Submitted:
21 October 2024
Posted:
21 October 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
The COVID-19 pandemic, caused by the Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2), is in its 5th year and is being maintained by the inability of current spike-alone based COVID-19 vaccines to prevent transmission leading to the continuous emergence of variants and sub-variants of concern (VOCs). This underscores the critical need for next-generation broad-spectrum pan-Coronavirus vaccines (pan-CoV vaccine) to break this cycle and end the pandemic. The development of a pan-CoV vaccine offering protection against a wide array of VOCs requires two key elements: (1) identifying protective antigens that are highly conserved between passed, current and future VOCs; and (2) developing a safe and efficient antigens deliver system for induction of broad-based B- and T-cell immunity. This review will (1) present the current state of antigen delivery platforms involving a multifaceted approach, including bioinformatics, molecular and structural biology, immunology, and advanced computational methods; (2) discuss the challenges facing development of safe and effective antigen delivery platforms; and (3) highlight the potential of nucleoside-modified mRNA encapsulated in lipid nanoparticles (LNP) as the platform that is well suited to the needs of a next-generation pan-CoV vaccine, such as the ability to induce broad-based immunity and amenable to large-scale manufacturing to safely provide durable protective immunity against current and future Coronavirus threats.
Keywords:
antigen delivery system
; antigen delivery platform
; SARS-CoV-2
; pan-coronavirus vaccine
; mRNA
; srRNA
; LNP
Introduction
Coronaviruses comprise a vast group of viruses capable of causing a spectrum of illnesses, ranging from mild conditions like the common cold to more serious diseases such as Middle East Respiratory Syndrome (MERS-CoV) and severe acute respiratory syndrome (SARS-CoV) [1]. The clinical manifestations of infections caused by these viruses are highly variable, spanning from asymptomatic cases to severe disease marked by pneumonia, respiratory distress, and fever. In extreme instances, the disease may advance to acute respiratory distress syndrome (ARDS), septic shock, and death resulting from multi-organ failure. Severe COVID-19, particularly in vulnerable populations like the elderly and individuals with underlying health conditions, has necessitated hospitalization and mechanical ventilation, overwhelming healthcare infrastructures and prompting national lockdowns and large-scale vaccination efforts.
Additionally, the long-term morbidity associated with COVID is significant, with up to 10% of individuals, regardless of initial disease severity, developing long COVID. This chronic condition is characterized by persistent, multisystemic symptoms such as muscle pain, fatigue, and cognitive impairment. The exact mechanisms and immunopathology causing long COVID-19 remain areas of intense investigation. Therefore, a pan-coronavirus vaccine capable of protecting individuals from disease and reducing community spread of virus could help mitigate the burden of disease caused by multiple coronaviruses, including SARS-CoV-2, MERS-CoV, and endemic HCoVs, potentially reducing severe illness, hospitalizations, and long-term complications such as long COVID.
The development of pan-coronavirus vaccines that protect from the current and future SARS-CoV-2 variants necessitates a multifaceted approach, incorporating molecular and structural biology, immunology, and advanced computational methods. A key to the design of these vaccines is the identification of conserved regions across coronavirus families that can serve as targets for cross-reactive neutralizing antibodies and CD4+ and CD8+ T-cell immunity [2]. The Spike (S) protein, especially its receptor-binding domain (RBD), has emerged as a primary target for neutralizing antibodies due to its critical role in virus entry into host cells [3]. However, identification of other conserved epitopes remains a significant challenge and an area of active research [2,4,5,6,7]. An effective pan-coronavirus vaccine, by definition, needs to prevent severe disease and/or infection caused by all viruses within the coronavirus family. The current widely employed SARS-CoV-2 vaccines based solely on the spike glycoprotein were very effective in blunting the severity of the pandemic in its early stages, but waning immunity and antigenic variation between emergent strains have limited their utility. As a result, frequent boosting and updating of the vaccine to better match circulating virus strains are being used to address this limitation. So far, this strategy has not been able to disrupt the transmission cycle, hence it is not a long-term solution to ending this pandemic or preventing future ones.
This article reviews clinical trial data gathered from public databases, scientific literature, and research announcements up to the current year, 2024. Focus is placed on the current state of antigen delivery platforms best suited for pan-coronavirus vaccines, emphasizing the challenges and innovations in developing these vaccines that can provide durable immunity against current and future coronavirus threats, evaluating their immunogenicity, efficacy, safety, and cross-reactive potential against various coronavirus strains.
Antigen delivery platforms for universal coronavirus vaccines: Research and development of pan-coronavirus vaccines have utilized technology platforms such as protein subunits, viral vectors, mRNA, and nanoparticle technologies to deliver the antigen of interest. These platforms have shown promise in eliciting broad and robust immune responses in preclinical models [8,9,10,11,12,13,14,15,16]. These delivery platforms being explored for pan-coronavirus vaccine development are at the forefront of immunological research, leveraging cutting-edge science to create vaccines that are not only effective against multiple strains of coronaviruses but can also be rapidly developed and deployed in response to new viral threats. Below we will highlight the delivery platforms that have been proposed for effective pan-coronavirus vaccines:
1. Protein Subunit to deliver next-generation PanCoVax vaccine candidates. Protein subunit vaccines include pieces (subunits) of the virus (e.g., whole antigens and/or conserved peptides) to stimulate an immune response without introducing the whole virus. These protein based-pan-coronavirus subunits vaccines are sufficient to teach the immune system to recognize and attack the virus but cannot cause disease since they are only a part of the virus proteome. In addition, immune responses can be targeted to specific viral proteins associated with protection and avoid responses against other proteins that could be ineffectual or deleterious.
A. protein + adjuvant to deliver next-generation PanCoVax vaccine candidates. Adjuvanted protein-based subunit vaccines constitute a significant category of vaccines. Subunit vaccines face specific challenges compared to other platforms, primarily due to the need for optimization in the production of each protein antigen. Among the most advanced COVID-19 subunit vaccines is Novavax’s NVX-CoV-2373 (marketed as Nuvaxovid), which is produced using insect cells and formulated with the Matrix-M adjuvant, derived from saponin [17]. This vaccine includes the stabilized, full-length spike (S) protein in its prefusion conformation, designed for optimal antigenicity. Similarly, Sanofi-GSK’s vaccine VidPrevtyn Beta contains prefusion-stabilized S protein trimers of SARS-CoV-2, also produced in insect cells and combined with the AS03 adjuvant [18]. Both vaccines have completed human clinical trials and have been authorized for use by the European Union. Recombinant protein vaccines are attractive candidates due to their excellent safety profiles, lack of genome integration risk, absence of live viral elements, and compatibility with immunocompromised individuals. Additionally, they exhibit high production efficiency and stability [17,19,20,21]. In recent studies, seven vaccine formulations containing various combinations of the RBD antigens from SARS-CoV, MERS-CoV, and SARS-CoV-2 XBB.1.5, along with Alum and CpG55.2 adjuvants, were tested in animal models. Mice immunized with the trivalent RBD-based vaccine developed strong antibody responses against all three antigens and efficiently neutralized the corresponding pseudo viruses [22]. To date, only a few protein-based pan-coronavirus vaccines are being evaluated in clinical trials (view section 5. Current Delivery platforms of pan-coronavirus vaccines in clinical trials).
B. Peptides to deliver next-generation PanCoVax vaccine candidates. Peptide-based vaccines with a carrier also constitute a significant category of vaccines. Through the combination of several peptides, the vaccines increase the likelihood of cross-reactive T and B cell responses, as well as creating the possibility of using specific peptides to omit immunopathological pro-inflammatory sequences from the vaccine construct and focus the immune response toward critical epitopes (Figure 2). This also allows for flexibility as the peptides can be adjusted to include new epitopes for emerging coronavirus strains [23]. Additionally, the use of peptides rather than the live virus means the vaccine is non-infectious. Since the components of the vaccine are also not redundant with the human genome, there a significant decrease in the risk of triggering allergic or autoimmune responses [24]. Compared to adjuvanted protein-based subunit vaccines, peptide vaccines have a faster development through automated peptide synthesizers, as well as a fast purification process [23]. The carriers are also straightforward to produce. Both combined allow for standardized mass production of peptide vaccines in an economically feasible manner without bio contaminants [23,24,25]. When properly adjuvanted, the peptide-based vaccines have been proved to be highly immunogenic, however, the vaccines could also potentially be produced without an adjuvant, utilizing carriers like liposomes. The peptide-based vaccines have also shown to have greatly improved stability, are water soluble, and have less of a need for cold storage [23,25]. The development is slower than mRNA vaccines though due to the time needed for the optimization of peptide epitopes and the carrier. Mosaic-8b from CalTech is a notable example. The mosaic vaccine was engineered using a multivalent protein domain known as SpyCatcher, enabling it to display receptor-binding domains (RBDs) from both human and animal coronaviruses. These RBDs include those from Bat CoV RaTG13, Bat CoV SHC014, Bat CoV Rs4081, Bat CoV RmYN02, Bat CoV Rf1, Bat CoV WIV1, Pangolin CoV Pang17, and SARS-CoV-2, and are presented on a nanoparticle carrier [26]. Additionally, SK Biosciences has advanced efforts toward developing a peptide-based vaccine by fusing RBDs from SARS-CoV-2, SARS-CoV-1, Bat CoV RaTG13, and Bat CoV WIV1 to a carrier protein, 153-50A [26,27].
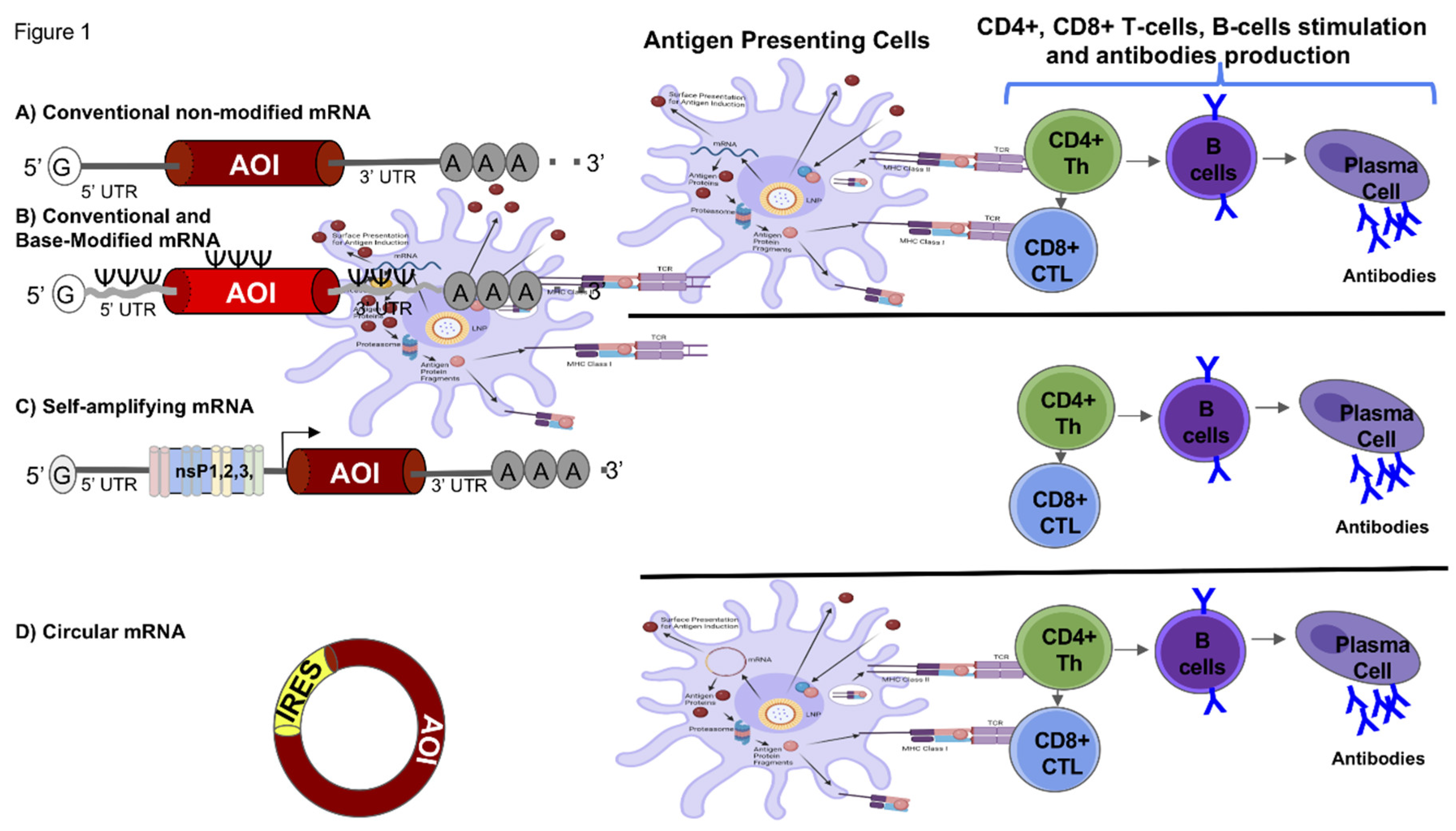
Figure 1.
Pan-coronavirus mRNA-LNP based vaccines. Pan-coronavirus vaccines involving the use of an mRNA transcript that encodes an antigen and LNP. Linear non-replicating mRNAs include a sequence for antigens of interest (AOI, such as the Spike, Nucleocapsid and other conserved proteins) flanked by 5′ and 3′ untranslated regions (UTRs), a cap structure at the 5′ end, and a poly(A) tail at the 3′ end. Depending on whether modified or native nucleosides are used during in vitro transcription, either (A) unmodified or (B) modified mRNAs or are produced; (C) Self-amplifying RNA (saRNA) has a similar sequence organization but additionally includes: (1) a sequence encoding four non-structural proteins (nsP1–4) that form a replicase to amplify the saRNA, and (2) a viral-origin sub genomic promoter (black arrow) that initiates antigen transcription; (D) Circular RNA (circRNA) based vaccines consists of a covalently closed single-stranded RNA containing the sequence of antigens of interest and an internal ribosome entry site (IRES) to initiate antigen translation. The antigens of interest are produced endogenously by the antigen-presenting cells (APCs) translational machinery (red circles), degraded by proteasomes (pink circles), and presented on major histocompatibility complex (MHC) class I molecules (pink circles), leading to a specific CD8+ cytotoxic T cell response against coronaviruses variant of concern. Additionally, the antigen protein can be exported from the cell, endocytosed by the same or another APC, degraded by endosomal proteases, and presented on MHC II molecules, resulting in a CD4+ helper T cell response. Immunization progresses with CD4+ helper T cells aiding in the activation of B cells to produce anti-coronavirus specific antibodies and neutralizing antibodies. CD8+ cytotoxic T cells specifically target and eliminate coronavirus-infected cells.
Figure 1.
Pan-coronavirus mRNA-LNP based vaccines. Pan-coronavirus vaccines involving the use of an mRNA transcript that encodes an antigen and LNP. Linear non-replicating mRNAs include a sequence for antigens of interest (AOI, such as the Spike, Nucleocapsid and other conserved proteins) flanked by 5′ and 3′ untranslated regions (UTRs), a cap structure at the 5′ end, and a poly(A) tail at the 3′ end. Depending on whether modified or native nucleosides are used during in vitro transcription, either (A) unmodified or (B) modified mRNAs or are produced; (C) Self-amplifying RNA (saRNA) has a similar sequence organization but additionally includes: (1) a sequence encoding four non-structural proteins (nsP1–4) that form a replicase to amplify the saRNA, and (2) a viral-origin sub genomic promoter (black arrow) that initiates antigen transcription; (D) Circular RNA (circRNA) based vaccines consists of a covalently closed single-stranded RNA containing the sequence of antigens of interest and an internal ribosome entry site (IRES) to initiate antigen translation. The antigens of interest are produced endogenously by the antigen-presenting cells (APCs) translational machinery (red circles), degraded by proteasomes (pink circles), and presented on major histocompatibility complex (MHC) class I molecules (pink circles), leading to a specific CD8+ cytotoxic T cell response against coronaviruses variant of concern. Additionally, the antigen protein can be exported from the cell, endocytosed by the same or another APC, degraded by endosomal proteases, and presented on MHC II molecules, resulting in a CD4+ helper T cell response. Immunization progresses with CD4+ helper T cells aiding in the activation of B cells to produce anti-coronavirus specific antibodies and neutralizing antibodies. CD8+ cytotoxic T cells specifically target and eliminate coronavirus-infected cells.
2. Viral Vector to deliver next-generation PanCoVax vaccine candidates. Viral vector vaccines utilize a harmless virus (distinct from the coronavirus) as a vehicle to transport genetic material from the target coronavirus into human cells. Within these cells, the genetic instructions prompt the production of a specific protein from the coronavirus, stimulating the immune system to mount a defense against the virus upon future encounters. This mechanism triggers a potent immune response. These vaccines are known for eliciting a strong and enduring immune reaction, closely resembling the response to a natural viral infection. Viral vectors offer versatility, as they can transport genes encoding multiple proteins, potentially broadening the range of immunity conferred. Among viral vectors, adenovirus stands out, attracting significant attention as a pivotal therapeutic vector due to its status as the first DNA virus to undergo extensive medical exploration (Figure 3). The benefits of adenoviruses as a vaccine platform arise from their advantageous biological properties, including genetic stability, high transduction efficiency, and ease of scalability for large-scale production [28,29]. Adenoviruses are non-enveloped, double-stranded DNA viruses, with genomes typically ranging between 34 and 43 kilobases. In humans, they are commonly associated with mild respiratory and ocular infections [30]. To date, over 150 primate adenoviruses (AdVs) have been identified, many of which are being explored for vaccine development. Unlike certain other viral vectors, adenoviruses do not integrate into the host genome but instead exist in an episomal form [31]. These viruses can be isolated from a variety of species, including humans and animals such as chimpanzees. In humans, approximately 50 adenovirus serotypes have been classified, grouped into seven subtypes (A–G) based on sequence similarities and their ability to induce erythrocyte agglutination [32,33].
A. Adenoviruses to deliver next-generation PanCoVax vaccine candidates.
The two primary approaches for developing coronavirus vaccines involve utilizing either entire viruses or genetically engineered vaccine antigens. These vaccine antigens can be administered through various methods [34]. Whole virus vaccines have the potential to elicit a robust immune response, providing protection against coronavirus infections. Genetically engineered vaccines, which target specific coronavirus proteins, are extensively employed to enhance vaccine safety and efficacy. Coronavirus proteins, such as Nucleocapsid (N), Spike (S), and Membrane (M), can be delivered through DNA recombinant and viral vector vaccines. [35]. A critical aspect of adenovirus-based vaccine development involves the incorporation of a transgene cassette into the adenoviral genome, achieved via homologous recombination or direct cloning [35,36]. This transgene cassette includes a robust promoter to drive sustained and high-level expression of the introduced genes. [35,36]. In the case of the coronavirus, host proteases cleave the S protein into two subunits, S1 and S2. The Receptor Binding Domain (RBD) within S1 is essential for binding to the Angiotensin Converting Enzyme-2 (ACE2) receptor, while the S2 subunit contains the fusion peptide necessary for membrane fusion and viral entry [37]. The S protein is particularly significant because it contains epitopes that elicit neutralizing antibodies, a focus of many vaccine strategies [38]. Under optimal transfection conditions, high-titer adenoviruses can be produced within five days. One prominent example is the AZD1222 vaccine, developed by the University of Oxford and AstraZeneca, which uses a ChAdOx1 adenoviral vector to deliver the gene encoding the coronavirus S protein. This vaccine, administered intramuscularly, expresses the S protein in a trimeric prefusion state, critical for its immunogenicity [39]. AZD1222 has undergone extensive clinical testing, with eight phase I, 30 phase II, and 11 phase III trials registered globally. Covishield, the Serum Institute of India’s version of AZD1222, has completed additional trials, including one phase II and one phase III trial. In phase I/II trials involving 1,090 participants aged 18–55, AZD1222 demonstrated a favorable safety profile and a strong immune response [40].
B. Adeno Associated Viruses to deliver next-generation PanCoVax vaccine candidates.
Adeno-associated viruses (AAVs) are frequently employed as viral vectors for gene therapy due to their low immunogenicity and ability to provide sustained gene expression after infection. AAVs have also been incorporated into a novel coronavirus vaccine. Prior research has demonstrated that AAV vector vaccines exhibit a favorable safety profile and elicit robust and long-lasting humoral and cellular immune responses in both mice and nonhuman primates [41,42,43,44]. The Spike protein-encoding gene from the original SARS-CoV-2 strain was incorporated into AAVrh32.33, resulting in decreased neutralizing antibody levels in treated mice lasting up to a year. This treatment also induced a sustained cellular immune response and conferred complete protection against SARS-CoV-2 in cynomolgus monkeys [45]. In another study, the AAV6 virus carrying the Spike protein-encoding gene of the original strain was compared with an approved inactivated vaccine in animal models. Findings revealed that a single dose of the AAV6-based vaccine produced higher IgG antibody titers that persisted longer than those generated by the inactivated vaccine [46].
3. mRNA to deliver next-generation universal coronavirus vaccines candidates. mRNA (messenger RNA) vaccines represent a revolutionary approach in vaccine development. Unlike certain traditional vaccines, mRNA vaccines do not use live viruses. Instead, they employ a synthetic strand of mRNA that encodes the instructions for cells to produce a viral protein, such as the Spike (S) protein found in coronaviruses. Once produced by the cells, these proteins stimulate an immune response, teaching the immune system to recognize and combat the virus without being exposed to the disease. mRNA present several advantages such as rapid development: mRNA vaccines can be developed swiftly, as they require only the genetic sequence of the virus; high flexibility: mRNA vaccines can be easily adapted to target new mutations or multiple strains by tweaking the mRNA sequence and highly safe, as no live virus is used, there is little to no risk of causing disease in the vaccinated individual [2,11,47,48,49,50].
Various types of mRNA have been suggested and employed for vaccine delivery. They have been categorized based on their mode of action into two types: conventional mRNA and self-amplifying (or self-replicating) mRNA vaccines [51,52,53]. Currently, there are five types of RNA vaccines: conventional and base-modified, non-amplifying mRNA, which incorporate chemically modified nucleotides and have been widely used for COVID-19 vaccines; self-replicating RNA (srRNA), which is based on an engineered viral genome but devoid of viral structural protein genes; Circular mRNA (circRNA) that are a class of single-stranded RNAs with covalently closed structures, which protects it from exonuclease-mediated degradation [54] and thermostable mRNA that use lyophilizable, thermostable nanostructured lipid carrier to effectively deliver mRNA [55,56].
The advantages of conventional and base-modified mRNA vaccines are: (1) high effectiveness in protein translation, (2) non-infectious and non-integrating making them low-risk for infection and/or genetic alteration, (3) can be rapidly produced, and (4) are adaptable to various pathogens. However, conventional, and base-modified mRNA vaccines require cold storage (-20°C to -70°C) to maintain their stability, and mRNA degrades quickly in cells (6-12 hours), which limits the duration of antigen expression. mRNA can be non-amplifying, consisting of straightforward mRNA sequences that directly encode the target antigen which offer direct translation into protein antigens without need for viral replicase components. However, antigen expression duration is limited (1-3 days), meaning they will potentially require booster doses. Interestingly, self-replicating (srRNA) based vaccines include both the mRNA for the target antigen and viral replicase machinery which allow self-replication within cells leading to longer antigen expression (1-2 months) and achieving effective immune response with lower doses and leading to a stronger and more sustained immune response. In contrast, srRNA based vaccines require cold storage (-20°C to -70°C) for stability and their larger size (10-15 kb) complicates delivery and manufacturing, which may introduce additional safety concerns and regulatory hurdles. Recently, circular mRNA (circRNA) based vaccines are presented as an alternative to linear conventional and base-modified mRNA; instead, they have a closed-ring structure. The advantages of this type of vaccine are that their circular mRNA is resistant to exonucleases, leading to increased stability and longer half-life in cells (1-3 weeks) which offer prolonged duration of antigen expression (1-3 months). Importantly, circular mRNA does not require cold storage to remain stable, they can be stable at room temperature. To overcome the mRNA stability in high temperature, thermostable mRNA vaccines were engineered with specific sequences and formulations that maintain the mRNA’s integrity and function in higher temperatures. Aside from their ability to withstand high temperature, some other advantages of this vaccine are that they are low risk because they are non-infectious and non-integrating, and they can be rapidly produced and distributed in response to outbreaks, especially in regions without cold chain infrastructure (Table 1).
Various methods have been designed for delivering mRNA, tailored to specific applications. Most common approaches include liposomes, electroporation, gene guns, viral vectors, penetrating peptides, and polymers. Although naked, unformulated mRNA has been shown to be expressed in cells, its cellular uptake is generally very low, around 0.01% [57,58,59,60]. It is important to note that many of these delivery methods are primarily suitable for in vitro use. For instance, liposomes, which were the first delivery method employed in Nano pharmaceuticals, serve as versatile carriers capable of transporting both hydrophobic and hydrophilic substances, including proteins, nucleic acids, and small molecules [61]. Liposomes usually range from 20 to 1000 nm in size and consist of one or more lipid bilayers encapsulating aqueous and hydrophobic compartments. Studies have shown a relationship between liposome size and uptake by macrophages in vivo, emphasizing the need for precise size control in liposome-based delivery systems [62]. This necessitates careful measurement of liposome size for accurate delivery purposes [63]. Currently, lipid nanoparticles (LNPs) are the most commonly used method for therapeutic mRNA delivery. LNPs consist of positively charged cationic and ionizable lipids that complex with the negatively charged mRNA, thereby enhancing cellular uptake, and facilitating endosomal escape [64,65]. Despite being less immunogenic and toxic compared to viral vectors, LNPs still exhibit some cytotoxicity, which can be influenced by the lipid composition or PEG modifications. This cytotoxicity may lead to membrane damage or vacuolization of cytoplasm and could potentially disrupt crucial cellular pathways and cell cycle stages. To mitigate these effects, new lipids are being developed to enhance delivery efficiency while reducing residence time in tissues and cytotoxicity. However, producing LNPs at large scale can be challenging, but solid lipid nanoparticles and nanostructured lipid carriers have been developed to address these limitations, offering improved stability, sterilizability, and bioavailability compared to traditional LNPs [61].
A. Conventional modified RNA (mRNA) to deliver next-generation PanCoVax vaccine candidates. Conventional mRNA vaccines utilize cellular machinery to translate the given protein. In contrast, self-amplifying mRNA vaccines include additional coding for accessory proteins, such as RNA-dependent RNA polymerase, capping enzymes, and proteases, which facilitate self-replication alongside the target RNA. [66,67,68,69]. Advantages of the mRNA technology include but not limited to: (1) expression of antigens in situ results in the native conformation of protein antigens, (2) it mimics a virus infection thereby inducing broad-based immunity, without the need for a virus vector, and (3) generic methods of synthetic production, purification and characterization of mRNA products lead to faster discovery, development, and implementation of vaccines. This last attribute is critically important for infectious disease outbreaks like COVID-19, enabling a much faster response than possible. Currently, two major Conventional mRNA-based vaccines developed by Moderna and Pfizer have been widely used during the COVID-19 pandemic [19,70,71]. Both vaccines employ a similar principle, utilizing mRNA encapsulated in lipid nanoparticles (LNPs) to encode the Spike protein. These vaccines have been administered to billions of individuals and have effectively provided protection against moderate and symptomatic SARS-CoV-2 infection. It should be noted, however, that immunity induced by these, which necessitates the use of booster vaccines [70,72].
B. Design and production of next-generation “pre-emptive” PanCoVax vaccine candidates: We have created PanCoVax vaccine constructs containing ten bioinformatically designed consensus sequences of SARS-CoV-2 antigenic regions that are carefully selected as being highly conserved across all human and animal CoV and are strongly immunogenic (“PanCoVax” candidates). Multiple lines of evidence demonstrate: (1) Three out of these ten T cell PanCoVax vaccine candidates alone conferred protection in the hamster model against infection with SARS-CoV-2 Delta variant, as measured by reduced weight loss and virus replication. (2) When combined with spike antigen, these T cell PanCoVax vaccine candidates provided an additive or synergistic protective efficacy against infection with Delta and re-infection with Omicron, by promoting cross-protective CD4+ and CD8+ T cells (against non-Spike antigens) and broadly neutralizing antibodies (nAb, against Spike) suggesting that multi-antigen PanCoVax vaccines have the potential to terminate infection before transmission and cross-protect against new variants, other HCoV, and future CoV zoonoses. This T-cell immunity would support efficacy in individuals with antibody defects and generate longer-lasting protection due to the inherent durability of T helper cell memory. We demonstrated that delivery of these multi-antigen PanCoVax vaccines boosts durable frontline mucosal immunity, by enhancing: (1) The frequency and function of tissue resident cross-reactive CD4+ and CD8+ TRM cells in the lungs; and (2) cross-reactive IgA/IgG antibodies levels in alveolar lavage that limited viral replication to abort infection in the earliest stages and block transmission.
A safe and effective PanCoVax vaccine inducing systemic and mucosal anti-viral B- and T-cell immunity against the emergence and rapid expansion of SARS-CoV-2 variants is currently not available [73]. For the past 3 years, we have made significant progress in the development of multi-antigen PanCoVax vaccine, identifying potential human B-cell, CD4+ T cells, and CD8+ T cell antigens and epitopes from the whole SARS-CoV-2 genome sequences spanning the virus’s multi-stage lifecycle. We used the sequences of the first SARS-CoV-2 strain immediately after it became publicly available in early January 2020 [74,75,76,77,78,79,80]. Using highly conserved large sequences comprised by ten antigens, we designed 10 PanCoVax vaccine candidates. The use of large protein antigenic sequences will provide broader coverage in the human population by allowing multiple CD4+ and CD8+ T cell epitopes to be presented by multiple HLA types and obviates the need to identify specific T cell epitopes. In addition, targeting CD4+ and CD8+ T cells to conserved regions that are subdominant in natural infection and/or that are not targeted by any currently licensed vaccines, is expected to provide an additional “layer” of protective immunity not currently seen in the infected and vaccinated population. Thus, the use of highly conserved antigenic regions will generate cross-reactive immunity that is expected to protect against new VOCs and zoonoses. The use of established vaccine platforms such as base-modified mRNA encapsulated in lipid nanoparticles (LNP) for which, toxicity, safety, scalability, and manufacturing are well-described substantially de-risks the program. The mRNA/LNP-based multi-antigen PanCoVax vaccine candidates incorporate common B-cell, CD4+ and CD8+ T-cell antigens that are conserved in human SARS-CoV and animal SARS-CoV-like strains. Examples of our mRNA/LNP-based multi-antigen PanCoVax vaccine candidates.
Our data demonstrate that the first three mRNA/LNP-based PanCoVax vaccine candidates (i.e., NSP-2, NSP14 and Nucleocapsid) induced protective CD4+ and CD8+ T-cell-mediated protection against infection with the Delta variant [2]. We selected the mRNA/LNP-based delivery system because it facilitates presentation of antigens to both MHC-I (endogenous) and MHC-II (exogenous) pathways via direct- and cross-presentation [81,82,83,84,85,86,87,88,89,90,91]. In addition, a prototype multi-antigen PanCoVax vaccine candidate that combines different molar ratios of T cell antigens together with Spike B cell antigen demonstrated a significant additive effect leading to better protection than each antigen alone against infection with Delta and re-infection with Omicron variant. These results demonstrate the successful design and construction of an mRNA/LNP-based multi-antigen PanCoVax vaccine candidate and confirm the utility of previous mRNA/LNP-based vaccines in other systems [2].
These ten highly conserved non-Spike antigens are common amongst 8.7 million SARS-CoV-2 strains, including twenty-one VOCs, SARS-CoV, MERS-CoV, common cold CoVs, and animal CoVs. Among these, seven antigens were preferentially recognized by CD8+ and CD4+ T-cells from unvaccinated asymptomatic COVID-19 patients, irrespective of VOC infection. Three of these conserved non-Spike T cell antigens (NSP2, NSP14, and Nucleocapsid) originate from the early-expressed Replication and Transcription Complex (RTC) region. When administered to golden Syrian hamsters in combination with the Spike protein as nucleoside-modified mRNA encapsulated in lipid nanoparticles (LNP)—forming a combined mRNA/LNP-based pan-CoV vaccine—they: (i) Induced high frequencies of lung-resident antigen-specific CXCR5+CD4+ T follicular helper (TFH) cells, GzmB+CD4+ and GzmB+CD8+ cytotoxic T cells (TCYT), and CD69+IFN-γ+TNFα+CD4+ and CD69+IFN-γ+TNFα+CD8+ effector T cells (TEFF); and (ii) Reduced viral load and COVID-19-like symptoms caused by various VOCs, including the highly pathogenic B.1.617.2 Delta variant and the highly transmissible, heavily Spike-mutated XBB1.5 Omicron sub-variant. This combined mRNA/LNP-based pan-CoV vaccine could be rapidly adapted for clinical use to provide broader cross-protective immunity against emerging, highly mutated, and pathogenic VOCs [1,2,92].
C. Self-replicating RNA (srRNA) vaccine platforms to deliver next-generation PanCoVax vaccine candidates: Compared to conventional mRNA, the srRNA vaccine delivery platform offers several additional and attractive features that makes this vaccine delivery system well-suited for application to our next-generation, broad spectrum Pan-CoVax vaccine project. These attributes include longer duration of transgene expression and an enhanced adjuvant effect leading to more potent antigen-specific adaptive immunity, particularly T cell responses, which is a key feature of our strategy to induce strong, cross-strain T cell responses and thereby provide broad protection. In addition, there is a higher potential for co-expressing multiple transgenes from the same srRNA molecule [69]. Moreover, due to their amplification ability, srRNA vaccines could be effective at lower RNA doses, which in turn would lead to higher manufacturing volumes, lower costs, and an improved therapeutic index compared to conventional mRNA vaccines [67]. Potent antigen-specific B and T cell responses can be induced by srRNA at doses as low as picograms in an animal model [69]. The best example of srRNA pan coronavirus vaccine is being developed by Gritstone, encoding viral antigens, including both Spike protein and conserved non-Spike CD8 epitopes aimed at eliciting T-cell responses [93]. Arcturus’ self-amplifying COVID-19 mRNA vaccine candidate (ARCT-154) employs a replicon based on the Venezuelan equine encephalitis virus (VEEV), where the VEEV structural proteins have been replaced with the stabilized, mutation-full-length ancestral Spike protein of SARS-CoV-2. Phase 1/2/3a/3b trials were conducted and demonstrated that two doses of ARCT-154 were well tolerated and immunogenic at the time of the delta (B.1.617.2) variant was circulating. [94]. ARCT-154 has been announced to meet primary efficacy endpoint in phase 3 study and recently approved regulators in Japan [95].
D. Circular mRNA vaccine platforms to deliver next-generation PanCoVax vaccine candidates: Although mRNA has shown promising efficacy in infectious disease and cancer vaccines, its application is hindered by challenges related to expression duration, stability, and immunogenicity. mRNA has a short lifespan and is quickly degraded by intracellular enzymes and the immune system, typically persisting for only 1–3 days [96]. Consequently, mRNA vaccines often result in relatively low levels of antigen expression, necessitating repeated inoculations to achieve sufficient immune responses. However, a new class of therapeutic mRNAs, circular RNAs (circRNAs), is emerging and may help overcome some of the drawbacks associated with linear mRNAs. CircRNAs offer enhanced stability and reduced autoimmunity, potentially providing a solution to these challenges [97,98]. In contrast to linear mRNA, circRNA boasts a notably high stability attributed to its covalently closed ring structure, which shields it from degradation mediated by exonucleases [99]. Studies have indicated that circRNAs possess a median half-life that is at least 2.5 times longer than their linear mRNA counterparts in mammalian cells [100,101]. This enhanced stability allows for protein translation from circRNA to persist for nearly a week, resulting in a significantly higher protein yield compared to linear mRNA, often increased by hundreds of times [96,97,101]. Therefore, CircRNA represents a superior alternative for sustained protein expression, providing enhanced antigenic stimulation and enabling robust antigen production for up to a week [96]. CircRNA vaccines hold promise for enhancing the stability and immunogenicity of mRNA vaccines due to their closed-loop structure, which confers increased stability both in vitro and in vivo, thereby facilitating extended expression of target genes [96]. Pancoronavirus circRNARBD vaccines have been shown to elicited high levels of broad-spectrum neutralizing antibodies against Delta, Omicron and against the current VOCs and long-lasting protective immunity against SARS-CoV-2 in mice and rhesus macaques [102].
E. Thermostable mRNA vaccine platforms to deliver next-generation PanCoVax vaccine candidates: One of the major obstacles facing current coronavirus vaccines is the challenge of widespread distribution during a pandemic [3,103,104,105]. Both authorized vaccines require deep cold chain storage (-70°C for Pfizer/BioNtech and -20°C for Moderna’s SARS-CoV-2 RNA vaccines). Even frozen shipping and storage under standard freezer conditions pose difficulties, especially in areas with limited medical infrastructure [106,107,108]. The instability of RNA vaccines presents a significant challenge, with the underlying physicochemical causes still lacking comprehensive research or understanding. However, some aspects are evident. Firstly, RNA vaccine molecules are prone to cleavage by common ribonucleases (RNases), despite attempts to engineer stabilization. Secondly, because of its size, negative charge, and hydrophilic nature, RNA faces difficulties in traversing cell membranes to reach target cells post-injection. Hence, formulations for RNA delivery become essential to stabilize and shield RNA molecules from degradation [58,60,109]. The primary approach employed for administering RNA vaccines, encompassing most SARS-CoV-2 vaccines under clinical investigation to date, revolves around the use of lipid nanoparticle (LNP) delivery systems, where the negatively charged RNA molecule is enclosed within an intricate lipid framework, yielding RNA/LNP complexes typically sized between 70 and 100 nm in diameter. These complexes serve to shield the RNA from degradation by RNases and facilitate successful endocytosis by the cell [110,111,112]. Nevertheless, both the stability of RNA and LNP remain problematic [108], as indicated by sensitivity to freezing temperatures, leading to adverse effects on their colloidal stability upon freeze/thaw cycles. Consequently, several alternative lipid-based delivery systems have been suggested and advanced for the administration of RNA vaccines [105,108,113,114,115]. However, a critical current need remains an effective and thermostable RNA vaccine delivery system. Research has demonstrated the effectiveness of a lyophilizable, thermostable nanostructured lipid carrier in delivering both mRNA- and self-replicating RNA-based vaccines via intramuscular injection. The liquid nanostructured lipid carrier alone remains stable for at least 1 year when stored at refrigerated temperatures. Additionally, lyophilized nanostructured lipid carrier-RNA complexes have been shown to maintain their biophysical properties and capacity to induce protein expression in vivo for at least 8 months at room temperature and at least 21 months under refrigerated conditions.
4. Nanoparticle Technologies to deliver next-generation PanCoVax vaccine candidates. Nanoparticle vaccines use tiny, engineered particles that can mimic the virus’s structure or used as vehicle platform of RNA and proteins. These nanoparticles can display multiple copies of viral RNAs, and proteins enhancing the immune system’s ability to recognize and remember the virus. The delivery of RNA and repetitive display of the antigen can trigger a stronger immune response. Nanoparticles can be engineered to improve the stability of the vaccine and enhance delivery to the immune system.
A. Self-Assembling Protein Nanoparticles successful in pre-clinical but not successful in clinical vaccine trials against infectious diseases. Self-Assembling Protein Nanoparticles (SAPNs) were discovered in years 2000s actively developed from 2006 [116] to 2024 [87,117,118,119,120,121] as a platform for vaccine delivery due to their ability to self-organize into structures with a potential to induce or boost B and T cell responses. Computer modeling has been used to design SAPNs composed of proteins as building blocks. Structure-based designs of nanoparticles with regular polyhedral symmetry and a diameter of about 16 nm, which self-assembles from single protein chains have been reported. Each protein chain is composed of two coiled-coil oligomerization domains with different oligomerization states joined by a short linker segment. In aqueous solution the proteins form nanoparticles of about 20 nm diameter. Reported mouse studies in 2015 [81] and 2017 [89] designed vaccines against Plasmodium falciparum malaria based on these SAPNs that consist of repetitive antigen display technology to display malaria-specific B cell and T cell epitopes. The resulting protein sequence, when assembled into a nanoparticle, induces strong, long-lived, and protective immune responses against infection with the parasite in mice.
However, to the best of our knowledge, although SAPNs platform has been discovered and extensively studied since 2006 [87,116,117,118,119,120,121]: (1) there are no reported peer-reviewed study demonstrating superiority of SAPNs platform compared to the most recent mRNA/LNP platform when it comes to safety, immunogenicity, and protective efficacy in animal models and against any infectious diseases, including SARS-CoV-2; and (2) Moreover, there are no successful clinical trials against any infectious diseases, including SARS-CoV-2, using this SAPNs technology. Although SAPNs platform-based vaccines were successful in mice in 2015 [81] and 2017 [89], a subsequent SAPNs platform-based malaria-vaccine that moved into a clinical trial appears to have been terminated for reasons that remain to be determined.
Therefore, the SAPN vaccine delivery platform still requires further development to create a safe, immunogenic, and protective antigen delivery platform that can be adapted to the clinic. As reviewed in 2022 by Morales-Hernández [118] and discussed by many other research groups [122,123,124,125,126,127,128,129,130], several challenges might be associated with using SAPNs for vaccine delivery in the clinic: (1) Compared to mRNA/LNP platform, SAPN platform, used selected expression systems that requires achieving consistent assembly and stability of SAPNs that is crucial for ensuring effective vaccine delivery and prolonged shelf life; (2) Compared to mRNA/LNP platform, potential spacial and conformational issues and protein display, orientation, denaturation and aggregation could affect the efficacy of SAPS-based vaccines; (3) Compared to mRNA/LNP platform, consistent and efficient load multiple antigens and control release from SAPNs platform for effective vaccine delivery remain a challenge. In addition, limitations in the amount of protein antigen that can be loaded on a single SAPNs make this delivery system not compatible with combination vaccines that use several protein antigens [123,124,125,127]; (4) Compared to mRNA/LNP platform, compared to mRNA/LNP platform, SAPNs efficacy to target and activate the desired T cell response and particularly neutralizing antibodies cannot be consistently achieved by SAPN system without adding built-in adjuvants that may present toxicity [128]; (5) Compared to mRNA/LNP platform, one must ensure that SAPNs do not induce unwanted immune responses or toxicity a critical point for safe use in the clinic [126]; and (6) Compared to mRNA/LNP technology platform, regulatory requirements for SAPNs-based vaccines may involve complex testing and compliance with standards [122,130].
B. Current Delivery platforms of pan-coronavirus vaccines in clinical trials.
The imperative for a universal coronavirus vaccine is driven by the continuous threat posed by multiple coronaviruses affecting humans, such as SARS-CoV-2, Middle Eastern respiratory syndrome coronavirus (MERS-CoV), and the four endemic human coronaviruses (HCoVs OC43, HKU1, 229E, and NL63) [131,132,133]. Prophylactic vaccines could significantly reduce the morbidity attributed to these pathogens. Although still in the early stages, clinical trials have provided encouraging data on the safety and potential efficacy of pan-coronavirus vaccines [134,135,136]. Some candidates have demonstrated the capacity to induce broad-spectrum neutralizing antibodies and T-cell responses, suggesting cross-protection against multiple coronavirus strains [137,138]. Developing vaccines that can stay ahead of virus mutation is a significant hurdle. A particular obstacle for pan-coronavirus vaccines is the diverse utilization of entry receptors by different coronaviruses. For instance, ACE2 is used by SARS-CoV-1, SARS-CoV-2, and NL63, while DDP4 serves as the entry receptor for MERS, and 9-O-acetylated sialic acid is utilized by OC43 and HKU1. Additionally, 229E employs the aminopeptidase N receptor. Neutralizing antibodies primarily act by blocking the interaction between the virus’s receptor binding domain and the host cell entry receptor. Consequently, developing pan-coronavirus vaccines would require focusing on epitopes that can universally inhibit binding to all coronavirus entry receptors. T cells inherently possess greater cross-reactivity compared to B cells, as they recognize short peptide sequences presented on major histocompatibility complexes (MHC) rather than complex structural epitopes. Moreover, T cells can target conserved internal viral proteins, whereas B cells predominantly recognize surface and structural proteins that are accessible to antibodies on extracellular viruses. Indeed, the interaction between T cells and MHC-peptide is highly adaptable which underscores the potency of T-cell immunity in recognizing and responding to a wide array of pathogenic challenges, including those posed by diverse coronaviruses [139]. Most SARS-CoV-2-specific T cells have been demonstrated to be at least pan-variant reactive, meaning they can recognize epitopes that are retained in currently circulating viruses [140,141,142,143]. As such, apart from a few key exceptions, instances of T-cell escape are rare. This suggests that T-cell-mediated immunity may play a crucial role in providing broad protection against various SARS-CoV-2 variants, contributing to the effectiveness of pan-coronavirus vaccine strategies [144,145,146]. Currently few clinical trials aim to protect against different subgroups of coronaviruses, using different delivery systems, and giving promising results in phase1/2 trials (Table 2):
- Pfizer BNT162b4: Pfizer’s vaccine candidate BNT162b4 (an mRNA vaccine encoding segments of the N and M proteins, and short segments from the ORF1ab polyprotein), developed to enhance T-cell responses against conserved non-spike antigens of SARS-CoV-2, encodes conserved, immunogenic segments of the nucleocapsid, membrane, and ORF1ab proteins targeting diverse HLA alleles. BNT162b4 elicits robust CD4+ and CD8+ T-cell responses to a wide array of epitopes in preclinical animal models, while preserving spike-specific immunity. Notably, BNT162b4 has been shown to protect animal models from severe disease and reduce viral loads following infection with various SARS-CoV-2 variants [147].
- Gritstone GRT-R910. GRT-R910 is an investigational vaccine that was designed to enhance the immunogenicity and protective efficacy against the current and future SARS-CoV-2 VOC. A randomized, double-blinded Phase 2b study has being conducted to assess the efficacy, safety, and immunogenicity of Gritstone bio’s next-generation COVID-19 vaccine candidate compared to an approved COVID-19 vaccine. Gritstone’s vaccine incorporates a self-replicating mRNA, encoding viral antigens, including both Spike protein (as seen in first-generation COVID-19 vaccines) and conserved non-Spike CD8 epitopes aimed at eliciting T-cell responses [93].
- OVX033 from Osivax: A protein subunit vaccine, aiming to protect against sarbecoviruses, the subgroup of coronaviruses from which SARS viruses come from. The vaccine contains the full-length nucleocapsid antigen of SARS-CoV-2 which is genetically fused to the self-assembling sequence OVX313, which is a “55-amino acids sequence, hybrid of the C-terminal fragments of two avian C4bp α-chain sequences, that naturally oligomerizes into heptamers.” The OVX033 aims to target the nucleocapsid (N) protein within SARS-CoV-2, which is highly conserved among the Sarbecoviruses [148]. OVX033 was tested either unadjuvanted or formulated with a squalene-in-water emulsion containing cholesterol and QS21 saponin [148]. After evaluating the efficiency of the OVX033 vaccine using a hamster model of SARS-CoV-2 infection, the vaccine proved effective against 3 different SARS-CoV-2 variants of concerns as seen through significant decrease in weight loss, lung viral loads, inflammation, lymphoplasmacytic perivascular infiltration, and pneumonia incidence. The vaccine also showed improved immunogenicity as T-cell responses were triggered within the hamsters due to the vaccine. These sufficient results supported further evaluation of the vaccine within human trials. Currently, participants are being recruited in Paris to test the safety and immunogenicity of three dosages – they’re aiming for 48 participants.
- PanCoV from LinkInVax, developed by INSERM: A protein subunit vaccine aiming at sarbecoviruses. It was developed by INSERM, the French national health agency similar to the US NIH. The vaccine targets “the RBD of the SARS-CoV-2 spike protein to the CD40 receptor” and includes “T- and B-cell epitopes spanning sequences from S and nucleocapsid (N) proteins from SARS-CoV-2 and highly homologous to 38 sarbecoviruses, including SARS-CoV-2 VOCs [149].” The vaccine was successful at eliciting SARS-CoV-2 Spike protein-specific IgG switched human B cells with a single injection adjuvanted with polyinosinic-polycytidylic acid [149]. There was also evidence of inducing human B-cell and T-cell responses in the humanized mice. Studies in Macaques demonstrated a “blockade” of new cell infections, the destruction of infected cells, and improved protection against SARS-CoV-2 reinfection [149,150] without the need for an adjuvant. Currently, a combined phase 1 and 2 trials has been registered to test this vaccine, with and without an adjuvant. They aim to recruit 240 people.
- CalTech: The California Institute of Technology (CalTech) is developing a Mosaic vaccine utilizing nanoparticles that display 60 randomly organized spike receptor-binding domains (RBDs) originated from the spike trimers of eight distinct sarbecoviruses (mosaic-8 RBD nanoparticles). The Mosaic-8b vaccine is designed to produce antibodies targeting against conserved and relatively hidden epitopes, as opposed to the more commonly targeted, variable, and prominently exposed epitopes. This vaccine includes RBDs from eight SARS-like betacoronaviruses (sarbecoviruses) that encompasses the RBD from the currently prevalent SARS-CoV-2 virus along with RBDs from seven other animal sarbecoviruses. This diverse RBD representation is intended to provide broad-spectrum protection against a variety of related viruses. Preclinical studies have demonstrated that immunization with mosaic-8 nanoparticles elicited anti-coronavirus antibodies with robust neutralizing capabilities and effectiveness against multiple SARS-CoV-2 variants, including the highly transmissible Omicron variant. Furthermore, the vaccination conferred protection against both SARS-CoV-2 and SARS-CoV infections in mouse and nonhuman primate [151,152].
- DIOSynVax: Cambridge University spin-off DIOSynVax (Digitally Immune Optimized Synthetic Vaccines) is utilizing a highly advanced approach in vaccine development. In collaboration with PharmaJet, they have introduced pEVAC-PS, a needle-free intradermal vaccine encoding their synthetic antigen T2_17, designed based on coronavirus receptor binding domain (RBD) sequences [153]. DIOSynVax employs a viral-genome-informed methodology to generate an antigen sequence that phylogenetically aligns with representative sequences from all known sarbecoviruses, ensuring the retention of key antibody epitopes. This synthetic antigen has been evaluated across multiple platforms, including DNA, Modified Vaccinia Ankara (MVA), and mRNA. The antigen has elicited cross-reactive neutralizing antibodies against Delta and Omicron and other tested sarbecoviruses in preclinical studies different animal models [153].
- VBI Vaccines (USA, Canada): VBI Vaccines is developing vaccines that emulate the natural presentation of viruses to stimulate innate immune responses, leveraging their proprietary enveloped virus-like particle (eVLP) technology. Utilizing this technology, VBI designed VBI-2901, a vaccine that expresses a modified prefusion form of spike proteins from SARS-CoV-2, SARS-CoV-1, and MERS-CoV. In preclinical studies, mice immunized with VBI-2901 exhibited a potent neutralizing response against all variants tested, including Bat RaTG13. VBI-2901 elicited a 2.5-fold stronger response to the ancestral strain and a ninefold stronger response against the bat coronavirus when compared to the VBI-2902 vaccine ( containing only the ancestral Wuhan-Hu-1 spike protein) [154]. VBI-2901 is currently being evaluated in Phase I clinical trials.
- Walter Reed Army Institute of Research (WRAIR, USA): The Walter Reed Army Institute of Research (WRAIR) has engineered a recombinant spike ferritin nanoparticle (SpFN) vaccine for SARS-CoV-2 WA-1, in combination with the Army Liposomal Formulation (ALFQ) adjuvant, which includes monophosphoryl lipid A and QS-21 (SpFN/ALFQ). This self-assembling ferritin nanoparticle is designed to present eight prefusion-stabilized SARS-CoV-2 WA-1 spike glycoprotein trimers in an ordered and symmetrical fashion. The SpFN vaccine is paired with a unilamellar liposomal adjuvant that incorporates monophosphoryl lipid A and the saponin QS-21 (ALFQ), reputed for its ability to enhance the immunogenicity of various protein vaccine candidates and its favorable tolerance in human trials. This immunogen-adjuvant combination has demonstrated the capacity to elicit broad immunity against sarbecoviruses and confer protection against SARS-CoV-2 in preclinical models [155].
Challenges and Future Directions
While each of these vaccine delivery platforms offer certain advantages, they also face various challenges, such as ensuring safety, manufacturing speed and scalability, vaccine stability, enhancing the breadth and duration of immune protection, and overcoming pre-existing immunity. As research and development advances, combining attributes of these platforms may offer paths to even more effective pan-coronavirus vaccines. The continued evolution of these platforms in the context of pan-coronavirus vaccine development is a testament to the rapid progress being made in immunology and virology, offering hope for durable, broad-spectrum protection against current and emergent coronavirus threats.
Conclusion
Pan-coronavirus vaccines represent a potentially bold step forward in the global fight against current and emerging coronavirus infectious diseases. While significant challenges remain, the advancement of several pan-coronavirus vaccine candidates employing various delivery system technologies into clinical trials would offer the opportunity to better understand the critical attributes needed for an effective vaccine. The ultimate success of these pan-coronavirus vaccines will depend not only on their clinical safety and efficacy but also on the ease with which vaccines can be manufactured and distributed globally at large scale. As pan-coronavirus vaccine clinical trial landscape evolves, so too will our hope for future resilience to coronavirus outbreaks.
Acknowledgments
Studies of this report were supported by Public Health Service Research grants AI158060, AI150091, AI143348, AI147499, AI143326, AI138764, AI124911, and AI110902 from the National Institutes of Allergy and Infectious Diseases (NIAID) to LBM and by R43AI174383 to TechImmune, LLC. LBM has an equity interest in TechImmune, LLC., a company that may potentially benefit from the research results and serves on the company’s Scientific Advisory Board. LBM’s relationship with TechImmune, LLC., has been reviewed and approved by the University of California, Irvine by its conflict-of-interest policies.
Conflict of Interest
Studies of this report were supported by Public Health Service Research grants AI158060, AI150091, AI143348, AI147499, AI143326, AI138764, AI124911, and AI110902 from the National Institutes of Allergy and Infectious Diseases (NIAID) to LBM and by R43AI174383 to TechImmune, LLC. LBM has an equity interest in TechImmune, LLC., a company that may potentially benefit from the research results and serves on the company’s Scientific Advisory Board. LBM’s relationship with TechImmune, LLC., has been reviewed and approved by the University of California, Irvine by its conflict-of-interest policies.
References
- Prakash, S. , et al., Genome-Wide Asymptomatic B-Cell, CD4 (+) and CD8 (+) T-Cell Epitopes, that are Highly Conserved Between Human and Animal Coronaviruses, Identified from SARS-CoV-2 as Immune Targets for Pre-Emptive Pan-Coronavirus Vaccines. bioRxiv 2020. [Google Scholar]
- Coulon, P.-G. , et al., High frequencies of alpha common cold coronavirus/SARS-CoV-2 cross-reactive functional CD4+ and CD8+ memory T cells are associated with protection from symptomatic and fatal SARS-CoV-2 infections in unvaccinated COVID-19 patients. Frontiers in Immunology 2024, 15. [Google Scholar] [CrossRef] [PubMed]
- Jackson, L.A. , et al., An mRNA Vaccine against SARS-CoV-2 - Preliminary Report. N Engl J Med 2020, 383, 1920–1931. [Google Scholar] [CrossRef] [PubMed]
- Ishimaru, H. , et al., Epitopes of an antibody that neutralizes a wide range of SARS-CoV-2 variants in a conserved subdomain 1 of the spike protein. J Virol 2024, e0041624. [Google Scholar] [CrossRef] [PubMed]
- Magazine, N. , et al., Immune Epitopes of SARS-CoV-2 Spike Protein and Considerations for Universal Vaccine Development. Immunohorizons 2024, 8, 214–226. [Google Scholar] [CrossRef]
- Tan, T.J.C. , et al., Evidence of antigenic drift in the fusion machinery core of SARS-CoV-2 spike. Proc Natl Acad Sci U S A 2024, 121, e2317222121. [Google Scholar] [CrossRef]
- Teng, S. , et al., SARS-CoV-2 spike-reactive naive B cells and pre-existing memory B cells contribute to antibody responses in unexposed individuals after vaccination. Front Immunol 2024, 15, 1355949. [Google Scholar] [CrossRef]
- Tanunliong, G. , et al., Persistence of Anti-SARS-CoV-2 Antibodies in Long Term Care Residents Over Seven Months After Two COVID-19 Outbreaks. Front Immunol 2021, 12, 775420. [Google Scholar] [CrossRef]
- Park, T. , et al., Vaccines against SARS-CoV-2 variants and future pandemics. Expert Rev Vaccines 2022, 21, 1363–1376. [Google Scholar] [CrossRef]
- Murdocca, M. , et al., Peptide Platform as a Powerful Tool in the Fight against COVID-19. Viruses 2021, 13. [Google Scholar] [CrossRef]
- Farlow, A. , et al., The Future of Epidemic and Pandemic Vaccines to Serve Global Public Health Needs. Vaccines (Basel) 2023, 11. [Google Scholar]
- Ali, A.M. , et al., Circulating cancer giant cells with unique characteristics frequently found in patients with myelodysplastic syndromes (MDS). Med Oncol 2023, 40, 204. [Google Scholar] [CrossRef] [PubMed]
- Baghban, R., A. Ghasemian, and S. Mahmoodi, Nucleic acid-based vaccine platforms against the coronavirus disease 19 (COVID-19). Arch Microbiol 2023, 205, 150. [Google Scholar] [CrossRef] [PubMed]
- Amano, T. , et al., Controllable self-replicating RNA vaccine delivered intradermally elicits predominantly cellular immunity. iScience 2023, 26, 106335. [Google Scholar] [CrossRef]
- Amano, T. , et al., Controllable self-replicating RNA vaccine delivered intradermally elicits predominantly cellular immunity. bioRxiv 2022. [Google Scholar]
- Abdelaziz, M.O. , et al., Early protective effect of a (“pan”) coronavirus vaccine (PanCoVac) in Roborovski dwarf hamsters after single-low dose intranasal administration. Front Immunol 2023, 14, 1166765. [Google Scholar] [CrossRef]
- Keech, C. , et al., Phase 1-2 Trial of a SARS-CoV-2 Recombinant Spike Protein Nanoparticle Vaccine. N Engl J Med 2020, 383, 2320–2332. [Google Scholar] [CrossRef]
- Francica, J.R. , et al., Vaccination with SARS-CoV-2 Spike Protein and AS03 Adjuvant Induces Rapid Anamnestic Antibodies in the Lung and Protects Against Virus Challenge in Nonhuman Primates. bioRxiv 2021. [Google Scholar]
- Kyriakidis, N.C. , et al., SARS-CoV-2 vaccines strategies: a comprehensive review of phase 3 candidates. NPJ Vaccines 2021, 6, 28. [Google Scholar] [CrossRef]
- Goepfert, P.A. , et al., Safety and immunogenicity of SARS-CoV-2 recombinant protein vaccine formulations in healthy adults: interim results of a randomised, placebo-controlled, phase 1-2, dose-ranging study. Lancet Infect Dis 2021, 21, 1257–1270. [Google Scholar] [CrossRef]
- Kleanthous, H. , et al., Scientific rationale for developing potent RBD-based vaccines targeting COVID-19. NPJ Vaccines 2021, 6, 128. [Google Scholar] [CrossRef] [PubMed]
- Thimmiraju, S.R. , et al., A trivalent protein-based pan-Betacoronavirus vaccine elicits cross-neutralizing antibodies against a panel of coronavirus pseudoviruses. NPJ Vaccines 2024, 9, 132. [Google Scholar] [CrossRef] [PubMed]
- Shalash, A.O. , et al., Peptide-Based Vaccine against SARS-CoV-2: Peptide Antigen Discovery and Screening of Adjuvant Systems. Pharmaceutics 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Skwarczynski, M. and I. Toth, Recent advances in peptide-based subunit nanovaccines. Nanomedicine (Lond) 2014, 9, 2657–69. [Google Scholar] [CrossRef]
- Bagwe, P.V. , et al., Peptide-Based Vaccines and Therapeutics for COVID-19. Int J Pept Res Ther 2022, 28, 94. [Google Scholar] [CrossRef]
- Cankat, S., M. U. Demael, and L. Swadling, In search of a pan-coronavirus vaccine: next-generation vaccine design and immune mechanisms. Cell Mol Immunol 2024, 21, 103–118. [Google Scholar] [CrossRef]
- Tan, C.W. , et al., Broad-spectrum pan-genus and pan-family virus vaccines. Cell Host Microbe 2023, 31, 902–916. [Google Scholar] [CrossRef]
- Lee, C.S. , et al., Adenovirus-Mediated Gene Delivery: Potential Applications for Gene and Cell-Based Therapies in the New Era of Personalized Medicine. Genes Dis 2017, 4, 43–63. [Google Scholar] [CrossRef]
- Crystal, R.G. , Adenovirus: the first effective in vivo gene delivery vector. Hum Gene Ther 2014, 25, 3–11. [Google Scholar] [CrossRef]
- Lukashev, A.N. and A.A. Zamyatnin, Jr., Viral Vectors for Gene Therapy: Current State and Clinical Perspectives. Biochemistry (Mosc) 2016, 81, 700–8. [Google Scholar] [CrossRef]
- Coughlan, L. , Factors Which Contribute to the Immunogenicity of Non-replicating Adenoviral Vectored Vaccines. Front Immunol 2020, 11, 909. [Google Scholar] [CrossRef] [PubMed]
- Ewer, K. , et al., Chimpanzee adenoviral vectors as vaccines for outbreak pathogens. Hum Vaccin Immunother 2017, 13, 3020–3032. [Google Scholar] [CrossRef] [PubMed]
- Ghebremedhin, B. , Human adenovirus: Viral pathogen with increasing importance. Eur J Microbiol Immunol (Bp) 2014, 4, 26–33. [Google Scholar] [CrossRef] [PubMed]
- See, R.H. , et al., Comparative evaluation of two severe acute respiratory syndrome (SARS) vaccine candidates in mice challenged with SARS coronavirus. J Gen Virol 2006, 87, 641–650. [Google Scholar] [CrossRef]
- Ong, E. , et al., COVID-19 Coronavirus Vaccine Design Using Reverse Vaccinology and Machine Learning. Front Immunol 2020, 11, 1581. [Google Scholar] [CrossRef]
- Ong, E. , et al., COVID-19 coronavirus vaccine design using reverse vaccinology and machine learning. bioRxiv 2020. [Google Scholar]
- Chavda, V.P., R. Pandya, and V. Apostolopoulos, DNA vaccines for SARS-CoV-2: toward third-generation vaccination era. Expert Rev Vaccines 2021, 20, 1549–1560. [Google Scholar] [CrossRef]
- Duan, L. , et al., The SARS-CoV-2 Spike Glycoprotein Biosynthesis, Structure, Function, and Antigenicity: Implications for the Design of Spike-Based Vaccine Immunogens. Front Immunol 2020, 11, 576622. [Google Scholar] [CrossRef]
- Liu, Y. and Q. Ye, Safety and Efficacy of the Common Vaccines against COVID-19. Vaccines (Basel) 2022, 10. [Google Scholar]
- Folegatti, P.M. , et al., Safety and immunogenicity of the ChAdOx1 nCoV-19 vaccine against SARS-CoV-2: a preliminary report of a phase 1/2, single-blind, randomised controlled trial. Lancet 2020, 396, 467–478. [Google Scholar] [CrossRef]
- Li, C. and R.J. Samulski, Engineering adeno-associated virus vectors for gene therapy. Nat Rev Genet 2020, 21, 255–272. [Google Scholar] [CrossRef] [PubMed]
- Li, X. , et al., Novel AAV-based genetic vaccines encoding truncated dengue virus envelope proteins elicit humoral immune responses in mice. Microbes Infect 2012, 14, 1000–7. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F. , et al., Novel adeno-associated virus-based genetic vaccines encoding hepatitis C virus E2 glycoprotein elicit humoral immune responses in mice. Mol Med Rep 2019, 19, 1016–1023. [Google Scholar] [PubMed]
- Lin, J. , et al., A new genetic vaccine platform based on an adeno-associated virus isolated from a rhesus macaque. J Virol 2009, 83, 12738–50. [Google Scholar] [CrossRef]
- Zabaleta, N. , et al., An AAV-based, room-temperature-stable, single-dose COVID-19 vaccine provides durable immunogenicity and protection in non-human primates. Cell Host Microbe 2021, 29, 1437–1453. [Google Scholar] [CrossRef]
- Wu, F. , et al., Single-shot AAV-vectored vaccine against SARS-CoV-2 with fast and long-lasting immunity. Acta Pharm Sin B 2023, 13, 2219–2233. [Google Scholar] [CrossRef]
- Woldemeskel, B.A. , et al., CD4+ T cells from COVID-19 mRNA vaccine recipients recognize a conserved epitope present in diverse coronaviruses. J Clin Invest 2022, 132. [Google Scholar] [CrossRef]
- Pack, S.M. and P.J. Peters, SARS-CoV-2-Specific Vaccine Candidates; the Contribution of Structural Vaccinology. Vaccines (Basel) 2022, 10. [Google Scholar]
- Longet, S. , et al., mRNA vaccination drives differential mucosal neutralizing antibody profiles in naive and SARS-CoV-2 previously-infected individuals. Front Immunol 2022, 13, 953949. [Google Scholar] [CrossRef]
- Gupta, S.L. , et al., Loss of Pfizer (BNT162b2) Vaccine-Induced Antibody Responses against the SARS-CoV-2 Omicron Variant in Adolescents and Adults. J Virol 2022, 96, e0058222. [Google Scholar] [CrossRef]
- Ni, L. , Advances in mRNA-Based Cancer Vaccines. Vaccines (Basel) 2023, 11. [Google Scholar] [CrossRef]
- Iavarone, C. , et al., Mechanism of action of mRNA-based vaccines. Expert Rev Vaccines 2017, 16, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Silva-Pilipich, N. , et al., Self-Amplifying RNA: A Second Revolution of mRNA Vaccines against COVID-19. Vaccines (Basel) 2024, 12. [Google Scholar]
- Xie, J. , et al., Circular RNA: A promising new star of vaccine. J Transl Int Med 2023, 11, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Voigt, E.A. , et al., A self-amplifying RNA vaccine against COVID-19 with long-term room-temperature stability. NPJ Vaccines 2022, 7, 136. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, A. , et al., A flexible, thermostable nanostructured lipid carrier platform for RNA vaccine delivery. Mol Ther Methods Clin Dev 2022, 25, 205–214. [Google Scholar] [CrossRef]
- Witten, J. , et al., Recent advances in nanoparticulate RNA delivery systems. Proc Natl Acad Sci U S A 2024, 121, e2307798120. [Google Scholar] [CrossRef]
- Parhiz, H., E. N. Atochina-Vasserman, and D. Weissman, mRNA-based therapeutics: looking beyond COVID-19 vaccines. Lancet 2024, 403, 1192–1204. [Google Scholar] [CrossRef]
- Kowalski, P.S. , et al., Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery. Mol Ther 2019, 27, 710–728. [Google Scholar] [CrossRef]
- Sahin, U., K. Kariko, and O. Tureci, mRNA-based therapeutics--developing a new class of drugs. Nat Rev Drug Discov 2014, 13, 759–80. [Google Scholar] [CrossRef]
- Tenchov, R. , et al., Lipid Nanoparticles horizontal line From Liposomes to mRNA Vaccine Delivery, a Landscape of Research Diversity and Advancement. ACS Nano 2021, 15, 16982–17015. [Google Scholar] [CrossRef] [PubMed]
- Harashima, H. , et al., Enhanced hepatic uptake of liposomes through complement activation depending on the size of liposomes. Pharm Res 1994, 11, 402–6. [Google Scholar] [CrossRef] [PubMed]
- Nagayasu, A. Uchiyama, and H. Kiwada, The size of liposomes: a factor which affects their targeting efficiency to tumors and therapeutic activity of liposomal antitumor drugs. Adv Drug Deliv Rev 1999, 40, 75–87. [Google Scholar] [PubMed]
- Oberli, M.A. , et al., Lipid Nanoparticle Assisted mRNA Delivery for Potent Cancer Immunotherapy. Nano Lett 2017, 17, 1326–1335. [Google Scholar] [CrossRef]
- Hou, X. , et al., Lipid nanoparticles for mRNA delivery. Nat Rev Mater 2021, 6, 1078–1094. [Google Scholar] [CrossRef]
- Magini, D. , et al., Self-Amplifying mRNA Vaccines Expressing Multiple Conserved Influenza Antigens Confer Protection against Homologous and Heterosubtypic Viral Challenge. PLoS One 2016, 11, e0161193. [Google Scholar] [CrossRef]
- Vogel, A.B. , et al., Self-Amplifying RNA Vaccines Give Equivalent Protection against Influenza to mRNA Vaccines but at Much Lower Doses. Mol Ther 2018, 26, 446–455. [Google Scholar] [CrossRef]
- Bloom, K., F. van den Berg, and P. Arbuthnot, Self-amplifying RNA vaccines for infectious diseases. Gene Ther 2021, 28, 117–129. [Google Scholar] [CrossRef]
- Blakney, A.K., S. Ip, and A.J. Geall, An Update on Self-Amplifying mRNA Vaccine Development. Vaccines (Basel) 2021, 9. [Google Scholar]
- Patel, R. , et al., A comprehensive review of SARS-CoV-2 vaccines: Pfizer, Moderna & Johnson & Johnson. Hum Vaccin Immunother 2022, 18, 2002083. [Google Scholar]
- Rabaan, A.A. , et al., A Comprehensive Review on the Current Vaccines and Their Efficacies to Combat SARS-CoV-2 Variants. Vaccines (Basel) 2022, 10. [Google Scholar]
- Pegu, A. , et al., Durability of mRNA-1273 vaccine-induced antibodies against SARS-CoV-2 variants. Science 2021, 373, 1372–1377. [Google Scholar] [CrossRef] [PubMed]
- Abbott, T.R. , et al., Development of CRISPR as an Antiviral Strategy to Combat SARS-CoV-2 and Influenza. Cell 2020, 181, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M. and L. Song, Novel antibody epitopes dominate the antigenicity of spike glycoprotein in SARS-CoV-2 compared to SARS-CoV. Cell Mol Immunol 2020, 17, 536–538. [Google Scholar] [CrossRef]
- Walls, A.C. , et al., Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef]
- Tilocca, B. , et al., Molecular basis of COVID-19 relationships in different species: a one health perspective. Microbes Infect 2020, 22, 218–220. [Google Scholar] [CrossRef]
- Tetro, J.A. , Is COVID-19 receiving ADE from other coronaviruses? Microbes Infect 2020, 22, 72–73. [Google Scholar] [CrossRef]
- Grifoni, A. , et al., A Sequence Homology and Bioinformatic Approach Can Predict Candidate Targets for Immune Responses to SARS-CoV-2. Cell Host Microbe 2020, 27, 671–680. [Google Scholar] [CrossRef]
- Bhattacharya, M. , et al., Development of epitope-based peptide vaccine against novel coronavirus 2019 (SARS-COV-2): Immunoinformatics approach. J Med Virol 2020, 92, 618–631. [Google Scholar] [CrossRef]
- Ahmed, S.F. A. Quadeer, and M.R. McKay, Preliminary Identification of Potential Vaccine Targets for the COVID-19 Coronavirus (SARS-CoV-2) Based on SARS-CoV Immunological Studies. Viruses 2020, 12. [Google Scholar] [CrossRef]
- Burkhard, P. and D.E. Lanar, Malaria vaccine based on self-assembling protein nanoparticles. Expert Rev Vaccines 2015, 14, 1525–7. [Google Scholar] [CrossRef] [PubMed]
- Doll, T.A. , et al., Optimizing the design of protein nanoparticles as carriers for vaccine applications. Nanomedicine 2015, 11, 1705–13. [Google Scholar] [CrossRef] [PubMed]
- El Bissati, K. , et al., Effectiveness of a novel immunogenic nanoparticle platform for Toxoplasma peptide vaccine in HLA transgenic mice. Vaccine 2014, 32, 3243–8. [Google Scholar] [CrossRef] [PubMed]
- El Bissati, K. , et al., Protein nanovaccine confers robust immunity against Toxoplasma. NPJ Vaccines 2017, 2, 24. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q. , et al., Expression, purification and refolding of a self-assembling protein nanoparticle (SAPN) malaria vaccine. Methods 2013, 60, 242–7. [Google Scholar] [CrossRef]
- Kaba, S.A. , et al., A nonadjuvanted polypeptide nanoparticle vaccine confers long-lasting protection against rodent malaria. J Immunol 2009, 183, 7268–77. [Google Scholar] [CrossRef]
- Kaba, S.A. , et al., Self-assembling protein nanoparticles with built-in flagellin domains increases protective efficacy of a Plasmodium falciparum based vaccine. Vaccine 2018, 36, 906–914. [Google Scholar] [CrossRef]
- Kaba, S.A. , et al., Protective antibody and CD8+ T-cell responses to the Plasmodium falciparum circumsporozoite protein induced by a nanoparticle vaccine. PLoS One 2012, 7, e48304. [Google Scholar] [CrossRef]
- Karch, C.P. , et al., The use of a P. falciparum specific coiled-coil domain to construct a self-assembling protein nanoparticle vaccine to prevent malaria. J Nanobiotechnology 2017, 15, 62. [Google Scholar] [CrossRef]
- McCoy, M.E. , et al., Mechanisms of protective immune responses induced by the Plasmodium falciparum circumsporozoite protein-based, self-assembling protein nanoparticle vaccine. Malar J 2013, 12, 136. [Google Scholar] [CrossRef]
- Seth, L. , et al., Development of a self-assembling protein nanoparticle vaccine targeting Plasmodium falciparum Circumsporozoite Protein delivered in three Army Liposome Formulation adjuvants. Vaccine 2017, 35, 5448–5454. [Google Scholar] [CrossRef] [PubMed]
- Prakash, S. , et al., Cross-protection induced by highly conserved human B, CD4(+), and CD8(+) T-cell epitopes-based vaccine against severe infection, disease, and death caused by multiple SARS-CoV-2 variants of concern. Front Immunol 2024, 15, 1328905. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.D. , et al., GRT-R910: a self-amplifying mRNA SARS-CoV-2 vaccine boosts immunity for >/=6 months in previously-vaccinated older adults. Nat Commun 2023, 14, 3274. [Google Scholar] [CrossRef] [PubMed]
- Ho, N.T. , et al., Safety, immunogenicity and efficacy of the self-amplifying mRNA ARCT-154 COVID-19 vaccine: pooled phase 1, 2, 3a and 3b randomized, controlled trials. Nat Commun 2024, 15, 4081. [Google Scholar] [CrossRef]
- First self-amplifying mRNA vaccine approved. Nat Biotechnol 2024, 42, 4.
- Chen, R. , et al., Engineering circular RNA for enhanced protein production. Nat Biotechnol 2023, 41, 262–272. [Google Scholar] [CrossRef]
- Wesselhoeft, R.A. , et al., RNA Circularization Diminishes Immunogenicity and Can Extend Translation Duration In Vivo. Mol Cell 2019, 74, 508–520. [Google Scholar] [CrossRef]
- Breuer, J. , et al., What goes around comes around: Artificial circular RNAs bypass cellular antiviral responses. Mol Ther Nucleic Acids 2022, 28, 623–635. [Google Scholar] [CrossRef]
- Wesselhoeft, R.A., P. S. Kowalski, and D.G. Anderson, Engineering circular RNA for potent and stable translation in eukaryotic cells. Nat Commun 2018, 9, 2629. [Google Scholar] [CrossRef]
- Enuka, Y. , et al., Circular RNAs are long-lived and display only minimal early alterations in response to a growth factor. Nucleic Acids Res 2016, 44, 1370–83. [Google Scholar] [CrossRef]
- Memczak, S. , et al., Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–8. [Google Scholar] [CrossRef] [PubMed]
- Qu, L. , et al., Circular RNA vaccines against SARS-CoV-2 and emerging variants. Cell 2022, 185, 1728–1744.e16. [Google Scholar] [CrossRef] [PubMed]
- Schoenmaker, L. , et al., mRNA-lipid nanoparticle COVID-19 vaccines: Structure and stability. Int J Pharm 2021, 601, 120586. [Google Scholar] [CrossRef] [PubMed]
- Guan, S. and J. Rosenecker, Nanotechnologies in delivery of mRNA therapeutics using nonviral vector-based delivery systems. Gene Ther 2017, 24, 133–143. [Google Scholar] [CrossRef]
- Hassett, K.J. , et al., Optimization of Lipid Nanoparticles for Intramuscular Administration of mRNA Vaccines. Mol Ther Nucleic Acids 2019, 15, 1–11. [Google Scholar] [CrossRef]
- Kumru, O.S. , et al., Vaccine instability in the cold chain: mechanisms, analysis and formulation strategies. Biologicals 2014, 42, 237–59. [Google Scholar] [CrossRef]
- Chen, D. and D. Zehrung, Desirable attributes of vaccines for deployment in low-resource settings. J Pharm Sci 2013, 102, 29–33. [Google Scholar] [CrossRef]
- Crommelin, D.J.A. , et al., Addressing the Cold Reality of mRNA Vaccine Stability. J Pharm Sci 2021, 110, 997–1001. [Google Scholar] [CrossRef]
- Wang, Y.S. , et al., mRNA-based vaccines and therapeutics: an in-depth survey of current and upcoming clinical applications. J Biomed Sci 2023, 30, 84. [Google Scholar] [CrossRef]
- Reichmuth, A.M. , et al., mRNA vaccine delivery using lipid nanoparticles. Ther Deliv 2016, 7, 319–34. [Google Scholar] [CrossRef]
- Bahl, K. , et al., Preclinical and Clinical Demonstration of Immunogenicity by mRNA Vaccines against H10N8 and H7N9 Influenza Viruses. Mol Ther 2017, 25, 1316–1327. [Google Scholar] [CrossRef] [PubMed]
- Bahl, K. , et al., Preclinical and Clinical Demonstration of Immunogenicity by mRNA Vaccines against H10N8 and H7N9 Influenza Viruses. Mol Ther 2022, 30, 2874. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P. , et al., Long-term storage of lipid-like nanoparticles for mRNA delivery. Bioact Mater 2020, 5, 358–363. [Google Scholar] [PubMed]
- Stitz, L. , et al., A thermostable messenger RNA based vaccine against rabies. PLoS Negl Trop Dis 2017, 11, e0006108. [Google Scholar] [CrossRef]
- Zhang, N.N. , et al., A Thermostable mRNA Vaccine against COVID-19. Cell 2020, 182, 1271–1283. [Google Scholar] [CrossRef]
- Raman, S. , et al., Structure-based design of peptides that self-assemble into regular polyhedral nanoparticles. Nanomedicine 2006, 2, 95–102. [Google Scholar] [CrossRef]
- Lopez-Sagaseta, J. , et al., Self-assembling protein nanoparticles in the design of vaccines. Comput Struct Biotechnol J 2016, 14, 58–68. [Google Scholar] [CrossRef]
- Morales-Hernandez, S., N. Ugidos-Damboriena, and J. Lopez-Sagaseta, Self-Assembling Protein Nanoparticles in the Design of Vaccines: 2022 Update. Vaccines (Basel) 2022, 10. [Google Scholar]
- Adams, K. , et al., Prevalence of SARS-CoV-2 and Influenza Coinfection and Clinical Characteristics Among Children and Adolescents Aged <18 Years Who Were Hospitalized or Died with Influenza - United States, 2021-22 Influenza Season. MMWR Morb Mortal Wkly Rep 2022, 71, 1589–1596. [Google Scholar]
- Karch, C.P. , et al., Production of E. coli-expressed Self-Assembling Protein Nanoparticles for Vaccines Requiring Trimeric Epitope Presentation. J Vis Exp 2019. [Google Scholar] [CrossRef]
- Yang, Y. , et al., The biodistribution of self-assembling protein nanoparticles shows they are promising vaccine platforms. J Nanobiotechnology 2013, 11, 36. [Google Scholar] [CrossRef] [PubMed]
- Tinkle, S. , et al., Nanomedicines: Addressing the Scientific and Regulatory Gap. Handbook of Clinical Nanomedicine: Law, Business, Regulation, Safety, and Risk 2016, 2, 413–470. [Google Scholar]
- Tan, P. , et al., pH-Triggered Size-Transformable and Bioactivity-Switchable Self-Assembling Chimeric Peptide Nanoassemblies for Combating Drug-Resistant Bacteria and Biofilms. Advanced Materials 2023, 35. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.S., Q. Luo, and J.Q. Liu, Protein self-assembly supramolecular strategies. Chemical Society Reviews 2016, 45, 2756–2767. [Google Scholar] [CrossRef]
- Lundahl, M.L.E. , et al., Aggregation of protein therapeutics enhances their immunogenicity: causes and mitigation strategies. Rsc Chemical Biology 2021, 2, 1004–1020. [Google Scholar] [CrossRef]
- Morales-Hernández, S., N. Ugidos-Damboriena, and J. López-Sagaseta, Self-Assembling Protein Nanoparticles in the Design of Vaccines: 2022 Update. Vaccines 2022, 10. [Google Scholar] [CrossRef]
- Dutta, K., J. M. Zhuang, and S. Thayumanavan, Templated self-assembly of a covalent polymer network for intracellular protein delivery and traceless release. Abstracts of Papers of the American Chemical Society 2018, 256. [Google Scholar]
- Tapia, D., A. Reyes-Sandoval, and J.I. Sanchez-Villamil, Protein-based Nanoparticle Vaccine Approaches Against Infectious Diseases. Archives of Medical Research 2023, 54, 168–175. [Google Scholar] [CrossRef]
- Blanco, E., H. Shen, and M. Ferrari, Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nature Biotechnology 2015, 33, 941–951. [Google Scholar] [CrossRef]
- Butkovich, N. , et al., Advancements in protein nanoparticle vaccine platforms to combat infectious disease. Wiley Interdisciplinary Reviews-Nanomedicine and Nanobiotechnology 2021, 13. [Google Scholar] [CrossRef]
- Chen, S. , et al., Viruses from poultry and livestock pose continuous threats to human beings. Proc Natl Acad Sci U S A 2021, 118. [Google Scholar]
- Edwards, C.E. , et al., Swine acute diarrhea syndrome coronavirus replication in primary human cells reveals potential susceptibility to infection. Proc Natl Acad Sci U S A 2020, 117, 26915–26925. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. , et al., Functional and genetic analysis of viral receptor ACE2 orthologs reveals a broad potential host range of SARS-CoV-2. Proc Natl Acad Sci U S A 2021, 118. [Google Scholar]
- Koff, W.C. and S.F. Berkley, A universal coronavirus vaccine. Science 2021, 371, 759. [Google Scholar] [CrossRef]
- Giurgea, L.T., A. Han, and M.J. Memoli, Universal coronavirus vaccines: the time to start is now. NPJ Vaccines 2020, 5, 43. [Google Scholar] [CrossRef]
- Morens, D.M., J. K. Taubenberger, and A.S. Fauci, Universal Coronavirus Vaccines - An Urgent Need. N Engl J Med 2022, 386, 297–299. [Google Scholar] [CrossRef]
- Xia, S. , et al., Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res 2020, 30, 343–355. [Google Scholar] [CrossRef]
- Vithani, N. , et al., SARS-CoV-2 Nsp16 activation mechanism and a cryptic pocket with pan-coronavirus antiviral potential. Biophys J 2021, 120, 2880–2889. [Google Scholar] [CrossRef]
- Sewell, A.K. , Why must T cells be cross-reactive? Nat Rev Immunol 2012, 12, 669–77. [Google Scholar] [CrossRef]
- Tarke, A. , et al., SARS-CoV-2 vaccination induces immunological T cell memory able to cross-recognize variants from Alpha to Omicron. Cell 2022, 185, 847–859.e11. [Google Scholar] [CrossRef]
- Keeton, R. , et al., Author Correction: T cell responses to SARS-CoV-2 spike cross-recognize Omicron. Nature 2022, 604, E25. [Google Scholar] [CrossRef] [PubMed]
- Keeton, R. , et al., T cell responses to SARS-CoV-2 spike cross-recognize Omicron. Nature 2022, 603, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y. , et al., Ancestral SARS-CoV-2-specific T cells cross-recognize the Omicron variant. Nat Med 2022, 28, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Flament, H. , et al., Outcome of SARS-CoV-2 infection is linked to MAIT cell activation and cytotoxicity. Nat Immunol 2021, 22, 322–335. [Google Scholar] [CrossRef]
- Dolton, G. , et al., Emergence of immune escape at dominant SARS-CoV-2 killer T cell epitope. Cell 2022, 185, 2936–2951. [Google Scholar] [CrossRef]
- de Silva, T.I. , et al., The impact of viral mutations on recognition by SARS-CoV-2 specific T cells. iScience 2021, 24, 103353. [Google Scholar] [CrossRef]
- Arieta, C.M. , et al., The T-cell-directed vaccine BNT162b4 encoding conserved non-spike antigens protects animals from severe SARS-CoV-2 infection. Cell 2023, 186, 2392–2409.e21. [Google Scholar] [CrossRef]
- Primard, C. , et al., OVX033, a nucleocapsid-based vaccine candidate, provides broad-spectrum protection against SARS-CoV-2 variants in a hamster challenge model. Front Immunol 2023, 14, 1188605. [Google Scholar] [CrossRef]
- Coleon, S. , et al., Design, immunogenicity, and efficacy of a pan-sarbecovirus dendritic-cell targeting vaccine. EBioMedicine 2022, 80, 104062. [Google Scholar] [CrossRef]
- Alexandre, M. , et al., Modelling the response to vaccine in non-human primates to define SARS-CoV-2 mechanistic correlates of protection. Elife 2022, 11. [Google Scholar] [CrossRef]
- Cohen, A.A. , et al., Mosaic RBD nanoparticles protect against multiple sarbecovirus challenges in animal models. bioRxiv 2022. [Google Scholar]
- Cohen, A.A. , et al., Mosaic sarbecovirus nanoparticles elicit cross-reactive responses in pre-vaccinated animals. bioRxiv 2024. [Google Scholar]
- Vishwanath, S. , et al., A computationally designed antigen eliciting broad humoral responses against SARS-CoV-2 and related sarbecoviruses. Nat Biomed Eng 2023. [Google Scholar] [CrossRef] [PubMed]
- Bozic, J. , et al., Use of eVLP-based vaccine candidates to broaden immunity against SARS-CoV-2 variants. bioRxiv 2021. [Google Scholar]
- Ober Shepherd, B.L. , et al., SARS-CoV-2 recombinant spike ferritin nanoparticle vaccine adjuvanted with Army Liposome Formulation containing monophosphoryl lipid A and QS-21: a phase 1, randomised, double-blind, placebo-controlled, first-in-human clinical trial. Lancet Microbe 2024, 5, e581–e593. [Google Scholar] [CrossRef]
- Pardi, N. , et al., mRNA vaccines - a new era in vaccinology. Nat Rev Drug Discov 2018, 17, 261–279. [Google Scholar] [CrossRef]
- Hashiba, K. , et al., Overcoming thermostability challenges in mRNA-lipid nanoparticle systems with piperidine-based ionizable lipids. Commun Biol 2024, 7, 556. [Google Scholar] [CrossRef]
Figure 2.
Peptides to deliver next-generation PanCoVax vaccine candidates. A portion of an antigenic protein such as peptide or polypeptide from coronavirus is used alone or combination of peptides from different proteins and/or different variant to make a vaccine, potentially with the addition of an adjuvant. After injection of the vaccine, the peptides can be presented to antigen presenting cells and loaded onto MHC class I for presentation to CD8 or through MHC II for presentation to CD4+ T cells. to This stimulates some B cells to differentiate into plasma cells which then release antibodies.
Figure 2.
Peptides to deliver next-generation PanCoVax vaccine candidates. A portion of an antigenic protein such as peptide or polypeptide from coronavirus is used alone or combination of peptides from different proteins and/or different variant to make a vaccine, potentially with the addition of an adjuvant. After injection of the vaccine, the peptides can be presented to antigen presenting cells and loaded onto MHC class I for presentation to CD8 or through MHC II for presentation to CD4+ T cells. to This stimulates some B cells to differentiate into plasma cells which then release antibodies.
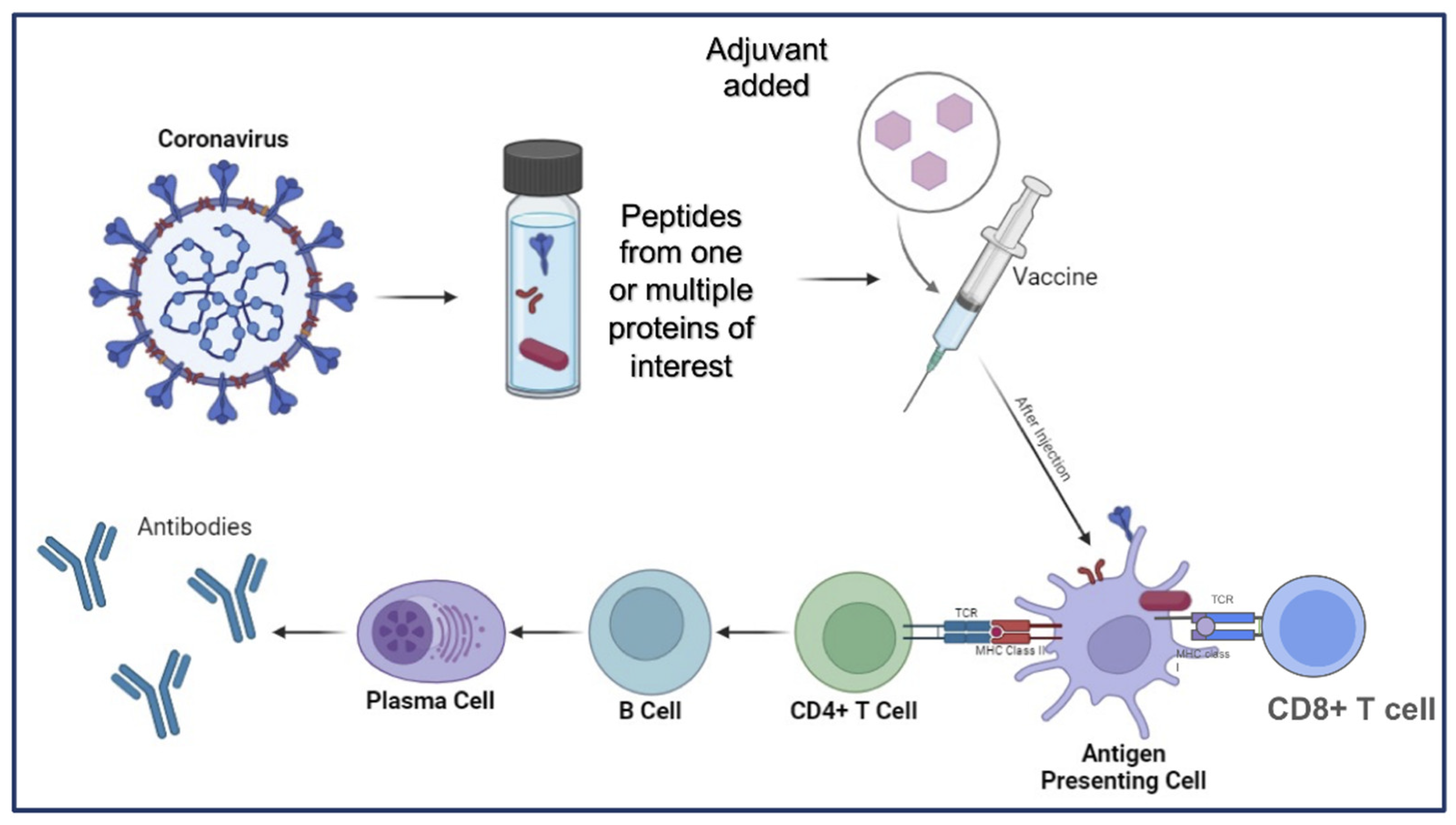
Figure 3.
Adenovirus to deliver next-generation PanCoVax vaccine candidates. The Adenovirus is non-enveloped and utilizes double-stranded DNA that encodes the antigen of interest from coronavirus. After construction of the viral vector vaccine, the following steps occur: (1) The vaccine is injected intramuscularly. (2) Muscle cells engulf the cells through endocytosis. (3) The vesicle around the Adenovirus breaks down. (4) The Adenovirus attaches to the nucleus and transcription begins. (5) The protein is created through translation. After this, the proteins peptides can either (a) be loaded onto MHC class I molecules for direct presentation to CD8+ T cells or (b) be presented to antigen presenting cells and loaded onto MHC II for presentation to CD4+ T cells, which stimulates some B cells to differentiate into plasma cells which then release antibodies.
Figure 3.
Adenovirus to deliver next-generation PanCoVax vaccine candidates. The Adenovirus is non-enveloped and utilizes double-stranded DNA that encodes the antigen of interest from coronavirus. After construction of the viral vector vaccine, the following steps occur: (1) The vaccine is injected intramuscularly. (2) Muscle cells engulf the cells through endocytosis. (3) The vesicle around the Adenovirus breaks down. (4) The Adenovirus attaches to the nucleus and transcription begins. (5) The protein is created through translation. After this, the proteins peptides can either (a) be loaded onto MHC class I molecules for direct presentation to CD8+ T cells or (b) be presented to antigen presenting cells and loaded onto MHC II for presentation to CD4+ T cells, which stimulates some B cells to differentiate into plasma cells which then release antibodies.
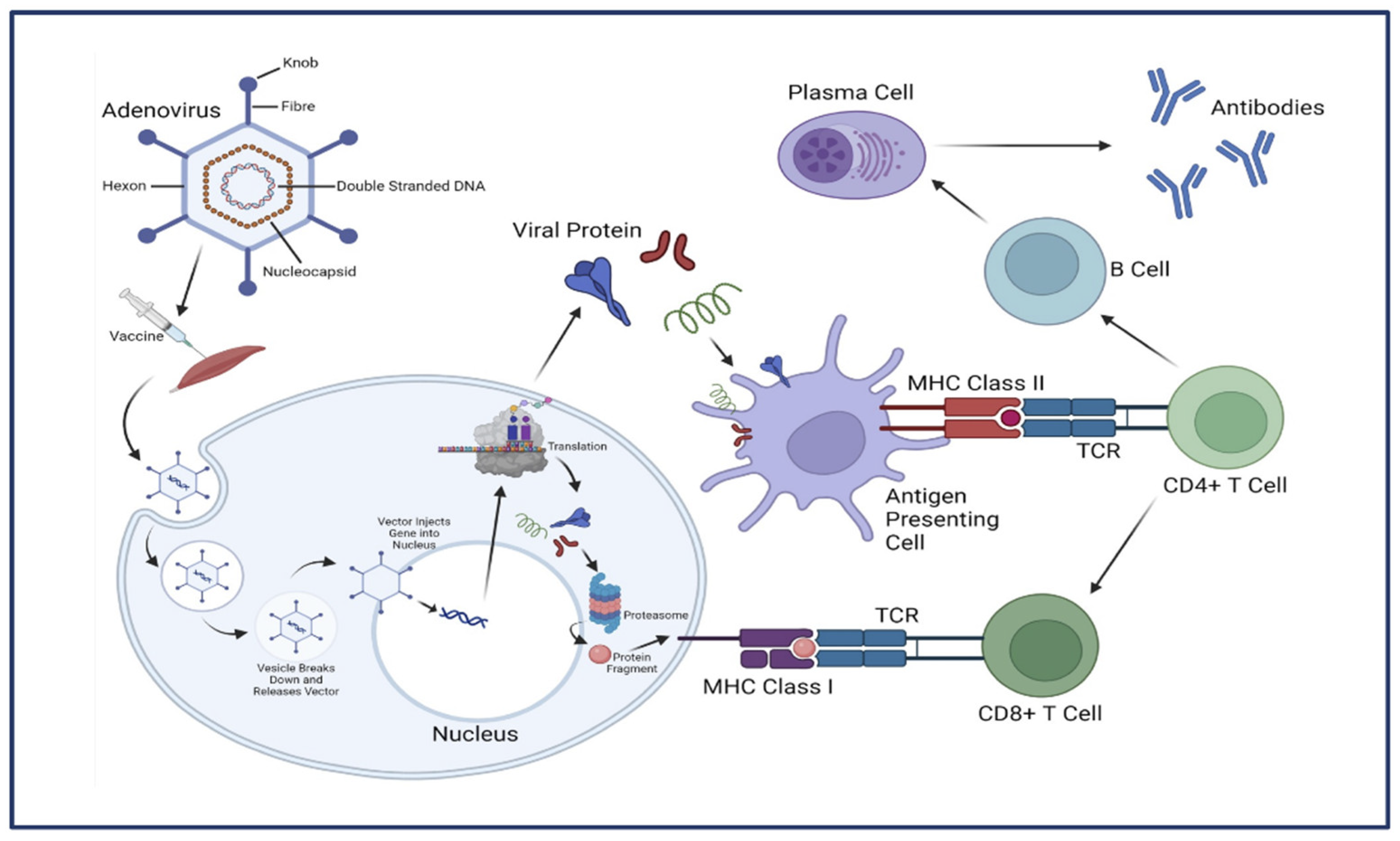

Table 1.
Characteristics of different types of mRNA used to deliver next-generation pan-coronavirus vaccines candidates.
Table 1.
Characteristics of different types of mRNA used to deliver next-generation pan-coronavirus vaccines candidates.
| Type of RNA Vaccine | Size of mRNA | Half-life in cells | Expression Duration | Cold Chain Required? | Mode of Action | Example of Vaccine | Sources |
|---|---|---|---|---|---|---|---|
| Conventional and base-modified mRNA, non-amplifying mRNA | Variable typically 1-5 kb | 6-12 hours | 1-3 days | Yes, typically requires cold storage (refrigeration) | Uses chemically modified nucleotides to enhance stability and translation, leading to protein antigen production and immune response induction. |
BioNTech/Pfizer. Highly effective, 95% efficacy, approved globally. Moderna. Highly effective, 94% efficacy, approved globally. |
[60,156] |
| Self-amplifying mRNA (saRNA) | 10-15 kb | 2-4 days | 1-2 months | Yes, typically requires cold storage (refrigeration) | Contains viral replicase machinery that allows the RNA to self-amplify within cells, producing a larger amount of antigen over an extended period. |
Arcturus Therapeutics. Ongoing, early results show strong immune response. Imperial College London. Phase 1 trial ongoing, safety and immune response being evaluated Gritstone. Phase I trials ongoing. |
[53,69,93] |
| Circular mRNA (circRNA) | Variable, typically 1-5 kb | 1-3 weeks | 1-3 months | No, can be stored at room temperature | Circular structure is resistant to exonucleases, leading to increased stability and prolonged antigen expression. | Laronde. Preclinical studies indicate potential for durability and stability | [54,99] |
| Thermostable mRNA | Variable typically 1-5 kb | 6-12 hours | 1-3 days | No, can be stored at room temperature | Engineered to maintain stability and function at higher temperatures. | CureVac. Moderate efficacy, 48% in Phase 3 trials | [115,157] |
Table 2.
Current pan-coronavirus vaccine in different stages of in clinical trials.
| Vaccine Name | Vaccine Type | Developer | Clinical Phase | Dose and Regimen | Antigen | Publications (PMID, Year) |
|---|---|---|---|---|---|---|
| BNT162b4 | mRNA | Pfizer | Phase I trial ongoing | N/A | segments of the SARS-CoV-2 nucleocapsid, membrane, and ORF1ab proteins, targeting diverse HLA alleles. | 37164012 (2023) |
| GRT-R910 | saRNA | Gritstone bio, Inc. | Phase 1, NCT05148962 | 2 different doses (10 or 30 mcg) | Full-length Spike and selected conserved non-Spike T cell epitopes. | 37280238 (2023 |
| Mosaic-8b | Protein subunit | California Institute of Technology (Caltech) | Yet to begin | N/A | 1 SARS-CoV-2 RBD + 7 sarbecovirus RBD | 38148330, (2024) 33436524 (2021) 35857620 (2022) 36370711 (2022) 36865256 (2023) |
| DIOS-CoVax/ pEVAC-PS |
mRNA | DIOSynvax | Phase I, (https://www.isrctn.com/ISRCTN87813400) |
1.2mg, 0.8mg 0.4mg, or 0.2mg, 2 doses, 30 days apart |
T2_17 (DIOSynVax Generated) | 38148330, (2024) https://doi.org/10.21203/rs.3.rs-995273/v1 (Under Review - Nature) |
| CD40.CoV2 | Protein subunit | Inserm Vaccine Research Institute | Yet to begin | N/A | Receptor-binding domain (RBD) of the SARS-CoV-2 spike protein | 38148330, (2024) 34471122 (2021) 35594660 (2022) 35801637 (2022) |
| OVX033 | Protein subunit | Osivax | Yet to begin | N/A | Nucleocapsid (N) Protein | 37409116 (2023) |
| Unnamed | mRNA, Viral vector and Protein subunit |
University of California Irvine/TechImmune LLC | Yet to begin | N/A | Multi-epitope | 33911008 (2021) 37292861 (2023) https://doi.org/10.1101/2023.05.23.542024 (Under Review - JV) |
| VBI-2901 | eVLP | VBI Vaccines | Phase I, NCT05548439 |
15-20 µg, 2 doses, 55 days apart | Spike trivalent (SARS- CoV-1, SARS-CoV-2, MERS-CoV) | 38148330, (2024) https://doi.org/10.1101/2021.09.28.462109 (Under Review) |
| SpFN 1B-06-PL | Protein subunit | Walter Reed Army Institute of Research (WRAIR) | Phase I, NCT04784767 |
25 µg or 50 µg, 2 doses | SARS-CoV-2 Spike Ferritin Nanoparticle | 34711815 (2021) 34903722 (2021) 34919799 (2021) 34914540 (2022) 35632473 (2022) 36934088 (2023) 37023746 (2023) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



