Submitted:
03 October 2024
Posted:
08 October 2024
You are already at the latest version
Abstract
Papillary tumors of the pineal region (PTPR) are extremely rare malignancies usually treated with surgery and adjuvant tumor bed radiotherapy (RT). We present the case of a 69-year-old man diagnosed with PTPR after presenting with two weeks of confusion and ataxia. Imaging the head with computed tomography (CT) and magnetic resonance imaging (MRI) showed hydrocephalus and a 2 cm pineal region mass. We review the presenting symptoms, investigations, and differential diagnosis for patients with pineal region masses. The pathological features, including immunohistochemistry, are described with exemplative images. We report how the patient’s hydrocephalus was managed with medications and neurosurgical interventions, as well as the curative treatment of his tumor with surgery and postoperative tumor bed RT with 54 Gray (Gy) in 30 fractions. We also review the PTPR literature, including prognostic features and the evidence for treatment modalities. Few publications outline adjuvant radiotherapy planning treatment volumes, which are reported here. The patient was followed with a brain MRI every three months and is asymptomatic with no evidence of recurrent disease 18 months after surgery. PTPR has very high local recurrence rates following treatment and further research is needed to find more effective interventions and improve patient outcomes.
Keywords:
papillary tumor
; papillary tumour
; pineal region
; surgery
; radiotherapy
1. Introduction
Pineal gland tumors make up less than 1% of primary brain tumors in adults and 3% in children [1]. Pineal parenchymal tumors are the second most common type of pineal gland tumors after germ cell tumors and include pineocytoma (World Health Organization (WHO) grade 1), pineal parenchymal tumors of intermediate differentiation (WHO grade 2-3), papillary tumor of the pineal region (PTPR; WHO grade 2-3), and pineoblastoma (WHO grade 4) [1,2]. PTPR was recognized as a distinct entity by the WHO in 2007 [3]. There is no established staging system. Less than 8% of pineal gland tumors are PTPR, under 0.08% of all primary brain tumors, making it very rare [4]. The median age at diagnosis is 29-33 years, and some studies report a slight female predominance, whereas others report a slight male predominance [1,5,6]. PTPR arises from the sub-commissural organ's third posterior ventricle ependyma cells near the pineal gland, not the pineal gland itself [1,5]. It has a similar histologic architecture to ependymoma and choroid plexus tumors [1,5]. PTPR usually has an epithelial-type growth pattern with layers of tumor cells covering vessels and creating perivascular pseudorosettes with a similar immunohistochemical (IHC) profile to choroid plexus tumors [5]. A review of the IHC features of 31 cases of PTPR found positive staining for vimentin (VIM), neuron-specific enolase (NSE), cytokeratin (CK) 18, S-100 protein P (S100-P), microtubule-associated protein 2 (MAP-2), neural cell adhesion molecule (NCAM/CD56), nestin, periodic acid Schiff (PAS) in most cases [5]. The same review found negative straining for CK 20, neurofilament (NF), CK5-6, CK7, Kir7.1, glial fibrillary acidic protein (GFAP), and epithelial cadherin (E-CAD) in most cases [5]. There are no known risk factors for PTPR [7]. Patients with pineal region tumors usually present with headaches (83%), vision abnormalities (67%), and gait changes (41.7%) [1,8]. Other common symptoms are lethargy, nausea, and vomiting (40%) [1]. Physical exams often reveal swelling of the optic disc (60%), impaired coordination (50%), impaired upward gaze (30%), upper or lower extremity tremors (20%), abnormal pupillary responses (17%), and hyperreflexia (13%) [1,8]. Initial investigations include magnetic resonance imaging (MRI) of the brain and entire spinal cord, cerebrospinal fluid cytology to exclude metastatic dissemination, and blood work including alpha-fetoprotein and beta-human chorionic gonadotropin (beta-HCG) to assess whether a germ cell tumor is present (the most common type of pineal gland tumor) [1]. PTPR usually appears as a heterogeneously enhancing mixed solid and cystic mass on MRI, with radiologic hydrocephalus present in up to 88.6% of patients [6,8]. Initial workup and treatment often co-occur with biopsy and cerebrospinal fluid diversion to manage acute obstructive hydrocephalus if present [1]. The most common pineal region tumors are germ cell tumors (59%), pineal parenchymal tumors (30%), and glioblastoma/astrocytoma (5%) [1]. The differential diagnosis for a pineal region mass also includes lymphoma, ganglioglioma, ependymoma, gliosarcoma, neuroendocrine carcinoma of the pineal parenchyma, schwannoma, choroid plexus papilloma, meningioma, metastases, lipomas, pineal cysts, arachnoid cysts, and vascular malformations [1,9]. Given the rarity of this malignancy, there is no standard treatment paradigm [2]. Surgery is almost always recommended for PTPR after the pathologic diagnosis is determined, with adjuvant tumor bed radiotherapy (RT) usually recommended post-operatively [6,8]. There are no established systemic therapies for PTPR [8]. Smaller tumor size and treatment with surgery (regardless of the extent of resection) were associated with improved prognosis in a meta-analysis [6]. PTPR has a very high risk of recurrence following treatment, with a 5-year progression-free survival of only 34.5% and a 5-year overall survival of <75% [2]. In this article, we present the case of a 69-year-old man with PTPR treated with surgery and adjuvant tumor bed radiotherapy. We discuss his presenting symptoms, investigations, and management, including RT volumes and dose fractionation, and review the PTPR treatment literature.
2. Case Presentation
A 69-year-old man was brought to the emergency department by his family following two weeks of confusion, anorexia, dehydration, and gait changes. He was known for myocardial infarction with stenting of the left anterior descending artery, cervical stenosis, chronic renal insufficiency, glaucoma, well-controlled hypertension, and dyslipidemia. Prior surgeries included iridectomy, appendectomy, and umbilical hernia repair. He took low-dose aspirin, ramipril, metoprolol, dorzolamide-timolol eye drops, and Lipitor daily. He had a 45-pack-year smoking history but had quit after his myocardial infarction seven years prior and did not drink alcohol or use recreational drugs. There was no noteworthy family history. The physical exam showed altered mental status (the patient was unaware of the reason he was brought to the hospital), right cranial nerve VI palsy, pupillary asymmetry (right pupil larger than left), decreased pupillary light reflexes in the right eye, and mild gait ataxia. Vital signs and the rest of the neurologic exam were unremarkable. CT and MRI of the head showed a 2.1 x 1.7 x 1.6 cm extra-axial mass in the pineal region with mixed solid and liquid components. A 9 mm anterior cystic component was hypointense on T1 and strongly hyperintense on T2 without post-gadolinium enhancement (Figure 1). A 7 mm posterior component of the mass was hypertense on T1, T2, and FLAIR imaging, and a hemorrhagic necrotic portion was suspected. Obstruction due to mass effect on the aqueduct of Sylvius was present, causing significant upstream hydrocephalus and signs of transependymal resorption. The third ventricle measured 4.9 x 1.8 cm (Figure 2). Foci of diffusion restriction were present, but the diffusion and SWAN studies were otherwise non-contributory. The initial diagnostic hypothesis was a pineocytoma or germ cell tumor.
Complete blood count, blood glucose, electrolytes, renal function tests, liver function tests, thyroid stimulating hormone, beta-HCG, and alpha-fetoprotein were normal. The patient was admitted to neurosurgery. Further workup with CT scans of the chest, abdomen, and pelvis and MRI of the entire spine were negative for suspicious extracranial findings. Four days after admission, the patient became disoriented and stopped speaking (Glasgow coma scale 6). An urgent CT scan of the head showed increased hydrocephalus with decreased high convexity sulci visibility. The patient was transferred to the intensive care unit, Diamox 50 mg po bid was started, mannitol 150 mg IV x 1 was given, and the head of his bed was elevated to 30 degrees. He was intubated and treated with urgent external ventricular drainage (EVD). Cerebrospinal fluid cytology was negative for neoplastic cells. The patient’s neurological status did not improve after EVD, and two days later, he was treated with a third ventriculostomy and biopsy of the pineal region mass. A post-operative CT scan of the head showed reduced hydrocephalus with decreased dilatation of the third ventricle and frontal horns. The patient’s neurologic status rapidly improved following ventriculostomy, and he was extubated on postoperative day 1, and the EVD was removed. Biopsy of the pineal region mass showed cellular proliferation with epithelial appearance, forming papillae lined by tall cylindrical cells with clear eosinophilic fibrillar cytoplasm and hyperchromatic oval nuclei (Figure 3). A few foci of necrosis were present, but there was no evidence of increased mitosis. The cells were strongly positive for Vimentin and CD56, focally positive for CK8/18, CKAE1/AE3, GFAP, and S100-P. The cells did not express ENU, neurofilament, synaptophysin, or E-CAD. Ki-67 was 1-2% (see Figure 4). The preferred diagnosis was a papillary tumor of the pineal region, WHO grade 2 or 3. The differential diagnosis included ependymoma, choroid plexus tumor, and less likely other pineal tumors or metastasis. The diagnosis was retained following a second opinion by a neuropathologist from a quaternary hospital.
The patient was transferred to a quaternary cancer center and treated with right suboccipital craniotomy and dissection of the pineal region mass using a supracerebellar infratentorial approach with minimal adverse effects. He had mild diplopia post-operatively that had been present before surgery and pain around the craniotomy site that was relieved with Tylenol. He recovered rapidly and was discharged three days after surgery. The pathology showed a proliferation of monotonous cells with solid and papillary patterns with focal hemorrhagic and necrotic zones. The neoplastic cells displayed moderately abundant cytoplasm with well-defined borders and epithelioid appearance, ovoid nuclei, and prominent nucleoli with papillary structures around the vessels suggestive of perivascular pseudorosettes. The tumor cells were highly expressive of MAP2, CD56, NSE, and CK18/8 around the papillary structures. C-MYC antibody was positive in most nuclei, and S100-P was weakly positive in a subpopulation of cells. The mitotic index was 2-3/mm2, and the Ki-67 was up to 15-20% in certain foci. NEUN, NF, synaptophysin, chromogranin, CD45, BRG1, GFAP, P53, IDH1 R132H, ATRX, OLIG2, BRAF V600E, CD34, EMA, E-CAD, and SMARCB1/INI1 were not significantly expressed. The pathology was consistent with a diagnosis of papillary tumor of the pineal region, WHO grade 3. The solid tumor panel (Ampli focus panel) was negative for clinically relevant DNA, RNA, or CNV alterations. The patient’s case was presented at neuro-oncology tumor board, and the consensus was to treat with adjuvant radiotherapy. The patient was treated with external beam radiotherapy 54 Gray (Gy) in 30 fractions to the tumor bed. Table 1 outlines the adjuvant RT planning volumes and dose fractionation used. The standard organ at-risk radiation dose constraints for conventional fractionation were respected (Table 2).
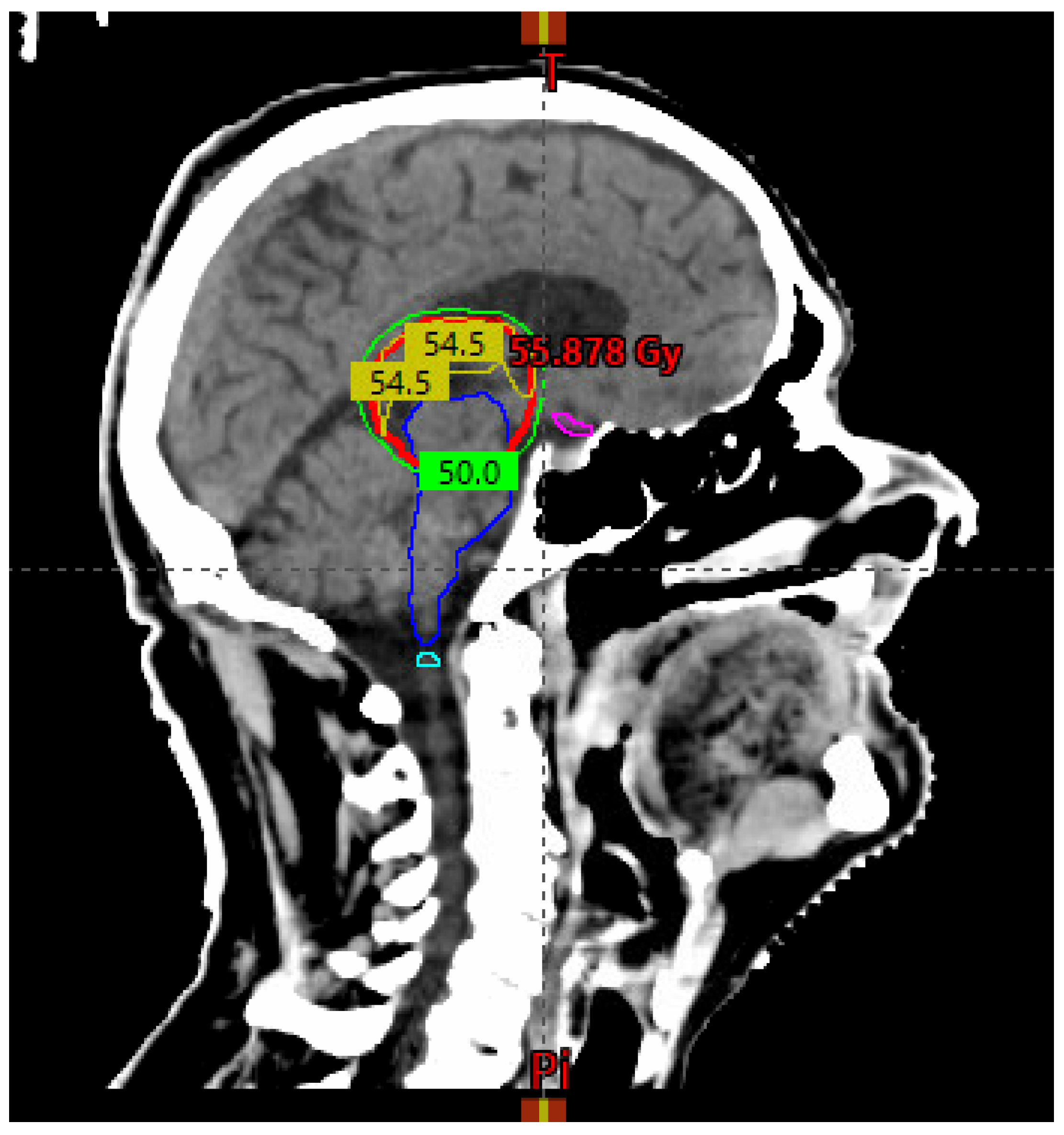
The treatment planning objective for the planning target volume (PTV) coverage was V50GY > 99% (see Figure 4 below).
Figure 4.
A sagittal view of the planning CT scan shows the 50 Gy isodose line (green) covering the PTV (red). The 54 Gy isodose line (yellow) curves superiorly to respect the brainstem (blue) and optic chiasm (magenta) radiation dose constraints.
Figure 4.
A sagittal view of the planning CT scan shows the 50 Gy isodose line (green) covering the PTV (red). The 54 Gy isodose line (yellow) curves superiorly to respect the brainstem (blue) and optic chiasm (magenta) radiation dose constraints.
After adjuvant RT, the patient was followed with a contrast-enhanced brain MRI every three months. At 18 months post-treatment, he reported no symptoms, and the MRI was negative for recurrent disease.
3. Discussion
PTPR is an exceedingly rare malignancy without high-quality evidence to guide treatment [2]. Patients are diagnostically challenging because they usually present with neurologic symptoms caused by obstructive hydrocephalus, and investigations and initial treatment happen concurrently [1]. Access to neurosurgery and radiation oncology care is not widely available outside of major cities, and patients living far away from tertiary care hospitals face additional access and financial burdens associated with having to travel, often very long distances, for treatment [12]. We conducted a literature review on PubMed to identify PTPR articles that evaluate the impact of treatment modalities and/or report treatment volumes for adjuvant conventionally fractionated radiotherapy. We used the search terms “papillary tumor/our of the pineal region” and “radiation or radiotherapy” or “surgery” or “chemotherapy”. Our search identified two systematic reviews, four review articles, one multicenter study, and seven case reports. We also identified seven articles that describe treatment volumes for adjuvant conventionally fractionated radiotherapy (Table 3). Yamaki and colleagues systematically reviewed the literature and identified 71 studies with 177 patients with PTPR [6]. They found that 36-month overall survival was improved when surgery was a part of treatment (hazard ratio (HR) 0.16, 95% confidence interval (CI) 0.05-0.45, p= 0.001) [6]. The only other significant prognostic factor was tumor size, with each additional centimeter associated with worse survival (HR 1.99, 05% CI 1.12-3.53 p=0.19) [6]. No additional benefit was found for gross total resection (versus subtotal resection or biopsy), adjuvant radiotherapy, or systemic therapies [6]. Another systematic review by Lancia and colleagues included 26 studies with 116 PTPR patients and reported that the majority of patients (52.6%) obtained gross total resection, and most patients (72.4%) received radiotherapy as part of their therapeutic strategy. 70% of patients treated with radiotherapy received adjuvant radiotherapy with 50.4-60 Gy in 1.8-2 Gy per fraction, the most common dose fractionation. 28.6% of patients were treated with curative radiotherapy, mostly with Gamma Knife stereotactic radiosurgery (SRS), using doses ranging from 12-36 Gy in 1 fraction, although conventional fractionation was also used for curative treatment [8]. Focal irradiation to the tumor bed was used in most cases, but whole brain or craniospinal irradiation with boost was also used in 9.8% of cases. 18.5% of patients received adjuvant chemotherapy using combinations of temozolomide, vincristine, cisplatin, etoposide, nimustine, and ifosfamide [8]. The authors did not analyze the impact on local control or survival associated with each treatment modality. A multicenter study by Fauchon and colleagues of 44 patients with histopathologically proven PTPR found that gross total resection was significantly associated with increased overall survival compared to subtotal resection or biopsy only (p=0.04) and that older age was associated with worse overall survival (p=0.03) [13]. The authors found no overall survival or progression-free survival benefit from adjuvant radiotherapy or chemotherapy [13]. Surgery is almost always recommended for PTPR after the pathologic diagnosis is confirmed and based on the available literature, is the most evidenced-based intervention [6,8]. After surgery, adjuvant radiotherapy (RT) is also usually given despite the lack of evidence of benefit and most often consists of external beam radiotherapy to the tumor bed and macroscopic disease with doses of 45-59.4 Gy in 1.8-2 Gy once daily fractions [6,8,13,14,15,16,17,18,19].
Whole brain radiotherapy and craniospinal irradiation are also sometimes used, particularly in cases with spinal dissemination, with doses of 30.4-36 Gy to the craniospinal axis with an 18-23.4 Gy boost to the tumor bed and gross disease [2,6,8,13,14]. Curative RT is reserved for cases where surgery isn’t possible [2,6,8]. Stereotactic radiosurgery (SRS) with doses ranging from 12-36 Gy in 1-3 fractions has also been used in curative and adjuvant settings with good local control and low toxicity [6,8]. Publications using brachytherapy are limited, but interstitial brachytherapy with Iodine-125 in the adjuvant setting has been reported [20]. Adjuvant chemotherapy is also used sometimes (18.5% in the systematic review by Lancia and colleagues) but the two available publications that attempted to quantify the impact reported no benefit [6,8,13). Toxicity rates following treatment are reassuringly low. Out of 116 patients with PTPR treated with surgery and RT analyzed by Lancia and colleagues, only three developed adverse effects, including one patient with grade 2 fatigue, one with grade 3 diplopia and hypersomnia, and another with grade 3 motor deficiency and Parinaud syndrome [8]. PTPR has a very high risk of local recurrence despite aggressive treatment that can be greater than 80% at 10 years [2,8]. The European Reference Network for all rare adult solid cancers (EUROCAN) reports an overall survival rate of < 75% at 5 years [2].
4. Conclusions
PTPR is a rare tumor with limited evidence to guide treatment that an oncologist might only encounter once in their career. The most common treatment is surgery followed by postoperative tumor bed RT with 45-59.4 Gy / 25-33 fractions and, sometimes, adjuvant chemotherapy. Smaller tumor size and treatment with surgery might be associated with better outcomes. There is limited evidence of benefit for treatments other than surgery. Recurrence rates are very high following treatment, and further research is needed to improve outcomes.
Author Contributions
Conceptualization, reviewing the literature, and writing the first draft, B.R.P.; M.B.; reviewing, editing, and writing.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
The patient gave written informed consent to publish their case details and diagnostic / treatment imaging.
Data Availability Statement
Further information can be provided upon request to the corresponding author.
Acknowledgments
Thank you, Dr. Milene Gonzalez-Verdecia, for your pathology expertise.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Moschovi M, Chrousos G. Pineal gland masses. In: UpToDate. Eichler AF (Deputy Editor), Wolters Kluwer. Accessed December 28th, 2023.
- Lombardi G, Poliani PL, Manara R, Berhouma M, Minniti G, Tabouret E, Razis E, Cerretti G, Zagonel V, Weller M, Idbaih A. Diagnosis and Treatment of Pineal Region Tumors in Adults: A EURACAN Overview. Cancers (Basel). 2022 Jul 27;14(15):3646. [CrossRef] [PubMed]
- Patil M, Karandikar M. Papillary tumor of pineal region: A rare entity. Asian J Neurosurg. 2016 Oct-Dec;11(4):453. [CrossRef] [PubMed]
- Mottolese C, Szathmari A, Beuriat PA. Incidence of pineal tumors. A review of the literature. Neurochirurgie. 2015 Apr-Jun;61(2-3):65-9. Epub 2014 Aug 10. [CrossRef] [PubMed]
- Fèvre-Montange M, Hasselblatt M, Figarella-Branger D, Chauveinc L, Champier J, Saint-Pierre G, Taillandier L, Coulon A, Paulus W, Fauchon F, Jouvet A. Prognosis and histopathologic features in papillary tumors of the pineal region: a retrospective multicenter study of 31 cases. J Neuropathol Exp Neurol. 2006 Oct;65(10):1004-11. [CrossRef] [PubMed]
- Yamaki VN, Solla DJF, Ribeiro RR, da Silva SA, Teixeira MJ, Figueiredo EG. Papillary Tumor of the Pineal Region: Systematic Review and Analysis of Prognostic Factors. Neurosurgery. 2019 Sep 1;85(3):E420-E429. [CrossRef] [PubMed]
- Gomez D, Parsons DW. Pineal Tumors. American Brain Tumor Association. October, 2023. Accessed August 4th, 2024. Available online: https://www.abta.org/tumor_types/pineal-tumors/.
- Lancia A, Becherini C, Detti B, Bottero M, Baki M, Cancelli A, Ferlosio A, Scoccianti S, Sun R, Livi L, Ingrosso G. Radiotherapy for papillary tumor of the pineal region: A systematic review of the literature. Clin Neurol Neurosurg. 2020 Mar;190:105646. Epub 2019 Dec 21. [CrossRef] [PubMed]
- Favero G, Bonomini F, Rezzani R. Pineal Gland Tumors: A Review. Cancers (Basel). 2021 Mar 27;13(7):1547. [CrossRef] [PubMed]
- Emami B, Lyman J, Brown A, Coia L, Goitein M, Munzenrider JE, Shank B, Solin LJ, Wesson M. Tolerance of normal tissue to therapeutic irradiation. Int J Radiat Oncol Biol Phys. 1991 May 15;21(1):109-22. [CrossRef] [PubMed]
- Marks LB, Yorke ED, Jackson A, Ten Haken RK, Constine LS, Eisbruch A, Bentzen SM, Nam J, Deasy JO. Use of normal tissue complication probability models in the clinic. Int J Radiat Oncol Biol Phys. 2010 Mar 1;76(3 Suppl):S10-9. [CrossRef] [PubMed]
- Harding L, McFarlane J, Honey CR, McDonald PJ, Illes J. Mapping the Landscape of Equitable Access to Advanced Neurotechnologies in Canada. Can J Neurol Sci. 2023 Jun;50(s1):s17-s25. Epub 2023 May 10. [CrossRef] [PubMed]
- Fauchon F, Hasselblatt M, Jouvet A, Champier J, Popovic M, Kirollos R, Santarius T, Amemiya S, Kumabe T, Frappaz D, Lonjon M, Fèvre Montange M, Vasiljevic A. Role of surgery, radiotherapy and chemotherapy in papillary tumors of the pineal region: a multicenter study. J Neurooncol. 2013 Apr;112(2):223-31. Epub 2013 Jan 12. [CrossRef] [PubMed]
- Nowicka E, Bobek-Billewicz B, Szymaś J, Tarnawski R. Late dissemination via cerebrospinal fluid of papillary tumor of the pineal region: a case report and literature review. Folia Neuropathol. 2016;54(1):72-9. [CrossRef] [PubMed]
- Buffenoir K, Rigoard P, Wager M, Ferrand S, Coulon A, Blanc JL, Bataille B, Listrat A. Papillary tumor of the pineal region in a child: case report and review of the literature. Childs Nerv Syst. 2008 Mar;24(3):379-84. Epub 2007 Oct 25. [CrossRef] [PubMed]
- Epari S, Bashyal R, Malick S, Gupta T, Moyadi A, Kane SV, Bal M, Jalali R. Papillary tumor of pineal region: report of three cases and review of literature. Neurol India. 2011 May-Jun;59(3):455-60. [CrossRef] [PubMed]
- Choque-Velasquez J, Colasanti R, Resendiz-Nieves J, Jahromi BR, Tynninen O, Collan J, Niemelä M, Hernesniemi J. Papillary Tumor of the Pineal Region in Children: Presentation of a Case and Comprehensive Literature Review. World Neurosurg. 2018 Sep;117:144-152. Epub 2018 Jun 12. [CrossRef] [PubMed]
- Pons-Escoda A, Sánchez Fernández JJ, de Vilalta À, Vidal N, Majós C. High Myoinositol on Proton MR Spectroscopy Could Be a Potential Signature of Papillary Tumors of the Pineal Region-Case Report of Two Patients. Brain Sci. 2022 Jun 19;12(6):802. [CrossRef] [PubMed]
- Mesny E, Lesueur P. Radiotherapy for rare primary brain tumors. Cancer Radiother. 2023 Sep;27(6-7):599-607. Epub 2023 Jul 21. [CrossRef] [PubMed]
- El Majdoub F, Blau T, Hoevels M, Bührle C, Deckert M, Treuer H, Sturm V, Maarouf M. Papillary tumors of the pineal region: a novel therapeutic option-stereotactic 125iodine brachytherapy. J Neurooncol. 2012 Aug;109(1):99-104. Epub 2012 Apr 17. [CrossRef] [PubMed]
Figure 1.
The sagittal T2 MRI head showed a pineal region mass with a hyperintense anterior cystic component.
Figure 1.
The sagittal T2 MRI head showed a pineal region mass with a hyperintense anterior cystic component.
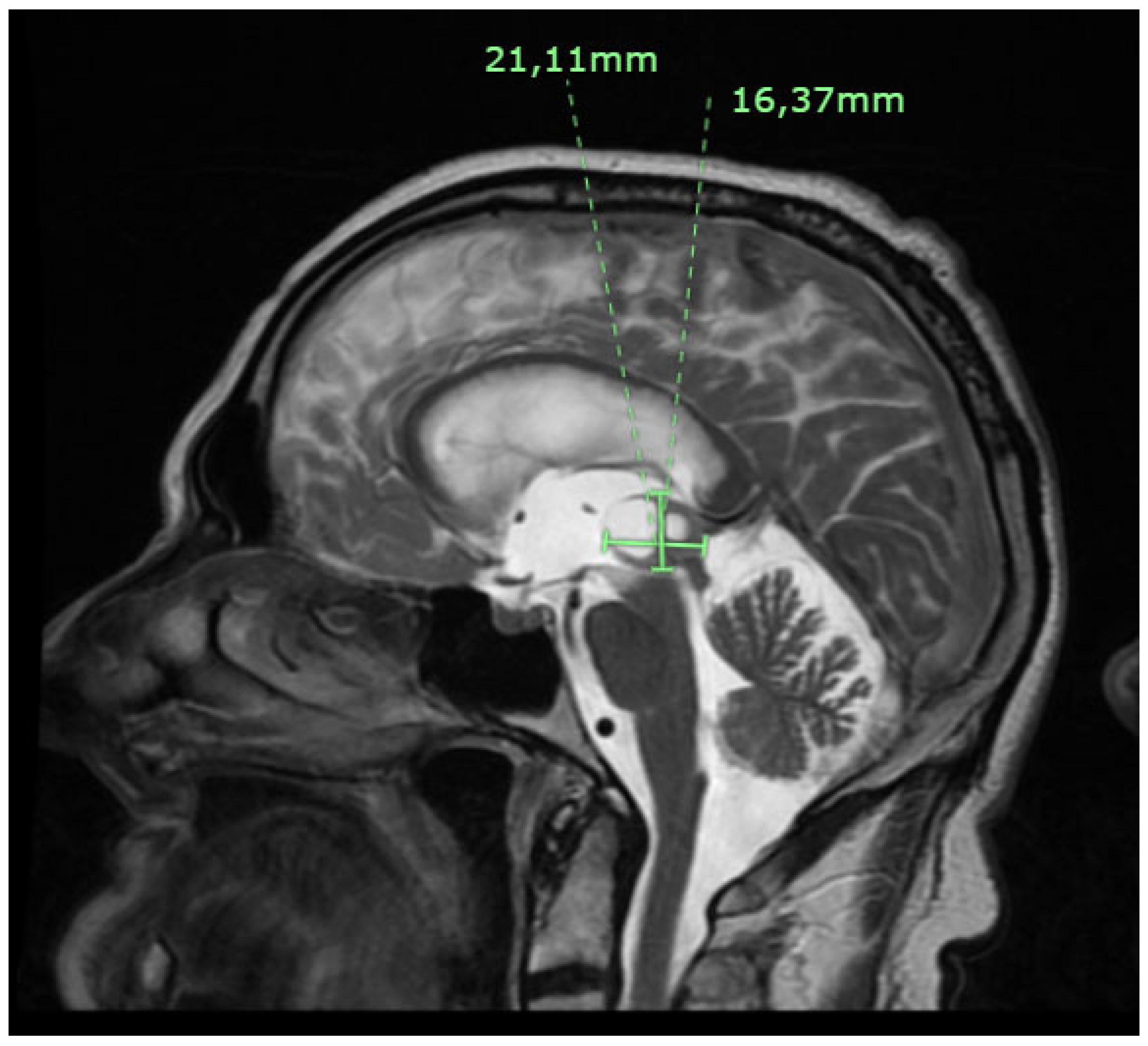
Figure 2.
Axial CT head with hydrocephalus / third ventricle dilatation secondary to the aqueduct of Sylvius obstruction by the pineal region mass.
Figure 2.
Axial CT head with hydrocephalus / third ventricle dilatation secondary to the aqueduct of Sylvius obstruction by the pineal region mass.

Figure 3.
The patient’s biopsy showed a papillary growth pattern. The papillae were covered with large columnar epithelial cells consistent with a papillary tumor of the pineal region.
Figure 3.
The patient’s biopsy showed a papillary growth pattern. The papillae were covered with large columnar epithelial cells consistent with a papillary tumor of the pineal region.
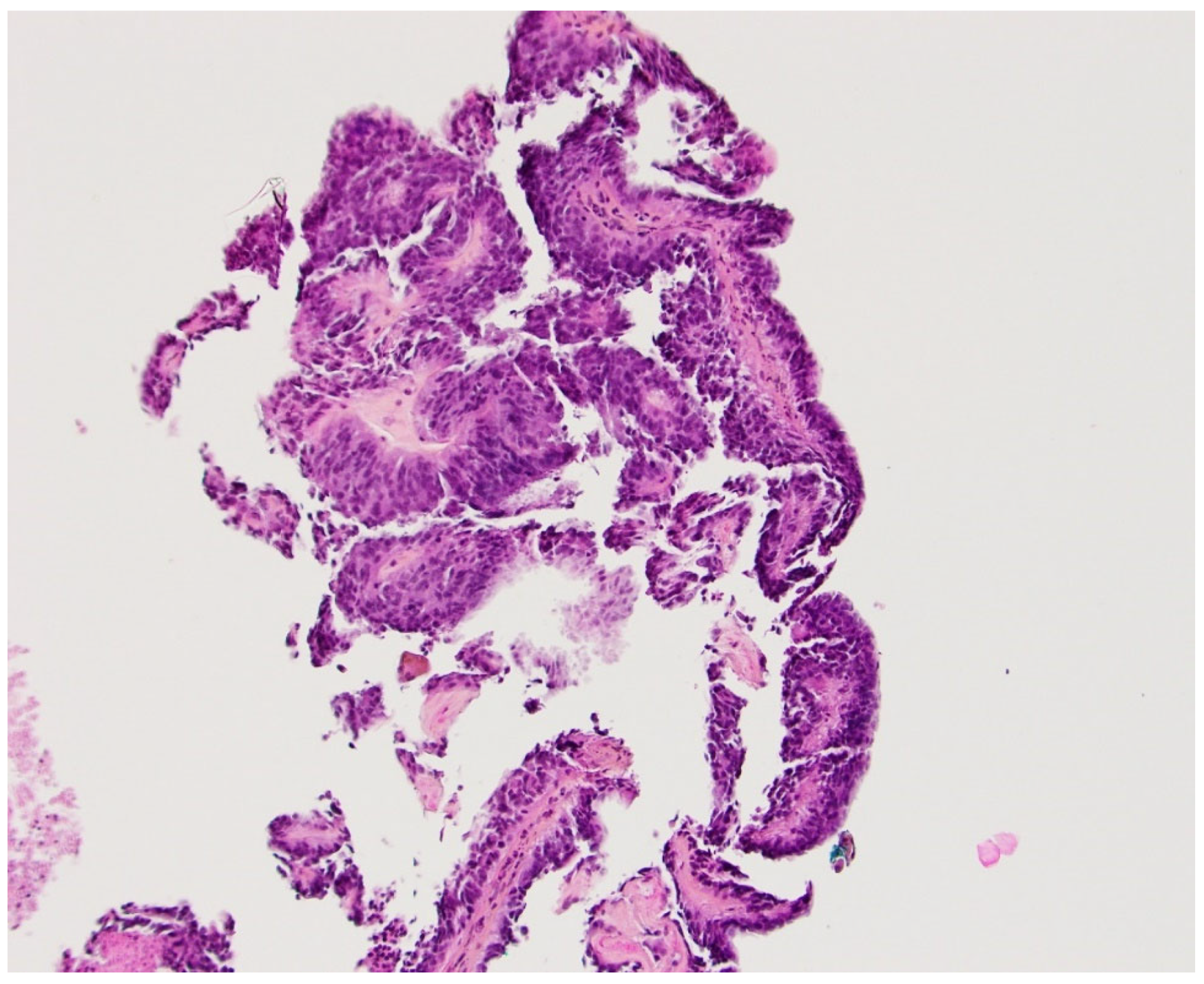
Figure 4.
Hematoxylin and eosin staining showed the tumor's papillary structure. CD56/NCAM was strongly expressed. CK 18/8 and GFAP were focally expressed. Ki-67 was low on the initial biopsy, showing a proliferation index between 1% and 2%. This tumor was negative for NF, epithelial membrane antigen (EMA), synaptophysin, chromogranin, and E-CAD, among others.
Figure 4.
Hematoxylin and eosin staining showed the tumor's papillary structure. CD56/NCAM was strongly expressed. CK 18/8 and GFAP were focally expressed. Ki-67 was low on the initial biopsy, showing a proliferation index between 1% and 2%. This tumor was negative for NF, epithelial membrane antigen (EMA), synaptophysin, chromogranin, and E-CAD, among others.
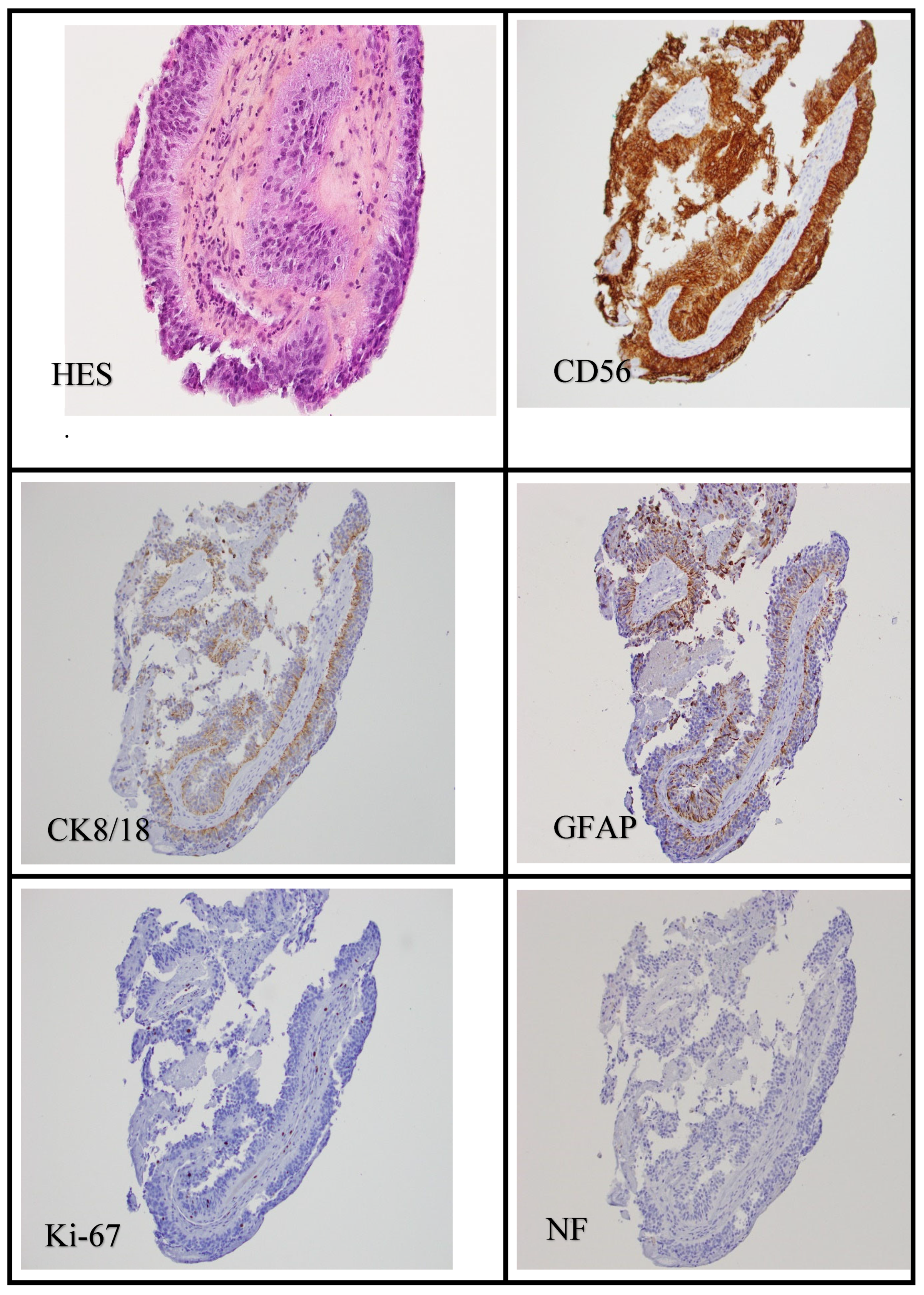

Table 1.
Radiotherapy treatment volumes and dose fractionation.
| Treatment Parameter | Definition |
|---|---|
| GTV | Residual disease based on pre-and post-op imaging |
| CTV | GTV + tumor bed on planning CT and MRI + 5 mm margin cropped to anatomic boundaries* |
| PTV | CTV + 5 mm concentric margin |
| Dose and fractionation | 54 Gy / 30 fractions delivered Monday to Friday over six weeks in 1.8 Gy once daily fractions |
Abbreviations: GTV, gross tumor volume; CTV, clinical target volume; PTV, planning target volume; Gy, Gray; CT, computed tomography; MRI, magnetic resonance imaging; pre-op, preoperative; post-op, post-operative. *Anatomic boundaries included brain parenchyma that the tumor had displaced.
Table 2.
Organ at risk radiation dose constraints (conventional fractionation*).10-11.
| Organs at risk | Dose constraint (to organ at risk + 3 mm concentric margin) |
|---|---|
| Brainstem | V54 Gy < 0.03 cc |
| Spinal cord | V45 Gy < 0.03 cc |
| Optic chiasm | V54 Gy < 0.03 cc |
| Optic nerves | V54 Gy < 0.03 cc |
| Brain | V60Gy < 33%, V50Gy < 66% |
| Eyes | V45 Gy < 0.03 cc |
Abbreviations: V54Gy, the volume of the organ at risk receiving 54 Gy or more; Gy, Gray; cc, cubic centimeter; mm, millimeter; V45Gy, the volume of the organ at risk receiving 45 Gy or more; V60Gy, the volume of the organ at risk receiving 60 Gy or more. *Dose constraints for RT delivered in 1.8-2 Gy, once daily, fractions.
Table 3.
Publications reporting adjuvant RT treatment volumes for PTPR.
| Author | Year | Publication Type | # Patients | Surgery Type | Adj RT Dose/Fractionation | RT Volumes |
|---|---|---|---|---|---|---|
| Buffenoir et al. [15] | 2008 | Case Report | 1 | GTR | 50 Gy / 1.86 Gy per fraction. Five fractions per week | "Tumor bed plus wall of third ventricle" |
| Epari et al. [16] | 2011 | Case Report | 2 | GTR (both patients) | 54 Gy / 30 fractions (both patients) | "Local radiation" |
| Lancia et al. [8] | 2017 | Case Report | 1 | Recurrent disease. STR | 59.4 Gy / 33 fractions. Five fractions per week | CTV = contrast-enhanced lesion + 5 mm margin, PTV = 3 mm expansion of CTV |
| Choque-Velasques et al. [17] | 2018 | Case Report | 1 | GTR | 54 Gy divided into daily doses of 1.8 Gy | "Focal RT to the pineal tumor bed" |
| Pons-Escoda et al. [18] | 2022 | Case Report | 1 | STR | 50 Gy / 25 fractions | "Enhancing area (on post-op MRI)" |
| Lombardi et al. [2] | 2022 | Review Article | NA | RT indicated for STR or recurrent tumors | 50.4-54 Gy in 1.8-2 Gy fractions | “Focal RT” |
| Mesny et al. [19] | 2023 | Review Article | NA | RT indicated for STR or recurrent tumors | 50.4-54 Gy in 1.8-2 Gy fractions | GTV = post-operative cavity and any residual lesion.CTV = margins are undefined |
Abbreviations: #, number; adj, adjuvant; RT, radiotherapy; GTR, gross total resection; STR, subtotal resection; GTV, gross tumor volume; CTV, clinical target volume; Gy, Gray; NA, not available.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



