
Submitted:
22 October 2024
Posted:
22 October 2024
You are already at the latest version
Abstract
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder with cognitive dysfunction, memory decline, and behavioral disturbance, and pathologically characterized by the accumulation of amyloid plaques and neurofibrillary tangles in the brain. Although various hypotheses have been proposed to explain the pathogenesis of AD, including the amyloid beta hypothesis, oxidative stress hypothesis, and abnormal phosphorylation of tau proteins, the exact pathogenic mechanisms underlying AD remain largely undefined. Furthermore, effective curative treatments are very limited. Epidemiologic studies provide convincing evidence for a significant association between type 2 diabetes and AD. Here, we showed energy metabolism using glucose, lactate and ketone bodies and lipids as energy substrates in normal brain, and changes in such energy metabolism due to type 2 diabetes. We also showed the influences of such altered energy metabolism due to type 2 diabetes on the pathology of AD. Furthermore, we comprehensively searched the risk factors related with type 2 diabetes for AD, and showed possible therapeutic interventions based on considering risk factors and altered brain energy metabolism due to type 2 diabetes for the development of AD.
Keywords:
Alzheimer’s disease
; amyloid beta
; glucagon-like peptide 1 receptor agonists
; insulin resistance
; oxidative stress
; type 2 diabetes
1. Introduction
Alzheimer’s disease (AD) is the leading cause of cognitive impairment in older individuals (aged 65 and above) and one of the key factors of disability and mortality in later life throughout the world [1]. AD is a progressive neurodegenerative disorder characterized by the accumulation of amyloid plaques and neurofibrillary tangles in the brain, and this disease is an incurably neurodegenerative disorder with cognitive dysfunction, memory decline, and behavioral disturbance [2]. Various hypotheses have been proposed to explain the pathogenesis of AD, including the amyloid beta (Aβ) hypothesis [3], oxidative stress hypothesis [4], and abnormal phosphorylation of tau proteins [5]. However, the exact pathogenic mechanisms underlying AD remain largely undefined. The accumulation of soluble and insoluble aggregated Aβ may initiate or potentiate pathologic processes in AD. Aβ vaccines that are significantly capable of reducing amyloid plaques were unable to prevent AD progression [6]. Unfortunately, to date, almost all the clinical trials targeting at eliminating Aβ deposit have done little to improve cognitive function [6,7]. Recently, lecanemab, a humanized IgG1 monoclonal antibody that binds with high affinity to Aβ soluble protofibrils, was developed and tested to treat patients with early AD [8]. Lecanemab reduced brain amyloid burden in patients with early AD and resulted in moderately less decline on measures of cognition and mental function than placebo at 18 months [8]. However, longer trials are warranted to determine the efficacy and safety of lecanemab in early AD. Furthermore, the effective therapy based on tau pathology is lack [9,10,11].
The Rotterdam Study, a prospective population-based cohort study among 6,370 elderly subjects showed that diabetes almost doubled the risk of AD (relative risk [RR], 1.9; 95% confidence interval [CI], 1.2 to 3.1) [12]. The Hisayama study, Japanese cohort study,
showed that the age- and sex-adjusted incidence of AD was significantly higher in subjects with diabetes than in those with normal glucose tolerance. This association remained robust even after adjustment for confounding factors for AD (adjusted hazard ratio [aHR], 2.05; 95% CI, 1.18 to 3.57, p = 0.01). The risk of developing AD significantly increased with elevated 2-hour post-load glucose levels, but no such association was observed for fasting plasma glucose levels [13]. These studies suggest that impaired glucose metabolism is associated with the development of AD. Here, we show energy metabolism using glucose, lactate and ketone bodies and lipids as energy substrates in normal brain, and changes in such energy metabolism due to type 2 diabetes. We showed the influences of such altered energy metabolism due to type 2 diabetes on the pathology of AD. Furthermore, we comprehensively searched the risk factors related with type 2 diabetes for AD, and showed possible therapeutic interventions based on considering risk factors and altered brain energy metabolism due to type 2 diabetes for the development of AD.
2. Glucose Metabolism and Insulin Signaling in Normal Brain
Glucose transporters (GLUTs), insulin receptor (IR), insulin receptor substrate (IRS), and monocarboxylate transporters (MCTs) in normal brain were shown in Figure 1.
2.1. Neurons
The fundamental unit of the nervous system is the neuron, which forms complex circuits that receive and integrate information and generate adaptive responses [14]. Each neuron is composed of an input domain consisting of multiple dendrites along with the cell body, which is also responsible for the majority of macromolecule synthesis for the cell. The output domain is the axon which is a singular extension from the cell body that propagates the action potential to the synapse, where signals pass from one neuron to another. The insulin-independent GLUT3 is the major glucose transporter in neurons. The density and distribution of GLUT3 in axons, dendrites and neuronal soma correlates with local cerebral energy demands [15]. Neurons also express GLUT1 [16]. Activation by insulin induces GLUT4 translocation to the neuron cell membrane via an AKT-dependent mechanism [17,18] and is thought to improve glucose flux into neurons during periods of high metabolic demand, such as during learning [19]. Neurons express IRS1 and IRS2, which are enriched in presynaptic and postsynaptic compartments [20]. Insulin has many roles in neurons, and these roles are mediated by signalling through its two major effector pathways: the insulin–IRS–AKT and Mitogen-activated protein kinase (MAPK) pathways [21,22]. Insulin receptors are highly enriched in synapses [23], and have important effects on neurosynaptic functioning [24,25,26,27]. Furthermore, insulin has a crucial role in the development and maintenance of excitatory synapses [28] and has been shown to promote dendritic spine formation and excitatory synapse development [29]. Insulin promotes neuronal survival by inhibiting apoptosis [30]. Facilitating functions of neurons are supported by astrocytes, oligodendrocytes and microglia.
2.2. Astrocytes
Astrocytes are a subtype of glial cells that make up the majority of cells in the human central nervous system (CNS). They perform metabolic, structural, homeostatic, and neuroprotective tasks such as clearing excess neurotransmitters, stabilizing and regulating the blood-brain barrier (BBB), and promoting synapse formation [31,32,33,34]. Glucose is transported across the BBB through GLUT1 expressed on endothelial cells. Astrocytes take up glucose via GLUT1 and can process glucose glycolytically and transport lactate to neurons as an alternative fuel source during hypoglycaemia in a process known as the astrocyte–neuron lactate shuttle [35,36]. Astrocyte-neuron lactate transport is required for long-term memory formation [37]. Like hepatocytes, astrocytes have been known for a long time to have a glucose-containing energy reserve in the form of glycogen [38]. It seems clear that it is mobilized during periods of high neuronal activity through the action of various neuroactive substances and that an energy substrate arising from glycogenolysis is released by astrocytes, most likely to be used by neurons. Unlike the situation in hepatocytes, however, this released energy substrate is not glucose but lactate. Astrocytes can export lactate through monocarboxylate transporter (MCT) 1/4 [39]. The lactate may be taken up by oligodendrocytes through MCT1, or by neurons through MCT2 [39]. Astrocytes bind insulin with high affinity and express IRS1, IRS2 and downstream signalling molecules AKT and MAPK [40]. Activation of such signalling is mediated by insulin or insulin-like growth factor 1 (IGF1) which also activates IR [41,42,43]. Glial IR are downregulated in response to chronically high levels of insulin whereas neuronal IR are not downregulated [44]. Astrocytes play a part in inflammatory responses in the brain, and insulin modulates astrocyte inflammatory cytokine secretion in response to inflammatory stimuli [41].
2.3. Oligodendrocytes
Oligodendrocytes form the myelin sheath which surrounds axons and enables rapid conduction of the nerve impulse [45]. They are the end product of a cell lineage which has to undergo a complex and precisely timed program of proliferation, migration, differentiation, and myelination to finally produce the insulating sheath of axons. Due to this complex differentiation program, and due to their unique metabolism/physiology, oligodendrocytes count among the most vulnerable cells of the CNS. GLUT1 exists in oligodendrocytes [46]. Glucose may also be taken up by oligodendrocytes through GLUT1 [39]. The lactate from astrocytes may be taken up by oligodendrocytes through MCT1 [39]. The lactate is converted to pyruvate, which can be used for ATP production in oligodendrocytes may be particularly important to produce lipids for myelination. Oligidendrocytes express IR, IRS1 and IRS2. AKT signalling is important for mediating oligodendrocyte proliferation, survival, differentiation and myelination. The activation of AKT signalling by IGF1 in oligodendrocytes is well established and is known to promote differentiation and axonal ensheathment [47,48].
2.4. Microglia
Microglia are the resident immune cells, providing immune surveillance and remodeling of neuronal circuits during development and trauma. Microglia predominantly express GLUT3 and GLUT5, but under inflammatory conditions, GLUT1 expression is upregulated to increase glucose uptake and promote glycolysis [49,50,51,52]. Miicroglia express IR and IRS1, and insulin modulates microglial inflammatory responses in a complex manner [40]. Depending on its concentration in culture, insulin can enhance the secretion of certain inflammatory cytokines and inhibit the production of others.
3. Lipid Metabolism in Normal Brain
The brain is a highly energy-demanding organ. Brain utilizes about 20% of the total O2 consumed by the resting body [53]. Lipids are components that make up over 50% of the brain, however, energy provision to the brain preferentially relies on a continuous circulatory supply of glucose to neural cells; the utilization of fatty acids (FAs) is less preferred [54]. About 20% of the total energy consumption of the adult brain originates from the oxidation of FAs [55,56], predominantly occurring in astrocytes and not neurons [55]. However, lipids play a crucial role in the maintenance of normal brain function as structural constituents of neuronal and neuroglial cell membranes and precursors of signaling molecules [57]. Lipid metabolism in normal brain was shown in Figure 2.
3.1. Neurons and Astrocytes
Neurons and astrocytes operate as a tightly coupled unit for energy metabolism in the brain. Furthermore, neuron-astrocyte metabolic coupling protects against activity-induced FAs toxicity [58]. While neurons expend a considerable amount of ATP on neurotransmission, astrocytes provide neurons with metabolic substrates such as lactate and ketone bodies via MCTs, and antioxidants [59]. FAs are stored within cells as triacylglycerides (TG) in lipid droplets. FAs in lipid droplets removes excess free FAs (FFA) from the cytoplasm, which are toxic and disrupt mitochondrial membrane integrity [60,61]. Lipid droplets deliver FAs into mitochondria for an alternative energy source during periods of energy depletion [62]. Neurons do not make lipid droplets and have a low capacity for FAs consumption in mitochondria for energy production [63]. Neurons are especially susceptible to detrimental activities of reactive oxygen species (ROS) due to their poor antioxidative equipment [64]. Spurning the β-oxidation pathway in mitochondria of neurons and donation of metabolites to neurons for synthesis of antioxidants by astrocytes protect neurons against FFA-linked lipotoxicity [64]. Neuronal autophagy of ROS-emitting mitochondria combined with the transfer of degradation-committed FFA for their disposal in astrocytes, is a potent protective strategy against ROS and harmful activities of FFA [64].
During enhanced neuronal activity, lactate produced by astrocytes is transported to neurons via MCTs. In neurons, lactate is used in oxidative metabolism for ATP production and/or for FAs snthesis [65]. To avoid FFA toxicity, neurons release FAs in lipoprotein-like particles containing apo E, which can enter astrocytes by endocytosis. FAs are released from lipoprotein particles and are incorporated into lipid droplets [65]. FAs released from lipid droplets can be used as a fuel in mitochondrial β-oxidation in astrocytes. During energy deprivation, astrocytes metabolize FAs by β-oxidation to produce ketone bodies and transfer ketone bodies via MCTs to neuronal mitochondria for ATP synthesis [65].
3.2. Oligidendrocytes
Oligodendrocytes have much lower energetic demands than neurons, which require substantial energy to support the execution of action potentials [66,67]. Oligodendroglial energy demands are required for myelination and providing metabolic support to neurons. Oligodendrocytes support the energetic needs of neurons, being responsible for supplying neurons with glycolytic products, lactate and pyruvate, providing extra fuel to maintain their intense activity [68,69,70].
FAs can be taken up from the blood by endothelial cells and passed through the BBB by astrocytes to oligodendrocytes. Both oligodendrocytes and oligodendrocyte precursor cells express fatty acid transport proteins (FATP) and fatty acid binding proteins (FABPs) [71]. Ablation of these proteins impairs oligodendrocyte precursor cells proliferation and oligodendrocyte differentiation [72]. FAs derived from the circulation can support myelination, although they cannot fully substitute for specialized lipids that are internally synthetized by oligodendrocytes [73]. Interestingly, oligodendrocyte myelination also relies on lipids supplied by astrocytes, and when this synthesis is impaired, FAs derived by oligodendrocytes from the circulation may compensate [74]. FAs are stored as lipid droplets, mainly in astrocytes, but also in oligodendrocytes during development; however, these stored lipids are mainly used for myelin production [65].
3.3. Microglia
Microglia are brain-resident macrophages that constitute the largest population of immune cells in the CNS. They can initiate, modulate, and resolve inflammation [75,76]. Microglial lipid metabolism is tightly regulated during development, damage, and disease [77]. Microglia facilitate primary myelination [78,79,80], by phagocytosis of apoptotic oligodendrocytes and myelin debris during early development [81]. Microglial clearance of myelin derived lipids is required for optimal remyelination following demyelination [82,83]. Demyelination causes release of myelin-derived lipids such as ceramides, cholesterol, phospholipids, and sphingolipids. Healthy microglia clear lipids via cell surface scavenger receptors [76].
Lipoprotein lipase (LPL) is predominantly expressed in the microglia [84,85]. Moreover, microglial LPL expression has been implicated in brain development, damage, and disease [86,87,88]. Patients with AD show reduced LPL abundance in the hippocampus, and reduced LPL enzymatic activity in their cerebrospinal fluid (CSF) [88]. LPL plays a key role in microglial metabolism and function [87,89,90,91]. LPL is a feature of phagocytic and “reparative” microglia, and that LPL may support remyelination by facilitating lipid uptake and promoting FAs oxidation while maintaining an anti-inflammatory microglial phenotype [89].
4. Risk Factors Related with Type 2 Diabetes for the Development of AD
Risk factors related with type 2 diabetes for the development of AD were shown in Figure 3.
4.1. Insulin Resistance
A meta-analysis including 47 qualified articles with 2,981 patients showed that
fasting blood glucose levels (p < 0.001), fasting plasma insulin levels (p < 0.001), and homeostatic model assessment of insulin resistance (HOMA-IR) (p < 0.001) were higher in AD patients than in controls [92]. HOMA-IR was negatively correlated with Mini-Mental State Examination scale score (MMSE) (p = 0.001). This result suggests a significant association between insulin resistance and AD. Another meta-analysis showed a significant association between metabolic syndrome and AD, mainly driven by large retrospective studies [93]. The pooled analysis revealed an increased risk of AD in metabolic syndrome (HR, 1.10; 95% CI, 1.05 to 1.15).
The reduction of expression of insulin, IGF-1 and IGF-2 as well as their receptors participates in the pathogenesis of AD. The expression of insulin and IGF-1/2 receptors were markedly reduced in AD brains, which is correlated with the pathological alterations, including increased glycogen synthase kinase-3 (GSK-3) activity and amyloid precursor protein (APP) mRNA level [94]. Insulin/IGF-1 signaling defects was associated with phosphatidyl-inositide 3-kinases (PI3K)/Akt pathway through producing harmful cascades in glucose metabolism [95]. Reduced expression and function of PI3K/Akt-mediated GLUTs in AD brain could lead to brain glucose hypometabolism and the subsequent decline in mitochondrial ATP production [96]. The comparison in the function of brain insulin-PI3K-Akt signaling pathway in the frontal cortices among subjects with AD, type 2 diabetes, type 2 diabetes and AD, and control subjects, showed that the deficiency of insulin-PI3K-Akt signaling was more significant in subjects with both type 2 diabetes and AD [95]. The levels and the activation of the insulin-PI3K-Akt signaling components were negatively correlated with the level of tau phosphorylation [95]. MAPK pathway has been shown to be significantly activated in AD patients, which is correlated with increased neuroinflammation, tau hyperphosphorylation, and Aβ trafficking [96]. The tau hyperphosphorylation is related to an excess activation of GSK-3β and MAPK, which are major tau kinases responsible for tau phosphorylation [97,98].
4.2. Dyslipidemia
A dose-response meta-analysis of prospective cohort studies showed that every 3-mmol/L increase in serum total cholesterol (TC) or triglyceride (TG) is linearly associated with a 9% or 12% increase in RR of AD [99]. The longitudinal cohort study showed that LDL-C was associated with all measures of AD neuropathology (neurofibrillary tangles, beta-amyloid, Braak stage, modified Consortium to Establish a Registry for AD [CERAD] score and global AD pathology) and cerebral amyloid angiopathy independent of Apo E after adjusting for age, sex, cholesterol-lowering medication use, body mass index, smoking and education [100]. A systematic search of meta-analyses using 100 primary studies and five meta-analyses demonstrated that LDL-C levels increased risk for AD [101]. A meta-analysis of a case-control study indicated that increased TC levels were associated with an elevated risk of AD and decreased HDL-C levels and increased LDL-C levels may be related with an elevated risk of AD in subjects aged 60-70 [102]. In the meta-analysis, serum LDL-C and TC levels were associated with the risk of AD in Asian individuals [103]. A wealth of experimental and epidemiological evidence has suggested that apolipoprotein A-I (apoA-I), the main protein constituent of HDL, may protect against AD. In the meta-analysis, compared to healthy controls, AD subjects had a lower serum apo A-I level. The pooled weighted mean difference from a random-effects model was -0.31 g/L (p < 0.0001) (95% CI, -0.62 to 0.01) [104].
The human Apo E gene is encoded on chromosome 19, and it has three allelic variants: ε2, ε3, and ε4 [105]. The individuals carrying the Apo E ε4 allele exhibit a high risk of sporadic AD [106]. Individuals with a single Apo E ε4 allele have a 3.2 times higher risk of developing AD, whereas, in those with two Apo E ε4 alleles, the risk of developing AD is increased by 8 to 10 folds [107]. This can be attributed to the influence of the Apo E ε4 allele on Aβ, either by reducing its clearance or by increasing its production in the brain [108]. The study aimed to investigate whether Apo E alleles have a distinct impact on serum lipids in AD was performed [109]. Higher TC and LDL-C levels were found in Apo E ε4 allele carriers compared with non-carriers, and the difference was more significant in patients with AD than in healthy controls.
4.3. Advanced Glycation End Products (AGEs) and the Receptor for AGEs (RAGE)
AGEs accumulation in the brain leads to AD. AGEs are a crucial contributing factor to the onset and development of AD [110]. The need for focused treatment strategies is emphasized by the harmful consequences of AGEs on the brain, which include oxidative stress, inflammation, the development of amyloid plaques, and the creation of neurofibrillary tangles. RAGE, a multiligand receptor found in both neurons and cerebral microvascular endothelia that bind Aβ, is also a substrate for A disintegrin and metalloproteinase 10 (ADAM10) [111,112]. The interaction of RAGE with Aβ has been implicated in the amplification of oxidative stress, mitochondrial dysfunction and inflammation, resulting in RAGE-induced Alzheimer-like pathophysiological changes that contribute to the development of AD [113]. Ectodomain shedding of RAGE by ADAM10 generates a soluble counterpart of RAGE, sRAGE, which lacks the cytosolic and transmembrane domains, and sRAGE acts as a decoy receptor that antagonizes RAGE-mediated adverse effects [111,114]. The circulating sRAGE levels decreased in the plasma of AD patients [115]. The ADAM10 polymorphism was associated with the risk and development of AD by decreasing the expression of ADAM10 and reducing the plasma levels of sRAGE, which may be correlated with the clinical progression of AD [116].
4.4. Oxidative Stress
Abnormal oxidative stress is an established feature of AD. A meta-analysis of in vivo magnetic resonance spectroscopy studies showed that neuroinflammation and oxidative stress may occur in the early stages of AD, and may likely precede neuron loss in its progression [117]. Isoprostanes are biomarkers of oxidative stress. They are produced in membranes by ROS-induced lipid peroxidation of polyunsaturated fatty acids (PUFAs) like arachidonic acid. A systematic review of observational studies on the associations of F2-isoprostanes and the specific biomarker 8-iso-prostaglandin F2α which is the most abundantly produced F2-isoprostane with AD were conducted [118]. F2-isoprostane levels were significantly associated with AD [118]. F2-isoprostane and 8-iso-prostaglandin F2α levels were significantly elevated in tissue samples of the frontal lobe of AD patients. Moreover, F2-isoprostane levels in CSF and 8-iso-prostaglandin F2α levels in blood samples of AD patients were significantly increased.
The markers of lipid peroxidation are elevated in blood in AD, and total antioxidant capacity is decreased [119]. Non-enzymatic antioxidants in blood (uric acid, vitamins A, E and C, α- and β-carotene) were significantly decreased [119]. There is significant oxidative damage in peripheral blood early in the process of neurodegeneration. Selenium is an essential trace element well recognized for its antioxidant role in humans. Random-effects meta-analysis indicated a decrease (standard mean difference [SMD], - 0.42) in brain tissue selenium levels in AD as compared to non-AD controls [120]. The selenium levels were decreased in the temporal, hippocampal, and cortex regions in AD.
4.5. Inflammtion
The Neutrophil-to-Lymphocyte Ratio (NLR) is a clinical indicator of peripheral inflammation that has been extensively studied and is easily accessible. It is a straightforward calculation that indicates the equilibrium between the innate (neutrophils) and adaptive (lymphocytes) immune responses in different diseases and situations [121,122]. The mean NLR was 0.59 higher in AD patients compared to healthy participants (95% CI, 0.38 to 0.80) [123]. Similarly, the mean NLR was higher in AD than mild cognitive impairment (MCI) patients (MD, 0.23; 95% CI, 0.13 to 0.33). The mean NLR was higher in individuals with MCI compared to healthy participants (MD, 0.37; 95% CI, 0.22 to 0.52).
The 18-kDa translocator protein (TSPO) is increasingly recognized as a molecular target for positron emission tomography (PET) imaging of inflammatory responses in various CNS disorders [124,125]. Regions known to be vulnerable in AD, such as posterior cingulate cortex and hippocampus, are consistently shown to display increased TSPO PET signal [126]. In fact, volume changes assessed on MRI in these two regions, designated as epicentres of the pathology, are considered the best predictors of AD progression [127].
4.6. Mitochondrial Dysfunction
Mitochondrial dysfunction has also been observed prior to amyloid plaque deposition [128]. Mitochondrial dysfunction induces overproduction of ROS [129,130]. In AD, mitochondrial abnormalities are considered the main source of oxidative stress [131]. The PET studies also indicated that mitochondrial dysfunction has shown in the early stage of AD, and the mitochondrial-related energy failure may precede glycolysis-related hypometabolism in regions with pathologically confirmed early neurodegeneration in AD [132]. Proteomics studies showed that mitochondrial dysfunction is an early biochemical event that might play a central role in driving AD pathogenesis [133].
4.7. Impaired Autophagy
Autophagy, also known as self-cleaning and self-eating, is a conserved biological process in eukaryotes [134]. Autophagy can provide nutrients to maintain cellular function by breaking down macromolecules, organelles, proteins, and end products, under starvation conditions. Moreover, it also helps in maintaining cellular homeostasis by eliminating damaged organelles, misfolded proteins, and lipid droplets [135]. Autophagy is intricately linked to glycolipid levels within the body, and it exerts a protective effect on pancreatic β-cells [136]. Studies have shown that a prolonged high-glucose-high-fat diet can hinder autophagy while fasting and restricting calorie intake can activate it [137].
In AD, defects in endocytosis and autophagy have emerged as key drivers of accumulation of cellular waste products, progressive neuronal dysfunction, and neurodegeneration [138]. A systematic review and meta-analysis suggested that dsregulation of proteins in the endosomal-lysosomal and autophagy pathway may play an important role in AD pathogenesis [139].
4.8. Overactivity of GSK-3
A reduced ability of insulin to activate glucose transport in skeletal muscle, termed insulin resistance, is a primary defect leading to the development of impaired glucose tolerance and type 2 diabetes. GSK-3 is a serine/threonine kinase with important roles in the regulation of glycogen synthesis, protein synthesis, gene transcription, and cell differentiation in various cell types [140]. Overexpression and overactivity of GSK-3 in skeletal muscle of rodent models of obesity and obese type 2 diabetic humans are associated with an impaired ability of insulin to activate glucose disposal and glycogen synthase [140]. Selective inhibition of GSK-3 in insulin-resistant skeletal muscle causes improvements in insulin-stimulated glucose transport activity that are likely caused by enhanced post-insulin receptor insulin signaling and GLUT4 translocation [140]. Inhibiting GSK-3 activity by pharmacological intervention has become an important strategy for the management of type 2 diabetes [141]. The inhibition of GSK-3 improves insulin signaling through IRS-1, PI3K and protein kinase B (PKB/Akt) pathways [141]. GSK-3 overactivation disrupts neural growth, development, and function. It directly promotes tau phosphorylation, regulates APP cleavage, leading to Aβ formation, and directly or indirectly triggers neuroinflammation and oxidative damage [142].
4.9. Islet Amyloid Polypeptide (IAPP, or Amylin)
The IAPP plays a role in glucose homeostasis but aggregates to form islet amyloid in type 2 diabetes [143]. Islet amyloid formation contributes to β-cell dysfunction and death. IAPP is asscoaited with cctivation of the inflammasome, defects in autophagy, endoplasmic reticulum (ER) stress, generation of ROS, membrane disruption in AD brain. AD and type 2 diabetes are considered protein misfolding disorders associated with the accumulation of protein aggregates; Aβ and tau in the brain during AD, and IAPP in pancreatic islets in type 2 diabetes. Misfolded IAPP produced in type 2 diabetes may potentiate AD pathology by cross-seeding Aβ, providing a molecular explanation for the link between these diseases [144]. Transgenic animals expressing both IAPP and APP exhibited exacerbated AD-like pathology compared with AD transgenic mice or AD transgenic animals with type 1 diabetes [144]. IAPP was colocalized with amyloid plaques in brain parenchymal deposits, which suggested that IAPP and APP may directly interact and aggravate AD [144]. Inoculation of pancreatic IAPP aggregates into the brains of AD transgenic mice resulted in more severe AD pathology and significantly greater memory impairments than untreated animals. IAPP and Aβ co-aggregation and cross-seeding might contribute to the cross-talk between two diseases.
Furthemore, the ion-mobility mass spectrometry study showed the direct molecular interaction between tau and IAPP [145]. The effect of IAPP on tau aggregation and the IAPP-tau fibrils were tested in vitro [146]. IAPP interacts with tau and accelerates the formation of a more toxic strain with enhanced seeding activity and neurotoxicity. Intrahippocampal injection of the IAPP-tau strain into the tau P301S transgenic mice substantially promoted the spreading of tau pathology and induced more severe synapse loss and cognitive deficits compared with tau fibrils.
5. Changes in Energy Metabolism by Type 2 Diabetes and AD
Changes in expression of GLUTs by type 2 diabetes and AD were shown in Table 1.
5.1. Changes in GLUTs by Type 2 Diabetes
GLUT1 is expressed in all brain cells including the endothelial cells and with very low neuronal expression, while GLUT3 is almost restricted to neurons [147]. Levels of the main BBB carrier GLUT1 were found reduced in the hippocampus of insulin resistant rats [148]. Lower levels of both GLUT1 and GLUT3 were found in the brain of mice under a diet rich in fat and sugar for 3 months [149]. Mice fed an high fat diet for 3 months also showed reduced density of the neuronal GLUT3, and of the insulin-dependent GLUT4 that is key for synaptic fueling, when compared to controls [150]. Hippocampal neurons of obese type 2 diabetic subjects displayed reduced GLUT4 expression and neuronal soma area, associated with an increased expression of NF-κB. [151]. Insulin resistance and type diabetes reduce GLUT3 and GLUT4 which are crucial GLUTs for neuron.
5.2. Changes in GLUTs by AD
AD is characterized by cerebral glucose hypometabolism. Post-mortem studies showed consistent reductions in GLUT1 and GLUT3 in the hippocampus and cortex of AD brains, areas of the brain closely associated with AD pathology [152]. Longitudinal rodent studies clearly indicate that changes in GLUT1 and GLUT3 only occur after Aβ pathology is present, and several studies indicate Aβ itself may be responsible for changes in GLUTs expression [152]. Uncontrolled microglial activation has been implicated in AD [153], glucose uptake in microglia is facilitated predominately by GLUT1, particularly under inflammatory conditions [154]. 2-Deoxy-2-[18F]fluoro-d-glucose PET (FDG-PET) is widely used to study cerebral glucose metabolism. AD patients exhibited a positive association between glucose uptake and microglial activity [155]. Increased glucose uptake by activated microglia may deprive glucose availability in neruon, astrocytes and oligodendrocytes. Oligomeric Aβ causes impaired hippocampal insulin signaling and reduced GLUT4 translocation, accompanied by cognitive impairment and hippocampal hypometabolism [156]. GLUT4 immunoreactivity colocalises with cholinergic markers [157], suggesting a role for GLUT4 dysregulation in AD which is characterised by damage to cholinergic neurons [158,159]. GLUT2 was significantly increased in the AD brain and brains of subjects with both AD and type 2 diabetes, possibly due to astrocyte overactivation [160]. Thus, the increase of GLUT2 level in AD brain homogenates is most likely due simply to astrocyte over-activation, which is a well known phenomenon in AD brain [161].
5.3. Changes in MCTs by Type 2 Diabetes
Astrocytes mainly express MCT1 and MCT4, whereas neurons mainly express MCT2 [162,163]. The activity of the astrocyte-neuron lactate shuttle is involved in brain energetic support, and the process of lactate transport from astrocytes to neurons, which are important in neuronal energy support, depends on the activity of MCTs [164,165]. MCT2 protein levels in type 2 diabetes model rats decreased significantly in the hippocampus and hypothalamus compared to their controls [166], sugegsting that type 2 diabetes reduces lactate or ketone bodies availability of neuron.
5.4. Changes in MCTs by AD
Alterations of cerebral lactate metabolism in the double-transgenic APP/presenilin 1 (APP/PS1) mouse model of AD was studied [167]. Lactate content and expressions of cerebral MCT1, MCT2, and MCT4 were decreased in APP/PS1 mice, suggesting that the decreases of cerebral lactate content and lactate transporters may lead to the blockage of lactate and ketone bodies transport from glia to neurons, resulting in neuronal enegy substrate deficit in neurons.
5.5. Changes in Lipid Metabolism by AD
The utilization of FAs is less preferred in neuron [54], however, neurons in AD patients cannot glucose, lactate and ketone bodies due to reduced expression of GLUTs and MCTs. Therefore, neurons in AD patients must use FAs. Brain-type FABP levels were elevated in serum of 29% of the patients with AD, and in 2% of the healthy donors [168], suggesting an increased FA use in brain in AD patients. CSF levels of FABP3 were elevated in AD patients compared with control subjects (p < 0.01) [169]. Another work showed that CSF FABP3 concentration distinguished between healthy controls and patients with AD with a sensitivity and specificity of 76% and 84%, respectively. Both patients with AD and MCI due to AD had higher FABP3 levels in CSF when compared with cognitively healthy controls [170]. An increased FA use in brain may be a crucial determinant of the development of AD.
FFAs accumulated in non-adipose tissues are metabolized through the de novo synthesis pathway into lipid derivatives, such as ceramides and other sphingolipids [171]. Excessive accumulation of ceramides triggers a series of cellular stress responses, leading to apoptosis in different tissues and contributing to the pathogenesis of type 2 diabetes and AD [172,173,174]. Ceramide accumulation is involved in the pathogenesis of AD. Ceramide accumulation in the brain could be directly due to the increase in FFAs levels or indirectly promoted by FFAs-induced neuroinflammation under lipotoxic conditions [175]. High levels of FFAs can enter and be metabolized in the brain, resulting in increased ceramide synthesis [176]. An increase in ceramide levels has been observed in the brains of patients with early stages of AD [177]. Healthy microglia clear ceramide via cell surface scavenger receptors [76], however, activated microglia cannot clear ceramide which may increase ceramide accumulation.
Astrocytes provide key neuronal support, and their phenotypic transformation is implicated in neurodegenerative diseases. Brain critically depends on astrocytic mitochondrial oxidative phosphorylation to degrade FAs. Astrocytic mitochondria dysfunction is observed in patients with type 2 diabetes [178]. Aberrant astrocytic mitochondrial oxidative phosphorylation induces lipid droplet accumulation followed by neurodegeneration that recapitulates key features of AD [179]. When FA load overwhelms astrocytic mitochondrial oxidative phosphorylation capacity, elevated acetyl-CoA levels induce astrocyte reactivity by enhancing signal transducer and activator of transcription 3 (STAT3) acetylation and activation [179]. Intercellularly, lipid-laden reactive astrocytes stimulate neuronal FA oxidation and oxidative stress, activate microglia through interluekin-3 signaling, and inhibit the biosynthesis of FAs and phospholipids required for myelin replenishment [179].
Microglia become increasingly dysfunctional with aging and contribute to the onset of AD through defective phagocytosis, attenuated cholesterol efflux, and excessive secretion of pro-inflammatory cytokines. High blood glucose drives microglial activation and M1 polarization, and M1 microglia release pro-inflammatory cytokines, causing neuronal damage [180]. Dysfunctional microglia also accumulate lipid droplets. Microglia lacking LPL showed excessive accumulation of lipid droplet-like structures, and displayed a pro-inflammatory lipidomic profile, increased cholesterol ester content, and reduced cholesterol efflux [181]. Peroxisome proliferator-activated receptor (PPAR) agonists rescued the lipid droplet-associated phenotype in microglia lacking LPL [181].
5.6. Effects of Changes in Energy Metabolim in Brain Due to Type 2 Diabetes on the Development of AD
Effects of changes in energy metabolism in brain due to type 2 diabetes on the development of AD were shown in Figure 4.
Neurons are provided with energy substrates by astrocytes. Insulin resistance and type 2 diabetes reduce expressions of GLUTs in neuron and astrocytes. Increased glucose uptake by activated microglia further deprives glucose availability in neuron and astrocytes. Dysfunctional astrocytes cannot supply glucose, lactate and antioxidants to neuron. Astrocytic dysfunctional mitochondria cannot oxidize FAs, resulting in reduced supply of ketone bodies to neuron. Neurons critically depends on astrocytic mitochondrial oxidation to degrade FAs. Dysfunctional activated astrocytes stimulate neuronal FA oxidation and increase neuronal oxidative stress, activate microglia, and inhibit the biosynthesis of FAs required for myelin replenishment in oligodendrocytes. Furthermore, activated astrocytes and microglia induce inflammation. Such phenotypic changes in neurons, astrocytes, microglia and oligodendrocytes due to changes in energy metabolism due to type 2 diabetes may be associated with the development of AD.
6. Possible Therapeutic Intervention for AD Considering AD Risk Factors
Possible therapeutic intervention for AD based on considering AD risk factors were shown in Table 2.
6.1. Exercise
The meta-analysis including 29 prospective cohort studies showed a favorable effect of physical activity on AD risk decline (HR, 0.72; 95% CI, 0.65 to 0.80). Subgroup analysis of physical activity intensity demonstrated an inverse dose-response relationship between physical activity and AD, effect sizes of which were significant in moderate (HR, 0.85; 95% CI, 0.80 to 0.93) and high physical activity (HR, 0.56; 95% CI, 0.45 to 0.68), but not in low physical activity (HR, 0.94; 95% CI, 0.77 to 1.15) [182].
The meta-analysis including 27 randomized controlled trials (RCTs) showed a nonlinear dose-response relationship between exercise and cognitive improvement in AD, with the optimal aerobic exercise dose identified at 660 METs-min/week for enhancing cognitive function in AD [183].
6.2. Diet
In the meta-analysis comprising 35 studies, individuals reporting the highest vs. the lowest fish consumption were associated with a lower likelihood of AD (RR [relative risk], 0.80; 95% CI, 0.67 to 0.96) [184]. The Mediterranean diet was reported to reduce the risk of development of AD (OR, 0.73; 95% CI, 0.62 to 0.85) [185].
6.3. Antioxidants
6.3.1. Vitamin E
Individuals with AD had lower circulatory concentrations of α-tocophenol compared with healthy controls [186]. However, levels of β-, γ- and δ-tocophenols did not significantly differ between groups of AD and age-related cognitive deficits compared to controls. Another meta-analysis also showed that AD patients had a lower concentration of serum vitamin E compared with healthy controls among older people (WMD [weighed mean difference], -6.811 μmol/L; 95% CI, -8.998 to -4.625; p < 0.001) [187]. High vitamin E intake from diet and supplements significantly reduces the risk of AD (OR, 0.78; 95% CI, 0.64 to 0.94) [188].
6.3.2. Vitamin C
A Mendelian randomization study to investigate the causality of vitamin C on the risk of AD found suggestive evidence that genetic liability to higher vitamin C levels was associated with a lower risk of AD (OR, 0.968; 95% CI; 0.946 to 0.991; p = 0.007) using the fixed-effects inverse-variance-weighted method [189]. The meta-analysis including 73 eligible cohort studies totaling > 28,257 participants showed that the pooled RR of AD was 0.70 (95% CI, 0.51 to 0.95) for the dietary plus supplemental intake of vitamin C. Moreover, pooled RRs of AD and vitamin C intake per 20 mg/day increase were 0.98 (95% CI, 0.97 to 0.99) via dietary plus supplemental intake, 0.98 (95% CI, 0.96 to 1.00) in the dietary only intake [190].
6.3.3. Resveratrol
Resveratrol is a potent Sirtuin1 (SIRT1) enhancer that facilitates neuroprotection and promotes neurogenesis in the hippocampus of the adult brain [191]. In the meta-analysis comprised five trials involving 271 AD patients, compared with placebo therapy, resveratrol treatment resulted in a significant improvement in AD Cooperative Study- Activities of Daily Living (ADAS-ADL) scores, CSF Aβ40 and plasma Aβ40 levels (SMD, 0.43; 95% CI, 0.07 to 0.79) [192].
6.4. Statins
3-hydroxyl 3-methylglutaryl CoA reductase (HMGCR)-based anti-AD drugs can provide lipid-lowering-targeted and anti-inflammatory effects and have thus been receiving extensive attention in recent years [193]. As a highly functional gene of AD, HMGCR provides multi-target effects while inducing lipid accumulation, oxidative stress, Aβ deposition, and microgliosis, which further aggravate damage to the CNS. In the pooled analyses of the meta-analysis, statins were associated with a decreased risk of AD [21 studies, OR 0.68 (CI 0.56-0.81)] [194].
6.5. PPARα Agonists
PPAR-α is expressed in the brain and plays a significant role in oxidative stress, energy homeostasis, mitochondrial FA metabolism and inflammation. PPAR-α takes part in regulation of genes coding proteins that are involved in glutamate homeostasis and cholinergic/dopaminergic signaling in the brain [195]. PPAR-α regulates expression of genes coding enzymes engaged in APP metabolism, and activates gene coding of α secretase, which is responsible for APP degradation. It also down regulates β secretase (BACE-1), the main enzyme responsible for Aβ peptide release in AD. In AD brain expression of genes of PPAR-α and PPAR-γ coactivator-1 alpha (PGC-1α) is significantly decreased [195]. Specific activators of PPAR-α may be important for improvement of brain cells metabolism and cognitive function in AD.
Bezafibrate treatment led to significant improvement of cognitive/memory function in AD mice accompanied by alleviation of amyloid pathology and neuronal loss as well as reduced oxidative stress and neuroinflammation [196]. Fenofibrate increased the expression of PPAR-α, and decreased BACE-1 mRNA and protein levels, and also reduced soluble APP and Aβ releases in APP/PS1 transgenic mice [197].
6.6. n-3-PUFA
Omega-3 fatty acids (n-3-PUFA) such as eicosatetraenoic acid (EPA) and docosahexaenoic acid (DHA) are essential to normal neural development and function. Comparing the highest to lowest category of fish intake, a higher intake of fish was associated with a 36% (95% CI, 8 to 56%) lower risk of AD [198]. Dose-response meta-analysis showed that an increment of 100g per week of fish intake was associated with an 11% lower risk of AD (RR, 0.89; 95% CI, 0.79 to 0.99). In the meta-analysis, the Clinical Dementia Scale showed reduced progression of cognitive decline among AD patients with n-3-PUFA supplemental interventions, with no differences between different n-3-PUFA supplements [199].
6.7. Anti-Diabetic Drugs
A systematic umbrella review and meta-analysis including 100 reviews and 27 cohort/case-control studies (n = 3046661) showed that metformin, pioglitazone, glucagon-like peptide 1 receptor agonists (GLP-1RAs) and sodium glucose co-transporter-2 inhibitors (SGLT2is) were associated with significant reduction in risk of dementia [200].
6.7.1. Pioglitazone
Pioglitazone have shown promising effects on neuroinflammation and homeostasis of amyloid plaques, but there is a lack of research papers providing conclusive evidence. In the meta-analysis, the Wechsler Memory Scale-Revised logical memory I scores had a significant improvement in the pioglitazone group (MD, 2.02; 95% CI, 0.09 to 3.95), suggesting that pioglitazone is a safe medication that has a promising effect in slowing the advancement of AD [201].
6.7.2. Metformin
The study was performed to comprehensively investigate the risk of AD associated with various antidiabetic classes. A total of 1,565,245 patients from 16 studies were included. AD risks were significantly lower with metformin and SGLT2is than other antidiabetic drugs [202]. However, two meta-analyses failed to show that metformin reduces risk of AD [203,204].
6.7.3. SGLT2is
Acetylcholine is one of the important neurotransmitters in the human brain, and participates in the electrophysiological activity between neurons, ensuring signal transmission between neurons [205]. The cholinergic hypothesis revealed the important role of acetylcholine in AD, emphasizing that synaptic loss and atrophy in AD impair neurotransmitter conduction [206]. Reduced binding of acetylcholine and cholinergic receptors is one of the important reasons for the emergence of psychiatric symptoms in AD patients. Therefore, acetylcholinesterase (AChE) inhibitors (such as donepezil and galantamine) are used to improve cognitive function in AD patients. SGLT2 inhibition significantly reduced AChE activity and increased monoamine levels, leading to an improvement in memory dysfunction in mice [207]. There is emerging data from murine studies that SGLT2is can cross the BBB and may have neuroprotective effects, such as increasing the brain-derived neurotrophic factor (BDNF), reducing the amyloid burden, inhibiting AChE [208]. Furthermore, SGLT2is have the multifaceted mechanisms of action for the treatments of AD, such as encompassing antioxidative stress, anti-neuroinflammation, upregulation of autophagy, anti-apoptosis, and protection of endothelial cells [209].
A very recent database containing information on medications prescribed to 233183 patients aged 50 years or older between 2018 and 2020 showed that GLP-1RAs and SGLT2is might be associated with lower odds of anti-AD drugs usage, while insulins might be linked to higher odds of using anti-AD drugs [210]. A retrospective examination of data from a cohort of 1,348,362 participants with type 2 diabetes (≥ 40 years) showed that SGLT2is use was associated with reduced risks of AD (aHR, 0.81; 95% CI, 0.76 to 0.87) [211].
6.7.4. GLP-1RAs
GLP-1 has been shown to have neuroprotective properties in vitro and in vivo. In AD mouse model, APP/PS1 mice, liraglutide prevented memory impairments in object recognition and water maze tasks, and prevented synapse loss and deterioration of synaptic plasticity in the hippocampus [212]. In liraglutide-treated APP/PS1 mice, overall β-amyloid plaque count in the cortex and dense-core plaque numbers were reduced by 40-50%, while levels of soluble amyloid oligomers were reduced by 25%. Activated microglia was significantly reduced by liraglutide. Liraglutide increased young neurons in the dentate gyrus in APP/PS1 mice.
A systematic review and meta-analysis of preclinical studies showed that GLP-1 RAs could improve the learning and memory abilities of AD rodents; in terms of pathology, GLP-1 RAs could reduce Aβ deposition and phosphorylated tau levels in the brains of AD rodents [213].
In a placebo-controlled double-blind phase II clinical trial testing liraglutide in 200 patients with AD (NCT01843075), liraglutide significantly slowed down the deterioration in cognitive impairments, and found that brain temporal lobe volumes and parietal lobe volumes shrank less, and the total grey matter cortical volume shrank less in the liraglutide group compared to the placebo group as shown in MRI brain scans [214].
SIRT1 was found to be closely related to expression of GLP-1R in hippocampus of AD model mice [214]. Semaglutide increased the expression levels of SIRT1 and GLUT4 in the hippocampus of AD model mice, accompanied by an improvement in learning and memory, decreased in Aβ plaques and neurofibrillary tangles [215]. Further, semaglutide improved glucose metabolism by promoting glycolysis, improving glycolytic disorders, and increasing the membrane translocation of GLUT4, in the brain of AD model mice. These effects were blocked by the SIRT1 inhibitor, suggesting that SIRT1/GLUT4 signaling pathway may be an important mechanism for GLP-1RAs to promote glucose metabolism in the brain. Two phase III clinical trial testing semaglutide (Wegovy, Ozempic, Rybelsus) in AD patients are underway (NCT04777396 and NCT04777409) [216].
6.7.5. Dual GLP-1 and Glucose Dependent Insulinotropic Polypeptide (GIP) Receptor Agonist (Dual GLP-1/GIP RA)
GIP is a peptide hormone of the incretin family. In AD, energy utilization is much reduced, and GIP has the potential to reverse this [217]. Furthermore, GIP can reduce the inflammation response in the brain and reduce levels of pro-inflammatory cytokines. In GIP-treated AD models, memory is rescued, synapse numbers and synaptic plasticity in the hippocampus is normalized, amyloid plaque load and the chronic inflammation is reduced [217]. Recently, dual GLP-1/GIP receptor agonists have been developed.
Dual GLP-1/GIP RA administration significantly prevented spatial learning deficits, and decreased phosphorylated tau levels in the rat cerebral cortex and hippocampus in streptozotocin-induced AD model rat [218]. Dual GLP-1/GIP RA reduced chronic inflammation and apoptosis, by reactivating insulin signaling pathways. Dual GLP-1/GIP RA improved cognitive impairment in a range of tests and relieved pathological features of APP/PS1/tau mice, enhanced long-term potentiation in the hippocampus, increased numbers of synapses and dendritic spines, normalized volume and numbers of mitochondria, while downregulating amyloid, phosphorylated tau protein [219]. Dual GLP-1/GIP RA was more effective in reversing memory loss, enhancing synaptic plasticity in the hippocampus, reducing amyloid plaques and lowering pro-inflammatory cytokine levels in the brain than liraglutide in the APP/PS1 mouse model of AD [220].
6.7.6. Imeglimin
Mitochondrial dysfunction is a prominent pathological feature of type 2 diabetes and AD. Apoptotic cell death has been shown to constitute the terminal process in AD. A decrease in mitochondrial membrane potential causing opening of the permeability transition pore (PTP) in mitochondrial membranes has been implicated as a critical effector of apoptosis [221]. Opening of the PTP leads to the release of so-called apoptosis initiation factors that induce the degradative events of apoptosis [221].
Imeglimin improves mitochondrial dysfunction and reduces ROS [222]. The effects of imeglimin on ischemia-induced brain damage, induced in rats by the occlusion of the cerebral artery, was investigated [223]. Imeglimin significantly reduced the size of cerebral infarction, cerebral edema, and the neurological defects of ischemia. Imeglimin protected against ischemia-induced neuronal loss, microglial proliferation and activation, and increased astrocytes and cells that produce anti-inflammatory cytokines. Further, imeglimin acutely prevented the opening of PTP in cultured neurons and astrocytes. Such properties of imeglimin can be promising treatment for AD.
7. Conclusions
The functions of neurons, astrocytes, oligodendrocytes and microglia are highly regulated by glucose and insulin signaling in normal brain. Changes in brain energy metabolism due to type 2 diabetes are significantly associated with the development of AD. We showed possible therapeutic interventions based on considering risk factors and altered brain energy metabolism due to type 2 diabetes for the development of AD.
Author Contributions
H.Y., M.H., H.A., and H.K. conceived the review; H. Y. wrote the paper; H.K. edited the paper and provided critical guidance. authors have read and agreed to the published version of the manuscript.
Funding
This review research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest in relation to the present review paper.
References
- Jia, L.; Quan, M.; Fu, Y.; Zhao, T.; Li, Y.; Wei, C.; Tang, Y.; Qin, Q.; Wang, F.; Qiao, Y.; et al. Dementia in China: epidemiology, clinical management, and research advances. Lancet. Neurol. 2020, 19, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Dalal, S.; Ramirez-Gomez, J.; Sharma, B.; Devara, D.; Kumar, S. MicroRNAs and synapse turnover in Alzheimer's disease. Ageing. Res. Rev. 2024, 99, 102377. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science. 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Maccioni, R.B.; Farías, G.; Morales, I.; Navarrete, L. The revitalized tau hypothesis on Alzheimer’s disease. Arch. Med. Res. 2010, 41, 226–231. [Google Scholar] [CrossRef]
- Terry, A.V. Jr.; Buccafusco, J.J. The cholinergic hypothesis of age and Alzheimer’s disease-related cognitive deficits: Recent challenges and their implications for novel drug development. J. Pharmacol. Exp. Ther. 2003, 306, 821–827. [Google Scholar] [CrossRef]
- Holmes, C.; Boche, D.; Wilkinson, D.; Yadegarfar, G.; Hopkins, V.; Bayer, A.; Jones, R.W.; Bullock, R.; Love, S.; Neal, J.W.; et al. Long-term effects of Abeta42 immunisation in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008, 372, 216–223. [Google Scholar] [CrossRef]
- Gravitz, L. Drugs: a tangled web of targets. Nature. 2011, 475, S9–S11. [Google Scholar] [CrossRef]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S. ; Lecanemab in Early Alzheimer's Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef]
- Gozes, I. Tau pathology and future therapeutics. Curr. Alzheimer. Res. 2010, 7, 685–696. [Google Scholar] [CrossRef]
- Medina, M. Recent developments in tau-based therapeutics for neurodegenerative diseases. Recent. Pat. CNS. Drug. Discov. 2011, 6, 20–30. [Google Scholar] [CrossRef]
- Pritchard, S.M.; Dolan, P.J.; Vitkus, A.; Johnson, G.V. The toxicity of tau in Alzheimer disease: turnover, targets and potential therapeutics. J. Cell. Mol. Med. 2011, 15, 1621–1635. [Google Scholar] [CrossRef] [PubMed]
- Ott, A.; Stolk, R.P.; van Harskamp, F.; Pols, H.A.; Hofman, A.; Breteler, M.M. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology. 1999, 53, 1937–1942. [Google Scholar] [CrossRef]
- Ohara, T.; Doi, Y.; Ninomiya, T.; Hirakawa, Y.; Hata, J.; Iwaki, T.; Kanba, S.; Kiyohara, Y. Glucose tolerance status and risk of dementia in the community: the Hisayama study. Neurology. 2011, 77, 1126–1134. [Google Scholar] [CrossRef] [PubMed]
- Bigbee, J.W. Cells of the Central Nervous System: An Overview of Their Structure and Function. Adv. Neurobiol. 2023, 29, 41–64. [Google Scholar]
- Jurcovicova, J. Glucose transport in brain - effect of inflammation. Endocr. Regul. 2014, 48, 35–48. [Google Scholar] [CrossRef]
- Maher, F.; Davies-Hill, T.M.; Lysko, P.G.; Henneberry, R.C.; Simpson, I.A. Expression of two glucose transporters, GLUT1 and GLUT3, in cultured cerebellar neurons: Evidence for neuron-specific expression of GLUT3. Mol. Cell. Neurosci. 1991, 2, 351–360. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.S.; Reagan, L.P. Glucose transporter expression in the central nervous system: relationship to synaptic function. Eur. J. Pharmacol. 2004, 490, 13–24. [Google Scholar] [CrossRef]
- Grillo, C.A.; Piroli, G.G.; Hendry, R.M.; Reagan, L.P. Insulin-stimulated translocation of GLUT4 to the plasma membrane in rat hippocampus is PI3-kinase dependent. Brain. Res. 2009, 1296, 35–45. [Google Scholar] [CrossRef]
- Pearson-Leary, J.; McNay, E.C. Novel roles for the insulin-regulated glucose transporter-4 in hippocampally dependent memory. J. Neurosci. 2016, 36, 11851–11864. [Google Scholar] [CrossRef]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef]
- Nelson, T.J.; Sun, M.K.; Hongpaisan, J.; Alkon, D.L. Insulin, PKC signaling pathways and synaptic remodeling during memory storage and neuronal repair. Eur. J. Pharmacol. 2008, 585, 76–87. [Google Scholar] [CrossRef] [PubMed]
- van der Heide, L.P.; Ramakers, G.M.; Smidt, M.P. Insulin signaling in the central nervous system: learning to survive. Prog. Neurobiol. 2006, 79, 205–221. [Google Scholar] [CrossRef] [PubMed]
- Werther, G.A.; Hogg, A.; Oldfield, B.J.; McKinley, M.J.; Figdor, R.; Mendelsohn, F.A. Localization and characterization of insulin-like growth factor-i receptors in rat brain and pituitary gland using in vitro autoradiography and computerized densitometry* A distinct distribution from insulin receptors. J. Neuroendocrinol. 1989, 1, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Mielke, J.G.; Wang, Y.T. Insulin, synaptic function, and opportunities for neuroprotection. Prog. Mol. Biol. Transl. Sci. 2011, 98, 133–186. [Google Scholar]
- Gralle, M. The neuronal insulin receptor in its environment. J. Neurochem. 2017, 140, 359–367. [Google Scholar] [CrossRef]
- Fadel, J.R.; Reagan, L.P. Stop signs in hippocampal insulin signaling: the role of insulin resistance in structural, functional and behavioral deficits. Curr. Opin. Behav. Sci. 2016, 9, 47–54. [Google Scholar] [CrossRef]
- De Felice, F.G. Alzheimer’s disease and insulin resistance: translating basic science into clinical applications. J. Clin. Invest. 2013, 123, 531–539. [Google Scholar] [CrossRef]
- Chiu, S.L.; Chen, C.M.; Cline, H.T. Insulin receptor signaling regulates synapse number, dendritic plasticity, and circuit function in vivo. Neuron. 2008, 58, 708–719. [Google Scholar] [CrossRef]
- Lee, C.C.; Huang, C.C.; Hsu, K.S. Insulin promotes dendritic spine and synapse formation by the PI3K/Akt/mTOR and Rac1 signaling pathways. Neuropharmacology. 2011, 61, 867–879. [Google Scholar] [CrossRef]
- Kim, S.J.; Han, Y. Insulin inhibits AMPA-induced neuronal damage via stimulation of protein kinase B (Akt). J. Neural. Transm (Vienna). 2005, 112, 179–191. [Google Scholar] [CrossRef]
- Vasile, F.; Dossi, E.; Rouach, N. Human astrocytes: structure and functions in the healthy brain. Brain. Struct. Funct. 2017, 222, 2017–2029. [Google Scholar] [CrossRef] [PubMed]
- Oberheim, N.A.; Goldman, S.A.; Nedergaard, M. Heterogeneity of astrocytic form and function. Methods. Mol. Biol. 2012, 814, 23–45. [Google Scholar]
- Ransom, B.R.; Ransom, C.B. Astrocytes: multitalented stars of the central nervous system. Methods. Mol. Biol. 2012, 814, 3–7. [Google Scholar] [PubMed]
- Benarroch, E.E. Neuron-astrocyte interactions: partnership for normal function and disease in the central nervous system. Mayo. Clin. Proc. 2005, 80, 1326–1338. [Google Scholar] [CrossRef]
- Wender, R.; Brown, A.M.; Fern, R.; Swanson, R.A.; Farrell, K.; Ransom, B.R. Astrocytic glycogen influences axon function and survival during glucose deprivation in central white matter. J. Neurosci. 2000, 20, 6804–6810. [Google Scholar] [CrossRef]
- Pellerin, L.; Pellegri, G.; Bittar, P.G.; Charnay, Y.; Bouras, C.; Martin, J.L.; Stella, N.; Magistretti, P.J. Evidence supporting the existence of an activity-dependent astrocyte-neuron lactate shuttle. Dev. Neurosci. 1998, 20, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Stern, S.A.; Bozdagi, O.; Huntley, G.W.; Walker, R.H.; Magistretti, P.J.; Alberini, C.M. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell. 2011, 144, 810–823. [Google Scholar] [CrossRef]
- Waitt, A.E.; Reed, L.; Ransom, B.R.; Brown, A.M. Emerging roles for glycogen in the CNS. Front. Mol. Neurosci. 2017, 10, 73. [Google Scholar] [CrossRef] [PubMed]
- Rinholm, J.E.; Hamilton, N.B.; Kessaris, N.; Richardson, W.D.; Bergersen, L.H.; Attwell, D. Regulation of oligodendrocyte development and myelination by glucose and lactate. J. Neurosci. 2011, 31, 538–548. [Google Scholar] [CrossRef]
- Albrecht, J.; Wroblewska, B.; Mossakowski, M.J. The binding of insulin to cerebral capillaries and astrocytes of the rat. Neurochem. Res. 1982, 7, 489–494. [Google Scholar] [CrossRef]
- Garwood, C.J.; Ratcliffe, L.E.; Morgan, S.V.; Simpson, J.E.; Owens, H.; Vazquez-Villaseñor, I.; Heath, P.R.; Romero, I.A.; Ince, P.G.; Wharton, S.B. Insulin and IGF1 signalling pathways in human astrocytes in vitro and in vivo; characterisation, subcellular localisation and modulation of the receptors. Mol. Brain. 2015, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Spielman, L.J.; Bahniwal, M.; Little, J.P.; Walker, D.G.; Klegeris, A. Insulin modulates in vitro secretion of cytokines and cytotoxins by human glial cells. Curr. Alzheimer. Res. 2015, 12, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Heni, M.; Hennige, A.M.; Peter, A.; Siegel-Axel, D.; Ordelheide, A.M.; Krebs, N.; Machicao, F.; Fritsche, A.; Häring, H.U.; Staiger, H. . Insulin promotes glycogen storage and cell proliferation in primary human astrocytes. PLoS. ONE. 2011, 6, e21594. [Google Scholar] [CrossRef]
- Clarke, D.W.; Boyd, F.T. Jr.; Kappy, M.S.; Raizada, M.K. Insulin binds to specific receptors and stimulates 2-deoxy-D-glucose uptake in cultured glial cells from rat brain. J. Biol. Chem. 1984, 259, 11672–11675. [Google Scholar] [CrossRef]
- Bradl, M.; Lassmann, H. Oligodendrocytes: biology and pathology. Acta. Neuropathol. 2010, 119, 37–53. [Google Scholar] [CrossRef]
- Yu, S.; Ding, W.G. The 45 kDa form of glucose transporter 1 (GLUT1) is localized in oligodendrocyte and astrocyte but not in microglia in the rat brain. Brain. Res. 1998, 797, 65–72. [Google Scholar] [CrossRef]
- Ye, P.; Li, L.; Lund, P.K.; D’Ercole, A.J. Deficient expression of insulin receptor substrate-1 (IRS-1) fails to block insulin-like growth factor-I (IGF-I) stimulation of brain growth and myelination. Brain. Res. Dev. Brain. Res. 2002, 136, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.L.; Fragoso, G.; Miron, V.E.; Darlington, P.J.; Mushynski, W.E.; Antel, J.; Almazan, G. Response of human oligodendrocyte progenitors to growth factors and axon signals. J. Neuropathol. Exp. Neurol. 2010, 69, 930–944. [Google Scholar] [CrossRef]
- Kalsbeek, M.J.; Mulder, L.; Yi, C.X. Microglia energy metabolism in metabolic disorder. Mol. Cell. Endocrinol. 2016, 438, 27–35. [Google Scholar] [CrossRef]
- Payne, J.; Maher, F.; Simpson, I.; Mattice, L.; Davies, P. Glucose transporter Glut 5 expression in microglial cells. Glia. 1997, 21, 327–331. [Google Scholar] [CrossRef]
- Douard, V.; Ferraris, R.P. Regulation of the fructose transporter GLUT5 in health and disease. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E227–37. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Pavlou, S.; Du, X.; Bhuckory, M.; Xu, H.; Chen, M. Glucose transporter 1 critically controls microglial activation through facilitating glycolysis. Mol. Neurodegener. 2019, 14, 2. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.J.; Attwell, D. The energetics of CNS white matter. J. Neurosci. 2012, 32, 356–371. [Google Scholar] [CrossRef]
- Schonfeld, P.; Reiser, G. Why does brain metabolism not favor burning of fatty acids to provide energy? Reflections on disadvantages of the use of free fatty acids as fuel for brain. J. Cereb. Blood Flow Metab. 2013, 33, 1493–1499. [Google Scholar] [CrossRef]
- Ebert, D.; Haller, R.G.; Walton, M.E. Energy contribution of octanoate to intact rat brain metabolism measured by 13c nuclear magnetic resonance spectroscopy. J. Neurosci. 2003, 23, 5928–5935. [Google Scholar] [CrossRef]
- Panov, A.; Orynbayeva, Z.; Vavilin, V.; Lyakhovich, V. Fatty acids in energy metabolism of the central nervous system. BioMed. Res. Int. 2014, 2014, 472459. [Google Scholar] [CrossRef] [PubMed]
- Barber, C.N.; Raben, D.M. Lipid metabolism crosstalk in the brain: Glia and neurons. Front. Cell. Neurosci. 2019, 13, 212. [Google Scholar] [CrossRef]
- Ioannou, M.S.; Jackson, J.; Sheu, S.H.; Chang, C.L.; Weigel, A.V.; Liu, H.; Pasolli, H.A.; Xu, C.S.; Pang, S.; Matthies, D.; et al. Neuron-Astrocyte Metabolic Coupling Protects against Activity-Induced Fatty Acid Toxicity. Cell. 2019, 177, 1522–1535. [Google Scholar] [CrossRef]
- Bélanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell. Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef]
- Unger, R.H.; Clark, G.O.; Scherer, P.E.; Orci, L. Lipid homeostasis, lipotoxicity and the metabolic syndrome. Biochim. Biophys. Acta. 2010, 1801, 209–214. [Google Scholar] [CrossRef]
- Nguyen, T.B.; Louie, S.M.; Daniele, J.R.; Tran, Q.; Dillin, A.; Zoncu, R.; Nomura, D.K.; Olzmann, J.A. DGAT1-Dependent Lipid Droplet Biogenesis Protects Mitochondrial Function during Starvation-Induced Autophagy. Dev. Cell. 2017, 42, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Rambold, A.S.; Cohen, S.; Lippincott-Schwartz, J. Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev. Cell. 2015, 32, 678–692. [Google Scholar] [CrossRef] [PubMed]
- Schönfeld, P.; Reiser, G. Why does brain metabolism not favor burning of fatty acids to provide energy? Reflections on disadvantages of the use of free fatty acids as fuel for brain. J. Cereb. Blood. Flow. Metab. 2013, 33, 1493–1499. [Google Scholar] [CrossRef] [PubMed]
- Schönfeld, P.; Reiser, G. How the brain fights fatty acids' toxicity. Neurochem. Int. 2021, 148, 105050. [Google Scholar] [CrossRef] [PubMed]
- Smolič, T.; Zorec, R.; Vardjan, N. Pathophysiology of Lipid Droplets in Neuroglia. Antioxidants (Basel). 2021, 11, 22. [Google Scholar] [CrossRef]
- Rone, M.B.; Cui, Q.L.; Fang, J.; Wang, L.C.; Zhang, J.; Khan, D.; Bedard, M.; Almazan, G.; Ludwin, S.K.; Jones, R.; et al. Oligodendrogliopathy in multiple sclerosis: Low glycolytic metabolic rate promotes oligodendrocyte survival. J. Neurosci. 2016, 36, 4698–4707. [Google Scholar] [CrossRef]
- Rosko, L.; Smith, V.N.; Yamazaki, R. , Huang, J.K. Oligodendrocyte bioenergetics in health and disease. Neuroscientist. 2019, 25, 334–343. [Google Scholar] [CrossRef]
- Fünfschilling, U.; Supplie, L.M.; Mahad, D.; Boretius, S.; Saab, A.S.; Edgar, J.; Brinkmann, B.G.; Kassmann, C.M.; Tzvetanova, I.D.; Möbius, W.; et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature. 2012, 485, 517–521. [Google Scholar] [CrossRef]
- Saab, A.S.; Tzvetanova, I.D.; Nave, K.A. The role of myelin and oligodendrocytes in axonal energy metabolism. Curr. Opin. Neurobiol. 2013, 23, 1065–1072. [Google Scholar] [CrossRef]
- Späte, E.; Zhou, B.; Sun, T.; Kusch, K.; Asadollahi, E.; Siems, S.B.; Depp, C.; Werner, H.B.; Saher, G.; Hirrlinger, J.; et al. Downregulated expression of lactate dehydrogenase in adult oligodendrocytes and its implication for the transfer of glycolysis products to axons. Glia. 2024, 72, 1374–1391. [Google Scholar] [CrossRef]
- Fernandes, M.G.F.; Pernin, F.; Antel, J.P.; Kennedy, T.E. From BBB to PPP: Bioenergetic requirements and challenges for oligodendrocytes in health and disease. J. Neurochem. 2024. [Google Scholar] [CrossRef] [PubMed]
- Poitelon, Y.; Kopec, A.M.; Belin, S. Myelin fat facts: An overview of lipids and fatty acid metabolism. Cells. 2020, 9, 812. [Google Scholar] [CrossRef] [PubMed]
- Dimas, P.; Montani, L.; Pereira, J.A.; Moreno, D.; Trötzmüller, M.; Gerber, J.; Semenkovich, C.F.; Köfeler, H.C.; Suter, U. CNS myelination and remyelination depend on fatty acid synthesis by oligodendrocytes. Elife. 2019, 8, e44702. [Google Scholar] [CrossRef]
- Camargo, N.; Goudriaan, A.; van Deijk, A.F.; Otte, W.M.; Brouwers, J.F.; Lodder, H.; Gutmann, D.H.; Nave, K.A.; Dijkhuizen, R.M.; Mansvelder, H.D.; et al. Oligodendroglial myelination requires astrocyte-derived lipids. PLoS Biol. 2017, 15, e1002605. [Google Scholar] [CrossRef]
- Shemer, A.; Erny, D.; Jung, S.; Prinz, M. Microglia Plasticity during Health and Disease: An Immunological Perspective. Trends Immunol. 2015, 36, 614–624. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Zhang, Y.Q.; Qadri, Y.J.; Serhan, C.N.; Ji, R.R. Microglia in Pain: Detrimental and Protective Roles in Pathogenesis and Resolution of Pain. Neuron. 2018, 100, 1292–1311. [Google Scholar] [CrossRef]
- Loving, B.A.; Bruce, K.D. Lipid and Lipoprotein Metabolism in Microglia. Front Physiol. 2020, 11, 393. [Google Scholar] [CrossRef]
- Jakovcevski, I.; Filipovic, R.; Mo, Z.; Rakic, S.; Zecevic, N. Oligodendrocyte development and the onset of myelination in the human fetal brain. Front. Neuroanat. 2009, 3, 5. [Google Scholar] [CrossRef]
- Harrison, J.K.; Jiang, Y.; Chen, S.; Xia, Y.; Maciejewski, D.; McNamara, R.K.; Streit, W.J.; Salafranca, M.N.; Adhikari, S.; Thompson, D.A.; et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc. Natl. Acad. Sci. 1998, 95, 10896–10901. [Google Scholar] [CrossRef]
- Barres, B.A.; Hart, I.K.; Coles, H.S.R.; Burne, J.F.; Voyvodic, J.T.; Richardson, W.D.; Raff, M.C. Cell death and control of cell survival in the oligodendrocyte lineage. Cell. 1992, 70, 31–46. [Google Scholar] [CrossRef]
- Li, Q.; Cheng, Z.; Zhou, L.; Darmanis, S.; Neff, N.F.; Okamoto, J.; Gulati, G.; Bennett, M.L.; Sun, L.O.; Clarke, L.E.; et al. Developmental Heterogeneity of Microglia and Brain Myeloid Cells Revealed by Deep Single-Cell RNA Sequencing. Neuron. 2019, 101, 207–223. [Google Scholar] [CrossRef] [PubMed]
- Bruce, K.D.; Gorkhali, S.; Given, K.; Coates, A.M.; Boyle, K.E.; Macklin, W.B.; Eckel, R.H. Lipoprotein Lipase Is a Feature of Alternatively-Activated Microglia and May Facilitate Lipid Uptake in the CNS During Demyelination. Front. Mol. Neurosci. 2018, 11, 57. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, A.F.; Miron, V.E. The pro-remyelination properties of microglia in the central nervous system. Nat. Rev. Neurol. 2019, 15, 447–458. [Google Scholar] [CrossRef]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.B.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron. 2016, 89, 37–53. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. J. Neurosci. 2014, 34, 11929. [Google Scholar] [CrossRef]
- Paradis, E.; Clavel, S.; Julien, P.; Murthy, M.R.; de Bilbao, F.; Arsenijevic, D.; Giannakopoulos, P.; Vallet, P.; Richard, D. Lipoprotein lipase and endothelial lipase expression in mouse brain: Regional distribution and selective induction following kainic acid-induced lesion and focal cerebral ischemia. Neurobiol. Dis. 2004, 15, 312–325. [Google Scholar] [CrossRef]
- Ma, Y.; Bao, J.; Zhao, X.; Shen, H.; Lv, J.; Ma, S.; Zhang, X.; Li, Z.; Wang, S.; Wang, Q.; et al. Activated cyclin-dependent kinase 5 promotes microglial phagocytosis of fibrillar beta-amyloid by up-regulating lipoprotein lipase expression. Mol. Cell Proteom. 2013, 12, 2833–2844. [Google Scholar] [CrossRef]
- Gong, H.; Dong, W.; Rostad, S.W.; Marcovina, S.M.; Albers, J.J.; Brunzell, J.D.; Vuletic, S. Lipoprotein lipase (LPL) is associated with neurite pathology and its levels are markedly reduced in the dentate gyrus of Alzheimer’s disease brains. J. Histochem. Cytochem. 2013, 61, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell. 2017, 169, 1276–1290. [Google Scholar] [CrossRef]
- Bruce, K.D.; Gorkhali, S.; Given, K.; Coates, A.M.; Boyle, K.E.; Macklin, W.B.; Eckel, R.H. Lipoprotein Lipase Is a Feature of Alternatively-Activated Microglia and May Facilitate Lipid Uptake in the CNS During Demyelination. Front. Mol. Neurosci. 2018, 11, 57. [Google Scholar] [CrossRef]
- Gao, Y.; Vidal-Itriago, A.; Kalsbeek, M.J.; Layritz, C.; Garcia-Caceres, C.; Tom, R.Z.; Eichmann, T.O.; Vaz, F.M.; Houtkooper, R.H.; van der Wel, N.; et al. Lipoprotein Lipase Maintains Microglial Innate Immunity in Obesity. Cell. Rep. 2017, 20, 3034–3042. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Dong, J.; Song, J.; Lvy, C.; Zhang, Y. Efficacy of Glucose Metabolism-Related Indexes on the Risk and Severity of Alzheimer's Disease: A Meta-Analysis. J. Alzheimers. Dis. 2023, 93, 1291–1306. [Google Scholar] [CrossRef] [PubMed]
- Zuin, M.; Roncon, L.; Passaro, A.; Cervellati, C.; Zuliani, G. Metabolic syndrome and the risk of late onset Alzheimer's disease: An updated review and meta-analysis. Nutr. Metab. Cardiovasc. Dis. 2021, 31, 2244–2252. [Google Scholar] [CrossRef] [PubMed]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer's disease--is this type 3 diabetes? J. Alzheimers. Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, F.; Grundke-Igbal, I.; Igbal, K.; Gong, C.X. Deficient brain insulin signalling pathway in Alzheimer's disease and diabetes. J. Pathol. 2011, 225, 54–62. [Google Scholar] [CrossRef]
- Bosco, D.; Fava, A.; Plastino, M.; Montalcini, T.; Pujia, A. Possible implications of insulin resistance and glucose metabolism in Alzheimer's disease pathogenesis. J. Cell. Mol. Med. 2011, 15, 1807–1821. [Google Scholar] [CrossRef]
- de la Monte, S.M. Insulin resistance and Alzheimer's disease. BMB. Rep. 2009, 42, 475–481. [Google Scholar] [CrossRef]
- Moreira, P.I.; Cardoso, S.M.; Pereira, C.M.; Santos, M.S.; Oliveira, C.R. Mitochondria as a therapeutic target in Alzheimer's disease and diabetes CNS. Neurol. Disord. Drug. Targets. 2009, 8, 492–511. [Google Scholar] [CrossRef]
- Zhu, Y.; Liu, X.; Zhu, R.; Zhao, J.; Wang, Q. Lipid levels and the risk of dementia: A dose-response meta-analysis of prospective cohort studies. Ann. Clin. Transl. Neurol. 2022, 9, 296–311. [Google Scholar] [CrossRef]
- Wingo, A.P.; Vattathil, S.M.; Liu, J.; Fan, W.; Cutler, D.J.; Levey, A.I.; Schneider, J.A.; Bennett, D.A.; Wingo, T.S. LDL cholesterol is associated with higher AD neuropathology burden independent of APOE. J. Neurol. Neurosurg. Psychiatry. 2022, 93, 930–938. [Google Scholar] [CrossRef]
- Sáiz-Vazquez, O.; Puente-Martínez, A.; Ubillos-Landa, S.; Pacheco-Bonrostro, J.; Santabárbara, J. Cholesterol and Alzheimer's Disease Risk: A Meta-Meta-Analysis. Brain. Sci. 2020, 10, 386. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Wang, F.; Yang, J.; Peng, H.; Li, Y.; Li, B.; Wang, S. Revealing a Novel Landscape of the Association Between Blood Lipid Levels and Alzheimer's Disease: A Meta-Analysis of a Case-Control Study. Front. Aging. Neurosci. 2020, 11, 370. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, Z.; Jia, X.; Zhang, H.; Zhang, H.; Li, J.; Zhang, K. Prediction of Alzheimer's disease with serum lipid levels in Asian individuals: a meta-analysis. Biomarkers. 2019, 24, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Zuin, M.; Cervellati, C.; Trentini, A.; Passaro, A.; Rosta, V.; Zimetti, F.; Zuliani, G. Association between Serum Concentrations of Apolipoprotein A-I (ApoA-I) and Alzheimer's Disease: Systematic Review and Meta-Analysis. Diagnostics (Basel). 2021, 11, 984. [Google Scholar] [CrossRef]
- Filippini, N.; MacIntosh, B.J.; Hough, M.G.; Goodwin, G.M.; Frisoni, G.B.; Smith, S.M.; Matthews, P.M.; Beckmann, C.F.; Mackay, C.E. Distinct patterns of brain activity in young carriers of the APOE-epsilon4 allele. Proc. Natl. Acad. Sci. USA. 2009, 106, 7209–7214. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer disease meta analysis consortium. JAMA. 1997, 278, 1349–1356. [Google Scholar] [CrossRef]
- Stalmans, P.; Parys-Vanginderdeuren, R.; De Vos, R.; Feron, E.J. ICG staining of the inner limiting membrane facilitates its removal during surgery for macular holes and puckers. Bull. Soc. Belge. Ophtalmol. 2001, 281, 21–26. [Google Scholar]
- Xu, H.; Fu, J.; Mohammed Nazar, R.B.; Yang, J.; Chen, S.; Huang, Y.; Bao, T.; Chen, X. Investigation of the Relationship between Apolipoprotein E Alleles and Serum Lipids in Alzheimer's Disease: A Meta-Analysis. Brain. Sci. 2023, 13, 1554. [Google Scholar] [CrossRef]
- Kothandan, D.; Singh, D.S.; Yerrakula, G.; D, B.; N, P.; Santhana Sophia, B.V.; A, R.; Ramya Vg, S.; S, K.; M, J. Advanced Glycation End Products-Induced Alzheimer's Disease and Its Novel Therapeutic Approaches: A Comprehensive Review. Cureus. 2024, 16, e61373.
- Park, I.H.; Yeon, S.I.; Youn, J.H.; Choi, J.E.; Sasaki, N.; Choi, I.H.; Shin, J.S. Expression of a novel secreted splice variant of the receptor for advanced glycation end products (RAGE) in human brain astrocytes and peripheral blood mononuclear cells. Mol. Immunol. 2004, 40, 1203–1211. [Google Scholar] [CrossRef] [PubMed]
- Emanuele, E.; D'Angelo, A.; Tomaino, C.; Binetti, G.; Ghidoni, R.; Politi, P.; Bernardi, L.; Maletta, R.; Bruni, A.C.; Geroldi, D. Circulating levels of soluble receptor for advanced glycation end products in Alzheimer disease and vascular dementia. Arch. Neurol. 2005, 62, 1734–1736. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Liu, N.; Wang, C.; Qin, B.; Zhou, Y.; Xiao, M.; Chang, L.; Yan, L.J.; Zhao, B. Role of RAGE in Alzheimer’s Disease. Cell. Mol. Neurobiol. 2016, 36, 483–495. [Google Scholar] [CrossRef]
- Yan, S.F.; Ramasamy, R.; Schmidt, A.M. Receptor for AGE (RAGE) and its ligands-cast into leading roles in diabetes and the inflammatory response. J. Mol. Med. 2009, 87, 235–247. [Google Scholar] [CrossRef]
- Xu, X.Y.; Deng, C.Q.; Wang, J.; Deng, X.J.; Xiao, Q.; Li, Y.; He, Q.; Fan, W.H.; Quan, F.Y.; Zhu, Y.P.; et al. Plasma levels of soluble receptor for advanced glycation end products in Alzheimer’s disease. Int. J. Neurosci. 2017, 127, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.H.; Chen, W.; Jiang, L.Y.; Yang, Y.X.; Yao, L.F.; Li, K.S. Influence of ADAM10 Polymorphisms on Plasma Level of Soluble Receptor for Advanced Glycation End Products and The Association With Alzheimer's Disease Risk. Front. Genet. 2018, 9, 540. [Google Scholar] [CrossRef]
- Song, T.; Song, X.; Zhu, C.; Patrick, R.; Skurla, M.; Santangelo, I.; Green, M.; Harper, D.; Ren, B.; Forester, B.P.; et al. Ageing. Res. Rev. 2021, 72, 101503.
- Trares, K.; Chen, L.J.; Schöttker, B. Association of F(2)-isoprostane levels with Alzheimer's disease in observational studies: A systematic review and meta-analysis. Ageing. Res. Rev. 2022, 74, 101552. [Google Scholar] [CrossRef] [PubMed]
- Schrag, M.; Mueller, C.; Zabel, M.; Crofton, A.; Kirsch, W.M.; Ghribi, O.; Squitti, R.; Perry, G. Oxidative stress in blood in Alzheimer's disease and mild cognitive impairment: a meta-analysis. Neurobiol. Dis. 2013, 59, 100–110. [Google Scholar] [CrossRef]
- Varikasuvu, S.R.; Prasad, V.S.; Kothapalli, J.; Manne, M. Brain Selenium in Alzheimer's Disease (BRAIN SEAD Study): a Systematic Review and Meta-Analysis. Biol Trace Elem Res. 2019, 189, 361–369. [Google Scholar] [CrossRef]
- Buonacera, A.; Stancanelli, B.; Colaci, M.; Malatino, L. Neutrophil to lymphocyte ratio: an emerging marker of the relationships between the immune system and diseases. Int. J. Mol. Sci. 2022, 23, 3636. [Google Scholar] [CrossRef] [PubMed]
- Zahorec, R. Neutrophil-to-lymphocyte ratio, past, present and future perspectives. Bratisl. Lek. Listy. 2021, 122, 474–488. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, A.; Mohammadi, M.; Almasi-Dooghaee, M.; Mirmosayyeb, O. Neutrophil to lymphocyte ratio in Alzheimer's disease: A systematic review and meta-analysis. PLoS. One. 2024, 19, e0305322. [Google Scholar] [CrossRef] [PubMed]
- Cumming, P.; Burgher, B.; Patkar, O.; Breakspear, M.; Vasdev, N.; Thomas, P.; Liu, G.J.; Banati, R. Sifting through the surfeit of neuroinflammation tracers. J. Cereb. Blood. Flow. Metab. 2018, 38, 204–224. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Hu, K.; Shao, T.; Hou, L.; Zhang, S.; Ye, W.; Josephson, L.; Meyer, J.H.; Zhang, M.R.; Vasdev, N.; et al. Recent developments on PET radiotracers for TSPO and their applications in neuroimaging. Acta. Pharm. Sin. B. 2021, 11, 373–393. [Google Scholar] [CrossRef]
- De Picker, L.J.; Morrens, M.; Branchi, I.; Haarman, B.C.M.; Terada, T.; Kang, M.S.; Boche, D.; Tremblay, M.E.; Leroy, C.; Bottlaender, M.; et al. TSPO PET brain inflammation imaging: A transdiagnostic systematic review and meta-analysis of 156 case-control studies. Brain. Behav. Immun. 2023, 113, 415–431. [Google Scholar] [CrossRef]
- Lee, P.L.; Chou, K.H.; Chung, C.P.; Lai, T.H.; Zhou, J.H.; Wang, P.N.; Lin, C.P. Posterior cingulate cortex network predicts Alzheimer’s disease progression. Front. Aging. Neurosci. 2020, 12, 608667. [Google Scholar] [CrossRef]
- Gillardon, F.; Rist, W.; Kussmaul, L.; Vogel, J.; Berg, M.; Danzer, K.; Kraut, N.; Hengerer, B.; et al. Proteomic and functional alterations in brain mitochondria from Tg2576 mice occur before amyloid plaque deposition. Proteomics. 2007, 7, 605–616. [Google Scholar] [CrossRef]
- Bhat, A.H.; Dar, K.B.; Anees, S.; Zargar, M.A.; Masood, A.; Sofi, M.A.; Ganie, S.A.; et al. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed. Pharmacother. 2015, 74, 101–110. [Google Scholar] [CrossRef]
- Calabrese, V.; Lodi, R.; Tonon, C.; D’Agata, V.; Sapienza, M.; Scapagnini, G.; Mangiameli, A.; Pennisi, G.; Stella, A.M.; Butterfield, D.A. Oxidative stress, mitochondrial dysfunction and cellular stress response in Friedreich’s ataxia. J. Neurol. Sci. 2005, 233, 145–162. [Google Scholar] [CrossRef]
- Perry, G.; Cash, A.D.; Smith, M.A. Alzheimer disease and oxidative stress. J. Biomed. Biotechnol. 2002, 2, 120–123. [Google Scholar] [CrossRef]
- Terada, T.; Obi, T.; Bunai, T.; Matsudaira, T.; Yoshikawa, E.; Ando, I.; Futatsubashi, M.; Tsukada, H.; Ouchi, Y. In vivo mitochondrial and glycolytic impairments in patients with Alzheimer disease. Neurology. 2020, 94, e1592–e1604. [Google Scholar] [CrossRef]
- Abyadeh, M.; Gupta, V.; Chitranshi, N.; Gupta, V.; Wu, Y.; Saks, D.; Wander Wall, R.; Fitzhenry, M.J.; Basavarajappa, D.; You, Y.; et al. Mitochondrial dysfunction in Alzheimer's disease - a proteomics perspective. Expert. Rev. Proteomics. 2021, 18, 295–304. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition) 1. Autophagy. 2021, 17, 1–382. [Google Scholar]
- Mizushima, N.; Levine, B. Autophagy in human diseases. N. Engl. J. Med. 2020, 383, 1564–1576. [Google Scholar] [CrossRef]
- Riahi, Y.; Wikstrom, J.D.; Bachar-Wikstrom, E.; Polin, N.; Zucker, H.; Lee, M.S.; Quan, W.; Haataja, L.; Liu, M.; Arvan, P.; et al. Autophagy is a major regulator of beta cell insulin homeostasis. Diabetologia. 2016, 59, 1480–1491. [Google Scholar] [CrossRef]
- Bagherniya, M.; Butler, A.E.; Barreto, G.E.; Sahebkar, A. The effect of fasting or calorie restriction on autophagy induction: A review of the literature. Ageing. Res. Rev. 2018, 47, 183–197. [Google Scholar] [CrossRef]
- Nixon, R.A. Amyloid precursor protein and endosomal-lysosomal dysfunction in Alzheimer’s disease: inseparable partners in a multifactorial disease. FASEB. J. 2017, 31, 2729–2743. [Google Scholar] [CrossRef]
- Krance, S.H.; Wu, C.Y.; Chan, A.C.Y.; Kwong, S.; Song, B.X.; Xiong, L.Y.; Ouk, M.; Chen, M.H.; Zhang, J.; Yung, A.; et al. Endosomal-Lysosomal and Autophagy Pathway in Alzheimer's Disease: A Systematic Review and Meta-Analysis. J. Alzheimers. Dis. 2022, 88, 1279–1292. [Google Scholar] [CrossRef]
- Henriksen, E.J.; Dokken, B.B. Role of glycogen synthase kinase-3 in insulin resistance and type 2 diabetes. Curr. Drug. Targets. 2006, 7, 1435–1441. [Google Scholar] [CrossRef]
- Bala, A.; Roy, S.; Das, D.; Marturi, V.; Mondal, C.; Patra, S.; Haldar, P.K.; Samajdar, G. Role of Glycogen Synthase Kinase-3 in the Etiology of Type 2 Diabetes Mellitus: A Review. Curr. Diabetes. Rev. 2022, 18, e300721195147. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wei, M.; Guo, M.; Wang, M.; Niu, H.; Xu, T.; Zhou, Y. GSK3: A potential target and pending issues for treatment of Alzheimer's disease. CNS. Neurosci. Ther. 2024, 30, e14818. [Google Scholar] [CrossRef] [PubMed]
- Akter, R.; Cao, P.; Noor, H.; Ridgway, Z.; Tu, L.H.; Wang, H.; Wong, A.G.; Zhang, X.; Abedini, A.; Schmidt, A.M.; et al. Islet Amyloid Polypeptide: Structure, Function, and Pathophysiology. J. Diabetes. Res. 2016, 2016, 2798269. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Gonzalez, I.; Edwards Iii, G.; Salvadores, N.; Shahnawaz, M.; Diaz-Espinoza, R.; Soto, C. Molecular interaction between type 2 diabetes and Alzheimer's disease through cross-seeding of protein misfolding. Mol. Psychiatry. 2017, 22, 1327–1334. [Google Scholar] [CrossRef]
- Arya, S.; Claud, S.L.; Cantrell, K.L.; Bowers, M.T. Catalytic Prion-Like Cross-Talk between a Key Alzheimer's Disease Tau-Fragment R3 and the Type 2 Diabetes Peptide IAPP. ACS. Chem. Neurosci. 2019, 10, 4757–4765. [Google Scholar] [CrossRef]
- Zhang, G.; Meng, L.; Wang, Z.; Peng, Q.; Chen, G.; Xiong, J.; Zhang, Z. Islet amyloid polypeptide cross-seeds tau and drives the neurofibrillary pathology in Alzheimer's disease. Mol. Neurodegener. 2022, 17, 12. [Google Scholar] [CrossRef]
- Simpson, I.A.; Carruthers, A.; Vannucci, S.J. Supply and demand in cerebral energy metabolism: the role of nutrient transporters. J. Cereb. Blood. Flow. Metab. 2007, 27, 1766–1791. [Google Scholar] [CrossRef]
- Soares, A.F.; Nissen, J.D.; Garcia-Serrano, A.M.; Nussbaum, S.; Waagepetersen, H.S.; Duarte, J.M.N. (2019). Glycogen metabolism is impaired in the brain of male type 2 diabetic Goto-Kakizaki rats. J. Neurosci. Res. 2019, 97, 1004–1017. [Google Scholar] [CrossRef]
- Kothari, V.; Luo, Y.; Tornabene, T.; O’Neill, A.M.; Greene, M.W.; Geetha, T. , Babu, J.R. High fat diet induces brain insulin resistance and cognitive impairment in mice. Biochim. Biophys. Acta. 2017, 1863, 499–508. [Google Scholar] [CrossRef]
- Liu, Z.; Patil, I.Y.; Jiang, T.; Sancheti, H.; Walsh, J.P.; Stiles, B.L. , Yin, F.; Cadenas, E. (2015). High-fat diet induces hepatic insulin resistance and impairment of synaptic plasticity. PLoS. One. 2015, 10, e0128274. [Google Scholar]
- Yonamine, C.Y.; Passarelli, M.; Suemoto, C.K.; Pasqualucci, C.A.; Jacob-Filho, W.; Alves, V.A.F.; Marie, S.K.N.; Correa-Giannella, M.L.; Britto, L.R.; Machado, U.F. Postmortem Brains from Subjects with Diabetes mellitus display reduced GLUT4 expression and soma area in hippocampal neurons: Potential involvement of inflammation. Cells. 2023, 12, 1250. [Google Scholar] [CrossRef] [PubMed]
- Kyrtata, N.; Emsley, H.C.A.; Sparasci, O.; Parkes, L.M.; Dickie, B.R. A Systematic Review of Glucose Transport Alterations in Alzheimer's Disease. Front. Neurosci. 2021, 15, 626636. [Google Scholar] [CrossRef]
- Cartier, N.; Lewis, C.A.; Zhang, R.; Rossi, FM. The role of microglia in human disease: therapeutic tool or target? Acta. Neuropathol. 2014, 128, 363–380. [Google Scholar] [CrossRef]
- Wang, L.; Pavlou, S.; Du, X.; Bhuckory, M.; Xu, H.; Chen, M. Glucose transporter 1 critically controls microglial activation through facilitating glycolysis. Mol. Neurodegener. 2019, 14, 2. [Google Scholar] [CrossRef]
- Xiang, X.; Wind, K.; Wiedemann, T.; Blume, T.; Shi, Y.; Briel, N.; Beyer, L.; Biechele, G.; Eckenweber, F.; Zatcepin, A.; et al. Microglial activation states drive glucose uptake and FDG-PET alterations in neurodegenerative diseases. Sci. Transl. Med. 2021, 13, eabe5640. [Google Scholar] [CrossRef] [PubMed]
- Pearson-Leary, J.; McNay, E.C. Intrahippocampal administration of amyloid-beta(l-42) oligomers acutely impairs spatial working memory, insulin signaling, and hippocampal metabolism. J. Alzheimers. Dis. 2012, 30, 413–422. [Google Scholar] [CrossRef]
- Choeiri, C.; Staines, W.; Messier, C. Immunohistochemical localization and quantification of glucose transporters in the mouse brain. Neuroscience. 2002, 111, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s Disease Is a Synaptic Failure. Science. 2002, 298, 789–791. [Google Scholar] [CrossRef]
- Davies, P.; Maloney, A.J.F. Selective loss of central cholinergic neurons in Alzheimer’s disease. The Lancet. 1976, 308, 1403–1403. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Brain glucose transporters, O-GlcNAcylation and phosphorylation of tau in diabetes and Alzheimer's disease. J. Neurochem. 2009, 111, 242–249. [Google Scholar] [CrossRef]
- Skaper, S.D. The brain as a target for inflammatory processes and neuroprotective strategies. Ann. N.Y. Acad. Sci. 2007, 1122, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Debernardi, R.; Pierre, K.; Lengacher, S.; Magistretti, P.J.; Pellerin, L. Cell-specific expression pattern of monocarboxylate transporters in astrocytes and neurons observed in different mouse brain cortical cell cultures. J. Neurosci. Res. 2003, 73, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Khatri, N.; Man, H.Y. Synaptic activity and bioenergy homeostasis: implications in brain trauma and neurodegenerative diseases. Front. Neurol. 2013, 4, 199. [Google Scholar] [CrossRef]
- Whitlock, J.R.; Heynen, A.J.; Shuler, M.G. , Bear, M.F. Learning induces long-term potentiation in the hippocampus. Science. 2006, 313, 1093–1097. [Google Scholar] [CrossRef]
- Herrero-Mendez, A.; Almeida, A.; Fernández, E.; Maestre, C.; Moncada, S.; Bolaños, J.P. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat. Cell Biol. 2009, 11, 747–752. [Google Scholar] [CrossRef]
- Shima, T.; Jesmin, S.; Matsui, T.; Soya, M.; Soya, H. Differential effects of type 2 diabetes on brain glycometabolism in rats: focus on glycogen and monocarboxylate transporter 2. J. Physiol. Sci. 2018, 68, 69–75. [Google Scholar] [CrossRef]
- Zhang, M.; Cheng, X.; Dang, R.; Zhang, W.; Zhang, J.; Yao, Z. Lactate Deficit in an Alzheimer Disease Mouse Model: The Relationship With Neuronal Damage. J. Neuropathol. Exp. Neurol. 2018, 77, 1163–1176. [Google Scholar] [CrossRef]
- Teunissen, C.E.; Veerhuis, R.; De Vente, J.; Verhey, F.R.; Vreeling, F.; van Boxtel, M.P.; Glatz, J.F.; Pelsers, M.A. Brain-specific fatty acid-binding protein is elevated in serum of patients with dementia-related diseases. Eur. J. Neurol. 2011, 18, 865–871. [Google Scholar] [CrossRef] [PubMed]
- Rosén, C.; Mattsson, N.; Johansson, P.M.; Andreasson, U.; Wallin, A.; Hansson, O.; Johansson, J.O.; Lamont, J.; Svensson, J.; Blennow, K.; et al. Discriminatory Analysis of Biochip-Derived Protein Patterns in CSF and Plasma in Neurodegenerative Diseases. Front. Aging. Neurosci. 2011, 3, 1. [Google Scholar] [CrossRef]
- Hu, W.T.; Chen-Plotkin, A.; Arnold, S.E.; Grossman, M.; Clark, C.M.; Shaw, L.M.; Pickering, E.; Kuhn, M.; Chen, Y.; McCluskey, L.; et al. Novel CSF biomarkers for Alzheimer's disease and mild cognitive impairment. Acta. Neuropathol. 2010, 119, 669–678. [Google Scholar] [CrossRef]
- Hammerschmidt, P.; Brüning, J.C. Contribution of Specific Ceramides to Obesity-Associated Metabolic Diseases. Cell. Mol. Life. Sci. 2022, 79, 395. [Google Scholar] [CrossRef] [PubMed]
- Boslem, E.; Meikle, P.J.; Biden, T.J. Roles of Ceramide and Sphingolipids in Pancreatic β-Cell Function and Dysfunction. Islets. 2012, 4, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Paumen, M.B.; Ishida, Y.; Muramatsu, M.; Yamamoto, M.; Honjo, T. Inhibition of Carnitine Palmitoyltransferase I Augments Sphingolipid Synthesis and Palmitate-Induced Apoptosis. J. Biol. Chem. 1997, 272, 3324–3329. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.; Akashi, H.; Drosatos, K.; Liao, X.; Jiang, H.; Kennel, P.J.; Brunjes, D.L.; Castillero, E.; Zhang, X.; Deng, L.Y.; et al. Increased de Novo Ceramide Synthesis and Accumulation in Failing Myocardium. JCI. Insight. 2017, 2, e82922. [Google Scholar] [CrossRef]
- Jazvinšćak Jembrek, M.; Hof, P.R.; Šimić, G. Ceramides in Alzheimer’s Disease: Key Mediators of Neuronal Apoptosis Induced by Oxidative Stress and Aβ Accumulation. Oxid. Med. Cell. Longev. 2015, 2015, 346783. [Google Scholar] [CrossRef]
- Patil, S.; Melrose, J.; Chan, C. Involvement of Astroglial Ceramide in Palmitic Acid-Induced Alzheimer-like Changes in Primary Neurons. Eur. J. Neurosci. 2007, 26, 2131–2141. [Google Scholar] [CrossRef]
- Filippov, V.; Song, M.A.; Zhang, K.; Vinters, H.V.; Tung, S.; Kirsch, W.M.; Yang, J.; Duerksen-Hughes, P.J. Increased Ceramide in Brains with Alzheimer’s and Other Neurodegenerative Diseases. J. Alzheimer’s. Dis. 2012, 29, 537–547. [Google Scholar] [CrossRef]
- Shen, Z.; Li, Z.Y.; Yu, M.T.; Tan, K.L.; Chen, S. Metabolic perspective of astrocyte dysfunction in Alzheimer's disease and type 2 diabetes brains. Biomed. Pharmacother. 2023, 158, 114206. [Google Scholar] [CrossRef]
- Mi, Y.; Qi, G.; Vitali, F.; Shang, Y.; Raikes, A.C.; Wang, T.; Jin, Y.; Brinton, R.D.; Gu, H.; Yin, F. Loss of fatty acid degradation by astrocytic mitochondria triggers neuroinflammation and neurodegeneration. Nat. Metab. 2023, 5, 445–465. [Google Scholar] [CrossRef]
- Tian, Y.; Jing, G.; Ma, M.; Yin, R.; Zhang, M. Microglial activation and polarization in type 2 diabetes-related cognitive impairment: A focused review of pathogenesis. Neurosci. Biobehav. Rev. 2024, 165, 105848. [Google Scholar] [CrossRef]
- Loving, B.A.; Tang, M.; Neal, M.C.; Gorkhali, S.; Murphy, R.; Eckel, R.H.; Bruce, K.D. Lipoprotein Lipase Regulates Microglial Lipid Droplet Accumulation. Cells. 2021, 10, 198. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, Q.; Cong, W.; Mu, S.; Zhan, R.; Zhong, S.; Zhao, M.; Zhao, C.; Kang, K.; Zhou, Z. Effect of physical activity on risk of Alzheimer's disease: A systematic review and meta-analysis of twenty-nine prospective cohort studies. Ageing. Res. Rev. 2023, 92, 102127. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Yang, Y.; Hu, X.; Zhang, L.; Xiong, Z.; Bai, Y.; Zeng, J.; Xu, F. Effective dosage and mode of exercise for enhancing cognitive function in Alzheimer's disease and dementia: a systematic review and Bayesian Model-Based Network Meta-analysis of RCTs. BMC. Geriatr. 2024, 24, 480. [Google Scholar] [CrossRef] [PubMed]
- Godos, J.; Micek, A.; Currenti, W.; Franchi, C.; Poli, A.; Battino, M.; Dolci, A.; Ricci, C.; Ungvari, Z.; Grosso, G. Fish consumption, cognitive impairment and dementia: an updated dose-response meta-analysis of observational studies. Aging. Clin. Exp. Res. 2024, 36, 171. [Google Scholar] [CrossRef]
- Nucci, D.; Sommariva, A.; Degoni, L.M.; Gallo, G.; Mancarella, M.; Natarelli, F.; Savoia, A.; Catalini, A.; Ferranti, R.; Pregliasco, F.E.; et al. Association between Mediterranean diet and dementia and Alzheimer disease: a systematic review with meta-analysis. Aging. Clin. Exp. Res. 2024, 36, 77. [Google Scholar] [CrossRef]
- Ashley, S.; Bradburn, S.; Murgatroyd, C. A meta-analysis of peripheral tocopherol levels in age-related cognitive decline and Alzheimer's disease. Nutr. Neurosci. 2021, 24, 795–809. [Google Scholar] [CrossRef]
- Dong, Y.; Chen, X.; Liu, Y.; Shu, Y.; Chen, T.; Xu, L.; Li, M.; Guan, X. Do low-serum vitamin E levels increase the risk of Alzheimer disease in older people? Evidence from a meta-analysis of case-control studies. Int. J. Geriatr. Psychiatry. 2018, 33, e257–e263. [Google Scholar] [CrossRef]
- Zhao, R.; Han, X.; Zhang, H.; Liu, J.; Zhang, M.; Zhao, W.; Jiang, S.; Li, R.; Cai, H.; You, H. Association of vitamin E intake in diet and supplements with risk of dementia: A meta-analysis. Front. Aging. Neurosci. 2022, 14, 955878. [Google Scholar] [CrossRef]
- Chen, L.; Sun, X.; Wang, Z.; Lu, Y.; Chen, M.; He, Y.; Xu, H.; Zheng, L. The impact of plasma vitamin C levels on the risk of cardiovascular diseases and Alzheimer's disease: A Mendelian randomization study. Clin. Nutr. 2021, 40, 5327–5334. [Google Scholar] [CrossRef]
- Zhou, F.; Xie, X.; Zhang, H.; Liu, T. Effect of antioxidant intake patterns on risks of dementia and cognitive decline. Eur. Geriatr. Med. 2023, 14, 9–17. [Google Scholar] [CrossRef]
- Surya, K.; Manickam, N.; Jayachandran, K.S.; Kandasamy, M.; Anusuyadevi, M. Resveratrol Mediated Regulation of Hippocampal Neuroregenerative Plasticity via SIRT1 Pathway in Synergy with Wnt Signaling: Neurotherapeutic Implications to Mitigate Memory Loss in Alzheimer's Disease. J. Alzheimers. Dis. 2023, 94, S125–S140. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Guan, X.; Min, D. Evidence of Clinical Efficacy and Pharmacological Mechanisms of Resveratrol in the Treatment of Alzheimer's Disease. Curr. Alzheimer. Res. 2023, 20, 588–602. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wu, X.; Wang, R.; Han, L.; Li, H.; Zhao, W. Mechanisms of 3-Hydroxyl 3-Methylglutaryl CoA Reductase in Alzheimer's Disease. Int. J. Mol. Sci. 2023, 25, 170. [Google Scholar] [CrossRef]
- Olmastroni, E.; Molari, G.; De Beni, N.; Colpani, O.; Galimberti, F.; Gazzotti, M.; Zambon, A.; Catapano, A.L.; Casula, M. Statin use and risk of dementia or Alzheimer's disease: a systematic review and meta-analysis of observational studies. Eur. J. Prev. Cardiol. 2022, 29, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Wójtowicz, S.; Strosznajder, A.K.; Jeżyna, M.; Strosznajder, J.B. The Novel Role of PPAR Alpha in the Brain: Promising Target in Therapy of Alzheimer's Disease and Other Neurodegenerative Disorders. Neurochem. Res. 2020, 45, 972–988. [Google Scholar] [CrossRef]
- Lu, Y.; Fujioka, H.; Wang, W.; Zhu, X. Bezafibrate confers neuroprotection in the 5xFAD mouse model of Alzheimer's disease. Biochim. Biophys. Acta. Mol. Basis. Dis. 2023, 1869, 166841. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, Y.; Qiao, P.F.; Zhao, F.L.; Yan, Y. Fenofibrate reduces amyloidogenic processing of APP in APP/PS1 transgenic mice via PPAR-α/PI3-K pathway. Int. J. Dev. Neurosci. 2014, 38, 223–231. [Google Scholar] [CrossRef]
- Wu, S.; Ding, Y.; Wu, F.; Li, R.; Hou, J.; Mao, P. Omega-3 fatty acids intake and risks of dementia and Alzheimer's disease: a meta-analysis. Neurosci. Biobehav. Rev. 2015, 48, 1–9. [Google Scholar] [CrossRef]
- Calderon Martinez, E.; Zachariah Saji, S.; Salazar Ore, J.V.; Borges-Sosa, O.A.; Srinivas, S.; Mareddy, N.S.R.; Manzoor, T.; Di Vanna, M.; Al Shanableh, Y.; Taneja, R.; et al. The effects of omega-3, DHA, EPA, Souvenaid in Alzheimer's disease: A systematic review and meta-analysis. Neuropsychopharmacol. Rep. 2024. [Google Scholar] [CrossRef]
- Kuate Defo, A.; Bakula, V.; Pisaturo, A.; Labos, C.; Wing, S.S.; Daskalopoulou, S.S. Diabetes, antidiabetic medications and risk of dementia: A systematic umbrella review and meta-analysis. Diabetes. Obes. Metab. 2024, 26, 441–462. [Google Scholar] [CrossRef]
- Basutkar, R.S.; Sudarsan, P.; Robin, S.M.; Bhaskar, V.; Viswanathan, B.; Sivasankaran, P. Drug Repositioning of Pioglitazone in Management and Improving the Cognitive Function among the Patients With Mild to Moderate Alzheimer's Disease: A Systematic Review and Meta-Analysis. Neurol. India. 2023, 71, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Sunwoo, Y.; Park, J.; Choi, C.Y.; Shin, S.; Choi, Y.J. Risk of Dementia and Alzheimer's Disease Associated With Antidiabetics: A Bayesian Network Meta-Analysis. Am. J. Prev. Med. 2024, 67, 434–443. [Google Scholar] [CrossRef]
- Zhang, J.H.; Zhang, X.Y.; Sun, Y.Q.; Lv, R.H.; Chen, M.; Li, M. Metformin use is associated with a reduced risk of cognitive impairment in adults with diabetes mellitus: A systematic review and meta-analysis. Front Neurosci. 2022, 16, 984559. [Google Scholar] [CrossRef]
- Luo, A.; Ning, P.; Lu, H.; Huang, H.; Shen, Q.; Zhang, D.; Xu, F.; Yang, L.; Xu, Y. Association Between Metformin and Alzheimer's Disease: A Systematic Review and Meta-Analysis of Clinical Observational Studies. J. Alzheimers. Dis. 2022, 88, 1311–1323. [Google Scholar] [CrossRef]
- Zhang, L.; Li, D.; Cao, F.; Xiao, W.; Zhao, L.; Ding, G.; Wang, Z.Z. Identification of Human Acetylcholinesterase Inhibitors from the Constituents of EGb761 by Modeling Docking and Molecular Dynamics Simulations. Comb. Chem. High. Throughput. Screen. 2018, 21, 41–49. [Google Scholar] [CrossRef]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer's disease. Brain. 2018, 141, 1917–1933. [Google Scholar] [CrossRef]
- Arafa, N.M.S.; Ali, E.H.A.; Hassan, M.K. Canagliflozin prevents scopolamine-induced memory impairment in rats: comparison with galantamine hydrobromide action. Chem. Biol. Interact. 2017, 277, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Stanciu, G.D.; Ababei, D.C.; Solcan, C.; Bild, V.; Ciobica, A.; Beschea Chiriac, S.I.; Ciobanu, L.M.; Tamba, B.I. Preclinical Studies of Canagliflozin, a Sodium-Glucose Co-Transporter 2 Inhibitor, and Donepezil Combined Therapy in Alzheimer's Disease. Pharmaceuticals (Basel). 2023, 16, 1620. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Liang, L.; Dai, Y.; Hui, S. The role and mechanism of dapagliflozin in Alzheimer disease: A review. Medicine (Baltimore). 2024, 103, e39687. [Google Scholar] [CrossRef]
- Zamora, M.G.; García-Lluch, G.; Moreno, L.; Pardo, J.; Pericas, C.C. Assessment of sodium-glucose cotransporter 2 inhibitors (SGLT2i) and other antidiabetic agents in Alzheimer's disease: A population-based study. Pharmacol. Res. 2024, 206, 107295. [Google Scholar] [CrossRef]
- Kim, H.K.; Biessels, G.J.; Yu, M.H.; Hong, N.; Lee, Y.H.; Lee, B.W.; Kang, E.S.; Cha, B.S.; Lee, E.J.; Lee, M. SGLT2 Inhibitor Use and Risk of Dementia and Parkinson Disease Among Patients With Type 2 Diabetes. Neurology. 2024, 103, e209805. [Google Scholar] [CrossRef]
- McClean, P.L.; Parthsarathy, V.; Faivre, E.; Hölscher, C. The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer's disease. J. Neurosci. 2011, 31, 6587–6594. [Google Scholar] [CrossRef]
- Kong, F.; Wu, T.; Dai, J.; Zhai, Z.; Cai, J.; Zhu, Z.; Xu, Y.; Sun, T. Glucagon-like peptide 1 (GLP-1) receptor agonists in experimental Alzheimer's disease models: a systematic review and meta-analysis of preclinical studies. Front. Pharmacol. 2023, 14, 1205207. [Google Scholar] [CrossRef] [PubMed]
- Femminella, G.D.; Frangou, E.; Love, S.B.; Busza, G.; Holmes, C.; Ritchie, C.; Lawrence, R.; McFarlane, B.; Tadros, G.; Ridha, B.H.; et al. Correction to: Evaluating the effects of the novel GLP-1 analogue liraglutide in Alzheimer's disease: study protocol for a randomised controlled trial (ELAD study). Trials. 2020, 21, 660. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.J.; Li, X.R.; Chai, S.F.; Li, W.R.; Li, S.; Hou, M.; Li, J.L.; Ye, Y.C.; Cai, H.Y.; Hölscher, C.; et al. Semaglutide ameliorates cognition and glucose metabolism dysfunction in the 3xTg mouse model of Alzheimer's disease via the GLP-1R/SIRT1/GLUT4 pathway. Neuropharmacology. 2023, 240, 109716. [Google Scholar] [CrossRef]
- Hölscher, C. Glucagon-like peptide-1 class drugs show clear protective effects in Parkinson's and Alzheimer's disease clinical trials: A revolution in the making? Neuropharmacology. 2024, 253, 109952. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Q.; Hölscher, C. GIP has neuroprotective effects in Alzheimer and Parkinson's disease models. Peptides. 2020, 125, 170184. [Google Scholar] [CrossRef]
- Shi, L.; Zhang, Z.; Li, L.; Hölscher, C. A novel dual GLP-1/GIP receptor agonist alleviates cognitive decline by re-sensitizing insulin signaling in the Alzheimer icv. STZ rat model. Behav. Brain. Res. 2017, 327, 65–74. [Google Scholar] [CrossRef]
- Cai, H.Y.; Yang, D.; Qiao, J.; Yang, J.T.; Wang, Z.J.; Wu, M.N.; Qi, J.S.; Hölscher, C. A GLP-1/GIP Dual Receptor Agonist DA4-JC Effectively Attenuates Cognitive Impairment and Pathology in the APP/PS1/Tau Model of Alzheimer's Disease. J. Alzheimers. Dis. 2021, 83, 799–818. [Google Scholar] [CrossRef]
- Maskery, M.; Goulding, E.M.; Gengler, S.; Melchiorsen, J.U.; Rosenkilde, M.M.; Hölscher, C. The Dual GLP-1/GIP Receptor Agonist DA4-JC Shows Superior Protective Properties Compared to the GLP-1 Analogue Liraglutide in the APP/PS1 Mouse Model of Alzheimer's Disease. Am. J. Alzheimers. Dis. Other. Demen. 2020, 35, 1533317520953041. [Google Scholar] [CrossRef]
- Tatton, W.G.; Chalmers-Redman, R.M. Mitochondria in neurodegenerative apoptosis: an opportunity for therapy? Ann. Neurol. 1998, 44, S134–S141. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Adachi, H.; Hakoshima, M.; Katsuyama, H. Glucose-Lowering Effects of Imeglimin and Its Possible Beneficial Effects on Diabetic Complications. Biology (Basel). 2023, 12, 726. [Google Scholar] [CrossRef] [PubMed]
- Zemgulyte, G.; Umbrasas, D.; Cizas, P.; Jankeviciute, S.; Pampuscenko, K.; Grigaleviciute, R.; Rastenyte, D.; Borutaite, V. Imeglimin Is Neuroprotective Against Ischemic Brain Injury in Rats-a Study Evaluating Neuroinflammation and Mitochondrial Functions. Mol. Neurobiol. 2022, 59, 2977–2991. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Glucose transporters, insulin receptor, insulin receptor substrate, and monocarboxylate transporters in normal brain. Red up and down arrows indicate increase in phenomenon, substances and expression of molecules. Black arrows indicate the flow of substances. BBB, blood brain barrier; G, glucose; GLUT, glucose transporter; Gly, glycogen; IR, insulin receptor; IRS, insulin receptor substrate; L, lactate; MCT, monocarboxylate transporter.
Figure 1.
Glucose transporters, insulin receptor, insulin receptor substrate, and monocarboxylate transporters in normal brain. Red up and down arrows indicate increase in phenomenon, substances and expression of molecules. Black arrows indicate the flow of substances. BBB, blood brain barrier; G, glucose; GLUT, glucose transporter; Gly, glycogen; IR, insulin receptor; IRS, insulin receptor substrate; L, lactate; MCT, monocarboxylate transporter.
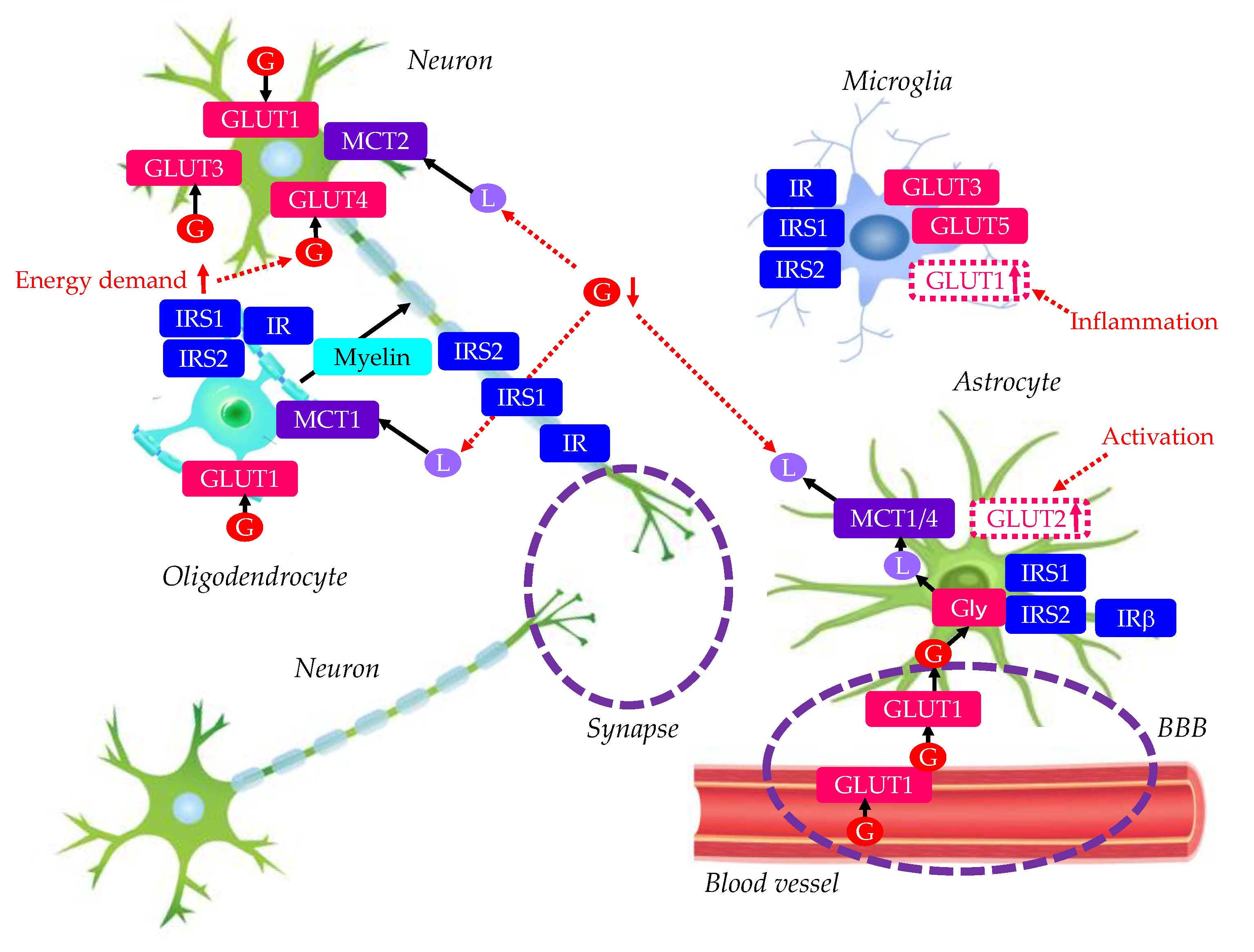
Figure 2.
Lipid metabolism in normal brain. C, cholesterol; Ce, ceramides; E, apo E; FA, fatty acid; FABP, FA binding protein; FATP, FA transport proteins; GLUT, K, ketone bodies; L, lactate; LP, lipoprotein-like particles; LPL, lipoprotein lipase; MCT, monocarboxylate transporter; P, phospholipids; S, sphingolipids; SR, scavenger receptor.
Figure 2.
Lipid metabolism in normal brain. C, cholesterol; Ce, ceramides; E, apo E; FA, fatty acid; FABP, FA binding protein; FATP, FA transport proteins; GLUT, K, ketone bodies; L, lactate; LP, lipoprotein-like particles; LPL, lipoprotein lipase; MCT, monocarboxylate transporter; P, phospholipids; S, sphingolipids; SR, scavenger receptor.
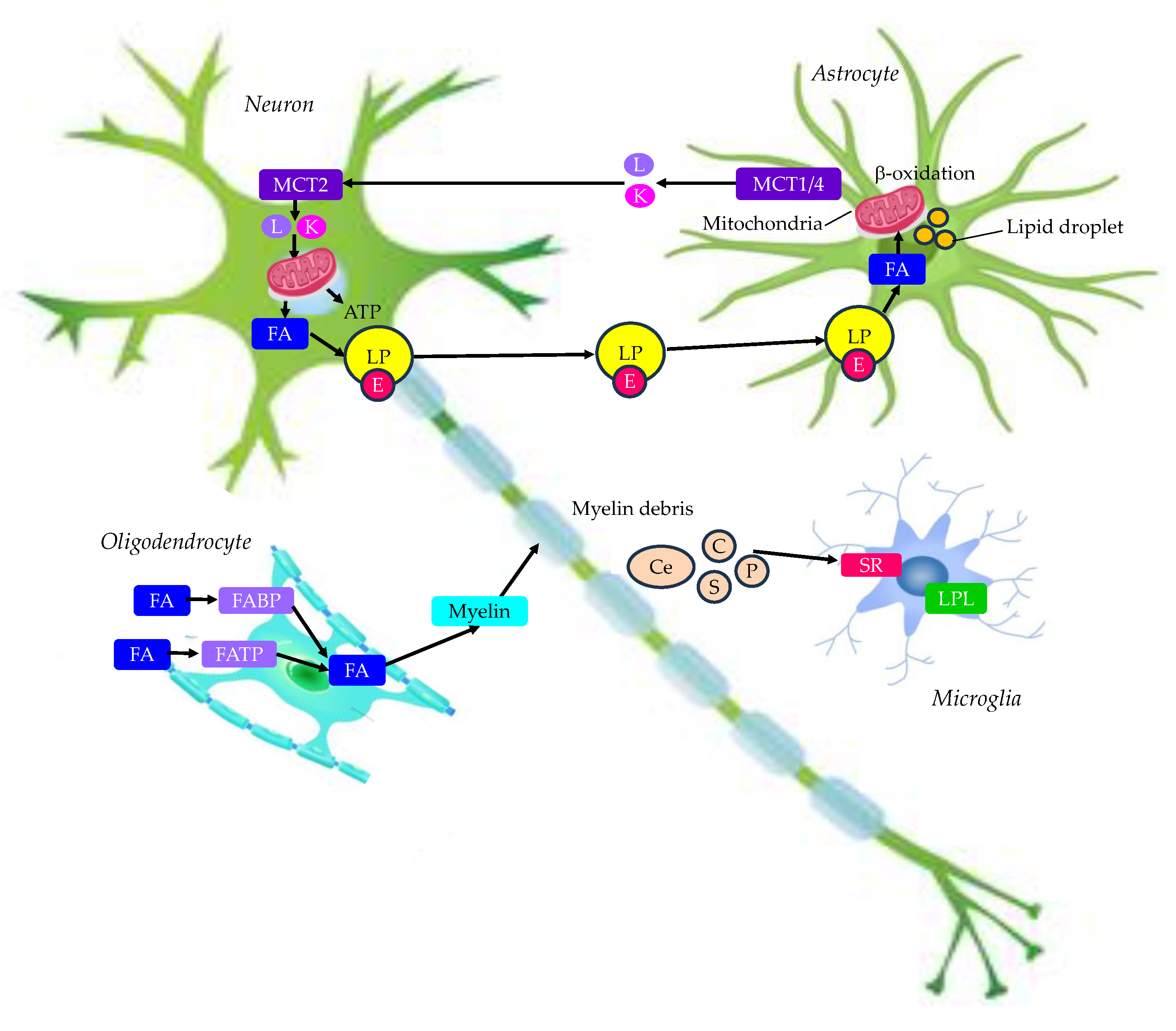
Figure 3.
Risk factors related with type 2 diabetes for the development of AD. Aβ, amyloid beta; AGEs, advanced glycation end products; APP, amyloid precursor protein; GSK-3, glycogen synthase kinase-3; HDL-C, high-density lipoprotein-cholesterol; IAPP, islet amyloid polypeptide; IGF, insulin-like growth factor; LDL-C, low-density lipoprotein-cholesterol; PI3K, phosphatidyl-inositide 3-kinases; RAGE, receptor for AGEs; TG, triglyceride.
Figure 3.
Risk factors related with type 2 diabetes for the development of AD. Aβ, amyloid beta; AGEs, advanced glycation end products; APP, amyloid precursor protein; GSK-3, glycogen synthase kinase-3; HDL-C, high-density lipoprotein-cholesterol; IAPP, islet amyloid polypeptide; IGF, insulin-like growth factor; LDL-C, low-density lipoprotein-cholesterol; PI3K, phosphatidyl-inositide 3-kinases; RAGE, receptor for AGEs; TG, triglyceride.
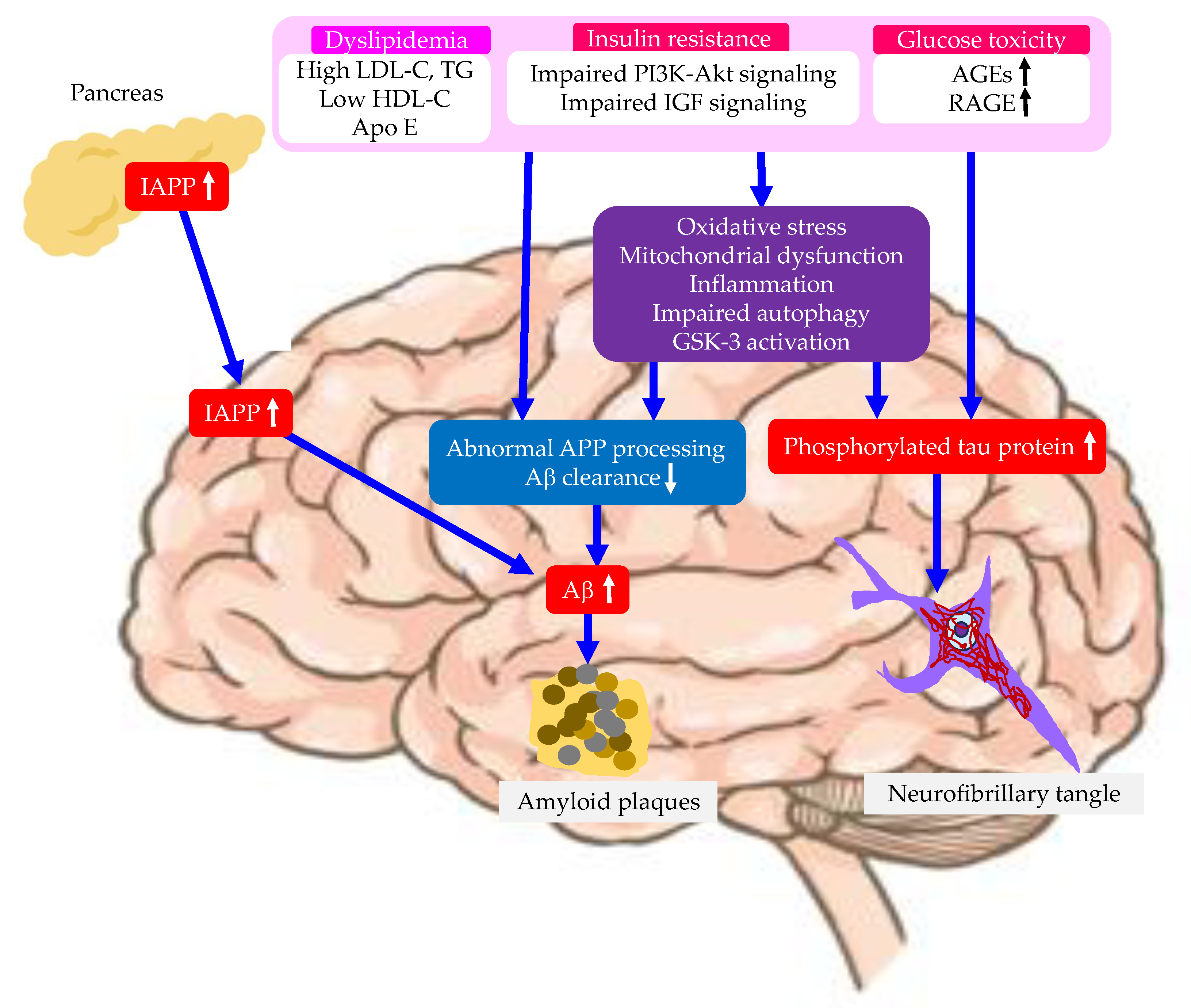
Figure 4.
Effects of changes in energy metabolism in brain due to type 2 diabetes on the development of AD. AO, antioxidants; E, apo E; FA, fatty acid; G, glucose; K, ketone bodies; L, lactate; LP, lipoprotein-like particles; LPL, lipoprotein lipase.
Figure 4.
Effects of changes in energy metabolism in brain due to type 2 diabetes on the development of AD. AO, antioxidants; E, apo E; FA, fatty acid; G, glucose; K, ketone bodies; L, lactate; LP, lipoprotein-like particles; LPL, lipoprotein lipase.
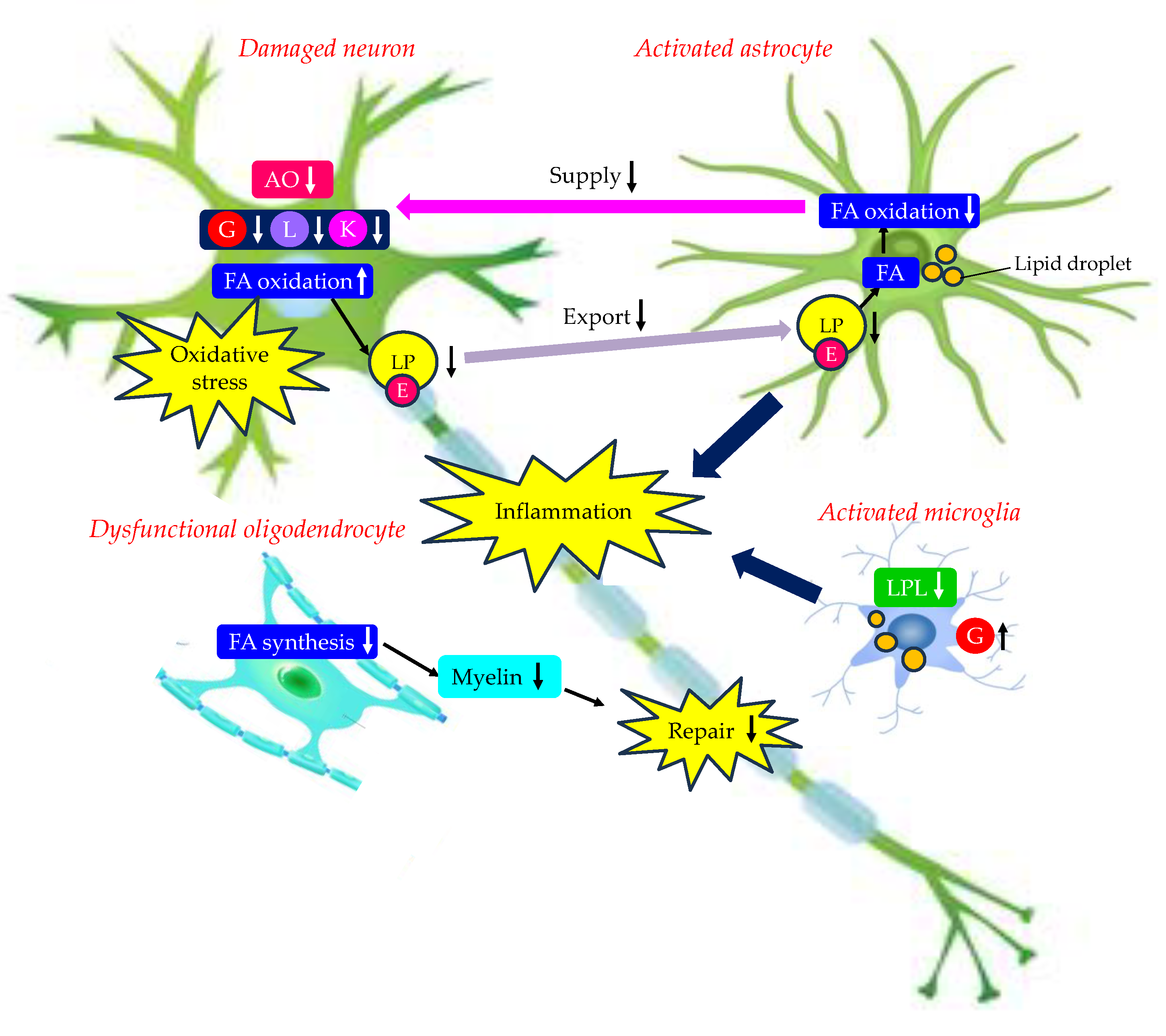

Table 1.
Changes in expression of glucose transporters by type 2 diabetes and AD.
| Glucose transporters | Type 2 diabetes | AD |
| GLUT1 | ↓ | ↓(↑ in microglia) |
| GLUT2 | Unknown | ↑(in astrocytes) |
| GLUT3 | ↓ | ↓ |
| GLUT4 | ↓ | Translocation↓ |
Table 2.
Possible therapeutic interventions for AD considering AD risk factors.
| Risk factors | Possible therapeutic interventions |
| High LDL-C | Diet, Exercise, Statin |
| High TG | Diet, Exercise, PPAR-a agonists, n3-PUFA |
| Low HDL-C | Diet, Exercise, Stain, PPAR-a agonists |
| Apo E abnormality | PPAR-a agonists |
| Insulin resistance | Diet, Exercise, Metformin, Pioglitazone, SGLT2is, GLP-1RAs |
| Oxidative stress | Antioxidants (Vitamin C and E, Polyphenols) |
| Mitochondrial dysfunction | Imeglimin |
AD, Alzheimer’s disease; GLP-1RAs, glucagon-like peptide 1 receptor agonists; HDL-C, high-density lipoprotein-cholesterol; LDL-C, low-density lipoprotein-cholesterol; PPAR, peroxisome proliferator-activated receptor; SGLT2is, sodium glucose co-transporter-2 inhibitors; TG, triglyceride.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



