Submitted:
12 August 2024
Posted:
13 August 2024
You are already at the latest version
Abstract
A major reason for failure of the immune system to detect tumor antigens (TA) is the insufficient uptake, processing, and presentation of TA by antigen-presenting-cells (APC). Immunogenicity of public and private TA of the individual patient can be markedly increased by in situ targeting of tumor cells for uptake by APC, without the need to identify and characterize the TA. This is feasible by intra-tumoral administration of alpha-gal micelles comprised of glycolipids presenting the carbohydrate-antigen “alpha-gal epitope” (Galalpha1-3Gal beta1-4GlcNAc-R). Humans produce a natural antibody called “anti-Gal” (constituting ~1% of immunoglobulins) which binds alpha-gal epitopes. Tumor injected alpha-gal micelles spontaneously insert into tumor cell membranes, so that multiple alpha-gal epitopes present on tumor cells. Anti-Gal binding to these epitopes activates the complement system, resulting in killing of tumor cells, and recruitment of multiple APC (dendritic cells and macrophages) into treated tumors by chemotactic complement cleavage peptides C5a and C3a. In this process of converting the treated tumor into personalized TA vaccine, recruited APC internalize anti-Gal opsonized tumor cells and cell membranes, process the internalized TA and transport them to regional lymph-nodes. TA peptides presented on APC activate TA specific T and B cells to proliferate and destroy metastatic tumor cells presenting the TA. Studies in anti-Gal producing mice demonstrated induction of effective protection against distant metastases of the highly tumorigenic B16 melanoma following injection of natural and synthetic alpha-gal micelles into primary tumors. This treatment was further found to synergize with anti-PD1 antibody. Phase-1 clinical-trials indicated that this immunotherapy is safe and can induce infiltration of CD4+ and CD8+ T cells into untreated distant metastases. It is suggested that in addition to converting treated metastases into autologous TA vaccine, this treatment should be considered as neo-adjuvant therapy administering alpha-gal micelles into primary tumors immediately following their detection. Such immunotherapy will convert tumors into personalized anti-TA vaccine for the period prior to their resection.
Keywords:
cancer immunotherapy
; anti-Gal antibody
; α-gal epitope
; α-gal micelles
; α-gal glycolipids
; AGI-134
; APC recruitment
; α-gal therapy
; tumor antigens
1. Introduction
Tumor antigens (TA), whether they are private antigens, or public shared antigens, can serve as the “Achilles heel” of malignancies in cancer immunotherapy. The TA in this review are known and unknown antigens presented uniquely on tumor cells, including: products of mutated genes, products of oncogenic viruses, aberrantly expressed cellular proteins, altered glycoproteins and glycolipids, and oncofetal antigens. Induction of a protective T cell and B cell responses against TA may result in successful immunotherapy. In a proportion of patients with solid tumors the immune system “succeeds” by itself to mount a protective immune response against the patient’s TA. This has been demonstrated in a variety of cancers including colon carcinoma [1,2], ovarian carcinoma [3] and breast carcinoma [4,5]. Retrospective studies in patients with these types of cancer indicated that the patients had much longer survival and absence of metastases if their resected tumors displayed infiltration of many T cells, in comparison to patients displaying low or no infiltration of T cells into the tested tumors. The high infiltration of T cells in tumors of patients with good prognosis was specific to the malignant tissues and not found in normal tissues. This implied that the protective immune response associated with the lack of metastases was the result of specific immune response to TA present on malignant cells and absent from normal cells. In contrast, the poor prognosis observed in patients with low or no T cell infiltration into tumors (also called “cold tumors”) suggested that in these patients the immune system failed to react effectively against the patient’s TA and thus, to prevent formation of metastases and progression of disease.
Activation of T cells against tumor cells presenting TA requires intra-tumoral uptake of TA by antigen presenting cells (APC) such as dendritic cells and macrophages. These APC process and present TA peptides on HLA molecules and transport the presented TA peptides to regional lymph-nodes for the activation of TA specific T cells. The absence of infiltrating T cells in cold tumors may be the result of various factors including an immunosuppressive microenvironment within the lesion, prevention of infiltration of APC into the tumor, poor presentation of TA by the tumor cells and immune tolerance to TA [6,7]. It has been further suggested that most cancer vaccines have not shown significant clinical benefit because of poor uptake of vaccinating TA by APC [7].
The accumulated experience in cancer immunotherapy led to the realization that a large proportion of the TA are formed by multiple mutations specific to the individual patient. These mutations are generated due to genomic instability in tumor cells [8,9,10]. Many of these mutations alter amino acids within various proteins and convert them into TA that may be targets for immunotherapy. The combination of “next generation sequencing” in tumors from individual cancer patients and algorithms developed in recent years, have enabled the identification of immunogenic TA peptides (i.e., peptides with high affinity to HLA molecules for effective presentation) that may be synthesized as “neoantigens” for the use as personalized vaccine in cancer patients [11,12,13,14]. It remains to determine the efficacy of this novel method in cancer immunotherapy.
The present review describes an alternative simple method for inducing protective immune response against all types of TA in the individual cancer patient. This method which uses the self-tumor as the source immunizing TA achieves the following objectives of personalized cancer vaccines including: 1. Use of all self-TA as vaccine without the need for identifying them, 2. Effective targeting of the immunizing TA for robust uptake by APC, 3. Presentation by APC of neoantigen peptides with high affinity to HLA, 4. Overcoming immune tolerance to self-TA. This vaccine is generated by injecting α-gal micelles into solid tumors. The α-gal glycolipids comprising these micelles spontaneously insert into the cell membrane of tumor cells in treated lesions. These glycolipids bind the abundant natural anti-Gal antibody [15] which targets tumor cells and cell membranes expressing the full range of TA for effective uptake by APC and their transport to regional lymph-nodes. The processing and presentation of TA peptides by these APC within lymph-nodes induces a protective immune response against metastatic tumor cells.
2. The Natural Anti-Gal Antibody and the α-Gal Epitope
Anti-Gal is an antibody that is naturally produced throughout life in humans [15,16] in response to antigenic stimulation by gastrointestinal bacteria [17,18,19]. Anti-Gal is produced in all humans who are not severely immunocompromised. It constitutes ∼1% of serum immunoglobulins [16] and as many as 1% of B cells can produce this antibody [20]. In fetal and newborn blood, anti-Gal is found as maternal IgG, whereas in children and adults, it is found as IgG, IgM, and IgA classes [16,21,22,23,24]. Anti-Gal is a polyclonal antibody, and its ligand is a carbohydrate antigen called the ”α-gal epitope” with the structure Galα1-3Galβ1-4GlcNAc-R on mammalian glycolipids, glycoproteins and proteoglycans [25,26,27,28]. The α-gal epitope is naturally expressed on cell surface glycans of nonprimate mammals, prosimians (lemurs), and New World monkeys, in which it is synthesized by the glycosylation enzyme α1,3galactosyltransferase (α1,3GT) [29,30]. The GGTA1 gene encoding α1,3GT is found only in mammals [31,32,33] and is absent in birds, reptiles, amphibians and fish, therefore, non-mammalian vertebrates lack α-gal epitopes. The GGTA1 gene was inactivated in ancestral Old World primates 20-30 million years ago [32,33,34], therefore, humans, apes, and Old World monkeys lack the ability to synthesize α-gal epitopes and produce the natural anti-Gal antibody [35].
Anti-Gal binding in vitro to cells presenting α-gal epitopes can mediate killing of the cells both by complement dependent cytolysis (CDC) and by antibody-dependent cell cytolysis (ADCC) [36,37,38]. The potential of this antibody to mediate in vivo CDC and ADCC has been demonstrated in xenotransplantation studies in which pig organs are transplanted into humans or monkeys. Binding of the recipients’ anti-Gal to α-gal epitopes on pig cells in the re-perfused organs leads to rapid (hyperacute) rejection due to cell destruction by these two mechanisms [36,39,40,41]. The in vivo anti-Gal/α-gal epitope interaction provides the possibility of harnessing the immunologic potential of this antibody in a number of α-gal associated therapies in several clinical settings including scar-free regeneration of injuries in the skin [42,43,44], regeneration of injured ischemic myocardium following myocardial infarction [45,46], regeneration of injured spinal cord [46,47] and conversion of self-TA into anti-tumor vaccines, discussed in the present review.
3. Anti-Gal Mediated Targeting of Tumor Cell Vaccines Presenting TA and α-Gal Epitopes to APC
As mentioned above, a major cause for lack of effective immune response to TA in cancer patients is the inability of APC to identify tumor cells presenting TA as cells or cell membranes that “ought” to be internalized for processing and presentation of TA peptides [7]. Similar to any other vaccine, also TA vaccines must be internalized by dendritic cells and macrophages in order to process the vaccinating antigens and present the TA peptides on HLA molecules of these APC. The APC further transport the presented peptides to regional lymph-nodes where they activate TA specific CD8+ T cells which proliferate, leave the lymph-node, circulate in the body and kill metastatic TA presenting tumor cells. In addition, these APC activate CD4+ T cells that provide help to B cells producing anti-TA antibodies. Thus, a key step in increasing immunogenicity of TA of tumor cells within the individual patient is the induction of robust uptake of many tumor cells and cell membranes by APC.
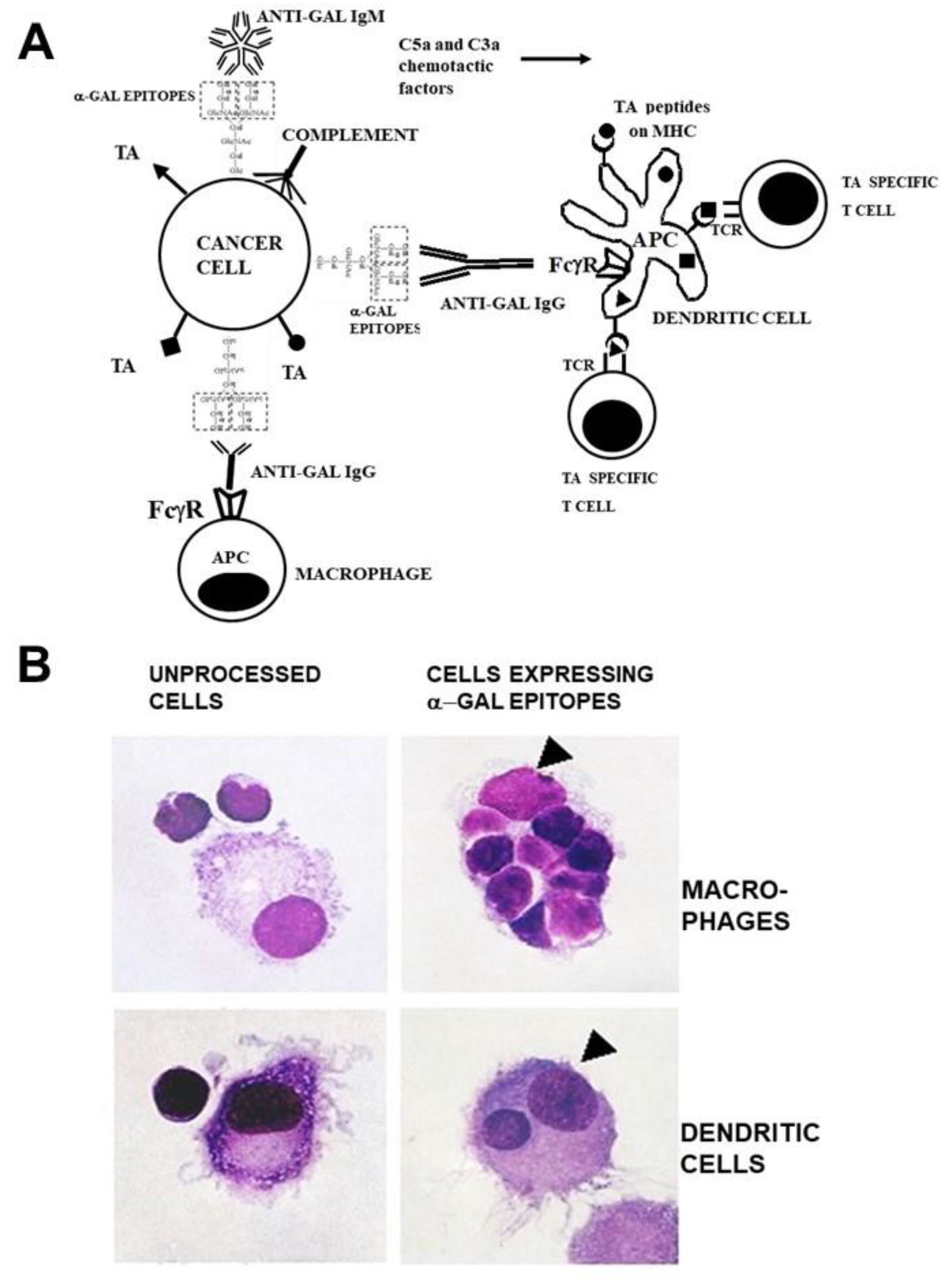
Since anti-Gal is ubiquitously produced in large amounts in all humans who are not severely immunosuppressed, it was hypothesized that cancer patients may be vaccinated with lethally irradiated self-tumor cells or cell membranes engineered to present α-gal epitopes [48,49]. In addition, it was suggested that self-TA in tumor lesions could be converted in situ into personalized anti-TA vaccines by inducing expression α-gal epitopes on the tumor cells within treated lesions [50]. As illustrated in Figure 1A, vaccinating tumor cells presenting α-gal epitopes bind the natural anti-Gal IgM and IgG antibody molecules and activate the complement system. In addition to tumor cell cytolysis, the chemotactic complement cleavage peptides C5a and C3a generated by the complement activation effectively recruit macrophages and dendritic cells to the vaccination site. These recruited APC effectively phagocytose tumor cells and cell membranes opsonized by anti-Gal IgG following binding of the Fc tail of this antibody to Fcγ receptors (FcγR) on APC and binding of C3b on the opsonized cells to C3b receptors (CR1) on APC. Due to this robust phagocytosis, many internalized tumor cells and cell membranes are transported by the APC to regional lymph-nodes. Private and public TA on internalized tumor cell membranes are processed by the APC and the TA peptides with high affinity to HLA molecules are presented by these molecules for effective activation of TA specific T cells. The activated T cells mount protective humoral and cellular immune responses against metastatic tumor cells presenting the immunizing TA. The increased immunogenicity of vaccines presenting α-gal epitopes could be quantified with influenza virus vaccine [51] and gp120 of HIV vaccine presenting α-gal epitopes [52] in anti-Gal producing mice. Presentation of α-gal epitopes on both vaccines resulted in ~100-fold higher titers of protective anti-viral antibodies than following vaccination with the corresponding vaccines lacking this epitope.
The high uptake efficacy of self-tumor cells presenting α-gal epitopes by APC could be demonstrated in vitro by co-incubation of B cell lymphoma cells from a patient with autologous macrophages or dendritic cells and in presence of autologous anti-Gal antibody. The lymphoma cells were enzymatically engineered to present α-gal [53]. Cells were incubated for 2 hrs at 37oC with macrophages and dendritic in presence of anti-Gal. Within this period as many as 8 tumor cells opsonized by anti-Gal were internalized by macrophages (Figure 1B). In contrast, tumor cells lacking α-gal epitopes were not phagocytosed by macrophages. Dendritic cells also internalized tumor cells opsonized by anti-Gal, albeit less than macrophages, whereas no phagocytosis was observed with lymphoma cells lacking α-gal epitopes (Figure 1B). A similar robust anti-Gal mediated uptake by macrophages of immunizing cells presenting α-gal epitopes could be demonstrated with human leukemia cells [48].
Figure 1.
Anti-Gal binding to α-gal epitopes on tumor cells targets them and their TA for uptake by APC and improves TA immunogenicity. A. Illustration of the hypothesis. Tumor cells presenting α-gal epitopes (dashed line rectangles) bind natural anti-Gal IgM and IgG antibodies. The activated complement system generates complement cleavage chemotactic peptides C5a and C3a that induce rapid recruitment of APC (dendritic cells and macrophages). Anti-Gal IgG opsonized tumor cells are phagocytosed by APC following Fc/FcγR interaction. The internalized TA (●, ■, ▲) are transported by the APC to regional lymph nodes and are processed. Immunogenic TA peptides are presented on class I and class II MHC molecules, thereby activating tumor specific cytotoxic and helper T cell clones with the corresponding T cell receptors (TCR). Modified with permission from ref. [54]. B. Anti-Gal mediated uptake by autologous APC of human B lymphoma cells engineered to present α-gal epitopes. Lymphoma cells with or without α-gal epitopes were incubated with autologous anti-Gal and autologous macrophages or dendritic cells. Triangles mark the nuclei of the APC. Note robust phagocytosis of lymphoma cells presenting α-gal epitopes by the macrophage and of one lymphoma cell by the dendritic cell. No uptake of lymphoma cells lacking α-gal epitopes is observed (x1000). Modified with permission from ref. [53].
Figure 1.
Anti-Gal binding to α-gal epitopes on tumor cells targets them and their TA for uptake by APC and improves TA immunogenicity. A. Illustration of the hypothesis. Tumor cells presenting α-gal epitopes (dashed line rectangles) bind natural anti-Gal IgM and IgG antibodies. The activated complement system generates complement cleavage chemotactic peptides C5a and C3a that induce rapid recruitment of APC (dendritic cells and macrophages). Anti-Gal IgG opsonized tumor cells are phagocytosed by APC following Fc/FcγR interaction. The internalized TA (●, ■, ▲) are transported by the APC to regional lymph nodes and are processed. Immunogenic TA peptides are presented on class I and class II MHC molecules, thereby activating tumor specific cytotoxic and helper T cell clones with the corresponding T cell receptors (TCR). Modified with permission from ref. [54]. B. Anti-Gal mediated uptake by autologous APC of human B lymphoma cells engineered to present α-gal epitopes. Lymphoma cells with or without α-gal epitopes were incubated with autologous anti-Gal and autologous macrophages or dendritic cells. Triangles mark the nuclei of the APC. Note robust phagocytosis of lymphoma cells presenting α-gal epitopes by the macrophage and of one lymphoma cell by the dendritic cell. No uptake of lymphoma cells lacking α-gal epitopes is observed (x1000). Modified with permission from ref. [53].
4. Protective Efficacy of Self-TA Vaccines Presenting α-gal Epitopes
4.1. The Experimental Animal Model
Studies on efficacy of α-gal presenting TA vaccines cannot be performed in standard experimental animal models such as mouse, rat, rabbit or guinea-pig. The reason is that these animals, like other nonprimate mammals, naturally synthesize the α-gal epitope and thus, are immunotolerant to it and cannot produce the anti-Gal antibody. This obstacle was overcome by the generation of knockout mice for the α1,3GT gene GGTA1 (GT-KO mice) [55,56]. These mice lack α-gal epitopes and can produce the anti-Gal antibody following immunization with xenogeneic cells or cell membranes presenting α-gal epitopes such as porcine mononuclear cells [57], porcine kidney membrane homogenate [43,45], rabbit red cells [58] or immunization with glycoproteins such as synthetic α-gal linked to bovine serum albumin (α-gal -BSA) [59]. The choice for tumor cells in such studies is also limited because most mouse cell lines naturally present α-gal epitopes and therefore they are lysed by anti-Gal in the presence of complement or by ADCC. One of the very few mouse cancer cell lines lacking the α-gal epitope is B16 melanoma which grows well in GT-KO mice and does not bind anti-Gal [58].
4.2. Immune Protection against B16 Melanoma by Tumor Cell Vaccines Presenting α-gal Epitopes
A subclone of B16 melanoma called “B16/BL6” (referred to here as B16), that is highly tumorigenic was used in the preliminary studies on efficacy of tumor vaccines presenting α-gal epitopes [58]. These cells were engineered to synthesize and present α-gal epitopes by stable transfection with the α1,3galactosyltransferase (α1,3GT) gene GGTA1. This transgenic cell line, designated B16α-gal expressed multiple α-gal epitopes and bound the anti-Gal antibody [58]. B16α-gal cells were lethally irradiated and 2x106 of these cells were used as a vaccine injected subcutaneously into anti-Gal producing GT-KO mice (i.e., mice receiving prior immunizations with rabbit red cells). Injections of 2x106 irradiated B16 cells (lacking α-gal epitopes) were used as vaccine control. Two weeks post immunization, the mice were challenged subcutaneously with 0.5x106 live B16 cells, and tumor development was monitored for 60 days. A third of the mice immunized with B16α-gal cells did not develop any tumor after the challenge whereas all mice immunized with the parental B16 cells developed tumors within 8-26 days post-challenge (Figure 2A). These findings strongly suggested that one immunization with irradiated engineered B16α-gal tumor cells elicited a protective immune response against TA of the tumor, which destroyed the challenging B16 cells. In contrast, vaccines consisting of the same cells which lacked α-gal epitopes failed in eliciting a protective anti-TA immune response [58].
Of particular interest were the histological characteristics of the developing tumors. Tumors developing in mice immunized with B16 cells displayed highly proliferative tumor cells, as indicated by the relatively small size of the cells and basophilic staining of the cytoplasm due to high concentration of ribosomes (Figure 2B). In contrast, tumors developing in mice vaccinated with B16α-gal cells were surrounded by multiple mononuclear cells (Figure 2C). Immunostaining analysis indicated that ~70% of these mononuclear cells were T cells stained by anti-CD3 antibodies, and 30% were macrophages [58]. No B cells were found in these infiltrates. Tumor cells adjacent to the mononuclear cell were large and melanin producing, with vacuolated cytoplasm, suggesting ongoing cytolysis. The melanin (dark granules) in these cells suggested that they stopped proliferating and matured into melanin producing cells due to cytokines produced by infiltrating T cells and macrophages. This α-gal therapy in mice immunized with B16α-gal was subsequently independently validated using B16α-gal cells that were generated by stable transfection with retrovirus vector containing the α1,3GT gene GGTA1 [60]. Similar protective results were also obtained by generating B16α-gal cell vaccines with adenovirus containing this gene (called AdαGT) and transduction of B16 cells with this virus [61].
Induction of a protective anti-TA immune response was also observed in GT-KO mice immunized with both pancreatic carcinoma cells [62] and ovarian carcinoma cells engineered to present α-gal epitopes [63] and with breast cancer cells engineered to present α-gal epitopes and fused with dendritic cells [64]. All these studies have suggested that vaccines made of tumor cells engineered to present α-gal epitopes can induce a protective immune response against self-TA without the need to identify these TA in the individual patient. This immune response can destroy metastatic cells and slow or eliminate the race between the expanding tumor mass and the immune response destroying tumor cells. However, as may be inferred from Figure 2C, if the vaccine is applied in advanced stages of the disease when tumor mass is large, the immune protective response against the various TA may not be effective enough for destroying all tumor cells but may be able to slow the progression of the disease.
4.3. Clinical Trials with Tumor Cells Engineered In Vitro to Present α-Gal Epitopes
The α-gal therapy with autologous tumor cells and cell membranes engineered to present α-gal epitopes was studied in phase I clinical trials with refractory patients at advanced stage of the disease, having hepatocellular carcinoma [65], pancreatic carcinoma [66] and lymphoma [67]. Homogenates of cell membranes from the two solid tumors [65,66] and lymphoma cell suspensions [67] were subjected to enzymatic synthesis of α-gal epitopes by recombinant α1,3GT, UDP-Gal and neuraminidase as previously detailed [48,49]. The tumor membrane homogenates and lymphoma cells presenting α-gal epitopes were incubated for several days with autologous dendritic cells and anti-Gal in order to target the vaccinating materials for robust uptake by APC. Subsequently, the whole mixture was administered by several injections of autologous vaccine to the patients. Among the hepatocellular carcinoma patients, an average of 70% increase in survival time and increase in positive delayed hypersensitivity were observed, with no serious side effects or autoimmune diseases [65]. Treated patients with pancreatic carcinoma [66] also displayed a strong delayed-type hypersensitivity to the autologous cancer cell lysate with no serious side effects. Among the 14 patients with lymphoma, complete and partial remission occurred in four and three patients, respectively [67]. The disease status remained unchanged in five patients, and disease progression was observed in two patients. Overall, these studies suggested that administration of self-tumor cells or cell membranes presenting α-gal epitopes is safe and in some of the treated patients this vaccination process may induce a protective immune response. However, at advanced stages of the disease the efficacy of this immunotherapy may be limited because of the large tumor mass in the treated patient.
5. Conversion of Self-TA into Vaccines by Natural α-Gal Micelles
The in vitro engineering of leukemia and lymphoma cells to express α-gal epitopes and to serve as self-TA vaccine is relatively easy and involves harvesting tumor cells from blood or lymphoid organs and use of recombinant α1,3GT, UDP-Gal and neuraminidase [48,49] or AdαGT transduction [61,68]. However, the technical difficulties involved in preparing α-gal vaccines against self-TA by homogenizing resected solid tumors and synthesizing α-gal epitopes on the fragmented cell membranes, prompted the development of methods for in situ induction of α-gal epitope expression on cells of solid tumors, thereby converting such tumors into anti self-TA vaccines, functioning as described in Figure 1A. Studies on expression of α-gal epitopes within the treated lesions following intra-tumoral injection of recombinant α1,3GT, AdαGT, or α-gal micelles indicated that α-gal micelles had the highest efficacy for achieving intra-tumoral expression of α-gal epitopes [50].
5.1. α-Gal Glycolipids and α-Gal Micelles
α-Gal micelles are micelles comprised of α-gal glycolipids (Figure 3A). These glycolipids are made of carbohydrate chains (glycans) with terminal α-gal epitopes which are linked at the reducing end to a ceramide composed of sphingosine and fatty acid joined by an amide bond. When α-gal glycolipids are suspended in an aqueous solution they form small spheric structures called micelles, in which hydrophobic chains of the ceramides form the inner area of the sphere whereas the hydrophilic glycans protrude into the aqueous solution surrounding the micelle (Figure 3B). Individual α-gal glycolipid molecules detach in a reversible manner from the micelles and get inserted back into it. However, when α-gal micelles are near cell membranes, the α-gal glycolipid molecules detaching from the micelles insert in an irreversible manner into the lipid bilayer of the cell membranes. The reason for this irreversible insertion is that energetically, the ceramides of glycolipids are much more stable when surrounded by the phospholipids of the cell membrane than in the micelles surrounded by water molecules. Therefore, injection of α-gal micelles into solid tumors results in their rapid insertion into the tumor cell membranes (Figure 3B), leading to presentation of multiple α-gal epitopes on the tumor cells. These epitopes bind anti-Gal and initiate the sequence of processes illustrated in Figure 1A.
5.2. Production of α-Gal Micelles
A rich source for natural α-gal glycolipids is membranes of rabbit red cells which express ∼2×106 α-gal epitopes per cell, many of which are of glycolipids and the rest are of glycoproteins [29]. The glycolipid example in Figure 3 has 10 carbohydrate units with two branches (antennae,) each with an α-gal epitope. The α-gal glycolipids in rabbit red cell membranes have a total of 1-8 α-gal epitopes per molecule, each on one branch [69,70,71,72,73,74]. α-Gal glycolipids were extracted together with phospholipids and cholesterol by incubation of rabbit red cell membranes (ghosts) in a solution of chlorophorm:methanol 1:2 and further purified by Folch partition which removes the phospholipids and cholesterol [50,75].
5.3. Insertion of α-Gal Glycolipids into Tumor Cell Membranes
Insertion of α-gal glycolipids into tumor cells incubated with α-gal micelles is demonstrated in Figure 3C. B16 melanoma cells in suspension were incubated for 2 h at 37°C with α-gal micelles at various concentrations of the micelles, then washed and immunostained with the monoclonal anti-Gal antibody M86 which binds specifically to α-gal epitopes [76]. Flow cytometry analysis demonstrated a correlation between the concentration of the α-gal micelles and the extent of α-gal glycolipids inserted into the B16 cell membranes. Binding of serum anti-Gal to inserted α-gal glycolipids could further mediate complement dependent cytolysis (CDC) of the B16 cells (Figure 3D).
5.4. In Vivo Recruitment of APC into Treated Tumor Lesions
In vivo effects of intra-tumoral injection of α-gal micelles were determined with B16 melanoma lesions generated in the skin of anti-Gal producing GT-KO mice. Tumor lesions were generated in mice that were injected intradermally in two sites, ~2.5 cm apart, with 1x106 live B16 cells. Within 5 days, cutaneous tumor lesions developed with a diameter of ~5.0 mm. One of the lesions in each mouse was injected at three points with 1.0 mg α-gal micelles and the other was injected with PBS as control. No infiltration of PMN or mononuclear cells was observed in histologic sections of PBS injected tumors 2- and 7- days post PBS injection (Figure 4A). In contrast, extensive infiltration of mononuclear cells was observed already within 48 hrs post-injection of α-gal micelles (Figure 4B). This infiltration, mediated by the complement cleavage chemotactic peptides, further increased after 7 days (Figure 4C). Analysis of the tumor infiltrating mononuclear cells in the α-gal micelles injected lesions indicated that they include dendritic cells, macrophages, NK cells, CD4+ and CD8+ T cells [50]. Inspection of the tumors 10 days after these injections indicated that the PBS injected tumors increased their size by ~3 fold (Figure 4D and 4E). In contrast, most tumors injected with α-gal micelles either stopped growing or decreased their size, some to the extent of near complete elimination (Figures 4D and 4E). These findings implied that the in vivo insertion of α-gal glycolipids into the B16 cell membranes resulted in complement activation and killing of varying proportions of the tumor cells within the treated lesions. Repeating this study in wild-type mice (WT mice) demonstrated no significant differences between α-gal micelles injected and PBS injected lesions (Figure 4E) [50]. This implied that in the absence of anti-Gal (WT mice synthesize α-gal epitopes and cannot produce this antibody) the α-gal micelles injection has no effect on the tumor growth. It should be stressed that the killing of tumor cells within lesions injected with α-gal micelles is not the main objective of this treatment. The main objective is to convert live or dead tumor cells into cells that are “palatable” to APC by their opsonization with anti-Gal. The extensive phagocytosis of anti-Gal opsonized tumor cells by the recruited APC achieves the aim of internalization of large amounts of TA into the APC without the need for identifying the TA in the individual patient.
5.5. α−Gal Micelles Mediated Increased Transport of Processed Surrogate TA Peptides by APC
The increase in uptake and processing of TA mediated by intra-tumoral injection of α-gal micelles could be demonstrated with B16 cells containing the ovalbumin (OVA) transgene and producing OVA as a surrogate TA (designated B16/OVA cells) [78]. Detection of APC internalizing B16/OVA cells was performed by their coincubation with the CD8+ hybridoma B3Z T cells which have the T cell receptor (TCR) for presentation of the peptide SIINFEKL [79,80,81]. This peptide is the immunodominant OVA peptide when presented on class I MHC molecules. When TCRs on B3Z hybridoma cells engage the peptide SIINFEKL presented on Kb MHC molecules of APC, the B3Z cells are activated and therefore, their β-galactosidase transgene (Lac-Z) with IL2 promoter is activated. This activation simulates activation of CD8+ T cells which results in production of IL2 following activation by the corresponding peptide. The catalytic activity of the produced β-galactosidase is detected by the fluorescence of a substrate cleaved by this enzyme. This fluorescence can be detected in the activated B3Z cells by flow cytometry .
Figure 5.
APC mediated transport of OVA as a surrogate TA from α-gal micelles treated B16/OVA lesions to inguinal lymph nodes in anti-Gal producing GT-KO mice. The lymph nodes were excised 2 weeks post treatment and APC presenting the OVA immunodominant SIINFEKL peptide were detected by flow cytometry following activation of lac-Z transgene under IL2 promoter in activated CD8+ B3Z T cell hybridoma cells. The percentage of activated B3Z cells was determined by double staining CD8+ cells (PerCP red) and di-galactoside hydrolyzed by β-galactosidase (FITC green). Reproduced with permission from ref. [50]. .
Figure 5.
APC mediated transport of OVA as a surrogate TA from α-gal micelles treated B16/OVA lesions to inguinal lymph nodes in anti-Gal producing GT-KO mice. The lymph nodes were excised 2 weeks post treatment and APC presenting the OVA immunodominant SIINFEKL peptide were detected by flow cytometry following activation of lac-Z transgene under IL2 promoter in activated CD8+ B3Z T cell hybridoma cells. The percentage of activated B3Z cells was determined by double staining CD8+ cells (PerCP red) and di-galactoside hydrolyzed by β-galactosidase (FITC green). Reproduced with permission from ref. [50]. .
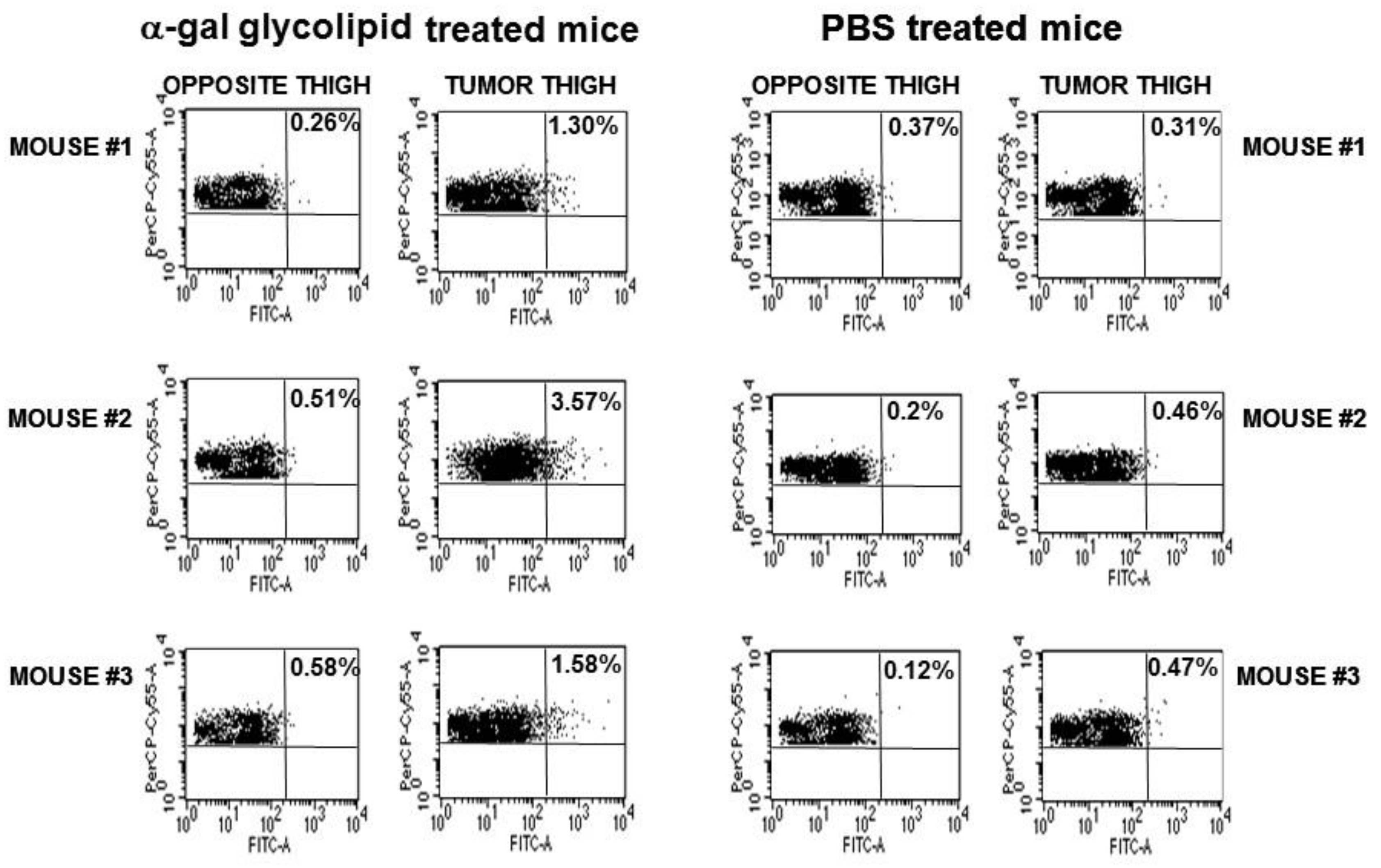
B16/OVA lesions with a diameter of ~5 mm on the right thigh of anti-Gal producing GT-KO mice were injected on Day 1 and Day 7 with 1.0 mg α-gal micelles or with PBS. The inguinal lymph-nodes (i.e., draining lymph-nodes of the lesion) in the right thigh and those in the left thigh were harvested and their cells were coincubated with B3Z cells. The proportion of APC presenting SIINFEKL as a surrogate TA peptide, which migrated from the treated lesion to the draining lymph-node could be deduced from the percentage of activated B3Z cells. The proportion of SIINFEKL presenting APC in the right leg inguinal lymph-nodes draining the B16/OVA lesion injected with α-gal micelles was on average 6-fold higher than that in the inguinal lymph-nodes draining PBS treated tumors. These findings strongly suggested a robust uptake of OVA as surrogate TA in B16/OVA cells, as well as transport and processing of the OVA peptide SIINFEKL by APC in α-gal micelles injected lesions in comparison to PBS injected control lesions.
5.6. Increased Protection against Distant Metastatic Cells by Intra-Tumoral Injection of α-Gal Micelles
The efficacy of α-gal micelles in inducing a systemic protective immune response which prevents the development of distant “established metastases” (abscopal effect) was evaluated in anti-Gal producing GT-KO mice by two methods. First the mice were injected subcutaneously with live 1x106 B16 melanoma cells in the right flank (simulating the “primary tumor”). When the tumors reached 5 mm (after ~5 days) the tumors were injected with 1 mg α-gal micelles in PBS. One and two weeks later, the mice received two additional injections of 1 mg α-gal micelles. Control mice received 3 injections of PBS instead of α-gal micelles. One week after the third injection the mice were challenged with 0.5x106 live B16 melanoma cells in the left flank, thereby simulating distant metastatic cells. Ten of the 15 mice treated with α-gal micelles displayed no growth of the challenging tumor cells into a melanoma lesion (Figure 6A). In contrast, the challenging tumor cells in all PBS treated mice developed into melanoma lesions (Figure 6B).
In a second experiment, simultaneously with the injection of 1x106 B16 melanoma cells in the right flank as “primary tumor”, the mice were injected with 1x104 tumor cells in the left flank, which simulated a “distant micrometastasis”. The primary tumor lesions reaching the size of ∼5 mm were injected with 1 mg α-gal micelles, or with PBS as control. The injections were repeated one week later and development of the “distant metastasis” in each of the mice was monitored. In this experiment, distant micrometastases were established for 5 days before the primary tumor was injected with α-gal micelles. In all PBS-treated mice, “distant metastasis” developed into visible 2–8 mm tumor lesions in the left flank by Day 15 after the first injection of the “primary” tumor (Figure 6D). However, in 50% of mice injected with α-gal micelles into the “primary tumor”, no tumor growth was observed at the “distant metastasis” site, even after 30 days. In the rest of the mice in this group, slower tumor growth was observed in comparison to most PBS treated mice (Figure 6C) [82]. These studies indicated that the injection of the melanoma lesions with α-gal micelles resulted in the development of a systemic anti-TA immune response which protected against distant metastases growth in a significant proportion of treated mice.
5.7. CD8+ T Cells Are the Main Protective Cells in Immunized Mice
Identification of the immune cells which mediate the immune protection against the development of distant B16 melanoma micrometastases into tumor lesions was evaluated by adoptive transfer studies of splenocytes from tumor bearing mice treated with α-gal micelles. B16 tumor lesions in GT-KO mice producing anti-Gal were injected twice with 1.0 mg α-gal micelles in one week interval. The spleens of the tumor bearing mice were harvested one week after the second injection. Forty millions of the harvested splenocytes were administered intravenously to naïve GT-KO mice who were also challenged 24 hrs earlier with 0.5×106 live B16 cells. The rest of the splenocytes were subjected to depletion of CD8+ T cells using magnetic micro-beads coated with anti-CD8+ antibody. Forty millions of the splenocytes depleted of CD8+ T cells were administered to paired naïve mice challenged with B16 cells 24 hrs earlier [82]. As shown in Figure 7, the transferred splenocytes prevented the development of challenging B16 cells into melanoma lesions in most of the recipients. However, all paired recipients of splenocytes depleted of CD8+ T cells displayed growth of tumors that reached the size of 25 mm within < 30 days. These findings clearly indicated that the main cell population which mediated destruction of challenging B16 cells in the naïve recipients was the TA specific CD8+ T cells that were activated following administration of α-gal micelles into the treated lesions of the donor mice [82].
Overall, studies with α-gal micelles in the pre-clinical model of anti-Gal producing GT-KO mice strongly suggested that administration of these micelles into solid tumors can convert them into in situ vaccines against TA on the treated tumor cells, without the need for identifying the TA. Such vaccines may induce a protective immune response against tumor cells that present both “private” and “public” TA in the treated patient. In the absence of α-gal micelles treatment, the patient’s TA may be “ignored” by the immune system because of insufficient uptake and processing of TA by APC and and thus, poor presentation of the immunogenic TA peptides for T cell activation.
6. Clinical Trials with Natural α-Gal Micelles
Based on the studies described above, two Phase 1 clinical trials were performed with α-gal micelles. The first was a dose escalation and safety study in patients with solid tumors at advanced stages of the disease [83]. The second trial had the same objectives and was performed in patients with advance cutaneous melanoma [84]. All patients were treated with standard therapy and experienced recurrence of the disease prior to each of the clinical trials.
In the first study, 11 patients with a variety of solid tumors were treated at the UMass Medical Center after approval by a local Human Investigations Committee and in accordance with an assurance approved by the Department of Health and Human Services, (FDA IND 12946). Eight patients had metastatic cancers including colon, pancreatic neuroendocrine, prostate, renal, ovarian, and mucinous appendiceal tumors [83]. The three patients, who did not have metastatic disease had locally advanced pancreatic adenocarcinoma. The patients received intra-tumorally 0.1, 1, or 10 mg α-gal micelles injected into the target tumor lesions under ultrasound guidance, CT guidance, or under manual and visual control in patients who had large palpable tumors. The patients were monitored at regular intervals for a period of 2 years. None of the patients developed clinical or laboratory signs of toxicity, symptoms of allergic responses, autoimmune conditions, anti-nuclear antibodies, or antibodies to normal tissue antigens [83]. Injected tumors did not regress, and patients developed evidence of disease progression ranging from minimal to significant at various time points after the 4-week endpoint for toxicity analysis. However, several patients were alive with disease for 13+ to 48+ months, even though disease progression was evident by imaging. In addition, two patients with advanced pancreatic adenocarcinoma who prior to the treatment were expected to survive only 4-6 months, survived following the α-gal micelle treatment for 18 and 23 months [83].
A second study was performed at the University of Wisconsin Carbone Cancer Center under FDA IND 12946, in 9 patients with unresectable melanoma (recurrent stage III or stage IV) that was refractory to therapy, with at least one cutaneous metastasis [84]. All patients received intra-tumorally two injections of 0.1, 1.0, or 10 mg α-gal micelles, administered 4 weeks apart. Prior to the second injection, the patients received intradermally 10 μg of α-gal micelles and then observed for one hour to confirm the absence of α-gal syndrome. The sizes of treated and untreated lesions were monitored during regular clinic visits. Two patients maintained stable disease for 8 and 7 months after treatment, and the remaining seven patients had disease progression. Three of the injected lesions were stable in size, 4 weeks after the second of α-gal micelles injection and the remaining six lesions had at least a 20% increase in that period. However, several findings suggested some response to the treatment. Increased necrosis of the tumor tissue was demonstrated in biopsies of the injected lesions in five of nine patients, and in nontreated tumor lesions in two of four evaluable patients. In addition, in three of eight patients an approximate two-fold to five-fold increase in the frequency of public TA pentamer+ cells were observed post treatment vs. pre-treatment [84].
Overall, these two Phase I clinical trials indicated that the treatment was safe, however, the treatment in the advanced stages of the disease did not induce a potent enough protective immune response to result in complete cure or in distinct regression of tumors. It is possible that increased doses may be more effective in eliciting a protective immune response in patients with large tumor burden. In addition, it is reasonable to assume that the treatment with α-gal micelles is likely to be more effective at early stages of the disease, when the tumor burden is lower and thus, the probability of the immune system for “catching up” with the developing metastases is higher. It is also important to administer this treatment to patients in whom the immune system is functional and not impaired by recent chemotherapy treatments.
7. Conversion of Self-TA into Vaccines by Synthetic α-gal micelles
The induction of a protective immune response against distant metastases in mice with B16 melanoma lesions injected with α-gal micelles [50,82], prompted the biotech companies Agalimmune Ltd. and BioLineRx Ltd. to determine whether a similar conversion of tumors into vaccine against self-TA is feasible with synthetic α-gal glycolipids [38]. For this purpose, a fully synthetic α-gal glycolipid-like molecule, called “AGI-134” was chosen (produced by Kode Biotech, Auckland, NZ). AGI-134 is comprised of a synthetic α-gal epitope linked via an adipate (di-carboxylate) linker to a lipid tail [85].
As detailed in ref. [38], in vitro studies with AGI-134 indicated that it can insert well into the cell membrane of both human and mouse tumor cells and bind human and mouse anti-Gal antibodies. This binding further activated the complement cascade and induced CDC as well as ADCC. AGI-134-treated B16 cells were opsonized by anti-Gal and phagocytosed by APC. In addition, ovalbumin (OVA) serving as a surrogate TA in cells treated with AGI-134 was effectively processed following uptake by APC and its immunodominant peptide SIINFEKL was cross-presented by the APC [38].
In vivo studies that were conducted in anti-Gal producing GT-KO mice with B16 melanoma cells, indicated that intra-tumoral administration of AGI-134 into B16 melanoma lesions resulted in regression of the subcutaneous tumors, similar to the regression observed with natural α-gal micelles in Figure 4 above. Moreover, in most mice, administration of AGI-134 into “primary” tumors induced a protective immune response that prevented development of distant metastases, simulated by 104 B16 cells injected into the contralateral flank [38]. No protective immune response was observed if AGI-134 was administered to tumors in mice that lacked the anti-Gal antibody. The efficacy of AGI-134 was found to be dose-dependent and displayed the maximal efficacy of distant metastases prevention at a 1-mg dose. At suboptimal doses 0.5 mg and 0.1 mg the efficacy was reduced, however significant protection was observed in comparison to PBS controls. It is of interest to note that AGI-134 at suboptimal doses synergized with the check point inhibitor anti-PD-1 antibody in preventing development of distant metastases [38].
Overall, the studies with AGI-134 suggest that it displays characteristics like those of the natural α-gal micelles in that its administration into tumors results in conversion of the treated lesions into vaccines that induce a protective immune response against self-TA, thereby preventing metastases formation.
8. Clinical Trial with Synthetic α-Gal Micelles
The pre-clinical studies with AGI-134 described above [38] were followed by a multicenter open-label Phase 1/2a clinical trial performed by BiolineRX Ltd, in order to evaluate safety and efficacy of AGI-134 immunotherapy. The study was performd with 38 patients who had a variety of solid tumor in the UK, Spain and Israel and was listed as ClinicalTrials.gov ID NCT03593226. The results of this trial were announced so far only as a press release by BioLineRX on December 20, 2022 [86]. That press release indicated that after an accelerated dose-escalation study in 5 patients to determine the maximum tolerated dose, the safety and tolerability of AGI-134 was evaluated in 33 patients. The treatment with AGI-134 was found to be well tolerated, and the adverse events were mostly transient, and mild to moderate in their severity. Clinical response evaluation according to RECIST1.1 criteria indicated that a stable disease (SD) was observed in 11 of the 38 patients (29%) and that 7 of these 11 patients had a prior checkpoint inhibitor therapy which failed. Biopsy studies indicated that following treatment with AGI-134, leukocyte infiltration including macrophages and dendritic cells was observed within few days in 59% of the treated tumors. One of the most significant indications for an anti-tumor immune response following AGI-134 treatment has been infiltration of T helper cells and of cytotoxic T cells in 47% of the un-injected tumor lesions. These findings support the assumption that α-gal glycolipids inserted into tumor cell membranes bind anti-Gal and target these cells and cell membranes for effective uptake by APC. These APC transport processed TA peptides to lymph-nodes for the activation of TA specific CD4+ and CD8+ T cells.
9. Conclusions Regarding the Possible Efficacy of α-Gal Micelles Immunotherapy at Various Stages of the Disease
The clinical trials with the natural and synthetic α-gal micelles in cancer patients with advanced disease indicated that these micelles are not toxic, and their use is safe. However, the ability of this therapy to induce a protective immune response against self-TA in the patients was found to be lower than that observed in the pre-clinical animal model. It is probable that the efficacy of this immunotherapy greatly decreases in advanced stages of the disease when the total tumor mass in the primary tumor and in metastases is high. All patients approved for participation in the Phase 1 studies described above were in advance stages of the disease and failed standard therapies. In addition, the immune system in some of the patients may have been suppressed to various degrees due to earlier chemotherapy treatments. The studies in mice suggest that when the tumor mass is large, even an effective immune response cannot “catch up” with the expanding tumor metastases and destroy them (Figure 2). However, in earlier stages of the disease, the treatment in mice succeeded in destroying tumor cells distant from the treated lesions (Figure 6 and Figure 7 and ref. [38]). These findings suggest that the α-gal micelles immunotherapy is likely to be most effective as a neo-adjuvant therapy. Since resection of tumors is performed in many patients several weeks after detection, it is suggested that intra-tumoral injection of α-gal micelles should be performed immediately upon detection of the tumor. Recruitment of APC into the treated lesion, the uptake of anti-Gal opsonized tumor cells and cell membranes by APC and migration of such APC out of the tumor to lymph nodes may occur within 1-3 weeks post treatment. Thus, at the time of resection of the treated lesion, the APC may be in the course of transporting processed TA peptides to lymph nodes for eliciting a protective immune response against metastatic cells presenting these TA. Tumor cell proliferation in humans is usually much slower than that of the B16 melanoma cells in mice. Therefore, α-gal micelles immunotherapy may be beneficial in patients with micro-metastases which are not visible at the time of detection of the primary tumor. It is of note that patients with α-gal syndrome and those suspected to have this syndrom because of allergy to meat, may also require anti-allergic treatment while being treated with α-gal micelles.
The abscopal effects of α-gal micelles treatment in the experimental model also suggest that injection of such natural or synthetic micelles into several of the accessible metastases may help in prevention of disease progression due to eliciting an anti-TA protective immune response. However, as indicated above, there seems to be an inverse relationship between the size of the tumor mass and the efficacy of the anti-TA immune response induced by the α-gal micelles treatment. In addition, patients with suppressed immune system due to recent chemotherapy treatments may not be able to mount an effectve anti-TA immune response following α-gal micelles treatment.
Theoretically, immunotherapy with α-gal micelles may be applicable also in patients with hematological malignancies. Spontaneous insertion of α-gal glycolipids following in vitro incubation of B16 melanoma cells with α-gal micelles was demonstrated in Figure 3C and 3D. Similar in vitro incubation of lethally irradiated human leukemia, lymphoma or myeloma cells with α-gal micelles at 37oC results in insertion of α-gal glycolipids into tumor cell membranes and presentation of multiple α-gal epitopes on the tumor cells. Immunization of the treated patient by such personalized anti-TA vaccines presenting α-gal epitopes may result in activation of the immune system against the self-TA according to the steps described in Figure 1A.
In conclusion, the studies described above suggest that α-gal micelles immunotherapy may be considered for use in cancer patients as a neo-adjuvant therapy or as an adjuvant treatment that enable effective activation of the immune system against the full range of TA in the individual patient without the need for identifying these TA.
Funding
This review received no funding.
Conflicts of Interest
The author declares no conflict of interest.
References
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Berger, A.; Bindea, G.; Meatchi, T.; Bruneval, P.; Trajanoski, Z.; Fridman, W.H.; Pagès, F.; et al. Histopathologic-based prognostic factors of colorectal cancers are associated with the state of the local immune reaction. J. Clin. Onco. 2011, 29, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intra-tumoral T cells, recurrence, and survival in epithelial ovarian cancer. New Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Otterlei Fjørtoft, M.; Huse, K.; Rye, I.H. The Tumor Immune Microenvironment in Breast Cancer Progression. Acta Oncol. 2024, 63, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Wood, S.J.; Gao, Y.; Lee, J.H.; Chen, J.; Wang, Q.; Meisel, J.L.; Li, X. High tumor infiltrating lymphocytes are significantly associated with pathological complete response in triple negative breast cancer treated with neoadjuvant KEYNOTE-522 chemoimmunotherapy. Breast Cancer Res Treat. 2024, 205, 193–199. [Google Scholar] [CrossRef]
- Lin, M.J.; Svensson-Arvelund, J.; Lubitz, G.S.; Marabelle, A.; Melero, I.; Brown, B.D.; Brody, J.D. Cancer vaccines: The next immunotherapy frontier. Nat. Cancer 2022, 3, 911–926. [Google Scholar] [CrossRef] [PubMed]
- Jeon, D.; Hill, E.; McNeel, D.G. Toll-like receptor agonists as cancer vaccine adjuvants. Hum. Vaccine Immunother. 2024, 20, 2297453. [Google Scholar] [CrossRef]
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjöblom, T.; Leary, R.J.; Shen, D.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The genomic landscapes of human breast and colorectal cancers. Science 2007, 318, 1108–1113. [Google Scholar] [CrossRef]
- Campbell, P.J.; Yachida, S.; Mudie, L.J.; Stephens, P.J.; Pleasance, E.D.; Stebbings, L.A.; Morsberger, L.A.; Latimer, C.; McLaren, S.; Lin, M.L.; et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 2010, 467, 1109–1113. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A. Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Efremova, M.; Finotello, F.; Rieder, D.; Trajanoski, Z. Neoantigens Generated by Individual Mutations and Their Role in Cancer Immunity and Immunotherapy. Front Immunol. 2017, 8, 1679. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.; Jordan, K.R.; Buhrman, J.D.; Lei, J.; Deng, H.; Marrack, P.; Dai, S.; Kappler, J.W.; Slansky, J.E.; Yin, L. Structures suggest an approach for converting weak self-peptide tumor antigens into superagonists for CD8 T cells in cancer. Proc. Natl. Acad. Sci. U S A. 2021, 118, e2100588118. [Google Scholar] [CrossRef] [PubMed]
- Lin MJ, Svensson-Arvelund J, Lubitz GS, Marabelle A, Melero I, Brown BD, Brody JD. Cancer vaccines: The next immunotherapy frontier. Nat Cancer. 2022, 3, 911–926.
- Millar, D.G.; Yang, S.Y.C.; Sayad, A.; Zhao, Q.; Nguyen, L.T.; Warner, K.; Sangster, A.G.; Nakatsugawa, M.; Murata, K.; Wang, B.X.; et al. Identification of antigenic epitopes recognized by tumor infiltrating lymphocytes in high grade serous ovarian cancer by multi-omics profiling of the auto-antigen repertoire. Cancer Immunol. Immunother. 2023, 72, 2375–2392. [Google Scholar] [CrossRef] [PubMed]
- Galili, U. Anti-Gal: An abundant human natural antibody of multiple pathogeneses and clinical benefits. Immunology 2013, 140, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Galili, U.; Rachmilewitz, E.A.; Peleg, A.; Flechner, I. A unique natural human IgG antibody with anti-α-galactosyl specificity. J. Exp. Med. 1984, 160, 1519–1531. [Google Scholar] [CrossRef] [PubMed]
- Galili, U.; Mandrell, R.E.; Hamadeh, R.M.; Shohet, S.B.; Griffiss, J.M. Interaction between human natural anti-α-galactosyl immunoglobulin G and bacteria of the human flora. Infect. Immun. 1988, 56, 1730–1737. [Google Scholar] [CrossRef] [PubMed]
- Posekany, K.J.; Pittman, H.K.; Bradfield, J.F.; Haisch, C.E.; Verbanac, K.M. Induction of cytolytic anti-Gal antibodies in α-1,3-galactosyltransferase gene knockout mice by oral inoculation with Escherichia coli O86:B7 bacteria. Infect. Immun. 2002, 70, 6215–6222. [Google Scholar] [CrossRef] [PubMed]
- Mañez, R.; Blanco, F.J.; Díaz, I.; Centeno, A.; Lopez-Pelaez, E.; Hermida, M.; Davies, H.F.; Katopodis, A. Removal of bowel aerobic gram-negative bacteria is more effective than immunosuppression with cyclophosphamide and steroids to decrease natural α-galactosyl IgG antibodies. Xenotransplantation 2001, 8, 15–23. [Google Scholar] [CrossRef]
- Galili, U.; Anaraki, F.; Thall, A.; Hill-Black, C.; Radic, M. One percent of circulating B lymphocytes are capable of producing the natural anti-Gal antibody. Blood 1993, 82, 2485–2493. [Google Scholar] [CrossRef]
- Hamadeh, R.M.; Galili, U.; Zhou, P.; Griffiss, J.M. Human secretions contain IgA, IgG and IgM anti-Gal [anti-α-galactosyl] antibodies. Clin. Diagn. Lab. Immunol. 1995, 2, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Minanov, O.P.; Itescu, S.; Neethling, F.A.; Morgenthau, A.S.; Kwiatkowski, P.; Cooper, D.K.; Michler, R.E. Anti-Gal IgG antibodies in sera of newborn humans and baboons and its significance in pig xenotransplantation. Transplantation 1997, 63, 182–186. [Google Scholar] [CrossRef]
- Doenz, U.; Nydegger, U.E.; Kueng, A.; Carrel, T.; Mohacsi, P. Anti-Galα1-3Gal IgM/IgG antibody levels in infants: Do they have a clinical relevance in pediatric xenotransplantation? J. Heart Lung Transpl. 2000, 19, 1108–1113. [Google Scholar] [CrossRef]
- Hamanova, M.; Chmelikova, M.; Nentwich, I.; Thon, V.; Lokaj, J. Anti-gal IgM, IgA and IgG natural antibodies in childhood. Immunol. Lett. 2015, 164, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Galili, U.; Macher, B.A.; Buehler, J.; Shohet, S.B. Human natural anti-α-galactosyl IgG. II. The specific recognition of α[1,3]-linked galactose residues. J. Exp. Med. 1985, 162, 573–582. [Google Scholar] [CrossRef]
- Galili, U.; Buehler, J.; Shohet, S.B.; Macher, B.A. The human natural anti-Gal IgG. III. The subtlety of immune tolerance in man as demonstrated by crossreactivity between natural anti-Gal and anti-B antibodies. J. Exp. Med. 1987, 165, 693–704. [Google Scholar]
- Towbin, H.; Rosenfelder, G.; Wieslander, J.; Avila, J.L.; Rojas, M.; Szarfman, A.; Esser, K.; Nowack, H.; Timpl, R. Circulating antibodies to mouse laminin in Chagas disease, American cutaneous leishmaniasis, and normal individuals recognize terminal galactosyl [α1-3]-galactose epitopes. J. Exp. Med. 1987, 166, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Teneberg, S.; Lönnroth, I.; Torres Lopez, J.F.; Galili, U.; Olwegard Halvarsson, M.; Angstrom, J.; Karlsson, K.A. Molecular mimicry in the recognition of glycosphingolipids by Galα3Galß4GlcNAcß-binding Clostridium difficile toxin A, human natural anti-α-galactosyl IgG and the monoclonal antibody Gal-13: Characterization of a binding-active human glycosphingolipid, non-identical with the animal receptor. Glycobiology 1996, 6, 599–609. [Google Scholar]
- Galili, U.; Clark, M.R.; Shohet, S.B.; Buehler, J.; Macher, B.A. Evolutionary relationship between the anti-Gal antibody and the Galα1-3Gal epitope in primates. Proc. Natl. Acad. Sci. USA. 1987, 84, 1369–1373. [Google Scholar] [CrossRef]
- Galili, U.; Shohet, S.B.; Kobrin, E.; Stults, C.L.M.; Macher, B.A. Man, apes, and Old-World monkeys differ from other mammals in the expression of α-galactosyl epitopes on nucleated cells. J. Biol. Chem. 1988, 263, 17755–17762. [Google Scholar] [CrossRef]
- Larsen, R.D.; Rajan, V.P.; Ruff, M.; Kukowska-Latallo, J.; Cummings, R.D.; Lowe, J.B. Isolation of a cDNA encoding murine UDP galactose: ßD-galactosyl-1,4-N-acetyl-D-glucosaminide α1,3-galactosyltransferase: Expression cloning by gene transfer. Proc. Natl. Acad. Sci. USA. 1989, 86, 8227–8231. [Google Scholar] [CrossRef] [PubMed]
- Joziasse, D.H.; Shaper, J.H.; Van den Eijnden, D.H.; Van Tunen, A.H.; Shaper, N.L. Bovine α1-3galactosyltransferase: Isolation and characterization of a cDNA clone. Identification of homologous sequences in human genomic DNA. J. Biol. Chem. 1989, 264, 14290–14297. [Google Scholar] [CrossRef] [PubMed]
- Galili, U.; Swanson, K. Gene sequences suggest inactivation of α1-3 galactosyltransferase in catarrhines after the divergence of apes from monkeys. Proc. Natl. Acad. Sci. USA. 1991, 88, 7401–7404. [Google Scholar] [CrossRef] [PubMed]
- Larsen, R.D.; Rivera-Marrero, C.A.; Ernst, L.K.; Cummings, R.D.; Lowe, J.B. Frameshift and nonsense mutations in a human genomic sequence homologous to a murine UDP-Gal:β-D-Gal[1,4]-D-GlcNAc α[1,3]-galactosyltransferase cDNA. J. Biol. Chem. 1990, 265, 7055–7061. [Google Scholar] [CrossRef] [PubMed]
- Galili, U. Paleo-immunology of human anti-carbohydrate antibodies preventing primate extinctions. Immunology 2023, 168, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Galili, U. Interaction of the natural anti-Gal antibody with α-galactosyl epitopes: A major obstacle for xenotransplantation in humans. Immunol. Today 1993, 14, 480–482. [Google Scholar] [CrossRef] [PubMed]
- Almeida, I.C.; Milani, S.R.; Gorin, P.A.J.; Travassos, L.R. Complement-mediated lysis of Trypanosoma cruzi trypomastigotes by human anti-α-galactosyl antibodies. J. Immunol. 1991, 146, 2394–2400. [Google Scholar] [CrossRef]
- Shaw, S.M.; Middleton, J.; Wigglesworth, K.; Charlemagne, A.; Schulz, O.; Glossop, M.S.; Whalen, G.F.; Old, R.; Westby, M.; Pickford, C; et al. AGI-134: A fully synthetic α-Gal glycolipid that converts tumors into in situ autologous vaccines, induces anti-tumor immunity and is synergistic with an anti-PD-1 antibody in mouse melanoma models. Cancer Cell Int. 2019, 19, 346. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.K.C.; Good, A.H.; Koren, E.; Oriol, R.; Malcolm, A.J.; Ippolito, R.M.; Neethling, F.A.; Ye, Y.; Romano, E.; Zuhdi, N. Identification of α-galactosyl and other carbohydrate epitopes that are bound by human anti-pig antibodies: Relevance to discordant xenografting in man. Transpl. Immunol. 1993, 1, 198–205. [Google Scholar] [CrossRef]
- Sandrin, M.S.; Vaughan, H.A.; Dabkowski, P.L.; McKenzie, I.F.C. Anti-pig IgM antibodies in human serum react predominantly with Gal (αl-3)Gal epitopes. Proc. Natl. Acad. Sci. USA. 1993, 90, 11391–11395. [Google Scholar] [CrossRef]
- Collins, B.H.; Cotterell, A.H.; McCurry, K.R.; Alvarado, C.G.; Magee, J.C.; Parker, W.; Platt, J.L. Cardiac xenografts between primate species provide evidence for the importance of the α-galactosyl. determinant in hyperacute rejection. J. Immunol. 1995, 154, 5500–5510. [Google Scholar] [PubMed]
- Galili, U.; Wigglesworth, K.; Abdel-Motal, U.M. Accelerated healing of skin burns by anti-Gal/alpha-gal liposomes interaction. Burns 2010, 36, 239–251. [Google Scholar] [CrossRef]
- Wigglesworth, K.M.; Raski, W.J.; Mishra, R.; Szomolanyi-Tsuda, E.; Greiner, D.L.; Galili, U. Rapid recruitment and activation of macrophages by anti-Gal/α-Gal liposome interaction accelerates wound healing. J. Immunol. 2011, 186, 4422–4432. [Google Scholar] [CrossRef] [PubMed]
- Galili, U. α-Gal Nanoparticles in Wound and Burn Healing Acceleration. Adv. Wound Care (New Rochelle). 2017, 6, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Galili, U.; Zhu, Z.; Chen, J.; Goldufsky, J.W.; Schaer, G.L. Near Complete Repair after Myocardial Infarction in Adult Mice by Altering the Inflammatory Response with Intramyocardial Injection of α-Gal Nanoparticles. Front. Cardiovasc. Med. 2021, 8, 719160. [Google Scholar] [CrossRef] [PubMed]
- Galili, U.; Li, J.; Schaer, G.L. Regeneration in Mice of Injured Skin, Heart, and Spinal Cord by α-Gal Nanoparticles Recapitulates Regeneration in Amphibians. Nanomaterials (Basel) 2024, 22, 730. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, B.; Galili, U.; Seanger, M.; Burket, N.J.; Koss, W.; Lokender, M.S.; Wolfe, K.M.; Husak, S.J.; Stark, C.J.; Solorioet, C.; et al. Alpha-Gal Nanoparticles in CNS Trauma: II. Immunomodulation Following Spinal Cord Injury (SCI) Improves Functional Outcomes. Tissue Eng. Regen. Med. 2024, 21, 437–453. [Google Scholar] [CrossRef] [PubMed]
- LaTemple, D.C.; Henion, T.R.; Anaraki, F.; Galili, U. Synthesis of alpha-galactosyl epitopes by recombinant alpha1,3galactosyl transferase for opsonization of human tumor cell vaccines by anti-galactose. Cancer Res. 1996, 56, 3069–3074. [Google Scholar] [PubMed]
- Galili, U.; LaTemple, D.C. Natural anti-Gal antibody as a universal augmenter of autologous tumor vaccine immunogenicity. Immunol Today 1997, 18, 281–285. [Google Scholar] [CrossRef]
- Galili, U.; Wigglesworth, K.; Abdel-Motal, U.M. Intra-tumoral injection of α-gal glycolipids induces xenograft-like destruction and conversion of lesions into endogenous vaccines. J. Immunol. 2007, 178, 4676–4687. [Google Scholar] [CrossRef]
- Abdel-Motal, U.M.; Guay, H.M.; Wigglesworth, K.; Welsh, R.M.; Galili, U. Increased immunogenicity of influenza virus vaccine by anti-Gal mediated targeting to antigen presenting cells. J. Virol. 2007, 81, 9131–9141. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Motal, U.M.; Wang, S.; Lu, S.; Wigglesworth, K.; Galili, U. Increased Immunogenicity of Human Immunodeficiency Virus gp120 Engineered to Express Galα1-3Galβ1-4GlcNAc-R Epitopes. J. Virol. 2006, 80, 6943–6951. [Google Scholar] [CrossRef] [PubMed]
- Manches, O.; Plumas, J.; Lui, G.; Chaperot, L.; Molens, J.P.; Sotto, J.J.; Bensa, J.C.; Galili, U. Anti-Gal-mediated targeting of human B lymphoma cells to antigen-presenting cells: A potential method for immunotherapy using autologous tumor cells. Haematologica. 2005, 90, 625–634. [Google Scholar] [PubMed]
- Galili, U. The natural anti-Gal antibody as foe turned friend in medicine; Academic Press/Elsevier Publishers: London; Amsterdam; New York, 2018. [Google Scholar]
- Thall, A.D.; Maly, P.; Lowe, J.B. Oocyte Gala1,3Gal epitopes implicated in sperm adhesion to the zona pellucida glycoprotein ZP3 are not required for fertilization in the mouse. J. Biol. Chem. 1995, 270, 21437–21440. [Google Scholar] [CrossRef] [PubMed]
- Tearle, R.G.; Tange, M.J.; Zannettino, Z.L.; Katerelos, M.; Shinkel, T.A.; Van Denderen, B.J.; Lonie, A.J.; Lyons, I.; Nottle, M.B.; Cox, T.; et al. The a-1,3-galactosyltransferase knockout mouse. Implications for xenotransplantation. Transplantation 1996, 61, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Cretin, N.; Bracy, J.; Hanson, K.; Iacomini, J. The role of T cell help in the production of antibodies specific for Gal alpha 1-3Gal. J. Immunol. 2002, 168, 1479–1483. [Google Scholar] [CrossRef] [PubMed]
- LaTemple, D.C.; Abrams, J.T.; Zhang, S.Y.; Galili, U. Increased immunogenicity of tumor vaccines complexed with anti-Gal: Studies in knockout mice for alpha1,3galactosyltransferase. Cancer Res. 1999, 59, 3417–3423. [Google Scholar] [PubMed]
- Benatuil, L.; Kaye, J.; Rich, R.F.; Fishman, J.A.; Green, W.R.; Iacomini, J. The influence of natural antibody specificity on antigen immunogenicity. Eur. J. Immunol. 2005, 35, 2638–2647. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.R.; Mautino, M.R.; Unfer, R.C.; Seregina, T.M.; Vahanian, N.; Link, C.J. Effective treatment of preexisting melanoma with whole cell vaccines expressing alpha(1,3)-galactosyl epitopes. Cancer Res. 2005, 65, 10555–10561. [Google Scholar] [CrossRef]
- Deriy, L.; Ogawa, H.; Gao, G.P.; Galili, U. In vivo targeting of vaccinating tumor cells to antigen-presenting cells by a gene therapy method with adenovirus containing the alpha1,3galactosyltransferase gene. Cancer Gene Ther. 2005, 12, 528–539. [Google Scholar] [CrossRef]
- Deguchi, T.; Tanemura, M.; Miyoshi, E.; Nagano, H.; Machida, T.; Ohmura, Y.; Kobayashi, S.; Marubashi, S.; Eguchi, H.; Takeda, Y.; et al. Increased immunogenicity of tumor-associated antigen, mucin 1, engineered to express alpha-gal epitopes: A novel approach to immunotherapy in pancreatic cancer. Cancer Res. 2010, 70, 5259–5269. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Dong, Z.; Zhang, Q.; Wang, Q.; Lai, D. Epithelial ovarian cancer stem-like cells expressing α-gal epitopes increase the immunogenicity of tumor associated antigens. BMC Cancer 2015, 15, 956. [Google Scholar] [CrossRef] [PubMed]
- Mo, F.; Xue, D.; Duan, S.; Liu, A.; Yang, X.; Hou, X.; Lu, X. Novel fusion cells derived from tumor cells expressing the heterologous α-galactose epitope and dendritic cells effectively target cancer. Vaccine 2019, 37, 926–936. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Xu, M.B.; Yun, M.M.; Wang, Y.Z.; Zhang, R.M.; Meng, X.K.; Ou-Yang, X.H.; Yun, S. Hepatocellular Carcinoma-specific Immunotherapy with Synthesized α1,3 Galactosyl Epitope-Pulsed Dendritic Cells and Cytokine-Induced Killer Cells. World J Gastroenterol. 2011, 17, 5260–5266. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Yun, M.M.; Xu, M.B.; Wang, Y.Z.; d Yun, S. Pancreatic Carcinoma-specific Immunotherapy Using Synthesised Alpha-Galactosyl Epitope-Activated Immune Responders: Findings from a Pilot Study. Int. J. Clin. Oncol. 2013, 18, 657–665. [Google Scholar] [CrossRef]
- Qiu, Y.; Yun, M.M.; Dong, X.; Xu, M.; Zhao, R.; Han, X.; Ou-Yang, X.H.; Yun, S. Combination of Cytokine-Induced Killer and Dendritic Cells Pulsed with Antigenic α-1,3 galactosyl Epitope-Enhanced Lymphoma Cell Membrane for Effective B-Cell Lymphoma Immunotherapy. Cytotherapy 2016, 18, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Deriy, L.; Chen, Z.-C.; Gao, G.-P.; Galili, U. Expression of α-gal Epitopes on HeLa Cells Transduced with Adenovirus Containing α1,3galactosyltransferase cDNA. Glycobiology 2002, 12, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Eto, T.; Iichikawa, Y.; Nishimura, K.; Ando, S.; Yamakawa, T. Chemistry of lipids of the posthemolytic residue or stroma of erythrocytes. XVI. Occurrence of ceramide pentasaccharide in the membrane of erythrocytes and reticulocytes in rabbit. J. Biochem. (Tokyo) 1968, 64, 205–213. [Google Scholar] [CrossRef]
- Stellner, K.; Saito, H.; Hakomori, S. Determination of aminosugar linkage in glycolipids by methylation. Aminosugar linkage of ceramide pentasaccharides of rabbit erythrocytes and of Forssman antigen. Arch. Biochem. Biophys. 1973, 133, 464–472. [Google Scholar] [CrossRef]
- Honma, K.; Manabe, H.; Tomita, M.; Hamada, A. Isolation and partial structural characterization of macroglycolipid from rabbit erythrocyte membranes. J. Biochem. 1981, 90, 1187–1196. [Google Scholar] [CrossRef]
- Dabrowski, U.; Hanfland, P.; Egge, H.; Kuhn, S.; Dabrowski, J. Immunochemistry of I/i-active oligo- and polyglycosylceramides from rabbit erythrocyte membranes. Determination of branching patterns of a ceramide pentadecasaccharide by 1H nuclear magnetic resonance. J. Biol. Chem. 1984, 259, 7648–7651. [Google Scholar] [PubMed]
- Egge, H.; Kordowicz, M.; Peter-Katalinic, J.; Hanfland, P. Immunochemistry of I/i-active oligo- and polyglycosylceramides from rabbit erythrocyte membranes. J. Biol. Chem. 1985, 260, 4927–4935. [Google Scholar] [CrossRef] [PubMed]
- Hanfland, P.; Kordowicz, M.; Peter-Katalinic, J.; Egge, H.; Dabrowski, J.; Dabrowski, J. Structure elucidation of blood group B-like and I-active ceramide eicosa- and pentacosasaccharides from rabbit erythrocyte membranes by combined gas chromatography-mass spectrometry; electron-impact and fast atom-bombardment mass spectrometry; and two-dimensional correlated, relayed-coherence transfer, and nuclear Overhauser effect 500-MHz 1H-n.m.r. spectroscopy. Carbohydr. Res. 1988, 178, 1–21. [Google Scholar] [PubMed]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipids from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Galili, U.; LaTemple, D.C.; Radic, M.Z. A sensitive assay for measuring α-gal epitope expression on cells by a monoclonal anti-Gal antibody. Transplantation 1998, 65, 1129–1132. [Google Scholar] [CrossRef] [PubMed]
- 77. Galili, U.; Albertini, M.R.; Sondel, P.M.; Wigglesworth, K.; Sullivan, M.; Whalen, G.F. In Situ Conversion of Melanoma Lesions into Autologous Vaccine by Intra-tumoral Injections of α-gal Glycolipids. Cancers (Basel). 2010, 2, 773–793. [Google Scholar] [CrossRef] [PubMed]
- Lugade, A.A.; Moran, J.P.; Gerber, S.A.; Rose, R.C.; Frelinger, J.G.; Lord, E.M. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J. Immunol. 2005, 174, 7516–7523. [Google Scholar] [CrossRef] [PubMed]
- Karttunen, J.; Sanderson, S.; Shastri, N. Detection of rare antigen-presenting cells by the lacZ T-cell activation assay suggests an expression cloning strategy for T-cell antigens. Proc. Natl. Acad. Sci. USA 1992, 89, 6020–6024. [Google Scholar] [CrossRef] [PubMed]
- Shastri, N.; Gonzalez, F. Endogenous generation and presentation of the ovalbumin peptide/Kb complex to T cells. J. Immunol. 1993, 150, 2724–2736. [Google Scholar] [CrossRef]
- Sanderson, S.; Shastri, N. LacZ inducible, antigen/MHC-specific T cell hybrids. Int. Immunol. 1994, 6, 369–376. [Google Scholar] [CrossRef]
- Abdel-Motal, U.M.; Wigglesworth, K.; Galili, U. Intra-tumoral injection of a-gal glycolipids induces a protective anti-tumor T cell response which overcomes Treg activity. Cancer Immunol. Immunother. 2009, 58, 1545–1556. [Google Scholar] [CrossRef] [PubMed]
- Whalen, G.F.; Sullivan, M.; Piperdi, B.; Wasseff, W.; Galili, U. Cancer immunotherapy by intra-tumoral injection of α-gal glycolipids. Anticancer Res. 2012, 32, 3861–3868. [Google Scholar] [PubMed]
- Albertini, M.R.; Ranheim, E.A.; Zuleger, C.L.; Sondel, P.M.; Hank, J.A.; Bridges, A.; Newton, M.A.; McFarland, T.; Collins, J.; Clements, E.; et al. Phase I study to evaluate toxicity and feasibility of intra-tumoral injection of α-gal glycolipids in patients with advanced melanoma. Cancer Immunol Immunother. 2016, 65, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Georgakopoulos, T.; Komarraju, S.; Henry, S.; Bertolini, J. An improved Fc function assay utilizing CMV antigen-coated red blood cells generated with synthetic function-spacer-lipid constructs. Vox Sang. 2012, 102, 72–78. [Google Scholar] [CrossRef]
- PR Newswire: BioLineRx Announces Results from Phase 1/2a Study of Investigational Anti-Tumor Vaccine AGI-134 in Metastatic Solid Tumors. Provided by BioLineRx Ltd. December 20, 2022.
Figure 2.
Protective anti-TA vaccination in GT-KO mice with B16α-gal cells. A. Mice free of melanoma lesions in the group immunized with 2x106 B16α-gal cells (●) or with B16 cells (○) and challenged 2 weeks later with 0.5x106 live B16 cells (n=27 mice per group). B. Representative tumor lesion (~10 mm diameter) from a mouse immunized with irradiated B16 cells, 2 weeks prior to challenge with live B16 cells, as (○) in (A). C. As in (B), but the mouse was immunized with B16α-gal cells 2 weeks prior to challenge, as the (●) mice which developed tumor in (A). Note the multiple mononuclear cells surrounding the tumor cells, the vacuoles and melanin granules in the tumor cells adjacent to the infiltrating mononuclear cells. Hematoxylin & Eosin. Modified from ref. [58] with permission.
Figure 2.
Protective anti-TA vaccination in GT-KO mice with B16α-gal cells. A. Mice free of melanoma lesions in the group immunized with 2x106 B16α-gal cells (●) or with B16 cells (○) and challenged 2 weeks later with 0.5x106 live B16 cells (n=27 mice per group). B. Representative tumor lesion (~10 mm diameter) from a mouse immunized with irradiated B16 cells, 2 weeks prior to challenge with live B16 cells, as (○) in (A). C. As in (B), but the mouse was immunized with B16α-gal cells 2 weeks prior to challenge, as the (●) mice which developed tumor in (A). Note the multiple mononuclear cells surrounding the tumor cells, the vacuoles and melanin granules in the tumor cells adjacent to the infiltrating mononuclear cells. Hematoxylin & Eosin. Modified from ref. [58] with permission.
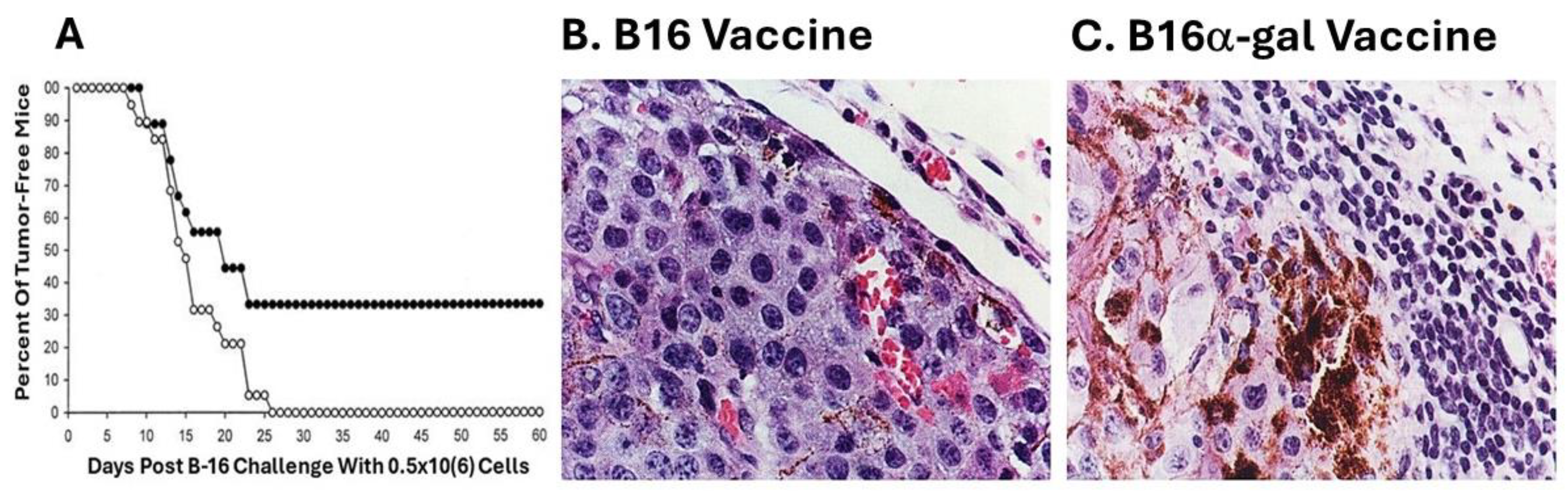
Figure 3.
α-Gal glycolipids in micelles are inserted into tumor cell membranes. A. A representative α-gal glycolipid- ceramide decahexoside (10 carbohydrate units) with two branches each carrying an α-gal epitope (marked by a dashed rectangle). B. Illustrated section in an α-gal micelle comprised of α-gal glycolipids. These glycolipids detach from the micelle and re-insert into it. However, if the micelles are near tumor cell membranes, the α-gal glycolipids spontaneously insert into these membranes in a stable manner, resulting in situ in subsequent binding of anti-Gal to the α-gal epitope on these glycolipids. C. Binding of monoclonal anti-Gal antibody to B16 cells following incubation of these cells for 2h at 37oC with various concentrations of α-gal micelles, as measured by flow cytometry. D. Complement dependent cytolysis (CDC) of B16 cells incubated for 2h with 1mg/ml α-gal micelles. Mouse serum containing anti-Gal and rabbit complement (●); mouse serum without rabbit complement (◯); fresh human serum (▲); control B16 cells lacking α-gal epitopes and incubated with mouse serum and rabbit complement, or with human serum (□) (mean of 4 experiments). Modified with permission from ref. [54]. .
Figure 3.
α-Gal glycolipids in micelles are inserted into tumor cell membranes. A. A representative α-gal glycolipid- ceramide decahexoside (10 carbohydrate units) with two branches each carrying an α-gal epitope (marked by a dashed rectangle). B. Illustrated section in an α-gal micelle comprised of α-gal glycolipids. These glycolipids detach from the micelle and re-insert into it. However, if the micelles are near tumor cell membranes, the α-gal glycolipids spontaneously insert into these membranes in a stable manner, resulting in situ in subsequent binding of anti-Gal to the α-gal epitope on these glycolipids. C. Binding of monoclonal anti-Gal antibody to B16 cells following incubation of these cells for 2h at 37oC with various concentrations of α-gal micelles, as measured by flow cytometry. D. Complement dependent cytolysis (CDC) of B16 cells incubated for 2h with 1mg/ml α-gal micelles. Mouse serum containing anti-Gal and rabbit complement (●); mouse serum without rabbit complement (◯); fresh human serum (▲); control B16 cells lacking α-gal epitopes and incubated with mouse serum and rabbit complement, or with human serum (□) (mean of 4 experiments). Modified with permission from ref. [54]. .
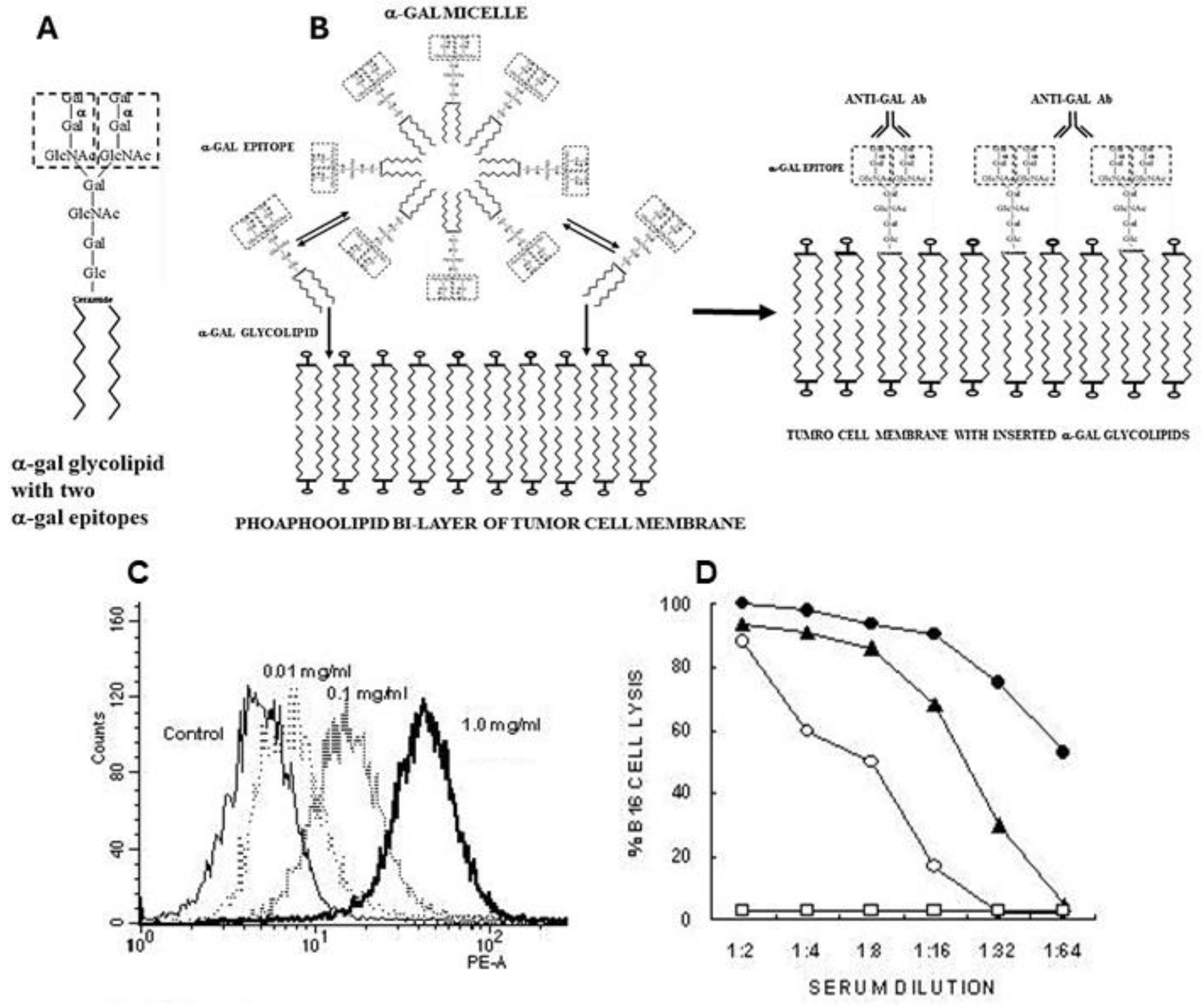
Figure 4.
Effects of 1 mg α-gal micelles injection into representative B16 tumor lesions (~5mm in diameter) in anti-Gal producing GT-KO mice (KO mice) or wild-type mice (WT mice). A. Injection of PBS into control lesions results in no infiltration of mononuclear cells as observed in B16 lesions, 7 days post-injection. B. Day 2 post-injection of α-gal micelles into lesions of anti-Gal producing GT-KO mice. Note the multiple mononuclear cells extravasating the blood vessel (BV). C. Day 7 post-injection of α-gal micelles. Note the marked increase in the number of extravasating mononuclear cells. D. Two melanoma lesions on a representative mouse, 10 days after intra-tumoral injection of PBS (left lesion) or of α-gal micelles (right lesion). Note the near complete regression of the right lesion vs. the increased size of the left lesion to ~12 mm. E. Comparison of paired B16 lesions diameter in 10 GT-KO mice or 5 WT mice as in (D), injected with α-gal micelles or with PBS, on Day 10 post-injection. Note the prevention of growth or decreased size in the lesions treated with α-gal micelles vs. the continuing growth of lesions injected with PBS in KO mice. In contrast, no differences in lesion growth post-injection were observed in WT mice lacking the anti-Gal antibody. Figures A-C H&E x100 reproduced from ref. [77] with permission and Figures D and E adapted from ref. [50] with permission.
Figure 4.
Effects of 1 mg α-gal micelles injection into representative B16 tumor lesions (~5mm in diameter) in anti-Gal producing GT-KO mice (KO mice) or wild-type mice (WT mice). A. Injection of PBS into control lesions results in no infiltration of mononuclear cells as observed in B16 lesions, 7 days post-injection. B. Day 2 post-injection of α-gal micelles into lesions of anti-Gal producing GT-KO mice. Note the multiple mononuclear cells extravasating the blood vessel (BV). C. Day 7 post-injection of α-gal micelles. Note the marked increase in the number of extravasating mononuclear cells. D. Two melanoma lesions on a representative mouse, 10 days after intra-tumoral injection of PBS (left lesion) or of α-gal micelles (right lesion). Note the near complete regression of the right lesion vs. the increased size of the left lesion to ~12 mm. E. Comparison of paired B16 lesions diameter in 10 GT-KO mice or 5 WT mice as in (D), injected with α-gal micelles or with PBS, on Day 10 post-injection. Note the prevention of growth or decreased size in the lesions treated with α-gal micelles vs. the continuing growth of lesions injected with PBS in KO mice. In contrast, no differences in lesion growth post-injection were observed in WT mice lacking the anti-Gal antibody. Figures A-C H&E x100 reproduced from ref. [77] with permission and Figures D and E adapted from ref. [50] with permission.
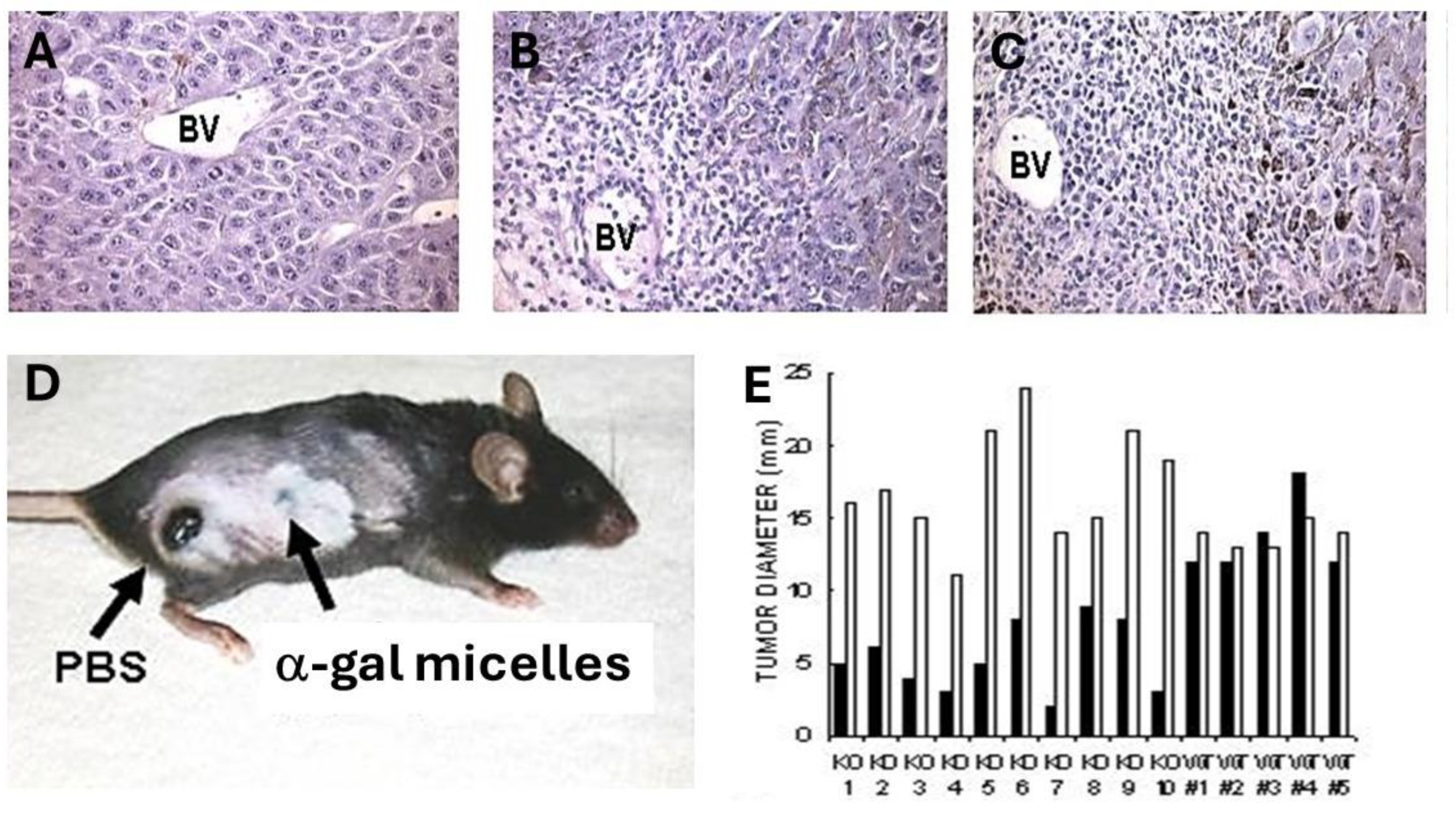
Figure 6.
Prevention of distant lesions development by treatment of primary tumor lesions with α-gal micelles in anti-Gal producing GT-KO mice. A. Mice received 3 weekly intra-tumoral injections of 1mg α-gal micelles into primary B16 melanoma lesions in the right flank. Subsequently, the mice were challenged with 0.5x106 B16 cells in the left flank, simulating distant metastatic cells. Tumor growth of the challenging B16 cells was daily monitored. Ten of 15 mice injected with α-gal micelles displayed no development of a distant metastasis . B. Control mice with primary tumor injected with PBS. All mice displayed metastatic tumor growth. C. One million B16 melanoma cells were administered subcutaneously into the right flank as “primary” tumor. At the same time 1x104 cells were administered into the left flank simulating a “distant” micrometastasis. The primary tumor was injected with α-gal micelles as in (A) and the injection was repeated a week later. Monitoring the development of a distant metastasis in the left flank indicated that the treatment prevented metastases growth in 4 of the 8 mice tested. D. Tumor challenges as in (C), however, the right flank tumor received two PBS injections (n=8 mice/group). All mice developed metastases. Adapated from ref. [82] with permission. .
Figure 6.
Prevention of distant lesions development by treatment of primary tumor lesions with α-gal micelles in anti-Gal producing GT-KO mice. A. Mice received 3 weekly intra-tumoral injections of 1mg α-gal micelles into primary B16 melanoma lesions in the right flank. Subsequently, the mice were challenged with 0.5x106 B16 cells in the left flank, simulating distant metastatic cells. Tumor growth of the challenging B16 cells was daily monitored. Ten of 15 mice injected with α-gal micelles displayed no development of a distant metastasis . B. Control mice with primary tumor injected with PBS. All mice displayed metastatic tumor growth. C. One million B16 melanoma cells were administered subcutaneously into the right flank as “primary” tumor. At the same time 1x104 cells were administered into the left flank simulating a “distant” micrometastasis. The primary tumor was injected with α-gal micelles as in (A) and the injection was repeated a week later. Monitoring the development of a distant metastasis in the left flank indicated that the treatment prevented metastases growth in 4 of the 8 mice tested. D. Tumor challenges as in (C), however, the right flank tumor received two PBS injections (n=8 mice/group). All mice developed metastases. Adapated from ref. [82] with permission. .
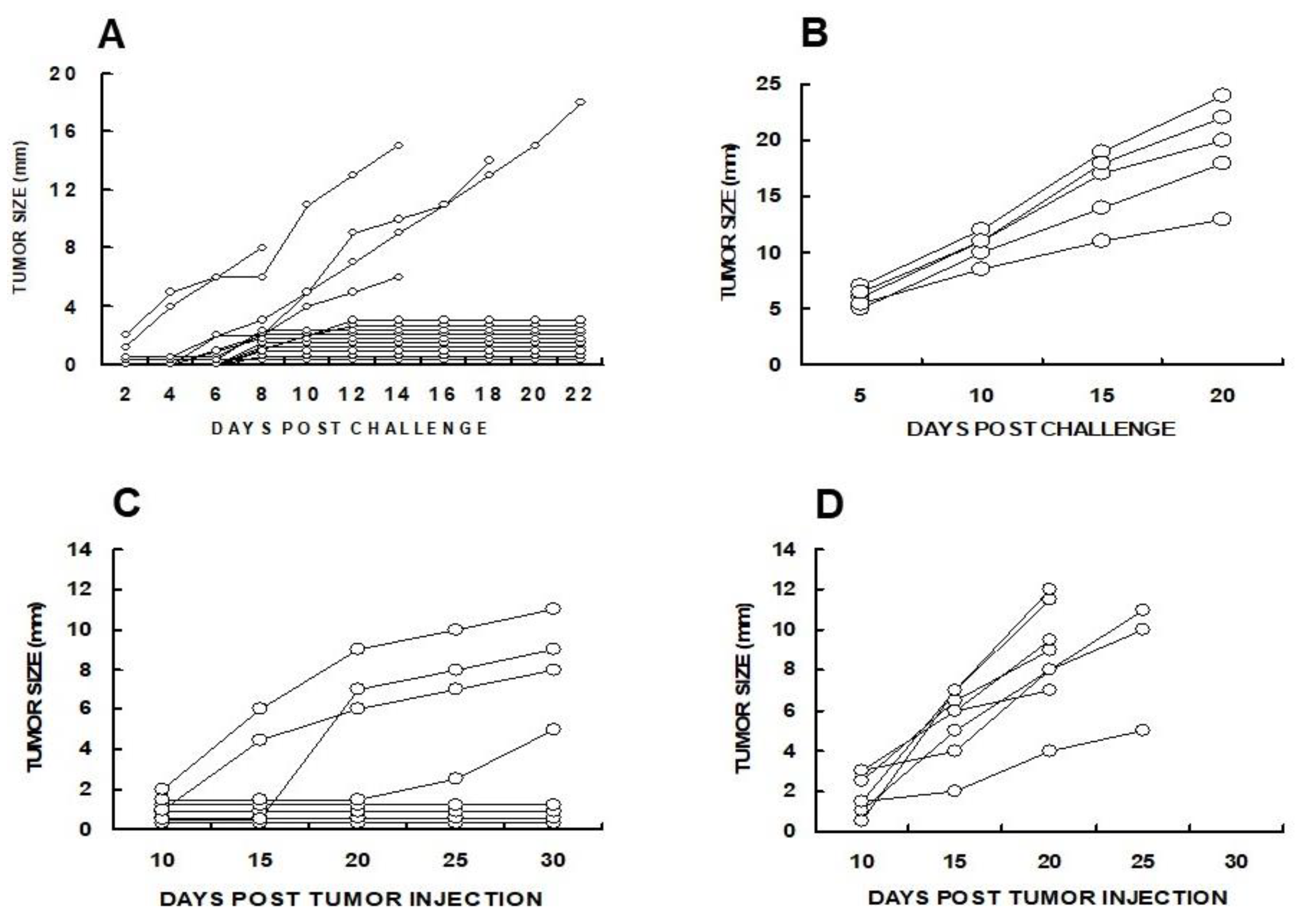
Figure 7.
Protection against B16 tumor growth following α-gal micelles treatment is mediated by CD8+ T cells. Forty million splenocytes were transferred from anti-Gal GT-KO mice with B16 lesions treated with α-gal micelles, to naïve GT-KO recipient mice. These recipients were challenged subcutaneously with 0.5x106 live B16 cells, 24h prior to receiving the splenocytes. Tumor growth was monitored for 30 days. (◯)- Transfer of total splenocytes, (●)- splenocytes depleted of CD8+ T cells. The size of the growing tumors is described at various time points post adoptive transfer. Reprinted with permission from ref. [82]. .
Figure 7.
Protection against B16 tumor growth following α-gal micelles treatment is mediated by CD8+ T cells. Forty million splenocytes were transferred from anti-Gal GT-KO mice with B16 lesions treated with α-gal micelles, to naïve GT-KO recipient mice. These recipients were challenged subcutaneously with 0.5x106 live B16 cells, 24h prior to receiving the splenocytes. Tumor growth was monitored for 30 days. (◯)- Transfer of total splenocytes, (●)- splenocytes depleted of CD8+ T cells. The size of the growing tumors is described at various time points post adoptive transfer. Reprinted with permission from ref. [82]. .
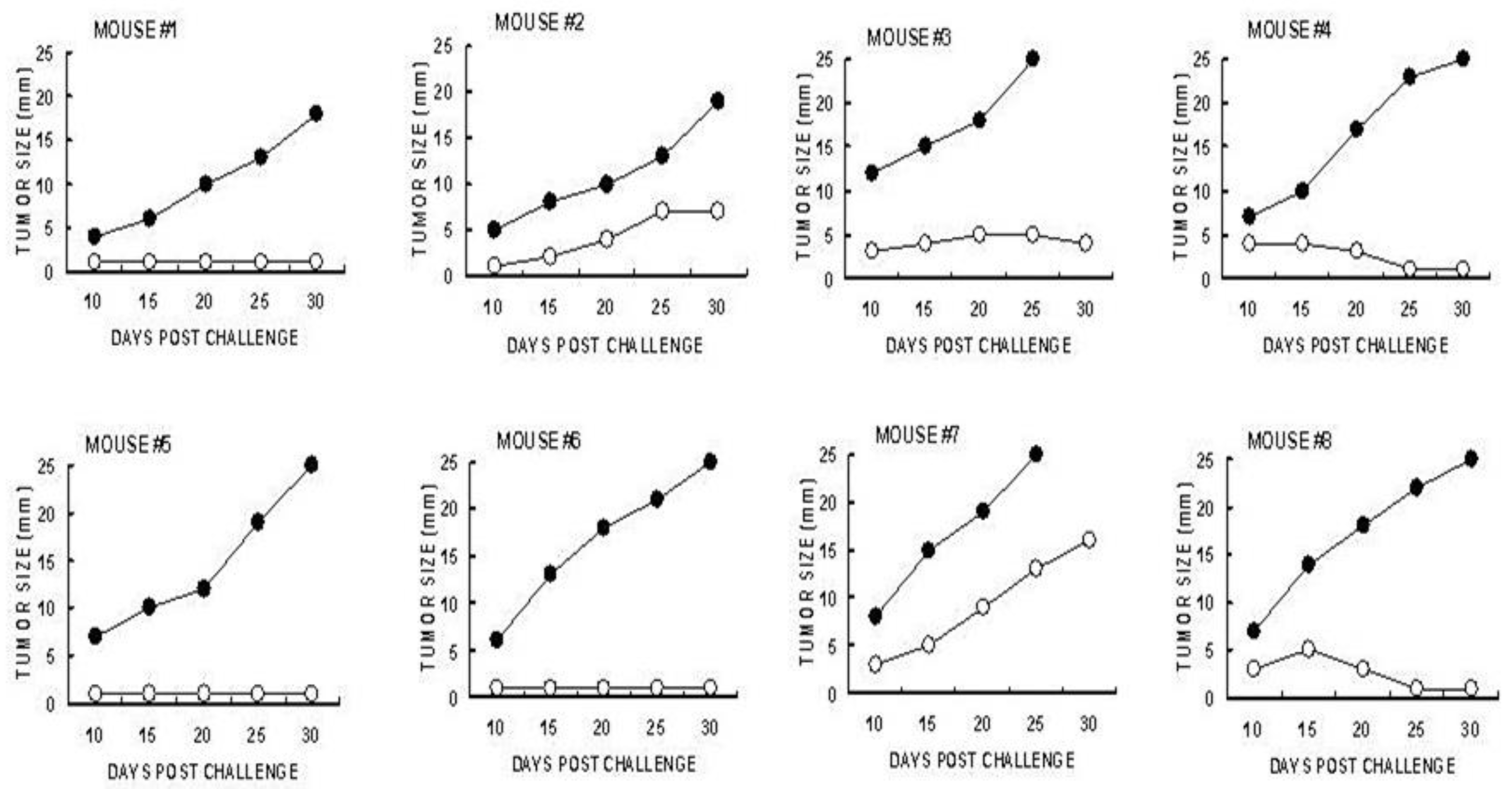
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



