
Submitted:
10 August 2024
Posted:
13 August 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
The high transmissibility, rapid evolution, and immune escape of SARS-CoV-2 variants can influence the course of infection and, in turn, morbidity and mortality in COVID-19, posing a challenge in controlling transmission rates and contributing to the emergence and spread of new variants. The factors that shape viral genetic variation are consequently essential for understanding the evolution and transmission of SARS-CoV-2, especially in vaccinated individuals where immune response plays a role in the progression and spread of this disease. This study assessed. In this context, we evaluated the impact of immunity induced by the CoronaVac vaccine (Butantan/Sinovac) on intra-host genetic diversity, analyzing 118 whole-genome sequences of SARS-CoV-2 from unvaccinated and vaccinated patients infected with the Gamma variant. Vaccination with CoronaVac favors negative selection at the intra-host level in different genomic regions and prevent greater genetic diversity of SARS-CoV-2, which may help reduce the emergence of new mutations, reinforcing the importance of vaccination in reducing virus transmission.
Keywords:
COVID-19
; vaccination
; Gamma lineage
; negative selection
; breakthrough infections
1. Introduction
Variants of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) emerged due to rapid virus evolution as well as high transmission rates, especially in Variants of Concern (VOCs) such as Alpha (B.1.1.7) [1], Beta (B.1.351) [2], Gamma [3], Delta (B.1.617.2) [4], and Omicron (B.1.1.529) [5]. These variants are characterized by specific mutations along the ~30 kilobase (kb) genome composed of 14 open reading frames (ORFs): ORF1a and ORF1b, which produce non-structural proteins (Nsps) Nsp1 to Nsp16; four structural proteins (spike [S], membrane [M], envelope [E], and nucleocapsid [N]); and some accessory proteins, including ORF3a, ORF6, ORF7a, ORF7b, ORF8, ORF9, and ORF10 [6].
The rapid evolution of RNA viruses promotes high mutation rates that can neutralize the host's immune response, which may be subject to inter- and intra-host selection pressures [7,8]. Furthermore, replication errors also combine with genome mutations favored by innate defense mechanisms to boost the viral transmissibility, infectivity, disease severity, ability to evade the human immune system, and the evolutionary rate of SARS-CoV-2 [6,9,10,11].
The COVID-19 pandemic spurred rapid vaccine development, since herd immunity can prevent severe disease in different age groups [12,13]. In Brazil, CoronaVac (Butantan Institute/Sinovac Biotech), an inactivated whole-virus vaccine, was first approved for emergency use in January 2021 [14], and its wide distribution helped decrease severe cases and deaths [14]. However, the rapid occurrence of mutations and the emergence of new VOCs after the distribution of this and other vaccines distributed in Brazil made controlling viral transmission a challenge, due to escape of natural or vaccine-acquired immunity [15,16].
Although the diversity of SARS-CoV-2 in viral populations has been demonstrated [8,17,18,19], little information is available on how specific vaccine types may exert selective pressures and shape virus evolution [20]. For this reason, to better understand the intra-host diversity of SARS-CoV-2 after vaccination in Brazil in early 2021, we investigated 118 whole genomes from unvaccinated and vaccinated patients who were diagnosed with COVID-19 from April to July 2021, in São José do Rio Preto and surrounding cities in the state of São Paulo, Brazil. Patients were divided into two equal groups: 1) unvaccinated individuals (infected with SARS-CoV-2 who did not receive any dose of vaccine) and 2) vaccinated individuals (infected with SARS-CoV-2 14 or more days after having received their second dose of the CoronaVac vaccine - breakthrough infections). We found that the CoronaVac vaccine favored negative selection in different regions of the SARS-CoV-2 genome, thus reducing genetic diversity at the intra-host level. This outcome reinforces the importance of vaccination as a tool to prevent the emergence of new variants that may enhance virus fitness and affect the progression of COVID-19 worldwide.
2. Materials and Methods
2.1. Samples
Nasopharyngeal swab samples were obtained from residents of São José do Rio Preto and surrounding cities who were positively diagnosed for COVID-19 between April 14 and July 15, 2021. Based on the immunization criteria, 118 samples were selected and divided into two groups: (1) 59 samples from unvaccinated patients with ages ranging from two to 65 years (average age: 31 years), and (2) 59 samples from fully immunized patients (individuals who received two doses of CoronaVac (Butantan Institute/Sinovac Biotech) who were diagnosed with COVID-19 more than 14 days after having completed the two-dose vaccination schedule), with ages ranging from 24 to 91 years (average: 60 years). Our sample groups were selected based on the vaccination schedule in the state of São Paulo, which prior to the time of sampling included: men and women over 37 years of age, pregnant women, indigenous people, Quilombolas (Afro-Brazilian descendants of escaped slaves living in settlement communities), immunocompromised individuals, patients with comorbidities, health professionals, public security and prison administration workers, public transport staff, and education professionals.
2.2. Ethics
This study was approved by the institutional review board of the Faculdade de Medicina de São José do Rio Preto (protocol: 31588920.0.0000.5415, approved on November 29, 2021). Informed consent was not required since all samples were collected for routine diagnosis and the data were analyzed anonymously, ensuring total confidentiality for all participants.
2.3. Molecular Investigation
Total RNA was extracted from 140 µL of nasopharyngeal swab samples using a QIAamp Viral RNA Mini Kit (QIAGEN, Hilden, Germany), following the manufacturer's instructions. SARS-CoV-2 RNA was investigated with a one-step real-time polymerase chain reaction (RT-qPCR) using primers and probes targeting the envelope (E), the nucleocapsid (N), and human RNAse P, using the GeneFinder COVID-19 Plus RealAmp Kit (OSANG Healthcare, KOR) [21]; RT-qPCR was conducted in a QuantStudio 3 Real-Time PCR System (Thermo Fisher Scientific, USA) according to the manufacturer's instructions. The results were interpreted based on cycle quantification value (Cq), in which samples presenting Cq less or equal to 40 were considered positive, as recommended in the kit instructions. Positive and negative controls included in the GeneFinder COVID-19 Plus RealAmp Kit (non-infectious DNA plasmids coding for the SARS-CoV-2 E gene and N gene) were used in the assay.
2.4. Whole-Genome Sequencing
Whole-genome sequencing was performed through cDNA synthesis, whole-genome amplification, and library preparation following the instructions provided with the Illumina CovidSeq Test (Illumina, USA). The quality and size of the libraries were verified using the Agilent 4150 TapeStation system (Agilent, Santa Clara, CA, USA). The libraries were pooled in equimolar concentrations, and sequencing was conducted using an Illumina MiSeq system with a MiSeq Reagent Kit v2 (2 x 150 cycles) (Illumina, San Diego, CA, USA).
2.5. Genome Assembling and Variant Analyses
The quality of raw reads was checked using the FastQC v.0.11.4 analysis tool [22]. Cutadapt v.4.6 [23] was used to filter out low-quality reads, low-quality bases, reads with a minimal length of 75 base pairs (bp), and for primer removal. The cleaned paired-end reads were mapped against the Wuhan-Hu-1 reference genome (NC_045512.2) using BWAmem 0.7.17 software [24]. Post-processing steps, PCR duplicate removal, and generation of consensus sequences were done using SAMtools v1.10 [24,25] and iVar [26]. The genomes were subjected to the Pangolin COVID-19 Lineage Assigner Tool version v.4.0.5 to confirm the variant classification [27].
After lineage identification, only sequences classified as Gamma lineage were selected for the subsequent analyses to avoid mutation bias from different SARS-CoV-2 variants. Intra-host single nucleotide variant (iSNV) analysis was carried out using LoFreq v.2.1.5 [28]. We identified iSNVs with the following criteria: minimum coverage of 100 mapped reads for each genome position, base quality >30, and minor alternative allele frequency (AAF) ≥5%. The biological effects of the identified iSNVs were annotated using SnpEff v.2.0.5, with default settings [29]. Plots to visualize the AAFs for all samples were generated in R v.3.6.1 software [30], employing the ggplot2 package [31].
2.6. Evolutionary Analyses
The inference of selective pressures on a particular region/gene can be manifested as diversifying or purifying selection. We used a combination of two evolutionary analyses to enhance detection of relevant sites in the SARS-CoV-2 genome in unvaccinated and vaccinated individuals. Fixed Effects Likelihood (FEL) was used to identify sites experiencing pervasive diversifying or purifying selection [32], and the Mixed Effects Model of Evolution (MEME) was used to detect sites undergoing both pervasive and episodic diversifying selection. Both methods were implemented in HyPhy v. 2.5.32 software [33] and used a Maximum Likelihood to infer non-synonymous (dN) and synonymous (dS) substitution rates on a per-site basis for given coding alignment and the corresponding phylogenetic tree [33].
2.7. Statistics
The collected data were entered into an Excel spreadsheet (Microsoft, Redmond, WA, USA) and imported into R [30]. The chi-square test was used to determine whether the expected frequency in the groups was met. A significance level of 0.05 (5%) was adopted for all statistical tests.
3. Results
The distribution of iSNVs along the SARS-CoV-2 genome was identified and normalized by gene size (Figure 1A, Tables S1 and S2). For both groups, the highest percentage of iSNVs was identified in the ORF6 gene (2.15% in the unvaccinated group and 2.69% in the vaccinated group), followed by the N gene (2.06% in the unvaccinated and 2.38% in the vaccinated group). No significant difference was identified between the groups (p>0.05). In general, the majority of gene coding structural proteins (E, M and N) showed a higher percentage of iSNVs in the vaccinated group than in the unvaccinated group, except for the S gene, that exhibited similar percentages of iSNVs in both groups. Likewise, ORF1ab, ORF3a, ORF7a, and ORF10 showed a higher percentage of iSNVs in the vaccinated group than in the unvaccinated group. The opposite was detected in ORF8. No statistical difference was identified in the non-structural proteins (p>0.05).

Table 1.
Number of non-synonymous (NS) and synonymous (S) iSNVs found in the SARS-CoV-2 genome from patients vaccinated and unvaccinated with CoronaVac. Percentage values represent the number of iSNVs normalized by gene size.
Table 1.
Number of non-synonymous (NS) and synonymous (S) iSNVs found in the SARS-CoV-2 genome from patients vaccinated and unvaccinated with CoronaVac. Percentage values represent the number of iSNVs normalized by gene size.
| Unvaccinated | Vaccinated | |||
| Region | NS (%) | S (%) | NS (%) | S (%) |
| ORF1ab | 79 (58.5%) | 74 (71.8%) | 92 (54.8%) | 96 (73.3%) |
| S | 20 (14.8%) | 12 (11.7%) | 27 (16.1%) | 6 (4.6%) |
| ORF3a | 9 (6.7%) | 0 (0.0%) | 13 (7.7%) | 3 (2.3%) |
| E | 0 (0.0%) | 2 (1.9%) | 0 (0.0%) | 3 (2.3%) |
| M | 0 (0.0%) | 2 (1.9%) | 3 (1.8%) | 7 (5.3%) |
| ORF6 | 3 (2.2%) | 1 (1.0%) | 3 (1.8%) | 2 (1.5%) |
| ORF7a | 2 (1.5%) | 2 (1.9%) | 4 (2.4%) | 3 (2.3%) |
| ORF7b | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) |
| ORF8 | 6 (4.4%) | 0 (0.0%) | 5 (3.0%) | 1 (0.8%) |
| N | 16 (11.9%) | 10 (9.8%) | 20 (11.9%) | 10 (7.6%) |
| ORF10 | 0 (0.0%) | 0 (0.0%) | 1 (0.5%) | 0 (0.0%) |
| TOTAL | 135 (100%) | 103 (100%) | 168 (100%) | 131 (100%) |
Next, we investigated whether the number of non-synonymous and synonymous iSNVs can be affected by the allele frequency, classifying all the detected iSNVs as major (iSNVs showing more than 50% of mapping reads to an alternative allele) or minor variants (iSNVs displaying frequency from 5–49% of mapping reads to an alternative allele). In the unvaccinated group, we found that 204/243 (83.9%) iSNVs were considered major variants, with 112/204 (54.9%) classified as non-synonymous and 92/204 (45.1%) synonymous. In this same group, 39/243 (16.1%) iSNVs were classified as minor (26/39, 66.7% non-synonymous and 13/39, 33.3% synonymous). In the vaccinated group, 269/309 (87.1%) iSNVs were considered major (151/269, 56.1% non-synonymous and 118/269, 43.9% synonymous) and 40/309 (12.9%) minor (26/40, 65% non-synonymous and 14/40, 35% synonymous) (Figure 1B, Tables S3 and S4).
We also analyzed the number of shared and exclusive iSNVs found in SARS-CoV-2 genomes from the vaccinated and unvaccinated groups (Tables S5 and S6). A total of 84 shared iSNVs were identified, 70 of which (83.3%) did not correspond to Gamma lineage-defining mutations. Most of the shared iSNVs were distributed through ORF1ab (44/84, 52.39%) and S (16/84, 19.05%) (Table S5). Moreover, it is worth noting that some Gamma lineage-defining mutations were lost in both groups, notably the amino acid substitutions S:Glu484Lys (lost in 20 sequences from both the unvaccinated and vaccinated groups), S:Asn501Tyr (absent in 19 and 20 sequences in the unvaccinated and vaccinated groups, respectively), S:His655Tyr (lost in 27 sequences in both groups) and ORF3a:Gly174Cys, which was lost in all the genome sequences analyzed (Table S5). We found 154 iSNVs that were exclusive to the unvaccinated group and 215 iSNVs exclusive to the vaccinated group; most of these were classified as non-synonymous mutations (Table S6). The number of shared or exclusive iSNVs identified in a single patient sample was also analyzed; we found a higher prevalence of iSNVs in only one sequence in the vaccinated group (n=221/299, 73.9%) compared with the unvaccinated group (n=161/238, 67.6%) (Table S1 and S2).
Analysis of the allele composition of each site showed that the most prevalent substitution was C>T in both groups (unvaccinated: n=118/238, 49.6%; vaccinated: n=141/299, 47.2%), for structural (24/118, 20.3% in the unvaccinated and 32/141, 22.7% in the vaccinated group) and non-structural proteins (94/118, 79.7% in the unvaccinated and 109/141, 77.3% in the vaccinated group) (Table S1, S2). However, by normalizing the number of nucleotide substitution by gene size we found a higher frequency of this transition in ORF6 than in other genomic regions in the unvaccinated group, while a higher density of C>T was displayed in ORF3a in the vaccinated group. After ORF6, the S gene presented the second-highest number of C>T substitution. The second most common substitution observed was G>T and G>A in sequences from unvaccinated (n=35/118, 29.7%, n=28/118, 23.7%, respectively) and vaccinated (n=44/141, 31.2%, n=27/141, 19.1%, respectively) patients.
Interestingly, for the non-synonymous mutations we detected two genomic sites with major differences in allele frequency within both groups. The non-synonymous iSNV in the S region at position 21,974 corresponds to a transversion (G>T) that represents an amino acid substitution (Asp138Tyr) observed in 23 unvaccinated and 39 vaccinated patients. This mutation displayed an allele frequency ranging from 19% to 100% and from 52% to 100% in SARS-CoV-2 sequences from the unvaccinated and vaccinated groups, respectively. Similarly, we identified a mutation at the 22,812 genomic position which corresponds to a transversion (A>C); this nucleotide substitution represents an amino acid change (Lys417Thr) found in 43 unvaccinated and 56 vaccinated patients, with an allele frequency ranging from 14% to 100% and from 12% to 100% in the unvaccinated and vaccinated groups, respectively (Figure 1B).
Finally, to better understand the selective pressures that shape intra-host evolution of SARS-CoV-2 in unvaccinated and vaccinated individuals, we used a combination of two tests to detect selection signatures across the SARS-CoV-2 genome. Our analyses identified 10 sites, located in five proteins, which are ORF1ab, S, M, ORF6and ORF7a, under negative selection, and 1 site located in ORF1ab under positive selection in unvaccinated patients (Table 2). Meanwhile, in vaccinated individuals we identified 26 sites under negative selection distributed across seven proteins (ORF1ab, ORF3a, E, ORF6, ORF7a, ORF8 and N), and three sites under positive selection located in ORF1ab and N (Table 2).
When we normalized the number of sites under selection by gene size, we noticed that the envelope protein showed the highest percentage (2.63%) of sites under negative selection in the vaccinated group compared to other genomic regions and compared to the unvaccinated group. The second protein with the most sites under negative selection was ORF6 (1.61%), for both groups (Figure 2). For sites under positive selection, N and ORF1ab were the only proteins found in the unvaccinated as well as vaccinated groups.
4. Discussion
Vaccines were the primary measure to reduce transmission of SARS-CoV-2 as well as severe cases of COVID-19 and resulting deaths [34], even though some studies have suggested that vaccination against this virus could increase intra-host diversity through selective pressures for vaccine escape mutations [35,36]. Our study of unvaccinated and vaccinated individuals infected with Gamma lineage SARS-CoV-2 demonstrated the opposite result. In fact, we found similar average numbers of iSNVs in samples from vaccinated and unvaccinated patients, corroborating reports in previous studies [20,37] and confirming that vaccination with CoronaVac did not increase intra-host genetic variation in patients with breakthrough infections. It is important to note that this result may not be the same for all SARS-CoV-2 lineages or different vaccines; for example, in 2023 Gu et al. reported that the incidence of iSNVs in patients infected with the Delta lineage who had received two doses of Comirnaty (BNT162b2) vaccine was significantly higher than in unvaccinated patients [20].
As it is important to evaluate the entire context of iSNVs (such as their allele frequency and impact on changing protein or gene function) to better understand the factors underlying virus evolution, we evaluated the number of iSNVs detected in only one sample in both groups. Interestingly, we found that although a greater number of iSNVs was distributed throughout the SARS-CoV-2 genome of the vaccinated group than in unvaccinated patients, most of these iSNVs (73.9%) ranged from 5% to 49% of allele frequency, meaning that these mutations are sporadic and may not be fixed in the viral population over the long term. Similarly, Gu et al. showed that over 70% of the iSNV sites identified in their samples were uniquely observed in a single patient [20]. These findings reinforce that minor iSNVs do not provide very relevant information for understanding the diversity of SARS-CoV-2 in vaccinated and unvaccinated individuals. In this way, the results of this study demonstrate that vaccination with the CoronaVac vaccine does not enhance the mutation rate or change the mutation profile of SARS-COV-2 Gamma lineage variants.
Additionally, most of the iSNVs we detected showed allele frequency >50%. Among these, we observed a large number of Gamma-defining mutations in both groups [3]. Notable among these were two non-synonymous mutations, S:D138Y and S:K417T, since they presented highly variable allele frequency among the samples from both groups. These two important locations are amino acid substitutions that can affect binding by monoclonal and polyclonal antibodies, influencing host cell entry and, in turn, transmissibility [3]. Even though we found iSNVs displaying different allele frequencies throughout the entire genome of SARS-CoV-2 from vaccinated and unvaccinated individuals, a wide difference in allele frequency was clearly identified in the S gene, reinforcing its mutational potential, which has already been mentioned in other studies [34,38,39].
Further analyses of iSNVs determined that the unvaccinated and vaccinated groups displayed similar percentages of non-synonymous and synonymous mutation, and we found no association between mutation class and vaccination status. These results are corroborated by previous studies showing that non-synonymous mutations were the most frequent type of alteration in SARS-CoV-2 samples from around the world [40]. Even so, synonymous mutations were detected in over 40% of all nucleotide substitutions and still require careful examination, since they can affect codon usage, maintenance of secondary RNA structure, and long-term translation efficiency [41]. Furthermore, we did not identify any significant genetic variation in any protein (including the spike); this ran counter to other studies, which showed that variations in the amino acid sequences of this protein can influence interaction with the host receptor, pathogenesis, viral replication, infectivity, and transmissibility [38,42]. We also analyzed the number of iSNVs normalized by protein size, and found no significant differences in the genetic diversity of structural and non-structural proteins in unvaccinated and vaccinated patients, suggesting that CoronaVac does not favor intra-host diversity of SARS-CoV-2 in any particular genomic hotspot, which was previously verified by other studies using different vaccine methodologies [20].
Additionally, we showed that C > T substitution was the most frequent SNP detected. This is in line with previous findings demonstrating that the C > T transition was responsible for 55.1% of all SARS-CoV-2 mutations identified in 2020 and the G > T transversion G>T (found in this study as the second most common in the S gene) was the most common nucleotide substitution in the SARS-CoV-2 genome worldwide [37,41]. The C > T mutational event has been implicated as important for controlling virus replication since the excessive occurrence of this transition is linked to a host APOBEC-like (apolipoprotein B mRNA editing) process that plays a role in antiviral defense against retroviruses and may drive several mutational hot spots in the SARS-CoV-2 genome without providing an adaptive advantage to the virus but still affecting its rate of evolution [41,43].
To better understand how vaccination with CoronaVac may influence genetic diversity in the SARS-CoV-2 genomes, we analyzed the selective pressures at genomic sites. In general, we identified a greater occurrence of negative selection throughout the SARS-CoV-2 genome, especially in the vaccinated group, showing that CoronaVac is able to modulate evolution of SARS-CoV-2 over a short period of time. Similar results have already been found for other RNA viruses [44], such as influenza [45], dengue [46], Chikungunya [47], and SARS-CoV-2 [17,45]. Here, however, we demonstrated that negative selection was favored in vaccinated individuals when this vaccine was the most widely distributed in Brazil in 2021. In fact, CoronaVac has been implicated in the production of T cells specific to several SARS-CoV-2 antigens [48], which may confer an advantage in clearing virus infection [49]. In this way, CoronaVac controls transmission and pathogenesis through humoral and cellular immune responses against different virus proteins, together with negative selection pressure against low-frequency variants on most viral proteins, suggesting its role in reducing virus diversity.
Although our findings clearly demonstrate CoronaVac’s influence in preventing intra-host diversity of SARS-CoV-2, it is important to note that this study has limitations. In an attempt to minimize potential bias caused by mutations of different variants of SARS-CoV-2, we did not compare vaccinated and unvaccinated patients presenting breakthrough infections with other lineages. Similarly, we were unable to assess whether the selective pressures verified in individuals who received CoronaVac would be the same if other vaccine technologies were used, because during the sampling period the Gamma lineage was the dominant circulating VOC (reaching more than 90%) [14]. Furthermore, CoronaVac was the first vaccine to be licensed and widely used in the country during this same period. Since that time, several lineages have been introduced [50] and a number of other licensed SARS-CoV-2 vaccines have been administered, complicating this kind of study. Moreover, it is important to emphasize that because we opted to analyze sequences from patients who did not receive any vaccine against SARS-CoV-2, our sampling time was limited to three months, which is a short period for observing the effects of selective pressures on the virus genome. Despite the limited sampling period, our results indicate that vaccination may prevent intra-host virus diversity, reinforcing that the evolutionary process is ongoing and must be continuously monitored.
Although SARS-CoV-2 vaccine efficacy decreases over time [51], especially against rapidly evolving viruses, and despite the demonstrated ability of SARS-CoV-2 lineages to escape neutralizing antibodies [52,53], our findings highlight that two doses of CoronaVac vaccine favor negative selection in structural and non-structural genes of SARS-CoV-2 obtained from patients infected with the Gamma lineage. This study suggests that vaccination is important to reduce the emergence of new variants at the intra-host level, preventing SARS-CoV-2 genetic diversity and the emergence of new and concerning mutations that may confer higher adaptative value to SARS-CoV-2 variants.
Supplementary Materials
All data used to perform the analyses and graphs are available in Table 1, Table 2, and Supplementary Tables S1-S7. All SARS-CoV-2 genomes generated and analyzed in this study are available in the GenBank database (https://www.ncbi.nlm.nih.gov/genbank/), and their respective access numbers are provided in Supplementary Table S7.
Author Contributions
Conceptualization: MLN, BdCM, CAB, LS. Methodology: BdCM, CAB, LS, AN. Investigation: BdCM, CAB, LS, AN. Visualization: BdCM, CAB, LS. Funding acquisition: NV, MLN. Project administration: NV, MLN. Supervision: NV, MLN. Writing original draft: BdCM, CAB, NV, LS. Writing review & editing: BdCM, CAB, LS, NV, MLN.
Funding
This study was funded in part by the National Institute of Allergy and Infectious Diseases (NIAID) through the Centers for Research in Emerging Infectious Diseases (CREID) “The Coordinating Research on Emerging Arboviral Threats Encompassing the Neotropics (CREATE- NEO)” grant U01 AI151807 (to NV); by INCT Viral Genomic Surveillance and One Health grant 405786/2022-0, and by the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) via grants 2022/03645-1 (MLN), 2019/21711-9 (BdCM), 2023/14670-0 (CAB), The Rede Corona-Ômica BR MCTI/FINEP, which is part of Rede Vírus/MCTI (FINEP 01.20.0029.000462/20, CNPq 404096/2020-4). The funders had no role in the study design, collection, analyses, interpretation of data, writing of the manuscript, or the decision to publish the results.
Institutional Review Board Statement
The study was approved by the institutional review board (Ethics Committee of the Faculdade de Medicina de São José do Rio Preto, FAMERP; protocol number: CAE 31588920.0.0000.5415, November 29, 2021). Informed consent was not required, given that all data were analyzed anonymously, maintaining total confidentiality of each participant.
Informed Consent Statement
The consent terms were waived by the Institutional Review Board.
Data Availability Statement
All data generated or analyzed during this study are available in the Mendeley Data repository, DOI: 10.17632/rcxddvxv3c.2.
Acknowledgments
We wish to thank the Hospital de Base de São José do Rio Preto for support provided during sample collection and the Laboratório Multiusuário (LMU) at Faculdade de Medicina de São José do Rio Preto for allowing us to use the equipment there.
Conflicts of Interest
The authors declare no competing interests.
References
- Volz, E.; Mishra, S.; Chand, M.; Barrett, J.C.; Johnson, R.; Geidelberg, L.; Hinsley, W.R.; Laydon, D.J.; Dabrera, G.; O’Toole, Á.; et al. Assessing Transmissibility of SARS-CoV-2 Lineage B.1.1.7 in England. Nature 2021, 593, 266–269. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 Variant of Concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef]
- Faria, N.R.; Mellan, T.A.; Whittaker, C.; Claro, I.M.; da Candido, D.S.; Mishra, S.; E Crispim, M.A.; S Sales, F.C.; Hawryluk, I.; McCrone, J.T.; et al. Genomics and Epidemiology of the P.1 SARS-CoV-2 Lineage in Manaus, Brazil; Vol.
- Pascarella, S.; Ciccozzi, M.; Zella, D.; Bianchi, M.; Benedetti, F.; Benvenuto, D.; Broccolo, F.; Cauda, R.; Caruso, A.; Angeletti, S.; et al. SARS-CoV-2 B.1.617 Indian Variants: Are Electrostatic Potential Changes Responsible for a Higher Transmission Rate? J Med Virol 2021, 93, 6551–6556. [Google Scholar] [CrossRef]
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Althaus, C.L.; Anyaneji, U.J.; Bester, P.A.; Boni, M.F.; Chand, M.; et al. Rapid Epidemic Expansion of the SARS-CoV-2 Omicron Variant in Southern Africa. Nature 2022, 603, 679–686. [Google Scholar] [CrossRef]
- Yang, H.; Rao, Z. Structural Biology of SARS-CoV-2 and Implications for Therapeutic Development. Nat Rev Microbiol 2021, 19, 685–700. [Google Scholar] [CrossRef]
- Toyoshima, Y.; Nemoto, K.; Matsumoto, S.; Nakamura, Y.; Kiyotani, K. SARS-CoV-2 Genomic Variations Associated with Mortality Rate of COVID-19. J Hum Genet 2020, 65, 1075–1082. [Google Scholar] [CrossRef]
- Karamitros, T.; Papadopoulou, G.; Bousali, M.; Mexias, A.; Tsiodras, S.; Mentis, A. SARS-CoV-2 Exhibits Intra-Host Genomic Plasticity and Low-Frequency Polymorphic Quasispecies. Journal of Clinical Virology 2020, 131. [Google Scholar] [CrossRef]
- Tonkin-Hill, G.; Martincorena, I.; Amato, R.; Lawson, A.R.; Gerstung, M.; Johnston, I.; Jackson, D.K.; Park, N.; Lensing, S. V.; Quail, M.A.; et al. Patterns of Within-Host Genetic Diversity in SARS-COV-2. Elife 2021, 10. [Google Scholar] [CrossRef]
- Markov, P. V.; Ghafari, M.; Beer, M.; Lythgoe, K.; Simmonds, P.; Stilianakis, N.I.; Katzourakis, A. The Evolution of SARS-CoV-2. Nat Rev Microbiol 2023, 21, 361–379. [Google Scholar] [CrossRef] [PubMed]
- Xi, B.; Zeng, X.; Chen, Z.; Zeng, J.; Huang, L.; Du, H. SARS-CoV-2 within-Host Diversity of Human Hosts and Its Implications for Viral Immune Evasion. mBio 2023, 14. [Google Scholar] [CrossRef] [PubMed]
- Kashte, S.; Gulbake, A.; El-Amin, S.F.; Gupta, A. COVID-19 Vaccines: Rapid Development, Implications, Challenges and Future Prospects. Hum Cell 2021, 34, 711–733. [Google Scholar] [CrossRef]
- Li, R.; Liu, J.; Zhang, H. The Challenge of Emerging SARS-CoV-2 Mutants to Vaccine Development. Journal of Genetics and Genomics 2021, 48, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Banho, C.A.; Sacchetto, L.; Campos, G.R.F.; Bittar, C.; Possebon, F.S.; Ullmann, L.S.; Marques, B. de C.; da Silva, G.C.D.; Moraes, M.M.; Parra, M.C.P.; et al. Impact of SARS-CoV-2 Gamma Lineage Introduction and COVID-19 Vaccination on the Epidemiological Landscape of a Brazilian City. Communications Medicine 2022, 2. [Google Scholar] [CrossRef]
- Hacisuleyman, E.; Hale, C.; Saito, Y.; Blachere, N.E.; Bergh, M.; Conlon, E.G.; Schaefer-Babajew, D.J.; DaSilva, J.; Muecksch, F.; Gaebler, C.; et al. Vaccine Breakthrough Infections with SARS-CoV-2 Variants. New England Journal of Medicine 2021, 384, 2212–2218. [Google Scholar] [CrossRef]
- Estofolete, C.F.; Banho, C.A.; Campos, G.R.F.; Marques, B.C.; Sacchetto, L.; Ullmann, L.S.; Possebon, F.S.; Machado, L.F.; Syrio, J.D.; Araújo Junior, J.P.; et al. Case Study of Two Post Vaccination SARS-CoV-2 Infections with P1 Variants in Coronavac Vaccinees in Brazil. Viruses 2021, 13. [Google Scholar] [CrossRef]
- Lythgoe, K.A.; Hall, M.; Ferretti, L.; de Cesare, M.; MacIntyre-Cockett, G.; Trebes, A.; Andersson, M.; Otecko, N.; Wise, E.L.; Moore, N.; et al. SARS-CoV-2 within-Host Diversity and Transmission. Science (1979) 2021, 372. [Google Scholar] [CrossRef] [PubMed]
- Armero, A.; Berthet, N.; Avarre, J.C. Intra-Host Diversity of Sars-Cov-2 Should Not Be Neglected: Case of the State of Victoria, Australia. Viruses 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Voloch, C.M.; Da Silva Francisco, R.; De Almeida, L.G.P.; Brustolini, O.J.; Cardoso, C.C.; Gerber, A.L.; Guimarães, A.P.D.C.; Leitão, I.D.C.; Mariani, D.; Ota, V.A.; et al. Intra-Host Evolution during SARS-CoV-2 Prolonged Infection. Virus Evol 2021, 7. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Quadeer, A.A.; Krishnan, P.; Ng, D.Y.M.; Chang, L.D.J.; Liu, G.Y.Z.; Cheng, S.M.S.; Lam, T.T.Y.; Peiris, M.; McKay, M.R.; et al. Within-Host Genetic Diversity of SARS-CoV-2 Lineages in Unvaccinated and Vaccinated Individuals. Nat Commun 2023, 14. [Google Scholar] [CrossRef]
- OSANG HEALTHCARE GeneFinderTM COVID-19 Fast RealAmp Kit Available online:. Available online: https://www.osanghc.com/en/products_en/molecular-diagnosis/# (accessed on 24 November 2023).
- Brabaham Bioinformatics FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 24 November 2023).
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet J 2011, 17, 10. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H. A Statistical Framework for SNP Calling, Mutation Discovery, Association Mapping and Population Genetical Parameter Estimation from Sequencing Data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [PubMed]
- Castellano, S.; Cestari, F.; Faglioni, G.; Tenedini, E.; Marino, M.; Artuso, L.; Manfredini, R.; Luppi, M.; Trenti, T.; Tagliafico, E. Ivar, an Interpretation-oriented Tool to Manage the Update and Revision of Variant Annotation and Classification. Genes (Basel) 2021, 12. [Google Scholar] [CrossRef]
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; et al. Assignment of Epidemiological Lineages in an Emerging Pandemic Using the Pangolin Tool. Virus Evol 2021, 7. [Google Scholar] [CrossRef] [PubMed]
- Wilm, A.; Aw, P.P.K.; Bertrand, D.; Yeo, G.H.T.; Ong, S.H.; Wong, C.H.; Khor, C.C.; Petric, R.; Hibberd, M.L.; Nagarajan, N. LoFreq: A Sequence-Quality Aware, Ultra-Sensitive Variant Caller for Uncovering Cell-Population Heterogeneity from High-Throughput Sequencing Datasets. Nucleic Acids Res 2012, 40, 11189–11201. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff: SNPs in the Genome of Drosophila Melanogaster Strain W1118; Iso-2; Iso-3. Fly (Austin) 2012, 6, 80–92. [Google Scholar] [CrossRef]
- R Core Team R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna. Available online: https://www.R-project.org (accessed on 24 November 2023).
- Wickham, H.; Chang, W.; Henry, L.; Pedersen, T.L.; Takahashi, K.; Wilke, C.; Woo, K.; Yutani, H.; Dunnington, D. Ggplot2: Elegant Graphics for Data Analysis. Available online: https://ggplot2.tidyverse.org/ (accessed on 24 November 2023).
- Kosakovsky Pond, S.L.; Frost, S.D.W. Not so Different after All: A Comparison of Methods for Detecting Amino Acid Sites under Selection. Mol Biol Evol 2005, 22, 1208–1222. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Poon, A.F.Y.; Velazquez, R.; Weaver, S.; Hepler, N.L.; Murrell, B.; Shank, S.D.; Magalis, B.R.; Bouvier, D.; Nekrutenko, A.; et al. HyPhy 2.5 - A Customizable Platform for Evolutionary Hypothesis Testing Using Phylogenies. Mol Biol Evol 2020, 37, 295–299. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.J.; et al. SARS-CoV-2 Variants, Spike Mutations and Immune Escape. Nat Rev Microbiol 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Zhou, D.; Dejnirattisai, W.; Supasa, P.; Liu, C.; Mentzer, A.J.; Ginn, H.M.; Zhao, Y.; Duyvesteyn, H.M.E.; Tuekprakhon, A.; Nutalai, R.; et al. Evidence of Escape of SARS-CoV-2 Variant B.1.351 from Natural and Vaccine-Induced Sera. Cell 2021, 184, 2348–2361. [Google Scholar] [CrossRef]
- Fontanet, A.; Cauchemez, S. COVID-19 Herd Immunity: Where Are We? Nat Rev Immunol 2020, 20, 583–584. [Google Scholar] [CrossRef] [PubMed]
- Mercatelli, D.; Giorgi, F.M. Geographic and Genomic Distribution of SARS-CoV-2 Mutations. Front Microbiol 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R.; et al. Spike Mutation D614G Alters SARS-CoV-2 Fitness. Nature 2021, 592, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Magazine, N.; Zhang, T.; Wu, Y.; McGee, M.C.; Veggiani, G.; Huang, W. Mutations and Evolution of the SARS-CoV-2 Spike Protein. Viruses 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Saldivar-Espinoza, B.; Garcia-Segura, P.; Novau-Ferré, N.; Macip, G.; Martínez, R.; Puigbò, P.; Cereto-Massagué, A.; Pujadas, G.; Garcia-Vallve, S. The Mutational Landscape of SARS-CoV-2. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P. Rampant C→U Hypermutation in the Genomes of SARS-CoV-2 and Other Coronaviruses: Causes and Consequences for Their Short- and Long-Term Evolutionary Trajectories. mSphere 2020, 5. [Google Scholar] [CrossRef]
- Fung, S.; Liu, D.X. Human Coronavirus: Host-Pathogen Interaction. 2019. [CrossRef]
- Di Giorgio, S.; Martignano, F.; Gabriella Torcia, M.; Mattiuz, G.; Conticello, S.G. Evidence for Host-Dependent RNA Editing in the Transcriptome of SARS-CoV-2; 2020; Vol.
- Hughes, A.L.; Hughes, M.A.K. More Effective Purifying Selection on RNA Viruses than in DNA Viruses. Gene 2007, 404, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Ghafari, M.; Du Plessis, L.; Raghwani, J.; Bhatt, S.; Xu, B.; Pybus, O.G.; Katzourakis, A. Purifying Selection Determines the Short-Term Time Dependency of Evolutionary Rates in SARS-CoV-2 and PH1N1 Influenza. Mol Biol Evol 2022, 39. [Google Scholar] [CrossRef] [PubMed]
- Holmes, E.C. Patterns of Intra- and Interhost Nonsynonymous Variation Reveal Strong Purifying Selection in Dengue Virus. J Virol 2003, 77, 11296–11298. [Google Scholar] [CrossRef] [PubMed]
- Riemersma, K.K.; Coffey, L.L. Chikungunya Virus Populations Experience Diversity- Dependent Attenuation and Purifying Intra-Vector Selection in Californian Aedes Aegypti Mosquitoes. PLoS Negl Trop Dis 2019, 13. [Google Scholar] [CrossRef]
- Bueno, S.M.; Abarca, K.; González, P.A.; Gálvez, N.M.S.; Soto, J.A.; Duarte, L.F.; Schultz, B.M.; Pacheco, G.A.; González, L.A.; Vázquez, Y.; et al. Safety and Immunogenicity of an Inactivated Severe Acute Respiratory Syndrome Coronavirus 2 Vaccine in a Subgroup of Healthy Adults in Chile. Clinical Infectious Diseases 2022, 75, E792–E804. [Google Scholar] [CrossRef] [PubMed]
- Duarte, L.F.; Gálvez, N.M.S.; Iturriaga, C.; Melo-González, F.; Soto, J.A.; Schultz, B.M.; Urzúa, M.; González, L.A.; Vázquez, Y.; Ríos, M.; et al. Immune Profile and Clinical Outcome of Breakthrough Cases After Vaccination With an Inactivated SARS-CoV-2 Vaccine. Front Immunol 2021, 12. [Google Scholar] [CrossRef]
- Giovanetti, M.; Slavov, S.N.; Fonseca, V.; Wilkinson, E.; Tegally, H.; Patané, J.S.L.; Viala, V.L.; San, E.J.; Rodrigues, E.S.; Santos, E.V.; et al. Genomic Epidemiology of the SARS-CoV-2 Epidemic in Brazil. Nat Microbiol 2022, 7, 1490–1500. [Google Scholar] [CrossRef] [PubMed]
- Feikin, D.R.; Higdon, M.M.; Abu-Raddad, L.J.; Andrews, N.; Araos, R.; Goldberg, Y.; Groome, M.J.; Huppert, A.; O’Brien, K.L.; Smith, P.G.; et al. Duration of Effectiveness of Vaccines against SARS-CoV-2 Infection and COVID-19 Disease: Results of a Systematic Review and Meta-Regression. The Lancet 2022, 399, 924–944. [Google Scholar] [CrossRef]
- Garcia-Beltran, W.F.; Lam, E.C.; St. Denis, K.; Nitido, A.D.; Garcia, Z.H.; Hauser, B.M.; Feldman, J.; Pavlovic, M.N.; Gregory, D.J.; Poznansky, M.C.; et al. Multiple SARS-CoV-2 Variants Escape Neutralization by Vaccine-Induced Humoral Immunity. Cell 2021, 184, 2372–2383. [Google Scholar] [CrossRef]
- Planas, D.; Bruel, T.; Grzelak, L.; Guivel-Benhassine, F.; Staropoli, I.; Porrot, F.; Planchais, C.; Buchrieser, J.; Rajah, M.M.; Bishop, E.; et al. Sensitivity of Infectious SARS-CoV-2 B.1.1.7 and B.1.351 Variants to Neutralizing Antibodies. Nat Med 2021, 27, 917–924. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Intra-host genetic diversity of SARS-COV-2 after two doses of CoronaVac. (A) Percentage of iSNVs across the SARS-CoV-2 genome from unvaccinated and vaccinated patients in relation to gene size. (B) Major and minor allele frequency for non-synonymous (blue) and synonymous (orange) variants in the unvaccinated and vaccinated groups. Coding regions of the SARS-CoV-2 genome, based on the reference genome (NC_045512.2), are shown at the bottom of the figure. ORF: open reading frame. S: spike. E: envelope. M: membrane. N: nucleocapsid.
Figure 1.
Intra-host genetic diversity of SARS-COV-2 after two doses of CoronaVac. (A) Percentage of iSNVs across the SARS-CoV-2 genome from unvaccinated and vaccinated patients in relation to gene size. (B) Major and minor allele frequency for non-synonymous (blue) and synonymous (orange) variants in the unvaccinated and vaccinated groups. Coding regions of the SARS-CoV-2 genome, based on the reference genome (NC_045512.2), are shown at the bottom of the figure. ORF: open reading frame. S: spike. E: envelope. M: membrane. N: nucleocapsid.
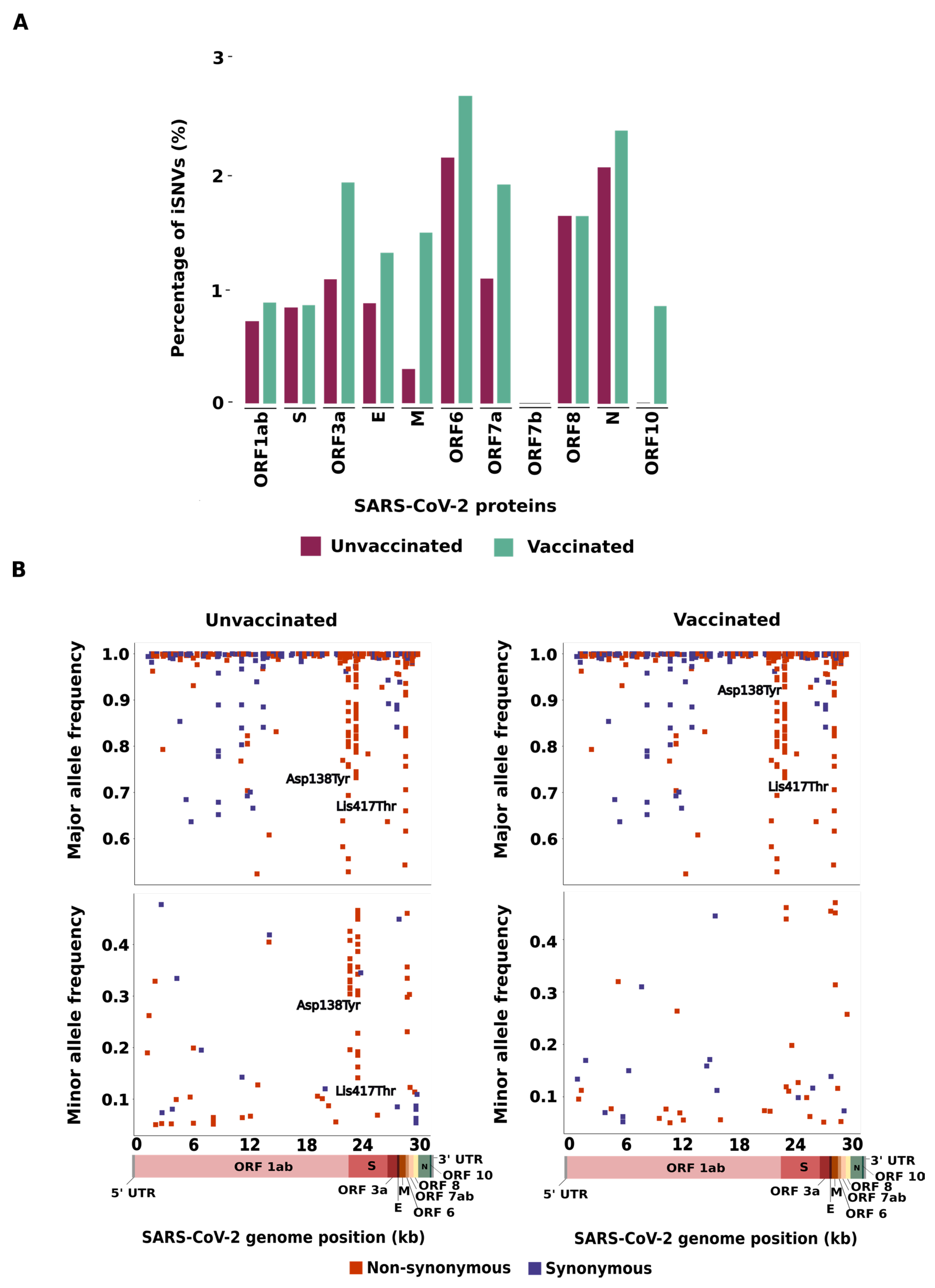
Figure 2.
Sites under negative and positive selection detected in the genomes of unvaccinated and vaccinated individuals with CoronaVac against SARS-CoV-2. ORF: open reading frame. S: spike. E: envelope. N: nucleocapsid.
Figure 2.
Sites under negative and positive selection detected in the genomes of unvaccinated and vaccinated individuals with CoronaVac against SARS-CoV-2. ORF: open reading frame. S: spike. E: envelope. N: nucleocapsid.
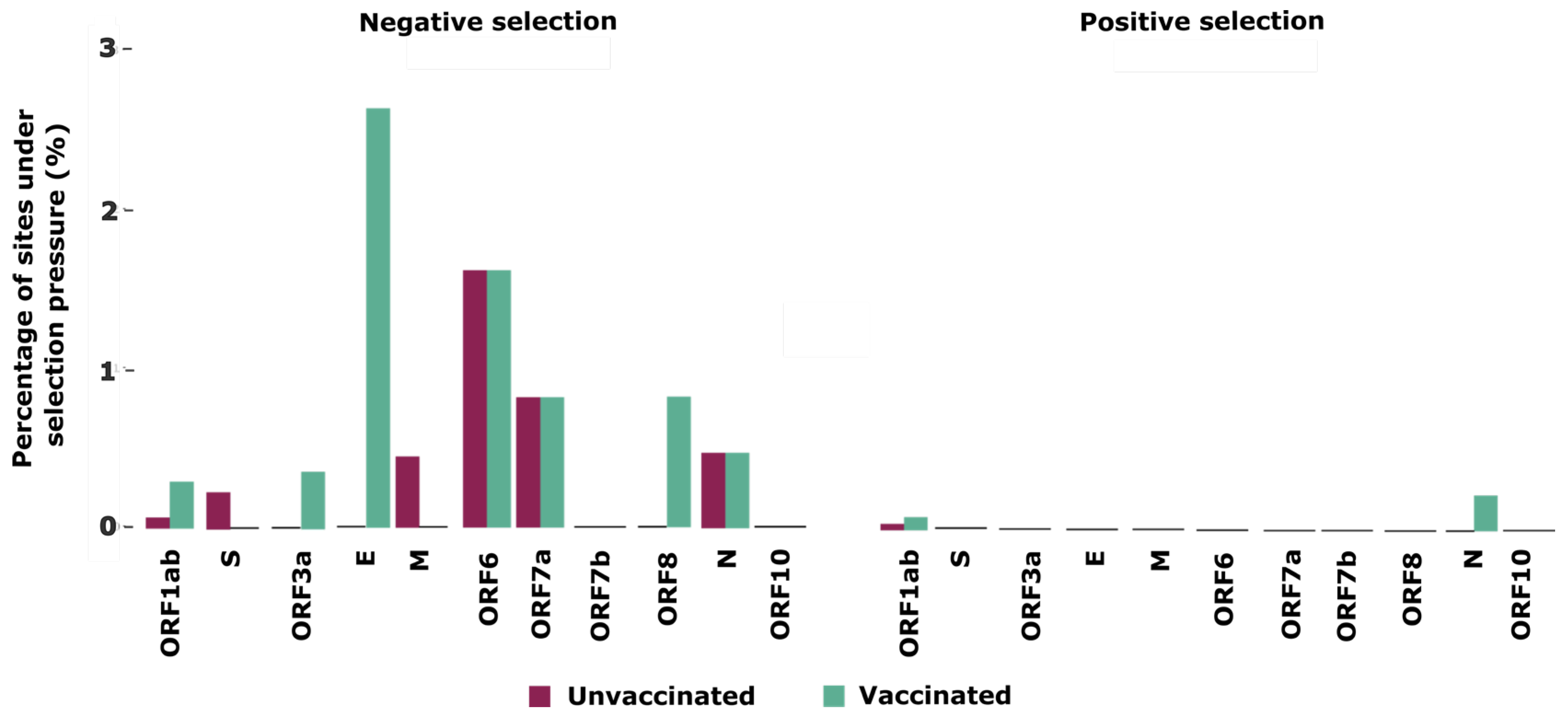
Table 2.
Sites under positive or negative selection for each SARS-CoV-2 coding region analyzed using MEME and FEL.
Table 2.
Sites under positive or negative selection for each SARS-CoV-2 coding region analyzed using MEME and FEL.
| Unvaccinated | Vaccinated | |||
| Locus | Positive | Negative | Positive | Negative |
| ORF1ab | NSP6 (106) | NSP3 (106, 681), NSP10 (82), NSP13 (495) | NSP3 (1303), NSP6 (107) | NSP2 (91,443), NSP3 (236, 394, 447, 662, 1092, 1121, 1742), NSP6 (76, 138), NSP10 (16), NSP13 (237, 356), NSP14 (302, 373), NSP15 (278), NSP16 (178) |
| S | 0 | 554, 995, 1065 | 0 | 0 |
| ORF3a | 0 | 0 | 0 | 43 |
| E | 0 | 0 | 0 | 8,23 |
| M | 0 | 53 | 0 | 0 |
| ORF6 | 0 | 49 | 0 | 61 |
| ORF7a | 0 | 88 | 0 | 11 |
| ORF8 | 0 | 0 | 0 | 75 |
| N | 0 | 0 | 200 | 194, 363 |
| ORF10 | 0 | 0 | 0 | 0 |
ORF: open reading frame. S: spike. E: envelope. N: nucleocapsid.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



