the series of these compounds against resistant C. tropicalis, and C. parapsilosis [21]. Among the series of 1-phenyl-N-tosyl-1H-tetrazole-5-carboxamide derivatives 4-fluorophenyl substituted one (H) additionally to good antibacterial properties has shown strong inhibition of several Candida strains, along with C. glabrata [22].
Among the series of (2R,3R)-3-((3-substitutied-phenyl-isoxazol-5-yl)methoxy)-2-(2,4-difluorophenyl)-1-(1H-tetrazol-1-yl)butan-2-ol derivatives, compound J displayed outstanding antifungal activity against fluconazole-resistant C. albicans, C. glabrata and C. auris [24]. And 1-(2,4-dihydroxythiobenzoyl)-2-undecyl-imidazole (K) was the most active in their group against C. glabrata [25].
Hence, the antifungal structure-activity relationship (SAR) on above-mentioned derivatives had provided some insights into the structural features that were important for their activity:
- Heterocyclic ring modifications. Incorporating heterocyclic rings, such as pyridine, piperazine, triazole, imidazole, or oxazole, in conjunction with the tetrazole moiety, can modulate antifungal activity and selectivity.
- Substitution pattern on the tetrazole ring. Generally, electron-withdrawing substituents such as halogens, trifluoromethyl or nitro groups, on the tetrazole ring or adjacent aromatic rings tends to enhance antifungal activity.
- Aryl substituents. Electron-rich aryl or hetaryl groups are often preferred.
- Steric effects. The introduction of bulky substituents, such as cyclohexyl or benzyl groups, can improve selectivity towards fungal cells over mammalian cells.
- Linker chain length and flexibility. The length and flexibility of the linker chain between the tetrazole moiety and other functional groups can improve the binding affinity to the target enzyme or receptor.
- Hydrophobicity and lipophilicity. Moderate hydrophobicity and lipophilicity of the tetrazole derivatives can enhance their ability to penetrate the fungal cell membrane and reach their target site: long undecyl chain, phenyl rings, etc. However, excessive hydrophobicity or lipophilicity may lead to poor solubility and bioavailability issues.
Moreover, understanding the virulence factors and antifungal resistance mechanisms of C. glabrata is crucial for developing effective treatment strategies [26,27,28]. So, the development of azole resistance has been primarily attributed to activating mutations in the pleiotropic drug resistance factor PDR1, leading to the overexpression of drug efflux pumps such as CDR1, PDH1, and SNQ2 [29,30]. Likewise, deletion of UPC2A results in increased susceptibility of C. glabrata. Consistently, disruption of CgCKB1 and CgCKB2 also attenuated the virulence in mouse models of invasive candidiasis [31]. It was demonstrated that a three-helix bundle KIX domain in the Med15a mediator subunit of C. glabrata (CgMed15a KIX), plays a crucial role in its growth inhibition by interacting with the PDR1 [32]. Furthermore, other inhibition pathways have been reported, including disruption of ergosterol biosynthesis and cell wall synthesis [10,15,16,17,30,33,34,35,36,37,38,39], targeting adhesin-like proteins [18], serine protease KEX2 [40], fructose-bisphosphate aldolase [41], calcineurin [42], squalene epoxidase [43], histidine kinase [44], proteasome [45], voltage-gated calcium channels [46], heat shock proteins [47], and the non-essential stress kinase YCK2 [48].
These details highlight the importance of carefully optimizing various structural features, physicochemical properties, and pharmacokinetic parameters to develop tetrazole derivatives with potent and selective antifungal activity while maintaining favorable drug-like properties. Overall, fused N-heterocyclic ring systems with electron-withdrawing groups, halogen substituents, aryl or heteroaryl substituents enhance antifungal potency across these diverse molecular scaffolds.
In this context, 5,6-dihydrotetrazolo[1,5-c]quinazolines (a-d, Figure 1) targeting C. glabrata is a promising research area. So, in this study, we aim to investigate their in vitro antifungal activity, in silico toxicity, molecular docking, and quantitative structure-activity relationship (QSAR) analysis against C. glabrata, providing insights into their potential as effective antifungal agents against this clinically relevant pathogen.
2. Results and Discussion
2.1. Synthesis
The synthetic procedures were reported in the previous study [23], namely the 2-(1H-tetrazol-5-yl)aniline undergoes condensation reactions with corresponding aldehydes and ketones under acidic conditions to form a series of substituted 5,6-dihydrotetrazolo[1,5-c]quinazolines (a-d) (Figure 1).
Among 15 chosen compounds for investigation there are two unreported before substances: c11 and c12. Hence, LC-MS, elemental analysis and IR spectra confirmed their structure, and the purity. In 13C NMR spectrum of c11, the carbon signal of C5 was observed at the 76.63 ppm. In 1H NMR spectrum the signal of quinazoline NH was registered at the 8.30 ppm for c11 and at the 7.88 ppm for c12; protons of an aromatic ring at the 8.21–7.13 ppm, and alkyl substituents at the 2.72–1.01 ppm with corresponding multiplicity.
2.2. Antifungal Studies
Previous computational techniques, such as molecular docking, and absorption, distribution, metabolism, excretion, and toxicity (ADMET) parameters by SwissADME [49,50,51] of c series, have provided valuable insights into the binding interactions and pharmacokinetic properties of these tetrazole derivatives, guiding the rational design of potent and selective antimicrobial agents: 4-(5-methyl-5,6-dihydrotetrazolo[1,5-c]quinazolin-5-yl)phenol (c6) along with 4-(5-methyl-5,6-dihydrotetrazolo[1,5-c]quinazolin-5-yl)benzoic acid (c10) as the most promising molecules for synthesis and drug purposeful search. Besides, the latter had Vina score stronger, than Tedizolid, towards ribosomal 50S protein L2P (PDB ID: 2QEX) [50] and to penicillin-binding protein 2X (PDB ID: 2ZC4) additionally with other 3 substances (c1, c5, and c7) [51]. A search for PAINS (pains interfering compounds, or frequent hitting compounds / promiscuous compounds), which are molecules containing substructures, that show a strong response in assays independent of the target protein, yielded no hits for all studied compounds [49].
Moreover, preliminary antifungal in vitro studies revealed that minimum inhibition concentration (MIC) of c10 was less than 2 mg/L against C. glabrata [52], despite resistance of C. kefyr (Kluyveromyces marxianus) and C. utilis (Cyberlindnera jadinii). And against C. albicans MIC for c1 and c6 was 128 mg/L, and for c9, c10, and d1 was 256 mg/L [23].
So, to obtain valuable results it was decided to choose 0.125-64 mg/L as the test concentration range against C. glabrata. In the result, the half of studied tetrazole derivatives exhibited varying degrees of antifungal inhibition properties (Table 1, Figure 2). Notably, compound c12 showed inhibition at a concentration of 16 mg/L, while compounds b1, c6, and a2 displayed inhibition at 8 mg/L, 4 mg/L, and 4 mg/L, respectively. Compound c10 exhibited inhibition at a concentration as low as 2 mg/L.
The most potent compounds were c1 and c5, which demonstrated inhibition at the remarkably low concentration of 0.125 mg/L. In comparison, the reference drugs amphotericin B and caspofungin exhibited inhibition at 8 mg/L, while micafungin showed inhibition at 4 mg/L. Due to their exceptional potency, compounds c1 and c5 were further studied by diluting them ten-fold, but fungal growth was observed, indicating their MICs were not lower. Also, it’s interesting, that compounds c9 and d1 were not active against C. glabrata as against C. albicans [23].
2.3. Antifungal Studies
Based on the obtained antifungal activity results, SAR against C. glabrata can be summarized as follows (Figure 2).
- R1 alkyl prolongation: extending the alkyl chain from methyl to propyl may introduce unfavorable steric clashes or conformational restrictions, leading to decreased activity.
- 4th Position substitution of phenyl ring: the preference for substitution at the 4th position over other positions on the bicyclic ring system suggests, that the steric and electronic environment at this specific site is optimal for binding to the target enzyme. Substituents at this position may participate in critical interactions like hydrogen bonding, π-stacking, or filling a hydrophobic pocket. Besides the presence of aromatic moiety in these compounds increased hydrophobicity, which improves their permeability into the cell membrane, therefore enhancing the antifungal activity.
- CH3 vs. Cl when R1 = H: the preference for a methyl group over chloro, when R1 is unsubstituted could be attributed to the more lipophilic nature of the methyl substituent, and to steric factors, where the smaller hydrogen atom allows for better accommodation, and binding within the target pocket. The chloro group, being larger and more electronegative, may experience unfavorable steric clashes or result in suboptimal binding interactions.
- Br/COOH/OH vs. CN when R1 = methyl: the preference for bromo, carboxyl, or hyd-roxyl substituents over a cyano group at R2 suggests, that the electron-withdrawing nature of the groups may be disfavored. The electron-rich bromine, carboxyl, and hydroxyl groups could form favorable hydrogen bonding or ionic interactions with the target.
- Change from phenyl to cyclohexyl or indolin-2-one substituents: this structural modification leads to a decrease in the biological activity, potentially indicating that the size of the rings is important for the desired activity.
- NO2 / COOH substitution into the 3rd position: the introduction of strongly electron-withdrawing nitro or carboxyl groups at the 3rd position may significantly alter the electronic distribution and potentially disrupt crucial binding interactions, leading to a complete loss of activity.
- Cl substitution into the 4th position: Similar to the 3rd position substitution, placing a chloro group at the 4th position of the R2 phenyl ring also leads to a complete lack of activity. This indicates that the specific substitution pattern on the phenyl ring is essential for the compound to exhibit the desired a antifungal effects.
- Change from phenyl to pyridine or substituted indolin-2-one: similar to the cyclohexyl and indolin-2-one modifications, changing the phenyl ring to a pyridine or substituted indolin-2-one moiety likely disrupts essential aromatic interactions or introduces steric hindrances, leading to a complete loss of activity.
In summary, the SAR analysis suggests that the R2 substituent plays a critical role in maintaining the desired biological activity. changing the phenyl ring to a cyclohexyl or indolin-2-one moiety likely disrupts crucial π-π stacking or aromatic interactions with the target binding site, resulting in decreased activity.
2.4. Molecular Docking
Using computational methods like molecular docking [53], it may be possible to explore binding modes and identify other substituents or scaffolds that can occupy different pockets within the enzyme active site. These in silico predictions can guide the design and synthesis of novel compounds to achieve better fit and higher binding affinity.
The Table 2 presents the results of online molecular docking calculations by CB-Dock2 website [54,55] for two lead-compounds c1 and c5, against 18 various antifungal protein targets (from RCSB Protein Data Bank (RCSB PDB) [56]) taken majorly from Nakaseomyces glabratus (C. glabrata), Saccharomyces cerevisiae, and C. albicans.
It’s worth to mention, that C. glabrata shares a recent common ancestor with several Saccharomyces species, and belongs to a clade different from that of other Candida species (namely those that recode the CUG codon to serine) [57]. Vina scores of the strongest affinities, cavities volumes, docking sizes and corresponding amino acid contact residues are presented in Supplementary Materials, Table S5.
The docking results indicate that both compounds c1 and c5 have the potential to interact with a diverse range of protein targets involved in various cellular processes in Candida and Saccharomyces species. The strongest predicted binding affinity (kcal/mol) was observed for c1 against importin subunit alpha (-9.8), sterol 14-alpha demethylase (-10.2), and sterol uptake control protein 2 (-10.4) (Table 2). While compound c5 showed the highest docking scores against 6,7-dimethyl-8-ribityllumazine synthase (-9.4), exo-β-(1,3)-glucanase (-9.7), and also sterol uptake control protein 2 (-9.9).
The strong predicted binding to key targets such as sterol biosynthesis enzymes, transcription factors, and cell wall-modifying enzymes suggests, that these compounds could be explored further in vitro against them as potential antifungal agents. Nevertheless, both compounds exhibited relatively lower docking scores against dihydrofolate or methylglyoxal reductase, and flavin mononucleotide adenylyltransferase, suggesting potentially weaker binding to these proteins.
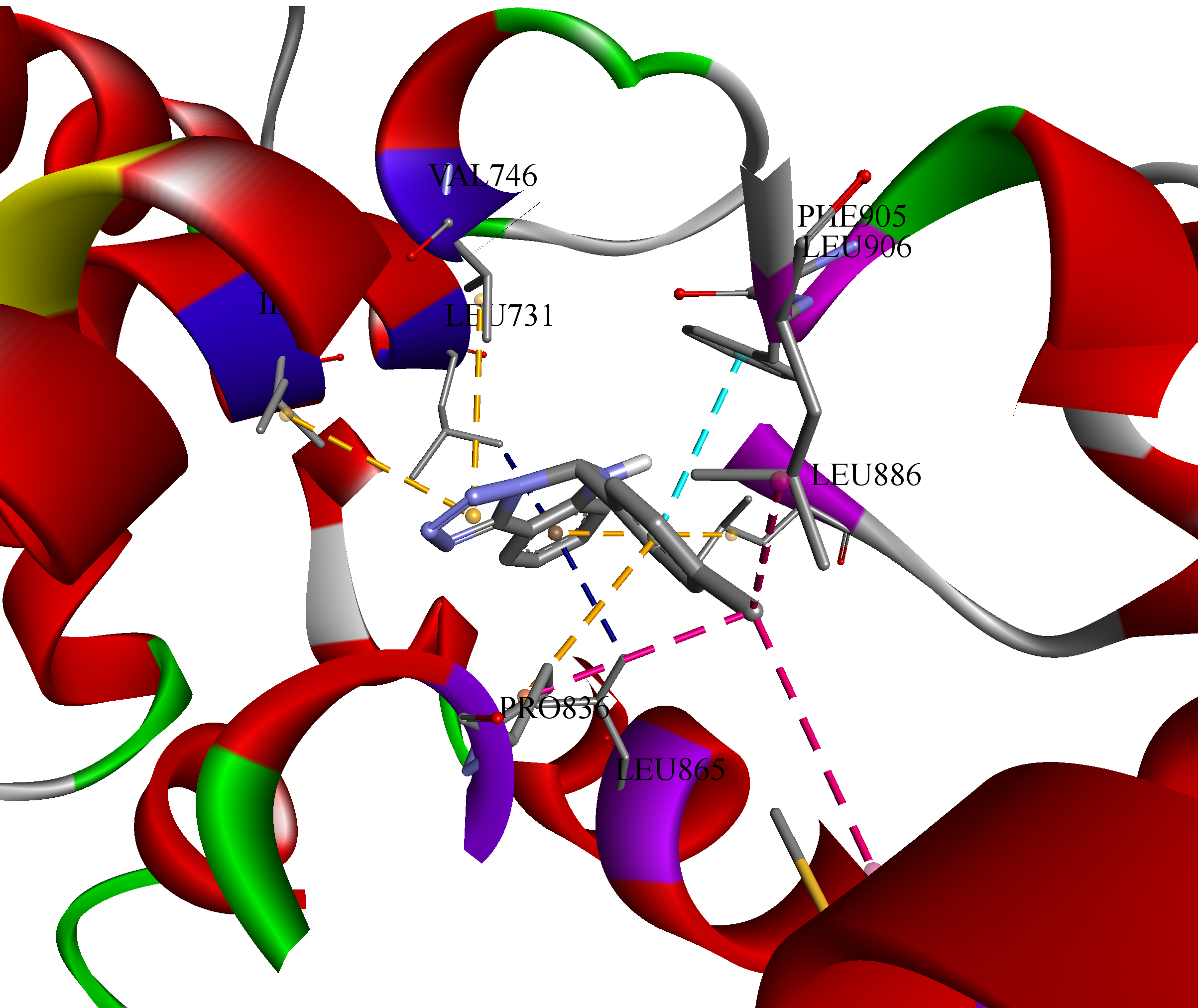
To inspect the binding poses and understand the interactions between the ligand and receptor, it was decided to 3D visualize the highest affinity result (-10.4 kcal/mol), namely, c1 towards sterol uptake control protein 2 (PDB ID: 7VPR) (Figure 3).
Hence, all 10 formed bonds (amino acid / distance in Ǻ) of c1 were hydrophobic: π-σ (LEU731 / 3.51, LEU865 / 3.48); π-π stacked (PHE905 / 4.40); alkyl (PRO836 / 4.52, MET858 / 5.00, LEU906 / 4.38); and π-alkyl (LEU886 / 5.28, ILE727 / 4.89, VAL746 / 4.77, PRO836 / 4.59), showing structure’s flexibility to fit into the cavity of protein D chain.
It’s interesting and worth to mention, that in the previous study [23] docking grid was centered at (71, 66, 4) with dimensions (14, 16, 14) into sterol 14-alpha demethylase by obtained X-ray results (PDB ID: 5TZ1), and AutoDock Vina scores for c1 and c5 were -8.2 and -8.3 kcal/mol. While now CB-Dock2 [54] identified a different best binding pocket for them centered at (66, 35, 41) with dimensions (32, 28, 19) with highest predicted affinities of -10.2 and -9.2 kcal/mol, respectively (Supplementary Materials, Table S5). Whereas, among 5 other cavities proposed by CB-Dock2 on 5TZ1, one was found with the same X-ray chosen coordinates, and affinity for c1 and c5 in this pocket were a bit stronger: -8.8 and -8.9 kcal/mol, correspondingly.
So, we decided to do one more additional molecular docking calculation to see what will be the difference of the grid and affinity scores on one more protein. Considering the same found in vitro MIC of c1 and c5 we have chosen metal binding protein enolase 1 (PDB ID: 7VRD), because their Vina scores we also predicted the same (-8.1 kcal/mol) by CB-Dock2 [54] (Table 2). Calculated RMSD (root mean square deviation) was obtained as 1.124 Å, so results were considered reliable [58,59] (Table 3).
The docking grid centered at (-35, -37, 4) with dimensions (22, 22, 22) was found according to position of reported ligand, and affinity of c1 and c5 was calculated of -7.8 kcal/mol off-site by AutoDock Vina. While CB-Dock2 proposed a different best binding pocket, centered at (-10, -18, 24) with dimensions (35, 31, 35). And the predicted score for each compound was -8.1 kcal/mol. Nevertheless, a second-best potential binding pocket by CB-Dock2 was proposed with the same coordinates as the reported X-ray grid, and affinity for c1 and c5 in this pocket were practically the same: -7.7 and -7.8 kcal/mol, as shown by us respectively.
Hence, online tools like CB-Dock2 can facilitate and guide enzymatic studies of potential biologically active compounds. And this multi-pronged strategy, integrating both in vitro and further in vivo studies with structural and mechanistic characterization, can provide a high level of confidence in the substance’s target-specific reactivity and its potential for development as a selective modulator of the enzyme of interest.
2.5. Quantitative Structure-Activity Relationship
Furthermore, the synergistic blend of organic synthesis, analytical chemistry, pharmaceutical chemistry, and molecular docking within the framework of Quantitative Structure-Activity Relationship (QSAR) significantly contributes to the advancement of novel antifungal drug development [60,61,62]. This integrative approach can help predict the biological activity of new tetrazole derivatives guiding the design of more potent antifungal agents. Thus, the calculated QSAR models of antifungal activity showed a high goodness-of-fit (r2 = 0.81-0.85, Figure 4) and predictive ability (Q2loo = 0.68-0.71), indicating its reliability in modeling.
Model-1, μM = -860.236(±487.862) * Mor26m + 3002.6084 (±1007.9199) * Mor29e + 1374.0919 (±311.2194). n = 15, r2 = 0.8474; s = 189.6460; F = 24.9931; p = 0.0001; RMSEtr = 164.2383; R2cv (Q2loo) = 0.6781; R2-R2cv = 0.1694; RMSEcv = 238.5710; MAEcv = 198.0963; PRESScv = 682993.3683; CCCcv = 0.8349; RMSEex = 871.0944; MAEex = 683.7867; PRESSext = 2276416.4022.
Where Mor26m and Mor29e: 3D-MoRSE descriptors, weighted by weighted by mass and Sanderson electronegativity (Supplementary Materials, Table S2) [63].
Model-2, mg/L = 158.3513 (±95.4438) * Mor10m + 871.3969 (±329.9217) * Mor29e + 294.6011 (±75.7156). n = 15, r2 = 0.8142; s = 61.3710; F = 19.7157; p = 0.0001; RMSEtr = 53.1489; R2cv (Q2loo) = 0.7114; R2-R2cv = 0.1028; RMSEcv = 66.2355; MAEcv = 53.7468; PRESScv = 52645.7556; CCCcv = 0.8516; RMSEex = 153.0649; MAEex = 138.7554; PRESSext = 70286.6325.
So, the significance of the models is supported by the low p-value of the F-statistic and low error metrics [64]. But, using only 15 data points for model validation can be a limitation, especially in the context of QSAR modeling, where having a larger dataset can improve the robustness and reliability of the model in the future studies. Nevertheless, it was found, that 3D-MoRSE descriptors, Mor26m and Mor29e for mass and Sanderson electronegativity, were important for inhibition C. glabrata pathway.
2.6. Toxicity Prediction
Furthermore, pharmaceuticals and agrochemicals have been linked to various undesirable negative impacts on health and the environment. To aid in identifying green fungicides, the cropCSM [65,66] provides an assessment of molecule’s impact on honey bee (A. mellifera) toxicity, as well as toxicity to mallards and flathead minnows (Figure 5).
Additionally, it includes measures of human health, such as AMES toxicity, rat LD50, and oral chronic toxicity (Figure 5, Supplementary Materials, Table S3). Hence, considering environmental toxicity, all the compounds show no herbicidal and no honey bee toxicity. Only compound a1 and a2 show avian toxicity, while the rest of the compounds do not. The aquatic minnow toxicity values (log LD50, mg/kg/day) range from -0.06 to 1.58, not of dangerous level. The presence of the nitro group may contribute to the increased minnow toxicity, as nitro groups are electron-withdrawing and increase the reactivity of the compounds.
Mentioning human toxicity, namely AMES toxicity: most of the compounds (except b2 and b3) show positive results, due to some functional groups, that might influence mutagenic potential of compounds. Nevertheless, the rat acute toxicity (LD50, mg/kg) values range from 487.1 to 1465.9 (Supplementary Materials, Table S2) of light level. The majority of rat chronic toxicity (LOAEL, mg/kg/day) values range from 10.7 to 51.9, also indicating medium toxicity.
From the provided data, it appears that the compounds exhibit varying levels of toxicity across different endpoints, still of moderate effect.
2.7. Pearson Correlations
By analyzing the relationships between the predicted toxicity, in silico antifungal affinity and found in vitro activities, it may be possible to identify key structural determinants that contribute to the observed toxicological profiles. And this information could be valuable for the design and development of new compounds with improved safety profiles or for the optimization of existing compounds to mitigate potential adverse effects. So, based on the Pearson correlation results presented in the Figure 6 (Supplementary Materials, Table S4), the following conclusions are found.
Minnow toxicity (MT) has a strong positive correlation (r2 = 0.72534) with CYP51 (sterol 14-alpha demethylase, PDB ID - 5TZ1) affinity [23]. This suggests that compounds with lower affinity for the CYP51 enzyme tend to have lower minnow toxicity. It could be caused by formation of less bonds with proteins, so less toxic. MT has a moderate negative correlation with rat acute toxicity (RAT) (r2 = -0.48236) and rat chronic toxicity (RCT) (r2 = -0.55494). This implies that compounds with lower minnow toxicity tend to have higher acute and chronic toxicity in rats due to different mechanisms of action. RCT has a strong negative correlation with CYP51 affinity (r2 = -0.87526), suggesting that compounds with higher rat chronic toxicity tend to have higher affinity for the CYP51 enzyme.
And there was found no statistically significant correlations of predicted toxicities with MIC (Supplementary Materials, Table S4). It is important to note that correlation does not necessarily imply causation, and further experimental validation and mechanistic studies may be needed to confirm these relationships and understand the underlying biological mechanisms.
3. Conclusions
Overall, the increasing prevalence of C. glabrata infections and the associated challenges of antifungal resistance have catalyzed extensive research efforts to discover novel antifungal substances. 5,6-Dihydrotetrazolo[1,5-c]quinazolines have emerged as promising candidates, exhibiting potent antifungal activities against C. glabrata through various predicted mechanisms of action. Notably, compounds c1 and c5 demonstrated remarkable inhibition at concentrations as low as 0.125 mg/L, outperforming reference drugs like amphotericin B and caspofungin. The SAR analysis provided insights into the structural features essential for antifungal activity: presence of heterocyclic rings, bulky aryl or heteroaryl groups were found to enhance hydrophobic interactions, and electron-rich bromine, carboxyl, and hydroxyl groups could form favorable hydrogen bonding or ionic interactions with the target. Computational docking studies predicted strong binding affinities of compounds c1 and c5 towards various antifungal targets, including sterol biosynthesis enzymes, transcription factors, protein transport, and cell wall-modifying enzymes in C. glabrata and related species. QSAR models were developed, demonstrating good predictive ability and identifying 3D-MoRSE descriptors related to mass and electronegativity as important for inhibiting fungal growth. In silico toxicity predictions suggested low to moderate toxicity levels for most compounds, with varying profiles across different endpoints.
Hence, the integration of synthetic chemistry, molecular hybridization strategies, computational techniques, along with further in vitro and in vivo experimental validations, and the exploration of alternative targets has paved the way for the development of more effective and selective antifungal agents against C. albicans.
4. Materials and Methods
4.1. Synthesis
4.1.1. General
Melting points were determined in open capillary tubes in a «Mettler Toledo МР 50» apparatus and were uncorrected. The elemental analyses (C, H, N) were performed using the ELEMENTAR vario EL cube analyzer (USA). Analyses were indicated by symbols of the elements or functions within ±0.3% of the theoretical values. 1H NMR spectra (400 MHz) and 13C NMR spectra (125 MHz) were recorded on a Varian-Mercury 400 (Varian Inc., Palo Alto, CA, USA) spectrometers with TMS as internal standard in DMSO-d6 solution. LC-MS were recorded using chromatography/mass spectrometric system which consists of high-performance liquid chromatography «Agilent 1100 Series» (Agilent, Palo Alto, CA, USA) equipped with diode-matrix and mass-selective detector «Agilent LC/MSD SL» (atmospheric pressure chemical ionization – APCI). Electron impact mass spectra (EI-MS) were recorded on a Varian 1200 L instrument at 70 eV (Varian, USA). The purity (>95% pure) of obtained compounds was checked by 1H, 13C-NMR and LC-MS.
4.1.2. Synthesis of the c11 and c12
2-(1H-Tetrazol-5-yl)aniline (1.0 g; 6 mM) was dissolved in propan-2-ol (10 mL). Then the corresponding aldehyde or ketone (6 mM) was added to the solution, and 1 drop of concentrated sulfuric acid was added. The mixture was refluxed for 1 h. and cooled. A formed precipitate was filtered and washed firstly with propan-2-ol (5 mL), then with cold water (100 mL).
5-Methyl-5-(3-nitrophenyl)-5,6-dihydrotetrazolo[1,5-c]quinazoline (c11). Beige solid; 84% yield, mp 233–235°C. 1H NMR (400 МHz): δ (ppm) 8.30 (s, 1H, NH), 8.21 (s, 1H, Ph-2), 8.10 (d, J = 8.2 Hz, 1H, Ph-4), 7.77 (d, J = 7.5 Hz, 1H, H-10), 7.57 (t, J = 8.0 Hz, 1H, Ph-5), 7.43 (d, 1H, d, J = 7.9 Hz, 1H, Ph-6), 7.35 (t, J = 8.0 Hz, 1H, H-8), 7.09 (d, J = 8.2 Hz, 1H, H-7), 6.88 (t, J = 7.5 Hz, 1H, H-9), 2.35 (s, 3H, CH3). 13С NMR (125 МHz): δ (ppm) 149.07, 148.46, 145.02, 142.68, 134.31, 131.72, 131.21, 125.66, 124.19, 120.41, 120.06, 116.16, 107.63, 76.63, 28.51. IR (cm−1) 1622, 1525, 1476, 1380, 1340, 1216, 1080, 898, 804, 749, 724, 693. LC-MS: m/z = 309 [M+Н]+. Anal. calcd. for C15H12N6O2: C, 58.44; H, 3.92; N, 27.26; O, 10.38. Found: C, 58.48; H, 3.87; N, 27.33; O, 10.36.
5-Phenyl-5-propyl-5,6-dihydrotetrazolo[1,5-c]quinazoline (c12). Beige solid; 48% yield, mp 164–166°C. 1H NMR (400 МHz): 7.88 (s, 1H, NH), 7.72 (d, J = 7.7 Hz, 1H, H-10), 7.34-7.13 (m, 5H, Ph), 7.14 (s, 1H), 7.08 (d, J = 8.2 Hz, 1H, H-7), 6.81 (t, J = 7.5 Hz, 1H, H-8), 2.72 (ddd, J = 15.7, 12.0, 4.4 Hz, 1H, CCH2), 2.35 (ddd, J = 15.0, 12.0, 4.4 Hz, 1H, CCH2), 1.77 – 1.62 (m, 1H, CCH2CH2), 1.43 (dt, J = 12.7, 6.6 Hz, 1H, CCH2CH2), 1.01 (t, J = 7.4 Hz, 3H, CH3). IR (cm−1): 1622, 1495, 861, 737, 705, 692. LC-MS: m/z = 292 [M+Н]+. Anal. calcd. for C17H17N5: C, 70.08; H, 5.88; N, 24.04. Found: C, 70.12; H, 5.84; N, 24.09.
4.2. Antifungal Studies
The method of serial dilutions (0.125–256 mg/L and 0.12 – 62.50 μg/L) of 5,6-dihydrotetrazolo[1,5-c]quinazolines (Figure 1, Table 1) on meat-peptone broth was carried out in the bacteriological laboratory of Zaporizhzhia Regional Clinical Hospital of Zaporizhzhia Regional Council (Ukraine) [67] against Candida glabrata (Nakaseomyces glabratus), C. kefyr (Kluyveromyces marxianus), C. utilis (Cyberlindnera jadinii), that were isolated from patients’ biological material, and identified by chromatic Candida media (Liofilchem, Italy). Microorganism strains didn’t reveal sensitivity towards the chosen solvent, namely DMSO (2.5%). All growth experiments were carried out in duplicate.
4.3. Molecular Docking Studies
Macromolecule from RCSB Protein Data Bank (PDB) [56] was used as biological target, namely metal binding protein enolase 1 (PDB ID: 7VRD). The 15 mol files of 5,6-dihydrotetrazolo[1,5-c]quinazoline derivatives were drawn by MarvinSketch 20.20.0 and saved in mol format; optimized by HyperChem 8.0.8, using molecular mechanical MM+ algorithm combined with semiempirical PM3 molecular modeling method with a maximum number of cycles and Polak-Ribiere (Conjugate Gradient) algorithm. The next step was a reoptimization of the MM+ optimized structures by applying semiempirical PM3 molecular modeling method. Obtained files were further used for calculations; mol files were converted to pdb by Open Babel GUI 2.3.2; pdb files were converted to pdbqt by AutoDocTools 1.5.6. Vina 1.1.2 was used to carry out docking studies [68]. The following grid box was used for 7VRD: center_x = -35, center_y = -37, center_z = -4, size_x = 22, size_y = 22, size_z = 22. Discovery Studio v17.2.0.16349 was used for visualization. To validate the docking method by the value of RMSD (root-mean-squared deviation), which characterizes the degree of reliable docking probability, the reference ligand (2-phosphoglyceric acid) was extracted and then reused for the redocking process [58]. If the found pose has a RMSD less than 2 Å relative to the X-ray conformation, then it is generally considered a reasonable docking [59]. So, RSMD value 1.124 Å between the experimental and the reference conformation ligand was calculated to be reliable.
Also CB-Dock2 [54], a protein-ligand auto blind docking tool, that inherits the curvature-based cavity detection procedure with AutoDock Vina, was used for calculations of tested substances’ affinity to 18 macromolecules from RCSB Protein Data Bank (PDB) [56] as a biological targets, namely 4N9N, 7VPR, 5TZ1, 5JLC, 4HOG, 7YMU, 1EQP, 7EKU, 7P43, 7O9Q, 4D3W, 7VPS, 2C1T, 4KQ6, 3FWK, , 7VRD, 7QP0, 7VPT online. Vina Scores, cavities volumes, docking sizes and corresponding amino acid contact residues are given in Supplementary Materials, Table S4.
4.4. QSAR Modeling
All structures were drawn by MarvinSketch 20.20.0 and saved in mol format; optimized by HyperChem 8.0.8 using molecular mechanical MM+ algorithm combined with semiempirical PM3 molecular modeling method with a maximum number of cycles and Polak-Ribiere (Conjugate Gradient) algorithm. The next step was a reoptimization of the MM+ optimized structures by applying semiempirical PM3 molecular modeling method. Obtained files were further used for calculations. Descriptors were calculated using Dragon 5.5 (> 1500 descriptors) (Dragon 5.5 for Windows, Talete S.r.l., Milano, Italy) by procedure described earlier. Validation of equations in order to confirm their predictive ability was carried out using a prediction set (external) and training set (internal). Cross-sleep validation was performed by the “leave-one-out” method. The optimal equation is one in which the standard error is minimal. The definition of all used molecular descriptors and the calculation procedures were summarized elsewhere [63,69]. The correlation coefficients for all pair of descriptor variables used in the models were evaluated to identify highly correlated descriptors in order to detect redundancy in the data set. Hence, descriptors with constant variables and near-constant variables were excluded from the further consideration (r2 ≥ 0.95). The genetic algorithm (GA) and multiple linear regression analysis (MLRA) were used to select the descriptors and to generate the correlation models that relate the structural features to the cell growth percent of different cancer cell lines. The combination of the GA-MLRA technique was applied to obtain the best QSAR models using the QSARINS 2.2.4. It splits compounds data as the following: random selection of 20% of compounds for prediction set and 80% for training set. For each obtained model, such random selection was different. Models, which showed statistical significance according to the parameters at a higher level (r2 ≥ 0.5), were selected for a more thorough rendering. For these models, the following options were given: the amount of generation algorithm setup was set until seven descriptors, and generation per size was established to the value of 10000.
4.5. Toxicity Studies
A tool CropCSM of Biosig Lab [65,66] was used for online prediction toxicities (Supplementary Materials, Table S2) of molecules to rapidly identify safe and effective herbicides on honey bee (A. mellifera), mallard and flathead minnow toxicity, in addition to measures of human health, including AMES toxicity, rat LD50 and oral chronic toxicity using SMILES of substances.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1: SMILES of studied substances; Table S2: Parameters of QSAR equation and their definition; Table S3: Predicted herbicide, environmental and human toxicity by CropCSM of Biosig Lab; Table S4: Pearson correlation results calculated in Origin 2018; Figures of IR, LC-MS, 1H and 13C spectra of c11 and c12; Table S5: CB-Dock2 website results of cavity detection and protein-ligand blind molecular docking.
Author Contributions
Conceptualization and methodology, L.A.; software, L.A. and O.A.; investigation, L.A., O.A., A.F., and I.K.; resources, L.A., A.F., S.K. and M.A.; original draft preparation, L.A.; writing- review and editing/visualization/project administration, L.A., O.A.; supervision, S.K. and M.A; funding acquisition, M.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by JSPS KAKENHI, grant number 24H01322, and JST FOREST Program, grant number JPMJFR205X.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All raw data from this study are available upon reasonable request to the corresponding author.
Acknowledgments
Authors gratefully acknowledge National University Corporation Kyushu University, Fukuoka, Japan, for opportunity to take part in the relief program for Ukrainian students and researchers; JSPS KAKENHI and JST FOREST Program; Armed Forces of Ukraine and Territorial Defense Forces of the Armed Forces of Ukraine for preparing this paper in the safe conditions of Zaporizhzhia, Ukraine; Enamine Ltd. (Kyiv, Ukraine) for technical support of synthetic work; large language model Claude 3 by Anthropic for assisting in English language.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Anderson, H.W. Yeast-Like Fungi of the Human Intestinal Tract. J. Infect. Dis. 1917, 21, 341–386. [Google Scholar] [CrossRef]
- Hazen, K.C. New and Emerging Yeast Pathogens. Clin. Microbiol. Rev. 1995, 8, 462–478. [Google Scholar] [CrossRef] [PubMed]
- Pappas, P.G.; Lionakis, M.S.; Arendrup, M.C.; Ostrosky-Zeichner, L.; Kullberg, B.J. Invasive Candidiasis. Nat. Rev. Dis. Primers 2018, 4, 18026. [Google Scholar] [CrossRef] [PubMed]
- Ryan, P.; Motherway, C.; Powell, J.; Elsaka, A.; Sheikh, A.A.; Jahangir, A.; O’Connell, N.H.; Dunne, C.P. Candidaemia in an Irish Intensive Care Unit Setting between 2004 and 2018 Reflects Increased Incidence of Candida glabrata. J. Hosp. Infect. 2019, 102, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Gülmez, D.; Sığ, A.K.; Akar, N.; Duyan, S.; Arıkan Akdağlı, S. Changing Trends in Isolation Frequencies and Species of Clinical Fungal Strains: What Do the 12-Years (2008-2019) Mycology Laboratory Data Tell About? Mikrobiyol. Bul. 2021, 55, 53–66. [Google Scholar] [CrossRef]
- Medeiros, M.A.; Melo, A.P.; Bento, A.D.; Souza, L.B.; Neto, F.D.; Garcia, J.B.; Zuza-Alves, D.L.; Francisco, E.C.; Melo, A.S.; Chaves, G.M. Epidemiology and Prognostic Factors of Nosocomial Candidemia in Northeast Brazil: A Six-Year Retrospective Study. PLoS ONE 2019, 14, e0221033. [Google Scholar] [CrossRef]
- Rodrigues, C.F.; Silva, S.; Henriques, M. Candida glabrata: A Review of Its Features and Resistance. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 673–688. [Google Scholar] [CrossRef]
- Domagk, D.; Bisping, G.; Poremba, C.; Fegeler, W.; Domschke, W.; Menzel, J. Common Bile Duct Obstruction Due to Candidiasis. Scand. J. Gastroenterol. 2001, 36, 444–446. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, Z.; Abolhasani, H.; Mirjafary, Z.; Najafi, G.; Heidari, F. Efficient Synthesis, Molecular Docking and ADMET Studies of New 5-Substituted Tetrazole Derivatives. J. Mol. Struct. 2023, 1277, 134867. [Google Scholar] [CrossRef]
- Break, T.J.; Desai, J.V.; Healey, K.R.; Natarajan, M.; Ferre, E.M.N.; Henderson, C.; Zelazny, A.; Siebenlist, U.; Cohen, O.J.; Schotzinger, R.J.; Garvey, E.P.; Lionakis, M.S. VT-1598 Inhibits the In Vitro Growth of Mucosal Candida Strains and Protects Against Fluconazole-Susceptible and -Resistant Oral Candidiasis in IL-17 Signaling-Deficient Mice. J. Antimicrob. Chemother. 2018, 73, 2089–2094. [Google Scholar] [CrossRef] [PubMed]
- Cleveland, A.A.; Harrison, L.H.; Farley, M.M.; Hollick, R.; Stein, B.; Chiller, T.M.; Lockhart, S.R.; Park, B.J. Declining Incidence of Candidemia and the Shifting Epidemiology of Candida Resistance in Two US Metropolitan Areas, 2008-2013: Results from Population-Based Surveillance. PLoS ONE 2015, 10, e0120452. [Google Scholar] [CrossRef] [PubMed]
- Arendrup, M.C.; Arikan-Akdagli, S.; Jørgensen, K.M.; Barac, A.; Steinmann, J.; Toscano, C.; Hoenigl, M. European Candidaemia Is Characterized by Notable Differential Epidemiology and Susceptibility Pattern: Results from the ECMM Candida III Study. J. Infect. 2023, 87, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.S.; Lee, S.S.; Chen, W.C.; Tseng, C.H.; Lee, N.Y.; Chen, P.L.; Li, M.C.; Syue, L.S.; Lo, C.L.; Ko, W.C.; Hung, Y.P. COVID-19-Associated Candidiasis and the Emerging Concern of Candida Auris Infections. J. Microbiol. Immunol. Infect. 2023, 56, 672–679. [Google Scholar] [CrossRef]
- Malani, A.N.; Psarros, G.; Malani, P.N.; Kauffman, C.A. Is Age a Risk Factor for Candida glabrata Colonisation? Mycoses 2011, 54, 531–537. [Google Scholar] [CrossRef]
- Gómez-García, O.; Andrade-Pavón, D.; Campos-Aldrete, E.; Ballinas-Indilí, R.; Méndez-Tenorio, A.; Villa-Tanaca, L.; Álvarez-Toledano, C. Synthesis, Molecular Docking, and Antimycotic Evaluation of Some 3-Acyl Imidazo[1,2-a]pyrimidines. Molecules 2018, 23, 599. [Google Scholar] [CrossRef]
- Osmaniye, D.; Baltacı Bozkurt, N.; Levent, S.; Benli Yardımcı, G.; Sağlık, B.N.; Ozkay, Y.; Kaplancıklı, Z.A. Synthesis, Antifungal Activities, Molecular Docking and Molecular Dynamic Studies of Novel Quinoxaline-Triazole Compounds. ACS Omega 2023, 8, 24573–24585. [Google Scholar] [CrossRef] [PubMed]
- Çevik, A.U.; Celik, I.; Işık, A.; Gül, Ü.D.; Bayazıt, G.; Bostancı, H.E.; Özkay, Y.; Kaplancıklı, Z.A. Synthesis, and Docking Studies of Novel Tetrazole-S-Alkyl Derivatives as Antimicrobial Agents. Phosphorus Sulfur Silicon Relat. Elem. 2023, 198, 137–144. [Google Scholar] [CrossRef]
- Atalay, E.V.; Asar, S. Determination of the Inhibition Effect of Hesperetin and Its Derivatives on Candida glabrata by Molecular Docking Method. Eur. Chem. Biotechnol. J. 2024, 1, 27–38. [Google Scholar] [CrossRef]
- De Groot, P.W.; Kraneveld, E.A.; Yin, Q.Y.; Dekker, H.L.; Groß, U.; Crielaard, W.; de Koster, C.G.; Bader, O.; Klis, F.M.; Weig, M. The Cell Wall of the Human Pathogen Candida glabrata: Differential Incorporation of Novel Adhesin-Like Wall Proteins. Eukaryot. Cell 2008, 7, 1951–1964. [Google Scholar] [CrossRef]
- Afsarian, M.H.; Farjam, M.; Zarenezhad, E.; Behrouz, S.; Rad, M.N.S. Synthesis, Antifungal Evaluation and Molecular Docking Studies of Some Tetrazole Derivatives. Acta Chim. Slov. 2019, 66, 874–887. [Google Scholar] [CrossRef]
- Upadhayaya, R.S.; Sinha, N.; Jain, S.; Kishore, N.; Chandra, R.; Arora, S.K. Optically Active Antifungal Azoles: Synthesis and Antifungal Activity of (2R,3S)-2-(2,4-Difluorophenyl)-3-(5-[2-[4-aryl-piperazin-1-yl]-ethyl]-tetrazol-2-yl/1-yl)-1-[1,2,4]-triazol-1-yl-butan-2-ol. Bioorg. Med. Chem. 2004, 12, 2225–2238. [Google Scholar] [CrossRef] [PubMed]
- Selvarasu, S.; Srinivasan, P.; Mannathusamy, G.; Maria Susai, B. Synthesis, Characterization, in Silico Molecular Modeling, Anti-Diabetic and Antimicrobial Screening of Novel 1-Aryl-N-tosyl-1H-tetrazole-5-carboxamide Derivatives. Chem. Data Collect. 2021, 32, 100648. [Google Scholar] [CrossRef]
- Antypenko, L.; Antypenko, O.; Karnaukh, I.; Rebets, O.; Kovalenko, S.; Arisawa, M. 5,6-Dihydrotetrazolo[1,5-c]Quinazolines: Toxicity Prediction, Synthesis, Antimicrobial Activity, Molecular Docking and Perspectives. Arch. Pharm. 2023, 356, e2300029. [Google Scholar] [CrossRef] [PubMed]
- Ni, T.; Chi, X.; Xie, F.; Li, L.; Wu, H.; Hao, Y.; Wang, X.; Zhang, D.; Jiang, Y. Design, Synthesis, and Evaluation of Novel Tetrazoles Featuring Isoxazole Moiety as Highly Selective Antifungal Agents. Eur. J. Med. Chem. 2024, 246, 115007. [Google Scholar] [CrossRef] [PubMed]
- Matysiak, J.; Niewiadomy, A.; Krajewska-Kułak, E.; Mącik-Niewiadomy, G. Synthesis of Some 1-(2,4-Dihydroxythiobenzoyl)Imidazoles, -Imidazolines and -Tetrazoles and Their Potent Activity Against Candida Species. Il Farmaco 2003, 58, 455–461. [Google Scholar] [CrossRef]
- Shantal, C.N.; Juan, C.C.; Sánchez, L.B.; Jerónimo, C.H.; Benitez, E.G. Candida glabrata is a Successful Pathogen: an Artist Manipulating the Immune Response. Microbiol. Res. 2022, 260, 127038. [Google Scholar] [CrossRef]
- Frías-De-León, M.G.; Hernández-Castro, R.; Conde-Cuevas, E.; García-Coronel, I.H.; Vázquez-Aceituno, V.A.; Soriano-Ursúa, M.A.; Farfán-García, E.D.; Ocharán-Hernández, E.; Rodríguez-Cerdeira, C.; Arenas, R.; Robledo-Cayetano, M.; Ramírez-Lozada, T.; Meza-Meneses, P.; Pinto-Almazán, R.; Martínez-Herrera, E. Candida glabrata Antifungal Resistance and Virulence Factors, a Perfect Pathogenic Combination. Pharmaceutics 2021, 13, 1529. [Google Scholar] [CrossRef]
- Nguyen, B.V.G.; Nguyen, H.H.N.; Vo, T.H.; Le, M.T.; Tran-Nguyen, V.K.; Vu, T.T.; Nguyen, P.V. Prevalence and Drug Susceptibility of Clinical Candida Species in Nasopharyngeal Cancer Patients in Vietnam. One Health 2023, 18, 100659. [Google Scholar] [CrossRef]
- Whaley, S.G. Contributions of the Major C. glabrata Triazole Resistance Determinants to the Novel Investigational Tetrazoles VT-1598 and VT-1161. Available online: https://doi.org/10.26226/morressier.5ac39997d462b8028d89a2ae. (accessed on 27 April 2024).
- Wiederhold, N.P.; Patterson, H.P.; Tran, B.H.; Yates, C.M.; Schotzinger, R.J.; Garvey, E.P. Fungal-Specific Cyp51 Inhibitor VT1598 Demonstrates In Vitro Activity Against Candida and Cryptococcus Species, Endemic Fungi, Including Coccidioides Species, Aspergillus Species and Rhizopus arrhizus. J. Antimicrob. Chemother. 2018, 73, 404–408. [Google Scholar] [CrossRef]
- Ni, Q.; Wu, X.; Su, T.; Jiang, C.; Dong, D.; Wang, D.; Chen, W.; Cui, Y.; Peng, Y. The Regulatory Subunits of CK2 Complex Mediate DNA Damage Response and Virulence in Candida Glabrata. BMC Microbiol. 2023, 23, 317. [Google Scholar] [CrossRef] [PubMed]
- Waseem, M.; Das, S.; Mondal, D.; Jain, M.; Thakur, J.K.; Subbarao, N. Identification of Novel Inhibitors Against Med15a KIX Domain of Candida glabrata. Int. J. Biol. Macromol. 2023, 253, 126720. [Google Scholar] [CrossRef]
- Qian, A.; Zheng, Y.; Wang, R.; Wei, J.; Cui, Y.; Cao, X.; Yang, Y. Design, Synthesis, and Structure-Activity Relationship Studies of Novel Tetrazole Antifungal Agents with Potent Activity, Broad Antifungal Spectrum and High Selectivity. Bioorg. Med. Chem. Lett. 2017, 27, 5741–5745. [Google Scholar] [CrossRef] [PubMed]
- Sheng, C.; Zhang, W.; Ji, H.; Zhang, M.; Song, Y.; Xu, H.; Zhu, J.; Miao, Z.; Jiang, Q.; Yao, J.; Zhou, Y.; Zhu, J.; Lü, J. Structure-Based Optimization of Azole Antifungal Agents by CoMFA, CoMSIA, and Molecular Docking. J. Med. Chem. 2006, 49, 2512–2525. [Google Scholar] [CrossRef] [PubMed]
- Andrade-Pavón, D.; Sánchez-Sandoval, E.; Tamariz, J.; Ibarra, J.A.; Hernández-Rodríguez, C.; Villa-Tanaca, L. Inhibitors of 3-Hydroxy-3-methylglutaryl Coenzyme A Reductase Decrease the Growth, Ergosterol Synthesis and Generation of petite Mutants in Candida glabrata and Candida albicans. Int. J. Mol. Sci. 2023, 24, 16868. [Google Scholar] [CrossRef]
- Warrilow, A.G.; Hull, C.M.; Parker, J.E.; Garvey, E.P.; Hoekstra, W.J.; Moore, W.R.; Schotzinger, R.J.; Kelly, D.E.; Kelly, S.L. The Clinical Candidate VT-1161 is a Highly Potent Inhibitor of Candida albicans CYP51 but Fails to Bind the Human Enzyme. Antimicrob. Agents Chemother. 2014, 58, 7121–7127. [Google Scholar] [CrossRef] [PubMed]
- Łukowska-Chojnacka, E.; Kowalkowska, A.; Gizińska, M.; Koronkiewicz, M.; Staniszewska, M. Synthesis of Tetrazole Derivatives Bearing Pyrrolidine Scaffold and Evaluation of Their Antifungal Activity Against Candida albicans. Eur. J. Med. Chem. 2019, 164, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Reithofer, V.; Fernandez-Pereira, J.; Alvarado, M.; de Groot, P.; Essen, L.O. A Novel Class of Candida glabrata Cell Wall Proteins with Beta-Helix Fold Mediates Adhesion in Clinical Isolates. PLoS Pathog. 2021, 17, e1009980. [Google Scholar] [CrossRef]
- Zhou, H.; Yang, N.; Li, W.; Peng, X.; Dong, J.; Jiang, Y.; Yan, L.; Zhang, D.; Jin, Y. Exploration of Baicalein-Core Derivatives as Potent Antifungal Agents: SAR and Mechanism Insights. Molecules 2023, 28, 6340. [Google Scholar] [CrossRef]
- Bondaryk, M.; Łukowska-Chojnacka, E.; Staniszewska, M. Tetrazole Activity against Candida albicans. The Role of KEX2 Mutations in the Sensitivity to (±)-1-[5-(2-Chlorophenyl)-2H-tetrazol-2-yl]propan-2-yl Acetate. Bioorg. Med. Chem. Lett. 2015, 25, 2657–2663. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-López, N.A.; Aguayo-Ortiz, R.; Cuéllar-Cruz, M. Molecular Modeling of the Phosphoglycerate Kinase and Fructose-Bisphosphate Aldolase Proteins from Candida glabrata and Candida albicans. Med. Chem. Res. 2023, 32, 2356–2369. [Google Scholar] [CrossRef]
- Juvvadi, P.R.; Lamoth, F.; Steinbach, W.J. Calcineurin as a Multifunctional Regulator: Unraveling Novel Functions in Fungal Stress Responses, Hyphal Growth, Drug Resistance, and Pathogenesis. Fungal Biol. Rev. 2014, 28, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Ryder, N.S. Terbinafine: Mode of Action and Properties of the Squalene Epoxidase Inhibition. Br. J. Dermatol. 1992, 126, 2–7. [Google Scholar] [CrossRef]
- Buschart, A.; Gremmer, K.; El-Mowafy, M.; van den Heuvel, J.; Mueller, P.P.; Bilitewski, U. A Novel Functional Assay for Fungal Histidine Kinases Group III Reveals the Role of HAMP Domains for Fungicide Sensitivity. J. Biotechnol. 2012, 157, 268–277. [Google Scholar] [CrossRef]
- Yau, K.P.S.; Weerasinghe, H.; Olivier, F.A.B.; Lo, T.L.; Powell, D.R.; Koch, B.; Beilharz, T.H.; Traven, A. The Proteasome Regulator Rpn4 Controls Antifungal Drug Tolerance by Coupling Protein Homeostasis with Metabolic Responses to Drug Stress. PLoS Pathog. 2023, 19, e1011338. [Google Scholar] [CrossRef] [PubMed]
- Sung, D.J.; Kim, J.G.; Won, K.J.; Kim, B.; Shin, H.C.; Park, J.Y.; Bae, Y.M. Blockade of K+ and Ca2+ channels by azole antifungal agents in neonatal rat ventricular myocytes. Biol. Pharm. Bull. 2012, 35, 1469–1475. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Li, T.; Yu, C.; Sun, S. Candida albicans Heat Shock Proteins and HSPS-Associated Signaling Pathways as Potential Antifungal Targets. Front. Cell. Infect. Microbiol. 2017, 7, 520. [Google Scholar] [CrossRef]
- Caplan, T.; Lorente-Macías, Á.; Stogios, P.J.; Evdokimova, E.; Hyde, S.; Wellington, M.A.; Liston, S.; Iyer, K.R.; Puumala, E.; Shekhar-Guturja, T.; Robbins, N.; Savchenko, A.; Krysan, D.J.; Whitesell, L.; Zuercher, W.J.; Cowen, L.E. Overcoming Fungal Echinocandin Resistance through Inhibition of the Non-Essential Stress Kinase YCK2. Cell Chem. Biol. 2020, 27, 269–282. [Google Scholar] [CrossRef]
- Antypenko, O.; Antypenko, L.; Kalnysh, D.; Kovalenko, S. ADME Properties Prediction of 5-Phenyl-5,6-Dihydrotetrazolo[1,5-c]Quinazolines. Grail Sci. 2022, 12-13, 684–692. [Google Scholar] [CrossRef]
- Antypenko, O.; Antypenko, L.; Kalnysh, D.; Kovalenko, S. Molecular Docking of 5-Phenyl-5,6-Dihydrotetrazolo[1,5-c]Quinazolines to Ribosomal 50S Protein L2P (2QEX). Grail Sci. 2022, 12-13, 693–698. [Google Scholar] [CrossRef]
- Antypenko, O.; Antypenko, L.; Rebets, O.; Kovalenko, S. Molecular Docking of 5-Phenyl-5,6-Dihydrotetrazolo[1,5-c]Quinazolines to Penicillin-Binding Protein 2X (PBP 2X) and Preliminary Results of Antifungal Activity. Grail Sci. 2022, 14-15, 615–620. [Google Scholar] [CrossRef]
- Antypenko, L.; Antypenko, O.; Fominichenko, A.; Karnaukh, I. Antifungal Activity of 5,6-Dihydrotetrazolo[1,5-c]Quinazoline Derivative against Several Candida Species. In Proceedings of the VI International Scientific and Practical Conference Theoretical and Empirical Scientific Research: Concept and Trends, Oxford, United Kingdom; 2024; pp. 416–418. [Google Scholar] [CrossRef]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular Docking: A Powerful Approach for Structure-Based Drug Discovery. CCADD 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- CB-Dock2. Cavity Detection Guided Blind Docking. Available online: https://cadd.labshare.cn/cb-dock2/index.php (accessed on 22 April 2024).
- Liu, Y.; Yang, X.; Gan, J.; Chen, S.; Xiao, Z.X.; Cao, Y. CB-Dock2: Improved Protein–Ligand Blind Docking by Integrating Cavity Detection, Docking and Homologous Template Fitting. Nucleic Acids Res. 2022, 50, W159–W164. [Google Scholar] [CrossRef] [PubMed]
- RCSB Protein Data Bank (RCSB PDB). Available online: https://www.rcsb.org/ (accessed on 22 April 2024).
- Roetzer, A.; Gabaldón, T.; Schüller, C. From Saccharomyces cerevisiae to Candida glabrata in a Few Easy Steps: Important Adaptations for an Opportunistic Pathogen. FEMS Microbiol. Lett. 2011, 314, 1–9. [Google Scholar] [CrossRef] [PubMed]
- DockRMSD. Docking Pose Distance Calculation. Available online: https://seq.2fun.dcmb.med.umich.edu//DockRMSD (accessed on 24 April 2024).
- Bell, E.W.; Zhang, Y. DockRMSD: An Open-Source Tool for Atom Mapping and RMSD Calculation of Symmetric Molecules through Graph Isomorphism. J. Cheminform. 2019, 11, 40. [Google Scholar] [CrossRef] [PubMed]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Tropsha, A. QSAR Modeling: Where Have You Been? Where Are You Going To? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [CrossRef] [PubMed]
- Gramatica, P. Principles of QSAR Modeling. IJQSPR 2020, 5, 61–97. [Google Scholar] [CrossRef]
- Gramatica, P.; Cassani, S.; Chirico, N. QSARINS-Chem: Insubria Datasets and New QSAR/QSPR Models for Environmental Pollutants in QSARINS. J. Comput. Chem. 2014, 35, 1036–1044. [Google Scholar] [CrossRef]
- Todeschini, R.; Consonni, V. Handbook of Molecular Descriptors; Mannhold, R. , Kubinyi, H., Timmerman, H., Eds.; Wiley-VCH: Weinheim, New York, 2000. [Google Scholar]
- Tropsha, A. Best Practices for QSAR Model Development, Validation, and Exploitation. Mol. Inform. 2010, 29, 476–488. [Google Scholar] [CrossRef]
- CropCSM: Identifying Safe and Potent Herbicides. Available online: https://biosig.lab.uq.edu.au/crop_csm/prediction (accessed on 24 April 2024).
- Douglas, E.V.; Pires, D.E.V.; Stubbs, K.A.; Mylne, J.S.; Ascher, D.B. CropCSM: Designing Safe and Potent Herbicides with Graph-Based Signatures. Brief. Bioinform. 2022, 23, bbac042. [Google Scholar] [CrossRef]
- Antypenko, L.; Rebets, O.; Karnaukh, I.; Antypenko, O.; Kovalenko, S. Step-by-Step Method for the Determination of Minimum Inhibitory Concentration of a Hydrophobic Biologically Active Substance by the Broth Dilution Method. Grail Sci. 2022, 17, 468–474. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Gramatica, P.; Chirico, N. QSARINS-chem: Insubria Datasets and New QSAR/QSPR Models for Environmental Pollutants in QSARINS. J. Comput. Chem. 2013, 34, 2121–2132. [Google Scholar] [CrossRef]
Figure 1.
Examples of reported antifungal compounds targeting Candida species, with a focus on the studied 5,6-dihydrotetrazolo[1,5-c]quinazolines (numbering follows a previous antimicrobial study [23], and includes investigated substances with two additional novel ones, c11 and c12, for continuity).
Figure 1.
Examples of reported antifungal compounds targeting Candida species, with a focus on the studied 5,6-dihydrotetrazolo[1,5-c]quinazolines (numbering follows a previous antimicrobial study [23], and includes investigated substances with two additional novel ones, c11 and c12, for continuity).
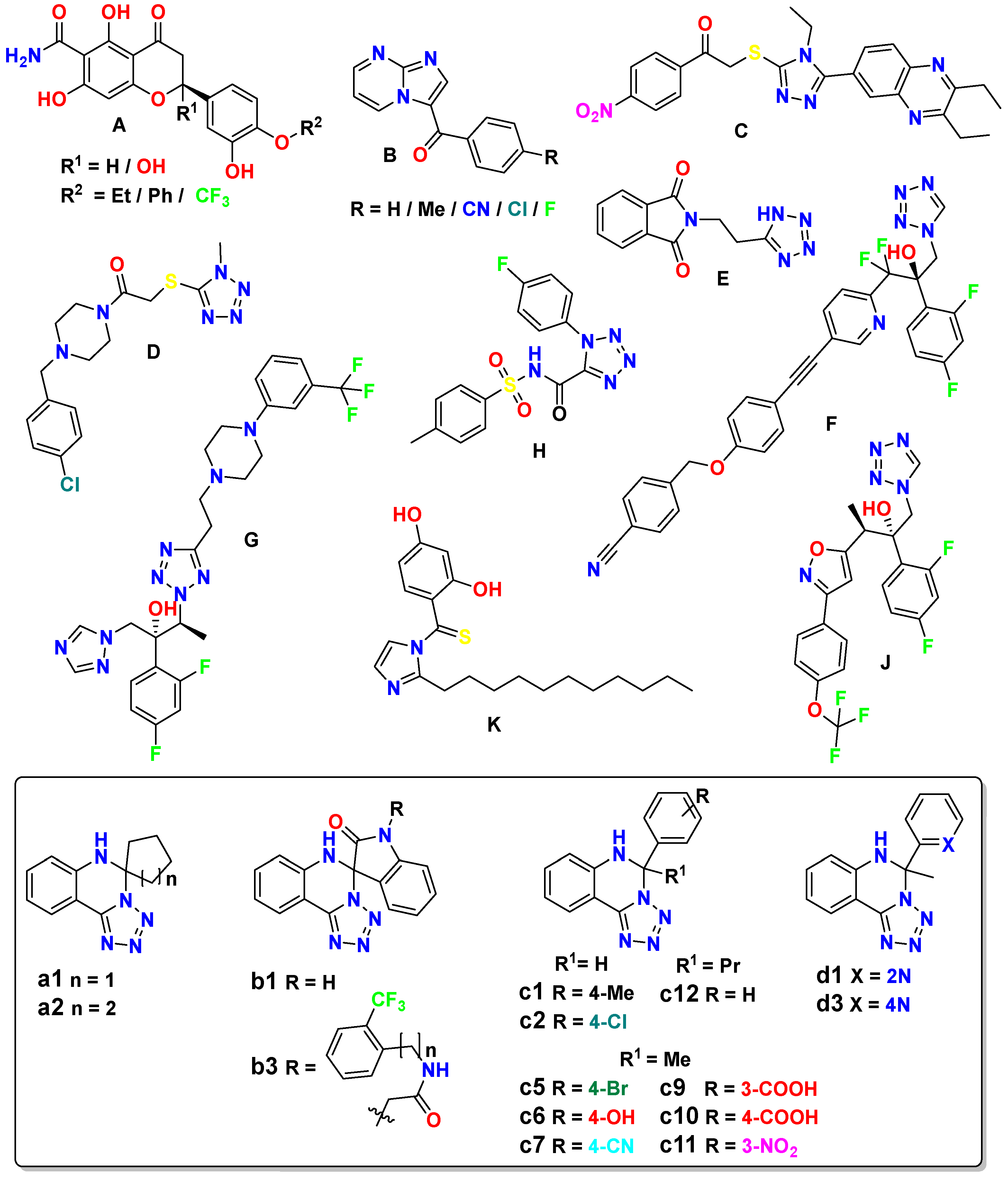
Figure 2.
Minimum inhibition concentration (μM) of 5,6-dihydrotetrazolo[1,5-c]quinazolines (yellow color) and references (Mf: micafungin, Cf: caspofungin, AfB: amphotericin B; blue color). And their structure-activity relationship against Candida glabrata. General molecular structure was optimized by HyperChem 8.0.8, and Discovery Studio v21.1.0.20298 was used for 3D visualization.
Figure 2.
Minimum inhibition concentration (μM) of 5,6-dihydrotetrazolo[1,5-c]quinazolines (yellow color) and references (Mf: micafungin, Cf: caspofungin, AfB: amphotericin B; blue color). And their structure-activity relationship against Candida glabrata. General molecular structure was optimized by HyperChem 8.0.8, and Discovery Studio v21.1.0.20298 was used for 3D visualization.
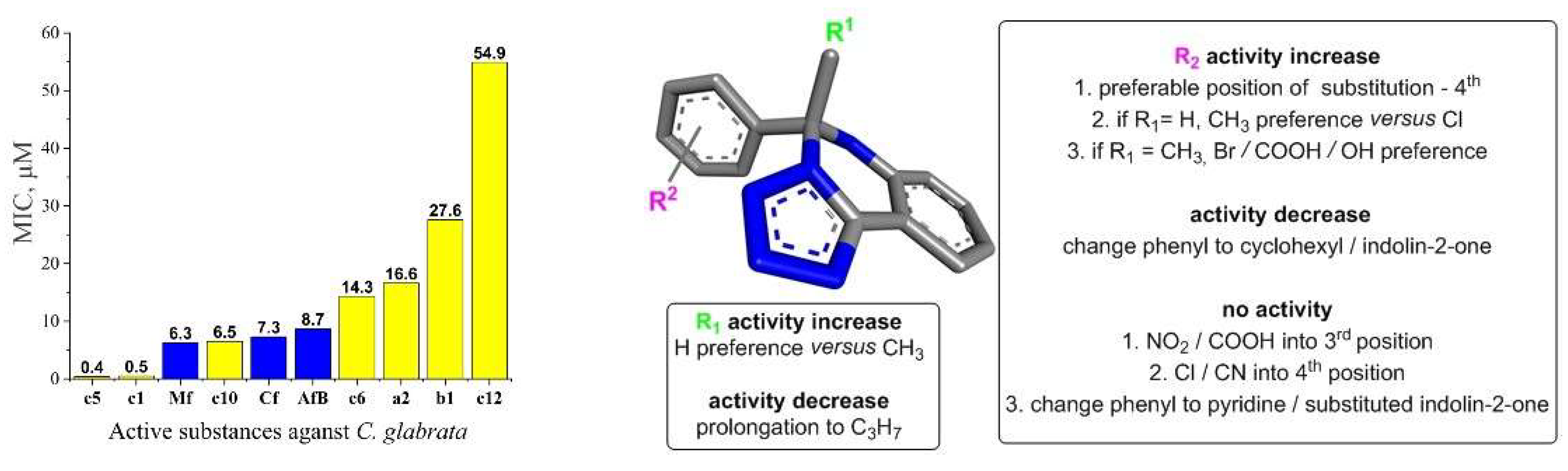
Figure 3.
Visual 3D representation of the sterol uptake control protein 2 (PDB ID: 7VPR) with lead-compound c1 (Vina score -10.4 kkal/mol), showing bonds formation in its cavity of chain D. All ten formed bonds were hydrophobic: π-σ in blue color; π-π stacked in light blue color; alkyl in pink color; π-alkyl in orange color. Discovery Studio v21.1.0.20298 was used for 3D visualization.
Figure 3.
Visual 3D representation of the sterol uptake control protein 2 (PDB ID: 7VPR) with lead-compound c1 (Vina score -10.4 kkal/mol), showing bonds formation in its cavity of chain D. All ten formed bonds were hydrophobic: π-σ in blue color; π-π stacked in light blue color; alkyl in pink color; π-alkyl in orange color. Discovery Studio v21.1.0.20298 was used for 3D visualization.
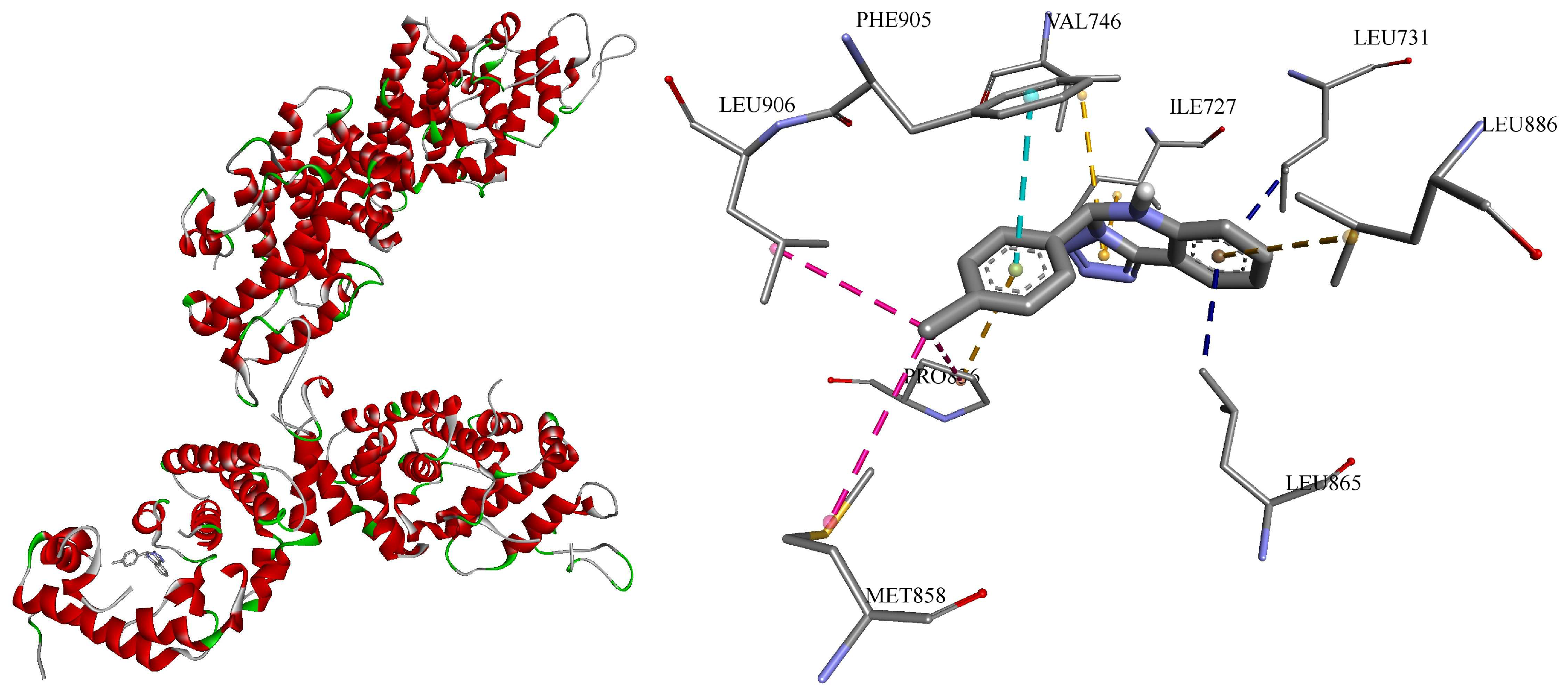
Figure 4.
Correlation graph of predicted vs. experimental MIC (μM / mg/L) of model equations.
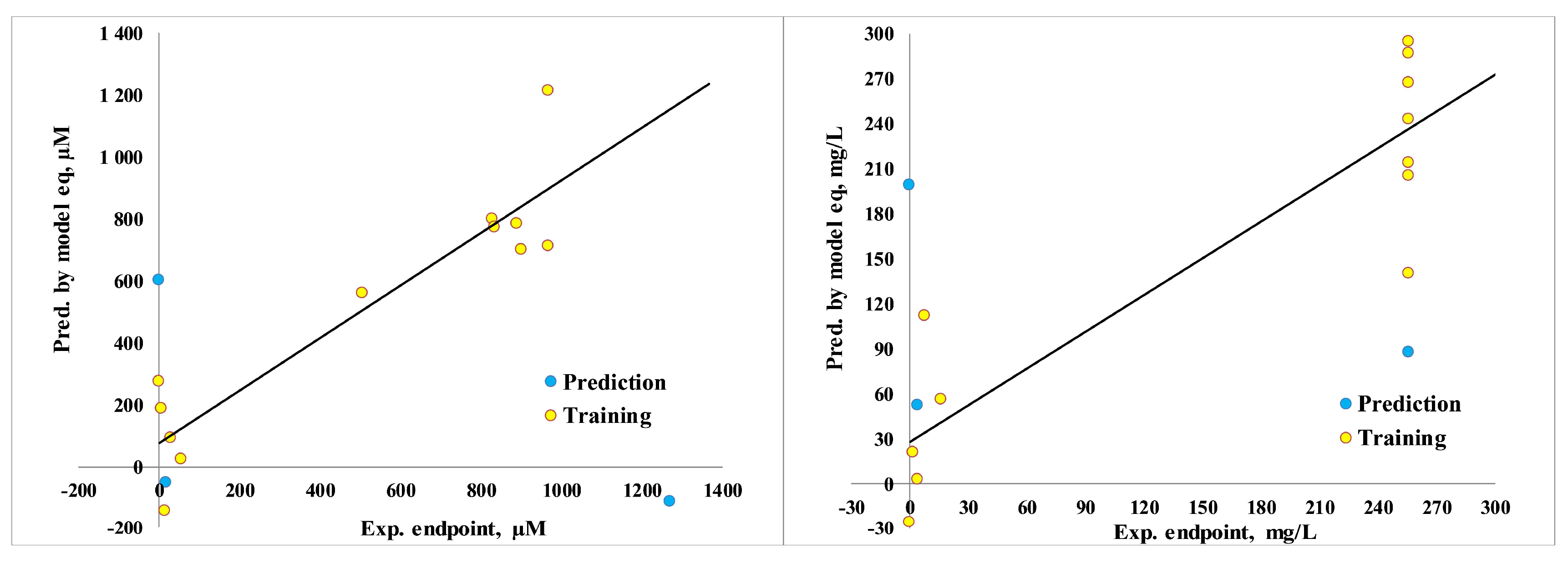
Figure 5.
Calculated minnow toxicity (log LD50, mg/kg/day, results were multiplied in 100 for the same scale; results below 30: high acute), rat acute toxicity (LD50, mg/kg; results were divided in 10 for the same scale; results under 5: strong; 5-50: moderate; 50-500: slightly; over 500: safe), and rat chronic toxicity (lowest observed adverse effect level (LOAEL), mg/kg/day; results under 10: strong; 10-50: medium; over 50: weak).
Figure 5.
Calculated minnow toxicity (log LD50, mg/kg/day, results were multiplied in 100 for the same scale; results below 30: high acute), rat acute toxicity (LD50, mg/kg; results were divided in 10 for the same scale; results under 5: strong; 5-50: moderate; 50-500: slightly; over 500: safe), and rat chronic toxicity (lowest observed adverse effect level (LOAEL), mg/kg/day; results under 10: strong; 10-50: medium; over 50: weak).
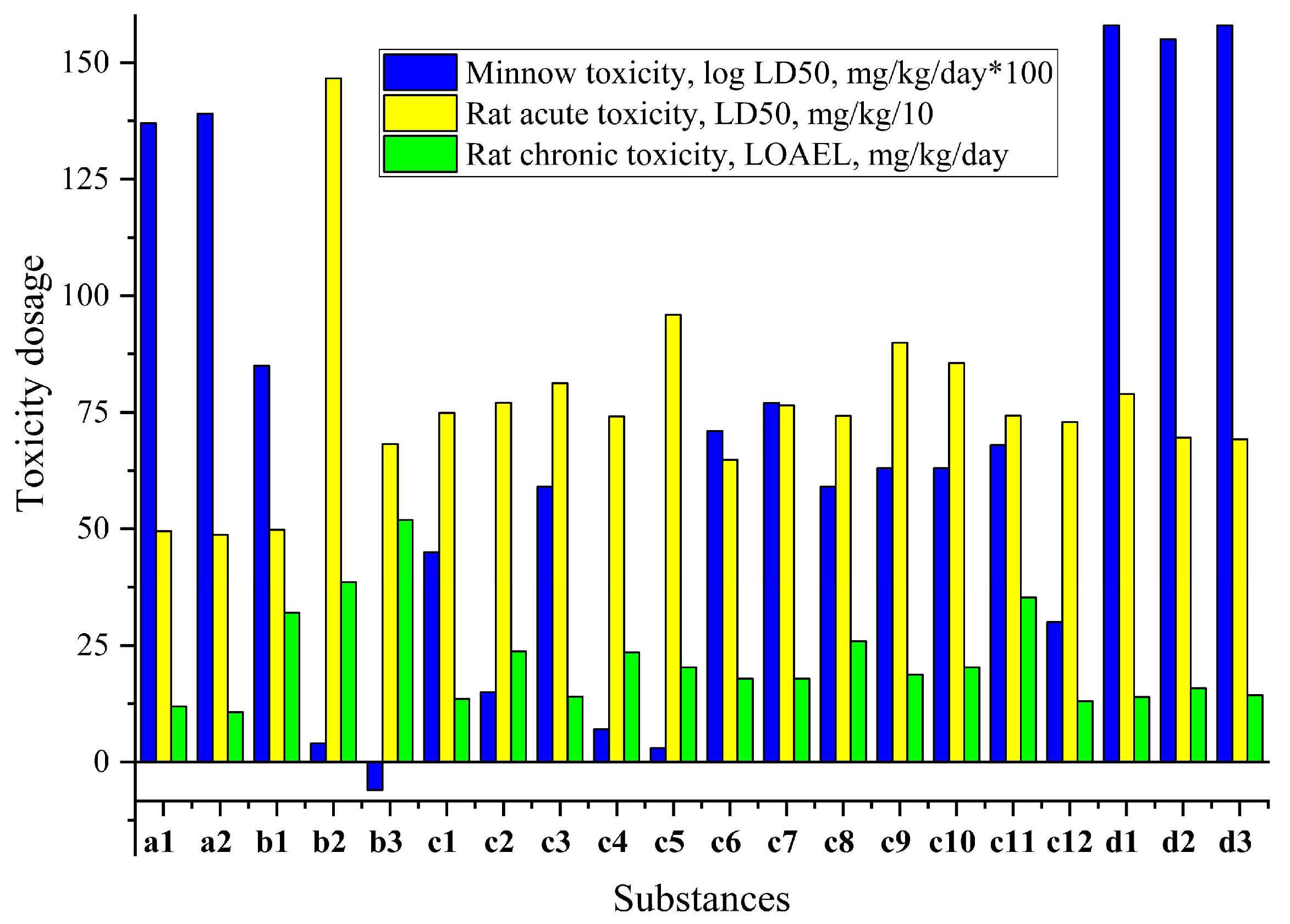
Figure 6.
Pearson coefficient of correlation between predicted affinity (Vina score, kcal/mol) to CYP51 (sterol 14-alpha demethylase, PDB ID - 5TZ1) [23] and toxicity (MT: minnow toxicity, log LD50, mg/kg/day), RAT: rat acute toxicity (LD50, mg/kg), RCT: rat chronic toxicity (LOAEL, mg/kg/day) [65].
Figure 6.
Pearson coefficient of correlation between predicted affinity (Vina score, kcal/mol) to CYP51 (sterol 14-alpha demethylase, PDB ID - 5TZ1) [23] and toxicity (MT: minnow toxicity, log LD50, mg/kg/day), RAT: rat acute toxicity (LD50, mg/kg), RCT: rat chronic toxicity (LOAEL, mg/kg/day) [65].

Table 1.
Antifungal activity results against C. glabrata by in vitro serial dilution method.
| Substance | Minimum inhibition concentration (64 – 0.125 mg/L), concentration of substance (μM) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 64 | 32 | 16 | 8 | 4 | 2 | 1 | 0.50 | 0.25 | 0.125 | |
| c1 | -* | - | - | - | - | - | - | - | - | 0.47 |
| c5 | - | - | - | - | - | - | - | - | - | 0.37 |
| c10 | - | - | - | - | - | 6.50 | + | + | + | + |
| a2 | - | - | - | - | 16.58 | + | + | + | + | + |
| c6 | - | - | - | - | 14.32 | + | + | + | + | + |
| b1 | - | - | - | 27.56 | + | + | + | + | + | + |
| c12 | - | - | 54.91 | + | + | + | + | + | + | + |
|
a1, b3, c2, c7, с9, c11, d1, d3 |
+ | + | + | + | + | + | + | + | + | + |
| Growth control | + | + | + | + | + | + | + | + | + | + |
| 2.5% DMSO control | + | + | + | + | + | + | + | + | + | + |
| Sterility control | - | - | - | - | - | - | - | - | - | - |
| *Absence (−) / presence (+) of opalescence. Minimum inhibition concentration of references: amphotericin B: 8 mg/L (8.66 μM), caspofungin: 8 mg/L (7.32 μM), and micafungin: 4 mg/L (6.30 μM). Repeated twice. | ||||||||||
Table 2.
The strongest calculated affinity to the various antifungal targets of c1 and c5, kcal/mol.
Table 2.
The strongest calculated affinity to the various antifungal targets of c1 and c5, kcal/mol.
| # | Strain* | Classification | Molecule ID | PDB ID** | # | Vina Score |
|---|---|---|---|---|---|---|
| 1 | SC S288C | transcription | sterol uptake control protein 2 | 4N9N | c1 | -9.6 |
| c5 | -9.9 | |||||
| 2 | NG CBS138 | transcription | sterol uptake control protein 2 | 7VPR | c1 | -10.4 |
| c5 | -7.9 | |||||
| 3 | CA | oxidoreductase / oxidoreductase inhibitor |
sterol 14-alpha demethylase | 5TZ1 | c1 | -10.2 |
| c5 | -9.2 | |||||
| 4 | NG CBS138 | oxidoreductase / oxidoreductase inhibitor |
lanosterol 14-alpha demethylase | 5JLC | c1 | -9.6 |
| c5 | -8.8 | |||||
| 5 | NG CBS138 | oxidoreductase / oxidoreductase inhibitor |
dihydrofolate reductase | 4HOG | c1 | -8.5 |
| c5 | -7.9 | |||||
| 6 | NG | oxidoreductase / oxidoreductase inhibitor |
NADPH-dependent methylglyoxal reductase GRE2 |
7YMU | c1 | -8.1 |
| c5 | -8.2 | |||||
| 7 | CA | hydrolase | exo-b-(1,3)-glucanase | 1EQP | c1 | -9.6 |
| c5 | -9.7 | |||||
| 8 | NG CBS138 | sugar binding protein | 4-alpha-glucanotransferase | 7EKU | c1 | -9.5 |
| c5 | -9.2 | |||||
| 9 | NG CBS138 | carbohydrate | 1,4-alpha-glucan-branching enzyme | 7P43 | c1 | -9.3 |
| c5 | -8.8 | |||||
| 10 | NG CBS138 | cell adhesion | adhesin-like wall protein 1 A-domain | 7O9Q | c1 | -9.4 |
| c5 | -9.1 | |||||
| 11 | NG CBS 138 | cell adhesion | epithelial adhesin 1 | 4D3W | c1 | -7.1 |
| c5 | -7.4 | |||||
| 12 | NG CBS138 | protein transport | importin subunit alpha | 7VPS | c1 | -9.8 |
| c5 | -9.3 | |||||
| 13 | SC | protein transport | importin alpha subunit | 2C1T | c1 | -9.2 |
| c5 | -8.7 | |||||
| 14 | NG CBS138 | transferase | 6,7-dimethyl-8-ribityllumazine synthase |
4KQ6 | c1 | -9.4 |
| c5 | -9.4 | |||||
| 15 | NG | transferase | flavin mononucleotide adenylyltransferase |
3FWK | c1 | -7.9 |
| c5 | -7.9 | |||||
| 16 | CA SC5314 | metal binding protein | enolase 1 | 7VRD | c1 | -8.1 |
| c5 | -8.1 | |||||
| 17 | NG | apoptosis | metacaspase-1 | 7QP0 | c1 | -7.6 |
| c5 | -7.8 | |||||
| 18 | NG CBS 138 | protein transport | importin alpha arm domain | 7VPT | c1 | -7.1 |
| c5 | -7.0 | |||||
| *SC - Saccharomyces cerevisiae, NG - Nakaseomyces glabratus (Candida glabrata), CA - Candida albicans. **Protein targets are taken from RCSB Protein Data Bank [56]. | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



