
Submitted:
23 August 2024
Posted:
23 August 2024
You are already at the latest version
Abstract
Esophageal cancer (EC) is one of the most common malignant tumors worldwide, and its two major types, esophageal adenocarcinoma (EAC) and esophageal squamous cell carcinoma (ESCC) present a severe global public health problem with an increasing incidence and mortality. Established risk factors include smoking, alcohol consumption, and dietary habits, but recent research highlights the substantial role of oral microbiota in EC pathogenesis. This review explores the intricate relationship between microbiome and esophageal carcinogenesis, focusing on eight significant mechanisms: chronic inflammation, microbial dysbiosis, production of carcinogenic metabolites, direct interaction with epithelial cells, epigenetic modifications, interaction with gastroesophageal reflux disease (GERD), metabolic changes, and angiogenesis. Certain harmful bacteria, such as Porphyromonas gingivalis and Fusobacterium nucleatum, are specifically implicated in sustaining irritation and tumor progression through pathways including NF-κB and NLRP3 inflammasome. Additionally, the review explores how microbial byproducts, including short-chain fatty acids (SCFAs) and reactive oxygen species (ROS), contribute to DNA harm and disease advancement. Furthermore, the impact of reflux on microbiota composition and its role in esophageal carcinogenesis is evaluated. By combining epidemiological data with mechanistic understanding, this review underscores the potential to target microbiota-immune system interplay for novel therapeutic and diagnostic strategies to prevent and treat esophageal cancer.
Keywords:
Esophageal cancer
; oral microbiota
; microbial dysbiosis
; carcinogenic metabolites
; chronic inflammation
1. Introduction
Upper gastrointestinal cancers including gastric and EC have high incidence and mortality worldwide. According to the World Health Organization, cancer is now the top or second leading cause of death among individuals under 70 in over 90 countries, rising to the third or fourth position in an additional 22 nations [1]. While the root causes of several cancers like smoking for lung cancer or alcohol combined with occupational hazards for upper gastrointestinal cancers are well established, the etiology of EC is multifactorial.
EC represents the seventh most common malignancy diagnosed worldwide, with more than 470000 new cases per year [2]. The two fundamental histological subtypes of EC are EAC and ESCC. EAC is predominantly found in developed countries and is strongly associated with GERD, Barrett’s esophagus (BE), obesity, low fruit and vegetable intake, and smoking [3]. On the contrary, ESCC is more common in developing countries, with risk factors including alcohol consumption, smoking, and poor oral health [4].
East Asia has one of the highest incidence and mortality rates for gastrointestinal cancers, with a great disease burden. In 2020, East Asia reported nearly half of the world’s newly diagnosed gastrointestinal cancer cases, with significant contributions from countries like Mongolia, Japan, China, South Korea, and North Korea [5]. The mortality rates reflect the high incidence, underscoring the urgent need for effective prevention, early detection, and treatment strategies. These divergences may be attributable to varying environmental, lifestyle, and dietary elements.
EC is generally diagnosed at a late stage, resulting in poor prognosis and low survival rates. The five-year survival rate of most countries is around 15 to 25 percent, thus there is an urgent need for new avenues of prevention, risk stratification, and early detection [6]. Emerging evidence further suggests the possible involvement of upper digestive tract microbiota in the etiology of EC, especially in the case of EAC’s rising incidence in developed countries.
The highly diversified microbiota of the upper digestive tract includes mutualists, commensals, and the pathogens that actively promote carcinogenesis by activating toll-like receptors (TLRs) or resisting carcinogenesis as the organisms can synthesize vitamins or provide barriers to carcinogenesis. The cross-sectional studies show that there is a significant difference in the microbiota of people with GERD, BE, EAC, esophageal squamous dysplasia, and ESCC compared to healthy controls [7]. Furthermore, diseases in the oral dysbiosis context such as periodontitis have been found to be related with a higher risk of EC [8].
Recent investigations have highlighted the crucial contribution of human microbiota in cancer development. The human mouth hosts a diverse array of microorganisms including bacteria, archaea, fungi and viruses. Numerous analyses demonstrated a close connection between oral microbes and gastrointestinal tumors. These microorganisms may contribute to carcinogenesis through mechanisms such as the production of carcinogenic substances, chronic inflammation, and altered cell metabolisms [8].
Despite advances in comprehending microbiota’s role in cancer, esophageal microbiota’s direct involvement in EC remains underexplored. High-throughput metagenomic DNA sequencing has improved our understanding of the human microbiome and its potential role in carcinogenesis directly through the immune system or indirectly through metabolites and toxins. Emerging evidence indicates microbiota can be manipulated to treat several diseases including cancer, suggesting a deeper understanding of microbiome-immune interactions could guide the development of future diagnostics and therapies for EC.
1.1. Risk Factors
1.1.1. Common Non-Modifiable Risk Factors
Ethnicity has a considerable influence on the risk of gastrointestinal cancers, and this varies between countries. For instance, in the USA, East Asians and Hispanics as well as Blacks experience a higher risk of stomach cancer than non-Hispanic white population [9]. East Asians and Black individuals are also at higher risk of liver cancer relative to white individuals with chronic hepatitis C virus infection and cirrhosis [10]. ESCC is predominant in East Asia, whereas EAC is common in the West [11]. Age is a substantial risk factor for gastrointestinal cancers, with almost all cases occurring in persons aged over 50 years. [12]. In addition, men are more at risk of getting most gastrointestinal cancers than women, implying that the cancer’s etiology is likely more frequent in some groups or occurs earlier in life for the males [13]. Also, a family history of gastrointestinal cancers increases the risk, probably due to the inheritance of genetic features and sharing the environmental risk factors. Specifically, mutations in tumor protein p53 (TP53), cyclin-dependent kinase inhibitor 2A (CDKN2A), and neurogenic locus notch homolog protein 1 (NOTCH1), increase the risk of EC [14].
1.1.2. Common Modifiable Risk Factors
Smoking is a major modifiable lifestyle risk factor for the occurrence of gastrointestinal cancers in East Asia and account significantly to colorectal cancer incidence and mortality burden among Chinese men [15]. Another major risk factor is drinking alcohol, which can lead to a rise in colorectal, stomach, oesophageal and gallbladder cancers [16,17,18]. The other risk factors are unhealthy dietary habits including low intake of dairy and whole grains, high consumption of salt along with processed foods. The positive associations with colorectal cancer are strong for red or processed meat but diets rich in fruits, vegetables, vitamins, and fiber are generally protective [19]. Physical inactivity and sedentary behavior have been linked positively to various cancers including colorectal, stomach, liver cell gall bladder and pancreatic while regular physical activity has been associated with decreased risk of different cancers [20]. In Asia, obesity is defined as a BMI of ≥25 kg/m² and associated with gastrointestinal cancers [15]. Metabolic disorders, such as type 2 diabetes and hypertension, are also associated with increased risks of these cancers in East Asia [21].
1.1.3. Highlighted Risk Factors in East Asia
In East Asia, the population is at higher risk of stomach cancer, with a higher prevalence of Helicobacter pylori infection compared to Western countries, as a significant cause of many gastric cancer cases [22]. A distinctive risk factor for biliary tract, liver, and gall duct cancer is the liver fluke Clonorchis sinensis, which is a significant risk in Guangdong and Guangxi provinces in Southern China and Heilongjiang Province in Northern China [23,24]. Among several risk factors for liver cancer, chronic hepatitis B and C virus infections are distinctive because they are highly prevalent in East Asia but not in Western countries [25,26]. ESCC is preceded by recurrent exposure to very hot substances, as a significant risk to the lining of the esophagus due to repeated thermal injuries [27,28]. A higher incidence of congenital biliary cysts and anomalous pancreaticobiliary duct junctions in East Asia increases the risks for gallbladder cancer through chronic inflammation [29].
1.1.4. Transition to Microbiota Focus
While these risk factors lay the groundwork in large accounts for understanding EC, recent studies have discovered a notable role of microbiota in cancer genesis, especially concerning EC. Microorganisms also live in the human mouth, a complex environment with places for bacteria, among archaea, fungi, and viruses. For example, the bacterial communities in the oral cavity are much less diverse and variable than at other body sites, like on the skin or within the gut. Many studies have shown that oral bacterium and gastrointestinal tumors have a tight relationship [30]. These microorganisms can incite cancer development through their ability to produce carcinogenic compounds, causing constant inflammation and alteration in host cell metabolism.
Environmental factors such as smoking and alcohol consumption, which are known to contribute to chronic inflammation and epithelial cell transformation, are also known to affect the composition of the oral microbiota [31]. These factors, by increasing the risk for a dysbiotic state in which harmful bacteria can colonize and beneficial species are lost, lead to disease, up to cancer [32]. This underlines the critical role of chronic inflammation in cancer genesis, urging us to be more aware and cautious.
New studies have indicated that changes in the esophageal microbiota order ratio to gram-negative bacteria will cause inflammation, TLR-4 activation with lipopolysaccharide (LPS), and increased reflux from relaxation of nitric oxide synthase (NOS)-mediated sphincter muscle [33]. However, the direct contribution of esophageal microbiota to EC is still poorly understood despite emerging insight into how these microbes interact with cancer.
The human microbiome has been established to play a crucial role in the pathogenesis of human cancers. Recent technological improvements, such as metagenomic DNA sequencing, provide higher-resolution insights, opening up potential for using microbiota as another means to treat numerous diseases, including cancer. However, to fully harness this potential, it is important to study more about the interaction between microbiome and the immune system. This will provide a novel strategies for EC therapy with relevant biomarkers, equipping us in the exciting frontier of cancer research [34].
This review highlights recent research indicating that EC and, more specifically, ESCC are closely linked to chronic inflammation and the composition of gut or esophageal microbiota. We further discuss several microbiota-mediated pathways as potential targets of EC therapy and explore strategies to enhance prevention, early diagnosis/screening, and treatments for this highly lethal disease.
2. Epidemiology Findings
Recent studies have begun to characterize the significant microbial variations linked to ESCC. Such modifications expose novel biomarkers useful for early diagnostics, the prediction of disease outcomes, and the response to therapeutic interventions. Different researchers have demonstrated that the over-abundance of specific microbes in ESCC tissues compared to non-tumor or healthy controls, respectively, suggested a specific role of microbiota during the pathogenesis of ESCC (Table 1).

Table 1.
Comparison of microbial populations in ESCC and control Samples.
| Sample | Microbes increased in ESCC | Microbes decreased in ESCC or increased in control samples | Reference |
|---|---|---|---|
| 67 paired samples (ESCC tissue vs non-tumor tissue) |
Fusobacteria phylum Fusobacterium genus |
Firmicutes phylum Streptococcus genus |
[35] |
| 32 ESCC samples vs 21 healthy controls |
Streptococcus genus Actinobacillus genus Peptostreptococcus genus Fusobacterium genus Prevotella genus |
Fusobacteria phylum Faecalibacterium genus Bacteroides genus Curvibacter genus Blautia genus |
[36] |
| 32 ESCC samples vs 15 esophagitis samples | Streptococcus genus |
Bacteroidetes genus Faecalibacterium genus Bacteroides genus Blautia genus |
[36] |
| 17 ESCC samples vs 16 healthy control samples |
Fusobacteria phylum Prevotella genus Pseudomonas genus |
Actinobacteria phylum Ralstonia genus Burkholderia-Caballeronia-Paraburkholderia genus |
[37] |
| 17 ESCC samples vs 15 post-op ESCC samples |
Fusobacteria phylum Bacteroidetes phylum Prevotella genus |
Pseudomonas genus |
[37] |
| 100 ESCC samples vs 100 adjacent tissue samples or 30 normal esophagus samples | P. gingivalis | [38] | |
| 18 ESCC samples vs 11 normal esophagus samples |
Fusobacteria phylum Bacteroidetes phylum Spirochaetes phylum T. amylovorum, S. infantis, P. nigrescens, P. endodontalis, V. dispar, A. segnis, P. melaninogenica, P. intermedia P. tannerae, P. nanceiensis, S. anginosus |
Proteobacteria phylum Thermi Phylum |
[39] |
| 120 ESCC samples vs adjacent tissue sample from same subjects |
R. mucilaginosa, P. endodontalis unclassified species in the genus Leptotrichia unclassified species in the genus Phyllobacterium unclassified species in the genus Sphingomonas |
class Bacilli N. subflava H. pylori A. parahaemolyticus A. rhizosphaerae, unclassified species in the genus Campylobacter unclassified species in the genus Haemophilus |
[40] |
| 60 ESCC samples vs paired adjacent normal tissue samples | F. nucleatum | [41] | |
| 54 ESCC samples vs 4 normal esophageal tissues |
Proteus genus Firmicutes genus Bacteroides genus Fusobacterium genus |
[42] | |
| 7 ESCC samples vs 70 normal control samples (together with 70 esophagitis, 70 low-grade intraepithelial neoplasia and 19 high-grade intraepithelial neoplasia) |
Streptococcus genus Haemophilus genus Neisseria genus Porphyromonas genus |
[43] | |
| 48 ESCC samples vs matched control samples | Staphylococcus genus | [44] | |
| 111 ESCC samples vs 41 normal samples |
Bacteroidetes phylum Fusobacteria phylum Spirochaetae phylum Streptococcus genus F. nucleatum |
Butyrivibrio genus Lactobacillus genus |
[45] |
| 31 ESCC samples vs matched controls |
Peptostreptococcaceae, Leptotrichia, Peptostreptococcus, Anaerovoracaceae, Filifactor, Anaerovoracaceae-Eubacterium_ brachygroup, Lachnoanaerobaculum, Dethiosulfatibacteraceae, Solobacterium, Johnsonella, Prevotellaceae UCG_001, and Tannerella (higher in N0 stage) Treponema and Brevibacillus (higher in N1 and N2 stages) Acinetobacter (higher in T3 stage) Corynebacterium, Aggregatibacter, Saccharimonadaceae-TM7x, and Cupriavidus (higher in T4 stage) |
[46] |
Numerous subsequent studies have consistently found a higher abundance of pathogenic organisms in ESCC tissues relative to non-tumor tissues or healthy controls. Fusobacterium nucleatum has been found to be more frequently enriched in ESCC tissues, with several studies by Jiang et al. (2021), Shao et al. (2019), and Yamamura et al. (2016). F. nucleatum abundance to the progression of tumor stages and worse clinical outcomes. This indicates a possible future biomarker for ESCC development and prognosis [35,36,41].
Porphyromonas gingivalis is one of the critical species in ESCC tissues. For example, Gao et al. (2016) demonstrated this bacterium in 61% of cancerous tissues and validated a significant association with ESCC development [38]. The presence of P. gingivalis was closely associated with differentiation status, metastasis, and overall survival rates in ESCC patients, suggesting that it may serve as a potential clinical target for ESCC treatment.
Recent studies have also investigated the microbial diversity and composition in ESCC tissues, showing a modest reduction of general bacterial richness. Jiang et al. (2021) and Li et al. (2020) found that all the alpha diversity indices were decreased in tissues of ESCC, suggesting a reduction in microbial taxa richness —moreover, dramatic differences in specific microbial phyla and genera community [36,37]. ESCC tissues generally showed up-regulated Fusobacteria, Bacteroidetes, and Firmicutes, while Actinobacteria and Proteobacteria were less than healthy controls [37,39].
Furthermore, additional studies of associations of specific microbial genera in clinicopathological phenotypes of ESCC have provided more proof for a relationship between this disorder and microbiota. For example, tissue samples from ESCC patients compared to normal esophagus showed that Streptococcus and Prevotella genera were enriched in ESCC and have been statistically associated as an event seen along disease progression [36,43]. Proteus in ESCC and change in Firmicutes and Bacteroides abundance among different morphological types of ESCC implies that microbial composition may affect tumor traits [42].
Studies on the prognostic value of these microorganisms have been done as well. The presence of F. nucleatum showed a statistically significant association with a shorter survival time, indicating that it might be a prognostic biomarker [41]. Kovaleva et al.(2021) concluded that Staphylococcus had a positive relationship with inflammatory tumor infiltrates and the resulting prognostic value in ESCC through the conducted two sets of patients [44].
The relationship between microbiota and the tumor microenvironment (TME) in ESCC was investigated in-depth. For example, Lin et al. (2022) revealed that the microbial co-occurrence networks were significantly denser and, by far, more complicated in tumor-adjacent tissues compared to tumor tissues, with differentially abundant microbiota being predominantly linked in tumor-adjacent tissues to signaling pathways involved in carcinogenesis [40].
Functional studies have also uncovered specific roles of certain microbiota in ESCC. For instance, Yang et al. (2021) found that ESCC-associated microbiota exhibited altered nitrate and nitrite reductase activity, suggesting that these functional changes may contribute to the development of esophageal cancer. [39].
In conclusion, the reviewed studies show significant changes to the esophageal microbiota associated with ESCC. The organisms diagnosed as changes include F. nucleatum and P. gingivalis. Some of these changes might be possible biomarkers for ESCC diagnosis and prognosis. The decreased microbiota diversity and specific functional changes have also been detailed. This can help in comprehending how microbiota causes esophagus cancer. Understanding the probable diagnoses and the mechanisms linking the various changes is crucial since it sluices epidemiologic studies. A total comprehension of microbiota concerning its carcinogenic role may help develop its-based therapy for ESCC.
3. Chronic Inflammation
3.1. Immune Regulation by Microbiota
The esophageal microbiota regulates inflammation and immune responses through interactions with mucosal immune cells (Table 2).
Table 2.
Microbiota and mechanisms contributing to chronic inflammation in EC.
| Bacteria | Mechanism | Impact on EC | References |
|---|---|---|---|
| P. gingivalis | Activates ERK1/2-Ets1 and PAR2/NF-κB pathways | Increased secretion of pro-inflammatory cytokines and chemokines reprogramming TME | [47,48,49] |
| Interacts with T cells and macrophages | Disrupts epithelial barrier, induces DNA damage, triggers pro-oncogenic signals | [50] | |
| LPS activates TLR-4 leading to NF-κB activation | Promotes cell proliferation, inhibits apoptosis, induces angiogenesis through VEGF expression | [51] | |
| Inhibits HDACs through SCFAs modulating Treg cell function | Supports tumor growth, metastasis, and resistance to therapy | [52,53,54] | |
| F. nucleatum | Activates NOD1/RIPK2/NF-κB and NLRP3 inflammasome pathways | Induces high levels of IL-6 and IL-8, driving inflammation-related carcinogenesis | [47,55] |
| LPS activates TLR-4 leading to NF-κB activation |
Recruits and reprograms immune cells within TME, supporting tumor progression and immune evasion | [56] | |
| Interacts with T cells and macrophages | Promotes cell proliferation, inhibits apoptosis, induces angiogenesis through VEGF expression | [51] | |
| E. coli | Upregulates TLRs 1-3, 6, 7, and 9 | Induces early carcinogenic molecular changes through TLR signalling pathway activation | [57] |
| Veillonella, Prevotella, Neisseria | Produces LPS, activates TLR-4 leading to NF-κB activation | Creates a pro-inflammatory environment, contributing to carcinogenesis | [58,59] |
| A. actinomycetemcomitans | Produces virulence factors such as leukotoxin and cytotoxic distension toxin | Exacerbates inflammation and cancer risk | [60] |
| Streptococcus | Increases prevalence with age, producing pro-inflammatory cytokines | Influences chronic inflammation and increases the risk of EC | [61] |
| Campylobacter | Enriched in GERD and BE tissues, associated with IL-18 expression | Associated with increased expression of carcinogenesis-related cytokines | [62] |
Dysbiosis disrupts this balance, leading to chronic inflammation and oncogenesis. Key pathogenic bacteria such as P. gingivalis and F. nucleatum significantly contribute to cancer development by activating inflammatory pathways, including nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), extracellular signal-related kinases 1 and 2-E26 transformation specific sequence 1 (ERK1/2-Ets1), and nucleotide-binding oligomerization domain-containing protein 1/receptor-interacting serine/threonine-protein kinase 2/NLR family pyrin domain containing 3 (NOD1/RIPK2/NLRP3) inflammasome [47,48,49,55]. These interactions lead to the production of pro-inflammatory cytokines such as interleukins IL-1β, IL-6, IL-8, and tumor necrosis factor-α (TNF-α). Microbial metabolites, such as SCFAs from beneficial microbiota, modulate regulatory T (Treg) cell function by inhibiting histone deacetylases (HDACs), impacting immune homeostasis [52,53,54]. Dysbiosis, characterized by harmful bacteria prevalence and reduced SCFA-producing bacteria, creates an inflammatory microenvironment conducive to ESCC. This chronic inflammation disrupts the epithelial barrier and reprograms immune cells within the TME, promoting tumor growth, metastasis, and therapy resistance [63] (Figure 1).
3.2. Activation of Inflammatory and Signalling Pathways
ESCC development has been linked to activating inflammatory pathways and producing cytokines, chemokines, and ROS. The LPS of gram-negative bacteria, such as P. gingivalis and F. nucleatum, binds to TLR-4 on immune cells and activates the MyD88-dependent pathway, leading to the activation of the NF-κB and the production of pro-inflammatory cytokines IL-1β, IL-6, IL-8, TNF-α in ESCC [56,64,65]. Additionally, P. gingivalis activates the ERK1/2-Ets1 and PAR2/NF-κB pathways, while the activation of the NOD1/RIPK2/NF-κB and NLRP3 inflammasome pathways acts as downstream effectors for F. nucleatum. Inflammatory pathways that become activated destabilize the epithelial barrier and activate the host’s DNA damage and pro-oncogenic signals, triggering the carcinogenesis process [50]. More so, the persistency of NF-kB activation is promoted by continuous exposure to microbial products for host immune cells, causing increased inflammation, cell proliferation, protection of cells from apoptosis, and inducing angiogenesis through vascular endothelial growth factor (VEGF) expression [51]. Chronic inflammation resulting from pathogenic microbiota increases the levels of pro-inflammatory cytokines, which contribute to ESCC development. TLRs are activated by Escherichia coli, the most prevalent organism in both BE and EAC, suggesting that this organism has an early role in carcinogenesis [57]. Bacteria such as Veillonella, Prevotella, and Neisseria, which are linked with GERD and BE, promote chronic inflammation and carcinogenesis [58,59]. In addition, A. actinomycetemcomitans generates factors that exaggerate inflammation and cancer hazard [60]. Age-related changes in the esophageal microbiota, including increased Streptococcus abundance, influence chronic inflammation and cancer development [61].
3.3. TME and Immune Reprogramming
The TME in EC is a complex biological environment that contains immune cells, endothelial cells, cancer-associated fibroblasts, adipocytes, extracellular matrix proteins, and secretory factors such as chemokines, cytokines, and growth factors. The TME is infiltrated with cells programmed to perform immunosuppressive functions, including tumor-associated macrophages, myeloid-derived suppressor cells, and Treg cells [66,67]. Pathogenic bacteria, notably P. gingivalis and F. nucleatum modulate TME by activating chemokines, growth factors, and cytokine production from tumor cells [73,74,75]. In the progression of EC, persistent chronic inflammation promoted via dysbiosis and pathogenic bacteria is crucial. Continuous stimulation of pro-inflammatory pathways, especially the signal transducer and activator of transcription 3 (STAT3) and NF-kB dependent cascades, maintains a prototypic inflamed tumor microenvironment, which can be itself driving the initial steps leading to oncogenesis. The IL-6 binds with its receptor (IL-6Rα), which uses signaling of the STAT3 pathway related to poor prognosis in EC [68]. The potential therapeutic value of treatment with selective inhibitors of STAT3 is being explored preclinically. P. gingivalis stimulates ERK1/2-Ets1, PAR2/NF-κB pathways, whereas F. nucleatum activates NOD signaling through RIPK2-dependent NF-κB and inflammasome to induce cytokines that drive chronic inflammatory immune-suppressive activity [47,55,69]. A. actinomycetemcomitans produces inflammatory and immunosuppressive virulence that helps with TME modulation [60]. Targeting pathogenic bacteria and signaling pathways in the TME shows promise as a therapeutic approach for EC.
3.4. Influence of GERD and Microbiota Changes
The esophageal microbiome was primarily affected by GERD, leading to changes that favor chronic inflammation and carcinogenesis. Patients with GERD harbor an increased proportion of Firmicutes and Proteobacteria and a greater gram-negative/gram-positive ratio [70]. This gram-positive to gram-negative bacteria transition is associated with reflex esophagitis and BE. This switch results in gram-negative bacteria, including Veillonella, Prevotella, Neisseria, and Fusobacteria [71,72]. This bacteria can produce LPS that, in turn, activates the TLR-4 receptor on either epithelial cells or immune cells. Consequently, NF-kB is activated, producing proinflammatory cytokines (IL-1β, IL-6, IL-8, and TNF-α) [58,73,74]. Studies demonstrate greater bacterial diversity and a gram-positive to gram-negative division in BE and reflux esophagitis. This redistribution of microbiota promotes an upregulation in the level of proinflammatory mediators, which maintains the system in a chronic state of inflammation.
Other bacteria, such as Veillonella and Prevotella, contribute to a more proinflammatory environment, whereas F. nucleatum activates NF-κB and enhances cytokine production. Enrichment of Campylobacter species in GERD and BE tissues upregulated carcinogenesis-related cytokines expression [62]. Aging-associated microbiota changes could escalate cancer risk, e.g., highlighting increased Streptococcus abundance in age and inflammatory contexts. The upregulation of TLRs by E. coli suggests that microbial involvement in early carcinogenic events was stimulated through increased proinflammatory cytokines. LPS, secreted by Gram-negative bacteria in BE, binds to TLR-4 and stimulates the activation of the NF-κB signal pathway, increasing the level of inflammatory cytokines, which all together promote chronic inflammation leading towards progression from adenocarcinoma. Understanding microbiota changes and mechanisms by which they influence inflammation and carcinogenesis may yield new tools to manage EC risk in GERD patients with targeted approaches.
4. Microbial Dysbiosis
The development of ESCC is related to microbial dysbiosis (Table 3), disrupted host-microbial interactions that alter the regular composition of the microbial community. An imbalance in microbial composition is linked with ESCC; however, multiple risk factors, such as hormonal imbalances, dietary compounds, toxins, and antibiotics, contribute to dysbiosis. Microbial dysbiosis promotes immune regulation disruption and the inclusion of chronic inflammation in causative factors associated with ESCC and oncogenesis. Reduced diversity of esophageal microbiota has been observed in patients with ESCC, including the reduced proportion of beneficial Streptococcus species and an increased number of pathogenic bacteria such as P. gingivalis and F. nucleatum that contribute to a tumor-promoting microenvironment [38,40,75]. Pathogenic bacteria are important as ESCC correlates with the progression of the disease, poorer prognosis, and a severe response to chemotherapy. F. nucleatum leads to shorter survival and increased tumor behavior by activating chemokines, such as CCL20 [41]. P. gingivalis is also related to ESCC severity with a decreased survival rate so that these bacteria may serve as biomarkers for ESCC targeting [38].
Table 3.
Microbial dysbiosis: Mechanisms and impact in EC.
| Bacteria | Mechanism | Impact on EC | References |
|---|---|---|---|
| P. gingivalis | - Activates NF-κB, ERK1/2-Ets1, and PAR2/NF-κB pathways | - Increased production of pro-inflammatory cytokines (IL-1β, IL-6), disruption of epithelial barriers, DNA damage | [47,48,49] |
| - Elicits chronic inflammation and immune evasion | - Promotes tumor growth and progression, poor clinical outcomes, potential biomarker for ESCC | [38] | |
| F. nucleatum | - Activates NF-κB, NOD1/RIPK2/NF-κB, and NLRP3 inflammasome pathways | - Induction of pro-inflammatory cytokines (IL-6, IL-8), creating a pro-tumorigenic environment | [47,55] |
| - Chemokine activation, specifically CCL20 | - Aggressive tumor behaviour, shorter survival, immune suppression, aiding in tumor progression and metastasis | [41] | |
| - Utilizes FadA adhesin/invasin to bind E-cadherin, activating β-catenin signalling | activation of pro-inflammatory cytokines, oncogenes, and stimulation of cancer cell proliferation | [76] | |
| T. denticola, S. anginosus | - Found in higher abundance in cancerous esophageal tissues | - Production of inflammatory mediators, promotion of an immunosuppressive microenvironment | [77] |
| E. coli | - Upregulates TLRs 1-3, 6, 7, and 9 | - Induces early carcinogenic molecular changes through TLR signalling pathway activation | [78] |
| Prevotella | - Produces LPS, activates TLR-4, leading to NF-κB activation | - Promotes chronic inflammation, mucosal barrier disruption, and enhancement of inflammatory milieu | [79] |
| Neisseria | - Produces LPS, activates TLR-4, leading to NF-κB activation | - Promotes chronic inflammation, mucosal barrier disruption, and enhancement of inflammatory milieu | [80,81] |
| Eikenella | - Associated with low fiber intake, leading to increased gram-negative bacteria | - Produces endotoxins that trigger inflammation and promote carcinogenesis | [82] |
| A. segnis, T. amylovorum, P. endodontalis, S. infantis, V. dispar, S. anginosus, P. intermedia, P. melaninogenica | - Identified in high-throughput profiling of ESCC | - Contributes to chronic inflammation and tumor-promoting microenvironment | [39] |
| Campylobacter | - Enriched in GERD and BE, associated with IL-18 expression | - Associated with increased expression of carcinogenesis-related cytokines | [83,84] |
| Parvimonas | - Associated with low fiber intake, leading to increased gram-negative bacteria | - Produces endotoxins that trigger inflammation and promote carcinogenesis | [82] |
| Leptotrichia | - Observed in GERD and BE patients | - Produces pro-inflammatory molecules, exacerbating mucosal damage and inflammation, contributing to progression to EAC | [85] |
| Lautropia, Bulleidia, Catonella, Corynebacterium, Moryella, Peptococcus, Cardiobacterium | - Lower carriage in ESCC patients compared to controls | - Altered saliva microbiota associated with higher risk of ESCC | [86] |
| Tannerella forsythia | - Increased levels in EC patients | - Associated with higher risk of EAC | [87] |
The adaptive immune system functions with the participation of the commensal bacteria from the human body. The dysbiosis of such bacteria disrupts the interaction and, thus, leads to immune deregulation and cancer progression. Some pathogens, such as P. gingivalis and F. nucleatum, avoid the immune response and induce a persistent inflammation and carcinogenesis by activating NF-κB, ERK1/2- Ets1, and NOD1/RIPK2/NLRP3 inflammasome pathways. Thus, these pathways provoke the secretion of pro-inflammatory cytokines and the onset of the barrier pathogen-induced disease [47,48,49,55]. At the same time, diet and many external factors profoundly impact microbial dysbiosis. For instance, it was clear that a high-fiber diet strengthens the beneficial bacteria Firmicutes. However, the absence of fiber in the diet, as well as alcohol consumption and tobacco usage, activate the harmful gram-negative bacteria, for example, Prevotella, Neisseria, and Eikenella. The secreted endotoxins target the epithelial cells, initiating their inflammation and carcinogenesis [82,88,89,90].
Microbial dysbiosis is obvious in GERD and BE, where increasing gram-negative bacteria intensifies mucosal damage and inflammation, driving the development from benign to malignant tissue [91]. Proton pump inhibitors reduce stomach acid production, which alters the esophageal microbiota by increasing gastric pH and decreasing exposure to acid, which can disrupt the balance of beneficial and harmful bacteria [92].
Motility disorder such as achalasia further adds to the dysbiosis state because food stasis provides a playground for bacteria growth by reducing esophageal clearance. The range of potential pathogens is much broader in cancer patients and can include Prophyromonas, Prevotella, or Fusobacterium, abundantly common among achalasia patients, which may act to promote inflammation and carcinogenesis [93,94].
Recent evidence has begun to explore the particular presence of pathogens, such as P. gingivalis and F. nucleatum, in ESCC. In order to thrive and continue growing, these bacteria work together to suppress chronic inflammation while promoting immune evasion, which allows for tumor survival and progression. Understanding these interactions can help develop targeted therapeutic strategies to manage dysbiosis and reduce the risk of EC.
5. Production of Carcinogenic Metabolites
The production of carcinogenic metabolites by various bacteria significantly contributes to the development and progression of EC (Table 4). Anaerobic bacteria such as Bacteroides, Clostridium, Faecalibacterium, and Ruminococcus produce SCFAs like butyrate, acetate, and propionate through the fermentation of dietary fibers [95,96,97,98]. These SCFAs are essential for maintaining intestinal barrier integrity and exerting anti-inflammatory effects. However, in EC patients, a reduction in SCFA production weakens the intestinal barrier and contributes to a pro-inflammatory environment, promoting carcinogenesis [99,100]. SCFAs inhibit histone deacetylases (HDACs), influencing Treg cell function and reducing inflammation; their deficiency thus fosters tumor development [52] (Figure 2).
Table 4.
Microbiota and production of carcinogenic metabolites in EC.
| Bacteria | Mechanism | Impact on EC | References |
|---|---|---|---|
| Bacteroides, Clostridium, Faecalibacterium, Ruminococcus | Produce SCFAs like butyrate, acetate, and propionate through dietary fiber fermentation | Reduced SCFA production contributes to a pro-inflammatory environment and weakened intestinal barrier, promoting carcinogenesis | [98] |
| Neisseria, Streptococcus, Candida | Metabolize alcohol into acetaldehyde, a highly toxic and carcinogenic substance | Causes DNA damage, mutagenesis, and gut microbiota disruption, increasing EC risk | [101] |
| P. gingivalis, H. pylori, E. coli | Produce ROS | Leads to DNA damage, cellular transformation, tumor survival, invasion, angiogenesis, and metastasis | [102,103] |
| S. oralis, S. mitis, S. sanguinis, S. gordonii, L. fermentum, L. jensenii, L. acidophilus, B. adolescentis | Produce RNS | Contribute to DNA damage and cancer progression through nitrosative stress | [104,105] |
| P. gingivalis, F. nucleatum | Overexpress MMPs; P. gingivalis produces gingipains to activate MMP-9; F. nucleatum stimulates MMP-9 and MMP-13 through p38 signaling | Degrade extracellular matrix, disrupt cell adhesion, facilitating cancer cell invasion and metastasis, critical in tumor progression | [49,106] |
| P. gingivalis, Prevotella intermedia, A. actinomycetemcomitans, F. nucleatum | Produce H2S, a genotoxic volatile sulfur compound | Induces genomic instability and cumulative mutations, promoting tumor growth and spread by activating various signaling pathways | [107,108] |
| Lactobacillus, Lactococcus, Bifidobacterium, Streptococcus, Leuconostoc, Pediococcus | Produce lactic acid through fermentation | Overproduction creates an acidic and hypoxic tumor microenvironment, suppressing immune responses and enhancing metastatic efficiency | [109] |
| E. coli | Secretes colibactin, a metabolic genetic toxic substance | Induces DNA double-strand breaks, leading to genomic instability and contributing significantly to carcinogenesis | [110] |
Alcohol is metabolized into acetaldehyde by bacteria such as Neisseria, Streptococcus, and the fungus Candida, which have high alcohol dehydrogenase (ADH) activity [101]. Acetaldehyde is a toxic and carcinogenic metabolite that causes DNA damage, mutagenesis, and disrupts the gut microbiota, significantly increasing the risk of EC through chronic exposure [111].
The production of ROS during microbiota-induced inflammation is another mechanism by which cancer development occurs. Microbes like P. gingivalis, H. pylori, and E. coli produce ROS to infect the host cells. Hence, P. gingivalis secretes nucleoside diphosphate kinase (NDK) that regulates ATP-mediated ROS release. ROS release does significant DNA damage and promotes the activation of transcription factors that cause inflammation and facilitate the progression of cancer [102,103,112,113]. In addition, reactive nitrogen species (RNS) are products of some microbes that promote activation of nitrosative stress, which causes DNA damage and contributes to cancer progression. RNS are products of S. oralis, S. mitis, S. sanguinis, S. gordonii, L. fermentum, L. jensenii, L. acidophilus, and B. adolescentis [104,105]. Matrix metalloproteinases (MMPs) are essential for the movement of cancer cells from the original site to other sites. P. gingivalis produces gingipains that activate MMP-9, while F. nucleatum stimulates p38 signaling and leads to the secretion of MMP-9 and -13 [49,106]. Hence, one of the primary roles of MMPs is to facilitate cancer metastasis through the degradation of the extracellular matrix [114].
Hydrogen sulfide (H2S) is a volatile sulfur compound produced by oral bacteria, such as P. gingivalis, P. intermedia, A. actinomycetemcomitans, and F. nucleatum, which is genotoxic and induces genomic instability. Moreover, the accumulation of mutations stimulated by H2Sadvances tumor growth and dissemination by activating several signaling pathways [107,108]. Another genus of aciduric bacteria, Lactobacillus, Lactococcus, Bifidobacterium, Streptococcus, Leuconostoc, and Pediococcus, compel the fermentation of glucose to lactic acid. Furthermore, excessive production of lactic acid initiates an acidic and hypoxic atmosphere of the tumor, suppresses immune responses, and enhances metastatic efficiency, facilitating tumor progression [109,115,116].
E. coli secretes colibactin, a metabolic genetic toxic substance with a potent carcinogenic effect, causing the emergence of DNA double-strand breaks and genomic instability, which contributes highly to carcinogenesis [110,117].
In conclusion, the microbial genera secreting carcinogenic metabolites, especially SCFAs and acetaldehyde, ROS, RNS, MMPs, H2S, and lactic acid, are closely associated with EC. Metabolites of microbial origin form a pro-inflammatory microenvironment that also damages DNA and indirectly stimulates growth and invasion processes characteristic of oncogenesis/metastasis via suppression of immune response. This understanding, therefore, opens up potential pharmacological targets for controlling and reducing the risk of EC due to microbial dysbiosis and metabolite production.
6. Direct Interaction with Epithelial Cells
Microbiota are directly involved in interactions with epithelial cells that contribute to the development and progression of EC (Table 5). P. gingivalis and F. nucleatum can also target epithelial cells directly, enhancing carcinogenesis using several different mechanisms. Breakdown of the state where commensal bacteria controls and maintains equilibrium within esophageal mucosa can cause diseases triggered by harmful bacterial strains. For example, P. gingivalis triggers the ERK1/2-Ets1 and PAR2/NF-κB axis; F. nucleatum activates NOD1/RIPK2 signaling to potentiate ESCC cell proliferation and migration [49,55]. Consequently, poor oral hygiene contributes to esophageal diseases by causatively facilitating colonization by pathogenic bacteria interacting directly with the squamous epithelium. F. nucleatum promotes malignant tumor progression through chemokine activation and influences the TME [118,119].
Bacteria such as Campylobacter, Leptotrichia, Fusobacterium, Rothia, and Capnocytophaga are enriched in patients with GERD and BE, indicating direct interaction with esophageal epithelial cells [61,84]. This leads to chronic inflammation and epithelial cell transformation. Long-term activation of TLR-2 and TLR-4 in BE and EAC promotes a pro-inflammatory environment conducive to cancer development [120,121].
Table 5.
Microbiota and their direct interaction mechanisms with epithelial cells in EC.
| Bacteria | Mechanism | Impact on EC | References |
|---|---|---|---|
| P. gingivalis | Activates ERK1/2-Ets1 and PAR2/NF-κB pathways | Promotes proliferation, migration, and invasion of epithelial cells | [47,48] |
| Induces antiapoptotic activity via JAK1/AKT/STAT3 pathway | Reduces apoptotic activity of epithelial cells | [122] | |
| Secretes NDK | Enhances BCL2 to BAX ratio | [112] | |
| Accelerates S-phase progression by manipulating CDK activity | Promotes cancer cell proliferation | [123] | |
| F. nucleatum | Activates NOD1/RIPK2/NF-κB pathway | Enhances ESCC cell growth and migration | [47,55] |
| Influences TME through chemokine activation | Associated with shorter survival times and aggressive tumor behavior | [118,119] | |
| Activates TLR-4 | Promotes β-catenin signaling leading to oncogene activation | [76,124] | |
| Binds to E-cadherin on carcinoma cells | Facilitates cancer cell proliferation | [76] | |
| Campylobacter, Leptotrichia, Rothia, Capnocytophaga | Enriched in GERD and BE | Contributes to chronic inflammation and epithelial cell transformation | [61,84] |
| A. actinomycetemcomitans | Produces virulence factors that interact with epithelial cells | Promotes cell transformation and carcinogenesis | [60] |
| T. denticola, S. mitis, S. anginosus | Dominates microbiota in cancerous esophageal tissues | Suggests direct interaction with epithelial cells contributing to disease progression | [77] |
| Candida, Neisseria | Metabolizes alcohol into acetaldehyde | Causes DNA damage, mutagenesis, and disrupts gut microbiota | [125,126] |
Microbes can promote malignant transformation of epithelial cells. For example, P. gingivalis and F. nucleatum can induce epithelial-mesenchymal transition, predisposing cells to malignant transformation [127]. P. gingivalis also induces antiapoptotic activity via the JAK1/AKT/STAT3 pathway, reduces apoptotic activity through phosphorylation of BCL-associated death protein (BAD), and enhances the B-cell lymphoma-2 (BCL2) to BCL-2 associated X-protein (BAX) ratio [122,128,129,130]. F. nucleatum activates TLR-4, upregulating autophagy and downregulating apoptosis, leading to a chemoresistance phenotype. It also binds to E-cadherin on carcinoma cells, activating β-catenin signaling, which promotes cancer cell proliferation [76,124].
Several virulence factors produced by A. actinomycetemcomitans can interact with epithelial cells, leading to neoplastic transformation and carcinogenesis [60]. F. nucleatum can establish a strong adherence with host cells through the presence of adhesins, such as FadA, thus increasing the risk for the development of tumors [131]. In particular, sustained P. gingivalis infection may lead to the acquisition of cancer stem cell properties, enhancing malignancy in non-neoplastic cells and promoting uncontrolled proliferation by modulating cyclin-dependent kinase (CDK) function [123].
Together, these data implicate direct bacterial interaction with epithelial cells in EC through modulation of inflammatory and cell processes towards a cancerous phenotype. These interactions not only contribute to a better understanding of the intricate pathways by which EC is developed, but also could potentially represent novel therapeutic targets that may provide new strategies in controlling and diminishing EC risk.
7. Epigenetic Modifications
Epigenetic modifications refer to the heritable changes in gene expression that occur without any changes to the DNA sequence. These modifications significantly affect the course of EC development and are mainly affected by microbiota alterations (Table 6). Microbiota produces SCFA substances, such as butyrate, that influence HDACs. The inhibition then affects Treg cells’ function, altering the immune reaction and local inflammation. An optimal proinflammatory milieu will create a favorable condition to initiate carcinogenesis [52,53,54]. In BE and EAC, activation of TLR-4 can directly affect the expression of cyclooxygenase-2 (COX-2), which occurs through NF-κB-independent mechanisms such as mitogen and stress-activated kinase (MSK) and mitogen-activated protein kinase (MAPK) that potentially involve altered epigenetic factors [121,132] (Figure 3).
Table 6.
Microbiota and epigenetic modifications in EC.
| Bacteria | Mechanism | Impact on EC | References |
|---|---|---|---|
| P. gingivalis | - Inhibits HDACs through SCFAs, modifying Treg cell function and numbers | - Creates a pro-inflammatory environment, contributing to carcinogenesis | [54,133] |
| - Upregulates miR-194 and Akt, downregulates GRHL3 and PTEN | - Enhances pro-proliferative and pro-migratory phenotype of esophageal tumors | [134] | |
| F. nucleatum | - Alters macrophage infiltration and methylation of the CDKN2A promoter | - Silences tumor suppressor genes and activates oncogenes, promoting cancer development | [135] |
| - Activates β-catenin signaling, leading to transcriptional activation of oncogenes | - Promotes cancer cell proliferation through activation of oncogenic pathways | [76,136] | |
| Microbiota in General | - Produces SCFAs that inhibit HDACs, impacting immune response and inflammation | - Creates a pro-inflammatory environment, contributing to carcinogenesis | [54] |
| - Interacts with epithelial cells, leading to genetic changes in mRNAs, miRNAs, and LncRNAs | - Disrupts normal cell regulatory mechanisms, promoting malignant transformation | [137,138] | |
| Microbiota in BE and EAC | - Activates TLR-4, influencing COX-2 expression through NF-κB-independent pathways like MSK and MAPK | - Leads to modifications in gene expression that promote inflammation and tumorigenesis | [121,132] |
P. gingivalis infection upregulates miR-194 and Akt and downregulates grainy head-like transcription factor 3 (GRHL3) and phosphatase and tensin homolog (PTEN), promoting esophageal tumors’ pro-proliferative and pro-migratory phenotype [134]. Thus, P. gingivalis modulates miRNA expression in cancer progression through epigenetic regulation [139].
Numerous genetic changes were characterized following the interaction of oral microbiota with epithelial cells. Specifically, mRNAs, miRNAs, and long non-coding RNAs (LncRNAs) were differentially expressed [137,138]. When P53 is downregulated as a tumor suppressor gene, several changes occur in epithelial cells, which is characteristic of malignant transformation [140]. These changes promote EC development by interfering with cellular regulation.
Additionally, F. nucleatum shifts macrophage infiltration and methylation status of the cyclin-dependent kinase inhibitor 2A (CDKN2A) promoter in cancerous lesions [135]. This interaction alters gene expression profiles, leading to malignant transformation by repressing tumor suppressor genes and inducing oncogenes. The transcriptional activation of oncogenes such as C-myc and cyclin D1 by F. nucleatum is mediated via the up-regulation of β-catenin signaling, suggesting a direct mechanistic link between microbial infection with subsequent epigenetic modifications leading to cancer induction [76,136].
To summarize, microbiota-induced epigenetic variation substantially contributes to the development of EC. Such changes involve the inhibition of HDAC by SCFAs, activation of TLR-4 on COX-2 expression and modulation of miRNA by P. gingivalis, changes in the genetic coding and DNA methylation patterns by F. nucleatum, and signaling in the EC with the other oncogenic pathway, such as β-catenin signaling. Indeed, all these processes cooperate to perform crucial roles in promoting EC development by facilitating the bidirectional relationship between microbiota and host cells.
8. Interaction with GERD
The crosstalk of GERD with EC development results from alterations or dysbiosis in the microbiome, chronic inflammation, and dietary consumption (Table 7). Symptomatic GERD exposes the esophagus to persistent acid and bile salts, which causes chronic inflammation and injury that predisposes to the development of BE, a premalignant condition leading then onwards toward EAC [141]. Metaplasia and the increased inflammatory cytokines regulated by chronic inflammation from GERD create a cancer-friendly environment.
As individuals age, changes in stomach microflora due to chronic inflammation and decreased acidity can modulate the esophageal microbiome [142]. GERD significantly alters the esophageal microbiome, increasing gram-negative bacteria and decreasing gram-positive bacteria [143]. LPS from gram-negative bacteria in GERD patients activate TLR-4, leading to inflammation, carcinogenesis, and DNA damage through ROS production [70,144,145].
Another consideration for a strong association between GERD and microbiome is aimed at particular microbiotas of GERD and BE patients, which draws a connection between GERD and cancer risk. The microbiotas of GERD and BE are enriched with Campylobacter, Leptotrichia, Fusobacterium, Rothia, and Capnocytophaga compared to controls and EAC [61,62]. All these bacteria cause persistent inflammation and enhance the transformation of epithelial cells, which further affects adenocarcinoma. In the case of GERD and BE, the effect of chronic inflammation also leads to the increased production of ROS, further promoting tumorigenesis [146].
Table 7.
Microbiota and interaction with GERD: Mechanisms and impact in EC.
| Bacteria | Mechanism | Impact on EC | References |
|---|---|---|---|
| H. pylori | - Causes chronic gastritis, leading to changes in gastric acid secretion and subsequent GERD | - Promotes the progression of GERD to BE and EAC | [147] |
| Campylobacter | - Enrichment in GERD and BE patients, associated with inflammatory responses | - Contributes to chronic inflammation and changes in the esophageal mucosa, promoting the progression to EAC | [148] |
| F. nucleatum | - Adheres to and invades epithelial cells, modulates immune response, and promotes inflammation | - Exacerbates progression of BE to EAC through TLR activation and promoting an oncogenic microenvironment | [149] |
| Prevotella | - Increased prevalence in the esophageal microbiota of GERD patients, known for its role in inflammatory processes | - Leads to chronic inflammation and mucosal damage, fostering conditions conducive to BE and EAC | [150] |
| S. anginosus | - Associated with the esophageal microbiota in GERD and BE, contributing to chronic inflammation | - Promotes epithelial cell alterations, facilitating progression from GERD to BE and EAC | [77,151] |
| Leptotrichia | - Enrichment in GERD and BE patients, associated with inflammatory responses | - Promotes chronic inflammation and epithelial cell transformation, contributing to carcinogenesis | [152,153] |
| Rothia | - Enrichment in GERD and BE patients, associated with inflammatory responses | - Contributes to chronic inflammation and mucosal damage, facilitating the progression to EAC | [154] |
| Capnocytophaga | - Enrichment in GERD and BE patients, associated with inflammatory responses | - Promotes chronic inflammation and changes in the esophageal mucosa, fostering conditions conducive to EAC | [155] |
Thus, high-fat diets are associated with reduced microbial and bile acid diversity that promotes BE and EC development. Obesity, commonly secondary to a high-fat diet, is also a significant risk factor for GERD and increases the prevalence of BE and EAC [156,157]. This diet may act together with GERD-induced inflammatory and microbial changes to increase cancer risk.
Sustained activation of TLR-2 and -4 in BE and EAC may mediate inflammation, cell proliferation, and carcinogenesis by recognizing dysbiotic microbial components, driving a pro-inflammatory milieu [121,158].
Specific bacteria play critical roles in this process:
- Campylobacter: It is overrepresented in GERD and BE patients, induction of chronic inflammation and mucosa alteration that might contribute to EAC emergence [148].
- F. nucleatum: It binds to or invades epithelial cells, modulates the immune response, and promotes inflammation, which enhances progression from BE to EAC through TLR activation [149].
- Prevotella: It is overrepresented in GERD, a precursor to BE and EAC, and it may facilitate chronic inflammation and mucosal damage [150].
- Rothia: It is increased in GERD and BE patients, causing chronic inflammation and mucosal damage that promotes progression to EAC [154].
- Capnocytophaga: It tends to be enriched in GERD and BE patients, mechanistically promoting chronic inflammation and esophageal mucosal changes, thereby creating conditions conducive to EAC [155].
Overall, the onset of EC via persistent inflammation and modifications in esophageal microflora, together with induced ROS creation, highlights the complex interplay of aging, diet, and obesity as pathways of interaction between GERD and the microbiome in individuals at risk for carcinogenesis.
9. Metabolic Changes and EC
The abiotic factors driving the gut microbiota-related metabolic changes accessible for development and progression are metabolite production, dysbiosis, dietary habits, and obesity. Table 8 presents the specific bacteria associated with these changes that promote carcinogenesis through various mechanisms. The metabolites function as SCFAs, bile acids, and branched-chained amino acids typically released by microorganisms, which help regulate immune function and digestion [95,96,97,98]. For instance, bacteria such as Bacteroides, Clostridium, Faecalibacterium, and Ruminococcus produce SCFAs that support immune function and gut homeostasis. However, a reduction in the levels or activity of these beneficial bacteria can lead to decreased SCFA production, creating a pro-inflammatory environment that contributes to an increased risk of cancer [161].
Dysbiosis is the imbalance of microbiota due to the environment, stress, or low immune function. It disrupts the microbiota’s average balance, resulting in various metabolic disorders, obesity, insulin resistance, and chronic inflammation. The specific reduced diversity of oral microbiota is highly associated with ESCC. H. pylori and Campylobacter are among the bacteria that cause these metabolic diseases [40,162]. A Western diet, which is high in fat and low in fiber, is associated with systemic low-grade inflammation and an increased load of various metabolic diseases. Furthermore, this diet disrupts the gut microbiome and bile acid metabolism, leading to the formation of BE and EAC [163,164]. Obesity, which is also associated with the Western diet, is additionally considered a risk factor for GERD, Barrett’s esophagus, and EAC.
Lactic acid-producing bacteria including Lactobacillus, Streptococcus, Bifidobacterium, and Leuconostoc, also facilitates the disease progression. They can create a low pH and hypoxic microenvironment conducive for tumor metastasis where they induce the Warburg effect [85] that supports cancer cell survival and proliferation, thereby promoting EC progression [165]. In addition, it has been reported that F. nucleatum can produce LPS that activates β-catenin signaling, thus enhancing oncogene expression (C-myc and cyclin D1), and promoting cancer cell proliferation [136].
P. gingivalis similarly modifies adenosine triphosphate/ P2X purinoceptor 7 (ATP/P2X7) signaling, which affects ROS and antioxidant response and, therefore, contributes to cancer development through ROS-induced DNA damage and inflammation [112]. Streptococci and Candida also metabolize alcohol to acetaldehyde, a toxic metabolite responsible for the development of DNA damage, leading to the risk of carcinogenesis [101].
In summary, metabolic changes driven by specific bacteria and environmental factors significantly contribute to the development and progression of EC. These changes include the production of functional metabolites, dysbiosis, dietary influences, lactate metabolism, LPS and cytokine production, and alcohol metabolism to acetaldehyde.
10. Angiogenesis
Angiogenesis, the generation of new blood vessels from pre-existing vasculatures, is fundamental to tumor progression as it supplies growth and metastasizing with essential nutrients and oxygen. In EC, the inflammatory microenvironment triggered by several bacteria promotes angiogenesis to a final extent through multiple pathways (Table 9).
Table 9.
Microbiota and mechanisms of angiogenesis in EC.
| Bacteria | Mechanism | Impact on EC | References |
|---|---|---|---|
| H. pylori | Increases ROS production through virulence factors | Activates angiogenesis and cancer development | [166] |
| Promotes hypoxic conditions stabilizing HIF-1α | Upregulates pro-angiogenic genes such as VEGF, contributing to tumor progression and poor prognosis | [167] | |
| F. nucleatum | Influences IL-8 production | Enhances angiogenesis and tumor invasiveness | [168] |
| Enhances IL-1β production | Creates a pro-inflammatory and pro-angiogenic microenvironment | [169] | |
| Increases TNF-α levels | Contributes to angiogenesis and tumor progression | [64] | |
| Activates β-catenin signaling, enhancing β-catenin, C-myc, and cyclin D1 expression | Enhances cancer cell proliferation and tumor growth | [76] | |
| P. gingivalis | Modulates inflammatory responses and cytokine production | Enhances tumor angiogenesis | [169] |
| Increases TNF-α levels | Promotes cancer cell proliferation and metastasis | [170] | |
| Produces H2S, activating proliferation, migration, and invasive signaling pathways | Contributes to a hypoxic, pro-angiogenic microenvironment | [107] | |
| Streptococcus species | Stimulates the production of angiogenic factors such as IL-8, VEGF, and bFGF | Promotes angiogenesis and cancer cell growth | [171] |
| General oral microbiota | Produces IL-1β, which activates endothelial cells to produce VEGF and other pro-angiogenic factors | Provides an inflammatory microenvironment conducive to angiogenesis and tumor progression | [172,173] |
ROS generated within the inflammatory microenvironment are implicated in cancer initiation and progression by inducing mutagenesis and enhancing angiogenesis. H. pylori and other bacteria may increase ROS production, thus activating angiogenesis and contributing to cancer development. In addition, this mechanism has been well-documented in gastric cancer with relevance to EC (Figure 4). Stabilizing hypoxia-inducible factor 1-alpha (HIF-1α) in the core of tumors upregulates the surrounding pro-angiogenic factor such as VEGF [174,175]. Serum levels of VEGF have been documented to increase in association with disease progression, with raised levels correlating with increased tumor burden and predicting poor prognosis in the case of ESCC [176]. At the same time, H. pylori may participate in this relation as it contributes to hypoxia, stabilizing HIF-1α [167].
Most importantly, IL-8 was determined to support angiogenesis and the proliferation and migration of cancer cells [177]. Angiogenesis is considered a hallmark of EAC development, as it ensures the growth of the tumor by creating a source of nutrients. H. pylori and F. nucleatum upregulates IL-8 levels in an effort to foster angiogenesis [58,168,178].
Inflammation changes the TME considerably and can aggravate the development of a disease by triggering the process of angiogenesis. Macrophages and dendritic cells found in tissues affected by EAC begin to produce VEGF and MMPs to support angiogenesis and make a particular tumor more invasive [179,180]. At the same time, P gingivalis and F. nucleatum are also involved in these processes, affecting an inflammatory response and cytokine generation [169]. IL-1β is produced after infection and is known to activate endothelial cells, trigger VEGF production, and other pro-angiogenic factors. As a result, the TME becomes inflammatory and more suitable for angiogenesis and tumor progression [181,182]. Bacteria such as H. pylori and F. nucleatum significantly impact this cytokine, exacerbating the processes of IL-1β production [183].
TNF-α enhances the expression of many angiogenic factors, such as IL-8, VEGF, and basic fibroblast growth factor (bFGF), which promote angiogenesis [184,185]. In addition, bacteria such as P. gingivalis and F. nucleatum increase the levels of TNF-α and, thus, intensify the process of angiogenesis [64,170]. Moreover, H2S, produced by oral bacteria and P. gingivalis, affects the development and spread of the tumor and the activation of the growth and migration of different signals, which are invasive pathways, promoting the process of tumor angiogenesis [107,186]. Consequently, H2S helps to produce the microenvironment, which is favorable for building new blood vessels.
To sum up, it is evident that the process of EC angiogenesis is significantly affected by some bacteria impacting the inflamed reactions and generating the pro-angiogenic factors. These include ROS formation, HIF-1α stabilization, cytokine and metabolite production, and H2S activity. All the effects of bacteria on the process of angiogenesis may be used for the further research of the possible tasks of therapy target for EC.
11. Future Directions
Microbiome involvement in the development and progression of EC has opened up new perspectives for research and treatment. Additional studies are indispensable to elucidate more intricate mechanisms of how particular bacteria, such as F. nucleatum and P. gingivalis, promote carcinogenesis. A combination of in vitro, in vivo, and clinical studies is necessary to reach this level. In vitro studies in co-culture systems of esophageal epithelial cells with these bacteria will provide insight into direct interactions, cellular responses, and pathway activations. Additionally, advanced tools such as RNA sequencing and metabolomics will uncover the mechanism of altered gene expression or metabolic changes induced by these bacteria; meanwhile, cell proliferation activity assays (apoptosis, migration capacity) would further establish their involvement in cancer development. The construction and usage of animal model systems, like xenografts or those with genetically altered mice for microbial-induced carcinogenesis, will be essential to establish the in vivo significance of cancer. Specifically engineered “Germ-free” mice with bacterial populations of interest could prove pivotal indirect evidence for microbiota’s role in tumor development, allowing additional studies on inflammation and immune responses and sensitivity to potential therapeutic intervention opportunities.
Correlating microbiome analysis with clinical data of EC, GERD, and healthy individuals will be vital in identifying the potential significance of these changes. Methods like 16S rRNA sequencing, metagenomics, and metatranscriptomics will be invaluable to profiling microbial communities and revealing architectural features of microorganisms that determine the function. Our next step is correlating these findings with clinical outcomes, which may provide the candidate biomarkers for early detection and prognosis. It will be necessary to validate candidate biomarkers found in primary studies in larger patient cohorts and develop sensitive and specific assays for their detection through a biological sample that could easily standardized, like blood, saliva, or biopsy tissue. It has helped this become clinically useful.
Consequently, machine learning algorithms that combine microbiome and metabolomics information with clinical variables can be implemented to build predictive models for the outcomes of EC patients. Moreover, drug screening for small molecule inhibitors of bacterial virulence factors and cancer-promoting pathways will open a new door to developing antisense-based therapeutics. Antibiotics, probiotics, and fecal microbiota transplantation (FMT) should be further investigated for their efficacy in manipulating the microbiome during cancer progression. The potential of these immunotherapeutic strategies through immune checkpoint inhibitors and vaccines against F. nucleatum and P. gingivalis is also discussed to improve anti-cancer immunity.
12. Conclusions
In this review, we discuss the mechanistic role of microbiota in EC development and progression. Our review emphasizes the role of a complex network within EC pathogenesis involving not only these pathogens but also further enhanced by other microbial metabolites and is in part driven by chronic inflammation and immune modulation. Targeting the microbiota opens new avenues for diagnosis, prognosis, and therapeutic development, but more importantly, their ability to drive these associations. The findings suggest that future studies should include more comprehensive microbiome profiling, investigating these mechanisms with a focus on gut-microbiota-targeted personalized medicine to identify candidate strategies for preventing and treating this highly aggressive form of human cancer.
Author Contributions
Conceptualization, K.T.M.; literature search, K.T.M. and K.S.T.; writing—original draft preparation, K.T.M. and K.S.T; writing—review and editing, K.T.M. and K.S.T; visualization, K.T.M. Both authors have read and agreed to published version of the manuscript.
Funding
This research was funded by the Yong Loo Lin School of Medicine, National University of Singapore, grant numbers A-8000685-00-00 and A-8000629-00-00 to K.S.T.
Acknowledgments
The images of immune cells and receptors were obtained from Clker.com, which provides free downloadable images.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA: A Cancer Journal for Clinicians 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Deboever, N.; Jones, C.M.; Yamashita, K.; Ajani, J.A.; Hofstetter, W.L. Advances in diagnosis and management of cancer of the esophagus. BMJ 2024, 385, e074962. [Google Scholar] [CrossRef]
- Coleman, H.G.; Xie, S.H.; Lagergren, J. The Epidemiology of Esophageal Adenocarcinoma. Gastroenterology 2018, 154, 390–405. [Google Scholar] [CrossRef] [PubMed]
- Lindkvist, B.; Johansen, D.; Stocks, T.; Concin, H.; Bjørge, T.; Almquist, M.; Häggström, C.; Engeland, A.; Hallmans, G.; Nagel, G.; et al. Metabolic risk factors for esophageal squamous cell carcinoma and adenocarcinoma: a prospective study of 580 000 subjects within the Me-Can project. BMC Cancer 2014, 14, 103. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Lucero-Prisno, D.E., 3rd; Zhang, L.; Xu, W.; Wong, S.H.; Ng, S.C.; Wong, M.C.S. Updated epidemiology of gastrointestinal cancers in East Asia. Nat Rev Gastroenterol Hepatol 2023, 20, 271–287. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Yang, Y.; Zhao, Y.; Huang, Y. The prognostic value of cyclooxygenase-2 expression in patients with esophageal cancer: evidence from a meta-analysis. Onco Targets Ther 2017, 10, 2893–2901. [Google Scholar] [CrossRef]
- Peters, B.A.; Wu, J.; Pei, Z.; Yang, L.; Purdue, M.P.; Freedman, N.D.; Jacobs, E.J.; Gapstur, S.M.; Hayes, R.B.; Ahn, J. Oral Microbiome Composition Reflects Prospective Risk for Esophageal Cancers. Cancer Research 2017, 77, 6777–6787. [Google Scholar] [CrossRef]
- Baima, G.; Ribaldone, D.G.; Romano, F.; Aimetti, M.; Romandini, M. The Gum–Gut Axis: Periodontitis and the Risk of Gastrointestinal Cancers. Cancers 2023, 15, 4594. [Google Scholar] [CrossRef]
- Dong, E.; Duan, L.; Wu, B.U. Racial and Ethnic Minorities at Increased Risk for Gastric Cancer in a Regional US Population Study. Clin Gastroenterol Hepatol 2017, 15, 511–517. [Google Scholar] [CrossRef]
- Nguyen, M.H.; Whittemore, A.S.; Garcia, R.T.; Tawfeek, S.A.; Ning, J.; Lam, S.; Wright, T.L.; Keeffe, E.B. Role of ethnicity in risk for hepatocellular carcinoma in patients with chronic hepatitis C and cirrhosis. Clin Gastroenterol Hepatol 2004, 2, 820–824. [Google Scholar] [CrossRef]
- Arnold, M.; Laversanne, M.; Brown, L.M.; Devesa, S.S.; Bray, F. Predicting the Future Burden of Esophageal Cancer by Histological Subtype: International Trends in Incidence up to 2030. Am J Gastroenterol 2017, 112, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Haggar, F.A.; Boushey, R.P. Colorectal cancer epidemiology: incidence, mortality, survival, and risk factors. Clin Colon Rectal Surg 2009, 22, 191–197. [Google Scholar] [CrossRef]
- Derakhshan, M.H.; Liptrot, S.; Paul, J.; Brown, I.L.; Morrison, D.; McColl, K.E. Oesophageal and gastric intestinal-type adenocarcinomas show the same male predominance due to a 17 year delayed development in females. Gut 2009, 58, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.B.; Chen, Z.L.; Li, J.G.; Hu, X.D.; Shi, X.J.; Sun, Z.M.; Zhang, F.; Zhao, Z.R.; Li, Z.T.; Liu, Z.Y.; et al. Genetic landscape of esophageal squamous cell carcinoma. Nat Genet 2014, 46, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.J.; Huang, Q.C.; Bao, C.Z.; Li, Y.J.; Li, X.Q.; Ye, D.; Ye, Z.H.; Chen, K.; Wang, J.B. Attributable causes of colorectal cancer in China. BMC Cancer 2018, 18, 38. [Google Scholar] [CrossRef]
- Li, Y.; Eshak, E.S.; Shirai, K.; Liu, K.; Dong, J.Y.; Iso, H.; Tamakoshi, A.; Group, J.S. Alcohol Consumption and Risk of Gastric Cancer: The Japan Collaborative Cohort Study. J Epidemiol 2021, 31, 30–36. [Google Scholar] [CrossRef]
- Yang, X.; Chen, X.; Zhuang, M.; Yuan, Z.; Nie, S.; Lu, M.; Jin, L.; Ye, W. Smoking and alcohol drinking in relation to the risk of esophageal squamous cell carcinoma: A population-based case-control study in China. Sci Rep 2017, 7, 17249. [Google Scholar] [CrossRef]
- Im, P.K.; Millwood, I.Y.; Kartsonaki, C.; Chen, Y.; Guo, Y.; Du, H.; Bian, Z.; Lan, J.; Feng, S.; Yu, C.; et al. Alcohol drinking and risks of total and site-specific cancers in China: A 10-year prospective study of 0.5 million adults. Int J Cancer 2021, 149, 522–534. [Google Scholar] [CrossRef]
- Azeem, S.; Gillani, S.W.; Siddiqui, A.; Jandrajupalli, S.B.; Poh, V.; Syed Sulaiman, S.A. Diet and Colorectal Cancer Risk in Asia--a Systematic Review. Asian Pac J Cancer Prev 2015, 16, 5389–5396. [Google Scholar] [CrossRef]
- Pham, N.M.; Mizoue, T.; Tanaka, K.; Tsuji, I.; Tamakoshi, A.; Matsuo, K.; Ito, H.; Wakai, K.; Nagata, C.; Sasazuki, S.; et al. Physical activity and colorectal cancer risk: an evaluation based on a systematic review of epidemiologic evidence among the Japanese population. Jpn J Clin Oncol 2012, 42, 2–13. [Google Scholar] [CrossRef]
- Naing, C.; Lai, P.K.; Mak, J.W. Immediately modifiable risk factors attributable to colorectal cancer in Malaysia. BMC Public Health 2017, 17, 637. [Google Scholar] [CrossRef] [PubMed]
- Eslick, G.D.; Lim, L.L.; Byles, J.E.; Xia, H.H.; Talley, N.J. Association of Helicobacter pylori infection with gastric carcinoma: a meta-analysis. Am J Gastroenterol 1999, 94, 2373–2379. [Google Scholar] [CrossRef]
- Hong, S.T.; Fang, Y. Clonorchis sinensis and clonorchiasis, an update. Parasitol Int 2012, 61, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.B.; Gan, X.Q.; Zhao, J.G.; Zheng, W.J.; Li, W.; Jiang, Z.H.; Zhu, T.J.; Zhou, X.N. Effectiveness of health education in improving knowledge, practice and belief related to clonorchiasis in children. Acta Trop 2020, 207, 105436. [Google Scholar] [CrossRef] [PubMed]
- de Martel, C.; Maucort-Boulch, D.; Plummer, M.; Franceschi, S. World-wide relative contribution of hepatitis B and C viruses in hepatocellular carcinoma. Hepatology 2015, 62, 1190–1200. [Google Scholar] [CrossRef] [PubMed]
- Ott, J.J.; Stevens, G.A.; Groeger, J.; Wiersma, S.T. Global epidemiology of hepatitis B virus infection: new estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine 2012, 30, 2212–2219. [Google Scholar] [CrossRef]
- Chen, Y.; Tong, Y.; Yang, C.; Gan, Y.; Sun, H.; Bi, H.; Cao, S.; Yin, X.; Lu, Z. Consumption of hot beverages and foods and the risk of esophageal cancer: a meta-analysis of observational studies. BMC Cancer 2015, 15, 449. [Google Scholar] [CrossRef]
- Tai, W.P.; Nie, G.J.; Chen, M.J.; Yaz, T.Y.; Guli, A.; Wuxur, A.; Huang, Q.Q.; Lin, Z.G.; Wu, J. Hot food and beverage consumption and the risk of esophageal squamous cell carcinoma: A case-control study in a northwest area in China. Medicine (Baltimore) 2017, 96, e9325. [Google Scholar] [CrossRef]
- Hundal, R.; Shaffer, E.A. Gallbladder cancer: epidemiology and outcome. Clin Epidemiol 2014, 6, 99–109. [Google Scholar] [CrossRef]
- Li, X.; Zhu, S.; Zhang, T.; Chen, X. Association between oral microflora and gastrointestinal tumors (Review). Oncol Rep 2021, 46, 160. [Google Scholar] [CrossRef]
- Sharma, T.; Gupta, A.; Chauhan, R.; Bhat, A.A.; Nisar, S.; Hashem, S.; Akhtar, S.; Ahmad, A.; Haris, M.; Singh, M.; et al. Cross-talk between the microbiome and chronic inflammation in esophageal cancer: potential driver of oncogenesis. Cancer Metastasis Rev 2022, 41, 281–299. [Google Scholar] [CrossRef] [PubMed]
- Jiménez De Nunzio, S.; Portal-Núñez, S.; Arias Macías, C.M.; Bruna Del Cojo, M.; Adell-Pérez, C.; Latorre Molina, M.; Macías-González, M.; Adell-Pérez, A. Does a Dysbiotic Oral Microbiome Trigger the Risk of Chronic Inflammatory Disease? Current Treatment Options in Allergy 2023, 10, 364–383. [Google Scholar] [CrossRef]
- Yang, L.; Lu, X.; Nossa, C.W.; Francois, F.; Peek, R.M.; Pei, Z. Inflammation and intestinal metaplasia of the distal esophagus are associated with alterations in the microbiome. Gastroenterology 2009, 137, 588–597. [Google Scholar] [CrossRef]
- Bhatt, A.P.; Redinbo, M.R.; Bultman, S.J. The role of the microbiome in cancer development and therapy. CA Cancer J Clin 2017, 67, 326–344. [Google Scholar] [CrossRef]
- Shao, D.; Vogtmann, E.; Liu, A.; Qin, J.; Chen, W.; Abnet, C.C.; Wei, W. Microbial characterization of esophageal squamous cell carcinoma and gastric cardia adenocarcinoma from a high-risk region of China. Cancer 2019, 125, 3993–4002. [Google Scholar] [CrossRef]
- Jiang, Z.; Wang, J.; Shen, Z.; Zhang, Z.; Wang, S. Characterization of Esophageal Microbiota in Patients With Esophagitis and Esophageal Squamous Cell Carcinoma. Front Cell Infect Microbiol 2021, 11, 774330. [Google Scholar] [CrossRef]
- Li, D.; He, R.; Hou, G.; Ming, W.; Fan, T.; Chen, L.; Zhang, L.; Jiang, W.; Wang, W.; Lu, Z.; et al. Characterization of the Esophageal Microbiota and Prediction of the Metabolic Pathways Involved in Esophageal Cancer. Front Cell Infect Microbiol 2020, 10, 268. [Google Scholar] [CrossRef]
- Gao, S.; Li, S.; Ma, Z.; Liang, S.; Shan, T.; Zhang, M.; Zhu, X.; Zhang, P.; Liu, G.; Zhou, F.; et al. Presence of Porphyromonas gingivalis in esophagus and its association with the clinicopathological characteristics and survival in patients with esophageal cancer. Infect Agent Cancer 2016, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Chen, C.H.; Jia, M.; Xing, X.; Gao, L.; Tsai, H.T.; Zhang, Z.; Liu, Z.; Zeng, B.; Yeung, S.J.; et al. Tumor-Associated Microbiota in Esophageal Squamous Cell Carcinoma. Front Cell Dev Biol 2021, 9, 641270. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Rao, W.; Xiang, Z.; Zeng, Q.; Liu, S.; Yu, K.; Zhou, J.; Wang, J.; Chen, W.; Chen, Y.; et al. Characteristics and interplay of esophageal microbiota in esophageal squamous cell carcinoma. BMC Cancer 2022, 22, 696. [Google Scholar] [CrossRef]
- Yamamura, K.; Baba, Y.; Nakagawa, S.; Mima, K.; Miyake, K.; Nakamura, K.; Sawayama, H.; Kinoshita, K.; Ishimoto, T.; Iwatsuki, M.; et al. Human Microbiome Fusobacterium Nucleatum in Esophageal Cancer Tissue Is Associated with Prognosis. Clin Cancer Res 2016, 22, 5574–5581. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Bai, W.; Zhao, C.; Wang, J. Distribution of esophagus flora in esophageal squamous cell carcinoma and its correlation with clinicopathological characteristics. Transl Cancer Res 2020, 9, 3973–3985. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Shao, D.; Zhou, J.; Gu, J.; Qin, J.; Chen, W.; Wei, W. Signatures within esophageal microbiota with progression of esophageal squamous cell carcinoma. Chin J Cancer Res 2020, 32, 755–767. [Google Scholar] [CrossRef]
- Kovaleva, O.; Podlesnaya, P.; Rashidova, M.; Samoilova, D.; Petrenko, A.; Mochalnikova, V.; Kataev, V.; Khlopko, Y.; Plotnikov, A.; Gratchev, A. Prognostic Significance of the Microbiome and Stromal Cells Phenotype in Esophagus Squamous Cell Carcinoma. Biomedicines 2021, 9. [Google Scholar] [CrossRef]
- Li, Z.; Shi, C.; Zheng, J.; Guo, Y.; Fan, T.; Zhao, H.; Jian, D.; Cheng, X.; Tang, H.; Ma, J. Fusobacterium nucleatum predicts a high risk of metastasis for esophageal squamous cell carcinoma. BMC Microbiol 2021, 21, 301. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Xiao, Q.; Chen, H.; Zhou, T.; Yin, Y. Comparison of tumor-associated and nontumor-associated esophageal mucosa microbiota in patients with esophageal squamous cell carcinoma. Medicine (Baltimore) 2022, 101, e30483. [Google Scholar] [CrossRef] [PubMed]
- Binder Gallimidi, A.; Fischman, S.; Revach, B.; Bulvik, R.; Maliutina, A.; Rubinstein, A.M.; Nussbaum, G.; Elkin, M. Periodontal pathogens Porphyromonas gingivalis and Fusobacterium nucleatum promote tumor progression in an oral-specific chemical carcinogenesis model. Oncotarget 2015, 6, 22613–22623. [Google Scholar] [CrossRef]
- Geng, F.; Liu, J.; Guo, Y.; Li, C.; Wang, H.; Wang, H.; Zhao, H.; Pan, Y. Persistent Exposure to Porphyromonas gingivalis Promotes Proliferative and Invasion Capabilities, and Tumorigenic Properties of Human Immortalized Oral Epithelial Cells. Front Cell Infect Microbiol 2017, 7, 57. [Google Scholar] [CrossRef]
- Inaba, H.; Sugita, H.; Kuboniwa, M.; Iwai, S.; Hamada, M.; Noda, T.; Morisaki, I.; Lamont, R.J.; Amano, A. Porphyromonas gingivalis promotes invasion of oral squamous cell carcinoma through induction of proMMP9 and its activation. Cell Microbiol 2014, 16, 131–145. [Google Scholar] [CrossRef]
- Tortora, S.C.; Agurto, M.G.; Martello, L.A. The oral-gut-circulatory axis: from homeostasis to colon cancer. Frontiers in Cellular and Infection Microbiology 2023, 13. [Google Scholar] [CrossRef]
- Okuyama, K.; Yanamoto, S. Oral Bacterial Contributions to Gingival Carcinogenesis and Progression. Cancer Prevention Research 2023, 16, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Rooks, M.G.; Garrett, W.S. Gut microbiota, metabolites and host immunity. Nat Rev Immunol 2016, 16, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; de Zoeten, E.F.; Ozkaynak, E.; Chen, C.; Wang, L.; Porrett, P.M.; Li, B.; Turka, L.A.; Olson, E.N.; Greene, M.I.; et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med 2007, 13, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- Rangan, P.; Mondino, A. Microbial short-chain fatty acids: a strategy to tune adoptive T cell therapy. Journal for ImmunoTherapy of Cancer 2022, 10, e004147. [Google Scholar] [CrossRef] [PubMed]
- Nomoto, D.; Baba, Y.; Liu, Y.; Tsutsuki, H.; Okadome, K.; Harada, K.; Ishimoto, T.; Iwatsuki, M.; Iwagami, S.; Miyamoto, Y.; et al. Fusobacterium nucleatum promotes esophageal squamous cell carcinoma progression via the NOD1/RIPK2/NF-κB pathway. Cancer Lett 2022, 530, 59–67. [Google Scholar] [CrossRef]
- Shi, T.; Wang, J.; Dong, J.; Hu, P.; Guo, Q. Periodontopathogens Porphyromonas gingivalis and Fusobacterium nucleatum and Their Roles in the Progression of Respiratory Diseases. Pathogens 2023, 12, 1110. [Google Scholar] [CrossRef]
- Zaidi, A.H.; Kelly, L.A.; Kreft, R.; Barlek, M.; Omstead, A.N.; Matsui, D.; Boyd, N.H.; Gazarik, K.E.; Heit, M.I.; Nistico, L.; et al. Associations of microbiota and toll-like receptor signaling pathway in esophageal adenocarcinoma. BMC Cancer 2016, 16. [Google Scholar] [CrossRef]
- Bakhti, S.Z.; Latifi-Navid, S. Oral microbiota and Helicobacter pylori in gastric carcinogenesis: what do we know and where next? BMC Microbiology 2021, 21, 71. [Google Scholar] [CrossRef]
- Asili, P.; Mirahmad, M.; Rezaei, P.; Mahdavi, M.; Larijani, B.; Tavangar, S.M. The Association of Oral Microbiome Dysbiosis with Gastrointestinal Cancers and Its Diagnostic Efficacy. Journal of Gastrointestinal Cancer 2023, 54, 1082–1101. [Google Scholar] [CrossRef]
- Belibasakis, G.N.; Maula, T.; Bao, K.; Lindholm, M.; Bostanci, N.; Oscarsson, J.; Ihalin, R.; Johansson, A. Virulence and Pathogenicity Properties of Aggregatibacter actinomycetemcomitans. Pathogens 2019, 8, 222. [Google Scholar] [CrossRef]
- Deshpande, N.P.; Riordan, S.M.; Castaño-Rodríguez, N.; Wilkins, M.R.; Kaakoush, N.O. Signatures within the esophageal microbiome are associated with host genetics, age, and disease. Microbiome 2018, 6, 227. [Google Scholar] [CrossRef] [PubMed]
- Blackett, K.L.; Siddhi, S.S.; Cleary, S.; Steed, H.; Miller, M.H.; Macfarlane, S.; Macfarlane, G.T.; Dillon, J.F. Oesophageal bacterial biofilm changes in gastro-oesophageal reflux disease, Barrett's and oesophageal carcinoma: association or causality? Alimentary Pharmacology and Therapeutics 2013, 37, 1084–1092. [Google Scholar] [CrossRef]
- Soler, M.F.; Abaurrea, A.; Azcoaga, P.; Araujo, A.M.; Caffarel, M.M. New perspectives in cancer immunotherapy: targeting IL-6 cytokine family. Journal for ImmunoTherapy of Cancer 2023, 11, e007530. [Google Scholar] [CrossRef] [PubMed]
- Yáñez, L.; Soto, C.; Tapia, H.; Pacheco, M.; Tapia, J.; Osses, G.; Salinas, D.; Rojas-Celis, V.; Hoare, A.; Quest, A.F.G.; et al. Co-Culture of P. gingivalis and F. nucleatum Synergistically Elevates IL-6 Expression via TLR4 Signaling in Oral Keratinocytes. International Journal of Molecular Sciences 2024, 25, 3611. [Google Scholar] [CrossRef] [PubMed]
- Kamarajan, P.; Ateia, I.; Shin, J.M.; Fenno, J.C.; Le, C.; Zhan, L.; Chang, A.; Darveau, R.; Kapila, Y.L. Periodontal pathogens promote cancer aggressivity via TLR/MyD88 triggered activation of Integrin/FAK signaling that is therapeutically reversible by a probiotic bacteriocin. PLOS Pathogens 2020, 16, e1008881. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat Med 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Yang, W.; Liu, S.; Mao, M.; Gong, Y.; Li, X.; Lei, T.; Liu, C.; Wu, S.; Hu, Q. T-cell infiltration and its regulatory mechanisms in cancers: insights at single-cell resolution. Journal of Experimental & Clinical Cancer Research 2024, 43, 38. [Google Scholar] [CrossRef]
- Hirano, T. IL-6 in inflammation, autoimmunity and cancer. International Immunology 2020, 33, 127–148. [Google Scholar] [CrossRef]
- Meng, F.; Li, R.; Ma, L.; Liu, L.; Lai, X.; Yang, D.; Wei, J.; Ma, D.; Li, Z. Porphyromonas gingivalis promotes the motility of esophageal squamous cell carcinoma by activating NF-kappaB signaling pathway. Microbes Infect 2019, 21, 296–304. [Google Scholar] [CrossRef]
- Liu, Y.; Yu, J.; Yang, Y.; Han, B.; Wang, Q.; Du, S. Investigating the causal relationship of gut microbiota with GERD and BE: a bidirectional mendelian randomization. BMC Genomics 2024, 25, 471. [Google Scholar] [CrossRef]
- Lv, J.; Guo, L.; Liu, J.J.; Zhao, H.P.; Zhang, J.; Wang, J.H. Alteration of the esophageal microbiota in Barrett’s esophagus and esophageal adenocarcinoma. World J Gastroenterol 2019, 25, 2149–2161. [Google Scholar] [CrossRef] [PubMed]
- Ajayi, T.A.; Cantrell, S.; Spann, A.; Garman, K.S. Barrett’s esophagus and esophageal cancer: Links to microbes and the microbiome. PLoS Pathog 2018, 14, e1007384. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Latif, M.M.; Kelleher, D.; Reynolds, J.V. Potential role of NF-kappaB in esophageal adenocarcinoma: as an emerging molecular target. J Surg Res 2009, 153, 172–180. [Google Scholar] [CrossRef]
- Rai, A.K.; Panda, M.; Das, A.K.; Rahman, T.; Das, R.; Das, K.; Sarma, A.; Kataki, A.C.; Chattopadhyay, I. Dysbiosis of salivary microbiome and cytokines influence oral squamous cell carcinoma through inflammation. Archives of Microbiology 2021, 203, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; He, R.; Hou, G.; Ming, W.; Fan, T.; Chen, L.; Zhang, L.; Jiang, W.; Wang, W.; Lu, Z.; et al. Characterization of the Esophageal Microbiota and Prediction of the Metabolic Pathways Involved in Esophageal Cancer. Frontiers in Cellular and Infection Microbiology 2020, 10. [Google Scholar] [CrossRef]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef]
- Narikiyo, M.; Tanabe, C.; Yamada, Y.; Igaki, H.; Tachimori, Y.; Kato, H.; Muto, M.; Montesano, R.; Sakamoto, H.; Nakajima, Y.; et al. Frequent and preferential infection of Treponema denticola, Streptococcus mitis, and Streptococcus anginosus in esophageal cancers. Cancer Sci 2004, 95, 569–574. [Google Scholar] [CrossRef]
- Zaidi, A.H.; Kelly, L.A.; Kreft, R.E.; Barlek, M.; Omstead, A.N.; Matsui, D.; Boyd, N.H.; Gazarik, K.E.; Heit, M.I.; Nistico, L.; et al. Associations of microbiota and toll-like receptor signaling pathway in esophageal adenocarcinoma. BMC Cancer 2016, 16, 52. [Google Scholar] [CrossRef]
- Larsen, J.M. The immune response to Prevotella bacteria in chronic inflammatory disease. Immunology 2017, 151, 363–374. [Google Scholar] [CrossRef]
- Dietrich, M.; Bartfeld, S.; Munke, R.; Lange, C.; Ogilvie, L.A.; Friedrich, A.; Meyer, T.F. Activation of NF-κB by Neisseria gonorrhoeae is associated with microcolony formation and type IV pilus retraction. Cellular Microbiology 2011, 13, 1168–1182. [Google Scholar] [CrossRef]
- Mikucki, A.; McCluskey, N.R.; Kahler, C.M. The Host-Pathogen Interactions and Epicellular Lifestyle of Neisseria meningitidis. Front Cell Infect Microbiol 2022, 12, 862935. [Google Scholar] [CrossRef] [PubMed]
- Nobel, Y.R.; Snider, E.J.; Compres, G.; Freedberg, D.E.; Khiabanian, H.; Lightdale, C.J.; Toussaint, N.C.; Abrams, J.A. Increasing Dietary Fiber Intake Is Associated with a Distinct Esophageal Microbiome. Clin Transl Gastroenterol 2018, 9, 199. [Google Scholar] [CrossRef] [PubMed]
- Hameed, A. Human Immunity Against Campylobacter Infection. Immune Netw 2019, 19, e38. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Ando, T.; Ishiguro, K.; Maeda, O.; Watanabe, O.; Funasaka, K.; Nakamura, M.; Miyahara, R.; Ohmiya, N.; Goto, H. Characterization of bacterial biota in the distal esophagus of Japanese patients with reflux esophagitis and Barrett’s esophagus. BMC Infect Dis 2013, 13, 130. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Shrestha, P.; Qiu, Z.; Harman, D.G.; Teoh, W.C.; Al-Sohaily, S.; Liem, H.; Turner, I.; Ho, V. Distinct Microbiota Dysbiosis in Patients with Non-Erosive Reflux Disease and Esophageal Adenocarcinoma. J Clin Med 2020, 9. [Google Scholar] [CrossRef]
- Chen, X.; Winckler, B.; Lu, M.; Cheng, H.; Yuan, Z.; Yang, Y.; Jin, L.; Ye, W. Oral Microbiota and Risk for Esophageal Squamous Cell Carcinoma in a High-Risk Area of China. PLoS One 2015, 10, e0143603. [Google Scholar] [CrossRef]
- Peters, B.A.; Wu, J.; Pei, Z.; Yang, L.; Purdue, M.P.; Freedman, N.D.; Jacobs, E.J.; Gapstur, S.M.; Hayes, R.B.; Ahn, J. Oral Microbiome Composition Reflects Prospective Risk for Esophageal Cancers. Cancer Res 2017, 77, 6777–6787. [Google Scholar] [CrossRef]
- Conteh, A.R.; Huang, R. Targeting the gut microbiota by Asian and Western dietary constituents: a new avenue for diabetes. Toxicol Res (Camb) 2020, 9, 569–577. [Google Scholar] [CrossRef]
- Kaakoush, N.O.; Lecomte, V.; Maloney, C.A.; Morris, M.J. Cross-talk among metabolic parameters, esophageal microbiota, and host gene expression following chronic exposure to an obesogenic diet. Sci Rep 2017, 7, 45753. [Google Scholar] [CrossRef]
- Lee, W.H.; Chen, H.M.; Yang, S.F.; Liang, C.; Peng, C.Y.; Lin, F.M.; Tsai, L.L.; Wu, B.C.; Hsin, C.H.; Chuang, C.Y.; et al. Bacterial alterations in salivary microbiota and their association in oral cancer. Sci Rep 2017, 7, 16540. [Google Scholar] [CrossRef]
- Osias, G.L.; Bromer, M.Q.; Thomas, R.M.; Friedel, D.; Miller, L.S.; Suh, B.; Lorber, B.; Parkman, H.P.; Fisher, R.S. Esophageal bacteria and Barrett’s esophagus: a preliminary report. Dig Dis Sci 2004, 49, 228–236. [Google Scholar] [CrossRef]
- Amir, I.; Konikoff, F.M.; Oppenheim, M.; Gophna, U.; Half, E.E. Gastric microbiota is altered in oesophagitis and Barrett’s oesophagus and further modified by proton pump inhibitors. Environ Microbiol 2014, 16, 2905–2914. [Google Scholar] [CrossRef]
- Geng, Z.H.; Zhu, Y.; Chen, W.F.; Fu, P.Y.; Xu, J.Q.; Wang, T.Y.; Yao, L.; Liu, Z.Q.; Li, X.Q.; Zhang, Z.C.; et al. The role of type II esophageal microbiota in achalasia: Activation of macrophages and degeneration of myenteric neurons. Microbiol Res 2023, 276, 127470. [Google Scholar] [CrossRef]
- Takahashi, K.; Sato, H.; Mizusawa, T.; Tominaga, K.; Ikarashi, S.; Hayashi, K.; Mizuno, K.I.; Hashimoto, S.; Yokoyama, J.; Terai, S. Comparison of Oral and Esophageal Microbiota in Patients with Achalasia Before and After Peroral Endoscopic Myotomy. Turk J Gastroenterol 2021, 32, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Fleming, S.E.; Yeo, S. Production and Absorption of Short-Chain Fatty Acids. In Dietary Fiber: Chemistry, Physiology, and Health Effects, Kritchevsky, D., Bonfield, C., Anderson, J.W., Eds.; Springer US: Boston, MA, 1990; pp. 301–315. [Google Scholar]
- Tomás-Pejó, E.; González-Fernández, C.; Greses, S.; Kennes, C.; Otero-Logilde, N.; Veiga, M.C.; Bolzonella, D.; Müller, B.; Passoth, V. Production of short-chain fatty acids (SCFAs) as chemicals or substrates for microbes to obtain biochemicals. Biotechnology for Biofuels and Bioproducts 2023, 16, 96. [Google Scholar] [CrossRef] [PubMed]
- Peterson, C.T.; Perez Santiago, J.; Iablokov, S.N.; Chopra, D.; Rodionov, D.A.; Peterson, S.N. Short-Chain Fatty Acids Modulate Healthy Gut Microbiota Composition and Functional Potential. Current Microbiology 2022, 79, 128. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Pramanik, S. Structural diversity, functional aspects and future therapeutic applications of human gut microbiome. Archives of Microbiology 2021, 203, 5281–5308. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Li, W.; Meng, H. A double-edged sword: Role of butyrate in the oral cavity and the gut. Mol Oral Microbiol 2021, 36, 121–131. [Google Scholar] [CrossRef]
- Deng, Y.; Tang, D.; Hou, P.; Shen, W.; Li, H.; Wang, T.; Liu, R. Dysbiosis of gut microbiota in patients with esophageal cancer. Microb Pathog 2021, 150, 104709. [Google Scholar] [CrossRef]
- Tagaino, R.; Washio, J.; Abiko, Y.; Tanda, N.; Sasaki, K.; Takahashi, N. Metabolic property of acetaldehyde production from ethanol and glucose by oral Streptococcus and Neisseria. Scientific Reports 2019, 9, 10446. [Google Scholar] [CrossRef]
- Handa, O.; Naito, Y.; Yoshikawa, T. Helicobacter pylori: a ROS-inducing bacterial species in the stomach. Inflammation Research 2010, 59, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Charoensaensuk, V.; Chen, Y.-C.; Lin, Y.-H.; Ou, K.-L.; Yang, L.-Y.; Lu, D.-Y. Porphyromonas gingivalis Induces Proinflammatory Cytokine Expression Leading to Apoptotic Death through the Oxidative Stress/NF-κB Pathway in Brain Endothelial Cells. Cells 2021, 10, 3033. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Torres, I.; Manzano-Pech, L.; Rubio-Ruíz, M.E.; Soto, M.E.; Guarner-Lans, V. Nitrosative Stress and Its Association with Cardiometabolic Disorders. Molecules 2020, 25. [Google Scholar] [CrossRef] [PubMed]
- Abranches, J.; Zeng, L.; Kajfasz, J.K.; Palmer, S.R.; Chakraborty, B.; Wen, Z.T.; Richards, V.P.; Brady, L.J.; Lemos, J.A. Biology of Oral Streptococci. Microbiol Spectr 2018, 6. [Google Scholar] [CrossRef]
- Uitto, V.J.; Baillie, D.; Wu, Q.; Gendron, R.; Grenier, D.; Putnins, E.E.; Kanervo, A.; Firth, J.D. Fusobacterium nucleatum increases collagenase 3 production and migration of epithelial cells. Infect Immun 2005, 73, 1171–1179. [Google Scholar] [CrossRef]
- Wu, D.-D.; Ngowi, E.E.; Zhai, Y.-K.; Wang, Y.-Z.; Khan, N.H.; Kombo, A.F.; Khattak, S.; Li, T.; Ji, X.-Y. Role of Hydrogen Sulfide in Oral Disease. Oxidative medicine and cellular longevity 2022, 2022, 1886277. [Google Scholar] [CrossRef]
- Attene-Ramos, M.S.; Wagner, E.D.; Plewa, M.J.; Gaskins, H.R. Evidence that hydrogen sulfide is a genotoxic agent. Mol Cancer Res 2006, 4, 9–14. [Google Scholar] [CrossRef]
- Karpiński, T.M.; Szkaradkiewicz, A.K. Characteristic of bacteriocines and their application. Pol J Microbiol 2013, 62, 223–235. [Google Scholar] [CrossRef]
- Faïs, T.; Delmas, J.; Barnich, N.; Bonnet, R.; Dalmasso, G. Colibactin: More Than a New Bacterial Toxin. Toxins (Basel) 2018, 10. [Google Scholar] [CrossRef]
- Nieminen, M.T.; Salaspuro, M. Local Acetaldehyde-An Essential Role in Alcohol-Related Upper Gastrointestinal Tract Carcinogenesis. Cancers (Basel) 2018, 10. [Google Scholar] [CrossRef]
- Choi, C.H.; Spooner, R.; DeGuzman, J.; Koutouzis, T.; Ojcius, D.M.; Yilmaz, Ö. Porphyromonas gingivalis-nucleoside-diphosphate-kinase inhibits ATP-induced reactive-oxygen-species via P2X7 receptor/NADPH-oxidase signalling and contributes to persistence. Cell Microbiol 2013, 15, 961–976. [Google Scholar] [CrossRef]
- Spooner, R.; Yilmaz, O. The role of reactive-oxygen-species in microbial persistence and inflammation. Int J Mol Sci 2011, 12, 334–352. [Google Scholar] [CrossRef]
- Niland, S.; Riscanevo, A.X.; Eble, J.A. Matrix Metalloproteinases Shape the Tumor Microenvironment in Cancer Progression. Int J Mol Sci 2021, 23. [Google Scholar] [CrossRef] [PubMed]
- Byun, J.-K. Tumor lactic acid: a potential target for cancer therapy. Archives of Pharmacal Research 2023, 46, 90–110. [Google Scholar] [CrossRef] [PubMed]
- Lunt, S.J.; Chaudary, N.; Hill, R.P. The tumor microenvironment and metastatic disease. Clin Exp Metastasis 2009, 26, 19–34. [Google Scholar] [CrossRef]
- Pleguezuelos-Manzano, C.; Puschhof, J.; Rosendahl Huber, A.; van Hoeck, A.; Wood, H.M.; Nomburg, J.; Gurjao, C.; Manders, F.; Dalmasso, G.; Stege, P.B.; et al. Mutational signature in colorectal cancer caused by genotoxic pks(+) E. coli. Nature 2020, 580, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Huang, L. Fusobacterium nucleatum carcinogenesis and drug delivery interventions. Advanced Drug Delivery Reviews 2024, 209, 115319. [Google Scholar] [CrossRef]
- Li, Y.; Xing, S.; Chen, F.; Li, Q.; Dou, S.; Huang, Y.; An, J.; Liu, W.; Zhang, G. Intracellular Fusobacterium nucleatum infection attenuates antitumor immunity in esophageal squamous cell carcinoma. Nat Commun 2023, 14, 5788. [Google Scholar] [CrossRef]
- Huhta, H.; Helminen, O.; Lehenkari, P.P.; Saarnio, J.; Karttunen, T.J.; Kauppila, J.H. Toll-like receptors 1, 2, 4 and 6 in esophageal epithelium, Barrett’s esophagus, dysplasia and adenocarcinoma. Oncotarget 2016, 7, 23658–23667. [Google Scholar] [CrossRef]
- Verbeek, R.E.; Siersema, P.D.; Ten Kate, F.J.; Fluiter, K.; Souza, R.F.; Vleggaar, F.P.; Bus, P.; van Baal, J.W. Toll-like receptor 4 activation in Barrett’s esophagus results in a strong increase in COX-2 expression. J Gastroenterol 2014, 49, 1121–1134. [Google Scholar] [CrossRef]
- Mao, S.; Park, Y.; Hasegawa, Y.; Tribble, G.D.; James, C.E.; Handfield, M.; Stavropoulos, M.F.; Yilmaz, O.; Lamont, R.J. Intrinsic apoptotic pathways of gingival epithelial cells modulated by Porphyromonas gingivalis. Cell Microbiol 2007, 9, 1997–2007. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Singh, A.K. Porphyromonas gingivalis in oral squamous cell carcinoma: a review. Microbes and Infection 2022, 24, 104925. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Guo, F.; Yu, Y.; Sun, T.; Ma, D.; Han, J.; Qian, Y.; Kryczek, I.; Sun, D.; Nagarsheth, N.; et al. Fusobacterium nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell 2017, 170, 548–563. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.G.S.; Kakodkar, P.; Nayanar, G. Capacity of Candida species to produce acetaldehyde at various concentrations of alcohol. J Oral Maxillofac Pathol 2022, 26, 161–165. [Google Scholar] [CrossRef]
- Muto, M.; Hitomi, Y.; Ohtsu, A.; Shimada, H.; Kashiwase, Y.; Sasaki, H.; Yoshida, S.; Esumi, H. Acetaldehyde production by non-pathogenic Neisseria in human oral microflora: implications for carcinogenesis in upper aerodigestive tract. Int J Cancer 2000, 88, 342–350. [Google Scholar] [CrossRef]
- Ma, Y.; Yu, Y.; Yin, Y.; Wang, L.; Yang, H.; Luo, S.; Zheng, Q.; Pan, Y.; Zhang, D. Potential role of epithelial-mesenchymal transition induced by periodontal pathogens in oral cancer. J Cell Mol Med 2024, 28, e18064. [Google Scholar] [CrossRef]
- Yilmaz, O.; Jungas, T.; Verbeke, P.; Ojcius, D.M. Activation of the phosphatidylinositol 3-kinase/Akt pathway contributes to survival of primary epithelial cells infected with the periodontal pathogen Porphyromonas gingivalis. Infect Immun 2004, 72, 3743–3751. [Google Scholar] [CrossRef]
- Yao, L.; Jermanus, C.; Barbetta, B.; Choi, C.; Verbeke, P.; Ojcius, D.M.; Yilmaz, O. Porphyromonas gingivalis infection sequesters pro-apoptotic Bad through Akt in primary gingival epithelial cells. Mol Oral Microbiol 2010, 25, 89–101. [Google Scholar] [CrossRef]
- Nakhjiri, S.F.; Park, Y.; Yilmaz, O.; Chung, W.O.; Watanabe, K.; El-Sabaeny, A.; Park, K.; Lamont, R.J. Inhibition of epithelial cell apoptosis by Porphyromonas gingivalis. FEMS Microbiol Lett 2001, 200, 145–149. [Google Scholar] [CrossRef]
- Ye, C.; Liu, X.; Liu, Z.; Pan, C.; Zhang, X.; Zhao, Z.; Sun, H. Fusobacterium nucleatum in tumors: from tumorigenesis to tumor metastasis and tumor resistance. Cancer Biol Ther 2024, 25, 2306676. [Google Scholar] [CrossRef]
- Ergun, P.; Kipcak, S.; Bor, S. Epigenetic Alterations from Barrett’s Esophagus to Esophageal Adenocarcinoma. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Rooks, M.G.; Garrett, W.S. Gut microbiota, metabolites and host immunity. Nature Reviews Immunology 2016, 16, 341–352. [Google Scholar] [CrossRef]
- Liang, G.; Wang, H.; Shi, H.; Zhu, M.; An, J.; Qi, Y.; Du, J.; Li, Y.; Gao, S. Porphyromonas gingivalis Promotes the Proliferation and Migration of Esophageal Squamous Cell Carcinoma through the miR-194/GRHL3/PTEN/Akt Axis. ACS Infect Dis 2020, 6, 871–881. [Google Scholar] [CrossRef]
- Park, H.E.; Kim, J.H.; Cho, N.Y.; Lee, H.S.; Kang, G.H. Intratumoral Fusobacterium nucleatum abundance correlates with macrophage infiltration and CDKN2A methylation in microsatellite-unstable colorectal carcinoma. Virchows Arch 2017, 471, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Peng, Y.; Yu, J.; Chen, T.; Wu, Y.; Shi, L.; Li, Q.; Wu, J.; Fu, X. Invasive Fusobacterium nucleatum activates beta-catenin signaling in colorectal cancer via a TLR4/P-PAK1 cascade. Oncotarget 2017, 8, 31802–31814. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Xing, J.; Jiang, Z.; Zhang, Z.; Zhang, H.; Wang, D.; Tang, D. Effects of Long Non-Coding RNAs Induced by the Gut Microbiome on Regulating the Development of Colorectal Cancer. Cancers (Basel) 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Nikolaieva, N.; Sevcikova, A.; Omelka, R.; Martiniakova, M.; Mego, M.; Ciernikova, S. Gut Microbiota-MicroRNA Interactions in Intestinal Homeostasis and Cancer Development. Microorganisms 2022, 11. [Google Scholar] [CrossRef]
- Olsen, I.; Yilmaz, Ö. Possible role of Porphyromonas gingivalis in orodigestive cancers. J Oral Microbiol 2019, 11, 1563410. [Google Scholar] [CrossRef]
- Hermeking, H. MicroRNAs in the p53 network: micromanagement of tumour suppression. Nat Rev Cancer 2012, 12, 613–626. [Google Scholar] [CrossRef]
- Chela, H.K.; Gangu, K.; Ertugrul, H.; Juboori, A.A.; Daglilar, E.; Tahan, V. The 8th Wonder of the Cancer World: Esophageal Cancer and Inflammation. Diseases 2022, 10. [Google Scholar] [CrossRef]
- Park, C.H.; Lee, S.K. Exploring Esophageal Microbiomes in Esophageal Diseases: A Systematic Review. J Neurogastroenterol Motil 2020, 26, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Yano, Y.; Etemadi, A.; Abnet, C.C. Microbiome and Cancers of the Esophagus: A Review. Microorganisms 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Quinn, P.J. Endotoxins: Lipopolysaccharides of Gram-Negative Bacteria. In Endotoxins: Structure, Function and Recognition, Wang, X., Quinn, P.J., Eds.; Springer Netherlands: Dordrecht, 2010; pp. 3–25. [Google Scholar]
- Li, Q.; von Ehrlich-Treuenstätt, V.; Schardey, J.; Wirth, U.; Zimmermann, P.; Andrassy, J.; Bazhin, A.V.; Werner, J.; Kühn, F. Gut Barrier Dysfunction and Bacterial Lipopolysaccharides in Colorectal Cancer. Journal of Gastrointestinal Surgery 2023, 27, 1466–1472. [Google Scholar] [CrossRef]
- Ranneh, Y.; Ali, F.; Akim, A.M.; Hamid, H.A.; Khazaai, H.; Fadel, A. Crosstalk between reactive oxygen species and pro-inflammatory markers in developing various chronic diseases: a review. Applied Biological Chemistry 2017, 60, 327–338. [Google Scholar] [CrossRef]
- Canzi Almada de Souza, R.; Hermênio Cavalcante Lima, J. Helicobacter pylori and gastroesophageal reflux disease: a review of this intriguing relationship. Diseases of the Esophagus 2009, 22, 256–263. [Google Scholar] [CrossRef]
- Jalanka, J.; Gunn, D.; Singh, G.; Krishnasamy, S.; Lingaya, M.; Crispie, F.; Finnegan, L.; Cotter, P.; James, L.; Nowak, A.; et al. Postinfective bowel dysfunction following <em>Campylobacter enteritis</em> is characterised by reduced microbiota diversity and impaired microbiota recovery. Gut 2023, 72, 451. [Google Scholar] [CrossRef] [PubMed]
- Engevik, M.A.; Danhof, H.A.; Ruan, W.; Engevik, A.C.; Chang-Graham, A.L.; Engevik, K.A.; Shi, Z.; Zhao, Y.; Brand, C.K.; Krystofiak, E.S.; et al. Fusobacterium nucleatum Secretes Outer Membrane Vesicles and Promotes Intestinal Inflammation. mBio 2021, 12. [Google Scholar] [CrossRef]
- Kawar, N.; Park, S.G.; Schwartz, J.L.; Callahan, N.; Obrez, A.; Yang, B.; Chen, Z.; Adami, G.R. Salivary microbiome with gastroesophageal reflux disease and treatment. Scientific Reports 2021, 11, 188. [Google Scholar] [CrossRef]
- Coker, O.O.; Dai, Z.; Nie, Y.; Zhao, G.; Cao, L.; Nakatsu, G.; Wu, W.K.; Wong, S.H.; Chen, Z.; Sung, J.J.Y.; et al. Mucosal microbiome dysbiosis in gastric carcinogenesis. Gut 2018, 67, 1024–1032. [Google Scholar] [CrossRef]
- Lory, S. The Family Leptotrichiaceae. In The Prokaryotes: Firmicutes and Tenericutes, Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer Berlin Heidelberg: Berlin, Heidelberg, 2014; pp. 213–214. [Google Scholar]
- Zeng, R.; Gou, H.; Lau, H.C.H.; Yu, J. Stomach microbiota in gastric cancer development and clinical implications. Gut 2024, gutjnl-2024-332815. [Google Scholar] [CrossRef]
- Rigauts, C.; Aizawa, J.; Taylor, S.L.; Rogers, G.B.; Govaerts, M.; Cos, P.; Ostyn, L.; Sims, S.; Vandeplassche, E.; Sze, M.; et al. R othia mucilaginosa is an anti-inflammatory bacterium in the respiratory tract of patients with chronic lung disease. Eur Respir J 2022, 59. [Google Scholar] [CrossRef] [PubMed]
- Chesdachai, S.; Tai, D.B.G.; Yetmar, Z.A.; Misra, A.; Ough, N.; Abu Saleh, O. The Characteristics of Capnocytophaga Infection: 10 Years of Experience. Open Forum Infect Dis 2021, 8, ofab175. [Google Scholar] [CrossRef]
- Bertona, S.; Monrabal Lezama, M.; Patti, M.G.; Herbella, F.A.M.; Schlottmann, F. Gastroesophageal Reflux Disease in Obese Patients. In Gastroesophageal Reflux Disease: From Pathophysiology to Treatment, Schlottmann, F., Herbella, F.A.M., Patti, M.G., Eds.; Springer Nature Switzerland: Cham, 2023; pp. 117–125. [Google Scholar]
- Lee, K.X.; Quek, K.F.; Ramadas, A. Dietary and Lifestyle Risk Factors of Obesity Among Young Adults: A Scoping Review of Observational Studies. Current Nutrition Reports 2023, 12, 733–743. [Google Scholar] [CrossRef] [PubMed]
- El-Zayat, S.R.; Sibaii, H.; Mannaa, F.A. Toll-like receptors activation, signaling, and targeting: an overview. Bulletin of the National Research Centre 2019, 43, 187. [Google Scholar] [CrossRef]
- Scida, S.; Russo, M.; Miraglia, C.; Leandro, G.; Franzoni, L.; Meschi, T.; De’ Angelis, G.L.; Di Mario, F. Relationship between Helicobacter pylori infection and GERD. Acta Biomed 2018, 89, 40–43. [Google Scholar] [CrossRef]
- Shiga, K.; Tateda, M.; Saijo, S.; Hori, T.; Sato, I.; Tateno, H.; Matsuura, K.; Takasaka, T.; Miyagi, T. Presence of Streptococcus infection in extra-oropharyngeal head and neck squamous cell carcinoma and its implication in carcinogenesis. Oncol Rep 2001, 8, 245–248. [Google Scholar] [CrossRef]
- Feitelson, M.A.; Arzumanyan, A.; Medhat, A.; Spector, I. Short-chain fatty acids in cancer pathogenesis. Cancer and Metastasis Reviews 2023, 42, 677–698. [Google Scholar] [CrossRef]
- Xie, F.J.; Zhang, Y.P.; Zheng, Q.Q.; Jin, H.C.; Wang, F.L.; Chen, M.; Shao, L.; Zou, D.H.; Yu, X.M.; Mao, W.M. Helicobacter pylori infection and esophageal cancer risk: an updated meta-analysis. World J Gastroenterol 2013, 19, 6098–6107. [Google Scholar] [CrossRef]
- Clemente-Suárez, V.J.; Beltrán-Velasco, A.I.; Redondo-Flórez, L.; Martín-Rodríguez, A.; Tornero-Aguilera, J.F. Global Impacts of Western Diet and Its Effects on Metabolism and Health: A Narrative Review. Nutrients 2023, 15. [Google Scholar] [CrossRef]
- Malesza, I.J.; Malesza, M.; Walkowiak, J.; Mussin, N.; Walkowiak, D.; Aringazina, R.; Bartkowiak-Wieczorek, J.; Mądry, E. High-Fat, Western-Style Diet, Systemic Inflammation, and Gut Microbiota: A Narrative Review. Cells 2021, 10. [Google Scholar] [CrossRef]
- San-Millán, I.; Brooks, G.A. Reexamining cancer metabolism: lactate production for carcinogenesis could be the purpose and explanation of the Warburg Effect. Carcinogenesis 2017, 38, 119–133. [Google Scholar] [CrossRef]
- Sah, D.K.; Arjunan, A.; Lee, B.; Jung, Y.D. Reactive Oxygen Species and H. pylori Infection: A Comprehensive Review of Their Roles in Gastric Cancer Development. Antioxidants (Basel) 2023, 12. [Google Scholar] [CrossRef]
- Valenzuela-Valderrama, M.; Cerda-Opazo, P.; Backert, S.; González, M.F.; Carrasco-Véliz, N.; Jorquera-Cordero, C.; Wehinger, S.; Canales, J.; Bravo, D.; Quest, A.F.G. The Helicobacter pylori Urease Virulence Factor Is Required for the Induction of Hypoxia-Induced Factor-1α in Gastric Cells. Cancers 2019, 11, 799. [Google Scholar] [CrossRef] [PubMed]
- McIlvanna, E.; Linden, G.J.; Craig, S.G.; Lundy, F.T.; James, J.A. Fusobacterium nucleatum and oral cancer: a critical review. BMC Cancer 2021, 21, 1212. [Google Scholar] [CrossRef] [PubMed]
- Aral, K.; Milward, M.R.; Gupta, D.; Cooper, P.R. Effects of Porphyromonas gingivalis and Fusobacterium nucleatum on inflammasomes and their regulators in H400 cells. Mol Oral Microbiol 2020, 35, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Shen, X.; Zhou, M.; Tang, B. Periodontal Pathogens Promote Oral Squamous Cell Carcinoma by Regulating ATR and NLRP3 Inflammasome. Frontiers in Oncology 2021, 11. [Google Scholar] [CrossRef]
- Brouwer, S.; Rivera-Hernandez, T.; Curren, B.F.; Harbison-Price, N.; De Oliveira, D.M.P.; Jespersen, M.G.; Davies, M.R.; Walker, M.J. Pathogenesis, epidemiology and control of Group A Streptococcus infection. Nature Reviews Microbiology 2023, 21, 431–447. [Google Scholar] [CrossRef]
- Shaw, M.H.; Kamada, N.; Kim, Y.G.; Núñez, G. Microbiota-induced IL-1β, but not IL-6, is critical for the development of steady-state TH17 cells in the intestine. J Exp Med 2012, 209, 251–258. [Google Scholar] [CrossRef]
- Gelfo, V.; Romaniello, D.; Mazzeschi, M.; Sgarzi, M.; Grilli, G.; Morselli, A.; Manzan, B.; Rihawi, K.; Lauriola, M. Roles of IL-1 in Cancer: From Tumor Progression to Resistance to Targeted Therapies. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
- Shimada, H.; Takeda, A.; Nabeya, Y.; Okazumi, S.I.; Matsubara, H.; Funami, Y.; Hayashi, H.; Gunji, Y.; Kobayashi, S.; Suzuki, T.; et al. Clinical significance of serum vascular endothelial growth factor in esophageal squamous cell carcinoma. Cancer 2001, 92, 663–669. [Google Scholar] [CrossRef]
- Malhotra, R.; Tyson, D.W.; Rosevear, H.M.; Brosius, F.C. Hypoxia-inducible factor-1alpha is a critical mediator of hypoxia induced apoptosis in cardiac H9c2 and kidney epithelial HK-2 cells. BMC Cardiovascular Disorders 2008, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Kong, Z.; Sun, F.; Meng, Q.; Zhou, M.; Yu, J.; Hu, L. Investigating the predictive value of vascular endothelial growth factor in the evaluation of treatment efficacy and prognosis for patients with non-surgical esophageal squamous cell carcinoma. Frontiers in Oncology 2022, 12. [Google Scholar] [CrossRef] [PubMed]
- Waugh, D.J.J.; Wilson, C. The Interleukin-8 Pathway in Cancer. Clinical Cancer Research 2008, 14, 6735–6741. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Ouyang, Y.; Lu, N.; Li, N. The NF-κB Signaling Pathway, the Microbiota, and Gastrointestinal Tumorigenesis: Recent Advances. Frontiers in Immunology 2020, 11. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Zhang, S.; Gong, Z.; Li, X.; Cao, K.; Deng, H.; He, Y.; et al. The role of microenvironment in tumor angiogenesis. Journal of Experimental & Clinical Cancer Research 2020, 39, 204. [Google Scholar] [CrossRef]
- Newman, A.C.; Hughes, C.C.W. Macrophages and angiogenesis: a role for Wnt signaling. Vascular Cell 2012, 4, 13. [Google Scholar] [CrossRef]
- Carmi, Y.; Dotan, S.; Rider, P.; Kaplanov, I.; White, M.R.; Baron, R.; Abutbul, S.; Huszar, M.; Dinarello, C.A.; Apte, R.N.; et al. The role of IL-1β in the early tumor cell-induced angiogenic response. J Immunol 2013, 190, 3500–3509. [Google Scholar] [CrossRef]
- Malkova, A.M.; Gubal, A.R.; Petrova, A.L.; Voronov, E.; Apte, R.N.; Semenov, K.N.; Sharoyko, V.V. Pathogenetic role and clinical significance of interleukin-1β in cancer. Immunology 2023, 168, 203–216. [Google Scholar] [CrossRef]
- El-Omar, E.M. The importance of interleukin 1β in<em>Helicobacter pylori</em> associated disease. Gut 2001, 48, 743. [Google Scholar] [CrossRef]
- Yoshida, S.; Ono, M.; Shono, T.; Izumi, H.; Ishibashi, T.; Suzuki, H.; Kuwano, M. Involvement of interleukin-8, vascular endothelial growth factor, and basic fibroblast growth factor in tumor necrosis factor alpha-dependent angiogenesis. Mol Cell Biol 1997, 17, 4015–4023. [Google Scholar] [CrossRef]
- Naserian, S.; Abdelgawad, M.E.; Afshar Bakshloo, M.; Ha, G.; Arouche, N.; Cohen, J.L.; Salomon, B.L.; Uzan, G. The TNF/TNFR2 signaling pathway is a key regulatory factor in endothelial progenitor cell immunosuppressive effect. Cell Communication and Signaling 2020, 18, 94. [Google Scholar] [CrossRef] [PubMed]
- Basic, A.; Blomqvist, M.; Dahlén, G.; Svensäter, G. The proteins of Fusobacterium spp. involved in hydrogen sulfide production from L-cysteine. BMC Microbiology 2017, 17, 61. [Google Scholar] [CrossRef]
Figure 1.
Role of inflammation, dysbiosis, and microbial interactions with epithelial cells in the development of esophageal cancer (EC). This figure illustrates the intricate interactions between pathogenic bacteria, microbial metabolites, and the host’s immune system, which together drive chronic inflammation, immune dysregulation, and the progression of esophageal cancer. Porphyromonas gingivalis and Fusobacterium nucleatum are central to this process, activating critical signaling pathways including NF-κB, ERK1/2-Ets1, JAK1/AKT/STAT3, and the NOD1/RIPK2/NLRP3 inflammasome. These pathways lead to the production of pro-inflammatory cytokines (IL-1β, IL-6, IL-8, TNF-α), promoting cell proliferation, inhibition of apoptosis, and inducing angiogenesis, thereby fostering a tumor-promoting microenvironment. The figure also highlights how microbial dysbiosis which is marked by an increase in harmful bacteria such as Prevotella, Neisseria, Eikenella, and Aggregatibacter actinomycetemcomitans, coupled with a decrease in beneficial SCFA-producing bacteria. Dysbiosis exacerbates inflammation and compromises the epithelial barrier. It is further exacerbated by factors such as a low-fiber diet, alcohol consumption, and tobacco use, which together create a milieu conducive to cancer development. Moreover, the direct interaction of these bacteria with the esophageal epithelium, through mechanisms such as TLR-4 activation and E-cadherin binding, promotes epithelial-mesenchymal transition, enhances cancer cell survival, and contributes to chemoresistance. The figure encapsulates the multifaceted role of microbiota in driving EC, from microbial-induced inflammation and immune modulation to direct epithelial transformation, presenting potential therapeutic targets for mitigating EC risk.
Figure 1.
Role of inflammation, dysbiosis, and microbial interactions with epithelial cells in the development of esophageal cancer (EC). This figure illustrates the intricate interactions between pathogenic bacteria, microbial metabolites, and the host’s immune system, which together drive chronic inflammation, immune dysregulation, and the progression of esophageal cancer. Porphyromonas gingivalis and Fusobacterium nucleatum are central to this process, activating critical signaling pathways including NF-κB, ERK1/2-Ets1, JAK1/AKT/STAT3, and the NOD1/RIPK2/NLRP3 inflammasome. These pathways lead to the production of pro-inflammatory cytokines (IL-1β, IL-6, IL-8, TNF-α), promoting cell proliferation, inhibition of apoptosis, and inducing angiogenesis, thereby fostering a tumor-promoting microenvironment. The figure also highlights how microbial dysbiosis which is marked by an increase in harmful bacteria such as Prevotella, Neisseria, Eikenella, and Aggregatibacter actinomycetemcomitans, coupled with a decrease in beneficial SCFA-producing bacteria. Dysbiosis exacerbates inflammation and compromises the epithelial barrier. It is further exacerbated by factors such as a low-fiber diet, alcohol consumption, and tobacco use, which together create a milieu conducive to cancer development. Moreover, the direct interaction of these bacteria with the esophageal epithelium, through mechanisms such as TLR-4 activation and E-cadherin binding, promotes epithelial-mesenchymal transition, enhances cancer cell survival, and contributes to chemoresistance. The figure encapsulates the multifaceted role of microbiota in driving EC, from microbial-induced inflammation and immune modulation to direct epithelial transformation, presenting potential therapeutic targets for mitigating EC risk.
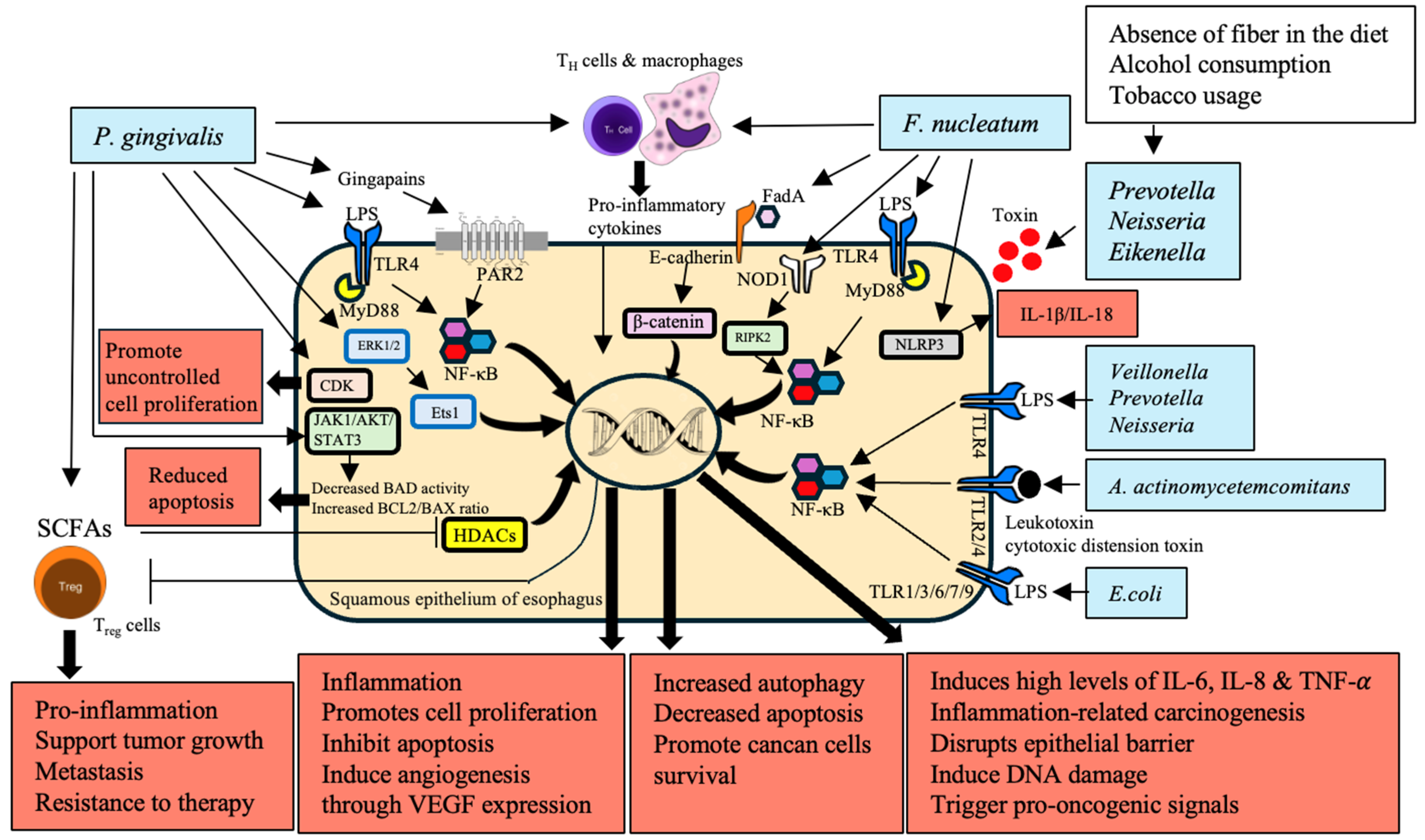
Figure 2.
Production of carcinogenic metabolites by microbiota and their role in esophageal cancer (EC) development. The figure illustrates how different types of microbial metabolites trigger the development and progression of esophageal cancer. Anaerobic bacteria such as Bacteroides, Clostridium, Faecalibacterium, and Ruminococcus produce short-chained fatty acids. SCFAs are typically helpful and functional in immune and intestinal systems. However, its reduction results in a pro-inflammatory environment, leaky intestine, and promotion of carcinogenesis. Escherichia coli generates colibactin, which causes DNA double-strand breaks, thereby affecting genomic instability and cancer. Other types of microorganisms, such as Neisseria, Streptococcus, and Candida, transform alcohol into acetaldehyde, which is a carcinogenic compound and triggers DNA damage and mutagenesis. Porphyromonas gingivalis, Helicobacter pylori, and E. coli produce reactive oxygen species (ROS), while Streptococcus oralis and S. mitis generate reactive nitrogen species (RNS). Both ROS and RNS irreversibly damage DNA and contribute to chronic inflammation, as well as cellular transformation. Moreover, P. gingivalis and F. nucleatum produce matrix metalloproteinases (MMP-9 and MMP-13), which degrade extracellular matrix and facilitate the invasion of cancer cells and metastasis. In addition, P. gingivalis produces H2S, which leads to genomic instability and provides a proper environment for tumor growth. Finally, lactic acid is produced by Lactobacillus, Streptococcus, and related genera. It complicates metabolism by creating an acidic, hypoxic tumor microenvironment, suppresses the immune system, and supports metastasis. Therefore, multiple microbial metabolites underlie the development of EC, thus serving as potential candidates for pharmacological targeting.
Figure 2.
Production of carcinogenic metabolites by microbiota and their role in esophageal cancer (EC) development. The figure illustrates how different types of microbial metabolites trigger the development and progression of esophageal cancer. Anaerobic bacteria such as Bacteroides, Clostridium, Faecalibacterium, and Ruminococcus produce short-chained fatty acids. SCFAs are typically helpful and functional in immune and intestinal systems. However, its reduction results in a pro-inflammatory environment, leaky intestine, and promotion of carcinogenesis. Escherichia coli generates colibactin, which causes DNA double-strand breaks, thereby affecting genomic instability and cancer. Other types of microorganisms, such as Neisseria, Streptococcus, and Candida, transform alcohol into acetaldehyde, which is a carcinogenic compound and triggers DNA damage and mutagenesis. Porphyromonas gingivalis, Helicobacter pylori, and E. coli produce reactive oxygen species (ROS), while Streptococcus oralis and S. mitis generate reactive nitrogen species (RNS). Both ROS and RNS irreversibly damage DNA and contribute to chronic inflammation, as well as cellular transformation. Moreover, P. gingivalis and F. nucleatum produce matrix metalloproteinases (MMP-9 and MMP-13), which degrade extracellular matrix and facilitate the invasion of cancer cells and metastasis. In addition, P. gingivalis produces H2S, which leads to genomic instability and provides a proper environment for tumor growth. Finally, lactic acid is produced by Lactobacillus, Streptococcus, and related genera. It complicates metabolism by creating an acidic, hypoxic tumor microenvironment, suppresses the immune system, and supports metastasis. Therefore, multiple microbial metabolites underlie the development of EC, thus serving as potential candidates for pharmacological targeting.
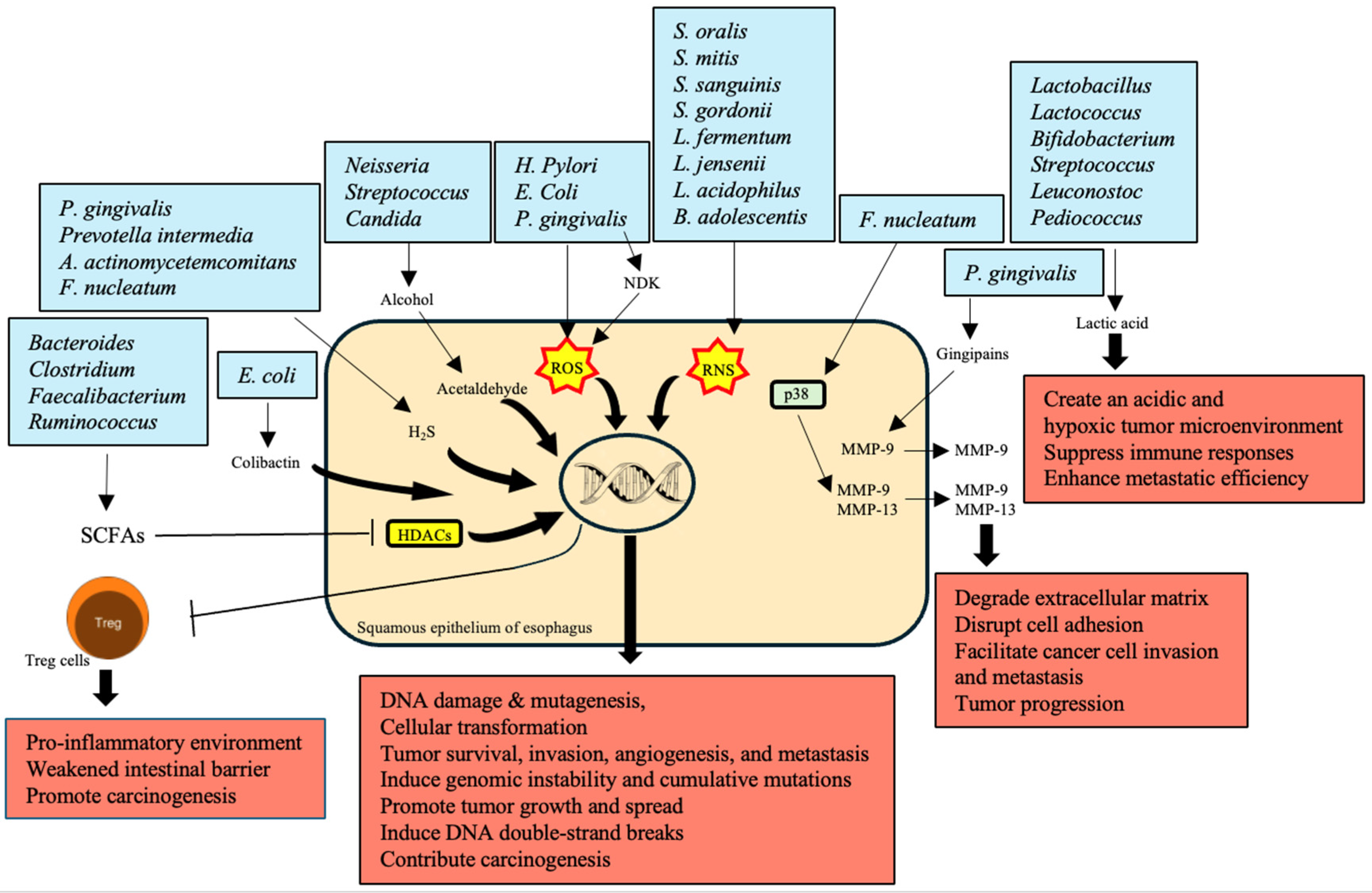
Figure 3.
Epigenetic modifications induced by microbiota in the development of esophageal cancer (EC). This figure illustrates how microbiota-induced epigenetic modifications contribute to the development and progression of EC. Short-chain fatty acids (SCFAs) produced by bacteria such as Porphyromonas gingivalis and others influence histone deacetylases (HDACs), which, in turn, affect regulatory T (Treg) cell function, promoting a pro-inflammatory environment that supports carcinogenesis. P. gingivalis infection also upregulates miR-194 and Akt, while downregulating tumor suppressor genes like grainy head-like transcription factor 3 (GRHL3) and phosphatase and tensin homolog (PTEN), thus enhancing cell proliferation and migration. Additionally, Fusobacterium nucleatum influences the methylation status of the cyclin-dependent kinase inhibitor 2A (CDKN2A) promoter, leading to the silencing of tumor suppressor genes and the activation of oncogenes. The activation of Toll-like receptor-4 (TLR-4) in the context of Barrett’s Esophagus and esophageal adenocarcinoma further drives inflammation and tumorigenesis through cyclooxygenase-2 (COX-2) expression and β-catenin signaling. These interactions collectively promote cancer development by altering gene expression profiles, leading to enhanced cancer cell proliferation and migration. This figure highlights the critical role of microbial-induced epigenetic changes in shaping the tumor microenvironment and facilitating esophageal carcinogenesis.
Figure 3.
Epigenetic modifications induced by microbiota in the development of esophageal cancer (EC). This figure illustrates how microbiota-induced epigenetic modifications contribute to the development and progression of EC. Short-chain fatty acids (SCFAs) produced by bacteria such as Porphyromonas gingivalis and others influence histone deacetylases (HDACs), which, in turn, affect regulatory T (Treg) cell function, promoting a pro-inflammatory environment that supports carcinogenesis. P. gingivalis infection also upregulates miR-194 and Akt, while downregulating tumor suppressor genes like grainy head-like transcription factor 3 (GRHL3) and phosphatase and tensin homolog (PTEN), thus enhancing cell proliferation and migration. Additionally, Fusobacterium nucleatum influences the methylation status of the cyclin-dependent kinase inhibitor 2A (CDKN2A) promoter, leading to the silencing of tumor suppressor genes and the activation of oncogenes. The activation of Toll-like receptor-4 (TLR-4) in the context of Barrett’s Esophagus and esophageal adenocarcinoma further drives inflammation and tumorigenesis through cyclooxygenase-2 (COX-2) expression and β-catenin signaling. These interactions collectively promote cancer development by altering gene expression profiles, leading to enhanced cancer cell proliferation and migration. This figure highlights the critical role of microbial-induced epigenetic changes in shaping the tumor microenvironment and facilitating esophageal carcinogenesis.
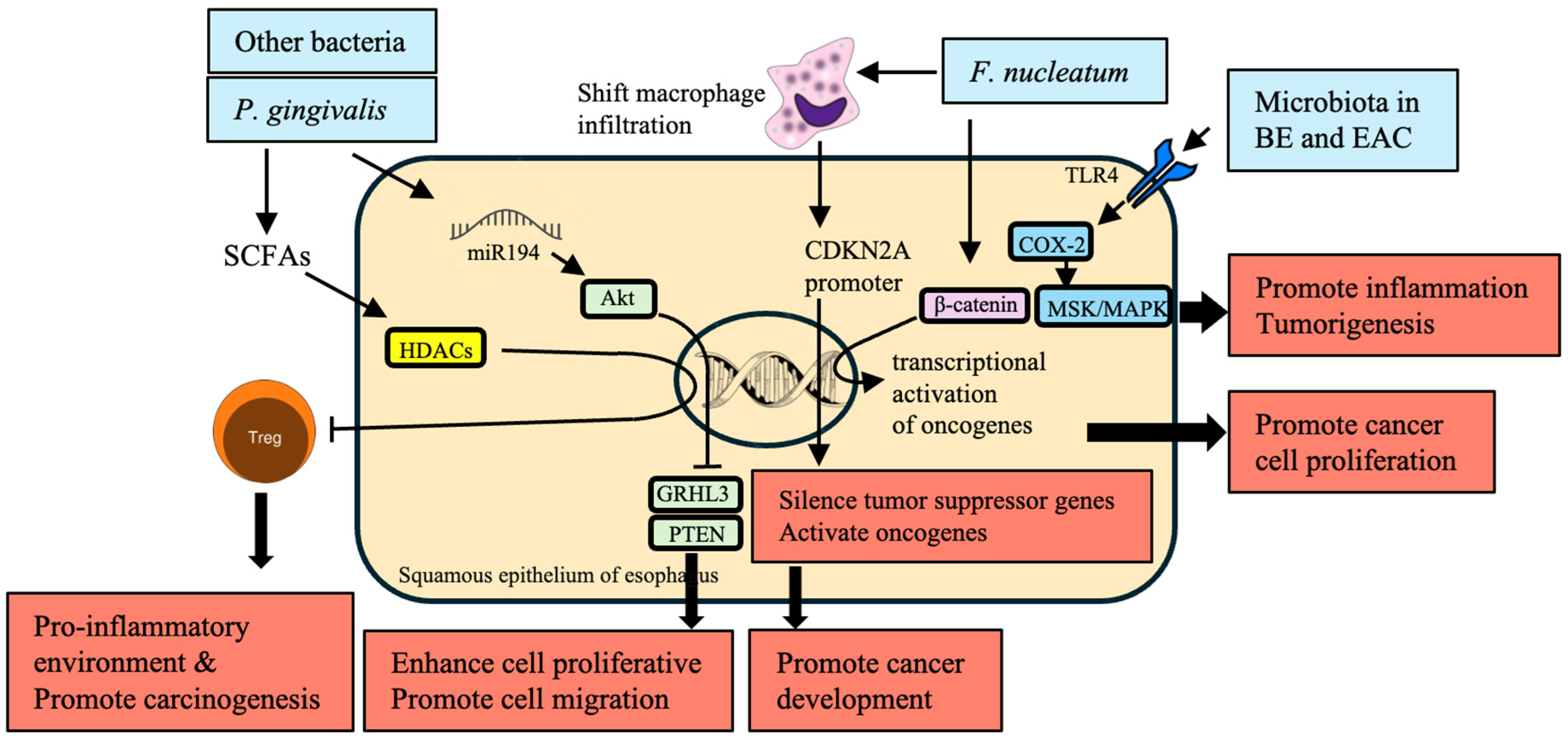
Figure 4.
Microbiota-driven angiogenesis in the development of esophageal cancer (EC). This figure illustrates the role of specific bacteria in promoting angiogenesis, a key process mediating the development and growth of EC. This process is mediated by the actions of Porphyromonas gingivalis, Fusobacterium nucleatum, and Helicobacter pylori, which include ROS production as well as stabilization of hypoxia-inducible factor 1-alpha (HIF-1α) that subsequently induces pro-angiogenic factors such as VEGF. The signaling pathways specified by these bacteria, in turn, result in the release of pro-inflammatory cytokines (IL-8, IL-1β, and TNF-α) that promote an even more tumor conducive environment. In the tumor microenvironment (TME), macrophages and dendritic cells promote new blood vessel formation through VEGF and the release of matrix metalloproteinases (MMPs) that break down the extracellular matrix. This angiogenic process is critical for tumor invasiveness, progression, and metastasis. P. gingivalis and F. nucleatum are highlighted to have a characteristic of enhancing the exacerbation response and accelerating angiogenesis by elevating TNF-α levels along with other angiogenic factors, Additionally, it illustrates the role of H2S released from oral bacteria in establishing a pro-angiogenic microenvironment that results in EC progression and dissemination. This figure highlights the intricate interplay between microbial-induced inflammation and angiogenesis that are instrumental to EC management as potential therapeutic targets.
Figure 4.
Microbiota-driven angiogenesis in the development of esophageal cancer (EC). This figure illustrates the role of specific bacteria in promoting angiogenesis, a key process mediating the development and growth of EC. This process is mediated by the actions of Porphyromonas gingivalis, Fusobacterium nucleatum, and Helicobacter pylori, which include ROS production as well as stabilization of hypoxia-inducible factor 1-alpha (HIF-1α) that subsequently induces pro-angiogenic factors such as VEGF. The signaling pathways specified by these bacteria, in turn, result in the release of pro-inflammatory cytokines (IL-8, IL-1β, and TNF-α) that promote an even more tumor conducive environment. In the tumor microenvironment (TME), macrophages and dendritic cells promote new blood vessel formation through VEGF and the release of matrix metalloproteinases (MMPs) that break down the extracellular matrix. This angiogenic process is critical for tumor invasiveness, progression, and metastasis. P. gingivalis and F. nucleatum are highlighted to have a characteristic of enhancing the exacerbation response and accelerating angiogenesis by elevating TNF-α levels along with other angiogenic factors, Additionally, it illustrates the role of H2S released from oral bacteria in establishing a pro-angiogenic microenvironment that results in EC progression and dissemination. This figure highlights the intricate interplay between microbial-induced inflammation and angiogenesis that are instrumental to EC management as potential therapeutic targets.

Table 8.
Microbiota and metabolic changes in EC.
| Bacteria | Mechanism | Impact on EC | References |
|---|---|---|---|
| Bacteroides, Clostridium, Faecalibacterium, Ruminococcus | Produce SCFAs, modulate inflammation | Maintain gut health; reduced SCFA production leads to a pro-inflammatory environment and cancer risk | [95,98] |
| H. pylori | Induces chronic gastritis, alters gastric acid secretion | Promotes GERD, BE, and EAC | [162] |
| Campylobacter | Induces inflammatory responses | Promotes chronic inflammation and progression to BEand adenocarcinoma | [40] |
| Lactobacillus, Streptococcus, Bifidobacterium, Leuconostoc | Produce lactic acid, create low pH hypoxic environment, induce Warburg effect | Immunosuppression, enhanced tumor metastasis, support cancer cell survival and proliferation | [85] |
| F. nucleatum | Produces LPS, activates β-catenin signaling, enhances oncogene expression (C-myc, cyclin D1) | Promotes cancer cell proliferation, chronic inflammation, and carcinogenesis | [136] |
| P. gingivalis | Modulates ATP/P2X7 signaling, affects ROS and antioxidant responses | Contributes to cancer development through ROS-mediated DNA damage and inflammatory responses | [112] |
| Streptococci, Candida yeasts | Metabolize alcohol to acetaldehyde via ADH activity | Causes DNA damage, increases carcinogenesis risk | [101] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



