Submitted:
05 August 2024
Posted:
06 August 2024
You are already at the latest version
Abstract
The bi- or multi-nucleated Reed-Sternberg cell (RS) is the diagnostic cornerstone of EBV-positive and EBV-negative classical Hodgkin lymphoma (cHL). cHL is a germinal center (GC) derived B-cell disease. Hodgkin cells (H) are the mononuclear precursors of RS. An experimental model has to fulfill three conditions to qualify as common pathogenic denominator: i) to be of germinal center-derived B-cell origin, ii) to be EBV-negative to avoid EBV latency III expression, and iii) to support permanent LMP1 expression upon induction. These conditions are unified in the EBV-, Diffuse Large B-Cell Lymphoma (DLBCL) cell line BJAB-tTA-LMP1. 3D reconstuctive nanotechnology revealed spatial, quantititive and qualitative disturbance of telomere/shelterin interactions in mononuclear H-like cells, further progression during transition to RS-like cells, including progressive complexity of the karytotype with every mitotic cycle, due to BBF (breakage/bridge/fusion) events. The findings of this model were confirmed on diagnostic patient samples and correlate with clincal outcome. Moreover, in vitro signficant disturbance of the lamin AC/telomere interaction progressively occured. In summary, our research over the past three decades identifed cHL as the first lymphoid maligancy driven by a disturbed telomere/shelterin/lamin AC interaction, generating the diagnostic RS. Our findings may act as trailblazer for tailored therapies in refractory cHL.
Keywords:
Hodgkin lymphoma
; Reed-Sternberg cell
; Nanotechnology
; 3D Q-FISH
; Telomere/shelterin
; TRF2
; Lamin A/C
; LMP1 oncogene
; Breakage-Bridge-Fusion (BBF) cycles
; t-stumps
1. Introduction
Hodgkin lymphoma (HL) was first described in 1832 [1] and its diagnostic tumor cell, the bi- or multinuclear Reed-Sternberg (RS) cell independently by an Austrian [2] and an American [3] pathologist in 1898 and 1902, respectively. Precursor of the RS cell is the mononuclear Hodgkin (H) cell, itself originating from germinal center (GC) B-lymphocytes [4,5,6,7]. As a particularity, circulating small clonotypic precursor B-cells are very rare or even undetectable [8]. In about 40-50% of HL cases H and RS express the Epstein-Barr-Virus (EBV) encoded latent membrane protein 1 (LMP1) or its deletion variants [9,10]. The WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues [11], divides HL into Nodular lymphocyte predominant Hodgkin lymphoma (NLPHL) and Classic Hodgkin lymphoma (cHL). Histologically, most cHL cases are characterized by a low number of H cells and even less frequent bi- or multinuclear RS cells in a mixed background of non-neoplastic T-lymphocytes, inflammatory and accessory cells. cHL is subdivided into four histological subtypes: Nodular sclerosis (NS), mixed cellularity (MC), lymphocyte-rich (LR) and lymphocyte-depleted (LD). Regardless of subtype, H and RS cells express an immune-profile that is typically CD30+, CD15+, CD45 LCA-, CD20-, PAX5+ (weaker than in nonneoplastic B-cells), MUM-1+, OCT-2-, BOB-1-. In EBV-associated cHL, H and RS cells are typically EBER+ and LMP1+ (Figure 1) and the postulated lymphoid precursor is a pre-apoptotic GC B-lymphocyte, rescued by EBV-encoded latent genes [6,7,11].
Though nearly all (98-99%) cHL cases are of B-cell genotype, very rare cases of cHL, maximal 1-2%, harbor a true T-cell genotype in their H and RS cells [13,14,15], pointing to a common pathogenic denominator, basically independent of a B – or T-cell genotype. The hypothesis of a basic molecular mechanism as common pathogenic denominator is also supported by the fact that transfection of the LMP1 oncogene in the EBV- Hodgkin cell line L-428 and the mononuclear human embryonic kidney cell line 293 induces RS and RS-like multinucleated cell formation, respectively [16]. H and RS cells present numerical chromosome aberrations, are in hyperploid range [15,17], exhibit a complex and instable karyotype [18,19]. However, despite recurrent translocations and gene amplifications [20,21], a cHL defining translocation has not been identified [22].
Thanks to a myriad of epidemiological, molecular and genetic approaches our understanding of the specific nature of cHL has considerably advanced in the past three decades and several, comprehensive reviews document this progress [23,24,25,26]. Already in the year 2000 we postulated the identification of a common pathogenic denominator, closely connected to targets of the LMP1 oncogene [27], based on the oncoprotein’s permanent activation of the NF-kappa-B, JAK3-STAT and c-Jun signaling pathways, mandatory for H and RS formation [5,22,25,26]. However, the identification of this common pathogenic denominator at the origin of the H and diagnostic RS ceels was only recently achieved. The development of a highly accurate 3D nanotechnology approach, applicable to cytospins and deparaffinized histologic diagnostic slides [28,29,30,31], and the molecular characterization of the telomerase/shelterin complex [32,33,34,35] allowed this progress.
2. Review
2.1. LMP1: the Golden Key to H and RS in 2D Restricted Research
The nucleotide sequence and protein structure of LMP1, encoded by the BNLF1 gene of EBV, were discovered in 1984 [36]. A year later LMP1 was identified as viral oncogene by its capacity to transform rodent fibroblasts and to render them tumorigenic in nude mice [37]. Experimental LMP1 expression in primary human GC B-cells induces transcriptional profiles characteristic for Hodgkin cell lines, in particular loss of B-cell antigens as CD19, CD20 and CD79 but induction of ID2 hampering B-cell differentiation [38]. In mononucleosis infectiosa (IM) latency II type B-lymphoid cells (EBNA2-, LMP1+) are regularly observed [39] and lymph node biopsies of IM contain few bi-nucleated LMP1+ cells with characteristic RS owl-eye cytomorphology [40]. Most importantly, the naturally occurring EBNA2-deleted EBV strain P3HR1, causes in a subset of infected cord-blood humanized mice Hodgkin-like type II latency B-cell lymphomas with numerous bi- and multi-nucleated LMP1+, CD30+, CD45-, RS-like cells [41]. From these findings it appears evident that permanent LMP1 expression is an inducer of multi-nuclearity and characteristic cytomorphology of RS. But which steps finally led to genomic instability, as numerical increase in chromosomes, increasingly complex cytogenetics and multi-nucleation, had still to be determined.
The LMP1 induced deregulation of key components of the shelterin complex, the safeguard of telomeres [32], appears as major element in this puzzle [42]. Lymphoblastoid cell lines [LCL] from EBV- healthy donors, infected with EBV strain 95.8, develop after 4 weeks deletions, fragments of chromosomes, dicentric chromosomes, unbalanced translocations and chromatid gaps, when karyotyped by SKY (spectral karyotyping) [42]. These structural chromosomal anomalies are not identifiable in normal, mitogen stimulated EBV- lymphocytes [42]. In LCL numerous foci of γH2AX and of MRE11, indicating DSB (double strand breaks) and DNA damage response, respectively, occur, but are not found in the mitogen stimulated control lymphocytes. Moreover, in about 75% of the analyzed LCL nuclei contain TRF2 (telomere repeat binding factor 2) free telomeres, consistent with telomere uncapping. Based on these 2D seminal papers (16,27,32,41,42] it is evident that the target genes of the multifunctional LMP1 oncoprotein are intrinsically associated with the generation of H and RS, as suspected already two decades ago [27,43]. To uncover the mechanisms of these interactions/steps, was only possible through the progressive development of 3D telomere q-FISH [28,29,44,45] over the last decade.
2.2. 3D Telomere-Shelterin Complex: The Railway Turntable in H and RS Morphogenesis
Small telomere and telomere poor Hodgkin cells were first identified by 2D telomere-PNA-FISH through Balta-Yildirim in cHL lymph node touch preparations and HDLM-2 cytospins [46]. Major progress was only achieved through 3D nanotechnology analysis of the nuclear spatial distribution and size of the telomeres and telomere aggregates (clusters of telomeres found in close association not identifiable as separate entities due to the optical resolution limit of 200 nm [47]) combined with immunohistochemistry in 3 H cell lines (HDLM-2, L-428, l-1236) and 3 diagnostic lymph node biopsies (2 NS, 1 MC) [48]. This work uncovered the mechanistic steps of the transition from H to RS cells [48]. RS cells of the 3 H cell lines show significantly shorter and significantly fewer telomeres in relation to the total nuclear volume, when compared to H [48]. (Cytospins allow analysis of the entire volume of H and RS cells by 3D technique, whereas 3D of 5μ of histologic sections is limited to a segment of the nucleus/nuclei, given the nuclear diameter of H and RS is much greater than 5μ). In RS of HDLM-2 multiple pairs of centromeres (anti-Centrin-2 antibody) and multiple atypical spindles (anti-γ-tubulin) are identified. γH2AX immune-staining, indicating double-strand DNA breaks, is most impressive in outré giant RS nuclei, but only faintly present in H nuclei. Multiple atypical spindles are also present in large RS of the diagnostic biopsy of the MC case. These centrosome and mitotic spindle abnormalities confirm our earlier 2D observations in L-428 cells [49] but add the 3D configuration, number and size of telomeres, as new essential element in the transition of H to RS. In all three H cell lines, but also in the diagnostic biopsies of one NS and the MC case, the transition from H to RS was characterized by a significant increase of short and very short telomeres, so called t-stumps [50] and the emergence of virtually telomere free “ghost” nuclei in RS [48]. Analogous findings were observed in a fourth H cell line, U-HO1, from a primary refractory Hodgkin lymphoma [51]. The 3D kinetics were partially reversible in U-HO1-PTPN1 (non-receptor-protein-tyrosine phosphatase N1), a stably transfected daughter H cell line of U-HO1. Culture of U-HO1-PTPN1 cells induced de-phosphorylation of STAT5 with consecutive lack of Akt/PKB activation and cellular arrest in G2, promoting induction of apoptosis. Contrary to U-HO1 with a high STAT5A expression, U-HO1-PTPN1 was characterized by nearly absent STAT5A expression, three times longer doubling time, accumulation of RS, prevention of “t-stump” formation and four fold increase of apoptotic H and RS. These findings confirm that STAT5 is essential for cHL [52] but also open the transition from H to RS as target for new therapeutic research.
When analyzing 3 LMP1+ and 3 EBV- cHL cases the primary 3D findings in HDLM-2, L-428, L-1236 and U-HO1 were further confirmed [53]. Our 3D telomere FISH findings on H cell lines and diagnostic lymph node biopsies define RS as end stage tumor cells because their telomere loss, increase of “t-stumps”, aggregate formation and generation of “ghost” nuclei, will no longer sustain chromosome segregation and the ability to divide further [48]. In our personal experience this limit for RS in diagnostic biopsies is up to 10-12 nuclei [10].
2.3. 3D nuclear Remodeling during the Transition from H to RS
Maintenance of proper nuclear architecture is essential for proper cell function including mitosis, transcription, translation and genomic stability [54,55,56]. Chromosomes are organized in a non-random manner to assess correct cellular function as convincingly shown by the different architecture of rod cell nuclei in animals with diurnal and nocturnal vision, respectively [57]. Significant 3D nuclear remodeling is also identified during the transition from H to RS [58]. Using a fixation protocol maintaining the 3D interphase nuclear structure [59], we analyzed the positions of chromosomes 9 and 22 in the H-cell lines HDLM-2, L-428 and L-1236 [58], using healthy lymphocytes (neighboring chromosomal territories) and CML cells (overlapping due to the t(9;22) as negative and positive controls, respectively. Significant differences in the chromosome territories were observed in H for both chromosomes and were progressing when transforming to RS (for example in a three-nucleated RS most chromosome 9 and 22 content present in one nucleus, the two others nearly free of 9 and 22 material consistent with “ghost” nuclei [49]). Chromosome painting of metaphase spreads in HDLM-2 identified presence of chromosomes with “zebra.”- stripe like pattern, typical for chromosomes having undergone several rounds of BBF. Spectral caryotyping (SKY) confirmed “zebra” chromosomes involving different chromosome partners in L-1236 [58]. The complexity of chromosomal rearrangements progresses from H to RS; for example a t(1;17) found in H, progresses to a t(1;17;13;18) or t(1;17;18;19).
Ongoing nuclear remodeling was confirmed by 3D SIM (structured illumination microscopy). Nuclei of RS were frequently linked to each other through inter-nuclear bridges, consisting of stretched DNA fibers and/or individual chromosomes. DNA free spaces “holes” increased from H to RS and progressed further from bi- to tetra-nuclear RS [60]. The disruption of nuclear architecture, progressing from H to RS, as suggested by our group earlier [48] was confirmed. Moreover, we documented complex chromosome dynamics during the transition from H to RS [58].
2.4. LMP1 Induces TRF2 de-Regulation as Essential Step for H Formation and Progression to RS
TRF2 was identified in 1992 independently by a French [61] and a American [62] group. Main functions of TRF2 are prevention of DSB repair activities, protection of eukaryotic chromosome ends, and nuclear envelope interactions [63,64,65]. Based on our previous clinical experience, immune-histologic 2D findings and 3D nanotechnology results, it was highly probable that the LMP1 oncoprotein targeted directly or indirectly key proteins of the shelterin complex, and, upon reactivation in GC derived B-cells, induced H and RS through permanent activation. To test this hypothesis we needed a long-term tet-off inducible LMP1 expression system in a GC derived B-cell line. The BJAB-tTA-LMP1 cell line and its negative control BJAB-tTA correspond to these prerequisites [66].
Indeed, permanent expression of LMP1 in the BJAB-tTA-LMP1 cell line induces significant down-regulation of telomere repeat binding factor 1 (TRF1), TRF2 and protection of telomere 1 (POT1) at both, the transcriptional and translational level (the most impressive down-regulation (>60%) was observed in TRF2), resulting in a highly significant increase of multinucleated, LMP1+ RS-like cells (p<0.0001) at days 14 and 21 [67]. Moreover, this LMP1 induced significant (p<0.05) down-regulation of TRF1, TRF2 and POT1 at the transcriptional and translational level was reversible upon re-suppression of LMP1 (tet-on) at day3, when measured at day7 and 14. The same was true for suppression at day7, when measured at day14 [67].
Since conditional TRF2 deletion elicits an ATM (ataxia-telangectasia) mediated telomere damage response with γH2AX up-regulation resulting in telomere fusions and giant chromosomes [68], we performed SKY on 20 metaphases of BJAB-tTA-LMP1 expressed (tet-off) on day 1 and on day 20, each, and on 20 metaphases of BJAB-tTA-LMP1 suppressed (tet-on) on day 20. Variation of chromosome number in LMP1 expressed was small at day 1 (44-58 chromosomes) but major at day 14 (19-316 chromosomes) indicating presence of “ghost” and giant RS-like cells. The number of “zebra” chromosomes increased by a factor of 3, indicating ongoing BBF cycles. Thus, LMP1 induced down-regulation of TRF2 results in complex cytogenetics, a hallmark of most RS cells.
To test the hypothesis that TRF2 was intrinsically associated with the generation of multinucleated RS-like cells, we established a stable BJAB-tTA-LMP1/mycTRF2 transfectant capable to produce Myc driven TRF2, independent of the LMP1 mediated suppression. And indeed, the myc driven TRF2 compensated the TRF2 suppression induced by LMP1 (tet-off). The formation of multinucleated RS-like cells was blocked, proving that TRF2 was essential to prevent multi-nucleation [67]. Again, LMP1 induced multinucleated RS-like cells showed uneven telomere distribution, nearly absent TRF2 at day14, and abundant “t-stumps” at day 21, when compared to LMP1suppressed. Thus, the generation of unprotected telomeres was induced by LMP1. Analogous findings were confirmed by combined 3D telomere FISH-TRF2 immunohistochemistry in primary H and RS cells of EBV- cHL [67] underscoring a common pathogenic denominator, mostly consistent with the telomere-shelterin complex.
2.5. 3D Disruption of Direct 3D Telomere-TRF2 Interaction Is a Hallmark of H and RS
In order to confirm our in vitro post GC B-cell model of EBV-associated cHL, where the LMP1 oncogene mediates multi-nuclearity through down-regulation of TRF2, we further developed and adapted our 3D combined quantitative TRF2-telomere immuno Q-FISH protocol (3D TRF2/Telo-Q-FISH) [29,45,47,67] to monolayers of H and RS including the surrounding lymphocytes [69]. Cytocentrifuged monolayers of lymph node suspensions from 14 diagnostic cHL biopsies (4 LMP1 expressing) were analyzed. The reactive surrounding lymphocytes as well as benign BJ-5ta fibroblasts served as controls. This approach allowed for the first time the 3D analysis of the entire nuclear content of H and RS [69]. Of note the entire nuclear analysis is not achieved with laser dissection, performed on 5 mu sections, given the nuclear diameter of H and RS generally >10 mu.
As expected, in all LMP1+ and EBV- cases, the internal control lymphocytes and the BJ-5ta fibroblasts showed a tight 1:1 telomere-TRF2 association, excluding technical pitfalls in the H and RS analysis. On the contrary, all H and RS (193 H and 122 RS) of all 14 cHL cases showed an unambiguous disruption of the direct, quantitative and qualitative telomere/TRF2 interaction [69]. Interestingly two, at first glance opposite appearing patterns, were observed. The 4 LMP1 expressing and 4 EBV- cases (total 8 cases) were characterized by a most significant attrition of TRF2 spots during the transition from H to RS, (for example ratio telomere/TRF2 signals from 2.5 in H to 3.8 in RS), leaving most telomeres de-protected in RS, designed as Pattern B. In Pattern B “Ghost” RS with virtually both, absent TRF2 and telomere signals were regularly observed, but “ghost” H cells with very large nuclei (15-24 um) were rare (Figure 2).
Six cases (all EBV negative) were characterized by significantlt more free TRF2 spots than telomere signals, identified as Pattern A. Some signals co-localized, but there were always also free telomere signals, consistent with de-protection [69]. Again, there was progression of the signal ratio telomere/TRF2 with transition from H to RS (for example 0.5 in H to 0.2 in RS). Most of the remaining telomere spots were “t-stumps”. Nuclei of RS with Pattern A had often intra-nuclear DNA bridges. Interestingly, in vitro overexpression of TRF2 leads to replication stalling, chromosome end to end fusions, and loss of telomeric sequences [70]. Ultrafine anaphase bridges with loss of telomeric sequences and telomere shortening occur rapidly. Significantly elevated TRF2 levels also mechanistically induce genomic instability [70] and very short telomeres are fused by both the classical and alternative NHEJ (non-homologous end joining) pathways [71]. To the best of our knowledge, the reason for this high TRF2 expression in H and RS of some EBV- cHL cases is not known as to our knowledge. Thus, beside TRF2 down-regulation also TRF2 overexpression leads to genomic instability, the driving force in refractory cHL. The defective 3D steric interaction of telomeres with shelterin proteins, especially TRF2, an essential element in the molecular pathology of cHL, was only recently recognized by other research groups [23,26,41]. Figure 3 summarizes the main events of the molecular, mechanistic pathogenesis of EBV positive and of some EBV negative cHL, based on our 3D experimental and translational findings.
2.6. Lamin Basics and Lamin A/C Overexpression in H and RS
Lamins are type V intermediate filament proteins, associate physically with the inner nuclear membrane, tether heterochromatin in the periphery, and exert important scaffolding functions within the nucleus [76,77,78]. In human cells two types of lamin have been identified: B-type lamins, encoded by the LMNB1 gene for lamin B1 [79] and the LMNB2 gene for lamin B2 [80], and A-type lamins, encoded by the LMNA gene, the alternative splicing of which produces lamin A and lamin C [81]. Recently 3D-SIM allowed to characterize a distinct fiber meshwork at the supra-molecular level for lamin A, C, B1 and B2 [82], and cryo-electron microscopic tomography identified a coil-coiled, 51 nm long and 3.5 nm thick rod-like dimer with two lateral globular domains as the building blocks of all lamins [83,84]. Lamin dimers form head-to-tail polymers, which laterally interact to form proto-filaments [84,85]. Stochastic optical reconstruction microscopy (STORM) further revealed that lamin B1 and lamin A/C form two concentric and overlapping nutshell-like networks adjacent to the inner nuclear membrane (INM) [86]. Lamin B1 preferentially localizes directly adjacent to the INM, whereas lamin A/C localizes closer to the nucleoplasm [86]. Lamin B1 and lamin B2 are constitutively expressed and appear to be necessary for cell survival [87], whereas lamin A/C, also distributed throughout the nuclear interior [77], is involved in transcription [88] and plays a crucial role in the regulation of mitotic spindle assembly and positioning [89]. Distinct mutations in the coding sequence of the latter result in severe congenital disorders, known as laminopathies [90], the most impressive being Hutchinson-Gilford progeria syndrome (HGPS) [91].
Already in 1997 Jansen and coworkers [92] documented through immunohistochemistry strong lamin A/C expression in H and RS but no expression in centrocytes, centroblasts and mantle zone lymphocytes of reactive lymph node follicles. Lamin A/C was also identified in stimulated blood lymphocytes [93,94] and its overexpression confirmed by 3D nanotechnology in H- and RS cells by our group [94]. The regular, spherically shaped lamin A/C organization, identified in activated lymphocytes, is disturbed in mononuclear H-cells and undergoes further structural changes during the transformation of H to binuclear and finally multinuclear RS [94]. This dynamic process of steadily increasing lamin A/C associated nuclear compartmentalization of individual nuclei was identified in both, H-cell lines (Supplementary Video S1 A and B in form of HDLM-2 Video Clip A H-cell, and Video Clip B RS-cell) and H and RS of diagnostic lymph node biopsies [31,94].
2.7. Progressive Disruption of the 3D Lamin A/C-TRF2-DNA Interaction from H to RS
There is increasing evidence of tight interactions of lamins with telomeres and the shelterin complex. Human fibroblasts with a homozygous nonsense mutation of the lamin A/C gene and thus completely devoid of lamin A/C expression, show increased nuclear plasticity and telomere mobility [95]. Chromatin immuno-precipitation (ChIP) assays performed with lamin A/C antibody revealed binding of lamin A/C to telomeres and expression of a dominant negative telomere repeat binding factor 2 (TRF2) mutant leads to loss of telomere integrity and to the formation of DNA damage foci at telomeres [96]. Indeed, TRF2 is not only essential for formation and maintenance of the configuration of functional telomeres at chromosomal ends [97], but also binds to interstitial telomeric sequences (ITS) [98]. The interaction of TRF2 with lamin A/C occurs at the long linker region of TRF2 [99]. On the other hand, lamin A/C is essential for maintaining the chromosomal nuclear territories and this occurs partially through direct interaction with TRF2 bound to t-loops at ITS [100], so-called interstitial t-loops (ITL). Impairment or loss of this 3D lamin A/C-TRF2 interaction at ITL results in increased chromatin dynamics [101], changes in gene expression, impaired chromosome stability and genomic integrity [102,103].
Considering the experimental data of TRF2 down-regulation [64,65,67] and overexpression [70,71] on cellular 3D structure we suspected the direct 3D interaction of lamin A/C with TRF2, localized at t-loops of telomeres and ITS, to undergo substantial changes during the formation of H and further transformation to RS. We applied our combined quantitative 3D TRF2-telomere immuno Q-FISH (3D TRF2/Telo-Q-FISH) protocol [45] and extended it to lamin A/C as a further target. Our findings revealed very high and abnormal lamin A/C protein expression and completely disrupted lamin A/C-TRF2-ITL/telomere interaction in H- and RS in contrast with BJ-5ta control fibroblasts (Figure 5) [24,31,104]. A significant increase of lamin A/C accumulation at RNA level and, even more impressively, at the protein level, occurred from normal lymphocytes (at 1) to H and finally RS (over 20) [94,104], consistent with progressive destruction of the nuclear architecture from H to bi- and finally multi-nuclear RS.
Considering the experimental data of TRF2 down-regulation [64,65,67] and overexpression [70,71] on cellular 3D structure, we suspected the direct 3D interaction of lamin A/C with TRF2, localized at t-loops of telomeres and ITS, to undergo substantial changes during the formation of H and further transformation to RS. We applied our combined quantitative 3D TRF2-telomere immuno Q-FISH (3D TRF2/Telo-Q-FISH) protocol [45] and extended it to lamin A/C as a further target. Our findings revealed very high and abnormal lamin A/C protein expression and a completely disrupted lamin A/C-TRF2-ITL/telomere interaction in H- and RS (abnormal internal lamin A/C structures within the nucleus) in contrast with BJ-5ta control fibroblasts (Figure 4) [24,31,104]. A significant increase of lamin A/C accumulation at the RNA level, and even more impressively, at the protein level, from normal lymphocytes (at 1) to H and finally RS (over 20) is observed [94,104]. This is consistent with progressive destruction of the nuclear architecture from H to bi- and finally multi-nuclear RS.
2.8. Prognostic and Future Potential Therapeutic Implications of the 3D Nanotechnology Findings
The international most commonly used chemotherapy regimen for cHL is ABVD (Adriamycin [doxorubicin], bleomycin, vinblastine, dacarbazine) [105,106,107,108]. In two large studies disease-free survival at 4 years was 85.8% [109] and progression free survival at 10 years still high with 69% [110], not inferior to more recently developed chemotherapy regimens. However, despite this high success rate, a reliable upfront biomarker to identify primary refractory and early relapsing patients is still an unmet need. We are confident that cHL patients with a high percentage of t-stumps in the diagnostic biopsy [111], heralding high risk for eary relapse or ABVD refractoriness [111] might upfront benefit from Brentuximab-Vedotin and/or recently developed check-point inhibitors like Pembrolizumab and Nivolumab [112,113] ore more costly treatments as allo-HSCT and cellular therapies [114].
fBALM (fluctuation-assisted binding-activated localization microscopy) may reprent a game-changing technology in the study of nanomolecular structures and drug design for a wide variety of pathological conditions. This technique has been successfully applied to H and RS [115] With a resolution of around 50 nm therapeutic effects of small selective molecules on nuclear sites of DNA/protein interactions could be visualized. Inhibition of telomerase using BIBR1532 followed by ALT (alternative lengthening of telomere) inhibition by Trabectedin, =the ALT pathway is activated in advanced cHL [116]-, caused a decrease of greater than 90% in cell viability in three patient-derived HL cell lines [117]. Thus, 3D shelterin/telomere nanotechnology approach and fBALM appear to be promising methods for future small selective molecule treatment evaluation [118].
3. Conclusions
In the present review we have focused on the 3D nanotechnology based progress in the elucidation of the molecular steps leading from an activated B-lymphocyte to the H and finally diagnostic RS formation in cHL. Our 3D analysis of the shelterin-telomere-lamin A/C complex over the past 2 decades was the trailblazer of this discovery. The “golden key” to this advancement was the LMP1 driven identification of TRF2 as the master protein in the transformation from H to RS. A new player in the field is lamin A/C interacting with TRF2. Our research also allowed identification of the telomere t-stumps quantification as upfront biomarker in cHL. We are confident that the sensitivity and specifity of this biomarker will increase in association with recently developed molecular biomarkers like ctDNA quantification [119] and be useful for clinical applications. Moreover, in the near future 3D analysis of recently discovered TRF2/telomere interacting proteins will foster progress [120].
References
- Hodgkin, T. On some Morbid Appearances of the Absorbent Glands and Spleen. Med. Chir. Trans. 1832, 17, 68–114. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, C. Über eine eigenartige unter dem Bilde der Pseudoleukämie verlaufende Tuberculosdes lymphatischen Apparates. Ztschr. Heilk. 1898, 119, 21–90. [Google Scholar]
- Reed, D.M. On the pathological changes in Hodgkin's disease, with especial reference to its relation to tuberculosis. Johns Hopkins Hosp. Rep. 1902, 10, 133–196. [Google Scholar]
- Küppers, R.; Rajewsky, K.; Zhao, M.; Simons, G.; Laumann, R.; Fischer, R.; Hansmann, M.L. Hodgkin disease: Hodgkin and Reed-Sternberg cells picked from histological sections show clonal immunoglobulin gene rearrangements and appear to be derived from B cells at various stages of development. Proc. Natl. Acad. Sci. USA 1994, 91, 10962–10966. [Google Scholar] [CrossRef] [PubMed]
- Küppers, R. Molecular biology of Hodgkin’s lymphoma. In Advances in Cancer Research; Academic Press: Elsevier, Amsterdam, Netherlands, 2002; Volume 84, pp. 277–312. [Google Scholar]
- Mancao, C.; Altmann, M.; Jungnickel, B.; Hammerschmidt, W. Rescue of “crippled” germinal center B cells from apoptosis by Epstein-Barr virus. Blood 2005, 106, 4339–4344. [Google Scholar] [CrossRef] [PubMed]
- Thorley-Lawson, D.A. EBV Persistence—Introducing the Virus. Curr. Top. Microbiol. Immunol. 2015, 390, 151–209. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.J.; Gocke, C.D.; Kasamon, Y.L.; Miller, C.B.; Perkins, B.; Barber, J.P.; Vala, M.S.; Gerber, J.M.; Gellert, L.L.; Siedner, M.; Lemas, M.V.; Brennan, S.; Ambinder, R.F.; Matsui, W. Circulating clonotypic B cells in classic Hodgkin lymphoma. Blood 2009, 113, 5920–6. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pallesen, G.; Hamilton-Dutoit, S.J.; Rowe, M.; Young, L.S. Expression of Epstein-Barr virus latent gene products in tumour cells of Hodgkin's disease. Lancet 1991, 337, 320–2. [Google Scholar] [CrossRef] [PubMed]
- Knecht, H.; Bachmann, E.; Brousset, P.; Sandvej, K.; Nadal, D.; Bachmann, F.; Odermatt, B.F.; Delsol, G.; Pallesen, G. Deletions within the LMP1 oncogene of Epstein-Barr virus are clustered in Hodgkin's disease and identical to those observed in nasopharyngeal carcinoma. Blood 1993, 82, 2937–42. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef]
- Massarweh, S.; Udden, M.M.; Shahab, I.; Kroll, M.; Sears, D.A.; Lynch, G.R.; Teh, B.S.; Lu, H.H. HIV-related Hodgkin's disease with central nervous system involvement and association with Epstein-Barr virus. Am J Hematol. 2003, 72, 216–9. [Google Scholar] [CrossRef] [PubMed]
- Seitz, V.; Hummel, M.; Marafioti, T.; Anagnostopoulos, I.; Assaf, C.; Stein, H. Detection of clonal T-cell receptor gamma-chain gene rearrangements in Reed-Sternberg cells of classic Hodgkin disease. Blood 2000, 95, 3020–4. [Google Scholar] [CrossRef] [PubMed]
- Tzankov, A.; Bourgau, C.; Kaiser, A.; Zimpfer, A.; Maurer, R.; Pileri, S.A.; Went, P.; Dirnhofer, S. (2005) Rare expression of T-cell markers in classical Hodgkin's lymphoma. Mod. Pathol. 2005, 18, 1542–1549. [Google Scholar] [CrossRef] [PubMed]
- Drexler, H.G.; Pommerenke, C.; Eberth, S.; Nagel, S. Hodgkin lymphoma cell lines: to separate the wheat from the chaff. Biol. Chem. 2018, 399, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Knecht, H.; McQuain, C.; Martin, J.; Rothenberger, S.; Drexler, H.G.; Berger, C.; Bachmann, E.; Kittler, E.L.; Odermatt, B.F.; Quesenberry, P.J. Expression of the LMP1 oncoprotein in the EBV negative Hodgkin's disease cell line L-428 is associated with Reed-Sternberg cell morphology. Oncogene 1996, 13, 947–53. [Google Scholar] [PubMed]
- Weber-Matthiesen, K.; Deerberg, J.; Poetsch, M.; Grote, W.; Schlegelberger, B. Numerical chromosome aberrations are present within the CD30+ Hodgkin and Reed-Sternberg cells in 100% of analyzed cases of Hodgkin's disease. Blood 1995, 86, 1464–8. [Google Scholar] [CrossRef] [PubMed]
- Deerberg-Wittram, J.; Weber-Matthiesen, K.; Schlegelberger, B. Cytogenetics and molecular cytogenetics in Hodgkin's disease. Ann. Oncol. 1996, 7 Suppl 4, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Janz, M.; Mathas, S. The pathogenesis of classical Hodgkin's lymphoma: what can we learn from analyses of genomic alterations in Hodgkin and Reed-Sternberg cells? Haematologica 2008, 93, 1292–5. [Google Scholar] [CrossRef] [PubMed]
- Martín-Subero JI, Klapper W, Sotnikova A, Callet-Bauchu E, Harder L, Bastard C, Schmitz R, Grohmann S, Höppner J, Riemke J, Barth TF, Berger F, Bernd HW, Claviez A, Gesk S, Frank GA, Kaplanskaya IB, Möller P, Parwaresch RM, Rüdiger T, Stein H, Küppers R, Hansmann ML, Siebert R; Deutsche Krebshilfe Network Project Molecular Mechanisms in Malignant Lymphomas. Chromosomal breakpoints affecting immunoglobulin loci are recurrent in Hodgkin and Reed-Sternberg cells of classical Hodgkin lymphoma. Cancer Res. 2006, 66, 10332–8. [CrossRef] [PubMed]
- Roemer, M.G.; Advani, R.H.; Ligon, A.H.; Natkunam, Y.; Redd, R.A.; Homer, H.; Connelly, C.F.; Sun, H.H.; Daadi, S.E.; Freeman, G.J.; Armand, P.; Chapuy, B.; de Jong, D.; Hoppe, R.T.; Neuberg, D.S.; Rodig, S.J.; Shipp, M.A. PD-L1 and PD-L2 Genetic Alterations Define Classical Hodgkin Lymphoma and Predict Outcome. J. Clin. Oncol. 2016, 34, 2690–7. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cuceu, C.; Hempel, W.M.; Sabatier, L.; Bosq, J.; Carde, P.; M'Kacher, R. Chromosomal Instability in Hodgkin Lymphoma: An In-Depth Review and Perspectives. Cancers (Basel) 2018, 10. [Google Scholar] [CrossRef]
- Piris MA, Medeiros LJ, Chang KC Hodgkin lymphoma: a review of pathological features and recent advances in pathogenesis. Pathology 2020, 52, 154–165. [CrossRef]
- Bienz, M.; Ramdani, S.; Knecht, H. Hodgkin’s lymphoma: Past, Present, Future. Int. J. Mol. Sci. 2020, 21, 6623. [Google Scholar] [CrossRef] [PubMed]
- Weniger, M.A.; Küppers, R. Molecular biology of Hodgkin lymphoma. Leukemia 2021, 35, 968–981. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Oliveira, L.O.D.; Costa, I.B.; Quaresma, J.A.S. Association between Epstein-Barr virus LMP-1 and Hodgkin lymphoma LMP-1 mechanisms in Hodgkin lymphoma development. Rev. Med. Virol. 2024, 34, e2561. [Google Scholar] [CrossRef] [PubMed]
- Knecht, H.; Berger, C.; Rothenberger, S.; Odermatt, B.F.; Brousset, P. The role of Epstein-Barr virus in neoplastic transformation. Oncology 2001, 60, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Vermolen, B.J.; Garini, Y.; Mai, S.; Mougey, V.; Fest, T.; Chuang, T.C.; Chuang, A.Y.; Wark, L.; Young, I.T. Characterizing the three-dimensional organization of telomeres. Cytometry A. 2005, 67, 144–50, Erratum in: Cytometry A. 2007 May;71(5):345. [Google Scholar] [CrossRef] [PubMed]
- Louis, S.F.; Vermolen, B.J.; Garini, Y.; Young, I.T.; Guffei, A.; Lichtensztejn, Z.; Kuttler, F.; Chuang, T.C.; Moshir, S.; Mougey, V.; et al. c-Myc induces chromosomal rearrangements through telomere and chromosome remodeling in the interphase nucleus. Proc. Natl. Acad. Sci. USA 2005, 102, 9613–9618. [Google Scholar] [CrossRef] [PubMed]
- Mai S, Knecht H, Gadji M, Olujohungbe A Hematological disorder diagnosis by 3D q-FISH 2018, US Patent 9,963,745.
- Sawan, B.; Petrogiannis-Haliotis, T.; Knecht, H. Molecular Pathogenesis of Hodgkin’s Lymphoma: Advances Through the Key Player LMP1 and 3D Nanotechnology. In: Interdisciplinary Cancer Research. Springer, Cham, 2022. [CrossRef]
- de Lange, T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–10. [Google Scholar] [CrossRef] [PubMed]
- de Lange, T. Shelterin-Mediated Telomere Protection. Annu. Rev. Genet. 2018, 52, 223–247. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.J.; Cech, T.R. Shaping human telomeres: from shelterin and CST complexes to telomeric chromatin organization. Nat. Rev. Mol. Cell. Biol. 2021, 22, 283–298. [Google Scholar] [CrossRef] [PubMed]
- Brankiewicz-Kopcinska, W.; Kallingal, A.; Krzemieniecki, R.; Baginski, M. Targeting shelterin proteins for cancer therapy. Drug Discov Today 2024, 29, 104056. [Google Scholar] [CrossRef] [PubMed]
- Fennewald, S.; van Santen, V.; Kieff, E. Nucleotide sequence of an mRNA transcribed in latent growth-transforming virus infection indicates that it may encode a membrane protein. J. Virol. 1984, 51, 411–9. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, D.; Liebowitz, D.; Kieff, E. An EBV membrane protein expressed in immortalized lymphocytes transforms established rodent cells. Cell 1985, 43(3 Pt 2), 831–40. [Google Scholar] [CrossRef] [PubMed]
- Vockerodt, M.; Morgan, S.L.; Kuo, M.; Wei, W.; Chukwuma, M.B.; Arrand, J.R.; Kube, D.; Gordon, J.; Young, L.S.; Woodman, C.B.; Murray, P.G. The Epstein-Barr virus oncoprotein, latent membrane protein-1, reprograms germinal centre B cells towards a Hodgkin's Reed-Sternberg-like phenotype. J Pathol. 2008, 216, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Niedobitek, G.; Agathanggelou, A.; Herbst, H.; Whitehead, L.; Wright, D.H.; Young, L.S. Epstein-Barr virus (EBV) infection in infectious mononucleosis: virus latency, replication and phenotype of EBV-infected cells. J. Pathol. 1997, 182, 151–9. [Google Scholar] [CrossRef] [PubMed]
- Knecht, H.; Brousset, P.; Bachmann, E.; Sandvej, K.; Odermatt, B.F. (1994) Latent membrane protein 1: a key oncogene in EBV-related carcinogenesis? Acta Haematol. 1993, 90, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Romero-Masters, J.C.; Huebner, S.; Ohashi, M.; Hayes, M.; Bristol, J.A.; Nelson, S.E.; Eichelberg, M.R.; Van Sciver, N.; Ranheim, E.A.; Scott, R.S.; Johannsen, E.C.; Kenney, S.C. EBNA2-deleted Epstein-Barr virus (EBV) isolate, P3HR1, causes Hodgkin-like lymphomas and diffuse large B cell lymphomas with type II and Wp-restricted latency types in humanized mice. PLoS Pathog. 2020, 16, e1008590. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lacoste, S.; Wiechec, E.; Dos Santos Silva, A.G.; Guffei, A.; Williams, G.; Lowbeer, M.; Benedek, K.; Henriksson, M.; Klein, G.; Mai, S. Chromosomal rearrangements after ex vivo Epstein-Barr virus (EBV) infection of human B cells. Oncogene 2010, 29, LCL503–15. [Google Scholar] [CrossRef] [PubMed]
- Knecht, H.; Berger, C.; McQuain, C.; Rothenberger, S.; Bachmann, E.; Martin, J.; Esslinger, C.; Drexler, H.G.; Cai, Y.C.; Quesenberry, P.J.; Odermatt, B.F. Latent membrane protein 1 associated signaling pathways are important in tumor cells of Epstein-Barr virus negative Hodgkin's disease. Oncogene 1999, 18, 7161–7. [Google Scholar] [CrossRef] [PubMed]
- Gadji, M.; Vallente, R.; Klewes, L.; Righolt, C.; Wark, L.; Kongruttanachok, N.; Knecht, H.; Mai, S. Nuclear remodeling as a mechanism for genomic instability in cancer. Adv. Cancer Res. 2011, 112, 77–126. [Google Scholar] [CrossRef] [PubMed]
- Knecht, H.; Mai, S. The Use of 3D Telomere FISH for the Characterization of the Nuclear Architecture in EBV-Positive Hodgkin's Lymphoma. Methods Mol. Biol. 2017, 1532, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Balta-Yildrim, Z. Untersuchungen zu Telomerlängen bei Hodgkin- und Reed-Sternberg-Zellen. Doctoral Thesis, RWTH Aachen. Nov. 15, 2006.
- Chuang, T.C.; Moshir, S.; Garini, Y.; Chuang, A.Y.; Young, I.T.; Vermolen, B.; van den Doel, R.; Mougey, V.; Perrin, M.; Braun, M.; Kerr, P.D.; Fest, T.; Boukamp, P.; Mai, S. The three-dimensional organization of telomeres in the nucleus of mammalian cells. BMC Biol 2004, 2, 12. [Google Scholar] [CrossRef] [PubMed]
- Knecht, H.; Sawan, B.; Lichtensztejn, D.; Lemieux, B.; Wellinger, R.J.; Mai, S. The 3D nuclear organization of telomeres marks the transition from Hodgkin to Reed-Sternberg cells. Leukemia 2009, 23, 565–573. [Google Scholar] [CrossRef]
- Pihan, G.A.; Purohit, A.; Wallace, J.; Knecht, H.; Woda, B.; Quesenberry, P.; Doxsey, S.J. Centrosome defects and genetic instability in malignant tumors. Cancer Res. 1998, 58, 3974–85. [Google Scholar] [PubMed]
- Xu, L.; Blackburn, E.H. Human cancer cells harbor T-stumps, a distinct class of extremely short telomeres. Mol. Cell 2007, 28, 315–27. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Knecht, H.; Brüderlein, S.; Wegener, S.; Lichtensztejn, D.; Lichtensztejn, Z.; Lemieux, B.; Möller, P.; Mai, S. 3D nuclear organization of telomeres in the Hodgkin cell lines U-HO1 and U-HO1-PTPN1: PTPN1 expression prevents the formation of very short telomeres including "t-stumps". BMC Cell Biol. 2010, 11, 99. [Google Scholar] [CrossRef] [PubMed]
- Scheeren, F.A.; Diehl, S.A.; Smit, L.A.; Beaumont, T.; Naspetti, M.; Bende, R.J.; Blom, B.; Karube, K.; Ohshima, K.; van Noesel, C.J.; Spits, H. IL-21 is expressed in Hodgkin lymphoma and activates STAT5: evidence that activated STAT5 is required for Hodgkin lymphomagenesis. Blood 2008, 111, 4706–15. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Knecht, H.; Sawan, B.; Lichtensztejn, Z.; Lichtensztejn, D.; Mai, S. 3D Telomere FISH defines LMP1-expressing Reed-Sternberg cells as end-stage cells with telomere-poor ‘ghost’ nuclei and very short telomeres. Lab. Investig. 2010, 90, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Cremer, T.; Cremer, M.; Dietzel, S.; Müller, S.; Solovei, I.; Fakan, S. Chromosome territories--a functional nuclear landscape. Curr. Opin. Cell Biol. 2006, 18, 307–16. [Google Scholar] [CrossRef] [PubMed]
- Lanctôt, C.; Cheutin, T.; Cremer, M.; Cavalli, G.; Cremer, T. Dynamic genome architecture in the nuclear space: regulation of gene expression in three dimensions. Nat. Rev. Genet. 2007, 8, 104–15. [Google Scholar] [CrossRef] [PubMed]
- Cremer, T.; Cremer, M.; Hübner, B.; Silahtaroglu, A.; Hendzel, M.; Lanctôt, C.; Strickfaden, H.; Cremer, C. The Interchromatin Compartment Participates in the Structural and Functional Organization of the Cell Nucleus. Bioessays 2020, 42, e1900132. [Google Scholar] [CrossRef] [PubMed]
- Solovei, I.; Kreysing, M.; Lanctôt, C.; Kösem, S.; Peichl, L.; Cremer, T.; Guck, J.; Joffe, B. Nuclear architecture of rod photoreceptor cells adapts to vision in mammalian evolution. Cell 2009, 137, 356–68. [Google Scholar] [CrossRef] [PubMed]
- Guffei, A.; Sarkar, R.; Klewes, L.; Righolt, C.; Knecht, H.; Mai, S. Dynamic chromosomal rearrangements in Hodgkin’s lymphoma are due to ongoing three-dimensional nuclear remodeling and breakage-bridge-fusion cycles. Haematologica 2010, 95, 2038–2046. [Google Scholar] [CrossRef] [PubMed]
- Guffei, A.; Lichtensztejn, Z.; Gonçalves Dos Santos Silva, A.; Louis, S.F.; Caporali, A.; Mai, S. c-Myc-dependent formation of Robertsonian translocation chromosomes in mouse cells. Neoplasia 2007, 9, 578–88. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Righolt, C.H.; Guffei, A.; Knecht, H.; Young, I.T.; Stallinga, S.; van Vliet, L.J.; Mai, S. Differences in nuclear DNA organization between lymphocytes, Hodgkin and Reed-Sternberg cells revealed by structured illumination microscopy. J. Cell. Biochem. 2014, 115, 1441–8. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bilaud, T.; Brun, C.; Ancelin, K.; Koering, C.E.; Laroche, T.; Gilson, E. Telomeric localization of TRF2, a novel human telobox protein. Nat. Genet. 1997, 17, 236–9. [Google Scholar] [CrossRef] [PubMed]
- Broccoli, D.; Smogorzewska, A.; Chong, L.; de Lange, T. Human telomeres contain two distinct Myb-related proteins, TRF1 and TRF2. Nat. Genet. 1997, 17, 231–5. [Google Scholar] [CrossRef] [PubMed]
- Feuerhahn, S.; Chen, L.Y.; Luke, B.; Porro, A. No DDRama at chromosome ends: TRF2 takes centre stage. Trends Biochem. Sci. 2015, 40, 275–85. [Google Scholar] [CrossRef] [PubMed]
- Rai, R.; Biju, K.; Sun, W.; Sodeinde, T.; Al-Hiyasat, A.; Morgan, J.; Ye, X.; Li, X.; Chen, Y.; Chang, S. Homology directed telomere clustering, ultrabright telomere formation and nuclear envelope rupture in cells lacking TRF2B and RAP1. Nat. Commun. 2023, 14, 2144, Erratum in: Nat. Commun. 2023, Jun 7;14(1), 3319. doi: 10.1038/s41467-023-39144-7. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rai, R.; Sodeinde, T.; Boston, A.; Chang, S. Telomeres cooperate with the nuclear envelope to maintain genome stability. Bioessays 2024, 46, e2300184. [Google Scholar] [CrossRef] [PubMed]
- Floettmann, J.E.; Ward, K.; Rickinson, A.B.; Rowe, M. Cytostatic effect of Epstein-Barr virus latent membrane protein-1 analyzed using tetracycline-regulated expression in B cell lines. Virology 1996, 223, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Lajoie, V.; Lemieux, B.; Sawan, B.; Lichtensztejn, D.; Lichtensztejn, Z.; Wellinger, R.; Mai, S.; Knecht, H. LMP1 mediates multinuclearity through downregulation of shelterin proteins and formation of telomeric aggregates. Blood 2015, 125, 2101–10. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Celli, G.B.; de Lange, T. DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat. Cell Biol. 2005, 7, 712–8. [Google Scholar] [CrossRef] [PubMed]
- Knecht, H.; Johnson, N.A.; Haliotis, T.; Lichtensztejn, D.; Mai, S. Disruption of direct 3D telomere-TRF2 interaction through two molecularly disparate mechanisms is a hallmark of primary Hodgkin and Reed-Sternberg cells. Lab. Investig. 2017, 97, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Nera, B.; Huang, H.S.; Lai, T.; Xu, L. Elevated levels of TRF2 induce telomeric ultrafine anaphase bridges and rapid telomere deletions. Nat. Commun. 2015, 6, 10132. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nera, B.; Huang, H.S.; Hendrickson, E.A.; Xu, L. Both the classical and alternative non-homologous end joining pathways contribute to the fusion of drastically shortened telomeres induced by TRF2 overexpression. Cell Cycle 2019, 18, 880–888. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bargou, R.C.; Emmerich, F.; Krappmann, D.; Bommert, K.; Mapara, M.Y.; Arnold, W.; Royer, H.D.; Grinstein, E.; Greiner, A.; Scheidereit, C.; et al. Constitutive nuclear factor-kappaB-RelA activation is required for proliferation and survival of Hodgkin’s disease tumor cells. J. Clin. Investig. 1997, 100, 2961–2969. [Google Scholar] [CrossRef] [PubMed]
- Rothenberger, S.; Bachmann, E.; Berger, C.; McQuain, C.; Odermatt, B.F.; Knecht, H. Natural 30 bp and 69 bp deletion variants of the LMP1 oncogene do stimulate NF-κB mediated transcription. Oncogene 1997, 14, 2123–2126. [Google Scholar] [CrossRef]
- Knecht, H.; Mai, S. LMP1 and Dynamic Progressive Telomere Dysfunction: A Major Culprit in EBV-Associated Hodgkin's Lymphoma. Viruses 2017, 9, 164. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- MacLeod, R.A.; Spitzer, D.; Bar-Am, I.; Sylvester, J.E.; Kaufmann, M.; Wernich, A.; Drexler, H.G. Karyotypic dissection of Hodgkin’s disease cell lines reveals ectopic subtelomeres and ribosomal DNA at sites of multiple jumping translocations and genomic amplification. Leukemia 2000, 14, 1803–1814. [Google Scholar] [CrossRef]
- Moir, R.D.; Yoon, M.; Khuon, S.; Goldman, R.D. Nuclear Lamins A and B1: Different Pathways of Assembly during Nuclear Envelope Formation in Living Cells. J. Cell. Biol. 2000, 151, 1155–68. [Google Scholar] [CrossRef] [PubMed]
- Naetar, N.; Ferraioli, S.; Foisner, R. Lamins in the nuclear interior - life outside the lamina. J. Cell. Sci. 2017, 130, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Dubik, N.; Mai, S. Lamin A/C: Function in Normal and Tumor Cells. Cancers (Basel) 2020, 12, 3688. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lin, F.; Worman, H.J. Structural organization of the human gene (LMNB1) encoding nuclear lamin B1. Genomics 1995, 27, 230–6. [Google Scholar] [CrossRef] [PubMed]
- Höger, T.H.; Zatloukal, K.; Waizenegger, I.; Krohne, G. (1990) Characterization of a second highly conserved B-type lamin present in cells previously thought to contain only a single B-type lamin. Chromosoma 1990, 99(6), 99(6),379–90. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.Z.; Chaudhary, N.; Blobel, G. cDNA sequencing of nuclear lamins A and C reveals primary and secondary structural homology to intermediate filament proteins. Proc. Natl. Acad. Sci. USA 1086, 83, 6450–6454. [Google Scholar] [CrossRef] [PubMed]
- Shimi, T.; Kittisopikul, M.; Tran, J.; Goldman, A.E.; Adam, S.A.; Zheng, Y.; Jaqaman, K.; Goldman, R.D. Structural organization of nuclear lamins A, C, B1, and B2 revealed by superresolution microscopy. Mol. Biol. Cell. 2015, 26, 4075–86. [Google Scholar] [CrossRef]
- Turgay, Y.; Eibauer, M.; Goldman, A.E.; Shimi, T.; Khayat, M.; Ben-Harush, K.; Dubrovsky-Gaupp, A.; Sapra, K.T.; Goldman, R.D.; Medalia, O. The molecular architecture of lamins in somatic cells. Nature 2017, 543, 261–264. [Google Scholar] [CrossRef]
- Makarov, A.A.; Zou, J.; Houston, D.R.; Spanos, C.; Solovyova, A.S.; Cardenal-Peralta, C.; Rappsilber, J.; Schirmer, E.C. Lamin A molecular compression and sliding as mechanisms behind nucleoskeleton elasticity. Nat. Commun. 2019, 10, 3056. [Google Scholar] [CrossRef] [PubMed]
- de Leeuw, R.; Gruenbaum, Y.; Medalia, O. Nuclear Lamins: Thin Filaments with Major Functions. Trends Cell 2018, 28, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Nmezi B, Xu J, Fu R et al. Concentric organization of A- and B-type lamins predicts their distinct roles in the spatial organization and stability of the nuclear lamina. Proc. Natl. Acad. Sci. USA 2019, 116, 4307–4315. [CrossRef] [PubMed]
- Yang SH, Jung HJ, Coffinier C, et al. Are B-type lamins essential in all mammalian cells? Nucleus 2019, 2, 562–9. [CrossRef]
- Hutchinson, C.J.; Worman, H.J. (2004) A-type lamins: Guardians of the soma? Nat. Cell Biol. 2004, 6, 1062–1067. [Google Scholar] [CrossRef] [PubMed]
- Qi, R.; Xu, N.; Wang, G.; et al. The lamin-A/C-LAP2α-BAF1 protein complex regulates mitotic spindle assembly positioning. J. Cell Sci. 2015, 128, 2830–2841. [Google Scholar] [CrossRef] [PubMed]
- Shimi, T.; Butin-Israeli, V.; Adam, S.A.; et al. Nuclear lamins in cell regulation and disease. Cold Spring Harb. Symp. Quant. Biol. 2010, 75, 75,525–31. [Google Scholar] [CrossRef] [PubMed]
- De Sandre-Giovannoli A, Bernard R Cau P et al. Lamin a truncation in Hutchinson-Gilford progeria. Science 2003, 300, 2055. [CrossRef] [PubMed]
- Jansen, M.P.; Machiels, B.M.; Hopman, A.H.; Broers, J.L.; Bot, F.J.; Arends, J.W.; Ramaekers, F.C.; Schouten, H.C. Comparison of A and B-type lamin expression in reactive lymph nodes and nodular sclerosing Hodgkin’s disease. Histopathology 1997, 31, 304–312. [Google Scholar] [CrossRef]
- Rocha-Perugini, V.; Gonzalez-Granado, J.M. Nuclear envelope lamin-A as a coordinator of T cell activation. Nucleus 2014, 5, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Contu, F.; Rangel-Pozzo, A.; Trokajlo, P.; Wark, L.; Klewes, L.; Johnson, N.A.; Petrogiannis-Haliotis, T.; Gartner, J.G.; Garini, Y.; Vanni, R.; et al. Distinct 3D Structural Patterns of Lamin A/C Expression in Hodgkin and Reed-Sternberg Cells. Cancers 2018, 10, 286. [Google Scholar] [CrossRef]
- De Vos, W.H.; Houben, F.; Hoebe, R.A.; Hoebe, R.A.; Hennekam, R.; van Engelen, B.; Manders, E.M.; Ramaekers, F.C.; Broers, J.L.; Van Oostveldt, P. Increased plasticity of the nuclear envelope and hypermobility of telomeres due to the loss of A–type lamins. Biochim. Biophys. Acta 2010, 1800, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Gonzales-Suarez I, Redwood AB, Perkins SM et al. Novel roles for A-type lamins in telomere biology and the DNA damage response pathway. EMBO J. 2009, 28, 2414–2427. [CrossRef] [PubMed]
- Doksani Y, Wu JY, de Lange T, Zhuang X (2013) Superresolution fluorescence imaging of telomeres reveals TRF2-dependent T-loop formation. Cell 2013, 155, 345–356. [CrossRef] [PubMed]
- Simonet T, Zaragosi LE, Philippe C et al. The human TTAGGG repeat factors 1 and 2 bind to a subset of interstitial telomeric sequences and satellite repeats. Cell Res. 2011, 21, 1028–1038. [CrossRef] [PubMed]
- Travina, A.O.; Ilicheva, N.V.; Mittenberg, A.G.; Shabelnikov, S.V.; Kotova, A.V.; Podgornaya, O.I. The Long Linker Region of Telomere-Binding Protein TRF2 Is Responsible for Interactions with Lamins. Int. J. Mol.Sci. 2021, 22, 3293. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wood, A.M.; Rendtlew Danielsen, J.M.; Lucas, C.A.; Rice, E.L.; Scalzo, D.; Shimi, T.; Goldman, R.D.; Smith, E.D.; Le Beau, M.M.; Kosak, S.T. (2014) TRF2 and lamin A/C interact to facilitate the functional organization of chromosome ends. Nat. Commun. 2014, 5, 5467. [Google Scholar] [CrossRef] [PubMed]
- Bronshtein I, Kepten E, Kanter I et al. Loss of lamin A function increases chromatin dynamics in the nuclear interior. Nat. Commun. 2015, 6, 8044. [CrossRef] [PubMed]
- Wood, A.M.; Laster, K.; Rice, E.L.; Kosak, S.T. A beginning of the end: new insights into the functional organization of telomeres. Nucleus 2015, 172–178. [Google Scholar] [CrossRef]
- Smith, E.D.; Garza-Gongora, A.G.; MacQuarrie, K.L.; Kosak, S.T. Interstitial telomeric loops and implications of the interaction between TRF2 and lamin A/C. Differentiation 2018, 102, 19–26. [Google Scholar] [CrossRef]
- Contu, F. Spatial organization of lamin A/C in Hodgkin’s lymphoma and multiple myeloma. Doctoral thesis, University of Cagliari, February 2020.
- Canellos, G.P.; Anderson, J.R.; Propert, K.J.; Nissen, N.; Cooper, M.R.; Henderson, E.S.; Green, M.R.; Gottlieb, A.; Peterson, B.A. Chemotherapy of advanced Hodgkin's disease with MOPP, ABVD, or MOPP alternating with ABVD. N Engl. J. Med. 1992, 327, 1478–84. [Google Scholar] [CrossRef] [PubMed]
- Kuruvilla, J. Standard therapy of advanced Hodgkin lymphoma. Hematology Am. Soc. Hematol. Educ. Program 2009, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Gordon, L.I.; Hong, F.; Fisher, R.I.; Bartlett, N.L.; Connors, J.M.; Gascoyne, R.D.; Wagner, H.; Stiff, P.J.; Cheson, B.D.; Gospodarowicz, M.; Advani, R.; Kahl, B.S.; Friedberg, J.W.; Blum, K.A.; Habermann, T.M.; Tuscano, J.M.; Hoppe, R.T.; Horning, S.J. Randomized phase III trial of ABVD versus Stanford V with or without radiation therapy in locally extensive and advanced-stage Hodgkin lymphoma: an intergroup study coordinated by the Eastern Cooperative Oncology Group (E2496). J Clin Oncol 2013, 31, 684–91. [Google Scholar] [CrossRef] [PubMed]
- Jacob, L.A.; Begum, T.; Halder, A.; Babu, M.C.S.; Lokesh, K.N.; Rudresha, A.H.; Rajeev, L.K.; Saldanha, S.C. Clinical Profile and Outcome of Adult Classical Hodgkin's Lymphoma: Real World Single Centre Experience. Indian J. Hematol. Blood Transfus. 2024, 40, 392–399. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Carde, P.; Karrasch, M.; Fortpied, C.; Brice, P.; Khaled, H.; Casasnovas, O.; Caillot, D.; Gaillard, I.; Bologna, S.; Ferme, C.; Lugtenburg, P.J.; Morschhauser, F.; Aurer, I.; Coiffier, B.; Meyer, R.; Seftel, M.; Wolf, M.; Glimelius, B.; Sureda, A.; Mounier, N. Eight Cycles of ABVD Versus Four Cycles of BEACOPPescalated Plus Four Cycles of BEACOPPbaseline in Stage III to IV, International Prognostic Score ≥ 3, High-Risk Hodgkin Lymphoma: First Results of the Phase III EORTC 20012 Intergroup Trial. J. Clin. Oncol. 2016, 34(17), 34(17),2028–36. [Google Scholar] [CrossRef] [PubMed]
- Merli, F.; Luminari, S.; Gobbi, P.G.; Cascavilla, N.; Mammi, C.; Ilariucci, F.; Stelitano, C.; Musso, M.; Baldini, L.; Galimberti, S.; Angrilli, F.; Polimeno, G.; Scalzulli, P.R.; Ferrari, A.; Marcheselli, L.; Federico, M. Long-Term Results of the HD2000 Trial Comparing ABVD Versus BEACOPP Versus COPP-EBV-CAD in Untreated Patients With Advanced Hodgkin Lymphoma: A Study by Fondazione Italiana Linfomi. J. Clin. Oncol. 2016, 34, 1175–81. [Google Scholar] [CrossRef] [PubMed]
- Knecht, H.; Kongruttanachok, N.; Sawan, B.; Brossard, J.; Prevost, S.; Turcotte, E.; Lichtensztejn, Z.; Lichtensztejn, D.; Mai, S. Three-dimensional Telomere Signatures of Hodgkin- and Reed-Sternberg Cells at Diagnosis Identify Patients with Poor Response to Conventional Chemotherapy. Transl. Oncol. 2012, 5, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Shanbhag, S.; Ambinder, R.F. Hodgkin lymphoma: A review and update on recent progress. CA Cancer J. Clin. 2018, 68, 116–132. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Williams, D.; Gray, C.; Picheca, L. Immune Checkpoint Inhibitors for Classical Hodgkin Lymphoma in Brentuximab Vedotin-naïve Patients: A Review of Clinical Effectiveness, Cost-Effectiveness, and Guidelines [Internet]. Ottawa (ON): Canadian Agency for Drugs and Technologies in Health; 2019 Jun 21. PMID: 31503428. Burton C, Allen P, Herrera AF. Paradigm Shifts in Hodgkin Lymphoma Treatment: From Frontline Therapies to Relapsed Disease. Am, Soc. Clin. Oncol. Educ. Book 2024, 44, e433502. [Google Scholar] [CrossRef] [PubMed]
- Grover, N.S.; Dittus, C.; Thakkar, A.; Beaven, A.W. The optimal management of relapsed and refractory Hodgkin lymphoma: post-brentuximab and checkpoint inhibitor failure. Hematology Am. Soc. Hematol, Educ. Program. 2023, 2023, 510–518. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Szczurek, A.; Klewes, L.; Xing, J.; Gourram, A.; Birk, U.; Knecht, H.; Dobrucki, J.W.; Mai, S.; Cremer, C. Imaging chromatin nanostructure with binding-activated localization microscopy based on DNA structure fluctuations. Nucleic Acids Res. 2017, 45, e56. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- M'kacher, R.; Cuceu, C.; Al Jawhari, M.; Morat, L.; Frenzel, M.; Shim, G.; Lenain, A.; Hempel, W.M.; Junker, S.; Girinsky, T.; Colicchio, B.; Dieterlen, A.; Heidingsfelder, L.; Borie, C.; Oudrhiri, N.; Bennaceur-Griscelli, A.; Moralès, O.; Renaud, S.; Van de Wyngaert, Z.; Jeandidier, E.; Delhem, N.; Carde, P. The Transition between Telomerase and ALT Mechanisms in Hodgkin Lymphoma and Its Predictive Value in Clinical Outcomes. Cancers (Basel) 2018, 10, 169. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lima, M.F.; Freitas, M.O.; Hamedani, M.K.; Rangel-Pozzo, A.; Zhu, X.D.; Mai, S. Consecutive Inhibition of Telomerase and Alternative Lengthening Pathway Promotes Hodgkin's Lymphoma Cell Death. Biomedicines 2022, 10(9), 2299. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Brankiewicz-Kopcinska, W.; Kallingal, A.; Krzemieniecki, R.; Baginski, M. Targeting shelterin proteins for cancer therapy. Drug Discov. Today 2024, 29, 104056. [Google Scholar] [CrossRef] [PubMed]
- Maco, M.; Kupcova, K.; Herman, V.; Ondeckova, I.; Kozak, T.; Mocikova, H.; Havranek, O.; Czech Hodgkin Lymphoma Study Group. Circulating tumor DNA in Hodgkin lymphoma. Ann. Hematol. 2022, 101, 2393–2403. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Braun, H.; Xu, Z.; Chang, F.; Viceconte, N.; Rane, G.; Levin, M.; Lototska, L.; Roth, F.; Hillairet, A.; Fradera-Sola, A.; Khanchandani, V.; Sin, Z.W.; Yong, W.K.; Dreesen, O.; Yang, Y.; Shi, Y.; Li, F.; Butter, F.; Kappei, D. ZNF524 directly interacts with telomeric DNA and supports telomere integrity. Nat. Commun. 2023, 14, 8252. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
Figure 1.
Very rare brain localization in a stage IVB case of EBV+ cHL, lymphocyte depleted, in an immune-compromised HIV+ patient. Note the nearly syncytial proliferation of H and RS cells, as described in a case report [12], and mainly absent reactive lymphocytes. Original magnification 40x. (A) Standard H&E staining. (B). Immune-staining with anti CD30 MoAb; (C) EBER in situ hybridization demonstrating the EBV positivity of H and RS cells but also some small lymphoid precursors. (D). Immune-staining with anti LMP1 MoAb. Note LMP1 expression is only observed in H and RS cells whereas small lymphoid precursors are still LMP1 -. EBV+ H and RS cells in addition expressed CD15, MUM-1, PAX-5, Ki-67 (not shown).
Figure 1.
Very rare brain localization in a stage IVB case of EBV+ cHL, lymphocyte depleted, in an immune-compromised HIV+ patient. Note the nearly syncytial proliferation of H and RS cells, as described in a case report [12], and mainly absent reactive lymphocytes. Original magnification 40x. (A) Standard H&E staining. (B). Immune-staining with anti CD30 MoAb; (C) EBER in situ hybridization demonstrating the EBV positivity of H and RS cells but also some small lymphoid precursors. (D). Immune-staining with anti LMP1 MoAb. Note LMP1 expression is only observed in H and RS cells whereas small lymphoid precursors are still LMP1 -. EBV+ H and RS cells in addition expressed CD15, MUM-1, PAX-5, Ki-67 (not shown).
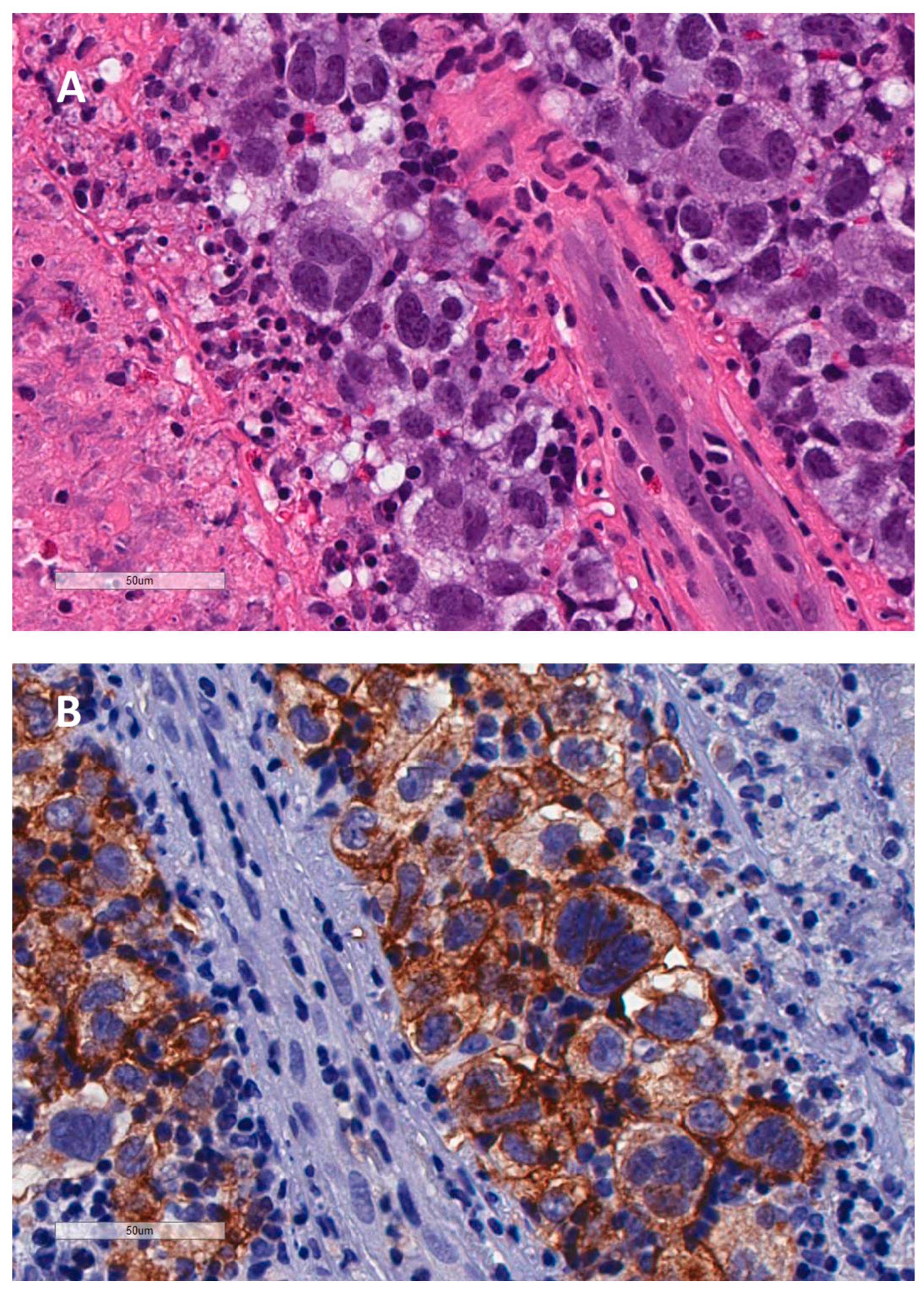
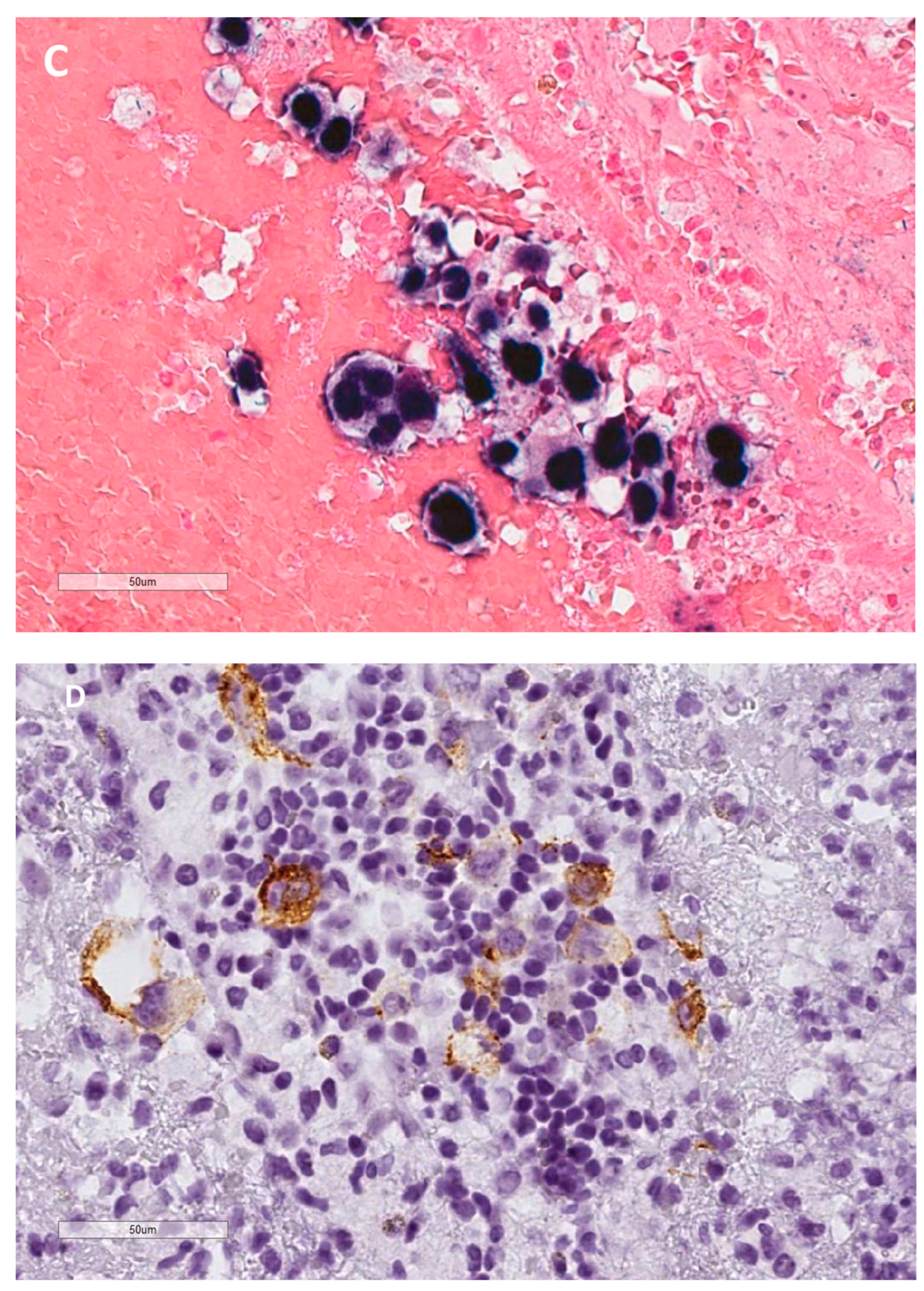
Figure 2.
There is a complete loss of telomeres and TRF2 in a large LMP1 expressing H cell whereas the surrounding lymphocytes show a normal, tight 1:1 3D telomere/TRF2 interaction. White arrows identify corresponding (identical) telomere/TRF2 spots. Lower right is a DAPI staining in transparency mode to optimize cyto-morphological aspects. Upper right (telomere) and middle right (TRF2) demonstrate that the surrounding lymphocytes contain mainly small to mid-sized telomere with a preserved 1:1 3D interaction with TRF2. The left panel (transparency mode) reveals a true “ghost” H cell virtually without any TRF2 and telomere signals. The lymphocyte corona serves as an internal control.
Figure 2.
There is a complete loss of telomeres and TRF2 in a large LMP1 expressing H cell whereas the surrounding lymphocytes show a normal, tight 1:1 3D telomere/TRF2 interaction. White arrows identify corresponding (identical) telomere/TRF2 spots. Lower right is a DAPI staining in transparency mode to optimize cyto-morphological aspects. Upper right (telomere) and middle right (TRF2) demonstrate that the surrounding lymphocytes contain mainly small to mid-sized telomere with a preserved 1:1 3D interaction with TRF2. The left panel (transparency mode) reveals a true “ghost” H cell virtually without any TRF2 and telomere signals. The lymphocyte corona serves as an internal control.
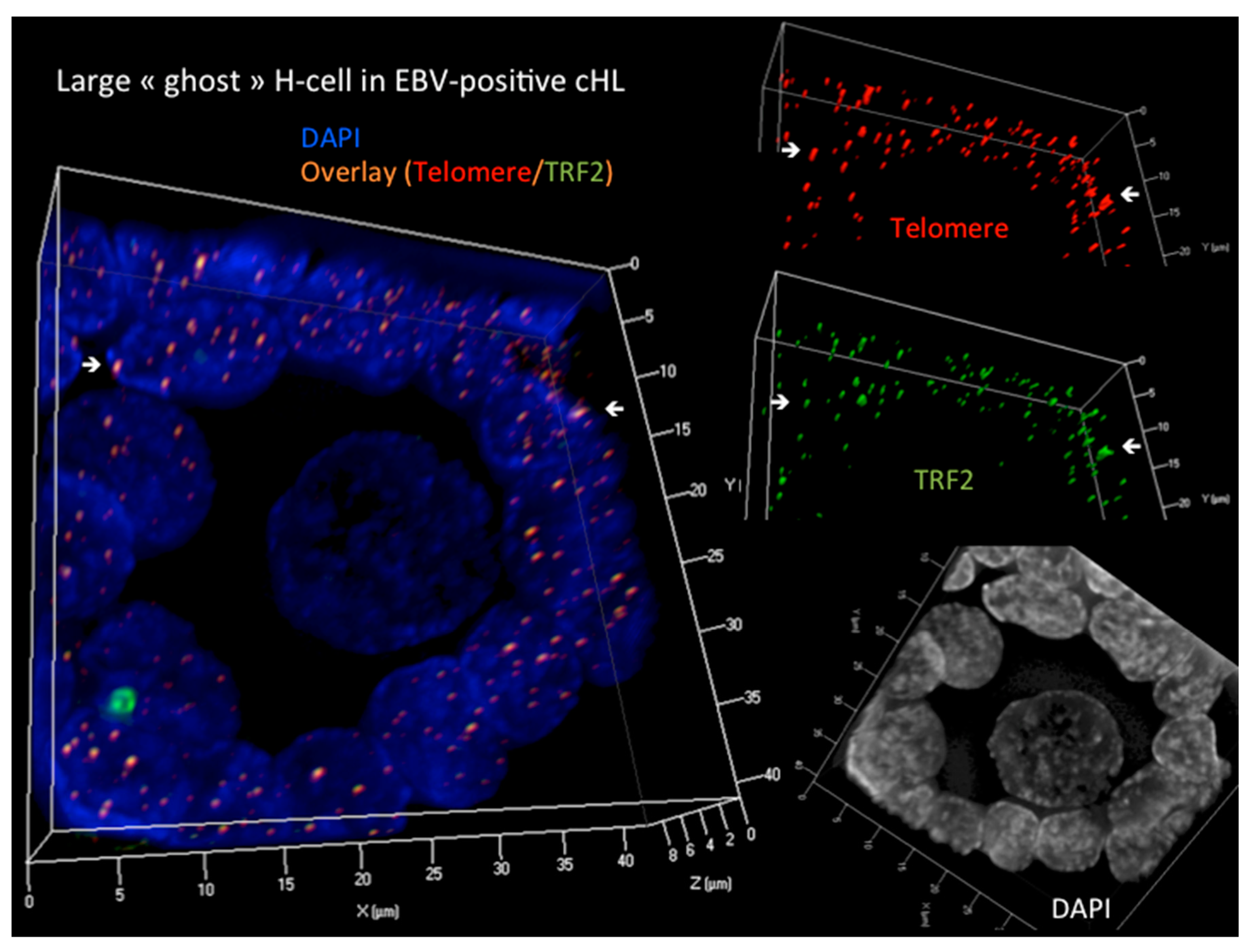
Figure 3.
Shown from left to the right a continuous transformation from a GC B-lymphocyte into a H cell and terminal RS cell. A “crippled” GCB-lymphocyte [6] or an EBV+ memory B-lymphocyte [7] re-entering GC reaction undergoes LMP1 oncoprotein induced transformation, targeting the telomere/shelterin complex through a “pincer attack”. NF-kB driven mitosis [72] combined with TRF2 down-regulation [48,67], lead to telomere shortening and de-protection, followed by sister chromatid fusion at the origin of repeated BBF-cycles, resulting first in H-cells, and, after several subsequent mitotic cycles, in the end-stage multi-faceted, often telomere poor RS-cells [46,48,53,58]. The constitutive NF-kB activation [72,73], the dysfunctional telomere-shelterin complex, in particular TRF2 [67,69] appears to emerge as a logical candidate for a common pathogenic denominator not only for EBV-positive [74] but also for many, TRF2 poor EBV-negative cHL [75].
Figure 3.
Shown from left to the right a continuous transformation from a GC B-lymphocyte into a H cell and terminal RS cell. A “crippled” GCB-lymphocyte [6] or an EBV+ memory B-lymphocyte [7] re-entering GC reaction undergoes LMP1 oncoprotein induced transformation, targeting the telomere/shelterin complex through a “pincer attack”. NF-kB driven mitosis [72] combined with TRF2 down-regulation [48,67], lead to telomere shortening and de-protection, followed by sister chromatid fusion at the origin of repeated BBF-cycles, resulting first in H-cells, and, after several subsequent mitotic cycles, in the end-stage multi-faceted, often telomere poor RS-cells [46,48,53,58]. The constitutive NF-kB activation [72,73], the dysfunctional telomere-shelterin complex, in particular TRF2 [67,69] appears to emerge as a logical candidate for a common pathogenic denominator not only for EBV-positive [74] but also for many, TRF2 poor EBV-negative cHL [75].
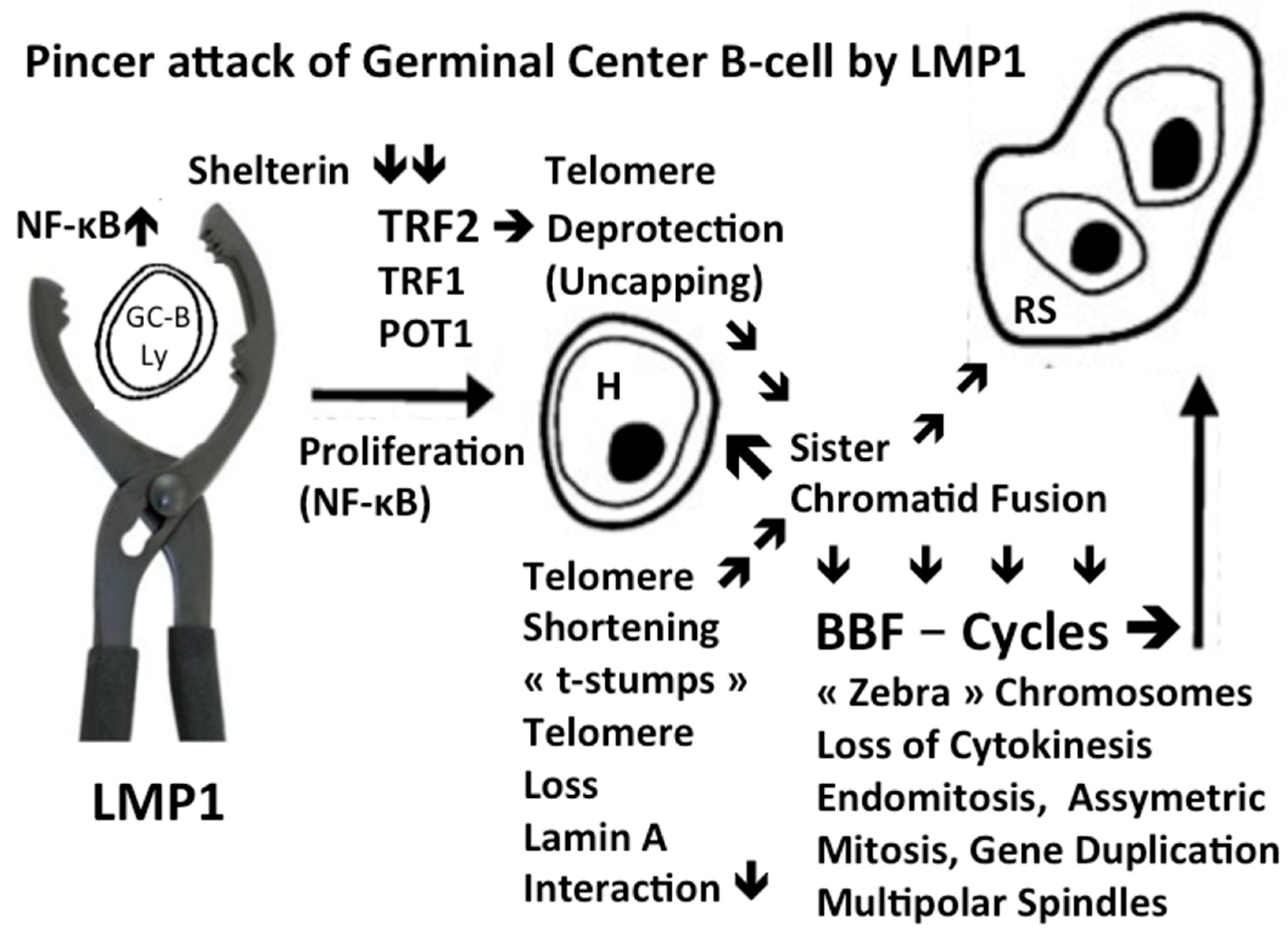
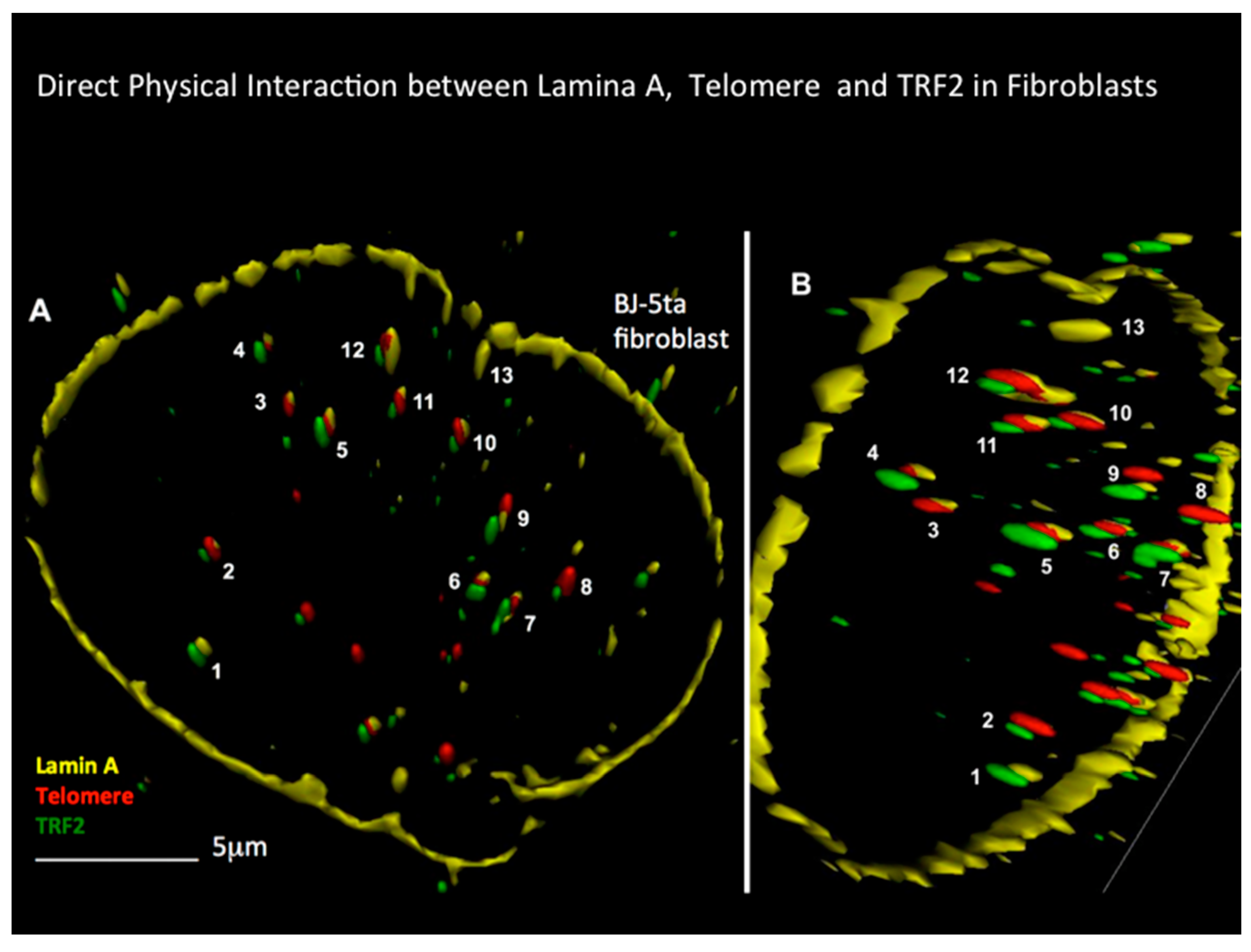
Figure 4.
3D Lamin A-telomere-TRF2 in surface mode reconstitution in a benign BJ-5ta fibroblast at different angles (A and B) is shown. 1-13 are corresponding spots. Direct 3D interaction of lamin A-telomere-TRF2 is seen at spots 2,5,6,7,10,11 and 12. Direct lamin A-telomere interaction is seen at spots 3 and 4, and direct lamin A-TRF2 interaction is seen at spot 1. This type of interaction is completely disrupted in H and RS [24,104].
Figure 4.
3D Lamin A-telomere-TRF2 in surface mode reconstitution in a benign BJ-5ta fibroblast at different angles (A and B) is shown. 1-13 are corresponding spots. Direct 3D interaction of lamin A-telomere-TRF2 is seen at spots 2,5,6,7,10,11 and 12. Direct lamin A-telomere interaction is seen at spots 3 and 4, and direct lamin A-TRF2 interaction is seen at spot 1. This type of interaction is completely disrupted in H and RS [24,104].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



