
Submitted:
08 August 2024
Posted:
09 August 2024
You are already at the latest version
Abstract
White adipose tissue (WAT) and gut are involved as inducers of neuroinflammation when they detect injury and trigger inflammation. The autonomous nervous system innervates both tissues, although the roles of the sympathetic, parasympathetic and enteric nervous systems have not been fully elucidated. Here, we revisited the participation of both types of inflamed peripheral tissue in neuroinflammation. We first analyzed how inflamed peripheral WAT plays a key role in neuroinflammation once metainflammation is installed. Second, we described the impact of insulin resistance (IR) on hypothalamic dysfunction, T2DM and neurodegenerative diseases. Finally, we analyzed the gut-brain axis, examining cell interactions, soluble factors, the sensing of microbes and the role of the intestinal microbiota and mucosal barriers in neuroinflammation. Since intestinal mucosa and secondary lymphoid organs are densely innervated by the sympathetic, parasympathetic, and enteric nervous systems, we analyzed bidirectional crosstalk between different tissues, neurons, and immune systems in healthy and pathological circumstances to restore normal tissues functions, and consequently homeostasis.
Keywords:
Neuroinflammation
; Hypertrophic White Adiposity
; T2DM
; Neurodegenerative Diseases
; Central & Autonomic Nervous Systems
; Immune System
; Gut-Brain Axis
; Microbiota
1. Introduction
Neuroinflammation is an inflammatory response disrupting central nervous system (CNS) homeostasis; it can be activated by broad etiologies (e.g., infection, traumatic brain injury, toxic metabolites, malnutrition, and autoimmunity). Neuroinflammation may take place throughout the entire life, with serious health consequences ranging from metainflammation (obesity-driven metabolic dysfunction and chronic low-grade inflammation) to neurodegenerative diseases (NDs). Neuroinflammation is known to involve disruption in signaling pathway proteins, receptor activities, and a variety of cell functions.
Moreover, the involvement of several key components is related to the development and maintenance of neuroinflammation, e.g., epigenetic marks, monocytes, activated microglia, infiltrating T cells, matrix metalloproteinases (MMP), ion channels, microRNAs, insulin receptors, and the autophagic process [1]. In fact, any kind of peripheral inflammation triggers neuroinflammation, wherein blood-brain barrier (BBB), glia, and neuron dysfunctions are involved [2]. This neuro-immune crosstalk is not one-way, but rather a complex, bidirectional system, wherein the CNS and the immune system constantly communicate and influence each other. Indeed, when the BBB is compromised, harmful substances enter the brain, modifying its undoubtedly delicate environment. After these substances enter the brain, brain-resident homeostatic microglia cells are activated and local inflammation is in consequence triggered. Symptoms of neuroinflammation result: slow metabolism, body weight gain, diabetes mellitus, dysbiosis, mood and neurodegenerative disorders [1,3]. Moreover, neuroinflammation is undoubtedly involved in the physiopathology of serious degenerative diseases (NDs) [e.g., Parkinson's Disease (PD), Alzheimer's Disease (AD), Huntington Disease (HD) and amyotrophic lateral sclerosis (ALS)] [1]. “Activated” microglia cells and elevated pro-inflammatory factors are clearly associated with CNS function damage. Microglia comprise immune cells in the CNS and play a key role in maintaining their homeostasis [3]. Astrocytes control blood flow and extracellular neurotransmitter levels to ensure that their endogenous microenvironment is optimum for neuron function. Without stimulation/injury, neurotransmitters, neurotrophic factors, anti-inflammatory cytokines, and intercellular contacts inhibit neuroglia activity [4,5]. However, when the brain’s environment is disrupted, microglia cells are stimulated and secrete an excess of pro-inflammatory factors. The brain compartment is highly preserved from an active immune function in physiological conditions. In fact, antigen presentation is actively inhibited, microglia cells are maintained “homeostatic”, and immune components are excluded from the brain by the BBB. However, once brain injury occurs, “activated” microglial cells produce pro-inflammatory factors, thereby inducing neuroinflammation. Whether microglia elicit detrimental or beneficial effects in brain neurons is dependent on the transition from their “homeostatic” to “activated” state [6,7].
White adipose tissue (WAT) and gut, as metabolic-endocrine organs, closely interact both peripherally and through the CNS. This interaction, facilitated by the blood and the vagus nerve, has significant implications for the CNS [8]. These endocrine organs establish a WAT-gut axis [9, from which their signals impact on the CNS, forming the brain-WAT-gut axis. The CNS, in response, plays a crucial role in enhancing the production and secretion of brain-derived neurotransmitters and neuropeptides, activating various hormone axes and, through the ANS-dependent function, regulating several peripheral mechanisms including those of WAT, the gastrointestinal tract, and the immune system [10]. The dynamic loop between the CNS and these tissues is not only vital for survival in both physiological and pathological conditions, but is also fascinating in its complexity and potential for further research.
This review centers on the pivotal role of injured tissues (WAT and gut) in promoting neuroinflammation. It delves into how these endogenous responses impact both local and distant tissues, and how the brain’s defense mechanisms are activated to restore homeostasis. The mechanistic and pathological observations presented here provide a deeper understanding of the consequences of WAT and gut participation in the neuroinflammatory process.
2. Relevance of WAT and Gastrointestinal Tract Functions in Neuroinflammation.
The functions of WAT and the gastrointestinal tract are highly relevant in the context of neuroinflammation. The BBB plays a key role in neuroinflammation. Indeed, changes in the BBB´s permeability [11] and function after systemic inflammation due to increased peripheral pro-inflammatory signals in blood have been reported. In turn, messages entering brain, originated by these molecules and pathogens, cause brain´s pathophysiology of neuroinflammation and neurodegeneration [12]. More recently, a gut vascular barrier (GVB) was described [13], as well as a connection between the BBB and the GVB that may interfere with cognitive behaviors [14]. The GVB is composed of interacting endothelial cells, glial cells and pericytes; this barrier prevents large molecule translocation from the gut lumen [15].
When excessive WAT (namely the visceral-mesenteric depot) accumulation occurs, such as occurs in different obese phenotypes, tissue dysfunctionality can activate immune cells, resulting in a chronic degree of peripheral inflammation [16]. In fact, obesity is associated with neuroendocrine, metabolic, and immune system disorders such as enhancement of inflammatory cytokine production and release, overall insulin resistance (IR), vascular endothelial inflammation and atherosclerosis, among others [10]. Consequently, the development of a deep dysmetabolic phenotype increases the individual’s susceptibility to further development of type 2 diabetes mellitus (T2DM).
The microbiota is also a key player in this loop, and interaction between intestinal microbes with immune cells and neurons has been extensively described to shape the physiology of intestinal and distant tissues in both homeostatic and pathological circumstances. Altered gut microbiota (GM) has been claimed to be critical in tumor development and other inflammatory disorders. However, the mechanisms whereby it occurs remains a matter of study [17]. In general, inflammatory bowel diseases (IBDs) are disorders associated with obesity among other factors playing a role in their development and course [16]. For instance, in Crohn's disease, a crucial role of mesenteric WAT in the pathophysiology of intestinal inflammation has been reported [18]. The involvement of large-size mesenteric adipocytes producing an excess of pro-inflammatory adipokines (e.g., leptin, TNF-α, PAI-1, and resistin) is implicated in the pathogenesis of IBDs [16]. As a result, the excess of WAT-secreted pro-inflammatory adipokines together with Immune System-derived pro-inflammatory cytokines (e.g., TNF-α, IL-1, IL-6, IFN-γ) at the gut level impact directly (through peripheral blood) and in the ANS, on the hypothalamus, inducing several CNS dysfunctions as occurs in neurodegenerative disorders (NDs) [19]. It has been reported that in AD, developed brain IR enhances CNS TNF-α and endothelin-1 production and IRS-1 signaling deficiency and reduces nitric oxide (NO) production. As a result, low brain blood flow and increased neuroinflammation characterize AD patients [20].
The main function of the gastrointestinal tract is food processing, digestion, and absorption of nutrients. To accomplish this complex task, the whole tract is colonized by many microbes that probably outnumber the quantity of cells in our body. To control this scenario, the organism coordinates the generation of nutrients, absorption and secretion of metabolites, gut motility, and secretion of enzymes and inflammation promoted by the continuous antigenic challenge in the extended intestinal mucosa. To deal with an environment of permanent exposure to different and dynamic stimuli (food components and environmental agents and microorganisms), the tissue must constantly adapt to maintain homeostasis. To achieve this end, the gastrointestinal tract is equipped with a highly specialized mucosal immune system in close contact with a dense network of neurons. The intricate interplay between the nervous and immune systems has evolved as an integrated network that continuously surveys the organism, tracking for internal and environmental stimuli that could disturb tissue homeostasis. This amalgamated circuitry communicates the peripheral tissues with the CNS to respond in different circumstances and play a substantial role in tissue physiology. Inflammation is the hallmark of immune response to harmful stimuli and acts to remove the harmful ones, followed by the healing process. The nervous and immune systems conduct sensory and effector mechanisms to maintain tissue homeostasis.
Sensing potential pathogenic or harmless stimuli is pivotal to the immune and nervous systems. It promotes a reaction when a microorganism or damaging perturbation is detected in the context of an infection. A delicate and regulated network of immune and neuron cells colocalize in multiple tissues, e.g., mucosal barriers (skin, lung and gut), secondary lymphoid organs, and adipose tissue, orchestrates host defense mechanisms, thereby restoring tissue physiology. This neuron-immune unit is an integrated circuit that exerts concerted actions in health and disease [21].
The complex neuro-immune crosstalk is bidirectional and relies on cell-cell contacts (direct communication) and soluble factors (indirect communication) that exert modulatory functions on proximal and distant cells. Neurons and immune cells express many common receptors for conserved and vital ligands on microbes, and also cytokine receptors to modulate cell activity. In addition, immune cells express receptors for neurotransmitters and neuropeptides secreted by neurons [22]. Neuro-immune units (anatomical locations wherein immune and neuronal cells colocalize and interact among them to lead tissue physiology), sensing peripheral internal/environmental stimuli, communicate the presence of noxious signals to the CNS. Then, immune cells are instructed by the CNS to respond to the tissue receiving the injury and thereafter to promote effective immunity and tissue homeostasis.
The neuro-immune unit can operate in the CNS (brain and spinal cord) or the peripheral nervous system (somatosensory and autonomic nervous systems). Neurons and glial cells in the CNS send efferent signals to peripheral tissues via neurotransmitters and neuropeptides and immune cells secrete cytokines that bind neuron receptors, either locally or systemically, in the CNS. Some nerves can also signal information back from the tissue to the CNS and then, through efferent nerves, instruct peripheral immune cells for an effective immune response. The vagus nerve is one of the twelve cranial nerves representing the main extrinsic parasympathetic nerve connecting the brain and the proximal colon. It is mainly implicated in modulation of intestinal immune response depending on the composition of intestinal microbiota or gut inflammation; the brain promotes, through vagal efferent function, a regulatory signal that controls pro-inflammatory cytokine release. The distal colon parasympathetic innervation originates from the sacral spinal nerves and both nerves coordinately act as a network [23]. The mucosa and secondary lymphoid organs are highly innervated by the ANS, which is composed of the sympathetic (SNS), parasympathetic (PNS) and enteric nervous system (ENS). Cell bodies of the SNS (spinal cord) and PNS (brain and sacral spinal cord) are localized in spinal cord ganglia (extrinsic intestinal neurons). In contrast, those from the ENS are entirely contained within the gastrointestinal tract wall (intrinsic intestinal neurons) and are organized in several plexuses throughout the entire intestinal wall (mucosa, submucosa and muscle layer). The ENS is the most significant neuron accumulation outside the CNS; it is organized in myenteric (between the circular and longitude muscles) and submucosal plexus [24] (Figure 1).
The ENS comprises sensory neurons that sense intestinal content, and motor neurons that drive secretory functions and peristalsis [25]. It is also interesting that the intestine harbors the body´s largest lymphoid cell compartment, concentrated mainly in the mucosa layer that lines with the intestinal lumen [26,27]. This neuro-immune unit allows a more integrated and concerted immune response because the two cell systems can sense different stimuli. It seems to be evolutionarily conserved since non-mammalian organisms (zebrafish, Caenorhabditis elegans, Drosophila, etc.) also have integrated mechanisms for protection [28,29].
3. Hypertrophic White Adiposity-Induced Insulin Resistance (IR) as a Main Instigator of Neuroinflammation.
3.1. Sympathetic Innervation of Adipose Tissue.
Although there is controversy regarding ANS innervation of WAT pad depots, sensory fibers seem to play a relevant regulatory role in WAT functionality. Indeed, as already accepted, WAT adipocytes are in contact with nerve fibers in the parenchyma [30].
Evidence has indicated that visceral WAT bidirectionally communicates with the brain through afferent sensory fibers and efferent sympathetic fibers [31,32]. The long-isoform of leptin receptor (Ob-Rb) was found on dorsal root ganglia neurons from WAT, thereby suggesting that leptin potentially communicates with sensory nerves in WAT through the dorsal root ganglia via [33]. Furthermore, SNS-stimulated lipolysis, and intra-WAT injection of free fatty acids can increase WAT afferent nerve activity [34]. Although largely hypothesized that these nerves were of SNS origin, irrefutable proof of SNS innervation of WAT came from a study by Youngstrom and Bartness demonstrating bidirectional innervation of WAT [35], determining that the SNS ganglia, at T13/L2-L3, innervate WAT pads directly upon adipocytes [36]. In addition, surgical denervation of WAT depots indicated that sympathetic neurons are synaptically connected and generate neuronal pathways from WAT to brain [37]. Sympathetic denervation of WAT was also proved to increase mass depot due to hyperplasia and decreased lipolysis [38]. Conversely, sensory denervation of visceral WAT increased pad mass throughout adipocyte hypertrophy, thereby providing differential ANS control of WAT function among sympathetic and sensory nerves [39,40].
3.2. Insulin-Resistant WAT Cells and Metainflammation Precede Neuroinflammation Development.
Visceral fat depots of large-size WAT adipocytes characterize obese phenotypes. These hypertrophic cells are seriously compromised in IR and highly secrete pro-inflammatory adipocytokines. In turn, hypertrophic WAT cells are not recognized as normal-host´s cells and are thus infiltrated by cells from the immune system, mainly macrophages. Then, those infiltrating cells from the immune system also release pro-inflammatory cytokines (e.g., TNF-, IL-1, IL-6), generating a vicious cycle of signals until the peripheral inflammatory state is solved or, conversely, chronically perpetuated. In the latter scenario, the individual is at a high risk of: 1. an established dysmetabolic state, accepted as metainflammation [41], resulting in the appearance of co-morbidities seriously dangerous for the individual´s health (Figure 2) and 2. if this condition is not rapidly counteracted, neuroinflammation will follow until it is resolved.
The obesity epidemic and T2DM, originally called twin, actually combines with a critical component: IR; now known as the triplet epidemic. This is not a minor concept because the astonishing growth of these problems seriously conditions global public health due to the difficulties of early diagnosis of the IR state. Metabolic abnormalities during a chronic insulin-resistant state of the organism end in cardiovascular disease-related death. They could therefore be called silent killers. Research has been essential to develop the idea of an association between obesity and diabetes. Thus, it has progressively been linked not only with obesity’s severity but also to weight gain and duration of weight catch-up. Numerous experimental works and epidemiological and clinical research have cemented an interesting story that, in part, shows the stormy (excessive release of pro-inflammatory cytokines) relationship between obesity and T2DM [42,43,44]. Indeed, changes in tissue and peripheral lipids could be considered the most prominent promoters of IR [45,46,47]. It is imperative to recognize that WAT is essential for regulating energy expenditure and therefore for life. However, the problem appears when this functionality is broken, and the adipocyte is no longer a friendly component but a foe. Hyperglycemia takes place, among others, through a disrupted adipo-insular axis function (Figure 3, panel a). Physiologically, a stimulatory effect of insulin on adipocyte leptin production (synthesis and release) operates; reciprocally, direct leptin inhibitory activity on β-cell insulin production operates [48]. Thus, when this endocrine axis becomes dysfunctional, compensatory activity through the ANS (vagus nerve) [49] could be triggered to rapidly modulate glycemia (Figure 3, panel b). Leptin circulating levels directly correlate with body fat mass and increase after food intake, whereas they decrease during fasting [50,51]. Physiologically, insulin acts directly on adipocytes, increasing ob expression and secretion [52]. In turn, leptin reduces pancreatic β-cell function, thus decreasing insulinemia. Leptin's in vivo role in glucose metabolism has been proposed to be mediated by the ANS [53].
White adipocyte multiple adipokine production (e.g., TNF-α, ΙL−1 and IL-6) [54,55] favors the increase in leptin WAT and other pro-inflammatory adipokines, [56]. Leptin is key in controlling food intake and in regulating energy expenditure by inducing satiety through hypothalamic Ob-Rb activity [57,58]; a mechanism mediated through the JANUS/STAT pathway [59]. Leptin inhibits lipogenesis and increases β-oxidation, adipocyte apoptosis, and UCP-1 expression through both autocrine mechanism and ANS efferent pathways [60]. Thus, as mentioned above, leptin inhibits insulin and thereby affects glucose homeostasis by combining with its hypothalamic effect [61]. Conversely, leptin inhibits glucagon production in pancreatic α−cells [62]. Therefore the adipose-insular axis plays a predominant regulatory role in energy balance, wherein insulin and leptin are the active peripheral messengers. Consequently, dysfunction of this axis facilitates both obesity and T2DM development [63]. LEP-R, mainly at hypothalamic level, is a characteristic in most obese phenotypes [59,64,65]. Energy homeostasis requires a precise balance between metabolic substrate utilization by the brain and peripheral organs and by exogenous (diet) or endogenous (liver, gut, adipose tissue, and kidney) arrangement. The main physiological objective of glycemia is to guarantee the appropriate supply of GLU to brain and other tissues, including gut [66]. Therefore, dysfunction of the adipose-insular axis may facilitate obesity-associated metainflammation (eg., dyslipidemia, Metabolic Syndrome, T2DM) [63].
3.3. Hypothalamic-Mediated Mechanisms Involved in Neuroinflammation
Leptin resistance is a characteristic in some obese phenotypes, and several hypothalamic mechanisms have been proposed to explain this process [59]. As mentioned above, energy homeostasis requires a precise balance between metabolic substrate utilization (mainly GLU and FFAs) by the brain and peripheral organs and exogenous (diet) or endogenous tissue arrangement. The main physiological objective is a GLU peripheral level that guarantees appropriate GLU supply to the brain and other organs. Once metainflammation is installed, it must be considered that the hypothalamus plays a key role in whole-body energy homeostasis and metabolism [65]. This communication is due to the arrival of peripheral signals (e.g., hormones, adipo/cytokines, metabolites) translating their activities into the CNS. Signals received by different hypothalamic nuclei (namely the ARC nucleus) induce many peripheral functions susceptible to being modified once neuroinflammation is triggered. Various reports indicate that several obese phenotypes, including that from high-fat diet (HFD) intake, modify CNS energy homeostasis [66,67,68,69]. In fact, HFD triggers hypothalamic neuroinflammation after activating the Toll-like receptor 4 (TLR4) [70]. Moreover, many undesirable cellular mechanisms merge, such as oxidative stress (OS), namely at the endoplasmic reticulum (ER) level, up-regulation of SOCS3 and IKKβ/NF-κB paths [71]. HFD-induced obesity also activates immune cells inside the CNS (microglia and astrocytes), thereby worsening neuroinflammation [71,72,73,74]. It has been demonstrated that HFD-induced hypothalamic neuroinflammation occurs earlier than developing peripheral inflammation and hypertrophic expansion of the WAT mass [66,72]. Furthermore, inhibition of microglia functionality and hypothalamic blocking of neuroinflammation-mediated pathways could prevent obesity-dependent dysmetabolism [66,72,73,74] to cope with neuroinflammation. Diet-induced obesity (DIO) activates the inflammatory process at the hypothalamic level; as a consequence, enhanced production of pro-inflammatory cytokines and impairment in both insulin and leptin signaling pathways mechanisms develop in an overall insulin- and leptin-resistant individual phenotype of neuroinflammation [75].
As mentioned, the BBB is an essential structure protecting the brain against peripheral inflammation. Therefore, a change in its function is relevant for developing and maintaining neuroinflammation. Modifying BBB permeability [11,76] can result in increased translocation of circulating inflammatory signals, such as soluble factors, cells or pathogens, resulting in one of the main causes of neuroinflammation and enhanced susceptibility to ND development. However, other brain structures are affected and responsible for neuroinflammation in combination with the injured hypothalamus. Pertaining to the limbic system, the hippocampus is a structure located in the inner area of the temporal lobe and is particularly important in regulating emotional responses. Indeed, hippocampal impairment can be found in the early phases of NDs. This structure is highly susceptible to damage by obesogenic factors, mainly saturated FFAs [76,77], which in turn contribute to hippocampal neuroinflammation and thereby to ND development [72,73,74]. Obesogenic diets can enhance the expression of pro-inflammatory cytokines in the hippocampus and, in turn activate the microglia [75]. As a result, hippocampal neuroinflammation alters neuronal communication and metabolism once triggered, similarly to what occurs in hypothalamic neuroinflammation [66,76,78], and notices the modification of BBB permeability [76]. The main cellular structure involved in regulating energy homeostasis is the mitochondria. In obese individuals, mitochondrial dysfunction (decreased energy metabolism) due to dysfunctional glucose transport is a key altered mechanism for retaining fat energy [79], cooperating greatly with the brain´s dysfunction and IR [80,81,82,83,84].
Moreover, mild enhanced chronic glucocorticoids in the periphery (from both hypertrophic adipocyte metabolism and established corticoadrenal leptin-resistance) could inactivate the counter-regulation of white to brown-like adipocyte trans-differentiation (browning) due to a glucocorticoid receptor-mediated mechanism [85], thereby cooperating with decreased dissipation of body energy. Indeed, ROS generation exceeds antioxidant activity, causing cellular oxidative damage. WAT OS is a major contributor to IR and cellular dysfunction [86] and is regulated in a depot-specific manner. Also, WAT OS varies with age; compared to younger mice, aged C57BL/6 mice exhibit increased ROS in visceral WAT (namely epididymal and epicardial WATs) [87,88]. Oxidized lipids and proteins accumulate in the visceral fat pad [89,90]. Consequently, significant challenges exist in defining the specifics of ROS-driven pathology and its connection with human obesity and T2DM (see Figure 2).
3.4. Insulin Resistance-Related Neuroinflammation in Neurodegenerative Diseases and the Role of Gut-Microbiota.
Brain neuroinflammation may be extremely dangerous for the development of NDs. Indeed, once the overall IR state is established, a high risk for development of AD by a mechanism mainly dependent upon poor brain insulin signaling could be established. This is supported by the fact that brain IR can promote local tau pathology and amyloidogenesis [91]. Moreover, an increased risk of developing T2DM has been reported in AD patients [92], which supports the interrelationship between brain lesions and metabolic disturbances in AD patients, including deterioration of cognitive function [93]. Functional insulin signaling has been reported to promote neuron plasticity and ameliorate memory in humans treated with intranasal insulin [94]. The IR developed in the AD brain appears to be related to Aβ plaques and tau pathologies [95], as supported by inhibited brain IRS-1 in patients showing taupathies [96], wherein Aβ oligomers promote insulin receptor internalization [97]. Also, enhanced activation of c-Jun N-terminal kinase, Protein kinase R, and TNF-α takes place and, in turn, inhibits IRS-1 function [95,98]. All these observations strongly support that impaired glucose homeostasis in AD patients seems to be closely related to brain (hypothalamic) damage due to local IR (abnormal insulin-signaling), thus augmenting brain TNF-α production and probably perpetuating neuroinflammation (Figure 4).
Pathophysiological causes of Parkinson’s disease (PD) are still unclear; however, IR, mitochondrial dysfunction, OS, and neuroinflammation seem to be the most critical disease-related dys-mechanisms [99,100,101]. Unfortunately, at present, dopaminergic neuron death and PD progression appear to be indomitable events only susceptible to palliative treatment for motor dysfunction through drug therapy, such as dopamine (DA) precursor (DOPA) and DA-agonists. As shown in Figure 2, hypertrophic adipocytes do highly secrete TNF-α and increased NFkβ expression augmenting their signaling. In the IR-established endogenous environment during hypertrophic expansion of WAT mass, the PI3K/AKT signaling path becomes disrupted. Then, reduced/lack of inhibition of NFkβ production and modification of other cell-function mediators (eg., GSk3β, FOX01, mTOR) occur. In turn, mitochondrial dysfunction and α-Synuclein (α-Syn) aggregation are leading causes of death of dopaminergic neurons [102], resulting in neuroinflammation. Moreover, pancreas-secreted islet amyloid polypeptide (IAPP) can cooperate with α-Syn aggregation, so that reduced neuroprotection aggravates PD progression [103] (Figure 5).
Supporting evidence in the international literature highlights the importance of the microbiota-gut-brain axis function in NDs [104,105,106]. Indeed, the interaction between gut microbiota and microglia in AD has been found. In homeostatic condition, the gut microbiome regulates microglial maturation and activation via short chain fatty acid (SCFAs) release [105]. In this study, authors found that germ-free and antibiotic-treated mice suffered from impaired microglial immune responses when challenged with bacterial lipopolysaccharide and lymphocytic choriomeningitis virus infection. However, microglial defects were partially restored by recolonization with complex microbiota and SCFA supplementation [105]. Similarly, regarding PD, gut microbiota appears to be a key factor in this ND. Gastro-intestinal symptoms and altered gut microbiota are found in PD patients [107]. Several pieces of evidence [108,109] demonstrated that the gut microbiome was influenced by development of α-Syn pathology, microglial activation, and motor deficits in α-Syn-overexpressing (ASO) mice that appeared to be influenced by gut microbiome. This became evident in experimentation with SPF ASO mice which exhibit greater PD pathological signs than their germen-free and antibiotic-treated counterparts. Remarkably, treatment with fecal microbes from PD patients for germen-free ASO mice restored the primary disease, α-Syn-mediated motor dysfunctionality [108]. Altogether, studies support the relevance of the microbiota-gut-brain axis in the pathogenesis of ND, such as AD and PD [109].
4. Gut and Neuroinflammation.
4.1. The Gut-Brain Axis: Role of the Hypothalamus.
The presence of a pathogen within gut mucosa triggers induction of mechanisms that may lead to excessive inflammation and permanent tissue damage with disturbance of tissue homeostasis. The gut-brain axis (GBA) (Figure 6) is a bidirectional communication network linking the ENS and CNS with intestinal cells, allowing the brain to influence intestinal functions, from gut organogenesis to effector immune mechanisms, and the gut, to influence mood, cognition, and mental health [110,111,112]. This complex GBA integrates emotional and cognitive brain centers with the intestinal function to fine-tune immune response to invading pathogens and tissue damage, restoring homeostasis.
Indeed, once external threats by pathogen invasion and other injuries are detected, neurons can directly sense them and rapidly communicate with immune cells, and vice versa. Soluble factors are essential in this crosstalk, while neurons can interact with many cells [macrophages, neutrophils, dendritic cells (DCs), epithelial cells, mast cells, innate lymphoid cells (ILCs), and adaptive cells (B and T lymphocytes)] to promote the antimicrobial response [113,114].
The ANS, the hypothalamo-pituitary-adrenal (HPA) axis, and nerves in the gastrointestinal tract (ENS) link gut and brain directly and indirectly. The way behavior and immunity are synchronized during infection to halt the spread of microorganisms (e.g., fever, lethargy, anorexia, social isolation) has been thoroughly described. Secreted pro-inflammatory cytokines from innate cells affect HPA axis function. Then, cortisol produced impacts the liver, promoting acute phase protein production (amyloid A protein, fibrinogen, C-RP, complement components, mannose-binding protein, coagulation proteins, etc.) that exert immune functions. A gut-vascular barrier (GVB) that controls the passage of different components and microorganisms from the intestinal lumen to the blood, preventing dissemination of bacteria to the liver and other tissues was recently described [13,14]. The GVB and the BBB are interconnected within the GBA, which is critical to understand how inflammation and psychosocial disruptions could affect homeostasis.
4.2. The Nervous System and Gut: Evidence of Cell Interaction.
As mentioned above, the intestine is continuously exposed to microorganisms and derived metabolites. Hence, the gastro-intestinal tract is densely innervated and contains most of the organism’s immune cells. This environmental antigenic challenge makes intestinal mucosa the main body sensory interface. Indeed, immune cells sense microorganisms, their metabolites, and stress and signals from damaged tissues through different pattern-recognition receptors (PRR). Exogenous pathogen-associated molecular patterns (PAMPs) (e.g., flagellin, LPS or CpG), endogenous membrane-bound, secreted stress molecules, damage-associated molecular patterns (DAMPs) (e.g., decreased intracellular K+, extracellular ATP, uric crystals, sustained hyperglycemia) activate cells bearing PRRs. Then, activated cells communicate with neighboring cells (immune and neurons, or innervating fibers) to promote inflammation that modifies gut motility, immune cell recruitment and activation, and gut permeability, thereby interfering with CNS function (energy homeostasis, food intake, etc.). Sensitive and enteric neurons can also directly detect microorganisms at barrier tissues by expressing different PRRs such as TLR7 or TLR2/TLR4 , and in turn secrete neurotransmitters and neuropeptides. Understanding neuro-immune interactions and their impact on barrier tissue physiology has led to the development of novel tools to characterize neuron and immune cells [114].
ANS components have been described to control the main inductive sites of the gut from embryonic life to control lymphocyte activation and differentiation [115,116,117,118]. The ENS is involved in developing Peyer´s patches, vital inductive tissues for generating mucosal immunity in the gut [117]. Moreover, sympathetic nerves located close to Peyer´s patch also innervate the submucosa and muscularis, and control blood flow and cell distribution. Although Peyer´s patches are distributed along the human small intestine, the highest density is found at the ileum, which is highly innervated compared to the other intestinal mucosa. This suggests the role of fibers and neurons in immune cell activation. Cells located in the follicle dome, B and T areas (macrophages, dendritic cells, B cells, T cells, and IgA-producing plasma cells) are proximal to projections from the submucosa plexus, a site that include intrinsic sensory and motor neurons, as well as extrinsic sympathetic neurons [118]. These cells also express type 2 muscarinic acetylcholine receptors to detect neurotransmitters [119]. This means that noradrenergic fibers modulate cytokine and chemokine secretion from B and T cells and also control immunoglobulins released by plasma cells through binding to adrenergic receptors [120,121]. At the muscularis site, neuron-macrophage interaction controls TNF-α secretion and phagocytosis [122], and parasympathetic innervation of the gut, mainly through the vagus nerve, also controls ENS, barrier function, inflammation, and immunity [123]. Intestinal macrophages, a heterogeneous cell population, possess differential functions depending on tissue location and are also highly concentrated and distributed in different intestinal wall layers. Lamina propria macrophages (LpMs) are located under the mucosa's epithelial layer and close to gut lumen. Macrophages and dendritic cells are sentinel cells that surveil luminal microorganisms. They adapt to environmental changes, respond to microorganisms that could translocate within intestinal tissue, and promote tolerogenic circuits for harmless antigens, such as food [124]. LpMs extend protrusions through the epithelium to capture bacteria directly from the intestinal lumen through the tight junctions [125] without disrupting barrier integrity. The chemokine fractalkine (CX3CL1), secreted by epithelial cells, controls this mechanism by interacting with the fractalkine receptor (CX3CR1) expressed in LpMs [126,127]. Then, interaction of the intestinal lamina propria resident CX3CR1+ LpMs with migrating CD103+ dendritic cells promotes the homing of CCR7+ CD103+ dendritic cells with antigen cargo to mesenteric lymph nodes [128,129]. This mechanism is critical for inducing specific tolerogenic circuits to harmless antigens [130,131].
Nevertheless, macrophages can be detected distant from the lumen, in the outer smooth muscle layer. Muscularis macrophages (MMs) have a stellate morphology, are positioned along nerve fibers adjacent to neuron bodies, and are endowed with a tissue-protective phenotype to counteract inflammation [132]. Interestingly, LpMs are sensitive to luminal changes, whereas MMs are sensitive to tissue changes. The environment’s perturbation triggers a differential activation gene profile in gut-resident macrophage subsets and promotes specialized functions. The expression of a higher level of membrane β2 adrenergic receptors in MMs than in LpMs may reflect the proximity of muscularis macrophages to the neuronal network. Gut extrinsic SNS innervation of the muscularis layer has amply been shown to primarily contribute to catecholaminergic response through NE secretion. MMs responded by inducing Arginase 1 (Arg1) expression, while Tnf-gene expression remains unchanged upon intestinal infection [133]. Arg1 is implicated in tolerogenic immune mechanisms and preserving neurons from apoptosis [134]. The neuron-immune unit counteracts intestinal over-inflammation to avoid tissue damage with neuron loss. In a steady state, MMs are also involved in critical intestinal functions, such as modulation of gastro-intestinal motility due to the secretion of bone morphogenetic protein 2 (BMP2), a factor that impacts enteric neurons expressing BMPR. Likewise, neurons respond by secreting the macrophage colony stimulatory factor (M-CSF). Commensal bacteria promote this bidirectional interaction, as germ-free [135] or antibiotic-treated individuals [128] who exhibited severe dysmotility, with a lower expression of Bmp2 and Csf1, and lower frequency of MMs, together with no changes in LpMs number, compared with normal individuals [136].
The interaction between ENS, SNS and PNS neurons innervating the gut muscularis and lamina propria with tissue-resident MMs and LpMs, respectively, is involved in the neuro-immune unit’s adaptation to different tissue niches in response to environmental perturbations and in maintaining resistance and tolerance balance to cope with allostasis [136].
The neuro-immune unit also contains mast cells proximal to nerve fibers, mainly in mucosa and submucosa [137]. Enteric neurons secrete the growth factor Kit ligand, a factor interacting with the tyrosine-kinase receptor Kit on mast cells, which is essential for cell maintenance [138,139]. Nociceptor neurons are sensory neurons containing several cytosolic neuropeptides (e.g., substance P, vasoactive intestinal peptide, calcitonin-gene related peptide) that can be released during inflammation, modulating immune and stromal cells. These peptides activate vascular smooth muscle, endothelial, epithelial, and mast cells. In particular, substance P triggers mast cell degranulation accompanied by visceral pain, diarrhea, and dysmotility. This crosstalk between neurons and mast cells is essential in steady state for gut homeostasis and in inflammatory conditions (such as parasite infection, irritable bowel syndrome, and food allergy). These circumstances can be accompanied by gastro-intestinal motility disturbance and promoted by mast cell degranulation at high frequency [140,141,142].
Nociceptor sensory neurons also control epithelial barrier integrity and homeostasis through calcitonin-gene-related peptide (CGRP) secretion and mucin production. Goblet cells are specialized epithelial cells that produce mucins and enzymes that form the outer mucus layer to prevent the contact of microbes with the apical face of epithelial cells and microbial penetration [143]. Commensal and food antigens have been described to promote neuronal CGRP secretion, which interacts with Ramp1 on goblet cells to drive mucus secretion. Deficiencies on the CGRP-Ramp1 axis show decreased mucin production and susceptibility to colitis [144].
Enteric neurons also coordinate the function of innate lymphoid cells (ILC) in the gut during early immunity. ILCs lack PRR but produce vast amounts of cytokines in response to tissue disturbances [145]. Tissue-resident ILCs react to alarmins and cytokines, hormones, prostaglandins, metabolites (e.g., aryl hydrocarbon receptor ligands, vitamin A) and are sensitive to neurotransmitters and neuropeptides [146]. The different subsets of ILC are located close to SNS, PNS and ENS neurons and crosstalk between cells regulates activation or suppression depending on the context [147]. Vagus nerve acetylcholine (ACh) acts as an anti-inflammatory factor suppressing pro-inflammatory type 2 cytokine release from ILC2 cells, via the α7-nicotinic Ach receptor and is involved in infection resolution, allergy and autoimmunity [148,149]. The anti-inflammatory and host-protective effect of Ach was also described in macrophages and ILC3 cells for coping with infections [150]. Norepinephrine (NE), a SNS-derived neurotransmitter, also inhibits ILC2 by binding to the β2-adrenergic receptors. The role of NE was described in activation, proliferation, and type 2 cytokine secretion in helminth infections and allergic gut and lung inflammation [151]. Moreover, neuromedin U, a neuropeptide derived from ENS neurons, rapidly and robustly stimulates ILC2 through neuromedin U receptors for type 2 pro-inflammatory cytokine secretion during gut and lung worm infection [146,151,152]. ILC1 cells are also sensitive to suppressive effects of glucocorticoids (GC) through GC-Rs; LPS-induced IFN-γ secretion can be limited in mice by a direct GC effect on NKp46+ ILC1 cells [153]. GCs also inhibit ILC2 and CD4+T cells in lung allergic inflammation [154]. Finally, the food-induced enteric neuron-derived vasoactive intestinal peptide (VIP) also stimulates ILC2 cells through VIP receptor 2 to trigger type 2 inflammation [155]. VIP also stimulates IL-22 secretion by ILC3 cells and this cytokine promotes barrier integrity and production of antimicrobial peptides by intestinal epithelial cells [156]. VIP production after food intake indicates that enteric neurons prepare intestinal mucosa for tissue preservation. In summary, ILCs are primary sensors of neuronal factors and crosstalk between ILCs and neurons is essential for tissue-homeostasis regulation.
Remarkably, neurons also modulate the adaptive immune response. The expression of Coh-/NE-gic or neuropeptide receptors on dendritic cells (DC), which act as a nexus between innate and adaptive immunity, suggests that neurons regulate DC function. Stimulation of β-2 adrenergic receptors abrogates cross-presentation with further Th1 suppression and Th2/Th17 induction [157]. Conversely, α-adrenergic receptor stimulation enhances antigen uptake and migration for T cell activation [158]. VIP showed a regulatory function through Treg differentiation [159] and catecholamines have been reported to impact B and T cell functions, promoting lymphocyte recruitment, Th2, Th17, Treg, and antibody responses [160,161].
These data demonstrate that neuron-immune crosstalk is coordinated to detect the presence of microorganisms or tissue injury, exerting an intricate circuit of activation and regulatory mechanisms to restore tissue homeostasis. This is critical in tissues where cells, such as neurons, have no or reduced migratory properties and proliferative potential to regenerate and re-populate. Mucosal neurons and immune cells have adopted common sensing strategies and are equipped with specific PRRs to detect microbes directly. After immune cell activation, effector protective mechanisms are induced and an anti-inflammatory response is promoted to preserve intestine homeostasis. Immune cells and neurons can develop different mechanisms to solve inflammation and tissue repair during restoration of homeostasis. Uncontrolled and chronic exaggerated inflammation drives many intestinal diseases, e.g., food allergy, intestinal BD, irritable bowel syndrome, and infectious disease with septic shock [113]. Animal models and human clinical trials have demonstrated that vagus nerve stimulation controls chronic inflammation and alleviates pathological conditions such as Crohn´s disease and rheumatoid arthritis (RA) in humans [162,163], and colitis, hypovolemic shock, endotoxemia, and RA in experimental models [163,164,165].
4.3. The Roles of the Microbiota and Barriers
The gut-brain axis was originally described as a unidirectional signaling network that top-down regulated intestinal functions from the CNS. Nevertheless, extensive studies on intestinal microbiota have described complex interaction between microbes and the mucosal immune system in the 21st century and revealed the bidirectional regulation of neurons and immune cells. In the last decade, intestinal microbes were included in the GBA, and we are starting to understand how enteric microbiota influences the gut-brain relationship (e.g., mental state, emotional regulation, neuromuscular function, regulation of HPA axis, development of mucosal immune structures, regulation of mucosal immune cells). Commensal microorganisms or their metabolites modulate the CNS and PNS, and sensing microbes is not always considered an injurious signal. GBA gut microbiota communicates with the CNS through the vagus nerve and systemic circulation (Figure 6). Different vascular and epithelial barriers, such as the intestinal epithelial barrier, gut-vascular barrier, blood-brain barrier, choroid plexus vascular barrier and blood-cerebrospinal fluid barrier, are also critical to maintaining a regulated network for local and systemic functions [166]. Maintaining epithelial barrier and vascular structures in the gut promotes correct exchange of metabolites between the lumen and the mucosal tissue to modulate selective permeability. Microbiota is implicated in preserving a functional GVB and, strikingly, the integrity and permeability of BBB. Germ-free mice exhibit a highly leaky BBB with impaired expression of tight-junction proteins, resulting in distal neurological disease [167]. During dysbiosis the GVB is disrupted, gut permeability is increased, bacteria are translocated, and metabolites enter the circulation, causing local and distant inflammation [166]. It has been documented that antibiotic-induced dysbiosis led to GVB dysfunction and liver disease [168], and GVB disruption in a colitis model provoked a leaky cerebral vascular barrier [14]. It can be assumed that the GVB-BBB axis represents a multifaceted additional alliance that connects the gut with the brain, and that microbiota is critically involved in controlling its integrity and local and distal inflammation. Microbial dysbiosis, which has been implicated in Inflammatory Bowel diseases and Irritable Bowel Syndrome, may be considered a comorbid condition for neuroinflammatory processes such as AD, PD and/or depression [169,170,171]
Enteric and extrinsic neurons can directly sense microbiota and microbiota can modulate neuronal functions by producing short-chain fatty acids (SCFA) such as propionate, acetate, butyrate, etc. [172]. An exaggerated HPA stress response described in germ-free models was partially mitigated during early exposure to microbial commensals [173]. This work revealed a link between microorganisms, cognitive processes, and neurological and psychological functions [172,174,175,176]. Also, microbial dysbiosis has been linked to the pathology of prevalent neurological conditions, including autism, depression, AD and PD [177,178]. Different gut microbial metabolites have been described to traffic to the brain through circulation. However, the vagus nerve is also deeply involved in this bidirectional interaction, linking bacteria to the brain. Microbes and microbial components trigger vagal responses whether they are pathogenic or commensal [179]. Early Citrobacter rodentium infection increased vagal activation and anxiety-like behavior in rodents [180], whereas exposure to commensal Lactobacillus johnsonii enhanced vagal nerve activity, lowering blood pressure [181]. LPS is also known to elicit broad neuroinflammatory effects, including alterations at the site of the CNS-blood interface, the BBB, following injection into the bloodstream or cerebral spinal fluid [182,183]. Early exposure to intestinal microorganisms also impacts BBB formation and integrity. Germen-free mice exhibit impaired BBB development and increased permeability. Microbial colonization or exposure to microbial-produced SCFAs increases expression of endothelial tight junction proteins, significantly improving BBB integrity [167]. In conventional mice, antibiotic treatment decreases microbial diversity and intestinal SCFA levels. Antibiotic treatment also alters the expression of tight junction proteins in the hippocampus and amygdala, further suggesting a link between SCFA and BBB integrity [176]. Propionate was recently reported to protect cultured BBB endothelial cells from inflammatory and oxidative stresses via the receptor FFAR3 [184,185,186,187,188,189,190].
As described above, different cells can synthesize and secrete neurotransmitters and neuropeptides, and specific bacteria from microbiota can generate or metabolize precursors, potentially limiting their availability to the host. Tryptophan (Trp) is an essential amino acid ingested with diet and microbes in the intestinal lumen metabolize it. Germ-free mice showed elevated levels of serum Trp and its metabolites kynurenine and serotonin, which were reduced following colonization [191]. Patients with IBD suffering from depression [192,193] showed low levels of Trp in serum [194]. Mice deprived of Trp also showed enhanced susceptibility to colitis [195], which was reverted with Trp administration [196].
Furthermore, serotonin is a crucial neurotransmitter involved in modulating intestinal peristalsis, innate and adaptive cell functions, anxiety, and depression. Depression-like behavior induced in rats was reverted with serotonin diet supplementation [197]. This means that microbiota is implicated in Trp availability and serotonin synthesis, and the balance between utilization and metabolization impacts Trp and serotonin availability. In conclusion, alterations in microbiota composition may lead to gut inflammation, depression, social-cognitive deficits, visceral pain, dysmotility, etc. Administration of probiotic Byfidobacterium capable of producing Trp metabolites or supplementation with Trp or food-derived bioactive molecules may be considered alternative therapeutic strategies to restore GBA homeostasis and protect against neurodegeneration [198,199].
5. Relation between Brain, Gut, and White Adiposity Processes.
Figure 7 schematically displays the intricate relationship between brain, gut, and WAT functions (the Brain-Gut-WAT axis). Firstly, due to the appearance of endocrine-metabolic dysfunction after WAT inflammation, pro-inflammatory adipokines (WAT-derived) and cytokines (Immune System-derived) increase neuroinflammation by affecting the brain. Indeed, as mentioned, these signals can enter the brain, specifically due to changes in BBB permeability, perpetuating visceral WAT accumulation because neuronal insulin and leptin sensing are disrupted (IR/LEP-R). As a result, brain dysfunction enhances appetite (incremented orexigenic signals, i.e., NPY and AgRP neuron production) and lowers satiety (decreased anorectic signals, i.e., POMC and CART neuron activities). These mechanisms, combined with accumulation of the already dysfunctional WAT (excess leptin and decreased adiponectin secretions by large-in-size, IR-resistant white adipocytes), aggravate this picture. Indeed, hypertrophic WAT adipocytes and immune system-infiltrating cells perpetuate the peripheral inflammatory process and also neuroinflammation until both (peripheral and brain) dysfunctions are solved. As mentioned, gut microbiota plays a relevant role in the interrelation between gut, white adiposity and brain functions. Indeed, once metainflammation is installed in an organism (eg. WAT mass hypertrophic expansion), BBB permeability ismodified. As a result, bacteria/virus may enter the brain, strongly cooperating with neuroinflammation development. Therefore, microbiome perturbations (dysbiosis) then influence both brain and WAT functionalities. All three compartments form part of a pathophysiological vicious circle of harmful signals. This vicious circle is summarized in Figure 7, where the concept is clearly represented in both physiological conditions (in blue) and in an obese/diabetic phenotype (in red).
WAT metabolism is also controlled by neuro-immune cells involving adipose tissue-resident macrophages (ATRMs). Interestingly, a population of white adipose tissue macrophages is colocalized with sympathetic fibers [200]. These cells are known as sympathetic neuro-associated macrophages (SAMs); they are in charge of clearing NE through both solute carrier family 6 member-2 (SLC6A2), a known NE transporter, and monoamine oxidase A (MAOA) activity. SAMs can interact with SNS fibers within WAT, are morphologically distinct from ATRMs, and possess a unique gene expression pattern [200], including expression of genes related to synaptic signaling, cell-cell adhesion, and neuron development. Unlike the circular morphology of ATRMs, SAMs wrap around SNS fibers and exhibit a stellate morphology. Interestingly, SAMs contained significant amounts of intracellular NE, but lack requisite enzymes for NE synthesis, as reported for macrophages [201]. These macrophages are CX3CR1+ cells and exhibit a pro-inflammatory shape similar to pro-inflammatory M1 rather than M2 macrophages. Although other macrophages have been shown to express MAOA [200], only SAMs express Slc6a2 [200]. SAMs are thought to act by catching-up with NE excess after SNS stimulation and thereby metabolizing it. SAMs seem to be recruited to WAT in different obese models (DIO and genetic) and may contribute to white adipocyte hypertrophy through a mechanism of NE over-degradation. Interestingly, ablation of Slc6a2 from SAMs in obese mice leads to obesity rescue by reestablishing NE levels, thereby enhancing lipolysis and energy expending processes. Remarkably, SAMs were also noticed in human SNS [200]. In most these studies, CX3CR1+ immune cells are implicated in the neuro-immune interaction in WAT. All together, these findings suggest that multiple subsets of CX3CR1+ macrophages act in concert to maintain energy homeostasis through interactions with SNS innervation of WAT depots.
As expected, due to the proximity between pancreas, liver, and gut, the origin of nerves innervating the gut is consistent with those innervating pancreas and liver. Gaskell pioneered the understanding of nerves innervating peripheral tissues, including the gut [202]. A pivotal study by Berthoud et al. laid the groundwork for further research in this area [203]. This report concluded that vagal innervation of the gut originates primarily from the gastric branch, celiac branch, while a small contribution from the hepatic branch innervates the distal stomach [203]. Current research is investigating nerve sub-types and their roles in proper gut function. Cholecystokinin receptor (CCKR), a known afferent nerve receptor in gut [204], increases expression in the nodose ganglia of the vagus in DIO rats [205]. Knockout for ob-Rb on sensory nerves in vagal afferents increases body weight gain, indicating inappropriate gut–brain signaling [206]. The latter is crucial evidence for the relevance of gut-brain communication through vagal afferent innervation of the gut and its relation to adipose tissue accumulation. This finding complements previous data on WAT regarding the presence of the Ob-Rb on nerves innervating WAT [33].
Adipose tissue innervation and brain-adipose neural communication offer an exciting area of research. New insights into innervation patterns, neuro-immune interactions, and regulation of nerve plasticity in WAT depots have merged. Other indicators of bi-directional communication between gut-resident nerves and immune cells have recently been studied. In this regard, an interesting role for neurotrophic factors has been implicated in the progression and severity of gut inflammatory responses. One example is nerve growth factor (NGF), secreted by a variety of immune cell types, including mast cells and macrophages, promoting a plethora of signaling pathways and encompassing anti-inflammation [207] and organism survival [207]. NGF-signaling pathway is linked to knowledge of sensory neuropeptide expression (i.e. Substance P) in the rat gut [208]. NGF also promotes the formation of colonic afferent central terminals localized at the dorsal horn of the spinal cord, and has been reported to increase visceral nociception in experimental colitis [209].
6. Concluding Remarks.
Due to the severe lack of precise background and information for both clinicians and the general public regarding the fast-growing epidemic condition of insulin resistance-related pathologies (i.e. Metabolic Syndrome, Obesity, T2DM and NDs), early diagnosis and treatment of these pathological conditions are imperative. Even by following WHO recommendations clinicians and patients could help to prevent further development of IR-dependent pathologies. Moreover, early diagnosis of an IR state will help public health systems by reducing future high-cost treatments (i.e. bariatric surgery). More importantly, it will improve insulin-resistant patients and their relatives´ quality of life. In addition to IR states, our body is continuously exposed to dangerous factors, such as malnutrition and microbial injury at mucosal sites. To cope with this favorable environment for further neuroinflammation development, once white hypertrophic adipocytes have developed and metainflammation is established, mucosal intestinal tissues are densely populated with immune cells (the mucosal immune system). However, the nervous system is intricately woven into the mucosal immune system. Together, they play a pivotal role in sensing dysbiosis, orchestrating an inflammatory response to counteract injury. The neuron-immune unit is constantly surveilling this complex antigenic context, with the objective of coping with allostasis and, in turn, restoring overall homeostasis through an integrated network of interactions, briefly known as the Brain-WAT-Gut axis. Microbiota can be considered an additional and key regulator of this axis functionality; indeed, microbiota is an integral part of this multi-directional axis, therefore influencing tissue physiology.
In consequence, any individual injury will result not only in disturbed local tissues but also in distant tissue (e.g. WAT and gut) functions. In turn, the harmful signals they secrete impact on peripheral blood in the brain. Thus, neuroinflammation becomes established until reward mechanisms are activated to cope with this detrimental allostasis.
Author Contributions
Conceptualizations by E.S. and G.D. Writing original draft preparation, editing and final writing by E.S. and G.D. Both authors have read and agreed to publish the final manuscript.
Funding
This review was supported by “Fondation Rolf C. Gaillard” (Switzerland; grant number FPREDM2023-2025, to E.S.) and a grant from the National Funding Agency for Science in Argentina (grant number PICT 2020 3166, to G.D.,).
Acknowledgments
Authors wish to thank Mrs. Susan Hale Rogers for her carefully correction of the English’s grammar and edition of this manuscript.
Conflicts of Interest
Both authors declare no any conflict of interest.
Abbreviations:
| Ab | Amyloid beta |
| Ach | Acetylcholine |
| AD | Alzheimer's Disease |
| AgRP | Aguti-related protein |
| AKT | Serine/Threonine Kinase |
| ALS | Amyotrophic Lateral Sclerosis |
| ANS | Autonomic Nervous System |
| Arg1 | Arginase 1 |
| ASO | α-Syn-Overexpressing |
| α-Syn | α-Synuclein |
| ATP | Adenosine Triphosphate |
| ATRM | Adipose Tissue-Resident Macrophage |
| BBB | Blood-Brain Barrier |
| BMP2 | Bone Morphogenetic Protein 2 |
| BMPR | Bone Morphogenetic Protein Receptor |
| CART | Cocaine-Amphetamine Related Transcript |
| CCKR | Cholecystokinin Receptor |
| CGRP | Calcitonin-Gene-Related Peptide |
| CNS | Central Nervous System |
| CpG ODN | CpG Oligodeoxynucleotide |
| C-RP | C Reactive Protein |
| CSF1 | Colony Stimulating Factor 1 |
| CX3CL1 | Chemokine Fractalkine |
| CX3CR1 | Fractalkine Receptor 1 |
| DA | Dopamine |
| DAMP | Damage-Associated Molecular Pattern |
| DC | Dendritic Cells |
| DIO | Diet-Induced Obesity |
| DOPA | Dihydroxy Phenyl Alanine |
| ENS | Enteric Nervous System |
| ER | Endoplasmic Reticulum |
| FFA | Free Fatty Acid |
| FFAR | Free Fatty Acid Receptor |
| GBA | Gut-Brain axis |
| GC | Glucocorticoid |
| GLU | Glucose |
| GM | Gut Microbiota |
| GR | Glucocorticoid Receptor |
| GRE | Glucocorticoid Response Element |
| GSK3b | Glycogen Synthase Kinase 3b |
| GVB | Gut-Vascular Barrier |
| HD | Huntington Disease |
| HFD | High Fat Diet |
| HPA | Hypotalamo-Pituitary-Adrenal |
| IAPP | Islet Amyloid Polypeptide |
| IAPP | Islet Amyloid Polypeptide |
| IBD | Inflammatory Bowel Disease |
| IDE | Insulin-Degrading Enzyme |
| IFN- | Interferon-gamma |
| IKKβ | Inhibitory Kappa beta Kinase |
| IL-1 | Interleukin-1 |
| IL-6 | Interleukin-6 |
| ILC | Innate Lymphoid Cell |
| Ins | Insulin |
| IR | Insulin Resistance |
| IRS-1 | Insulin Receptor Susbtrate-1 |
| JAK-STAT | Janus Kinase-Signal Transducer and Activator of Transcription |
| L2-3 | Spinal Lumbar Segments 2 & 3 |
| LEP-R | Leptin Receptor |
| LPM | Lamina Propria Macrophage |
| LPS | Lipopolysaccharide |
| MAOA | Monoamine Oxidase A |
| microRNA | micro Ribonucleic Acid |
| MM | Muscularis Macrophage |
| MMP | Matrix Metalloproteinases |
| MR | Mineralocorticoid Receptor |
| mTOR | Mechanistic Target of Rapamycin |
| ND | Neurodegenerative Disease |
| NE | Norepinephrine |
| NF | Neuroinflammation |
| NF-κB | Nuclear Factor-Kappa B |
| NGF | Nerve Growth Factor |
| NO | Nitric Oxide |
| NPY | Neuropeptide Y |
| Ob-Rb | Leptin Receptor form b |
| OS | Oxidative Stress |
| PAI-1 | Plasminogen Activator Inhibitor type 1 |
| PAMP | Pathogen-Associated Molecular Pattern |
| PD | Parkinson's Disease |
| PI3K | Phosphatidylinositol 3-Kinase |
| PNS | Parasympathetic Nervous System |
| POMC | Pro-opiomelanocortin |
| PRR | Pattern-Recognition Receptors |
| RA | Rheumatoid Arthritis |
| Ramp1 | Receptor Activity Modifying Protein 1 |
| ROS | Reactive Oxygen Substances |
| SAM | Sympathetic Neuro-Associated Macrophage |
| SCFA | Short Chain Fatty Acid |
| SLC6A2 | Solute Carrier Family 6 Member-2 |
| SNS | Sympathetic Nervous System |
| SOCS3 | Suppressor of Cytokine Signaling 3 |
| SPF | Specific Pathogen Free |
| T13 | Spinal Thoracic Segment 13 |
| T2DM | Type 2 Diabetes Mellitus |
| Th | T Helper Lymphocytes |
| TLR7 | Toll-Like Receptor |
| TNF- | Tumor Necrosis-alpha |
| TOX01 | Forkhead Box Protein 01 |
| Trp | Tryptophan |
| UCP-1 | Uncoupling Protein-1 |
| VIP | Vasoactive Intestinal Peptide |
| WAT | White Adipose Tissue |
| WHO | World Health Organization |
References
- Niranjan, R. Recent advances in the mechanisms of neuroinflammation and their roles in neurodegeneration. Neurochem. Int. 2018, 120, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Lyman, M.; Lloyd, D.G.; Ji, X.; Vizcaychipi, M.P.; Ma, D. Neuroinflammation: The role and consequences. J. Neuroscience. Research. 2014, 79, 1–12. [Google Scholar] [CrossRef]
- Subhramanyam, C.S.; Wang, C.; Hu, Q.; Dheen, S.T. Microglia-mediated neuroinflammation in neurodegenerative diseases. J. Semin. Cell. Dev. Biol. 2019, 94, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.C.; Chiu, K.; Ho, Y.S.; So, K.F. Modulation of neuroimmune responses on glia in the central nervous system: implication in therapeutic intervention against neuroinflammation. J. Cell. Mol. Immunol. 2009, 6, 317–326. [Google Scholar] [CrossRef]
- Lyons, A.; McQuillan, K.; Deighan, B.F.; O'Reilly, J.A.; Downer, E.J.; Murphy, A.C.; Watson, M.; Piazza, A.; O'Connell, F.; Griffin, R.; et al. Decreased neuronal CD200 expression in IL-4-deficient mice results in increased neuroinflammation in response to lipopolysaccharide. J. Brain. Behavior. Immunity. 2009, 23, 1020–1027. [Google Scholar] [CrossRef] [PubMed]
- Bessis, A.; Biber, K.; Bilbo, S.; Blurton-Jones, M.; Boddeke, E.; Brites, D.; et al. Microglia states and nomenclature: a field at its crossroads. Neuron. 2022, 110, 3458–3483. [Google Scholar]
- Paolicelli, R.C.; Sierra, A.; Stevens, B.; Tremblay, M.E.; Aguzzi, A.; Ajami, B.; Amit, I.; Audinat, E.; Bechmann, I.; Bennett, M.; et al. Microglia states and nomenclature: a field at its crossroads. Neuron. 2022, 110, 3458–3483. [Google Scholar] [CrossRef] [PubMed]
- Bazoukis, G.; Stavrakis, S.; Armoundas, A.A. Vagus Nerve Stimulation and Inflammation in Cardiovascular Disease: A State-of-the-Art Review. J. Am. Heart Assoc. 2023, 2, e030539. [Google Scholar] [CrossRef]
- Sun, Z. , Wang, X., Feng, S.; Xie, C., Xing, Y., Guo, L., et al. A review of neuroendocrine immune system abnormalities in IBS based on the brain–gut axis and research progress of acupuncture intervention. Front Neurosci. 2023, 17, 934341. [Google Scholar] [CrossRef]
- Choe, S.S.; Huh, J.Y.; Hwang, I.J.; Kim, J.I.; Kim, J.B. Adipose Tissue Remodeling: its Role in energy Metabolism and Metabolic Disorders. Front. Endocrinol. 2016, 7, 30. [Google Scholar] [CrossRef]
- Candelario-Jalil, E.; Dijkhuizen, R.M.; Magnus, T. ; Neuroinflammation, Stroke, Blood-Brain Barrier: Dysfunction, and Imaging Modalities. Stroke. 2022, 53, 1473–1486. [Google Scholar] [CrossRef]
- Maldonado-Ruiz, R.; Fuentes-Mera, L.; Camacho, A. Central Modulation of Neuroinflammation by Neuropeptides and Energy-Sensing Hormones during Obesity. Biomed. Res. Int. 2017, 2017, 7949582. [Google Scholar] [CrossRef]
- Bertocchi, A.; Carloni, S.; Ravenda, P.S.V.; Bertalot, G.; Spadoni, I.; Lo Cascio, A.; Gandini, S.; Lizier, M.; Braga, D.; Asnicar, F.; et al. Gut vascular barrier impairment leads to intestinal bacteria dissemination and colorectal cancer metastasis to liver. Cancer Cell. 2021, 39, 708–724. [Google Scholar] [CrossRef]
- Carloni, S.; Bertocchi, A.; Mancinelli, S.; Bellini, M.; Erreni, M.; Borreca, A.; Braga, D.; Giugliano, S.; Mozzarelli, A.M.; Manganaro, D.; et al. Identification of a choroid plexus vascular barrier closing during intestinal inflammation. Science. 2021, 374, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Fehervari, Z. A gut vascular barrier. Nat. Immunol. 2016, 17, 47. [Google Scholar] [CrossRef]
- Bilski, J.; Mazur-Bialy, A.; Wojcik, D.; Surmiak, M.; Magierowski, M. , Sliwowski. Z., Pajdo, R.; Kwiecien, S.; Danielak, A.; Ptak-Belowska, A.; et al. Role of Obesity, Mesenteric Adipose Tissue, and Adipokines in Inflammatory Bowel Diseases. Biomolecules. 2019; 9, 780. [Google Scholar]
- Rautmann, A.W.; de La Serre, C.B. Microbiota's Role in Diet-Driven Alterations in Food Intake: Satiety, Energy Balance, and Reward. Nutrients. 2021, 13, 3067. [Google Scholar] [CrossRef] [PubMed]
- Sartor, R.B. Mechanisms of disease: Pathogenesis of Crohn’s disease and ulcerative colitis. Nat. Clin. Pract. Gastroenterol. Hepatol. 2006, 3, 390–407. [Google Scholar] [CrossRef]
- Więckowska-Gacek, A.; Mietelska-Porowska, A.; Wydrych, M.; Wojda, U. Western diet as a trigger of Alzheimer's disease: From metabolic syndrome and systemic inflammation to neuroinflammation and neurodegeneration. Ageing. Res. Rev. 2021, 70, 101397. [Google Scholar] [CrossRef] [PubMed]
- Spinedi, E.; Cardinali, D.P. Neuroendocrine-Metabolic Dysfunction and Sleep Disturbances in Neurodegenerative Disorders: Focus on Alzheimer's Disease and Melatonin. Neuroendocrinology. 2019, 108, 354–364. [Google Scholar] [CrossRef]
- Ordovas-Montanes, J.; Rakoff-Nahoum, S.; Huang, S.; Riol-Blanco, L.; Barreiro, O.; von Andrian, U.H. The Regulation of Immunological Processes by Peripheral Neurons in Homeostasis and Disease. Trends. Immunol. 2015, 36, 578–604. [Google Scholar] [CrossRef]
- Godinho-Silva, C.; Cardoso, F.; Veiga-Fernandes, H. . Neuro-Immune Cell Units: A New Paradigm in Physiology. Ann. Rev. Immunol. 2019, 26, 19–46. [Google Scholar] [CrossRef]
- Shepherd, A.J.; Beresford, L.J.; Bell, E.B.; Miyan, J.A. . Mobilisation of specific T cells from lymph nodes in contact sensitivity requires substance P. J. Neuroimmunol. 2005, 164, 115–23. [Google Scholar] [CrossRef]
- Veiga-Fernandes, H.; Pachnis, V. Neuroimmune regulation during intestinal development and homeostasis. Nat. Immunol. 2017, 18, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Furness, J.B.; Kunze, W.A.; Clerc, N. Nutrient tasting and signaling mechanisms in the gut. II. The intestine as a sensory organ: neural, endocrine, and immune responses. Am. J. Physiol. 1999, 277, G922–G928. [Google Scholar]
- Neunlist, M.; Van Landeghem, L.; Mahé, M.M.; Derkinderen, P.; des Varannes, S.B.; Rolli-Derkinderen, M. The digestive neuronal-glial-epithelial unit: a new actor in gut health and disease. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.A.; Chung, Y.C.; Pan, S.T.; Shen, M.Y.; Hou, Y.C.; Peng, S.J.; Pasricha, P.J.; Tang, S.C. 3-D imaging, illustration, and quantitation of enteric glial network in transparent human colon mucosa. SC. Neurogastroenterol. Motil. 2013, 25, e324–e338. [Google Scholar] [CrossRef] [PubMed]
- Meisel, J.D.; Panda, O.; Mahanti, P.; Schroeder, F.C.; Kim, D.H. Chemosensation of bacterial secondary metabolites modulates neuroendocrine signaling and behavior of C. elegans. Cell. 2014, 159, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Makhijani, K.; Alexander, B.; Rao, D.; Petraki, S.; Herboso, L.; Kukar, K.; Batool, I.; Wachner, S.; Gold, K.S.; Wong, C.; et al. Regulation of Drosophila hematopoietic sites by Activin-β from active sensory neurons. Nat. Commun. 2017, 8, 15990. [Google Scholar] [CrossRef]
- Zeng, W.; Pirzgalska, R.M.; Pereira, M.M.; Kubasova, N.; Barateiro, A.; Seixas, E.; Lu, Y.H.; Kozlova, A.; Voss, H.; Martins, G.G.; et al. Sympathetic neuro-adipose connections mediate leptin-driven lipolysis. Cell. 2015, 163, 84–94. [Google Scholar] [CrossRef]
- Fishman, R.B.; Dark, J. Sensory innervation of white adipose tissue. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1987, 253, R942–R944. [Google Scholar] [CrossRef]
- Song, C.K.; Schwartz, G.J.; Bartness, T.J. Anterograde transneuronal viral tract tracing reveals central sensory circuits from white adipose tissue. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R501–R511. [Google Scholar] [CrossRef]
- Murphy, K.T.; Schwartz, G.J.; Nguyen, N.L.T.; Mendez, J.M.; Ryu, V.; Bartness, T.J. Leptin-sensitive sensory nerves innervate white fat. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E1338–E1347. [Google Scholar] [CrossRef] [PubMed]
- Garretson, J.T.; Szymanski, L.A.; Schwartz, G.J.; Xue, B.; Ryu, V.; Bartness, T.J. Lipolysis sensation by white fat afferent nerves triggers brown fat thermogenesis. Mol. Metab. 2016, 5, 626–634. [Google Scholar] [CrossRef] [PubMed]
- Youngstrom, T.G.; Bartness, T.J. Catecholaminergic innervation of white adipose tissue in Siberian hamsters. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1995, 268, R744–R751. [Google Scholar] [CrossRef] [PubMed]
- Bartness, T.J.; Liu, Y.; Shrestha, Y.B.; Ryu, V. Neural innervation of white adipose tissue and the control of lipolysis. Front. Neuroendocrinol. 2014, 35, 473–493. [Google Scholar] [CrossRef] [PubMed]
- Bamshad, M.; Aoki, V.T.; Adkison, M.G.; Warren, W.S.; Bartness, T.J. Central nervous system origins of the sympathetic nervous system outflow to white adipose tissue. Am. J. Physiol. 1998, 275, R291–R299. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.B.S. Denervation as a tool for testing sympathetic control of white adipose tissue. Physiol. Behav. 2018, 190, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Song, C.K.; Giordano, A.; Cinti, S.; Bartness, T.J. Sensory or sympathetic white adipose tissue denervation differentially affects depot growth and cellularity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R1028–R1037. [Google Scholar] [CrossRef] [PubMed]
- Foster, M.T.; Bartness, T.J. Sympathetic but not sensory denervation stimulates white adipocyte proliferation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 291, R1630–R1637. [Google Scholar] [CrossRef]
- Herradon, G.; Ramos-Alvarez, M.P.; \Gramage, E. Connecting Metainflammation and Neuroinflammation Through the PTN-MK-RPTPβ/ζ Axis: Relevance in Therapeutic Development Front. Pharmacol. 2019, 10, 377. [Google Scholar]
- Reaven, G.M. The insulin resistance syndrome. Curr Atheroscler Rep. 2003, 5, 364–371. [Google Scholar] [CrossRef]
- McGarry, J.D. What if Minkowski had been ageusic? Diabetes. 1992, 41, 826–834. [Google Scholar]
- Wozniak, S.E.; Gee, L.L.; Wachtel, M.S.; Frezza, E.E. Adipose tissue: the new endocrine organ? Dig. Dis. Sci. 2009, 54, 1847–1856. [Google Scholar] [CrossRef] [PubMed]
- Lelliot, C.; Vidal Puig, A.J. Lipotoxicity, an imbalance between lipogenesis de novo and fatty acid-oxidation. Int. J. Obesity. 2004, 28, S22–S28. [Google Scholar] [CrossRef] [PubMed]
- Olefsky, J.M. Decreased insulin binding to adipocytes and circulating monocytes from obese subjects. J. Clin. Invest. 1976, 57, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.M.; Ciaraldi, T.P.; Brady, D.; Olefsky, J.M. Decreased activation rate of insulin-stimulated glucose transport in adipocytes from obese subjects. Diabetes. 1989, 38, 991–995. [Google Scholar] [CrossRef]
- Carnie, J.A.; Smith, D.G.; Mavris-Vavayannis, M. Effects of insulin on lipolysis and lipogenesis in adipocytes from genetically obese (ob/ob) mice. Biochem. J. 1979, 184, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Pocai, A.; Lam, T.K.; Gutierrez-Juarez, R.; Obici, S.; Schwartz, G.J.; Bryan, J.; Aguilar-Bryan, L.; Rossetti, L. . Hypothalamic K(ATP) channels control hepatic glucose production. Nature. 2005, 434, 1026–1031. [Google Scholar] [CrossRef]
- Kieffer, T.J.; Habener, J.F. The adipoinsular axis: effects of leptin on pancreatic beta-cells. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E1–E14. [Google Scholar] [CrossRef]
- Frühbeck, G.; Gómez-Ambrosi, J.; Muruzábal, F.J.; Burrell, M.A. The adipocyte: a model for integration of endocrine and metabolic signaling in energy metabolism regulation. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E827–E84747. [Google Scholar] [CrossRef]
- Banks, A.S.; Davis, S.M.; Bates, S.H.; Myers, M.G. Jr. Activation of downstream signals by the long form of the leptin receptor. J. Biol. Chem. 2000, 275, 14563–14572. [Google Scholar] [CrossRef]
- Mizuno, A.; Murakami, T.; Otani, S.; Kuwajima, M.; Shima, K. Leptin affects pancreatic endocrine functions through the sympathetic nervous system. Endocrinology. 1998, 139, 3863–3870. [Google Scholar] [CrossRef] [PubMed]
- Huan, J.N.; Li, J.; Han, Y.; Chen, K.; Wu, N.; Zhao, A.Z. Adipocyte-selective reduction of the leptin receptors induced by antisense RNA leads to increased adiposity, dyslipidemia, and insulin resistance. J. Biol. Chem. 2003, 278, 45638–45650. [Google Scholar] [CrossRef] [PubMed]
- Kern, P.A.; Saghizadeh, M.; Ong, J.M.; Bosch, R.J.; Deem, R.; Simsolo, R.B. The expression of tumor necrosis factor in human adipose tissue. Regulation by obesity, weight loss, and relationship to lipoprotein lipase. J. Clin. Invest. 1995, 95, 2111–2119. [Google Scholar] [CrossRef]
- Fain, J.N.; Madan, A.K.; Hiler, M.L.; Cheema, P.; Bahouth, S.W. Comparison of the release of adipokines by adipose tissue, adipose tissue matrix, and adipocytes from visceral and subcutaneous abdominal adipose tissues of obese humans. Endocrinology. 2004, 145, 2273–2282. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Kihara, S.; Funahashi, T.; Matsuzawa, Y.; Libby, P. Adiponectin: A key adipocytokine in metabolic syndrome. Clin. Sci. (Lond). 2006, 110, 267–278. [Google Scholar] [CrossRef]
- Niswender, K.D.; Schwartz, M.W. Insulin and leptin revisited: adiposity signals with overlapping physiological and intracellular signaling capabilities. Front. Neuroendocrinol. 2003, 24, 1–10. [Google Scholar] [CrossRef]
- Anubhuti Arora, S. Leptin and its metabolic interactions: an update. Diabetes. Obes. Metab. 2008, 10, 973–993. [Google Scholar] [CrossRef] [PubMed]
- Frühbeck, G. Intracellular signalling pathways activated by leptin. Biochem. J. 2006, 393, 7–20. [Google Scholar] [CrossRef]
- Uyama, N.; Geerts, A.; Reynaert, H. Neural connections between the hypothalamus and the liver. Anat. Rec. A. Discov. Mol. Cell. Evol. Biol. 2004, 280, 808–820. [Google Scholar] [CrossRef]
- Flak, J.N.; Myers, M.G. Jr. CNS Mechanisms of Leptin Action. Mol. Endocrinol. 2016, 30, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Unger, R.H. Minireview: Weapons of lean body mass destruction: The role of ectopic lipids in the metabolic syndrome. Endocrinology. 2003, 144, 5159–5165. [Google Scholar] [CrossRef] [PubMed]
- Morioka, T.; Asilmaz, E.; Hu, J.; Dishinger, J.F.; Kurpad, A.J.; Elias, C.F.; et al. Disruption of leptin receptor expression in the pancreas directly affects beta cell growth and function in mice. J. Clin. Invest. 2007, 117, 2860–2868. [Google Scholar] [CrossRef] [PubMed]
- Timper, K.; Bruning, J.C. Hypothalamic circuits regulating appetite and energy homeostasis: Pathways to obesity. Dis. Model. Mech. 2017, 10, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Thaler, J.P.; Yi, C.X.; Schur, E.A.; Guyenet, S.J.; Hwang, B.H.; Dietrich, M.O.; et al. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Invest. 2012, 122, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Velloso, L.A.; Schwartz, M.W. Altered hypothalamic function in diet-induced obesity. Int. J. Obes. 2011, 35, 1455–1465. [Google Scholar] [CrossRef] [PubMed]
- Bergen, H.T.; Mizuno, T.; Taylor, J.; Mobbs, C.V. Resistance to diet-induced obesity is associated with increased proopiomelanocortin mRNA and decreased neuropeptide Y mRNA in the hypothalamus. Brain. Res. 1999, 851, 198–203. [Google Scholar] [CrossRef]
- Briggs, D.I.; Lemus, M.B.; Kua, E.; Andrews, Z.B. Diet-induced obesity attenuates fasting-induced hyperphagia. J. Neuroendocrinol. 2011, 23, 620–626. [Google Scholar] [CrossRef]
- Milanski, M.; Degasperi, G.; Coope, A.; Morari, J.; Denis, R.; Cintra, D.E.; Tsukumo, D.M.; Anhe, G.; Amaral, M.E.; Takahashi, H.K.; et al. Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: Implications for the pathogenesis of obesity. J. Neurosci. 2009, 29, 359–370. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, G.; Zhang, H.; Karin, M.; Bai, H.; Cai, D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell. 2008, 135, 61–73. [Google Scholar] [CrossRef]
- Mendes, N.F.; Gaspar, J.M.; Lima-Junior, J.C.; Donato, J., Jr.; Velloso, L.A.; Araujo, E.P. TGF-beta1 down-regulation in the mediobasal hypothalamus attenuates hypothalamic inflammation and protects against diet-induced obesity. Metabolism. 2018, 85, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Kim, K.K.; Park, B.S.; Kim, D.H.; Jeong, B.; Kang, D.; Lee, T.H.; Park, J.W.; Kim, J.G.; Lee, B.J. Function of astrocyte MyD88 in high-fat-diet-induced hypothalamic inflammation. J. Neuroinflammation 2020, 17, 195. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, J.M.; Mendes, N.F.; Correa-da-Silva, F.; Lima-Junior, J.C.; Gaspar, R.C.; Ropelle, E.R.; Araujo, E.P.; Carvalho, H.M.; Velloso, L.A. Downregulation of HIF complex in the hypothalamus exacerbates diet-induced obesity. Brain. Behav. Immun. 2018, 73, 550–561. [Google Scholar] [CrossRef] [PubMed]
- De Souza, C.T.; Araujo, E.P.; Bordin, S.; Ashimine, R.; Zollner, R.L.; Boschero, A.C.; Saad, M.J.; Velloso, L.A. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology. 2005, 146, 4192–4199. [Google Scholar] [CrossRef] [PubMed]
- de Paula, G.C.; Brunetta, H.S.; Engel, D.F.; Gaspar, J.M.; Velloso, L.A.; Engblom, D.; de Oliveira, J.; de Bem, A.F. Hippocampal Function Is Impaired by a Short-Term High-Fat Diet in Mice: Increased Blood-Brain Barrier Permeability and Neuroinflammation as Triggering Events. Front. Neurosci. 2021, 15, 734158. [Google Scholar] [CrossRef] [PubMed]
- Hsu, T.M.; Konanur, V.R.; Taing, L.; Usui, R.; Kayser, B.D.; Goran, M.I.; Kanoski, S.E. Effects of sucrose and high fructose corn syrup consumption on spatial memory function and hippocampal neuroinflammation in adolescent rats. Hippocampus. 2015, 25, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Carraro, R.S.; Souza, G.F.; Solon, C.; Razolli, D.S.; Chausse, B.; Barbizan, R.; Victorio, S.C.; Velloso, L.A. Hypothalamic mitochondrial abnormalities occur downstream of inflammation in diet-induced obesity. Mol. Cell. Endocrinol. 2017, 460, 238–22. [Google Scholar] [CrossRef]
- Wardzinski, E.K.; Kistenmacher, A.; Melchert, U.H.; Jauch-Chara, K.; Oltmanns, K.M. Impaired brain energy gain upon a glucose load in obesity. Metabolism. 2018, 85, 90–96. [Google Scholar] [CrossRef]
- Valdearcos, M.; Douglass, J.D.; Robblee, M.M.; Dorfman, M.D.; Stifler, D.R.; Bennett, M.L.; Gerritse, I.; Fasnacht, R.; Barres, B.A.; Thaler, J.P.; et al. Microglial Inflammatory Signaling Orchestrates the Hypothalamic Immune Response to Dietary Excess and Mediates Obesity Susceptibility. Cell. Metab. 2017, 26, 185–197. [Google Scholar] [CrossRef]
- Davidson, T.L.; Hargrave, S.L.; Swithers, S.E.; Sample, C.H.; Fu, X.; Kinzig, K.P.; Zheng, W. Inter-relationships among diet, obesity and hippocampal-dependent cognitive function. Neuroscience. 2013, 253, 110–122. [Google Scholar] [CrossRef]
- Kratz, M.; Baars, T.; Guyenet, S. The relationship between high-fat dairy consumption and obesity, cardiovascular, and metabolic disease. Eur. J. Nutr. 2013, 52, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhang, H.; Yin, Y.; Li, J.; Tang, Y.; Purkayastha, S.; Li, L.; Cai, D. Obesity- and aging-induced excess of central transforming growth factor-beta potentiates diabetic development via an RNA stress response. Nat. Med. 2014, 20, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Yuan, L.; Yu, H.; Xi, Y.; Xiao, R. Mitochondrial dysfunction and oxidative damage in the brain of diet-induced obese rats but not in diet-resistant rats. Life. Sci. 2014, 110, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Martín, M.F.; Alzamendi, A.; Harnichar, A.E.; Castrogiovanni, D.; Zubiría, M.G.; Spinedi, E.; Giovambattista, A. Role of glucocorticoid receptor (GR) in white adipose tissue beiging. Life. Sci. 2023, 322, 121681. [Google Scholar] [CrossRef] [PubMed]
- Houstis, N.; Rosen, E.D.; Lander, E.S. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006, 440, 944–948. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ebenezer, P.J.; Dasuri, K.; Fernandez-Kim, S.O.; Francis, J.; Mariappan; N. ; Gao, Z.; Ye, J., Bruce-Keller, A.J.; Keller, J.N. Aging is associated with hypoxia and oxidative stress in adipose tissue: implications for adipose function. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E599–E607. [Google Scholar] [CrossRef] [PubMed]
- Salgado-Somoza, A.; Teijeira-Fernández, E.; Fernández, A.L.; GonzálezJuanatey, J.R.; Eiras, S. 2010 Proteomic analysis of epicardial and subcutaneous adipose tissue reveals differences in proteins involved in oxidative stress. Am. J. Physiol. Heart. Circ. Physiol. 2010, 299, H202–H209. [Google Scholar] [CrossRef] [PubMed]
- Long, E.K.; Olson, D.M.; Bernlohr, D.A. High-fat diet induces changes in adipose tissue trans-4-oxo-2-nonenal and trans-4-hydroxy-2-nonenal levels in a depot-specific manner. Free. Radic. Biol. Med. 2013, 63, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Hauck, A.K.; Zhou, T.; Hahn, W.; Petegrosso, R.; Kuang, R.; Chen, Y. , Bernlohr, D.A. Obesity-induced protein carbonylation in murine adipose tissue regulates the DNA-binding domain of nuclear zinc finger proteins. J. Biol. Chem. 2018, 293, 13464–13476. [Google Scholar] [CrossRef]
- Stanley, M.; MacAuley, S.L.; Holtzman, D.M. Changes in insulin and insulin signaling in Alzheimer’s disease: cause or consequence? J. Exp. Med. 2016, 213, 1375–1385. [Google Scholar] [CrossRef]
- Janson, J.; Laedtke, T.; Parisi, J.E.; O’Brien, P.; Petersen, R.C.; Butler, P.C. Increased risk of type 2 diabetes in alzheimer disease. Diabetes. 2004, 53, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation and cognitive decline. J. Clin. Invest. 2012, 122, 1316–1338. [Google Scholar] [CrossRef] [PubMed]
- Grillo, C.A.; Piroli, G.G.; Lawrence, R.C.; Wrighten, S.A.; Green, A.J.; Wilson, S.P.; Sakai, R.R.; Kelly, S.J.; Wilson, M.A.; Mott, D.D.; et al. Hippocampal insulin resistance impairs spatial learning and synaptic plasticity. Diabetes. 2015, 64, 3927–3936. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.R.; Lyra, E.; Silva, N.M.; Figueiredo, C.P.; Frozza, R.L.; Ledo, J.H.; Beckman, D.; Katashima, C.K.; Razolli, D.; Carvalho, B.M.; et al. Alzheimer-associated Ab oligomers impact the central nervous system to induce peripheral metabolic deregulation. Mol. Med. 2015, 7, 190–210. [Google Scholar]
- Yarchoan, M.; Toledo, J.B.; Lee, E.B.; Arvanitakis, Z.; Kazi, H.; Han, L.Y.; Louneva, N.; Lee, V.M.; Kim, S.F.; Trojanowski, J.Q.; et al. Abnormal serine phosphorylation of insulin receptor substrate 1 is associated with tau pathology in Alzheimer’s disease and tauopathies. Acta. Neuropathol. 2014, 128, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.Q.; De Felice, F.G.; Fernandez, S.; Chen, H.; Lambert, M.P.; Quon, M.J.; Krafft, G.A.; Klein, W.L. Amyloid b oligomers induce impairment of neuronal insulin receptors. FASEB. J. 2008, 22, 246–260. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, M.V.; Ferreira, S.T.; De Felice, F.G. Neuronal stress signaling and eIF2a phosphorylation as molecular links between Alzheimer’s disease and diabetes. Prog. Neurobiol. 2015, 129, 37–57. [Google Scholar] [CrossRef]
- Aviles-Olmos, I.; Limousin, P.; Lees, A.; Foltynie, T. Parkinson’s disease, insulin resistance and novel agents of neuroprotection. Brain. 2013, 136, 374–384. [Google Scholar] [CrossRef]
- Perry, V.H. The influence of systemic inflammation on inflammation in the brain: implications for chronic neurodegenerative disease. Brain. Behav. Immun. 2004, 18, 407–413. [Google Scholar] [CrossRef]
- Stark, R.; Roden, M. ESCI Award 2006. Mitochondrial function and endocrine diseases. Eur. J. Clin. Invest. 2007, 37, 236–248. [Google Scholar] [CrossRef]
- Rantham Prabhakara, J.P.; Feist, G.; Thomasson, S.; Thompson, A.; Schommer, E.; Ghribi, O. Differential effects of 24-hydroxycholesterol and 27-hydroxycholesterol on tyrosine hydroxylase and alpha-synuclein in human neuroblastoma SH-SY5Y cells. J. Neurochem. 2008, 107, 1722–1729. [Google Scholar] [CrossRef]
- Li, L.-Y.; Liu, S.-F.; Zhuang, J.L.; Li, M.M.; Huang, Z.-P.; Chen, Y.H.; Chen, X.-R.; Chen, C.-N.; Li-Chao Ye, S.L. Recent research progress on metabolic syndrome and risk of Parkinson's disease. Rev. Neurosci. 2022, 34, 719–735. [Google Scholar] [CrossRef]
- Miraglia, F.; Colla, E. ; Microbiome, Parkinson’s disease and molecular mimicry. Cells. 2019, 8, 222. [Google Scholar] [CrossRef]
- Erny, D.; Hrabě de Angelis, A.L.; Jaitin, D.; Wieghofer. P. ; Staszewski, O. ; David, E. ; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Chen, C. , Liao, J., Xia, Y., Liu, X., Jones, R., Haran, J., McCormick, B.; Sampson, T.R.; Alam, A.; Ye, K. Gut microbiota regulate Alzheimer’s disease pathologies and cognitive disorders via PUFA-associated neuroinflammation. Gut. 2022, 71, 2233–2252. [Google Scholar] [CrossRef] [PubMed]
- Warnecke, T. , Schäfer, K.-H., Claus, I., Del Tredici, K., Jost, W.H. Gastrointestinal involvement in Parkinson’s disease: pathophysiology, diagnosis, and management. NPJ. Parkinsons. Dis. 2022, 8, 31. [Google Scholar] [CrossRef]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E. , Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell. 2016, 167, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Seo, D.-o. Holtzman, D.M. Current understanding of the Alzheimer’s disease-associated microbiome and therapeutic strategies. Exp. Mol. Med. 2024, 56, 86–94. [Google Scholar] [CrossRef] [PubMed]
- van de Pavert, S.; Oliver, B.J.; Goverse, G.; Vondenhoff, M.F.; Greuter, M.; Beke, P.; Kusser, K.; Höpken, U.E.; Lipp, M.; Niederreither, K.; Blomhoff, R. Chemokine CXCL13 is essential for lymph node initiation and is induced by retinoic acid and neuronal stimulation. Nat. Immunol. 2009, 10, 1193–1199. [Google Scholar] [CrossRef]
- Patel, A.; Harker, N.; Moreira-Santos, L.; Ferreira, M.; Alden, K.; Timmis, J.; Foster, K.; Garefalaki, A.; Pachnis, P.; Andrews, P.; et al. Differential RET signaling pathways drive development of the enteric lymphoid and nervous systems. Sci. Signal. 2012, 5, ra55. [Google Scholar] [CrossRef]
- van de Pavert, S.A.; Ferreira, M.; Domingues, R.G.; Ribeiro, H.; Molenaar, R.; Moreira-Santos, L.; Almeida, F.F.; Ibiza, S.; Barbosa, I.; Goverse, G. Maternal retinoids control type 3 innate lymphoid cells and set the offspring immunity. Nature. 2014, 508, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Arti,s D. ; Chiu, I.M. Neuro-immune Interactions in the Tissues. Immunity. 2020, 52, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Veiga-Fernandes, H.; Mucida, D. Neuro-Immune Interactions at Barrier Surfaces. Cell. 2016, 165, 801–811. [Google Scholar] [CrossRef]
- Kouassi, E.; Li, S.Y.; Boukhris, W.; Millet, I.; Revillard, J.P. Opposite effects of the catecholamines dopamine and norepinephrine on murine polyclonal B-cell activation. Immunopharmacology. 1988, 16, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Kruszewska, B.; Felten, S.Y.; Moynihan, J.A. Alterations in cytokine and antibody production following chemical sympathectomy in two strains of mice. J. Immunol. 1995, 155, 4613–4620. [Google Scholar] [CrossRef] [PubMed]
- Veiga-Fernandes, H.; Coles, M.C.; Foster, K.E.; Patel, A.; Williams, A.; Natarajan, D.; Barlow, A.; Pachnis, V.; Dimitris, Kioussis. Tyrosine kinase receptor RET is a key regulator of Peyer's patch organogenesis. Nature. 2007, 446, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Vulchanova, L.; Casey, M.A.; Crabb, G.W.; Kennedy, W.R.; Brown, D.R. Anatomical evidence for enteric neuroimmune interactions in Peyer's patches. J Neuroimmunol. 2007, 185, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; von Wasielewski, R.; Lindenmaier, W.; Dittmar, K.E.J. Immmunohistochemical study of the blood and lymphatic vasculature and the innervation of mouse gut and gut-associated lymphoid tissue. Anat. Histol. Embryol. 2007, 36, 62–74. [Google Scholar] [CrossRef]
- Kasprowicz, D.J. Immmunohistochemical study of the blood and lymphatic vasculature and the innervation of Kohm AP, Berton MT, Chruscinski AJ, Sharpe A, V Sanders VM. Stimulation of the B cell receptor, CD86 (B7-2), and the beta 2-adrenergic receptor intrinsically modulates the level of IgG1 and IgE produced per B cell. J. Immunol. 2000, 165, 680–690. [Google Scholar]
- Sitkauskiene, B.; Sakalauskas, R. The role of beta(2)-adrenergic receptors in inflammation and allergy. Curr. Drug. Targets. Inflamm Allergy 2005, 4, 157–162. [Google Scholar] [CrossRef]
- Straub, R.H.; Pongratz, G.; Weidler, C.; Linde, H-J. ; Kirschning, C.J.; Glück, T.; Schölmerich, J.; Falk, W. Ablation of the sympathetic nervous system decreases gram-negative and increases gram-positive bacterial dissemination: key roles for tumor necrosis factor/phagocytes and interleukin-4/lymphocytes. J. Infect. Dis 2005, 192, 560–572. [Google Scholar] [CrossRef] [PubMed]
- Cheadle, G.A.; Costantini, T.W.; Bansal, V.; Eliceiri, B.P.; Coimbra, R. Cholinergic signaling in the gut: a novel mechanism of barrier protection through activation of enteric glia cells. Surg. Infect. (Larchmt). 2014, 15, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, D.; Miller, J.; Merad, M. Dendritic cell and macrophage heterogeneity in vivo. Immunity. 2011, 35, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Rescigno, M.; Urbano, M.; Valzasina, B.; Francolini, M.; Rotta, G.; Bonasio, R.; Granucci, F.; Kraehenbuhl, J.P.; Ricciardi-Castagnoli, P. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat. Immunol. 2001, 2, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Niess, J.H.; Brand, S.; Gu, X.; Landsman, L.; Jung, S.; McCormick, B.A.; Vyas, J.M.; Boes, M.; Ploegh, H.L.; Fox, J.G.; Littman, D.R.; Reinecker, H.C. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance, Science. 2005, 307, 254–258.
- Mazzini, E.; Massimiliano, L.; Penna, G.; Rescigno, M. Oral tolerance can be established via gap junction transfer of fed antigens from CX3CR1 macrophages to CD103 dendritic cells. Immunity. 2011, 35, 323–335. [Google Scholar] [CrossRef]
- Rescigno, M. Intestinal dendritic cells. Adv. Immunol. 2010, 107, 109–138. [Google Scholar]
- Scott, C.L.; Aumeunier, A.M.; Mowat, A.M. Intestinal CD103+ dendritic cells: master regulators of tolerance? Trends. Immunol. 2011, 32, 412–419. [Google Scholar] [CrossRef]
- Hadis, U.; Wahl, B.; Schulz, O.; Hardtke-Wolenski, M.; Schippers, A.; Wagner, N.; Müller, W. .; Sparwasser, T.; Förster, R.; Pabst, O. Intestinal tolerance requires gut homing and expansion of FoxP3+ regulatory T cells in the lamina propria. Immunity. 2011, 34, 237–246. [Google Scholar] [CrossRef]
- Shakhar, G.; Kolesnikov, M. Intestinal macrophages and DCs close the gap o tolerance. Immunity. 2014, 40, 171–173. [Google Scholar] [CrossRef]
- De Schepper, S.; Stakenborg, N.; Matteoli, G.; Verheijden, S.; Boeckxstaens, G.E. Muscularis macrophages: Key players in intestinal homeostasis and disease. Cell. Immunol. 2018, 330, 142–150. [Google Scholar] [CrossRef]
- Gabanyi, I.; Muller, P.A.; Feighery, L.; Oliveira, T.Y.; Costa-Pinto, F.A.; Mucida, D. Neuro-immune Interactions Drive Tissue Programming in Intestinal Macrophages. Cell. 2016, 164, 378–391. [Google Scholar] [CrossRef]
- Estévez, A.G.; Sahawneh, M.A.; Lange, P.S.; Bae, N.; Egea, M.; Ratan, R.R. Arginase 1 regulation of nitric oxide production is key to survival of trophic factor-deprived motor neurons. J. Neurosci. 2006, 26, 8512–8516. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, B.E.; Midtvedt, T.; Strandberg, K. Effects of microbial contamination on the cecum enlargement of germfree rats. Scand. J. Gastroenterol. 1970, 5, 309–314. [Google Scholar] [CrossRef]
- Muller, P.A.; Koscsó, B.; Rajani, G.M.; Stevanovic, K.; Berres, M.L.; Hashimoto, D.; Mortha, A.; Leboeuf, M.; Li, X.-M.; Mucida, D.; et al. Crosstalk between muscularis macrophages andentericneurons regulates gastrointestinal motility. Cell. 2014, 158, 300–313. [Google Scholar] [CrossRef] [PubMed]
- Stead, R.H.; Dixon, M.F.; Bramwell, N.H.; Riddell, R.H.; Bienenstock, J. Mast cells are closely apposed to nerves in the human gastrointestinal mucosa. Gastroenterology. 1989, 97, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.J.; Rothman, T.P.; Gershon, M.D. Development of the interstitial cell of Cajal: origin, kit dependence and neuronal and nonneuronal sources of kit ligand. J. Neurosci. Res. 2000, 59, 384–401. [Google Scholar] [CrossRef]
- Brandt EB, Strait RT, Hershko D, Wang Q, Muntel EE, Scribner TA, Zimmermann N, Finkelman FD, Rothenberg ME. Mast cells are required for experimental oral allergen-induced diarrhea. J. Clin. Invest. 2003, 112, 1666–1677. [Google Scholar] [CrossRef]
- De Winter BY, van den Wijngaard RM, de Jonge WJ. Intestinal mast cells in gut inflammation and motility disturbances. Biochim. Biophys. Acta. 2012, 1822, 66–73. [Google Scholar] [CrossRef]
- Barbara, G. : Wang, B.; Stanghellini, V.; de Giorgio, R.; Cremon, C.; Di Nardo, G.; Trevisani, M.; Campi, B.; Geppetti, P.; Tonini, M.; et al. Mast cell-dependent excitation of visceral-nociceptive sensory neurons in irritable bowel syndrome. Gastroenterology. 2007, 132, 26–37. [Google Scholar] [CrossRef]
- Cheng, L. , Luo QQ, Chen SL. The role of intestinal mast cell infiltration in irritable bowel syndrome. J. Dig. Dis. 2021, 22, 143–151. [Google Scholar] [CrossRef]
- Nystrom, E.E.L.; Birchenough, G.M.H.; van der Post, S.; Arike, L.; Gruber, A.D.; Hansson, G.C.; Johansson, M.E.V. Calcium-activated Chloride Channel Regulator 1 (CLCA1) Controls Mucus Expansion in Colon by Proteolytic Activity. EBioMedicine. 2018, 33, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Jacobson, A.; Meerschaert, K.A.; Sifakis, J.J.; Wu, M.; Chen, X.; Yang, T.; Zhou, Y. .; Anekal, P.V.; Rucker, R.A; et al.. Nociceptor neurons direct goblet cells via a CGRP-RAMP1 axis to drive mucus production and gut barrier protection. Cell. 2022, 185, 4190–4205.e25. [Google Scholar] [CrossRef] [PubMed]
- Bennstein, S.B.; Uhrberg, M. Biology and therapeutic potential of human innate lymphoid cells. FEBS. J. 2022, 289, 3967–3981. [Google Scholar] [CrossRef] [PubMed]
- Klose, C.S.N.; Mahlakõiv, T.; Moeller, J.B.; Rankin, L.C.; Flamar, A.L.; Kabata, H.; Monticelli, L.A.; Moriyama, S.; Putzel, G.G.; Rakhilin, N.; et al. The neuropeptide neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nature. 2017, 549, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Tracey, K.J. Physiology and immunology of the cholinergic antiinflammatory pathway. J. Clin. Invest. 2007, 117, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Matteoli, G.; Boeckxstaens, G.E. The vagal innervation of the gut and immune homeostasis. Gut 2013, 62, 1214–1222. [Google Scholar] [CrossRef] [PubMed]
- Galle-Treger, L.; Suzuki, Y.; Patel, N.; Sankaranarayanan, I.; Aron, J.L.; Hadi Maazi, H.; Chen, L.; Akbari, O. Nicotinic acetylcholine receptor agonist attenuates ILC2-dependent airway hyperreactivity. Nat. Comm. 2016, 7, 13202. [Google Scholar] [CrossRef] [PubMed]
- Dalli, J.; Colas, R.A.; Arnardottir, H.; Serhan, C.N. Vagal Regulation of Group 3 Innate Lymphoid Cells and the Immunoresolvent PCTR1 Controls Infection Resolution. Immunity. 2017, 46, 92–105. [Google Scholar] [CrossRef]
- Moriyama, S.; Brestoff, J.R.; Flamar, A-L. ; Moeller, J.B.; Klose, C.S.N.; Rankin, L.C.; Yudanin, L.A.; Monticelli, L.A.; Putzel, G.B.; Rodewald, H-R.; et al. β2-adrenergic receptor-mediated negative regulation of group 2 innate lymphoid cell responses. Science. 2018, 359, 1056–1061. [Google Scholar] [CrossRef]
- Wallrapp, A.; Riesenfeld, S.J.; Burkett, P.R.; Abdulnour, R.E.; Nyman, J.; Dionne, D.; Hofree, M.; Cuoco, M.S.; Rodman, C.; Farouq, D.; et al. The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature. 2017, 549, 351–356. [Google Scholar] [CrossRef]
- Quatrini, L.; Wieduwild. E.; Guia, S.; Bernat, C.; Glaichenhaus. N.; Vivier, E.; Ugolini, S. Host resistance to endotoxic shock requires the neuroendocrine regulation of group 1 innate lymphoid cells. J. Exp. Med. 2017, 214, 3531–3541. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.N.; Guo, Y.B.; Li, X.; Li, C.L.; Tan, W.P.; Fan, X.L.; Qin, Z.L.; Chen, D.; Wen, W.P.; Zheng, S.G.; et al. ILC2 frequency and activity are inhibited by glucocorticoid treatment via STAT pathway in patients with asthma. Allergy. 2018, 73, 1860–1870. [Google Scholar] [CrossRef] [PubMed]
- Talbot, S.; Abdulnour, R.E.; Burkett. P.R ; Lee, S. ; Cronin, S.J. ; Pascal, M.A.; Laedermann, C.; Foster, S.L.; Tran, J.V.; Lai, N. et al. Silencing Nociceptor Neurons Reduces Allergic Airway Inflammation. Neuron. 2015, 87, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Seillet, C.; Luong, K.; Tellier, J.; Jacquelot, N.; Shen, R.D.; Hickey, P.; Wimmer, V.C.; Whitehead, L.; Rogers, K.; Smyth, G.K; et al. The neuropeptide VIP confers anticipatory mucosal immunity by regulating ILC3 activity. Nat. Immunol. 2020, 21, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, M.C.; Araujo, L.P.; Maricato, J.T.; Nascimento, V.M.; Guereschi, M.G.; Rezende, R.M.; Quintana, F.J.; Basso, A.S. Norepinephrine Controls Effector T Cell Differentiation through beta2-Adrenergic Receptor-Mediated Inhibition of NF-kappaB and AP-1 in Dendritic Cells. J. Immunol. 2016, 196, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Maestroni, G.J. Dendritic cell migration controlled by alpha 1b-adrenergic receptors. J. Immunol. 2000, 165, 6743–6747. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M.; Chorny, A.; Gonzalez-Rey, E.; Ganea, D. Vasoactive intestinal peptide generates CD4+CD25+ regulatory T cells in vivo. J. Leukoc. Biol. 2005, 78, 1327–1338. [Google Scholar] [CrossRef] [PubMed]
- Padro, C.J.; Sanders, V.M. Neuroendocrine regulation of inflammation. Semin. Immunol. 2014, 26, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Nakai, A.; Hayano, Y.; Furuta, F.; Noda, M.; Suzuki, K. Control of lymphocyte egress from lymph nodes through β2-adrenergic receptors. J. Exp. Med. 2014, 211, 2583–2598. [Google Scholar] [CrossRef]
- Bonaz, B.; Sinniger, V.; Hoffmann, D.; Clarençon, D.; Mathieu, N.; Dantzer, C.; Vercueil, L.; Picq, C.; Trocmé, C.; Faure, P.; et al. Chronic vagus nerve stimulation in Crohn's disease: a 6-month follow-up pilot study. Neurogastroenterol. Motil. 2016, 28, 948–53. [Google Scholar] [CrossRef]
- Koopman, F.A.; Chavan, S.S.; Miljko, S.; Grazio, S.; Sokolovic, S.; Schuurman, P.R.; Mehta, A.D.; Levine, Y.A.; Faltys, M.; Zitnik. R.; et al. Vagus nerve stimulation inhibits cytokine production and attenuates disease severity in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA. 2016, 113, 8284–8289. [Google Scholar] [CrossRef] [PubMed]
- Guarini, S.; Altavilla, D.; Cainazzo, M.M.; Giuliani, D.; Bigiani, A.; Marini, H.; Squadrito, G.; Minutoli, L.; Bertolini, A.; Marini. R.; et al. Efferent vagal fibre stimulation blunts nuclear factor-kappaB activation and protects against hypovolemic hemorrhagic shock. Circulation. 2003, 107, 1189–1194. [Google Scholar] [CrossRef]
- Levine, Y.A.; Koopman, F.A.; Faltys, M.; Caravaca, A.; Bendele, A.; Zitnik, R.; Margriet, J.; Vervoordeldonk, M.J.; Tak, P. P, Neurostimulation of the cholinergic anti-inflammatory pathway ameliorates disease in rat collagen-induced arthritis. PLoS. One. 2014, 9, e104530. [Google Scholar] [CrossRef]
- Carloni, S.; Rescigno, M. The gut-brain vascular axis in neuroinflammation. Semin. Immunol. 2023, 69, 101802. [Google Scholar] [CrossRef] [PubMed]
- Braniste, V.; Al-Asmakh, M.; Kowal, C.; Anuar, F.; Abbaspour, A.; Tóth, M.; Korecka, A.; Bakocevic, N.; Ng, L.G.; Kundu, P. The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl. Med. 2014, 19, 6:263ra158. [Google Scholar] [CrossRef] [PubMed]
- Mouries, J.P.; Silvestri, A.; Spadoni, I.; Sorribas, M.; Wiest, R.; Mileti, E.; Galbiati, M.P.; Adorini, L.; Penna, G.; Rescigno, M. Microbiota-driven gut vascular barrier disruption is a prerequisite for non-alcoholic steatohepatitis development. J. Hepatol. 2019, 71, 1216–1228. [Google Scholar] [CrossRef]
- Häuser, W.; Janke, K-H. ; Klump, B.; Hinz, A. Anxiety and depression in patients with inflammatory bowel disease: comparisons with chronic liver disease patients and the general population. Inflamm. Bowel. Dis. 2011, 17, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Panara, A.J.; Yarur, A.J.; Rieders, B.; Proksell, S.; Deshpande, A.R.; Abreu, M.T.; Sussman, D.A. The incidence and risk factors for developing depression after being diagnosed with inflammatory bowel disease: a cohort study. Aliment. Pharmacol. Ther. 2014, 39, 802–810. [Google Scholar] [CrossRef]
- Di Tommaso, N.; Santopaolo, F.; Gasbarrini, A.; Ponziani, F.R. The Gut-Vascular Barrier as a New Protagonist in Intestinal and Extraintestinal Diseases. Int. J. Mol. Sci. 2023, 24, 1470. [Google Scholar] [CrossRef] [PubMed]
- Obata, Y.; Castaño, A.; Boeing, S.; Bon-Frauches, A.C.; Fung, C.; Fallesen, T.; Gomez de Agüero, N.; Yilmaz, B.; Lopes, R.; Huseynova, A. Neuronal programming by microbiota regulates intestinal physiology. Nature. 2020, 578, 284–289. [Google Scholar] [CrossRef]
- Sudo, N.; Chida, Y.; Aiba, Y.; Sonoda, J.; Oyama, N.; Yu, X-N. ; Kubo, C.; Koga, Y. Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J Physiol 2004, 558, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Farzi, A.; Reichmann, F.; Meinitzer, A.; Mayerhofer, R.; Jain, P.; Hassan, A.M.; Fröhlich, E.E.; Wagner, K.E.; Rinner, B.; Holzer, P. Synergistic effects of NOD1 or NOD2 and TLR4 activation on mouse sickness behavior in relation to immune and brain activity markers. Brain. Behav. Immun. 2015, 44, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Savignac, H.M.; Tramullas, M.; Kiely, B.; Dinan, T.G.; Cryan, J.F. Bifidobacteria modulate cognitive processes in an anxious mouse strain. Behav. Brain. Res. 2015, 287, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, E.E.; Farzi, A.; Mayerhofer, R.; Reichmann, F.; Jačan, A.; Wagner, B.; Zinser, E.; Bordag, N.; Magnes, C.; Fröhlich, E.; et al. Cognitive impairment by antibiotic-induced gut dysbiosis: Analysis of gut microbiota-brain communication. Brain. Behav. Immun. 2016, 56, 140–155. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.C.; Olson, C.A.; Hsiao, E.Y. Interactions between the microbiota, immune and nervous systems in health and disease. Nat Neurosci. 2017, 20, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Sherwin, E.; Dinan, T.G.; Cryan, J.F. Recent developments in understanding the role of the gut microbiota in brain health and disease. Ann. N. Y. Acad. Sci. 2018, 1420, 5–25. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, P.; Bienenstock, J.; Kunze, W.A. Vagal pathways for microbiome-brain-gut axis communication. Adv. Exp. Med. Biol. 2014, 817, 115–133. [Google Scholar]
- Lyte, M.; Li, W.; Opitz, N.; Gaykema, R.P.A.; Goehler, L.E. Induction of anxiety-like behavior in mice during the initial stages of infection with the agent of murine colonic hyperplasia Citrobacter rodentium. Physiol. Behav. 2006, 2006. 89, 350–357. [Google Scholar] [CrossRef]
- Tanida, M.; Yamano, T.; Maeda, K.; Okumura, N.; Fukushima, Y.; Nagai, K. Effects of intraduodenal injection of Lactobacillus johnsonii La1 on renal sympathetic nerve activity and blood pressure in urethane-anesthetized rats. Neurosci. Lett. 2005, 389, 109–114. [Google Scholar] [CrossRef]
- Takagi, H.; Kuruma, I. Effect of bacterial lipopolysaccharide on the content of serotonin and norepinephrine in rabbit brain. Jpn. J. Pharmacol. 1966, 16, 478–479. [Google Scholar] [CrossRef]
- Gaillard, P.J.; de Boer, A.B.G.; Breimer, D.D. Pharmacological investigations on lipopolysaccharide-induced permeability changes in the blood-brain barrier in vitro. Microvasc. Res. 2003, 65, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, E.E.; Farzi, A.; Mayerhofer, R.; Reichmann, F.; Jačan, A.; Wagner, B.; Zinser, E.; Bordag, N.; Magnes, C.; Fröhlich, E.; et al. Cognitive impairment by antibiotic-induced gut dysbiosis: Analysis of gut microbiota-brain communication. Brain. Behav. Immun. 2016, 56, 140–155. [Google Scholar] [CrossRef] [PubMed]
- Hoyles, L.; Snelling, T.; Umlai, U-K. ; Jeremy, KV.; Nicholson, J.K.; Carding, S.R.; Glem, R.C Microbiome-host systems interactions: protective effects of propionate upon the blood-brain barrier McArthur, S. Microbiome-host systems interactions: protective effects of propionate upon the blood-brain barrier. Microbiome. 2018, 6, 55. [Google Scholar]
- Clarke, G.; Grenham, S.; Scully, P.; Fitzgerald, P.; Moloney, R.D.; Shanahan, F.; Dinan, T.G.; Cryan, J.F. The microbiome-gut-brain axis during early life regulates the hippocampal serotonergic system in a sex-dependent manner. Mol. Psychiatry. 2013, 18, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Mikocka-Walus, A.; Knowles, S.R.; Keefer, L.; Graff, L. Controversies revisited: a systematic review of the comorbidity of depression and anxiety with inflammatory bowel diseases. Inflamm. Bowel. Dis. 2016, 22, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Neuendorf, R.; Harding, A.; Stello, N.; Hanes, D.; Wahbeh, H. Depression and anxiety in patients with inflammatory bowel diseases: a systematic review. J. Psychosom. Res. 2016, 87, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Nikolaus, S.; Schulte, B.; Al-Massad, N.; Thieme, F.; Schulte, D.M.; Bethge, J.; Rehman, A.; Tran, F.; Aden, K.; Häsler, R.; et al. Increased tryptophan metabolism is associated with activity of inflammatory bowel diseases. Gastroenterology. 2017, 153, 1504–1516. [Google Scholar] [CrossRef]
- Sobesky, J.L.; Barrientos, R.M.; De May, H.S.; Thompson, B.M.; Weber, M.D.; Watkins, L.R.; Maier, S.F. High-fat diet consumption disrupts memory and primes elevations in hippocampal IL-1β, an effect that can be prevented with dietary reversal or IL-1 receptor antagonism. Brain. Behav. Immun. 2014, 42, 22–32. [Google Scholar] [CrossRef]
- Gannon, O.J.; Robison, L.S.; Salinero, A.E.; Abi-Ghanem, C.; Mansour, F.M.; Kelly, R.D.; Tyagi, A.; Brawley, R.R.; Ogg, J.D.; Zuloaga, K.L. High-fat diet exacerbates cognitive decline in mouse models of Alzheimer’s disease and mixed dementia in a sex-dependent manner. J. Neuroinflammation. 2022, 19, 110. [Google Scholar] [CrossRef]
- Jones, N.S.; Watson, K.Q.; Rebeck, G.W. High-fat diet increases gliosis and immediate early gene expression in APOE3 mice, but not APOE4 mice. J. Neuroinflammation. 2021, 18, 214. [Google Scholar] [CrossRef]
- Vega-Torres, J.D.; Ontiveros-Angel, P.; Stuffle, E.C.; Solak, S.; Terrones, E.; Tyner, E.; Oropeza, M.; Dela Peña, I.; Obenaus, A.; Ford, B.D.; et al. Short-term exposure to an obesogenic diet during adolescence elicits anxiety-related behavior and neuroinflammation: Modulatory effects of exogenous neuregulin-1. Transl. Psychiatry. 2022, 12, 83. [Google Scholar] [CrossRef] [PubMed]
- Clarke, G.; Grenham, S.; Scully, P.; Fitzgerald, P.; Moloney, R.D.; Shanahan, F.; Dinan, T.G.; Cryan, J.F. The microbiome-gut-brain axis during early life regulates the hippocampal serotonergic system in a sex-dependent manner. Mol Psychiatry 2013, 18, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Mikocka-Walus, A.; Knowles, S.R.; Keefer, L.; Graff, L. Controversies revisited: a systematic review of the comorbidity of depression and anxiety with inflammatory bowel diseases. Inflamm Bowel Dis. 2016, 22, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Neuendorf, R.; Harding, A.; Stello, N.; Hanes, D.; Wahbeh, H. Depression and anxiety in patients with inflammatory bowel diseases: a systematic review. J Psychosom Res. 2016, 87, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Nikolaus, S.; Schulte, B.; Al-Massad, N.; Thieme, F.; Schulte, D.M.; Bethge, J.; et al. Increased tryptophan metabolism is associated with activity of inflammatory bowel diseases. Gastroenterology 2017, 153, 1504–1516.e2. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Choi, G.M.; Sur, B. Silibinin prevents depression-like behaviors in a single prolonged stress rat model: the possible role of serotonin. BMC Complement Med Ther 2020, 20, 70. [Google Scholar] [CrossRef] [PubMed]
- O'Mahony, S.M.; Clarke, G.; Borre, Y.E.; Dinan, T.G.; Cryan, J.F. Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav Brain Res 2015, 277, 32–48. [Google Scholar] [CrossRef]
- Pirzgalska, R.M.; Seixas, E.; Seidman, J.S.; Link, V.M.; Sánchez, N.M. , Mahú, I.; Mendes, R.; Gres, V.; Kubasova, N.; Morris, I.; et al. Sympathetic neuron associated macrophages contribute to obesity by importing and metabolizing norepinephrine. Nat. Med. 2017, 23, 1309–1318. [Google Scholar] [CrossRef]
- Fischer, K.; Ruiz, H.H.; Jhun, K.; Finan, B.; Oberlin, D.J.; van der Heide, V.; Kalinovich, A.V.; Petrovic, N.; Wolf, Y.; Clemmensen, C. Alternatively activated macrophages do not synthesize catecholamines or contribute to adipose tissue adaptive thermogenesis. Nat. Med. 2017, 23, 623. [Google Scholar] [CrossRef]
- Gaskell, W.H. On the structure, distribution and function of the nerves which innervate the visceral and vascular systems. J Physiol. 1886, 7, 1–80. [Google Scholar] [CrossRef]
- Berthoud, H.; Carlson, N.; Powley, T. Topography of efferent vagal innervation of the rat gastrointestinal tract. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1991, 260, R200–R207. [Google Scholar] [CrossRef] [PubMed]
- Bi, S.; Chen, J.; Behles, R.R.; Hyun, J.; Kopin, A.S.; Moran, T.H. Differential body weight and feeding responses to high-fat diets in rats and mice lacking cholecystokinin 1 receptors. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R55–R63. [Google Scholar] [CrossRef] [PubMed]
- Paulino, G.; Barbier de la Serre, C.; Knotts, T.A.; Oort, P.J.; Newman, J.W.; Adams, S.H.; Raybould, H.E. Increased expression of receptors for orexigenic factors in nodose ganglion of diet-induced obese rats. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E898–E903. [Google Scholar] [CrossRef] [PubMed]
- de Lartigue, G.; Ronveaux, C.C.; Raybould, H.E. Deletion of leptin signaling in vagal afferent neurons results in hyperphagia and obesity. Mol. Metab. 2014, 3, 595–607. [Google Scholar] [CrossRef]
- Minnone, G.; De Benedetti, F.; Bracci-Laudiero, L. NGF and its receptors in the regulation of inflammatory response. IJMS. 2017, 18, 1028. [Google Scholar] [CrossRef]
- Reinshagen, M.; Rom, H.; Steinkamp, M.; Lieb, K.; Geerling, I.; Von Herbay, A.; Flamig, G.; Eysselein, V.E.; Adler, G. Protective role of neurotrophins in experimental inflammation of the rat gut. Gastroenterology. 2000, 119, 368–376. [Google Scholar] [CrossRef]
- Harrington, A.M.; Brierley, S.M.; Isaacs, N.; Hughes, P.A.; Castro, J.; Blackshaw, L.A. Sprouting of colonic afferent central terminals and increased spinal mitogen-activated protein kinase expression in a mouse model of chronic visceral hypersensitivity. J. Comp. Neurol. 2012, 520, 2241–2255. [Google Scholar] [CrossRef]
Figure 1.
Innervation of the gastrointestinal tract by autonomous neurons. The extrinsic sympathetic neurons (orange) originate from the spinal cord and sympathetic ganglia; the extrinsic parasympathetic neurons (green) originate from the brain stem; and the enteric intrinsic neurons (blue) are distributed in the submucosa and myenteric plexus as a ganglionated circuit. All layers of the intestinal wall are innervated and interconnected. Extrinsic sensory neurons (red) transduce signals from the gut to the brain stem and spinal cord.
Figure 1.
Innervation of the gastrointestinal tract by autonomous neurons. The extrinsic sympathetic neurons (orange) originate from the spinal cord and sympathetic ganglia; the extrinsic parasympathetic neurons (green) originate from the brain stem; and the enteric intrinsic neurons (blue) are distributed in the submucosa and myenteric plexus as a ganglionated circuit. All layers of the intestinal wall are innervated and interconnected. Extrinsic sensory neurons (red) transduce signals from the gut to the brain stem and spinal cord.
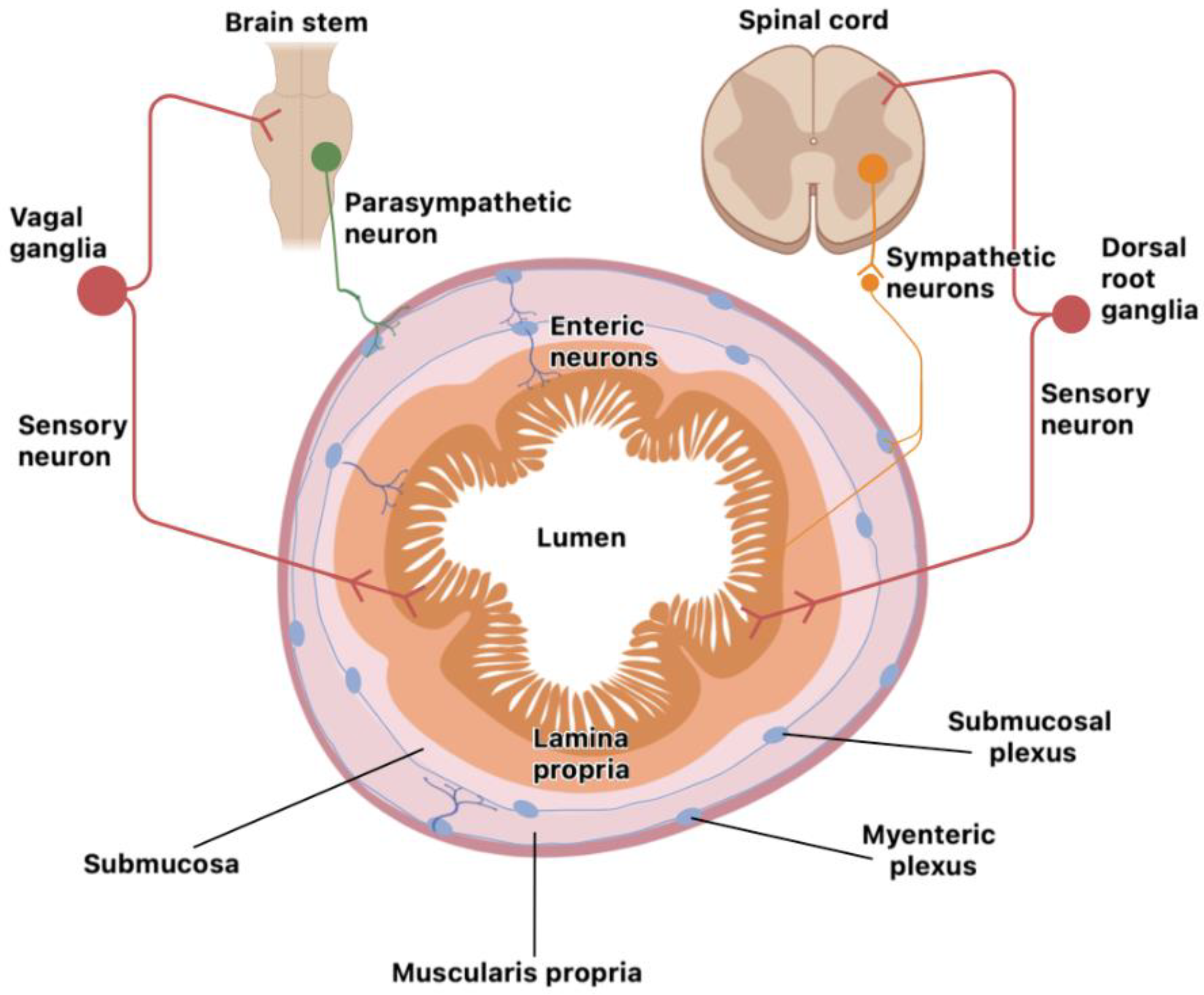
Figure 2.
Inflamed white adipose tissue (WAT) is characterized by large insulin-resistant (IR) adipocytes. WAT mass is now highly infiltrated by macrophages; pro-inflammatory adipocytokines and free fatty acids (FFA) will be released in excess. Thus, a chronic overall IR state induces several dysfunctional tissues: enhanced hepatic glucose production, hyperglycemia, diminished muscle glucose uptake, pancreatic β-cell apoptosis, type 2 diabetes mellitus and consequently, neuroinflammation (NF) is established.
Figure 2.
Inflamed white adipose tissue (WAT) is characterized by large insulin-resistant (IR) adipocytes. WAT mass is now highly infiltrated by macrophages; pro-inflammatory adipocytokines and free fatty acids (FFA) will be released in excess. Thus, a chronic overall IR state induces several dysfunctional tissues: enhanced hepatic glucose production, hyperglycemia, diminished muscle glucose uptake, pancreatic β-cell apoptosis, type 2 diabetes mellitus and consequently, neuroinflammation (NF) is established.
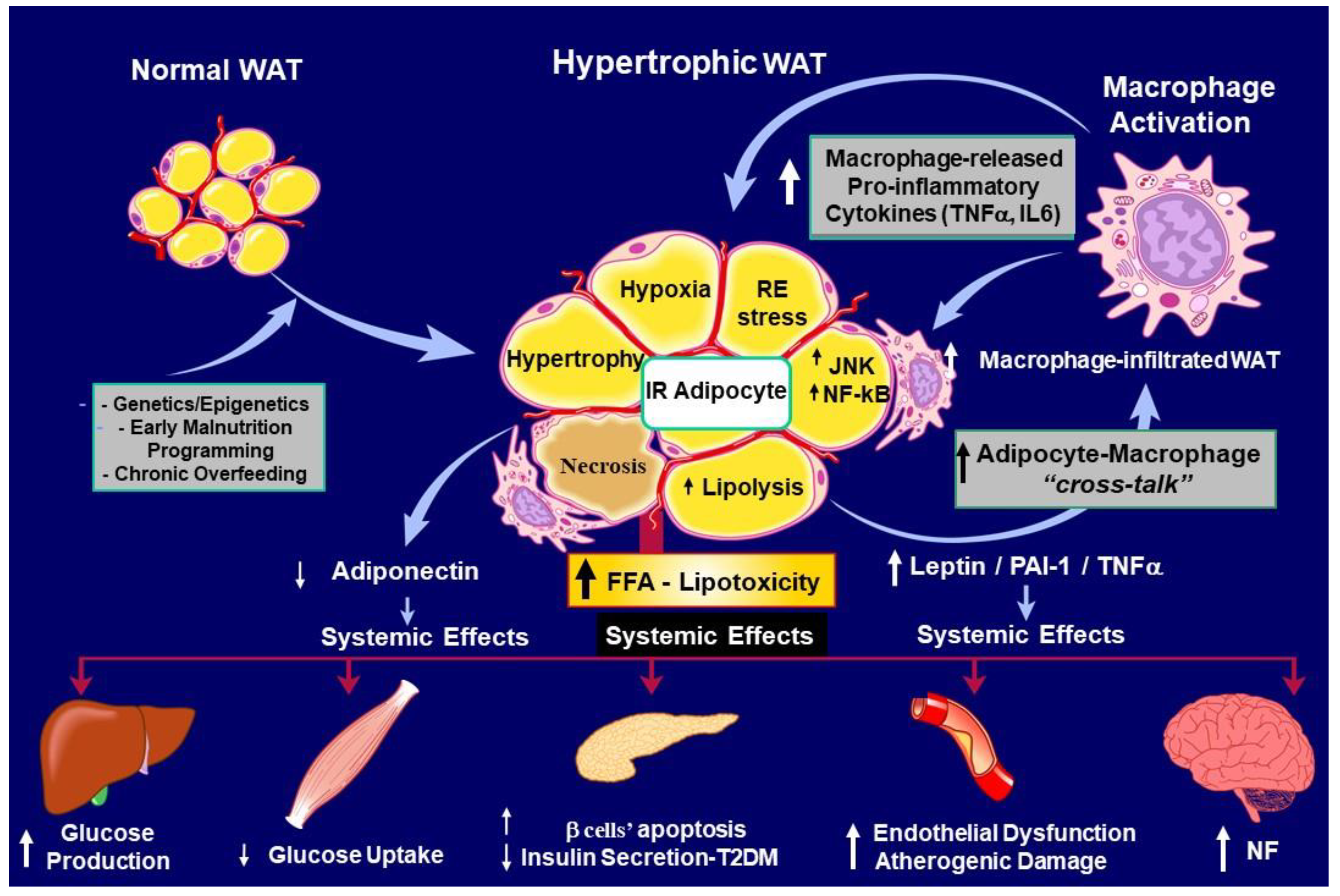
Figure 3.
Panel a) shows a disrupted adipo-insular axis function during leptin-resistantce (LEP-R) (Lep, leptin; Ins, insulin; β-cell, pancreatic β cell). The rapid, vagus-mediated, regulation of hyperglycemia shortly after triggering the insulin-resistant (IR) state is displayed in panel b). (BBB, brain blood barrier; IRS, insulin-receptor substrate; MAPK, mitogen-activated protein kinase; PIK3, phosphatidylinositol 3-kinase; K-ATP, potassium-dependent ATP; AN, arcuate nucleus; TNS, nucleus tractus solitarius; DMX, dorsal motor nucleus X).
Figure 3.
Panel a) shows a disrupted adipo-insular axis function during leptin-resistantce (LEP-R) (Lep, leptin; Ins, insulin; β-cell, pancreatic β cell). The rapid, vagus-mediated, regulation of hyperglycemia shortly after triggering the insulin-resistant (IR) state is displayed in panel b). (BBB, brain blood barrier; IRS, insulin-receptor substrate; MAPK, mitogen-activated protein kinase; PIK3, phosphatidylinositol 3-kinase; K-ATP, potassium-dependent ATP; AN, arcuate nucleus; TNS, nucleus tractus solitarius; DMX, dorsal motor nucleus X).
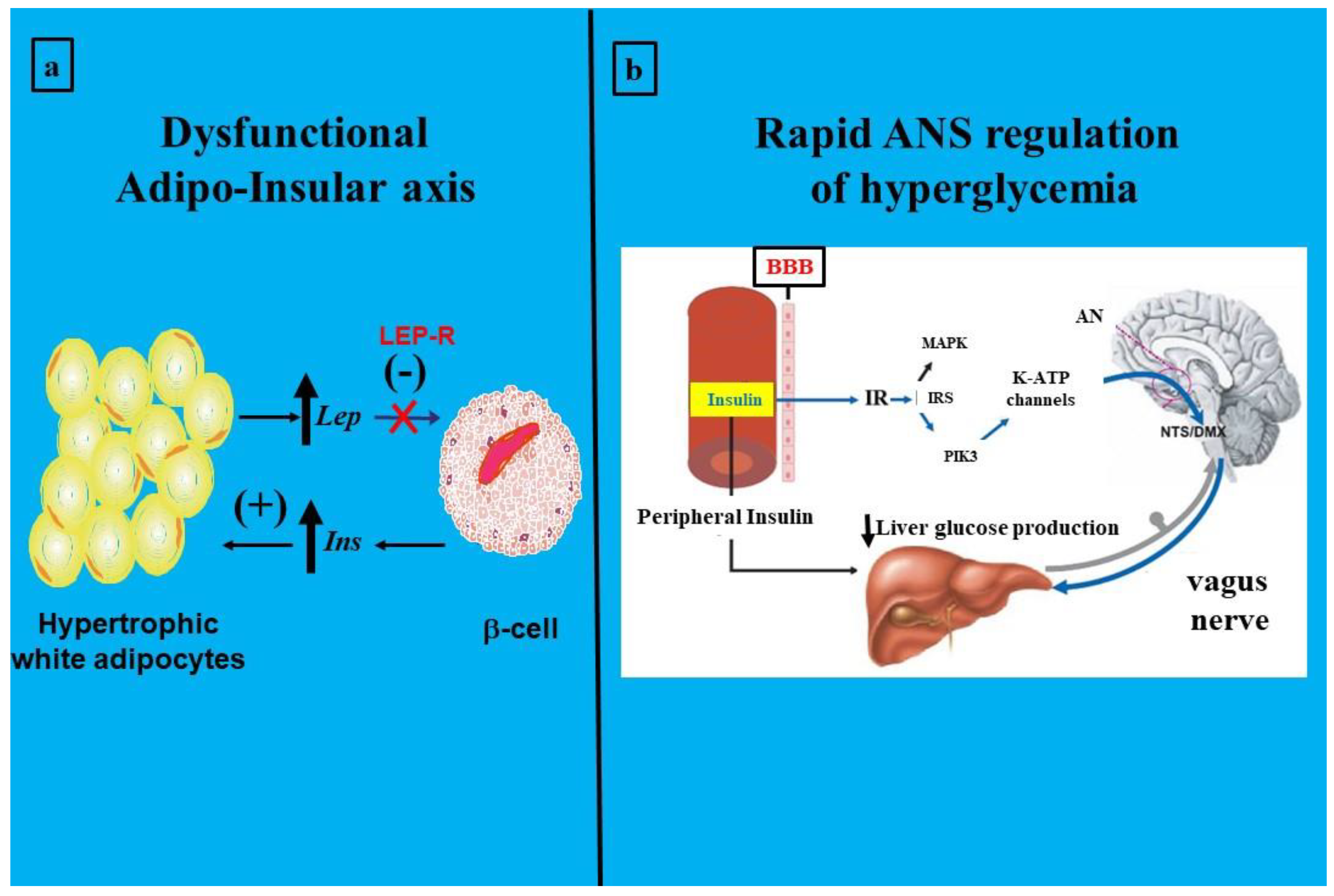
Figure 4.
Disrupted brain insulin-signaling in Alzheimer’s Disease. Amyloid-β (Aβ) accumulation increases TNF-α levels and activates c-Jun N-terminal kinase (JNK). Consequently, IRS-1 becomes inhibited. IR decreases insulin-degrading enzyme (IDE) expression, thereby diminishing IDE-induced Aβ degradation. Reduced brain insulin signaling decreases the inhibition of glycogen synthase kinase-3β (GSK-3β) activity on tau protein (TP) phosphorylation, resulting in hyperphosphorylated TP (PP-TP) production and microtubule depolymerization (MTDP). Consequently, neurofilament tangles (NFTs) deposition are augmented. Therefore, impaired insulin signaling induces neuron degeneration and modifies learning and memory (AD). Additionally, IRS-1 deficiency reduces nitric oxide (NO) production and up-regulates endothelin-1 production, therebyus decreasing brain blood flow and increasing neuroinflammation (NF). (Reproduced with Karger AG’s permission from Spinedi & Cardinali [20]).
Figure 4.
Disrupted brain insulin-signaling in Alzheimer’s Disease. Amyloid-β (Aβ) accumulation increases TNF-α levels and activates c-Jun N-terminal kinase (JNK). Consequently, IRS-1 becomes inhibited. IR decreases insulin-degrading enzyme (IDE) expression, thereby diminishing IDE-induced Aβ degradation. Reduced brain insulin signaling decreases the inhibition of glycogen synthase kinase-3β (GSK-3β) activity on tau protein (TP) phosphorylation, resulting in hyperphosphorylated TP (PP-TP) production and microtubule depolymerization (MTDP). Consequently, neurofilament tangles (NFTs) deposition are augmented. Therefore, impaired insulin signaling induces neuron degeneration and modifies learning and memory (AD). Additionally, IRS-1 deficiency reduces nitric oxide (NO) production and up-regulates endothelin-1 production, therebyus decreasing brain blood flow and increasing neuroinflammation (NF). (Reproduced with Karger AG’s permission from Spinedi & Cardinali [20]).
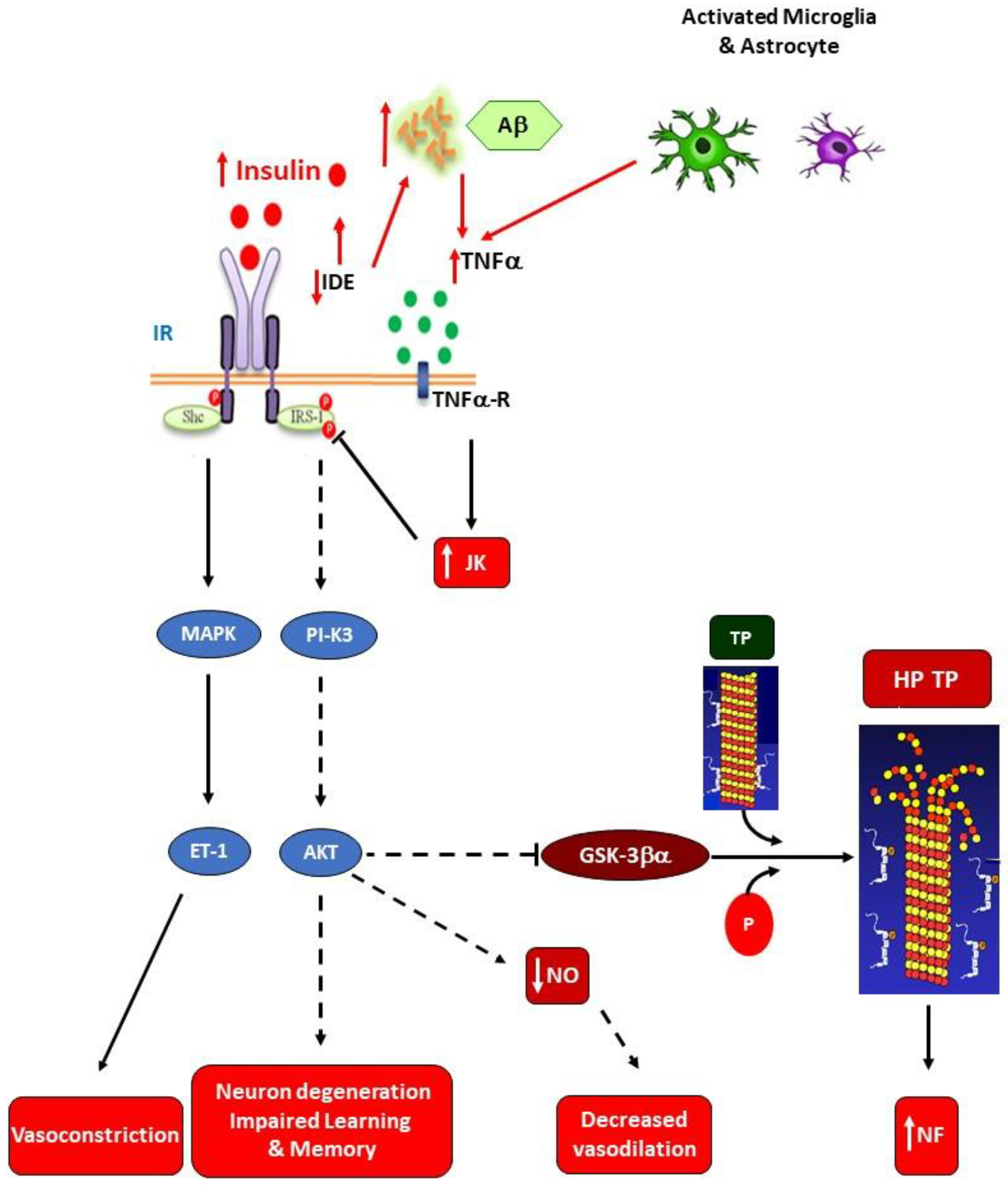
Figure 5.
Disrupted brain insulin-signaling in Parkinsos’s Disease. Amyloid-β (Aβ) accumulation increases TNFα levels and activates c-Jun N-terminal kinase (JNK). Consequently, IRS-1 becomes inhibited. IR decreases insulin-degrading enzyme (IDE) expression, thereby diminishing IDE-induced Aβ degradation. Diminished brain insulin signaling decreases inhibition of NFkβ activity on α-Synuclein (α-Syn) aggregation (panel b), thereby accumulating it. Consequently, impaired insulin signaling induces neuron degeneration and modifies motor activity. IRS-1 deficiency also reduces nitric oxide (NO) production and up-regulates endothelin-1 production, thereby decreasing brain blood flow and increasing neuroinflammation (NF). (Adapted, with Karger AG’s permission, from Spinedi & Cardinali [20]).
Figure 5.
Disrupted brain insulin-signaling in Parkinsos’s Disease. Amyloid-β (Aβ) accumulation increases TNFα levels and activates c-Jun N-terminal kinase (JNK). Consequently, IRS-1 becomes inhibited. IR decreases insulin-degrading enzyme (IDE) expression, thereby diminishing IDE-induced Aβ degradation. Diminished brain insulin signaling decreases inhibition of NFkβ activity on α-Synuclein (α-Syn) aggregation (panel b), thereby accumulating it. Consequently, impaired insulin signaling induces neuron degeneration and modifies motor activity. IRS-1 deficiency also reduces nitric oxide (NO) production and up-regulates endothelin-1 production, thereby decreasing brain blood flow and increasing neuroinflammation (NF). (Adapted, with Karger AG’s permission, from Spinedi & Cardinali [20]).
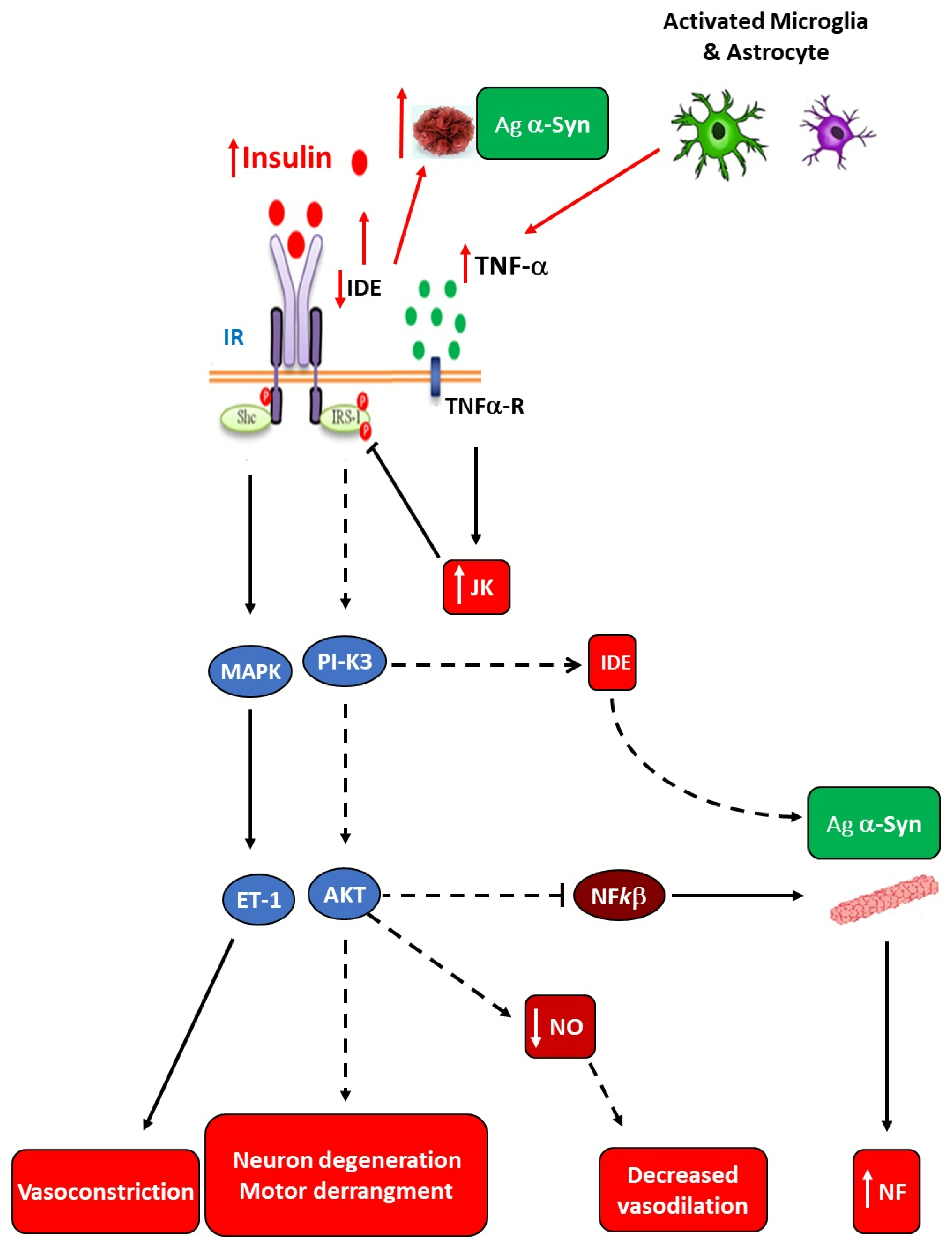
Figure 6.
Schematic representation of the gut-brain axis (GBA) including components of the intestinal microbiome, which interacts with nervous and immune cells. SCFA: short-chain fatty acid.
Figure 6.
Schematic representation of the gut-brain axis (GBA) including components of the intestinal microbiome, which interacts with nervous and immune cells. SCFA: short-chain fatty acid.
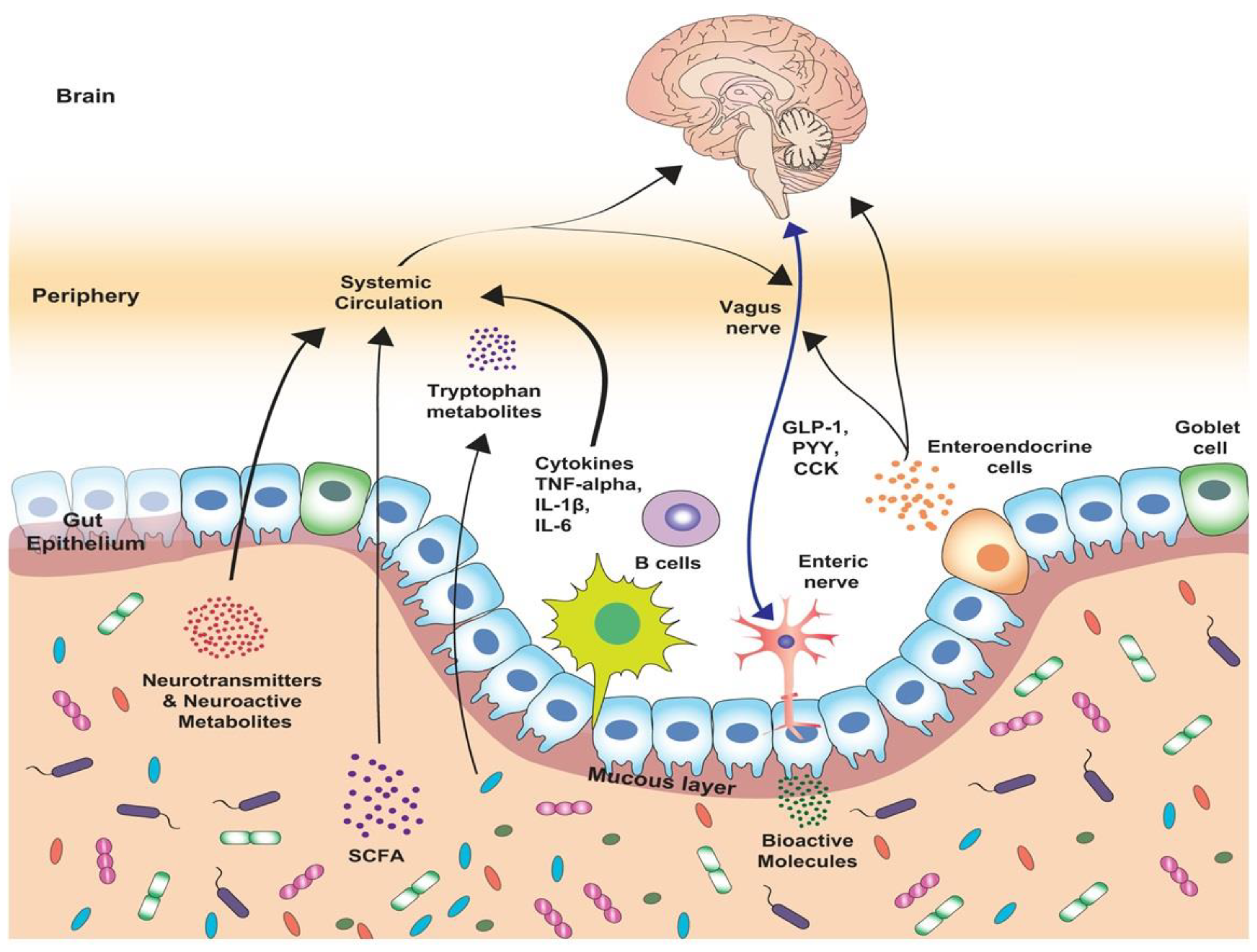
Figure 7.
A loop comprised by brain, , and white adipocyte intercommunication in both physiological (in blue) and pathophysiological (in red) conditions. Any disruption of this communication, such as hypertrophic WAT mass expansion, dysbiosis, and altered ANS function, causes metainflammation (Obesity/Diabetes), and thereafter, neuroinflammation development. (AgRP, Aguti-related protein; NPY, Neopeptide Y, CART, Cocaine-Amphetamine Related Transcript; POMC, Pro-opiomelanocortin).
Figure 7.
A loop comprised by brain, , and white adipocyte intercommunication in both physiological (in blue) and pathophysiological (in red) conditions. Any disruption of this communication, such as hypertrophic WAT mass expansion, dysbiosis, and altered ANS function, causes metainflammation (Obesity/Diabetes), and thereafter, neuroinflammation development. (AgRP, Aguti-related protein; NPY, Neopeptide Y, CART, Cocaine-Amphetamine Related Transcript; POMC, Pro-opiomelanocortin).
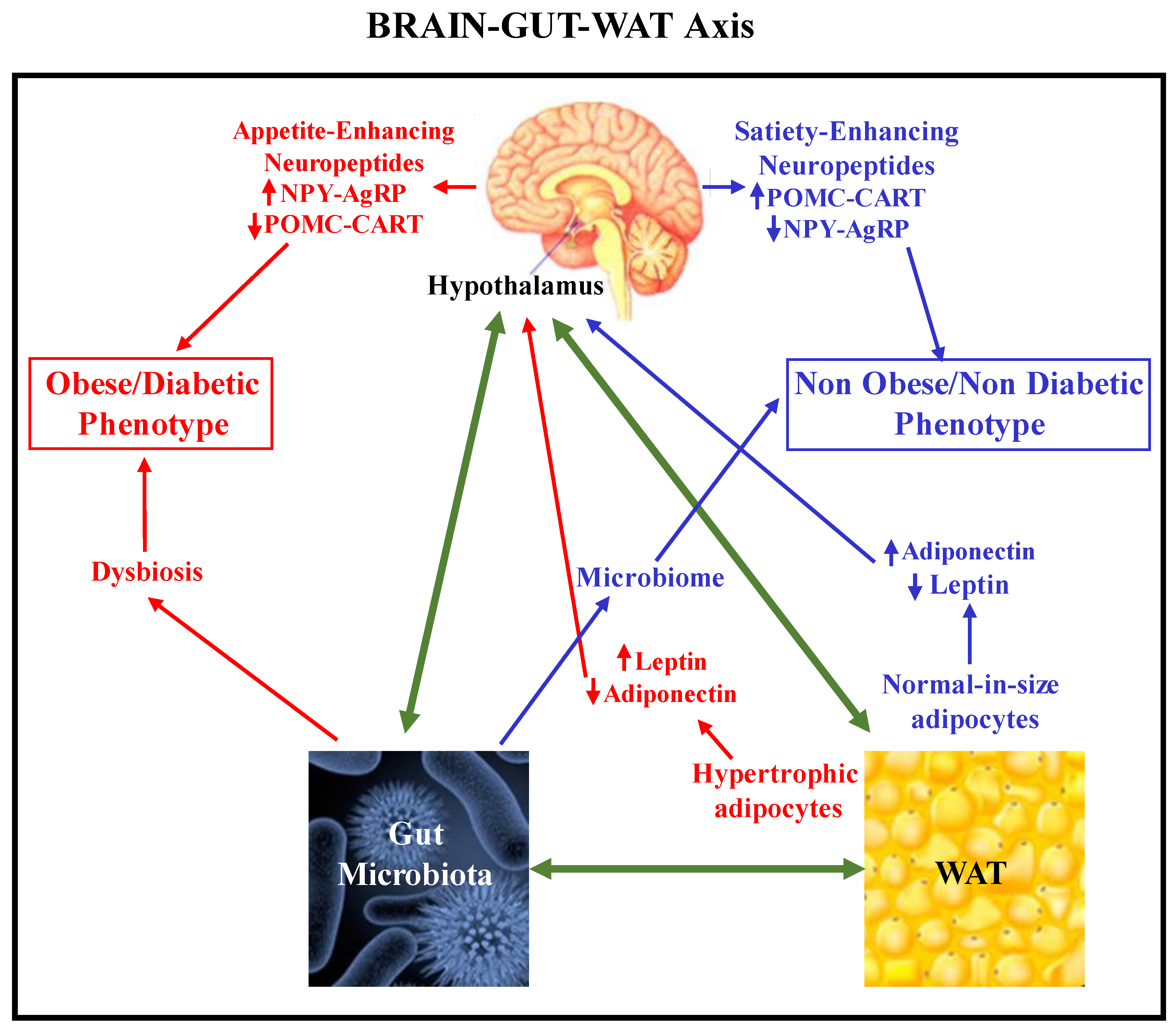
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



