Submitted:
18 September 2024
Posted:
18 September 2024
You are already at the latest version
Abstract
A chiral sodium glicerophosphate is successfully exploited as catalyst in Michael addition of methyl malonate to a number of chalcones. The reactions furnished the target adducts in satisfactory yields and good enantiomeric excesses. A tentative of computational study is presented aiming to understand the reaction mechanism.
Keywords:
conjugate addition
; organocatalyst
; chiral catalyst
1. Introduction
The employment of alkali and alkaline earth metal phosphates (Figure 1) in asymmetric catalysis [1,2,3], even though strictly not classifiable as asymmetric organocatalysts, displays many of its advantages, namely handiness of the catalyst, absence of costly and toxic heavy transition metals and easiness of preparation from the corresponding phosphoric acids [4,5].
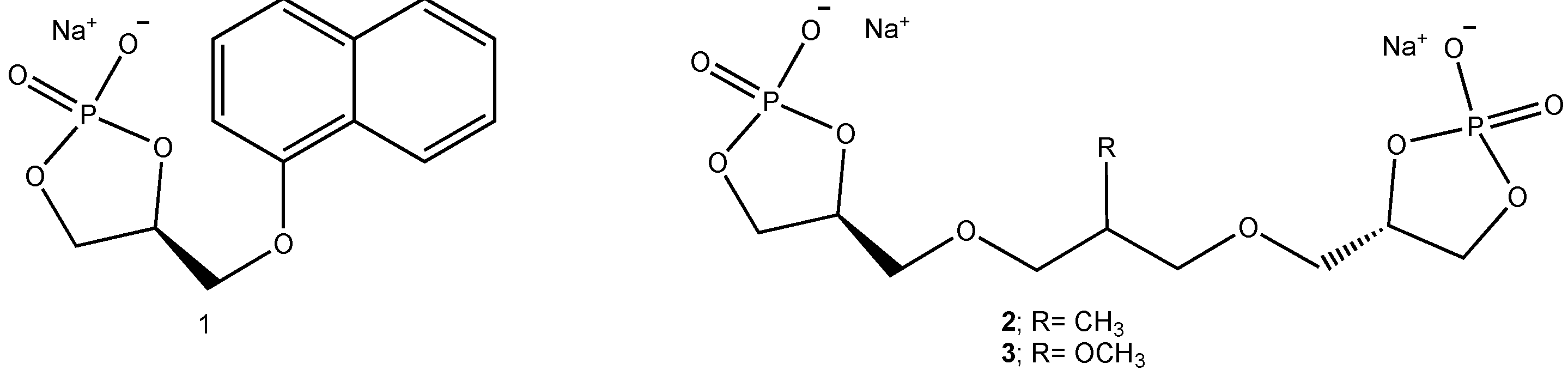
On this ground, we recently proposed a new class of sodium phosphate catalysts, namely cycloglycerophosphates (Figure 2) prepared from enantiopure solketal (the acetonide of glycerol) [6]. One of these adducts, namely (S)-4-((naphthalen-1-yloxy)methyl)-1,3,2-dioxaphospholan-2-olate 2-oxide sodium (Figure 2; 1) was employed as catalyst in addition of TMSCN to aldehydes at room temperature, to afford gem-cyanohydrins [6]. Unfortunately, no enantioselectivity was observed. This behaviour can be explained with an intrinsically low asymmetric induction exerted by the 5-membered phosphatidic acid scaffold, having the stereogenic center far from the catalyst active site and not supported by steric hindrance effects, because of the free rotation around the exocyclic bond bearing the bulky α-naphthyloxy group. On the other hand, in order to introduce C2-symmetry we prepared adducts 2 and 3 (Figure 2) [7]. They resulted into an outstanding enantiocontrol of the reaction, namely addition of TMSCN to aldehydes and ketones. The reaction mechanism was investigated on a minimal model of the catalyst with a DFT method. A calculation of the most stable conformation of the model catalyst for both C2-symmetric cycloglycerodiphosphates instead, accounts for the enantioselectivity, since the two cyclic phosphatidic moieties are syn, creating a confined pocket for the reactants through electrostatic interactions involving the sodium cations [7].
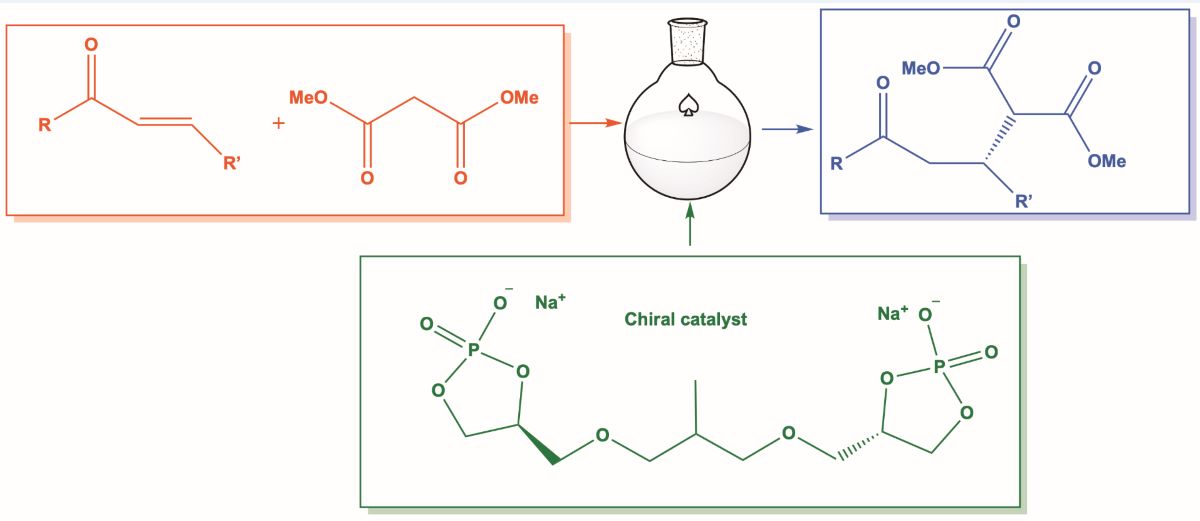
In light of the interesting results previously obtained and with the aim of expanding the synthetic uses of this class of catalysts, we studied 1,4 addition reactions of methyl malonate (4) to a number of chalcones 5 in the presence of 2 as chiral catalyst (Scheme 1). It must be stressed that the catalytic asymmetric conjugate addition of ß,-unsaturated carbonyl compounds is one of the most powerful carbon–carbon bond-forming reactions [8,9]. In fact, its complete atom economy, wide substrate scope, susceptibility to many classes of catalysts, and easily accessible starting materials render it one of the most modern of classical reactions. However, on the other hand, remain difficult to execute via either stoichiometric or catalytic approaches, despite recent advances [10].
The chiral catalysts used for this reaction are most varied [11]. However, the literature shows few examples catalysed by chiral phosphates. The most significant are three. In particular, in 2010 Chen reported an enantioselective addition of TMSCN to aromatic enones catalysed by sodium salt of (R)-3,3’-di(1-adamantyl)-1,1’-binaphthyl-2,2’-diylphosphoric acid [12] and Antilla in 2011 described a chiral vapol phosphate catalysed Michael reaction of 3-substituted indoles with methyl vinyl ketone [13]. Finally, in 2019 Zhu and Niemeyer reported that heterobifunctional rotaxanes featuring an amine-based thread and a chiral 1,1′-binaphthylphosphoric acid-based macrocycle efficiently catalysed the addition of malonates to Michael acceptors [14].
2. Results and Discussion
As a model reaction, we decided to begin the study by reacting methyl malonate (4) with E-1,3-diphenylpropen-3-one (5a) in the presence of catalyst 2 at room temperature and with a number of solvents (Table 1; entries 8,9,10,11). Only traces of target compound dimethyl 2-(3-oxo-1,3-diphenylpropyl)malonate) 6a were detected in MeOH (Table 1; entry 2) or 2-MeTHF (Table 1; entry 5) . Also increasing the amount of catalyst to 10 mol% (Table 1; entries 3,6) and carrying out the reaction at 60°C (Table 1; entries 4,7) no appreciable results were obtained. Since the catalyst 2 is not basic enough to deprotonate the malonate and therefore to allow nucleophilic attack to 5a, in order to transform the malonate into the corresponding more nucleophilic anion, we added an equimolar amount of sodium methoxide to 4 dissolved in methanol.
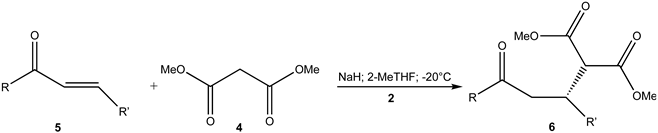
Under these condition (Table 1; entry 12), fairly good yield of target 6a was obtained (52%), although with low enantiomeric excess (34%). Changing solvent and using 2-MeTHF (Table 1; entry 15) the yield of the target 6a significantly increased up to 90% but the enantiomeric excess was similar (37%) to that previously obtained. It must be stressed that, in this case (Table 1; entry 15), the anion of malonate was formed adding NaH.
By lowering the temperature to 0°, enantiomeric excess of 57% (Table 1; entry 16) was obtained. By further lowering the temperature to -20°C, to our delight, the enantiomeric excess was 91.5% (Table 1; entry 17). A further lowering of the temperature to -45°C did not lead to changes (Table 1; entry 18). Moreover, this reaction was not completed after 8 hours. On the other hand, it is necessary to underline that the reaction occurred between the anion of 4 and 5a even in the absence of the catalyst 2 (Table 1; entries 19,20).
In order to explore the applicability of this optimized procedure, we tested other reactions between aromatic or heteroaromatic chalcones, variously substituted with electron withdrawing or electron donating substituents (Table 2; entries 1–20). The yields were always good and good enantiomeric excesses were obtained, always between 87 and 94%. The average enantiomeric excess for 20 examples was 90.2%.
Moving on to examine the reactivity of aliphatic chalcones (or similar compounds), quite surprisingly, no reactions occurred with methyl vinyl ketone (5x; Table 2, entry 23) methyl acrylate (5y; Table 2, entry 24) or acrylaldehyde (5z; Table 2, entry 25). We believe that, due to the lack of an electron-donating and activating group linked to the C atom in ß of CO, the nucleophilic attack of malonate cannot occur. Instead, the compounds 5u, 5v, 5w showed the same reactivity as the corresponding aromatic ones (Table 2; entries 21–23) even if the enantiomeric excesses obtained were lower (average enantiomeric excess for tree examples was about 83.2%)
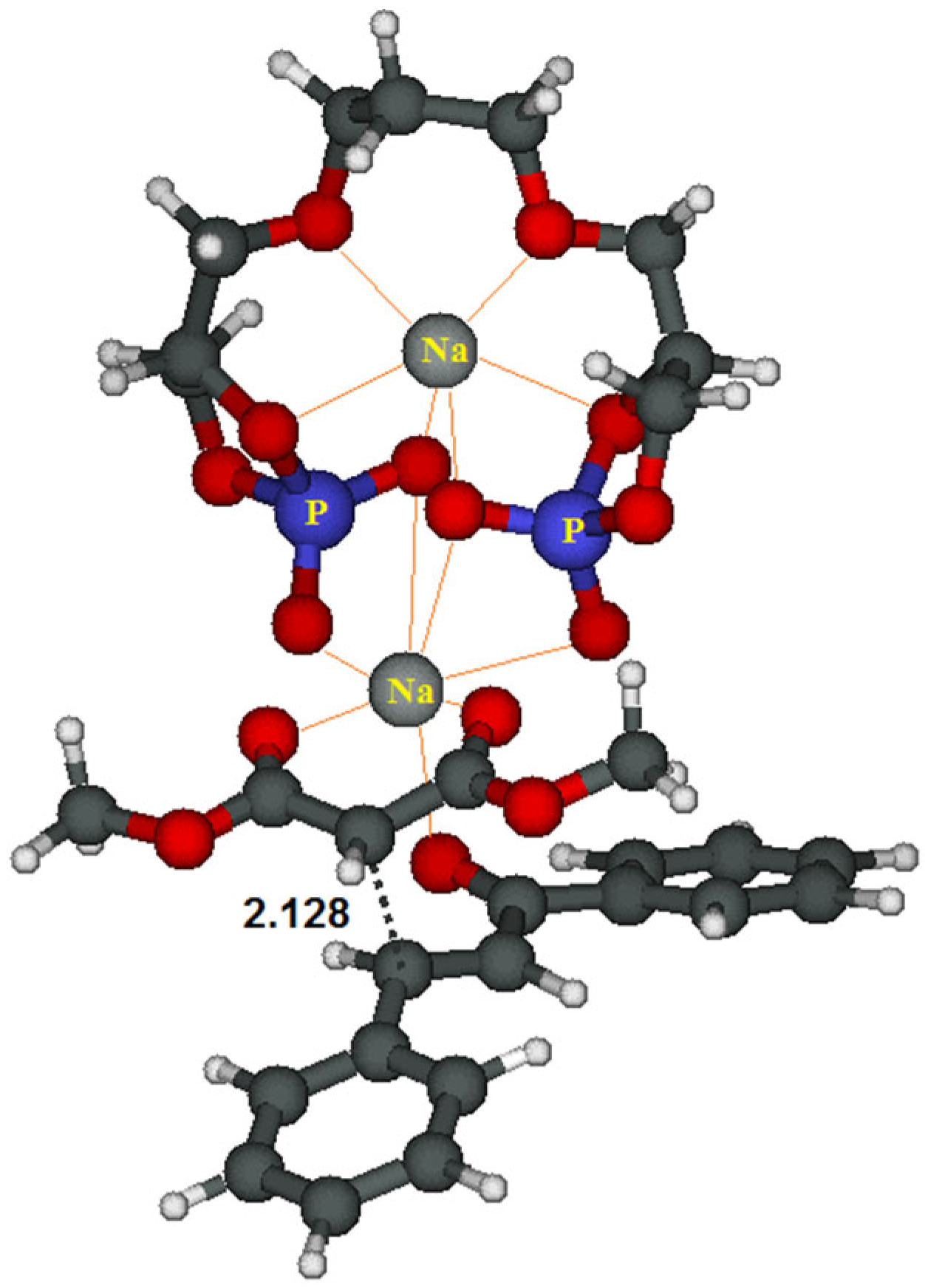
In an attempt to rationalize the experimental outcomes, we performed a computational study on the reaction with chalcone 5a (see SI-CD for details on the method and a more extended discussion). In the transition structure (TS) for the nucleophilic addition (Figure 3), the catalyst is depicted on the upper part of the figure; the upper sodium cation is fully solvated by the oxygen atoms (in red) of the catalyst while the second on the lower part interacts also with the oxygen atoms of the malonate (in the middle) and of the chalcone (in the lower part).
We believe that, thanks to these interactions in TS, one of the prochiral faces of the chalcone is shielded. So, the nucleophilic attack of the anion of 4 on 5 can occur mainly from a single direction, allowing 6 to be obtained with a good enantiomeric, as depicted in Scheme 2 and qualitatively reproduced by the calculation. As already mentioned previously (Table 1, entry 19), the reaction occurs even in the absence of 2. Therefore, its essential function is to create a chiral fashion.
We also tried to model the effect of the catalyst on the kinetics of the reaction. A small increase in the catalyzed reaction rate was observed, but the computations failed to fully reproduce it. However, the computed energy difference between the non-catalyzed and the catalyzed reactions was very small (0.5 kcal mol-1) and strongly dependent on the solvent effects which are very difficult to fully reproduce.
3. Materials and Methods
3.1. General
Catalyst 2 [7] and chalcones 5 [15] were prepared as reported in the literature. Analytical grade reagents and solvents (purchased from Merck, Carlo Erba, Alfa Aesar and Fluorochem) were used and reactions were monitored by GC, GC-MS. Column chromatography and TLC were performed on Merck silica gel 60 (70-230 mesh ASTM) and GF 254, respectively. Petroleum ether (PE) refers to the fraction boiling in the range 40–70 °C. GC analyses were performed on a PerkinElmer AutoSystem XL GC with a methylsilicone capillary column. Mass spectra were recorded on an HP5989B mass selective detector connected to an HP 5890 GC with a cross-linked methyl silicone capillary column. Chiral analyses were performed on a HPLC using a Daicel CHIRALPAK-IG (250 x 4.6 mm, 5 mm) on Essential LC- 16 series and eluting with i-PrOH and n-hexane. For the determination of optical rotation a Jasco P-2000 polarimeter was used. 1H NMR and 13C NMR spectra were recorded on a Brucker spectrometer at 400 and 100 MHz respectively. IR spectra were recorded on a IR Perkin-Elmer UATR-two spectrometer. Structures and purity of all the adducts 6 obtained in this research were confirmed by their spectral (NMR, GC-MS, IR) data. Satisfactory microanalyses were obtained for all new compounds.
3.2. As a Catalyst in Michael Reactions. Typical Procedure: Synthesis of (R)(-)-Dimethyl 2-(3-oxo-1,3-diphenylpropyl)Malonate (6a)
NaH (60% dispersion in paraffin liquid; 44 mg, 1.1 mmol) was added as to a stirring solution of methyl malonate (4; 132 mg, 1 mmol) in 2-MeTHF (5 mL). A white suspension was immediately formed. After 10 min of further stirring, the mixture was cooled to -20°C. At this point, chiral catalyst (2; 5 mol%, 21 mg, 0.05 mmol) and E- 1,3-diphenylprop-2-en-1-one (5a; 208 mg, 1 mmol) were added. The mixture was stirred for 8 h, until TLC (eluent PE/EE 4:1) GC and GC-MS analyses showed the complete disappearance of 4 and 5a and the complete formation of dimethyl 2-(3-oxo-1,3-diphenylpropyl)malonate (6a; MS (70eV) m/z : 340 (37) [M]+). Then, H2O (5 mL) was added to the reaction mixture, which was extracted with Et2O (10 mL). The organic phase was washed with H2O (5 mL) and dried over sodium sulfate. The solvent was evaporated under reduced pressure. The crude residue was purified in a short chromatographic column (eluent: PE/EE 4:1). The resulting white solid was virtually pure title compound (6a; 305 mg; 90% yield). After the above work-up, Ee was measured.
4. Conclusions
We have reported the enantioselective conjugate addition of methyl malonate (4) to chalcones 5 catalyzed by a C2-symmetric cycloglycerodiphosphate 2. The target adducts 6 were obtained in satisfactory yields with good enantiomeric excess. In light of this and in light of its ease of preparation and purification, this catalyst turns to be very competitive for this type of asymmetric protocol since results comparable to those obtained with other catalytic systems dedicated to this purpose and already cited in the literature were obtained.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Physical data of malonates 6: page S-2; NMR Spectra and chiral analyses of malonates 6: page S-7; Computational method: page S-54; Schemes, tables, pictures and Cartesian coordinates for the uncatalyzed reaction: page S-56; for the catalyzed reaction: page S-67.
Author Contributions
Conceptualization, S.D. and G.G.; methodology, S.D.; software, G.G.; validation, S.D. and G.G.; formal analysis, S.D.; investigation, S.D., G.G., J.R. and A.R.B.; resources, S.D.; data curation, S.D.; writing—original draft preparation, S.D and G.G.; writing—review and editing, S.D and G.G.; visualization, S.D.; supervision, S.D.; project administration, S.D.; funding acquisition, S.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data reported in this article can be obtained from the authors upon reasonable request. Samples of the compounds 6 are available from the authors.
Acknowledgments
This work has been supported by the University of Torino and by Ministero dell’Università e della Ricerca Scientifica (MIUR). The authors acknowledge support from the Project CH4.0 of the Chemistry Department of UNITO under MIUR program “Dipartimenti di Eccellenza 2—3–2027” (CUP: D13 C22003520001).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Book: Akiyama, T.; Ojima, I. Catalytic asymmetric synthesis. 4th Edition, John Wiley and Sons USA, 2022.
- Book: R. A. Aitken, S. N. Kilényi, Asymmetric Synthesis, Springer Netherlands, 2012.
- Garg, A.; Rendina, D.; Bendale, H.; Akiyama, T.; Ojima, I. Recent advances in catalytic asymmetric synthesis. Front. Chem. 2024, 12, 1–28. [CrossRef]
- Cocco, E.; Antenucci, A.; Carlone, A.; Manini, P.; Pesciaioli, F.; Dughera, S. Stereoselective Reactions Promoted by Alkali Metal Salts of Phosphoric Acid Organocatalysts. ChemCatChem, 2024, e202400328. [CrossRef]
- Brodt, N.; Niemeyer, J. Chiral organophosphates as ligands in asymmetric metal catalysis Org. Chem. Front., 2023, 10, 3080–3109. [CrossRef]
- Antenucci, A.; Messina, M.; Bertolone, M.; Bella, M.; Carlone, A.; Salvio, R.; Dughera, S. Turning renewable feedstocks into a valuable and efficient chiral phosphate salt catalyst. Asian J. Org. Chem., 2021, 10, 3279–3284. [CrossRef]
- Antenucci, A.; Ghigo, G.; Cassetta, D.; Alcibiade, M.; Dughera, S. Design, synthesis and application of C2-symmetric cycloglycerodiphosphate. Adv. Synth. Catal., 2023, 365, 1170–1178. [CrossRef]
- Tsogoeva, S. B. Recent Advances in Asymmetric Organocatalytic 1,4-Conjugate Additions. Eur. J. Org. Chem., 2007, 1701–1716. [CrossRef]
- Zheng, K.; Liu, X.; Feng, X. Recent advances in metal-catalyzed asymmetric 1,4-conjugate addition (ACA) of nonorganometallic nucleophiles. Chem. Rev.,2018, 118, 7586–7656. [CrossRef]
- Rachwalski, M.; Buchcic-Szychowska, A.; Lesniak, S. Recent advances in selected asymmetric reactions promoted by chiral catalysts: cyclopropanations, Friedel–Crafts, Mannich, Michael and other zinc-mediated processes. An update. Symmetry 2021, 13, 1762–1786. [CrossRef]
- Hayashi, M.; Matsubara, R. Recent topics on catalytic asymmetric 1,4-addition. Tetrahedron Lett., 2017, 58, 1793–1805. DOI:.
- Yang, J.; Wu, S.; Chen, F.-X. Chiral sodium phosphate catalyzed enantioselective 1,4-addition of TMSCN to aromatic enones. Synlett, 2010, 18, 2725–2728. [CrossRef]
- Zheng, W.; Zhang, Z.; Kaplan, M. J.; Antilla, J. C. Chiral calcium VAPOL phosphate mediated asymmetric chlorination and Michael reactions of 3-Substituted oxindoles. J. Am. Chem. Soc. 2011, 133, 3339–3341. [CrossRef]
- Pairault, N.; Zhu, H.; Jansen, D.; Huber, A.; Daniliuc, C. G.; Grimme, S.; Niemeyer, J. Heterobifunctional rotaxanes for asymmetric Catalysis. Angew. Chem. Int. Ed., 2020, 59, 5102 – 5107. [CrossRef]
- Kohler, E. P.; Chadwell, H. M. Benzalacetophenone. Org. Synth. 1922, 2, 1. [CrossRef]
- Capreti, N. M. R.; Jurberg, I.D. Michael addition of soft carbon nucleophiles to alkylidene isoxazol-5-ones: a divergent entry to β-branched carbonyl compounds. Org.Lett., 2015, 17, 2490−2493. [CrossRef]
- Espinosa, M.; Blay, G.; Cardona, L.; Pedro, J. R. Asymmetric conjugate addition of malonate esters to α,β-unsaturated N-sulfonyl imines: an expeditious route to chiral δ-aminoesters and piperidones. Chem. Eur. J., 2013, 19, 14861–14866. [CrossRef]
- Cao, D.; Fang, G.; Zhang, J.; Wang, H.; Zheng, C.; Zhao, G. Enantioselective Michael addition of malonates to chalcone derivatives catalyzed by dipeptide-derived multifunctional phosphonium salts. J. Org. Chem., 2016, 81, 9973–9982. [CrossRef]
- Mao, Z.; Jia, Y.; Li, W.; Wang, R. Water-compatible iminium activation: highly enantioselective organocatalytic Michael addition of malonates to α,β-unsaturated enones. J. Org. Chem., 2010, 75, 7428–7430. [CrossRef]
- Kohler, E. P.; Hill, G. A.; Bigelow, L. A. Studies in the cyclopropane series. Third paper. J. Am. Chem. Soc., 1917, 39, 2405–2418. [CrossRef]
- De Simone, N. A.; Meninno, S.; Talotta, C.; Gaeta, C.; Neri, P.; Lattanzi, A. Solvent-free enantioselective Michael reactions catalyzed by a calixarene-based primary amine thiourea. J. Org. Chem., 2018, 83, 10318–10325. [CrossRef]
- Ueda, A.; Umeno, T.; Doi, M.; Agakawa, K.; Kudo, K.; Tanaka, M. Helical-peptide-catalyzed enantioselective Michael addition reactions and their mechanistic insights. J. Org. Chem. 2016, 81, 6343–6356. [CrossRef]
- Dudzinski, K.; Pakulska, A. M.; Kwiatkowski, P. An efficient organocatalytic method for highly enantioselective Michael addition of malonates to enones catalyzed by readily accessible primary amine-thiourea. Org. Lett. 2012, 14, 4222–4225. [CrossRef]
- Jiricek, J.; Blechert, S. Enantioselective synthesis of (--)-gilbertine via a cationic cascade cyclization. J. Am. Chem. Soc. 2004, 126, 3534–3538. [CrossRef]
- Parr, R.G. Density Functional Theory of Atoms and Molecules, in: Horizons Quantum Chem., Springer Netherlands, 1980: pp. 5–15. [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [CrossRef]
- Zhao, Y.; Truhlar, D.G. Density functionals with broad applicability in chemistry. Acc. Chem. Res. 2008,41,157–167. [CrossRef]
- Schaefer, A.; Horn, H.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys.1992, 97, 2571–2577. [CrossRef]
- Foresman, J.; Frisch, A. Exploring chemistry with electronic structure methods, 1996, Gaussian Inc, Pittsburgh, PA, 1996. https://gaussian.com/expchem3/(accessed June 4, 2021).
- Ribeiro, R.F.; Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Use of solution-phase vibrational frequencies in continuum models for the free energy of solvation. J. Phys. Chem. B. 2011, 115, 14556–14562. [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions J. Phys. Chem. B, 2009, 113, 6378–6396. [CrossRef]
- Truhlar, D.G.; Garrett, B.C.; Klippenstein, S.J. Current status of transition-state theory J. Phys. Chem. 1996, 100, 12771–. [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; Li, X.; Caricato, M.; Marenich, A.V.; Bloino, J.; Janesko, B.G.; Gomperts, R.; Mennucci, B.; Hratch, Gaussian 16, Revision A.03, 2016.
- Schaftenaar, G.; Noordik, J.H. Molden: a pre- and post-processing program for molecular and electronic structures. J. Comput. Aided. Mol. Des. 2000, 14, 123–134. [CrossRef]
Figure 1.
Methal phosphates.
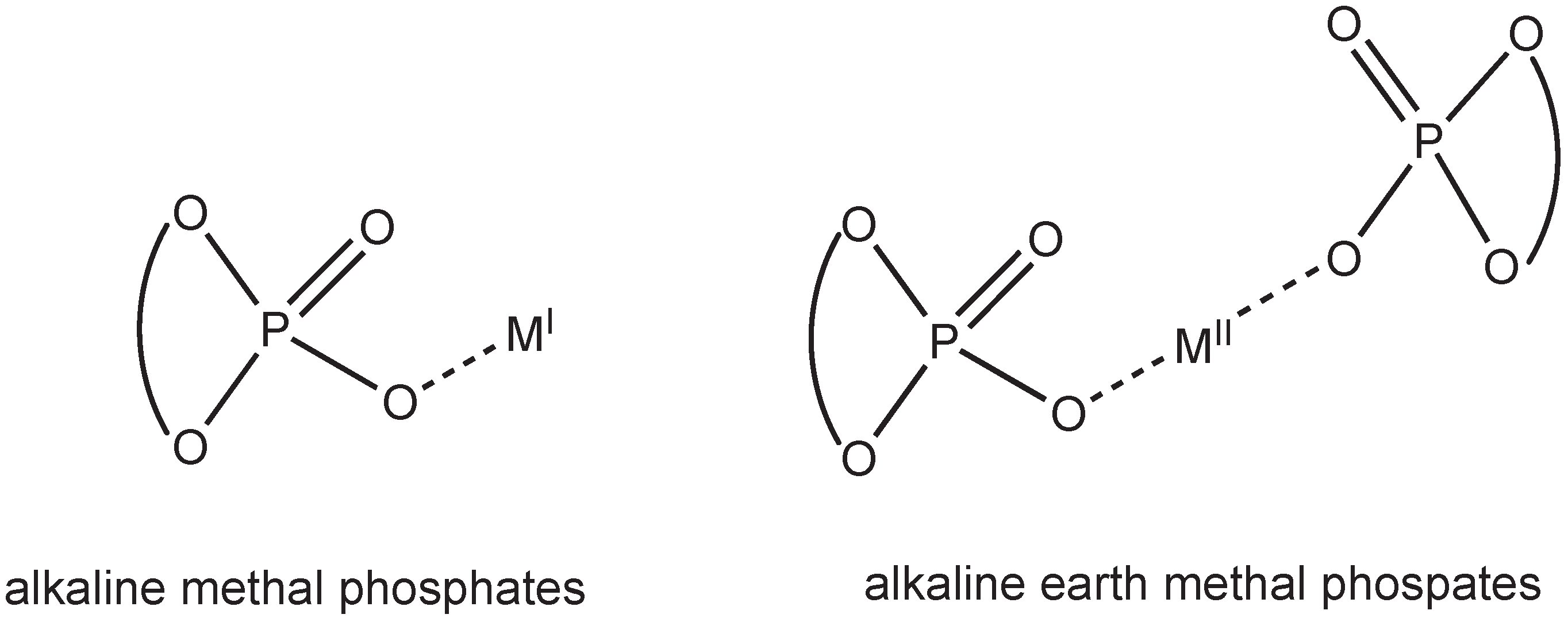
Figure 2.
Chiral cycloglycerophosphates.
Scheme 1.
1,4-Addition of methyl malonate 4 to chalcones 5 in the presence of chiral glycerophosphate 2.
Scheme 1.
1,4-Addition of methyl malonate 4 to chalcones 5 in the presence of chiral glycerophosphate 2.
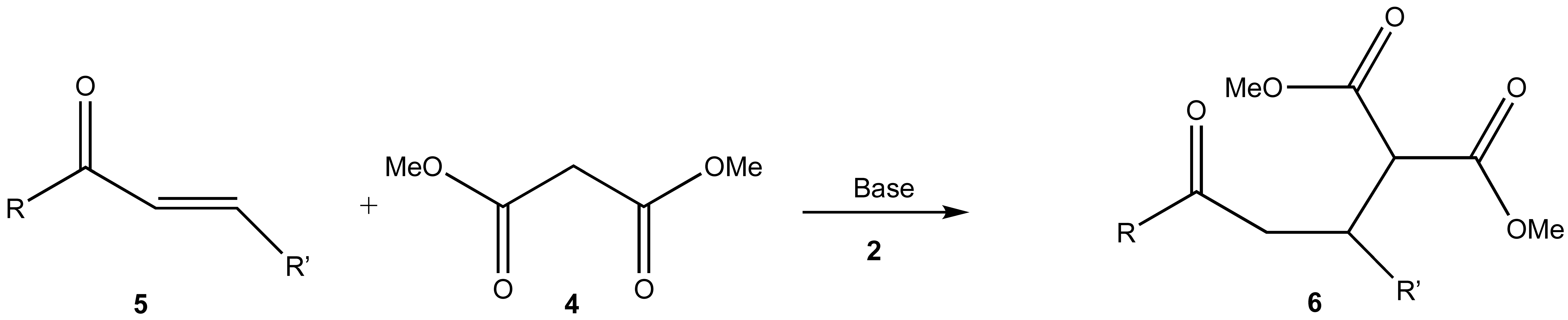
Figure 3.
Transition structure (TS) for the nucleophilic addition.
Scheme 2.
Hypothesized mechanism.
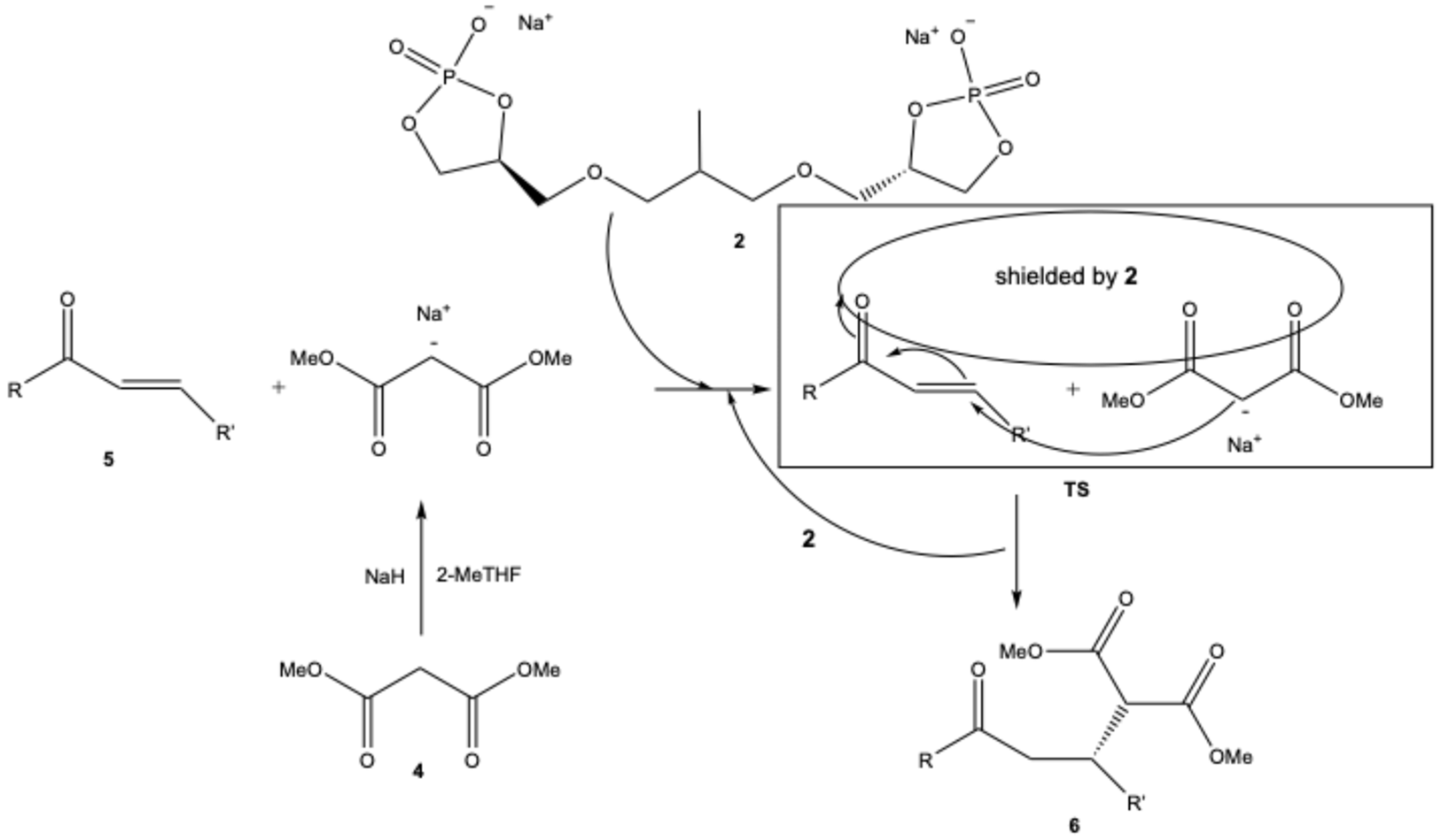

Table 1.
Trial reactions.
 | |||||||
| Entry | Catalyst 2 (mmol%) | Base | Solvent | T (°C) | Time (h) | Yield (%)1,2 | Ee (%)3 |
| 1 | - | - | MeOH | rt | 24 | traces | - |
| 2 | 5 | - | MeOH | rt | 24 | traces | - |
| 3 | 10 | - | MeOH | rt | 24 | traces | - |
| 4 | 10 | MeOH | 60 | 24 | traces | - | |
| 5 | 5 | - | 2-MeTHF | rt | 24 | traces | - |
| 6 | 10 | - | 2-MeTHF | rt | 24 | traces | - |
| 7 | 10 | - | 2-MeTHF | 60 | 24 | traces | - |
| 8 | 5 | - | Toluene | rt | 24 | - | - |
| 9 | 5 | - | DCM | rt | 24 | - | - |
| 10 | 5 | - | DMSO | rt | 24 | - | - |
| 11 | 5 | - | MeCN | rt | 24 | - | - |
| 12 | 5 | CH3ONa | MeOH | rt | 8 | 52 | 34.3 |
| 13 | 10 | CH3ONa | MeOH | rt | 6 | 51 | 32.8 |
| 14 | 5 | CH3ONa | Toluene | rt | 24 | - | - |
| 15 | 5 | NaH | 2-MeTHF | rt | 3 | 92 | 37.5 |
| 16 | 5 | NaH | 2-MeTHF | 0 | 5 | 90 | 57.2 |
| 17 | 5 | NaH | 2-MeTHF | -20 | 8 | 90 | 91.5 |
| 18 | 5 | NaH | 2-MeTHF | -45 | 8 | 21 | 90.8 |
| 19 | - | NaH | 2-MeTHF | rt | 3 | 91 | - |
| 20 | - | NaH | 2-MeTHF | -20 | 8 | 90 | - |
1 Reactants 1c, 2 and MeONa/NaH are in ratio 1:1:1. 2 Yields are referred to pure and isolated 6a, purified in a short chromatographic column (eluent petroleum ether/diethyl ether 3:2). 3 Ee was determined by HPLC analyses on a chiral stationary phase.
Table 2.
Synthesis of adducts 6.
 | |||||
| Entry | R in 5,6 | R’ in 5,6 | Time (h) | Yield (%) of 6 1,2,3 | Ee (%)4 |
| 1 | C6H5 | C6H5 | 8 | 6a; 90 | 91.5 |
| 2 | C6H5 | 4-NO2C6H4 | 10 | 6b; 87 | 88.9 |
| 3 | C6H5 | 4-ClC6H4 | 6 | 6c; 89 | 92.9 |
| 4 | C6H5 | 4-MeC6H4 | 4 | 6d; 92 | 90.0 |
| 5 | C6H5 | 4-CNC6H4 | 10 | 6e; 92 | 90.3 |
| 6 | C6H5 | 2-NO2C6H4 | 12 | 6f; 77 | 90.5 |
| 7 | C6H5 | 3-NO2C6H4 | 8 | 6g; 90 | 89.9 |
| 8 | C6H5 | Thiophen-2-yl | 6 | 6h; 82 | 89.6 |
| 9 | C6H5 | Furan-2-yl | 6 | 6i; 77 | 89.3 |
| 10 | Me | C6H5 | 6 | 6j; 82 | 92.5 |
| 11 | Me | 2-MeC6H4 | 8 | 6k; 90 | 94.4 |
| 12 | Me | 3-MeOC6H4 | 6 | 6l; 85 | 88.8 |
| 13 | C6H5 | Me | 4 | 6m; 87 | 89.5 |
| 14 | 4-CF3C6H4 | C6H5 | 6 | 6n; 92 | 89.7 |
| 15 | 2-MeOC6H4 | C6H5 | 6 | 6o; 79 | 87.0 |
| 16 | 3-MeC6H4 | 4-ClC6H4 | 8 | 6p; 82 | 89.9 |
| 17 | 3-MeC6H4 | 4-NO2C6H4 | 10 | 6q; 91 | 87.0 |
| 18 | 4-CF3C6H4 | 4-MeC6H4 | 6 | 6r; 90 | 92.4 |
| 19 | 4-ClC6H4 | C6H5 | 6 | 6s; 85 | 89.0 |
| 20 | 4-NO2C6H4 | C6H5 | 6 | 6t; 85 | 90.5 |
| 21 | Et | Me | 4 | 6u; 85 | 81.8 |
| 22 | 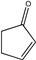 |
6 | 6v; 83 | 87.5 | |
| 23 | 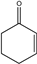 |
6 | 6w; 91 | 70.2 | |
| 24 | CH3 | H | 8 | 6x; - | - |
| 25 | OCH3 | H | 8 | 6y; - | - |
| 26 | H | H | 8 | 6z; - | - |
1 Reactants 1c, 2 and NaH are in ratio 1:1:1.2 Temperature was -20°C. 3 Yields are referred to pure and isolated 6, purified in a short chromatographic column (eluent petroleum ether/diethyl ether 3:2). 4 Ee was determined by HPLC analyses on a chiral stationary phase.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



