Submitted:
04 September 2024
Posted:
06 September 2024
You are already at the latest version
Abstract
Background: Studying patients carrying identical by descent (IBD) pathogenic gene variants allows to control for the disease-causing genetic background and to more accurately document the impact of modifiers. Familial hypercholesterolemia (FH) is characterized by elevated LDL-c levels, premature atherosclerosis and is often caused by defects in the LDLR gene. There is a high prevalence of FH in French Canada carry IBD FH-causing due to a founder effect from France in the 17th Century. Several FH patients currently living in French Canada (founder population) and in France (colonizing population) carry IBD FH-causing variants. The expression of FH is affected by environmental and genetic modifiers and patients with IBD variants may present different characteristics. Methods: In this study, we compared FH clinical expression patients carrying IBD LDLR pathogenic variants living in France or Canada. Four IBD variants, namely c.259T>G p.(Trp87Gly); c.2000G>A p.(Cys667Tyr); c.682G>A p.(Glu228Lys); and c.1048C>T p.(Arg350*) were selected. Untreated plasma lipid profiles, APOE genotype, cardiovascular risk factors and occurrence of symptomatic ASCVD were compared in 105 adult carriers (30 from France and 75 from French Canada). Results: All parameters were similar between the two populations, except for untreated total cholesterol (10.14 ± 1.89 mmol/L vs 8.65 ± 1.84 mmol/L, p=0.0006) and LDL-c concentrations (7.94 ± 1.86 mmol/L vs 6.93 ± 1.78 mmol/L, p=0.016) that were significantly higher in FH patients living in France, a trend that was observed in all studied LDLR variants. Conclusion: This study illustrates that FH patients sharing IBD pathogenic LDLR variants that have evolved in different geographic, cultural and socio-economic environments for hundreds of years, differ in term of cholesterol levels highlighting the importance of better understanding the interplay between genetic and environmental modulators of FH expression.
Keywords:
Familial hypercholesterolemia
; Founder effect
; Identical by descent variant
Introduction
Familial hypercholesterolemia (FH) is characterized by the accumulation of low-density lipoprotein (LDL) particles and elevated LDL-cholesterol (LDL-c) levels due to lack of functionality or availability of LDL receptors (LDLR). FH is most often caused by pathogenic LDLR gene variants or by variants in genes having deleterious effects on LDLR functions (1, 2). Patients affected by FH generally have elevated LDL-c levels from birth (above 5 mmol/L in adulthood) and are at risk of developing atherosclerosis early in life (3, 4). FH prevalence is estimated at 1:250 globally, although its prevalence is significantly higher in some isolates or founder populations (5).
FH expression is affected by multiple modifiers and varies from one individual to another, even among carriers of the same FH-causing pathogenic variant (4). Environmental and genetic modifiers can directly affect LDL-c concentration and an increasing number of evidence show that these modifiers can also impact the epigenome and modulate FH clinical expression (6). Unlimited combinations of genes and environmental factors therefore contribute to each person’s unique blend of traits, health, and identity, making it difficult to precisely predict the individual trajectory of FH expression and response to treatment.
Identical by descent (IBD) pathogenic gene variants have emerged as interesting candidates to study the genetic underpinnings of atherosclerotic cardiovascular disease (ASCVD) as they are shared by individuals via common ancestors. Studying patients with IBD gene variants allows to control for the disease-causing genetic background and to more accurately document the impact of modifiers (genetic, clinical, environmental or socio-economic). Many research efforts have indeed been supporting the idea that studying IBD segments consists of a powerful method for controlling genetic background, while providing a more nuanced understanding of the role of various modifiers in disease expression an susceptibility. Not to mention interesting advances in IBD detection methods that have been recently developed, such as RaPID tool, that aims to improve the ability to identify segments with high accuracy and speed, despite having large biobank-scale databases. In addition to controlling the genetic background, these tools are helpful for investigating the impact of environmental and other modifiers on disease expression, while detecting population structure and familial relationships in specific therapeutic areas (https://academic.oup.com/gigascience/article/doi/10.1093/gigascience/giac111/6874525; https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1754-8).
French-Canadians provides an interesting example of a founder population where documented IBD pathogenic LDLR variants are more prevalent and originate from a common ancestral source from France in the 17th century (Figure 1) (7). Consequently, some French and French Canadians patients, today living on different continents, carry the same IBD FH-causing gene variants that have evolved for 4 centuries in different environments. In this proof-of-concept study, we compared LDL-c levels and other clinical characteristics in heterozygous FH (HeFH) patients from France and French Canada carrying IBD gene variants.
Methods
Four IBD FH-causing LDLR variants, namely c.259T>G p.(Trp87Gly); c.2000G>A p.(Cys667Tyr); c.682G>A p.(Glu228Lys); and c.1048C>T p.(Arg350*) originating from France with a proven French-Canadian founder effect were selected (7, 8). During their initial visit to the lipid clinics, FH patients were assessed by a multidisciplinary team. Data on lipid profiles, FH-related genotypes, treatment regimen, metabolic syndrome-associated parameters and cardiovascular risks were collected from those who consented to participate in this study, as detailed elsewhere (Bouhali T, Brisson D, St-Pierre J, et al. Low plasma adiponectin exacerbates the risk of premature coronary artery disease in familial hypercholesterolemia. Atherosclerosis 2008;196:262-9.). Plasma lipid profiles at baseline (untreated), ApoE genotype, cardiovascular risk factors and occurrence of symptomatic ASCVD were compared in 105 adult carriers (30 from France and 75 from French Canada). For the most frequent variant p.(Trp87Gly), patients in both groups were matched for age (+/-1 year) and sex in a 3:1 ratio. For the other variants, matching was depending on the number of subjects. Data comparisons were made using Chi-square, Fisher’s exact test, Student’s t-tests and Wilcoxon-Mann-Whitney for independent samples. Statistical analyses were performed using SPSS package version 25 (IBM Corp., Armonk, N.Y., USA).
This research study (French National Agency for the Safety of Medicines and Health Products, protocol reference 2014-A01549-38 and its Canadian counterpart, protocol reference ECO HyperTG-Hyperchol) were conducted in accordance with the principles of the Declaration of Helsinki, consistent with the Good Clinical Practice guidelines of the International Conference on Harmonization. Ethical approval was obtained in France from the CCTIRS (Comité Consultatif sur le Traitement de l’Information en matière de Recherche dans le domaine de la Santé) and the CNIL (Commission National de l’Informatique et des libertés) in 2015 and in Canada from Advarra IRB in 2014. All subjects were screened at their respective lipid clinics and agreed to participate in this study. An informed consent was obtained from all participants, and a code that systematically de-identifies all clinical data was assigned to each subject (Gaudet D, Arsenault S, Belanger C, et al. Procedure to protect confidentiality of familial data in community genetics and genomic research. Clin Genet 1999; 55 : 259-64). Participants were included in this analysis based on the availability of lipid-associated parameters data.
Results
Subjects from both populations were comparable in terms of sex, age, smoking status (current or not), statin intolerance, diabetes, obesity and hypertension (Table 1). Although not statistically significant, the prevalence of ApoE4 in this study was almost 2-fold more elevated among French Canadians (29% vs 17%). Mean baseline untreated total cholesterol (10.14 ± 1.89 mmol/L vs 8.65 ± 1.84 mmol/L, p=0.0006) and LDL-c concentrations (7.94 ± 1.86 mmol/L vs 6.93 ± 1.78 mmol/L, p=0.016) were significantly higher in the French cohort (Figure 1) and this was observed across all FH-causing variants. The coronary artery anatomy of those who had a coronarography will be the object of another publication.
Discussion
This study illustrates that FH patients of similar age, sex and risk profile, originating from France and French Canada, sharing well documented IBD pathogenic LDLR variants having evolved in different geographic, cultural and socio-economic environments for hundreds of years, differ in term of cholesterol levels (Figure 1), highlighting the importance of better understanding the interplay between genetic and environmental modulators of FH expression. Higher plasma total cholesterol and LDL-c concentrations were observed in FH patients from France despite the fact that the ApoE4 allele (generally associated with higher cholesterol concentration) was 2-fold more prevalent among French Canadians who presented lower LDL-c than their French IBD FH counterpart.
Since the colonization of New France in the 17th century, the dissemination of FH causing gene variants in French Canada was important due to large pedigrees (often > 12 children per nuclear family) and limited population migration movements due to the harshness of transport routes (8). The combination of these factors led to a phenomenon called endogamy justifying the high prevalence of IBD FH in French Canada (1:80), which is much higher than France and the rest of the world (1:250). In addition, most cases of FH in French-Canada were mainly caused by a small number of IBD LDLR pathogenic variants (8). The historical high prevalence of FH in French-Canada is not a consequence of consanguinity but of endogamy (9). Comparing the expression of IBD genetic diseases in a founder population and in the colonizing population offers a unique opportunity to explain differences in the clinical expression or the response to interventions, controlling for the effect of the disease-causing variant. Indeed, IBD pathogenic variants that have evolved for hundreds of years in different geographic environments allow to control for the variance of FH clinical expression that is specifically due to the pathogenic variant, which is the case of LDL-c, the main feature of FH. One advantage of IBD pathogenic variants is that differences in the clinical expression of the disease are not determined by the pathogenic variant itself but rather by other modifying factors. This constitutes a real advantage in identifying key genetic, epigenetic or environmental factors affecting FH expression, risk trajectory or response to treatment. Specifically, when controlling for the genetic cause of FH, observed differences in LDL-c could contribute to the identification of genetic variants that increase susceptibility to LDL accumulation and atherogenicity beyond LDLR.
For instance, it is well documented that other genes, including APOE, could highly impact the lipid profile or ASCVD risk, and that environmental factors, nutritional habits, physical activity, bacterial exposure, stress and other factors affect the epigenome, gene expression and microbiota (10). By controlling for the FH-causing gene variant (using IBD variants), the analysis of modifiers is simplified, potentially uncovering novel pathways that influence FH expression, risk trajectory or response to current emerging therapies. Indeed, recent findings have also revealed the disproportionate prevalence between French-Canadians and French-Americans due to the proposed endogamy within these populations, resulting in a high prevalence of FH-causing genetic mutations. In alignment with our study, Mszar R. et al. suggested that the investigation of patients carrying IBD pathogenic gene variants is pivotal for addressing the underdiagnosed rate of FH in specific geographical territories, while identifying CV risks in these genetically self-contained communities (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7242505/). Similar findings were also described within the Indian population, showing the high prevalence of FH and unique genetic architecture caused by consanguineous marriages practices as well as multiple endogamous groups (https://www.sciencedirect.com/science/article/pii/S2773044123001110) (https://www.sciencedirect.com/science/article/pii/S0019483223004571).
In this proof-of-concept study we used LDL-cholesterol as a probe to highlight differences in FH expression between patients from two populations sharing IBD LDLR pathogenic variants. The next steps involve leveraging the advantages of identifying IBD pathogenic variants across various founder populations and multiple countries. Planned analyses include exome sequencing, DNA methylation studies, as well as comprehensive endophenotypic, clinical and environmental investigations. These efforts aim to better document modifiers of FH, uncover potential new targets for preventing ASCVD, and identify factors that modulate responses to emerging therapies. Integrating these diverse approaches within the clinical pathway will enhance our understanding of FH and help improving treatment strategies for affected populations on different worldwide continents.
Also, populations from other countries, such as Lebanon, USA, UK, Turkey, India and South Africa, will be involved as these communities sharing FH-causing IBD variants (11-13). These populations importantly differ in terms of life habits and access to accurate diagnosis and treatment. Therefore, access issues and quality of life will also be assessed through this project as part of the SMASH initiative (System and Molecular Approaches of Severe Dyslipidemias) (www.smash-access.org).
References
- Saadatagah S, Jose M, Dikilitas O, Alhalabi L, Miller AA, Fan X, et al. Genetic basis of hypercholesterolemia in adults. NPJ Genom Med. 2021;6(1):28. [CrossRef]
- Goldstein JL, Hobbs HH, Brown MS. Familial Hypercholesterolemia. In: Valle DL, Antonarakis S, Ballabio A, Beaudet AL, Mitchell GA, eds. The Online Metabolic and Molecular Bases of Inherited Disease. New York, NY: McGraw-Hill Education; 2019.
- Cuchel M, Raal FJ, Hegele RA, Al-Rasadi K, Arca M, Averna M, et al. 2023 Update on European Atherosclerosis Society Consensus Statement on Homozygous Familial Hypercholesterolaemia: new treatments and clinical guidance. Eur Heart J. 2023;44(25):2277-91. [CrossRef]
- Santos RD, Gidding SS, Hegele RA, Cuchel MA, Barter PJ, Watts GF, et al. Defining severe familial hypercholesterolaemia and the implications for clinical management: a consensus statement from the International Atherosclerosis Society Severe Familial Hypercholesterolemia Panel. Lancet Diabetes Endocrinol. 2016;4(10):850-61. [CrossRef]
- Safarova MS, Santos RD, Moriarty PM. 34 - Special Patient Populations: Familial Hypercholesterolemia and Other Severe Hypercholesterolemias. In: Ballantyne CM, ed. Clinical Lipidology (Third Edition). New Delhi: Elsevier; 2024:320-35.e2.
- Block T, El-Osta A. Epigenetic programming, early life nutrition and the risk of metabolic disease. Atherosclerosis. 2017;266:31-40. [CrossRef]
- Couture P, Morissette J, Gaudet D, Vohl MC, Gagne C, Bergeron J, et al. Fine mapping of low-density lipoprotein receptor gene by genetic linkage on chromosome 19p13.1-p13.3 and study of the founder effect of four French Canadian low-density lipoprotein receptor gene mutations. Atherosclerosis. 1999;143(1):145-51. [CrossRef]
- Gaudet D, Tremblay G, Perron P, Gagne C, Ouadahi Y, Moorjani S. [Familial hypercholesterolemia in eastern Quebec: a public health problem? The experience of the hyperlipidemia clinic of Chicoutimi]. Union Med Can. 1995;124(2):54-60.
- Davignon J, Roy M. Familial hypercholesterolemia in French-Canadians: taking advantage of the presence of a "founder effect". Am J Cardiol. 1993;72(10):6D-10D. [CrossRef]
- Zhang M, Hu T, Ma T, Huang W, Wang Y. Epigenetics and environmental health. Front Med. 2024. [CrossRef]
- Abifadel M, Rabes JP, Jambart S, Halaby G, Gannage-Yared MH, Sarkis A, et al. The molecular basis of familial hypercholesterolemia in Lebanon: spectrum of LDLR mutations and role of PCSK9 as a modifier gene. Hum Mutat. 2009;30(7):E682-91. [CrossRef]
- Leitersdorf E, Tobin EJ, Davignon J, Hobbs HH. Common low-density lipoprotein receptor mutations in the French Canadian population. J Clin Invest. 1990;85(4):1014-23. [CrossRef]
- Ashavaid TF, Altaf AK, Nair KG. Molecular basis of familial hypercholesterolemia: An Indian experience. Indian J Clin Biochem. 2000;15(Suppl 1):11-9. [CrossRef]
Figure 1.
Baseline (untreated) total cholesterol and LDL-cholesterol levels in FH carriers of identical by descent (IBD) pathogenic LDLR gene variants in 75 FH patients from the French-Canadian founder population and 30 patients from France (colonizing population). Patients from both countries carried either the c.259T>G p.(Trp87Gly); c.2000G>A p.(Cys667Tyr); c.682G>A p.(Glu228Lys); or c.1048C>T p.(Arg350*) LDLR gene variants, all pathogenic, having a proven French founder effect and having evolved in a different environment for 4 centuries. Both groups were comparable for ApoE genotype, smoking habits, anthropometric, diabetes and other cardiovascular risk factors. Patients were matched for age and sex (3:1 for the p.(Trp87Gly) variant). Baseline cholesterol levels were significantly lower among French-Canadians in all genotypes (see text).
Figure 1.
Baseline (untreated) total cholesterol and LDL-cholesterol levels in FH carriers of identical by descent (IBD) pathogenic LDLR gene variants in 75 FH patients from the French-Canadian founder population and 30 patients from France (colonizing population). Patients from both countries carried either the c.259T>G p.(Trp87Gly); c.2000G>A p.(Cys667Tyr); c.682G>A p.(Glu228Lys); or c.1048C>T p.(Arg350*) LDLR gene variants, all pathogenic, having a proven French founder effect and having evolved in a different environment for 4 centuries. Both groups were comparable for ApoE genotype, smoking habits, anthropometric, diabetes and other cardiovascular risk factors. Patients were matched for age and sex (3:1 for the p.(Trp87Gly) variant). Baseline cholesterol levels were significantly lower among French-Canadians in all genotypes (see text).
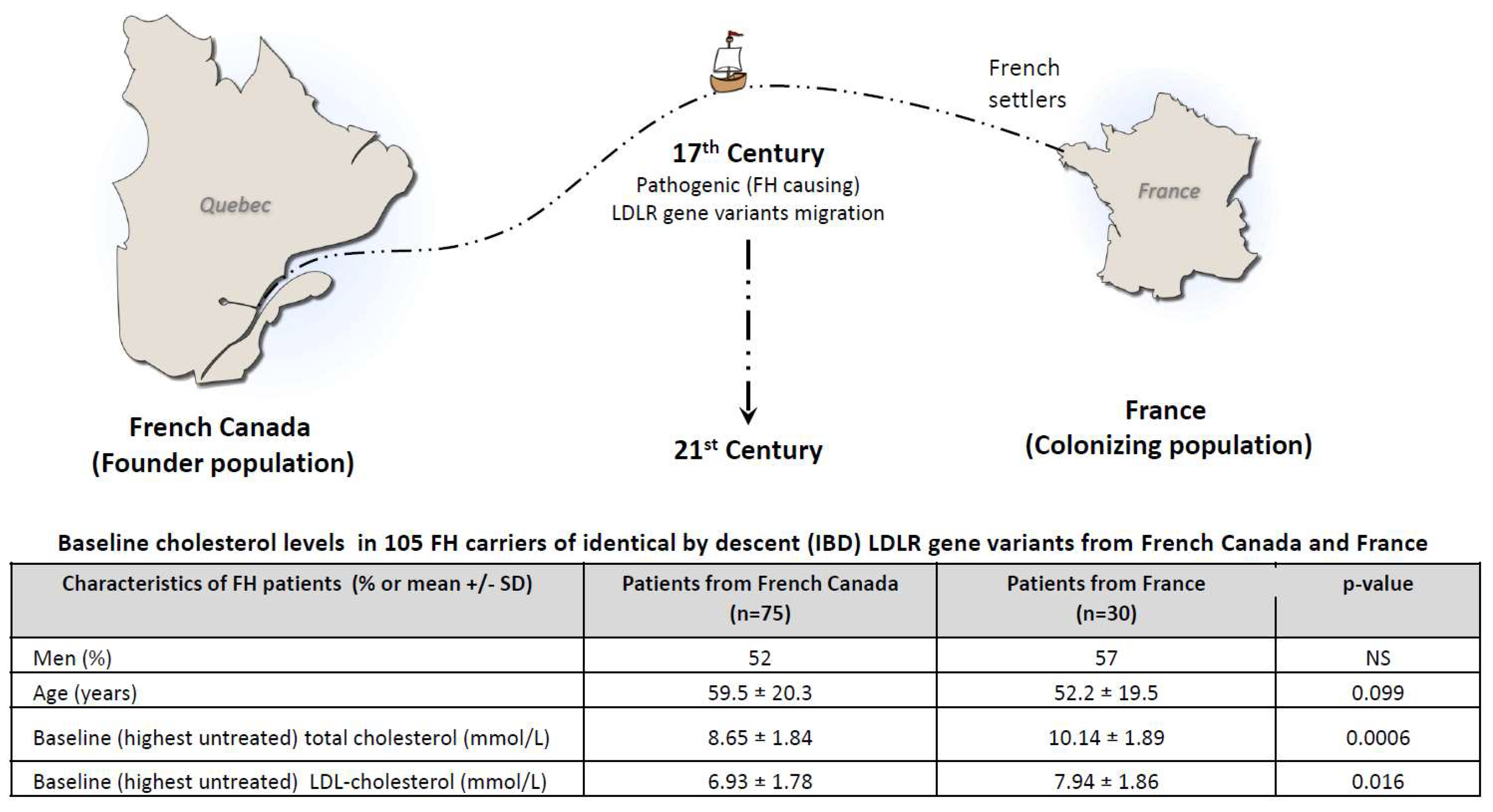

Table 1.
Characteristics of HeFH patients from France and French-Canada.
| Characteristics (% or mean ± SD) |
France (n=30) | French-Canada (n=75) | p-value |
| Men (%) | 57 | 52 | NS |
| Age (years) | 52.2 ± 19.5 | 59.5 ± 20.3 | NS |
| ApoE2 carriers (%) | 4 | 14 | NS |
| ApoE4 carriers (%) | 17 | 29 | NS |
| Diabetes (%) | 3 | 7 | NS |
| Obesity (%) | 11 | 19 | NS |
| Hypertension (%) | 17 | 22 | NS |
| Current smokers (%) | 31 | 22 | NS |
| Statin intolerance (%) | 4 | 19 | NS |
| ASCVD (%) | 33 | 33 | NS |
| Age at first event (years) | 47.7 ± 11.3 | 46.2 ± 11.1 | NS |
NS = p>0.05. Continuous variables are mean ± SD. ApoE: apolipoprotein E, ASCVD: atherosclerotic cardiovascular disease.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



